Embed Size (px)
Citation preview

Review
Central role of peroxisomes in isoprenoid biosynthesis§
Werner J. Kovacs, Lisa M. Olivier, Skaidrite K. Krisans*
Department of Biology, San Diego State University, San Diego, CA 92182, USA
Abstract
Peroxisomes contain enzymes catalyzing a number of indispensable metabolic functions mainly relatedto lipid metabolism. The importance of peroxisomes in man is stressed by the existence of genetic disordersin which the biogenesis of the organelle is defective, leading to complex developmental and metabolicphenotypes. The purpose of this review is to emphasize some of the recent findings related to the localiza-tion of cholesterol biosynthetic enzymes in peroxisomes and to discuss the impairment of cholesterol bio-synthesis in peroxisomal deficiency diseases. # 2002 Elsevier Science Ltd. All rights reserved.
0163-7827/02/$ - see front matter # 2002 Elsevier Science Ltd. All rights reserved.
PI I : S0163-7827(02 )00002-4
Progress in Lipid Research 41 (2002) 369–391
www.elsevier.com/locate/plipres
Contents
1. Introduction ........................................................................................................................................................... 3702. The cholesterol biosynthetic pathway: conversion of acetyl-CoA to FPP ............................................................. 373
2.1. Acetoacetyl-CoA thiolase .............................................................................................................................. 3732.2. HMG-CoA synthase...................................................................................................................................... 3742.3. HMG CoA reductase .................................................................................................................................... 3742.4. Mevalonate kinase......................................................................................................................................... 375
2.5. Conversion of mevalonate to FPP ................................................................................................................ 3752.5.1. Phosphomevalonate kinase................................................................................................................ 3762.5.2. Mevalonate diphosphate decarboxylase ............................................................................................ 376
2.5.3. Isopentenyl diphosphate isomerase ................................................................................................... 3762.6. Farnesyl diphosphate synthase ...................................................................................................................... 376
3. Conversion of FPP to cholesterol .......................................................................................................................... 378
4. Discussion:peroxisomal targeting of cholesterol biosynthetic enzymes.................................................................. 3785. Isoprenoid biosynthesis in peroxisomal biogenesis disorders................................................................................. 380
5.1. Plasma cholesterol levels ............................................................................................................................... 380
5.2. Measurement of cholesterol biosynthesis enzyme activities using fibroblasts of PBD patients..................... 3805.3. Cholesterol biosynthesis in peroxisome-deficient fibroblasts ......................................................................... 380
§ Authors contributed equally to the manuscript.* Corresponding author. Tel.: +1-619-594-5368; fax: +1-619-594-5676.
E-mail address: [email protected] (S. Krisans).

1. Introduction
Cholesterol biosynthesis has been the subject of extensive study. A number of years ago it wasdiscovered that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate limitingenzyme in the synthesis of cholesterol, can be found in peroxisomes in addition to its previouslyknown location, the endoplasmic reticulum [1]. Additional experiments corroborated this finding,which for the first time linked the biosynthesis of cholesterol to peroxisomes [2]. Since these initial
5.4. Measurement of cholesterol biosynthesis enzyme activities in peroxisome-deficient tissues.......................... 381
5.5. Peroxisome-deficient rodent cells................................................................................................................... 3815.6. Isoprenoids in mevalonic aciduria ................................................................................................................. 3815.7. Further non-sterol isoprenoids ...................................................................................................................... 382
5.8. LDL-receptor pathway.................................................................................................................................. 3826. Neurodevelopmental aspects of peroxisome biogenesis disorders.......................................................................... 3837. Mouse models for peroxisome biogenesis disorders............................................................................................... 3858. Embryonic development and cholesterol ............................................................................................................... 385
9. Conclusion ............................................................................................................................................................. 386Acknowledgements...................................................................................................................................................... 387References ................................................................................................................................................................... 387
Nomenclature
AA-CoA acetoacetyl-CoACDPX2 conradi-huenermann syndromeCHILD congenital hemidysplasia with ichthyosiform erythroderma and limb
defectsCHO chinese hamster ovaryCNS central nervous systemDHAP dihydroxyacetonephosphateER endoplasmic reticulumFPP farnesyl diphosphateGFP green fluorescent proteinHA hemagglutininHIDS hyperimmunoglobulinemia D and periodic fever syndromeHMG-Co 3-hydroxy-3-methylglutaryl coenzymeIPP isopentenyl diphosphateIRD infantile refsum diseaseLDL low density lipoproteinLPDS lipoprotein deficient serumMPD mevalonate diphosphate decarboxylaseMvK mevalonate kinase
370 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

findings, data has been accumulating which clearly demonstrate that all of the enzymes requiredto generate farnesyl diphosphate (FPP) from acetyl-CoA are localized in peroxisomes.Peroxisomes were first biochemically characterized in the 1960s by Christian de Duve [3] but
only in recent years their biogenesis and cellular biochemical contributions have been moreextensively studied. Peroxisomes are involved in biosynthesis of ether phospholipids, bile acids,cholesterol and cholesterol intermediates, the oxidation of very long chain fatty acids, and thecatabolism of phytanic acid and glyoxylate [4].Peroxisomes are bound by a single membrane containing a number of membrane proteins
required for biogenesis of the organelle, substrate transport and docking complexes for theimport of matrix proteins [for recent reviews see 5 and 6]. Peroxisomal matrix proteins aretranslated on free polyribosomes [4] and post-translationally translocated into the matrix of theorganelle by specific targeting signals. Matrix protein import is believed to occur in three steps;(1) binding of import receptors to targeting signals, (2) binding of the complex to the peroxisomesand (3) an ATP dependent translocation of the protein across the membrane [7].Currently two classes of peptide signals for peroxisomal matrix protein targeting have been
identified. The first characterized peroxisomal targeting signal (PTS-1) is a tripeptide with theconsensus sequence (S/A/C)(K/H/R)(L/M) found at the extreme carboxy terminus of most per-oxisomal proteins [8,9]. The second peroxisomal targeting signal, PTS-2, is a nine amino acidbipartite sequence with the consensus sequence (R/K) (L/V/I) (x5) (H/Q) (L/A) [10–12]. The PTS-2 can be found at variable distances from the amino terminus and, in some cases, is cleaved afterimport [10]. Although the consensus sequence has been defined there is evidence that the PTS-2may not simply be a required sequence of amino acids; the targeting information may consist of astructural or charge based motif [13]. Whereas most known peroxisomal proteins have an iden-tifiable PTS-1 or a PTS-2, there are other peroxisomal proteins in which a targeting sequence hasnot been identified. For such proteins it is hypothesized that import may occur (1) through theuse of nonconventional PTS-1 or PTS-2 sequences which are not currently recognized, (2) byusing a third type of targeting pathway which has yet to be identified or (3) may multimerize in
NALD neonatal adrenoleukodystrophyPBD peroxisome biogenesis disorderPEX peroxinPAHX phytanoyl-CoA hydroxylasePLP proteolipid proteinPTS peroxisomal targeting signalPMvK phosphomevalonate kinaseRCDP rhizomelic chondrodysplasia punctataSHH sonic hedgehogSLOS smith-lemli-opitz syndromeSREBP sterol regulatory element-binding proteinVLCFA very long chain fatty acidsZS zellweger syndrome
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 371

the cytosol with other proteins destined for matrix import, gaining import by ‘‘piggybacking’’ asa heteromeric complex [14,15]. While such protein complexes and folded proteins cannot gain accessto the endoplasmic reticulum (ER), mitochondria, or chloroplasts, several lines of evidence indicatethat these structures can enter peroxisomes without changing shape or conformation [16].The indispensable role peroxisomes play in normal cellular function is most apparent through
studies of the fatal human peroxisomal biogenesis disorders. Twenty-five genetically determinedhuman peroxisomal disorders have been described and are subdivided into two major categories:(1) disorders of peroxisome biogenesis (PBD) and (2) disorders that involve single peroxisomalmetabolic enzymes (Table 1) [17,18]. The disorders of the first category are comprised clinically oftwo distinct groups: the Zellweger spectrum disorders and rhizomelic chondrodysplasia punctata(RCDP). The PBDs are severe autosomal recessive disorders caused by a defect in any of at least12 different peroxin (PEX) genes that encode proteins necessary for peroxisome biogenesis andperoxisomal protein import [19]. The Zellweger spectrum ranges from the severe Zellweger cere-bro-hepato-renal syndrome (ZS) through the clinically less involved neonatal adrenoleukody-strophy (NALD), infantile Refsum disease (IRD), to still milder variants. Several studies haveestablished that the phenotypic heterogeneity within the Zellweger spectrum is explained, in part,by functional differences of the mutant protein encoded by various alleles rather than by theidentity of the mutated gene [19,20]. Since peroxisomes contain more than 60 enzyme activities, thelack of functional intact peroxisomes that lack matrix enzymes creates severe biochemical abnorm-alities, leading to a variety of clinical symptoms (Table 2) [17,18,21,22]. Many malformations arealready evident in biochemically affected 20–24 week (postmenstrual) fetuses [23]. Zellwegersyndrome patients rarely survive their first year. By contrast, NALD and IRD patients showsimilar though less severe clinical phenotypes than those of ZS patients, with NALD patientssurviving up to a decade and many IRD patients beyond their third decade [24]. RCDP, bycontrast, is a partial PBD affecting either the PTS-2-specific protein import or a number of
Table 1Peroxisomal disorders
Peroxisome biogenesis disorders Single peroxisomal enzyme deficiencies
Zellweger spectrum disordersa Adrenoleukodystrophya/Adrenomyeloneuropathya
Zellweger syndrome (ZS)a Acyl-CoA oxidase deficiencya
Neonatal adrenoleukodystrophy (NALD)a 3-Ketoacyl-CoA thiolase deficiencya
Infantile Refsum disease (IRD)a D-Bifunctional enzyme deficiencya
Rhizomelic chondrodysplasia punctata (RCDP)a Classical Refsum diseasea
Hyperpipecolic acidemiaa Dihydroxyacetonephosphate (DHAP)acyltransferase deficiencya
Alkyl-DHAP synthase deficiencya
Mevalonate kinase deficiencya
a-Methylacyl-CoA racemase deficiencya
Acatalasemia
Glutaric aciduria type IIIa
Hyperoxaluria type I
a Nervous system affected.
372 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

