Embed Size (px)
Citation preview

1
Aryl Hydrocarbon Receptor Modulation of TNFαααα-induced Apoptosis and Lysosomal Disruption ina Hepatoma Model that is Caspase-8 Independent
Joseph A. Caruso‡, Patricia A. Mathieu‡, Aby Joiakim‡, Hong Zhang¶ and John J. Reiners, Jr.‡
‡Institute of Environmental Health Sciences, Wayne State University, Detroit, Michigan, 48201 and¶Gene Trove Division, Isis Pharmaceuticals, Inc., Carlsbad, California, 92008
Running Title: AhR modulation of TNF-induced Apoptosis
Address correspondence to: John J. Reiners, Jr., Institute of Environmental Health Sciences, Wayne StateUniversity, 2727 Second Ave., Detroit, Michigan, Tel. 313 963-7662; Fax. 313 577-0082; E-Mail:[email protected]
Recent studies suggest that the arylhydrocarbon receptor (AhR) modulatessusceptibilities to some pro-apoptotic agents.AhR-containing murine hepatoma 1c1c7cultures underwent apoptosis followingexposure to TNFαααα + cycloheximide (CHX). Incontrast, Tao cells, an AhR-deficient variant ofthe 1c1c7 line, were refractory to thistreatment. AhR sense/antisense transfectionstudies demonstrated that AhR contentsinfluenced susceptibility to the pro-apoptoticeffects of TNFαααα + CHX. 1c1c7 cells and allvariants expressed comparable amounts ofTNF receptor-1 and TRADD. However, no cellline expressed FADD, and consequently pro-caspase-8 was not activated. AhR content didnot influence JNK and NF-κκκκB activation.However, Bid, pro-caspase-9, -3 and –12processing occurred only in AhR-containingcells. Analyses of cathepsin B and D activities indigitonin-permeabilized cultures, and themonitoring of cathepsin B/D co-localizationwith Lamp-1, indicated that TNFαααα + CHXdisrupted late endosomes/lysosomes in onlyAhR-containing cells. Stabilization of acidicorganelles with 3-O-methylsphingomyelininhibited TNFαααα + CHX induced apoptosis. Thecathepsin D inhibitor pepstatin A suppressed invitro cleavage of Bid by 1c1c7 lysosomalextracts. It also delayed the induction ofapoptosis, and partially prevented Bid cleavageand pro-caspases-3/7 activation in culturestreated with TNFαααα + CHX. Similar suppressiveeffects occurred in cultures transfected withmurine Bid antisense oligonucleotides. Thesestudies show that in cells where pro-caspase-8 isnot activated, TNFαααα + CHX can initiateapoptosis through lysosomal disruption.
Released proteases such as cathepsin D triggerthe apoptotic program by activating Bid.Furthermore, in the absence of exogenousligand, the AhR modulates lysosomaldisruption/permeability.
INTRODUCTION
The late endosomal / lysosomal network(referred to hereafter as ‘lysosomes’) consists of aseries of dynamic and interactive acidicorganelles. Lysosomes are involved in numerousintracellular processes, including plasmamembrane and receptor recycling, cholesteroltrafficking, antigen processing, autophagy,sphingolipid metabolism, and protein degradation(1-3). Recent studies have also implicated roles forlysosomal constituents in the induction ofapoptosis (reviewed in 4). Specifically, manyexogenous and endogenous agents facilitatelysosomal membrane permeabilization, causingrelease of resident proteases into the cytosol andinduction of apoptosis. Amongst such agents arehydrogen peroxide (5,6), generators of reactiveoxygen species (7-9), sphingosine (10), amyloid βpeptide (11), α -tocopheryl succinate (12),methylmercury (6), VP-16 (13) and tumor necrosisfactor alpha (TNFα1, 14-19). Studies employingmolecular and pharmacological approaches toregulate the expressions or the activities of thelysosomal proteases cathepsins B and D (8,13-18),or employing cell lines having acidic organellesdeficient in these proteases due to a traffickingdisorder (20,21), have clearly implicated a role forthese two proteases in the pro-apoptotic effects ofseveral of the above agents.
The mechanism by which lysosomalproteases induce apoptosis is speculative. Papershave appeared suggesting that lysates of partially
http://www.jbc.org/cgi/doi/10.1074/jbc.M508383200The latest version is at JBC Papers in Press. Published on January 30, 2006 as Manuscript M508383200
Copyright 2006 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

2
purified lysosomes are capable of directlyactivating pro-caspase-3 (22,23). However,recombinant cathepsins have failed to catalyzesuch a cleavage (24). Alternatively, several groupshave demonstrated the in vitro cleavage of Bid byextracts from highly purified preparations oflysosomes (11,21,25), and by individualrecombinant-derived cathepsins (24,25). Severalagents that induce lysosomal damage lead to Bidcleavage in vivo, and do so prior to the activationof the apoptosome (11,18,25,26). Given the abilityof tBid, the cleavage product of Bid, to inducecytochrome c release, and the role of the latter inthe activation of the apoptosome, Bid cleavagerepresents a viable mechanism by whichlysosomal disruption could induce apoptosis.Indeed, in the case of TNFα, the pro-apoptoticeffects of the cytokine are muted in cells treatedwith small molecule inhibitors of Bid (27), orhaving reduced Bid contents (19). However, thereis a confounder in most TNFα studies.Specifically, the cytokine generally activates pro-caspase-8, and caspase-8 also processes Bid totBid. Hence, in principal, Bid could potentially beactivated by TNFα exposure via multiple routes.
The aryl hydrocarbon receptor (AhR) is aligand-activated transcription factor. Over the pastthree decades the ligand-activated AhR has beendocumented to both positively and negativelyregulate a variety of genes having dioxin responseelements in their promoters (28-31). The productsof such genes are involved in diverse functionssuch as phase I and II metabolism, cell cycleregulation, apoptosis, and development (28,29,32).In addition, recent studies suggest that the AhRmay have ligand-independent activities.Specifically, homologs of the mammalian AhRreceptor lacking a ligand-binding domain havebeen implicated in the developmental regulation oflower eukaryotes (33,34). Secondly, analyses ofAhR-deficient cell lines suggest that AhR content,in the absence of exogenous ligands, regulates cellmorphology (35), progression through G1 (35,36),and susceptibility to the pro-apoptotic effects ofceramide (37), Fas ligand and CD95 cross-linkingantibody (38), and the lysosomal photosensitizerN-aspartyl chlorin e6 (NPe6, 25).
To date, only a handful of proteins havebeen implicated as regulators of lysosomalfragility (15,39-41). One such protein may be theAhR. Specifically, we have demonstrated that theAhR-containing murine hepatoma cell line 1c1c7undergoes apoptosis in photodynamic therapy(PDT) protocols employing the lysosomal
sensitizer NPe6 (9,25). In PDT protocols light-activated photosensitizers generate a pulse ofsinglet oxygen in the immediate vicinity of thesensitizer. In the case of 1c1c7 cells NPe6 isexclusively sequestered in lysosomes (9).Irradiation of NPe6-loaded cells causes lysosomaldisruption and the chronologically orderedcleavage of Bid, release of cytochrome c andactivation of the apoptosome (9). In contrast,although variants of the 1c1c7 line deficient in theAhR accumulated comparable levels of sensitizerin their acidic organelles, the latter were markedlyresistant to lysosomal disruption followingirradiation (25). Since restoration of AhRexpression restored susceptibility to PDT-inducedacidic organelle disruption and apoptosis, itappeared that AhR content regulated lysosomefragility (25).
In the current study the effects of AhRcontent on TNFα -induced apoptosis wereinvestigated in cells of the 1c1c7 lineage. In suchcells AhR content regulated susceptibilities to bothTNFα-induced apoptosis and acidic organelledisruption. Knock-down of cellular Bid contentswith Bid antisense oligonucleotides demonstratedthat the BH3 protein mediated a portion of the pro-apoptotic effects of TNFα. Unexpectedly, pro-caspase-8 was not activated by TNFα due to theabsence of the adapter protein FADD, and did notcontribute to Bid activation. Instead, lysosomal-derived cathepsin D was partially responsible forBid cleavage, and a portion of the apoptoticprogram induced by TNFα. Lysosomal proteaserelease also led to an activation of pro-caspase-12.Hence, the 1c1c7 model proved to be a uniquesystem for investigating the contributions of non-caspase-8 pathways to TNFα -mediated Bidcleavage and apoptosis, and for identifying theAhR as a putative modifier of lysosomalpermeability.
EXPERIMENTAL PROCEDURES
Materials - The fluorescent molecule HO33342was purchased from Molecular Probes (Eugene,OR). Ac-DEVD-AMC was obtained from BDBiosciences (San Diego, CA). AMC, GW4869,pepstatin A, cathepsin D substrate MOCAc-GKPILFFRLK(Dnp)-D-R-NH2, cathepsin Bsubstrate Z-RR-AMC, and recombinant humanTNFα were purchased from Calbiochem (La Jolla,CA). Bicinchoninic acid, leupeptin, andcycloheximide were purchased from Sigma-
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

3
A l d r i c h ( S t . L o u i s , M O ) . 3 - O -methylsphingomyelin was obtained fromBIOMOL (Plymouth Meeting, PA). Cathepsin Dinhibitors diazoacetyl-DL-2-aminohexanoic acid-methyl ester and acetyl pepstatin A were obtainedfrom Bachem (King of Prussia, PA). CA-074, CA-074-Me, E-64, and E-64d were obtained fromPeptide Institute, Inc. (Louisville, KY). Z-FA-FMK was obtained from Enzyme SystemsProducts (Livermore, CA). Recombinant murineBid was from R&D Systems, Inc. (Minneapolis,MN). Lipofectin was purchased from Invitrogen(Carlsbad, CA). HA14-1 was obtained from RyanScientific, Inc. (Isle of Palms, SC).
Cell Culture and Viability Measurements -Murine hepatoma 1c1c7, Tao, TCMV, TAHR,WCMV and WARV cells lines were obtainedfrom J. Whitlock, Jr. (Stanford University, PaloAlto, CA). The origins and characterizations ofthese cell lines have been described (seereferences in 37). All cell lines were cultured at37˚C in α-minimal essential medium (αMEM)containing 5% fetal bovine serum and 100 µg/mlstreptomycin and 100 units/ml penicillin. TheTCMV, TAHR, WARV and WCMV lines weremaintained in medium that also contained 500µg/ml Geneticin.
Viability was assessed by counting cellswith a hemocytometer, and scoring bothmorphology and trypan blue permeability. In theTNFα and TNFα + CHX treatment groups a smallpercentage of cells expressed apoptoticmorphological features (i.e., shrunken, blebbed), atthe time of analyses, without being trypan bluepermeable. These cells, if allowed additional time,eventually became trypan blue permeable. Hence,they were scored as being non-viable.
DEVDase Assay - Activations ofprocaspase-3 and –7 were analyzed by monitoringthe generation of 7-amino-4-methylcoumarin(AMC) from the caspase substrate Ac-DEVD-AMC. The procedures used for the harvesting ofcells and measurements of DEVDase activity havebeen described in detail (25). Changes influorescence over time were converted into pmolof product by comparison to a standard curvemade with AMC. DEVDase specific activities arereported as nmol product per min per mg protein.The bicinchoninic acid assay, using BSA as astandard, was used to estimate proteinconcentrations.
Western Blot Analyses - The reagents andprocedures used for the preparation and processingof cell lysates, SDS gel electrophoresis, andimmuno detection of caspases-9 and -3 have beendescribed in detail (25). Caspase-12 was detectedin a similar manner using an affinity purifiedrabbit polyclonal antibody raised against a KLH-coupled synthetic peptide of murine caspase-12 asthe primary antibody (Cell Signaling Technology,Inc., Beverly, MA). The pro- and processed formsof caspase-8 were detected with either a rabbitpolyclonal antibody raised to a recombinantprotein corresponding to amino acids 217-350 ofhuman caspase-8 (Santa Cruz Biotechnology,Santa Cruz, CA) or a rabbit polyclonal antibodyraised to a synthetic peptide comprising aminoacids 2-20 of human caspase-8 (BD PharMingen,San Diego, CA).
Immunofluorescence Detection of Lamp-1And Cathepsins B and D - Cultures grown onpoly-L-lysine coated coverslips were washed 3xwith PBS and fixed in 4% paraformaldehyde/PBSsolution for 30 min. Thereafter, the fixed cellswere washed 3x with PBS and permeabilized withmethanol for 5 min on ice. After washing (3xPBS), non-specific autofluorescence wasquenched with 100 mM glycine/PBS for 30 min.Subsequent washes were carried out 3x withPBS/0.1% saponin. The coverslips were washedand then incubated with 5% BSA/PBS/0.1%saponin for 1 h at 37ºC to block non-specificimmunoglobin binding. The coverslips werewashed and incubated with a 1:20 dilution ofrabbit anti-human cathepsin D antibody(Oncogene Research Products, San Diego), or a1:500 dilution of rabbit anti-mouse cathepsin Bantibody (gift of Dr. B. Sloane, Wayne StateUniversity School of Medicine) and a 1:1000dilution of the 1D4B rat anti-mouse Lamp-1antibody (Development Studies Hybridoma Bank,Department of Biological Sciences, University ofIowa, Iowa City, IA) in blocking buffer for 2 h at37ºC. After washing, secondary detectionconsisted of incubation with 1:200 dilutions ofAlexaFluor 488 goat-anti-rabbit IgG andAlexaFluor 546 goat anti-rat IgG (MolecularProbes) in blocking buffer for 1 h at 37ºC. Thecoverslips were washed and incubated for 5 min atroom temperature with 500 nM HO33342/PBS.After a final wash, the coverslips were invertedonto a drop of SlowFade solution (MolecularProbes) on glass slides and sealed with acrylic nailpolish. Digital images were captured using an
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

