Embed Size (px)
Citation preview

A Role of Autoantibody-Mediated Platelet Destructionin Thrombocytopenia in Patients With Cirrhosis
Mikio Kajihara,1 Shinzo Kato,1 Yuka Okazaki,2 Yutaka Kawakami,2 Hiromasa Ishii,1
Yasuo Ikeda,3 and Masataka Kuwana2
Thrombocytopenia is a common manifestation in patients with liver cirrhosis (LC), but itsunderlying mechanism remains controversial. This study examined the role of anti-plateletautoimmunity in cirrhotic thrombocytopenia by determining the autoantibody response toGPIIb-IIIa, a major platelet surface autoantigen recognized by anti-platelet antibodies inpatients with idiopathic thrombocytopenic purpura (ITP). Circulating B cells producinganti–GPIIb-IIIa antibodies as well as platelet-associated and plasma anti–GPIIb-IIIa anti-bodies were examined in 72 patients with LC, 62 patients with ITP, and 52 healthy controls.In vitro anti–GPIIb-IIIa antibody production was induced in cultures of peripheral bloodmononuclear cells (PBMCs) by stimulation with GPIIb-IIIa. The frequency of anti–GPIIb-IIIa antibody–producing B cells in patients with LC was significantly greater than in healthycontrols (10.9 � 6.2 vs. 0.4 � 0.3/105 PBMCs; P < .0001) and was even higher than thefrequency in patients with ITP (8.2 � 5.2; P � .007). Anti–GPIIb-IIIa antibodies in thepatients with LC and ITP were mainly present on the surfaces of circulating platelets ratherthan in the plasma in an unbound form. Furthermore, PBMCs from patients with LC andITP produced anti–GPIIb-IIIa antibodies on antigenic stimulation with GPIIb-IIIa in vitro,and the specific antibodies produced had the capacity to bind normal platelet surfaces. Inconclusion, the similar profile of the anti–GPIIb-IIIa autoantibody response in patients withLC and ITP suggests that autoantibody-mediated platelet destruction may contribute at leastin part to cirrhotic thrombocytopenia. (HEPATOLOGY 2003;37:1267-1276.)
Thrombocytopenia is a major hematologic disor-der commonly observed in patients with liver cir-rhosis (LC). Historically, the condition was
principally attributed to hypersplenism, which is accom-panied by an increase in sequestration and the accelerateddestruction of platelets in the pathologically enlarged andcongested spleen.1-3 The human spleen physiologicallyaccommodates about one third of the total platelet mass,1
and a classic study by Aster, using radiolabeled platelets,showed that up to 90% of platelets localized within thepathologically swollen spleen.1 However, consistent im-provements in thrombocytopenia have not been achievedby portal decompression procedures such as portosys-temic shunting,1,4-6 and thrombocytopenia may even per-sist after splenectomy in some cases.7 On the other hand,insufficient production of thrombopoietin in the liver hasbeen proposed to be associated with thrombocytopenia inpatients with LC. Because thrombopoietin, a principalregulator of megakaryothrombopoiesis, is predominantlyproduced by the liver,8-10 its production is assumed to beimpaired in the state of advanced liver failure. In thisregard, it has been reported that the reduced serumthrombopoietin concentration in patients with LC is re-stored after orthotopic liver transplantation in conjunc-tion with an increase in platelet count.11,12 However,several independent studies have shown that serumthrombopoietin levels are maintained in patients withLC.13,14 In addition, the number of megakaryocytes in thebone marrow is reported to be preserved in patients withLC.1 Therefore, thrombocytopenia in patients with LCcannot be fully explained by current theories, including
Abbreviations: LC, liver cirrhosis; IgG, immunoglobulin G; PAIgG, platelet-associated immunoglobulin G; ITP, idiopathic thrombocytopenic purpura; HBV,hepatitis B virus; HCV, hepatitis C virus; ALD, alcoholic liver disease; PBMC,peripheral blood mononuclear cell; ELISA, enzyme-linked immunosorbent assay.
From the Divisions of 1Gastroenterology and 3Hematology, Department of In-ternal Medicine, and 2Institute for Advanced Medical Research, Keio UniversitySchool of Medicine, Tokyo, Japan.
Received December 17, 2002; accepted March 4, 2003.Supported by the Keio University Medical Science Fund; the Ministry of Health,
Welfare, and Labour in Japan; and the Mochida Memorial Foundation.Address reprint requests to: Masataka Kuwana, M.D., Ph.D., Institute for Ad-
vanced Medical Research, Keio University School of Medicine, 35, Shinanomachi,Shinjuku, Tokyo 160-8582, Japan. E-mail: [email protected]; fax: (81)3-5362-9259.
Copyright © 2003 by the American Association for the Study of Liver Diseases.0270-9139/03/3706-0010$30.00/0doi:10.1053/jhep.2003.50209
1267

splenic sequestration of platelets and impaired plateletproduction arising from insufficient thrombopoietin se-cretion.
On the other hand, several reports have shown an in-creased level of immunoglobulin G (IgG) bound to plate-lets (platelet-associated IgG [PAIgG]) in patients withchronic liver disease,15-17 suggesting the presence of auto-antibodies reactive with platelets in these patients. How-ever, such increases in PAIgG are known to be rathernonspecific and are often found in patients with nonau-toimmune thrombocytopenia.18,19 Further studies exam-ining the potential role of anti-platelet autoantibodies incirrhotic thrombocytopenia have not been performed;thus, this remains an open question.
A typical autoimmune disease characterized by in-creased platelet destruction mediated by anti-platelet au-toantibodies is idiopathic thrombocytopenic purpura(ITP).20 Several distinct platelet surface autoantigens, in-cluding GPIIb-IIIa, GPIb-IX, and GPIa-IIa, have beenidentified in patients with ITP.21 Of these, GPIIb-IIIa(also designated as �IIb�3 integrin or CD41/CD61),which is a calcium-dependent heterodimeric membranereceptor for fibrinogen, is the most common autoantigentargeted by anti-platelet antibodies.21-23 Several assays todetect anti–GPIIb-IIIa antibodies have been devel-oped,23-25 but these assays require complicated proceduresbecause the pathogenic antibodies are mainly present onthe surfaces of circulating platelets but not in the plasmaor serum. We have recently established a convenient andsensitive assay for identifying patients with autoantibody-mediated thrombocytopenia by detecting circulating Bcells that produce anti–GPIIb-IIIa antibodies.26,27 More-over, we have developed an in vitro culture system inwhich pathogenic anti–GPIIb-IIIa antibodies are pro-duced by antigen-specific collaboration between GPIIb-IIIa–reactive T and B cells from patients with ITP.28,29 Inthis study, these sophisticated assay systems were used toevaluate the potential role of an anti-platelet autoanti-body response in thrombocytopenia in patients with LC.Patients with ITP were also analyzed as a control for theautoantibody-mediated thrombocytopenia.
