Embed Size (px)
Citation preview

Angiotensin II Type 2 Receptor Expression AfterVascular Injury
Differing Effects of Angiotensin-Converting Enzyme Inhibitionand Angiotensin Receptor Blockade
Thomas A. Barker, Michael P. Massett, Vyacheslav A. Korshunov, Amy M. Mohan,Amy J. Kennedy, Bradford C. Berk
Abstract—It has been suggested that the effects of angiotensin II type 1 receptor (AT1R) blockers are in part because ofangiotensin II type 2 receptor (AT2R) signaling. Interactions between the AT2R and kinins modulate cardiovascularfunction. Because AT2R expression increases after vascular injury, we hypothesized that the effects on vascularremodeling of the AT1R blocker valsartan and the ACE inhibitor benazepril require AT2R signaling through thebradykinin 1 and 2 receptors (B1R and B2R). To test this hypothesis, Brown Norway rats were assigned to 8 treatments(n�16): valsartan, valsartan�PD123319 (AT2R inhibitor), valsartan�des-arg9-[Leu8]-bradykinin (B1R inhibitor),valsartan�HOE140 (B2R inhibitor), benazepril, benazepril�HOE140, amlodipine, and vehicle. After 1 week of treatment,carotid balloon injury was performed. Two weeks later, carotids were harvested for morphometry and analysis ofreceptor expression by immunohistochemistry and Western blotting. Valsartan and benazepril significantly reduced theintima:media ratio compared with vehicle. Blockade of AT2R, B1R, or B2R in the presence of valsartan prevented thereduction seen with valsartan alone. B2R blockade inhibited the effect of benazepril. Injury increased AT1R, AT2R, B1R,and B2R expression. Treatment with valsartan but not benazepril significantly increased intima AT2R expression 2-foldcompared with vehicle, which was not reversed by inhibition of AT2R, B1R, and B2R. Functionally, valsartan increasedintimal cGMP levels compared with vehicle, and this increase was inhibited by blocking the AT2R, B1R, and B2R.Results suggest that AT2R expression and increased cGMP represent a molecular mechanism that differentiates AT1Rblockers, such as valsartan, from angiotensin-converting enzyme inhibitors like benazepril. (Hypertension. 2006;48:942-949.)
Key Words: valsartan � angiotensin � AT2R � restenosis � rat
Angiotensin II (Ang II) exerts its effects primarily through2 functionally distinct receptors, the angiotensin type 1
receptor (AT1R) and angiotensin type 2 receptor (AT2R). TheAT1R is widely distributed in adults, whereas the AT2R isonly highly expressed in the fetus.1 However, AT2R expres-sion increases in pathological conditions, such as vascularinjury.2 These 2 receptors exert effects that are generallyconsidered antagonistic. Blocking Ang II activity with angio-tensin-converting enzyme (ACE) inhibitors reduces the riskof mortality in patients at high risk for cardiovascular events.3
Recently, AT1R blockers (ARBs) have been shown to beeffective in reducing stroke in patients with left ventricularhypertrophy4 and mortality in heart failure patients.5
Kinins exert diverse physiological actions, including vasodi-lation,6 increased capillary permeability, and inflammation.7 Themajor kinins, which are agonists at the bradykinin type 2receptor (B2R), are metabolized to produce the des-Arg9-kinins,
which are agonists at bradykinin type 1 receptors (B1Rs).Although the B2R is widely and constitutively expressed, theB1R is normally expressed at low levels, although it can beinduced by growth factors and cytokines.8
The renin–angiotensin and kinin systems are intimately linked,because ACE metabolizes both angiotensin I and bradykinin.Recently, an AT2R pathway has been shown to signal via the B2Rto produce vasodilation in resistance vessels9 and coronary mi-crovessels,10 to regulate blood pressure,11–13 to mediate renalproduction of NO,14 and to promote cardiac fibrosis.15
Treatment of hypertensive patients with ARBs and ACEinhibitors decreases cardiovascular events and renal failure toan extent greater than predicted by the reduction in bloodpressure.4,16,17 Because there is increased Ang II duringblockade of the AT1R with ARBs, it has been suggested thatsome beneficial effects of ARBs may be because of actions ofAng II at the AT2R.18 As discussed above, there may be an
Received June 5, 2006; first decision June 20, 2006; revision accepted August 7, 2006.From the Cardiovascular Research Institute (T.A.B., M.P.M., V.A.K., A.M.M., B.C.B.) and Departments of Medicine and Biostatistics (A.J.K.),
University of Rochester, Rochester, NY.Correspondence to Bradford C. Berk, University of Rochester, Box 679, 601 Elmwood Ave, Rochester, NY 14642. E-mail bradford_berk@urmc.
rochester.edu© 2006 American Heart Association, Inc.
Hypertension is available at http://www.hypertensionaha.org DOI: 10.1161/01.HYP.0000241061.51003.b7
942 by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

