Embed Size (px)
Citation preview

Exercise training combined with angiotensin IIreceptor blockade limits post-infarct ventricularremodelling in rats
Xiaohua Xu1, Wenhan Wan1, Lisa Ji2, Shunhua Lao1, Anthony S. Powers1, Weiyan Zhao1,John M. Erikson3, and John Q. Zhang1*
1Laboratory of Cardiovascular Research, University of Texas at San Antonio, 1 UTSA Circle, San Antonio, TX 78249, USA;2Department of Pharmacology, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA;and 3Division of Cardiology, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA
Received 8 November 2007; revised 31 December 2007; accepted 25 January 2008; online publish-ahead-of-print 5 February 2008
Time for primary review: 32 days
Aims Our aim was to test the hypothesis that angiotensin II receptor blockade combined with exercisetraining after myocardial infarction (MI) could attenuate post-MI left ventricular remodelling and pre-serve cardiac function.Methods and results Sprague–Dawley rats underwent ligation of the left descending coronary artery,resulting in MI, or a sham operation. Losartan treatment and exercise training were initiated 1 weekafter infarction and continued for 8 weeks, either as a single intervention or combined. Collagenvolume fraction in the sedentary MI (MISed) group was significantly higher than other MI groupstreated with exercise training and/or losartan. Compared with MISed group, hearts of rats receivingexercise and/or losartan treatment had lower tissue inhibitor of matrix metalloproteinase (TIMP)1. Matrix metalloproteinase (MMP) 2 or MMP-9 did not differ among all groups. Additionally, the levelof angiotensin II receptor type 1 (AT1) protein significantly decreased in response to exercise training.Furthermore, angiotensin converting enzyme (ACE) binding was markedly lower in hearts receiving exer-cise training than in the MISed hearts. Cardiac function was preserved in rats receiving exercise training,and the beneficial effect was further improved by exercise combined with losartan treatment in com-parison to the MISed group.Conclusion Our results suggest that post-MI exercise training and/or AngII receptor blockade reducesTIMP-1 expression and mitigates the expressions of ACE and AT1 receptor. These improvements, inturn, attenuate myocardial fibrosis and preserve post-MI cardiac function.
KEYWORDSMyocardial infarction;
Remodelling;
Exercise;
Angiotensin;
Metalloproteinases
1. Introduction
Myocardial infarction (MI) results in structural and molecularalterations to both cardiac myocytes and the extracellularmatrix (ECM).1 Left ventricular (LV) remodelling after MIoriginates as an adaptive process when the heart attemptsto compensate for acute loss in contractile function. Overtime, however, LV remodelling becomes maladaptive and arisk for the development of congestive heart failure (CHF).2
The cardiac renin-angiotensin system (RAS) is activatedduring the process of post-MI remodelling.3 Local generationof angiotensin II (AngII) and Ang II receptors have beenreported to be increased in the infarcted hearts.3,4 Inaddition, many studies have demonstrated that inhibitionof cardiac RAS with angiotensin converting enzyme (ACE)
inhibitors or angiotensin II receptor blockers (ARBs)improves LV function, prevents geometric remodelling, andprolongs survival, suggesting that AngII plays an importantrole in post-MI remodelling.5–8
The dynamic synthesis and breakdown of ECM proteinsplay a pivot role in post-MI LV remodelling. Matrix metallo-proteinases (MMPs) are a family of extracellular proteasesthat are responsible for the ECM degradation during tissueremodelling under normal and pathological conditions,whereas MMP activity is tightly controlled by the endogenoustissue inhibitors of metalloproteinases (TIMPs).9–12 In par-ticular, the imbalance between MMP and TIMP expression isassociated with myocardial matrix collagen disruption andcardiac remodelling.13,14 Four types of TIMPs have beenidentified. Among these, TIMP-1 is produced by differentcell types, including most types of connective tissue cellsand those involved in the inflammatory processes. It hasbeen demonstrated that AngII increases TIMP-1 expression
* Corresponding author. Tel: þ1 210 458 7390; fax: þ1 210 458 5873.E-mail address: [email protected]
Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2008.For permissions please email: [email protected].
Cardiovascular Research (2008) 78, 523–532doi:10.1093/cvr/cvn028
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from

in rat heart endothelial cells and rat aortic smooth musclecells, whereas TIMP-1 inhibits MMPs.15,16
Exercise training is emerging as an important complemen-tary intervention in heart failure.17,18 Previous studies havedemonstrated that exercise enhances aerobic capacity,attenuates LV dilation, regresses cellular hypertrophy, andimproves cardiomyocyte contractility and myofilament func-tion.2,19,20 These beneficial effects may be, in part, due tothe exercise training-induced attenuation of RAS since it iswell known that inhibition of RAS improves cardiac remodel-ling.5–7 Indeed, exercise training not only normalized circu-lating renin-angiotensin-aldosterone system in animals withMI,21,22 but also lowered plasma AngII in patients with heartfailure.23 However, the impact of exercise training onfactors contributing to myocardial fibrosis has not been elu-cidated. It seems plausible to hypothesize that the exercisetraining-induced improvement of cardiac function is due tothe ameliorated myocardial fibrosis and remodelling. There-fore, the present study was designed to assess the effect ofcombination of AngII blockade and exercise training on myo-cardial remodelling and function after myocardial infarc-tion. We investigated the changes in MMP-2, MMP-9,TIMP-1, ACE, and AT1 at both gene and protein levels afterMI. Our aim was to test the hypothesis that exercise trainingwould beneficially improve factors involving in post-MI fibro-sis, reduce collagen content, thereby attenuating deleter-ious cardiac remodelling and preserving cardiac function.Such effects could be potentiated by addition of angiotensinII type 1 (AT1) receptor blocker, losartan, in conjunctionwith exercise training.
2. Methods
2.1 Animals and myocardial infarction
Animals used in these experiments were treated in accordance withNational Institutes of Health Guide for the Care and Use of Labora-tory Animals, and the study protocols were approved by the Insti-tutional Animal Care and Use Committee at University of Texas atSan Antonio. Seven-week-old male Sprague–Dawley rats (HarlanIndustries, Indianapolis, IN, USA) were housed at constant tempera-ture (22+28C) on a 12 h light/dark cycle. They were fed ad libitumon standard laboratory rat chow and had free access to tap water. MIwas made by ligation of the left anterior descending coronary arteryas described previously.24
2.2 Experimental groups
Echocardiography was performed on the surviving rats 1 week aftersurgery. Rats were matched by cardiac functions using echocardio-graphy and randomly assigned to the following experimentalgroups: Sham-operated control (Sham), sedentary MI (MISed), MI
plus exercise (MI þ Ex), MI plus losartan (MISed þ Los), and MI plusexercise and losartan (MI þ Ex þ Los).
2.3 Drug treatment and exercise training
Losartan treatment (20 mg/kg/day) was initiated 1 week post-MI.The determination of losartan dosage was based on the previousstudies,25,26 which demonstrated positive effect in improvingcardiac function and attenuating cardiac hypertrophy without symp-toms of side effect. The drug administration was performed viagastric gavage twice a day for 8 weeks. Tap water was also givenby gastric gavage to rats without losartan treatment to avoid thepossible physiological alterations associated with gavage-inducedstress. Rats assigned to the exercise groups started exercising at 1week post-MI using a motorized rodent treadmill (Figure 1),whereas the sham and sedentary groups remained sedentarythroughout the experiment period. To allow gradual adaptation toexercise stress, exercise training was initiated at 10 m/min, 58incline for 10 min per session. The speed and duration were gradu-ally increased to 16 m/min and 50 min per session (including a 5 minwarm-up at 10 m/min) and maintained constant throughout theexperiment. Exercise training was performed 5 days per week for8 weeks.
