Embed Size (px)
Citation preview

Gediminas Cepinskas, Cameron W. Lush and Peter R. KvietysMediated Phenomenon−BκAdhesion to Cultured Endothelial Cells : A Nuclear Factor-
Anoxia/Reoxygenation-Induced Tolerance With Respect to Polymorphonuclear Leukocyte
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 1999 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.84.1.1031999;84:103-112Circ Res.
http://circres.ahajournals.org/content/84/1/103World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

Anoxia/Reoxygenation-Induced Tolerance With Respect toPolymorphonuclear Leukocyte Adhesion to
Cultured Endothelial CellsA Nuclear Factor-kB–Mediated Phenomenon
Gediminas Cepinskas, Cameron W. Lush, Peter R. Kvietys
Abstract—Exposing human umbilical vein endothelial cells (HUVECs) to anoxia/reoxygenation (A/R) results in anincrease in polymorphonuclear leukocyte (PMN) adhesion to HUVECs. This A/R-induced hyperadhesion is completelyprevented by a previous (24 hours earlier) exposure of HUVECs to A/R. This phenomenon has been termed “A/Rtolerance.” Exposing HUVECs to A/R induces an increase in nuclear factorkB (NF-kB) in HUVEC nuclei within 4hours. Interfering with either NF-kB activation (proteasome inhibitor) or translocation (double-stranded oligonucleo-tides containing NF-kB binding sequence) prevents the development of A/R tolerance (ie, the increase in A/R-inducedPMN adhesion to HUVECs is the same after the first and second A/R challenges). NO production by HUVECs isincreased after the second A/R challenge, but not after the first A/R challenge. Inhibition of NO synthase (NOS) duringthe second A/R challenge prevents the development of A/R tolerance with respect to PMN adhesion. However, whileHUVECs contained endothelial NOS protein, no inducible NOS was detected in either tolerant or nontolerant cells.Further studies indicated that inhibition of GTP-cyclohydrolase I (an enzyme involved in de novo synthesis of animportant cofactor for NOS activity, tetrahydrobiopterin) prevented the generation of NO in A/R-tolerant cells.Extracellular generation of NO (NO donor) did not effect the hyperadhesion response induced by the initial A/Rchallenge. A/R also induced an oxidant stress in naive HUVECs, but not in A/R-tolerant HUVECs. Inhibition of NOSduring the second A/R insult results in the generation of an oxidant stress similar to that observed after the first A/Rchallenge. Taken together, the findings of the present study are consistent with a role for NF-kB in the development ofA/R tolerance (with respect to PMN adhesion), perhaps by transcriptional regulation of GTP-cyclohydrolase. Theincreased NO production during the second A/R insult reduces PMN adhesion most likely by reducing the intracellularoxidant stress induced by A/R.(Circ Res. 1999;84:103-112.)
Key Words: nitric oxiden nitric oxide synthasen GTP-cyclohydrolase In tetrahydrobiopterin
Reperfusion of previously ischemic tissues results in aseries of events reminiscent of an acute inflammatory
response.1,2 Intravital microscopy approaches have shownthat the microvasculature, particularly the postcapillaryvenules, is the initial target of ischemia/reperfusion (I/R)–induced dysfunction. I/R results in an increase in leukocyteadhesion to postcapillary venules and emigration into theinterstitium, as well as an increase in the microvascularpermeability to albumin. Antioxidants, antagonists of lipidmediators (platelet-activating factor [PAF] and leukotriene B4
[LTB 4]), and immunoneutralization of adhesion molecules onneutrophils (CD11/CD18) or endothelial cells (intercellularadhesion molecule-1) have all been shown to reduce both theneutrophil-endothelial cell adhesion interactions and the in-crease in vascular permeability noted after I/R.
In vitro models have been developed to mimic the micro-vascular dysfunction elicited by I/R. This approach involves
exposing cultured endothelial cells to anoxia (or hypoxia) andsubsequently reoxygenating them (anoxia/reoxygenation;A/R). Challenging endothelial cells with A/R results inincreases in (1) oxidant production, (2) activation of nucleartranscription factors (nuclear factorkB [NF-kB] and activatorprotein-1), (3) adhesion molecule expression, (4) increasedadhesivity to neutrophils, and (5) permeability to macromol-ecules.1 These in vitro approaches have allowed for mecha-nistic studies not readily performed using in vivo models. Forexample, supernatants obtained from A/R-conditioned endo-thelial cells can increase surface expression of CD11/CD18on neutrophils and promote neutrophil adhesion to naiveendothelial monolayers, an effect inhibited by antioxidantsand a PAF receptor antagonist.3 In addition, in vitro ap-proaches have allowed for the characterization of the contri-bution of various adhesion molecules on endothelial cells andneutrophils at different times after reoxygenation.4 Taken
Received June 2, 1998; accepted October 10, 1998.From the Vascular Cell Biology Laboratory, London Health Sciences Centre-Research Inc, London, Ontario, Canada.Correspondence to Peter R. Kvietys, PhD, Vascular Cell Biology Laboratory, London Health Sciences Centre-Research Inc, 375 South St, Room C210,
London, Ontario, Canada N6A 4G5. E-mail [email protected]© 1999 American Heart Association, Inc.
Circulation Researchis available at http://www.circresaha.org
103 by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

together, the in vitro model of A/R very closely simulates thesituation in vivo after I/R and allows for a more in-depthanalysis of the cellular and molecular events involved in thispathology.
Although I/R induces a leukocyte-mediated microvascularand parenchymal cell dysfunction in affected tissue, I/R canalso induce a series of events that renders the tissue moreresistant to a subsequent I/R insult. This phenomenon, termed“I/R tolerance” has been demonstrated in the heart,5,6 kid-neys,7 brain,8 and intestine.9 Typically, I/R tolerance requiresseveral hours or days to develop, indicating that sometranscriptional event may be involved. Studies in the intestineindicate that the development of I/R tolerance is not due to anadaptational response of the parenchymal cells but to somecell type in the lamina propria.9 Endothelial cells are aresident cell population of the lamina propria of a variety oforgan systems. They can also mount an adaptational responseto an oxidant stress that renders them less susceptible to theinjurious effects of a second oxidant challenge imposedseveral hours later.10 Thus, to study the mechanisms involvedin the development of I/R tolerance, we focused our attentionon endothelial cells and used our in vitro model of A/R todefine some of the key cellular events involved in thisphenomenon.
The present study is the first to demonstrate that endothe-lial cells can, indeed, develop A/R tolerance with respect toneutrophil adhesion to endothelium (ie, the typical A/R-induced increase in neutrophil adhesion is abolished if theendothelial cells are pretreated with an A/R insult). We alsopresent evidence indicating that activation and translocationof the nuclear transcription factor (NF-kB) to HUVEC nucleiplays a critical role in the development of this tolerance.Furthermore, we have dissected out some of the criticalpathways involved in the NF-kB-mediated development ofA/R tolerance in endothelial cells and propose a workinghypothesis for future studies of this phenomenon.