4
Axioplan 2 Imaging Microscope equipped with anApoTome optical sectioning device (Carl ZeissAG, Germany).
Quantitative analyses of fluorescent imageswere performed with MetaMorph software(Version 6.3r2, Molecular Devices, Sunnyvale,CA). Within each experiment, time settings forevery exposure were held constant, andcontrast/brightness settings were unaltered. Theimages captured by Axiovision were exported asTIFF format and then imported into MetaMorph asa dual channel image (i.e. Channel A=Red=Lamp-1 staining, Channel B=Green=Cathepsin B/Dstaining). Individual cells were digitally isolatedand the inclusive threshold value was set at 50.Area measurements were used for co-localizationanalysis, which are expressed as percentage areaof Channel A above threshold that does notoverlap Channel B, and vice versa. Thismeasurement is independent of fluorescenceintensity. For relative intensity measurements, theintegrated value for Channel A was divided by theintegrated value for Channel B. The integratedvalue represents the sum of all grayscale values forevery pixel that is above threshold within aselected region.
Digitonin Permeabilization – Culturesestablished in 35 mm dishes were fed with freshmedium 16-20 h prior to treatment. Cultures weretreated with nothing, CHX, TNFα or CHX +TNFα for 0.5, 1, 2 or 3 h at 37˚C prior to beingwashed 3x with release buffer (1 mM PIPES, pH7.0, 80 µM L-cysteine in HBSS). Washed cultureswere incubated at 37˚C with 1 ml of release bufferwith, or without 0.1% Triton X-100 or variedconcentrations of digitonin. After a 30 minincubation culture fluids were removed andcentrifuged at 13800xg for 30 min at 4˚C.Supernatant fluids were removed and transferredto a new tube, and stored on ice until used inenzymatic assays.
Lactate dehydrogenase and cathepsin assays- Supernatant fluids isolated from nontreated,digitonin-treated, and Triton X-100-treatedcultures were diluted with release buffer, andtriplicate 100 µl samples were dispensed into a 96well plate. The lactate dehydrogenase assay wasinitiated by adding 100 µl of a buffer containing200 mM Tris-HCL, pH 7.3, 10 mM sodiumpyruvate and 0.5 mM β-NADH. The oxidation ofNADH was monitored for 20 min at 37˚C on a
SpectraMAXPlus (Molecular Devices, Sunnyvale,CA) microplate reader at 340 nm. Supernatantfluids were diluted with release buffer to assurelinearity.
For assay of cathepsin B/L activity,triplicate 100 µl samples were dispensed into a 96well, black flat-bottomed, microfluor plate(Thermo Labsystems, Milford, MA). The assaywas initiated by addition of 100 µl of a solutioncontaining 100 mM sodium acetate, pH 5.5, 200mM NaCl, 4 mM EDTA, 10 mM DTT and 100µM Z-RR-AMC. The production of AMC wascontinuously monitored for 20 min at 37˚C on aSpectraMAX Gemini microplate reader employingexcitation/emission wavelengths of 355/460 nm,respectively. Analyses performed in the presenceof 5 µM CA-074 or 5 µM E-64 were used toestimate contributions of cathepsin B or cathepsinsB + L, respectively, to Z-RR-AMC cleavage.Cathepsin B accounted for >95% of the releasedAMC.
For assay of cathepsin D, triplicate 75 µlsamples were dispensed into a 96 well, black flat-bottomed microfluor plate. The assay was initiatedby addition of 25 µl of solution containing 160mM sodium formate, pH 3.5, and 40 µM of theinternally quenched substrate MOCAc-GKPILFFRLK(Dnp)-D-R-NH2. Cleavage of thesubstrate Phe-Phe bond results in fluorescence thatwas monitored at 37˚C on a SpectraMAX Geminimicroplate reader employing excitation/emissionwavelengths of 328/393 nm, respectively.Analyses performed in the presence of 1 µMpepstatin A were used to estimate contributions ofcathepsin D (≥98%) to the monitored reaction.
Activities measured in the supernatants ofcultures incubated with only release buffer wereused to correct for spontaneous release ofenzymes. Activities measured in supernatants fromcultures treated with release buffer + digitoninwere divided by the activities measured in TritonX-100-lysed cultures, and multiplied by 100 toestimate a % of Total Activity. Cell countinganalyses indicated that cell numbers per platewithin a group of 3-6 plates were within ± 5% ofthe mean. Hence, we assumed that all plates withina treatment group had similar cell densities, andcalculated the % of Total Activity based upon themeans of 3 – 6 plates per treatment.
Endosome/Lysosome Isolation - Theprocedures used for the isolation and disruption of
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

5
lysosomes, and assessment of their purity, havebeen described in detail (9).
In vitro Bid Cleavage Assay - Theprocedures used for in vitro cleavage ofrecombinant Bid by lysosomal extracts, andWestern blot analyses of Bid cleavage products,have been described in detail (25).
Oligonucleotide Transfection - 1c1c7 cellswere suspended in αMEM containing 5% FBS(without antibiotics), and plated in 35 mm culturedishes. Approximately 20 h after plating thecultures were washed 3x with PBS and themedium was replaced with 0.8 ml of αMEM.Cultures were transfected with 0.2 ml of αMEMcontaining 5 µg Lipofectin, and in some cases 100pmol of either murine Bid antisense or scrambles e n s e o l i g o n u c l e o t i d e s . T h elipofectin/oligonucleotide mixtures were preparedaccording to the instructions provided by themanufacturer. Six h after transfection the cultureswere washed 3x with αMEM, refed with 1.5 ml ofαMEM containing 2% FBS, and returned to ahumidified CO2 chamber. Cultures were used 24-80 h after transfection. The oligonucleotides usedin these studies are 20-mer 2′-O-MOE chimericantisense oligonucleotides. These antisenseo l i g o n u c l e o t i d e s c o n t a i n 2′-O-MOE/phosphorothioate residues flanking a 2′-oligodeoxynucleotide/phosphorothioate centralregion that supports RNase H-mediated cleavageof target mRNAs (42). The sequence of the murineBid-ant i sense ( Is i s 119935) i s 5′-GACCATGTCCTGGCCAGAAA-3′. Bolded andunderlined residues indicate 2′-O-MOE-modifiedresidues. The control oligonucleotide used in thecurrent studies is Isis 29848, which has a randommixture of modified bases at each position. Adescription of the synthesis and characterization ofthese oligos has been published (42).
Electrophoretic Mobility Shift Assay -Nuclear extracts were prepared by a publishedprocedure (43). A complementary pair of syntheticDNA oligonucleotides containing the sequences5′-AGTTGAGGGGACTTTCCCAGGC-3′ and 3′-TCAACTCCCCTGAAAGGGTCCG-5′ w e r eannealed and 5′ labeled with T4 polynucleotidekinase and [γ 32P]ATP. The EMSA bindingreaction contained 10 µg of nuclear protein, 1 µgof poly(dI-dC) and 1x105 DPM of labeled DNAprobe in a final volume of 25 µl of binding buffer
(10 mM Tris, pH 7.5, 50 mM NaCl, 1 mM DTT, 1mM EDTA and 5% glycerol). DNA-proteincomplexes were resolved by electrophoresisthrough 4% polyacrylamide gels. The gels weredried and protein/DNA complexes were visualizedby autoradiography.
[3H]leucine Incorporation – The methodused for monitoring the incorporation of[3H]leucine into protein has been described indetail (44).
Statistical analyses – Data were analyzed byone-way ANOVA analysis of variance followedby Tukey’s Multiple Comparison Test (Prism,GraphPad Software, San Diego, CA). Differencesbetween/amongst groups were scored asstatistically significant if p<0.01.
RESULTS
Optimization of TNFα / C H X - i n d u c e dapoptosis - Exposure of 1c1c7 cultures to 30 pg/mlT N F α resulted in an ~10-fold increase inDEVDase within 4-8 h of treatment (Fig. 1A), anda small decrease in cell viability (Fig. 1C).Increasing the concentration of TNFα did notincrease the proportion of dying cells or furtherelevate DEVDase activities2. CHX by itself neitheractivated DEVDase (Fig. 1B) nor caused trypanblue permeability (Fig. 1C) over the time courseinvestigated. However, TNFα + CHX co-treatment markedly decreased cell viability andelevated DEVDase activities above what occurredwith only TNFα or CHX (Figs. 1A,B and C).Potentiation by CHX only occurred with doses(0.5 - 1 µg/ml) that potently suppressed theincorporation of [3H]leucine into acid precipitablematerial (Fig. 1D).
TNFα + CHX-induced lysosome disruptionin 1c1c7 cultures - Several studies have implicateda role for lysosomal proteases in TNFα-inducedapoptosis (14-19). Preliminary studies usingacridine orange to monitor lysosomal membraneintegrity indicated that TNFα + CHX co-treatmentaffected these organelles in 1c1c7 cultures2.Although acridine orange is commonly used tomonitor acidic organelle intactness, it haslimitations. Specifically, its fluorescence is pH-dependent. Conditions that cause acid organellealkylation without membrane disruption cannot bedistinguished from conditions that cause
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

6
membrane permeability. In order to circumventthis caveat we monitored the co-localization of thelate endosomal/lysosomal luminal proteasescathepsins B and D with the lateendosomal/lysosomal integral membrane proteinLamp-1 (1). Figures 2 and 3 are representative ofthe effects of TNFα, CHX and TNFα + CHX in1c1c7 cultures on cathepsin B and D fluorescencestaining patterns, respectively. Lamp-1 andcathepsins B and D exhibited punctate staining inboth non-treated 1c1c7 cultures (Figs. 2A,B and3A,B). Both cathepsins extensively co-localizedwith Lamp-1 in non-treated 1c1c7 cultures (Figs.2C and 3C). A visual inspection of the panelspresented in Figs. 2 and 3 suggest that CHX orTNFα treatments either have no, or only subtle,effects on Lamp-1 and cathepsin B/D staining, andLamp-1/cathepsin co-localization. However, co-treatment of 1c1c7 cultures with CHX + TNFαmarkedly reduced cathepsin B (Fig. 2M) andcathepsin D (Fig. 3M) staining, without havingperceivable affects on Lamp-1 staining (Figs. 2Nand 3N). Indeed, the predominant red coloration ofthe cathepsin B/D + Lamp-1 overlay emphasizesthe preferential losses of cathepsins occurring inTNFα + CHX-treated cultures (Figs. 2O and 3O).
MetaMorph analyses of individual cellswere used to quantify the effects of CHX andT N F α treatments on Lamp-1 and cathepsinstaining and co-localization in 1c1c7 cultures(Table I). Three parameters were measured. First,the accumulative intensities of Lamp-1 andcathepsin B/D spots in individual cells weredetermined, and expressed as the ratio of Lamp-1to cathepsin staining. An increase in this ratiocould reflect either an overall increase in Lamp-1staining, and/or losses of cathepsin staining.Second, an estimate was made of how manyLamp-1 (+) spots did not co-localize withcathepsin (+) spots. An increase in the percentageof Lamp-1 (+) spots having this phenotype,coupled with an increase in the Lamp-1/cathepsinintensity ratio, would be indicative of losses of co-localization due to the release of cathepins fromLamp-1 (+) vesicles. Third, an estimate was madeof how many cathepsin (+) spots did not co-localize with Lamp-1. Presumably, such apopulation is non-endosomal/lysosomal in nature.It should not be affected if our treatmentsexclusively target endosomes or lysosomes.
MetaMorph analyses of 1c1c7 culturesindicated that TNFα + CHX co-treatmentdramatically increased both the Lamp-1/cathepsin
D intensity ratio, and the % of Lamp-1 (+) spotsnot containing cathepsin D (Table I). Similarresults, although of lesser magnitude, occurredwith cathepsin B. TNFα + CHX co-treatment of1c1c7 cultures neither increased nor decreased the% of cathepsin (+) spots that did not containLamp-1 (Table I). Collectively, these findingssuggest that lysosomal/endosomal cathepsin D,and to a lesser degree B, are released into thecytosols of 1c1c7 cells following TNFα + CHXco-treatment.
3-O-MeSM regulation of lysosome fragility -3-O-methylsphingomyelin (3-O-MeSM) is asynthetic analog of sphingomyelin in which the C3hydroxyl group has been replaced with a methoxygroup. We recently reported that 3-O-MeSM israpidly incorporated into the acidic organelles of1c1c7 cells, and prevents the rupture of lysosomesoccurring in photodynamic therapy protocolsemploying the lysosomal photosensitizer NPe6(45). Strong protection was afforded by 50 µM 3-O-MeSM in the PDT protocol (45). Pre-treatmentwith 50 µM 3-O-MeSM protected 1c1c7 culturesagainst the pro-apoptotic effects of TNFα + CHXco-treatment, as assessed by morphology (Fig.4A), DEVDase activation (Fig. 4B), measurementsof trypan blue permeability (Fig. 4C) and pro-caspases-3, -9 and –12 processing (Fig. 4D).Treatment of 1c1c7 cultures with 50 µM 3-O-MeSM did not alter cathepsin B (Fig. 2D),cathepsin D (Fig. 3D) or Lamp-1 (Figs. 2E and3E) staining, or cathepsin co-localization withLamp-1 (Figs. 2F and 3F, Table I). However, pre-treatment with 3-O-MeSM prevented TNFα +CHX mediated disruption of lysosomes.Specifically, cathepsins B and D remained co-localized with Lamp-1 in 1c1c7 cultures that hadbeen treated with 3-O-MeSM prior to TNFα +CHX exposure (Figs. 2R and 3R, Table I).
3-O-MeSM was originally described in theliterature as a weak inhibitor of neutralsphingomyelinase (SMase, 46). Because theneutral SMase inhibitor GW4869 has beenreported to suppress TNFα-induced apoptosis inMCF-7 cells (47), we thought it prudent todetermine if the effects of 3-O-MeSM in oursystem were related to neutral SMase. To examinethis possibility 1c1c7 cultures were pretreated withGW4869 prior to co-treatment with TNFα + CHX.Pre- and co-treatment with concentrations ofGW4869 approaching its solubility limit in our
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