enzymes of plasmalogen biosynthesis. It is characterized by unique skeletal abnormalities and amore limited set of peroxisomal deficiencies [22,24].
2. The cholesterol biosynthetic pathway: conversion of acetyl-CoA to FPP
2.1. Acetoacetyl-CoA thiolase
Acetoacetyl-CoA thiolase (AA-CoA thiolase) catalyzes the conversion of acetyl-CoA to acet-oacetyl-CoA in the first reaction of the cholesterol biosynthetic pathway. AA-CoA thiolase isencoded by two genes which encode a mitochondrially localized enzyme (mt AA-CoA thiolase)[25] and a cytosolic enzyme (cyt AA-CoA thiolase) [26]. However, purified peroxisomes can alsosynthesize acetoacetyl-CoA from acetyl-CoA [27,28]. Analysis of the protein sequences of bothAA-CoA thiolases revealed a consensus PTS-1 (QKL) at the carboxy terminus of the mt AA-CoA thiolase, in addition to its amino terminal mitochondrial targeting sequence, whereas noperoxisomal targeting sequence was detected in the cytosolic AA-CoA thiolase. Myc epitopefusion constructs for full length, amino terminal truncated and carboxy terminal truncated AA-CoA thiolase were expressed in chinese hamster ovary (CHO) cells and by double-label immu-nofluorescence it has been shown that the identified PTS-1 can direct mt AA-CoA thiolase toperoxisomes [29].The dual targeting of a single gene product is an interesting topic for speculation and study. How
is the localization to two subcellular compartments regulated? Are the needs of one compartmentmet primarily with the excess passively entering the second compartment or are there cellular cuesindicating what proportions of newly synthesized enzyme transit to each compartment? Areadditional chaperones or receptors required to mask or enhance a targeting signal? Can anenzyme exit a organelle and transfer to another after it enters mitochondria or peroxisomes?
Table 2
Features of disorders of peroxisome assembly
Clinical Features Biochemical abnormalities Neurological abnormalities
Dysmorphic features Accumulation of very long Abnormal neuronal migration
High forehead chain fatty acids (VLCFA) Cerebral hemispheresLarge fontanelles Accumulation of phytanic acid, CerebellumEpicanthus pristanic acid and pipecolic acid Inferior olivary complex
Abnormal ears Depletion of plasmalogens Abnormal Purkinje cellHypoplastic supraorbital ridges Accumulation of abnormal bile dendritic arborization
Hypotonia acids (trihydroxycholestanoic Abnormal white matter
Hepatomegaly and dihydroxycholestanoic acid) DemyelinationRenal cysts Accumulation of bioactive DysmyelinationRetinopathy compounds like eicosanoids, HypomyelinationCataracts retinoids Postdevelopmental
Impaired hearing Depletion of docosahexaenoic acid neuronal degenerationsChondrodysplasia punctata (DHA)Psychomotor delay Hypocholesterolemia
Neonatal seizures
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 373

Whatever the cellular mechanism of dual targeting may be, it is already clear that mt AA-CoAthiolase is not alone in this regard. Dual localization to peroxisomes and mitochondria has beenidentified for at least three other enzymes in recent years: (1) HMG-CoA lyase contains a 27amino acid amino terminal mitochondrial targeting sequence and a C-terminal PTS-1 (CKL)[30,31], (2) the mammalian �3,5- �2,4-dienoyl-CoA isomerase [32] has both a 40 amino acidamino terminal mitochondrial targeting sequence and a C-terminal PTS-1 (SKL) and (3) humana-methylacyl-CoA racemase has both an amino terminal mitochondrial targeting signal and acarboxy terminal PTS-1 (-kASL) [33].
2.2. HMG-CoA synthase
HMG-CoA synthase catalyzes the second reaction in the cholesterol biosynthetic pathway,converting acetoacetyl-CoA to HMG-CoA. Similar to AA-CoA thiolase, two genes for HMG-CoA synthase have been identified. One gene encodes a mitochondrial enzyme while the otherencodes an enzyme originally believed to be localized to the cytosol [34]. However, subcellularfractionation studies demonstrated that HMG-CoA synthase activity was present in rat liverperoxisomes [35]. Furthermore, by immunoelectron microscopy with antibodies made againstcytosolic HMG-CoA synthase significant labeling was detected in the peroxisomal matrix withonly a small amount of labeling in the cytosol. In addition, by double-label immunofluoresence ithas been shown that the immunofluorescence pattern obtained with the cytosolic HMG-CoAsynthase antibody is superimposable over that obtained with the antibody to the peroxisomalmarker catalase, indicating that these two enzymes are colocalized [29]. Taken together, thesebiochemical and immunological data indicate that a significant amount of HMG-CoA synthase isfound in the peroxisomes.Analysis of the protein sequences of the two HMG-CoA synthase proteins revealed neither a
consensus PTS-1 nor PTS-2. However, the cytosolic HMG-CoA synthase does contain asequence similar to a PTS-2. This PTS-2-like sequence diverges from the consensus amino acids,with the basic amino acids, either arginine or lysine, being replaced with a serine [(SV) �5 (QL)].Through the use of myc-epitope expression vectors and immunofluorescence studies it wasdetermined that HMG-CoA synthase is a peroxisomal protein which requires the sequence (SV)�5 (QL) for peroxisomal import [29].
2.3. HMG CoA reductase
HMG-CoA reductase, the rate limiting enzyme of the cholesterol biosynthetic pathway, cata-lyzes the conversion of HMG-CoA into mevalonate. A number of studies including enzymeassays and western blotting on subcellular fractions, immunofluorescence and immunoelectronmicroscopy indicate that HMG-CoA reductase is located in both the endoplasmic reticulum (ER)and in peroxisomes [1,2,36,37]. In a peroxisome biogenesis strain of Saccharomyces cerevisiae inwhich the PEX1 gene was disrupted, reduced HMG-CoA reductase and FPP synthase activitieswere observed [38]. These results indicate that functional peroxisomes are necessary for thenormal activities of HMG-CoA reductase and FPP synthase and suggest that they are localized inperoxisomes [38]. Furthermore, peroxisomal reductase is regulated in an independent mannerfrom the ER reductase as demonstrated by (1) the peroxisomal HMG-CoA reductase is more
374 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

resistant to inhibition by statins (2) compared to the ER HMG-CoA reductase, (2) the rate ofperoxisomal HMG-CoA reductase degradation is unaffected by the presence of mevalonate and(3) the peroxisomal reductase is not phosphorylated [39]. These data indicate that ER andperoxisomal HMG-CoA reductase may have separate functions in isoprenoid biosynthesis [39].Furthermore, two genes for mammalian HMG-CoA reductase have been suggested [36]. How-ever, efforts to clone the peroxisomal HMG-CoA reductase and sequence analysis from thepublished human genome have yet to lead to its discovery. In yeast and plants, two HMG-CoAreductase genes have been identified [40,41], suggesting that two HMG-CoA reductase genescould be possible in other species.
2.4. Mevalonate kinase
Mevalonate kinase (MvK) phosphorylates mevalonate in the fourth reaction of the cholesterolbiosynthetic pathway. The peroxisomal localization of MvK has been conclusively demonstrated[42,43]. Subcellular fractionation studies show that MvK and the peroxisomal marker enzymecatalase colocalize both within cytosolic and peroxisomal fractions [43]. Furthermore, immunoe-lectronmicroscopy and indirect immunofluoresence of endogenous and transfected MvK indicatethat the enzyme is predominantly localized in peroxisomes [43]. Analysis of the amino acidsequence revealed a consensus PTS-2 (KV) �5 (HA) in the amino terminal region of MvK.Although several expression constructs were generated in our lab to verify this sequence as theperoxisomal targeting signal, no fusion vector containing full length or truncated MvK attachedto epitope tags or reporter genes has been imported into peroxisomes [29]. These results arepuzzling given that other PTS-2 sequences attached similarly to these epitopes or reporter geneswere targeted to peroxisomes. However, given the biochemical and microscopy data indicatingthat endogenous MvK is peroxisomal, it is possible that by adding sequences to the MvK codingregion the natural protein folding may be altered and as a consequence the binding of the PTS-2receptor may be prevented.
2.5. Conversion of mevalonate to FPP
Three enzymes catalyze the reactions required to convert mevalonate into isopentenyl diphos-phate (IPP). Originally these enzymes, phosphomevalonate kinase (PMvK), mevalonate diphos-phate decarboxylase (MPD) and isopentenyl diphosphate (IPP) isomerase, were believed to becytosolic; however, recent studies showed that they are localized to peroxisomes. Biardi et al.showed that the activities of MvK, PMvK and MPD were equal in intact and digitonin per-meabilized CV-1 cells [44]. Digitonin permeabilizes membranes by forming complexes with themembrane cholesterol and a selective permeabilization is achieved by incubation of cells withdigitonin since the plasma membrane is rich in cholesterol, while membranes of internalorganelles are almost devoid of cholesterol. Therefore digitonin-treatment depletes cells ofcytosolic components while organelle-bounded enzymes are retained [44]. Therefore these dataindicate that the enzyme activities were contained in membrane-bound organelles and not in thecytosol. Moreover, there is a significant decrease in enzymatic activities of HMG-CoA reductase,MvK, PMvK, MPD, IPP isomerase and FPP synthase in liver tissue from individuals afflictedwith PBDs, whereas squalene synthase and marker enzymes for the ER and mitochondria are not
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 375