2.4 Echocardiographic measurements
Echocardiographic measurements were performed the day beforethe initiation of post-MI exercise training and after 8 weeks of exer-cise training using an echocardiographic system equipped with a10 MHz transducer (SonoHeart Elite, SonoSite Bothell, WA, USA).Rats were anaesthetized with 2% isoflurane mixed with oxygen,and a two-dimensional short-axis view of the LV was obtained atthe level of the papillary muscle to record M-mode tracings. Wemeasured LV anterior wall thickness; LV end-diastolic dimension(LVEDd) and end-systolic LV dimension (LVESd); LV endocardial frac-tional shortening (FS) was calculated as (LVEDd 2 LVESd)/LVEDd andwas expressed as a percentage.27 All measurements were averagedover three consecutive cardiac cycles.
2.5 In vivo haemodynamic measurements
Following 8 weeks of exercise training, all rats were anaesthetizedas described in the previous section, and the right carotid artery wasexposed. A pressure transducer (Model SPR-838, Millar instruments)was inserted retrograde from the carotid artery to LV cavity. Haemo-dynamic parameters measured were LV systolic pressure (LVSP), LVend-diastolic pressure (LVEDP), aortic systolic pressure, aortic dias-tolic pressure, and peak velocities of contraction and relaxation(dP/dtmax). After measuring, hearts were harvested, frozen in iso-pentane with dry ice, and stored at 2808C until use.
2.6 Determination of infarct size and collagencontent
Cryostat sections (6 mm) of the hearts were stained with collagen-specific picrosiriusred (PSR) for fibrillar collagen measurements at
Figure 1 Experiment flow chart.
X. Xu et al.524
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from
four levels from the apex to the base. Infarct size was calculated bydividing the sum of the planimetered endocardial and epicardial cir-cumferences of the infarcted area by the sum of the total epicardialand endocardial circumferences of the LV.28 Total epicardial andendocardial lengths occupied by the infarct as identified by PSRstaining were measured using computer software (Image Pro Plus,Media Cybernetics, Silver Spring, MD, USA). For collagen volumemeasurement, the PSR stained cross-sections were imaged usingpolarized light which causes clear demarcation of the fibrillar col-lagens.29 The LV cross-sections were digitized using a 20� objectivein the non-infarcted myocardium. Collagen volume in the non-infarcted myocardium was measured using Image Pro Plus program.
2.7 Quantitative real-time polymerase chainreaction
Total RNA was extracted from non-infarcted LV with TRIzol. AfterDNase treatment, 1 mg of total RNA samples was reversed-transcribed with oligo (dT) primers and MMLV reverse transcriptase(Promega, Madison, WI, USA). Quantification of cardiac geneexpression was determined by real-time polymerase chain reaction(PCR). The relative expression of TIMP-1, MMP-2, MMP-9, AT1, andACE mRNA was normalized to the amount of b-actin in the samecDNA using the standard curve method. The primers and probesused in the study were Assay-on-Demand gene expression products(Applied Biosystems). Because of proprietary issues and the policyof Applied Biosystems, the exact primer sequences used for thereal-time PCR experiments are not provided but can be requestedfrom the company based on the information shown in Table 1.
2.8 Western blot
Twenty micrograms of protein was separated by sodium dodecylsulfate-polyacrylamide gel and then transferred to PVDF mem-branes (Bio-Rad, Hercules, CA, USA). Membranes were incubatedwith primary antibodies overnight at 48C. Primary antibodies usedwere anti-MMP-2 (Chemicon), anti-MMP-9 (Sigma), anti-TIMP-1(Chemicon), anti-AT1 receptor (Santa Cruz), and GAPDH (SantaCruz). Membranes were then washed and incubated with horse-radish peroxidase-conjugated secondary antibodies. The mem-branes were detected with enhanced chemiluminescence(Amersham, Little Chalfont, Buckinghamshire, UK) followed byexposure to X-ray film. The protein bands on the X-ray film werescanned, and band density was calculated by Quantity One software(Bio-Rad).
2.9 Angiotensin converting enzyme binding
[125I]351A, a tyrosyl derivative of lisinopril and potent competitiveinhibitor of ACE was used as the radioligand to label ACE. 351A wasiodinated by the chloramines T method and separated from free125I by SP Sephadex C25 column chromatography. ACE binding wasperformed as described previously.30
2.10 Statistical analyses
One-way analyses of variance (ANOVA) were carried out to deter-mine whether there were significant mean differences among theexperiment groups followed by Student–Newman–Keuls post hoccomparisons. A P-value of ,0.05 was considered statistically signifi-cant. Values are expressed as mean+ SEM.
3. Results
3.1 General characteristics and post-infarctsurvival
The exercise training regimen was well tolerated by rats ofboth the MI þ Ex and MI þ Ex þ Los groups. MI was associ-ated with �50% mortality during the first 48 h post-MI. Allthe animals used in our study survived the post-MI trainingperiod for 8 weeks accompanied by twice a day gastricgavage. Table 2 presents the characteristics of animalsincluded in the studies. Only animals with an infarct sizeof more than 30% were included for further analysis. Thus,the population of the five groups was as follows: Sham(n ¼ 10), MISed (n ¼ 9), MI þ Ex (n ¼ 10), MISed þ Los (n ¼12), and MI þ Ex þ Los (n ¼ 13). The infarct size wassimilar in all MI groups with an average range of 40.80–41.67% (Table 2). There was no significant differencefor the body weight among the experimental groups(P . 0.05). The ratio of heart weight to body weight wassignificantly higher in the MI þ Ex (P , 0.05) and MISed(P , 0.01) groups compared with the Sham group, whereasthe ratio was markedly decreased in the MI þ Ex þ Losgroup in comparison to both the MI þ Ex (P , 0.05) andMISed (P , 0.001) groups.
