Embed Size (px)
DESCRIPTION
Structural variation calling with the SVMerge pipeline. see http://svmerge.sourceforge.net
Citation preview

Vertebrate Resequencing Informatics 22nd March, 2011
Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly
Kim Wong/Thomas Keane Vertebrate Resequencing Informatics
http://svmerge.sourceforge.net

Vertebrate Resequencing Informatics 22nd March, 2011
Genomic Structural Variation
Large DNA rearrangements (>100bp) Frequent causes of disease Referred to as genomic disorders Mendelian diseases or complex traits such as behaviors
E.g. increase in gene dosage due to increase in copy number Prevalent in cancer genomes
Many types of genomic structural variation (SV) Insertions, deletions, copy number changes, inversions,
translocations & complex events Comparative genomic hybridization (CGH) traditionally used to for copy number discovery CNVs of 1–50 kb in size have been under-ascertained
Next-gen sequencing revolutionised field of SV discovery Parallel sequencing of ends of large numbers of DNA fragments Examine alignment distance of reads to discover presence of
genomic rearrangments Resolution down to ~100bp

Vertebrate Resequencing Informatics 22nd March, 2011
Simple types of Structural Variation
Vertebrate Resequencing Informatics 22nd March, 2011
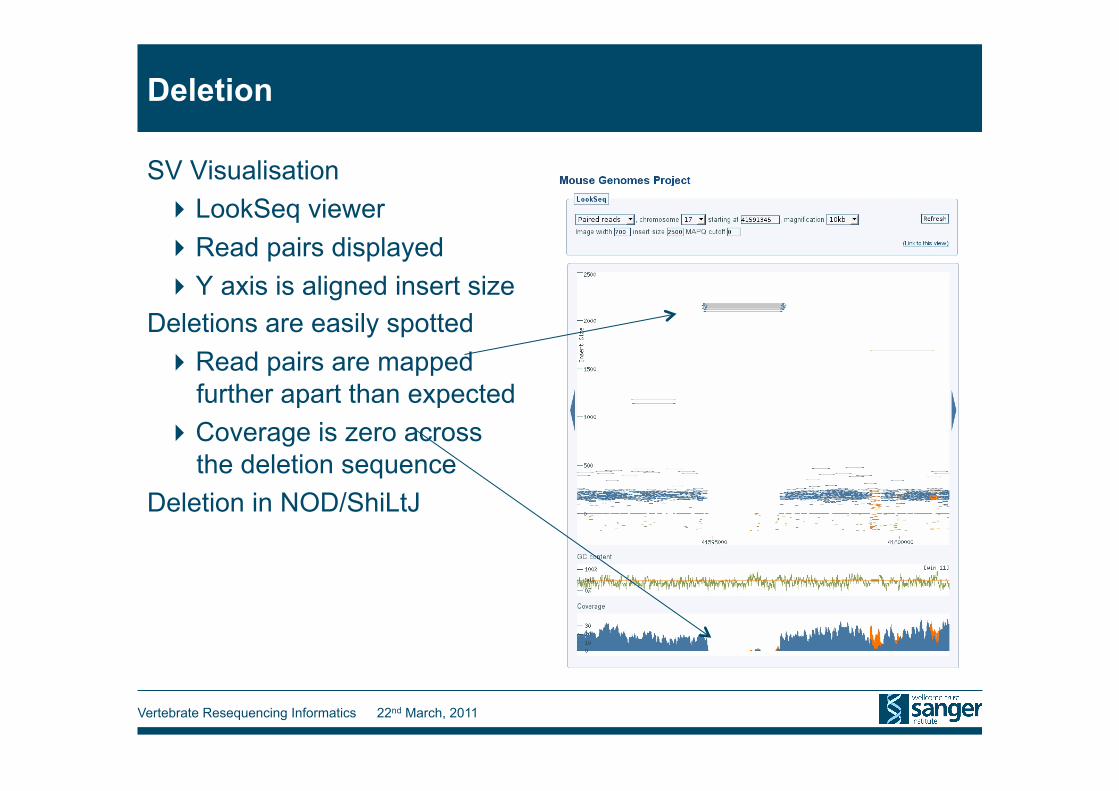
Deletion
SV Visualisation LookSeq viewer Read pairs displayed Y axis is aligned insert size
Deletions are easily spotted Read pairs are mapped
further apart than expected Coverage is zero across
the deletion sequence Deletion in NOD/ShiLtJ

Vertebrate Resequencing Informatics 22nd March, 2011
Inversion
Mate pairs align in the same orientation
Coverage zero at breakpoints

Vertebrate Resequencing Informatics 22nd March, 2011
Insertion
One end mapped reads
Coverage zero at breakpoint

Vertebrate Resequencing Informatics 22nd March, 2011
Insertion Insertion
Inversion
Complex SV Events

Vertebrate Resequencing Informatics 22nd March, 2011
Human Examples
Stankiewicz and Lupski (2010) Ann. Rev. Med.