affected [45]. Taken together, the data are highly suggestive of the peroxisomal localization ofPMvK, MPD and IPP isomerase. Subsequent experiments that confirmed their peroxisomallocalization and determined the peroxisomal targeting signals required for import are:
2.5.1. Phosphomevalonate kinasePMvK catalyzes the conversion of mevalonate 5-phosphate into mevalonate 5-diphosphate as
the fifth reaction of the cholesterol biosynthetic pathway. Through the use of green fluorescentprotein (GFP) fusion constructs it has been shown that this enzyme requires a functional PTS-1import system and the carboxy terminal tripeptide,-SRL, a consensus sequence for a PTS-1, tolocalize to peroxisomes. In studies where this tripeptide was removed the protein was mislocalizedto the cytosol [46].
2.5.2. Mevalonate diphosphate decarboxylaseMPD catalyzes the sixth reaction of the cholesterol biosynthetic pathway in which the six car-
bon mevalonate diphosphate is dehydrated and decarboxylated to form isopentenyl diphosphate.Immunofluorescence studies utilizing hemagglutinin (HA)-epitope tagged MPD constructs indi-cate that MPD is a peroxisomal protein which requires a functional PTS-2 receptor for importinto the organelle. Furthermore, the newly identified PTS-2 sequence of HMG-CoA synthase,(SV) �5 (QL), is the peroxisomal targeting signal [29].
2.5.3. Isopentenyl diphosphate isomeraseIPP isomerase, which reversibly isomerizes the double bond of IPP, contains both a putative
carboxy terminal PTS-1 (-YRM in human and -HRM in hamster) and an amino terminal PTS-2[(HL) �5 (QL), human and hamster]. Subcellular localization studies using HA-epitope taggedexpression constructs indicate that IPP isomerase is targeted to peroxisomes through the use of itscarboxy terminal tripeptide. When this tripeptide was removed from HA-tagged IPP isomerasethe expressed protein remained in the cytosol [47]. These studies suggest that the putative PTS-2 isnot necessary to target IPP isomerase to peroxisomes.
2.6. Farnesyl diphosphate synthase
FPP synthase catalyzes two sequential 1–4 condensation reactions of isopentenyl diphosphatewith the allylic diphosphates dimethylallyl diphosphate and geranyl diphosphate [48]. The productFPP is utilized in the synthesis of cholesterol, farnesylated and geranylgeranylated proteins,dolichols, coenzyme Q, and the isoprenoid moiety of heme a [49]. FPP synthase has been identi-fied as being predominantly peroxisomal based on enzyme assays of subcellular rat liver fractions,immunofluorescence and immunoelectron microscopy studies [45]. These studies have clearlydemonstrated that the majority, if not all of the endogenous FPP synthase protein is colocalizedwith catalase in hepatic H35 cells as well as in CHO cells [50]. Moreover, FPP synthase iscytosolic in peroxisome-deficient CHO cells [50]. Curiously however, FPP synthase does notcontain any sequence related to either of the two known peroxisomal targeting signals.Immunofluorescence studies of myc tagged FPP synthase demonstrated that FPP synthase uti-
lizes the PTS-2 import pathway and requires a sequence found within the amino terminal 20amino acids for localization. This region however does not contain a consensus PTS nor are the
376 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

amino acids conserved when comparing human FPP synthase [51] to rat FPP synthase [52]. Incontrast, the peroxisomal targeting signals of human and rodent AA-CoA thiolase [53,54],HMG-CoA synthase [55,56], MvK [57,58], PMvK [46,59], MPD [60,61], and IPP isomerase[44,62] are conserved (Table 3). Therefore, this apparent lack of sequence conservation of FPPsynthase between mammalian species might indicate that the identified amino terminal sequenceis not a specific receptor binding site. However, it is clear that FPP synthase is a peroxisomalprotein in a variety of mammalian cell lines including Hep G2 (human), CV-1 (monkey), SKB-R3(human), H35 (rat) and CHO (hamster) [45,50]. It is believed that peroxisomal proteins withouttargeting signals can gain access to the matrix by piggybacking or through a third import path-way. Therefore, while the data show that FPP synthase utilizes the PTS-2 import pathway, it isunclear if the sequence identified as being necessary for peroxisomal import is a degenerate PTS-2like sequence or a sequence required for the piggybacking of FPP synthase with another PTS-2containing protein.In a recent study, Gupta et al. [50] verified the peroxisomal localization of FPP synthase. This
study also analyzed the leakage of peroxisomal FPP synthase to the cytosol through digitoninpermeabilization experiments. Interestingly, in the rat hepatoma cell line H35 they observed afterpermeabilization what appears to be two pools of FPP synthase, one which easily diffuses fromperoxisomes and one which remains peroxisomal. The authors suggest that FPP synthase isstored in peroxisomes and then transported to different cytosolic locations to generate FPP.Furthermore, they hypothesize that the localization and storage of FPP synthase in peroxisomesmay be due to a FPP synthase peroxisomal ‘‘carrier’’ protein. The suggestion that FPP synthaseinteracts with another protein for localization is consistent with the possibility that FPP synthasepiggybacks with another peroxisomal protein to gain entry to peroxisomes.
Table 3Peroxisomal targeting signals of the cholesterol biosynthetic enzymes
Enzyme Signal Sequence (species)
Acetoacetyl-CoA thiolase (mito) PTS-1 -QKL (human)
-QKL (rat)HMG-CoA synthase PTS-2 V(X5)QL (human)
SV(X5)QL (human)HMG-CoA reductase unknown
Mevalonate kinase PTS-2 KV(X5)QL (human)KV(X5)QL (rat)
Phosphomevalonate kinase PTS-1 -SRL (human)
-AKL (rat)Mevalonate-PP decarboxylase PTS-2 SV(X5)QL (human)
SV(X5)QL (rat)
Isopentenyl-PP isomerase PTS-1 -YRM (human)-HRM (rat)
Farnesyl-PP synthase PTS-2 MNGDQNSDVYAQEKQDFVQH (human)
MNGDQKLDVHNQEKQNFIQH (rat)
Consensus PTS-1 (S/A/C)(K/H/R)(L/M)PTS-2 (R/K)(L/I/V)(X5)(H/Q)(L/A)
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 377

