Embed Size (px)
Citation preview

Neurobiology of Disease 41 (2011) 249–260
Contents lists available at ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r.com/ locate /ynbd i
Altered adult hippocampal neurogenesis in the YAC128 transgenic mouse model ofHuntington disease
Jessica M. Simpson a,1, Joana Gil-Mohapel a,⁎,1, Mahmoud A. Pouladi b,c, Mohamed Ghilan a, Yuanyun Xie b,c,Michael R. Hayden b,c, Brian R. Christie a,d,e
a Division of Medical Sciences and Department of Biology, University of Victoria, Victoria, BC, Canadab Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canadac Center for Molecular Medicine and Therapeutics, Department of Medical Genetics and Family Research Institute, University of British Columbia, Vancouver, BC, Canadad The Brain Research Center and Program in Neuroscience, University of British Columbia, Vancouver, BC, Canadae The Department of Cellular and Physiological Sciences, University of British Columbia, Vancouver, BC, Canada
⁎ Corresponding author. Division of Medical ScienUniversity of Victoria, Victoria, BC, V8W 2Y2, Canada. Fa
E-mail address: [email protected] (J. Gil-Mohapel).1 The two authors contributed equally to this work.
Available online on ScienceDirect (www.scienced
0969-9961/$ – see front matter. Crown Copyright © 20doi:10.1016/j.nbd.2010.09.012
a b s t r a c t
a r t i c l e i n f oArticle history:Received 10 May 2010Revised 16 September 2010Accepted 20 September 2010Available online 25 September 2010
Keywords:Huntington diseaseAdult neurogenesisCell proliferationDentate gyrusSubventricular zoneYAC128 transgenic miceHippocampus
Perturbations in neurogenesis in the adult brain have been implicated in impaired learning and memory. Inthe present study, we investigated which stages of the neurogenic process are affected in the transgenicYAC128 mouse model of Huntington disease (HD). Hippocampal neuronal proliferation was altered inthe dentate gyrus (DG) of YAC128 mice as compared with wild-type (WT) littermate controls in earlysymptomatic to end-stage mice. In addition, we detected a significantly lower number of immature neuronsin the DG of young, pre-symptomatic YAC128 mice. This decrease in neuronal differentiation persistedthrough the progression of the disease, and resulted in an overall reduction in the number of new matureneurons in the DG of YAC128 mice. There were no changes in cell proliferation and differentiation in thesubventricular zone (SVZ). In this study, we demonstrate decreases in neurogenesis in the DG of YAC128mice,and these deficits may contribute to the cognitive abnormalities observed in these animals.
Crown Copyright © 2010 Published by Elsevier Inc. All rights reserved.
Introduction
Huntington disease (HD) is an autosomal dominant neurodegen-erative disorder that is caused by the expansion of CAG repeats in thegene that encodes the protein huntingtin (The Huntington's DiseaseCollaborative Research Group, 1993). This causes a toxic gain offunction in the huntingtin protein, aberrant proteolysis, and thesubsequent appearance of N-terminal mutant huntingtin fragmentsthat precipitate and form neuronal intranuclear inclusions (DiFigliaet al., 1997). There is a significant cell loss in the striatum and certainlayers of the cortex initially, followed bymore diffuse cell loss in otherregions of the brain, including the hippocampus and the hypothala-mus (Vonsattel and DiFiglia, 1998; Rosas et al., 2003; Petersen et al.,2005). Some of the earliest behavioural symptoms include moodswings, depression, as well as impaired learning and memory. As thedisease progresses, concentration on intellectual tasks becomesincreasingly difficult and dementia is commonly observed. During
ces, Island Medical Program,x: +1 250 772 5505.
irect.com).
10 Published by Elsevier Inc. All rig
this time the affected individuals also develop severe motor impair-ments, including loss of voluntary motor coordination or chorea.Weight loss, muscle wasting, and other peripheral manifestations arealso common (for review see Gil and Rego, 2008).
Experimental studies of neuronal damage in HD have beenfacilitated by the development of the yeast artificial chromosome(YAC) 128 transgenic mouse model, which expresses the full-lengthhuman mutant HD gene with 128 CAG repeats (Slow et al., 2003) andfaithfully recapitulates many features of the human condition (forreview see Heng et al., 2008; Ehrnhoefer et al., 2009). The YAC128mice display a uniform phenotype with age-dependent striatal andsubsequent cortical neurodegeneration and cell loss (Slow et al.,2003) and show progressive motor and cognitive deficits, such asimpairments in procedural learning and memory as well as strategyshifting (Van Raamsdonk et al., 2005; Slow et al., 2003). In particular,the motor deficits follow a biphasic phenotype, with hyperactivityfirst observed at 3 months of age, followed by hypoactivity by12 months of age (Slow et al., 2003), which replicates the progressionfrom chorea and bradykinesia to rigidity that is observed in humanHD patients (for review see Gil and Rego, 2008).
Throughout adulthood, select regions of the mammalian brainretain the ability to generate new neurons, a process calledneurogenesis. These regions include the subventricular zone (SVZ)
hts reserved.

250 J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
and the subgranular zone (SGZ) of the hippocampal dentate gyrus(DG). In the SGZ, new granule neurons functionally integrate into theexisting cytoarchitecture (van Praag et al., 2005) and extend axonalprojections into the CA3 region of the hippocampus (for review seeCameron and McKay, 1998). Adult neurogenesis occurs in the humanSGZ (Eriksson et al., 1998), and these adult neuronal stem cells havebeen proposed as an endogenous source of healthy cells for thetreatment of neurodegenerative diseases (for review see Lie et al.,2004; Mohapel and Brundin, 2004; Gil-Mohapel et al., 2010). Theprocess of adult neurogenesis appears vital for some forms of normalcognitive functioning, such as spatial learning and memory, andrecent studies have demonstrated that neurogenesis is disrupted in anumber of neurodegenerative disorders that include: Alzheimer'sdisease (Jin et al., 2004; Li et al., 2008); Parkinson's disease (Höglingeret al., 2004; Crews et al., 2008); and HD (Curtis et al., 2003; Gil et al.,2004, 2005; Lazic et al., 2004, 2006). In postmortem HD brains, anincrease in proliferation in the SVZ has been documented (Curtis et al.,2003). However, these results are in contrast with the findingsobtained in R6/1 (Lazic et al., 2004, 2006) and R6/2 (Gil et al., 2004,2005) HD transgenic mouse models, which show no differences inSVZ cell proliferation but display a dramatic decrease in neurogenesisin the DG. In the R6/2mice, this reduction in the DG can be detected asearly as two-weeks of age, when no other behavioural symptoms arepresent (Gil et al., 2005). However, the fast progression of the diseasein the R6/1 and R6/2 transgenic mouse lines, and the severity of theirphenotype, have been suggested to be more representative of thejuvenile-onset form of HD (for review see Gil and Rego, 2009). On theother hand, the YAC128 mice seem to display a disease progressionmore akin to the human condition and have also been shown todisplay cognitive (Slow et al., 2003) and depressive-like symptoms(Pouladi et al., 2009) that precede the onset of motor deficits.
Importantly, both a decrease in spatial learning and memory andan increase in depression have been correlated with a reduction inadult hippocampal neurogenesis (Gould et al., 1999; Malberg et al.,2000; Manev et al., 2001; reviewed by Mirescu and Gould, 2006),suggesting a link between the development of these phenotypes andan altered neurogenic process in the HD brain. Thus, in the presentstudy we investigated whether adult neurogenesis is compromised bythe progression of the disease in the YAC128 HD transgenic mousemodel.
Materials and methods
Transgenic mice
Transgenic HD mice expressing human huntingtin with approx-imately 128 CAG repeats (YAC128) and their wild-type (WT)littermates were used for these experiments. The colony wasmaintained at the Department of Medical Genetics, University ofBritish Columbia (Vancouver, BC, Canada). Briefly, a well-character-ized YAC (353G6) spanning the entire HD gene including itspromoter region was used to create these mice (Slow et al., 2003).Homologous recombination was used to incorporate 128 CAGrepeats obtained from the DNA of a juvenile-onset HD patient intothe YAC following a previously described method (Duff et al., 1994).Mice were maintained on the FVB/N background strain (CharlesRiver, Wilmington, MA, USA). These experiments used 3- (12WT, 12YAC128), 9- (20 WT, 22 YAC128), 12- (22 WT, 21 YAC128), and 18-month-old (9 WT, 8 YAC128) mice (equal numbers of males andfemales were used for each group). The mice were housed in groupswith ad libitum access to food and water with a normal 12 h light/dark cycle with ambient temperature and humidity. All experimentalprocedures were conducted in accordance with the University ofVictoria, the University of British Columbia, and the Canadian Councilfor Animal Care policies.
Treatments and tissue processing
Generation of newborn cells was assessed by injecting 200 mg/kgof bromodeoxyuridine (BrdU, in 0.1 M TBS, pH 7.2; Sigma-Aldrich,St. Louis, MO, USA) intraperitoneally (i.p.) every 12 h for 3 consec-utive days. BrdU is a thymidine analogue that is incorporated intothe DNA helix during the S-phase of the cell cycle and is commonlyused as an exogenously administered marker for cell proliferation(Cooper-Kuhn and Kuhn, 2002). To examine cell proliferation, micewere sacrificed at 3, 9, 12, and 18 months of age, 12 h after the lastBrdU injection. To analyze both the differentiation and survival ofthe newborn cells that incorporated BrdU, a separate set of 8-month(9 WT, 11 YAC128) and 11-month (10WT, 9 YAC128) old mice weresacrificed 4 weeks after receiving the last BrdU injection (i.e., at 9and 12 months of age, respectively). Mice were deeply anesthetizedwith avertin (2.5%, i.p.) and transcardially perfused with 0.9% salinefollowed by 4% paraformaldehyde (PFA). The brains were removedand left in 4% PFA overnight at 4 °C and then transferred to 30%sucrose. Following saturation in sucrose, serial coronal sections werecut on a vibratome (Leica VT1000S, Nussloch, Germany) at a 30 μmthickness. Sections were collected in a 1/6 section sampling fractionand stored in a cryoprotectant solution (0.04 M TBS, 30% ethyleneglycerol, 30% glycerol) at −20 °C.
