Embed Size (px)
Citation preview

Progress in Neurobiology 97 (2012) 152–172
Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases
Fabio Sambataro a,b, Maria Pennuto a,*a Department of Neuroscience and Brain Technologies, Istituto Italiano di Tecnologia, Genova 16163, Italyb Brain Center for Social and Motor Cognition @UniPr, Istituto Italiano di Tecnologia, Parma 43100, Italy
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
2. Spinal and bulbar muscular atrophy (SBMA) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
2.1. Clinical features of SBMA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
2.2. Pathogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
2.3. Cell-autonomous toxicity in SBMA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
2.3.1. Toxicity in motor neurons. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
2.3.2. Toxicity in skeletal muscle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
2.4. Non-cell-autonomous toxicity in SBMA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
2.4.1. Altered neurotrophic support to motor neurons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
2.4.2. Growth factor support to motor neurons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
2.4.3. Spinal nucleus of the bulbocavernosus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
2.4.4. Altered spermatogenesis in SBMA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
3. Huntington’s disease (HD). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
3.1. Clinical features of HD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
3.2. Pathogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
A R T I C L E I N F O
Article history:
Received 31 May 2011
Received in revised form 21 October 2011
Accepted 26 October 2011
Available online 2 November 2011
Keywords:
Polyglutamine diseases
Neurons
Skeletal and heart muscle
Glia
Spermatogenesis
Adipose tissue
Pancreas
A B S T R A C T
Polyglutamine diseases are neurodegenerative disorders caused by expansion of polyglutamine tracts in
the coding regions of specific genes. One of the most important features of polyglutamine diseases is that,
despite the widespread and in some cases ubiquitous expression of the polyglutamine proteins, specific
populations of neurons degenerate in each disease. This finding has led to the idea that polyglutamine
diseases are cell-autonomous diseases, in which selective neuronal dysfunction and death result from
damage caused by the mutant protein within the targeted neuronal population itself. Development of
animal models for conditional expression of polyglutamine proteins, along with new pharmacologic
manipulation of polyglutamine protein expression and toxicity, has led to a remarkable change of the
current view of polyglutamine diseases as cell-autonomous disorders. It is becoming evident that toxicity
in the neighboring non-neuronal cells contributes to selective neuronal damage. This observation implies
non-cell-autonomous mechanisms of neurodegeneration in polyglutamine diseases. Here, we describe
cell-autonomous and non-cell-autonomous mechanisms of polyglutamine disease pathogenesis, including
toxicity in neurons, skeletal muscle, glia, germinal cells, and other cell types.
� 2011 Elsevier Ltd. All rights reserved.
Abbreviations: PolyQ, polyglutamine; SBMA, spinal and bulbar muscular atrophy; SCA, spinocerebellar ataxia; AR, androgen receptor; Hsp, heat shock protein; GnRH,
gonadotropin-releasing hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone; JNK, cJun N-terminal kinase; MRF, myogenic regulatory factors; NT-3,
neurotrophin-3; BDNF, brain-derived neurotrophin factor; CNTF, ciliary neurotrophic factor; GDNF, glial cell-derived neurotrophic factor; VEGF, vascular endothelial growth
factor; IGF-1, insulin-like growth factor 1; SNB, spinal nucleus of bulbocavernosus; DLN, dorsolateral nucleus; HD, Huntington’s disease; GABA, gamma-aminobutyric acid;
PGC-1a, peroxisome proliferator-activated receptor gamma co-activator 1-alpha; GFAP, glial fibrillary acidic protein; GLP-1, glucagon-like peptide-1; FDA, food and drug
Contents lists available at SciVerse ScienceDirect
Progress in Neurobiology
jo u rn al ho m epag e: ww w.els evier . c om / lo cat e/pn eu ro b io
administration; PPARg2, peroxisome proliferator-activated receptor g2; C/EBPa, CAAT enhancer binding protein a; UCP-1, uncoupling protein 1; NMDA, N-methyl-d-
aspartate; HAP-1, huntingtin-interacting protein-1; ERAD, endoplasmic reticulum-associated protein degradation; TBP, TATA-binding protein.
* Corresponding author at: Department of Neuroscience and Brain Technologies, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy.
Tel.: +39 010 71781793; fax: +39 010 71781230.
E-mail address: [email protected] (M. Pennuto).
0301-0082/$ – see front matter � 2011 Elsevier Ltd. All rights reserved.
doi:10.1016/j.pneurobio.2011.10.003

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 153
3.3. Cell-autonomous toxicity in HD. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
3.3.1. Toxicity in medium-sized projection spiny neurons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
3.3.2. Toxicity in HD skeletal and heart muscle. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
3.3.3. Toxicity in glial cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
3.3.4. PolyQ-huntingtin toxicity in pancreatic beta-cells. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
3.3.5. Metabolic abnormalities: toxicity in adipose tissue. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
3.3.6. Altered spermatogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
3.4. Non-cell-autonomous toxicity in HD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
3.4.1. Excitotoxicity and cortico-striatal axis dysfunction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
3.4.2. Nigro-striatal axis dysfunction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
3.4.3. Hypothalamic-endocrine axis in HD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
4. Spinocerebellar ataxia (SCA) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
4.1. Clinical features of SCA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
4.2. Cell-autonomous and non-cell-autonomous toxicity in SCA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
5. Mechanisms underlying cell-autonomous and non-cell-autonomous toxicity in polyQ diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
5.1. Expression levels of disease proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
5.2. Subcellular localization of mutant protein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
5.3. Accumulation of polyQ proteins in inclusions and micro-aggregates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
5.4. Alteration of polyQ protein function . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
6. Concluding remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
1. Introduction
Polyglutamine (polyQ) diseases represent a family of nineneurodegenerative disorders, which include Huntington’s disease,dentatorubral-pallidoluysian atrophy, spinal and bulbar muscularatrophy, and spinocerebellar ataxia type 1, 2, 3, 6, 7, and 17. Thesedisorders are caused by expansion of the trinucleotide CAG tandemrepeat, encoding a polyQ tract, in the exonic regions of specificgenes; these genes are huntingtin, atrophin-1, androgen receptor,ataxin-1, ataxin-2, ataxin-3, CACNA1A, ataxin-7, and the TATA-binding protein, respectively.
PolyQ diseases share several features. Even if the mutantprotein is expressed beginning in early development, polyQdiseases are late-onset disorders. The length of the polyQ tractsinfluences disease presentation and predicts greater severity andyounger age of onset with increasing repeat lengths (Andrew et al.,1993; Snell et al., 1993). Similar to other tandem repeat disorders,polyQ diseases show ‘‘genetic anticipation,’’ with the followinggeneration likely to inherit a longer repeat than the previous one,thereby resulting in increased disease severity with earlier onset.Expanded polyQ tracts confer to the mutant protein the tendencyto accumulate as insoluble material, which appears in the form ofinclusions and micro-aggregates or oligomers. Despite polyQproteins being expressed in both neuronal and non-neuronal cells,neurons are extremely and selectively sensitive to the accumula-tion of expanded polyQ proteins. Furthermore, only specific typesof neurons degenerate in each polyQ disease. Selective neuronalvulnerability has long been interpreted to be the result of cell-autonomous toxicity due to the expression of mutant protein,possibly exacerbated by age-dependent generation of a toxicenvironment. However, this scenario has recently been challengedby the discovery that toxic pathways that lead to neuronal damageare influenced by damage occurring in non-neuronal cells. Thisfinding suggests that in addition to cell-autonomous toxicity inneuronal cells, damage in non-neuronal cells, such as muscle andglial cells, are likely to play a critical role in the pathogenesis ofpolyQ diseases. Non-cell-autonomous pathways of degenerationhave been described in neurodegenerative conditions such asamyotrophic lateral sclerosis and Parkinson’s disease (reviewed byIlieva et al., 2009; Lobsiger and Cleveland, 2007). Here, we describecell-autonomous and non-cell-autonomous mechanisms of neu-rodegeneration in spinal and bulbar muscular atrophy andHuntington’s disease as models of polyQ diseases.
2. Spinal and bulbar muscular atrophy (SBMA)
2.1. Clinical features of SBMA
SBMA is characterized by the degeneration and loss of lowermotor neurons in the brainstem and spinal cord, which manifestclinically as progressive weakness, with atrophy and fasciculationof proximal limb and bulbar muscles (Kennedy et al., 1968). Distalmuscle weakness and atrophy are observed in the arms more thanthe legs. The exordium of the disease usually manifests withcramps, hand tremor and fatigue, followed several years later bymuscle weakness, which disrupts patients’ ability to walk withoutassistance. Patients show fasciculations in the face with contrac-tion of muscles around the mouth and chin, fasciculation of thetongue, and in some cases dysarthria and dysphagia. In addition tothe neuromuscular phenotype, SBMA patients also show signs ofmild androgen insensitivity, including gynecomastia, reducedfertility, and testicular atrophy. In the family of polyQ diseases,SBMA is unique in that it is a sex-specific disease, with fullmanifestations occurring only in men. Women, even if homozy-gous for the mutation, present with subclinical disease manifesta-tions (Schmidt et al., 2002).
2.2. Pathogenesis
It is now well established that SBMA is the consequence of theexpansion of the polyQ tract in the androgen receptor (AR) (LaSpada et al., 1991). In normal individuals, the polyQ tract has alength that ranges between 9 and 36 residues, and its expansionover 38 residues causes disease. AR is a transcription factoractivated by the sex hormone testosterone and its more potentderivative dihydrotestosterone. In its inactive state, AR localizesto the cytosol in association with heat shock proteins (Hsps) suchas Hsp90, Hsp70, and Hsp40. Binding of AR to its natural ligandsresults in dissociation from the Hsps and translocation to thenucleus. Ligand binding also induces a conformational change,which makes AR competent to bind DNA and interact withtranscription co-regulators (co-activators and co-repressors) toregulate the expression of androgen-responsive genes. Theligand-dependent nature of SBMA is well recapitulated in animalmodels of disease: in transgenic and knock-in mice expressingpolyQ-expanded AR (polyQ-AR), the disease fully manifests onlyin males (Chevalier-Larsen et al., 2004; Katsuno et al., 2002;

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172154
Yu et al., 2006a). Transgenic female mice are asymptomatic unlessthey are administered testosterone. Transgenic fruit flies expressingpolyQ-AR develop neurodegeneration only when reared in ahormone-containing medium (Pandey et al., 2007; Takeyama etal., 2002). Conversely, reduction of testosterone levels in the serumof SBMA mice by castration prevents disease manifestations(Chevalier-Larsen et al., 2004; Katsuno et al., 2002). Theandrogen-dependent nature of SBMA suggests that reduction ofandrogen levels in the serum of SBMA patients might betherapeutically relevant. Serum androgen levels are regulated bythe hypothalamic-pituitary-testicular axis (Fig. 1). The hypothala-mus releases the gonadotropin-releasing hormone (GnRH), which inturn stimulates the anterior pituitary to release luteinizing hormone(LH) and follicle-stimulating hormone (FSH). LH stimulates Leydigcells to secrete testosterone, while FSH stimulates Sertoli cells topromote spermatogenesis (see Section 2.4.4). The hypothalamic-pituitary-testicular axis is regulated by negative feedback loops.Inhibin and testosterone, which are secreted by Sertoli cells andLeydig cells, respectively, inhibit the hypothalamus and the anteriorpituitary, thereby negatively affecting the net release of testosteronein the serum. Testosterone levels are elevated in some SBMApatients, suggesting that alterations in the hypothalamic-pituitary-testicular axis may be responsible at least in part for the endocrineabnormalities observed in SBMA patients (Dejager et al., 2002).Androgen levels in the serum can be reduced by treatment with theGnRH analog leuprorelin (Fig. 1). Such an approach has beensuccessfully pursued in a mouse model of SBMA (Katsuno et al.,2003) and has shown promising results in a phase II clinical trial(Banno et al., 2009). Another promising approach to limit the effectsof androgens in SBMA patients is to inhibit 5-alpha-reductase, theenzyme converting testosterone to dihydrotestosterone, usingdutasteride (Fernandez-Rhodes et al., 2011). Interestingly, treat-ment with testosterone does not exacerbate phenotype in bothSBMA patients (Goldenberg and Bradley, 1996; Neuschmid-Kasparet al., 1996) and SBMA mice (Chevalier-Larsen and Merry, 2011),
Fig. 1. The hypothalamic-pituitary-testicular axis. The hypothalamus releases gonadotr
luteinizing hormone (LH) and the follicle-stimulating hormone (FSH) that target testis ce
testosterone, which trigger negative feedback on the hypothalamus and the anterior pi
serum testosterone levels, followed by a reduction due to negative feedback loops.
suggesting that physiological levels of testosterone are sufficient fordeveloping full-blown disease. Exacerbation of clinical phenotypefollowing testosterone treatment was reported only in one case andthis occurrence was probably due to side effects of concomitantmedications (Kinirons and Rouleau, 2008).
2.3. Cell-autonomous toxicity in SBMA
The classical view of SBMA pathogenesis is that, similar to othermotor neuron diseases, the disease is caused by cell-autonomousmotor neuron degeneration, which in turn results in muscleatrophy. However, there is emerging evidence that damagetriggered by the interaction between androgens and polyQ-ARtargets not only motor neurons, but also non-neuronal cells, suchas skeletal muscle cells, suggesting that damage in the skeletalmuscle may have a primary role in disease pathogenesis.
2.3.1. Toxicity in motor neurons
Although AR is expressed in a variety of neuronal populations inthe central nervous system, including spinal cord, olfactory bulb,hippocampus, cerebellum, cortex, and hypothalamus (Kerr et al.,1995; Roselli et al., 1989; Simerly et al., 1990), expansion of thepolyQ tract in AR causes selective loss of lower motor neurons fromthe brainstem and spinal cord. Lower motor neurons in thebrainstem and anterior (ventral) horn of the spinal cord expressvery high levels of AR compared to other neuronal populations, andthis may be responsible for cell-autonomous neurodegeneration(Tetzlaff et al., 2007). Furthermore, motor neurons represent adirect target of androgen action, and this can contribute toselectivity of neuronal toxicity. Androgens affect the survival,morphology, and physiology of motor neurons from the spinal cordand brainstem. In mouse organotypic spinal cord cultures,androgens promote neurite extension (Hauser et al., 1987) andincrease motor neuron survival (Hauser and Toran-Allerand,1989). Importantly, these effects do not require the presence of
opin-releasing hormone (GnRH). GnRH stimulates the anterior pituitary to secrete
lls. FSH and LH stimulate Sertoli and Leydig cells, respectively, to release inhibin and
tuitary. Treatment with leuprorelin, a GnRH analog, results in an initial increase of
F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 155
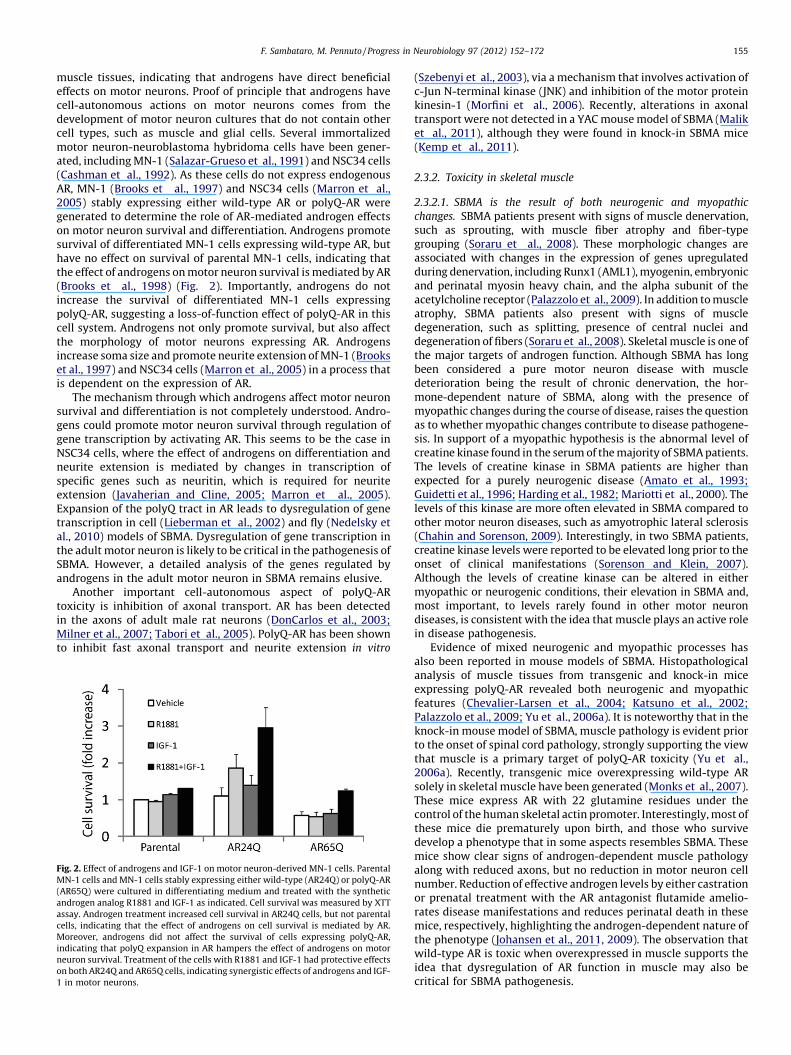
muscle tissues, indicating that androgens have direct beneficialeffects on motor neurons. Proof of principle that androgens havecell-autonomous actions on motor neurons comes from thedevelopment of motor neuron cultures that do not contain othercell types, such as muscle and glial cells. Several immortalizedmotor neuron-neuroblastoma hybridoma cells have been gener-ated, including MN-1 (Salazar-Grueso et al., 1991) and NSC34 cells(Cashman et al., 1992). As these cells do not express endogenousAR, MN-1 (Brooks et al., 1997) and NSC34 cells (Marron et al.,2005) stably expressing either wild-type AR or polyQ-AR weregenerated to determine the role of AR-mediated androgen effectson motor neuron survival and differentiation. Androgens promotesurvival of differentiated MN-1 cells expressing wild-type AR, buthave no effect on survival of parental MN-1 cells, indicating thatthe effect of androgens on motor neuron survival is mediated by AR(Brooks et al., 1998) (Fig. 2). Importantly, androgens do notincrease the survival of differentiated MN-1 cells expressingpolyQ-AR, suggesting a loss-of-function effect of polyQ-AR in thiscell system. Androgens not only promote survival, but also affectthe morphology of motor neurons expressing AR. Androgensincrease soma size and promote neurite extension of MN-1 (Brookset al., 1997) and NSC34 cells (Marron et al., 2005) in a process thatis dependent on the expression of AR.
The mechanism through which androgens affect motor neuronsurvival and differentiation is not completely understood. Andro-gens could promote motor neuron survival through regulation ofgene transcription by activating AR. This seems to be the case inNSC34 cells, where the effect of androgens on differentiation andneurite extension is mediated by changes in transcription ofspecific genes such as neuritin, which is required for neuriteextension (Javaherian and Cline, 2005; Marron et al., 2005).Expansion of the polyQ tract in AR leads to dysregulation of genetranscription in cell (Lieberman et al., 2002) and fly (Nedelsky etal., 2010) models of SBMA. Dysregulation of gene transcription inthe adult motor neuron is likely to be critical in the pathogenesis ofSBMA. However, a detailed analysis of the genes regulated byandrogens in the adult motor neuron in SBMA remains elusive.
Another important cell-autonomous aspect of polyQ-ARtoxicity is inhibition of axonal transport. AR has been detectedin the axons of adult male rat neurons (DonCarlos et al., 2003;Milner et al., 2007; Tabori et al., 2005). PolyQ-AR has been shownto inhibit fast axonal transport and neurite extension in vitro
Fig. 2. Effect of androgens and IGF-1 on motor neuron-derived MN-1 cells. Parental
MN-1 cells and MN-1 cells stably expressing either wild-type (AR24Q) or polyQ-AR
(AR65Q) were cultured in differentiating medium and treated with the synthetic
androgen analog R1881 and IGF-1 as indicated. Cell survival was measured by XTT
assay. Androgen treatment increased cell survival in AR24Q cells, but not parental
cells, indicating that the effect of androgens on cell survival is mediated by AR.
Moreover, androgens did not affect the survival of cells expressing polyQ-AR,
indicating that polyQ expansion in AR hampers the effect of androgens on motor
neuron survival. Treatment of the cells with R1881 and IGF-1 had protective effects
on both AR24Q and AR65Q cells, indicating synergistic effects of androgens and IGF-
1 in motor neurons.
(Szebenyi et al., 2003), via a mechanism that involves activation ofc-Jun N-terminal kinase (JNK) and inhibition of the motor proteinkinesin-1 (Morfini et al., 2006). Recently, alterations in axonaltransport were not detected in a YAC mouse model of SBMA (Maliket al., 2011), although they were found in knock-in SBMA mice(Kemp et al., 2011).
2.3.2. Toxicity in skeletal muscle
2.3.2.1. SBMA is the result of both neurogenic and myopathic
changes. SBMA patients present with signs of muscle denervation,such as sprouting, with muscle fiber atrophy and fiber-typegrouping (Soraru et al., 2008). These morphologic changes areassociated with changes in the expression of genes upregulatedduring denervation, including Runx1 (AML1), myogenin, embryonicand perinatal myosin heavy chain, and the alpha subunit of theacetylcholine receptor (Palazzolo et al., 2009). In addition to muscleatrophy, SBMA patients also present with signs of muscledegeneration, such as splitting, presence of central nuclei anddegeneration of fibers (Soraru et al., 2008). Skeletal muscle is one ofthe major targets of androgen function. Although SBMA has longbeen considered a pure motor neuron disease with muscledeterioration being the result of chronic denervation, the hor-mone-dependent nature of SBMA, along with the presence ofmyopathic changes during the course of disease, raises the questionas to whether myopathic changes contribute to disease pathogene-sis. In support of a myopathic hypothesis is the abnormal level ofcreatine kinase found in the serum of the majority of SBMA patients.The levels of creatine kinase in SBMA patients are higher thanexpected for a purely neurogenic disease (Amato et al., 1993;Guidetti et al., 1996; Harding et al., 1982; Mariotti et al., 2000). Thelevels of this kinase are more often elevated in SBMA compared toother motor neuron diseases, such as amyotrophic lateral sclerosis(Chahin and Sorenson, 2009). Interestingly, in two SBMA patients,creatine kinase levels were reported to be elevated long prior to theonset of clinical manifestations (Sorenson and Klein, 2007).Although the levels of creatine kinase can be altered in eithermyopathic or neurogenic conditions, their elevation in SBMA and,most important, to levels rarely found in other motor neurondiseases, is consistent with the idea that muscle plays an active rolein disease pathogenesis.