Materials and MethodsEndothelial CellsHuman umbilical vein endothelial cells (HUVECs) were harvestedfrom umbilical cords by collagenase treatment (Worthington Bio-chemical, Inc, Freehold, NJ) as previously described.3 The cells weregrown in medium M199 (GIBCO) supplemented with 10% heat-in-activated FCS (Intergen, Purchase, NY), thymidine (2.4 mg/L),heparin sodium (10 IU/mL), penicillin (100 IU/mL), streptomycin(100mg/mL) (Sigma Chemical Co), and endothelial cell mitogen (80mg/mL) (Biomedical Technologies, Stoughton, Mass). The cellcultures were incubated at room air with 5% CO2, 37°C, and 95%humidity and expanded by brief trypsinization with 0.25% trypsin inPBS containing 0.025% EDTA. First through third–passageHUVECs were seeded into fibronectin-coated 48-well tissue cultureplates (Costar) and used for experiments when confluent.
NeutrophilsHuman neutrophilic polymorphonuclear leukocytes (PMNs) wereisolated from venous blood of healthy adults using standard dextransedimentation and gradient separation on Histopaque-1077 (SigmaChemical Co).3 This procedure yields a PMN population that is 95%to 98% viable (trypan blue exclusion) and 98% pure (acetic acid-crystal violet staining).
A/R ProtocolThe in vitro model of I/R used in the present study is similar to thatdescribed previously.3 Briefly, confluent HUVECs monolayers wereexposed to anoxia by incubation in a Plexiglas chamber that wascontinuously purged (1 L/min) with an anoxic gas mixture (93% N2,5% CO2, and 2% H2). To ensure an oxygen-free environment, the gasmixture was passed through a catalytic deoxygenator (Fisher Chem-ical) before entry into the chamber. Temperature in the chamber wasmaintained at 37°C by a heating pad. After a 30-minute period ofanoxia, reoxygenation was initiated by exposing the endothelial cellsto room air in a CO2-cell culture incubator (A/R). As a control,HUVECs were identically treated except that they were exposed tonormoxia (21% O2, 5% CO2, and 74% N2) instead of anoxia(normoxic controls; normoxia/reoxygenation, N/R).
PMN Adhesion AssayIsolated neutrophils were suspended in PBS buffer and radiolabeledby incubating the cells at 53107 cells/mL with a 50 mCiNa51CrO4/mL PMN suspension at 37°C for 60 minutes. Subse-quently, the cells were washed with cold PBS to remove unincorpo-rated radioactivity. Radiolabeled neutrophils (13106/well) wereadded to HUVEC monolayers, and 30 minutes later the percentage ofadded PMNs that remained adherent after a wash procedure wasquantitated using a standard approach.3,11
Electrophoretic Mobility Shift AssayHUVECs grown in 48-well plates were scraped for preparation ofnuclear extracts for electrophoretic mobility shift assay (EMSA) aspreviously described.12 The double-stranded oligonucleotide con-taining consensus (59-AGGGACTTTCCGCTGGGGACTTTCC-39)binding sites for NF-kB (provided by Dr T. Archer) was labeled with[g-32P[ATP (Amersham Canada Ltd) by using T4 polynucleotidekinase (MBI Fermentas Inc) as previously described.13 One pico-mole of the labeled oligonucleotide was incubated with 5mg ofnuclear protein in the presence or absence of a 50-fold excess of coldoligonucleotide for 30 minutes, and the reaction mixture was thenloaded onto native 5% polyacrylamide gel and electrophoresed at250 V in 0.53Tris-borate EDTA buffer. The dried gels then wereexposed to x-ray films (Kodak) for 16 hours in cassettes withintensifying screens.
Nitrite/Nitrate (NO 2–/NO3
–)HUVEC production of NO was determined indirectly by measuringNO2
–/NO3– concentration in HUVEC supernatants as previously
described.14 Briefly, supernatants (100mL) obtained from HUVECsafter treatments (using M199 medium without phenol red) werecollected and incubated for 30 minutes at 37°C in the presence of 0.2U/mL of Aspergillusnitrate reductase (Boehringer Mannheim Can-ada), 50 mmol/L HEPES buffer, (pH 7.4), 5mmol/L flavin adeninedinucleotide, and 0.1 mmol/L NADPH. Subsequently, lactate dehy-drogenase (Boehringer Mannheim Canada) and sodium pyruvate(Sigma Chemical Co) were added to a final concentration of 10U/mL and 10 mmol/L, respectively, and the samples were incubatedfor an additional 10 minutes at 37°C. The Griess reagent was addedto the samples (100mL), and after an additional 15-minute incuba-tion at room temperature, absorbance was read at 540 nm. As astandard, sodium nitrite solution (1 to 50mmol/L) in M199 was used.
Western Blot of NOSAfter treatments, HUVEC monolayers were lysed using a hot 23concentrated electrophoresis sample buffer (135125 mmol/L Tris-HCl, pH 6.8, 2% SDS, and 5% glycerol). Protein concentration of thesamples was measured using a Bio-Rad detergent-compatible proteinassay. Subsequently,b-mercaptoethanol was added to the samples ata final concentration of 1% vol/vol, and they were denatured byboiling for 5 minutes. Ten micrograms of protein was electropho-resed on 7% SDS-polyacrylamide gel and transferred to reinforcednitrocellulose membranes (Schleicher & Schuell, Inc) using anelectrophoretic transfer unit (Hoefer, Inc) at a constant current of 1A for 3 hours in a transfer buffer containing 25 mmol/L Tris,
104 Anoxia/Reoxygenation and Neutrophil Adhesion
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

190 mmol/L glycine, and 20% methanol. Blots were incubatedovernight in a blocking solution (3% BSA and 0.05% Tween 20 inPBS). Subsequently, the membranes were treated with mouse anti-human endothelial NOS (eNOS) monoclonal antibody (mAb) or amouse anti-macrophage NOS antibody (inducible NOS [iNOS]mAb) (Transduction Laboratories Inc). The NOS mAb binding wasdetected using biotinylated anti-mouse IgG and Vectastain Eliteavidin-biotin complex–peroxidase detection system (Vector Labora-tories Inc).
Reactive Oxygen IntermediatesTo assess oxidant production within endothelial cells, we used aquantitative measure of H2O2-derived oxidant formation by monitor-ing the oxidation of dihydrorhodamine 123 (DHR 123) (MolecularProbes, Inc), an oxidant-sensitive fluorochrome.15 HUVEC mono-layers were treated with DHR 123 (5mmol/L) in phenol red-freeM199 for 1 hour before experiments. After treatments, supernatantswere removed, and cells were washed with PBS and lysed in a buffercontaining 0.1% CHAPS, 50 mmol/L K2HPO4 (pH 7.0), and0.1 mmol/L EDTA. The cell lysates were sonicated at a 30% poweroutput for 1 minute and centrifuged at 2000g for 10 minutes at 4°C.The clarified cell sonicates were diluted 40 times and analyzed forDHR 123 oxidation (rhodamine fluorescence) at excitation andemission wavelengths of 502 and 523 nm, respectively. The fluo-rescence intensity was expressed as optical units permg of cellprotein.