Immunohistochemistry
One series of free-floating sections was processed for BrdUimmunohistochemistry as previously described (Gil et al., 2005).Briefly, after thorough rinsing, the sectionswere incubated in 2 NHClat 65 °C for 30 min in order to denature the DNA. The sections werethen pre-incubated for 1 h in 5% normal horse serum (NHS) and0.25% Triton X-100 in 0.1 M TBS and then incubated for 48 h at 4 °Cwith a mouse monoclonal antibody against BrdU (1:50; Dako,Glostrup, Denmark) in TBS containing 5% NHS. After incubationwith a biotinylated horse anti-mouse IgG secondary antibody(1:200; Vector Laboratories, Burlingame, CA, USA) for 2 h, thebound antibodieswere visualized using an avidin–biotin–peroxidasecomplex system (Vectastain ABC Elite Kit, Vector Laboratories) withdiaminobenzidine (DAB; Vector Laboratories) as a chromogen. Thesections were mounted onto 2% gelatin-coated microscope slides,dehydrated in a series of ethanol solutions of increasing concentra-tions, and coverslipped with Permount mounting medium (FisherScientific, Fair Lawn, NJ, USA).
Adjacent series of sections were also processed for detection of theendogenous proliferative markers Ki-67, a nuclear protein that isexpressed during all active phases of the cell cycle, but is absent fromcells at rest (Kee et al., 2002; for review see Christie and Cameron,2006), and proliferating cell nuclear antigen (PCNA), which isexpressed during all active phases of the cell cycle and for a shortperiod of time after cells become post-mitotic (for review see Christieand Cameron, 2006). Briefly, after thorough rinsing, the sections wereincubated in 10 mM sodium citrate buffer (in 0.1 M TBS, pH 6.0) at95 °C for 5 min. This step was repeated twice in order to completelyunmask the antigens. After quenching with 3% H2O2/10% methanol in0.1 M TBS for 15 min and pre-incubating with 5% normal goat serum(NGS) for 1 h, the sections were incubated for 48 h at 4 °C with arabbit polyclonal anti-Ki-67 primary antibody (1:500; Vector Labora-tories) or a rabbit polyclonal antibody against PCNA (1:100; SantaCruz Biotechnology, Santa Cruz, CA, USA). After thorough rinsing thesections were then incubated for 2 h with the secondary antibody(biotin-conjugated goat anti-rabbit IgG, 1:200; Vector Laboratories),and visualized as described earlier.
Finally, two additional series of sections were processed for eitherdoublecortin (DCX) immunohistochemistry, a marker for newlydifferentiated and migrating neuroblasts (Brown et al., 2003), or theneurogenic differentiation protein (NeuroD), a basic helix–loop–helix

251J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
transcription factor involved in neuronal differentiation (Miyata et al.,1999). Briefly, after quenching and pre-incubation with NHS, thesections were incubated for 48 h at 4 °C with a goat anti-DCX primaryantibody (1:400; c-18, Santa Cruz Biotechnology) or a goat anti-NeuroD primary antibody (1:200; Santa Cruz Biotechnology), respec-tively. The sections were then incubated for 2 h with the secondaryantibody (biotin-conjugated horse anti-goat IgG, 1:200; VectorLaboratories), and visualized as described earlier.
Neuronal differentiation was also assessed by BrdU/neuronalnuclei (NeuN) double-labeling. Briefly, after DNA denaturation in2 N HCl at 65 °C for 30 min and pre-incubation with the proper sera,the sections were incubated for 48 h at 4 °C in rat anti-BrdU (1:100;Harlan Sera-Lab, Belton, UK) andmouse anti-NeuN (1:100; Chemicon,Billerica, MA, USA) primary antibodies. The sections were thenincubated with the secondary antibodies Cy3-conjugated donkeyanti-rat IgG (1:200; Jackson ImmunoResearch, West Grove, PA, USA)and biotinylated horse anti-mouse (1:200; Vector Laboratories) for2 h, followed by incubation with Alexa-488-conjugated streptavidin(1:200; Molecular Probes, Leiden, The Netherlands) for 2 h. Thesections were mounted onto 2% gelatin-coated microscope slides,coverslipped with PVA mounting medium with DABCO antifading(Sigma-Aldrich), and stored in the dark at 4 °C.
Morphological quantification
All morphological analyses were performed on coded slides, withthe experimenter blinded, using an Olympus microscope (OlympusBX51, Center Valley, PA, USA) with 10×, 40× and 100× objectives.Image Pro-Plus software (version 5.0 for WindowsTM, MediaCybermetic Inc., Silver Spring, MD, USA) and a Cool Snap HQ camera(Photometrics, Tucson, AZ, USA) were used for image capture. Aprofile analysis was performed to estimate the total numbers of BrdU-,Ki-67-, PCNA-, DCX- and NeuroD-immunopositive cells in the SGZ ofthe DG of the hippocampus by manually counting all positive cellspresent within two–three nuclear diameters below the granule celllayer (GCL). All sections containing the DG region of the hippocampus(from 1.34 mm posterior to Bregma to 3.52 mm posterior to Bregma;Paxinos and Franklin, 2001) were used for the analysis. To estimateany volumetric discrepancy between the DG of WT and YAC128 mice,we compared the volume of the GCL between 18 month-old WT andYAC128 mice, using adjacent coronal sections throughout the entirerostral/caudal extent of the DG region of the hippocampus. Every sixth30 μm-thick section from the NeuroD immunostained brains wasimaged. Using Image Pro-Plus software (Media Cybermetic Inc.), theGCL was outlined and the area obtained. Volume was estimated usingthe formula∑A×T× I, where∑A is the sum of the areameasured oneach section, T is the section thickness (30 μm), and I is the number ofsection intervals (6). There were no significant volumetric differencesfound between the GCL of WT (0.251±0.013 mm3, n=9) andYAC128 (0.227±0.011 mm3, n=8; Student's t-test p=0.16) miceat 18 months of age, a time-point when YAC128 are severely diseasedand show pronounced striatal atrophy and cell loss (Slow et al., 2003).Thus, the results were expressed as the total number of labeled cells inthe DG sub-region of the hippocampus by multiplying the averagenumber of labeled cells/DG section by the total number of 30 μmthick-sections that contain the DG (estimated as 73 sections). Thetotal numbers of Ki-67-, PCNA- and DCX-positive cells in the SVZwerealso assessed by manually counting all positive cells in all sectionscontaining this region (from 1.42 mm anterior to Bregma to 0.58 mmposterior to Bregma; Paxinos and Franklin, 2001). The total number oflabeled cells in the SVZ was calculated by multiplying the averagenumber of labeled cells/SVZ section by the total number of 30 μmthick-sections that contain the SVZ (estimated as 48 sections). As wedid not observe significant differences between sexes (data notshown), cell counts from both males and females were pooled withingenotypes.
For sections that were processed for BrdU/NeuN immunohisto-chemistry, a maximum of 50 BrdU-labeled cells per mouse wererandomly selected for analysis of co-labeling with NeuN, using aconfocal laser-scanning microscope (Olympus BX61WI) connectedto a PC running the Olympus FluoView FV10-ASW 1.7c Software,at 1 μm optical thickness. BrdU and NeuN were considered to be co-localized if nuclear co-localization was observed over the extent ofthe nucleus in consecutive 1 μm z-stacks, if profiles of green (NeuN)and red (BrdU) fluorescence coincided, and when co-localizationwas confirmed in x–y, x–z and y–z cross-sections produced byorthogonal reconstructions from z-series.
Statistical analysis
Differences between mean values of experimental groups werecompared using Student's t-test or two-factor analysis of variance(ANOVA), followed by Tukey's post-hoc tests as appropriate, usingR 2.10.1 (R Project for Statistical Computing, Free Software Foundation,Boston, MA, USA). Data are presented as mean±standard error of themean (SEM). A p value of b0.05 was considered statistically significant.
Results
YAC128 mice exhibit altered hippocampal cell proliferation
We used BrdU to label cells in the S-phase of the cell cycle byinjecting YAC128 and WT mice with this exogenous proliferationmarker every 12 h during three consecutive days and sacrificing theanimals 12 h after the last injection. We observed an age-relateddecline in the number of proliferating cells in mice of both genotypesafter 3 months of age. This decrease reached a plateau at 9 months ofage, which is consistent with previous literature (Drapeau et al.,2007; Ben Abdallah et al., 2010). We also observed a small butsignificant decrease in the number of BrdU-positive cells betweenYAC128 mice and their WT controls (two-factor ANOVA: genotype:p=0.04, F(1,77)=4.32; age: pb0.0001, F(3,77)=171.67; age×gen-otype: p=0.14, F(3,77)=1.89). Thus, at 18 months of age YAC128mice exhibit 26% less BrdU-positive cells (i.e., proliferating cellsin the S-phase) when compared with their WT littermate controls(Fig. 1).
In order to further analyze the DG cell proliferation in theYAC128 HD mouse model we used the endogenous proliferationmarker Ki-67, which is expressed during all active phases of the cellcycle (Kee et al., 2002; for review see Christie and Cameron, 2006).Again, we observed an age-related decline in the number ofproliferating cells in mice of both genotypes, as well as a small butsignificant decrease in the number of Ki-67-positive cells betweenYAC128 mice and WT controls (two-factor ANOVA: genotype:p=0.04, F(1,71)=4.57; age: pb0.0001, F(3,71)=52.16; age×gen-otype: p=0.88, F(3,71)=0.22). Moreover, similarly to the resultsobtained with BrdU, at 18 months of age YAC128 mice exhibit 26%less Ki-67-positive cells when compared with their WT littermatecontrols (Figs. 2A–C). Since the cell cycle marker PCNA, which isexpressed during all phases of the cell cycle (for review see Christieand Cameron, 2006), has been used in previous studies assessing cellproliferation in both human HD tissue (Curtis et al., 2003) and R6/2mice (Gil et al., 2005), we also used this marker to evaluate cellproliferation in YAC128 mice. Again, we found an age-relateddecline in the number of PCNA-positive cells. However, nosignificant differences in the number of mitotic cells betweengenotypes was detected with this marker (two-factor ANOVA:genotype: p=0.28, F(1,76)=1.19; age: pb0.0001, F(3,76)=49.35;age×genotype: p=0.91, F(3,76)=0.17) (Figs. 2D–F). The fact thatmore cells were labeled with PCNA than with Ki-67 is a commonfinding; levels of Ki-67 expression appear to change across the cellcycle, which may cause Ki-67 to be undetectable during the early

Fig. 1. Decrease in the number of BrdU-labeled cells in the dentate gyrus of YAC128 mice. (A) Cells in the S-phase of the cell cycle were assessed by immunohistochemistry for theexogenous marker BrdU. An age-related decline in the number of BrdU-labeled cells was observed in the DG after 3 months of age. A small but significant overall decrease in thenumber of BrdU-positive cells in the DG of YAC128mice as compared to theirWT controls was also observed (two-factor ANOVA: genotype: p=0.04, F(1,77)=4.32; age: pb0.0001,F(3,77)=171.67; age×genotype: p=0.14, F(3,77)=1.89). Representative sections of the DG processed for BrdU immunohistochemistry in WT (B) and YAC128 (C) mice at3 months of age. Arrows indicate BrdU-positive cells in the insets. Data are presented as means±SEM. Scale bars=20 μm, scale bars (insets)=5 μm.