important link between AT2R signaling and B2R pathways.11
This interaction might be particularly important in vascularinjury where both the AT2R and B1R are known to modulatethe tissue response as shown by pharmacological2,19 andgenetic20 inhibition of these receptors. Previous work showedthat ACE inhibition and the ARB valsartan decreased neoin-tima formation after rabbit carotid injury.21 Specifically, bothtreatments reduced circulating endothelin-1, thromboxane B2,and 6-keto-prostaglandin F1�, but changes in the vessel wallitself were not studied. Here we hypothesize that the benefi-cial effects in vascular injury of AT1R blockade and ACEinhibition are mediated, in part, by a pathway involving AT2Rsignaling via the B1R and B2R, which increases vascularcGMP levels. We measured expression of these receptorsquantitatively in the injured vessel, as well as the effects ofreceptor blockade on intima formation. Our results show thatthe valsartan reduction in intima formation correlates withexpression of the AT2R and cGMP in the intima and media.In contrast, the ACE inhibitor benazepril also decreasedintima formation but did not increase AT2R expression orintima and media cGMP levels, suggesting a different mech-anism of action.
MethodsAnimal Surgery, Drug Treatment, and AnalysesFourteen-week-old male Brown Norway rats purchased from CharlesRiver (Wilmington, MA) were used in accordance with the guide-lines of the National Institutes of Health and American HeartAssociation for the care and use of laboratory animals (approved bythe University of Rochester Committee on Animal Resources). Rats(250 to 300 g) were randomly assigned to 8 treatments (n�16 pergroup): (1) valsartan at 30 mg/kg per day (AT1R inhibitor, NovartisPharmaceuticals), (2) valsartan at 30 mg/kg per day plus PD123319at 30 mg/kg per day (AT2R inhibitor); (3) valsartan at 30 mg/kg/dplus des-arg9-[Leu8]-bradykinin at 10 mg/kg per day (B1R inhibitor,Bachem USA); (4) valsartan at 30 mg/kg per day plus HOE140 at500 �g/kg per day (B2R inhibitor, Bachem USA); (5) benazepril at20 mg/kg per day (Novartis Pharmaceuticals); (6) benazepril at 30mg/kg per day plus HOE140 at 500 �g/kg per day; (7) amlodipine at20 mg/kg per day (Novartis Pharmaceuticals); and (8) vehicle (0.15mol/L NaOH). In preliminary experiments, we compared the bloodpressure reduction obtained with several doses of valsartan andbenazepril and chose doses that gave an equivalent reduction after 2weeks. We did not have groups with individual AT2R, B1R, and B2Rblockers, because previous work already showed the effects of thesecompounds alone on neointima formation.19,22,23 Drugs were admin-istered daily by intraperitoneal injection at 9:00 AM. We did notinclude HOE140 or des-arg9-[Leu8]-bradykinin alone, because pre-vious studies showed no effect on intima formation.24,25
All of the surgical procedures were performed after intraperitonealinjection of 50 mg/kg of ketamine (Hospira) and 5 mg/kg of xylazine(Butler). Five rats from each group underwent continuous arterialblood pressure assessment by radiotelemetry (TA11PA-C20, DataScience) using devices inserted 2 weeks before drug treatment.26
Left common carotid balloon injury was performed after 7 days ofdrug treatment. A Fogarty 2F balloon catheter was inserted andwithdrawn 3 times.27 Buprenorphine (0.5 mg/kg) was administeredimmediately after the procedure and 6 hours later. Fourteen daysafter injury, half of the rats (n�8) were perfused with 10%formaldehyde (100 mm Hg), and carotids were paraffin embedded.Sections (4 �m thick) at the midpoint of the common carotid and0.5 mm proximal and distal to the midpoint were obtained. Carotidsfrom the remaining animals were frozen in liquid nitrogen forbiochemical analysis.
Morphometry and ImmunohistochemistryAfter staining with hematoxylin/eosin, sections were analyzed byMicroComputer Imaging Device (MCID) and software (ImagingResearch Incorporated). Measurements were made for each of the 3sites sampled and averaged. Antibodies included: AT1R (1:200polyclonal rabbit, Santa Cruz Biotechnology), AT2R (1:2500 poly-clonal goat, Santa Cruz Biotechnology), B1R (1:2000 polyclonal goatSanta Cruz Biotechnology), B2R (1:100 polyclonal mouse BDBiosciences), and cGMP (1:2000 polyclonal sheep, kindly donatedby Dr J de Vente, Maastricht University, Maastricht, the Nether-lands). With the exception of the cGMP, antigen retrieval with10 mmol/L of citrate buffer at pH 6.0 heated to 120°C and pressuredto 10 PSI for 20 minutes was used. For quantitation, thresholds wereset based on no primary antibody and converted to grayscale forMCID software. No counterstain was used. Grayscale quantitationhad an interobserver correlation of 0.97. After analysis, coverslipswere removed and slides counterstained with hematoxylin.
Immunoblot AnalysisCarotids were lysed and sonicated, nonsolubilized proteins removedby centrifugation, and the supernatant subjected to electrophoresis ona 7.5% sodium dodecyl sulfate polyacrylamide gel. Immunoblotswere performed at dilutions of 1:100 (AT1R, AT2R, and B1R) or1:500 (B2R). The dominant band on Western blot chosen forquantitative analysis was of the following molecular mass (based onexpression of receptor cDNAs in Chinese hamster ovary cells, vesselwall, and the manufacturer’s information): AT1R 60 kDa, AT2R 43kDa, B1R 45 kDa, and B2R 70 kDa. It is likely that the 60-kDa formof the AT1R is the fully glycosylated form. A limitation of theimmunoblot analysis is the unavailability of the immunizing peptidesfor preadsorption control studies to demonstrate specificity. Odysseyinfrared imaging system (LiCOR) was used to visualize proteins andOdyssey software used for quantification.
Statistical AnalysisAnalyses were performed using SAS Version 9.1 (SAS Institute).Because of sample sizes and number of comparisons, only compar-isons with P�0.01 were classified as significant (0.01 set a priori).Baseline mean arterial pressure (MAP), heart rate (HR), heartweight:body weight ratio, and Western blot group comparisons werecomputed using PROC GLM. If group was found to be a significantpredictor, then the LSMEANS statement was added to compute themultiple comparisons. The last 3 MAP and HR measurements wereaveraged for each subject and then modeled in the same fashion asthe baseline MAP measurements.
The morphometry measures were analyzed using repeated measuresANOVA. If the group was found to be significant, then the LSMEANSstatement was used again for the multiple comparisons, and P�0.01 wasclassified as significant. When comparing the morphometry measure-ments for right carotid artery (RCA) versus left carotid artery (LCA), thedifference between the 2 measurements was analyzed to remove thecorrelation of the measurements using PROC GENMOD with the samepredictors as for RCA and LCA alone.
The immunohistochemistry receptor data were all paired; there-fore, all of the analyses were completed using a paired t test, if thedata were normal. For Western blot RCA–LCA data, the differencebetween the measurements was calculated, and a nonparametricsigned rank test was performed.
ResultsPhysiological ParametersCompared with vehicle, amlodipine-treated rats had a 10-mm Hg reduction in MAP after 1 week of treatment (FigureIA, available online at http://hyper.ahajournals.org). Valsar-tan and benazepril significantly reduced MAP to an extentsimilar to amlodipine. There was no significant difference inthe MAP between amlodipine and valsartan or benazeprilgroups. The effect of valsartan on MAP was unaltered by the
Barker et al AT2R Expression Decreases Neointima 943
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