InhibitorsA double-stranded (ds) phosphorothioate oligonucleotide (Bio-Synthesis, Inc) containing a consensus NF-kB binding site (sequenceof the sense strand is 59-AGGGACTTTCCGCTGGGGACTTTCC-39, and the antisense strand represents the reverse complement) wasused to inhibit NF-kB translocation to HUVEC nuclei.16 As acontrol, mutant ds-phosphorothioate oligonucleotide (sense strand59-ACTCACTTTCCGCTGCTCACTTTCC-39) was used underidentical conditions (mutated sites are underlined). In addition, aproteasome inhibitor, MG 132 (MyoGenics, Inc) was used to blockNF-kB activation.17 Standard NOS inhibitors were used to assess therole of NO. Nv-nitro-L-arginine methyl ester (L-NAME) or itsinactive entionomer, D-NAME (Sigma Chemical Co) was applied toHUVECs under various experimental conditions usingL-arginine-free M199 medium.L-Arginine was used to counteract the effects ofL-NAME. An inhibitor of GTP-cyclohydrolase I, 2,4-diamino-6-hydroxypyrimidine (DAHP) (Sigma Chemical Co), was used insome experiments.
Statistical AnalysisAll values are presented as mean6SEM. Each experiment wasperformed in triplicate. Statistical analysis was performed usingANOVA and Student’s t test (with Bonferroni corrections formultiple comparisons).P,0.05 was considered statisticallysignificant.
ResultsIn Vitro Model of I/R ToleranceWe have adapted our in vitro model of A/R-induced inflam-mation3 to assess some of the cellular mechanisms involvedin the development of A/R tolerance in endothelial cells.Exposing HUVECs to anoxia for 30 minutes and addingneutrophils (PMNs) upon reoxygenation resulted in an in-crease in PMN adhesion to HUVECs (Figure 1A). Thisobservation is consistent with our previous studies3 andclosely mimics the situation in vivo, in which leukocytesadhere to venular endothelium after reperfusion of ischemictissues.1,2
To mimic the in vivo models of I/R tolerance, HUVECswere exposed to a 30-minute period of anoxia and then
reoxygenated for different periods of time. Subsequently,HUVECs were exposed to a second A/R challenge, at whichtime PMN adhesion to HUVECs was assessed. As shown inFigure 1A, when HUVECs were exposed to an A/R insult and
Figure 1. Development of A/R tolerance with respect to PMNadhesion to HUVECs. A, Initially (0 hours), HUVECs were exposedto N/R or A/R and 24 hours later challenged with either N/R orA/R. 51Cr-labeled PMNs were added after the second N/R or A/Rchallenge, and PMN adhesion was assessed 30 minutes later.Compared with normoxic controls (N/R, N/R), a significant increasein PMN adhesion was observed after an A/R challenge (N/R, A/R).If HUVECs were initially exposed to A/R and subsequently chal-lenged with a second A/R insult (A/R, A/R), PMN adhesion was nodifferent from that observed in normoxic controls (A/R tolerance).n54. *P,0.05 as compared with N/R, A/R; #P,0.05 as comparedwith N/R, N/R. B, If the hiatus between A/R insults (A/R, A/R) wasreduced to 1 or 6 hours, A/R tolerance was not evident (ie, thePMN adhesion was the same as that observed after a single A/Rchallenge [N/R, A/R]). n54. *P,0.05 as compared with N/R, A/R.
Cepinskas et al January 8/22, 1999 105
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

then subsequently challenged with a second A/R insult (24hours later), the level of PMN adhesion to HUVECs after thesecond A/R insult was similar to that observed in normoxiccontrols. This decrease in PMN adhesion was not due to anadverse effect on endothelial cell viability (trypan blueexclusion). These findings indicate that HUVECs mounted anadaptational response to the first A/R insult, which preventedthe hyperadhesion of PMNs to HUVECs after the secondchallenge. We have termed this adaptational response “A/Rtolerance.”
Further characterization experiments indicated that the A/Rtolerance (with respect to PMN adhesion to HUVECs) did notoccur if the hiatus between the 2 A/R challenges was reducedto 1 or 6 hours (Figure 1B). These latter observationssuggested that a transcriptional event may be involved in thedevelopment of A/R tolerance in our model. We focused ourattention on the nuclear transcription factor NF-kB, whichhas been implicated in the regulation of number of geneswhose products modulate inflammation.18,19
Role of NF-kB in the Development ofA/R ToleranceTo assess whether A/R activates NF-kB and, thereby, pro-motes its translocation to the nuclei of HUVECs, we used anEMSA to detect NF-kB in nuclear extracts of HUVECsobtained at different times after the initial A/R insult. Asshown in Figure 2A, there was no significant increase inHUVEC nuclear NF-kB at 30 minutes after reoxygenation.Since PMN adhesion increased within 30 minutes afterreoxygenation, these findings indicate that it is unlikely thatNF-kB plays a role in the hyperadhesive response to theinitial A/R challenge in our model. However, NF-kB waspresent in the nuclei of HUVECs by 4 hours after the initialA/R insult and, therefore, may be involved in the develop-ment of A/R tolerance.
To assess whether activation of NF-kB plays a role in thedevelopment of A/R tolerance, we used 2 experimentalapproaches. First, we pretreated HUVECs with a proteasomeinhibitor (MG 132) that has previously been shown to inhibitNF-kB activation.17 MG 132 was added to HUVECs 30minutes before the initial A/R insult and removed 30 minutesafter reoxygenation. As shown in Figure 2B, this maneuverprevented the development of A/R tolerance; ie, PMN adhe-sion to HUVECs after the second A/R insult was similar tothat observed after an A/R insult imposed on nontolerant cells(N/R, A/R in Figure 2B). Pretreatment of HUVECs with MG132 alone (no initial A/R challenge) did not affect theincrease in PMN adhesion induced by an A/R challengeimposed 24 hours later (data not shown). Another approachwas to pretreat HUVECs with a phosphorothioate oligonu-cleotide, (pt)NF-kB, containing a consensus binding se-quence for NF-kB for 3 hours before the initial A/R chal-lenge. This maneuver has previously been shown to inhibitNF-kB translocation to HUVEC nuclei.16 As shown in Figure2B, (pt)NF-kB was just as effective as the proteasomeinhibitor in preventing the development of A/R tolerance.Pretreatment with a mutated (pt)NF-kB oligonucleotide wasineffective in preventing the development of A/R tolerance.These 2 approaches have previously been shown to be
effective in preventing the translocation of NF-kB to nucleiof HUVECs exposed to A/R.4 In addition, in the present studythese 2 inhibitors prevented NF-kB appearance in HUVECnuclei 4 hours after the initial A/R challenge (data notshown).