Evidence of mixed neurogenic and myopathic processes hasalso been reported in mouse models of SBMA. Histopathologicalanalysis of muscle tissues from transgenic and knock-in miceexpressing polyQ-AR revealed both neurogenic and myopathicfeatures (Chevalier-Larsen et al., 2004; Katsuno et al., 2002;Palazzolo et al., 2009; Yu et al., 2006a). It is noteworthy that in theknock-in mouse model of SBMA, muscle pathology is evident priorto the onset of spinal cord pathology, strongly supporting the viewthat muscle is a primary target of polyQ-AR toxicity (Yu et al.,2006a). Recently, transgenic mice overexpressing wild-type ARsolely in skeletal muscle have been generated (Monks et al., 2007).These mice express AR with 22 glutamine residues under thecontrol of the human skeletal actin promoter. Interestingly, most ofthese mice die prematurely upon birth, and those who survivedevelop a phenotype that in some aspects resembles SBMA. Thesemice show clear signs of androgen-dependent muscle pathologyalong with reduced axons, but no reduction in motor neuron cellnumber. Reduction of effective androgen levels by either castrationor prenatal treatment with the AR antagonist flutamide amelio-rates disease manifestations and reduces perinatal death in thesemice, respectively, highlighting the androgen-dependent nature ofthe phenotype (Johansen et al., 2011, 2009). The observation thatwild-type AR is toxic when overexpressed in muscle supports theidea that dysregulation of AR function in muscle may also becritical for SBMA pathogenesis.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172156
2.3.2.2. Muscle as primary target of polyQ-AR toxicity. Androgenshave remarkable anabolic effects on skeletal muscle. As mentionedabove, testosterone levels are regulated by the hypothalamic-pituitary-testicular axis (Fig. 1). Testosterone is mainly released bytesticular Leydig cells under the regulation of LH stimulation.Reduction of testosterone levels in the serum by administration ofGnRH analogs results in a decrease in muscle strength withconcomitant increase in fat body mass (Mauras et al., 1998).Conversely, testosterone administration in healthy individuals andin hypogonadal men results in enhanced muscle strength and size,increased lean body mass, and improved performance (Bhasinet al., 1997; Brodsky et al., 1996; Sinha-Hikim et al., 2002; Wang etal., 2000). Indeed, androgens and anabolic steroids are widely usedas performance-enhancing agents (reviewed by Hartgens andKuipers, 2004). The hypertrophic effect of androgens and anabolicsteroids on muscle is observed under both short-term and long-term use. Skeletal muscle is mainly composed of post-mitoticmultinucleated muscle fibers (myofibers) (Fig. 3). Myofibers canbe slow-twitch (type I) or fast-twitch (type IIa and b). Androgenspromote hypertrophy of both type I and type II muscle fibers andincrease muscle strength by stimulating new protein synthesis(Brodsky et al., 1996; Kadi et al., 1999).
During development, myofibers originate from migration ofmuscle precursor cells from somites to the nascent muscle. Inadulthood, myofibers form from satellite cells (Fig. 3). In skeletalmuscle, several cell types express AR, including satellite cells,fibroblasts, and mast cells (Sinha-Hikim et al., 2004). Interestingly,satellite cells are the cells that express the highest levels of AR.Since in satellite cells the expression of AR is regulated byandrogens, these cells are likely targets for polyQ-AR toxicity.Satellite cells are mononucleated cells located between musclefibers and the basal lamina (Mauro, 1961). Satellite cells representthe major reservoir for generation of new muscle fibers (Moss andLeblond, 1971). In response to specific mechanical, hormonal, andgrowth factor signaling stimulation, satellite cells are activated to
Fig. 3. Adult myogenesis. Skeletal muscle is composed of multiple fascicles, which in
multinucleated myotubes. Between the myofibers and the basal lamina are the satellite
response to injury or pathological processes, satellite cells are activated and become proli
fate. Differentiated myoblasts then fuse into myotubes, which can give origin to new
huntingtin may alter the process of activation and differentiation of satellite cells, the
become myoblasts, which are cells committed towards themyogenic fate. Myoblasts fuse with each other to generatemyotubes, which mature to form a new muscle fiber or fuse withan existing myofiber. During active proliferation, some satellitecells return to quiescence instead of entering the differentiationprocess, thereby providing muscle with new satellite cells. Satellitecell activation and differentiation is controlled by a specific geneexpression program, which is regulated by the myogenicregulatory factors (MRF) Myf5, MRF4, myogenin, and MyoD(reviewed by Kang and Krauss, 2010). Activated satellite cellsexpress Myf5. MyoD is required for the determination tomyoblasts, and myogenin and MRF4 are essential for terminaldifferentiation into myotubes.
Androgens have been shown to promote satellite cellproliferation, which may account for the hypertrophic effect ofandrogens on muscle (Sinha-Hikim et al., 2003). Evidence showsthat androgens activate Notch signaling (Brown et al., 2009),which stimulates the proliferation of satellite cells (reviewed byLuo et al., 2005). Androgens promote not only proliferation, butalso differentiation of satellite cells. The myoblast cell line C2C12differentiates into myotubes under androgen stimulation (Diel etal., 2008). In these cells, AR expression is regulated by androgens(Diel et al., 2008). The effect of androgens on C2C12 cells ismediated by AR, as suggested by its blockage by flutamide (Diel etal., 2008). Androgens influence satellite cell differentiationthrough the regulation of expression of key regulators of satellitecell activity (Lee, 2002). Androgens induce the expression ofmyogenin, which may in turn promote myoblast differentiation tomyotubes, along with markers of differentiation, such as creatinekinase, Pax7, SOX8, and Notch, and decrease the expression ofSOX9 and Delta. In both undifferentiated and differentiated cells,androgens stimulate the expression of the growth hormonemyostatin, which is a key regulator of muscle mass (Diel et al.,2008; Mendler et al., 2007). It would be relevant to determinewhether expression of these genes is altered by expansion of
turn are formed by myofibers surrounded by a basal lamina. Myofibers consist of
cells, which are the stem cells of muscle. Satellite cells are normally quiescent. In
ferative myoblasts. Proliferation is followed by differentiation towards the myogenic
myofibers or fuse with preexisting myofibers. Expression of polyQ-AR and polyQ-
reby affecting muscle regeneration in adulthood.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 157
polyQ in AR, and if this in turn plays a role in SBMA pathogenesis.Soraru et al. have proposed that myopathic changes in SBMAmuscle may be caused by defects in the activation of satellite cells(Soraru et al., 2008). It is possible that satellite cells expressingpolyQ-AR do not efficiently respond to androgens. Conversely, it ispossible that androgens trigger toxic responses in satellite cellsexpressing polyQ-AR. In any case, if satellite cells fail to repairmuscle during chronic denervation, this may contribute to muscledeterioration in SBMA. Androgens and AR function are alsoimportant in myofibers. Selective genetic ablation of AR inmyofibers results in a switch towards type I fibers, loss of bodyweight, and reduction of lean body mass, without altering musclestrength and resistance to fatigue (Ophoff et al., 2009). Thesefindings clearly indicate that a functional AR in myofibers isrequired for proper muscle homeostasis.
While androgens and AR have remarkable hypertrophic effectson skeletal muscle, they have detrimental effects in SBMA muscle.PolyQ expansion in AR has been proposed to cause toxicity inmuscle through altered gene expression (Mo et al., 2010). TheSBMA phenotype elicited by overexpression of either wild-type orpolyQ-AR in mice is associated with dysregulation of genetranscription in muscle. Another important pathway of toxicityin SBMA muscle is alteration of RNA splicing (Yu et al., 2009).Expression of polyQ-AR in the muscle of SBMA mice alters RNAprocessing in a hormone-dependent fashion. It remains to beestablished how these toxic pathways contribute to diseasepathogenesis and whether the changes in gene expression dueto altered gene transcription or altered RNA processing are primaryor secondary to disease pathogenesis.
Evidence from aging studies supports the role of androgens inSBMA muscle. During aging, muscle undergoes a progressive loss ofmuscle mass, a phenomenon known as sarcopenia (reviewed byGlass and Roubenoff, 2010; Sakuma and Yamaguchi, 2010).Histologically, this process is characterized by the loss of musclefibers, especially type II fibers, together with a decrease ofneuromuscular junctions and number of motor units. The reductionof the regenerative properties of the muscle tissue has beenidentified as the main cause of this age-dependent phenomenon.The regenerative properties of muscle rely on the presence ofsatellite cells, the number of which declines with age (Kadi et al.,2004). This reduction has been attributed to an age-related decreaseof the Notch signaling pathway, which is essential for satellite cellactivation (Conboy et al., 2003). In addition, the aging muscle showsincreased cell death by apoptosis (reviewed by Marzetti et al., 2008;Marzetti and Leeuwenburgh, 2006). Testosterone has been shown tocounteract sarcopenia in aging muscle by inducing musclehypertrophy (Sinha-Hikim et al., 2002, 2006, 2003). This effect ismediated by the inhibition of JNK and myostatin and by thestimulation of Notch and Akt signaling pathways (Brown et al., 2009;Kovacheva et al., 2010). As SBMA is an age-related disorder, it ispossible that muscle damage is the result of cumulative toxic effectscoming from pathological processes induced by expression ofpolyQ-AR in muscle cells and concomitant degeneration processesoccurring during aging. Moreover, polyQ-AR alteration of theregenerative potential of satellite cells may contribute to acceleratethe aging of muscle. The literature reviewed in this section supportsthe idea that muscle cells, especially satellite cells, can be directlydamaged by polyQ-AR toxicity. Generation of animal models ofSBMA with muscle-restricted expression of polyQ-AR will help inclarifying whether muscle plays a primary or secondary role indisease pathogenesis.
2.4. Non-cell-autonomous toxicity in SBMA
Damage to motor neurons can also result from non-cell-autonomous toxic processes occurring in other cell types, such as
muscle and glial cells. These cells provide motor neurons withtrophic support, which is essential for motor neuron maintenancein adulthood. Neurotrophins and growth factors are two classes oftrophic factors that protect neurons by activating signalingpathways that prevent initiation of apoptotic pathways andpromote activation of pro-survival pathways. The expression ofneurotrophins and growth factors is altered in mouse models ofSBMA (Sopher et al., 2004; Yu et al., 2006a). This observationhighlights the idea that altered trophic support to motor neuronsfrom neighboring tissues may be responsible for non-cell-autonomous damage in SBMA. In addition to motor neuron loss,other non-neuronal cell types are vulnerable to polyQ-AR, such asSertoli and Leydig cells in testis, which may account for theendocrine abnormalities described in patients.
2.4.1. Altered neurotrophic support to motor neurons
The neurotrophin family of trophic factors includes neurotro-phin-3 (NT-3), brain-derived neurotrophic factor (BDNF), ciliaryneurotrophic factor (CNTF), and glial cell-derived neurotrophicfactor (GDNF). NT-3, CNTF, and GDNF have been shown to protectmotor neurons from degeneration and death after axotomy(Baumgartner and Shine, 1997; Gravel et al., 1997; Houenouet al., 1996; Ikeda et al., 1995; Oppenheim et al., 1995; Tan et al.,1996) and in motor neuron disease (Haase et al., 1997, 1998). CNTFdelays disease progression in a mouse model of motor neurondisease, the wobbler mouse (Mitsumoto et al., 1994a). Among theneurotrophin factors, GDNF has been shown to have the highestneuroprotective action on motor neurons after nerve injury(Henderson et al., 1994; Yan et al., 1995; Zurn et al., 1994). Glialcells produce neurotrophic factors including GDNF and CNTF. AsGDNF expression is decreased in the muscle of SBMA patients(Yamamoto et al., 1999), it is possible that toxicity in glial cellscontributes to SBMA pathogenesis via a mechanism that involvesaltered trophic support to motor neurons.
Muscle-secreted BDNF is retrogradely transported to theinnervating motor neurons, promoting survival and maintenance(DiStefano et al., 1992; Funakoshi et al., 1993). BDNF protectsmotor neurons from death after axotomy (Kishino et al., 1997;Sendtner et al., 1992). Similar to CNTF, BDNF also delays diseaseprogression in wobbler mice (Ikeda et al., 1995). It is noteworthythat a combined treatment of the wobbler mice with CNTF andBDNF protects motor neurons from degeneration rather thandelaying disease progression, suggesting that in motor neurondisease combined neurotrophin treatment may have therapeuticvalue (Mitsumoto et al., 1994b).
2.4.2. Growth factor support to motor neurons
Among growth factors, vascular endothelial growth factor(VEGF) and insulin-like growth factor 1 (IGF-1) have been shown tohave remarkable protective actions on motor neurons and havebeen implicated in motor neuron diseases. VEGF is a cytokine withangiogenic effects and neuroprotective actions on motor neuronsboth in vitro and in vivo (reviewed by Bogaert et al., 2006;Lambrechts and Carmeliet, 2006). Targeted deletion of the vegf
gene in the spinal cord results in selective motor neurondegeneration (Oosthuyse et al., 2001). VEGF protects NSC34 cellsand primary motor neurons from hypoxia and oxidative stress(Oosthuyse et al., 2001; Van Den Bosch et al., 2004). VEGF can alsohave indirect effects on motor neurons. Indeed, glial cells respondto this cytokine, releasing trophic factors that promote motorneuron survival. Moreover, vascular abnormalities due to alteredlevels of VEGF can contribute to motor neuron degeneration. SBMAmice show decreased levels of VEGF (Sopher et al., 2004).Importantly, restoration of VEGF levels ameliorates diseasemanifestations, indicating that dysregulation of VEGF is criticalto disease pathogenesis. VEGF has been shown to promote motor

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172158
neuron survival in other motor neuron diseases, such as amyo-trophic lateral sclerosis, as its reduction exacerbates phenotypes inmouse models of disease (Lambrechts et al., 2003), whereasaugmentation of VEGF levels ameliorates disease manifestations(Azzouz et al., 2004; Storkebaum et al., 2005). These observationsindicate VEGF as a critical factor for motor neuron diseases.
IGF-1 is a powerful pro-survival factor for many different celltypes, including neurons (Reviewed by Trejo et al., 2004). IGF-1promotes sprouting, axonal growth, and survival of embryonicmotor neurons in both normal (Caroni and Grandes, 1990) andpathological conditions (Hughes et al., 1993; Neff et al., 1993). IGF-1protects MN-1 cells expressing polyQ-AR from death (Palazzolo etal., 2007) (Fig. 2). In addition to a direct effect on motor neurons, IGF-1 can have positive effects on innervated muscles. IGF-1 has beenshown to induce muscle hypertrophy and to inhibit muscle atrophy(Coleman et al., 1995; Musaro et al., 2001) through activation ofphosphoinositide 3-kinase/Akt pathway (Bodine et al., 2001;Rommel et al., 2001). IGF-1/Akt signaling promotes musclehypertrophy by inducing novel protein synthesis through inhibitionof glycogen synthase kinase 3b and activation of mammalian targetof rapamycin (mTOR) (Bodine et al., 2001; Rommel et al., 2001). Thecontribution of these pathways to SBMA pathogenesis remains to beelucidated. Akt is known to inhibit FOXO, which induces muscleatrophy by stimulating protein degradation via activation of theubiquitin-proteasome system (Sandri et al., 2004) and autophagy(Zhao et al., 2007). FOXO activates transcription of the ubiquitinligases atrogin1/MAFbx and MuRF1 (Stitt et al., 2004). However, thispathway is not likely to be relevant to SBMA pathogenesis, asexpression of these genes is not upregulated in SBMA mice (Mo et al.,2010; Palazzolo et al., 2009). It remains to be established whetherFOXO-dependent activation of autophagy is responsible for degen-eration of SBMA muscle. Recently, Lieberman’s group has shown thatthere is an induction of the unfolded protein response in the muscleof both patients and mouse models of SBMA, and that this stressresponse leads to the activation of macro-autophagy and to muscleatrophy, further supporting a pathogenic role for autophagy in SBMAmuscle (Yu et al., 2011). IGF-1 exists in several isoforms (Musaro etal., 2007). A muscle-specific IGF-1 isoform, which we will refer tohere as mIGF-1, has been shown to inhibit muscle degenerationduring aging by promoting muscle regeneration (Musaro et al.,2001). We have previously shown that overexpression of mIGF-1selectively in the muscle of SBMA mice reduces muscle and spinalcord pathology, ameliorates disease manifestations, and attenuatesmotor dysfunction (Palazzolo et al., 2009). The mechanism throughwhich mIGF-1 protects SBMA mice from neurodegeneration involvesactivation of Akt and phosphorylation of polyQ-AR, an event thatleads to mutant protein degradation by the proteasome. Besides adirect role on muscle, muscle-specific overexpression of mIGF-1 maystimulate the secretion of growth factors and neurotrophins frommuscle, which in turn can have beneficial effects on motor neurons.Nonetheless, this finding indicates that muscle represents animportant therapeutic target for SBMA and supports the idea thatmuscle damage is critical for disease pathogenesis.
2.4.3. Spinal nucleus of the bulbocavernosus
Evidence shows that motor neuron survival is critically depen-dent on non-cell-autonomous trophic support in some tissues, suchas the rodent spinal nucleus of bulbocavernosus (SNB) and thedorsolateral nucleus (DLN). The motor neurons of these nucleiinnervate the perineal muscles bulbocavernosus/levator ani andischiocavernosus, respectively (reviewed by Sengelaub and Forger,2008). The SNB and the DLN are sexually dimorphic pools of motorneurons present in the lumbar spinal cord, with higher numbers inmale rats compared to females (Breedlove and Arnold, 1980; Jordanet al., 1982). Although the number of motor neurons in the SNB issimilar in males and females at birth, it declines in females during
development because of cell death (Nordeen et al., 1985). Androgentreatment can prevent this process in female rodents, providingevidence that androgens are required for motor neuron survival inthe SNB (Jordan et al., 1982; Nordeen et al., 1985). Conversely,prenatal treatment with flutamide results in a feminine-likedevelopment of SNB, further indicating that androgens are criticalfor maintenance of these motor neurons. Males with loss-of-function mutations in AR show motor neuron death similar to thatobserved in females, indicating that androgens require a functionalAR to exert their trophic effect on these motor neurons (Breedloveand Arnold, 1981). In the SNB, androgens also regulate motor neuronsoma size, which is larger in male rats compared to females(Breedlove and Arnold, 1980, 1981). This effect is mediated by AR, asrodents with inactive AR show feminine body size of these motorneurons (Breedlove and Arnold, 1981). Conversely, perinataltreatment of females with androgens results in male soma size(Ward et al., 1996). These observations clearly highlight a role forandrogens and AR in the maintenance of both the number andmorphology of motor neurons in the SNB during perinatal life.
The effect of androgens on the SNB is likely to be non-cell-autonomous with involvement of the innervated muscles as a directtarget of androgen action. Target muscles (Fishman et al., 1990;Johansen et al., 2007; Monks et al., 2004), but not motor neurons(Fishman et al., 1990), of the SNB express AR during the perinatal andearly post-natal age, when the effect of androgens on motor neuronsurvival is observed. Furthermore, local application of flutamide onmuscle prevents motor neuron survival (Fishman and Breedlove,1992). The mechanism through which muscles promote androgen-dependent motor neuron survival is not known. The observation thatperinatal treatment of rodent females with CNTF prevents motorneuron degeneration and perineal muscle atrophy suggests thatinnervated muscles provide the motor neuron with trophic support(Forger et al., 1993). Consistent with this hypothesis, treatment ofthese muscles with antagonists of neurotrophins results in motorneuron death, providing further indication that primary trophicsupport from muscle is critical to motor neuron survival in the SNB(Xu et al., 2001). Interestingly, androgens regulate the production oftrophic factors, such as BDNF, and its receptors in the SNB motorneurons and target muscles (Osborne et al., 2007; Ottem et al., 2007;Verhovshek et al., 2010). In turn, BDNF regulates the expression of ARin the SNB-innervated muscles (Al-Shamma and Arnold, 1997).There is a synergistic effect of BDNF and androgens in themaintenance of the dendritic arbor of the SNB (Yang et al., 2004).These observations support the idea that there is a tight relationshipbetween muscle and motor neurons in the SNB, and that muscleactively promotes motor neuron survival during development.
In adulthood, androgens regulate soma size of the motor neuronsin the SNB, as reduction of serum androgen levels by castrationresults in decreased soma size (Breedlove and Arnold, 1981).Conversely, treatment of adult female rats with testosteroneincreases soma size (Breedlove and Arnold, 1981). Interestingly,in adulthood AR is expressed in motor neurons and in innervatedmuscles of the SNB (Breedlove and Arnold, 1980; Dube et al., 1976).Its expression in motor neurons is crucial for mediating the effect ofandrogens on soma size, indicating the adult motor neuron as thecell-autonomous target of androgen action (Watson et al., 2001).Nevertheless, in adulthood the effect of androgens seems to bemediated at least in part by muscle, as androgens have poor effectson the soma size of axotomized motor neurons (Araki et al., 1991).
The human anatomical correlate of the SNB is Onuf’s nucleus. Itis noteworthy that, although the motor neurons that form Onuf’snucleus express high levels of AR, these cells are spared in SBMA(Rusmini et al., 2010; Sobue, 1995). Interestingly, cytoskeletal andmorphological abnormalities in the absence of motor neuron losshave been reported in Onuf’s nucleus in amyotrophic lateralsclerosis, indicating that these motor neurons are vulnerable to the

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 159
accumulation of toxic proteins (Bergmann et al., 1995; Kihira et al.,1997; Okamoto et al., 1991). It remains to be established whethersimilar changes are present in Onuf’s nucleus in SBMA patients.