Materials and Methods
Patients and Controls. Seventy-two patients (51men and 21 women) with LC who were being followedup at Keio University Hospital were enrolled in this study.LC was diagnosed based on clinical history, a physicalexamination, laboratory test results, and ultrasonographicand/or computed tomographic imaging studies with orwithout liver biopsy.30,31 Patients with LC with concom-itant autoimmune disease were excluded. The etiology of
LC was hepatitis B virus (HBV) infection in 15 patients,hepatitis C virus (HCV) infection in 37, and excessivealcohol intake (alcoholic liver disease [ALD]) in 20. De-mographic and clinical parameters recorded included age,sex, Child’s grade, platelet count, serum alanine amino-transferase level, serum IgG level, and the presence orabsence of splenomegaly (as determined by the ultra-sound splenic index) and hepatocellular carcinoma. Sixty-two adult patients with ITP (17 men and 45 women) and52 healthy individuals (32 men and 20 women) were alsoanalyzed as controls. ITP was defined as thrombocytope-nia persisting for longer than 6 months, normal or in-creased bone marrow megakaryocytes without morphologicevidence of dysplasia, and no secondary immune or nonim-mune diseases that could account for the thrombocytopenicstate.20,32 At the time of blood examination, all patients withITP had a platelet count less than 100 � 109/L and receivedno treatment (n � 32) or low-dose corticosteroid (�5 mgprednisolone daily) therapy (n � 30). The study protocolconformed to the ethical principles of the World MedicalAssociation Declaration of Helsinki as reflected in a prioriapproval from the Keio University Institutional ReviewBoard.
Cell Preparation. Heparinized peripheral blood sam-ples were obtained from all subjects. After the platelet-richplasma was isolated, the residual cell components wereapplied to Lymphoprep (Nycomed Pharma AS, Oslo,Norway) density gradient centrifugation to isolate the pe-ripheral blood mononuclear cells (PBMCs). Freshly iso-lated PBMCs were resuspended in RPMI 1640 containing10% heat-inactivated fetal bovine serum, 2 mmol/L L-glu-tamine, 50 U/mL penicillin, and 50 �g/mL streptomycinand immediately used in the following experiments. Theplatelets were isolated from the platelet-rich plasma by cen-trifugation, and the supernatant was used as plasma.
Preparation of GPIIb-IIIa Antigen. HumanGPIIb-IIIa was purified from outdated platelet concen-trates using affinity chromatography.28 Purified GPIIb-IIIa was dialyzed against phosphate-buffered salinecontaining 0.5 mmol/L CaCl2 and stored in aliquots at�80°C until use. For use in T-cell stimulation, GPIIb-IIIa was modified by treatment with porcine trypsin asdescribed elsewhere.28
Detection and Quantification of Anti–GPIIb-IIIaAntibody–Producing B Cells. B cells producing IgGanti–GPIIb-IIIa antibodies were detected and quantifiedusing an enzyme-linked immunospot assay as describedpreviously.26,27 In brief, polyvinylidene difluoride–bot-tomed 96-well plates were coated with 30 �g/mL of pu-rified GPIIb-IIIa in phosphate-buffered saline containing0.5 mmol/L CaCl2. PBMCs (105 cells/well) were culturedin GPIIb-IIIa–coated wells at 37°C in a humidified
1268 KAJIHARA ET AL. HEPATOLOGY, June 2003

atmosphere of 5% CO2 for 4 hours and subsequentlyincubated with alkaline phosphatase–conjugated goat an-ti-human IgG (ICN/Cappel, Aurora, OH) diluted 1,000-fold in phosphate-buffered saline containing 0.5 mmol/LCaCl2 at room temperature for 2 hours. Antibodiesbound to the membranes were subsequently visualized asdistinctive spots by incubation with nitro blue tetrazo-lium and 5-bromo-4-chloro-indolyl phosphate, and thenumber of spots was counted under a dissecting micro-scope. Each experiment was conducted in 10 independentwells, and the results represent the mean of the 10 values.The frequency of circulating anti–GPIIb-IIIa antibody–producing B cells was presented as the number per 105
PBMCs, and a cutoff value was defined as 2.0.26 Thefrequency was not adjusted by the number of PBMCs perblood volume; it was not different between patients withLC and patients with ITP.
Platelet-Associated and Plasma Anti–GPIIb-IIIaAntibodies. IgG anti–GPIIb-IIIa antibodies in plateleteluates (from 5 � 107 platelets) and plasma were mea-sured using an enzyme-linked immunosorbent assay(ELISA) as described elsewhere.23,27 Platelet eluates wereprepared by incubating the platelets with 0.1 mol/L HClfollowed by immediate neutralization with the additionof 0.2 mol/L NaOH. Antibody units were calculatedfrom the OD450 results, based on a standard curve ob-tained from serial concentrations of anti–GPIIb-IIIamonoclonal antibody (clone HPL1; Harlan Sera Labora-tory, Leicester, England). All samples were examined induplicate, and the results were calculated as the mean of 2values. The cutoff values for platelet-associated andplasma IgG anti–GPIIb-IIIa antibodies were 3.3 and 5.0units, respectively.27
Anti–GPIIb-IIIa Antibody Production in PBMCCultures With or Without Antigenic Stimulation.The in vitro production of IgG anti–GPIIb-IIIa antibod-ies in cultures of PBMCs was evaluated as described pre-viously.28,29 Briefly, PBMCs (2 � 106) were cultured withor without a combination of modified GPIIb-IIIa (5 �g/mL) and pokeweed mitogen (1 �g/mL) for 10 days. Lev-els of IgG anti–GPIIb-IIIa antibodies in undilutedculture supernatants were directly quantified by an anti–GPIIb-IIIa ELISA as previously described, and the resultswere expressed as OD450. All samples were examined induplicate, and the results were calculated as the mean ofduplicate values. In some experiments, PBMCs depletedof CD4� or CD8� T cells were prepared by incubationwith anti-CD4 or anti-CD8 monoclonal antibody–cou-pled magnetic beads (Dynal, Oslo, Norway) and used inthe assay for in vitro anti–GPIIb-IIIa antibody produc-tion.28
To evaluate the capacity of anti–GPIIb-IIIa antibodiesproduced in vitro to bind normal platelet surfaces, thefollowing assays were performed. First, PBMC culturesupernatants containing anti–GPIIb-IIIa antibodies wereincubated with serially diluted platelets (105 to 108) ob-tained from a healthy donor to absorb the antibodies thatbind to platelet surfaces, and the pretreated supernatantswere subsequently applied to anti–GPIIb-IIIa ELISA.28
Erythrocytes (108) and PBMCs (107) from the same do-nor were used as controls. Second, platelets from a healthydonor were incubated with the PBMC culture superna-tants that had or had not been stimulated with GPIIb-IIIa. The platelets were subsequently incubated with goatanti-human IgG F(ab�)2 conjugated to fluorescein iso-thiocyanate (Sigma Chemical Co., St. Louis, MO) for 30minutes at room temperature and then analyzed on aFACS Calibur flow cytometer (BD Pharmingen, San Di-ego, CA) using CellQuest software (BD Pharmingen). Insome experiments, intensity of the antibody binding wassemiquantitatively expressed as mean fluorescent inten-sity.