Role of NOS in the Development of A/R ToleranceOne anti-inflammatory gene that is activated by NF-kB is theone encoding iNOS. NOS is responsible for the production ofNO, a potent endogenous inhibitor of PMN adhesion toendothelium.20 To assess whether NO may play a role in thedevelopment of A/R tolerance, we measured the concentra-tion of NO2
–/NO3– (Griess reaction) in supernatants obtained
from HUVECs 30 minutes after an A/R challenge. As shownin Figure 3A, the NO2
–/NO3– concentration in supernatants of
HUVECs exposed to an initial A/R challenge were notdifferent from those observed in normoxic controls. How-ever, NO2
–/NO3– levels were increased in response to an A/R
Figure 2. Role of NF-kB in the development of A/R tolerancewith respect to PMN adhesion to HUVECs. A, Effect of A/R onnuclear appearance of NF-kB in HUVECs. HUVECs wereexposed to A/R, and 0.5, 4, and 12 hours after reoxygenation,nuclear extracts were obtained for EMSA. Five micrograms ofextracted protein were incubated with 1 pmol of g-32P-labeledoligonucleotide containing consensus NF-kB–binding sites andsubjected to 5% PAGE under nondenaturing conditions (seeMaterials and Methods for details). A representative experimentis shown; 2 additional experiments yielded similar results.NF-kB in nuclear extracts of HUVECs became apparent by 4hours after reoxygenation. B, Effects of NF-kB inhibitors onPMN adherence to A/R-tolerant HUVECs. The experimentalconditions are the same as those described in Figure 1. In someexperiments, HUVECs were pretreated for 3 hours with20 mmol/L ds-phosphorothioate oligonucleotide containing bind-ing sites for NF-kB, (pt)NF-kB, or a mutated oligonucleotide,D(pt)NF-kB, before the initial A/R. In other experiments,HUVECs were treated with 30 mmol/L MG 132, a proteasomeinhibitor, during the initial A/R and removed 30 minutes afterreoxygenation. Both (pt)NF-kB and MG 132 prevented thedevelopment of A/R tolerance with respect to PMN adhesion.n55. *P,0.05 as compared with N/R, A/R.
106 Anoxia/Reoxygenation and Neutrophil Adhesion
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

challenge imposed on tolerant HUVECs (exposed to an initialA/R challenge 24 hours previously).
Since NO production was increased in tolerant HUVECs,we assessed the role of NO in the development of A/Rtolerance. L-NAME, an NOS inhibitor, was added toHUVECs before the A/R challenges. As shown in Figure 3B,L-NAME had no effect on basal PMN adhesion to HUVECs(normoxic controls) or the hyperadhesion induced after theinitial A/R insult. However, L-NAME prevented the A/Rtolerance with respect to PMN adhesion (ie, the decreasedadhesion response usually noted after the second A/R chal-lenge was no longer observed). The coadministration ofL-arginine prevented this effect of L-NAME. D-NAME had
no effect on the A/R-induced PMN adhesion to HUVECs intolerant cells. Taken together, these findings indicate that thegeneration of NO by HUVEC NOS plays an important role inthe development of tolerance with respect to PMN adhesion.
To determine whether extracellularly generated NO couldbe as effective as intracellularly generated NO in preventingPMN adhesion to HUVECs, we used a NO donor. SpermineNONOate (Molecular Probes; 0.1 mmol/L final concentra-tion) was added along with the PMNs after the initial A/Rchallenge, and PMN adhesion was assessed. Although sperm-ine NONOate increased the concentration of NO2
–/NO3– in the
supernatant (Griess reaction) to levels (16mmol/L) greaterthan those observed after the second A/R challenge (Figure3), the NO donor did not prevent the A/R-induced increase inPMN adhesion to HUVECs (Figure 4).
Western blot analyses of HUVECs under basal conditions(normoxic controls) or after A/R challenges indicated thatHUVECs lack iNOS (data not shown). This observationconfirms previous reports that HUVECs do not containiNOS.21 By contrast, constitutive eNOS was present bothunder basal conditions and after the A/R challenges (Figure5). Interestingly, eNOS protein was increased after an A/Rchallenge to both nontolerant and tolerant HUVECs. Fromthe data presented in Figure 5, it appears that the increase ineNOS protein induced by the initial A/R insult had returnedto near basal levels within 24 hours and that the second A/Rchallenge induced a comparable increase in eNOS protein asobserved after the initial A/R insult.
Role of Tetrahydrobiopterin (BH 4) in theDevelopment of A/R TolerancePrevious studies have indicated that BH4 is an importantco-factor for eNOS activity in HUVECs.21,22 BH4 is synthe-sized by 2 pathways, which are de novo synthesis from GTPand a pterin salvage pathway that regenerates BH4 from
Figure 3. Role of NO in the development of A/R tolerance. A,Generation of NO in A/R-tolerant HUVECs. NO production wasassessed by measuring the concentration of NO2
–/NO3– (Griess
reaction) in the supernatants obtained from HUVECs after expo-sure to normoxic conditions (N/R, N/R), a first A/R challenge(N/R, A/R), and 2 A/R challenges separated by a 24-hour hiatus(A/R, A/R). An increase in NO production above normoxic con-trols was found in A/R-tolerant HUVECs (A/R, A/R). n55.*P,0.05 as compared with N/R, A/R. B, Effects of an NOS in-hibitor (L-NAME) on PMN adhesion to A/R-tolerant HUVECs.The experimental conditions are the same as those described inFigure 1. HUVECs were exposed to normoxic conditions (N/R,N/R), a first A/R challenge (N/R, A/R), and a second A/R chal-lenge administered 24 hours after the first A/R challenge (A/R,A/R). HUVECs were treated with L-NAME (100 mmol/L) duringthe second challenge. The specificity of L-NAME was assessedby introducing L-arginine (300 mmol/L) to the cells along withL-NAME or treating HUVECs with D-NAME (100 mmol/L). n56.L-NAME prevented the development of A/R tolerance withrespect to PMN adhesion. *P,0.05 as compared with N/R, A/R.
Figure 4. Effects of an NO donor, spermine NONOate (Spm-NONO), on PMN adhesion to HUVECs under control conditions(N/R) and after an A/R challenge (A/R). SpmNONO was addedon reoxygenation, and PMN adhesion was assessed 30 minuteslater. The NO donor did not affect PMN adhesion to HUVECs.n53.
Cepinskas et al January 8/22, 1999 107
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

dihydrobiopterin.22,23 The rate-limiting step in the de novosynthesis pathway involves the enzyme GTP-cyclohydrolaseI. Thus, we assessed the effects of DAHP, an inhibitor ofGTP-cyclohydrolase I, on NO production by HUVECs. Asshown in Figure 6, pretreatment of HUVECs with DAHP(5 mmol/L) dramatically reduced NO production byHUVECs under basal conditions (normoxic controls) andafter an A/R insult imposed on naive or tolerant HUVECs.The reduced NO production was not due to a DAHP-inducedadverse effect on HUVEC viability (trypan blue exclusion).These findings indicate that NO production under basalconditions and after the initial A/R insult are dependent onBH4 synthesis by GTP-cyclohydrolase I. More importantly,the increase in NO production usually observed after thesecond A/R challenge was completely abolished.