addition of PD123319, des-arg9-[Leu8]-bradykinin, or HOE140.The effect of benazepril on MAP was partially inhibited byHOE140. There was no significant difference in HR at baselineor after treatment between groups (Figure IB). Heart weight-:body weight ratio also was not significantly different betweengroups (data not shown).
Effect of Injury and Treatment onVessel MorphometryMorphometry of the uninjured RCA was not altered signifi-cantly by drug treatment. The lumen, media, and external elasticlamina (EEL) areas for the RCA did not differ across groups(Table IS). An intima developed 2 weeks after balloon injury ofthe LCA (Figure 1B). Comparison of vessel morphology be-tween injured LCA and uninjured RCA (Table and Table IS,available online at http://hyper.ahajournals.org) showed the ex-pected changes in the vehicle group (Figure 2A and 2B).Specifically, there was a significant 50% reduction in lumen area
of the LCA (LCA: 137�10 versus RCA: 256�13�103 �m2;P�0.0001) that was primarily because of intima formation in theLCA (134�13�103 �m2). There were small increases in mediaand EEL area in the LCA that did not differ significantly from theRCA.
Treatment with valsartan had no effect on the RCA butdramatically altered morphology in the injured LCA (Figure1C). Valsartan significantly decreased intima area by 51%compared with the vehicle (Figure 1C and Table; P�0.0001).There was no significant difference in lumen areas among thevalsartan treatment groups (Table), although there was a trendtoward valsartan alone having a larger lumen than vehicle (32%increase). Blocking AT2R with PD123319 in the presenceof valsartan significantly increased the intima area comparedwith valsartan alone. Similarly, valsartan plus des-arg9-[Leu8]-bradykinin had significantly greater intima than valsartan alone.Blocking B2R with HOE140 in the presence of valsartan alsoincreased intima area compared with valsartan alone, but thiswas of borderline significance (P�0.03). Amlodipine did notinhibit intima formation, suggesting that the effect of valsartanwas not primarily because of lowering blood pressure. AlthoughAT2R, B1R, and B2R blockade in association with valsartanreversed the effect of valsartan alone on intima, these groupsshowed a trend for smaller intima than the vehicle-treated group.This result is not expected because it is unlikely that anyindividual receptor completely mediates the valsartan effect(Table).
Valsartan alone also increased the media area comparedwith vehicle (99�6 versus 75�6 � 103 �m2; P�0.003).Inhibiting the AT2R, B1R, or B2R completely blocked theeffect of valsartan on media area. Other measurements ofvascular remodeling showed no significant changes amonggroups.
To correct for variation in vessel size, the intima:media (I:M)ratio was calculated for each group (Figure 1E). Compared withvehicle, amlodipine showed no reduction in I:M ratio, whereasvalsartan significantly reduced the I:M ratio. When PD123319was given with valsartan, the I:M ratio returned to vehicle levels.In addition, the B1R blocker (des-arg9-[Leu8]-bradykinin) and theB2R blocker (HOE140) also reversed the valsartan decrease in
Figure 1. Representative hematoxylin/eosin-stained cross-sections of (A) vehicle-treated RCA, (B) vehicle-treated LCA, (C)valsartan-treated RCA, and (D) valsartan-treated LCA. (E) Com-parison between treatment groups of LCA I:M ratio. *Signifi-cantly different from the other treatment groups. For all morpho-logical analyses, results are the means from 3 to 4 individualrats using 2 to 3 sections per vessel.
Morphometry Measurements of Injured LCA Treatment Groups
GroupLumen
�103 �m2Intima
�103 �m2Media
�103 �m2EEL
�103 �m2
Vehicle 137�10 134�13 75�6 347�12
Amlodipine 129�16 146�14 81�7 356�18
Valsartan 182�18 68�10* 99�6† 351�27
Val�PD 150�10 113�13 73�4 338�9
Val�des-BK 162�23 121�11 76�6 359�22
Val�HOE 155�22 118�20 67�8 341�24
Benazepril 219�26† 55�15† 92�7 366�17
Ben�HOE 156�18 128�18 82�6 366�22
All data shown as mean�SEM. Val indicates valsartan; PD, PD123319;des-BK, des-arg9-�Leu8�-bradykinin.
*Significantly different from other groups except valsartan plus HOE140,P�0.01.
†Significantly different from other groups (P�0.01).
944 Hypertension November 2006
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

I:M ratio. These data strongly suggest that the AT2R, B1R, andB2R participate in the valsartan effect to decrease intima formation.
Benazepril significantly decreased the intima area by 69% andthe I:M ratio compared with vehicle (Figure 1D and 1E andTable; P�0.0001). Benazepril significantly increased the lumenarea by 59% compared with vehicle (Table). The changes inintima, I:M ratio, and lumen were inhibited by the addition ofHOE140. Media and EEL area were unchanged by treatmentwith benazepril (Table).
Immunohistochemistry of Ang II and BradykininReceptors in Vehicle-Treated RatsTo quantitate changes in receptor expression, we performedimmunohistochemistry and used the MCID system. Controlswithout primary antibody were optimized to minimize back-ground staining. In the uninjured RCA of the vehicle group,there was substantial expression of both AT1R and B2R inmedia but not in adventitia (Figure IIA and IID). In contrast,there was little AT2R expression or B1R expression anywhere(Figure IIB and IIC). A previous study by Hutchinson et al2
showed that after injury, AT2R expression peaked at 48 to 72hours, whereas AT1R expression was slower, peaking at 96 to144 hours.
After injury, AT2R was highly expressed in intima, with noexpression in media or adventitia (Figure 2A). The AT1R andB2R were both highly expressed in intima and media (FigureIIIA and IIIC) but not adventitia. The B1R was expressedprimarily in the intima after injury but was also present to alesser extent in media (Figure IIIB).
Immunohistochemistry of Ang II and BradykininReceptors in Drug-Treated RatsIn the uninjured RCA, there was no change in receptor expres-sion by any of the drugs compared with vehicle (data notshown). In the LCA, amlodipine did not alter expression of the4 receptors, including the AT2R (Figure 2B and Figures IV toVIB). This finding suggests that altered Ang II and kininreceptor expression are primarily regulated by tissue injury,rather than by blood pressure.
In rats treated with valsartan, the most impressive changein the LCA was increased AT2R expression in the intima thatwas significantly greater compared with vehicle or amlodip-ine (�2 fold increase; P�0.002; Figures 2C and 3). Therewas also a small increase in AT2R expression in the media.There were no differences in AT1R, B1R, and B2R expressionwith valsartan compared with vehicle or amlodipine in theLCA (Figures IV to VI).
Surprisingly, the addition of AT2R, B1R, or B2R blockers tovalsartan did not affect the increase in AT2R, which was2-fold increased for all of the groups compared with vehicle(Figure 2D through 2F and 2I). Together these results suggestthat the effect of valsartan on AT2R expression after injury isrelated to elevated Ang II levels, because blockade of theAT2R did not reverse this effect. Finally, there was nosignificant difference in expression of AT1R, B1R, and B2R inthese groups compared with vehicle, amlodipine, or valsartan(Figure IV through VI). There was no nonspecific immuno-reactivity for the receptor antibodies (Figure VII). Benazeprildid not alter AT1R, AT2R, B1R, or B2R expression compared
with vehicle (Figures 2G and 2H, 3, and IV through VI),suggesting that the mechanism by which ACE inhibitorsreduce intima formation differs from ARBs, especially inrelation to AT2R expression.
Immunoblot AnalysisThe increase in expression of AT1R, AT2R, B1R, and B2Rafter balloon injury was confirmed by Western blotting(Figure 4A). Ponceau staining showed equal protein loading(data not shown). Increased AT2R expression in the injuredLCA of valsartan-treated groups was apparent by immuno-blot (Figure 4B). Consistent with the immunohistochemistry,the increase in AT2R with valsartan differed significantlyfrom vehicle and amlodipine (Figure 4C), whereas there wasno difference in AT2R protein levels in the benazepril group(Figure 4B and 4C). Valsartan in the absence of injury causedno change in expression of AT1R, AT2R, B1R, and B2R (datanot shown).
Correlation Analysis of I:M Ratio andReceptor ExpressionTo evaluate the relationship between receptor expression andintima formation, correlation analyses were performed usingquantitative measurements of immunohistochemical receptorabundance. Among multiple comparisons, the only signifi-cant correlation between I:M ratio and receptor expressionwas a negative correlation between AT2R and I:M ratio(details in Figure 5). Equally important is the obviousdifference between benazepril and valsartan; both drugssignificantly reduced I:M ratio, but only valsartan increasedAT2R expression, suggesting that their mechanism of actionis different.
Immunohistochemistry of cGMP inDrug-Treated RatsThe valsartan group had a 2.6�1.1-fold increase in cGMP inthe intima (Figure 6C and 6E, normalized to media) com-pared with vehicle-treated rats (Figure 6A and 6E). Theincrease in intima cGMP was completely blocked by bothPD123319 and HOE140 (Figure 6E). Benazepril increasedcGMP in the endothelium (Figure 6D, arrows), but intima andmedia cGMP did not differ from vehicle. These findingsfurther support the concept that valsartan and benazeprilinhibit intima formation by different mechanisms.
DiscussionThe 4 major findings of this study are that the valsartan-mediated reduction in intima formation after rat carotidballoon injury: (1) differs mechanistically from benazepril;(2) correlates with AT2R and cGMP expression in the intima;(3) depends on receptor-mediated events that require theAT2R, B1R, and B2R; and (4) is not mediated by loweringblood pressure. We confirmed previous findings that expres-sion levels of the AT2R, AT1R, B1R, and the B2R wereincreased by balloon injury. The present study is the first tomeasure receptor expression in response to both injury anddrug treatment. Our results are supported by previous workshowing that both captopril and valsartan decreased intimaformation after rabbit carotid injury,21 although this article
Barker et al AT2R Expression Decreases Neointima 945
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