7
culture medium (~6 µM) offered no protectionagainst the pro-apoptotic effects of TNFα + CHX2.
Lysosomal protease release is not a generalconsequence of apoptosis – The agent HA14-1activates the intrinsic apoptotic pathway viadisruption of the mitochondrial respiratory chain(48) and neutralization of the anti-apoptoticfunctions of Bcl-2/Bcl-XL (49). Exposure of 1c1c7cultures to 15-25 µM HA14-1 induces apoptosiswithin 1 h (25,45). Very recent studies suggest thatthis apoptotic response is unrelated toe n d o s o m a l / l y s o s o m a l d a m a g e ( 4 5 ) .Supplementary Fig. 1 depicts representative panelsof Lamp-1 and cathepsin D staining in 1c1c7cultures harvested 15, 30 and 60 min afterexposure to 20 µM HA14-1. MetaMorph analysesof individual cells indicated that HA14-1treatment, unlike TNFα + CHX treatment, neitherincreased the relative intensity of Lamp-1/cathepsin D staining, nor increased the % ofLamp-1 (+) spots not containing cathepsin D(Table II). Hence the apoptotic program initiatedby HA14-1 did not trigger the release of cathepsinD from lysosomes.
Role of Bid in TNFα-induced apoptosis -Recent studies suggest that Bid is a mediator ofthe pro-apoptotic effects of TNFα (18,19). Inorder to assess the contribution of Bid to theapoptotic program induced in our model by TNFα+ CHX, 1c1c7 cultures were treated with eitherBid antisense or scramble sense oligonucleotides.The Bid antisense oligo reduced Bid proteincontents by >85% within 24-48 h of transfection(Fig. 5A). Bid contents began to recoverthereafter, but were still reduced 72 h aftertransfection by ~50%. In contrast, Bid proteincontents were not affected by transfection ofscramble sense oligonucleotides or by lipofectintreatment (Fig. 5A).
Shrunken cells with apoptotic blebs wereevident within 4.5 h of TNFα + CHX treatment incultures that had been pretreated 48 h earlier withscramble sense oligonucleotides or lipofectin (Fig.5C). In contrast, no or few apoptotic cells wereobserved in cultures having reduced Bid levelsduring the same time period, or even 4 h later (Fig.5C). DEVDase activities in the various treatmentgroups correlated with the morphological data(Fig. 5B). The kinetics of DEVDase activationfollowing TNFα + CHX exposure, as well asactual activities, were identical for scramble sense
transfected and lipofectin-treated 1c1c7 cultureson each of the examined days. In contrast, thekinetics of DEVDase activation were delayed incultures having reduced Bid levels. Furthermore,DEVDase specific activities were lower in cultureshaving markedly reduced Bid contents (24 and 48h post transfection cultures).
Role of cathepsin D in TNFα-mediated Bidcleavage and apoptosis - Bid activation viaproteolytic cleavage can be catalyzed by caspase-8or lysosomal proteases (24). Bid cleavageoccurred in a time-dependent manner in 1c1c7cultures co-treated with TNFα + CHX (Fig. 6A).Extracts of 1c1c7 lysosomes cleaved Bid in an invitro assay (Fig. 6B). Bid cleavage was notaffected by supplementing extracts with thecathepsin B inhibitor CA-074, the broad spectrumcysteine protease inhibitor E-64, the cathepsin Band L inhibitor Z-FA-FMK, or the serine proteaseinhibitor leupeptin. However, cleavage activitywas lost in extracts pretreated with the cathepsin Dinhibitor pepstatin A (Fig. 6B).
Pretreatment of 1c1c7 cultures with acetylpepstatin A suppressed TNFα + CHX mediatedDEVDase activation in a concentration- (Fig. 6C)and time-dependent (Fig. 6D) fashion. Maximalinhibition occurred with 1 µM acetyl pepstatin A.Similar results were observed in 1c1c7 culturespretreated with 100-400 µM of the cathepsin Dinhibitor diazoacetyl-DL-2-aminohexanoic acid-methyl ester (DAME, Figs. 6C,D). Lightmicroscopy indicated that concentrations ofDAME > 400 µM caused cell stress by itself, andhence were not employed2. The protectionmediated by the two cathepsin D inhibitors washighly reproducible, but partial. Optimalconcentrations of the two inhibitors suppressedoverall DEVDase activities by ~50% (n = 6experiments), and markedly improved cell survival(Fig. 6F). Pretreatment of 1c1c7 cultures with 1µM acetyl pepstatin A also partially suppressedBid cleavage and procaspase-3 cleavage (Fig. 6A).In contrast to the effects of the cathepsin Dinhibitors, pre-treatment of 1c1c7 cultures with thecell permeable protease inhibitors CA-074-Me, E-64d, and Z-FA-FMK suppressed neither DEVDaseactivation (Fig. 6E) nor cell killing (Fig. 6F)following TNFα + CHX addition.
AhR-dependent modulation of sensitivity toTNFα/CHX-induced apoptosis - The Tao cell linewas derived from 1c1c7 cells (see references in
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

8
37). It contains greatly reduced levels of AhR (Fig.7A). Relative to the parental line, Tao cultureswere refractory to the pro-apoptotic effects ofTNFα ± CHX (Figs. 8A,B). Specifically, almostno activation of DEVDase occurred in Taocultures following exposure to TNFα, and onlymarginal activation occurred in cultures co-treatedwith TNFα and CHX (Fig. 8A). Differences inDEVDase activities were mirrored by dramaticdisparities in the activations of pro-caspases-9, -12and –3 in the two cell lines (Fig. 8B). Whereascoordinate losses and appearances of the pro- andprocessed forms of caspases-9, -12 and -3 weredetectable in 1c1c7 cultures within 4-8 h of TNFα+ CHX co-treatment, similar processing did notoccur in Tao cultures.
Transfection of Tao cells with an Ah senseexpression vector and subsequent selection ofGeneticin-resistant cells gave rise to the TAHRcell line (35), which has AhR contents similar to1c1c7 cells (Fig. 7A). Increased AhR expressionrestored sensitivity to the pro-apoptotic effects ofTNFα + CHX (Figs. 8C,D). The magnitude andkinetics of DEVDase activation and pro-caspase-9and –3 processing in TAHR cultures werecomparable to what were observed in wild type1c1c7 cells (compare Figs. 8A with 8C, and 8Bwith 8D). In contrast, AhR content was notaffected by stable introduction of the emptyexpression plasmid into Tao cells (resulting linedesignated TCMV, Fig 7A). TCMV culturesresponded to TNFα ± CHX in a fashioncomparable to Tao cells (compare Figs. 8A with8C, and 8B with 8D).
Transfection of 1c1c7 cells with an A hantisense expression vector gave rise to a stablecell line (35, designated WARV) havingdiminished AhR content (Fig. 7A). The WARVline, relative to either wild type 1c1c7 cells or itsvector-only cognate partner (WCMV cell line, Fig.7A), was notably resistant to the pro-apoptoticeffects of TNFα + CHX, as monitored by analysesof either DEVDase activities or pro-caspaseprocessing (Figs. 8E,F).
The elevated DEVDase activities/enhancedpro-caspase-3 processing occurring in TNFα +CHX co-treated AhR-containing cell typescorrelated with decreases in viability (SupplementTable I). Specifically, whereas trypan bluepermeability in 1c1c7 and WCMV culturesfollowing singular CHX or TNFα treatment wasnot markedly different than that measured incontrol cultures (4-6%), permeability increased to
~40-50% within 10-13 h of TNFα + CHX co-treatment. Similarly, the absence of pro-caspaseactivation in TNFα + CHX co-treated AhR-deficient Tao and WARV cell lines was paralleledby no changes in trypan blue permeability(Supplement Table I).
Recent studies have established that TNFαtriggers the recruitment of the adapter proteinTRADD to TNFr1, and the formation of asignaling complex capable of activating NF-κBand JNKs (50,51). Subsequently, components ofthe complex dissociate and re-associate withFADD, forming a complex that facilitates therecruitment, oligomerization and activation of pro-caspase-8 (50,51). The 1c1c7 cell line and all of itsvariants expressed comparable levels of pro-caspase-8, TNFr1 and TRADD (Fig. 7A). AhRdeficiency did not suppress formation of the initialsignaling complex. Specifically, ligandengagement of the TNFr1 activated JNKs and NF-κB in both 1c1c7 and Tao cultures (Figs. 7B andC, respectively). Exposure to TNFα + CHXcaused rapid, but transient activations of JNK1/2in the two cell lines (Fig. 7B). Whereas, JNKactivation in Tao cultures with TNFα + CHX wascomparable to what occurred with anisomycin, alesser response was noted with 1c1c7 cultures(Fig. 7B). TNF ± CHX exposure also caused therapid proteolytic degradation of IκBα2 andsubsequent NF-κB activation, as assessed byEMSA, in both cell lines (Fig. 7C). TNFα + CHXtreatment stimulated NF-κB activation above whatoccurred with TNFα alone. Similar to the trendobserved for JNK, a slightly stronger NF-κBEMSA signal was consistently observed with Taocells (Fig. 7C and 2 additional experiments).
Unexpectedly, we were unable to detecteither the disappearance of pro-caspase-8, or theappearance of processed caspase-8 in 1c1c7,TAHR or WCMV cultures treated with pro-apoptotic concentrations of TNFα ± CHX (Figs.8B,D,F). In addition, we were unable to eithersuppress the development of apoptosis by co-treatment with the caspase-8 inhibitor IETD-FMK,or detect the generation of an activity capable ofcleaving the caspase-8 substrate Ac-IETD-AFC in1c1c7 cells2. Additional analyses revealed that the1c1c7 cell line and its variants lacked the adapterprotein FADD (Fig. 7A). Given the role of FADDin the recruitment, oligomerization and activationof pro-caspase-8 (50,51), its absence provides areasonable explanation for why pro-caspase-8processing did not occur in 1c1c7, TAHR and
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

9
WCMV cultures treated with TNFα + CHX.Furthermore, the absence of caspase-8 activationeliminates one of the major potential activators ofBid, and emphasizes the importance of lysosomaldisruption for Bid activation in our model system.
AhR regulation of lysosome disruption byTNFα + CHX – Recent studies suggest that thelysosomes of Tao cultures are resistant todisruption by the oxidants generated in anNPe6/PDT protocol (25). Figures 9 and 10 arerepresentative of the effects of TNFα, CHX andTNFα + CHX in Tao cultures on cathepsin B andD fluorescence staining patterns, respectively.Like 1c1c7 cultures, Lamp-1 and cathepsins B andD exhibited punctate staining in non-treated Taocultures (Figs. 9A,B and 10A,B). Both cathepsinsextensively co-localized with Lamp-1 in non-treated Tao cultures (Figs. 9C and 10C). CHX orTNFα treatments either had no, or only subtle,effects on Lamp-1 and cathepsin B/D staining, andLamp-1/cathepsin co-localization in Tao cultures(Figs. 9 and 10, Table I). In marked contrast towhat occurred in 1c1c7 cultures, the intensities ofcathepsin B (Fig. 9J) and cathepsin D (Fig. 10J),and their co-localization with Lamp-1 (Figs. 9Land 10L), were not significantly altered followingco-treatment of Tao cultures with TNFα and CHX(Table I).
In order to confirm that the effects noted inTao cultures were AhR-related, Lamp-1 andcathepsin B/D staining intensities and co-localization were also performed in a second AhR-containing and -deficient cell pair. SupplementaryFigs. 2-5 depict representative panels of WCMVand WARV cultures stained after 4 h of singularor combined TNFα + CHX treatment. MetaMorphanalyses of images are summarized in Table I.CHX + TNFα co-treatment of AhR containingWCMV cells increased the Lamp-1/cathepsin Band D intensity ratios, and the % of Lamp-1 (+)spots not containing cathepsin B or D (Table I).Like 1c1c7 cells, the % of Lamp-1 (+) spots notcontaining cathepsin D in WCMV cells wasgreater than the % of Lamp-1 (+) spots notcontaining cathepsin B (Table I). Conversely, co-treatment of AhR-deficient WARV cultures withTNFα + CHX affected neither Lamp-1/cathespinB and D intensity ratios, nor the % of Lamp-1 (+)spots not containing cathepsins B or D (Table I).Hence, AhR-deficiency suppressed the release ofendosomal/lysosomal cathepsins by TNFα + CHXco-treatment in two different cell pairs.
Cathepsin release in digitonin permeabilizedcultures - As a complement to theimmunofluorescence studies we developed anadditional assay to monitor the release oflysosomal proteases into the cytosol. We reasonedthat released lysosomal proteases might bemeasurable in the extracellular fluids of culturespermeabilized with digitonin. Lactatedehydrogenase (LDH) was used as a cytosolicmarker protein. Treatment of 1c1c7 and Taocultures with 1-4 µ M digitonin causedconcentration-dependent releases of LDH (Fig.11A), cathepsin B (Fig. 11B) and cathepsin D(Fig. 11C). Concentrations of digitonin > 4 µMfacilitated additional releases of LDH, but hadminimal effects on cathepsins B and D. Digitonincaused comparable releases of LDH in 1c1c7 andTao cultures throughout the concentration rangeinvestigated. However, comparable effects werenot seen for cathepsins B and D. Greater releasesof both cathepsins were observed in digitonin-treated Tao cultures (Figs. 11B,C). In addition, theorganelles containing cathepsins B and Dresponded differentially to digitonin. Specifically,cathepsin D was preferentially released in bothcell lines following digitonin treatment (Figs.11B,C). This effect was particularly evident atconcentrations of digitonin ≥ 2 µM.
In order to assess the effects of TNFα, CHXand TNFα + CHX on lysosomal integrity wemonitored the releases of LDH and cathepsins Band D in cultures permeabilized with 2 µMdigitonin, over a 3 h time period. Two µMdigitonin was chosen because it represented asubmaximal releasing concentration for all 3enzymatic activities, and caused comparable LDHrelease in both 1c1c7 and Tao cells.
T N F α + CHX co-treatment of 1c1c7cultures did not trigger the release of LDH overthe 3 h exposure period (Fig. 12A). However, co-treatment did trigger releases of cathepsins B andD (Figs. 12B,C). Increases were reproduciblydetectable within an h, and statistically significantwithin 2 h of treatment. In contrast, TNF alone didnot stimulate the release of any of the threeenzymes. Similarly, CHX alone did not triggerreleases of LDH and cathepsin B throughout thetime course. However, CHX alone did stimulatestatistically significant releases of cathepsin D in1c1c7 cultures that were less than that observed inco-treated cultures.
Co-treatment of Tao cultures with TNFα +CHX did not trigger the release of LDH (Fig.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