252 J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
portion of the G1 phase. In contrast, PCNA appears to continue beingexpressed for a short time just after cells have become post-mitotic(for review see Christie and Cameron, 2006). Moreover, it has beensuggested that PCNA is expressed in cells undergoing DNA repair(Tomasevic et al., 1998), whichmight also account for the discrepantresults obtainedwith these two cell cycle markers. Regarding the factthat a significant reduction in the number of proliferating cellsbetween YAC128 mice and their WT littermate controls wasobserved with Ki-67 and BrdU but not with PCNA might reflectdifferences in cell cycle kinetics between WT and YAC128 mice.
Fig. 2. Evaluation of cell proliferation in the dentate gyrus of YAC128 and controlimmunohistochemistry for the endogenous cell cycle markers Ki-67 (A–C) and PCNA (D–F)proliferating cells with increasing age in the DG of both YAC128 and WT mice (A,D). (A) ThYAC128 mice compared to their controls (two-factor ANOVA: genotype: p=0.04, F(1,71)=significant differences were found between the genotypes with PCNA immunohistochemist49.35; age×genotype: p=0.91, F(3,76)=0.17). Representative sections of the DG processemice at 3 months of age. Arrows indicate immunopositive cells in the insets. Data are prese
YAC128 mice exhibit decreased hippocampal neuronal differentiation
The endogenousmarkerDCX, amicrotubule-binding protein that isexpressed in newly differentiated and migrating neuroblasts (Brownet al., 2003),was used to determine if therewas an overall difference inthe number of immature neurons between the two genotypes(Figs. 3A–C). Similar to the results obtained using BrdU, there was anage-related decline in DCX-positive neuroblasts after 3 months of age,at which time a plateau was reached. There was also a strikingsignificant decrease in the number of cells immunopositive for DCX in
mice using endogenous cell cycle markers. Cell proliferation was examined byat 3-, 9-, 12- and 18-months of age. There was a progressive decline in the number ofere was a small but significant overall decrease in the number of Ki-67-positive cells in4.57; age: pb0.0001, F(3,71)=52.16; age×genotype: p=0.88, F(3,71)=0.22), but nory (D; two-factor ANOVA: genotype: p=0.28, F(1,76)=1.19; age: pb0.0001, F(3,76)=d for Ki-67 (B,C) and PCNA (E,F) immunohistochemistry in WT (B,E) and YAC128 (C,F)nted as means±SEM. Scale bars=20 μm, scale bars (insets)=5 μm.
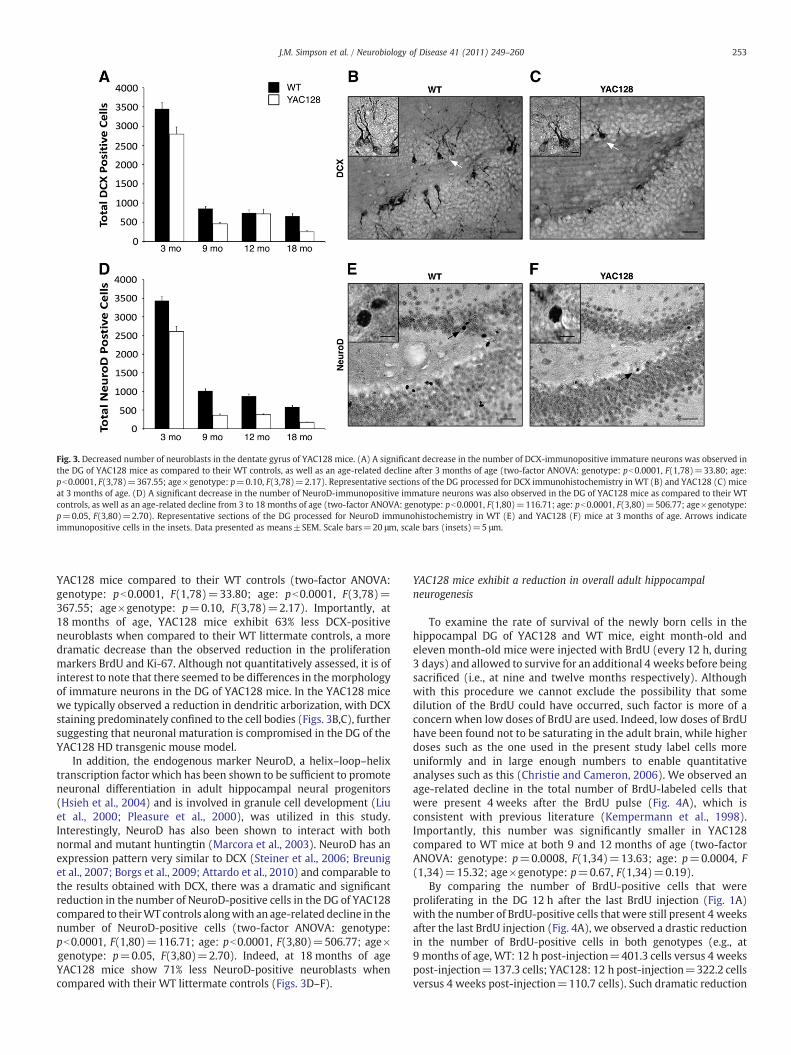
Fig. 3. Decreased number of neuroblasts in the dentate gyrus of YAC128 mice. (A) A significant decrease in the number of DCX-immunopositive immature neurons was observed inthe DG of YAC128 mice as compared to their WT controls, as well as an age-related decline after 3 months of age (two-factor ANOVA: genotype: pb0.0001, F(1,78)=33.80; age:pb0.0001, F(3,78)=367.55; age×genotype: p=0.10, F(3,78)=2.17). Representative sections of the DG processed for DCX immunohistochemistry in WT (B) and YAC128 (C) miceat 3 months of age. (D) A significant decrease in the number of NeuroD-immunopositive immature neurons was also observed in the DG of YAC128 mice as compared to their WTcontrols, as well as an age-related decline from 3 to 18 months of age (two-factor ANOVA: genotype: pb0.0001, F(1,80)=116.71; age: pb0.0001, F(3,80)=506.77; age×genotype:p=0.05, F(3,80)=2.70). Representative sections of the DG processed for NeuroD immunohistochemistry in WT (E) and YAC128 (F) mice at 3 months of age. Arrows indicateimmunopositive cells in the insets. Data presented as means±SEM. Scale bars=20 μm, scale bars (insets)=5 μm.
253J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
YAC128 mice compared to their WT controls (two-factor ANOVA:genotype: pb0.0001, F(1,78)=33.80; age: pb0.0001, F(3,78)=367.55; age×genotype: p=0.10, F(3,78)=2.17). Importantly, at18 months of age, YAC128 mice exhibit 63% less DCX-positiveneuroblasts when compared to their WT littermate controls, a moredramatic decrease than the observed reduction in the proliferationmarkers BrdU and Ki-67. Although not quantitatively assessed, it is ofinterest to note that there seemed to be differences in themorphologyof immature neurons in the DG of YAC128 mice. In the YAC128 micewe typically observed a reduction in dendritic arborization, with DCXstaining predominately confined to the cell bodies (Figs. 3B,C), furthersuggesting that neuronal maturation is compromised in the DG of theYAC128 HD transgenic mouse model.
In addition, the endogenous marker NeuroD, a helix–loop–helixtranscription factor which has been shown to be sufficient to promoteneuronal differentiation in adult hippocampal neural progenitors(Hsieh et al., 2004) and is involved in granule cell development (Liuet al., 2000; Pleasure et al., 2000), was utilized in this study.Interestingly, NeuroD has also been shown to interact with bothnormal and mutant huntingtin (Marcora et al., 2003). NeuroD has anexpression pattern very similar to DCX (Steiner et al., 2006; Breuniget al., 2007; Borgs et al., 2009; Attardo et al., 2010) and comparable tothe results obtained with DCX, there was a dramatic and significantreduction in the number of NeuroD-positive cells in the DG of YAC128compared to theirWT controls alongwith an age-related decline in thenumber of NeuroD-positive cells (two-factor ANOVA: genotype:pb0.0001, F(1,80)=116.71; age: pb0.0001, F(3,80)=506.77; age×genotype: p=0.05, F(3,80)=2.70). Indeed, at 18 months of ageYAC128 mice show 71% less NeuroD-positive neuroblasts whencompared with their WT littermate controls (Figs. 3D–F).
YAC128 mice exhibit a reduction in overall adult hippocampalneurogenesis
To examine the rate of survival of the newly born cells in thehippocampal DG of YAC128 and WT mice, eight month-old andeleven month-old mice were injected with BrdU (every 12 h, during3 days) and allowed to survive for an additional 4 weeks before beingsacrificed (i.e., at nine and twelve months respectively). Althoughwith this procedure we cannot exclude the possibility that somedilution of the BrdU could have occurred, such factor is more of aconcern when low doses of BrdU are used. Indeed, low doses of BrdUhave been found not to be saturating in the adult brain, while higherdoses such as the one used in the present study label cells moreuniformly and in large enough numbers to enable quantitativeanalyses such as this (Christie and Cameron, 2006). We observed anage-related decline in the total number of BrdU-labeled cells thatwere present 4 weeks after the BrdU pulse (Fig. 4A), which isconsistent with previous literature (Kempermann et al., 1998).Importantly, this number was significantly smaller in YAC128compared to WT mice at both 9 and 12 months of age (two-factorANOVA: genotype: p=0.0008, F(1,34)=13.63; age: p=0.0004, F(1,34)=15.32; age×genotype: p=0.67, F(1,34)=0.19).