2.4.4. Altered spermatogenesis in SBMA
In addition to the neuromuscular phenotype, SBMA patientsalso show signs of mild androgen insensitivity (Battaglia et al.,2003). These symptoms include gynecomastia, hypogonadism,progressive loss of libido, erectile dysfunction, oligospermia, andazoospermia, suggesting that expansion of polyQ in AR causestesticular abnormalities and alters the process of spermatogenesis.A functional AR is a prerequisite for the development of normalmale sexual organs, as demonstrated by the lack of male primaryand secondary sexual characteristics in patients with androgeninsensitivity syndrome and in mice with AR gene deletion (Yeh etal., 2002). Spermatogenesis is a stepwise process of generation ofmature sperm cells from germ cells that occurs in seminiferoustubules in the testis (Fig. 4). Each germ cell or spermatogoniumdivides to generate two cells, one spermatogonium and oneprimary spermatocyte. This cell further divides into two secondaryspermatocytes, each of which generates two spermatids orimmature spermatozoa. Immature spermatozoa then mature intosperm cells. Spermatogenesis is regulated by tight cooperationamong various cell types, including peritubular myoid cells, Sertolicells, and Leydig cells (Eddy, 2002). All of these cell types and germcells express AR (Kimura et al., 1993; Vornberger et al., 1994), withits expression varying in a stage-specific fashion during spermato-genesis (Vornberger et al., 1994). Expression of functional AR ingerm cells is not a prerequisite for spermatogenesis, as its selective
Fig. 4. Spermatogenesis. Spermatogenesis occurs in seminiferous tubules of the tes
spermatogonium and one primary spermatocyte. Each primary spermatocyte undergoes
meiosis to generate two spermatids or immature spermatozoa. Spermatids undergo a de
then released in the lumen of the seminiferous tubule. This process is regulated by the co
AR and huntingtin, may affect the function of these cells leading to altered spermatog
ablation in these cells neither hampers spermatogenesis norreduces fertility (Tsai et al., 2006). Myoid cells are mesenchymalcells that together with Sertoli cells form the basement membraneof the seminiferous tubule. The function of myoid cells includes theproduction of paracrine factors important for Sertoli cell function.In addition, myoid cells induce peristaltic waves that stimulatemature spermatids to move towards the epididymis. Lack offunctional AR in myoid cells results in reduced testis size andoligospermia with no effects on fertility, indicating that ARfunction in this cell type is important, but not critical for normalspermatogenesis (Zhang et al., 2006). In contrast, AR function isabsolutely required for normal spermatogenesis in Sertoli andLeydig cells. Sertoli cells are the nursery cells that providestructural, functional, and nutritional support to germ cellsthrough their process of maturation to spermatids. Sertoli cellsdivide the interior of the seminiferous tubules into two compart-ments: the basal compartment and the luminal compartment. ARfunction in these cell types is absolutely required for spermatogen-esis, as selective deletion of AR in these cells results in testicularabnormalities, reduced levels of testosterone in the serum, andarrested spermatogenesis at very early stages, thereby resulting ininfertility (Chang et al., 2004; De Gendt et al., 2004). Leydig cells arelocated in the interstitial space or between seminiferous tubules.Testosterone released by Leydig cells is necessary for spermatogen-esis (Sharpe et al., 1988). Similar to Sertoli cells, lack of AR in Leydigcells results in testicular atrophy, reduced testosterone levels in theserum, and infertility (Xu et al., 2007).
Endocrine abnormalities are also evident in murine models ofSBMA (Thomas et al., 2006; Yu et al., 2006b). Mice expressing
tis. The germ cells/spermatogonia divide by mitosis to generate two cells, one
meiosis to generate two secondary spermatocytes, each of which divides by a second
velopmental process that results in the generation of mature sperm cells, which are
mbined actions of myoid cells, Leydig cells, and Sertoli cells. PolyQ proteins, such as
enesis.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172160
mutant AR have reduced fertility and develop progressivetesticular atrophy with alterations in germline maturation andSertoli cell cytoskeleton. The lack of similarity of these abnormali-ties with those observed in mice expressing a non-functional AR ingerm and Sertoli cells suggests that altered spermatogenesis inSBMA is likely to be the result of a toxic gain of function, ratherthan a pure loss of function, conferred by polyQ expansion of AR. Itis possible that expression of polyQ-AR in Sertoli cells and Leydigcells causes damage to germ cells in a non-cell-autonomousfashion, thereby altering the process of spermatogenesis. On theother hand, direct toxic effects of polyQ-AR in germ cells may alsocontribute to alter spermatogenesis in a cell-autonomous manner.The mechanism(s) through which polyQ expansion in AR altersspermatogenesis remains to be clarified.
3. Huntington’s disease (HD)
3.1. Clinical features of HD
The most common polyQ disease, HD is an autosomaldominant neurodegenerative disorder that manifests in bothmen and women. The disease is characterized by motordysfunction, which initiates with chorea, dystonia, and move-ment incoordination, and culminates in loss of the ability to move,bradykinesia, and rigidity in the final stages of the disease(Paradisi et al., 2008; Rao et al., 2008; Thompson et al., 1988).These symptoms are associated with impairment of cognitivefunctions, such as attention, memory, executive function, as wellas with psychiatric symptoms, such as change in personality,depression, psychosis, and dementia, which often precede theonset of motor symptoms (Dewhurst et al., 1970; Folstein et al.,1983). Other symptoms include body weight loss, muscle atrophy,
Fig. 5. HD neuropathology. Striatal medium-sized projection spiny neurons (MSNs), the
nigra pars compacta (SNc) and glutamatergic inputs from cerebral cortex and project to
MSNs can be grouped into two subpopulations: one located in the direct pathway (indi
levels of D1 dopamine receptors, and the other in the indirect pathway (indicated in y
receptors. Neurons containing Enk and projecting to the external segment of the glo
substance P and projecting to the internal segment of the globus pallidus (GPi, direct p
cardiac dysfunction, testicular atrophy, and endocrine abnormal-ities, which all become particularly evident as disease progresses(Aziz et al., 2008; Sanberg et al., 1981).
Neuroimaging studies in preclinical HD individuals indicatethat early structural and functional changes are present in thestriatum as well as in several cortical regions years before the onsetof motor symptoms (Bohanna et al., 2008). A recent study by thecollaborative project ‘‘PREDICT-HD’’ demonstrated that significantreductions of striatal volume may predict the onset of manifest HDwithin a time frame of 1–4 years (Aylward et al., 2011).Prefrontocortico-striatal networks have been reported to befunctionally altered during working memory processing in bothpre-symptomatic and symptomatic HD patients (Wolf et al.,2008a,b, 2011), and these alterations are evident also in theabsence of behavioral performance or structural differences (Wolfet al., 2007; Zimbelman et al., 2007). Interestingly, cerebral flow incortico-striatal circuits is reduced also during resting state andpredicts disease onset (Wolf et al., 2011), thus suggesting thatchanges in these brain regions may be an intrinsic hallmark ofpreclinical HD condition.
3.2. Pathogenesis
In the nervous system, the region primarily affected in HD is thestriatum, including the caudate nucleus and the putamen (Fig. 5).The degree of striatal degeneration correlates with severity andprogression of disease. Disease progression is also associated withdegeneration of other brain regions, including cerebral cortex(layers III, V, and VI), globus pallidus, thalamus, subthalamicnucleus, and substantia nigra. The most severely affected cells arestriatal medium-sized projection spiny neurons, which correspondto 95% of striatal neurons, and cortical pyramidal neurons
most severely affected cells in HD, receive dopaminergic inputs from the substantia
the globus pallidus, substantia nigra pars reticulata (SNr), and ventral pallidum. The
cated in green) that contains substance P and dynorphin (Dyn) and expresses high
ellow) that contains enkephalin (Enk) and expresses high levels of D2 dopamine
bus pallidus (GPe, indirect pathway) degenerate earlier than neurons containing
athway). Excitatory fibers, red; inhibitory fibers, black; STN, subthalamic nucleus.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 161
(Vonsattel and DiFiglia, 1998). Other neuronal populations,including medium-sized aspiny cholinergic interneurons contain-ing somatostatin, neuropeptide Y, or NADPH diaphorase (or nitricoxide synthase), are relatively spared. Medium-sized projectionspiny neurons are g-aminobutyric acid (GABA)-ergic cells thatreceive dopaminergic inputs from the substantia nigra parscompacta and glutamatergic inputs from cerebral cortex andproject to the globus pallidus, substantia nigra pars reticulata, andventral pallidum. The medium-sized spiny neurons can be groupedinto two subpopulations: one located in the direct pathway thatcontains substance P and dynorphin and expresses high levels ofD1 dopamine receptors, and the other in the indirect pathway thatcontains enkephalin and expresses high levels of D2 dopaminereceptors. Striatal medium-sized spiny neurons show differentialvulnerability in HD. Neurons containing enkephalin and projectingto the external segment of the globus pallidus (indirect pathway)degenerate earlier than neurons containing substance P andprojecting to the internal segment of the globus pallidus (directpathway). As disease progresses, all projecting neurons degener-ate, including large pyramidal projection neurons in cortical layersIII, V, and VI.
The causative mutation in HD is expansion of the polyQ tract inthe gene coding for huntingtin (Macdonald et al., 1993). The CAGrepeat is in exon 1 of the huntingtin gene, and expansion over 36residues causes disease. Huntingtin is a ubiquitous proteinexpressed at low levels during early development and at highlevels in both brain and testis in adulthood (Li et al., 1993; Stronget al., 1993). At the subcellular level, huntingtin predominantlylocalizes to the cytosol, but it has also been detected in severalother compartments, including the nucleus, endoplasmic reticu-lum/Golgi apparatus, mitochondria, axons, and the synapticcompartment. PolyQ-huntingtin is the substrate of several cellularproteases, whose activity results in the generation of amino-terminal fragments that accumulate in the nucleus and that seemto be more toxic than the full-length protein (reviewed by Pennutoet al., 2009). The function of huntingtin remains nebulous andgenetic manipulation of huntingtin expression in vivo has onlymarginally helped to elucidate its function. Huntingtin has beenimplicated in several cellular processes (reviewed by Zuccato et al.,2010), including synthesis of neurotransmitters (Zuccato et al.,2003), axonal transport (Gauthier et al., 2004; Szebenyi et al., 2003;Trushina et al., 2004), and spindle orientation during mitosis(Godin et al., 2010). Deletion of huntingtin during developmentresults in embryonic lethality, indicating that it is essentialthroughout development (Duyao et al., 1995; Nasir et al., 1995;Zeitlin et al., 1995). Its deletion during adulthood in the forebrainand testis results in neuronal degeneration and sterility, highlight-ing a critical role of the protein in the maintenance and function ofthese tissues throughout adulthood (Dragatsis et al., 2000).
3.3. Cell-autonomous toxicity in HD
Huntingtin is expressed in several neuronal and non-neuronaltissues. PolyQ-huntingtin accumulates in cells in form ofamyloidogenic aggregates and inclusions not only in neurons,but also in non-neuronal cells. Neuronal cells are particularlyvulnerable to the accumulation of polyQ-huntingtin compared toother cell types. Selective neuronal vulnerability might be at leastin part due to the fact that neurons are post-mitotic cells. Celldivision may reduce the accumulation of misfolded protein,thereby preventing degeneration. Moreover, the cellular systemsdevoted to degradation of misfolded proteins, such as theubiquitin-proteasome system and the autophagic system, mayfunction less efficiently with increasing age. An intriguing aspect ofneuronal vulnerability is that, within the neuronal populations,striatal neurons are the most affected cells in HD. The molecular
details for selective striatal degeneration remain to be elucidated.In addition to neuronal damage, there is emerging evidence thatpolyQ-huntingtin might be primarily toxic to other cell types,raising the idea that HD is a multi-system disease.
3.3.1. Toxicity in medium-sized projection spiny neurons
There is a large body of evidence in support of cell-autonomoustoxicity of mutant huntingtin in striatal medium-sized projectionspiny neurons, as expression of mutant huntingtin in the striatumis sufficient to cause GABAergic cell dysfunction and death.Lentiviral-mediated expression of the disease protein in the ratstriatum results in the formation of intranuclear inclusions andselective GABAergic cell degeneration and death, whose severitycorrelates with repeat length and expression level of mutantprotein (de Almeida et al., 2002). Why striatal neurons die in HD isstill an unresolved question. Striatal neurons degenerating in HDexpress levels of polyQ-huntingtin similar to other neuronalpopulations spared in the early stages of disease, such ascerebellum, hippocampus, and cortex, thereby excluding the ideathat selective neuronal degeneration results from higher levels ofmutant protein expression in the affected neurons. Another factorthat may account for selectivity of striatal degeneration in HD isthe subcellular localization of mutant protein. Indeed, polyQ-huntingtin accumulates in the nuclei of the medium-sized spinyneurons more than in other neuronal populations (Van Raamsdonket al., 2007b). Selective enrichment of the disease protein in thenucleus of striatal neurons can exert cell-autonomous toxicity inHD (Peters et al., 1999; Saudou et al., 1998). It remains to beclarified why polyQ-huntingtin preferentially localizes to thenucleus of striatal neurons as compared to other neuronalpopulations.
One mechanism underlying selective neuronal vulnerability inHD is transcription dysregulation. Gene expression changes aredetected very early in HD and involve caudate nucleus more thanother brain areas (Hodges et al., 2006). Gene expression changesare evident also in the brain of mouse models of HD (Luthi-Carteret al., 2002). Changes in gene expression similar to those observedin vivo can be detected in isolated primary striatal neuronscultured in vitro, further supporting the cell-autonomous aspect ofthis phenomenon (Runne et al., 2008). Generation of mice thatexpress mutant huntingtin solely in medium-sized spiny neuronsreveals transcriptional dysregulation, providing genetic evidencethat these changes are cell-autonomous (Brown et al., 2008;Thomas et al., 2011). The expression of several genes is altered inHD neurons, including genes involved in mitochondrial functionand oxidative stress (reviewed by Chen, 2011; Jin and Johnson,2010). Evidence that mitochondrial dysfunction is critical tostriatal function is supported by the observation that systemicadministration of the mitochondrial toxin 3-nitropropionic acidcauses selective striatal degeneration, which results in symptomsthat resemble HD (Guyot et al., 1997).
3.3.2. Toxicity in HD skeletal and heart muscle
Skeletal muscle wasting, atrophy, and motor dysfunction areevident early in HD patients and, importantly, occur before bodyweight loss. HD patients develop energy metabolism defects inmuscle (Ciammola et al., 2011; Lodi et al., 2000) and show reducedmuscle strength in lower limbs (Busse et al., 2008). Skeletal musclepathology is also evident in animal models of HD. The R6/2 mousemodel, which expresses human huntingtin exon 1 with more than120 CAG repeats (Mangiarini et al., 1996), develops motor deficitsstarting around 5–6 weeks of age, before appearance of an overtphenotype that occurs around 8–9 weeks of age (Carter et al.,1999). Similar to HD patients, muscle deterioration progresseswith age in the R6/2 mice, with severe muscle atrophy andneuromuscular junction abnormalities being evident at late stages

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172162
of disease (Ribchester et al., 2004). The mechanisms underlyingmuscle atrophy in HD are not known. Huntingtin is highlyexpressed in both differentiating myoblasts and myotubes. PolyQ-huntingtin- and ubiquitin-positive inclusions can be detected inthe nucleus of differentiated myotubes from R6/2 mice (Orth et al.,2003) and HD patients (Ciammola et al., 2006). In addition,increased activation of pro-apoptotic pathways and mitochondrialdepolarization are detected in primary myoblasts derived from HDpatients (Ciammola et al., 2006). It would be of interest todetermine whether expression of polyQ-huntingtin in satellitecells, myoblasts, or myotubes exerts cell-autonomous toxicityduring muscle regeneration processes occurring in adulthood, andwhether these alterations affect muscle homeostasis during aging(Fig. 3). As described in SBMA muscle, gene expression has beenshown to be altered in HD muscle both in patients and mousemodels of disease (Luthi-Carter et al., 2002; Strand et al., 2005).These changes in gene expression in muscle are detected in veryearly stages of the disease, suggesting that polyQ-huntingtin isprimarily toxic to muscle. One gene whose expression and functionare altered in HD muscle is the peroxisome proliferator-activatedreceptor gamma co-activator 1-alpha (PGC-1a) (Chaturvedi et al.,2009). Because PGC-1a function is critical in HD pathogenesis (Cuiet al., 2006; Weydt et al., 2006), these changes in gene expressionare likely to be primary to disease pathogenesis. Changes in geneexpression in muscle can be the result of cell-autonomous toxicity,but it cannot be excluded that they also result from alterations inother cell types. For instance, altered trophic support from neuronsmay contribute to the abnormalities observed in muscle. Anotherfactor that can contribute to muscle wasting is altered muscleenergy metabolism, which has been reported in pre-symptomaticHD patients (Lodi et al., 2000). Interestingly, the severity of energymetabolism abnormalities correlates with the length of the polyQtract of polyQ-huntingtin.
In addition to skeletal muscle, cardiac muscle is also damaged inHD. Cardiac failure is the second leading cause of death in HDpatients. In the R6/2 mouse model of disease, huntingtin aggregationand mitochondrial abnormalities have been detected in cardiacmyocytes, together with altered heart morphology, weight, andfunction (Mihm et al., 2007). Generation of transgenic mice thatexpress amyloidogenic oligomers selectively in cardiac myocytesprovides proof-of-principle that accumulation of polyQ oligomers isresponsible for cell-autonomous cardiac abnormalities and prema-ture death (Pattison et al., 2008). This finding suggests that thecardiac abnormalities present in HD patients arise from cell-autonomous toxicity of polyQ-huntingtin in cardiac muscle.
3.3.3. Toxicity in glial cells
Glial cells, i.e. astrocytes, microglia, and oligodendrocytes,represent 90% of the cells present in the brain. All glial cell typesexpress huntingtin. Astrocytes play a critical role in HD pathogen-esis. Expression of mutant huntingtin selectively in astrocytesresults in altered expression of glutamate transporters, resulting inneuronal death through excitotoxicity (see Section 3.4.1). Damagein the CNS leads to activation of astrocytes, a process known asgliosis. Gliosis consists of astrocyte proliferation, upregulation ofglial fibrillary acidic protein (GFAP), and morphologic changes(reviewed by Chvatal et al., 2008). Reactive astrocytes are presentin the striatum of HD and their number correlates with diseaseprogression.
Microglia are the resident macrophages in the brain and spinalcord. Under normal conditions, microglia are present in restingstate and become activated upon injury (Moller, 2010). Activatedmicroglia are present in the striatum, cortex, and globus pallidus inHD brain and can be detected very early in disease, suggesting thatthis process may be primary to disease pathogenesis (Sapp et al.,2001; Tai et al., 2007). Microglia activation has also been detected
in mouse models of HD (Simmons et al., 2007). Followingmicroglia activation, cytotoxic substances including oxygenradicals, nitric oxide, glutamate, and neurotoxic cytokines, as wellas cytoprotective agents, including growth factors and cytopro-tective cytokines, are released. These substances can affectneuronal function and survival in a non-cell-autonomous fashion.As microglia express mutant huntingtin, it is possible that cell-autonomous alterations of microglia contribute to diseasepathogenesis.
Finally, oligodendrocytes have recently been found to beinvolved in HD pathogenesis (Xiang et al., 2011). Alterations ofPGC-1a expression and function in oligodendrocytes have beenreported in the striatum of HD mice, which correlated with defectsin myelination, suggesting that cell-autonomous dysfunction ofoligodendrocytes contributes to HD pathogenesis.
3.3.4. PolyQ-huntingtin toxicity in pancreatic beta-cells
Clinical studies indicate that HD patients are at high risk fordeveloping diabetes mellitus (Farrer, 1985; Podolsky et al., 1972)and often present with decreased glucose tolerance (Podolsky andLeopold, 1977). Recently, analysis of non-diabetic HD patientsrevealed altered insulin sensitivity and decreased insulin secretion(Lalic et al., 2008). However, an association between HD anddiabetes has recently been debated (Boesgaard et al., 2009) andhistological analysis of pancreatic autopsy specimens from HDindividuals detected no morphological abnormalities or amyloi-dogenic deposition in pancreatic islet cells, and no changes ininsulin transcript levels (Bacos et al., 2008). Glucose levels in theserum are negatively and positively regulated by the pancreatichormones insulin and glucagon, respectively. The endocrineportion of the pancreas is formed by clusters of cells called isletsof Langerhans. These regions include different cells: alpha-cellswhich secrete glucagon, beta-cells which produce insulin, anddelta-cells which release somatostatin, a modulator of the activityof alpha- and beta-cells. Diabetes mellitus and altered glucosetolerance have also been observed in mouse models of HD,including R6/2 mice (Hurlbert et al., 1999) and N171-82Q mice,which express the amino-terminal portion of mutant huntingtinwith 82 glutamine residues (Martin et al., 2009). The R6/1 mousemodel of HD, which expresses a shorter polyQ tract than the R6/2model, does not develop diabetes, yet it shows impaired glucosetolerance (Josefsen et al., 2008). Not only insulin, but also glucagonand somatostatin levels have been reported to be altered in HDmice (Andreassen et al., 2002). Although pancreas mass is notaltered in HD mice, the content of insulin in beta-cells and insulinsecretion are remarkably decreased. Importantly, progressiveaccumulation of mutant huntingtin-positive nuclear inclusionswas found in R6/2 beta-cells, suggesting cell-autonomous toxicityin this cell type (Bjorkqvist et al., 2005). Consistent with thishypothesis, beta-cell mass was decreased in R6/2 mice comparedto control mice. Moreover, exocytosis was altered specifically inbeta-cells, but not in alpha-cells. Interestingly, reduction of insulinsecretion was associated with a dramatic decrease in the numberof insulin-containing secretory vesicles in beta-cells. Mechanisti-cally, insulin deficiency has been proposed to be due to cell-autonomous alterations of gene transcription in beta-cells(Andreassen et al., 2002). In addition to cell-autonomous toxicityin pancreatic beta-cells, disruption of glucose homeostasis mayalso have non-cell-autonomous effects on the brain, as alteredglucose levels may cause neuronal dysfunction and may contributeto neuronal death in HD. It is noteworthy that SBMA andspinocerebellar ataxia 1 patients also develop diabetes, indicatingthat pancreatic dysfunction is common to polyQ diseases and, assuch, should be considered an important therapeutic target.Unfortunately, treatment of mice with anti-diabetic agents, such asglibenclamide to stimulate insulin secretion and rosiglitazone to

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 163
increase insulin sensitization, did not show any benefit in R6/2mice (Hunt and Morton, 2005). On the other hand, treatment of theR6/2 mice with the FDA-approved drug metformin, which haspleiotropic anti-diabetic effects, gave promising results (Ma et al.,2007). Secretion of insulin by beta-cells is regulated by glucagon-like peptide-1 (GLP-1), which is secreted by intestinal cells inresponse to food ingestion. Treatment of N171-82Q mice with theFDA-approved GLP-1 receptor agonist exendin-4 reduced bothserum glucose levels and morphological abnormalities in the isletsof Langerhans, ameliorated motor dysfunction, and extendedlifespan (Martin et al., 2009). As exendin-4 acts on both neuronaland peripheral tissues, the beneficial effects of this compound onthe HD phenotype in mouse is likely to involve direct protectiveeffects in neural as well as in peripheral tissues. It remains to beestablished whether pancreatic dysfunction in HD results fromcell-autonomous or non-cell-autonomous toxicity or both.