10
12D). Although small releases of cathepsin B (Fig.12E) and cathepsin D (Fig. 12F) occurred in co-treated cultures, the measured activities were notstatistically different than the zero time control. Itshould be noted that the zero time cathepsin valuesobserved in these studies are a consequence of thedigitonin concentration (2 µM) used to render thecells permeable (Figs. 11B,C), and do not reflectactual basal cytosolic activities.
DISCUSSION
TNFα engagement of the TNFr stimulatespro-survival and/or pro-apoptotic pathways inseveral cell types (50,51). In the case of the latter,the pro-apoptotic effects of TNFα are oftenattributed to its activation of pro-caspase-8.Caspase-8 can either activate pro-caspase-3directly or indirectly via its cleavage of Bid to tBidand the eventual activation of pro-caspase-9.Although 1c1c7 cells contained pro-caspase-8, itdid not undergo processing following exposure ofcultures to TNFα + CHX. This deficiencyprobably reflects the observed absence of FADDin 1c1c7 cultures. Without FADD, pro-caspase-8cannot be recruited to the activated TNFr andundergo the oligomerization and conformationalchanges necessary for activation. Hence, TNFα +CHX induced apoptosis in 1c1c7 cultures ismediated by a non-caspase-8 pathway.
Several studies, including the current one,demonstrate that TNFr activation leads tolysosomal permeabilization in cultured cells(14,16,18,19). Molecular and pharmacologicalapproaches have implicated several lysosomal-derived cathepsins as mediators of the pro-apoptotic effects of TNFα (13-18). Indeed, in thecurrent study pre-treatment with 3-O-MeSMprevented TNFα + CHX-mediated acidicorganelle destabilization, protease release, and thesubsequent induction of apoptosis. In principal,released proteases could contribute to non-caspase-8-mediated apoptosis via severalmechanisms. Ishisaka et al. (22,23) reported anactivation of pro-caspase-3 in cytosolic fractionsupon incubation with lysosomal extacts. Althoughthe identity of the activating enzyme is not known,in vitro cleavage assays have ruled out cathepsinsas the species responsible for pro-caspase-3activation (24). Second, inhibitor studies suggestthat cathespin D is capable of activating Bax (52).Such activation could facilitate cytochrome crelease and activation of the apoptosome. Third,
lysosomal extracts (9,24-26) and some cathepsins(18,24,26) are capable of converting Bid to tBid.Bid cleavage has been documented in several celltypes following disruption of lysosomes (18,26,this study). Activation of Bid, and the induction ofapoptosis, can be inhibited to variable degrees byco-treatment with cathepsin inhibitors (18,26, thisstudy). Finally, released lysosomal constituentsmay directly, or indirectly, damage or stress otherorganelles capable of triggering an apoptoticprogram. Incubation of isolated mitochondria withcathepsins B and D in vitro has been reported toprovoke the generation of reactive oxygen speciesand the release of cytochrome c (53).Alternatively, released lysosomal proteases maytarget the endoplasmic reticulum and facilitatepro-caspase-12 activation. Indeed, in the currentstudy, TNFα + CHX co-treatment of 1c1c7cultures activated pro-caspase-12. This activation,as well as the disruption of lysosomes, wassuppressed by pretreatment with 3-O-MeSM. Werecently reported that 3-O-MeSM suppresses therupture of lysosomes in NPe6/PDT protocols, butpromotes or has no effects on the pro-apoptoticactivities of HA14-1, staurosporine, tunicamycinand thapsigargin in 1c1c7 cultures (45). Becausethe latter two agents cause endoplasmic reticulumstress and pro-caspase-12 activation (54-56), itseems likely that the protective effects of 3-O-MeSM in our studies are upstream of theendoplasmic reticulum. Although TNFα-mediatedactivation of pro-caspase-12 has been reported(57), this is the first study that we are aware toimplicate a lysosomal factor in that process.
At least two lines of research indicate thatBid is an important mediator of the pro-apoptoticeffects of TNFα . First, Werneburg et al. (19)reported that cultured fibroblasts deficient in Bidare more resistant than their Bid-containingcounterparts to the pro-apoptotic effects of TNFα.Second, the pro-apoptotic effects of TNFα can besuppressed by co-incubation with small moleculeinhibitors of Bid (27). In the current study the pro-apoptotic effects of TNFα were both delayed andattenuated in 1c1c7 cultures having reduced Bidcontents as a consequence of pretreatment withBid antisense oligonucleotides. Bid depletionreduced DEVDase activation (a measure ofcaspase-3/7 activities) by ~50% in TNFα + CHX-treated cultures. Comparable effects were alsoachieved by treating cultures with BI-C692, asmall molecule inhibitor of Bid (27). Hence, Bidplays a role in TNFα-induced apoptosis in 1c1c7
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

11
cultures. However, it cannot be the sole factor.Although others have reached a similar conclusionabout Bid, in the current study we have been ableto obviate caspase-8 contributions to TNFα -induced Bid cleavage and apoptosis.
The extended loop of Bid, which containsthe caspase-8 cleavage site, is cleaved in vitro byextracts of purified lysosomes (9,24,25), and bycathepsins D (18), B, H, K, L and S (24,26).Although there are conflicting reports on theability of cathepsin D to cleave Bid in vitro(18,26), the lack of cleavage noted in one studymay be the consequence of performing the assay atpH 7.2 (26), which was subsequently reported tobe suboptimal (18). Nevertheless, studies byHeinrich et al. (18) and Bidere et al. (52) stronglysuggest that cathepsin D, but not papain-likecysteine protease (cathepsins B, L, H & K),mediates a portion of TNFα-induced apoptosisand Bid cleavage in some cell types. Similarly, ourstudies show that pepstatin A was the onlycathepsin inhibitor able to block lysosomal-mediated Bid cleavage in an in vitro assay, andsuppress TNF + CHX induced apoptosis. It shouldbe noted that other investigators have implicatedcathepsin B in both Bid cleavage (24,26) andTNFα-mediated apoptosis (14,15,16). The basisfor the differences amongst studies is not known.It is conceivable that the cell types used in thevarious studies contain different ratios of therelevant cathepsins, or have different cytoplasmiccontents of cystatins or stefins. High levels of thetwo latter cysteine protease inhibitors could negatecontributions by cathepsin B to Bid cleavage.
The pro-apoptotic effects of TNFα are oftenoffset by its ability to activate NF-κB signalingand induce the expressions of proteins involved incell survival (50,51). The protein synthesisinhibitor CHX is commonly used to negate thepro-survival activities of TNFα . Indeed, co-treatment of 1c1c7 cultures with TNFα and CHXpotentiated the pro-apoptotic response to TNFα,and this potentiation occurred at concentrations ofCHX that suppressed protein translation. Althoughthe effects of CHX are generally attributed to itssuppression of the translation of pro-survivalproteins such as cFLIP or cIAP, there may be asecond mechanism whereby CHX amplifies thepro-apoptotic effects of TNFα. Specifically, boththe cathepsin/Lamp-1 co-localization studies, andthe analyses of cathepsin activities in digitoninpermeabilized cultures, suggested that TNF +CHX treatment promoted the release of lysosomal
proteases. When used singularly, neither agentpromoted the release of cathepsins in the co-localization assay. Similarly, TNFα did notstimulate cathepsin release in the digitoninpermeabilization assay. However, some releasedid occur in CHX-treated cultures. These findingsemphasize that CHX many facilitate apoptosis inT N F α + CHX co-treatment protocols bymechanisms other than suppressing the synthesisof pro-survival proteins.
Previous analyses of 1c1c7 cells and itsAhR-containing and -deficient variants indicatedthat AhR content differentially influencessusceptibilities to subclasses of apoptotic inducers.Specifically, the 1c1c7, TAHR and WCMV celllines, and their respective AhR-deficientcounterparts (Tao, TCMV and WARV), exhibitcomparable susceptibilities to the pro-apoptoticeffects of HA14-1, staurosporine, and doxorubicin(25,37). In contrast, the three AhR-deficient lines,relative to their AhR-containing counterparts, areless sensitive to the pro-apoptotic effects of C2-ceramide (37), NPe6/PDT (25) and TNF + CHX(current study). It should be noted that ourobservations regarding AhR-deficiency andsusceptibility to pro-apoptotic agents are notunique. Park et al. (38) reported that FasL-mediated apoptosis in AhR-deficient rat BP8hepatoma cells required AhR expression.Similarly, Ah null mice, unlike wild type mice,were refractory to the cytotoxic effects of infusedCD95 cross-linking antibody (38).
A potential link between TNF + CHX andNPe6/PDT are their abilities to permeabilizelysosomes (this study, 25). Recent studies indicatethat C2-ceramide also triggers cathepsin releaseinto the cytosols of 1c1c7 cells prior to thedevelopment of apoptosis2. Furthermore, severalstudies have documented lysosomal permeabilityand a role for lysosomal proteases in CD95-mediated apoptosis (21,58). Hence, the resistanceof AhR-deficient cells to the pro-apoptotic effectsof these agents may relate to either a property oftheir acidic organelles, or their inability to activateprocesses causing lysosomal permeability.Although the current TNFα + CHX study does notdistinguish between the two possibilities, Nilssonet al. (59) reported that lysosomes are susceptibleto disruption by oxidants, and TNFα in some celltypes generates significant levels of oxidants(60,61). In the case of the NPe6/PDT protocol thephotosensitizer is sequestered in lysosomes (9).Analyses of AhR-containing and –deficient cellsdemonstrated that the acidic organelles of the
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

12
latter were resistant to the oxidant damage causedby singlet oxygen generated within the organellesfollowing photoactivation of NPe6 (25). Giventhat the oxidants were generated within theorganelles, and the amounts of sensitizer in AhR-deficient cells were comparable to, if not greaterthan their AhR-containing counterparts (25), itseems likely that AhR-deficiency affectsproperties of the lysosomes themselves (seeadditional discussion below).
A handful of proteins may function asmodifiers of lysosome permeability. Amongstsuch proteins are p53 (41), the phosphorylatedform of Bcl-2 (39), cathepsin B (15,17), andHsp70 (40). Whereas, cathepsin B and activatedp53 have been implicated in facilitating thedisruption of acidic organelles (15,17,41),phosphorylated Bcl-2 and Hsp70 have beenassigned protective roles (39,40). It is not knownwhether any of these proteins are related to howAhR content influences lysosomal fragility in ourcell lines.
Molecular and pharmacological approacheshave implicated roles for SMases in TNFα-induced apoptosis (47,62,63). Ceramide, theproduct of SMases, is converted by ceramidases tosphingosine. At least two studies suggest thatsphingosine functions as a lysosomal detergent(10,14). 3-O-MeSM was originally described as avery weak neutral SMase inhibitor (46), and waseffective in the current studies at suppressing bothTNFα + CHX-induced lysosome permeability andapoptosis. Although the neutral SMase inhibitorGW4869 did not suppress TNFα + CHX inducedapoptosis in our model, we have not performedcomparable studies with acidic SMase inhibitors.However, we think it is unlikely that SMases orsphingosine are responsible for lysosomepermeability and the apoptosis occurring in oursystem. Specifically, we have been unable todetect acidic or neutral SMase activation in 1c1c7cells following a 0.5 – 180 min exposure to TNF2.Furthermore, whereas cathepsin B can be readilyreleased from highly purified preparations of1c1c7 lysosomes by in vitro exposure to Triton X-100, no release occurs following exposure to 1 –10 µM sphingosine2.
Our studies suggest that lysosomal lipidcomposition may differ in AhR-containing and -deficient cell lines. Specifically, digitonintreatment released greater percentages of totalcathepsin B and D activities in the AhR-deficientTao line. Digitonin permeabilizes membranes byinteracting with cholesterol. Filipin staining of our
cell lines indicates massive accumulations ofunesterified cholesterol in punctate structures inthe AhR-deficient lines2. Whether this unestrifiedcholesterol is enriched in lysosomes has yet to bedetermined. However, we (25) recently noted thatour AhR-deficient cell lines share many of thecharacteristics of cell lines derived from patientssuffering from the lysosomal storage diseasemucolipidosis II (inclusion cell disease or I celldisease), including resistance to agents causinglysosomal breakage (20,21). I cells, as well asmany other lysosomal storage diseases,accumulate unesterified cholesterol in theirlysosomes (64-66). Unesterified cholesterol andsphingomyelin largely determine membranefluidity (67,68). Increases in unestrifiedmembrane cholesterol content are often paralleledby increases in sphingomyelin content (67).Increases in the two favor a liquid ordered state(67) . Although speculat ive, lysosomalaccumulation of unesterified cholesterol may altersusceptibility to agents causing lysosomaldestabilization. Indeed, recent studies indicate thatlysosomal incorporation of a sphingomyelinanalog increases resistance to oxidant-inducedlysosomal permeability (45).
In summary, analyses of the TNFα-inducedapoptotic program in cells of the 1c1c7 lineagehave yielded novel information about the role oflysosomal proteases in apoptosis, and functions ofthe AhR. Specifically, it is unlikely that caspase-8functions as an initiating caspase in 1c1c7 cellsbecause they are FADD deficient (Fig. 13). TNFαwill initiate apoptosis in 1c1c7 cells if cultures areco-treated with CHX. This activity of CHXreflects minimally two effects. First, CHX maysuppress the translation of mRNAs encodingsurvival proteins induced as a consequence ofTNFα activation of the NF-κB pathway. Second,C H X + T N Fα f a c i l i t a t e sdestabilization/permeability of lysosomes. Thispermeabilization does not occur in AhR deficientcells, or cultures pretreated with 3-O-MeSM (Fig13). Because AhR-deficiency also prevents NPe6disruption of lysosomes in PDT protocols (24), itappears that AhR content influences lysosomepermeability. As such, this represents a new andnovel function for the AhR. Released lysosomalfactors can initiate apoptosis by cleaving Bid, oractivating pro-caspase-12 (Fig 13). Once formed,caspase-12 can activate pro-caspase-9 directly(54,55), whereas tBid stimulates cytochrome crelease and apoptosome formation (69,70). The1c1c7 model represents a useful system for
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

