Embed Size (px)
Citation preview

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
Autocrine human growth hormone increases sensitivity of mammarycarcinoma cell to arsenic trioxide-induced apoptosis
Ali Zekri a,b, Seyed H. Ghaffari a,⇑, Meysam Yousefi a, Samad Ghanizadeh-Vesali c, Majid Mojarrad d,Kamran Alimoghaddam a, Ardeshir Ghavamzadeh a
a Hematology, Oncology and Stem Cell Transplantation Research Center, Tehran University of Medical Sciences, Tehran, Iranb Department of Medical Genetics, Tehran University of Medical Sciences, Tehran, Iranc Department of Hematology, School of Allied Medicine, Tehran University of Medical Sciences, Tehran, Irand Department of Medical Genetics, Mashhad University of Medical Sciences, Mashhad, Iran
a r t i c l e i n f o
Article history:Received 25 April 2013Received in revised form 1 July 2013Accepted 2 July 2013Available online 10 July 2013
Keywords:Autocrine human growth hormoneArsenic trioxideMammary adenocarcinomaBreast cancerc-MycApoptosis
a b s t r a c t
Human growth hormone (hGH) has been increasingly implicated in a variety of cancers; its up-regulationis observed in breast cancer and correlates with a poor outcome. Autocrine hGH promotes mammary car-cinoma cell survival, proliferation, immortalization; it confers an invasive phenotype as a result of an epi-thelial–mesenchymal transition and contributes to chemoresistance and radioresistance. Arsenic trioxide(ATO) is being successfully used as a first and second line therapy for the treatment of patients with acutepromyelocytic leukemia. It also inhibits tumor cell growth and induces apoptosis in a broad range of solidtumors. In the present study, we investigated the effect of hGH on sensitivity of a mammary adenocar-cinoma cell to ATO, using a stable hGH-transfectant MCF-7 cell line, MCF7-hGH. Our results demon-strated for the first time that the overexpression of hGH increased sensitivity of the breast cancer cellline MCF-7 to ATO through apoptotic and anti-proliferative mechanisms. The effect of ATO on the tran-scriptional level of genes involved in survival (Bcl-2, Bax and Survivin), self-sufficiency in growth signals(c-Myc, ARF, Cdc25A, p53 and Bax), immortalization (hTERT) and invasion and metastasis (MMP-2 andMMP-9, uPA and uPAR and E-cadherin) was more pronounced in MCF7-hGH compared with its parentalMCF-7 line. Our study may highlight the potential application of ATO for the treatment of patients withbreast cancer, especially in those who have metastatic and chemoresistant tumor phenotype possibly dueto the over expression of hGH.
� 2013 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
Breast cancer is the most prevalent malignancy found in wo-men (Siegel et al., 2012). Despite considerable advances in earlydetection, diagnosis, and treatment, breast cancer is among theleading causes of cancer-related deaths in women because of thedevelopment of resistance to a wide variety of drugs and meta-static spread of cancer cells to distant organs. Virtually, all theroughly 40,000 annual breast cancer related deaths in the UnitedStates can be said to have occurred because the chemotherapyfailed. Therefore, understanding the molecular factors responsiblefor drug resistance and metastasis of breast cancer is urgentlyneeded to develop novel therapeutic approaches.
An accumulating number of evidence reveals that humangrowth hormone (hGH) has a pivotal role in the development
and progression of tumors of the female reproductive system. Ina recent pathway analysis of mammary cancer genome-wide asso-ciation study, the growth hormone signaling pathway was identi-fied as the pathway third most significantly correlated withsusceptibility to develop mammary carcinoma (Menashe et al.,2010). In addition to the secretion of hGH from the pituitary, it isnow widely accepted that this hormone is also produced locallyin the mammary gland and functions in an autocrine or paracrinemanner (Harvey, 2010) for the proliferation and differentiation ofcells and tissues. Pathological roles of autocrine/paracrine hGHhas been postulated in mammary carcinoma (Thijssen, 2009; Perryet al., 2008). It has been demonstrated that autocrine hGH pro-motes mammary carcinoma cell growth and survival; migrationand invasion; induces tumor angiogenesis; chemoresistance andradioresistance; protects against oxidative cell stress; and its over-expression is sufficient to oncogenically transform the humanmammary epithelial cell to a mesenchymal morphology which isassociated with acquisition of migratory and invasive phenotypesduring carcinoma progression (Brunet-Dunand et al., 2009; Thiery,2002; Perry et al., 2008; Mukhina et al., 2004).
0303-7207/$ - see front matter � 2013 Elsevier Ireland Ltd. All rights reserved.http://dx.doi.org/10.1016/j.mce.2013.07.002
⇑ Corresponding author. Address: Hematology, Oncology and Stem Cell Trans-plantation Research Center, Shariati Hospital, Tehran University of MedicalSciences, Tehran, Iran. Tel.: +98 21 84902665; fax: +98 21 88004140.
E-mail address: [email protected] (S.H. Ghaffari).
Molecular and Cellular Endocrinology 377 (2013) 84–92
Contents lists available at SciVerse ScienceDirect
Molecular and Cellular Endocrinology
journal homepage: www.elsevier .com/locate /mce

Author's personal copy
Up-regulation of hGH is observed in breast cancer and corre-lates with a poor outcome. Also, a high level of hGH expressionhas been reported in metastatic breast cancer [26]. In a recentstudy investigating the potential association of hGH expressionwith the clinicopathological features of mammary carcinoma,hGH was expressed in a large number of tumor specimens(52.8%), and over expression level of hGH was significantly associ-ated with lymph node metastasis, tumor grade, tumor stage, andproliferative index in mammary carcinoma. Also, the upregulationof hGH was associated with a significant worse relapse-free sur-vival and overall survival in patients with mammary or endome-trial carcinoma (Wu et al., 2011). So, it seems reasonable toestablish different therapeutic agents for improving the outcomesof treatment and for long-term survival of breast cancer patients.
Arsenic trioxide (ATO) is a highly efficacious agent for thetreatment of both newly diagnosed and all-trans-retinoic acid(ATRA)-refractory acute promyelocytic leukemia (APL) patients(Ghavamzadeh et al., 2006; Ghavamzadeh et al., 2011). An over-whelming number of studies imply that ATO induces apoptosisin a variety of tumor cells including acute myeloid leukemia (Xuet al., 2009; Momeny et al., 2010; Ghaffari et al., 2012b), multiplemyeloma (Gazitt and Akay, 2005), glioblastoma (Dizaji et al.,2012) and neuroblastoma (Pettersson et al., 2007). The pleiotropiceffects of ATO include modulation of the intracellular glutathioneredox system and oxidative injury (Jing et al., 1999), induction ofmitotic arrest due to inhibition of spindle apparatus and microtu-buline formation (Li and Broome, 1999), DNA damage and inhibi-tion of DNA repair (Yoo et al., 2009) which finally leads toapoptosis. Additionally, there are evidences that ATO might sup-press growth and proliferation of tumor cells through the inhibi-tion of telomerase and shortening of telomere length (Zhanget al., 2003; Ghaffari et al., 2012b). Moreover, a recent study showsthat ATO by modulation of tumor and metastatic suppressor miR-NAs may elicit cell cycle arrest and apoptosis in APL cell (Ghaffariet al., 2012a). In the present study, we investigated the effect ofhGH on sensitivity of a mammary adenocarcinoma cell to ATO,using a stable hGH-transfectant MCF-7 cell line, MCF7-hGH. Ourresults showed for the first time that overexpression of hGH in amammary adenocarcinoma cell line not only did not have inhibi-tory effects against the cytotoxicity of ATO, but also it increasedthe sensitivity of the breast cancer cell line MCF-7 to ATO throughapoptotic and anti-proliferative mechanisms. The result of thisstudy designates that ATO treatment may be beneficial for thetreatment of patients with hGH positive tumors.