3.3.5. Metabolic abnormalities: toxicity in adipose tissue
Alteration of glucose homeostasis may also be a consequenceof metabolic dysfunction. One of the main symptoms of HD isbody weight loss (Kremer and Roos, 1992; Sanberg et al., 1981;Stoy and McKay, 2000), which is overt even at early stages ofdisease, before the onset of chorea (Djousse et al., 2002). Thedegree of weight loss correlates with the length of the polyQ tract(Aziz et al., 2008) and occurs even with a high-caloric dietaryregime (Sanberg et al., 1981). The observation that weight lossoccurs in the absence of hyperkinesia excludes that thissymptom is a consequence of excessive involuntary bodymovements and suggests that it might rather be a consequenceof dysfunctional fat metabolism and of direct toxicity of polyQ-huntingtin in adipose tissue. Adipose tissue dysfunction has beenreported in R6/2 mice (Fain et al., 2001). R6/2 mice undergo aninitial phase characterized by weight increase around 8 weeks ofage, which is followed by wasting. Importantly, in both HDpatients and mice, wasting is associated with increased energymetabolism, which leads to altered energy balance (Goodmanet al., 2008; van der Burg et al., 2008).
There are two types of adipose tissue, white and brown.Adipose tissue is composed of adipocytes or fat cells. White fatcells secrete hormones, such as adiponectin and leptin. Thesehormones play a crucial role in the regulation of energymetabolism, as food intake and energy balance are regulated byleptin at the level of the hypothalamus and by adiponectin at thelevel of muscle and liver. The levels of leptin and adiponectin aredecreased in HD patients (Popovic et al., 2004) and mouse modelsof disease (Phan et al., 2009). Importantly, in HD mice thesealterations are detectable before the changes in body weight,suggesting that adipose tissue dysfunction may be primary tobody weight loss. These defects are associated with alterations inthe expression of genes that regulate adipocyte differentiation,such as the transcription factors peroxisome proliferator-activat-ed receptor g2 (PPARg2) and CAAT enhancer binding protein a (C/EBPa), and of their target genes, diacylglycerol acyltransferaseand lipin-1. The mechanism proposed for dysregulation of geneexpression in adipocytes involves the PPARg co-activator PGC-1a.PGC-1a expression and function were found to be hampered inHD adipocytes by polyQ-huntingtin, further implicating PGC-1aas a critical player in HD pathogenesis.
HD mice also show defects in brown adipose tissue (Weydt etal., 2006). Brown adipose tissue regulates body temperature inrodents (reviewed by Cannon and Nedergaard, 2004; Cypess andKahn, 2010). A key regulator of thermogenesis in the brownadipose tissue is uncoupling protein 1 (UCP-1), whose expressionis regulated by PGC-1a in this tissue (Lin et al., 2005). HD micehave been shown to suffer from hypothermia due to a failure inthe activation of brown adipose tissue. Mechanistically, this is
due to dysfunction of PGC-1a in brown adipose tissue, which inturn results in altered UCP-1 expression levels. As UCP-1expression in this tissue is restricted to mitochondria, thispathway links mitochondrial dysfunction with transcriptiondysregulation in HD and again highlights the key role of PGC-1ain HD pathogenesis. Early signs of metabolic dysfunction in HDstrongly support the idea that polyQ-huntingtin toxicity in bothwhite and brown adipose tissue occurs through a cell-autono-mous mechanism.
3.3.6. Altered spermatogenesis
Huntingtin is highly expressed in testis. Recently, it has beenproposed that expression of polyQ-huntingtin in this tissue mayexert direct toxic effects in HD. Levels of testosterone and LH arealtered in HD patients, and testosterone levels in the seruminversely correlate with severity of disease (Markianos et al.,2005). These abnormalities suggest a broader hypothalamic-pituitary-testicular axis involvement in HD (Fig. 1). Moreover,testicular pathology has been reported in HD patients (VanRaamsdonk et al., 2007a). Patients present with reduced numbersof spermatocytes and spermatids (Fig. 4). Moreover, testes showaltered morphology of the seminiferous tubules, and severity ofthis phenotype correlates with the length of the polyQ tract.Deficits in the reproductive organs have also been observed inanimal models of HD. YAC128 and R6/2 mice present withtesticular atrophy and sterility very early during disease progres-sion (Van Raamsdonk et al., 2006). Notably, this phenotype isassociated with a reduction in the number of GnRH-positiveneurons and a decrease in the levels of testosterone in the serumand testis (Papalexi et al., 2005). Huntingtin is highly expressedboth in germ cells and Sertoli cells. Although it remains to beestablished whether the defects in spermatogenesis are cell-autonomous (primary to germ cell toxicity) or non-cell-autono-mous (secondary to Sertoli cell dysfunction), these observationssuggest that polyQ expansion in huntingtin exerts toxic effects thattarget the process of spermatogenesis.
3.4. Non-cell-autonomous toxicity in HD
The ubiquitous expression and critical function of huntingtinraises the question as to whether HD is a cell-autonomous or non-cell-autonomous disorder. Development of mouse models forconditional expression of polyQ-huntingtin has been critical toaddress this question. Expression of polyQ-huntingtin solely ineither medium-sized projection spiny neurons (Gu et al., 2007) orcortical pyramidal neurons (Gu et al., 2005) in rodents results incell-autonomous nuclear accumulation of mutant protein andaggregation. Confined expression of mutant protein in striatalneurons also causes cell-autonomous alteration of N-methyl-d-aspartate (NMDA) receptor function. However, restricted expres-sion of mutant huntingtin to these neuronal populations failed toelicit motor dysfunction and striatal or cortical neuropathologyand gliosis. Disease manifestations, including locomotor dysfunc-tion, gliosis, and neurodegeneration, are evident when mutantprotein is expressed in both neuronal and glial cells. Theseobservations provide evidence that expression of polyQ-hunting-tin in a subset of neurons that degenerate in HD is not sufficient toeffectively recapitulate the HD phenotype in mouse. Rather, thesefindings provide proof-of-principle that some aspects of diseasepathogenesis result from non-cell-autonomous toxicity. Altera-tions of the interactions between different neuronal populationsand between neuronal and non-neuronal (e.g. glial) cells can leadto dysfunction in neuronal circuitry, which ultimately leads toneuronal death. Dysfunctions of the cortico-striatal and nigro-striatal axis as well as the hypothalamic-endocrine axis arethought to represent major pathways of toxicity in HD.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172164
3.4.1. Excitotoxicity and cortico-striatal axis dysfunction
As the striatum receives glutamatergic inputs from the cerebralcortex (Fig. 5), the selective vulnerability of the striatum in HDmay be caused by aberrant glutamatergic stimulation. Glutamateis an excitatory neurotransmitter that activates both NMDA andnon-NMDA ionotropic glutamate receptors. Excitotoxicity is aphenomenon that results from aberrant NMDA receptor functionand/or excessive stimulation of glutamate receptors, which resultsin increased intracellular levels of calcium and consequentneurotoxicity (reviewed by Han et al., 2010; Zuccato et al.,2010). Medium-sized spiny neurons are particularly sensitive toglutamate stimulation, as demonstrated by treatment of rodentswith NMDA and non-NMDA receptor agonists, such as quinolinicacid and kainic acid (Coyle and Schwarcz, 1976; McGeer andMcGeer, 1976). This observation led to the idea that excitotoxicitycan contribute to HD pathogenesis. Increased susceptibility toexcitotoxicity has been reported in animal models of HD thatexpress full-length polyQ-huntingtin (Zeron et al., 2002).However, the R6/2, R6/1, and N171-82Q mice expressing exon 1of polyQ-huntingtin show striatal resistance to excitotoxicity,which may be the result of an adaptive response to neurotoxicity(Hansson et al., 1999; Morton and Leavens, 2000). Interestingly,the increased susceptibility of HD neurons to excitotoxicity seemsto be non-cell-autonomous, whereas the resistance to excitotoxi-city observed in symptomatic mice is cell-autonomous (Ho Kim etal., 2011). Alterations in glutamatergic transmission have beenattributed to aberrant cell-autonomous NMDA receptor expressionand function in HD medium-sized spiny neurons. These neuronsexpress high levels of the NMDA receptor NR2B subunit comparedto striatal interneurons, which are spared in HD. The observationthat expression and function of this NMDA receptor subunit isaltered in HD mice may at least in part explain the selectivevulnerability of medium-sized spiny neurons in HD. Similarly, theexpression of the metabotropic glutamate receptor mGluR2 isaltered in HD, leading to excitotoxicity. Overactivation of NMDAreceptors can alter downstream signaling pathways, leading toelevated intracellular calcium levels, mitochondrial dysfunction,and activation of apoptotic pathways in striatal neurons. Inaddition to cell-autonomous pathways of excitotoxicity, aberrantglutamate receptor stimulation can result from increased releaseof glutamate from cortical afferents, as observed in animal modelsof HD. Moreover, reduced uptake of glutamate from glial cells canalso increase glutamate levels. Astrocytes are crucial for glutamateuptake. This uptake process is altered in glial cells expressingpolyQ-huntingtin (Lievens et al., 2001; Shin et al., 2005).Expression of the sodium-dependent glutamate transporter GLT-1 is decreased in animal models of HD and in autopsy brainspecimens from HD patients (Lievens et al., 2001). Pharmacologicinduction of GLT-1 expression in R6/2 mice reduced diseasemanifestations, indicating a critical role for glutamate uptake inHD (Miller et al., 2008). Importantly, expression of GLT-1 isdecreased even when polyQ-huntingtin is expressed selectively inastrocytes, suggesting that excitotoxicity in HD is non-cell-autonomous (Bradford et al., 2009).
3.4.2. Nigro-striatal axis dysfunction
In addition to glutamatergic afferent fibers from the cortex, thestriatum receives dopaminergic inputs from the substantia nigrapars compacta (reviewed by Gil and Rego, 2008; Han et al., 2010)(Fig. 5). In the striatum, dopamine content exhibits a dorsal toventral gradient, which correlates with the progression of HDpathology and thereby suggests that alterations of dopaminesignaling can be central to disease. Consistent with this idea, nigro-striatal pathology has been described in HD (Bohnen et al., 2000;Ferrante and Kowall, 1987; Ginovart et al., 1997; Suzuki et al.,2001) with loss of dopaminergic neurons in the substantia nigra
(Huot et al., 2007; Oyanagi et al., 1989). Importantly, there is amarked reduction in expression of the enzyme tyrosine hydroxy-lase, which is a pivotal enzyme for dopamine biosynthesis(Yohrling et al., 2003), as well as the dopamine transporter andthe D1 and D2 dopamine receptors (Ginovart et al., 1997).Dopamine exerts excitatory signals when it binds to the D1receptor, and inhibitory signals when it binds to the D2 receptor.The medium-sized spiny neurons expressing high levels of D2receptors are more sensitive to polyQ-huntingtin compared tothose expressing high levels of D1 receptors (see Section 3.2).Alteration of the nigro-striatal axis may be responsible for bothmotor and cognitive dysfunction, and this may represent themolecular correlate of HD neuronal dysfunction. Reduction ofdopamine signaling can lead to activation of pro-apoptoticpathways, which involve activation of c-Jun, production of reactiveoxygen species, and initiation of JNK-dependent signaling.
3.4.3. Hypothalamic-endocrine axis in HD
Several symptoms developed by HD patients, such as weightloss, sleep dysfunction, and muscle wasting, can be secondary todamage occurring in CNS brain areas, such as the hypothalamus(reviewed by Petersen and Bjorkqvist, 2006). The hypothalamus isthe brain region that regulates metabolism and sleep. Signs ofatrophy in the hypothalamus have been reported in HD patients(Kassubek et al., 2004). Atrophy of the lateral tuberal nucleus andloss of somatostatin-containing neurons have been reported in HD(Kremer et al., 1990, 1991; Timmers et al., 1996). In the lateralhypothalamic area, there is loss of orexin-containing neurons bothin HD patients and in mouse models of disease (Petersen et al.,2005). Hypothalamic dysfunction may originate from aberrantinteraction of polyQ-huntingtin with the huntingtin-interactingprotein-1 (HAP-1) (Li et al., 1995). HAP-1 is highly expressed in thehypothalamus and binds with higher affinity to mutant ascompared to wild-type huntingtin, which may lead to a loss ofprotein function. HAP-1 expression is decreased in HD patients andmice, suggesting that HAP-1 may have a central role in HDpathogenesis.
4. Spinocerebellar ataxia (SCA)
4.1. Clinical features of SCA
Expansions of polyQ tracts in six different genes, known asataxin-1 (Orr et al., 1993), ataxin-2 (Imbert et al., 1996), ataxin-3(Kawaguchi et al., 1994), CACNA1A (Zhuchenko et al., 1997),ataxin-7 (David et al., 1997), and the TATA-binding protein (TBP)(Nakamura et al., 2001), are responsible for six types of autosomaldominant SCAs, designated SCA1, SCA2, SCA3, SCA6, SCA7, andSCA17, respectively. SCAs are late-onset and progressive neuro-logical disorders characterized by the loss of motor coordinationand balance, pyramidal and extra-pyramidal signs, peripheralneuropathy, cognitive dysfunction, slurred speech, and difficulty inswallowing. SCA patients develop atrophy of the cerebellum,which is primarily due to loss of Purkinje cells.
4.2. Cell-autonomous and non-cell-autonomous toxicity in SCA
SCAs are caused by cell-autonomous damage from polyQ-expanded proteins in Purkinje cells. Expression of either polyQ-ataxin-1 (Burright et al., 1995) or polyQ-ataxin-7 (Yvert et al.,2000) solely in Purkinje cells results in neuronal death anddevelopment of an ataxic phenotype in mouse. Interestingly, inaddition to cell-autonomous toxicity in these disorders, non-cell-autonomous Purkinje cell degeneration has been described inSCA7. Expression of mutant ataxin-7 in several neuronal types andglial cells, but not Purkinje cells, results in Purkinje cell loss as well

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 165
as ataxia (Garden et al., 2002). A critical role for glial cells in SCA7pathogenesis was established with the development of animalmodels of SCA7, in which expression of mutant ataxin-7 wasrestricted to Bergmann glia, the astrocytes that surround andsupport the Purkinje cell dendritic tree in the cerebellum (Custeret al., 2006). Specific expression of polyQ-ataxin-7 in Bergmannglia leads to Purkinje cell degeneration and results in a phenotypethat resembles SCA7 in mice. The mechanism through which glialcells lead to Purkinje cell degeneration involves glutamatetransport defects with consequent excitotoxicity.
Similar to SBMA and HD, patients suffering from SCA1 presentwith endocrine abnormalities (Lalic et al., 2010). SCA1 patientshave been described to have altered insulin secretion, decreasedinsulin sensitivity, and increased insulin resistance. It remains tobe established whether these defects are cell-autonomous or non-cell-autonomous in SCA1.
5. Mechanisms underlying cell-autonomous and non-cell-autonomous toxicity in polyQ diseases
The reason why neurons are primarily damaged by theexpression of polyQ protein remains an enigma, as the expressionof the majority of polyQ proteins is not restricted to neurons.Several factors can contribute to selective neuronal vulnerability,including specific changes in expression levels, subcellularlocalization, and alteration of folding and function of polyQproteins.
5.1. Expression levels of disease proteins
A critical factor that can contribute to selective neuronalvulnerability is the level of expression of mutant protein in thecentral nervous system. Huntingtin (Ferrante et al., 1997),atrophin-1 (Knight et al., 1997; Onodera et al., 1995), AR (Simerlyet al., 1990), and the SCA-related genes are expressed in a varietyof tissues in addition to the nervous system, but their expression isparticularly enriched in neurons and, in some cases, is higher in thedegenerating neurons compared to unaffected neurons. ARexpression levels are higher in the motor neurons of the anteriorhorn of the spinal cord, which degenerate in SBMA, compared toother neuronal populations (Poletti, 2004). Different expressionlevels of polyQ proteins modulate disease manifestations in mousemodels of polyQ diseases. For instance, severity of diseasecorrelates with the expression levels of mutant huntingtin inmouse models of HD (Hodgson et al., 1999). However, theexpression levels of polyQ-huntingtin do not correlate withselective neuronal degeneration, as striatal neurons degeneratingin HD express levels of mutant protein comparable to, if not lowerthan, other neuronal populations spared during the course ofdisease (Fusco et al., 1999). It is possible that expression of thedisease proteins over a certain threshold triggers disease, asneuronal cells may not be able to cope with the accumulation ofmutant protein throughout life. However, this cannot be the soledeterminant of selective neuronal vulnerability.
5.2. Subcellular localization of mutant protein
The subcellular localization of polyQ proteins is an importantaspect that can contribute to neuronal dysfunction and death. It ispossible that polyQ expansion alters the subcellular localization ofmutant protein, leading to its aberrant accumulation in specific cellareas, where it exerts toxic effects. In this regard, the nucleus hasbeen shown to represent a critical compartment for polyQ-mediated toxicity, as localization of expanded polyQ proteins inthe nucleus of neurons is often necessary for toxicity. Ataxin-1localizes to the nucleus of neurons and the cytosol of non-neuronal
cells (Servadio et al., 1995), and nuclear accumulation of polyQ-ataxin-1 is critical for disease pathogenesis (Klement et al., 1998).Nuclear accumulation of polyQ-huntingtin is required for neuronaldamage (Saudou et al., 1998). Ataxin-3 localizes mostly in thecytosol (Paulson et al., 1997). However, when ataxin-3 has anexpanded polyQ tract, it localizes to the nucleus of degeneratingneurons (Schmidt et al., 1998), and, similar to the other polyQdiseases, nuclear localization of ataxin-3 is a critical determinant ofdisease pathogenesis (Bichelmeier et al., 2007). Nuclear localiza-tion of polyQ-AR, which is induced by binding to testosterone, iscritical for toxicity (Montie et al., 2009; Nedelsky et al., 2010).
Although it is clear that the nucleus represents a criticalcompartment for disease pathogenesis, there is evidence thatcytoplasmic localization of polyQ proteins is also important fordisease pathogenesis. Ataxin-2 and ataxin-6 mainly localize to thecytosol. In YAC mice expressing mutant huntingtin, cytoplasmicdysfunction is evident prior to the onset of accumulation of mutantprotein in the nucleus and neurodegeneration (Hodgson et al.,1999). Rather, cytoplasmic dysfunction has been proposed to occurfirst and to be followed by cleavage of mutant huntingtin intofragments that translocate to the nucleus and cause nucleardysfunction (reviewed by Hackam et al., 1999).
5.3. Accumulation of polyQ proteins in inclusions and micro-
aggregates
A hallmark of polyQ diseases is the accumulation of polyQprotein in the forms of micro-aggregates and inclusions. Micro-aggregates or micro-oligomers are species that can be detected bybiochemical techniques, while inclusions can be detected byimmunohistochemical techniques (Palazzolo et al., 2009).Immunohistochemical analysis of brain specimens from patientsreveals that polyQ proteins accumulate in neurons in the form ofnuclear inclusions. Inclusions are present not only in degeneratingneuronal cells, but also in the neurons of brain areas that do notdegenerate. Importantly, neurodegeneration does not alwayscorrelate with the formation of nuclear inclusions, as in somecases neuronal damage can be detected prior to the onset ofnuclear inclusion formation (Hodgson et al., 1999). Moreover,neuronal survival has been shown to positively correlate with thepresence of inclusions (Arrasate et al., 2004). This observation hasled to the idea that inclusions may be protective species, and thatmicro-aggregates or oligomers may be the toxic species. This ideais supported by the observation that treatment of HD and SBMAcells with compounds that induce inclusion formation of polyQ-huntingtin and polyQ-AR decrease toxicity (Bodner et al., 2006;Palazzolo et al., 2010).
5.4. Alteration of polyQ protein function
Another aspect that makes it difficult to understand whyneurons are extremely and selectively vulnerable to the presenceof polyQ proteins is that, in addition to having ubiquitousdistribution throughout tissues, several polyQ proteins havecellular housekeeping functions. There is emerging evidence thatexpansion of the polyQ tract results in the alteration of the nativefunction of the polyQ protein, and that this is a critical aspect ofdisease pathogenesis. Ataxin-3 is a ubiquitin-binding protein withdeubiquitinating activity that regulates the degradation of proteinsvia the ubiquitin-proteasome system (Burnett et al., 2003). Thisfunction has been correlated with the ability of wild-type ataxin-3to suppress the toxicity of polyQ proteins (Warrick et al., 2005).Ataxin-3 has also been proposed to regulate protein degradationvia endoplasmic reticulum-associated protein degradation (ERAD)(Zhong and Pittman, 2006) and transcriptional repression (Evert etal., 2003). It is possible that expansion of the polyQ tract alters

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172166
ataxin-3 function, thereby affecting the pathway of proteindegradation via ERAD and the proteasome. Ataxin-7 is a subunitof the GCN5 histone acetyltransferase of the STAGA transcriptionco-activator complex (Helmlinger et al., 2004). PolyQ expansion inataxin-7 interferes with the normal function of the protein, therebyresulting in the dysregulation of histone acetylation, which maycontribute to the transcriptional dysregulation involved in polyQpathogenesis (McMahon et al., 2005; Palhan et al., 2005). TBP is acomponent of the core transcription complex TFIID, which plays acritical role in the initiation of transcription, as its binding to DNAis required for subsequent assembly of the initiation transcriptioncomplex and for expression of genes whose transcription isdependent on RNA polymerase II. Expansion of polyQ in TBPreduces homodimerization, but enhances its interaction withtranscription factor IIB (TFIIB), resulting in transcription dysre-gulation (Friedman et al., 2007). PolyQ expansion in ataxin-1interferes with the native function of the protein, in that it reducesthe proportion of the disease protein present in a complexcontaining the protein RBM17, while it enhances its interactionwith the complex containing capicua (Lim et al., 2008). Similarly,polyQ expansion in AR alters the ability of mutant protein tointeract with transcription co-regulators, which is critical fordisease pathogenesis (Nedelsky et al., 2010). These observationssupport the emerging concept that polyQ expansion causesselective neuronal vulnerability by altering (enhancing ordecreasing) the interaction of the disease proteins with nativeinteractors. It remains to be established why modification ofnative housekeeping protein function is critical to neurons andnot to other cell types.