Interaction of NO With OxidantsSupernatants from A/R-conditioned HUVECs can increasePMN adhesion to naive HUVECs, an effect prevented bycoadministration of catalase in the adhesion assay.3 Further-more, exogenous administration of oxidants to endothelialcells can promote PMN adhesion.2 Taken together theseobservations indicated that exposing HUVECs to A/R in-
duces an oxidant stress within HUVECs, which may promotePMN adhesion to HUVECs. Thus, 1 possible explanation forwhy HUVECs become tolerant to a second A/R challengemay be that HUVECs are subjected to less oxidant stressduring the second challenge. To test this hypothesis,HUVECs were preloaded with DHR 123 for 1 hour beforechallenge with A/R. As shown in Figure 7A, the first A/Rchallenge was associated with an increase in DHR 123oxidation, indicating that an intracellular oxidant stress oc-curred in HUVECs. By contrast, DHR 123 oxidation was notincreased by the second A/R challenge imposed 24 hoursafter the initial A/R insult. DHR 123 uptake by HUVECs wasthe same when loaded before the first or second A/Rchallenge. These findings indicate that the development ofA/R tolerance is associated with a decrease in the ability ofA/R to generate an oxidant stress in HUVECs.
Previous studies indicate that NO can interact with oxi-dants.24,25 Thus, we next tested the possibility that NOgenerated during the second A/R challenge was involved inreducing the oxidant stress incurred during the second A/Rchallenge. As shown in Figure 7B, administration ofL-NAME during the initial A/R insult had no effect on DHR123 oxidation. However, the presence of L-NAME during thesecond A/R challenge prevented the decrease in DHR 123oxidation typically seen after the second challenge (comparewith Figure 7A).
To assess whether the increased NO production and thesuppression of oxidative stress observed in HUVECs after thesecond A/R challenge was mediated by NF-kB, we usedthe proteasome inhibitor MG 132. The proteasome inhibitorwas added to HUVECs 30 minutes before the initial A/Rinsult and removed 30 minutes after reoxygenation. After thesecond A/R challenge either NO2
–/NO3– levels in the super-
natant were measured (Griess reaction) or oxidant stress in
Figure 5. Role of NOS in the development of A/R tolerance. A,Analysis of eNOS protein in HUVECs (Western blot). Cell lysateswere obtained from HUVECs exposed to normoxia (N/R, N/R),an A/R challenge (N/R, A/R), or 2 A/R challenges (A/R, A/R) orfrom HUVECs challenged with an initial A/R insult and reoxy-genated for 24 hours (A/R, N/R). Ten micrograms of total proteinwere subjected to 7% SDS-PAGE and transferred to nitrocellu-lose membranes. The blots were treated with mouse anti-humaneNOS mAb, a biotinylated secondary antibody, and evaluatedusing an avidin-biotin complex–peroxidase detection system.Shown is a representative assay of 8 separate experiments. B,Densitometric analysis of eNOS Western blots. The experimentalconditions are the same as those described in panel A. Valuesare averages of 8 independent experiments. The results indicatethat A/R increases eNOS protein in both A/R-tolerant and-nontolerant HUVECs. *P,0.05 as compared with N/R, N/R.#P,0.05 as compared with A/R, N/R.
Figure 6. The effects of DAHP on the production of NO in A/R-tolerant HUVECs. The experimental conditions are the same asthose in Figure 3A. HUVECs were challenged with normoxia(N/R, N/R), A/R (N/R, A/R), or 2 A/R episodes (A/R, A/R). Insome experiments 5 mmol/L DAHP, a GTP-cyclohydrolase I in-hibitor, was applied to HUVECs 30 minutes before the first chal-lenge. n54. DAHP decreased NO production under all condi-tions. *P,0.05 as compared with respective treatments withoutDAHP.
108 Anoxia/Reoxygenation and Neutrophil Adhesion
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from
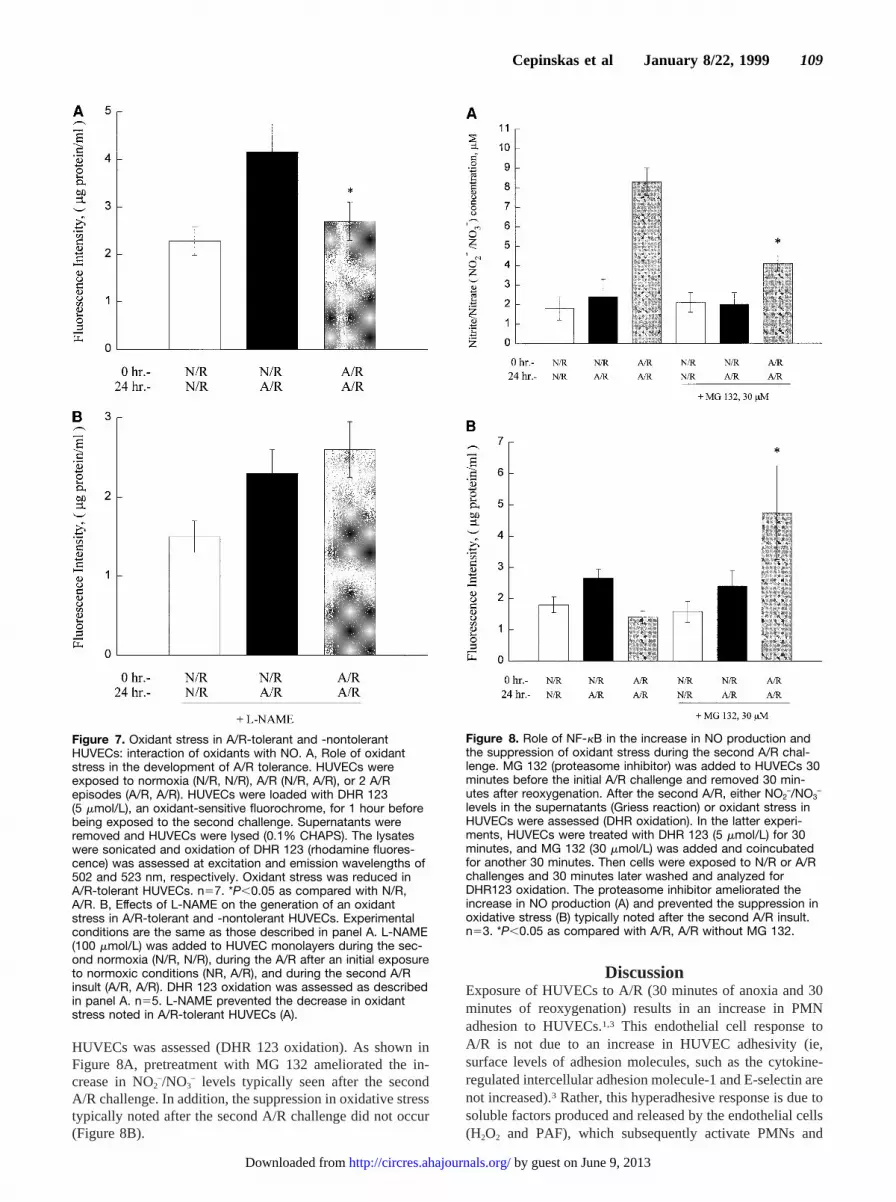
HUVECs was assessed (DHR 123 oxidation). As shown inFigure 8A, pretreatment with MG 132 ameliorated the in-crease in NO2–/NO3
– levels typically seen after the secondA/R challenge. In addition, the suppression in oxidative stresstypically noted after the second A/R challenge did not occur(Figure 8B).