3.2 Echocardiography
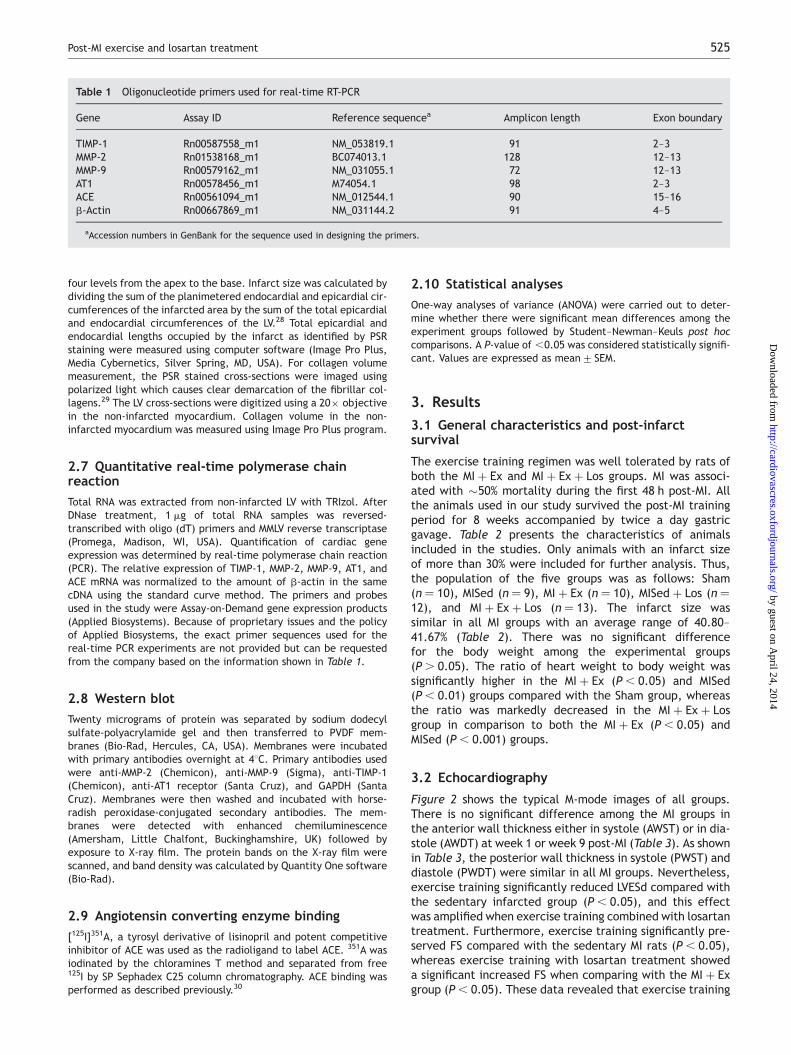
Figure 2 shows the typical M-mode images of all groups.There is no significant difference among the MI groups inthe anterior wall thickness either in systole (AWST) or in dia-stole (AWDT) at week 1 or week 9 post-MI (Table 3). As shownin Table 3, the posterior wall thickness in systole (PWST) anddiastole (PWDT) were similar in all MI groups. Nevertheless,exercise training significantly reduced LVESd compared withthe sedentary infarcted group (P , 0.05), and this effectwas amplified when exercise training combined with losartantreatment. Furthermore, exercise training significantly pre-served FS compared with the sedentary MI rats (P , 0.05),whereas exercise training with losartan treatment showeda significant increased FS when comparing with the MI þ Exgroup (P , 0.05). These data revealed that exercise training
Table 1 Oligonucleotide primers used for real-time RT-PCR
Gene Assay ID Reference sequencea Amplicon length Exon boundary
TIMP-1 Rn00587558_m1 NM_053819.1 91 2–3MMP-2 Rn01538168_m1 BC074013.1 128 12–13MMP-9 Rn00579162_m1 NM_031055.1 72 12–13AT1 Rn00578456_m1 M74054.1 98 2–3ACE Rn00561094_m1 NM_012544.1 90 15–16b-Actin Rn00667869_m1 NM_031144.2 91 4–5
aAccession numbers in GenBank for the sequence used in designing the primers.
Post-MI exercise and losartan treatment 525
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from
appreciably preserved cardiac function after MI withoutcausing scar thinning.
3.3 Haemodynamics
As shown in Table 2, MI caused a significant decrease in LVSPand dP/dtmax, whereas exercise training and losartan treat-ment could maintain them at a higher level compared withthe MISed hearts (P , 0.05). Both systolic aortic pressureand diastolic aortic pressure were significantly higher inthe Sham group than in the MI groups (P , 0.05). Althoughexercise alone to some extent reduced LVEDP in comparisonto the MISed group, the most pronounced effect occurredwhen exercise training was combined with losartan treat-ment (P , 0.05). These results indicate that both systolic
and diastolic functions were well preserved in the MI þ Exgroup after MI.
3.4 Collagen content in the non-infarcted LVmyocardium
Figure 5 shows the representative examples of PSR stainednon-infarcted LV sections under polarized light, as well asthe statistical analysis of PSR staining as the total collagenvolume fraction. The results showed that infarction causedan increase in cardiac fibrosis in the LV non-infarcted myo-cardium compared with the Sham group. Furthermore, thecollagen volume fraction in the MISed group was significantlyhigher than other infarcted groups (P , 0.05). These datarevealed that either exercise training or losartan treatment
Table 2 In vivo haemodynamics and general characteristics
Group Sham (n ¼ 10) MISed (n ¼ 9) MI þ Ex (n ¼ 10) MISed þ Los (n ¼ 12) MI þ Ex þ Los (n ¼ 13)
Infarct size (%) — 41.43+1.51 40.80+1.29 41.67+1.12 41.00+0.86BW (g) 394.0+10.7 393.6+7.4 390.7+8.7 390.5+6.3 384.9+4.3Heart (g)/BW (kg) 3.34+0.05 3.93+0.16** 3.82+0.09* 3.56+0.10 3.38+0.09#‡
LVSP (mmHg) 107.34+2.28 90.84+4.26** 99.24+1.59*† 96.41+0.67* 98.16+1.29*†
LVEDP (mmHg) 4.63+0.76 10.05+1.26** 7.52+1.12 7.35+2.19 5.20+0.67†
LV þ dP/dtmax (mmHg/s) 6482+260.0 3818+567.6** 4933+269.5**† 4720+189.9** 4818.9+125.4**†
LV 2 dP/dtmax (mmHg/s) 6583+419.1 3127+647.6** 4485+254.4**† 4482+408.3**† 4374+257.6**†
Systolic aortic pressure (mmHg) 112.60+1.57 99.78+4.66** 101.76+2.69** 101.46+1.66** 101.12+2.29**Diastolic aortic pressure (mmHg) 84.66+0.78 75.89+3.22** 77.77+2.27* 77.70+2.00* 78.56+1.71*
Values are expressed as mean+ SEM. BW, body weight at sacrificed; LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure;dP/dtmax, peak velocities of contraction and relaxation. *P , 0.05 compared with Sham; **P , 0.01 compared with Sham; †P , 0.05 compared with MISed;‡P , 0.001 compared with MISed; #P , 0.05 compared with MI þ Ex.
Figure 2 M-mode images of echocardiogram in left ventricular, the day before the initiation of post-MI exercise training (pre) and after 8 weeks of exercisetraining (post). These are typical M-mode images presenting each group. LVEDd, left ventricular end-diastolic dimension; LVESd, left ventricular end-systolicdimension.
X. Xu et al.526
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from

attenuated post-MI myocardial fibrosis, and the combinedtreatment of exercise and losartan consolidates the effect.