By comparing the number of BrdU-positive cells that wereproliferating in the DG 12 h after the last BrdU injection (Fig. 1A)with the number of BrdU-positive cells that were still present 4 weeksafter the last BrdU injection (Fig. 4A), we observed a drastic reductionin the number of BrdU-positive cells in both genotypes (e.g., at9 months of age, WT: 12 h post-injection=401.3 cells versus 4 weekspost-injection=137.3 cells; YAC128: 12 h post-injection=322.2 cellsversus 4 weeks post-injection=110.7 cells). Such dramatic reduction

Fig. 4. Decreased hippocampal neurogenesis in YAC128 mice. 8- and 11-month-old mice were sacrificed 4 weeks after the last BrdU injection (i.e., at 9- and 12-months of agerespectively) to allow for differentiation of the proliferating cells that incorporated BrdU. (A) Significant effects of age (9 versus 12 months of age) and genotype (WT versus YAC128)were found regarding the number of BrdU-labeled cells present in the DG 4 weeks after the last BrdU administration (two-factor ANOVA: genotype: p=0.0008, F(1,34)=13.63; age:p=0.0004, F(1,34)=15.32; age×genotype: p=0.67, F(1,34)=0.19). (B) The percentage of newly born cells (that incorporated BrdU) that co-express the mature neuronal markerNeuN is significantly lower in YAC128 mice when compared to their WT littermates (two-factor ANOVA: genotype: p=0.02, F(1,36)=5.83; age: p=0.11, F(1,36)=2.61;age×genotype: p=0.68, F(1,36)=0.18). (C) An estimation of overall neurogenesis based on the total number of BrdU-positive cells that survived the 4-week period multipliedby the proportion of BrdU-positive cells that acquired a mature neuronal phenotype indicates a significant decrease in the overall number of new neurons produced at both 9and 12 months of age in YAC128 mice (two-factor ANOVA: genotype: p=0.003, F(1,32)=10.45; age: p=0.016, F(1,32)=6.47; age×genotype: p=0.90, F(1,32)=0.02).(D) Representative confocal image of the YAC128 DG showing mature granule neurons labeled with NeuN (green) and BrdU-positive cells (red) that survived over 4 weeks. Arrowsindicate BrdU+/NeuN− immunopositive cells; arrowhead indicates a BrdU+/NeuN+immunopositive cell. (E) Representative example of a cell immunopositive for both BrdU(red) and NeuN (green) present in the DG of a YAC128 mouse at 9 months of age. Data presented as means±SEM. Scale bars=50 μm (D), 5 μm (E).
254 J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
is thought to be mainly due to cell death. Thus, by calculating the ratiobetween the number of BrdU-labeled cells 12 h post-injection and thenumber of BrdU-labeled cells 4 weeks post-injection, it was possibleto estimate an approximate rate of cell survival for both the 9- and12-month time-points (assuming that factors such as cell cyclelength and proliferation rate of the cells that incorporated BrdU arerelatively constant among mice of the same genotype). Identicalsurvival rateswere present in bothWT (34%) and YAC128 (34%)miceat 9 months of age, as well as at 12 months of age (WT=23%;YAC128=23%). Thus, although the total number of BrdU-positivecells present in YAC128mice both at 12 h and 4 weeks after the BrdUpulse was significantly smaller than that of their age-matched WTcontrols (Figs. 1A and 4A), the ratio between these two measure-mentswas the same between the two genotypes. This result indicatesthat although the pool of proliferating cells is significantly smallerin YAC128 mice, the rate of survival of the existing proliferating cellsis approximately the same in both WT and YAC128 mice.
By waiting 4 weeks after the last BrdU injection to perform ourhistological analysis, we were also able to determine the phenotypes
of the new cells that incorporated BrdU. Using confocal analysis, wefound that NeuN, a widely used marker of terminally differentiatedneurons (for review see Kempermann et al., 2004), and BrdUwere co-expressed in 18% of the cells from WT animals and 13% of cells fromYAC128 mice at 9 months of age, while about 15% in WT and 7% inYAC128 mice co-expressed NeuN At 12 months of age. A two-factorANOVA revealed that the differences between the percentages ofBrdU/NeuN co-labeled neurons in YAC128mice and their WT controlswere statistically significant (two-factor ANOVA: genotype: p=0.02,F(1,36)=5.83; age: p=0.11, F(1,36)=2.61; age×genotype:p=0.68, F(1,36)=0.18) (Figs. 4B,D,E). Overall neurogenesis, calcu-lated by multiplying the numbers of BrdU-positive cells that survivedthe 4-week period by the proportion of BrdU-positive cells that co-expressed NeuN, was significantly reduced with age and also betweengenotypes (two-factor ANOVA: genotype: p=0.003, F(1,32)=10.45;age: p=0.016, F(1,32)=6.47; age×genotype: p=0.90, F(1,32)=0.02) (Fig. 4C). Therefore, although the rate of survival was similarbetween genotypes, the reduction in the number of proliferating cells(Figs. 1 and 2A–C) and immature neurons (Fig. 3) in the YAC128 mice

255J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
translated into an overall decrease in the number of new neuronsreaching a mature stage.
YAC128 mice exhibit normal proliferation in the subventricular zone
In order to address if there are any significant differences in cellproliferation in the SVZ (the second neurogenic region in the adultbrain), immunohistochemistry for the endogenous cell proliferationmarkers Ki-67 (Fig. 5A) and PCNA (Fig. 5D) was performed in both 9-and 12-month-old YAC128 mice and their WT controls. Consistentwith previous literature (for review see Zhao et al., 2008), therewas anage-related decline in the total number of proliferating cells in the SVZof both YAC128 and WT mice. However, no significant differences incell proliferation were observed between genotypes at either 9 or12 months of age with both Ki-67 (two-factor ANOVA: genotype:p=0.73, F(1,41)=0.13; age: pb0.0001, F(1,41)=109.52; age×gen-otype: p=0.92, F(1,41)=0.01) and PCNA (two-factor ANOVA:genotype: p=0.25, F(1,41)=1.39; age: pb0.0001, F(1,41)=19.36;age×genotype: p=0.67, F(1,41)=0.18). The number of neuroblastsin the SVZ was also quantified by immunohistochemistry for theimmatureneuronalmarkerDCX, and at 12 months of age YAC128micedisplayed similar numbers of neuroblasts within the SVZ as their WTcontrols (WT: 7879.4±831.2 DCX-positive cells, YAC128: 8515.3±441.9 DCX-positive cells, Student's t-test p=0.51). Therefore, bothproliferation and differentiation appear unaltered in the SVZ ofYAC128 HD mice compared to WT littermates.
Fig. 5. No differences in cell proliferation in the subventricular zone between YAC128 mice atheir WT littermates was assessed by immunohistochemistry for the endogenous cell cycle mproliferating cells with increasing age in the SVZ of both YAC128 and WT mice (A,D). Howe(two-factor ANOVA: genotype: p=0.73, F(1,41)=0.13; age: pb0.0001, F(1,41)=109.52; aggenotype: p=0.25, F(1,41)=1.39; age: pb0.0001, F(1,41)=19.36; age×genotype: p=0.67(B,C) and PCNA (E,F) immunohistochemistry in WT (B,E) and YAC128 mice (C,F) at 9 mont
Discussion
Neurogenesis continues tooccur robustly in the adult brain primarilyin two specific regions, the SVZ and the SGZ of the DG in thehippocampus (for review see Zhao et al., 2008). Adult neurogenesis inthe hippocampus has been implicated in various cognitive processessuch as learning andmemory (for review see Kempermann, 2008), andit is possible that alterations in theneurogenic processmay contribute tosome of the cognitive deficits that characterize several neurodegener-ative disorders (for review see Lie et al., 2004; Mohapel and Brundin,2004; Gil-Mohapel et al., 2010). Thus, the possibility of restoring brainfunction by promoting neurogenesis in HD brains has emerged as apotential therapeutic option (for reviews see Mohapel and Brundin,2004; Gil and Rego, 2009; Gil-Mohapel et al., 2010).
In this study, we report an age-related decline in hippocampal cellproliferation, both in YAC128 mice and in WT littermates. Theprogressive reduction in hippocampal cell proliferation in WT miceis in agreement with previous reports showing reduced cellproliferation in the rodent DG with ageing (Bizon et al., 2004;Drapeau et al., 2007; Ahlenius et al., 2009; Ben Abdallah et al., 2010).However, YAC128 mice exhibited a small but significant reduction inhippocampal proliferation when compared to WT mice as assessedwith BrdU and Ki-67. The fact that this significant reduction in thenumber of proliferating cells was observed with these two markersbut not with PCNA (e.g., at 18 months of age we observed a 26%significant reduction in the number of BrdU-positive cells, a 26%significant reduction in the number of Ki-67-positive cells, and a 6%
nd WT controls. Cell proliferation in the SVZ of 9- and 12-month-old YAC128 mice andarkers Ki-67 (A–C) and PCNA (D–F). There was a progressive decline in the number of
ver, no significant differences between the genotypes in the number of both (A) Ki-67-e×genotype: p=0.92, F(1,41)=0.01) and (D) PCNA-positive cells (two-factor ANOVA:, F(1,41)=0.18) were observed. Representative sections of the SVZ processed for Ki-67hs of age. Data presented as means±SEM. Scale bars=50 μm.

256 J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
non-significant trend towards a reduction in the number of PCNA-positive cells) might reflect differences in cell cycle kinetics betweenWT and YAC128 mice. Indeed, it is possible that the ratio between thenumber of cells in G0 and the number of cells in all phases of the cellcycle is higher in YAC128 mice when compared with their WTcontrols. Thus, even though YAC128 mice have significantly less cellsin the active phases of the cell cycle (as assessed by Ki-67 and BrdU),an increase in the number of cells in the G0 phase mightcounterbalance this reduction and account for the non-significantdifferences in the number of PCNA-positive cells that were observedbetween YAC128 and WT mice. In support of this hypothesis, wild-type huntingtin is known to interact with the cell cycle machinery (forreview see Li and Li, 2004), as well as with the centrosome andmicrotubules (Gutekunst et al., 1995; Hoffner et al., 2002), the latterthrough interactions with ß-tubulin. Moreover, the HEAT (huntingtin,elongation factor 3, protein phosphatase 2A, target of rapamycin 1)repeat motifs present in the huntingtin sequence (Andrade andBork, 1995) are also found in a number of proteins that are involvedin mitotic progression, chromosomal dynamics and segregation(Neuwald and Hirano, 2000) and several of these proteins exerttheir functions via microtubules. Furthermore, a recent study hasshown that in vitro RNA interference-mediated silencing of wild-typehuntingtin in cells disrupts spindle orientation by mislocalizing thep150(Glued) subunit of dynactin, dynein, and the large nuclearmitotic apparatus (NuMA) protein, resulting in increased apoptosis.On the other hand, in vivo inactivation of wild-type huntingtin by RNAinterference or by ablation of the Hdh gene affects spindle orientationand cell fate of cortical progenitors of the ventricular zone in mouseembryos (Godin et al., 2010). Therefore, it is possible that mutanthuntingtin might induce a similar dysregulation of the mitoticmachinery thus underlying the proposed alteration in cell cyclekinetics observed in YAC128 mice. Future studies are thus warrantedin order to confirm this hypothesis. Importantly, a reduction in cellproliferation has also been observed in the R6 lines of HD transgenicmice (Gil et al., 2004; Lazic et al., 2004), with a 66% reduction in thenumber of BrdU-labeled cells being reported in end-stage R6/2 mice(Gil et al., 2005), which might reflect the severity of the phenotypecharacteristic of this truncated model.