2. Materials and methods
2.1. Stable cell line production, cell culture and ATO treatment
Stable MCF7-hGH cell line expressing hGH was constructed aspreviously report. Briefly; the coding region of hGH was amplifiedby reverse transcription PCR of human cDNA. PCR products werecloned into pCDNA3.1 (+) expression plasmid. MCF-7 cell wastransfected with pCDNA-hGH, and transfected cells stably express-ing transgene were selected by G418. The expression of hGHprotein was confirmed using immunocytochemistry. An emptypcDNA3 vector was used as a control.
All cell lines were cultured in RPMI 1640 medium supple-mented with 10% fetal bovine serum (Invitrogen, Gaithersburg,MD, USA), 2 mM L-glutamine (Invitrogen), 100 U/ml penicillin G(Invitrogen), and 100 lg/ml streptomycin (Invitrogen). Cells wereincubated at 37 �C in a humidified atmosphere containing 5%CO2. For ATO treatment, the cells were cultured in the absenceand presence of varying concentrations of ATO, ranging 1, 2, 3and 4 lM concentrations.
2.2. Microculture tetrazolium test (MTT assay)
The inhibitory effect of ATO on cell viability was measured byuptake of thiazolyl blue tetrazolium bromide (MTT, Sigma). Cellswere seeded into a 96-well plate at a density of 7000 cells/well;then they were treated with ATO at 1, 2, 3 and 4 lM for 24, 48and 72 h. The media were replaced by MTT solution (0.5 mg/ml),and after 3 h, with DMSO. The color absorbance was measured ata wavelength of 570 nm in an ELISA reader. The intensity of dis-solved formazan crystal was measured at 570 nm. The percentagecell viability was calculated as (ODexp/ODcon) � 100, where ODexp
and ODcon are the optical densities of treated and untreated cells,respectively.
2.3. Colony formation assay
Colony formation assay was used to assess in vitro cell survival.Cells were seeded onto 6-well plates with a density of 100 cells/well. After treatment with desired concentrations of ATO for48 h, the media were replaced by fresh media with no drug and fol-lowed for 3 weeks. Thereafter, the plates were rinsed with phos-phate-buffered saline (PBS) solution and stained with crystalviolet solution containing crystal violet (0.5% w/v) and glutaralde-hyde (6% v/v). Ultimately, colonies were counted by naked eye andthe survival fraction (SF) was estimated as: (mean colony counts)/(cells plated) � (plating efficiency), where plating efficiency (PE)was determined as (mean colony counts)/(cells plated for un-treated controls).
2.4. BrdU cell proliferation assay
The DNA synthesis of cells was measured by quantification ofincorporated BrdU via the colorimetric bromodeoxyuridine (BrdU)ELISA kit (Roche, Germany) according to the manufacturer’sinstructions. Briefly, cells (4000 cell/well) were treated with vari-ous concentrations of ATO for 48 h and then were incubated withthe BrdU labeling solution at 37 C for 8 h. The cells were then fixedand DNA was denatured using 200 ll of FixDenat solution. Follow-ing incubation with the peroxidase-conjugated anti-BrdU antibody(anti-BrdU-POD) at room temperature for 1 h, plates were exposedto 100 ll of substrate tetramethyl-benzidine (TMB). Lastly, plateswere read at 370 nm in an ELISA reader. The effect of ATO on theDNA synthesis was measured applying the succeeding formula:BrdU incorporation (%) = ODexp/ODcon � 100, where ODexp andODcon are the optical densitometries of the treated and untreatedcontrol cells, respectively.
2.5. Quantification of apoptosis using flow cytometry
For the detection of apoptosis induced by ATO, the Hoechst33342/Propidium iodide (PI) double stain apoptosis detectionwas used by a flow cytometry analysis. Briefly, about 1 � 106 cellswere treated with desired concentrations of ATO for 48 h, col-lected, washed twice with cold PBS, and fixed in 70% ethanol over-night. The cells were then incubated with 1 ml PBS containingHoechst 33,342 stock solution (5 mg/ml) and 1 ll of PI stock solu-tion (1 mg/ml). The fluorescence intensity of Hoechst 33,342 and PIwere analyzed by a flow cytometry instrument (Partec PasIII, Ger-many), using excitation/emission �350/461 and �535/617 nm forHoechst 33,342 and PI, respectively. Then the data was analyzedusing the FlowMax software.
2.6. Analysis of gene expression by quantitative real-time RT-PCR
Total RNA was extracted using TriPure Isolation Reagent (RocheApplied Science, Germany) from cultured cells and was quantified
A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92 85

Author's personal copy
by Nanodrop ND-1000 (Nanodrop Technologies, Wilmington, Del-aware, USA). Then, 1000 ng of extracted RNA was reverse tran-scribed into cDNA using the RevertAid First Strand cDNASynthesis kit (Fermentas UAB, Lithuania) according to the manu-facture’s protocol. Real time RT-PCR assay was performed usingRotor-Gene 6000 (Corbett Life Science, USA) thermal cycler detec-tion system using the primers shown in Table 1. Each reaction con-sisted of 10 ll Precision™ 2X qPCR Mastermix with SYBR green(PrimerDesign Ltd., Southampton, UK), 2 ll cDNA, 0.5 ll of eachprimer (10 pmol) and 7 ll of nuclease-free water (Qiagen, Hilden,Germany) to conduct PCR in a 20 ll of reaction mixture. Thermalcycling conditions were: 10 min at 90 �C for initial activation; fol-lowed by 40 cycles including a denaturation step at 94 �C for 10 s,annealing at 60 �C for 15 s, and extension at 70 �C for 20 s. Hypo-xanthine–guanine phosphoribosyl transferase1 (HPRT1) andbeta-2 microglobulin (B2M) were amplified as housekeeping genesand a fold change in relative expression of each target gene wascalculated based on the comparative Ct (2�DDCt) method. Analysisof melting curves was applied to approve whether all PCR productsare single. The efficiency of the target gene primers and the refer-ence gene primers were calculated by LinReg software (Table 1).
2.7. Gelatin zymography to measure MMPs enzymatic activity
To evaluate the effect of ATO on the gelatinolytic activity ofMMP-2 and MMP-9, zymography was applied according to thepreviously described method (Yousefi et al., 2012). Briefly, equalaliquots of conditioned media were fractionated by SDS–PAGE on7% polyacrylamide gels containing 2 mg/ml of gelatin A and gelatinB. After electrophoresis, gels were rinsed in 2.5% TritonX-100 to re-move the SDS followed by an incubation of the gels at 37 �C for 7 hin an incubation buffer. Then, the gels were stained with Coomas-sie Blue (0.5%) and destained with a buffer containing Tris–HCl(50 mM) and CaCl2 (5 mM). To quantify MMP-2 and MMP-9 zymo-grams, the gels were scanned and the bands intensity wasanalyzed by Bio-Rad Multi-Analyst and Quantity One software(Bio-Rad).
2.8. Statistical analysis
The results were expressed as mean ± SD. All the experimentswere carried out in triplicate except for the gelatin zymographyand the colony formation assay, which were performed in dupli-cate. Student’s t-test and one-way analysis of variance (one-wayANOVA) were used to determine the statistical significances ofdifference. P values <0.05 were considered significant.