3. Conversion of FPP to cholesterol
The conversion of FPP to lanosterol is believed to occur in the ER. Squalene synthase, cata-lyzing the first committed step of cholesterol biosynthesis, has been shown in two independentstudies to be exclusively an ER protein. The data include enzyme assays on subcellular fractionsas well as immunoelectronmicroscopy in which labeling was evident only on ERmembranes [63,64].In addition, isolated microsomes are able to synthesize cholesterol from squalene whereas isolatedperoxisomes can not [65]. The subsequent conversion of lanosterol to cholesterol is proposed to takeplace both in peroxisomes and the ER. This hypothesis is based on several individual observationsincluding: (1) subcellular fractionation studies revealed that the enzymes required for this con-version, dihydrolanosterol oxidase, �14-sterol reductase, c-4-sterol demethylase and �8- �7-sterolisomerase are localized in peroxisomes [66] (2) peroxisomes have also been shown to accumulatebiochemical intermediates between the conversion of lanosterol to cholesterol [67] and (3) choles-terol is synthesized from dihydrolanosterol in isolated peroxisomes at levels equivalent to synthesisfrom isolated microsomes [65]. While these results are intriguing and the data suggest peroxisomallocalization, further studies are necessary to delineate the subcellular localization of these proteins.
4. Discussion:peroxisomal targeting of cholesterol biosynthetic enzymes
Upon review of the current data it is clear that all of the enzymes required for the synthesis ofFPP from acetyl-CoA are localized in peroxisomes and the enzymes catalyzing the conversion ofmevalonate to FPP appear to be exclusively peroxisomal (Fig. 1). The data show that both per-oxisomal import pathways are utilized by these enzymes (Table 3) and PTS-1 and PTS-2 arefound in an alternating order. This oscillation is curious since in addition to the cholesterolbiosynthetic enzymes there are only three other known PTS-2 containing mammalian proteins3-ketoacyl-CoA thiolase [10,11], dihydroxyacetonephosphate (DHAP) synthase [68] and phytanoyl-CoA hydroxylase (PAHX) [69]. Interestingly, the first two reactions in the biosynthesis ofplasmalogens are peroxisomal and the enzymes have alternating targeting signals, DHAP acyl-transferase contains a PTS-1 [70] and DHAP synthase contains a PTS-2 [68]. Recently it has beenshown that the complete phytanic acid a-oxidation pathway is localized to peroxisomes [71]. Thefirst two enzymes in this pathway have been cloned and peroxisomal targeting signals have beenidentified: PAHX contains a N-terminal PTS-2 [69] and 2-hydroxyphytanoyl-CoA lyase containsa C-terminal PTS-1 [72]. From these observations several questions arise. What function doesalternating import pathways play in these reactions? Does alternating import allow a tighterregulation of the pathway? Do these enzymes form complexes prior to import? Does thismechanism of alternating protein import pathways improve the formation of products even in theevent of mutations in one of the pathways? In examining the b-oxidation pathway five enzymes,acyl-CoA synthetase, carnitine octanoyltransferase, acyl-CoA oxidase, the bifunctional protein,and 3-ketoacyl-CoA thiolase are peroxisomal. Of this pathway the first four enzymes containsequences similar to the PTS-1 consensus, although targeting has not been analyzed, and 3-ketoacyl-CoA thiolase contains a PTS-2. While this pathway does contain enzymes that seem touse both import pathways, they do not alternate. Perhaps in order to better understand the benefitof alternating pathways one needs to compare the biochemistry and regulation of cholesterol
378 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391
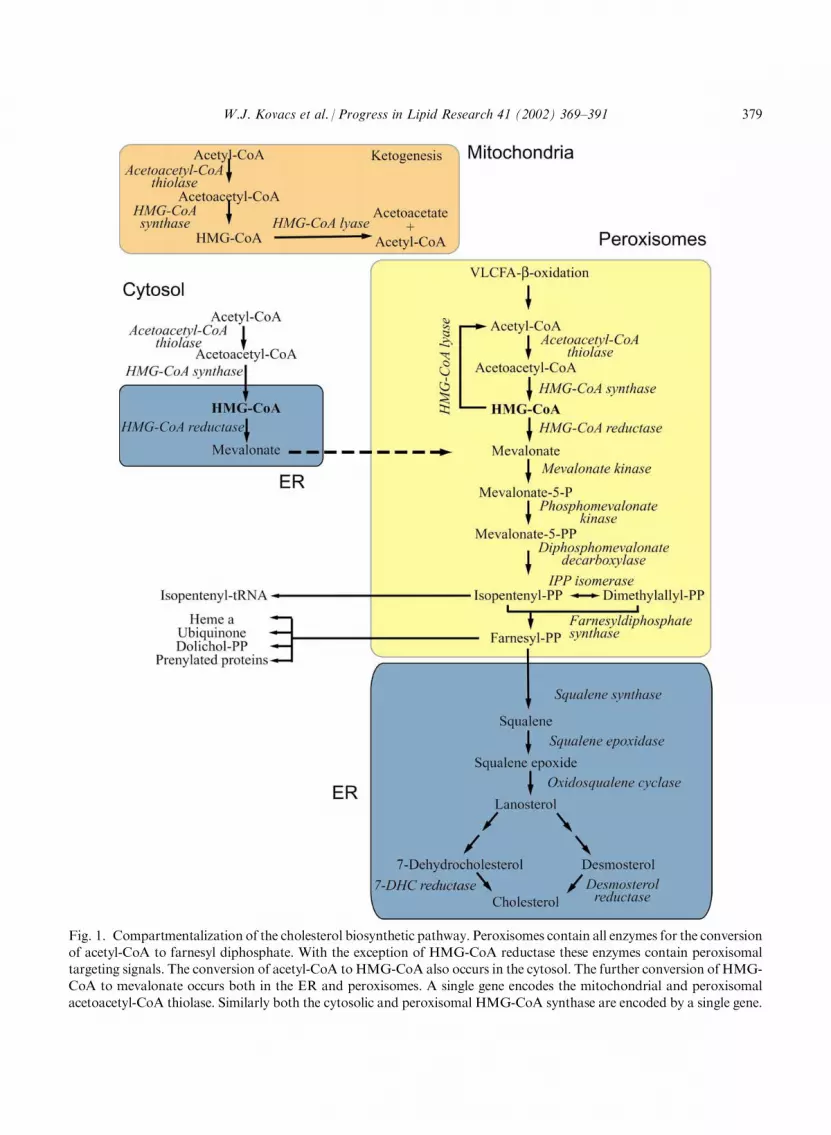
Fig. 1. Compartmentalization of the cholesterol biosynthetic pathway. Peroxisomes contain all enzymes for the conversionof acetyl-CoA to farnesyl diphosphate. With the exception of HMG-CoA reductase these enzymes contain peroxisomaltargeting signals. The conversion of acetyl-CoA to HMG-CoA also occurs in the cytosol. The further conversion of HMG-CoA to mevalonate occurs both in the ER and peroxisomes. A single gene encodes the mitochondrial and peroxisomal
acetoacetyl-CoA thiolase. Similarly both the cytosolic and peroxisomal HMG-CoA synthase are encoded by a single gene.
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 379

biosynthesis, plasmalogen biosynthesis and a-oxidation, which do alternate, with the b-oxidationpathway, which does not alternate.
5. Isoprenoid biosynthesis in peroxisomal biogenesis disorders
As several of the central steps of isoprenoid biosynthesis are predominantly located inperoxisomes it can be anticipated that the pathway is affected in peroxisomal biogenesis disordersthat prevent the formation of functional peroxisomes. While this hypothesis seems straight-forward, the answer remains inconclusive. This hypothesis has been tested through the followingobservations.
5.1. Plasma cholesterol levels
Patients suffering from infantile Refsum disease, the mildest form of peroxisomal biogenesisdisorders, have unusually low plasma cholesterol concentrations that are on average 30% ofcontrol levels [73–77]. Very low total cholesterol levels (47–89 mg/dl) were also found in sixpatients with more severe forms of the Zellweger spectrum [78].
5.2. Measurement of cholesterol biosynthesis enzyme activities using fibroblasts of PBD patients
MvK, MPD and IPP isomerase activity was normal in fibroblasts of eight different patientsdiagnosed with Zellweger syndrome [79]. MvK activity was significantly lowered in fibroblasts ofthree other Zellweger and NALD patients [43]. Activity and protein levels of squalene synthase,an enzyme exclusively localized in the ER [63,64], were unchanged in fibroblasts and liver tissueobtained from Zellweger and NALD patients.
5.3. Cholesterol biosynthesis in peroxisome-deficient fibroblasts
Analysis of cholesterol biosynthesis from acetate in cultured skin fibroblasts from two normal indi-viduals, 16 patients with Zellweger syndrome (seven different complementation groups [80] and threepatients with RCDP [81] demonstrated that the average cholesterol synthesis in all the 16 peroxisome-deficient fibroblast cell cultures was below control values (by 16–98%). In contrast, cholesterolsynthesis in the three RCDP cell lines was not significantly different from control values. There wasno obvious correlation between cholesterol biosynthesis levels and decreased plasmalogen biosyn-thetic capacity or a correlation with cholesterol biosynthesis and severity of the disease. In a study byCollins et al. [78] using fibroblasts of patients with Zellweger syndrome (n=2), NALD (n=1) andinfantile Refsum disease (n=2) incorporation of 3H-acetate into free cholesterol and cholesterol esterwas reduced by 50–90% as compared to control cells. These results were confirmed by a study ofMandel et al. [82], which demonstrated a significantly lower cellular cholesterol mass in fibroblastsfrom three PBD patients, as compared to control cells (41–59% of controls). The rate of cholesterolsynthesis was also reduced in all three PBD cell lines, being 16–20%of the control value. In contrast tothese results, two studies utilizing dermal fibroblasts from patients with Zellweger syndrome (n=2),NALD (n=3), adrenoleukodystrophy (n=2), adrenomyeloneuropathy (n=1) and RCDP (n=2)
380 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