Statistical Analyses. All continuous results were ex-pressed as the mean � SD. To assess the clinical charac-teristics among patients with LC with different etiologies,the �2 test was used to compare the frequency of categor-ical variables and the Kruskal-Wallis test to compare thecontinuous variables. Experimental results between 2groups were compared using the Mann-Whitney U test orWilcoxon signed rank test as appropriate. Potential asso-ciations of demographic and laboratory indices with theanti–GPIIb-IIIa antibody–producing B-cell frequencywere assessed using Pearson’s simple correlation coeffi-cients or Spearman’s rank correlation coefficients. Finally,a stepwise multiple regression analysis was conducted toidentify the independent variables associated with theplatelet count. P values less than .05 were considered sig-nificant. All statistical procedures were performed usingStatView software (SAS Institute, Cary, NC).
Results
Demographic and Clinical Characteristics of Pa-tients With LC. The mean platelet count of the patientswith LC was 97.3 � 54.8 � 109/L, and 61 patients (85%)were thrombocytopenic (�150 � 109/L). The plateletcount in the patients with LC was significantly higherthan in the patients with ITP (52.4 � 27.8 � 109/L; P �.0001). Table 1 shows the demographic and clinical char-acteristics of the patients with LC according to their eti-ologies. The sex distribution and age at examination weresignificantly different among the HBV, HCV, and ALDsubgroups. The patients with LC with HBV or ALD were
HEPATOLOGY, Vol. 37, No. 6, 2003 KAJIHARA ET AL. 1269
predominantly men, whereas men and women werenearly equally distributed among the HCV-positive pa-tients. The patients with LC with HCV tended to be olderthan those with HBV or ALD, and the difference betweenthe HCV and ALD subgroups was significant (P � .04).The platelet count was not significantly different amongthese 3 groups. The serum alanine aminotransferase andIgG levels were statistically different among the 3 LCsubgroups, and patients with ALD tended to have lowerlevels compared with the HBV- or HCV-positive pa-tients.
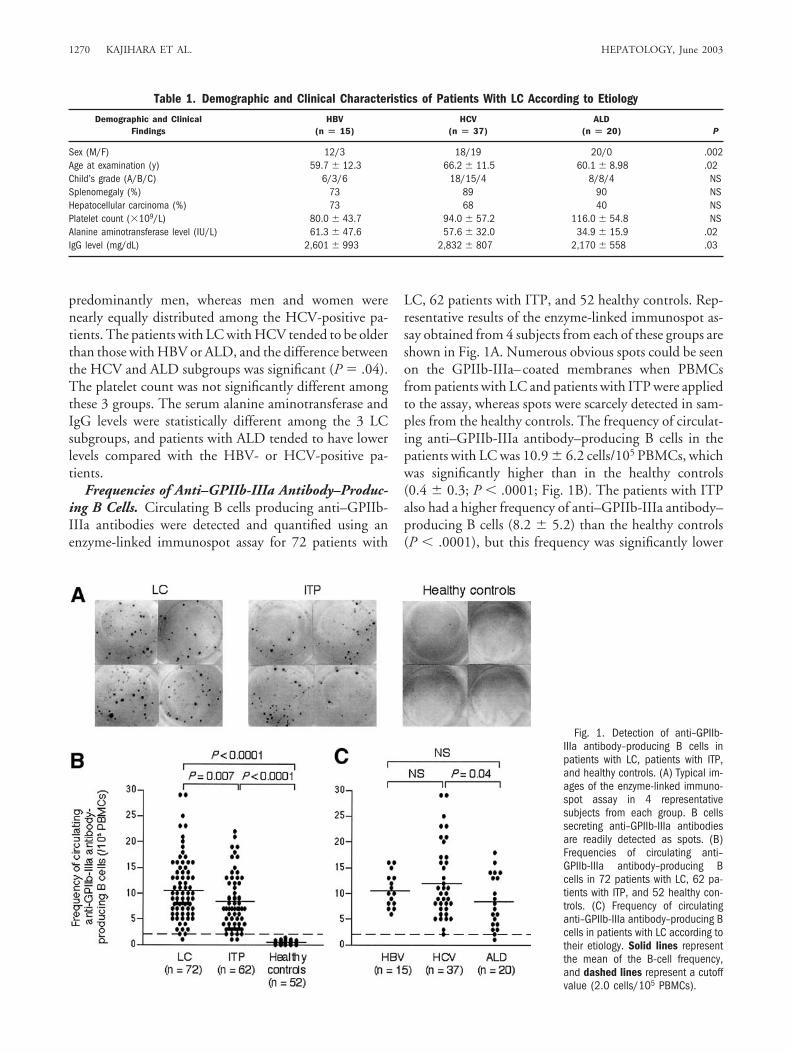
Frequencies of Anti–GPIIb-IIIa Antibody–Produc-ing B Cells. Circulating B cells producing anti–GPIIb-IIIa antibodies were detected and quantified using anenzyme-linked immunospot assay for 72 patients with
LC, 62 patients with ITP, and 52 healthy controls. Rep-resentative results of the enzyme-linked immunospot as-say obtained from 4 subjects from each of these groups areshown in Fig. 1A. Numerous obvious spots could be seenon the GPIIb-IIIa–coated membranes when PBMCsfrom patients with LC and patients with ITP were appliedto the assay, whereas spots were scarcely detected in sam-ples from the healthy controls. The frequency of circulat-ing anti–GPIIb-IIIa antibody–producing B cells in thepatients with LC was 10.9 � 6.2 cells/105 PBMCs, whichwas significantly higher than in the healthy controls(0.4 � 0.3; P � .0001; Fig. 1B). The patients with ITPalso had a higher frequency of anti–GPIIb-IIIa antibody–producing B cells (8.2 � 5.2) than the healthy controls(P � .0001), but this frequency was significantly lower
Table 1. Demographic and Clinical Characteristics of Patients With LC According to Etiology
Demographic and ClinicalFindings
HBV(n � 15)
HCV(n � 37)
ALD(n � 20) P
Sex (M/F) 12/3 18/19 20/0 .002Age at examination (y) 59.7 � 12.3 66.2 � 11.5 60.1 � 8.98 .02Child’s grade (A/B/C) 6/3/6 18/15/4 8/8/4 NSSplenomegaly (%) 73 89 90 NSHepatocellular carcinoma (%) 73 68 40 NSPlatelet count (�109/L) 80.0 � 43.7 94.0 � 57.2 116.0 � 54.8 NSAlanine aminotransferase level (IU/L) 61.3 � 47.6 57.6 � 32.0 34.9 � 15.9 .02IgG level (mg/dL) 2,601 � 993 2,832 � 807 2,170 � 558 .03
Fig. 1. Detection of anti–GPIIb-IIIa antibody–producing B cells inpatients with LC, patients with ITP,and healthy controls. (A) Typical im-ages of the enzyme-linked immuno-spot assay in 4 representativesubjects from each group. B cellssecreting anti–GPIIb-IIIa antibodiesare readily detected as spots. (B)Frequencies of circulating anti–GPIIb-IIIa antibody–producing Bcells in 72 patients with LC, 62 pa-tients with ITP, and 52 healthy con-trols. (C) Frequency of circulatinganti–GPIIb-IIIa antibody–producing Bcells in patients with LC according totheir etiology. Solid lines representthe mean of the B-cell frequency,and dashed lines represent a cutoffvalue (2.0 cells/105 PBMCs).