13
assessing the roles of lysosomal proteases inapoptosis, and deciphering the factors affecting
lysosomal fragility/permeability.
REFERENCES
1. Eskelinen, E.-L., Tanaka, Y., and Saftig, P. (2003) Trends Cell Biol. 13, 137-1452. Mullins, C., and Bonifacino, J. S. (2001) BioEssays 23, 333-3433. Pillay, C. S., Elliott, E., and Dennison, C. (2002) Biochem. J. 363, 417-4294. Guicciardi, M. E., Leist, M., and Gores, G. J. (2004) Oncogene 23, 2881-28905. Antunes, F., Cadenas, E., and Brunk, U. T. (2001) Biochem. J. 356, 549-5556. Dare, E., Li, W., Zhivotovsky, B., Yuan, S., and Ceccatelli, S. (2001) Free Rad. Biol. Med. 30, 1347-
13567. Roberg, K., Johansson, U., and Ollinger, K. (1999) Free Rad. Biol. Med. 27, 1228-12378. Ollinger, K. (2000) Arch. Biochem. Biophys. 373, 346-3519. Reiners, Jr., J. J., Caruso, J. A., Mathieu, P., Chelladurai, B., Yin, X.-M., and Kessel, D. (2002) Cell
Death Differ. 9, 934-94410. Kagedal, K., Zhao, M., Svensson, I., and Brunk, U. T. (2001) Biochem. J. 359, 335-34311. Ji, Z. S., Miranda, R. D., Newhouse, Y. M., Weisgraber, K. H., Huang, Y., and Mahley, R. W. (2002)
J. Biol. Chem. 277, 21821-2182812. Neuzil, J., Zhao, M., Ostermann, G., Sticha, M., Gellert, N., Weber, C., Eaton, J. W., and Brunk, U.
T. (2002) Biochem. J. 362, 709-71513. Emert-Sedlak, L., Shangary S., Rabinovitz, A., Miranda, M. B., Delach, S. M., and Johnston, D. E.
(2005) Mol. Cancer Ther. 4, 733-74214. Werneburg, N. W., Guicciardi, M. E., Bronk, S. F., and Gores, G. J. (2002) Am. J. Physiol.
Gastrointest. Liver Physiol. 283, G947-G95615. Guicciardi, M. E., Deussing, J., Miyoshi, H., Bronk, S. F., Svingen, P. A., Peters, C., Kaufmann, S.
H., and Gores, G. J. (2000) J. Clin. Invest. 106, 1127-113716. Foghsgaard, L., Wissing, D., Mauch, D., Lademann, U., Bastholm, L., Boes, M., Elling, F., Leist, M.,
and Jäättelä, M. (2001) J. Cell Biol. 153, 999-100917. Guicciardi, M. E., Miyoshi, H., Bronk, S. F., and Gores, G. J. (2001) Am. J. Pathol. 159, 2045-205418. Heinrich, M., Neumeyer, J., Jakob, M., Hallas, C., Tchikov, V., Winoto-Morbach, S., Wickel, M.,
Schneider-Brachert, W., Trauzold, A., Hethke, A., and Schulze, S. (2004) Cell Death Differ. 11, 550-563
19. Werneburg, N., Guicciardi, M. E., Yin, X.-M., and Gores, G. J. (2004) Am. J. Physiol. Gastrointest.Liver Physiol. 287, G436-443
20. Terman, A., Neuzil, J., Kagedal, K., Öllinger, K., and Brunk, U. T. (2002) Exp. Cell Res. 274, 9-1521. Tardy, C., Autefage, H., Garcia, V., Levade, T., and Andrieu-Abadie, N. (2004) J. Biol. Chem. 279,
52914-5292322. Ishisaka, R., Utsumi, T., Yabuki, M., Kanno, T., Furuno, T., Inoue, M., and Utsumi, K. (1998) FEBS
Lett. 18, 233-23623. Ishisaka, R., Kanno, T., Akiyama, J., Yoshioka, T., Utsumi, K., and Utsumi, T. (2001) J. Biochem.
(Tokyo) 129, 34-4124. Stoka, V., Turk, B., Schendel, S. L., Kim, T.-H., Cirman, T., Snipas, S. J., Ellerby, L. M., Bredesen,
D., Freeze, H., Abrahamson, M., Brommer, D., Krajewski, S., Reed, J. C., Yin, X.-M., Turk, V., andSalvesen, G. S. (2001) J. Biol. Chem. 276, 3149-3157
25. Caruso, J. A., Mathieu, P. A., Joiakim, A., Leeson, B., Kessel, D., Sloane, B. F., and Reiners, Jr., J. J.(2004) Mol. Pharmacol. 65, 1016-1028
26. Cirman, T., Oresic, K., Mazovec, G. D., Turk, V., Reed, J. C., Meyers, R. M., Salvesen, G. S., andTurk, B. (2004) J. Biol. Chem. 279, 3578-3587
27. Becattini, B., Sareth, S., Zhai, D., Crowell, K. J., Leone, M., Reed, J. C., and Pellecchia, M. (2004)Chem. Biol. 11, 1107-1117
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

14
28. Nebert, D. W., Roe, A. L., Dieter, M. Z., Solis, W. A., Yang, Y., and Dalton, T. P. (2000) Biochem.Pharmacol. 59, 65-85
29. Schmidt, J. V., and Bradfield, C. A. (1996) Annu. Rev. Cell Dev. Biol. 12, 55-8930. Sun, Y. V., Boverhof, D. R., Burgoon, L. D., Fielden, M. R., and Zacharewski, T. R. (2004) Nucl.
Acids Res. 32, 4512-452331. Boverhof, D. R., Burgoon, L. D., Tashiro, C., Chittim, B., Harkema, J. R., Jump, D. B., and
Zacharewski T.R. (2005) Tox. Sci. 85, 1048-106332. Puga, A., Tomlinson, C. R., and Xia, Y. (2005) Biochem. Pharm. 69, 199-20733. Duncan, D. M., Burgess, E. A., and Duncan, I. (1998) Genes Develop. 12, 1290-130334. Qin, H., and Powell-Coffman, J. A. (2004) Dev. Biol. 270, 64-7535. Ma, Q., and Whitlock, Jr., J. P. (1996) Mol. Cell Biol. 16, 2144-215036. Abdelrahim, M., Smith, III, R., and Safe, S. (2003) Mol. Pharm. 63, 1373-138137. Reiners, Jr., J. J., and Clift, R. E. (1999) J. Biol. Chem. 274, 2502-251038. Park, K-T, Mitchell, K. A., Huang, G., and Elferink, C. J. (2005) Mol. Pharm. 67, 612-62239. Zhao, M., Eaton, J. W., and Brunk, U. T. (2001) FEBS Lett. 509, 405-41240. Nylandsted, J., Gyrd-Hansen, M., Danielewicz, A., Fehrenbacher, N., Lademann, U., Høyer-Hansen,
M., Weber, E., Multhoff, G., Rohde, M., and Jäättelä, M. (2004) J. Exp. Med. 200, 425-43541. Yuan, X.-M., Li, W., Dalen, H., Lotem, J., Kama, K., Sachs, R., and Brunk, U. T. (2002) Proc. Natl.
Acad. Sci. USA, 99, 6286-629142. Zhang, H., Taylor, J., Luther, D., Johnston, J., Murray, S., Wyatt, J. R., Watt, A. T., Koo, S., York-
Defalco, C., Stecker, K., and Dean, N. M. (2003) J. Pharm. Exp. Ther. 307, 24-3343. Osborn, L., Kunkel, S., and Nabel, G. J. (1989) Proc. Natl. Acad. Sci. USA 86, 2336-234044. Joiakim, A., Mathieu, P. A., Elliot, A. A., and Reiners, Jr., J. J. (2004) Mol. Pharm. 66, 936-94745. Caruso, J. A., Mathieu, P. A., and Reiners, Jr., J. J. (2005) Biochem. J., 392, 325-334.46. Lister, M. D., Ruan, Z. S., and Bittman, R. (1995) Biochim. Biophys. Acta. 1256, 25-3047. Luberto, C., Hassler, D. F, Signorelli, P., Okamato, Y., Sawai, H., Boros, E., Martin, D. J., Obeid, L.
M., Hannun, Y. A., and Smith, G. K. (2002) J. Biol. Chem. 277, 41128-4113948. An, J., Chen, Y., and Huang, Z. (2004) J. Biol. Chem. 279, 19133-19140.49. Wang, J. L., Liu, D., Zhang, Z. J., Shan, S., Han, X., Srinivasula, S. M., Croce C. M., Alnemri, E. S.,
and Huang, Z. (2000) Proc. Natl. Acad. Sci. USA 97, 7123-712950. Micheau, O., and Tschopp, J. (2003) Cell 114, 181-19051. Muppidi, J. R., Tschopp, J., and Siegel, R. M. (2004) Immunity 21, 461-46552. Bidere, N., Lorenzo, H. K., Carmona, S., Laforge, M., Harper, F., Dumont, C., and Senik, A. (2003)
J. Biol. Chem. 278, 31401-3141153. Zhao, M., Antunes, F., Eaton, J.W., and Brunk, U.T. (2003) Eur. J. Biochem. 270, 3778-378654. Rao, R. V., Hermel, E., Castro-Obregon, S., del Rio, G., Ellerby, L. M., Ellerby, H. M., and
Bredesen, D. E. (2001) J. Biol. Chem. 276, 33869-3387455. Morishima, N., Nakanishi, K., Takenouchi, H., Shibata, T., and Yasuhiko, Y. (2002) J. Biol. Chem.
277, 34287-3429456. Breckenridge, D. G., Germain, M., Mathai, J. P., Nguyen, M., and Shore, G. C. (2003) Oncogene 22,
8608-861857. Kalai, M., Lamkanfi, M., Denecker, G., Boogmans, M., Lippens, S., Meeus, A., Declercq, W., and
Vandenabeele, P. (2003) J. Cell Biol. 162, 457-46758. Brunk, U. T., and Svensson, I. (1999) Redox. Rep. 4, 3-1159. Nilsson, E., Ghassemifar, R., and Brunk, U. T. (1997) Histochem J. 29, 857-86560. Xue, X., Piao, J-H., Nakajima, A., Sakon-Komazawa, S., Kojima, Y., Mori, K., Yagita, H., Okumura,
K., Harding, H., and Nakano, H. (2005) J. Biol. Chem. 280, 33917-3392561. Kamata, H., Honda, S-i., Maeda, S., Chang, L., Hirata, H., and Karin, M. (2005) Cell 120, 649-66162. Ségui, B., Cuvillier, O., Adam-Klages, S., Garcia, V., Malagarie-Cazenave, S., Lévêque, S, Caspar-
Bauguil, S., Coudert, J., Salvayre, R., Krönke, M., and Levade, T. (2001) J. Clin. Invest. 108, 143-151
63. Colell, A., Morales, A., Fernandez-Checa, J. C., and Garcia-Ruiz, C. (2002) FEBS Lett. 526, 135-14164. Simons, K., and Gruenberg, J. (2000) Trends Cell Biol. 10, 459-46265. Harzer, K., Massenkeil, G., and Frohlich, E. (2003) FEBS Lett. 537, 177-181
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