found that cholesterol biosynthesis rates were equivalent or higher than those in control fibro-blasts (n=5) when incubated with various radiolabeled substrates (acetate, octanoate, mevalonate)[83,84].
5.4. Measurement of cholesterol biosynthesis enzyme activities in peroxisome-deficient tissues
Studies by Krisans et al. [45] analyzing eight normal liver samples and six samples from patientsdiagnosed either with Zellweger syndrome or NALD have shown that enzymatic activities ofHMG-CoA reductase, MvK, PMvK, MPD, IPP isomerase and FPP synthase are consistentlyreduced (individual enzymes ranging from 16 to 61% of normal on average), whereas squalenesynthase and marker enzymes for the ER, mitochondria and cytosol were not affected. Further-more, MvK activity was absent in three of four biopsied livers from Zellweger patients [79] and intwo livers from RCDP type 1 patients [85].
5.5. Peroxisome-deficient rodent cells
In peroxisome-deficient PEX2�/� hamster cells (CHO-ZR-78 and ZR-82) total HMG-CoAreductase activity was reduced by 46 and 66%, respectively, when compared to wild-type cells (CHO-K1) [86]. The rates of both sterol (cholesterol) and non-sterol (dolichol) biosynthesis were sig-nificantly lower in the peroxisome-deficient cells, when either [3H]acetate or [3H]mevalonate was usedas substrate, and unchanged when cells were incubated with [3H]farnesol or [3H]dihydrolanosterol[86]. Furthermore, the study showed that lanosterol was clearly accumulated in the peroxisome-deficient PEX2�/� hamster cells [86]. On the contrary, in another study in the same cell linesHMG-CoA reductase activity was increased 2–3-fold as compared to wild-type cells and the incor-poration of [3H]acetate into sterols (cholesterol+lanosterol) was 3-fold (CHO-82) respectively 2-fold(CHO-78) higher than in wild-type cells [87]. However, in their procedure, as stated by the authors,3H-labeled sterols very similar to [3H]cholesterol were not resolved and their values could representthe sum of cholesterol and lanosterol, thus resulting in higher rates of total sterol biosynthesis.
5.6. Isoprenoids in mevalonic aciduria
Reduced sterol biosynthesis within the fetus is extremely deleterious, and results in abnormaldevelopment [for review see 88,89]. To date, seven distinct inherited disorders have been linked todefects in cholesterol biosynthesis (Table 4) [90–93]. Only two disorders result from an enzymedefect in the peroxisomal localized presqualene segment of the pathway, namely mevalonicaciduria and the hyperimmunoglobulinemia D and periodic fever syndrome (HIDS). Mevalonicaciduria is a rare autosomal recessive disorder characterized by a virtually complete deficiency ofmevalonate kinase [94,95], when measured in cultured skin fibroblasts or lymphoblasts of patients[95,96]. HIDS is characterized by recurrent episodes of fever associated with increased excretionof mevalonic acid during crisis and reduced activities of MvK. Surprisingly, squalene, cholesteroland bile acid levels are normal in patients with mevalonic aciduria [95,97]. Furthermore, incor-poration of radiolabeled acetate into cholesterol by cultured skin fibroblasts of mevalonic acid-uria patients is comparable with controls [98]. Abnormalities related to isoprenoid biosynthesisobserved in cells derived from mevalonic aciduria are increased activity of both HMG-CoA
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 381

reductase and the low density lipoprotein (LDL) receptor pathway [99,100] and decreased bio-synthesis of dolichol, ubiquinone and glycosylated macromolecules [98,100].
5.7. Further non-sterol isoprenoids
The ubiquinone level in liver was even higher in peroxisome-deficient PEX5�/� mice than inwild-type or heterozygous mice [101]. Dolichol-dependent N-linked glycosylation of proteins isnormal in the same mouse model, indirectly arguing for sufficient levels of the isoprenoiddolichol-phosphate [102].
5.8. LDL-receptor pathway
Downregulation of cholesterol biosynthesis by LDL is identical in PBD and control cells [81,83]suggesting that regulation of cholesterol synthesis via the LDL-receptor pathway [49] is operativein PBD cells. In addition, it has been shown that both in wild-type and in peroxisome-deficientCHO cells HMG-CoA reductase activity was repressed when cells were grown in medium con-taining regular fetal calf serum or when LDL, mevalonate or 25-hydroxycholesterol was added tolipoprotein deficient serum (LPDS) medium [87].One main conclusion seems to be justified from the current data: the presence of cholesterol
biosynthesis enzymes in peroxisomes, the low serum cholesterol levels in patients suffering fromPBDs, the reduced cholesterol biosynthetic capacity of cells lacking peroxisomes and decreasedactivities of cholesterol biosynthesis enzymes in tissues from PBD patients argue quite stronglythat peroxisomes are essential for normal cholesterol/isoprenoid biosynthesis. Still, we do notknow whether this deficiency contributes to the phenotypic characteristics observed in infantswith PBDs. The deficiency of MvK, PMvK, MPD, IPP isomerase and FPP synthase that isobserved in some experiments is either due to an accelerated degradation of proteins that arestable in the peroxisome but not in the cytosol [103], or perhaps to misregulatory effects of thegreatly disturbed lipid metabolism. At the moment we do not know what happens to the dis-tribution of the metabolic flux through the isoprenoid pathway if certain enzyme activitiesdecrease by x% in the case of PBDs, what the significance of compartmentalization is or how wellthe enzymes are able to carry out their function outside the peroxisome. The importance ofcompartmentalization is questioned when looking at data from RCDP patient fibroblast cells,
Table 4Enzyme deficiencies in the sterol biosynthesis pathway and resultant diseases
Enzyme deficiency Syndrome
Mevalonate kinase Mevalonic aciduriaHyperimmunoglobulinemia D and periodic fever syndrome
�8-�7-sterol isomerase Conradi–Huenermann syndrome (CDPX2)C-4-sterol demethylase CHILD syndrome (congenital hemidysplasia with
ichthyosiform erythroderma and limb defects)
�7-sterol reductase Smith–Lemli–Opitz syndrome�24-sterol reductase Desmosterolosis�14-sterol reductase Greenberg skeletal dysplasia
382 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

which should mistarget four of the enzymes of the pathway (HMG-CoA synthase, MvK, MPD,FPP synthase) and nonetheless show no deficiency of cholesterogenesis. A possible hypothesistaking these results into account is that the combined effect of upregulating individual enzymeexpression in the cholesterol biosynthetic pathway by sterol regulatory element-binding proteins(SREBPs) and accumulation of substrates may allow the pathway to function at almost normallevels. A recent study demonstrated that SREBPs activate every step of the cholesterol biosyn-thetic pathway, thereby contributing to an efficient cholesterol synthesis [104]. Furthermore, ithas been shown by metabolic-control analysis that complex metabolic pathways are not con-trolled by single rate-limiting steps and that control is shared among all steps, with more than onestep having significant influence on pathway flux [105]. This idea and the importance of seques-tration of cholesterol biosynthesis enzymes in the peroxisome is corroborated by the absence ofcholesterol deficiency even in mevalonic aciduria patients which show negligible MvK activity invitro. However, unpublished data from our lab indicate that the in vivo MvK activity in meva-lonic aciduria patients is higher than that measured in cell extracts, which could be due to ahigher fragility of the mutant MvK protein. This conclusion was drawn from experiments wherethe cholesterol biosynthesis rate was determined in intact cells and cell extracts. Interestingly,initial rates of cholesterol synthesis in cultured fibroblasts from Smith–Lemli–Opitz syndrome(SLOS) patients predicted on the basis of their mutations to have no 7-dehydrocholesterolreductase activity, the final step in the Kandutsch–Russell cholesterol biosynthetic pathway, mayalso be as high as 50% of all sterols [90].
6. Neurodevelopmental aspects of peroxisome biogenesis disorders
All of the peroxisomal disorders with neurologic involvement (15 of the 17 peroxisomal dis-orders) may show abnormalities in white matter of the central nervous system (CNS). The chan-ges in CNS white matter vary greatly between these diseases, but can be divided into three typesof lesions: (1) inflammatory demyelination, (2) non-inflammatory dysmyelination, and (3) a non-specific reduction in myelin volume or myelin-staining [106]. Within the brain, cholesterol isfound predominantly in white matter. White matter consists mainly of myelin, which is enrichedin cholesterol (28% dry wt.), as well as very long chain fatty acids (VLCFA) and plasmalogen,and it is estimated that 70% of the cholesterol in the whole brain is incorporated in myelin [107].Cholesterol in the central nervous system is derived almost entirely from in situ synthesis [107–112] and does not pass the blood-brain barrier to a significant extent. There is currently littleevidence for the net transfer of sterol from the plasma into the brain of the fetus, newborn oradult [113,114]. Little is known about the regulation of cholesterol biosynthesis in the CNS andmany other aspects of brain sterol biosynthesis remain unexplored.The non-inflammatory white matter lesions in disorders of peroxisome biogenesis are probably
multifactorial. The lesions appear primarily hypomyelinative/dysmyelinative and are probablyrelated to several factors [for review see 106,115]: (1) defects in neuronal migration or neurogen-esis that result in decreases in the normal complement of myelinated axons; (2) gliopathy due tothe accumulation of abnormal lipids, e.g. VLCFAs; (3) abnormal axonal gangliosides lacking theproper signalling characteristics for myelination; (4) marked deficiencies in myelin-associatedplasmalogens.
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 383