1270 KAJIHARA ET AL. HEPATOLOGY, June 2003
than the frequency in the patients with LC (P � .007).This difference might be explained by the fact that somepatients with ITP were on low-dose corticosteroids at ex-amination. When the subjects were categorized as havinga frequency below or above the cutoff level (2.0 cells/105
PBMCs), a positive result was detected in 71 patients withLC (99%), 58 patients with ITP (94%), and none of thehealthy controls.
When the patients with LC were subgrouped accord-ing to etiology (Fig. 1C), all 3 subgroups showed a higherfrequency of anti–GPIIb-IIIa antibody–producing B cellscompared with healthy controls (P � .0001 for all com-parisons), indicating that B cells producing anti–GPIIb-IIIa antibodies are present in the patients with LCirrespective of the etiology of the disease. However, thepatients with LC with HCV had a higher frequency ofanti–GPIIb-IIIa antibody—producing B cells than thosewith other etiologies, and the frequency between theHCV and ALD subgroups was significantly different(12.4 � 7.2 vs. 8.5 � 5.2; P � .04).
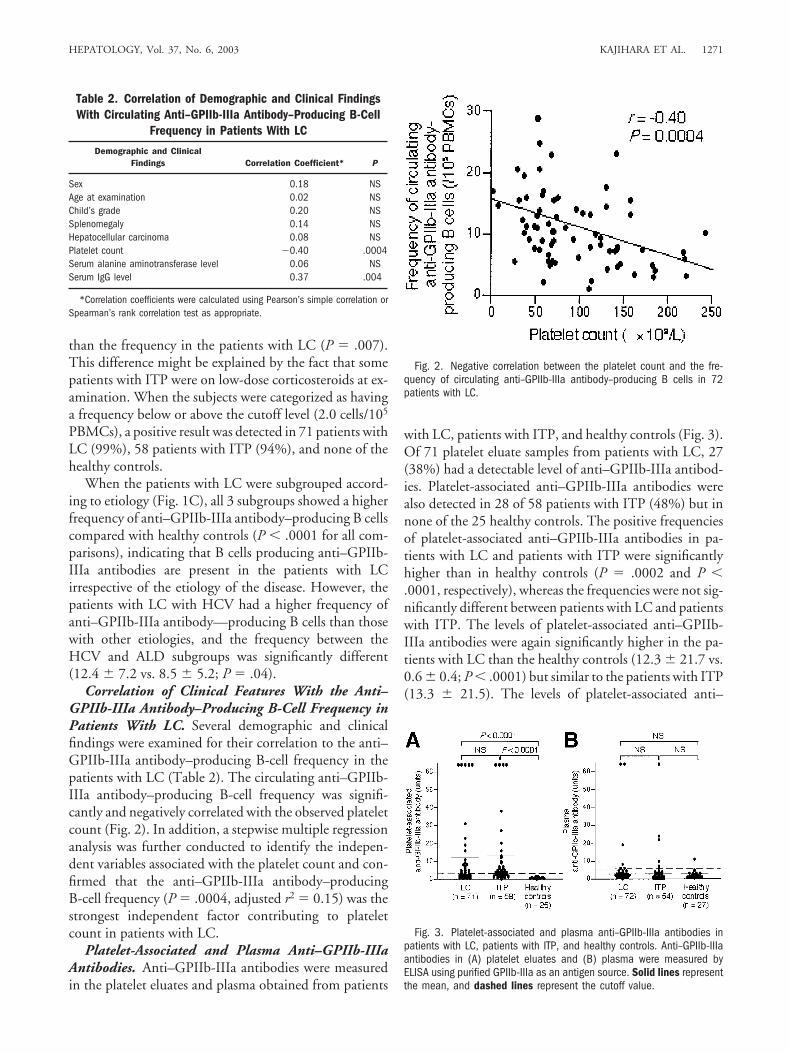
Correlation of Clinical Features With the Anti–GPIIb-IIIa Antibody–Producing B-Cell Frequency inPatients With LC. Several demographic and clinicalfindings were examined for their correlation to the anti–GPIIb-IIIa antibody–producing B-cell frequency in thepatients with LC (Table 2). The circulating anti–GPIIb-IIIa antibody–producing B-cell frequency was signifi-cantly and negatively correlated with the observed plateletcount (Fig. 2). In addition, a stepwise multiple regressionanalysis was further conducted to identify the indepen-dent variables associated with the platelet count and con-firmed that the anti–GPIIb-IIIa antibody–producingB-cell frequency (P � .0004, adjusted r2 � 0.15) was thestrongest independent factor contributing to plateletcount in patients with LC.
Platelet-Associated and Plasma Anti–GPIIb-IIIaAntibodies. Anti–GPIIb-IIIa antibodies were measuredin the platelet eluates and plasma obtained from patients
with LC, patients with ITP, and healthy controls (Fig. 3).Of 71 platelet eluate samples from patients with LC, 27(38%) had a detectable level of anti–GPIIb-IIIa antibod-ies. Platelet-associated anti–GPIIb-IIIa antibodies werealso detected in 28 of 58 patients with ITP (48%) but innone of the 25 healthy controls. The positive frequenciesof platelet-associated anti–GPIIb-IIIa antibodies in pa-tients with LC and patients with ITP were significantlyhigher than in healthy controls (P � .0002 and P �.0001, respectively), whereas the frequencies were not sig-nificantly different between patients with LC and patientswith ITP. The levels of platelet-associated anti–GPIIb-IIIa antibodies were again significantly higher in the pa-tients with LC than the healthy controls (12.3 � 21.7 vs.0.6 � 0.4; P � .0001) but similar to the patients with ITP(13.3 � 21.5). The levels of platelet-associated anti–
Table 2. Correlation of Demographic and Clinical FindingsWith Circulating Anti–GPIIb-IIIa Antibody–Producing B-Cell
Frequency in Patients With LC
Demographic and ClinicalFindings Correlation Coefficient* P
Sex 0.18 NSAge at examination 0.02 NSChild’s grade 0.20 NSSplenomegaly 0.14 NSHepatocellular carcinoma 0.08 NSPlatelet count �0.40 .0004Serum alanine aminotransferase level 0.06 NSSerum IgG level 0.37 .004
*Correlation coefficients were calculated using Pearson’s simple correlation orSpearman’s rank correlation test as appropriate.
Fig. 2. Negative correlation between the platelet count and the fre-quency of circulating anti–GPIIb-IIIa antibody–producing B cells in 72patients with LC.
Fig. 3. Platelet-associated and plasma anti–GPIIb-IIIa antibodies inpatients with LC, patients with ITP, and healthy controls. Anti–GPIIb-IIIaantibodies in (A) platelet eluates and (B) plasma were measured byELISA using purified GPIIb-IIIa as an antigen source. Solid lines representthe mean, and dashed lines represent the cutoff value.