3.5 Gene expressions in the non-infarcted leftventricular myocardium
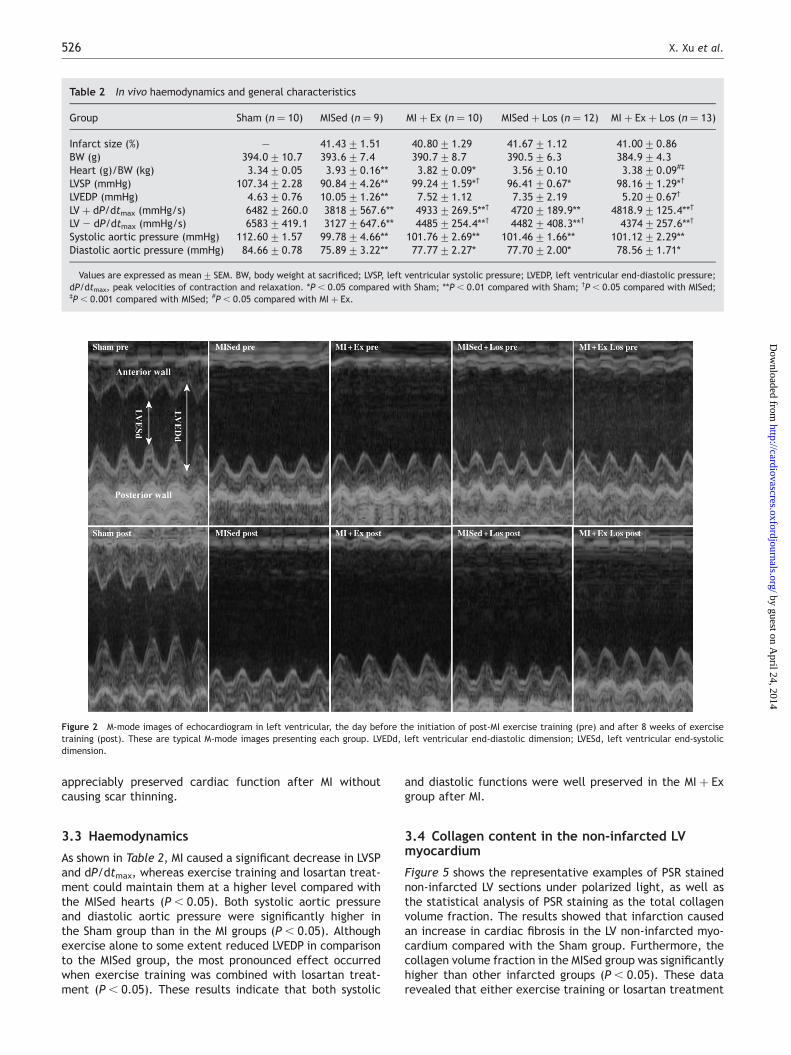
As seen in Figure 4, we did not find any significant differencein the mRNA level of MMP-2, MMP-9, or AT1 among theinfarctedgroups. Interestingly, TIMP-1mRNA levels decreasedrobustly in the MI þ Ex, MI þ Ex þ Los and MISed þ Los groupscompared with the MISed group (P , 0.05). This indicatesthat both exercise training and losartan treatment lowerTIMP-1 expression after 8 weeks of exercise training or losar-tan treatment. In addition, Figure 4 also showed that ACEexpression was significantly lower in the MI þ Ex group thanin the MISed group (P , 0.05), whereas the expression signi-ficantly increased in response to losartan treatment comparedwith their corresponding groups (P , 0.05).
3.6 Protein expressions in the non-infarcted leftventricular myocardium
As shown in Figure 3, we did not find any significant differ-ence in the protein expressions of MMP-2 or MMP-9 amongall the experimental groups. Consistent with mRNA levels,there was significantly less TIMP-1 protein expression inthe MI þ Ex, MI þ Ex þ Los, and MISed þ Los groups com-pared with the MISed group (P , 0.05). Thus, the resultsindicated that both exercise training and losartan treatmenthad TIMP-1 lowering effect. In addition, the expressions ofAT1 receptor protein in the MISed and the MISed þ Losgroups were significantly higher than the Sham group(P , 0.05), whereas the expressions in the MI þ Ex andMI þ Ex þ Los groups were significantly lower than in theMISed and MISed þ Los groups (P , 0.05).
3.7 Angiotensin converting enzyme binding
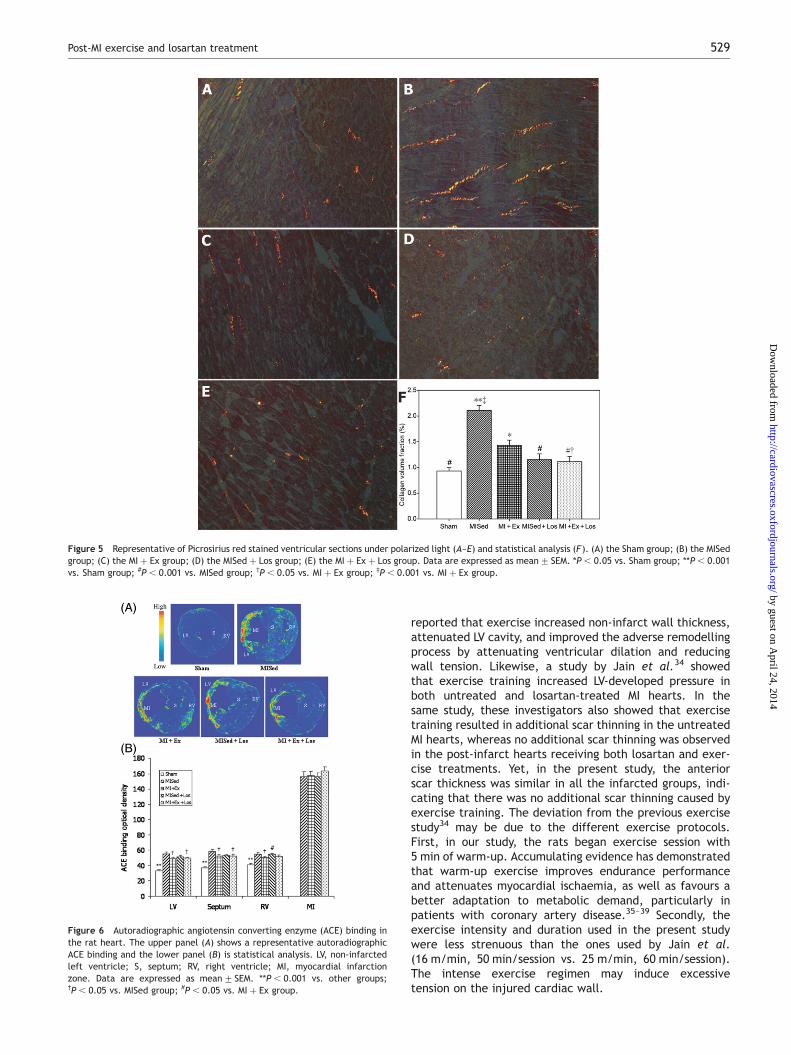
As illustrated in Figure 6, ACE binding density is low in thesham-operated heart, whereas in the infarcted rat heart,it is tremendously increased (P , 0.05) at the site of MI,septum, non-infarcted LV, and right ventricle. When com-pared with other infarcted groups, ACE binding density inthe MISed group was significantly increased in the non-infarcted LV and septum (P , 0.05). In addition, ACEbinding density markedly higher in RV in the MISed þ Losgroup than in the MI þ Ex group (P , 0.05). There was nosignificant difference in ACE binding density at the site ofMI among the four infarcted groups.
4. Discussion
In the present study, we demonstrated that exercise trainingafter MI provides beneficial effect on post-MI cardiac func-tion and LV remodelling by altering the gene and proteinexpressions that regulate myocardial fibrosis, whereas sucheffects only slightly improved by the combination of exer-cise and losartan. These data provide further insights intothe mechanisms underlying the improvement in morbidityand mortality produced by exercise training and ARB inpatients with MI. There are five major findings in thecurrent study. First, both ARB and exercise training signifi-cantly attenuated the expression of TIMP-1 at both geneand protein levels 9 weeks after MI. Secondly, exercise train-ing after MI attenuated the expression of AT1 receptorprotein. Thirdly, exercise training decreased the ACEbinding. Fourthly, both ARB and exercise training after MIsignificantly decreased collagen content and mitigatedcardiac fibrosis. Fifthly, exercise training with or withoutlosartan significantly preserved cardiac function.