DiscussionExposure of HUVECs to A/R (30 minutes of anoxia and 30minutes of reoxygenation) results in an increase in PMNadhesion to HUVECs.1,3 This endothelial cell response toA/R is not due to an increase in HUVEC adhesivity (ie,surface levels of adhesion molecules, such as the cytokine-regulated intercellular adhesion molecule-1 and E-selectin arenot increased).3 Rather, this hyperadhesive response is due tosoluble factors produced and released by the endothelial cells(H2O2 and PAF), which subsequently activate PMNs and
Figure 7. Oxidant stress in A/R-tolerant and -nontolerantHUVECs: interaction of oxidants with NO. A, Role of oxidantstress in the development of A/R tolerance. HUVECs wereexposed to normoxia (N/R, N/R), A/R (N/R, A/R), or 2 A/Repisodes (A/R, A/R). HUVECs were loaded with DHR 123(5 mmol/L), an oxidant-sensitive fluorochrome, for 1 hour beforebeing exposed to the second challenge. Supernatants wereremoved and HUVECs were lysed (0.1% CHAPS). The lysateswere sonicated and oxidation of DHR 123 (rhodamine fluores-cence) was assessed at excitation and emission wavelengths of502 and 523 nm, respectively. Oxidant stress was reduced inA/R-tolerant HUVECs. n57. *P,0.05 as compared with N/R,A/R. B, Effects of L-NAME on the generation of an oxidantstress in A/R-tolerant and -nontolerant HUVECs. Experimentalconditions are the same as those described in panel A. L-NAME(100 mmol/L) was added to HUVEC monolayers during the sec-ond normoxia (N/R, N/R), during the A/R after an initial exposureto normoxic conditions (NR, A/R), and during the second A/Rinsult (A/R, A/R). DHR 123 oxidation was assessed as describedin panel A. n55. L-NAME prevented the decrease in oxidantstress noted in A/R-tolerant HUVECs (A).
Figure 8. Role of NF-kB in the increase in NO production andthe suppression of oxidant stress during the second A/R chal-lenge. MG 132 (proteasome inhibitor) was added to HUVECs 30minutes before the initial A/R challenge and removed 30 min-utes after reoxygenation. After the second A/R, either NO2
–/NO3–
levels in the supernatants (Griess reaction) or oxidant stress inHUVECs were assessed (DHR oxidation). In the latter experi-ments, HUVECs were treated with DHR 123 (5 mmol/L) for 30minutes, and MG 132 (30 mmol/L) was added and coincubatedfor another 30 minutes. Then cells were exposed to N/R or A/Rchallenges and 30 minutes later washed and analyzed forDHR123 oxidation. The proteasome inhibitor ameliorated theincrease in NO production (A) and prevented the suppression inoxidative stress (B) typically noted after the second A/R insult.n53. *P,0.05 as compared with A/R, A/R without MG 132.
Cepinskas et al January 8/22, 1999 109
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

enhance their ability to adhere to HUVECs. In the presentstudy, we provide evidence that HUVECs can develop A/Rtolerance with respect to A/R-induced PMN adhesion toHUVECs. Our findings indicate that the A/R-induced in-crease in PMN adhesion to HUVECs can be completelyabolished by pretreating HUVECs with an A/R challenge 24hours previously (Figure 1). This adaptational response re-quires more than 6 hours to develop (Figure 1B), indicatingthat some transcriptional event may be involved.
We focused our attention on the nuclear transcriptionfactor NF-kB, since it regulates the activity of a variety ofgenes whose products modulate inflammation.18,19,26 Forexample, activation of NF-kB leads to the transactivation ofproinflammatory genes, such as those encoding (1) endothe-lial adhesion molecules involved in PMN adhesion to endo-thelial cells and (2) cytokines and growth factors.18 However,more relevant to the development of A/R tolerance in ourmodel are the anti-inflammatory genes activated by NF-kB.The potential anti-inflammatory effects of NF-kB activation,coupled to the ability of PAF and H2O2 (factors implicated inthe increased PMN adhesion to HUVECs in our model ofA/R) to activate NF-kB, prompted us to assess the potentialrole of NF-kB in the development of A/R tolerance in oursystem.
In endothelial cells, NF-kB exists as a heterodimer con-sisting of subunits designated as p50 and p65. It is localizedto the cytoplasm in an inactive form by virtue of itsassociation with a monomeric inhibitory protein, IkB. Acti-vation of NF-kB occurs by phosphorylation, ubiquitination,and subsequent degradation of IkB by the proteasome, amulticatalytic protease.18,19The loss of IkB allows the NF-kBheterodimer to translocate to the nucleus and initiate thetranscription of genes regulating the inflammatory process.As shown in Figure 2A, NF-kB is activated by the initial A/Rinsult, since it appears within HUVEC nuclei within 4 hours.Furthermore, a proteasome inhibitor (MG 132), which hasbeen shown to inhibit activation of NF-kB in HUVECs,17
prevented the development of A/R tolerance with respect toPMN adhesion to HUVECs (Figure 2B). Finally, interferingwith NF-kB translocation to the nuclei by using a double-stranded oligonucleotide containing NF-kB binding sites16
also prevented the development of A/R tolerance (Figure 2B).Taken together, our findings indicate that activation andtranslocation of NF-kB to the nuclei of HUVECs is animportant component of the development of A/R tolerance.Interestingly, NF-kB was detected in HUVEC nuclei at 4hours after the initial A/R challenge (Figure 2A), whereas thedevelopment of A/R tolerance with respect to PMN adhesionwas not detected as late as 6 hours after the initial A/Rchallenge (Figure 1B). The reason for the delay in thedevelopment of A/R tolerance is not clear, but delay may bedue to the time required for NF-kB to induce transactivationof relevant gene(s) and subsequent protein synthesis.
The gene(s) that NF-kB transactivates within HUVECs inorder for the anti-inflammatory phenotype to be manifested inA/R-tolerant cells (decreased PMN adhesion) is unclear.Since NO is an endogenous antiadhesive molecule, genesregulating NO production are possible candidates. Thus, wefirst examined the possibility that an increased production of
NO was involved in the development of A/R tolerance. Ourfindings indicate that, while the first A/R insult is notassociated with increased NO production, the second A/Rchallenge is (Figure 3A). Furthermore, inhibition of NOSactivity with L-NAME during the second A/R challengeprevented the development of A/R tolerance with respect toPMN adhesion to HUVECs (Figure 3B). These findingsindicate that an increase in NO production during the secondA/R challenge contributes to the manifestation of A/Rtolerance.