HEPATOLOGY, Vol. 37, No. 6, 2003 KAJIHARA ET AL. 1271
GPIIb-IIIa antibodies were not different among the pa-tients with LC with HBV, those with HCV, and thosewith ALD (11.8 � 21.9, 12.9 � 21.5, and 11.4 � 23.0,respectively). The platelet-associated anti–GPIIb-IIIa an-tibody level tended to be correlated with the circulatinganti–GPIIb-IIIa antibody–producing B-cell frequency inthe patients with LC (r � 0.23; P � .06).
On the other hand, anti–GPIIb-IIIa antibodies inplasma were detected in only 6 patients with LC (8%), 6patients with ITP (11%), and one healthy control (4%),and these frequencies were not statistically different.These findings indicate that the anti–GPIIb-IIIa antibod-ies in LC and patients with ITP are predominantly oncirculating platelets, rather than in the circulation in anunbound form, strongly suggesting that the anti–GPIIb-IIIa antibodies function as anti-platelet antibodies in vivo.
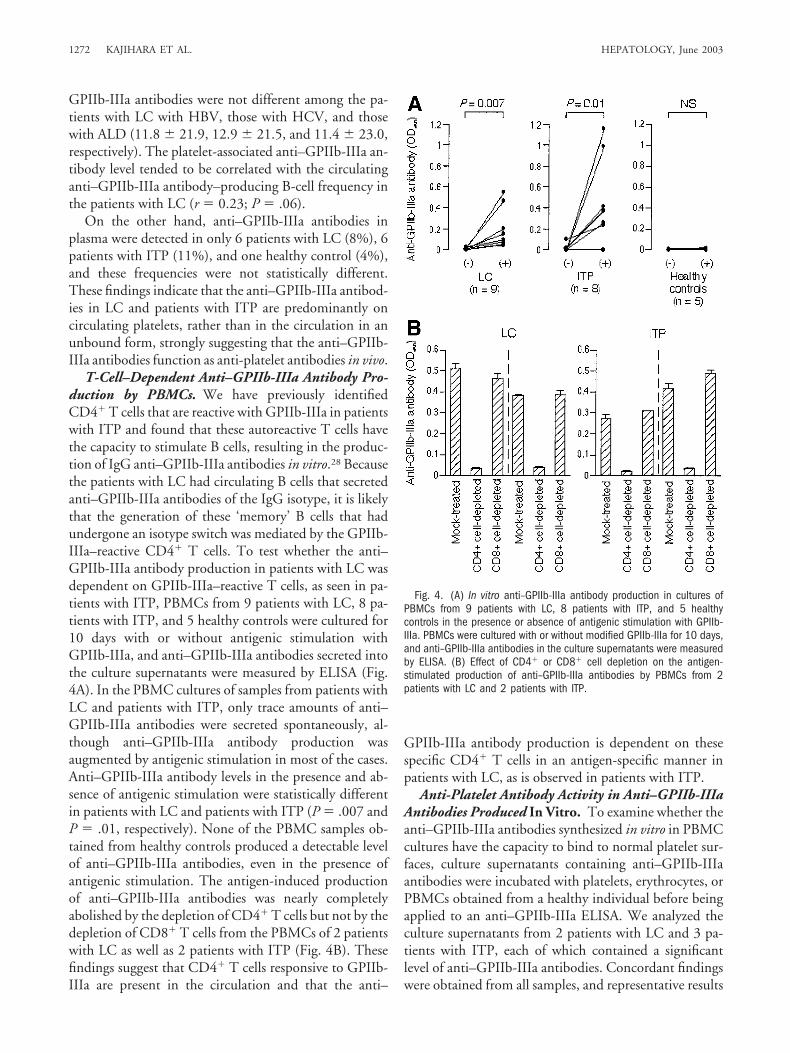
T-Cell–Dependent Anti–GPIIb-IIIa Antibody Pro-duction by PBMCs. We have previously identifiedCD4� T cells that are reactive with GPIIb-IIIa in patientswith ITP and found that these autoreactive T cells havethe capacity to stimulate B cells, resulting in the produc-tion of IgG anti–GPIIb-IIIa antibodies in vitro.28 Becausethe patients with LC had circulating B cells that secretedanti–GPIIb-IIIa antibodies of the IgG isotype, it is likelythat the generation of these ‘memory’ B cells that hadundergone an isotype switch was mediated by the GPIIb-IIIa–reactive CD4� T cells. To test whether the anti–GPIIb-IIIa antibody production in patients with LC wasdependent on GPIIb-IIIa–reactive T cells, as seen in pa-tients with ITP, PBMCs from 9 patients with LC, 8 pa-tients with ITP, and 5 healthy controls were cultured for10 days with or without antigenic stimulation withGPIIb-IIIa, and anti–GPIIb-IIIa antibodies secreted intothe culture supernatants were measured by ELISA (Fig.4A). In the PBMC cultures of samples from patients withLC and patients with ITP, only trace amounts of anti–GPIIb-IIIa antibodies were secreted spontaneously, al-though anti–GPIIb-IIIa antibody production wasaugmented by antigenic stimulation in most of the cases.Anti–GPIIb-IIIa antibody levels in the presence and ab-sence of antigenic stimulation were statistically differentin patients with LC and patients with ITP (P � .007 andP � .01, respectively). None of the PBMC samples ob-tained from healthy controls produced a detectable levelof anti–GPIIb-IIIa antibodies, even in the presence ofantigenic stimulation. The antigen-induced productionof anti–GPIIb-IIIa antibodies was nearly completelyabolished by the depletion of CD4� T cells but not by thedepletion of CD8� T cells from the PBMCs of 2 patientswith LC as well as 2 patients with ITP (Fig. 4B). Thesefindings suggest that CD4� T cells responsive to GPIIb-IIIa are present in the circulation and that the anti–
GPIIb-IIIa antibody production is dependent on thesespecific CD4� T cells in an antigen-specific manner inpatients with LC, as is observed in patients with ITP.
Anti-Platelet Antibody Activity in Anti–GPIIb-IIIaAntibodies Produced In Vitro. To examine whether theanti–GPIIb-IIIa antibodies synthesized in vitro in PBMCcultures have the capacity to bind to normal platelet sur-faces, culture supernatants containing anti–GPIIb-IIIaantibodies were incubated with platelets, erythrocytes, orPBMCs obtained from a healthy individual before beingapplied to an anti–GPIIb-IIIa ELISA. We analyzed theculture supernatants from 2 patients with LC and 3 pa-tients with ITP, each of which contained a significantlevel of anti–GPIIb-IIIa antibodies. Concordant findingswere obtained from all samples, and representative results
Fig. 4. (A) In vitro anti–GPIIb-IIIa antibody production in cultures ofPBMCs from 9 patients with LC, 8 patients with ITP, and 5 healthycontrols in the presence or absence of antigenic stimulation with GPIIb-IIIa. PBMCs were cultured with or without modified GPIIb-IIIa for 10 days,and anti–GPIIb-IIIa antibodies in the culture supernatants were measuredby ELISA. (B) Effect of CD4� or CD8� cell depletion on the antigen-stimulated production of anti–GPIIb-IIIa antibodies by PBMCs from 2patients with LC and 2 patients with ITP.