3. Results
3.1. Inhibitory effects of ATO on survival of MCF-7 and MCF7-hGH cells
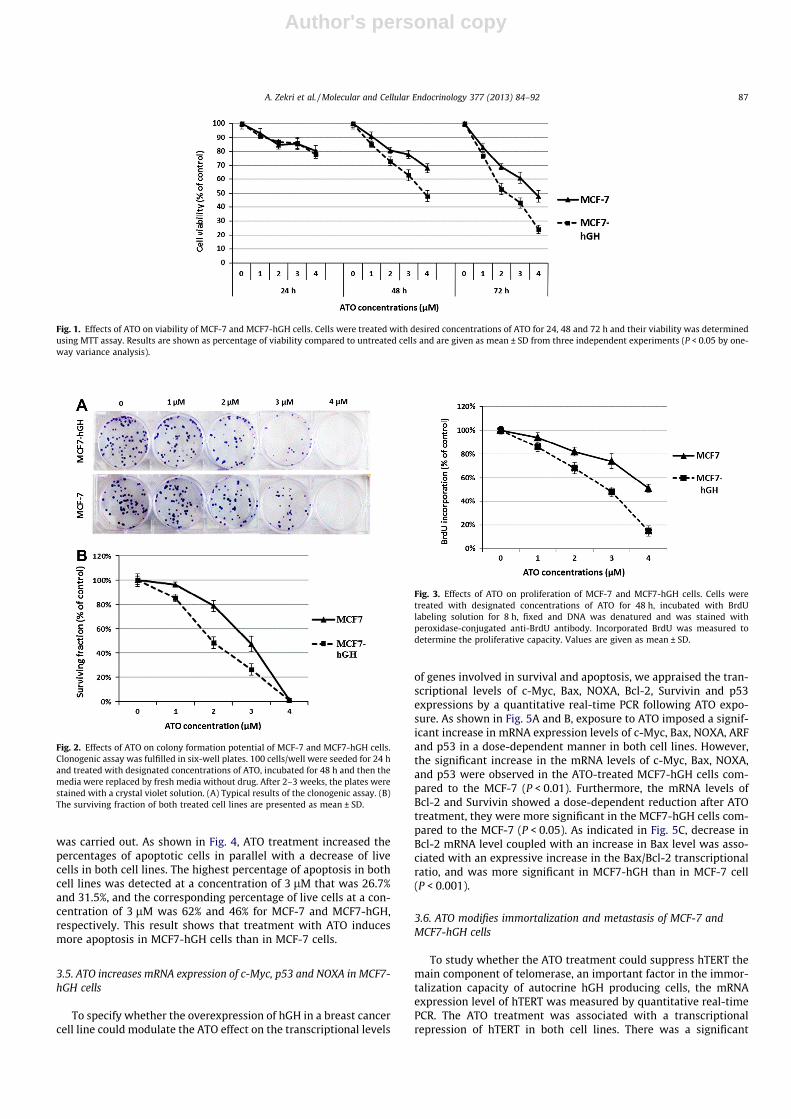
MTT assay was used to determine the inhibitory effect ofvarious concentrations of ATO at different time intervals (24, 48and 72 h) on the metabolic activity and viability of MCF-7 andMCF7-hGH cell lines. Our results demonstrated that ATO decreasedviability of both cell lines in a dose-dependent and time-dependentmanner. As shown in Fig. 1, ATO treatment for 24 h decreasedviability of both cell lines in an almost similar manner with nosignificant change in the reductions of viability between them.Treatment of the cell lines with ATO at 3 and 4 lM after 48 hreduced viability of MCF-7 cells to 78% and 69%; while viabilityof MCF7-hGH was decreased to 68% and 48%, respectively. Thereby,ATO treatment for 48 h and 72 h lead to more reduction in theviability of MCF7-hGH compare to MCF-7.
3.2. ATO decreases colony formation potentials of MCF-7 and MCF7-hGH cells
To explore whether exposure to ATO could suppress the surviv-ing fraction of MCF-7 and MCF7-hGH cells, colony formation assaywas applied. As shown in Fig. 2, treatments with 3 lM of ATO sig-nificantly reduced the surviving fraction of MCF-7 and MCF7-hGHby 47% and 26%, respectively. In addition, 4 lM of ATO diminishedall viable colonies and cells in both cell lines. Overall, the survivingfraction in the ATO treated MCF7-hGH cells was lower than in thetreated MCF-7 cells.
3.3. ATO inhibits proliferative potential of MCF-7 and MCF7-hGH cells
To study whether ATO treatment could suppress DNA synthesiswhich is proportional to the cell proliferation in MCF-7 and MCF7-hGH cells, a colorimetric BrdU incorporation assay was carried out.BrdU is incorporated into new strands of DNA during the S phase ofthe cell cycle. As shown in Fig. 3, a concentration-dependent de-crease in proliferation of both cell lines was observed after ATOtreatment for 48 h. As Fig. 2 depicts, ATO treatment at 4 lM for48 h reduced the proliferative capacity of MCF-7 and MCF7-hGHcells to 52% and 15% of the untreated control, respectively.
3.4. ATO induces apoptosis in MCF-7 and MCF7-hGH cells
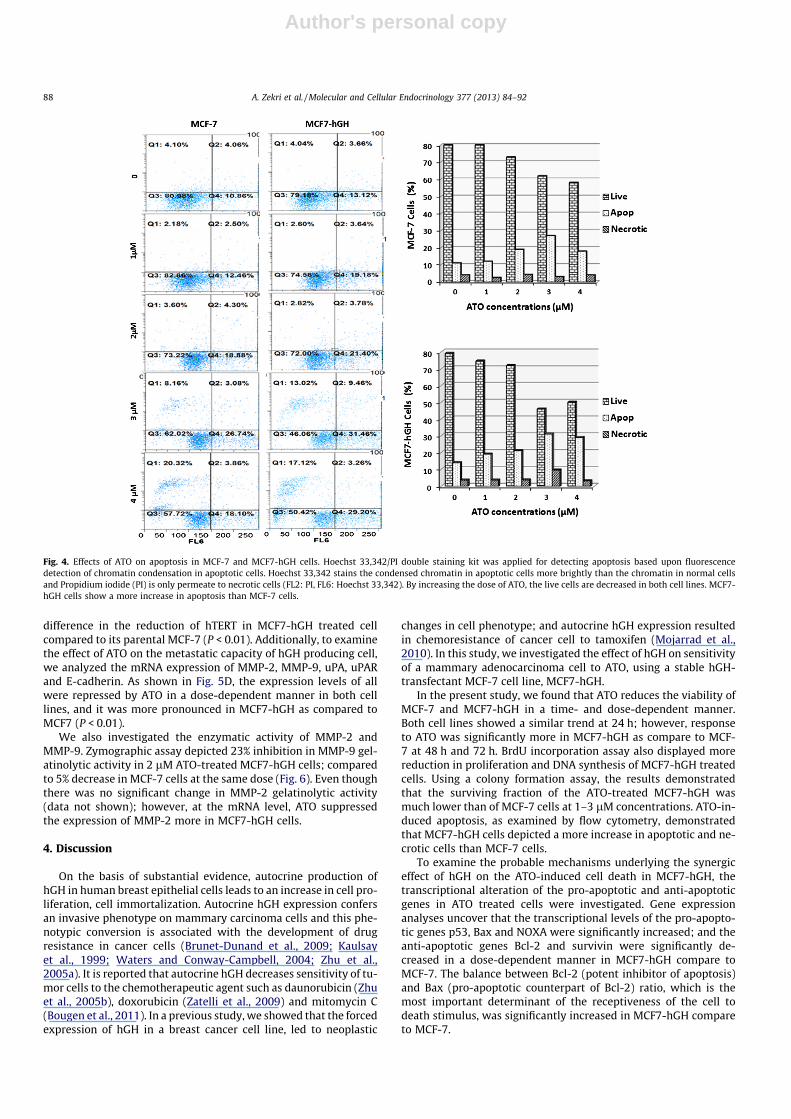
To investigate the effects of ATO on apoptosis of MCF-7 andMCF7-hGH mammary adenocarcinoma cells, a flow cytometryanalysis by the chromatin condensation/dead cell apoptosis assay
Table 1Nucleotide sequences of the primers used for real-time RT-PCR.