6. Concluding remarks
It is now widely accepted that polyQ diseases result fromcombined cell-autonomous and non-cell-autonomous pathways oftoxicity, which ultimately lead to neuronal dysfunction and death.In addition to neuronal toxicity, non-neuronal cells are alsosubjected to polyQ protein toxicity, in a cell-autonomous and non-cell-autonomous fashion. Clarification of the contribution of cell-autonomous and non-cell-autonomous toxicity in polyQ disease isneeded from a therapeutic point of view. Indeed, if maintenance ofa healthy nervous system is tightly linked to proper functioning ofperipheral tissues, therapeutic intervention in peripheral tissuesmay have a critical impact on the central nervous system.
Conflict of interest
The authors have no conflict of interest to declare.
Acknowledgments
We apologize to those authors whose work was not cited in thisreview due to space limitations. We thank N. Nedelsky forcomments and editing of the manuscript. This work was supportedby Marie Curie Reintegration grants (FP7-256448 to M.P. and FP7-276981 to F.S.), Telethon-Italy (GGP10037), the Kennedy’s DiseaseAssociation, Fondation Thierry Latran (AAP091102), and theMuscular Dystrophy Association (196646).
References
Al-Shamma, H.A., Arnold, A.P., 1997. Brain-derived neurotrophic factor regulatesexpression of androgen receptors in perineal motoneurons. Proc. Natl. Acad. Sci.U.S.A. 94, 1521–1526.
Amato, A.A., Prior, T.W., Barohn, R.J., Snyder, P., Papp, A., Mendell, J.R., 1993.Kennedy’s disease: a clinicopathologic correlation with mutations in the an-drogen receptor gene. Neurology 43, 791–794.
Andreassen, O.A., Dedeoglu, A., Stanojevic, V., Hughes, D.B., Browne, S.E., Leech, C.A.,Ferrante, R.J., Habener, J.F., Beal, M.F., Thomas, M.K., 2002. Huntington’s disease
of the endocrine pancreas: insulin deficiency and diabetes mellitus due toimpaired insulin gene expression. Neurobiol. Dis. 11, 410–424.
Andrew, S.E., Goldberg, Y.P., Kremer, B., Telenius, H., Theilmann, J., Adam, S., Starr, E.,Squitieri, F., Lin, B., Kalchman, M.A., et al., 1993. The relationship betweentrinucleotide (CAG) repeat length and clinical features of Huntington’s disease.Nat. Genet. 4, 398–403.
Araki, I., Harada, Y., Kuno, M., 1991. Target-dependent hormonal control of neuronsize in the rat spinal nucleus of the bulbocavernosus. J. Neurosci. 11, 3025–3033.
Arrasate, M., Mitra, S., Schweitzer, E.S., Segal, M.R., Finkbeiner, S., 2004. Inclusionbody formation reduces levels of mutant huntingtin and the risk of neuronaldeath. Nature 431, 805–810.
Aylward, E.H., Liu, D., Nopoulos, P.C., Ross, C.A., Pierson, R.K., Mills, J.A., Long, J.D.,Paulsen, J.S., 2011. Striatal volume contributes to the prediction of onset ofHuntington disease in incident cases. Biol. Psychiatry Sep 8. [Epub ahead of print]
Aziz, N.A., van der Burg, J.M., Landwehrmeyer, G.B., Brundin, P., Stijnen, T., Roos, R.A.,2008. Weight loss in Huntington disease increases with higher CAG repeatnumber. Neurology 71, 1506–1513.
Azzouz, M., Ralph, G.S., Storkebaum, E., Walmsley, L.E., Mitrophanous, K.A., Kings-man, S.M., Carmeliet, P., Mazarakis, N.D., 2004. VEGF delivery with retrogradelytransported lentivector prolongs survival in a mouse ALS model. Nature 429,413–417.
Bacos, K., Bjorkqvist, M., Petersen, A., Luts, L., Maat-Schieman, M.L., Roos, R.A.,Sundler, F., Brundin, P., Mulder, H., Wierup, N., 2008. Islet beta-cell area andhormone expression are unaltered in Huntington’s disease. Histochem. CellBiol. 129, 623–629.
Banno, H., Katsuno, M., Suzuki, K., Takeuchi, Y., Kawashima, M., Suga, N., Takamori,M., Ito, M., Nakamura, T., Matsuo, K., Yamada, S., Oki, Y., Adachi, H., Mina-miyama, M., Waza, M., Atsuta, N., Watanabe, H., Fujimoto, Y., Nakashima, T.,Tanaka, F., Doyu, M., Sobue, G., 2009. Phase 2 trial of leuprorelin in patients withspinal and bulbar muscular atrophy. Ann. Neurol. 65, 140–150.
Battaglia, F., Le Galudec, V., Cossee, M., Tranchant, C., Warter, J.M., Echaniz-Laguna,A., 2003. Kennedy’s disease initially manifesting as an endocrine disorder. J.Clin. Neuromuscul. Dis. 4, 165–167.
Baumgartner, B.J., Shine, H.D., 1997. Targeted transduction of CNS neurons withadenoviral vectors carrying neurotrophic factor genes confers neuroprotectionthat exceeds the transduced population. J. Neurosci. 17, 6504–6511.
Bergmann, M., Volpel, M., Kuchelmeister, K., 1995. Onuf’s nucleus is frequentlyinvolved in motor neuron disease/amyotrophic lateral sclerosis. J. Neurol. Sci.129, 141–146.
Bhasin, S., Storer, T.W., Berman, N., Yarasheski, K.E., Clevenger, B., Phillips, J., Lee,W.P., Bunnell, T.J., Casaburi, R., 1997. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J. Clin. Endocrinol. Metab. 82,407–413.
Bichelmeier, U., Schmidt, T., Hubener, J., Boy, J., Ruttiger, L., Habig, K., Poths, S.,Bonin, M., Knipper, M., Schmidt, W.J., Wilbertz, J., Wolburg, H., Laccone, F., Riess,O., 2007. Nuclear localization of ataxin-3 is required for the manifestation ofsymptoms in SCA3: in vivo evidence. J. Neurosci. 27, 7418–7428.
Bjorkqvist, M., Fex, M., Renstrom, E., Wierup, N., Petersen, A., Gil, J., Bacos, K.,Popovic, N., Li, J.Y., Sundler, F., Brundin, P., Mulder, H., 2005. The R6/2 transgenicmouse model of Huntington’s disease develops diabetes due to deficient beta-cell mass and exocytosis. Hum. Mol. Genet. 14, 565–574.
Bodine, S.C., Stitt, T.N., Gonzalez, M., Kline, W.O., Stover, G.L., Bauerlein, R., Zlotch-enko, E., Scrimgeour, A., Lawrence, J.C., Glass, D.J., Yancopoulos, G.D., 2001. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and canprevent muscle atrophy in vivo. Nat. Cell Biol. 3, 1014–1019.
Bodner, R.A., Outeiro, T.F., Altmann, S., Maxwell, M.M., Cho, S.H., Hyman, B.T.,McLean, P.J., Young, A.B., Housman, D.E., Kazantsev, A.G., 2006. Pharmacologicalpromotion of inclusion formation: a therapeutic approach for Huntington’s andParkinson’s diseases. Proc. Natl. Acad. Sci. U.S.A. 103, 4246–4251.
Boesgaard, T.W., Nielsen, T.T., Josefsen, K., Hansen, T., Jorgensen, T., Pedersen, O.,Norremolle, A., Nielsen, J.E., Hasholt, L., 2009. Huntington’s disease doesnot appear to increase the risk of diabetes mellitus. J. Neuroendocrinol. 21,770–776.
Bogaert, E., Van Damme, P., Van Den Bosch, L., Robberecht, W., 2006. Vascularendothelial growth factor in amyotrophic lateral sclerosis and other neurode-generative diseases. Muscle Nerve 34, 391–405.
Bohanna, I., Georgiou-Karistianis, N., Hannan, A.J., Egan, G.F., 2008. Magneticresonance imaging as an approach towards identifying neuropathologicalbiomarkers for Huntington’s disease. Brain Res. Rev. 58, 209–225.
Bohnen, N.I., Koeppe, R.A., Meyer, P., Ficaro, E., Wernette, K., Kilbourn, M.R., Kuhl,D.E., Frey, K.A., Albin, R.L., 2000. Decreased striatal monoaminergic terminals inHuntington disease. Neurology 54, 1753–1759.
Bradford, J., Shin, J.Y., Roberts, M., Wang, C.E., Li, X.J., Li, S., 2009. Expression ofmutant huntingtin in mouse brain astrocytes causes age-dependent neurologi-cal symptoms. Proc. Natl. Acad. Sci. U.S.A. 106, 22480–22485.
Breedlove, S.M., Arnold, A.P., 1980. Hormone accumulation in a sexually dimorphicmotor nucleus of the rat spinal cord. Science 210, 564–566.
Breedlove, S.M., Arnold, A.P., 1981. Sexually dimorphic motor nucleus in the ratlumbar spinal cord: response to adult hormone manipulation, absence inandrogen-insensitive rats. Brain Res. 225, 297–307.
Brodsky, I.G., Balagopal, P., Nair, K.S., 1996. Effects of testosterone replacement onmuscle mass and muscle protein synthesis in hypogonadal men – a clinicalresearch center study. J. Clin. Endocrinol. Metab. 81, 3469–3475.
Brooks, B.P., Merry, D.E., Paulson, H.L., Lieberman, A.P., Kolson, D.L., Fischbeck, K.H.,1998. A cell culture model for androgen effects in motor neurons. J. Neurochem.70, 1054–1060.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 167
Brooks, B.P., Paulson, H.L., Merry, D.E., Salazar-Grueso, E.F., Brinkmann, A.O., Wilson,E.M., Fischbeck, K.H., 1997. Characterization of an expanded glutamine repeatandrogen receptor in a neuronal cell culture system. Neurobiol. Dis. 3, 313–323.
Brown, D., Hikim, A.P., Kovacheva, E.L., Sinha-Hikim, I., 2009. Mouse model oftestosterone-induced muscle fiber hypertrophy: involvement of p38 mitogen-activated protein kinase-mediated Notch signaling. J. Endocrinol. 201, 129–139.
Brown, T.B., Bogush, A.I., Ehrlich, M.E., 2008. Neocortical expression of mutanthuntingtin is not required for alterations in striatal gene expression or motordysfunction in a transgenic mouse. Hum. Mol. Genet. 17, 3095–3104.
Burnett, B., Li, F., Pittman, R.N., 2003. The polyglutamine neurodegenerative proteinataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity.Hum. Mol. Genet. 12, 3195–3205.
Burright, E.N., Clark, H.B., Servadio, A., Matilla, T., Feddersen, R.M., Yunis, W.S.,Duvick, L.A., Zoghbi, H.Y., Orr, H.T., 1995. SCA1 transgenic mice: a model forneurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82,937–948.
Busse, M.E., Hughes, G., Wiles, C.M., Rosser, A.E., 2008. Use of hand-held dynamom-etry in the evaluation of lower limb muscle strength in people with Hunting-ton’s disease. J. Neurol. 255, 1534–1540.
Cannon, B., Nedergaard, J., 2004. Brown adipose tissue: function and physiologicalsignificance. Physiol. Rev. 84, 277–359.
Caroni, P., Grandes, P., 1990. Nerve sprouting in innervated adult skeletal muscleinduced by exposure to elevated levels of insulin-like growth factors. J. Cell Biol.110, 1307–1317.
Carter, R.J., Lione, L.A., Humby, T., Mangiarini, L., Mahal, A., Bates, G.P., Dunnett, S.B.,Morton, A.J., 1999. Characterization of progressive motor deficits in micetransgenic for the human Huntington’s disease mutation. J. Neurosci. 19,3248–3257.
Cashman, N.R., Durham, H.D., Blusztajn, J.K., Oda, K., Tabira, T., Shaw, I.T., Dahrouge,S., Antel, J.P., 1992. Neuroblastoma � spinal cord (NSC) hybrid cell lines resem-ble developing motor neurons. Dev. Dyn. 194, 209–221.
Chahin, N., Sorenson, E.J., 2009. Serum creatine kinase levels in spinobulbarmuscular atrophy and amyotrophic lateral sclerosis. Muscle Nerve 40, 126–129.
Chang, C., Chen, Y.T., Yeh, S.D., Xu, Q., Wang, R.S., Guillou, F., Lardy, H., Yeh, S., 2004.Infertility with defective spermatogenesis and hypotestosteronemia in malemice lacking the androgen receptor in Sertoli cells. Proc. Natl. Acad. Sci. U.S.A.101, 6876–6881.
Chaturvedi, R.K., Adhihetty, P., Shukla, S., Hennessy, T., Calingasan, N., Yang, L.,Starkov, A., Kiaei, M., Cannella, M., Sassone, J., Ciammola, A., Squitieri, F., Beal,M.F., 2009. Impaired PGC-1alpha function in muscle in Huntington’s disease.Hum. Mol. Genet. 18, 3048–3065.
Chen, C.M., 2011. Mitochondrial dysfunction, metabolic deficits, and increasedoxidative stress in Huntington’s disease. Chang Gung Med. J. 34, 135–152.
Chevalier-Larsen, E.S., Merry, D.E., 2011. Testosterone treatment fails to acceleratedisease in a transgenic mouse model of spinal and bulbar muscular atrophy. Dis.Model. Mech. Oct 4. [Epub ahead of print]
Chevalier-Larsen, E.S., O’Brien, C.J., Wang, H., Jenkins, S.C., Holder, L., Lieberman,A.P., Merry, D.E., 2004. Castration restores function and neurofilament altera-tions of aged symptomatic males in a transgenic mouse model of spinal andbulbar muscular atrophy. J. Neurosci. 24, 4778–4786.
Chvatal, A., Anderova, M., Neprasova, H., Prajerova, I., Benesova, J., Butenko, O.,Verkhratsky, A., 2008. Pathological potential of astroglia. Physiol. Res. 57 (Suppl.3), S101–S110.
Ciammola, A., Sassone, J., Alberti, L., Meola, G., Mancinelli, E., Russo, M.A., Squitieri,F., Silani, V., 2006. Increased apoptosis Huntingtin inclusions and altereddifferentiation in muscle cell cultures from Huntington’s disease subjects. CellDeath Differ. 13, 2068–2078.
Ciammola, A., Sassone, J., Sciacco, M., Mencacci, N.E., Ripolone, M., Bizzi, C., Colciago,C., Moggio, M., Parati, G., Silani, V., Malfatto, G., 2011. Low anaerobic thresholdand increased skeletal muscle lactate production in subjects with Huntington’sdisease. Mov. Disord. 26, 130–137.
Coleman, M.E., DeMayo, F., Yin, K.C., Lee, H.M., Geske, R., Montgomery, C., Schwartz,R.J., 1995. Myogenic vector expression of insulin-like growth factor I stimulatesmuscle cell differentiation and myofiber hypertrophy in transgenic mice. J. Biol.Chem. 270, 12109–12116.
Conboy, I.M., Conboy, M.J., Smythe, G.M., Rando, T.A., 2003. Notch-mediated resto-ration of regenerative potential to aged muscle. Science 302, 1575–1577.
Coyle, J.T., Schwarcz, R., 1976. Lesion of striatal neurones with kainic acid provides amodel for Huntington’s chorea. Nature 263, 244–246.
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C.N., Tanese, N., Krainc, D., 2006. Tran-scriptional repression of PGC-1alpha by mutant huntingtin leads to mitochon-drial dysfunction and neurodegeneration. Cell 127, 59–69.
Custer, S.K., Garden, G.A., Gill, N., Rueb, U., Libby, R.T., Schultz, C., Guyenet, S.J.,Deller, T., Westrum, L.E., Sopher, B.L., La Spada, A.R., 2006. Bergmann gliaexpression of polyglutamine-expanded ataxin-7 produces neurodegenerationby impairing glutamate transport. Nat. Neurosci. 9, 1302–1311.
Cypess, A.M., Kahn, C.R., 2010. The role and importance of brown adipose tissue inenergy homeostasis. Curr. Opin. Pediatr. 22, 478–484.
David, G., Abbas, N., Stevanin, G., Durr, A., Yvert, G., Cancel, G., Weber, C., Imbert, G.,Saudou, F., Antoniou, E., Drabkin, H., Gemmill, R., Giunti, P., Benomar, A., Wood,N., Ruberg, M., Agid, Y., Mandel, J.L., Brice, A., 1997. Cloning of the SCA7 genereveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70.
de Almeida, L.P., Ross, C.A., Zala, D., Aebischer, P., Deglon, N., 2002. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces aselective neuropathology modulated by polyglutamine repeat size, huntingtinexpression levels, and protein length. J. Neurosci. 22, 3473–3483.
De Gendt, K., Swinnen, J.V., Saunders, P.T., Schoonjans, L., Dewerchin, M., Devos, A.,Tan, K., Atanassova, N., Claessens, F., Lecureuil, C., Heyns, W., Carmeliet, P.,Guillou, F., Sharpe, R.M., Verhoeven, G., 2004. A sertoli cell-selective knockout ofthe androgen receptor causes spermatogenic arrest in meiosis. Proc. Natl. Acad.Sci. U.S.A. 101, 1327–1332.
Dejager, S., Bry-Gauillard, H., Bruckert, E., Eymard, B., Salachas, F., LeGuern, E.,Tardieu, S., Chadarevian, R., Giral, P., Turpin, G., 2002. A comprehensive endo-crine description of Kennedy’s disease revealing androgen insensitivity linkedto CAG repeat length. J. Clin. Endocrinol. Metab. 87, 3893–3901.
Dewhurst, K., Oliver, J.E., McKnight, A.L., 1970. Socio-psychiatric consequences ofHuntington’s disease. Br. J. Psychiatry 116, 255–258.
Diel, P., Baadners, D., Schlupmann, K., Velders, M., Schwarz, J.P., 2008. C2C12myoblastoma cell differentiation and proliferation is stimulated by androgensand associated with a modulation of myostatin and Pax7 expression. J. Mol.Endocrinol. 40, 231–241.
DiStefano, P.S., Friedman, B., Radziejewski, C., Alexander, C., Boland, P., Schick, C.M.,Lindsay, R.M., Wiegand, S.J., 1992. The neurotrophins BDNF NT-3, and NGFdisplay distinct patterns of retrograde axonal transport in peripheral andcentral neurons. Neuron 8, 983–993.
Djousse, L., Knowlton, B., Cupples, L.A., Marder, K., Shoulson, I., Myers, R.H., 2002.Weight loss in early stage of Huntington’s disease. Neurology 59, 1325–1330.
DonCarlos, L.L., Garcia-Ovejero, D., Sarkey, S., Garcia-Segura, L.M., Azcoitia, I., 2003.Androgen receptor immunoreactivity in forebrain axons and dendrites in therat. Endocrinology 144, 3632–3638.
Dragatsis, I., Levine, M.S., Zeitlin, S., 2000. Inactivation of Hdh in the brain and testisresults in progressive neurodegeneration and sterility in mice. Nat. Genet. 26,300–306.
Dube, J.Y., Lesage, R., Tremblay, R.R., 1976. Androgen and estrogen binding in ratskeletal and perineal muscles. Can. J. Biochem. 54, 50–55.
Duyao, M.P., Auerbach, A.B., Ryan, A., Persichetti, F., Barnes, G.T., McNeil, S.M., Ge, P.,Vonsattel, J.P., Gusella, J.F., Joyner, A.L., et al., 1995. Inactivation of the mouseHuntington’s disease gene homolog Hdh. Science 269, 407–410.
Eddy, E.M., 2002. Male germ cell gene expression. Recent Prog. Horm. Res. 57,103–128.
Evert, B.O., Vogt, I.R., Vieira-Saecker, A.M., Ozimek, L., de Vos, R.A., Brunt, E.R.,Klockgether, T., Wullner, U., 2003. Gene expression profiling in ataxin-3 expres-sing cell lines reveals distinct effects of normal and mutant ataxin-3. J. Neu-ropathol. Exp. Neurol. 62, 1006–1018.
Fain, J.N., Del Mar, N.A., Meade, C.A., Reiner, A., Goldowitz, D., 2001. Abnormalities inthe functioning of adipocytes from R6/2 mice that are transgenic for theHuntington’s disease mutation. Hum. Mol. Genet. 10, 145–152.
Farrer, L.A., 1985. Diabetes mellitus in Huntington disease. Clin. Genet. 27,62–67.
Fernandez-Rhodes, L.E., Kokkinis, A.D., White, M.J., Watts, C.A., Auh, S., Jeffries, N.O.,Shrader, J.A., Lehky, T.J., Li, L., Ryder, J.E., Levy, E.W., Solomon, B.I., Harris-Love,M.O., La Pean, A., Schindler, A.B., Chen, C., Di Prospero, N.A., Fischbeck, K.H.,2011. Efficacy and safety of dutasteride in patients with spinal and bulbarmuscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 10,140–147.
Ferrante, R.J., Gutekunst, C.A., Persichetti, F., McNeil, S.M., Kowall, N.W., Gusella, J.F.,MacDonald, M.E., Beal, M.F., Hersch, S.M., 1997. Heterogeneous topographic andcellular distribution of huntingtin expression in the normal human neostria-tum. J. Neurosci. 17, 3052–3063.
Ferrante, R.J., Kowall, N.W., 1987. Tyrosine hydroxylase-like immunoreactivity isdistributed in the matrix compartment of normal human and Huntington’sdisease striatum. Brain Res. 416, 141–146.
Fishman, R.B., Breedlove, S.M., 1992. Local perineal implants of anti-androgen blockmasculinization of the spinal nucleus of the bulbocavernosus. Brain Res. Dev.Brain Res. 70, 283–286.
Fishman, R.B., Chism, L., Firestone, G.L., Breedlove, S.M., 1990. Evidence for androgenreceptors in sexually dimorphic perineal muscles of neonatal male rats absence ofandrogen accumulation by the perineal motoneurons. J. Neurobiol. 21, 694–704.
Folstein, S., Abbott, M.H., Chase, G.A., Jensen, B.A., Folstein, M.F., 1983. The associa-tion of affective disorder with Huntington’s disease in a case series and infamilies. Psychol. Med. 13, 537–542.
Forger, N.G., Roberts, S.L., Wong, V., Breedlove, S.M., 1993. Ciliary neurotrophicfactor maintains motoneurons and their target muscles in developing rats. J.Neurosci. 13, 4720–4726.
Friedman, M.J., Shah, A.G., Fang, Z.H., Ward, E.G., Warren, S.T., Li, S., Li, X.J., 2007.Polyglutamine domain modulates the TBP-TFIIB interaction: implications for itsnormal function and neurodegeneration. Nat. Neurosci. 10, 1519–1528.
Funakoshi, H., Frisen, J., Barbany, G., Timmusk, T., Zachrisson, O., Verge, V.M.,Persson, H., 1993. Differential expression of mRNAs for neurotrophinsand their receptors after axotomy of the sciatic nerve. J. Cell Biol. 123, 455–465.