1272 KAJIHARA ET AL. HEPATOLOGY, June 2003

from one patient with LC and one patient with ITP areshown in Fig. 5. Anti–GPIIb-IIIa antibody reactivity wasabsorbed by preincubation of the culture supernatantswith normal platelets in a dose-dependent manner butnot by preincubation with erythrocytes or PBMCs.
Next, the PBMC culture supernatants from 5 patientswith LC, 2 patients with ITP, and 2 healthy controls withand without antigenic stimulation were incubated withthe platelets obtained from a healthy individual, and IgGbound to the platelet surfaces was detected by flow cytom-etry. The results obtained from all of the patients with LCand patients with ITP were similar, and a representativeresult is shown in Fig. 6. In a patient with LC, the inten-sity of IgG binding to the platelet surfaces was increasedwhen normal platelets were incubated with an antigen-stimulated PBMC culture supernatant (mean fluorescentintensity, 44.6) but not with the unstimulated culturesupernatant (mean fluorescent intensity, 13.2). In con-trast, no IgG binding was observed in platelets treatedwith the culture supernatants of healthy controls, irre-spective of antigenic stimulation.
DiscussionThe present study showed that nearly all patients with
LC exhibited an autoantibody response to GPIIb-IIIa, amajor platelet autoantigen in patients with ITP, based onthe presence of circulating B cells producing anti–GPIIa-IIIa antibodies, platelet-associated anti–GPIIb-IIIa anti-
bodies, and CD4� T cells with the ability to promoteanti–GPIIb-IIIa antibody production in B cells. All ofthese features are also detected in patients with ITP,27,28
in which enhanced platelet destruction is mediated byautoantibodies that bind to platelet surfaces. Surprisingly,the frequency of anti–GPIIb-IIIa antibody–producing Bcells and the levels of platelet-associated anti–GPIIb-IIIaantibodies in patients with LC were comparable to thosein patients with ITP. A negative correlation between thecirculating anti–GPIIb-IIIa antibody–producing B-cellfrequency and the observed platelet count and an inde-pendent effect of the circulating anti–GPIIb-IIIa anti-body–producing B-cell frequency on platelet count mightbe explained by the involvement of anti–GPIIb-IIIa anti-bodies in the process of platelet destruction in patientswith LC. Taken together, our present findings indicate acommon pathogenic process in LC and ITP and supportthe idea that the antibodies may contribute at least in partto thrombocytopenia in cirrhosis. This mechanism is notnecessarily incompatible with other proposed theories(i.e., splenic sequestration and insufficient thrombopoi-etin production) because cirrhotic thrombocytopeniacould be multifactorial.
The concept that an autoantibody-mediated process isinvolved in cirrhotic thrombocytopenia is not new. Thisidea was proposed based on an increased level ofPAIgG15-17 as well as a negative correlation betweenPAIgG and the platelet count in patients with LC.33
However, it is now widely recognized that the specificity
Fig. 5. Absorption of anti–GPIIb-IIIa antibodies in PBMC culture su-pernatants by incubation with normal platelets. Culture supernatants ofGPIIb-IIIa–stimulated PBMCs in (A) a patient with LC and (B) a patientwith ITP were incubated with serial numbers of platelets (105 to 108),erythrocytes (108), or PBMCs (107) obtained from a healthy donor.Anti–GPIIb-IIIa antibody levels in untreated or treated supernatants weremeasured by anti–GPIIb-IIIa ELISA. The results shown are representativeof 2 independent experiments.
Fig. 6. Flow cytometric analysis of normal platelets that were incu-bated with PBMC culture supernatants with or without antigenic stimu-lation with GPIIb-IIIa in a patient with LC (upper panels) and a healthycontrol (lower panels). Platelets obtained from a healthy donor wereincubated with the supernatants of PBMC cultures with or withoutGPIIb-IIIa and subsequently with fluorescein isothiocyanate–conjugatedanti-human IgG F(ab�)2. Cell staining is shown in shaded histograms, andopen histograms represent control platelets incubated with the secondaryantibody alone. Results shown are representative of at least 2 indepen-dent experiments. MFI, mean fluorescent intensity.
HEPATOLOGY, Vol. 37, No. 6, 2003 KAJIHARA ET AL. 1273

of PAIgG for autoantibody-mediated thrombocytopeniais quite low,18,19,34 and PAIgG is regarded as an inappro-priate test for the diagnosis of ITP in a practice guidelinerecently developed by the American Society of Hematol-ogy.35 In addition, an increase in PAIgG can be explainedby the presence of hypergammaglobulinemia, a conditionthat is frequently found in patients with LC, given thatPAIgG levels are positively correlated with circulatingIgG levels.36 Therefore, the autoantibody-mediatedmechanism for cirrhotic thrombocytopenia has remainedunproven because of a lack of convincing evidence show-ing a specific immune response to platelets in patientswith LC. In this regard, there is one report describing anincreased prevalence of platelet-associated anti–GPIIb-IIIa and anti–GPIb-IX antibodies in patients withchronic liver disease and thrombocytopenia, although thenumber of patients examined in that study was relativelysmall and patients with autoimmune liver disease wereincluded among the study subjects.37 Another study byPockros et al. also reported a small number of patientswith chronic HCV infection and thrombocytopenia asso-ciated with platelet-specific autoantibodies.38
It could be argued that the anti–GPIIb-IIIa antibodyproduction in patients with LC is merely a consequence ofthe sequestration and destruction of platelets in the en-larged spleen and that the anti–GPIIb-IIIa antibodiesthemselves have no pathogenic anti-platelet antibody ac-tivity in vivo. In support of this idea, anti–GPIIb-IIIaantibodies that are reactive with a cytoplasmic domain ofGPIIIa, which is not expressed on the platelet surfaces,were reported in the sera from some patients with ITP.39
However, we believe that the anti–GPIIb-IIIa antibodiesin patients with LC can bind to circulating platelets invivo based on the following findings: (1) anti–GPIIb-IIIaantibodies were present predominantly on the plateletsurfaces rather than in the plasma and (2) anti–GPIIb-IIIaantibodies produced in vitro by PBMCs bound to normalplatelet surfaces. Therefore, in patients with LC, it islikely that circulating platelets opsonized by IgG anti–GPIIb-IIIa antibodies are destroyed through phagocyto-sis by macrophages in the reticuloendothelial systemand/or through complement-dependent cytotoxicity viaFc receptors in vivo. To further verify the pathogenic roleof anti–GPIIb-IIIa antibodies, it is necessary to testwhether thrombocytopenia can be induced by injectinganti–GPIIb-IIIa antibodies derived from patients withLC into animals, although anti–GPIIb-IIIa antibodiesfrom patients with ITP have been shown to have a lowlevel of cross-reactivity with mouse platelets.40
The anti–GPIIb-IIIa antibody response was detectedin patients with LC independent of the etiology, indicat-ing that platelet-specific autoantibody production is asso-
ciated with a pathogenic process shared by diverseetiologies of LC. Our previous studies on patients withITP showed that the spleen is a primary site for bothanti-platelet antibody production and platelet destruc-tion.27 GPIIb-IIIa–reactive CD4� T cells are activated onthe recognition of antigenic peptides of GPIIb-IIIa pre-sented by antigen-presenting cells in the spleen and helpspecific B cells in the white pulp to produce pathogenicanti–GPIIb-IIIa antibodies. The positive correlation be-tween the frequency of anti–GPIIb-IIIa antibody–pro-ducing B-cell and serum IgG levels in patients with LCsuggests that a nonspecific immune activation may en-hance the autoimmune response to platelets. In this pro-cess, splenic macrophages that capture a large number ofopsonized platelets play a primary role in the activation ofGPIIb-IIIa–reactive T and B cells by efficiently present-ing the antigenic determinants of GPIIb-IIIa. Because theincreased sequestration and accelerated destruction ofplatelets in the congested spleen are noted in patients withLC irrespective of the etiology,1-3 it is possible that thepresentation of antigenic GPIIb-IIIa determinants bysplenic macrophages that nonspecifically phagocytoseplatelets in the splenic pool may trigger the activation ofGPIIb-IIIa–reactive T and B cells present in the normalimmune repertoire.28 In this regard, it has been reportedthat portal decompression or splenectomy improvesthrombocytopenia in some patients with LC.4-7 Thismight be explained by loss of antigenic presentation byfunctional antigen-presenting cells in the spleen after suchtreatment.