Data from previous studies lead to the following hypothesis for the etiology of most of theneuronal lesions [106]: abnormal fatty acids, particularly VLCFA and phytanic acid, accumulatein the peroxisomal disorders (due to a deficiency in peroxisomal a- and b-oxidation) and areincorporated into cell membranes resulting in a perturbation of the membrane microenviron-ments and the dysfunction, atrophy and death of vulnerable [116,117]. Defects of the peroxisomalfatty acid b-oxidation system per se would play a major role in migration disorders and demye-lination process, since isolated deficiencies of peroxisomal b-oxidation enzymes manifest theseneuropathologies [118]. This is especially true for D-bifunctional protein deficiency since patientsoften show disordered neuronal migration [18]. The incorporation of VLCFA into myelin, par-ticularly as proteolipid protein (PLP), may lead to myelin instability and dysmyelination; perhapsa direct cytotoxic effect on oligodendrocytes also occurs. Considering the plasmalogens are such aprominent component of myelin, it seems plausible that the deficiency of plasmalogens is anothercause of abnormal myelination. Indeed, peroxisomal activity is increased with the onset of mye-lination, and ethanolamine plasmalogens are actively synthesized during this period [119,120].Since cholesterol is such an important component of the CNS and derived from de novo
synthesis, it is tempting to speculate that reduced cholesterogenesis contributes to the brain-spe-cific lesions in PBDs such as dysmyelination or delayed myelination. However, there is currentlyno experimental evidence to support this hypothesis. In an initial approach to investigate theperoxisomal population in the brain we have found that activities for catalase, a marker enzymefor peroxisomes, 20,30-cyclic nucleotide 30-phosphodiesterase, a myelin/oligodendrocyte markerenzyme, IPP isomerase and HMG-CoA reductase show a similar postnatal development, withactivities reaching a maximum between 10 and 15 days [37]. These results are in agreement withprevious histochemical studies in rats showing an increase in peroxisomes in the first 2 weeks ofpostnatal life [121], a time when myelination is most pronounced in rodents [122]. On the con-trary, the dissimilarity between Zellweger syndrome and the recently characterized genuine cho-lesterol deficiency disorders [SLOS, Conradi-Huenermann syndrome (CDPX2), congenitalhemidysplasia with ichthyosiform erythroderma and limb defects (CHILD)] [90–92] are argu-ments against a significant role of peroxisomal cholesterogenesis in the etiology of PBDs. How-ever, although a deficiency of any of the enzymes in these disorders should inhibit endogenouscholesterol biosynthesis, the �7-sterol reductase (SLOS) [123,124], �8-�7-sterol isomerase (tat-tered mouse) [125] and C-4-sterol demethylase (bare patches mouse and striated mouse) [126]mutant mice all have distinct phenotypes. This may be due to variable teratogenic effects of differentprecursor molecules or variable ability of the sterols that are synthesized to substitute for cholesterol.Several authors have reported maturational abnormalities of the white matter in SLOS [for reviewsee 89]; however, most cranial MRI studies do not show white matter abnormalities.Enormous progress has been achieved during the last few years in identifying and under-
standing the biochemical and genetic defects that underlie the peroxisomal disorders. In spite ofthe definition of the gene defects, the mechanism through which these defects lead to abnormalmorphogenesis, defective white matter or neuronal degenerations, remains unknown in mostinstances. Investigations in human patients, in particular static morphologic examinations of tis-sues at autopsy, have contributed immensely to the recognition and understanding of these dis-eases, but such studies always have significant limitations, e.g. it is almost impossible to deducethe timecourse of changes in the brain: which changes came first or early in the disease process,and which followed as a result of the damage caused by the disease.
384 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

7. Mouse models for peroxisome biogenesis disorders
The pathogenesis of organ malformations and dysfunctions in patients with PBDs is notknown. However, in order to study these mechanisms two animal models of Zellweger syndromehave been generated by targeted inactivation of PEX2 [127] and PEX5 [128] genes. The functionof the PEX2 protein in the assembly of peroxisomes is still unclear, but mutations in this genewere shown to abolish PTS-1 and PTS-2 mediated import of peroxisomal proteins [129,130]. ThePEX5 gene encodes Pex5p, the cytoplasmic shuttle receptor for the import of most peroxisomalmatrix proteins (those targeted by a PTS-1 peptide) [19,131]. Both PEX2 and PEX5 knock-outmice have a severe peroxisomal import defect, lack functional peroxisomes, and exhibit all majorsigns, pathological defects, and biochemical abnormalities of ZS patients [127,128]. Therefore,these animal models provide an excellent model system to study the pathogenesis of organ dys-functions, including the characteristic defects in neuropathologic lesions, in peroxisome biogen-esis disorders and to investigate potential therapeutic strategies. A major advantage of the mousemodels is that the consequences of peroxisome deficiency can be analyzed at different stages ofembryonic development. In the case of the PEX2 knock-out mouse one long-living strain is alsoavailable, which allows even postnatal studies [132].Recently it was reported that in immortalized as well as primary PEX5�/� mouse fibroblast cul-
tures no difference in the cholesterol biosynthesis rate was observed, using [14C]mevalonate as sub-strate [101]. However, in the same study the cholesterol biosynthesis rate in fibroblasts from oneZellweger patient was shown to be 50% below control values, corresponding with previouslymentioned studies [101]. Cholesterol was not reduced in plasma, brain or liver of newborn peroxi-some-deficient PEX5�/� mice in comparison to wild-type or heterozygous littermates [101]. In con-trast to these findings our preliminary studies in PEX2�/� mice show that plasma total cholesterollevels are reduced by 40% as compared to wild-type or heterozygous littermates (unpublished data).
8. Embryonic development and cholesterol
Individuals with PBDs have abnormalities that already occur during fetal development. Since sev-eral studies have shown the importance of cholesterol in the fetal development [133] and peroxisomescontain all enzymes in the cholesterol biosynthetic pathway for the conversion of acetyl-CoA toFPP, it is tempting to speculate that disturbed cholesterol biosynthesis in the fetus contributes tothe etiology of PBDs. PEX2 and PEX5 mouse models for ZS now provide the opportunity to studythe putative involvement of peroxisomal cholesterol/isoprenoid biosynthesis in fetal development.The fetus has two potential sources of cholesterol, derived from endogenous and exogenous
origins. The endogenous source of cholesterol is comprised of that synthesized within the fetusitself. The rates of sterol synthesis are much greater in the fetus than in the adult and can accountfor a large proportion of the fetal cholesterol accrued [134]. The ability to regulate fetal sterolsynthesis rates is yet uncharacterized, but is dependent on the ability to transport regulatoryfactors to the fetus and on expression profiles of proteins that are involved in the regulation ofcholesterol synthesis.The inborn errors of cholesterol biosynthesis involve anabolic pathways in which the
endproduct deficiency cannot be adequately compensated for by maternal sources. Normal
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 385

development may be disrupted both by the accumulation of abnormal amounts of precursorsterols and deficiency of cholesterol. Whereas all evidence indicates that little or no cholesterolcrosses the placenta during the second and third trimesters, critical aspects of embryonic tissuedifferentiation may be sensitive to maternal blood cholesterol and LDL levels in the first trimester[135,136]. In the brain however, both in later fetal life and after birth, most if not all cholesterolis synthesized locally, not transported from blood lipoproteins. A deficiency of cholesterol inthe early weeks and months of fetal development may be an important determinant of thedysmorphic features and skeletal dysplasia of cholesterol biosynthesis disorders.Is cholesterol deficient in embryonic tissue of PBDs? Can a deficiency of cholesterol biosynth-
esis in PBD embryos lead to cholesterol levels below the known threshold for the development ofdysmorphogenesis? Lovastatin, an inhibitor of the key regulatory enzyme HMG-CoA reductase,cannot reduce cholesterol levels below that threshold [137]. Even the severe and diverse prenatalmetabolic disturbances of ZS have few effects on the embryonic body plan as seen in syndromescaused by mutations of homeobox genes and other transcriptional factors. But is a possibledecrease of cholesterol biosynthesis in the embryo sufficient to affect morphogenesis?A variety of cellular processes including gene transcription and signal transduction are regu-
lated by the cholesterol content of the membrane [138–140]. Considering the multiple roles ofcholesterol in the function of hedgehog proteins, it seems likely that besides the importance ofcholesterol in the autoprocessing of sonic hedgehog (SHH) [141] an interference could occur alsoat the level of signal transduction [142]. Cholesterol plays a key role in the assembly and functionof lipid rafts and caveolae [140,143]. Changes in cholesterol levels may modify the raft domains,and it has been shown recently that cholesterol-modified hedgehog is associated specifically withraft domains in Drosophila. Further examples include LDL receptor family members, as well asthe activation of some tyrosine kinase and MAP kinase pathways that involve cholesterol-richmicrodomains on the cell surface such as caveolae or rafts.Other less specific disturbances may contribute to the abnormal morphogenesis of PBDs.
Cholesterol is a major lipid component of the plasma membrane and changes in cholesterolcontent would be expected to affect membrane fluidity. If cholesterol is severely deficient in thefetus, such a severe disturbance in membrane sterol composition may affect developmental pro-cesses that involve cell-cell interactions, as suggested by Dehart et al. [144]. Studies by Lanoue etal. [145] suggested that in embryonic tissue a deficiency of cholesterol itself contributes to theabnormal embryological signaling of SLOS. Because of the necessity to integrate cholesterolbiosynthesis with other cellular processes, a general role of sterol intermediates in the control ofcell viability, proliferation or differentiation is conceivable. Decreased cholesterol levels couldalso influence development by affecting steroid hormone biosynthesis. Impaired synthesis ofneurosteroids could affect central nervous system development or function [146].
9. Conclusion
The data presented in this review illustrate a new model for the compartmentalization of thecholesterol biosynthetic pathway. As shown in Fig. 1 acetyl-CoA is derived in peroxisomes by theb-oxidation of very long chain fatty acids. This acetyl-CoA may then be utilized as substrate inthe synthesis of FPP within peroxisomes, since all of the enzymes required for these reactions are
386 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