Gene Accession number Forward primer (50–30) Reverse primer (50–30) Efficiency Size
HPRT1 NM_000194 CCTGGCGTCGTGATTAGTGAT AGACGTTCAGTCCTGTCCATAA 1.85 131c-Myc NM_002467 GTCAAGAGGCGAACACACAAC TTGGACGGACAGGATGTATGC 1.87 162ARF NM_001195132 ATGGAGCCTTCGGCTGACT GTAACTATTCGGTGCGTTGGG 1.90 108Cdc25A NM_001789 GGCAGTGATTATGAGCAACCA CAACAGCTTCTGAGGTAGGGA 1.95 174P53 NM_1126118 CAGCACATGACGGAGGTTGT TCATCCAAATACTCCACACGC 1.84 125Bax NM_138761 CGAGAGGTCTTTTTCCGAGTG GTGGGCGTCCCAAAGTAGG 1.91 242Bcl-2 NM_000633 CGGTGGGGTCATGTGTGTG CGGTTCAGGTACTCAGTCATCC 1.83 90NOXA NM_021127 CAAGAACGCTCAACCGAG GGAAGTTCAGTTTGTCTCC 1.88 95Survivin NM_001168 CCAGATGACGACCCCATAGAG TTGTTGGTTTCCTTTGCAATTTT 1.92 152uPA NM_002658 TCAAAAACCTGCTATGAGGGGA GGGCATGGTACGTTTGCTG 1.85 121uPAR NM_002659 TATTCCCGAAGCCGTTACCTC TCGTTGCATTTGGTGGTGTTG 1.81 275MMP-2 NM_004530 CTTCCAAGTCTGGAGCGATGT TACCGTCAAAGGGGTATCCAT 1.92 119MMP-9 NM_004994 GGGACGCAGACATCGTCATC TCGTCATCGTCGAAATGGGC 1.91 139hTERT NM_001193376 AACCTTCCTCAGCTATGCCC GCGTGAAACCTGTACGCCT 1.89 210E-Cadherin NM_004360 CGAGAGCTACACGTTCACGG GGGTGTCGAGGGAAAAATAGG 1.85 119
86 A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92
Author's personal copy
was carried out. As shown in Fig. 4, ATO treatment increased thepercentages of apoptotic cells in parallel with a decrease of livecells in both cell lines. The highest percentage of apoptosis in bothcell lines was detected at a concentration of 3 lM that was 26.7%and 31.5%, and the corresponding percentage of live cells at a con-centration of 3 lM was 62% and 46% for MCF-7 and MCF7-hGH,respectively. This result shows that treatment with ATO inducesmore apoptosis in MCF7-hGH cells than in MCF-7 cells.
3.5. ATO increases mRNA expression of c-Myc, p53 and NOXA in MCF7-hGH cells
To specify whether the overexpression of hGH in a breast cancercell line could modulate the ATO effect on the transcriptional levels
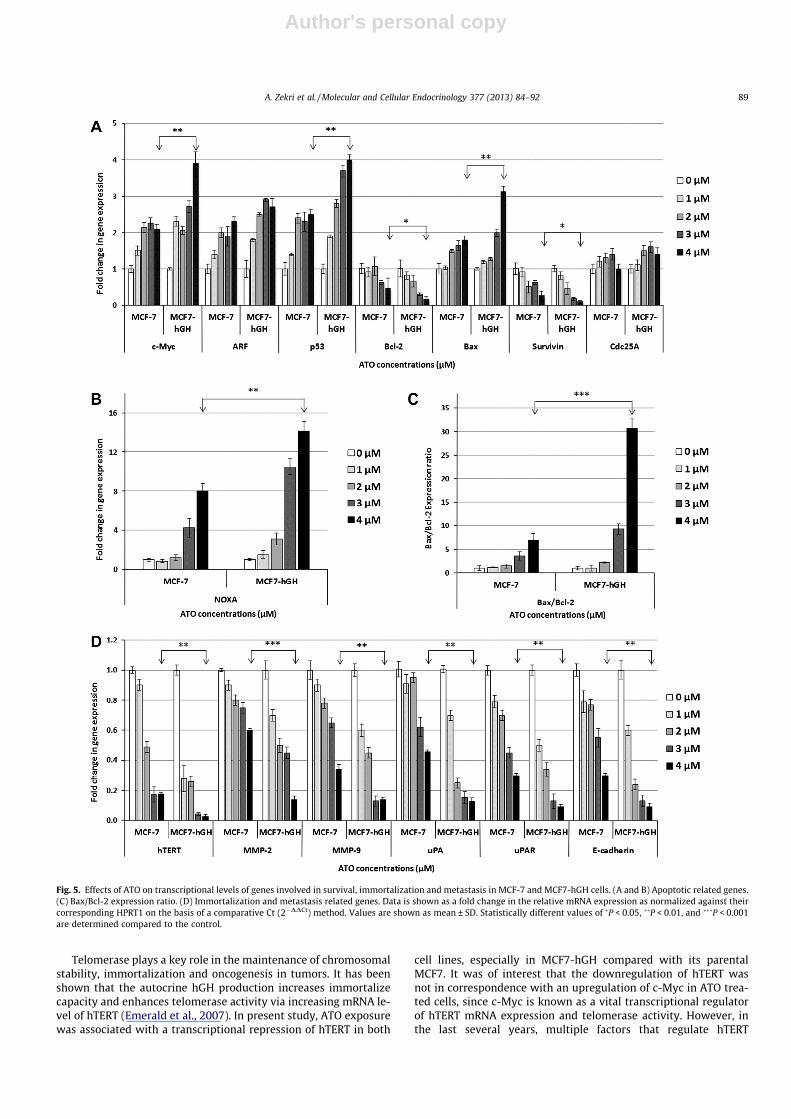
of genes involved in survival and apoptosis, we appraised the tran-scriptional levels of c-Myc, Bax, NOXA, Bcl-2, Survivin and p53expressions by a quantitative real-time PCR following ATO expo-sure. As shown in Fig. 5A and B, exposure to ATO imposed a signif-icant increase in mRNA expression levels of c-Myc, Bax, NOXA, ARFand p53 in a dose-dependent manner in both cell lines. However,the significant increase in the mRNA levels of c-Myc, Bax, NOXA,and p53 were observed in the ATO-treated MCF7-hGH cells com-pared to the MCF-7 (P < 0.01). Furthermore, the mRNA levels ofBcl-2 and Survivin showed a dose-dependent reduction after ATOtreatment, they were more significant in the MCF7-hGH cells com-pared to the MCF-7 (P < 0.05). As indicated in Fig. 5C, decrease inBcl-2 mRNA level coupled with an increase in Bax level was asso-ciated with an expressive increase in the Bax/Bcl-2 transcriptionalratio, and was more significant in MCF7-hGH than in MCF-7 cell(P < 0.001).