did not measure receptor expression. Importantly, we showfor the first time that treatment with valsartan further in-creased AT2R protein expression, primarily in the intima.Increased expression of the AT2R was not affected by AT2Rblockade or by blockade of B1R or B2R. Because AT1R, B1R,and B2R expression were not affected by treatment withvalsartan, our results suggest a critical role for an AT2R-cGMP pathway in the ability of valsartan to decrease intima.This concept is supported by the inverse relationship betweenexpression of AT2R and cGMP with the I:M ratio forvalsartan (Figure 5). Another novel finding is the differencebetween valsartan and benazepril with respect to changes inAT2R expression and cGMP. Valsartan increased both AT2Rexpression and cGMP in the neointima, whereas benazeprilhad no effect on the neointima and increased cGMP only inthe endothelium. This association suggests that increasedAT2R expression by valsartan may explain increased cGMP,as proposed by Carey and colleagues,14,28 for the kidney.
Based on these results, we propose the following mecha-nism for valsartan. Valsartan blocks the AT1R and increasesAng II levels. Valsartan increases AT2R expression that nowincreases kininogenase activity and generates bradykinin.Bradykinin-mediated activation of B1R and B2R inhibitsintima formation, in part via endothelial cell generation ofNO and increased cGMP in vascular smooth muscle cell(VSMC). Although it is possible that the effects on AT2R areparallel to the B1R and B2R, this seems unlikely, becauseindividual blockade would result in only a partial reversal ofthe effects of valsartan. Finally, there may be direct effectsmediated by the AT2R, especially via heterodimerization withthe B1R and B2R, to inhibit intima formation. Data to supportthe model include reports that valsartan increases Ang IIlevels,29 thus increasing AT2R activation. The importance ofincreased Ang II has been demonstrated in the angioten-sinogen transgenic mouse in which AT2R signaling inducesangiogenesis.30 We propose that a similar mechanism exists
Figure 2. Representative carotid arterycross-sections stained with AT2R antibody(brown) for (A) vehicle RCA, (B) amlodipine,(C) valsartan, (D) valsartan plus PD123319,(E) valsartan plus des-arg9-[Leu8]-bradykinin,(F) valsartan plus HOE140, (G) benazepril,and (H) benazepril plus HOE140.
946 Hypertension November 2006
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