localized in peroxisomes. The concentration of HMG-CoA may be crucial for the peroxisomalreactions since peroxisomes also contain HMG-CoA lyase, which can convert HMG-CoA toacetoacetate and acetyl-CoA, thus reversing the conversion by acetoacetyl-CoA thiolase andHMG-CoA synthase. Furthermore, it has been shown that peroxisomes contain an acyl-CoAthioesterase (PTE-2) [147] which hydrolyzes HMG-CoA, adding another possible level of reg-ulation to the pathway. In this compartmentalization model mevalonate is generated either inperoxisomes or comes to peroxisomes from the ER. Unpublished data from our group suggestthat the two HMG-CoA reductases yield two pools of mevalonate which have separate functionsin isoprenoid metabolism and regulation; mevalonate derived from the ER HMG-CoA reductasebeing used for sterol biosynthesis, while mevalonate derived from peroxisomal HMG-CoA lead-ing to the biosynthesis of the non-sterol products. Our goals therefore, for forthcoming researchis to address the challenging task of integrating the knowledge of the regulation of the cholesterolbiosynthetic pathway with the new levels of regulation which are possible due to the compart-mentalization of the pathway.
Acknowledgements
We wish to thank Daun Clizbe and Rainer Breitling for their helpful discussions on theseresearch topics. This work was supported in part by National Institute of Health grants DK58238and DK58040 to S.K. and by Max Kade Postdoctoral Exchange Grant to W. J. K.
References
[1] Keller GA, Barton MC, Shapiro DJ, Singer SJ. Proc Natl Acad Sci USA 1985;82:770–4.
[2] Keller GA, Pazirandeh M, Krisans SK. J Cell Biol 1986;103:875–86.
[3] de Duve C. Ann N Y Acad Sci 1996;804:1–10.
[4] Lazarow PB, Fujiki Y. Annu Rev Cell Biol 1985;1:489–530.
[5] Subramani S, Koller A, Snyder WB. Annu Rev Biochem 2000;69:399–418.
[6] Holroyd C, Erdmann R. FEBS Lett 2001;501:6–10.
[7] Hettema EH, Distel B, Tabak HF. Biochim Biophys Acta 1999;1451:17–54.
[8] Gould SJ, Keller GA, Subramani S. J Cell Biol 1987;105:2923–31.
[9] Elgersma Y, Vos A, van den Berg M, van Rosermund CWT, van ser Sluijs P, Distel B, et al. J Biol Chem 1996;
271:26375–82.
[10] Swinkles BW, Gould SJ, Bodnar AG, Rachubinski RA, Subramani S. EMBO J 1991;10:3255–62.
[11] Tsukamoto T, Hata S, Yokota S, Mirura S, Fujiki Y, Hijikata M, et al. J Biol Chem 1994;269:6001–60010.
[12] Gietl C, Faber KN, van der Klei IJ, Veenhuis M. Proc Natl Acad Sci USA 1994;91:3151–5.
[13] Flynn CR, Mullen RT, Trelease RN. The Plant Journal 1998;16:709–20.
[14] McNew JA, Goodman JM. J Cell Biol 1994;127:1245–57.
[15] Lee MS, Mullen RT, Trelease RN. Plant Cell 1997;9:185–97.
[16] Walton PA, Hill PE, Subramani S. Mol Biol Cell 1995;6:675–83.
[17] Gould SJ, Raymond GV, Valle D. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler K, Vogel-
stein B, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001.
p. 3181–217.
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 387

[18] Wanders RJA, Barth PG, Heymans HSA. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler K,
Vogelstein B, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001.
p. 3219–56.
[19] Gould SJ, Valle D. Trends Genet 2000;16:340–5.
[20] Moser HW. Mol Genet Metab 1999;68:316–27.
[21] Wanders RJA, Tager JM. Molec Aspects Med 1998;19:71–154.
[22] Lazarow PB, Moser HW. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular
bases of inherited disease. New York: McGraw-Hill, 1995. p. 2287–324.
[23] Powers JM, Moser HW, Moser AB, Upshur JK, Bradford BF, Pai SG, et al. Human Pathol 1995;16:610–20.
[24] Moser AB, Rasmussen M, Naidu S, Watkins PA, McGuinness M, Hajra AK, et al. J Pediatr 1995;127:13–22.
[25] Fukao T, Yamaguchi S, Kano M, Orii T, Fujiki Y, Osumi T, et al. J Clin Invest 1990;86:2086–92.
[26] Song XQ, Fukao T, Yamaguchi S, Miyazawa S, Hashimoto T, Orii T. Biochem Biophys Res Commun 1994;
201:478–85.
[27] Thompson SL, Krisans SK. J Biol Chem 1990;25:5731–5.
[28] Hovik R, Brodal B, Bartlett K, Osmundsen H. J Lipid Res 1991;32:993–9.
[29] Olivier LM, Kovacs W, Masuda K, Keller GA, Krisans SK. J Lipid Res 2000;41:1921–35.
[30] Ashmarina LI, Robert MF, Elsliger MA, Mitchell GA. Biochem J 1996;315:71–5.
[31] Ashmarina LI, Pshezhetsky AV, Branda S, Isaya G, Mitchell GA. J Lipid Res 1999;40:70–5.
[32] Filppula SA, Yagi AI, Kilpelainen SH,Novikov D, FitzpatrickD, VihinenM, et al. J Biol Chem 1998;273:349–55.
[33] Amery L, Fransen M, DeNys K, Mannaerts G, Van Veldhoven P. J Lipid Res 2000;41:1752–9.
[34] Ayte J, Gil-Gomez G, Haro D, Marrero PF, Hegardt FG. Proc Natl Acad Sci USA 1990;87:3874–8.
[35] Krisans SK, Rusnak N, Keller GA, Edwards PA. J Cell Biol 1988;107:122.
[36] Engfelt WH, Shackelford JE, Aboushadi N, Jessani N, Masuda K, Paton VG, et al. J Biol Chem 1997;
272:24579–87.
[37] Kovacs WJ, Faust PL, Keller GA, Krisans SK. Eur J Biochem 2001;268:4850–9.
[38] Szkopinska A, Swiezewska E, Skoneczny M. Biochimie 2001;83:427–32.
[39] Aboushadi N, Krisans SK. Biochemistry 2000;39:237–47.
[40] Basson ME, Thorsness M, Rine J. Proc Natl Acad Sci USA 1986;83:5563–7.
[41] Enjuto M, Balcells L, Campos N, Caelles C, Arro M, Boronat A. Proc Natl Acad Sci USA 1994;91:927–31.
[42] Stamellos KD, Shackelford JE, Tanaka RD, et al. J Biol Chem 1992;267:5560–8.
[43] Biardi L, Sreedhar A, Zokaei A, Vartak NB, Bozeat RL, Shackelford JE, et al. J Biol Chem 1994;269:1197–206.
[44] Biardi L, Krisans SK. J Biol Chem 1996;271:1784–8.
[45] Krisans SK, Ericsson J, Edwards PA, Keller GA. J Biol Chem 1994;269:14165–9.
[46] Olivier LM, Chambliss KL, Gibson KM, Krisans SK. J Lipid Res 1999;40:672–9.
[47] Paton VG, Shackelford JE, Krisans SK. J Biol Chem 1997;272:18945–50.
[48] Rilling HC, Chayet LT. In: Danielsson H, Stovall J, editors. Sterols and Bile Acids. New York: Elsevier Sci-
entific Publishing, 1985. p. 17–23.
[49] Goldstein JL, Brown MS. Nature 1990;343:425–30.
[50] Gupta SD, Mehan RS, Tansey TR, Chen HT, Goping G, Goldberg I, et al. J Lipid Res 1999;40:1572–84.
[51] Wilkin DJ, Kutsunai SY, Edwards PA. J Biol Chem 1990;265:4607–14.
[52] Ashby MN, Edwards PA. J Biol Chem 1989;264:635–40.
[53] FukaoT, Yamaguchi S, Kano M, Orii T, Fujiki Y, Osumi T, et al. J Clin Invest 1990;86:2086–92.
[54] Bonaldo MF, Lennon G, Soares MB. Genome Res 1996;6:791–806.
[55] Russ AP, Ruzicka V, Maerz W, Appelhans H, Gross W. Biochim Biophys Acta 1992;1132:329–31.
[56] Ayte J, Gil-Gomez G, Hegardt FG. Nucleic Acids Res 1990;18:3642.
[57] Tanaka RD, Lee LY, Schafer BL, Kratunis VJ, Mohler WA, Robinson GW, et al. Proc Natl Acad Sci USA
1990;87:2872–86.
[58] Schafer BL, Bishop RW, Kratunis VJ, Kalinowski SS, Mosley ST, Gibson KM, et al. J Biol Chem 1992;
267:13229–38.
388 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