In the present study we found a dramatic reduction in the numberof immature neurons in the YAC128 DG (e.g., at 18 months of age weobserved a 63% significant reduction in the number of DCX-positivecells and a 71% significant reduction in the number of NeuroD-positivecells). Similar results were observed in the R6/2 mouse model of HD,in which a 65% decrease in the number of DCX-positive cells wasobserved in the DG of 10 week-old mice (Gil et al., 2005). In thepresent study, the survival rate of BrdU-labeled cells was similarbetween YAC128 and WT mice (34% at 9 months and 23% at12 months of age). These low survival rates are in agreement withthe massive reduction in the number of BrdU-labeled cells commonlyreported after the first days up to more than 4 weeks post-injection,and is thought to be due to cell death (Dayer et al., 2003; Gould et al.,1999). Nevertheless, we found a significant reduction in the totalnumber of surviving BrdU-labeled cells in YAC128 mice whencompared to their WT littermates and this resulted in a decrease inthe proportion of surviving newly born cells that became neurons.The study by Gil et al. (2005) found no significant differences betweenR6/2 and WT mice in the proportion of surviving BrdU-labeled cellsthat developed a neuronal phenotype, and Lazic et al. (2004) failed todetect any cells co-labeled for BrdU and NeuN in R6/1 mice.Interestingly, in agreement with the observations made by Gil et al.(2005), we also noted morphological differences between YAC128and WT immature neurons in the DG of the hippocampus. Indeed,YAC128 neuroblasts appear atrophied and show a decrease indendritic arborization when compared with those present in WTmice. Striatal and cortical morphological changes have also beenreported in other mouse models of HD, including loss of spines and
reduction of somatic areas and dendritic fields (Spires et al., 2004;Laforet et al., 2001), and similar features have also been observed inhuman HD brains (Vonsattel and DiFiglia, 1998).
Taken together, our results strongly suggest that although adecrease in the pool of proliferating cells may contribute to the overallreduction in hippocampal neurogenesis, an intrinsic impairment inthe actual differentiation stage is probably a major contributor to theobserved changes in neurogenesis in the YAC128 mice. Indeed, therelatively small decrease in cell proliferation (observedwith BrdU andKi-67) alone cannot account for the much greater reduction in celldifferentiation (assessed with DCX and NeuroD) that was observed inYAC128mice as comparedwith theirWT controls. It can be speculatedthat this deficit would correspond to an abnormal transition fromType-2a to Type-2b progenitor cells, when the first markers of theneuronal lineage (such as NeuroD and DCX) start being expressed(reviewed in Kempermann et al., 2004). However, we cannot excludethe possibility that the transitions from Type-2b to Type-3 progenitorcells and from Type-3 progenitor cells to early post-mitotic immatureneurons are also compromised, as NeuroD and DCX continue beingexpressed throughout these stages of the neurogenic process(reviewed in Kempermann et al., 2004). A proper quantification ofthe percentages of Types-2a, -2b, and -3 progenitor cells and of post-mitotic immature neurons will provide further insight on the stage(s)of the neurogenic process that are directly affected by mutanthuntingtin in the YAC128 transgenic mice.
Differences in adult hippocampal neurogenesis between truncated andfull-length HD transgenic mouse models
The discrepancies in the changes in hippocampal neurogenesisobserved in the R6 lines (Gil et al., 2004; Lazic et al., 2004) and theYAC128mice are likely due todifferences between themodels used. TheR6 lines are truncated HD models that only express exon 1 of the HDgene (i.e., approximately 3% of the entire gene) with a long tail of CAGrepeats (approximately 115 and 150 repeats in the R6/1 and R6/2 linesrespectively) (Mangiarini et al., 1996). As a result of their truncated HDconstruct, the R6 mice only express N-terminal huntingtin fragments,which have been suggested to be the toxic component of the mutantprotein (for review see Gil and Rego, 2008). Consequently, these micedisplay an accelerated phenotype (Mangiarini et al., 1996) with limitedstriatal cell loss (Iannicola et al., 2000; Turmaine et al., 2000; Yu et al.,2003; Stack et al., 2005). Therefore, the R6 lines may not reflect theactual adult-onset human condition, being more representative of thejuvenile and infantile cases ofHD(for reviewseeGil andRego, 2009). Onthe other hand, the YAC128 model expresses the full-length human HDgene under the control of its endogenous promoter and regulatoryelements and presents a slower progression of the HD phenotype withmarked striatal cell loss in the end-stages of the disease (Slow et al.,2003). Interestingly, a recent studyhas shown that YACmice expressingfull-length mutant huntingtin that is resistant to cleavage by caspase-6do not develop motor deficits and striatal neurodegeneration and areresistant to excitotoxicity induced by quinolinic acid (Graham et al.,2006), further suggesting that the generation of the N-terminalfragments likely represents the initial step in the degeneration process.Furthermore, the depressive-like phenotype of the YAC128 mice wasalso completely rescued in these caspase-6 resistant YAC128 mice(Pouladi et al., 2009). On the other hand, R6 mice only express thetruncated N-terminal fragments, therefore bypassing the caspase- andcalpain-mediated proteolysis of full-length mutant huntingtin, a factorthatmight contribute to the faster disease progression observed in thesemice (Mangiarini et al., 1996). Thus, the aforementioned differencesbetween the truncated R6 and the full-length YAC128 models mayaccount for the observeddiscrepancies in hippocampal cell proliferationbetween these two HD transgenic mouse models (i.e., a markedreduction in the case of R6 mice and a less severe decrease in the caseof the YAC128 mice). However, the fact that both transgenic models

257J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
show a severe reduction in the number of immature neurons in the DGstrongly suggests that an impairment of the differentiation stage of theneurogenic process might be a common feature in HD. Studies in thehippocampalDGof humanHDpostmortem tissue are thuswarranted inorder to confirm this hypothesis.
Given the neurodegenerative nature of HD, there is concern thatthe observed decrease in the number of immature neurons could bedue to an overall loss of DG granule cells. Volume loss in thehippocampus of HD patients has been reported in a few cases (Rosaset al., 2003), while no significant differences in hippocampal neuronalnumerical densities between HD patients and controls were observedby Spargo et al. (1993). In addition, no neuronal loss in the DG of R6/2HD mice was observed as assessed by counting the total number ofmature neurons by stereology (Gil et al., 2004). In the YAC models,there is no obvious difference in gross organization (Hodgson et al.,1999) or change in hippocampal volume or the number of estimatedhippocampal neurons compared to WT mice (Van Raamsdonk et al.,2005). Nonetheless, there is strong electrophysiological and beha-vioural evidence for hippocampal dysfunction in the transgenictruncated HD mouse models (Murphy et al., 2000). Disturbedneurogenesis could thus contribute, at least in part, to this dysfunction.
Possible mechanisms underlying the deficits in adult hippocampalneurogenesis observed in HD transgenic mouse models
YAC128 mice have been found to display cognitive deficits and adepressive-like behaviour as early as 3 months of age (Pouladi et al.,2009). Importantly, both cognitive disturbances (specifically learningand memory) and depression have been correlated with a reductionin hippocampal neurogenesis (Gould et al., 1999; reviewed byMirescu and Gould, 2006). Chronic treatment with selective serotoninreuptake inhibitor (SSRI) antidepressants has been shown to lead toincreased brain-derived neurotrophic factor (BDNF) levels (Nibuya etal., 1995), and BDNF signaling has been shown to enhance the survivalof newborn neurons in the DG (Zigova et al., 1998). On the other hand,brain BDNF levels are decreased in HD patients (Zuccato and Cattaneo,2007), as well as R6 mice (Luthi-Carter et al., 2002; Canals et al., 2004;Spires et al., 2004), and YAC mice expressing the full-length humanHD gene with 72 CAG repeats (Zuccato et al., 2001). Future studies arewarranted in order to determine whether the same reduction is alsoobserved in YAC128 mice. Given the aforementioned evidence, SSRIsmay be neuroprotective in HD. In fact, treatment with an SSRI hasrecently been found to increase the survival, improve motor deficitsand delay brain atrophy in the N171-82Q mouse model of HD (Duanet al., 2008). Furthermore, R6/1 mice were shown to develop adepressive-like phenotype concomitant with a reduction in hippo-campal neurogenesis, which were both rescued by treatment with theSSRI fluoxetine (Grote et al., 2005; Pang et al., 2009), while theantidepressant sertraline was shown to improve the phenotype,rescue the deficit in hippocampal neurogenesis, increase hippocampaland striatal BDNF levels, and improve the survival of R6/2 mice (Penget al., 2008). However, antidepressant treatment had no effect on thedevelopment of the YAC128 depressive-like behaviour (Pouladi et al.,2009). As stated earlier, differences in the huntingtin constructsexpressed as well as in the background strain may account for thediscrepant results obtained with SSRIs in R6 and YAC128 mice. Insupport, behavioural effects and increased SGZ proliferation inresponse to fluoxetine have been found to depend on the mousegenetic background (Miller et al., 2008). Moreover, given thattreatment with SSRIs has been shown to ameliorate depression andneurogenesis in some transgenic models of HD, serotonin dysregula-tion may also play a role in these processes. In fact, reduced levels ofserotonin have been shown in R6/2 mice (Reynolds et al., 1999) anddecreased serotonin receptor binding sites have been reported inpatients with HD (Steward et al., 1993). Also of interest, recent meta-analysis data on antidepressant medication indicates that the
magnitude of benefit increases with the severity of depression, andmay be minimal in patients with mild or moderate symptoms(Fournier et al., 2010). Therefore, this therapeutic approach couldstill have beneficial results in HD patients showing a severe depressivephenotype.