Previous studies have demonstrated significant benefitsfrom exercise training post-MI.31,32 Orenstein et al.33
Table 3 Doppler echocardiographic assessment of left ventricular geometry and function
Sham MISed MI þ Ex MISed þ Los MI þ Ex þ Los
1 week post-MIHeart rate 354+6.33 349+7.23 347+6.73 357+6.06 359+5.37AWDT (mm) 1.62+0.09** 0.45+0.03 0.45+0.03 0.45+0.01 0.45+0.02AWST (mm) 2.72+0.07** 0.54+0.04 0.55+0.05 0.53+0.02 0.55+0.02PWDT (mm) 1.90+0.07 1.79+0.06 1.78+0.06 1.80+0.07 1.79+0.04PWST (mm) 2.77+0.08** 2.55+0.08 2.51+0.08 2.52+0.05 2.53+0.05LVEDd (mm) 6.77+0.14** 8.89+0.16 8.87+0.10 8.80+0.14 8.87+0.12LVESd (mm) 3.86+0.15** 7.17+0.18 7.20+0.12 7.14+0.09 7.12+0.11FS (%) 43.17+1.24** 20.56+0.96 19.42+0.60 20.85+0.80 21.31+0.48
9 week post-MIHeart rate 317+6.86 320+6.68 312+5.65 319+4.83 318+4.92AWDT (mm) 1.60+0.07** 0.50+0.05 0.55+0.05 0.50+0.02 0.51+0.02AWST (mm) 2.98+0.09** 0.62+0.06 0.64+0.05 0.60+0.02 0.63+0.02PWDT (mm) 2.04+0.09 2.10+0.14 2.04+0.07 2.00+0.07 1.97+0.06PWST (mm) 2.70+0.17 2.57+0.10 2.57+0.06 2.56+0.05 2.59+0.07LVEDd (mm) 7.77+0.19** 11.41+0.17 11.31+0.11 11.51+0.12 11.25+0.23LVESd (mm) 4.47+0.15** 10.01+0.20‡# 9.52+0.11 9.85+0.12 9.22+0.23FS (%) 42.56+1.09** 12.32+0.85‡ 15.84+0.80†§ 14.38+0.82† 18.17+0.80
LVEDd, left ventricular end-diastolic dimension; LVESd, left ventricular end-systolic dimension; FS, left ventricular fractional shorting; AWST, anterior wallsystolic thickness; AWDT, anterior wall diastolic thickness; PWST, posterior wall systolic thickness; PWDT, posterior wall diastolic thickness. **P , 0.001 com-pared with other MI groups; †P , 0.05 compared with the MI þ Ex þ Los group; ‡P , 0.01 compared with the MI þ Ex þ Los group; #P , 0.05 compared withthe MI þ Ex group; §P , 0.05 compared with the MISed group.
Post-MI exercise and losartan treatment 527
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from
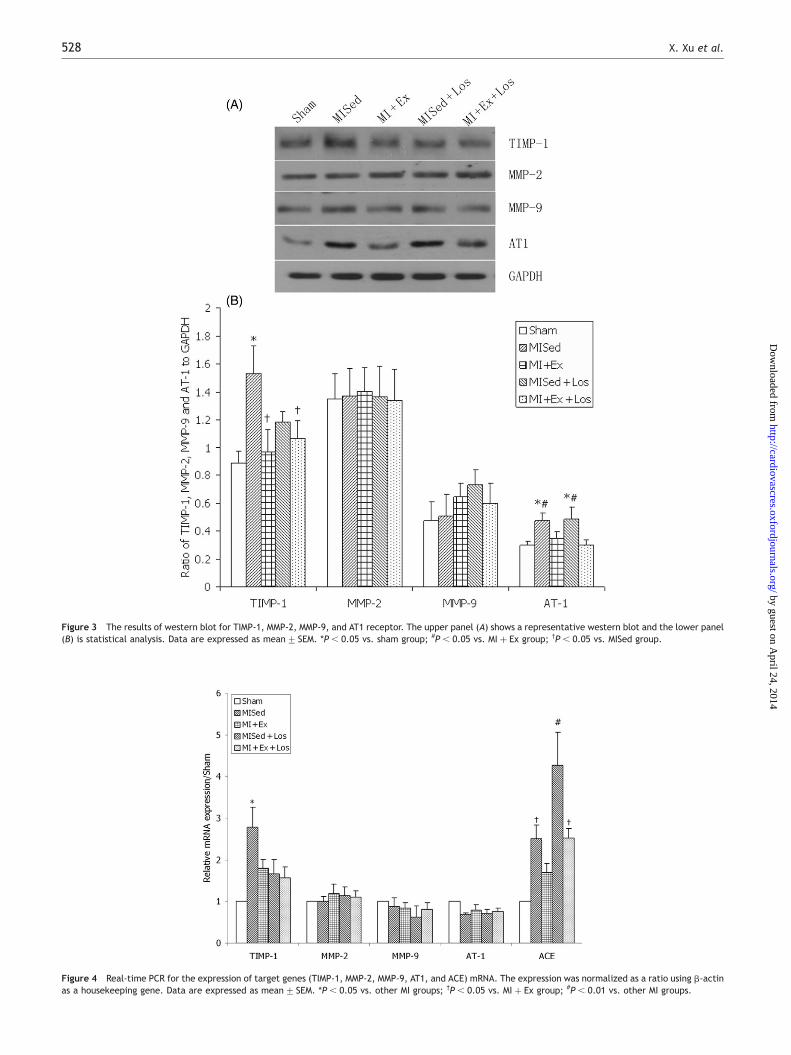
Figure 3 The results of western blot for TIMP-1, MMP-2, MMP-9, and AT1 receptor. The upper panel (A) shows a representative western blot and the lower panel(B) is statistical analysis. Data are expressed as mean+ SEM. *P , 0.05 vs. sham group; #P , 0.05 vs. MI þ Ex group; †P , 0.05 vs. MISed group.
Figure 4 Real-time PCR for the expression of target genes (TIMP-1, MMP-2, MMP-9, AT1, and ACE) mRNA. The expression was normalized as a ratio using b-actinas a housekeeping gene. Data are expressed as mean+ SEM. *P , 0.05 vs. other MI groups; †P , 0.05 vs. MI þ Ex group; #P , 0.01 vs. other MI groups.
X. Xu et al.528
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from
reported that exercise increased non-infarct wall thickness,attenuated LV cavity, and improved the adverse remodellingprocess by attenuating ventricular dilation and reducingwall tension. Likewise, a study by Jain et al.34 showedthat exercise training increased LV-developed pressure inboth untreated and losartan-treated MI hearts. In thesame study, these investigators also showed that exercisetraining resulted in additional scar thinning in the untreatedMI hearts, whereas no additional scar thinning was observedin the post-infarct hearts receiving both losartan and exer-cise treatments. Yet, in the present study, the anteriorscar thickness was similar in all the infarcted groups, indi-cating that there was no additional scar thinning caused byexercise training. The deviation from the previous exercisestudy34 may be due to the different exercise protocols.First, in our study, the rats began exercise session with5 min of warm-up. Accumulating evidence has demonstratedthat warm-up exercise improves endurance performanceand attenuates myocardial ischaemia, as well as favours abetter adaptation to metabolic demand, particularly inpatients with coronary artery disease.35–39 Secondly, theexercise intensity and duration used in the present studywere less strenuous than the ones used by Jain et al.(16 m/min, 50 min/session vs. 25 m/min, 60 min/session).The intense exercise regimen may induce excessivetension on the injured cardiac wall.