in vascular injury. AT2R mRNA is induced by vascularinjury,2 and we confirm increased AT2R protein using immu-nohistochemistry and Western blotting. Overexpression ofthe AT2R has been shown to reduce intima formation in theballoon-injured rat.31 Here we show by pharmacologicalinhibition that the AT2R is required for valsartan inhibition ofintima formation.
Because AT2R expression is normally low in adults, theincreased expression in pathological situations is likely animportant mechanism by which this receptor contributes to
the tissue response to injury.2 Crosstalk between the AT1Rand AT2R potentially occurs by several mechanisms, the bestcharacterized being generation of regulatory peptides, such asbradykinin and antagonism of intracellular signaling, whereasheterodimerization remains more speculative. Thus, the ef-fects of ARBs in this situation are likely because of increasedAT2R and bradykinin signaling, as well as AT1R inhibition.11
Our study confirms that AT2R expression is important aftervascular injury and shows that AT2R expression occursprimarily in the intima.
We show for the first time that AT2R expression is inducedby valsartan in blood vessels. Because AT2R, B1R, and B2Rinhibition in association with AT1R blockade did not changethis effect, the most likely explanation is that increased AngII levels upregulate AT2R expression. Increased Ang II levelswere shown previously to induce AT2R expression.32 Ourdata are consistent with a report that valsartan increases AT2Rexpression in the heart after ischemia-reperfusion injury,which is important in limiting infarct size.33
Previous reports have shown that AT2R-dependent eventsinvolve increased B2R signaling, including renal natriuresis,28
regulation of blood pressure,34 and vascular relaxation.9,11,13
An important signaling mechanism for the AT2R is crosstalkwith bradykinin via increased bradykinin generation, pre-sumed because of inhibition of the Na�-H� exchanger caus-ing intracellular acidosis and activation of kininogenaseactivity, which generates bradykinin.11 The B1R has beenshown to reduce intima formation after balloon injury in therat.19 Our study showed that both the B1R and the B2R wereincreased by injury and were required for valsartan to reduceintima formation. Combining these results with previousreports of AT2R signaling through increased bradykinin andkinin receptors, we propose that the effect of valsartan onintima formation after balloon injury is initially through the
Intim
al A
T 2R
sta
inin
g
AmlVeh Val Val+PD
Val+des-BK
Val+HOE
Ben Ben+HOE
****
0
0.5
1
1.5
2
2.5
3
Figure 3. Intimal AT2R immunoreactivity. The relative intimalarea stained by AT2R antibody was quantitated relative to thevehicle group, which was arbitrarily set to 1.0. *Significantly dif-ferent from vehicle.
Figure 4. Immunoblot analysis of receptor expression. (A) Com-parison of expression for AT1R, AT2R, B1R, and B2R proteins inthe vehicle group. (B) Comparison of LCA AT2R protein expres-sion between the treatment groups. (C) The relative AT2R immu-noreactivty was quantitated relative to the vehicle group, whichwas arbitrarily set to 1.0. *Significantly different from vehicle andamlodipine.
I:M ra
tio
AT2R expression
0
0.5
1
1.5
2
2.5
0 0.02 0.04 0.06 0.08 0.1 0.12
VehAmlVal
Val + PDVal + des-BKVal + HOEBen
Ben + HOE
Figure 5. Correlation analysis of AT2R expression and I:M ratio.Shown is a plot of means for I:M ratio and AT2R for each treat-ment group. Correlation analysis was performed using linearregression. Significant correlations between I:M ratio and AT2Rwere obtained for combined control, valsartan, and amlodipinegroups (R2�0.47; P�0.013). No significant correlation wasobserved for combined control, benazepril, benazepril�HOE,and amlodipine groups (R2�0.05) or for combined control,valsartan�PD, valsartan�HOE, valsartan�des-bradykinin, andamlodipine groups (R2�0.00).
Barker et al AT2R Expression Decreases Neointima 947
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

AT2R, which signals downstream through both the B1R andthe B2R, most likely by altering cGMP levels in the injuredvessels. A more speculative mechanism may be heterodimer-ization of the AT2R with the B1R and B2R, which mayaugment signaling by AT2R. This mechanism would explainwhy increased AT2R in the intima would inhibit VSMCproliferation and why blockade of either the B1R or B2Rblunted the effect of valsartan on intima formation.
Previous studies of intima formation in the mouse carotidafter ligation showed no effect of B1R or B2R antagonists onintima.35 In cultured VSMCs, bradykinin was shown to bothstimulate36 and inhibit growth.23 The fact that blocking eitherB1R or B2R abrogated the ability of valsartan to limit intimaformation is somewhat surprising, especially because block-ade did not decrease AT2R expression. However, it is reason-able to speculate that bradykinin receptor–specific signalsfrom both receptors (either in the same cell or different cells)are required to limit VSMC proliferation.
ARBs clearly exert effects through mechanisms that differfrom ACE inhibitors. Benazepril did not alter AT2R receptorexpression level (Figure 5), suggesting that ACE inhibitorsprimarily reduce intima formation by affecting bioavailabilityof receptor ligands (ie, decreased Ang II and increasedbradykinin). In addition, ACE inhibitors may also directlyaffect intracellular signaling as shown by Kohlstedt et al.37
Previous studies found that HOE140 reversed ACE inhibitor-mediated reduction of intima, suggesting a key role for B2R.Our results (Figure 1) support this concept.
PerspectivesThe present study provides insights into the similarities anddifferences in the vascular protective mechanisms of ACEinhibitors and ARBs. Our data suggest that the major similaritybetween ACE inhibition and AT1R blockade is via bradykininactions at the B2R. An important mechanistic difference is therole of increased AT2R expression in the actions of ARBs.Despite the fact that both ACE inhibition and AT1R blockaderequire B2R function, the increase in vascular cGMP was muchgreater in the presence of AT1R blockade and increased AT2Rexpression. These results suggest that heterodimerization of theAT2R with the B1R and B2R or a unique feature of AT2Rsignaling increases cGMP. An interesting possibility for futurestudy is the effect of AT2R on vascular cGMP phosphodiester-ases, which, if downregulated, would increase cGMP levels.Another concept that could be explored is the effect of agoniststhat stimulate increase AT2R expression or increase its activityon the vascular response to injury. Finally, our results providefurther mechanistic insight into why combined ARB–ACEinhibitor therapy may be clinically beneficial in some situations.
AcknowledgmentsWe thank Sarah Mack and David Nagel for help with histology.
Sources of FundingThis work was supported by an unrestricted grant from NovartisPharmaceuticals to B.C.B. and by a Fulbright Distinguished ScholarAward to T.A.B.
DisclosuresNone.
References1. Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM.
Expression of the subtype 2 angiotensin (AT2) receptor protein in ratkidney. Hypertension. 1997;30:1238–1246.
2. Hutchinson HG, Hein L, Fujinaga M, Pratt RE. Modulation of vasculardevelopment and injury by angiotensin II. Cardiovasc Res. 1999;41:689–700.
3. Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects ofan angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascularevents in high-risk patients. The Heart Outcomes Prevention EvaluationStudy Investigators. N Engl J Med. 2000;342:145–153.
4. Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, Faire U,Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, LindholmLH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular mor-bidity and mortality in the Losartan Intervention For Endpoint reductionin hypertension study (LIFE): a randomised trial against atenolol. Lancet.2002;359:995–1003.
5. Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I,Deedwania PC, Ney DE, Snavely DB, Chang PI. Randomised trial of
Figure 6. cGMP immunoreactivity is increased in valsartan-treated vessels. Representative injured LCA cross-sectionsstained with cGMP antibody for (A) vehicle, (B) amlodipine, (C)valsartan, and (D) benazepril-treated groups. (E) A comparisonof I:M cGMP staining between groups. The I:M ratios were cal-culated to enable comparison across vessels with differentintima sizes. The relative cGMP immunoreactivity was quanti-tated relative to the vehicle group, which was arbitrarily set to1.0. *Significantly different from valsartan; †significantly differentfrom vehicle.
948 Hypertension November 2006
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