An alternative mechanism that might contribute to the observedreduction in hippocampal neurogenesis in YAC128 mice involvesthe regulation of the transcription factor NeuroD by huntingtin.Normal huntingtin and the huntingtin-associated proteins hunting-tin-associated protein 1 (HAP1) and mixed-lineage kinase 2 (MLK2)modulate and stimulate the activity of NeuroD (Marcora et al., 2003),which is crucial for the development of a functional DG (Liu et al.,2000). Interestingly, we found a drastic decrease in the number ofNeuroD-positive cells in the DG of YAC128 mice. Interestingly, asimilar decrease in the expression of NeuroD has been recently foundin the DG of 11 week-old R6/2 HD mice (Fedele et al., 2010). Furtherexperiments are needed in order to conclude if a compromisedNeuroD pathway (due, for instance, to its abnormal interaction withmutant huntingtin) is contributing to a reduction in the generation ofnew neurons. In agreement with this hypothesis, the reduction inNeuroD expression that was observed in the R6/2 DG was notaccompanied by a decrease in the number of granule neuronalprogenitors (Fedele et al., 2010). These results strongly suggest that acompromised transition to the neuroblast stage is indeed underlyingthe reduction in hippocampal neurogenesis observed in these HDtransgenic mice.
In summary, mutant huntingtin-induced disturbances in keyregulators of the local DG neurogenic niche (i.e., a dysregulation ofthe pathways involving BDNF and/or NeuroD) may result in anenvironment not conducive to the production of new neurons in theHD brain.
Differences in adult SVZ neurogenesis between HD transgenic mousemodels and human HD
In contrast to the decrease in neurogenesis observed in the DG, wenoted no significant differences in both SVZ proliferation anddifferentiation in YAC128 mice, which is consistent with previousstudies in R6/2 (Gil et al., 2005; Phillips et al., 2005) and R6/1 mice(Lazic et al., 2006). The reasons for this specific impairment inhippocampal neurogenesis are currently not clear. An interestingexplanation involves a reduction in the mitochondrial enzyme alpha-ketoglutarate-dehydrogenase complex (KGDHC), which has beenimplicated in the pathology of HD in humans (Klivenyi et al., 2004).Indeed, a recent study found that mice deficient in various subunits ofKGDHC show increased levels of lipid peroxidation as well as reducednumbers of neuroblasts and proliferating cells in the adult DG,although no differences in SVZ proliferation were found (Calingasanet al., 2008). In agreement with these findings, it was recently shownthat the deficiencies in KGDHC observed in the malonate lesionmodel of HD, did not alter the lesion-induced increase in migrationof DCX-positive cells from the SVZ into the ipsilateral striatum(Calingasan et al., 2008). These findings might explain, at least in part,why a deficit in neurogenesis is specifically observed in the DG (andnot in the SVZ) of transgenic mouse models of HD. Further studies arethus warranted in order to determine whether this mitochondrialenzyme is also compromised in R6 and YAC mice.
However, the absence of alterations in SVZ neurogenesis that hasbeen consistently reported in HD transgenic mouse models is incontrast to the findings in human HD postmortem tissue, where anincrease in cell proliferation estimated by PCNA immunohistochem-istry has been reported (Curtis et al., 2003). This discrepancy couldbe due to factors such as higher numbers of CAG repeats andfrequency of inclusions in the transgenic mouse models as comparedto those in HD patients. Indeed, the human HD striatum ischaracterized by a lower frequency of inclusions than that observed

258 J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
in R6/2mice (DiFiglia et al., 1997; Meade et al., 2002). This differencemight suggest that the amount of free N-terminal toxic huntingtinfragments present in the human striatum is higher than in the R6/2mice (where the N-terminal fragments are primarily recruited intoinclusions). This may underlie the striatal cell loss that is character-istically observed in the human HD brain. Thus, the increase in cellproliferation that was observed in the human SVZ might occur inresponse to the greater number of degenerating neurons that ispresent in the adjacent striatum (Curtis et al., 2003). In agreement,no overt cell loss is found in the striatum of R6 mice (Iannicola et al.,2000; Turmaine et al., 2000; Yu et al., 2003; Stack et al., 2005), whichmight account for the absence of increased SVZ proliferation in thesemice. However, YAC128 mice replicate the striatal loss that occurs inthe HD brain, and yet no significant differences in SVZ proliferationwere observed in these HD mice. Although the reasons for thisdiscrepancy are currently unclear, the increase in SVZ proliferationthat was observed in the human HD brain was inferred based on anincrease in the number of PCNA-positive cells present in this region.While PCNA is indeed a marker of cell proliferation (for review seeChristie and Cameron, 2006), it has also been suggested that cellsundergoing DNA repair will express this protein (Tomasevic et al.,1998). Therefore caution should be taken when interpreting cellproliferation results that are exclusively based on PCNA immuno-histochemistry and the use of alternative markers of cell prolifera-tion is recommended to further validate these results.
Conclusions
In conclusion, the progressive decline in hippocampal neurogen-esis observed in the YAC128 mice is a novel neuropathological featurethat occurs early in the progression of the disease, before theappearance of overt motor and cognitive symptoms. Since neurogen-esis may play a role in hippocampal function, it is possible thattherapies aimed at restoring hippocampal neurogenesis may bebeneficial in alleviating the cognitive deficits characteristic of thesemice and the human condition.
Acknowledgments
J.M.S. is supported by scholarships from the Natural Sciences andEngineering ResearchCouncil of Canada (NSERC) and theMichael SmithFoundation for Health Research (MSFHR). J.G.M. acknowledges post-doctoral funding from the Portuguese Foundation for Science andTechnology (FCT) (SFRH/BPD/28799/2006) and NSERC. M.A.P. is therecipient of the BC Innovation Council Ripples of Hope Award inBiotechnology & Entrepreneurship, and doctoral awards from theCanadian Institute of Health Research (CIHR) and MSFHR. M.R.H. issupported by grants from the CIHR, the Huntington Society of Canada,the Huntington's Disease Society of America, and the CHDI Foundation.M.R.H. is a Killam University Professor and holds a Canada ResearchChair in Human Genetics. B.R.C. is a Michael Smith Senior Scholar and issupported by grants from CIHR and NSERC.
References
Ahlenius, H., Visan, V., Kokaia, M., Lindvall, O., Kokaia, Z., 2009. Neural stem andprogenitor cells retain their potential for proliferation and differentiation intofunctional neurons despite lower number in aged brain. J. Neurosci. 29, 4408–4419.
Andrade, M.A., Bork, P., 1995. HEAT repeats in the Huntington's disease protein. Nat.Genet. 11, 115–116.
Attardo, A., Fabel, K., Krebs, J., Haubensak, W., Huttner, W.B., Kempermann, G., 2010.Tis21 expression marks not only populations of neurogenic precursor cells but alsonew postmitotic neurons in adult hippocampal neurogenesis. Cereb. Cortex 20,304–314.
BenAbdallah,N.M., Slomianka, L., Vyssotski, A.L., Lipp,H.P., 2010. Early age-related changesin adult hippocampal neurogenesis in C57 mice. Neurobiol. Aging 31, 151–161.
Bizon, J.L., Lee, H.J., Gallagher, M., 2004. Neurogenesis in a rat model of age-relatedcognitive decline. Aging Cell 3, 227–234.
Borgs, L., Beukelaers, P., Vandenbosch, R., Nguyen, L., Moonen, G., Maquet, P., Albrecht,U., Belachew, S., Malgrange, B., 2009. Period 2 regulates neural stem/progenitor cellproliferation in the adult hippocampus. BMC Neurosci. 10, 30.
Breunig, J.J., Silbereis, J., Vaccarino, F.M., Sestan, N., Rakic, P., 2007. Notch regulates cellfate and dendrite morphology of newborn neurons in the postnatal dentate gyrus.Proc. Natl Acad. Sci. USA 104, 20558–20563.
Brown, J.P., Couillard-Després, S., Cooper-Kuhn, C.M., Winkler, J., Aigner, L., Kuhn, H.G.,2003. Transient expression of doublecortin during adult neurogenesis. J. Comp.Neurol. 467, 1–10.
Calingasan, N.Y., Ho, D.J., Wille, E.J., Campagna, M.V., Ruan, J., Dumont, M., Yang, L., Shi,Q., Gibson, G.E., Beal, M.F., 2008. Influence of mitochondrial enzyme deficiency onadult neurogenesis in mouse models of neurodegenerative diseases. Neuroscience153, 986–996.
Cameron, H.A., McKay, R., 1998. Stem cells and neurogenesis in the adult brain. Curr.Opin. Neurobiol. 8, 677–680.
Canals, J.M., Pineda, J.R., Torres-Peraza, J.F., Bosch, M., Martín-Ibañez, R., Muñoz, M.T.,Mengod, G., Ernfors, P., Alberch, J., 2004. Brain-derived neurotrophic factorregulates the onset and severity of motor dysfunction associated with enkepha-linergic neuronal degeneration in Huntington's disease. J. Neurosci. 24, 7727–7739.
Christie, B.R., Cameron, H.A., 2006. Neurogenesis in the adult hippocampus.Hippocampus 16, 199–207.
Cooper-Kuhn, C.M., Kuhn, H.G., 2002. Is it all DNA repair? Methodological considera-tions for detecting neurogenesis in the adult brain. Brain Res. Dev. Brain Res. 134,13–21.
Crews, L., Mizuno, H., Desplats, P., Rockenstein, E., Adame, A., Patrick, C., Winner, B.,Winkler, J., Masliah, E., 2008. Alpha-synuclein alters Notch-1 expression andneurogenesis in mouse embryonic stem cells and in the hippocampus of transgenicmice. J. Neurosci. 28, 4250–4260.
Curtis, M.A., Penney, E.B., Pearson, A.G., van Roon-Mom, W.M., Butterworth, N.J.,Dragunow, M., Connor, B., Faull, R.L., 2003. Increased cell proliferation andneurogenesis in the adult human Huntington's disease brain. Proc. Natl Acad. Sci.USA 100, 9023–9027.
Dayer, A.G., Ford, A.A., Cleaver, K.M., Yassaee, M., Cameron, H.A., 2003. Short-term andlong-term survival of new neurons in the rat dentate gyrus. J. Comp. Neurol. 460,563–572.
DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P., Aronin, N.,1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophicneurites in brain. Science 277, 1990–1993.