Figure 5 Representative of Picrosirius red stained ventricular sections under polarized light (A–E) and statistical analysis (F). (A) the Sham group; (B) the MISedgroup; (C) the MI þ Ex group; (D) the MISed þ Los group; (E) the MI þ Ex þ Los group. Data are expressed as mean+ SEM. *P , 0.05 vs. Sham group; **P , 0.001vs. Sham group; #P , 0.001 vs. MISed group; †P , 0.05 vs. MI þ Ex group; ‡P , 0.001 vs. MI þ Ex group.
Figure 6 Autoradiographic angiotensin converting enzyme (ACE) binding inthe rat heart. The upper panel (A) shows a representative autoradiographicACE binding and the lower panel (B) is statistical analysis. LV, non-infarctedleft ventricle; S, septum; RV, right ventricle; MI, myocardial infarctionzone. Data are expressed as mean+ SEM. **P , 0.001 vs. other groups;†P , 0.05 vs. MISed group; #P , 0.05 vs. MI þ Ex group.
Post-MI exercise and losartan treatment 529
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from

Our study showed that LVSP was markedly higher in theMI þ Ex and MI þ Ex þ Los groups, whereas LVEDP wasnotably lower in the MI þ Ex þ Los group compared withthat in the MISed group. FS was well maintained in theMI þ Ex group after 8 weeks of exercise training and dete-riorated in the MISed group over the same period of time.Furthermore, the beneficial effect was further improvedby the joined treatment of exercise training with losartan.In addition, heart to body weight ratio in the MI þ Ex þ Losgroup was lower than both the MISed and MI þ Ex groups.Thus, these data indicate that exercise training after MIsignificantly preserves LV function without additional scarthinning in this study, and AngII receptor blockade in con-junction with exercise training might make the lattermore beneficial.
The ECM forms a structural network between adjoiningcardiomyocytes, provides the mechanical supports, andmaintains the structural alignment throughout the cardiaccycle. However, after MI, alterations in the collagen matrixcause cardiac muscle stiffness and impair myocytere-lengthening, thus leading to progressive dysfunctionand heart failure.40 In the present study, collagen volumefraction in the MISed group was significantly higher than inboth exercise training and losartan treated groups. Thisfinding suggests that exercise training as well as losartantreatment may attenuate myocardial fibrosis in the infarctedheart, which in turn improves the cardiac function.
Matrix turnover is crucial to tissue repair, while MMPs andTIMPs are the key elements involved in matrix degradationand contribute to myocardial remodelling after MI.Increased MMPs expression or decreased TIMPs expressioncould result in enhanced proteolytic activity and degra-dation of ECM molecules.41 Webb et al.42 demonstratedthat TIMP-1 levels were higher at day 1 post-MI and remainsubstantially elevated through day 180 in patient with MI.The elevated TIMP-1 level may contribute to the accumu-lation of collagen content in the infarcted heart, leadingto myocardial fibrosis.43 Similarly, the present studyrevealed that TIMP-1 level increased significantly in theMISed rats after 9 weeks post-MI. Interestingly, for the firsttime, our study revealed that both exercise training andlosartan notably attenuates TIMP-1 expression at bothgene and protein levels. However, no significant changeswere detected in MMP-2 and MMP-9 among the five exper-imental groups, suggesting that it was just a basalexpression despite exercise training and losartan treatment.Hence, based on our results, we speculate that exercisetraining and losartan treatment after MI may reduce theTIMP-1 expression, improve the balance between MMPsand TIMPs, and enhance the proteolytic activity 9 weekspost-MI, thus decrease the collagen accumulation, therebyreduce the cardiac stiffness, preserve LVSP and dP/dtmax,and reduce LVEDP significantly in the late phase post-MI.
During post-MI remodelling, the circulating RAS is acti-vated to maintain blood volume, blood pressure, andorgan perfusion, but it also adversely affects the injuredheart.44,45 Evidence has shown that RAS not only inducescollagen accumulation globally in the infarct heart causingstiffness of myocardium, but also increases salt and waterretention and total peripheral resistance, eventuallyleading to chronic heart failure.46 Similarly, ACE inhibitionis well known to attenuate remodelling after MI, and its ben-eficial effects have been attributed, in part, to inhibition of
an activated RAS.47 AngII is one of the key factors regulatingcardiac remodelling following MI. Although AT1-receptorappears to mediate many of the deleterious effects ofchronic RAS activity, attenuating the harmful effects of sus-tained AngII stimulation can be achieved by direct antagon-ism of AT receptors.4 Fraccarollo et al.48 reported that LVcollagen expression and accumulation were comparablyreduced by AT1 receptor blockade, thus the reduction ofmyocardial AT1 receptor expression may contribute todecrease cardiac fibrosis. The possible mechanisms bywhich losartan influenced LV remodelling post-MI include areduction in afterload and preload, as well as directcardiac effects. Losartan causes a reduction in afterload,which, in turn, beneficially contributes to the improvementof LV function.8 Also, through the induction of natriuresisand diuresis, losartan has been shown to reduce in vivo LVdiastolic pressure, thereby reducing preload in post-MIhearts and possibly causing some of the beneficial effectsof AngII antagonism.8,49 In addition, some studies havereported that AT1 antagonists decrease mortality, decreasefibrosis in post-MI hearts to a similar extent as ACE inhibitiontherapy, and attenuate overall LV remodelling.5,34 It is worthnoting that our data demonstrate that the expression of AT1receptor significantly decreased at protein levels in responseto exercise training independent of losartan treatment 9weeks after MI. Furthermore, ACE mRNA expression signifi-cantly decreased in the MI þ Ex group when comparedwith MISed group, whereas the expression markedlyincreased in the losartan treated groups compared withthe corresponding untreated groups. The elevated ACEmRNA may be resulted from the tonic negative feedbackregulation induced by ARBs.50,51 On the other hand, ourresults also showed that the ACE binding density is largelyincreased in the infarcted hearts compared with the Shamgroup, which is consistent with the investigation by Sunet al.30 However, the ACE binding density is significantlylower in the exercise training groups than in the MISedgroup. Accordingly, we provide additional mechanismswhereby exercise training may exert beneficial effects onpost-MI myocardial remodelling through the attenuation ofcardiac renin-angiotensin system. To our knowledge, thepresent study is the first to demonstrate the effect ofpost-MI exercise training in conjunction with losartan treat-ment on cardiac ACE and AT1 receptors.
Although the exact mechanisms of post-MI exercisetraining-induced beneficial effects on myocardial remodel-ling are not fully elucidated, several studies have suggestedthat the effect of exercise training may be due to increasedbaroreflex sensitivity, reduced sympathetic activity, andenhanced vagal tone.22,52,53 In addition, a reduction in cir-culating AngII by exercise training may act favourably onbaroreflex control of sympathetic activity.54,55 Furthermore,early decrease of TIMP-1 in the infarcted heart coincideswith collagen degradation in the necrotic myocardium,whereas the subsequent increase of TIMP-1 in the infarctedheart might contribute to collagen accumulation at the latephase of post-MI remodelling.56 Losartan has been shown todecrease neurohormonal factors, such as AngII, norepi-nephrine, aldosterone, atrial natriuretic peptide, brainnatriuretic peptide (BNP), and ET-1.57,58 Therefore, itexerts protective effects on myocardial remodelling byblocking RAS.59 Although we did not measure the BNP inthis study, a significant decrease in plasma BNP along with
X. Xu et al.530
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from

an increase in LV ejection fraction has been observed byKinugawa et al.57 in chronic heart failure patients treatedwith Losartan and subjected to exercise. In addition,based on our study, it is conceivable to speculate thatdecreased expressions of TIMP-1, ACE, and AT1 receptorare involved in the mechanisms by which losartan and exer-cise would attenuate fibrosis and preserve cardiac function.