losartan versus captopril in patients over 65 with heart failure (Evaluationof Losartan in the Elderly Study, ELITE). Lancet. 1997;349:747–752.
6. Batenburg WW, Popp R, Fleming I, de Vries R, Garrelds IM, Saxena PR,Danser AH. Bradykinin-induced relaxation of coronary microarteries:S-nitrosothiols as EDHF? Br J Pharmacol. 2004;142:125–135.
7. Bandeira-Melo C, Calheiros AS, Silva PM, Cordeiro RS, Teixeira MM,Martins MA. Suppressive effect of distinct bradykinin B2 receptor an-tagonist on allergen-evoked exudation and leukocyte infiltration in sen-sitized rats. Br J Pharmacol. 1999;127:315–320.
8. Phagoo SB, Reddi K, Anderson KD, Leeb-Lundberg LM, Warburton D.Bradykinin B1 receptor up-regulation by interleukin-1beta and B1 agonistoccurs through independent and synergistic intracellular signaling mech-anisms in human lung fibroblasts. J Pharmacol Exp Ther. 2001;298:77–85.
9. Katada J, Majima M. AT(2) receptor-dependent vasodilation is mediatedby activation of vascular kinin generation under flow conditions. Br JPharmacol. 2002;136:484–491.
10. Batenburg WW, Garrelds IM, Bernasconi CC, Juillerat-Jeanneret L, VanKats JP, Saxena PR, Danser AH. Angiotensin II type 2 receptor-mediatedvasodilation in human coronary microarteries. Circulation. 2004;109:2296–2301.
11. Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S,Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N,Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A,Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpressionactivates the vascular kinin system and causes vasodilation J Clin Invest.1999;104:925–935.
12. Israel A, Cierco M, Sosa B. Angiotensin AT(2) receptors mediate vaso-depressor response to footshock in rats. Role of kinins, nitric oxide andprostaglandins. Eur J Pharmacol. 2000;394:103–108.
13. Sosa-Canache B, Cierco M, Gutierrez CI, Israel A. Role of bradykininsand nitric oxide in the AT2 receptor-mediated hypotension. J HumHypertens. 2000;14(suppl 1):S40–S46.
14. Siragy HM, Carey RM. The subtype 2 (AT2) angiotensin receptormediates renal production of nitric oxide in conscious rats. J Clin Invest.1997;100:264–269.
15. Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, MatsuuraH, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H. Cardiacangiotensin II type 2 receptor activates the kinin/NO system and inhibitsfibrosis. Hypertension. 2003;41:99–107.
16. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, ParvingHH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartanon renal and cardiovascular outcomes in patients with type 2 diabetes andnephropathy. N Engl J Med. 2001;345:861–869.
17. Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, RitzE, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2diabetes. N Engl J Med. 2001;345:851–860.
18. Barber MN, Sampey DB, Widdop RE. AT(2) receptor stimulationenhances antihypertensive effect of AT(1) receptor antagonist in hyper-tensive rats. Hypertension. 1999;34:1112–1116.
19. Agata J, Miao RQ, Yayama K, Chao L, Chao J. Bradykinin B(1) receptormediates inhibition of neointima formation in rat artery after balloonangioplasty. Hypertension. 2000;36:364–370.
20. Akishita M, Horiuchi M, Yamada H, Zhang L, Shirakami G, Tamura K,Ouchi Y, Dzau VJ. Inflammation influences vascular remodeling throughAT2 receptor expression and signaling. Physiol Genomics. 2000;2:13–20.
21. Feng TC, Ying WY, Hua RJ, Ji YY, de Gasparo M. Effect of valsartanand captopril in rabbit carotid injury. Possible involvement of bradykininin the antiproliferative action of the renin-angiotensin blockade. J ReninAngiotensin Aldosterone Syst. 2001;2:19–24.
22. van Kleef EM, Fingerle J, Daemen MJ. Angiotensin II-induced pro-gression of neointimal thickening in the balloon-injured rat carotid arteryis AT1 receptor mediated. Arterioscler Thromb Vasc Biol. 1996;16:857–863.
23. Murakami H, Yayama K, Miao RQ, Wang C, Chao L, Chao J. Kallikreingene delivery inhibits vascular smooth muscle cell growth and neointimaformation in the rat artery after balloon angioplasty. Hypertension. 1999;34:164–170.
24. Farhy RD, Ho KL, Carretero OA, Scicli AG. Kinins mediate the antipro-liferative effect of ramipril in rat carotid artery. Biochem Biophys ResCommun. 1992;182:283–288.
25. Farhy RD, Carretero OA, Ho K-L, Scicli AG. Role of kinins and nitricoxide in the effects of angiotension converting enzyme inhibitors onneointima formation. Circ Res. 1993;72:1202–1210.
26. Brockway BP, Mills PA, Azar SH. A new method for continuous chronicmeasurement and recording of blood pressure, heart rate and activity inthe rat via radio-telemetry. Clin Exp Hypertens A. 1991;13:885–895.
27. Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis afterarterial injury. Lab Invest. 1983;49:208–215.
28. Carey RM, Jin X, Wang Z, Siragy HM. Nitric oxide: a physiologicalmediator of the type 2 (AT2) angiotensin receptor. Acta Physiol Scand.2000;168:65–71.
29. Siragy HM, Xue C, Abadir P, Carey RM. Angiotensin subtype-2receptors inhibit renin biosynthesis and angiotensin II formation. Hyper-tension. 2005;45:133–137.
30. Walther T, Menrad A, Orzechowski HD, Siemeister G, Paul M, SchirnerM. Differential regulation of in vivo angiogenesis by angiotensin IIreceptors. FASEB J. 2003;17:2061–2067.
31. Nakajima M, Hutchinson HG, Fujinaga M, Hayashida W, Morishita R,Zhang L, Horiuchi M, Pratt RE, Dzau VJ. The angiotensin II type 2 (AT2)receptor antagonizes the growth effects of the AT1 receptor: Gain-of-function study using gene transfer. Proc Natl Acad Sci U S A. 1995;92:10663–10667.
32. Bonnet F, Cooper ME, Carey RM, Casley D, Cao Z. Vascular expressionof angiotensin type 2 receptor in the adult rat: Influence of angiotensin IIinfusion. J Hypertens. 2001;19:1075–1081.
33. Jugdutt BI, Menon V. AT1 receptor blockade limits myocardial injuryand upregulates AT2 receptors during reperfused myocardial infarction.Mol Cell Biochem. 2004;260:111–118.
34. Moore AF, Heiderstadt NT, Huang E, Howell NL, Wang ZQ, Siragy HM,Carey RM. Selective inhibition of the renal angiotensin type 2 receptorincreases blood pressure in conscious rats. Hypertension. 2001;37:1285–1291.
35. Emanueli C, Bonaria Salis M, Figueroa C, Chao J, Chao L, Gaspa L,Capogrossi MC, Madeddu P. Participation of kinins in the captopril-induced inhibition of intimal hyperplasia caused by interruption of carotidblood flow in the mouse. Br J Pharmacol. 2000;130:1076–1082.
36. Yau L, Wilson DP, Werner JP, Zahradka P. Bradykinin receptor antag-onists attenuate neointimal proliferation postangioplasty. Am J PhysiolHeart Circ Physiol. 2001;281:H1648–H1656.
37. Kohlstedt K, Brandes RP, Muller-Esterl W, Busse R, Fleming I. Angio-tensin-converting enzyme is involved in outside-in signaling in endothe-lial cells. Circ Res. 2004;94:60–67.
Barker et al AT2R Expression Decreases Neointima 949
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