Drapeau, E., Montaron, M.F., Aguerre, S., Abrous, D.N., 2007. Learning-induced survivalof new neurons depends on the cognitive status of aged rats. J. Neurosci. 27,6037–6044.
Duan, W., Peng, Q., Masuda, N., Ford, E., Tryggestad, E., Ladenheim, B., Zhao, M., Cadet, J.L.,Wong, J., Ross, C.A., 2008. Sertraline slows disease progression and increasesneurogenesis in N171-82Q mouse model of Huntington's disease. Neurobiol. Dis. 30,312–322.
Duff, K., McGuigan, A., Huxley, C., Schulz, F., Hardy, J., 1994. Insertion of a pathogenicmutation into a yeast artificial chromosome containing the human amyloidprecursor protein gene. Gene Ther. 1, 70–75.
Ehrnhoefer, D.E., Butland, S.L., Pouladi, M.A., Hayden, M.R., 2009. Mouse models ofHuntington disease: variations on a theme. Dis Model Mech 2, 123–129.
Eriksson, P.S., Perfilieva, E., Björk-Eriksson, T., Alborn, A.M., Nordborg, C., Peterson, D.A.,Gage, F.H., 1998. Neurogenesis in the adult human hippocampus. Nat. Med. 4,1313–1317.
Fedele, V., Roybon, L., Nordström, U., Li, J.Y., Brundin, P., 2010. Neurogenesis in R6/2mouse model of Huntington's disease is impaired at the level of NeuroD1.Neuroscience [Electronic publication ahead of print].
Fournier, J.M., DeRubeis, R.J., Hollon, S.D., Dimidjian, S., Amsterdam, J.D., Shelton, R.C.,Fawcett, J., 2010. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA 303, 47–53.
Gil, J.M., Leist, M., Popovic, N., Brundin, P., Petersén, A., 2004. Asialoerythropoietin is noteffective in the R6/2 line of Huntington's disease mice. BMC Neurosci. 5, 17.
Gil, J.M., Mohapel, P., Araújo, I.M., Popovic, N., Li, J.Y., Brundin, P., Petersén, A., 2005.Reduced hippocampal neurogenesis in R6/2 transgenic Huntington's disease mice.Neurobiol. Dis. 20, 744–751.
Gil, J.M., Rego, A.C., 2008. Mechanisms of neurodegeneration in Huntington's disease.Eur. J. Neurosci. 27, 2803–2820.
Gil, J.M., Rego, A.C., 2009. The R6 lines of transgenic mice: a model for screening newtherapies for Huntington's disease. Brain Res. Rev. 59, 410–431.
Gil-Mohapel, J., Simpson, J., Christie, B.R., 2010. Modulation of adult neurogenesis byphysical exercise and environmental enrichment: insights for the treatment ofneurological disorders. In: Kunlin, J. (Ed.), Adult Neurogenesis and Central NervousSystem Diseases. Transworld Research Network, Kerala, India, pp. 125–150.
Godin, J.D., Colombo, K., Molina-Calavita, M., Keryer, G., Zala, D., Charrin, B.C., Dietrich,P., Volvert, M.L., Guillemot, F., Dragatsis, I., Bellaiche, Y., Saudou, F., Nguyen, L.,Humbert, S., 2010. Huntingtin is required for mitotic spindle orientation andmammalian neurogenesis. Neuron 67, 392–406.
Gould, E., Beylin, A., Tanapat, P., Reeves, A., Shors, T.J., 1999. Learning enhances adultneurogenesis in the hippocampal formation. Nat. Neurosci. 2, 260–265.
Graham, R.K., Deng, Y., Slow, E.J., Haigh, B., Bissada, N., Lu, G., Pearson, J., Shehadeh, J.,Bertram, L., Murphy, Z., Warby, S.C., Doty, C.N., Roy, S., Wellington, C.L., Leavitt, B.R.,Raymond, L.A., Nicholson, D.W., Hayden, M.R., 2006. Cleavage at the caspase-6 siteis required for neuronal dysfunction and degeneration due to mutant huntingtin.Cell 125, 1179–1191.
Grote, H.E., Bull, N.D., Howard, M.L., van Dellen, A., Blakemore, C., Bartlett, P.F., Hannan,A.J., 2005. Cognitive disorders and neurogenesis deficits in Huntington's diseasemice are rescued by fluoxetine. Eur. J. Neurosci. 22, 2081–2088.

259J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
Gutekunst, C.A., Levey, A.I., Heilman, C.J., Whaley, W.L., Yi, H., Nash, N.R., Rees, H.D.,Madden, J.J., Hersch, S.M., 1995. Identification and localization of huntingtin inbrain and human lymphoblastoid cell lines with anti-fusion protein antibodies.Proc. Natl Acad. Sci. USA 92, 8710–8714.
Heng, M.Y., Detloff, P.J., Albin, R.L., 2008. Rodent genetic models of Huntington disease.Neurobiol. Dis. 32, 1–9.
Hodgson, J.G., Agopyan,N.,Gutekunst, C.A., Leavitt, B.R., LePiane, F., Singaraja, R., Smith,D.J.,Bissada, N., McCutcheon, K., Nasir, J., Jamot, L., Li, X.J., Stevens, M.E., Rosemond, E.,Roder, J.C., Phillips, A.G., Rubin, E.M., Hersch, S.M., Hayden, M.R., 1999. A YAC mousemodel for Huntington's disease with full-length mutant huntingtin, cytoplasmictoxicity, and selective striatal neurodegeneration. Neuron 23, 181–192.
Hoffner, G., Kahlem, P., Djian, P., 2002. Perinuclear localization of huntingtin as aconsequence of its binding to microtubules through an interaction with beta-tubulin: relevance to Huntington's disease. J. Cell Sci. 115, 941–948.
Höglinger, G.U., Rizk, P., Muriel, M.P., Duyckaerts, C., Oertel, W.H., Caille, I., Hirsch, E.C.,2004. Dopamine depletion impairs precursor cell proliferation in Parkinsondisease. Nat. Neurosci. 7, 726–735.
Hsieh, J., Nakashima, K., Kuwabara, T., Mejia, E., Gage, F.H., 2004. Histone deacetylaseinhibition-mediated neuronal differentiation of multipotent adult neural progen-itor cells. Proc. Natl Acad. Sci. USA 101, 16659–16664.
Iannicola, C., Moreno, S., Oliverio, S., Nardacci, R., Ciofi-Luzzatto, A., Piacentini, M., 2000.Early alterations in gene expression and cell morphology in a mouse model ofHuntington's disease. J. Neurochem. 75, 830–839.
Jin, K., Peel, A.L., Mao, X.O., Xie, L., Cottrell, B.A., Henshall, D.C., Greenberg, D.A., 2004.Increased hippocampal neurogenesis in Alzheimer's disease. Proc. Natl Acad. Sci.USA 101, 343–347.
Kee N, Sivalingam S, Boonstra R, Wojtowicz JM (2002) The utility of Ki-67 and BrdU asproliferative markers of adult neurogenesis.
Kempermann, G., 2008. The neurogenic reserve hypothesis: what is adult hippocampalneurogenesis good for? Trends Neurosci. 31, 163–169.
Kempermann, G., Kuhn, H.G., Gage, F.H., 1998. Experience-induced neurogenesis in thesenescent dentate gyrus. J. Neurosci. 18, 3206–3212.
Kempermann, G., Wiskott, L., Gage, F.H., 2004. Functional significance of adultneurogenesis. Curr. Opin. Neurobiol. 14, 186–191.
Klivenyi, P., Starkov, A.A., Caligasan, N.Y., Gardian, G., Browne, S.E., Yang, L., Bubber, P.,Gibson, G.E., Patel, M.S., Beal, M.F., 2004. Mice deficient in dihydrolipoamidedehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropro-pionic acid neurotoxicity. J. Neurochem. 88, 1352–1360.
Laforet, G.A., Sapp, E., Chase, K., McIntyre, C., Boyce, F.M., Campbell, M., Cadigan, B.A.,Warzecki, L., Tagle, D.A., Reddy, P.H., Cepeda, C., Calvert, C.R., Jokel, E.S., Klapstein, G.J.,Ariano,M.A., Levine,M.S., DiFiglia,M., Aronin, N., 2001. Changes in cortical and striatalneurons predict behavioral and electrophysiological abnormalities in a transgenicmurine model of Huntington's disease. J. Neurosci. 21, 9112–9123.
Lazic, S.E., Grote, H., Armstrong, R.J., Blakemore, C., Hannan, A.J., van Dellen, A., Barker,R.A., 2004. Decreased hippocampal cell proliferation in R6/1 Huntington's mice.NeuroReport 15, 811–813.
Lazic, S.E., Grote, H.E., Blakemore, C., Hannan, A.J., van Dellen, A., Phillips, W., Barker, R.A.,2006. Neurogenesis in the R6/1 transgenic mouse model of Huntington's disease:effects of environmental enrichment. Eur. J. Neurosci. 23, 1829–1838.
Li, S.H., Li, X.J., 2004. Huntingtin–protein interactions and the pathogenesis ofHuntington's disease. Trends Genet. 20, 146–154.
Li, B., Yamamori, H., Tatebayashi, Y., Shafit-Zagardo, B., Tanimukai, H., Chen, S., Iqbal, K.,Grundke-Iqbal, I., 2008. Failure of neuronal maturation in Alzheimer diseasedentate gyrus. J. Neuropathol. Exp. Neurol. 67, 78–84.
Lie, D.C., Song, H., Colamarino, S.A., Ming, G.L., Gage, F.H., 2004. Neurogenesis in theadult brain: new strategies for central nervous system diseases. Annu. Rev.Pharmacol. Toxicol. 44, 399–421.
Liu, M., Pleasure, S.J., Collins, A.E., Noebels, J.L., Naya, F.J., Tsai, M.J., Lowenstein, D.H.,2000. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus andepilepsy. Proc. Natl Acad. Sci. USA 97, 865–870.
Luthi-Carter, R., Hanson, S.A., Strand, A.D., Bergstrom, D.A., Chun, W., Peters, N.L.,Woods, A.M., Chan, E.Y., Kooperberg, C., Krainc, D., Young, A.B., Tapscott, S.J., Olson,J.M., 2002. Dysregulation of gene expression in the R6/2 model of polyglutaminedisease: parallel changes in muscle and brain. Hum. Mol. Genet. 11, 1911–1926.
Malberg, J.E., Eisch, A.J., Nestler, E.J., Duman, R.S., 2000. Chronic antidepressanttreatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20,9104–9110.