3.6. ATO modifies immortalization and metastasis of MCF-7 andMCF7-hGH cells
To study whether the ATO treatment could suppress hTERT themain component of telomerase, an important factor in the immor-talization capacity of autocrine hGH producing cells, the mRNAexpression level of hTERT was measured by quantitative real-timePCR. The ATO treatment was associated with a transcriptionalrepression of hTERT in both cell lines. There was a significant
Fig. 1. Effects of ATO on viability of MCF-7 and MCF7-hGH cells. Cells were treated with desired concentrations of ATO for 24, 48 and 72 h and their viability was determinedusing MTT assay. Results are shown as percentage of viability compared to untreated cells and are given as mean ± SD from three independent experiments (P < 0.05 by one-way variance analysis).
Fig. 2. Effects of ATO on colony formation potential of MCF-7 and MCF7-hGH cells.Clonogenic assay was fulfilled in six-well plates. 100 cells/well were seeded for 24 hand treated with designated concentrations of ATO, incubated for 48 h and then themedia were replaced by fresh media without drug. After 2–3 weeks, the plates werestained with a crystal violet solution. (A) Typical results of the clonogenic assay. (B)The surviving fraction of both treated cell lines are presented as mean ± SD.
Fig. 3. Effects of ATO on proliferation of MCF-7 and MCF7-hGH cells. Cells weretreated with designated concentrations of ATO for 48 h, incubated with BrdUlabeling solution for 8 h, fixed and DNA was denatured and was stained withperoxidase-conjugated anti-BrdU antibody. Incorporated BrdU was measured todetermine the proliferative capacity. Values are given as mean ± SD.
A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92 87
Author's personal copy
difference in the reduction of hTERT in MCF7-hGH treated cellcompared to its parental MCF-7 (P < 0.01). Additionally, to examinethe effect of ATO on the metastatic capacity of hGH producing cell,we analyzed the mRNA expression of MMP-2, MMP-9, uPA, uPARand E-cadherin. As shown in Fig. 5D, the expression levels of allwere repressed by ATO in a dose-dependent manner in both celllines, and it was more pronounced in MCF7-hGH as compared toMCF7 (P < 0.01).
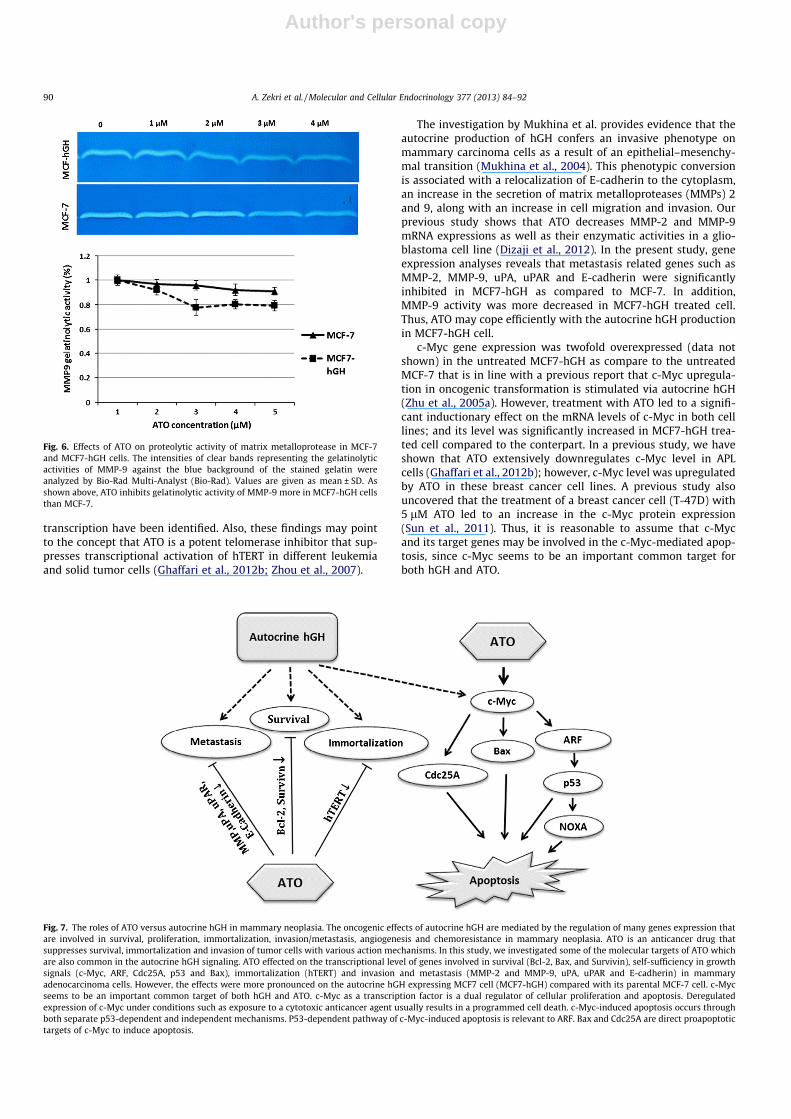
We also investigated the enzymatic activity of MMP-2 andMMP-9. Zymographic assay depicted 23% inhibition in MMP-9 gel-atinolytic activity in 2 lM ATO-treated MCF7-hGH cells; comparedto 5% decrease in MCF-7 cells at the same dose (Fig. 6). Even thoughthere was no significant change in MMP-2 gelatinolytic activity(data not shown); however, at the mRNA level, ATO suppressedthe expression of MMP-2 more in MCF7-hGH cells.
4. Discussion
On the basis of substantial evidence, autocrine production ofhGH in human breast epithelial cells leads to an increase in cell pro-liferation, cell immortalization. Autocrine hGH expression confersan invasive phenotype on mammary carcinoma cells and this phe-notypic conversion is associated with the development of drugresistance in cancer cells (Brunet-Dunand et al., 2009; Kaulsayet al., 1999; Waters and Conway-Campbell, 2004; Zhu et al.,2005a). It is reported that autocrine hGH decreases sensitivity of tu-mor cells to the chemotherapeutic agent such as daunorubicin (Zhuet al., 2005b), doxorubicin (Zatelli et al., 2009) and mitomycin C(Bougen et al., 2011). In a previous study, we showed that the forcedexpression of hGH in a breast cancer cell line, led to neoplastic
changes in cell phenotype; and autocrine hGH expression resultedin chemoresistance of cancer cell to tamoxifen (Mojarrad et al.,2010). In this study, we investigated the effect of hGH on sensitivityof a mammary adenocarcinoma cell to ATO, using a stable hGH-transfectant MCF-7 cell line, MCF7-hGH.
In the present study, we found that ATO reduces the viability ofMCF-7 and MCF7-hGH in a time- and dose-dependent manner.Both cell lines showed a similar trend at 24 h; however, responseto ATO was significantly more in MCF7-hGH as compare to MCF-7 at 48 h and 72 h. BrdU incorporation assay also displayed morereduction in proliferation and DNA synthesis of MCF7-hGH treatedcells. Using a colony formation assay, the results demonstratedthat the surviving fraction of the ATO-treated MCF7-hGH wasmuch lower than of MCF-7 cells at 1–3 lM concentrations. ATO-in-duced apoptosis, as examined by flow cytometry, demonstratedthat MCF7-hGH cells depicted a more increase in apoptotic and ne-crotic cells than MCF-7 cells.
To examine the probable mechanisms underlying the synergiceffect of hGH on the ATO-induced cell death in MCF7-hGH, thetranscriptional alteration of the pro-apoptotic and anti-apoptoticgenes in ATO treated cells were investigated. Gene expressionanalyses uncover that the transcriptional levels of the pro-apopto-tic genes p53, Bax and NOXA were significantly increased; and theanti-apoptotic genes Bcl-2 and survivin were significantly de-creased in a dose-dependent manner in MCF7-hGH compare toMCF-7. The balance between Bcl-2 (potent inhibitor of apoptosis)and Bax (pro-apoptotic counterpart of Bcl-2) ratio, which is themost important determinant of the receptiveness of the cell todeath stimulus, was significantly increased in MCF7-hGH compareto MCF-7.