Kennedy and Bradford C. BerkThomas A. Barker, Michael P. Massett, Vyacheslav A. Korshunov, Amy M. Mohan, Amy J.
Angiotensin-Converting Enzyme Inhibition and Angiotensin Receptor BlockadeAngiotensin II Type 2 Receptor Expression After Vascular Injury: Differing Effects of
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2006 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/01.HYP.0000241061.51003.b7
2006;48:942-949; originally published online September 18, 2006;Hypertension.
http://hyper.ahajournals.org/content/48/5/942World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2006/09/28/01.HYP.0000241061.51003.b7.DC1.htmlData Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 23, 2016http://hyper.ahajournals.org/Downloaded from

AT2R expression following vascular injury:
Differing effects of ACE inhibition and angiotensin receptor blockade
Thomas A. Barker, Michael P. Massett, Vyacheslav A. Korshunov, Amy M. Mohan, Amy J.
Kennedy*, Bradford C. Berk.
Online Data Supplement
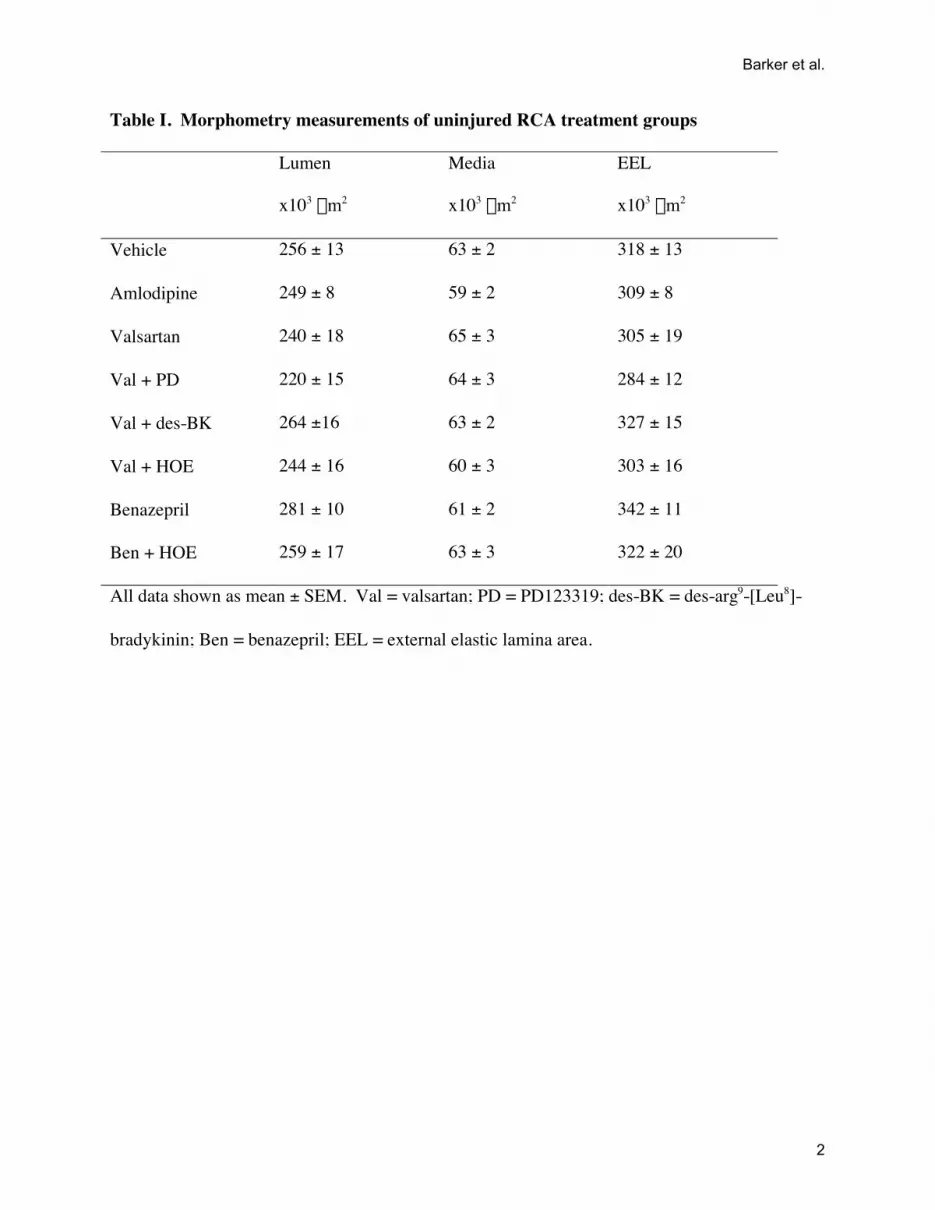
Table I. Morphometry measurements of uninjured RCA treatment groups
Lumen
x103 mm2
Media
x103 mm2
EEL
x103 mm2
Vehicle 256 ± 13 63 ± 2 318 ± 13
Amlodipine 249 ± 8 59 ± 2 309 ± 8
Valsartan 240 ± 18 65 ± 3 305 ± 19
Val + PD 220 ± 15 64 ± 3 284 ± 12
Val + des-BK 264 ±16 63 ± 2 327 ± 15
Val + HOE 244 ± 16 60 ± 3 303 ± 16
Benazepril 281 ± 10 61 ± 2 342 ± 11
Ben + HOE 259 ± 17 63 ± 3 322 ± 20
All data shown as mean ± SEM. Val = valsartan; PD = PD123319; des-BK = des-arg9-[Leu8]-
bradykinin; Ben = benazepril; EEL = external elastic lamina area.
Barker et al.
2