In the present study, however, patients with LC withHCV tended to have a higher frequency of anti–GPIIb-IIIa antibody–producing B cells compared with patientswith LC with other etiologies. Regarding this point, hepa-totropic HCV infection is often accompanied by positiveserologic tests for a variety of autoantibodies (e.g., anti-nuclear antibodies, anti–smooth muscle antibodies, andrheumatoid factor)41 and autoimmune disorders.42 More-over, several investigators have reported that chronicHCV infection itself is directly associated with autoim-mune thrombocytopenia.43,44 Therefore, it is possiblethat HCV infection enhances the autoantibody responseto GPIIb-IIIa in patients with LC.
Although the frequency of circulating B cells secretinganti–GPIIb-IIIa antibodies and platelet-associated anti–GPIIb-IIIa antibody levels were similar in patients withLC and patients with ITP, the degree of thrombocytope-nia was more prominent in the patients with ITP com-pared with the patients with LC. In fact, it is wellrecognized that thrombocytopenia in patients with LC israrely severe and usually falls within a range of 60 to 90 �109/L.31 In addition, an increased frequency of anti–
1274 KAJIHARA ET AL. HEPATOLOGY, June 2003

GPIIb-IIIa antibody–producing B cells has even been ob-served in patients with LC without thrombocytopenia.The precise reason for this difference is not clear, but thisphenomenon might be explained by a difference in thecapacity to process opsonized platelets via Fc receptors inthe reticuloendothelial system. Because the scavengerfunction of the reticuloendothelial system is reported tobe impaired in patients with LC,45 the clearance of opso-nized platelets might be less efficient in patients with LCcompared with patients with ITP. Another possibility isthat there is a difference in the subclass distribution of IgGanti–GPIIb-IIIa autoantibodies, because the IgG1 andIgG3 isotypes have a higher affinity for Fc receptors andare responsible for clearing most protein antigens whereasthe IgG2 and IgG4 isotypes have a low affinity and arepoor opsonins.46 In this regard, McMillan et al. reporteda patient with anti–GPIIb-IIIa antibodies exclusively ofthe IgG4 isotype who showed a normal platelet count buta significant bleeding tendency due to blocking of theligand binding to GPIIb-IIIa.47 Additional studies inves-tigating the differences in mechanisms for the autoanti-body-mediated thrombocytopenia between patients withLC and patients with ITP may be useful in clarifying thepathogenic process of cirrhotic thrombocytopenia as wellas in the development of novel therapeutic strategies forrefractory ITP.
References1. Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of
“hypersplenic” thrombocytopenia. J Clin Invest 1966;45:645-657.2. Harker LA, Finch CA. Thrombokinetics in man. J Clin Invest 1969;48:
963-974.3. Toghill PJ, Green S, Ferguson F. Platelet dynamics in chronic liver disease
with special reference to the role of the spleen. J Clin Pathol 1977;30:367-371.
4. McAllister E, Goode S, Cordista AG, Rosemurgy A. Partial portal decom-pression alleviates thrombocytopenia of portal hypertension. Am Surg1995;61:129-131.
5. Alvarez OA, Lopera GA, Patel V, Encarnacion CE, Palmaz JC, Lee M.Improvement of thrombocytopenia due to hypersplenism after transjugu-lar intrahepatic portosystemic shunt placement in cirrhotic patients. Am JGastroenterol 1996;91:134-137.
6. Sanyal AJ, Freedman AM, Purdum PP, Shiffman ML, Luketic VA. Thehematologic consequences of transjugular intrahepatic portosystemicshunts. HEPATOLOGY 1996;23:32-39.
7. Crane C. The choice of shunt procedure for cirrhotic patients with varicealbleeding, ascites and hypersplenism. Surg Gynecol Obstet 1962;115:12-28.
8. Kaushansky K. Thrombopoietin: the primary regulator of megakaryocyteand platelet production. Thromb Haemost 1995;74:521-525.
9. Shimada Y, Kato T, Ogami K, Horie K, Kokubo A, Kudo Y, Maeda E, etal. Production of thrombopoietin (TPO) by rat hepatocytes and hepatomacell lines. Exp Hematol 1995;23:1388-1396.
10. Eaton DL, de Sauvage FJ. Thrombopoietin: the primary regulator ofmegakaryocytopoiesis and thrombopoiesis. Exp Hematol 1997;25:1-7.
11. Martin TG III, Somberg KA, Meng YG, Cohen RL, Heid CA, de SauvageFJ, Shuman MA. Thrombopoietin levels in patients with cirrhosis beforeand after orthotopic liver transplantation. Ann Intern Med 1997;127:285-288.
12. Goulis J, Chau TN, Jordan S, Mehta AB, Watkinson A, Rolles K, Bur-roughs AK. Thrombopoietin concentrations are low in patients with cir-rhosis and thrombocytopenia and are restored after orthotopic livertransplantation. Gut 1999;44:754-758.
13. Shimodaira S, Ishida F, Ichikawa N, Tahara T, Kato T, Kodaira H, Ito T,et al. Serum thrombopoietin (c-Mpl ligand) levels in patients with livercirrhosis. Thromb Haemost 1996;76:545-548.
14. Stockelberg D, Andersson P, Bjornsson E, Bjork S, Wadenvik H. Plasmathrombopoietin levels in liver cirrhosis and kidney failure. J Intern Med1999;246:471-475.
15. Barrison IG, Knight ID, Viola L, Boots MA, Murray-Lion IM, MitchellTR. Platelet associated immunoglobulins on chronic liver disease. Br JHaematol 1981;48:347-350.
16. Leone G, Agostini A, Mango G, Landolfi R, Valori VM, Bizzi B. Mega-thrombocytes, platelet regeneration time and platelet associated IgG inidiopathic thrombocytopenic purpura and in thrombocytopenia associ-ated with chronic liver disease. Acta Haematol 1981;65:40-47.