Fig. 4. Effects of ATO on apoptosis in MCF-7 and MCF7-hGH cells. Hoechst 33,342/PI double staining kit was applied for detecting apoptosis based upon fluorescencedetection of chromatin condensation in apoptotic cells. Hoechst 33,342 stains the condensed chromatin in apoptotic cells more brightly than the chromatin in normal cellsand Propidium iodide (PI) is only permeate to necrotic cells (FL2: PI, FL6: Hoechst 33,342). By increasing the dose of ATO, the live cells are decreased in both cell lines. MCF7-hGH cells show a more increase in apoptosis than MCF-7 cells.
88 A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92
Author's personal copy
Telomerase plays a key role in the maintenance of chromosomalstability, immortalization and oncogenesis in tumors. It has beenshown that the autocrine hGH production increases immortalizecapacity and enhances telomerase activity via increasing mRNA le-vel of hTERT (Emerald et al., 2007). In present study, ATO exposurewas associated with a transcriptional repression of hTERT in both
cell lines, especially in MCF7-hGH compared with its parentalMCF7. It was of interest that the downregulation of hTERT wasnot in correspondence with an upregulation of c-Myc in ATO trea-ted cells, since c-Myc is known as a vital transcriptional regulatorof hTERT mRNA expression and telomerase activity. However, inthe last several years, multiple factors that regulate hTERT
Fig. 5. Effects of ATO on transcriptional levels of genes involved in survival, immortalization and metastasis in MCF-7 and MCF7-hGH cells. (A and B) Apoptotic related genes.(C) Bax/Bcl-2 expression ratio. (D) Immortalization and metastasis related genes. Data is shown as a fold change in the relative mRNA expression as normalized against theircorresponding HPRT1 on the basis of a comparative Ct (2�DDCt) method. Values are shown as mean ± SD. Statistically different values of �P < 0.05, ��P < 0.01, and ���P < 0.001are determined compared to the control.
A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92 89
Author's personal copy
transcription have been identified. Also, these findings may pointto the concept that ATO is a potent telomerase inhibitor that sup-presses transcriptional activation of hTERT in different leukemiaand solid tumor cells (Ghaffari et al., 2012b; Zhou et al., 2007).
The investigation by Mukhina et al. provides evidence that theautocrine production of hGH confers an invasive phenotype onmammary carcinoma cells as a result of an epithelial–mesenchy-mal transition (Mukhina et al., 2004). This phenotypic conversionis associated with a relocalization of E-cadherin to the cytoplasm,an increase in the secretion of matrix metalloproteases (MMPs) 2and 9, along with an increase in cell migration and invasion. Ourprevious study shows that ATO decreases MMP-2 and MMP-9mRNA expressions as well as their enzymatic activities in a glio-blastoma cell line (Dizaji et al., 2012). In the present study, geneexpression analyses reveals that metastasis related genes such asMMP-2, MMP-9, uPA, uPAR and E-cadherin were significantlyinhibited in MCF7-hGH as compared to MCF-7. In addition,MMP-9 activity was more decreased in MCF7-hGH treated cell.Thus, ATO may cope efficiently with the autocrine hGH productionin MCF7-hGH cell.
c-Myc gene expression was twofold overexpressed (data notshown) in the untreated MCF7-hGH as compare to the untreatedMCF-7 that is in line with a previous report that c-Myc upregula-tion in oncogenic transformation is stimulated via autocrine hGH(Zhu et al., 2005a). However, treatment with ATO led to a signifi-cant inductionary effect on the mRNA levels of c-Myc in both celllines; and its level was significantly increased in MCF7-hGH trea-ted cell compared to the conterpart. In a previous study, we haveshown that ATO extensively downregulates c-Myc level in APLcells (Ghaffari et al., 2012b); however, c-Myc level was upregulatedby ATO in these breast cancer cell lines. A previous study alsouncovered that the treatment of a breast cancer cell (T-47D) with5 lM ATO led to an increase in the c-Myc protein expression(Sun et al., 2011). Thus, it is reasonable to assume that c-Mycand its target genes may be involved in the c-Myc-mediated apop-tosis, since c-Myc seems to be an important common target forboth hGH and ATO.
Fig. 6. Effects of ATO on proteolytic activity of matrix metalloprotease in MCF-7and MCF7-hGH cells. The intensities of clear bands representing the gelatinolyticactivities of MMP-9 against the blue background of the stained gelatin wereanalyzed by Bio-Rad Multi-Analyst (Bio-Rad). Values are given as mean ± SD. Asshown above, ATO inhibits gelatinolytic activity of MMP-9 more in MCF7-hGH cellsthan MCF-7.
Fig. 7. The roles of ATO versus autocrine hGH in mammary neoplasia. The oncogenic effects of autocrine hGH are mediated by the regulation of many genes expression thatare involved in survival, proliferation, immortalization, invasion/metastasis, angiogenesis and chemoresistance in mammary neoplasia. ATO is an anticancer drug thatsuppresses survival, immortalization and invasion of tumor cells with various action mechanisms. In this study, we investigated some of the molecular targets of ATO whichare also common in the autocrine hGH signaling. ATO effected on the transcriptional level of genes involved in survival (Bcl-2, Bax, and Survivin), self-sufficiency in growthsignals (c-Myc, ARF, Cdc25A, p53 and Bax), immortalization (hTERT) and invasion and metastasis (MMP-2 and MMP-9, uPA, uPAR and E-cadherin) in mammaryadenocarcinoma cells. However, the effects were more pronounced on the autocrine hGH expressing MCF7 cell (MCF7-hGH) compared with its parental MCF-7 cell. c-Mycseems to be an important common target of both hGH and ATO. c-Myc as a transcription factor is a dual regulator of cellular proliferation and apoptosis. Deregulatedexpression of c-Myc under conditions such as exposure to a cytotoxic anticancer agent usually results in a programmed cell death. c-Myc-induced apoptosis occurs throughboth separate p53-dependent and independent mechanisms. P53-dependent pathway of c-Myc-induced apoptosis is relevant to ARF. Bax and Cdc25A are direct proapoptotictargets of c-Myc to induce apoptosis.
90 A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92

Author's personal copy
Recent studies demonstrated that c-Myc possesses a dual rolein promoting cellular proliferation and apoptosis. C-Myc has twoopposing functions in the induction of DNA damage response(DDR). It has been shown that c-Myc, as a DDR transcription factoris increased in a damaged cell and sensitizes the cell to apoptosis(Guerra et al., 2010; Supino et al., 2001). This effect of DDR relieson the p53 activation (Campaner and Amati, 2012). In fact, the ele-vated level of Myc sensitizes a variety of cell types to treatmentswith DNA-damaging drugs by promoting a strong DDR that trig-gers a potent apoptotic response (Guerra et al., 2010; Supinoet al., 2001; Campaner and Amati, 2012; Hermeking, 2003). Previ-ous studies reveal that ATO exposure induces DNA damage anddisturbs DNA repair (Li et al., 2010; Stevens et al., 2010). Thus, itseems that c-Myc, as a DNA damage-responsive transcription fac-tor is increased in the ATO-induced DNA damaged cell and sensi-tized the cell to apoptosis.
c-Myc is currently regarded as a central transcriptional onco-genic switch that regulates a large variety of cellular functionsthrough altering genes expression. Cdc25A and Bax are importantproapoptotic targets of c-Myc. Cdc25A as CDK-activating phospha-tase is a proto-oncogene that could promote apoptosis (Galaktio-nov et al., 1995); as shown in Fig. 5, Cdc25A is slightly increasedin both treated cell lines. It has been reported that cdc25A is essen-tial but not sufficient for c-Myc-induced apoptosis (Galaktionovet al., 1996). Another study revealed that the overexpression ofc-Myc upregulates the proapoptotic Bax expression (Mitchellet al., 2000). Our result shows that Bax was overexpressed in bothcell lines, but more in MCF7-hGH than in MCF-7. Moreover, it wasshown that Bax-null cells were deficient for the c-Myc-inducedapoptosis as compared with the wild-type Bax in embryonic fibro-blasts. They uncovered that Bax is a direct proapoptotic target ofc-Myc (Mitchell et al., 2000).