Supplement Figure Legends
Figure I. Comparison of (A) mean arterial blood pressure (n = 5 per group) and (B) heart rate (n
= 5 per group) between groups. * Significantly different from vehicle (p<0.01).
Figure II. Representative vehicle treated uninjured RCA cross-sections stained (brown) with
antibodies for (A) AT1R, (B) AT2R, (C) B1R and (D) B2R.
Figure III. Representative injured LCA cross-sections stained (brown) with antibody for (A)
AT1R, (B) B1R and (C) B2R.
Figure IV. Representative injured LCA cross-sections stained (brown) with AT1R antibody for
(A) vehicle, (B) amlodipine, (C) valsartan, (D) valsartan plus PD123319, (E) valsartan plus des-
arg9-[Leu8]-bradykinin, (F) valsartan plus HOE140, (G) benazepril, and (H) Benazepril plus
HOE140.
Figure V. Representative injured LCA cross-sections stained (brown) with B1R antibody for (A)
vehicle, (B) amlodipine, (C) valsartan, (D) valsartan plus PD123319, (E) valsartan plus des-arg9-
[Leu8]-bradykinin, (F) valsartan plus HOE140, (G) benazepril, and (H) Benazepril plus
HOE140.
Barker et al.
3

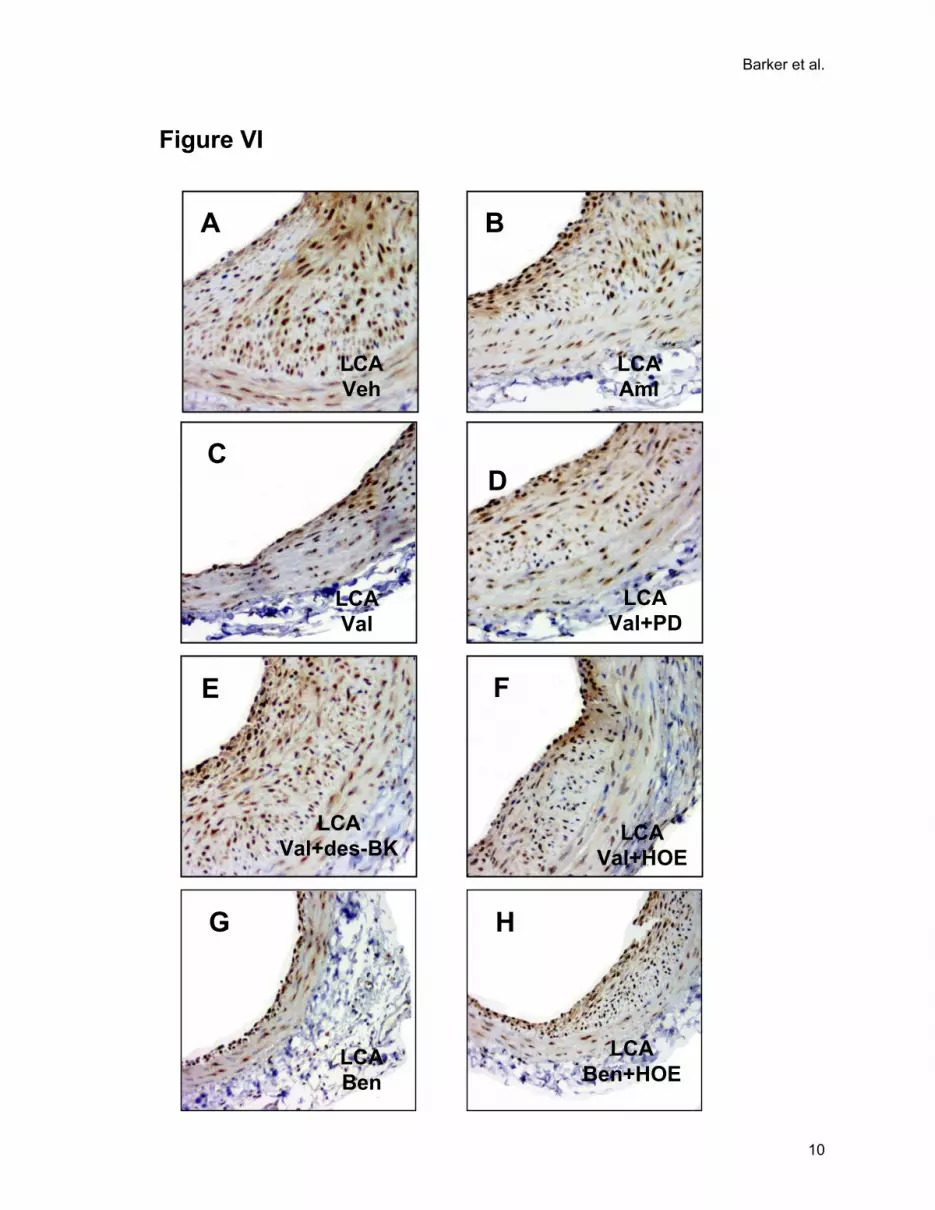
Figure VI. Representative injured LCA cross-sections stained (brown) with B2R antibody for
(A) vehicle, (B) amlodipine, (C) valsartan, (D) valsartan plus PD123319, (E) valsartan plus des-
arg9-[Leu8]-bradykinin, (F) valsartan plus HOE140, (G) benazepril, and (H) Benazepril plus
HOE140.
Figure VII. Immunohistochemistry negative controls for (A) AT1R, (B) AT2R, (C) B1R and (D)
B2R.
Barker et al.
4

A
MA
P (
mm
Hg
)H
R (
BP
M)
B
0
20
40
60
80
100
120BaselineTreatment
BenVeh Aml Val Val +PD
Val +HOE
Val +des-BK
Ben +HOE
***** *
A
Veh Aml Val Val +PD
Val +HOE
Val +des-BK
Ben +HOE
Ben
Figure I
0
50
100
150
200
250
300
350
400
Barker et al.
5

Figure II
B2R RCAVehicle
B1R RCAVehicle
C
AT1R RCAVehicle
A
D
AT2R RCAVehicle
B
Barker et al.
6

AT1R LCAVehicle
A
B
B1R LCAVehicle
C
B2R LCAVehicle
Figure III
Barker et al.
7

A
LCAVeh
C
LCAVal
B
LCAAml
Figure IV
LCAVal+des-BK
E
LCAVal+HOE
F
LCAVal+PD
D
LCABen
G
LCABen+HOE
H
Barker et al.
8

A
LCAVeh
C
LCAVal
B
LCAAml
Figure V
E
LCAVal+des-BK
D
LCAVal+PD
F
LCAVal+HOE
LCABen
LCABen+HOE
G H
Barker et al.
9
A
LCAVeh
C
LCAVal
B
LCAAml
Figure VI
LCAVal+HOE
F
LCAVal+PD
D
LCAVal+des-BK
E
G H
LCABen
LCABen+HOE
Barker et al.
10

A
AT1R
C
B1R
B
AT2R
Figure VII
B2R
D
Barker et al.
11





