15
66. Pagano, R. E. (2003) Phil. Trans. R. Soc. Lond. 358, 885-89167. Ridway, N. D. (2000) Biochim. Biopsy. Acta. 1484, 129-14168. Simons, K., and Ikonen, E. (2000) Science 290, 1721-172669. Wei, M.C., Lindsten, T., Nootha, V. K., Weiler, S., Gross, A., Ashiya, M., Thompson, C.B., and
Korsmeyer, S. J. (2000) Genes Dev. 14, 2060-207170. Korsmeyer, S. J., Wei, M. C., Saito, M., Weiler, S., Oh, K. J., and Schlesinger, P. H. (2000) Cell
Death Differ. 7, 1166-1173
FOOTNOTES
Acknowledgements: This work was supported by NIH-NIEHS grant ES009392, and was assisted by theCell Culture, Imaging and Cytometry, and Protein and Proteomics Facility Cores, which are supported byNIEHS grant P30 ES06639.
1The abbreviations used are: αMEM, α-minimal essential medium; Ac-DEVD-AMC, acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin; AhR, aryl hydrocarbon receptor; AMC, 7-amino-4-methylcoumarin; CA-074, L-3-trans-(propylcarbamoyl)oxirane-2-carbonyl-L-isoleucyl-L-proline; CA-074-Me, methyl ester of CA-074; CHX, cycloheximide; DAME, diazoacetyl-DL-2-aminohexanoic acid-methyl ester; E-64, (2S,3S)-trans-epoxysuccinyl-L-leucylamido-3-methylbutane; E-64d, ethyl ester of E-64; HA14-1, ethyl 2-amino-6-bromo-4-(1-cyano-2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate;EMSA, electrophoretic mobility shift assay; LDH, lactate dehydrogenase; HO, HO33342; NPe6, N-aspartyl chlorin e6; PDT, photodynamic therapy; 3-O-MeSM, 3-O-methylsphingomyelin; SMase,sphingomyelinase; TNFα, tumor necrosis factor alpha; TNFr, tumor necrosis factor receptor; Z-FA-FMK,benzyl-oxycarbonyl-Phe-Ala-fluoromethyl ketone.
2Caruso and Reiners, Jr., unpublished data
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

16
FIGURE LEGENDS
Fig. 1. CHX potentiation of TNFαααα-induced apoptosis. (A) 1c1c7 cultures were treated with nothing, 30pg/ml TNFα ± various amounts of CHX for different lengths of time before being harvested for analysesof DEVDase. In co-treatment protocols CHX was added 45-60 min prior to TNFα. Data represent means± S.D. of triplicate assays. Similar results were obtained in an additional study. Symbols are: no treatment(X), TNF (�), TNF + 1 µg/ml (�), 0.5 µg/ml (�), 0.1 µg/ml (�) or 0.05 µg/ml CHX (�). (B) 1c1c7cultures were treated with nothing (X) or 1 µg/ml (�), 0.5 µg/ml (�), or 0.1 µg/ml (�) CHX beforebeing harvested for analyses of DEVDase. Data represent means ± S.D. of triplicate assays. Similarresults were obtained in an additional study. (C) 1c1c7 cultures were treated with nothing (X), TNFα (�),various doses of CHX (�), or TNFα + CHX (�) as described in panel A, and were harvested 9-11 h afterTNFα addition for analyses of viability. Data represent means ± S.D. of 3-4 culture dishes (D) 1c1c7cultures were co-treated with [3H]leucine and various doses of CHX for 1 h prior to being processed foranalyses of [3H]leucine incorporation into protein. Data represent means ± S.D. of 4 plates per treatmentgroup. Similar results were obtained in two additional experiments.
Fig. 2. Cathepsin B and Lamp-1 staining and co-localization in 1c1c7 cultures treated with TNFαααα,,,,CHX and/or 3-O-MeSM. Cultures of 1c1c7 cells were treated with 30 pg/ml TNFα, 1 µg/ml CHX, or 50µM 3-O-MeSM prior to being harvested and processed for immunofluorescence analyses of cathepsin Band Lamp-1 staining and co-localization. In these studies 3-O-MeSM and CHX were added 90 and 30min, respectively, prior to TNFα. Cultures were harvested 4 h after TNFα addition. Fields arerepresentative of data generated in 4 independent experiments. Bar in panel A represents 20 µ.
Fig. 3. Cathepsin D and Lamp-1 staining and co-localization in 1c1c7 cultures treated with TNFαααα,,,,CHX and/or 3-O-MeSM. Treatment conditions are identical to those described in Fig. 2 except thatfixed cells were stained with antibodies to cathepsin D. The pictures in this Fig. are from the experimentreported in Fig. 2. Bar in panel A represents 20 µ.
Fig. 4. 3-O-MeSM suppression of TNFαααα + CHX induced apoptosis. Cultures of 1c1c7 cells weretreated with 30 pg/ml TNFα, 1 µg/ml CHX and/or varied concentrations of 3-O-MeSM. In co-treatmentprotocols 3-O-MeSM and CHX were added 90 and 30 min, respectively, prior to TNFα. Times indicatedin the panels represent h after TNFα addition. (A) 1c1c7 cultures were treated with TNFα, CHX, and/or50 µM 3-O-MeSM and photographed 9.5, 8.5 and 8 h after 3-O-MeSM, CHX and TNFα additions,respectively. (B) Cultures were treated with TNFα, CHX and various concentrations of 3-O-MeSM priorto being harvested for analyses of DEVDase activities at different times. Data represent means ± S.D. oftriplicate analyses. Similar results were obtained in two additional experiments. Symbols are: notreatment (X), CHX (�), 50 µM 3-O-MeSM (�), TNFα + CHX (�), CHX + TNF + 40 µM (�), or50µM (�) or 60 µM (�) 3-O-MeSM. (C) Cultures were treated with nothing (X), TNFα (�), CHX (�)3-O-MeSM (�), or TNFα + CHX + 3-O-MeSM (�) for 8-10 h prior to being harvested for analyses ofviability. Data represent means ± S.D. of 3 plates. (D) Cultures were treated with TNFα, CHX and 50 µM3-O-MeSM prior to being harvested for analyses of pro-caspase-3, –9 and –12 processing by western blotanalyses, using 25 µg of protein per lane.
Fig. 5. Role of Bid in TNFαααα-induced apoptosis. Subconfluent 1c1c7 cultures were treated for 6 h withnothing, lipofectin, Bid antisense oligonucleotides, or scramble sense oligonucleotides before beingwashed and refed. Approximately 24, 48 and 72 h after treatment the cultures were harvested for analysesof Bid contents by western blot analyses (A), or treated with 30 pg/ml TNFα + 1 µg/ml CHX for differentlengths of time prior to harvesting for analyses of DEVDase activities (B). Western blot analysesemployed 20 µg of protein per lane. DEVDase data represent means ± S.D. of triplicate assays. Similardata for each of the three days were obtained in a second independent experiment. Symbols are forcultures treated with: lipofectin (∆), scramble sense oligos (�), Bid antisense oligos (�), lipofection +TNFα + CHX (�), scramble sense oligo treated + TNFα + CHX (�), and Bid antisense oligo treated +
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

17
TNFα + CHX (�). (C) 1c1c7 cultures that had been treated 48 h prior with Bid antisense, scramble senseor lipofectin were exposed to 30 pg/ml TNFα and 1 µg/ml CHX. Pictures were taken immediately beforeTNFα + CHX addition, and at various times afterwards. Data in panel C are from the same experimentdepicted in panel B, day 2.
Fig. 6. Role of cathepsin D in TNFαααα-induced apoptosis. (A) 1c1c7 cultures were treated with 30 pg/mlTNFα, 1 µg/ml CHX and/or 1 µM acetyl pepstatin A for various lengths of time before being harvestedfor western blot analyses of Bid and caspase-3. In co-treatment protocols acetyl pepstatin A and CHXwere added the evening before and 45 min, respectively, prior to TNFα. Western blotting used 40 µg ofprotein per lane. (B) 40 ng of recombinant murine Bid was incubated with 0.5 µg of 1c1c7endosomal/lysosomal extract for 1 h at 37˚C in the presence of 5 µM CA-074, 10 µM E-64, 1 µM Z-FA-FMK, 100 µM leupeptin or 0.5 µM pepstatin A before being processed for western blot analyses of Bid.Similar results were obtained in a second independent experiment. (C) 1c1c7 cultures were treated with30 pg/ml TNFα, 1 µg/ml CHX, or various concentrations of acetyl pepstatin A or diazoacetyl-DL-2-aminohexanoic acid-methyl ester (DAME) prior to being harvested for analyses of DEVDase activities. Inco-treatment protocols acetyl pepstatin A and DAME were added the evening before TNFα. CHX wasadded 45 min prior to TNFα. Cultures were harvested for analyses 6 h after TNFα addition. (D) Similarto panel C except that cultures were treated with 1 µM acetyl pepstatin A and/or 400 µM DAME andharvested for analyses of DEVDase activities at various times after TNFα treatment. Symbols for panelsD and E: solvent (X), acetyl pepstatin A (∆), DAME (�), TNFα + CHX (�), TNFα + CHX + acetylpepstatin A (�), TNFα + CHX + DAME (�), TNFα + CHX + acetyl pepstatin A + DAME (�). (E)1c1c7 cultures were treated with 30 pg/ml TNFα, 1 µg/ml CHX, 5 µM CA-074-Me, 5 µM E-64d or 1 µMZ-FA-FMK for 8 h prior to being harvested for analyses of DEVDase activities. Protease inhibitors wereadded 1 h prior to TNFα. Symbols for panel E: solvent (X), TNFα + CHX (�), TNFα + CHX + CA-074-Me (�), TNFα + CHX + Z-FA-FMK (∆), and TNFα + CHX + E-64d (�). (F) 1c1c7 cultures weretreated as described in panels D and E and harvested 8.5 - 11 h after TNFα addition for analyses ofviability. Data represent means ± S.D. of 3 culture plates. *Greater than TNFα + CHX treatment group,but less than solvent control, p<0.01.
Fig. 7. Characterization of AhR contents, TNF signaling components, JNK and NF-κκκκB activationsin 1c1c7 variant cell lines. (A) Extracts from non-treated 1c1c7 cells, and 5 of its variants, were analyzedby Western blotting for the presence of AhR, pro-caspase-8, TNFr1, TRADD, and FADD. Analyses areof 25 µg of whole cell lysate per lane. A 5 µg sample of murine liver extract was used as a positiveFADD control. Each lane represents extract prepared from a different culture dish. (B) 1c1c7 and Taocultures were treated with 1 µM anisomycin or 30 pg/ml TNFα + 1 µg/ml CHX for varying lengths oftime before being harvested and processed for Western blot analyses of JNK activation. Analyses are of25 µg of whole cell lysate per lane. CHX was added 45 min prior to TNFα. (C) 1c1c7 and Tao cultureswere treated with 30 pg/ml TNF ± 1 µg/ml CHX for different lengths of time before being harvested andprocessed for EMSA analyses of NF-κB activation. Analyses are of 25 µg of nuclear lysate per lane.CHX was added 45 min prior to TNFα. The non-complexed, radiolabeled oligonucleotide has run off thegel.
Fig. 8. Effects of AhR content on the induction of apoptosis by TNFαααα ± CHX. AhR-containing celllines (1c1c7, TAHR, WCMV) and their respective AhR-deficient counterparts (Tao, TCMV, WARV)were treated with nothing, 30 pg/ml TNFα, 1 µg/ml CHX, or TNFα + CHX for different lengths of timebefore being harvested and processed for analyses of DEVDase activities (panels A,C,E) or pro-caspase-3, -8, -9, and -12 processing by western blot analyses (panels B,D,F). CHX was added 45 min prior toTNFα. DEVDase data represent means ± S.D. of four 1c1c7 experiments, and triplicate analysesperformed on a single extract for all other cell lines. Similar results for the latter 5 cells lines wereconfirmed in minimally a second experiment. Western blot analyses employed 25 µg protein per lane.Symbols are: no treatment (�), TNFα (�), CHX (�), and TNFα + CHX (�).
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

18
Fig. 9. Cathepsin B and Lamp-1 staining and co-localization in Tao cultures treated with TNFααααand/or CHX. Cultures of Tao cells were treated with 30 pg/ml TNFα and/or 1 µg/ml CHX prior to beingharvested and processed for subsequent immunofluorescence analyses of cathepsin B and Lamp-1staining and co-localization. In these studies CHX was added 30 min prior to TNFα. Cultures wereharvested 4 h after TNFα addition. Fields are representative of data generated in 3 independentexperiments. Bar in panel A represents 20 µ.
Fig. 10. Cathepsin D and Lamp-1 staining and co-localization in Tao cultures treated with TNFααααand/or CHX. Treatment conditions are identical to those described in Fig. 9 except that fixed cells werestained with antibodies to cathepsin D. The pictures in this Fig. are from the experiment reported in Fig.9. Bar in panel A represents 20 µ.
Fig. 11. Digitonin-mediated releases of lactate dehydrogenase and cathepsins B and D. Cultures of1c1c7 (�) and Tao (�) cells were incubated with increasing concentrations of digitonin, as described inMaterials and Methods, prior to harvesting of extracellular fluids for subsequent analyses of lactatedehydrogenase (panel A), cathepsin B (panel B) and cathepsin D (panel C) activities. Data representmeans ± S.D. of 5 – 15 experiments, in which the value for an individual experiment was determined byanalyses of 3-6 plates per treatment group. *significantly greater than corresponding 1c1c7 value, P<0.05.
Fig.12. TNFαααα and CHX triggered LDH and cathepsin B and D releases. Cultures of 1c1c7 (A,B,C)and Tao (D,E,F) cells were treated with 30 pg/ml TNFα (�), or 1 µg/ml CHX (�), or TNFα + CHX (�)for varying lengths of time before being permeabilized with digitonin, and collection of extracellularfluids for analyses of lactate dehydrogenase (A,D), cathepsin B (B,E) and cathepsin D (C,F) activities.*significantly greater than the zero time value, P<0.05.
Fig. 13. Schematic of TNF + CHX induced apoptosis in murine hepatoma cells of the 1c1c7 lineage.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