The c-Myc-induced apoptosis occurs through both p53-depen-dent and independent mechanisms. The p53-dependent model ofc-Myc-mediated apoptosis is relevant to ARF-p53 axis. The tumorsuppressor ARF protein is transcribed by the INK4a locus that en-codes Cdk inhibitor p16, but it is translated in an alternate readingframe (ARF) (Gregory et al., 2005). The ARF protein plays a role inan ‘oncogenic checkpoint’ that prevents an increased-cellular pro-liferation in response to aberrant signals from c-Myc, Ras and E2F(Bates et al., 1998; Palmero et al., 1998; Zindy et al., 1998). Our re-sult revealed a significant increase in ARF and p53 mRNA levels inMCF7-hGH treated cell compared with its parental MCF-7 line.
In conclusion, we showed that the autocrine expression of hGHin a mammary adenocarcinoma cell line not only did not haveinhibitory effects against the cytotoxicity of ATO, but also it in-creased the sensitivity of the breast cancer cell line MCF-7 toATO through apoptotic and anti-proliferative mechanisms. Ourfinding suggests that ATO treatment may be beneficial for thetreatment of patients with breast cancer patients, especially inthose who have metastatic and chemoresistant tumor phenotypepossibly due to the over expression of hGH. Based on experimentaldata, we propose a model showing the interaction and counterac-tion of hGH and ATO in a breast cancer cell (Fig. 7). We investigatedsome of the molecular targets of ATO which are also common inthe autocrine hGH signaling. ATO counteracts with hGH at tran-scriptional levels of genes involved in survival, proliferation,immortalization and invasion and metastasis; also it interacts toincrease the c-Myc expression level more in MCF7-hGH comparedwith its parental MCF-7 line.
Acknowledgment
This study was supported by Hematology, Oncology and StemCell Transplantation Research Center, Tehran University of MedicalSciences, Tehran, Iran.
References
Bates, S., Phillips, A.C., Clark, P.A., Stott, F., Peters, G., Ludwig, R.L., Vousden, K.H.,1998. P14ARF links the tumor suppressors RB and p53. Nature 395, 124–125.
Bougen, N.M., Yang, T., Chen, H., Lobie, P.E., Perry, J.K., 2011. Autocrine humangrowth hormone reduces mammary and endometrial carcinoma cell sensitivityto mitomycin C. Oncol. Rep. 26, 487–493.
Brunet-Dunand, S.E., Vouyovitch, C., Araneda, S., Pandey, V., Vidal, L.J., Print, C.,Mertani, H.C., Lobie, P.E., Perry, J.K., 2009. Autocrine human growth hormonepromotes tumor angiogenesis in mammary carcinoma. Endocrinology 150,1341–1352.
Campaner, S., Amati, B., 2012. Two sides of the Myc-induced DNA damage response:from tumor suppression to tumor maintenance. Cell Div. 7 (6).
Dizaji, M.Z., Malehmir, M., Ghavamzadeh, A., Alimoghaddam, K., Ghaffari, S.H., 2012.Synergistic effects of arsenic trioxide and silibinin on apoptosis and invasion inhuman glioblastoma U87MG cell line. Neurochem. Res. 37, 370–380.
Emerald, B.S., Chen, Y., Zhu, T., Zhu, Z., Lee, K.O., Gluckman, P.D., Lobie, P.E., 2007.AlphaCP1 mediates stabilization of hTERT mRNA by autocrine human growthhormone. J. Biol. Chem. 282, 680–690.
Galaktionov, K., Lee, A.K., Eckstein, J., Draetta, G., Meckler, J., Loda, M., Beach, D., 1995.CDC25 phosphatases as potential human oncogenes. Science 269, 1575–1577.
Galaktionov, K., Chen, X., Beach, D., 1996. Cdc25 cell-cycle phosphatase as a targetof c-myc. Nature 382, 511–517.
Gazitt, Y., Akay, C., 2005. Arsenic trioxide: an anti cancer missile with multiplewarheads. Hematology 10, 205–213.
Ghaffari, S.H., Bashash, D., Dizaji, M.Z., Ghavamzadeh, A., Alimoghaddam, K., 2012a.Alteration in miRNA gene expression pattern in acute promyelocytic leukemiacell induced by arsenic trioxide: a possible mechanism to explain arsenic multi-target action. Tumor. Biol. 33, 157–172.
Ghaffari, S.H., Momeny, M., Bashash, D., Mirzaei, R., Ghavamzadeh, A.,Alimoghaddam, K., 2012b. Cytotoxic effect of arsenic trioxide on acutepromyelocytic leukemia cells through suppression of NFkbeta-dependentinduction of hTERT due to down-regulation of Pin1 transcription. Hematology17, 198–206.
Ghavamzadeh, A., Alimoghaddam, K., Ghaffari, S.H., Rostami, S., Jahani, M., Hosseini,R., Mossavi, A., Baybordi, E., Khodabadeh, A., Iravani, M., et al., 2006. Treatmentof acute promyelocytic leukemia with arsenic trioxide without ATRA and/orchemotherapy. Ann. Oncol. 17, 131–134.
Ghavamzadeh, A., Alimoghaddam, K., Rostami, S., Ghaffari, S.H., Jahani, M., Iravani,M., Mousavi, S.A., Bahar, B., Jalili, M., 2011. Phase II study of single-agent arsenictrioxide for the front-line therapy of acute promyelocytic leukemia. J. Clin.Oncol. 29, 2753–2757.
Gregory, M.A., Qi, Y., Hann, S.R., 2005. The ARF tumor suppressor: keeping Myc on aleash. Cell Cycle 4, 249–252.
Guerra, L., Albihn, A., Tronnersjo, S., Yan, Q., Guidi, R., Stenerlow, B., Sterzenbach, T.,Josenhans, C., Fox, J.G., Schauer, D.B., et al., 2010. Myc is required for activationof the ATM-dependent checkpoints in response to DNA damage. PLoS One 5,e8924.
Harvey, S., 2010. Extrapituitary growth hormone. Endocrine 38, 335–359.Hermeking, H., 2003. The MYC oncogene as a cancer drug target. Curr. Cancer Drug
Targets 3, 163–175.Jing, Y., Dai, J., Chalmers-Redman, R.M., Tatton, W.G., Waxman, S., 1999. Arsenic
trioxide selectively induces acute promyelocytic leukemia cell apoptosis via ahydrogen peroxide-dependent pathway. Blood 94, 2102–2111.
Kaulsay, K.K., Mertani, H.C., Tornell, J., Morel, G., Lee, K.O., Lobie, P.E., 1999.Autocrine stimulation of human mammary carcinoma cell proliferation byhuman growth hormone. Exp. Cell. Res. 250, 35–50.