Vertebrate Resequencing Informatics 22nd March, 2011
Example 2: Transposable element insertion in mice

Vertebrate Resequencing Informatics 22nd March, 2011
SVMerge
Initially developed for mouse genomes project Several software packages currently available to discover SVs
Various approaches using information from anomalously mapped read pairs OR read depth analysis No single SV caller is able to detect the full range of structural variants Paired-end mapping information, for example, cannot detect SVs where the
read pairs do not flank the SV breakpoints Insertion calls made using the split-mapping approach are also size-limited
because the whole insertion breakpoint must be contained within a read Read-depth approaches can identify copy number changes without the need
for read-pair support, but cannot find copy number neutral events SVMerge, a meta SV calling pipeline, which makes SV predictions with a collection of SV callers Input is a BAM file per sample Run callers individually + outputs sanitized into standard BED format SV calls merged, and computationally validated using local de novo assembly Primarily a SV discovery/calling + validation tool

Vertebrate Resequencing Informatics 22nd March, 2011
SVMerge Workflow
Wong et al (2010)

Vertebrate Resequencing Informatics 22nd March, 2011
SV Callers
Wong et al (2010)

Vertebrate Resequencing Informatics 22nd March, 2011
Local Assembly Validation
Key to the approach is the computational validation step Local assembly and breakpoint refinement All SV calls (except those lacking read
pair support e.g. CNG/CNL) Algorithm Gather mapped reads, and any
unmapped mate-pairs (<1kb of a insertion breakpoint, <2kb of all other SV types)
Run local velvet assembly Realign the contigs produced with
exonerate Detect contig breaks proximal to the
breakpoint(s)

Vertebrate Resequencing Informatics 22nd March, 2011
Breakpoint Improvement (simulated)

Vertebrate Resequencing Informatics 22nd March, 2011
Breakpoint Improvement (Real data)
Yalchin and Wong et al, in prep

Vertebrate Resequencing Informatics 22nd March, 2011
Application to HapMap trio dataset
High-depth HapMap trio (NA18506, NA18507, NA18508) 42x, 42x and 40x
Reads processed through Vert. Reseq. Pipeline Aligned to the GRCh37 human reference using BWA Single BAM file for each individual
BreakDancerMax, Pindel, RDXplorer, SECluster, and RetroSeq Exclude calls 600 bp from a reference sequence gap 1 Mb from a centromere or telomere
Computational validation of raw candidate calls

Vertebrate Resequencing Informatics 22nd March, 2011
NA18506 Results

Vertebrate Resequencing Informatics 22nd March, 2011
Does multiple callers discover more SVs?

Vertebrate Resequencing Informatics 22nd March, 2011
How do the calls measure up?
Compared the overlap of the deletion, gain, and inversion calls against the curated Database of Genomic Variants Overlapped with calls in DGV at a rate significantly higher than
expected by random chance Deletions in DGV: 71% (NA18506), 81% (NA18507), and 71%
(NA18508) Copy number gains in DGV: 29% (NA18506), 32% (NA18507),
and 36% (NA18508) Inversions in DGV: 47% (NA18506), 69% (NA18507), and 51%
(NA18508) Child calls not in DGV also called in the parents Further 18% deletions, 32% inversions, 54% duplications Estimated max. false positive rate of 11%, 21%, and 17%
All child-only SV calls comprise 11% of the child's final SV call Considerable improvement from 'merged raw’ (50% unique)

Vertebrate Resequencing Informatics 22nd March, 2011
Complex SV Types
Yalchin and Wong et al, in prep

Vertebrate Resequencing Informatics 22nd March, 2011
Future Work
SVMerge primarily a discovery and validation tool Extensible pipeline so that calls from any method to be easily
incorporated Developed primarily for mouse genomes project Successfully applied to human trio dataset Computationally validation approach reduces false positives
Complex SVs Cataloging repeating combinations of multiple SV events in small
loci 2011 development Low coverage cross-population SV discovery Genotyping existing SVs in new samples Better support for heterozygous calls Integration of SVMerge into Vert. Reseq. pipeline for UK10K





