19
TABLES
Table ICathepsin and Lamp-1 co-localization in hepatoma cultures as a function of CHX, TNFα and/or 3-O-MeSM
treatmentAhR-containing (1c1c7, WCMV) and –deficient (Tao, WARV) cells were treated for 4 h with nothing
(NT), or singularly, or with combinations of 1 µg/ml CHX (C), 30 pg/ml TNFα (T), or 50 µM 3-O-MeSM (3OM)prior to being processed for Lamp-1 and cathepsins B and D staining and co-localization. Pre-treatment times aredescribed in the legend to Fig. 4. Data were generated by MetaMorph analyses of n numbers of cells, and representmeans ± S.D.
Cell type Treatment Type ofCathepsin
Relative intensityof
Lamp-1/Cathepsin
Lamp-1 (+)spots not
containingCathepsin (%)
Cathepsin (+) spotsnot containingLamp-1 (%)
n
1c1c7 NT B 1.24 ± 0.40 43.5 ± 10.5 32.1 ± 11.1 15C B 1.11 ± 0.57 45.5 ± 11.2 43.5 ± 14.3* 20T B 1.13 ± 0.43 38.8 ± 11.4 39.7 ± 12.2 24
C+T B 1.48 ± 0.81* 46.4 ± 14.7* 33.8 ± 14.0 213OM B 1.19 ± 0.27 33.1 ± 10.7 23.4 ± 6.6 21
C+T+3OM B 1.15 ± 0.46 30.4 ± 13.6 27.3 ± 12.8 21
1c1c7 NT D 1.19 ± 0.26 45.9 ± 10.2 32.1 ± 14.7 25C D 1.11 ± 0.50 46.2 ± 14.9 48.0 ± 18.6* 20T D 1.13 ± 0.55 41.3 ± 14.3 39.7 ± 17.7 22
C+T D 3.16 ± 1.82* 71.5 ± 7.3* 26.2 ± 9.0 283OM D 1.40 ± 0.53 47.4 ± 14.7 31.1 ± 13.1 22
C+T+3OM D 1.52 ± 0.37 57.6 ± 7.2 33.4 ± 9.6 20
Tao NT B 1.16 ± 0.20 45.9 ± 6.6 35.8 ± 7.3 9C B 1.30 ± 0.27 47.2 ± 18.0 37.2 ± 13.0 10T B 1.54 ± 0.40 51.5 ± 7.8 30.1 ± 6.0 10
C+T B 1.23 ± 0.77 46.5 ± 12.8 41.0 ± 18.3 11
Tao NT D 1.02 ± 0.26 48.8 ± 12.2 46.8 ± 9.0 11C D 1.03± 0.26 48.4 ± 8.8 43.9 ± 12.7 8T D 1.43 ± 0.78 53.1 ±14.6 40.1 ± 19.3 13
C+T D 1.50 ± 0.66 55.6 ± 9.1 38.1 ± 15.7 12
WCMV NT B 1.11 ± 0.38 35.3 ± 9.9 28.5 ± 9.0 9C B 1.14 ± 0.32 55.7 ± 9.5* 50.3 ± 20.4* 9T B 1.83 ± 0.81 48.0 ± 11.5 18.1 ± 10.6 9
C+T B 2.36 ± 1.09* 57.4 ± 11.5* 18.0 ± 10.8 9
WCMV NT D 0.92 ± 0.35 39.37 ± 15.4 43.5 ± 8.4 8C D 1.57 ± 0.77 49.5 ± 19.3 32.4 ± 9.7 8T D 1.82 ± 0.47 62.0 ± 11.4* 35.7 ± 8.0 9
C+T D 2.32 ± 0.80* 69.8 ± 12.8* 35.2 ± 6.7 9
WARV NT B 1.55 ± 0.46 42.6 ± 13.9 14.4 ± 7.7 8C B 1.02 ± 0.17 34.5 ± 5.7 30.1 ± 6.4 9T B 1.30 ± 0.39 36.7 ± 11.3 24.0 ± 10.4 8
C+T B 1.43 ± 0.68 45.4 ± 16.8 26.9 ± 12.6 9
WARV NT D 1.14 ± 0.56 40.1 ± 10.8 32.9 ± 15.3 8C D 0.86 ± 0.42 44.4 ± 15.7 52.8 ± 15.6 8T D 0.99 ± 0.30 35.2 ± 9.8 37.3 ± 12.4 8
C+T D 1.14 ± 0.46 37.1 ± 15.5 32.0 ± 10.2 9*Greater than non-treated group, p < 0.01
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
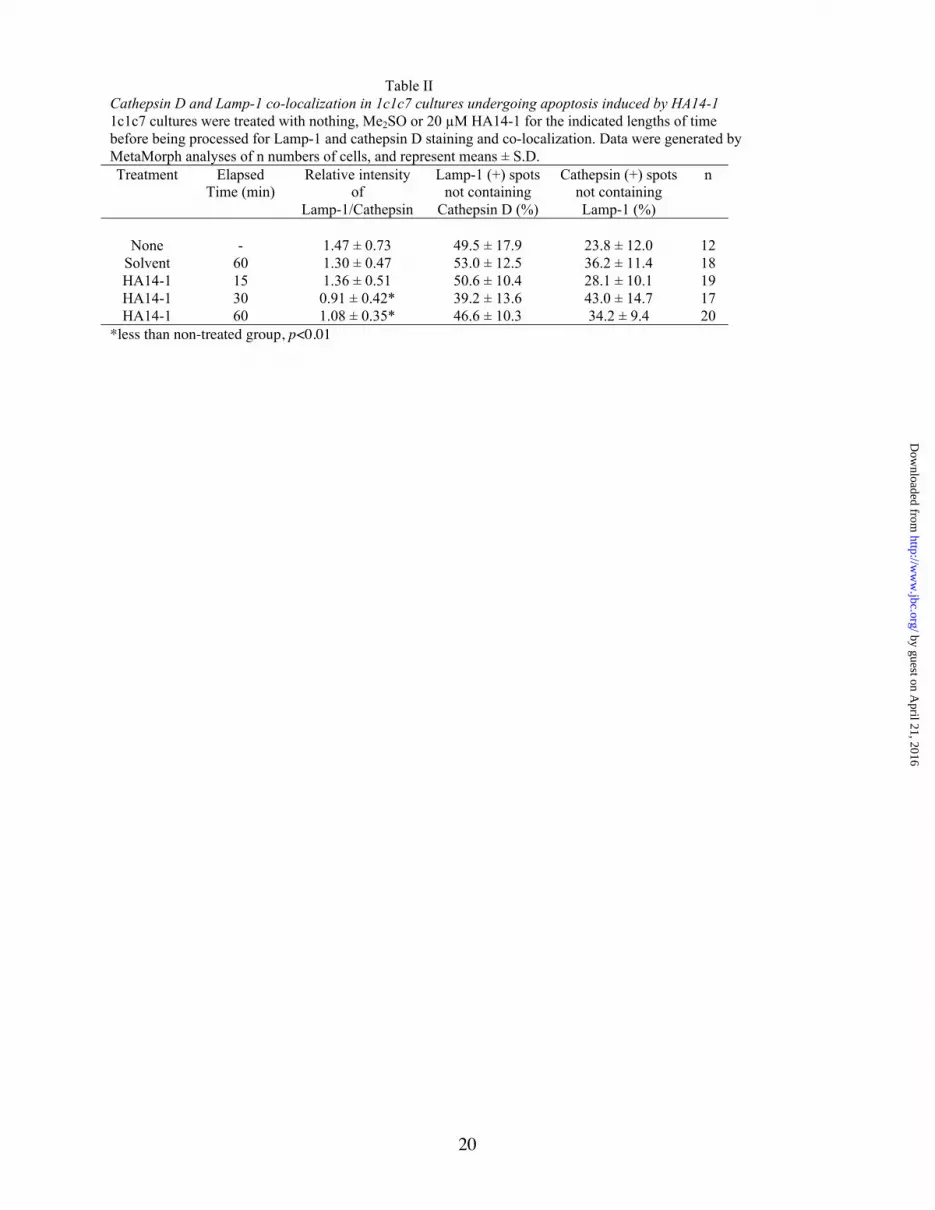
20
Table IICathepsin D and Lamp-1 co-localization in 1c1c7 cultures undergoing apoptosis induced by HA14-11c1c7 cultures were treated with nothing, Me2SO or 20 µM HA14-1 for the indicated lengths of timebefore being processed for Lamp-1 and cathepsin D staining and co-localization. Data were generated byMetaMorph analyses of n numbers of cells, and represent means ± S.D.Treatment Elapsed
Time (min)Relative intensity
ofLamp-1/Cathepsin
Lamp-1 (+) spotsnot containing
Cathepsin D (%)
Cathepsin (+) spotsnot containingLamp-1 (%)
n
None - 1.47 ± 0.73 49.5 ± 17.9 23.8 ± 12.0 12Solvent 60 1.30 ± 0.47 53.0 ± 12.5 36.2 ± 11.4 18HA14-1 15 1.36 ± 0.51 50.6 ± 10.4 28.1 ± 10.1 19HA14-1 30 0.91 ± 0.42* 39.2 ± 13.6 43.0 ± 14.7 17HA14-1 60 1.08 ± 0.35* 46.6 ± 10.3 34.2 ± 9.4 20
*less than non-treated group, p<0.01
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Figure 1, Caruso et al.
% S
urvi
val
0 0.1 0.5 1.050
60
70
80
90
100
C
CHX (µg/ml)D
0.001 0.01 0.1 1 10
CHX (µg/ml)
0
2
4
6
8
10
DP
M (
x 10
)
-3
A
0.01
0.1
1
10
DE
VD
ase
(nm
ol /
min
/ m
g)
B
0 2 4 6 8Time (h)
0.01
0.1
1
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
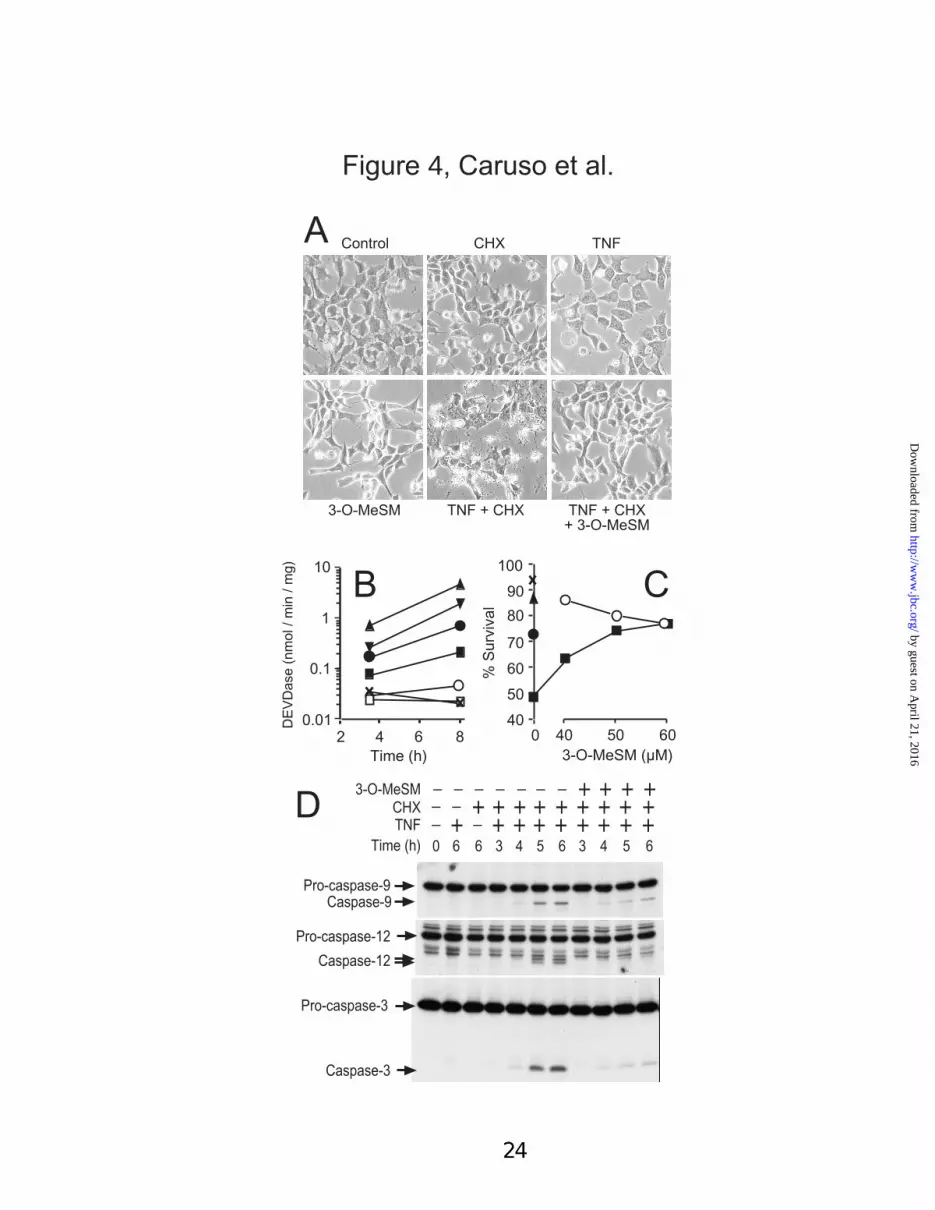
Figure 4, Caruso et al.
TNF + CHX+ 3-O-MeSM
CHXA Control TNF
TNF + CHX3-O-MeSM
+ ++ +_ __ ___ _
D
Caspase-3
Pro-caspase-3
Time (h)+_+ ++ ++ ++ +_TNF
0 3 4 63 4 5 566 6
CHX +_ + ++ ++ ++ +_
Caspase-12
Caspase-9
Pro-caspase-12
Pro-caspase-9
3-O-MeSM
DE
VD
ase
(nm
ol /
min
/ m
g)
Time (h)
B
20.01
0.1
1
10
4 6 8 0
% S
urvi
val
3-O-MeSM (µM)
40
50
60
70
80
90
100
40 50 60
C
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Bid antisense Scramble sense Lipofectin
0
4.5
8.5
Hours
B
C
Day 1 Day 2 Day 3
2 4 6 80
2
4
6
2 4 6 8
8
10
0
246
2 4 6 80
2
4
6
Hours After TNF + CHX Addition
DE
VD
ase
Act
ivity
Bid
Actin
Lipof
ectin
Scram
ble
anti-
Bid
No tre
atm
ent
Lipof
ectin
Scram
ble
anti-
Bid
No tre
atm
ent
Lipof
ectin
Scram
ble
anti-
Bid
No tre
atm
ent
Day 1 Day 2 Day 3A
Days After Transfection
Figure 5, Caruso et al.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Figure 6, Caruso et al.
Time (h)
E
0 2 4 6 80
2
4
6
8Time (h)
DE
VD
ase
(nm
ol /
min
/ m
g)D
2 4 6 80
2
1
C
0
2
1
5
4
3
None 0.1 1 10 100 1000Concentration (µM)
A CHX
AcPep AHours
+_ ++ +_ __35 73 5 70
Casp-3
Pro-casp-3
Bid
tBid
777
__TNF + ++ ++ +__ _++ ++ ++ +__ _+
B+_ +++Lysosomes ++
Bid
CA-074
E-64
Leup
eptin
Pepst
atin
A
Z-FA
-FM
K
% Viable
AcPep A + TNFα + CHXAcPep A (1 µM)
DAME + TNFα + CHXDAME (400 µM)
CA-075 + TNFα + CHXCA-074-Me (5 µM)
E-64d + TNFα + CHXE-64d (5 µM)
Z-FA-FMK + TNFα + CHXZ-FA-FMK (1 µM)
50 60 70 80 90 100
TNFα + CHX (1 µg/mL)Solvent
F
*
*
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Figure 7, Caruso et al
1c1c7 Tao TCMV TAHR WCMV WARV
AhR
TRADD
FADD
Pro-caspase-8
TNFr1
Mur
ine
liver
A
B
C
JNK2
CHX + TNF
AnisomycinTime (h)
pJNK1pJNK2
JNK1
_ ++ +++_ + ______0
0.5 0.51
230.2
_ ++ +++_ + ______0
0.5 0.51
230.2
1c1c7 Tao
0.5 2 0.5 2 0.5 2 0.5 200Time (h)
__ ++++++++TNF
+__ ___ _+ + +CHX
1c1c7 Tao
NF-kB + oligocomplex
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
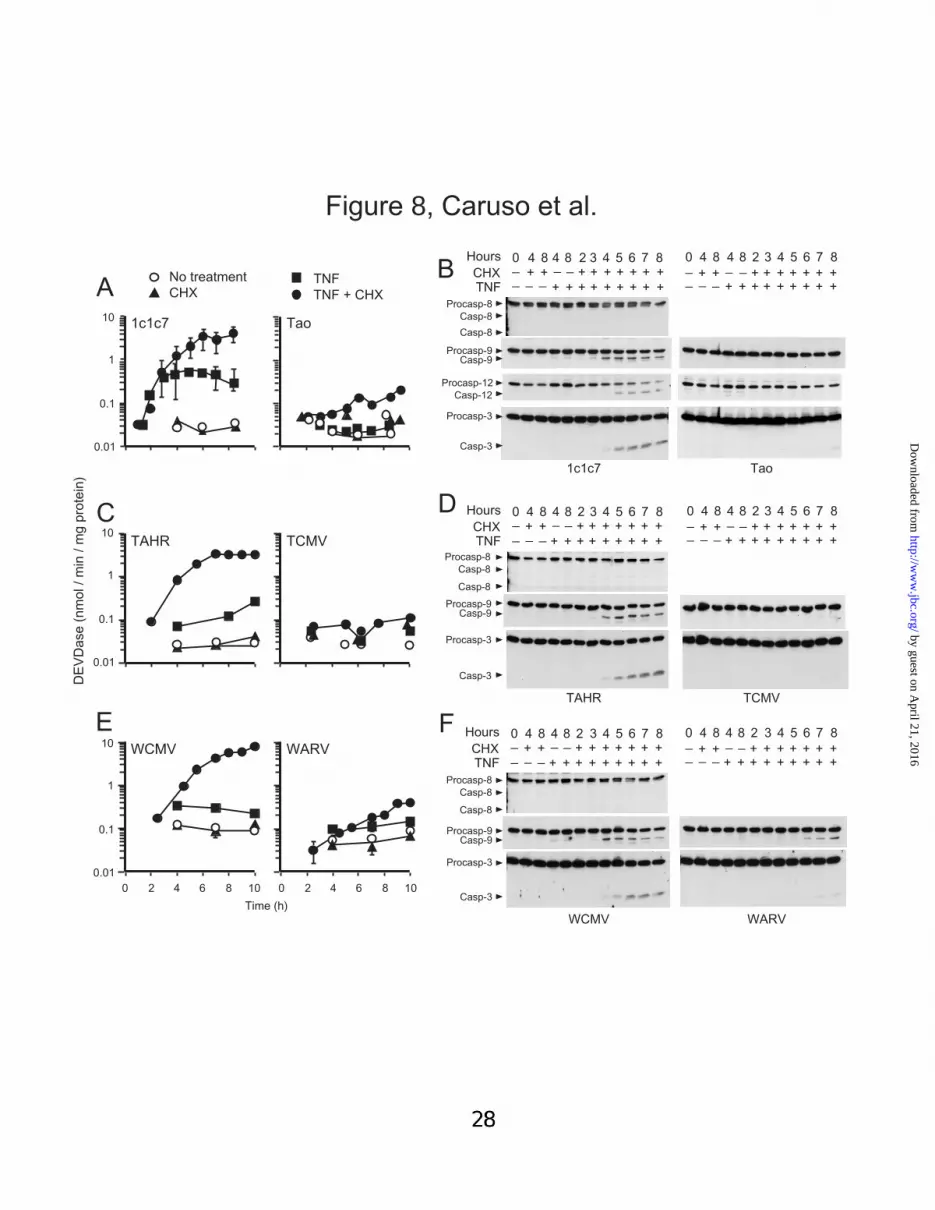
Figure 8, Caruso et al.
Time (h)
0.01
0.1
1
10
0 2 4 6 8 10 0 2 4 6 8 10
WCMV WARV
E
A
0.01
0.1
1
10 1c1c7 Tao
TNF + CHXTNF
CHXNo treatment
DE
VD
ase
(nm
ol /
min
/ m
g pr
otei
n)
0.01
0.1
1
10TCMVTAHR
C
Casp-3
Procasp-3
Procasp-9Casp-9
WARVWCMV
TNFCHX
Hours 0 4 8 4 8 2 3 4 5 6 7 8
+ +++++ + ++__ _+ + _ +++++ ++_ _
4 8 4 8 2 3 4 5 6 7 8
+ +++++ + ++__ _+ + _ +++++ ++_
Procasp-8Casp-8
Casp-8
F 0_
D
TAHR TCMV
Casp-3
Procasp-3
Procasp-9Casp-9
Procasp-8Casp-8
Casp-8
TNFCHX
0 4 8 4 8 2 3 4 5 6 7 8 0 4 8 4 8 2 3 4 5 6 7 8
+ +++++ + ++__ _+ +++++ + ++__ _+ + _ +++++ ++_ _
+ + _ +++++ ++_ _Hours
Procasp-12
1c1c7 Tao
BHours 0 4 8 4 8 2 3 4 5 6 7 8
+ +++++ + ++__ _+ + _ +++++ ++_ _
TNFCHX
0 4 8 4 8 2 3 4 5 6 7 8
+ +++++ + ++__ _+ + _ +++++ ++_ _
Casp-12
Casp-3
Procasp-3
Procasp-9Casp-9
Procasp-8Casp-8
Casp-8
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