Li, Y.M., Broome, J.D., 1999. Arsenic targets tubulins to induce apoptosis in myeloidleukemia cells. Cancer Res. 59, 776–780.
Li, Z., Piao, F., Liu, S., Wang, Y., Qu, S., 2010. Subchronic exposure to arsenic trioxide-induced oxidative DNA damage in kidney tissue of mice. Exp. Toxicol. Pathol.62, 543–547.
Menashe, I., Maeder, D., Garcia-Closas, M., Figueroa, J.D., Bhattacharjee, S., Rotunno,M., Kraft, P., Hunter, D.J., Chanock, S.J., Rosenberg, P.S., et al., 2010. Pathwayanalysis of breast cancer genome-wide association study highlights threepathways and one canonical signaling cascade. Cancer Res. 70, 4453–4459.
Mitchell, K.O., Ricci, M.S., Miyashita, T., Dicker, D.T., Jin, Z., Reed, J.C., El-Deiry, W.S.,2000. Bax is a transcriptional target and mediator of c-myc-induced apoptosis.Cancer Res. 60, 6318–6325.
Mojarrad, M., Momeny, M., Mansuri, F., Abdolazimi, Y., Tabrizi, M.H., Ghaffari, S.H.,Tavangar, S.M., Modarressi, M.H., 2010. Autocrine human growth hormoneexpression leads to resistance of MCF-7 cells to tamoxifen. Med. Oncol. 27, 474–480.
Momeny, M., Zakidizaji, M., Ghasemi, R., Dehpour, A.R., Rahimi-Balaei, M.,Abdolazimi, Y., Ghavamzadeh, A., Alimoghaddam, K., Ghaffari, S.H., 2010.Arsenic trioxide induces apoptosis in NB-4, an acute promyelocytic leukemiacell line, through up-regulation of p73 via suppression of nuclear factor kappaB-mediated inhibition of p73 transcription and prevention of NF-kappaB-mediated induction of XIAP, cIAP2, BCL-XL and survivin. Med. Oncol. 27, 833–842.
Mukhina, S., Mertani, H.C., Guo, K., Lee, K.O., Gluckman, P.D., Lobie, P.E., 2004.Phenotypic conversion of human mammary carcinoma cells by autocrinehuman growth hormone. Proc. Natl. Acad. Sci. USA 101, 15166–15171.
Palmero, I., Pantoja, C., Serrano, M., 1998. P19ARF links the tumor suppressor p53 toRas. Nature 395, 125–126.
A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92 91

Author's personal copy
Perry, J.K., Mohankumar, K.M., Emerald, B.S., Mertani, H.C., Lobie, P.E., 2008. Thecontribution of growth hormone to mammary neoplasia. J. Mammary GlandBiol. Neoplasia 13, 131–145.
Pettersson, H.M., Karlsson, J., Pietras, A., Ora, I., Pahlman, S., 2007. Arsenic trioxideand neuroblastoma cytotoxicity. J. Bioenerg. Biomembr. 39, 35–41.
Siegel, R., Naishadham, D., Jemal, A., 2012. Cancer statistics. CA Cancer J. Clin. 62,10–29.
Stevens, J.J., Graham, B., Walker, A.M., Tchounwou, P.B., Rogers, C., 2010. The effectsof arsenic trioxide on DNA synthesis and genotoxicity in human colon cancercells. Int J Environ Res Public Health 7, 2018–2032.
Sun, R.C., Board, P.G., Blackburn, A.C., 2011. Targeting metabolism with arsenictrioxide and dichloroacetate in breast cancer cells. Mol. Cancer 10, 142.
Supino, R., Perego, P., Gatti, L., Caserini, C., Leonetti, C., Colantuono, M., Zuco, V.,Carenini, N., Zupi, G., Zunino, F., 2001. A role for c-myc in DNA damage-inducedapoptosis in a human TP53-mutant small-cell lung cancer cell line. Eur. J.Cancer 37, 2247–2256.
Thiery, J.P., 2002. Epithelial–mesenchymal transitions in tumor progression. Nat.Rev. Cancer 2, 442–454.
Thijssen, J.H., 2009. On the possible role of mammary-derived growth hormone inhuman breast cancer. Maturitas 65 (Suppl 1), S13–S16.
Waters, M.J., Conway-Campbell, B.L., 2004. The oncogenic potential of autocrinehuman growth hormone in breast cancer. Proc. Natl. Acad. Sci. USA 101, 14992–14993.
Wu, Z.S., Yang, K., Wan, Y., Qian, P.X., Perry, J.K., Chiesa, J., Mertani, H.C., Zhu, T.,Lobie, P.E., 2011. Tumor expression of human growth hormone and humanprolactin predict a worse survival outcome in patients with mammary orendometrial carcinoma. J. Clin. Endocrinol. Metab. 96, E1619–E1629.
Xu, S.N., Chen, J.P., Liu, J.P., Xia, Y., 2009. Efficacy of arsenic trioxide for acutepromyelocytic leukemia: a systematic review and meta-analysis. Zhong Xi Yi JieHe Xue Bao 7, 801–808.
Yoo, D.R., Chong, S.A., Nam, M.J., 2009. Proteome profiling of arsenic trioxide-treated human hepatic cancer cells. Cancer Genomics Proteomics 6, 269–274.
Yousefi, M., Ghaffari, S.H., Soltani, B.M., Nafissi, S., Momeny, M., Zekri, A.,Behmanesh, M., Alimoghaddam, K., Ghavamzadeh, A., 2012. Therapeuticefficacy of silibinin on human neuroblastoma cells: Akt and NF-kappaBexpressions may play an important role in silibinin-induced response.Neurochem. Res. 37, 2053–2063.
Zatelli, M.C., Minoia, M., Mole, D., Cason, V., Tagliati, F., Margutti, A., Bondanelli, M.,Ambrosio, M.R., degli Uberti, E., 2009. Growth hormone excess promotes breastcancer chemoresistance. J. Clin. Endocrinol. Metab. 94, 3931–3938.
Zhang, X., Multani, A.S., Zhou, J.H., Shay, J.W., McConkey, D., Dong, L., Kim, C.S.,Rosser, C.J., Pathak, S., Benedict, W.F., 2003. Adenoviral-mediatedretinoblastoma 94 produces rapid telomere erosion, chromosomal crisis, andcaspase-dependent apoptosis in bladder cancer and immortalized humanurothelial cells but not in normal urothelial cells. Cancer Res. 63, 760–765.
Zhou, C., Boggess, J.F., Bae-Jump, V., Gehrig, P.A., 2007. Induction of apoptosis andinhibition of telomerase activity by arsenic trioxide (As2O3) in endometrialcarcinoma cells. Gynecol. Oncol. 105, 218–222.
Zhu, T., Starling-Emerald, B., Zhang, X., Lee, K.O., Gluckman, P.D., Mertani, H.C.,Lobie, P.E., 2005a. Oncogenic transformation of human mammary epithelialcells by autocrine human growth hormone. Cancer Res. 65, 317–324.
Zhu, Z., Mukhina, S., Zhu, T., Mertani, H.C., Lee, K.O., Lobie, P.E., 2005b. P44/42 MAPkinase-dependent regulation of catalase by autocrine human growth hormoneprotects human mammary carcinoma cells from oxidative stress-inducedapoptosis. Oncogene 24, 3774–3785.
Zindy, F., Eischen, C.M., Randle, D.H., Kamijo, T., Cleveland, J.L., Sherr, C.J., Roussel,M.F., 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes. Dev. 12, 2424–2433.
92 A. Zekri et al. / Molecular and Cellular Endocrinology 377 (2013) 84–92





