In summary, this study demonstrates that exercise trainingand/or AngII receptor blockade after MI is likely playing animportant role in cardiac remodelling by attenuatingTIMP-1 expression, improving the balance between MMPsand TIMPs, decreasing ACE and AT1 receptor expression,and thereby decreasing the collagen content. Theseimprovements, in turn, attenuate myocardial fibrosis andcardiac stiffness, and preserve post-MI cardiac function.Exercise training and AngII receptor blockade both havestrong beneficial effects, although the benefit of combiningexercise and AngII receptor blockade appears to be verymodest.
Acknowledgements
The authors thank Bryan Wilson for collagen measurement.
Conflict of interest: none declared.
Funding
This study was supported in part by grants from NationalHeart, Lung, and Blood Institute (RO1-HL074273) andAmerican Heart Association (0430058N).
References1. Gallagher G, Menzie S, Huang Y, Jackson C, Hunyor SN. Regional cardiac
dysfunction is associated with specific alterations in inflammatory cyto-kines and matrix metalloproteinases after acute myocardial infarctionin sheep. Basic Res Cardiol 2007;102:63–72.
2. de Waard MC, van der Velden J, Bito V, Ozdemir S, Biesmans L,Boontje NM et al. Early exercise training normalizes myofilament func-tion and attenuates left ventricular pump dysfunction in mice with alarge myocardial infarction. Circ Res 2007;100:1079–1088.
3. Sun Y, Weber KT. Angiotensin II receptor binding following myocardialinfarction in the rat. Cardiovasc Res 1994;28:1623–1628.
4. Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M. Regulation of genetranscription of angiotensin II receptor subtypes in myocardial infarction.J Clin Invest 1995;95:46–54.
5. Schieffer B, Wirger A, Meybrunn M, Seitz S, Holtz J, Riede UN et al. Com-parative effects of chronic angiotensin-converting enzyme inhibition andangiotensin II type 1 receptor blockade on cardiac remodeling after myo-cardial infarction in the rat. Circulation 1994;89:2273–2282.
6. Raya TE, Fonken SJ, Lee RW, Daugherty S, Goldman S, Wong PC et al.Hemodynamic effects of direct angiotensin II blockade compared to con-verting enzyme inhibition in rat model of heart failure. Am J Hypertens1991;4:334S–340S.
7. Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I et al. Ran-domised trial of losartan versus captopril in patients over 65 with heartfailure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet1997;349:747–752.
8. Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E et al. Effectsof angiotensin-converting enzyme inhibitors and angiotensin II type 1receptor antagonists in rats with heart failure. Role of kinins and angio-tensin II type 2 receptors. J Clin Invest 1997;99:1926–1935.
9. Mott JD, Werb Z. Regulation of matrix biology by matrix metalloprotei-nases. Curr Opin Cell Biol 2004;16:558–564.
10. Spinale FG. Matrix metalloproteinases: regulation and dysregulation inthe failing heart. Circ Res 2002;90:520–530.
11. Peterson JT, Hallak H, Johnson L, Li H, O’Brien PM, Sliskovic DR et al.Matrix metalloproteinase inhibition attenuates left ventricular
remodeling and dysfunction in a rat model of progressive heart failure.Circulation 2001;103:2303–2309.
12. Creemers EE, Davis JN, Parkhurst AM, Leenders P, Dowdy KB, Hapke Eet al. Deficiency of TIMP-1 exacerbates LV remodeling after myocardialinfarction in mice. Am J Physiol Heart Circ Physiol 2003;284:H364–H371.
13. Spinale FG, Coker ML, Thomas CV, Walker JD, Mukherjee R, Hebbar L.Time-dependent changes in matrix metalloproteinase activity andexpression during the progression of congestive heart failure: relationto ventricular and myocyte function. Circ Res 1998;82:482–495.
14. Jayasankar V, Woo YJ, Bish LT, Pirolli TJ, Berry MF, Burdick J et al. Inhi-bition of matrix metalloproteinase activity by TIMP-1 gene transfer effec-tively treats ischemic cardiomyopathy. Circulation 2004;110:II180–II186.
15. Chua CC, Hamdy RC, Chua BH. Angiotensin II induces TIMP-1 production inrat heart endothelial cells. Biochim Biophys Acta 1996;1311:175–180.
16. Castoldi G, Di Gioia CR, Pieruzzi F, D’Orlando C, Van De Greef WM,Busca G et al. ANG II increases TIMP-1 expression in rat aortic smoothmuscle cells in vivo. Am J Physiol Heart Circ Physiol 2003;284:H635–H643.
17. Kavanagh T, Mertens DJ, Hamm LF, Beyene J, Kennedy J, Corey P et al.Prediction of long-term prognosis in 12 169 men referred for cardiacrehabilitation. Circulation 2002;106:666–671.
18. Lee IM, Sesso HD, Oguma Y, Paffenbarger RS Jr. Relative intensity of phys-ical activity and risk of coronary heart disease. Circulation 2003;107:1110–1116.
19. Kemi OJ, Hoydal MA, Haram PM, Garnier A, Fortin D, Ventura-Clapier Ret al. Exercise training restores aerobic capacity and energy transfersystems in heart failure treated with losartan. Cardiovasc Res 2007;76:91–99.
20. Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O. Aerobic exer-cise reduces cardiomyocyte hypertrophy and increases contractility,Ca2þ sensitivity and SERCA-2 in rat after myocardial infarction. Cardio-vasc Res 2002;54:162–174.
21. Wan W, Powers AS, Li J, Ji L, Erikson JM, Zhang JQ. Effect of post-myocardial infarction exercise training on therenin-angiotensin-aldosterone system and cardiac function. Am J MedSci 2007;334:265–273.
22. Liu JL, Irvine S, Reid IA, Patel KP, Zucker IH. Chronic exercise reducessympathetic nerve activity in rabbits with pacing-induced heart failure:a role for angiotensin II. Circulation 2000;102:1854–1862.
23. Braith RW, Welsch MA, Feigenbaum MS, Kluess HA, Pepine CJ. Neuroendo-crine activation in heart failure is modified by endurance exercise train-ing. J Am Coll Cardiol 1999;34:1170–1175.
24. Sun Y, Zhang J, Zhang JQ, Weber KT. Renin expression at sites of repair inthe infarcted rat heart. J Mol Cell Cardiol 2001;33:995–1003.
25. Wang X, Sentex E, Saini HK, Chapman D, Dhalla NS. Upregulation ofbeta-adrenergic receptors in heart failure due to volume overload. AmJ Physiol Heart Circ Physiol 2005;289:H151–H159.
26. Dent MR, Aroutiounova N, Dhalla NS, Tappia PS. Losartan attenuatesphospholipase C isozyme gene expression in hypertrophied hearts dueto volume overload. J Cell Mol Med 2006;10:470–479.
27. Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regardingquantitation in M-mode echocardiography: results of a survey of echocar-diographic measurements. Circulation 1978;58:1072–1083.