Fusco, F.R., Chen, Q., Lamoreaux, W.J., Figueredo-Cardenas, G., Jiao, Y., Coffman, J.A.,Surmeier, D.J., Honig, M.G., Carlock, L.R., Reiner, A., 1999. Cellular localization ofhuntingtin in striatal and cortical neurons in rats: lack of correlation withneuronal vulnerability in Huntington’s disease. J. Neurosci. 19, 1189–1202.
Garden, G.A., Libby, R.T., Fu, Y.H., Kinoshita, Y., Huang, J., Possin, D.E., Smith, A.C.,Martinez, R.A., Fine, G.C., Grote, S.K., Ware, C.B., Einum, D.D., Morrison, R.S.,Ptacek, L.J., Sopher, B.L., La Spada, A.R., 2002. Polyglutamine-expanded ataxin-7promotes non-cell-autonomous purkinje cell degeneration and displays pro-teolytic cleavage in ataxic transgenic mice. J. Neurosci. 22, 4897–4905.
Gauthier, L.R., Charrin, B.C., Borrell-Pages, M., Dompierre, J.P., Rangone, H., Corde-lieres, F.P., De Mey, J., MacDonald, M.E., Lessmann, V., Humbert, S., Saudou, F.,

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172168
2004. Huntingtin controls neurotrophic support and survival of neurons byenhancing BDNF vesicular transport along microtubules. Cell 118, 127–138.
Gil, J.M., Rego, A.C., 2008. Mechanisms of neurodegeneration in Huntington’sdisease. Eur. J. Neurosci. 27, 2803–2820.
Ginovart, N., Lundin, A., Farde, L., Halldin, C., Backman, L., Swahn, C.G., Pauli, S.,Sedvall, G., 1997. PET study of the pre- and post-synaptic dopaminergic markersfor the neurodegenerative process in Huntington’s disease. Brain 120 (Pt 3),503–514.
Glass, D., Roubenoff, R., 2010. Recent advances in the biology and therapy of musclewasting. Ann. N. Y. Acad. Sci. 1211, 25–36.
Godin, J.D., Colombo, K., Molina-Calavita, M., Keryer, G., Zala, D., Charrin, B.C.,Dietrich, P., Volvert, M.L., Guillemot, F., Dragatsis, I., Bellaiche, Y., Saudou, F.,Nguyen, L., Humbert, S., 2010. Huntingtin is required for mitotic spindleorientation and mammalian neurogenesis. Neuron 67, 392–406.
Goldenberg, J.N., Bradley, W.G., 1996. Testosterone therapy and the pathogenesis ofKennedy’s disease (X-linked bulbospinal muscular atrophy). J. Neurol. Sci. 135,158–161.
Goodman, A.O., Murgatroyd, P.R., Medina-Gomez, G., Wood, N.I., Finer, N., Vidal-Puig, A.J., Morton, A.J., Barker, R.A., 2008. The metabolic profile of early Hun-tington’s disease – a combined human and transgenic mouse study. Exp. Neurol.210, 691–698.
Gravel, C., Gotz, R., Lorrain, A., Sendtner, M., 1997. Adenoviral gene transfer of ciliaryneurotrophic factor and brain-derived neurotrophic factor leads to long-termsurvival of axotomized motor neurons. Nat. Med. 3, 765–770.
Gu, X., Andre, V.M., Cepeda, C., Li, S.H., Li, X.J., Levine, M.S., Yang, X.W., 2007.Pathological cell-cell interactions are necessary for striatal pathogenesisin a conditional mouse model of Huntington’s disease. Mol. Neurodegener. 2, 8.
Gu, X., Li, C., Wei, W., Lo, V., Gong, S., Li, S.H., Iwasato, T., Itohara, S., Li, X.J., Mody, I.,Heintz, N., Yang, X.W., 2005. Pathological cell-cell interactions elicited by aneuropathogenic form of mutant Huntingtin contribute to cortical pathogene-sis in HD mice. Neuron 46, 433–444.
Guidetti, D., Vescovini, E., Motti, L., Ghidoni, E., Gemignani, F., Marbini, A.,Patrosso, M.C., Ferlini, A., Solime, F., 1996. X-linked bulbar and spinal mus-cular atrophy, or Kennedy disease: clinical, neurophysiological, neuropatho-logical, neuropsychological and molecular study of a large family. J. Neurol.Sci. 135, 140–148.
Guyot, M.C., Hantraye, P., Dolan, R., Palfi, S., Maziere, M., Brouillet, E., 1997.Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-likestriatal lesions in rats chronically treated with 3-nitropropionic acid. Neuro-science 79, 45–56.
Haase, G., Kennel, P., Pettmann, B., Vigne, E., Akli, S., Revah, F., Schmalbruch, H.,Kahn, A., 1997. Gene therapy of murine motor neuron disease using adenoviralvectors for neurotrophic factors. Nat. Med. 3, 429–436.
Haase, G., Pettmann, B., Vigne, E., Castelnau-Ptakhine, L., Schmalbruch, H., Kahn, A.,1998. Adenovirus-mediated transfer of the neurotrophin-3 gene into skeletalmuscle of pmn mice: therapeutic effects and mechanisms of action. J. Neurol.Sci. 160 (Suppl. 1), S97–S105.
Hackam, A.S., Hodgson, J.G., Singaraja, R., Zhang, T., Gan, L., Gutekunst, C.A., Hersch,S.M., Hayden, M.R., 1999. Evidence for both the nucleus and cytoplasm assubcellular sites of pathogenesis in Huntington’s disease in cell culture and intransgenic mice expressing mutant huntingtin. Philos. Trans. R. Soc. Lond. BBiol. Sci. 354, 1047–1055.
Han, I., You, Y., Kordower, J.H., Brady, S.T., Morfini, G.A., 2010. Differential vulnera-bility of neurons in Huntington’s disease: the role of cell type-specific features.J. Neurochem. 113, 1073–1091.
Hansson, O., Petersen, A., Leist, M., Nicotera, P., Castilho, R.F., Brundin, P., 1999.Transgenic mice expressing a Huntington’s disease mutation are resistant toquinolinic acid-induced striatal excitotoxicity. Proc. Natl. Acad. Sci. U.S.A. 96,8727–8732.
Harding, A.E., Thomas, P.K., Baraitser, M., Bradbury, P.G., Morgan-Hughes, J.A.,Ponsford, J.R., 1982. X-linked recessive bulbospinal neuronopathy: a reportof ten cases. J. Neurol. Neurosurg. Psychiatry 45, 1012–1019.
Hartgens, F., Kuipers, H., 2004. Effects of androgenic-anabolic steroids in athletes.Sports Med. 34, 513–554.
Hauser, K.F., MacLusky, N.J., Toran-Allerand, C.D., 1987. Androgen action in fetalmouse spinal cord cultures: metabolic and morphologic aspects. Brain Res. 406,62–72.
Hauser, K.F., Toran-Allerand, C.D., 1989. Androgen increases the number of cells infetal mouse spinal cord cultures: implications for motoneuron survival. BrainRes. 485, 157–164.
Helmlinger, D., Hardy, S., Sasorith, S., Klein, F., Robert, F., Weber, C., Miguet, L., Potier,N., Van-Dorsselaer, A., Wurtz, J.M., Mandel, J.L., Tora, L., Devys, D., 2004. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum.Mol. Genet. 13, 1257–1265.
Henderson, C.E., Phillips, H.S., Pollock, R.A., Davies, A.M., Lemeulle, C., Armanini, M.,Simmons, L., Moffet, B., Vandlen, R.A., Simpson, L.C., et al., 1994. GDNF: a potentsurvival factor for motoneurons present in peripheral nerve and muscle. Science266, 1062–1064.
Ho Kim, S., Thomas, C.A., Andre, V.M., Cummings, D.M., Cepeda, C., Levine, M.S.,Ehrlich, M.E., 2011. Forebrain striatal-specific expression of mutant huntingtinprotein in vivo induces cell-autonomous age-dependent alterations in sensi-tivity to excitotoxicity and mitochondrial function. ASN Neuro 3, e00060,doi:10.1042/AN20110009.
Hodges, A., Strand, A.D., Aragaki, A.K., Kuhn, A., Sengstag, T., Hughes, G., Elliston, L.A.,Hartog, C., Goldstein, D.R., Thu, D., Hollingsworth, Z.R., Collin, F., Synek, B.,Holmans, P.A., Young, A.B., Wexler, N.S., Delorenzi, M., Kooperberg, C., Augood,
S.J., Faull, R.L., Olson, J.M., Jones, L., Luthi-Carter, R., 2006. Regional and cellulargene expression changes in human Huntington’s disease brain. Hum. Mol.Genet. 15, 965–977.
Hodgson, J.G., Agopyan, N., Gutekunst, C.A., Leavitt, B.R., LePiane, F., Singaraja, R.,Smith, D.J., Bissada, N., McCutcheon, K., Nasir, J., Jamot, L., Li, X.J., Stevens, M.E.,Rosemond, E., Roder, J.C., Phillips, A.G., Rubin, E.M., Hersch, S.M., Hayden, M.R.,1999. A YAC mouse model for Huntington’s disease with full-length mutanthuntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration.Neuron 23, 181–192.
Houenou, L.J., Oppenheim, R.W., Li, L., Lo, A.C., Prevette, D., 1996. Regulation ofspinal motoneuron survival by GDNF during development and following injury.Cell Tissue Res. 286, 219–223.
Hughes, R.A., Sendtner, M., Thoenen, H., 1993. Members of several gene familiesinfluence survival of rat motoneurons in vitro and in vivo. J. Neurosci. Res. 36,663–671.
Hunt, M.J., Morton, A.J., 2005. Atypical diabetes associated with inclusion formationin the R6/2 mouse model of Huntington’s disease is not improved by treatmentwith hypoglycaemic agents. Exp. Brain Res. 166, 220–229.
Huot, P., Levesque, M., Parent, A., 2007. The fate of striatal dopaminergic neurons inParkinson’s disease and Huntington’s chorea. Brain 130, 222–232.
Hurlbert, M.S., Zhou, W., Wasmeier, C., Kaddis, F.G., Hutton, J.C., Freed, C.R., 1999.Mice transgenic for an expanded CAG repeat in the Huntington’s disease genedevelop diabetes. Diabetes 48, 649–651.
Ikeda, K., Klinkosz, B., Greene, T., Cedarbaum, J.M., Wong, V., Lindsay, R.M., Mitsu-moto, H., 1995. Effects of brain-derived neurotrophic factor on motor dysfunc-tion in wobbler mouse motor neuron disease. Ann. Neurol. 37, 505–511.
Ilieva, H., Polymenidou, M., Cleveland, D.W., 2009. Non-cell autonomous toxicity inneurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772.
Imbert, G., Saudou, F., Yvert, G., Devys, D., Trottier, Y., Garnier, J.M., Weber, C., Mandel,J.L., Cancel, G., Abbas, N., Durr, A., Didierjean, O., Stevanin, G., Agid, Y., Brice, A.,1996. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with highsensitivity to expanded CAG/glutamine repeats. Nat. Genet. 14, 285–291.
Javaherian, A., Cline, H.T., 2005. Coordinated motor neuron axon growth andneuromuscular synaptogenesis are promoted by CPG15 in vivo. Neuron 45,505–512.
Jin, Y.N., Johnson, G.V., 2010. The interrelationship between mitochondrial dys-function and transcriptional dysregulation in Huntington disease. J. Bioenerg.Biomembr. 42, 199–205.
Johansen, J.A., Breedlove, S.M., Jordan, C.L., 2007. Androgen receptor expression inthe levator ani muscle of male mice. J. Neuroendocrinol. 19, 823–826.
Johansen, J.A., Troxell-Smith, S.M., Yu, Z., Mo, K., Monks, D.A., Lieberman, A.P.,Breedlove, S.M., Jordan, C.L., 2011. Prenatal flutamide enhances survival in amyogenic mouse model of spinal bulbar muscular atrophy. Neurodegener. Dis.8, 25–34.
Johansen, J.A., Yu, Z., Mo, K., Monks, D.A., Lieberman, A.P., Breedlove, S.M., Jordan,C.L., 2009. Recovery of function in a myogenic mouse model of spinal bulbarmuscular atrophy. Neurobiol. Dis. 34, 113–120.
Jordan, C.L., Breedlove, S.M., Arnold, A.P., 1982. Sexual dimorphism and the influ-ence of neonatal androgen in the dorsolateral motor nucleus of the rat lumbarspinal cord. Brain Res. 249, 309–314.
Josefsen, K., Nielsen, M.D., Jorgensen, K.H., Bock, T., Norremolle, A., Sorensen, S.A.,Naver, B., Hasholt, L., 2008. Impaired glucose tolerance in the R6/1 transgenicmouse model of Huntington’s disease. J. Neuroendocrinol. 20, 165–172.
Kadi, F., Charifi, N., Denis, C., Lexell, J., 2004. Satellite cells and myonuclei in youngand elderly women and men. Muscle Nerve 29, 120–127.
Kadi, F., Eriksson, A., Holmner, S., Thornell, L.E., 1999. Effects of anabolic steroids on themuscle cells of strength-trained athletes. Med. Sci. Sports Exerc. 31, 1528–1534.
Kang, J.S., Krauss, R.S., 2010. Muscle stem cells in developmental and regenerativemyogenesis. Curr. Opin. Clin. Nutr. Metab. Care 13, 243–248.
Kassubek, J., Gaus, W., Landwehrmeyer, G.B., 2004. Evidence for more widespreadcerebral pathology in early HD: an MRI-based morphometric analysis. Neurol-ogy 62, 523–524 (author reply 524).
Katsuno, M., Adachi, H., Doyu, M., Minamiyama, M., Sang, C., Kobayashi, Y., Inukai,A., Sobue, G., 2003. Leuprorelin rescues polyglutamine-dependent phenotypesin a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med.9, 768–773.
Katsuno, M., Adachi, H., Kume, A., Li, M., Nakagomi, Y., Niwa, H., Sang, C., Kobayashi,Y., Doyu, M., Sobue, G., 2002. Testosterone reduction prevents phenotypicexpression in a transgenic mouse model of spinal and bulbar muscular atrophy.Neuron 35, 843–854.
Kawaguchi, Y., Okamoto, T., Taniwaki, M., Aizawa, M., Inoue, M., Katayama, S.,Kawakami, H., Nakamura, S., Nishimura, M., Akiguchi, I., et al., 1994. CAGexpansions in a novel gene for Machado-Joseph disease at chromosome14q32.1. Nat. Genet. 8, 221–228.
Kemp, M.Q., Poort, J.L., Baqri, R.M., Lieberman, A.P., Breedlove, S.M., Miller, K.E.,Jordan, C.L., 2011. Impaired motoneuronal retrograde transport in two mod-els of SBMA implicates two sites of androgen action. Hum. Mol. Genet. 20,4475–4490.
Kennedy, W.R., Alter, M., Sung, J.H., 1968. Progressive proximal spinal and bulbarmuscular atrophy of late onset. A sex-linked recessive trait. Neurology 18,671–680.
Kerr, J.E., Allore, R.J., Beck, S.G., Handa, R.J., 1995. Distribution and hormonalregulation of androgen receptor (AR) and AR messenger ribonucleic acid inthe rat hippocampus. Endocrinology 136, 3213–3221.
Kihira, T., Yoshida, S., Yoshimasu, F., Wakayama, I., Yase, Y., 1997. Involvement ofOnuf’s nucleus in amyotrophic lateral sclerosis. J. Neurol. Sci. 147, 81–88.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 169
Kimura, N., Mizokami, A., Oonuma, T., Sasano, H., Nagura, H., 1993. Immunocyto-chemical localization of androgen receptor with polyclonal antibody in paraf-fin-embedded human tissues. J. Histochem. Cytochem. 41, 671–678.
Kinirons, P., Rouleau, G.A., 2008. Administration of testosterone results in reversibledeterioration in Kennedy’s disease. J. Neurol. Neurosurg. Psychiatry 79, 106–107.
Kishino, A., Ishige, Y., Tatsuno, T., Nakayama, C., Noguchi, H., 1997. BDNF preventsand reverses adult rat motor neuron degeneration and induces axonal out-growth. Exp. Neurol. 144, 273–286.
Klement, I.A., Skinner, P.J., Kaytor, M.D., Yi, H., Hersch, S.M., Clark, H.B., Zoghbi, H.Y.,Orr, H.T., 1998. Ataxin-1 nuclear localization and aggregation: role in poly-glutamine-induced disease in SCA1 transgenic mice. Cell 95, 41–53.
Knight, S.P., Richardson, M.M., Osmand, A.P., Stakkestad, A., Potter, N.T., 1997.Expression and distribution of the dentatorubral-pallidoluysian atrophy geneproduct (atrophin-1/drplap) in neuronal and non-neuronal tissues. J. Neurol.Sci. 146, 19–26.
Kovacheva, E.L., Hikim, A.P., Shen, R., Sinha, I., Sinha-Hikim, I., 2010. Testosteronesupplementation reverses sarcopenia in aging through regulation of myostatin,c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology151, 628–638.
Kremer, H.P., Roos, R.A., 1992. Weight loss in Huntington’s disease. Arch. Neurol.49, 349.
Kremer, H.P., Roos, R.A., Dingjan, G., Marani, E., Bots, G.T., 1990. Atrophy of thehypothalamic lateral tuberal nucleus in Huntington’s disease. J. Neuropathol.Exp. Neurol. 49, 371–382.
Kremer, H.P., Roos, R.A., Dingjan, G.M., Bots, G.T., Bruyn, G.W., Hofman, M.A., 1991.The hypothalamic lateral tuberal nucleus and the characteristics of neuronalloss in Huntington’s disease. Neurosci. Lett. 132, 101–104.
La Spada, A.R., Wilson, E.M., Lubahn, D.B., Harding, A.E., Fischbeck, K.H., 1991.Androgen receptor gene mutations in X-linked spinal and bulbar muscularatrophy. Nature 352, 77–79.
Lalic, N.M., Dragasevic, N., Stefanova, E., Jotic, A., Lalic, K., Milicic, T., Petrovic, I.,Macesic, M., Kostic, V.S., 2010. Impaired insulin sensitivity and secretion innormoglycemic patients with spinocerebellar ataxia type 1. Mov. Disord. 25,1976–1980.
Lalic, N.M., Maric, J., Svetel, M., Jotic, A., Stefanova, E., Lalic, K., Dragasevic, N., Milicic,T., Lukic, L., Kostic, V.S., 2008. Glucose homeostasis in Huntington disease:abnormalities in insulin sensitivity and early-phase insulin secretion. Arch.Neurol. 65, 476–480.
Lambrechts, D., Carmeliet, P., 2006. VEGF at the neurovascular interface: therapeuticimplications for motor neuron disease. Biochim. Biophys. Acta 1762, 1109–1121.
Lambrechts, D., Storkebaum, E., Morimoto, M., Del-Favero, J., Desmet, F., Marklund,S.L., Wyns, S., Thijs, V., Andersson, J., van Marion, I., Al-Chalabi, A., Bornes, S.,Musson, R., Hansen, V., Beckman, L., Adolfsson, R., Pall, H.S., Prats, H., Vermeire,S., Rutgeerts, P., Katayama, S., Awata, T., Leigh, N., Lang-Lazdunski, L.,Dewerchin, M., Shaw, C., Moons, L., Vlietinck, R., Morrison, K.E., Robberecht,W., Van Broeckhoven, C., Collen, D., Andersen, P.M., Carmeliet, P., 2003. VEGF is amodifier of amyotrophic lateral sclerosis in mice and humans and protectsmotoneurons against ischemic death. Nat. Genet. 34, 383–394.
Lee, D.K., 2002. Androgen receptor enhances myogenin expression and acceleratesdifferentiation. Biochem. Biophys. Res. Commun. 294, 408–413.
Li, S.H., Schilling, G., Young 3rd, W.S., Li, X.J., Margolis, R.L., Stine, O.C., Wagster, M.V.,Abbott, M.H., Franz, M.L., Ranen, N.G., et al., 1993. Huntington’s disease gene(IT15) is widely expressed in human and rat tissues. Neuron 11, 985–993.
Li, X.J., Li, S.H., Sharp, A.H., Nucifora Jr., F.C., Schilling, G., Lanahan, A., Worley, P.,Snyder, S.H., Ross, C.A., 1995. A huntingtin-associated protein enriched in brainwith implications for pathology. Nature 378, 398–402.
Lieberman, A.P., Harmison, G., Strand, A.D., Olson, J.M., Fischbeck, K.H., 2002. Alteredtranscriptional regulation in cells expressing the expanded polyglutamineandrogen receptor. Hum. Mol. Genet. 11, 1967–1976.
Lievens, J.C., Woodman, B., Mahal, A., Spasic-Boscovic, O., Samuel, D., Kerkerian-LeGoff, L., Bates, G.P., 2001. Impaired glutamate uptake in the R6 Huntington’sdisease transgenic mice. Neurobiol. Dis. 8, 807–821.
Lim, J., Crespo-Barreto, J., Jafar-Nejad, P., Bowman, A.B., Richman, R., Hill, D.E., Orr,H.T., Zoghbi, H.Y., 2008. Opposing effects of polyglutamine expansion on nativeprotein complexes contribute to SCA1. Nature 452, 713–718.
Lin, J., Handschin, C., Spiegelman, B.M., 2005. Metabolic control through the PGC-1family of transcription coactivators. Cell Metab. 1, 361–370.
Lobsiger, C.S., Cleveland, D.W., 2007. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 10, 1355–1360.
Lodi, R., Schapira, A.H., Manners, D., Styles, P., Wood, N.W., Taylor, D.J., Warner, T.T.,2000. Abnormal in vivo skeletal muscle energy metabolism in Huntington’sdisease and dentatorubropallidoluysian atrophy. Ann. Neurol. 48, 72–76.
Luo, D., Renault, V.M., Rando, T.A., 2005. The regulation of Notch signaling in musclestem cell activation and postnatal myogenesis. Semin. Cell Dev. Biol. 16, 612–622.
Luthi-Carter, R., Hanson, S.A., Strand, A.D., Bergstrom, D.A., Chun, W., Peters, N.L.,Woods, A.M., Chan, E.Y., Kooperberg, C., Krainc, D., Young, A.B., Tapscott, S.J.,Olson, J.M., 2002. Dysregulation of gene expression in the R6/2 model ofpolyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Genet.11, 1911–1926.