Manev, H., Uz, T., Smalheiser, N.R., Manev, R., 2001. Antidepressants alter cell proliferationin the adult brain in vivo and in neural cultures in vitro. Eur. J. Pharmacol. 411, 67–70.
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M,Trottier Y, Lehrach H, Davies SW, Bates GP (1996). Exon 1 of the HD gene with anexpanded CAG repeat is sufficient to cause a progressive neurological phenotype intransgenic mice.
Marcora, E., Gowan, K., Lee, J.E., 2003. Stimulation of NeuroD activity by huntingtin andhuntingtin-associated proteins HAP1 and MLK2. Proc. Natl Acad. Sci. USA 100,9578–9583.
Meade, C.A., Deng, Y.P., Fusco, F.R., Del Mar, N., Hersch, S., Goldowitz, D., Reiner, A.,2002. Cellular localization and development of neuronal intranuclear inclusions instriatal and cortical neurons of R6/2 transgenic mice. J. Comp. Neurol. 449,241–269.
Miller, B.H., Schultz, L.E., Gulati, A., Cameron,M.D., Pletcher, M.T., 2008. Genetic regulationof behavioral and neuronal responses to fluoxetine. Neuropsychopharmacology 33,1312–1322.
Mirescu, C., Gould, E., 2006. Stress and adult neurogenesis. Hippocampus 16, 233–238.Miyata, T., Maeda, T., Lee, J.E., 1999. NeuroD is required for differentiation of the granule
cells in the cerebellum and hippocampus. Genes Dev. 13, 1647–1652.
Mohapel, P., Brundin, P., 2004. Harnessing endogenous stem cells to treat neurode-generative disorders of the basal ganglia. Parkinsonism Relat. Disord. 10, 259–264.
Murphy, K.P., Carter, R.J., Lione, L.A., Mangiarini, L., Mahal, A., Bates, G.P., Dunnett, S.B.,Morton,A.J., 2000. Abnormal synaptic plasticity and impaired spatial cognition inmicetransgenic for exon 1 of the human Huntington's disease mutation. J. Neurosci. 20,5115–5123.
Neuwald, A.F., Hirano, T., 2000. HEAT repeats associated with condensins, cohesins,and other complexes involved in chromosome-related functions. Genome Res. 10,1445–1452.
Nibuya, M., Morinobu, S., Duman, R.S., 1995. Regulation of BDNF and trkB mRNA in ratbrain by chronic electroconvulsive seizure and antidepressant drug treatments.J. Neurosci. 15, 7539–7547.
Pang, T.Y., Du, X., Zajac, M.S., Howard, M.L., Hannan, A.J., 2009. Altered serotoninreceptor expression is associated with depression-related behavior in the R6/1transgenic mouse model of Huntington's disease. Hum. Mol. Genet. 18, 753–766.
Paxinos, G., Franklin, K., 2001. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.Academic Press Inc., Elsevier, San Diego, CA, USA.
Peng, Q., Masuda, N., Jiang, M., Li, Q., Zhao, M., Ross, C.A., Duan, W., 2008. Theantidepressant sertraline improves the phenotype, promotes neurogenesis andincreases BDNF levels in the R6/2 Huntington's disease mouse model. Exp. Neurol.210, 154–163.
Petersen, Å., Gil, J., Maat-Schieman, M.L.C., Björkqvist, M., Tanila, H., Araújo, I.M., Smith,R., Popovic, N., Wierup, N., Norlén, P., Li, J.Y., Roos, R.A.C., Sundler, F., Mulder, H.,Brundin, P., 2005. Orexin loss in Huntington's disease. Hum. Mol. Genet. 14, 39–47.
Phillips, W., Morton, A.J., Barker, R.A., 2005. Abnormalities of neurogenesis in the R6/2mousemodel ofHuntington's disease are attributable to the invivomicroenvironment.J. Neurosci. 25, 11564–11576.
Pleasure, S.J., Collins, A.E., Lowenstein, D.H., 2000. Unique expression patterns of cell fatemolecules delineate sequential stages of dentate gyrus development. J. Neurosci. 20,6095–6105.
Pouladi, M.A., Graham, R.K., Karasinska, J.M., Xie, Y., Santos, R.D., Petersén, A., Hayden,M.R., 2009. Prevention of depressive behaviour in the YAC128 mouse model ofHuntington disease by mutation at residue 586 of huntingtin. Brain 132, 919–932.
Reynolds, G.P., Dalton, C.F., Tillery, C.L., Mangiarini, L., Davies, S.W., Bates, G.P., 1999.Brain neurotransmitter deficits in mice transgenic for the Huntington's diseasemutation. J. Neurochem. 72, 1773–1776.
Rosas, H.D., Koroshetz, W.J., Chen, Y.I., Skeuse, C., Vangel, M., Cudkowicz, M.E., Caplan,K., Marek, K., Seidman, L.J., Makris, N., Jenkins, B.G., Goldstein, J.M., 2003. Evidencefor more widespread cerebral pathology in early HD: an MRI-based morphometricanalysis. Neurology 60, 1615–1620.
Slow, E.J., van Raamsdonk, J., Rogers, D., Coleman, S.H., Graham, R.K., Deng, Y., Oh, R.,Bissada, N., Hossain, S.M., Yang, Y.Z., Li, X.J., Simpson, E.M., Gutekunst, C.A., Leavitt,B.R., Hayden, M.R., 2003. Selective striatal neuronal loss in a YAC128 mouse modelof Huntington disease. Hum. Mol. Genet. 12, 1555–1567.
Spargo, E., Everall, I.P., Lantos, P.L., 1993. Neuronal loss in the hippocampus inHuntington's disease: a comparison with HIV infection. J. Neurol. Neurosurg.Psychiatry 56, 487–491.
Spires, T.L., Grote, H.E., Garry, S., Cordery, P.M., VanDellen, A., Blakemore, C., Hannan, A.J.,2004. Dendritic spine pathology and deficits in experience-dependent dendriticplasticity in R6/1 Huntington's disease transgenic mice. Eur. J. Neurosci. 19,2799–2807.
Stack, E.C., Kubilus, J.K., Smith, K., Cormier, K., Del Signore, S.J., Guelin, E., Ryu, H.,Hersch, S.M., Ferrante, R.J., 2005. Chronology of behavioral symptoms andneuropathological sequela in R6/2 Huntington's disease transgenic mice. J. Comp.Neurol. 490, 354–370.
Steiner, B., Klempin, F., Wang, L., Kott, M., Kettenmann, H., Kempermann, G., 2006.Type-2 cells as link between glial and neuronal lineage in adult hippocampalneurogenesis. Glia 54, 805–814.
Steward, L.J., Bufton, K.E., Hopkins, P.C., Davies, W.E., Barnes, N.M., 1993. Reduced levelsof 5-HT3 receptor recognition sites in the putamen of patients with Huntington'sdisease. Eur. J. Pharmacol. 242, 137–143.
The Huntington's Disease Collaborative Research Group, 1993. A novel gene containinga trinucleotide repeat that is expanded and unstable on Huntington's diseasechromosomes. Cell 72, 971–983.
Tomasevic, G., Kamme, F., Wieloch, T., 1998. Changes in proliferating cell nuclearantigen, a protein involved in DNA repair, in vulnerable hippocampal neuronsfollowing global cerebral ischemia. Brain Res. Mol. Brain Res. 60, 168–176.
Turmaine, M., Raza, A., Mahal, A., Mangiarini, L., Bates, G.P., Davies, S.W., 2000.Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington'sdisease. Proc. Natl Acad. Sci. USA 97, 8093–8097.
van Praag, H., Shubert, T., Zhao, C., Gage, F.H., 2005. Exercise enhances learning andhippocampal neurogenesis in aged mice. J. Neurosci. 25, 8680–8685.
Van Raamsdonk, J.M., Pearson, J., Slow, E.J., Hossain, S.M., Leavitt, B.R., Hayden, M.R., 2005. Cognitive dysfunction precedes neuropathology and motor abnor-malities in the YAC128 mouse model of Huntington's disease. J. Neurosci. 25,4169–4180.
Vonsattel, J.P., DiFiglia, M., 1998. Huntington disease. J. Neuropathol. Exp. Neurol. 57,369–384.
Yu, Z.X., Li, S.H., Evans, J., Pillarisetti, A., Li, H., Li, X.J., 2003. Mutant huntingtin causescontext-dependent neurodegeneration inmicewithHuntington's disease. J. Neurosci.23, 2193–2202.
Zhao, B., Zhong, M., Jin, K., 2008. Neurogenesis and neurodegenerative diseases inhuman. Panminerva Med. 50, 55–64.
Zigova, T., Pencea, V., Wiegand, S.J., Luskin, M.B., 1998. Intraventricular administrationof BDNF increases the number of newly generated neurons in the adult olfactorybulb. Mol. Cell. Neurosci. 11, 234–245.

260 J.M. Simpson et al. / Neurobiology of Disease 41 (2011) 249–260
Zuccato, C., Cattaneo, E., 2007. Role of brain-derived neurotrophic factor inHuntington's disease. Prog. Neurobiol. 81, 294–330.
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B.R., Goffredo, D., Conti, L., MacDonald, M.E., Friedlander, R.M., Silani, V., Hayden, M.R., Timmusk, T., Sipione, S., Cattaneo, E.,2001. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease.Science 293, 493–498.
Glossary
ANOVA: analysis of varianceBDNF: brain-derived neurotrophic factorBrdU: bromodeoxyuridineDA: dopamineDAB: diaminobenzidineDCX: doublecortinDG: dentate gyrusGCL: granule cell layerHAP1: huntingtin-associated protein 1
HD: Huntington diseaseHEAT: huntingtin, elongation factor 3, protein phosphatase 2A, target of rapamycin 1i.p.: intraperitoneallyKGDHC: alpha-ketoglutarate-dehydrogenase complexMLK2: mixed-lineage kinase 2NeuroD: neurogenic differentiation proteinNeuN: neuronal nucleiNGS: normal goat serumNHS: normal horse serumNuMA: nuclear mitotic apparatusPCNA: proliferating cell nuclear antigenPFA: paraformaldehydeSEM: standard error of the meanSGZ: subgranular zoneSSRI: selective serotonin reuptake inhibitorSVZ: subventricular zoneWT: wild-typeYAC: yeast artificial chromosome





