0
20
40
60
0 2 4 6 80
20
40
60
0 2 4 6 8 0 2 4 6 80
20
40
60
80
100
% o
f Tot
al R
elea
se
Digitonin (µM)
LDH Cathepsin B Cathepsin D
A B C
*
* **
**
*
Figure 11, Caruso et al.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

0
10
20
30
40
0 1 2 3
CHX TNFα CHX + TNFα
% R
elea
se o
f Tot
al%
Rel
ease
of T
otal
% R
elea
se o
f Tot
al
0
10
20
30
40
0 1 2 3
A
B
0 1 2 30
10
20
30
40
50
Time (h)
C
0 1 2 30
5
10
15
20
25 D
0 1 2 30
10
20
30
40E
0
10
20
30
40
50
0 1 2 3
Time (h)
F
**
**
**
*
1c1c7 Tao
Figure 12, Caruso et al.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

FADDDeficiency
Casp-8 Procasp-8
cIAP, cFlipCHX
3-O-MeSMAhR deficiency
Endo/LysoEndo/Lyso
Cat D
Bid tBid
Pepstatin ADAME
Procasp-12
Casp-12Procasp-9Casp-9
Procasp-3Casp-3
TNFα
TNFr
Figure 13, Caruso et al.
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

SUPPLEMENTAL TABLES
Supplement Table IViability of hepatoma cell lines co-treated with CHX and TNFα
AhR-containing (1c1c7, WCMV) and –deficient (Tao, WARV) cultures were treated singularlyor with combinations of 1 µg/ml CHX and/or 30 pg/ml TNFα for 9-11 h prior to beingharvested for analyses of trypan blue exclusion. Data represent means ± S.D. of 3 or 4 culturedishes.
Treatment % Viable1c1c7 Tao WCMV WARV
No treatment 96.1 ± 1.5 91.3 ± 3.8 94.4 ± 1.9 94.4 ± 2.1CHX (1 µg/ml) 91.0 ± 1.8 89.4 ± 0.3 91.1 ± 1.9 95.3 ± 1.6
TNF 82.5 ± 3.9* 85.8 ± 2.8 92.3 ± 4.1 92.8 ± 3.6CHX + TNF 52.3 ± 5.0* 82.0 ± 3.3 60.2 ± 2.7* 90.4 ± 3.4
*less than no treatment control, p < 0.01.
SUPPLEMENTAL FIGURES
Supplement Fig. 1. Cathepsin D and Lamp-1 staining and co-localization in 1c1c7 cultures treatedwith HA14-1. 1c1c7 cultures were treated with nothing, Me2SO or 20 µM HA14-1 for 15, 30 or 60 minprior to being processed for subsequent immunofluorescence analyses of cathepsin D and Lamp-1staining and co-localization. Bar in panel A represents 20 µ.
Supplement Fig. 2. Cathespin B and Lamp-1 staining and co-localization in WCMV cultures treatedwith TNFαααα and CHX. Cultures of WCMV cells were treated with 30 pg/ml TNFα and/or 1 µg/ml CHXprior to being harvested and processed for subsequent immunofluorescence analyses of cathepsin B andLamp-1 staining and co-localization. In these studies CHX was added 30 min prior to TNFα. Cultureswere harvested 4 h after TNFα addition. Bar in panel A represents 20 µ.
Supplement Fig. 3. Cathepsin D and Lamp-1 staining and co-localization in WCMV cultures treatedwith TNFαααα and CHX. Treatment conditions are identical to those described in Supplement Fig. 2 exceptthat fixed cells were stained with antibodies to cathepsin D. The pictures in this Fig. are from theexperiment reported in Supplement Fig. 2. Bar in panel A represents 20 µ.
Supplement Fig. 4. Cathepsin B and Lamp-1 staining and co-localization in WARV cultures treatedwith TNFαααα and CHX. Cultures of WARV cells were treated with 30 pg/ml TNFα and/or 1 µg/ml CHXprior to being harvested and processed for subsequent immunofluorescence analyses of cathepsin B andLamp-1 staining and co-localization. In these studies CHX was added 30 min prior to TNFα. Cultureswere harvested 4 h after TNFα addition. Bar in panel A represents 20 µ.
Supplement Fig. 5. Cathepsin D and Lamp-1 staining and co-localization in WARV cultures treatedwith TNFαααα and CHX. Treatment conditions are identical to those described in Supplement Fig. 4 exceptthat fixed cells were stained with antibodies to cathepsin D. The pictures in this Fig. are from theexperiment reported in Supplement Fig. 4. Bar in panel A represents 20 µ.






Joseph A. Caruso, Patricia A. Mathieu, Aby Joiakim, Hong Zhang and John J. Reiners, Jrdisruption in a hepatoma model that is caspase-8 independent
-induced apoptosis and lysosomalαAryl hydrocarbon receptor modulation of TNF
published online January 30, 2006J. Biol. Chem.
10.1074/jbc.M508383200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2006/01/31/M508383200.DC1.html
http://www.jbc.org/content/early/2006/01/30/jbc.M508383200.citation.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on April 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from





