One likely gene target for NF-kB that would enhance NOproduction during the second A/R challenge is iNOS, whichcontains NF-kB–binding sites in its promoter region27 and istransactivated by NF-kB.18 However, Western blot analysesindicated that iNOS was not present in HUVECs under basalconditions or after challenge with A/R. These observationsare consistent with a previous report indicating that HUVECsdo not contain iNOS.21 On the other hand, eNOS was presentunder basal conditions and was increased by both the initialand second A/R challenges (Figure 5). The mechanismsinvolved in the A/R-induced increase in eNOS protein ob-served in the present study are unclear. Previous reports havenoted that eNOS message and protein can be increased bymechanical,28 shear,29 and oxidative30 stresses applied toendothelial cells. The modulation of eNOS message andprotein levels appear to involve transcriptional28–30 and/orposttranscriptional28 events. Thus, it could be argued that thedevelopment of A/R tolerance involves NF-kB-induced tran-scription of eNOS. That this is most likely not the case isbased on the following lines of evidence. First, although SP-1and activator protein-1–binding elements have been identi-fied in the promoter region of the human eNOS gene,31,32
there does not appear to be an NF-kB–binding site on thegene encoding eNOS. Second, while eNOS protein is in-creased within 30 minutes of reoxygenation (Figure 5), thereis no detectable NF-kB in the nucleus at this time (Figure 2).Third, although eNOS protein was increased during the initialA/R (Figure 5), there was no corresponding increase in NOproduction (Figure 3A). Thus, the NF-kB-mediated develop-ment of A/R tolerance (with respect to NO production andPMN adhesion) most likely involves some other transcrip-tional event besides induction of eNOS.
BH4 is an important cofactor for NOS activity. In thepresent study, inhibition of GTP-cyclohydrolase I, the en-zyme responsible for the de novo synthesis of BH4, com-pletely abolished NO synthesis by HUVECs (Figure 6). Ofparticular relevance to the present study is the observationthat the gene encoding for GTP-cyclohydrolase I contains anNF-kB–binding site in the promoter region.33 In addition,cytokine-induced NO production by endothelial cells,21 myo-cytes,34 and smooth muscle cells23 is dependent on GTP-cyclohydrolase I activity. Thus, taken together, our findingssuggest that NF-kB may contribute to the development ofA/R tolerance by transactivating the gene encoding GTP-cyclohydrolase I. Further studies are warranted to moredirectly address this possibility.
One interesting observation in the present study was thatthere appears to be an interaction between NO and oxidants inHUVECs during the development of A/R tolerance. The
110 Anoxia/Reoxygenation and Neutrophil Adhesion
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

initial A/R induced an oxidant stress, while the subsequentA/R challenge did not (Figure 7A). Furthermore, with addi-tion of the NOS inhibitor (L-NAME) during the second A/Rchallenge, the degree of A/R-induced oxidant stress wassimilar to that observed after the initial A/R challenge. Thesefindings indicate that the increased NO production during thesecond A/R challenge reduces the oxidant stress typicallyinduced by A/R. This contention is consistent with otherreports showing that, when NO production exceeds oxidantproduction, oxidant-mediated lipid peroxidation25 or hy-droxylation of benzoate24 is substantially inhibited. It is notentirely clear whether the decreased PMN adhesion to A/R-tolerant HUVECs is due directly to an increased NO produc-tion or the fact that NO decreases the oxidant stress. How-ever, since exogenous generation of NO (NO donor; spermineNONOate) during the initial A/R challenge did not preventthe hyperadhesion response (Figure 4), it is unlikely that NOdirectly inhibits PMN adhesion to HUVECs. Thus, the mostlikely explanation for our findings is that intracellularlygenerated NO inhibits PMN adhesion during the second A/Rchallenge by reducing the oxidant stress.
Figure 9 schematically depicts our working hypothesis onthe mechanisms involved in the development of A/R toler-ance in HUVECs. We propose that after the initial A/R insultan oxidant stress is generated within HUVECs that leads to anincrease in PMN adhesion to HUVECs (Figures 1 and 7). Onthe basis of our previous studies and those of others, it islikely that H2O2 plays an important role in this hyperadhesionvia generation of PAF.1,2 In addition, the initial A/R insultinduces a transcriptional event that leads to the developmentof A/R tolerance with respect to A/R-induced PMN adhesionto HUVECs. Specifically, NF-kB is activated and translo-cates to the nucleus (Figure 2). NF-kB then promotes thetranscription of a relevant gene(s), which results in anincrease in NO production by eNOS during the second A/Rchallenge. Our preliminary findings (inhibitor studies) indi-cate that NF-kB may transactivate the gene encoding GTP-cyclohydrolase I, which subsequently results in the synthesisof BH4, an important cofactor for eNOS activity (Figure 6).The increase in NO production during the second A/Rchallenge prevents the oxidant stress within HUVECs (Fig-
ures 7 and 8). The decrease in PMN adhesion to HUVECsafter the second A/R challenge may be a result of the lack ofan oxidant stress and associated PAF production. In thepresent study we also provide evidence that NF-kB isinvolved in the increased NO production and the suppressionof the A/R-induced oxidant stress (Figure 8). Although theresults of the present study support various aspects of thisworking hypothesis, further studies are necessary to firmlyestablish the relative roles of various proposed factors in thedevelopment of A/R tolerance.
AcknowledgmentsThis work was supported by grants MT-13668 and MT-12902 fromthe Medical Research Council of Canada. We thank Dr TrevorArcher, London Regional Cancer Center, for the synthesis of NF-kBoligonucleotides and Ronald Noseworthy for technical assistance.
References1. Kvietys PR, Granger DN. Endothelial cell monolayers as a tool for
studying microvascular pathophysiology.Am J Physiol. 1997;273:G1–G10.
2. Kvietys PR, Cepinskas G, Granger DN. Neutrophil-endothelial cell inter-actions during ischemia/reperfusion. In: Kamada T, Shiga T, McCuskeyRS, eds.Tissue Perfusion and Organ Function: Ischemia/ReperfusionInjury. Amsterdam, the Netherlands: Elsevier; 1996:179–191.
3. Yoshida N, Granger DN, Anderson DC, Rothlein R, Lane C, Kvietys PR.Anoxia/reoxygenation-induced neutrophil adherence to cultured endothe-lial cells. Am J Physiol. 1992;262:H1891–H1898.
4. Ischikawa H, Kvietys PR, Wolf RE, Yohikawa T, Granger DN, Aw TY.Molecular mechanisms of anoxia/reoxygenation-induced neutrophiladherence to cultured endothelial cells.Circ Res. 1997;81:922–931.
5. Yellon DM, Baxter GF. A “second window of protection” or delayedpreconditioning phenomenon: future horizons for myocardial protection.J Mol Cell Cardiol. 1995;27:1023–1034.