17. Graber D, Giuliani D, Leevy CM, Morse BS. Platelet-associated IgG inhepatitis and cirrhosis. J Clin Immunol 1984;4:108-11.
18. Mueller-Eckhardt C, Kayser W, Mersch-Baumert K, Mueller-Eckhardt G,Breidenbach M, Kugel HG, Graubner M. The clinical significance ofplatelet-associated IgG: a study on 298 patients with various disorders. Br JHaematol 1980;46:123-131.
19. Kelton JG, Powers PJ, Carter CJ. A prospective study of the usefulness ofthe measurement of platelet-associated IgG for the diagnosis of idiopathicthrombocytopenic purpura. Blood 1982;60:1050-1053.
20. Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N EnglJ Med 2002;346:995-1008.
21. He R, Reid DM, Jones CE, Shulman NR. Spectrum of Ig classes, speci-ficities, and titers of serum antiglycoproteins in chronic idiopathic throm-bocytopenic purpura. Blood 1994;83:1024-1032.
22. McMillan R, Tani P, Millard F, Berchtold P, Renshaw L, Woods VL Jr.Platelet-associated and plasma anti-glycoprotein autoantibodies in chronicITP. Blood 1987;70:1040-1045.
23. Hurlimann-Forster M, Steiner B, von Felten A. Quantitation of platelet-specific autoantibodies in platelet eluates of ITP patients measured by anovel ELISA using the purified glycoprotein complexes GPIIb/IIIa andGPIb/IX as antigens. Br J Haematol 1997;98:328-335.
24. Woods VL Jr, Oh EH, Mason D, McMillan R. Autoantibodies against theplatelet glycoprotein IIb/IIIa complex in patients with chronic ITP. Blood1984;63:368-375.
25. Kiefel V, Santoso S, Weisheit M, Mueller-Eckhardt C. Monoclonal anti-body—specific immobilization of platelet antigens (MAIPA): a new toolfor the identification of platelet-reactive antibodies. Blood 1987;70:1722-1726.
26. Kuwana M, Okazaki Y, Kaburaki J, Ikeda Y. Detection of circulating Bcells secreting platelet-specific autoantibody is useful in the diagnosis ofautoimmune thrombocytopenia. Am J Med 2003;114:322-325.
27. Kuwana M, Okazaki Y, Kaburaki J, Kawakami Y, Ikeda Y. Spleen is aprimary site for activation of platelet-reactive T and B cells in patients withimmune thrombocytopenic purpura. J Immunol 2002;168:3675-3682.
28. Kuwana M, Kaburaki J, Ikeda Y. Autoreactive T cells to platelet GPIIb-IIIain immune thrombocytopenic purpura. Role in production of anti-plateletautoantibody. J Clin Invest 1998;102:1393-1402.
29. Kuwana M, Kaburaki J, Kitasato H, Kato M, Kawai S, Kawakami Y, IkedaY. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by au-toreactive T cells in patients with immune thrombocytopenic purpura.Blood 2001;98:130-139.
30. Erlinger S, Benhamou JP. Cirrhosis: clinical aspects. In: Bircher J, Ben-hamou JP, McIntyre N, Rizetto M, Rodes J, eds. Oxford Textbook ofClinical Hepatology. Volume 1. 2nd ed. Oxford: Oxford University Press,1999:629-641.
31. Sherlock S, Dooley J. Diseases of the liver and biliary system. Oxford:Blackwell Sciences, 2002.
32. Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lan-cet 1997;349:1531-1536.
HEPATOLOGY, Vol. 37, No. 6, 2003 KAJIHARA ET AL. 1275

33. de Noronha R, Taylor BA, Wild G, Triger DR, Greaves M. Inter-relation-ships between platelet count, platelet IgG, serum IgG, immune complexesand severity of liver disease. Clin Lab Haematol 1991;13:127-135.
34. Chong BH, Keng TB. Advances in the diagnosis of idiopathic thrombo-cytopenic purpura. Semin Hematol 2000;37:249-260.
35. George JN, Woolf SH, Raskob GE, Wasser JS, Aledort LM, Ballem PJ,Blanchette VS, et al. Idiopathic thrombocytopenic purpura: a practiceguideline developed by explicit methods for the American Society of He-matology. Blood 1996;88:3-40.
36. McGrath KM, Stuart JJ, Richards F II. Correlation between serum IgG,platelet membrane IgG, and platelet function in hypergammaglobulinae-mic states. Br J Haematol 1979;42:585-591.
37. Pereira J, Accatino L, Alfaro J, Brahm J, Hidalgo P, Mezzano D. Plateletautoantibodies in patients with chronic liver disease. Am J Hematol 1995;50:173-178.
38. Pockros PJ, Duchini A, McMillan R, Nyberg LM, McHutchison J, Vier-nes E. Immune thrombocytopenic purpura in patients with chronic hep-atitis C virus infection. Am J Gastroenterol 2002;97:2040-2045.
39. Fujisawa K, O’Toole TE, Tani P, Loftus JC, Plow EF, Ginsberg MH,McMillan R. Autoantibodies to the presumptive cytoplasmic domain ofplatelet glycoprotein IIIa in patients with chronic immune thrombocyto-penic purpura. Blood 1991;77:2207-2213.
40. Dekel B, Marcus H, Shenkman B, Shimoni A, Shechter Y, Canaan A,Berrebi A, et al. Human/BALB radiation chimera engrafted with spleno-
cytes from patients with idiopathic thrombocytopenic purpura producehuman platelet antibodies. Immunology 1998;94:410-416.
41. Clifford BD, Donahue D, Smith L, Cable E, Luttig B, Manns M, Bonk-ovsky HL. High prevalence of serological markers of autoimmunity inpatients with chronic hepatitis C. HEPATOLOGY 1995;21:613-619.
42. Pawlotsky JM, Ben Yahia M, Andre C, Voisin MC, Intrator L, Roudot-Thoraval F, Deforges L, et al. Immunological disorders in C virus chronicactive hepatitis: a prospective case-control study. HEPATOLOGY 1994;19:841-848.
43. Bauduer F, Marty F, Larrouy M, Ducout L. Immunologic thrombocyto-penic purpura as presenting symptom of hepatitis C infection. Am J He-matol 1998;57:338-340.
44. Hernandez F, Blanquer A, Linares M, Lopez A, Tarin F, Cervero A. Au-toimmune thrombocytopenia associated with hepatitis C virus infection.Acta Haematol 1998;99:217-220.
45. Rimola A, Soto R, Bory F, Arroyo V, Piera C, Rodes J. Reticuloendothelialsystem phagocytic activity in cirrhosis and its relation to bacterial infec-tions and prognosis. HEPATOLOGY 1984;4:53-58.
46. Turner MI. Antibodies. In: Roitt I, Brostoff J, Male D, eds. Immunology.6th ed. Edinburgh: Mosby, 2001:65-85.
47. McMillan R, Bowditch RD, Tani P, Anderson H, Goodnight S. A non-thrombocytopenic bleeding disorder due to an IgG4-kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747-749.
1276 KAJIHARA ET AL. HEPATOLOGY, June 2003





