28. Mulder P, Boujedaini H, Richard V, Henry JP, Renet S, Munter K et al.Long-term survival and hemodynamics after endothelin-a receptor antag-onism and angiotensin-converting enzyme inhibition in rats with chronicheart failure: monotherapy versus combination therapy. Circulation2002;106:1159–1164.
29. Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assess-ment of myocardial collagen with picrosirius red staining and circularlypolarized light. Basic Res Cardiol 1994;89:397–410.
30. Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growthfactor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol 1998;30:1559–1569.
31. Sullivan MJ, Higginbotham MB, Cobb FR. Exercise training in patients withsevere left ventricular dysfunction. Hemodynamic and metabolic effects.Circulation 1988;78:506–515.
32. Coats AJ, Adamopoulos S, Radaelli A, McCance A, Meyer TE, Bernardi Let al. Controlled trial of physical training in chronic heart failure. Exer-cise performance, hemodynamics, ventilation, and autonomic function.Circulation 1992;85:2119–2131.
33. Orenstein TL, Parker TG, Butany JW, Goodman JM, Dawood F, Wen WHet al. Favorable left ventricular remodeling following large myocardialinfarction by exercise training. Effect on ventricular morphology andgene expression. J Clin Invest 1995;96:858–866.
Post-MI exercise and losartan treatment 531
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from

34. Jain M, Liao R, Ngoy S, Whittaker P, Apstein CS, Eberli FR. Angiotensin IIreceptor blockade attenuates the deleterious effects of exercise trainingon post-MI ventricular remodelling in rats. Cardiovasc Res 2000;46:66–72.
35. Noel M, Jobin J, Marcoux A, Poirier P, Dagenais GR, Bogaty P. Can pro-longed exercise-induced myocardial ischaemia be innocuous? Eur HeartJ 2007;28:1559–1565.
36. Bogaty P, Poirier P, Boyer L, Jobin J, Dagenais GR. What induces thewarm-up ischemia/angina phenomenon: exercise or myocardial ische-mia? Circulation 2003;107:1858–1863.
37. Bogaty P, Kingma JG Jr, Robitaille NM, Plante S, Simard S, Charbonneau Let al. Attenuation of myocardial ischemia with repeated exercise in sub-jects with chronic stable angina: relation to myocardial contractility,intensity of exercise and the adenosine triphosphate-sensitive potassiumchannel. J Am Coll Cardiol 1998;32:1665–1671.
38. Bishop D. Warm up II: performance changes following active warm up andhow to structure the warm up. Sports Med 2003;33:483–498.
39. Arai AE, Grauer SE, Anselone CG, Pantely GA, Bristow JD. Metabolic adap-tation to a gradual reduction in myocardial blood flow. Circulation 1995;92:244–252.
40. Weber KT. Extracellular matrix remodeling in heart failure: a role for denovo angiotensin II generation. Circulation 1997;96:4065–4082.
41. Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloprotei-nase inhibition after myocardial infarction: a new approach to preventheart failure? Circ Res 2001;89:201–210.
42. Webb CS, Bonnema DD, Ahmed SH, Leonardi AH, McClure CD, Clark LLet al. Specific temporal profile of matrix metalloproteinase releaseoccurs in patients after myocardial infarction: relation to left ventricularremodeling. Circulation 2006;114:1020–1027.
43. Lubos E, Schnabel R, Rupprecht HJ, Bickel C, Messow CM, Prigge S et al.Prognostic value of tissue inhibitor of metalloproteinase-1 for cardiovas-cular death among patients with cardiovascular disease: results from theAtheroGene study. Eur Heart J 2006;27:150–156.
44. Persson H, Andreasson K, Kahan T, Eriksson SV, Tidgren B, Hjemdahl Pet al. Neurohormonal activation in heart failure after acute myocardialinfarction treated with beta-receptor antagonists. Eur J Heart Fail2002;4:73–82.
45. Sun Y, Weber KT. RAS and connective tissue in the heart. Int J BiochemCell Biol 2003;35:919–931.
46. Hodsman GP, Kohzuki M, Howes LG, Sumithran E, Tsunoda K, Johnston CI.Neurohumoral responses to chronic myocardial infarction in rats. Circula-tion 1988;78:376–381.
47. Wollert KC, Studer R, von Bulow B, Drexler H. Survival after myocardialinfarction in the rat. Role of tissue angiotensin-converting enzyme inhi-bition. Circulation 1994;90:2457–2467.
48. Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive ame-lioration of left ventricular remodeling and molecular alterations by com-bined aldosterone and angiotensin receptor blockade after myocardialinfarction. Cardiovasc Res 2005;67:97–105.
49. Sladek T, Sladkova J, Kolar F, Papousek F, Cicutti N, Korecky B et al. Theeffect of AT1 receptor antagonist on chronic cardiac response to coronaryartery ligation in rats. Cardiovasc Res 1996;31:568–576.
50. Dandona P, Dhindsa S, Ghanim H, Chaudhuri A. Angiotensin II and inflam-mation: the effect of angiotensin-converting enzyme inhibition andangiotensin II receptor blockade. J Hum Hypertens 2007;21:20–27.
51. Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels inhypertensive humans. Circulation 2005;111:315–320.
52. Pliquett RU, Cornish KG, Patel KP, Schultz HD, Peuler JD, Zucker IH. Ame-lioration of depressed cardiopulmonary reflex control of sympatheticnerve activity by short-term exercise training in male rabbits withheart failure. J Appl Physiol 2003;95:1883–1888.
53. Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced peripheral chemoreflexfunction in conscious rabbits with pacing-induced heart failure. J ApplPhysiol 1999;86:1264–1272.
54. Townend JN, al-Ani M, West JN, Littler WA, Coote JH. Modulation ofcardiac autonomic control in humans by angiotensin II. Hypertension1995;25:1270–1275.
55. Grassi G, Cattaneo BM, Seravalle G, Lanfranchi A, Pozzi M, Morganti Aet al. Effects of chronic ACE inhibition on sympathetic nerve trafficand baroreflex control of circulation in heart failure. Circulation 1997;96:1173–1179.
56. Sun Y, Zhang JQ, Zhang J, Lamparter S. Cardiac remodeling by fibroustissue after infarction in rats. J Lab Clin Med 2000;135:316–323.
57. Kinugawa T, Kato M, Ogino K, Osaki S, Igawa O, Hisatome I et al. Effectsof angiotensin II type 1 receptor antagonist, losartan, on ventilatoryresponse to exercise and neurohormonal profiles in patients withchronic heart failure. Jpn J Physiol 2004;54:15–21.
58. Dahlof B, Zanchetti A, Diez J, Nicholls MG, Yu CM, Barrios V et al. Effectsof losartan and atenolol on left ventricular mass and neurohormonalprofile in patients with essential hypertension and left ventricular hyper-trophy. J Hypertens 2002;20:1855–1864.
59. Schiffrin EL. Remodeling of resistance arteries in essential hypertensionand effects of antihypertensive treatment. Am J Hypertens 2004;17:1192–1200.
X. Xu et al.532
by guest on April 24, 2014
http://cardiovascres.oxfordjournals.org/D
ownloaded from











![[Dissecting aortic aneurysm simulating an acute myocardial infarct]](https://img.pdfslide.net/doc/110x75/63558f23922cbb7c550ca86c/dissecting-aortic-aneurysm-simulating-an-acute-myocardial-infarct.jpg)

