6. Marber MS, Yellon DM. Myocardial adaptation, stress proteins, and thesecond window of protection.Ann N Y Acad Sci. 1996;793:123–141.
7. Yoshioka T, Bills T, Moore-Jarrett T, Greene H, Burr IM, Ichikawa I.Role of intrinsic antioxidant enzymes in renal oxidant injury.Kidney Int.1990;38:282–288.
8. Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R,Ueda H, Handa N, Kimura K, Kamada T. “Ischemic tolerance” phe-nomenon detected in various brain regions.Brain Res. 1991;561:203–211.
9. Osborne DL, Aw TY, Cepinskas G, Kvietys PR. Development of ische-mia/reperfusion tolerance in the rat small intestine: an epithelium-independent event.J Clin Invest. 1994;94:1910–1918.
10. Lu D, Maulik N, Moraru I, Kreutzer DL, Das DK. Molecular adaptationof vascular endothelial cells to oxidative stress.Am J Physiol. 1993;264:C715–C722.
11. Cepinskas G, Noseworthy R, Kvietys PR. Transendothelial neutrophilmigration: role of neutrophil-derived proteases and relationship to trans-endothelial protein movement.Circ Res. 1997;81:618–626.
12. Scheinman RI, Beg AA, Baldwin ASJ. NF-kappa B p100 (Lyt-10) is acomponent of H2TF1 and can function as an I kappa B-like molecule.Mol Cell Biol. 1993;13:6089–6101.
13. Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have oppositeeffects on activation of NF-kappa B and AP-1 in intact cells: AP-1 assecondary antioxidant-responsive factor.EMBO J. 1993;12:2005–2015.
14. Grisham MB, Johnson GG, Gautreaux MD, Berg RD. Measurement ofnitrate and nitrite in extracellular fluids: a window to systemic nitric oxidemetabolism.Methods: Companion Methods Enzymol. 1997;7:84–90.
15. Royall JA, Ischiropoulos H. Evaluation of 29-79-dichlorofluorescin anddihydrorhodamine 123 as fluorescent probes for intracellular H2O2 incultured endothelial cells.Arch Biochem Biophys. 1993;302:348–355.
16. Bielinska A, Shivdasani RA, Zhang L, Nabel GJ. Regulation of geneexpression with double-stranded phosphorothioate oligonucleotides.Science. 1990;250:997–1000.
17. Read MA, Neroh AS, Luscinskas FW, Palombella VJ, Maniatis T, CollinsT. The proteasome pathway is required for cytokine-induced endotheli-al-leukocyte adhesion molecule expression.Immunity. 1995;2:493–506.
Figure 9. Schematic of a working hypothesis illustrating thepotential mechanisms involved in A/R tolerance with respect toPMN adhesion to HUVECs. ROIs indicate reactive oxygenintermediates.
Cepinskas et al January 8/22, 1999 111
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from

18. Siebenlist U, Franzoso G, Brown K. Structure, regulation and function ofNF-kB. Annu Rev Cell Biol. 1994;10:405–455.
19. Thanos D, Maniatis T. NF-kB: a lesson in family values.Cell. 1995;80:529–532.
20. Kubes P, Granger DN. Leukocyte-endothelial cell interactions evoked bymast cells.Cardiovasc Res. 1996;32:699–708.
21. Rosenkranz-Weiss P, Sessa WC, Milstien S, Kaufman S, Watson CA, PoberJS. Regulation of nitric oxide synthesis by proinflammatory cytokines inhuman umbilical vein endothelial cells.J Clin Invest. 1994;93:2236–2243.
22. Werner-Felmayer G, Werner ER, Fuchs D, Hausen A, Reibnegger G,Schmidt K, Weiss G, Wachter H. Pteridine biosynthesis in human endo-thelial cells.J Biol Chem. 1993;268:1842–1846.
23. Nakayama DK, Geller DA, Di Silvio M, Bloomgarden G, Davies P, PittBR, Hatakeyama K, Kagamiyama H, Simmons RL, Billiar TR. Tetrahy-drobiopterin synthesis and inducible nitric oxide production in pulmonaryartery smooth muscle.Am J Physiol. 1994;266:L455–L460.
24. Miles AM, Bohle DS, Glassbrenner PA, Hansert B, Winks DA, GrishamMB. Modulation of superoxide-dependent oxidation and hydroxylationreactions by nitric oxide.J Biol Chem. 1996;271:40–47.
25. Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, KirkM, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation: formation of novel nitrogen-containingoxidized lipid derivatives.J Biol Chem. 1994;269:26066–26075.
26. Collins T. Biology of disease: endothelial nuclear factor-kB and theinitiation of the atherosclerotic lesion.Lab Invest. 1993;68:499–508.
27. Kim YM, Lee BS, Yi KY, Paik SG. Upstream NF-kappa B site is requiredfor the maximal expression of mouse inducible nitric oxide synthase gene
in interferon-gamma plus lipopolysaccharide-induced RAW 264.7 mac-rophages.Biochem Biophys Res Commun. 1998;236:655–660.
28. Awolesi MA, Sessa WC, Sumpio BE. Cyclic strain upregulates nitricoxide synthase in cultured bovine aortic endothelial cells.J Clin Invest.1995;96:1449–1454.
29. Xiao Z, Zhang Z, Diamond SL. Shear stress induction of the endothelialnitric oxide synthase gene is calcium dependent but not calcium activated.J Cell Physiol. 1997;171:205–211.
30. North AJ, Lau KS, Brannon TS, Wu LC, Wells LB, German Z, Shaul PW.Oxygen upregulates nitric oxide synthase gene expression in ovine fetalpulmonary artery endothelial cells.Am J Physiol. 1996;270:L643–L649.
31. Tang J-L, Zembowicz A, Xu X-M, Wu KK. Role of SP1 in transcriptionalactivation of human nitric oxide synthase type III gene.Biochem BiophysRes Commun. 1995;213:673–680.
32. Janssens SP, Shimouchi A, Quertermous T, Bloch DB, Bloch KD.Cloning and expression of a cDNA encoding human endothelium-derivedrelaxing factor/nitric oxide synthase.J Biol Chem. 1992;267:14519–14522.
33. Witter K, Werner T, Blusch JH, Schneider E-M, Riess O, Ziegler I, RodlW, Bacher A, Gutlich M. Cloning, sequencing and functional studies ofthe gene encoding human GTP cyclohydrolase I.Gene. 1996;171:285–290.
34. Kasai K, Hattori Y, Banba N, Hattori S, Motohashi S, Shimoda S-I,Nakanishi N, Gross SS. Induction of tetrahydrobiopterin synthesis in ratcardiac myocytes: impact on cytokine-induced NO generation.Am JPhysiol. 1997;273:H665–H672.
112 Anoxia/Reoxygenation and Neutrophil Adhesion
by guest on June 9, 2013http://circres.ahajournals.org/Downloaded from





