Ma, T.C., Buescher, J.L., Oatis, B., Funk, J.A., Nash, A.J., Carrier, R.L., Hoyt, K.R., 2007.Metformin therapy in a transgenic mouse model of Huntington’s disease.Neurosci. Lett. 411, 98–103.
Macdonald, M.E., Ambrose, C.M., Duyao, M.P., Myers, R.H., Lin, C., Srinidhi, L., Barnes,G., Taylor, S.A., James, M., Groot, N., Macfarlane, H., Jenkins, B., Anderson, M.A.,Wexler, N.S., Gusella, J.F., Bates, G.P., Baxendale, S., Hummerich, H., Kirby, S.,North, M., Youngman, S., Mott, R., Zehetner, G., Sedlacek, Z., Poustka, A., Frischauf,
A.M., Lehrach, H., Buckler, A.J., Church, D., Doucettestamm, L., Odonovan, M.C.,Ribaramirez, L., Shah, M., Stanton, V.P., Strobel, S.A., Draths, K.M., Wales, J.L.,Dervan, P., Housman, D.E., Altherr, M., Shiang, R., Thompson, L., Fielder, T.,Wasmuth, J.J., Tagle, D., Valdes, J., Elmer, L., Allard, M., Castilla, L., Swaroop,M., Blanchard, K., Collins, F.S., Snell, R., Holloway, T., Gillespie, K., Datson, N.,Shaw, D., Harper, P.S., 1993. A novel gene containing a trinucleotide repeat that isexpanded and unstable on Huntingtons-disease chromosomes. Cell 72, 971–983.
Malik, B., Nirmalananthan, N., Bilsland, L.G., La Spada, A.R., Hanna, M.G., Schiavo, G.,Gallo, J.M., Greensmith, L., 2011. Absence of disturbed axonal transport in spinaland bulbar muscular atrophy. Hum. Mol. Genet. 20, 1776–1786.
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C.,Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W., Bates, G.P., 1996. Exon 1 of theHD gene with an expanded CAG repeat is sufficient to cause a progressiveneurological phenotype in transgenic mice. Cell 87, 493–506.
Mariotti, C., Castellotti, B., Pareyson, D., Testa, D., Eoli, M., Antozzi, C., Silani, V.,Marconi, R., Tezzon, F., Siciliano, G., Marchini, C., Gellera, C., Donato, S.D., 2000.Phenotypic manifestations associated with CAG-repeat expansion in the an-drogen receptor gene in male patients and heterozygous females: a clinical andmolecular study of 30 families. Neuromuscul. Disord. 10, 391–397.
Markianos, M., Panas, M., Kalfakis, N., Vassilopoulos, D., 2005. Plasma testosteronein male patients with Huntington’s disease: relations to severity of illness anddementia. Ann. Neurol. 57, 520–525.
Marron, T.U., Guerini, V., Rusmini, P., Sau, D., Brevini, T.A., Martini, L., Poletti, A.,2005. Androgen-induced neurite outgrowth is mediated by neuritin in motorneurones. J. Neurochem. 92, 10–20.
Martin, B., Golden, E., Carlson, O.D., Pistell, P., Zhou, J., Kim, W., Frank, B.P., Thomas,S., Chadwick, W.A., Greig, N.H., Bates, G.P., Sathasivam, K., Bernier, M., Maudsley,S., Mattson, M.P., Egan, J.M., 2009. Exendin-4 improves glycemic control,ameliorates brain and pancreatic pathologies, and extends survival in a mousemodel of Huntington’s disease. Diabetes 58, 318–328.
Marzetti, E., Lawler, J.M., Hiona, A., Manini, T., Seo, A.Y., Leeuwenburgh, C., 2008.Modulation of age-induced apoptotic signaling and cellular remodeling byexercise and calorie restriction in skeletal muscle. Free Radic. Biol. Med. 44,160–168.
Marzetti, E., Leeuwenburgh, C., 2006. Skeletal muscle apoptosis, sarcopenia andfrailty at old age. Exp. Gerontol. 41, 1234–1238.
Mauras, N., Hayes, V., Welch, S., Rini, A., Helgeson, K., Dokler, M., Veldhuis, J.D.,Urban, R.J., 1998. Testosterone deficiency in young men: marked alterations inwhole body protein kinetics, strength, and adiposity. J. Clin. Endocrinol. Metab.83, 1886–1892.
Mauro, A., 1961. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9,493–495.
McGeer, E.G., McGeer, P.L., 1976. Duplication of biochemical changes of Hunting-ton’s chorea by intrastriatal injections of glutamic and kainic acids. Nature 263,517–519.
McMahon, S.J., Pray-Grant, M.G., Schieltz, D., Yates 3rd, J.R., Grant, P.A., 2005.Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SA-GA and SLIK histone acetyltransferase activity. Proc. Natl. Acad. Sci. U.S.A. 102,8478–8482.
Mendler, L., Baka, Z., Kovacs-Simon, A., Dux, L., 2007. Androgens negatively regulatemyostatin expression in an androgen-dependent skeletal muscle. Biochem.Biophys. Res. Commun. 361, 237–242.
Mihm, M.J., Amann, D.M., Schanbacher, B.L., Altschuld, R.A., Bauer, J.A., Hoyt, K.R.,2007. Cardiac dysfunction in the R6/2 mouse model of Huntington’s disease.Neurobiol. Dis. 25, 297–308.
Miller, B.R., Dorner, J.L., Shou, M., Sari, Y., Barton, S.J., Sengelaub, D.R., Kennedy, R.T.,Rebec, G.V., 2008. Up-regulation of GLT1 expression increases glutamate uptakeand attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuro-science 153, 329–337.
Milner, T.A., Hernandez, F.J., Herrick, S.P., Pierce, J.P., Iadecola, C., Drake, C.T., 2007.Cellular and subcellular localization of androgen receptor immunoreactivityrelative to C1 adrenergic neurons in the rostral ventrolateral medulla of maleand female rats. Synapse 61, 268–278.
Mitsumoto, H., Ikeda, K., Holmlund, T., Greene, T., Cedarbaum, J.M., Wong, V.,Lindsay, R.M., 1994. The effects of ciliary neurotrophic factor on motor dys-function in wobbler mouse motor neuron disease. Ann. Neurol. 36, 142–148.
Mitsumoto, H., Ikeda, K., Klinkosz, B., Cedarbaum, J.M., Wong, V., Lindsay, R.M.,1994. Arrest of motor neuron disease in wobbler mice cotreated with CNTF andBDNF. Science 265, 1107–1110.
Mo, K., Razak, Z., Rao, P., Yu, Z., Adachi, H., Katsuno, M., Sobue, G., Lieberman, A.P.,Westwood, J.T., Monks, D.A., 2010. Microarray analysis of gene expression byskeletal muscle of three mouse models of Kennedy disease/spinal bulbarmuscular atrophy. PLoS One 5, e12922.
Moller, T., 2010. Neuroinflammation in Huntington’s disease. J. Neural. Transm. 117,1001–1008.
Monks, D.A., Johansen, J.A., Mo, K., Rao, P., Eagleson, B., Yu, Z., Lieberman, A.P.,Breedlove, S.M., Jordan, C.L., 2007. Overexpression of wild-type androgenreceptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci.U.S.A. 104, 18259–18264.
Monks, D.A., O’Bryant, E.L., Jordan, C.L., 2004. Androgen receptor immunoreactivityin skeletal muscle: enrichment at the neuromuscular junction. J. Comp. Neurol.473, 59–72.
Montie, H.L., Cho, M.S., Holder, L., Liu, Y., Tsvetkov, A.S., Finkbeiner, S., Merry, D.E.,2009. Cytoplasmic retention of polyglutamine-expanded androgen receptorameliorates disease via autophagy in a mouse model of spinal and bulbarmuscular atrophy. Hum. Mol. Genet. 18, 1937–1950.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172170
Morfini, G., Pigino, G., Szebenyi, G., You, Y., Pollema, S., Brady, S.T., 2006. JNKmediates pathogenic effects of polyglutamine-expanded androgen receptoron fast axonal transport. Nat. Neurosci. 9, 907–916.
Morton, A.J., Leavens, W., 2000. Mice transgenic for the human Huntington’sdisease mutation have reduced sensitivity to kainic acid toxicity. Brain Res.Bull. 52, 51–59.
Moss, F.P., Leblond, C.P., 1971. Satellite cells as the source of nuclei in muscles ofgrowing rats. Anat. Rec. 170, 421–435.
Musaro, A., Dobrowolny, G., Rosenthal, N., 2007. The neuroprotective effects of alocally acting IGF-1 isoform. Exp. Gerontol. 42, 76–80.
Musaro, A., McCullagh, K., Paul, A., Houghton, L., Dobrowolny, G., Molinaro, M.,Barton, E.R., Sweeney, H.L., Rosenthal, N., 2001. Localized Igf-1 transgeneexpression sustains hypertrophy and regeneration in senescent skeletal muscle.Nat. Genet. 27, 195–200.
Nakamura, K., Jeong, S.Y., Uchihara, T., Anno, M., Nagashima, K., Nagashima, T.,Ikeda, S., Tsuji, S., Kanazawa, I., 2001. SCA17, a novel autosomal dominantcerebellar ataxia caused by an expanded polyglutamine in TATA-binding pro-tein. Hum. Mol. Genet. 10, 1441–1448.
Nasir, J., Floresco, S.B., O’Kusky, J.R., Diewert, V.M., Richman, J.M., Zeisler, J.,Borowski, A., Marth, J.D., Phillips, A.G., Hayden, M.R., 1995. Targeted disruptionof the Huntington’s disease gene results in embryonic lethality and behavioraland morphological changes in heterozygotes. Cell 81, 811–823.
Nedelsky, N.B., Pennuto, M., Smith, R.B., Palazzolo, I., Moore, J., Nie, Z., Neale, G.,Taylor, J.P., 2010. Native functions of the androgen receptor are essential topathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron67, 936–952.
Neff, N.T., Prevette, D., Houenou, L.J., Lewis, M.E., Glicksman, M.A., Yin, Q.W.,Oppenheim, R.W., 1993. Insulin-like growth factors: putative muscle-derivedtrophic agents that promote motoneuron survival. J. Neurobiol. 24, 1578–1588.
Neuschmid-Kaspar, F., Gast, A., Peterziel, H., Schneikert, J., Muigg, A., Ransmayr, G.,Klocker, H., Bartsch, G., Cato, A.C., 1996. CAG-repeat expansion in androgenreceptor in Kennedy’s disease is not a loss of function mutation. Mol. Cell.Endocrinol. 117, 149–156.
Nordeen, E.J., Nordeen, K.W., Sengelaub, D.R., Arnold, A.P., 1985. Androgens preventnormally occurring cell death in a sexually dimorphic spinal nucleus. Science229, 671–673.
Okamoto, K., Hirai, S., Ishiguro, K., Kawarabayashi, T., Takatama, M., 1991. Lightand electron microscopic and immunohistochemical observations of theOnuf’s nucleus of amyotrophic lateral sclerosis. Acta Neuropathol. 81, 610–614.
Onodera, O., Oyake, M., Takano, H., Ikeuchi, T., Igarashi, S., Tsuji, S., 1995. Molecularcloning of a full-length cDNA for dentatorubral-pallidoluysian atrophy andregional expressions of the expanded alleles in the CNS. Am. J. Hum. Genet. 57,1050–1060.
Oosthuyse, B., Moons, L., Storkebaum, E., Beck, H., Nuyens, D., Brusselmans, K., VanDorpe, J., Hellings, P., Gorselink, M., Heymans, S., Theilmeier, G., Dewerchin, M.,Laudenbach, V., Vermylen, P., Raat, H., Acker, T., Vleminckx, V., Van Den Bosch,L., Cashman, N., Fujisawa, H., Drost, M.R., Sciot, R., Bruyninckx, F., Hicklin, D.J.,Ince, C., Gressens, P., Lupu, F., Plate, K.H., Robberecht, W., Herbert, J.M., Collen,D., Carmeliet, P., 2001. Deletion of the hypoxia-response element in the vascularendothelial growth factor promoter causes motor neuron degeneration. Nat.Genet. 28, 131–138.
Ophoff, J., Van Proeyen, K., Callewaert, F., De Gendt, K., De Bock, K., Vanden Bosch, A.,Verhoeven, G., Hespel, P., Vanderschueren, D., 2009. Androgen signaling inmyocytes contributes to the maintenance of muscle mass and fiber type regula-tion but not to muscle strength or fatigue. Endocrinology 150, 3558–3566.
Oppenheim, R.W., Houenou, L.J., Johnson, J.E., Lin, L.F., Li, L., Lo, A.C., Newsome, A.L.,Prevette, D.M., Wang, S., 1995. Developing motor neurons rescued fromprogrammed and axotomy-induced cell death by GDNF. Nature 373, 344–346.
Orr, H.T., Chung, M.Y., Banfi, S., Kwiatkowski Jr., T.J., Servadio, A., Beaudet, A.L.,McCall, A.E., Duvick, L.A., Ranum, L.P., Zoghbi, H.Y., 1993. Expansion of anunstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet.4, 221–226.
Orth, M., Cooper, J.M., Bates, G.P., Schapira, A.H., 2003. Inclusion formation inHuntington’s disease R6/2 mouse muscle cultures. J. Neurochem. 87, 1–6.
Osborne, M.C., Verhovshek, T., Sengelaub, D.R., 2007. Androgen regulates trkBimmunolabeling in spinal motoneurons. J. Neurosci. Res. 85, 303–309.
Ottem, E.N., Beck, L.A., Jordan, C.L., Breedlove, S.M., 2007. Androgen-dependentregulation of brain-derived neurotrophic factor and tyrosine kinase B in thesexually dimorphic spinal nucleus of the bulbocavernosus. Endocrinology 148,3655–3665.
Oyanagi, K., Takeda, S., Takahashi, H., Ohama, E., Ikuta, F., 1989. A quantitativeinvestigation of the substantia nigra in Huntington’s disease. Ann. Neurol. 26,13–19.
Palazzolo, I., Burnett, B.G., Young, J.E., Brenne, P.L., La Spada, A.R., Fischbeck, K.H.,Howell, B.W., Pennuto, M., 2007. Akt blocks ligand binding and protects againstexpanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet. 16,1593–1603.
Palazzolo, I., Nedelsky, N.B., Askew, C.E., Harmison, G.G., Kasantsev, A.G., Taylor, J.P.,Fischbeck, K.H., Pennuto, M., 2010. B2 attenuates polyglutamine-expandedandrogen receptor toxicity in cell and fly models of spinal and bulbar muscularatrophy. J. Neurosci. Res. 88, 2207–2216.
Palazzolo, I., Stack, C., Kong, L., Musaro, A., Adachi, H., Katsuno, M., Sobue, G., Taylor,J.P., Sumner, C.J., Fischbeck, K.H., Pennuto, M., 2009. Overexpression of IGF-1 in
muscle attenuates disease in a mouse model of spinal and bulbar muscularatrophy. Neuron 63, 316–328.
Palhan, V.B., Chen, S., Peng, G.H., Tjernberg, A., Gamper, A.M., Fan, Y., Chait, B.T., LaSpada, A.R., Roeder, R.G., 2005. Polyglutamine-expanded ataxin-7 inhibitsSTAGA histone acetyltransferase activity to produce retinal degeneration. Proc.Natl. Acad. Sci. U.S.A. 102, 8472–8477.
Pandey, U.B., Nie, Z., Batlevi, Y., McCray, B.A., Ritson, G.P., Nedelsky, N.B., Schwartz,S.L., DiProspero, N.A., Knight, M.A., Schuldiner, O., Padmanabhan, R., Hild, M.,Berry, D.L., Garza, D., Hubbert, C.C., Yao, T.P., Baehrecke, E.H., Taylor, J.P., 2007.HDAC6 rescues neurodegeneration and provides an essential link betweenautophagy and the UPS. Nature 447, 859–863.
Papalexi, E., Persson, A., Bjorkqvist, M., Petersen, A., Woodman, B., Bates, G.P.,Sundler, F., Mulder, H., Brundin, P., Popovic, N., 2005. Reduction of GnRHand infertility in the R6/2 mouse model of Huntington’s disease. Eur. J. Neurosci.22, 1541–1546.
Paradisi, I., Hernandez, A., Arias, S., 2008. Huntington disease mutation in Vene-zuela: age of onset, haplotype analyses and geographic aggregation. J. Hum.Genet. 53, 127–135.
Pattison, J.S., Sanbe, A., Maloyan, A., Osinska, H., Klevitsky, R., Robbins, J., 2008.Cardiomyocyte expression of a polyglutamine preamyloid oligomer causesheart failure. Circulation 117, 2743–2751.
Paulson, H.L., Das, S.S., Crino, P.B., Perez, M.K., Patel, S.C., Gotsdiner, D., Fischbeck,K.H., Pittman, R.N., 1997. Machado-Joseph disease gene product is a cyto-plasmic protein widely expressed in brain. Ann. Neurol. 41, 453–462.
Pennuto, M., Palazzolo, I., Poletti, A., 2009. Post-translational modifications ofexpanded polyglutamine proteins: impact on neurotoxicity. Hum. Mol. Genet.18, R40–R47.
Peters, M.F., Nucifora Jr., F.C., Kushi, J., Seaman, H.C., Cooper, J.K., Herring, W.J.,Dawson, V.L., Dawson, T.M., Ross, C.A., 1999. Nuclear targeting of mutantHuntingtin increases toxicity. Mol. Cell. Neurosci. 14, 121–128.
Petersen, A., Bjorkqvist, M., 2006. Hypothalamic-endocrine aspects in Huntington’sdisease. Eur. J. Neurosci. 24, 961–967.
Petersen, A., Gil, J., Maat-Schieman, M.L., Bjorkqvist, M., Tanila, H., Araujo, I.M.,Smith, R., Popovic, N., Wierup, N., Norlen, P., Li, J.Y., Roos, R.A., Sundler, F.,Mulder, H., Brundin, P., 2005. Orexin loss in Huntington’s disease. Hum. Mol.Genet. 14, 39–47.
Phan, J., Hickey, M.A., Zhang, P., Chesselet, M.F., Reue, K., 2009. Adipose tissuedysfunction tracks disease progression in two Huntington’s disease mousemodels. Hum. Mol. Genet. 18, 1006–1016.
Podolsky, S., Leopold, N.A., 1977. Abnormal glucose tolerance and arginine tolerancetests in Huntington’s disease. Gerontology 23, 55–63.
Podolsky, S., Leopold, N.A., Sax, D.S., 1972. Increased frequency of diabetes mellitusin patients with Huntington’s chorea. Lancet 1, 1356–1358.
Poletti, A., 2004. The polyglutamine tract of androgen receptor: from functions todysfunctions in motor neurons. Front. Neuroendocrinol. 25, 1–26.
Popovic, V., Svetel, M., Djurovic, M., Petrovic, S., Doknic, M., Pekic, S., Miljic, D., Milic,N., Glodic, J., Dieguez, C., Casanueva, F.F., Kostic, V., 2004. Circulating andcerebrospinal fluid ghrelin and leptin: potential role in altered body weightin Huntington’s disease. Eur. J. Endocrinol. 151, 451–455.
Rao, A.K., Muratori, L., Louis, E.D., Moskowitz, C.B., Marder, K.S., 2008. Spectrum ofgait impairments in presymptomatic and symptomatic Huntington’s disease.Mov. Disord. 23, 1100–1107.
Ribchester, R.R., Thomson, D., Wood, N.I., Hinks, T., Gillingwater, T.H., Wishart, T.M.,Court, F.A., Morton, A.J., 2004. Progressive abnormalities in skeletal muscle andneuromuscular junctions of transgenic mice expressing the Huntington’s dis-ease mutation. Eur. J. Neurosci. 20, 3092–3114.
Rommel, C., Bodine, S.C., Clarke, B.A., Rossman, R., Nunez, L., Stitt, T.N., Yanco-poulos, G.D., Glass, D.J., 2001. Mediation of IGF-1-induced skeletal myotubehypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. CellBiol. 3, 1009–1013.
Roselli, C.E., Handa, R.J., Resko, J.A., 1989. Quantitative distribution of nuclearandrogen receptors in microdissected areas of the rat brain. Neuroendocrinolo-gy 49, 449–453.
Runne, H., Regulier, E., Kuhn, A., Zala, D., Gokce, O., Perrin, V., Sick, B., Aebischer, P.,Deglon, N., Luthi-Carter, R., 2008. Dysregulation of gene expression in primaryneuron models of Huntington’s disease shows that polyglutamine-relatedeffects on the striatal transcriptome may not be dependent on brain circuitry.J. Neurosci. 28, 9723–9731.
Rusmini, P., Bolzoni, E., Crippa, V., Onesto, E., Sau, D., Galbiati, M., Piccolella, M.,Poletti, A., 2010. Proteasomal and autophagic degradative activities in spinaland bulbar muscular atrophy. Neurobiol. Dis. 40, 361–369.
Sakuma, K., Yamaguchi, A., 2010. Molecular mechanisms in aging and currentstrategies to counteract sarcopenia. Curr. Aging Sci. 3, 90–101.
Salazar-Grueso, E.F., Kim, S., Kim, H., 1991. Embryonic mouse spinal cord motorneuron hybrid cells. Neuroreport 2, 505–508.
Sanberg, P.R., Fibiger, H.C., Mark, R.F., 1981. Body weight and dietary factors inHuntington’s disease patients compared with matched controls. Med. J. Aust. 1,407–409.
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., Walsh, K.,Schiaffino, S., Lecker, S.H., Goldberg, A.L., 2004. Foxo transcription factors inducethe atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atro-phy. Cell 117, 399–412.
Sapp, E., Kegel, K.B., Aronin, N., Hashikawa, T., Uchiyama, Y., Tohyama, K., Bhide, P.G.,Vonsattel, J.P., DiFiglia, M., 2001. Early and progressive accumulation of reactivemicroglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 60, 161–172.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172 171
Saudou, F., Finkbeiner, S., Devys, D., Greenberg, M.E., 1998. Huntingtin acts in thenucleus to induce apoptosis but death does not correlate with the formation ofintranuclear inclusions. Cell 95, 55–66.
Schmidt, B.J., Greenberg, C.R., Allingham-Hawkins, D.J., Spriggs, E.L., 2002. Expres-sion of X-linked bulbospinal muscular atrophy (Kennedy disease) in twohomozygous women. Neurology 59, 770–772.
Schmidt, T., Landwehrmeyer, G.B., Schmitt, I., Trottier, Y., Auburger, G., Laccone, F.,Klockgether, T., Volpel, M., Epplen, J.T., Schols, L., Riess, O., 1998. An isoform ofataxin-3 accumulates in the nucleus of neuronal cells in affected brain regionsof SCA3 patients. Brain Pathol. 8, 669–679.
Sendtner, M., Holtmann, B., Kolbeck, R., Thoenen, H., Barde, Y.A., 1992. Brain-derivedneurotrophic factor prevents the death of motoneurons in newborn rats afternerve section. Nature 360, 757–759.
Sengelaub, D.R., Forger, N.G., 2008. The spinal nucleus of the bulbocavernosus: firstsin androgen-dependent neural sex differences. Horm. Behav. 53, 596–612.
Servadio, A., Koshy, B., Armstrong, D., Antalffy, B., Orr, H.T., Zoghbi, H.Y., 1995.Expression analysis of the ataxin-1 protein in tissues from normal and spino-cerebellar ataxia type 1 individuals. Nat. Genet. 10, 94–98.
Sharpe, R.M., Fraser, H.M., Ratnasooriya, W.D., 1988. Assessment of the role ofLeydig cell products other than testosterone in spermatogenesis and fertility inadult rats. Int. J. Androl. 11, 507–523.
Shin, J.Y., Fang, Z.H., Yu, Z.X., Wang, C.E., Li, S.H., Li, X.J., 2005. Expression of mutanthuntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 171,1001–1012.
Simerly, R.B., Chang, C., Muramatsu, M., Swanson, L.W., 1990. Distribution ofandrogen and estrogen receptor mRNA-containing cells in the rat brain: anin situ hybridization study. J. Comp. Neurol. 294, 76–95.
Simmons, D.A., Casale, M., Alcon, B., Pham, N., Narayan, N., Lynch, G., 2007. Ferritinaccumulation in dystrophic microglia is an early event in the development ofHuntington’s disease. Glia 55, 1074–1084.
Sinha-Hikim, I., Artaza, J., Woodhouse, L., Gonzalez-Cadavid, N., Singh, A.B., Lee, M.I.,Storer, T.W., Casaburi, R., Shen, R., Bhasin, S., 2002. Testosterone-inducedincrease in muscle size in healthy young men is associated with muscle fiberhypertrophy. Am. J. Physiol. Endocrinol. Metab. 283, E154–E164.
Sinha-Hikim, I., Cornford, M., Gaytan, H., Lee, M.L., Bhasin, S., 2006. Effects oftestosterone supplementation on skeletal muscle fiber hypertrophy and satel-lite cells in community-dwelling older men. J. Clin. Endocrinol. Metab. 91,3024–3033.
Sinha-Hikim, I., Roth, S.M., Lee, M.I., Bhasin, S., 2003. Testosterone-induced musclehypertrophy is associated with an increase in satellite cell number in healthy,young men. Am. J. Physiol. Endocrinol. Metab. 285, E197–E205.
Sinha-Hikim, I., Taylor, W.E., Gonzalez-Cadavid, N.F., Zheng, W., Bhasin, S., 2004.Androgen receptor in human skeletal muscle and cultured muscle satellitecells: up-regulation by androgen treatment. J. Clin. Endocrinol. Metab. 89,5245–5255.
Snell, R.G., MacMillan, J.C., Cheadle, J.P., Fenton, I., Lazarou, L.P., Davies, P., MacDo-nald, M.E., Gusella, J.F., Harper, P.S., Shaw, D.J., 1993. Relationship betweentrinucleotide repeat expansion and phenotypic variation in Huntington’s dis-ease. Nat. Genet. 4, 393–397.
Sobue, G., 1995. X-linked recessive bulbospinal neuronopathy (SBMA). Nagoya J.Med. Sci. 58, 95–106.
Sopher, B.L., Thomas Jr., P.S., LaFevre-Bernt, M.A., Holm, I.E., Wilke, S.A., Ware, C.B.,Jin, L.W., Libby, R.T., Ellerby, L.M., La Spada, A.R., 2004. Androgen receptor YACtransgenic mice recapitulate SBMA motor neuronopathy and implicateVEGF164 in the motor neuron degeneration. Neuron 41, 687–699.
Soraru, G., D’Ascenzo, C., Polo, A., Palmieri, A., Baggio, L., Vergani, L., Gellera, C.,Moretto, G., Pegoraro, E., Angelini, C., 2008. Spinal and bulbar muscular atrophy:skeletal muscle pathology in male patients and heterozygous females. J. Neurol.Sci. 264, 100–105.
Sorenson, E.J., Klein, C.J., 2007. Elevated creatine kinase and transaminases inasymptomatic SBMA. Amyotroph. Lateral Scler. 8, 62–64.
Stitt, T.N., Drujan, D., Clarke, B.A., Panaro, F., Timofeyva, Y., Kline, W.O., Gonzalez, M.,Yancopoulos, G.D., Glass, D.J., 2004. The IGF-1/PI3K/Akt pathway preventsexpression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXOtranscription factors. Mol. Cell. 14, 395–403.
Storkebaum, E., Lambrechts, D., Dewerchin, M., Moreno-Murciano, M.P., Appel-mans, S., Oh, H., Van Damme, P., Rutten, B., Man, W.Y., De Mol, M., Wyns, S.,Manka, D., Vermeulen, K., Van Den Bosch, L., Mertens, N., Schmitz, C., Robber-echt, W., Conway, E.M., Collen, D., Moons, L., Carmeliet, P., 2005. Treatment ofmotoneuron degeneration by intracerebroventricular delivery of VEGF in a ratmodel of ALS. Nat. Neurosci. 8, 85–92.
Stoy, N., McKay, E., 2000. Weight loss in Huntington’s disease. Ann. Neurol. 48,130–131.
Strand, A.D., Aragaki, A.K., Shaw, D., Bird, T., Holton, J., Turner, C., Tapscott, S.J.,Tabrizi, S.J., Schapira, A.H., Kooperberg, C., Olson, J.M., 2005. Gene expression inHuntington’s disease skeletal muscle: a potential biomarker. Hum. Mol. Genet.14, 1863–1876.
Strong, T.V., Tagle, D.A., Valdes, J.M., Elmer, L.W., Boehm, K., Swaroop, M., Kaatz,K.W., Collins, F.S., Albin, R.L., 1993. Widespread expression of the human andrat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 5,259–265.
Suzuki, M., Desmond, T.J., Albin, R.L., Frey, K.A., 2001. Vesicular neurotransmittertransporters in Huntington’s disease: initial observations and comparison withtraditional synaptic markers. Synapse 41, 329–336.
Szebenyi, G., Morfini, G.A., Babcock, A., Gould, M., Selkoe, K., Stenoien, D.L., Young,M., Faber, P.W., MacDonald, M.E., McPhaul, M.J., Brady, S.T., 2003. Neuropatho-
genic forms of huntingtin and androgen receptor inhibit fast axonal transport.Neuron 40, 41–52.
Tabori, N.E., Stewart, L.S., Znamensky, V., Romeo, R.D., Alves, S.E., McEwen, B.S.,Milner, T.A., 2005. Ultrastructural evidence that androgen receptors arelocated at extranuclear sites in the rat hippocampal formation. Neuroscience130, 151–163.
Tai, Y.F., Pavese, N., Gerhard, A., Tabrizi, S.J., Barker, R.A., Brooks, D.J., Piccini, P., 2007.Microglial activation in presymptomatic Huntington’s disease gene carriers.Brain 130, 1759–1766.
Takeyama, K., Ito, S., Yamamoto, A., Tanimoto, H., Furutani, T., Kanuka, H., Miura,M., Tabata, T., Kato, S., 2002. Androgen-dependent neurodegeneration bypolyglutamine-expanded human androgen receptor in Drosophila. Neuron35, 855–864.
Tan, S.A., Deglon, N., Zurn, A.D., Baetge, E.E., Bamber, B., Kato, A.C., Aebischer, P.,1996. Rescue of motoneurons from axotomy-induced cell death by polymerencapsulated cells genetically engineered to release CNTF. Cell Transplant. 5,577–587.
Tetzlaff, J., Tanzer, L., Jones, K.J., 2007. Cellular localization of androgen and estrogenreceptors in mouse-derived motoneuron hybrid cells and mouse facial moto-neurons. Dev. Neurobiol. 67, 1362–1370.
Thomas, E.A., Coppola, G., Tang, B., Kuhn, A., Kim, S., Geschwind, D.H., Brown, T.B.,Luthi-Carter, R., Ehrlich, M.E., 2011. In vivo cell-autonomous transcriptionalabnormalities revealed in mice expressing mutant huntingtin in striatal but notcortical neurons. Hum. Mol. Genet. 20, 1049–1060.
Thomas Jr., P.S., Fraley, G.S., Damian, V., Woodke, L.B., Zapata, F., Sopher, B.L.,Plymate, S.R., La Spada, A.R., 2006. Loss of endogenous androgen receptorprotein accelerates motor neuron degeneration and accentuates androgeninsensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy.Hum. Mol. Genet. 15, 2225–2238.
Thompson, P.D., Berardelli, A., Rothwell, J.C., Day, B.L., Dick, J.P., Benecke, R.,Marsden, C.D., 1988. The coexistence of bradykinesia and chorea in Hunting-ton’s disease and its implications for theories of basal ganglia control ofmovement. Brain 111 (Pt 2), 223–244.
Timmers, H.J., Swaab, D.F., van de Nes, J.A., Kremer, H.P., 1996. Somatostatin 1-12immunoreactivity is decreased in the hypothalamic lateral tuberal nucleus ofHuntington’s disease patients. Brain Res. 728, 141–148.
Trejo, J.L., Carro, E., Garcia-Galloway, E., Torres-Aleman, I., 2004. Role of insulin-likegrowth factor I signaling in neurodegenerative diseases. J. Mol. Med. 82, 156–162.
Trushina, E., Dyer, R.B., Badger 2nd, J.D., Ure, D., Eide, L., Tran, D.D., Vrieze, B.T.,Legendre-Guillemin, V., McPherson, P.S., Mandavilli, B.S., Van Houten, B., Zeitlin,S., McNiven, M., Aebersold, R., Hayden, M., Parisi, J.E., Seeberg, E., Dragatsis, I.,Doyle, K., Bender, A., Chacko, C., McMurray, C.T., 2004. Mutant huntingtinimpairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol.Cell. Biol. 24, 8195–8209.
Tsai, M.Y., Yeh, S.D., Wang, R.S., Yeh, S., Zhang, C., Lin, H.Y., Tzeng, C.R., Chang, C.,2006. Differential effects of spermatogenesis and fertility in mice lackingandrogen receptor in individual testis cells. Proc. Natl. Acad. Sci. U.S.A. 103,18975–18980.
Van Den Bosch, L., Storkebaum, E., Vleminckx, V., Moons, L., Vanopdenbosch, L.,Scheveneels, W., Carmeliet, P., Robberecht, W., 2004. Effects of vascular endothelialgrowth factor (VEGF) on motor neuron degeneration. Neurobiol. Dis. 17, 21–28.
van der Burg, J.M., Bacos, K., Wood, N.I., Lindqvist, A., Wierup, N., Woodman, B.,Wamsteeker, J.I., Smith, R., Deierborg, T., Kuhar, M.J., Bates, G.P., Mulder, H.,Erlanson-Albertsson, C., Morton, A.J., Brundin, P., Petersen, A., Bjorkqvist, M.,2008. Increased metabolism in the R6/2 mouse model of Huntington’s disease.Neurobiol. Dis. 29, 41–51.
Van Raamsdonk, J.M., Gibson, W.T., Pearson, J., Murphy, Z., Lu, G., Leavitt, B.R.,Hayden, M.R., 2006. Body weight is modulated by levels of full-length hun-tingtin. Hum. Mol. Genet. 15, 1513–1523.
Van Raamsdonk, J.M., Murphy, Z., Selva, D.M., Hamidizadeh, R., Pearson, J., Petersen,A., Bjorkqvist, M., Muir, C., Mackenzie, I.R., Hammond, G.L., Vogl, A.W., Hayden,M.R., Leavitt, B.R., 2007. Testicular degeneration in Huntington disease. Neu-robiol. Dis. 26, 512–520.
Van Raamsdonk, J.M., Warby, S.C., Hayden, M.R., 2007. Selective degeneration inYAC mouse models of Huntington disease. Brain Res. Bull. 72, 124–131.
Verhovshek, T., Cai, Y., Osborne, M.C., Sengelaub, D.R., 2010. Androgen regulatesbrain-derived neurotrophic factor in spinal motoneurons and their targetmusculature. Endocrinology 151, 253–261.
Vonsattel, J.P., DiFiglia, M., 1998. Huntington disease. J. Neuropathol. Exp. Neurol.57, 369–384.
Vornberger, W., Prins, G., Musto, N.A., Suarez-Quian, C.A., 1994. Androgen receptordistribution in rat testis: new implications for androgen regulation of sper-matogenesis. Endocrinology 134, 2307–2316.
Wang, C., Swerdloff, R.S., Iranmanesh, A., Dobs, A., Snyder, P.J., Cunningham, G.,Matsumoto, A.M., Weber, T., Berman, N., 2000. Transdermal testosterone gelimproves sexual function, mood, muscle strength, and body compositionparameters in hypogonadal men. J. Clin. Endocrinol. Metab. 85, 2839–2853.
Ward, O.B., Wexler, A.M., Carlucci, J.R., Eckert, M.A., Ward, I.L., 1996. Critical periodsof sensitivity of sexually dimorphic spinal nuclei to prenatal testosteroneexposure in female rats. Horm. Behav. 30, 407–415.
Warrick, J.M., Morabito, L.M., Bilen, J., Gordesky-Gold, B., Faust, L.Z., Paulson, H.L.,Bonini, N.M., 2005. Ataxin-3 suppresses polyglutamine neurodegeneration inDrosophila by a ubiquitin-associated mechanism. Mol. Cell. 18, 37–48.
Watson, N.V., Freeman, L.M., Breedlove, S.M., 2001. Neuronal size in the spinalnucleus of the bulbocavernosus: direct modulation by androgen in rats withmosaic androgen insensitivity. J. Neurosci. 21, 1062–1066.

F. Sambataro, M. Pennuto / Progress in Neurobiology 97 (2012) 152–172172
Weydt, P., Pineda, V.V., Torrence, A.E., Libby, R.T., Satterfield, T.F., Lazarowski, E.R.,Gilbert, M.L., Morton, G.J., Bammler, T.K., Strand, A.D., Cui, L., Beyer, R.P., Easley,C.N., Smith, A.C., Krainc, D., Luquet, S., Sweet, I.R., Schwartz, M.W., La Spada, A.R.,2006. Thermoregulatory and metabolic defects in Huntington’s disease trans-genic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration.Cell Metab. 4, 349–362.
Wolf, R.C., Sambataro, F., Vasic, N., Schonfeldt-Lecuona, C., Ecker, D., Landwehr-meyer, B., 2008. Aberrant connectivity of lateral prefrontal networks in pre-symptomatic Huntington’s disease. Exp. Neurol. 213, 137–144.
Wolf, R.C., Sambataro, F., Vasic, N., Schonfeldt-Lecuona, C., Ecker, D., Landwehr-meyer, B., 2008. Altered frontostriatal coupling in pre-manifest Huntington’sdisease: effects of increasing cognitive load. Eur. J. Neurol. 15, 1180–1190.
Wolf, R.C., Sambataro, F., Vasic, N., Wolf, N.D., Thomann, P.A., Landwehrmeyer, G.B.,Orth, M., 2011. Longitudinal functional magnetic resonance imaging of cogni-tion in preclinical Huntington’s disease. Exp. Neurol. 231, 214–222.
Wolf, R.C., Vasic, N., Schonfeldt-Lecuona, C., Landwehrmeyer, G.B., Ecker, D., 2007.Dorsolateral prefrontal cortex dysfunction in presymptomatic Huntington’sdisease: evidence from event-related fMRI. Brain 130, 2845–2857.
Xiang, Z., Valenza, M., Cui, L., Leoni, V., Jeong, H.K., Brilli, E., Zhang, J., Peng, Q., Duan,W., Reeves, S.A., Cattaneo, E., Krainc, D., 2011. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelinationin experimental models of Huntington’s disease. J. Neurosci. 31, 9544–9553.
Xu, J., Gingras, K.M., Bengston, L., Di Marco, A., Forger, N.G., 2001. Blockade ofendogenous neurotrophic factors prevents the androgenic rescue of rat spinalmotoneurons. J. Neurosci. 21, 4366–4372.
Xu, Q., Lin, H.Y., Yeh, S.D., Yu, I.C., Wang, R.S., Chen, Y.T., Zhang, C., Altuwaijri, S.,Chen, L.M., Chuang, K.H., Chiang, H.S., Yeh, S., Chang, C., 2007. Infertility withdefective spermatogenesis and steroidogenesis in male mice lacking androgenreceptor in Leydig cells. Endocrine 32, 96–106.
Yamamoto, M., Mitsuma, N., Inukai, A., Ito, Y., Li, M., Mitsuma, T., Sobue, G., 1999.Expression of GDNF and GDNFR-alpha mRNAs in muscles of patients withmotor neuron diseases. Neurochem. Res. 24, 785–790.
Yan, Q., Matheson, C., Lopez, O.T., 1995. In vivo neurotrophic effects of GDNF onneonatal and adult facial motor neurons. Nature 373, 341–344.
Yang, L.Y., Verhovshek, T., Sengelaub, D.R., 2004. Brain-derived neurotrophic factorand androgen interact in the maintenance of dendritic morphology in a sexuallydimorphic rat spinal nucleus. Endocrinology 145, 161–168.
Yeh, S., Tsai, M.Y., Xu, Q., Mu, X.M., Lardy, H., Huang, K.E., Lin, H., Yeh, S.D., Altuwaijri,S., Zhou, X., Xing, L., Boyce, B.F., Hung, M.C., Zhang, S., Gan, L., Chang, C., 2002.Generation and characterization of androgen receptor knockout (ARKO) mice:an in vivo model for the study of androgen functions in selective tissues. Proc.Natl. Acad. Sci. U.S.A. 99, 13498–13503.
Yohrling 4th, G.J., Jiang, G.C., DeJohn, M.M., Miller, D.W., Young, A.B., Vrana, K.E.,Cha, J.H., 2003. Analysis of cellular, transgenic and human models of Hunting-ton’s disease reveals tyrosine hydroxylase alterations and substantia nigraneuropathology. Brain Res. Mol. Brain Res. 119, 28–36.
Yu, Z., Dadgar, N., Albertelli, M., Gruis, K., Jordan, C., Robins, D.M., Lieberman, A.P.,2006. Androgen-dependent pathology demonstrates myopathic contribution to
the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Invest. 116,2663–2672.
Yu, Z., Dadgar, N., Albertelli, M., Scheller, A., Albin, R.L., Robins, D.M., Lieberman, A.P.,2006. Abnormalities of germ cell maturation and sertoli cell cytoskeleton inandrogen receptor 113 CAG knock-in mice reveal toxic effects of the mutantprotein. Am. J. Pathol. 168, 195–204.
Yu, Z., Wang, A.M., Adachi, H., Katsuno, M., Sobue, G., Yue, Z., Robins, D.M., Lieber-man, A.P., 2011. Macroautophagy is regulated by the UPR-Mediator CHOP andAccentuates the Phenotype of SBMA Mice. PLoS Genet. 7, e1002321.
Yu, Z., Wang, A.M., Robins, D.M., Lieberman, A.P., 2009. Altered RNA splicingcontributes to skeletal muscle pathology in Kennedy disease knock-in mice.Dis. Model. Mech. 2, 500–507.
Yvert, G., Lindenberg, K.S., Picaud, S., Landwehrmeyer, G.B., Sahel, J.A., Mandel, J.L.,2000. Expanded polyglutamines induce neurodegeneration and trans-neuronalalterations in cerebellum and retina of SCA7 transgenic mice. Hum. Mol. Genet.9, 2491–2506.
Zeitlin, S., Liu, J.P., Chapman, D.L., Papaioannou, V.E., Efstratiadis, A., 1995. Increasedapoptosis and early embryonic lethality in mice nullizygous for the Hunting-ton’s disease gene homologue. Nat. Genet. 11, 155–163.
Zeron, M.M., Hansson, O., Chen, N., Wellington, C.L., Leavitt, B.R., Brundin, P.,Hayden, M.R., Raymond, L.A., 2002. Increased sensitivity to N-methyl-D-aspar-tate receptor-mediated excitotoxicity in a mouse model of Huntington’s dis-ease. Neuron 33, 849–860.
Zhang, C., Yeh, S., Chen, Y.T., Wu, C.C., Chuang, K.H., Lin, H.Y., Wang, R.S., Chang, Y.J.,Mendis-Handagama, C., Hu, L., Lardy, H., Chang, C., 2006. Oligozoospermia withnormal fertility in male mice lacking the androgen receptor in testis peritubularmyoid cells. Proc. Natl. Acad. Sci. U.S.A. 103, 17718–17723.
Zhao, J., Brault, J.J., Schild, A., Cao, P., Sandri, M., Schiaffino, S., Lecker, S.H., Goldberg,A.L., 2007. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6,472–483.
Zhong, X., Pittman, R.N., 2006. Ataxin-3 binds VCP/p97 and regulates retrotranslo-cation of ERAD substrates. Hum. Mol. Genet. 15, 2409–2420.
Zhuchenko, O., Bailey, J., Bonnen, P., Ashizawa, T., Stockton, D.W., Amos, C.,Dobyns, W.B., Subramony, S.H., Zoghbi, H.Y., Lee, C.C., 1997. Autosomaldominant cerebellar ataxia (SCA6) associated with small polyglutamineexpansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet.15, 62–69.
Zimbelman, J.L., Paulsen, J.S., Mikos, A., Reynolds, N.C., Hoffmann, R.G., Rao, S.M.,2007. fMRI detection of early neural dysfunction in preclinical Huntington’sdisease. J. Int. Neuropsychol. Soc. 13, 758–769.
Zuccato, C., Tartari, M., Crotti, A., Goffredo, D., Valenza, M., Conti, L., Cataudella, T.,Leavitt, B.R., Hayden, M.R., Timmusk, T., Rigamonti, D., Cattaneo, E., 2003.Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 35, 76–83.
Zuccato, C., Valenza, M., Cattaneo, E., 2010. Molecular mechanisms and potentialtherapeutical targets in Huntington’s disease. Physiol. Rev. 90, 905–981.
Zurn, A.D., Baetge, E.E., Hammang, J.P., Tan, S.A., Aebischer, P., 1994. Glial cell line-derived neurotrophic factor (GDNF), a new neurotrophic factor for motoneur-ones. Neuroreport 6, 113–118.





