[59] Chambliss KL, Slaughter CA, Schreiner R, Hoffmann GF, Gibson KM. J Biol Chem 1996;271:17330–4.
[60] Toth MJ, Huwyer L. J Biol Chem 1996;271:7895–8.
[61] Toth MJ, Huwyler L, Park J. Prep Biochem Biotechnol 1996;26:47–51.
[62] Xuan JW, Kowalski J, Chambers AF, Denhardt DT. Genomics 1994;20:129–31.
[63] Stamellos KD, Shackelford JE, Shechter I, Jiang G, Conrad D, Keller GA, et al. J Biol Chem 1993;268:12825–
36.
[64] Cohen LH, Griffioen M, van Roermund CW, Wanders RJ. Biochim Biophys Acta 1992;1126:114–8.
[65] Krisans SK. Ann NY Acad Sci 1996;804:142–64.
[66] Appelkvist EL, Reinhart M, Fischer R, Billheimer J, Dallner G. Arch Biochem Biophys 1990;282:318–25.
[67] Hashimoto F, Hayahi H. Biochim Biophys Acta 1994;1214:11–19.
[68] de Vet EC, Ijlst L, Oostheim W, Wanders RJ, van den Bosch H. J Biol Chem 1998;273:10296–301.
[69] Jansen GA, Ofman R, Ferdinandusse S, Ijlst L, Muijsers AO, Skjeldal OH, et al. Nat Genet 1997;17:190–3.
[70] Orfman R, Hettema EH, Hogenhout EM, Caruso U, Muijsers AO, Wanders RJ. Hum Mol Genet 1998;7:847–
53.
[71] Jansen GA, van den Brink DM, Ofman R, Draghici O, Dacremont G, Wanders RJ. Biochem Biophys Res
Commun 2001;283:674–9.
[72] Foulon V, Antonenkov VD, Croes K, Waelkens E, Mannaerts GP, Van Veldhoven PP, et al. Proc Natl Acad
Sci USA 1999;96:10039–44.
[73] Scotto JM, Hadchouel M, Odievre M, Laudat MH, Saudubray JM, Dulac O, et al. J Inherit Metab Dis 1982;
5:83–90.
[74] Poll-The BT, Saudubray JM, Ogier HA, Odievre M, Scotto JM, Monnens L, et al. Eur J Pediatr 1987;146:477–
83.
[75] Wanders RJ, Boltshauser E, Steinmann B, Spycher MA, Schutgens RB, et al. J Neurol Sci 1990;98:1–11.
[76] Mandel H, Meiron D, Schutgens RB, Wanders RJ, Berant M. J Pediatr Gastroenterol Nutr 1992;14:83–5.
[77] Mandel H, Berant M, Meiron D, Aizin A, Oiknine J, Brook JG, et al. J Inherit Metab Dis 1992;15:774–84.
[78] Collins JC, Keyserman F, Rumsey SC, Deckelbaum RJ. American Heart Association 1993 (Abstract).
[79] Wanders RJA, Romeijn GJ. J Inherit Metab Dis 1996;19:193–6.
[80] Lazarow PB, Moser HW. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic basis of
inherited disease. New York: McGraw-Hill, 1989. p. 1479–509.
[81] Hodge VJ, Gould SJ, Subramani S, Moser HW, Krisans SK. Biochem Biophys Res Commun 1991;181:537–41.
[82] Mandel H, Getsis M, Rosenblat M, Berant M, Aviram M. J Lipid Res 1995;36:1385–91.
[83] Malle E, Oettl K, Sattler W, Hoefler G, Kostner GM. Eur J Clin Invest 1995;25:59–67.
[84] Oettl K, Malle E, Grillhofer H, Sattler W, Kostner GM. Clin Chim Acta 1996;251:131–43.
[85] Wanders RJA, Romeijn GJ. Biochem Biophys Res Commun 1998;247:663–7.
[86] Aboushadi N, Krisans SK. J Lipid Res 1998;39:1781–91.
[87] Van Heusden GPH, van Beckhoven JRCM, Thieringer R, Raetz CRH, Wirtz KWA. Biochim Biophys Acta
1992;1126:81–7.
[88] Kelley RI. Adv Pediatr 2000;47:1–52.
[89] Roux C, Wolf C, Mulliez N, Gaoua W, Cormier V, Chevy F, et al. Am J Clin Nutr 2000;71:1270S–9s.
[90] Kelley RI, Hennekam RCM. J Med Genet 2000;37:321–35.
[91] Waterham HR, Wanders RJA. Biochim Biophys Acta 2000;1529:340–56.
[92] Moebius FF, Fitzky BU, Glossmann H. Trends Endocrinol Metab 2000;11:106–14.
[93] Nwokoro NA, Wassif CA, Porter FD. Mol Genet Metab 2001;74:105–19.
[94] Berger R, Smit GPA, Schierbeek H, Bijsterveld K, le Coultre R. Clin Chim Acta 1985;152:219–22.
[95] Hoffmann GF, Charpentier C, Mayatepek E, Mancini J, Leichsenring M, Gibson KM, et al. Pediatrics 1993;
91:915–21.
[96] Houten SM, Romeijn GJ, Koster J, Gray RGF, Darbyshire P, Smit GPA, et al. Hum Mol Genet 1999;8:1523–
8.
[97] Keller RK, Simonet WS. Biochem Biophys Res Commun 1988;152:857–61.
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 389

[98] Huebner C, Hoffmann GF, Charpentier C, Gibson KM, Finckh B, Puhl H, et al. Pediatr Res 1993;34:129–33.
[99] Gibson KM, Hoffmann GF, Schwall A, Broock RL, Aramaki S, Sweetman L, et al. J Lipid Res 1990;31:515–
21.
[100] Hoffmann GF, Wiesmann UN, Brendel S, Keller RK, Gibson KM. Pediatr Res 1997;41:541–6.
[101] Vanhorebeek I, Baes M, Declercq PE. Biochim Biophys Acta 2001;1532:28–36.
[102] Gressens P, Baes M, Leroux P, Lombet A, van Veldhoven P, Janssen A, et al. Ann Neurol 2000;48:336–43.
[103] Rachubinski RA, Subramani S. Cell 1995;83:525–8.
[104] Sakakura Y, Shimano H, Sone H, Takahashi A, Inoue K, Toyoshima H, et al. Biochem Biophys Res Commun
2001;286:176–83.
[105] Krauss S, Quant PA. J Theor Biol 1996;182:381–8.
[106] Powers JM, Moser HW. Brain Pathol 1998;8:101–20.
[107] Snipes GJ, Suter U. In: Bittman R, editor. Subcellular biochemistry, vol. Cholesterol: its functions and meta-
bolism in biology and medicine. New York: Plenum Press, 1997. p. 173–204.
[108] Jurevics H, Morell P. J Neurochem 1995;64:895–901.
[109] Jurevics H, Kidwai FZ, Morell P. J Lipid Res 1997;38:723–33.
[110] Turley SD, Burns DK, Dietschy JM. Am J Physiol 1998;274:E1099–0E1105.
[111] Spady DK, Dietschy JM. J Lipid Res 1983;24:303–15.
[112] Dietschy JM, Kita T, Suckling KE, et al. J Lipid Res 1983;24:469–80.
[113] Osono Y, Woollett LA, Herz J, Dietschy JM. J Clin Invest 1995;95:1124–32.
[114] Turley SD, Burns DK, Rosenfeld CR, Dietschy JM. J Lipid Res 1996;37:1953–61.
[115] Powers JM. J Neuropathol Exp Neurol 1995;5:710–9.
[116] Powers JM, Schaumburg HH, Johnson AB, Raine CS. Invest Cell Pathol 1980;3:353–76.
[117] Ho JK, Moser H, Kishimoto Y, Hamilton JA. J Clin Invest 1995;96:1455–63.
[118] Watkins PA, McGuinness MC, Raymond GV, Hicks BA, Sisk JM, Moser AB, et al. Ann Neurol 1995;38:472–
7.
[119] Lazo O, Singh AK, Singh I. J Neurochem 1991;56:1343–53.
[120] Korey SR, Orchen M. Arch Biochem Biophys 1959;83:381–9.
[121] Arnold G, Holtzman E. Brain Res 1978;155:1–17.
[122] Hinse CH, Shah N. J Neurochem 1971;18:1989–98.
[123] Wassif CA, Zhu P, Kratz L, Krakowiak PA, Battaile KP, Weight FF, et al. Hum Mol Gen 2001;10:555–64.
[124] Fitzky BU, Moebius FF, Asaoka H, Waage-Baudet H, Xu L, Xu G, et al. J Clin Invest 2001;108:905–15.
[125] Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, et al. Nat Genet 1999;22:286–90.
[126] Liu XY, Dangel AW, Kelley RI, Zhao W, Denny P, Botcherby M, et al. Nat Genet 1999;22:182–7.
[127] Faust PL, Hatten ME. J Cell Biol 1997;139:1293–305.
[128] Baes M, Gressens P, Baumgart E, Carmeliet P, Casteels M, Fransen M, et al. Nat Genet 1997;17:49–57.
[129] Tsukamoto T, Miura S, Fujiki Y. Nature 1991;350:77–81.
[130] Slawecki ML, Dodt G, Steinberg S, Moser AB, Moser HW, Gould SJ. J Cell Sci 1995;108:1817–29.
[131] Dammai V, Subramani S. Cell 2001;105:187–96.
[132] Faust PL, Su H, Moser A, Moser HW. J Mol Neurosci 2001;16:289–97.
[133] Farese RV, Herz J. Trends Genet 1998;14:115–20.
[134] Dietschy JM, Turley SD, Spady DK. J Lipid Res 1993;34:1637–59.
[135] Willnow TE, Hilpert J, Armstrong SA, Rohlmann A, Hammer RE, Burns DK, et al. Proc Natl Acad Sci USA
1996;93:8460–4.
[136] Howell BW, Herz J. Curr Opin Neurobiol 2001;11:74–81.
[137] Barbu V, Roux C, Dupuid R, Gardette J, Maziere J. Proc Soc Exp Biol Med 1984;176:54–9.
[138] Brown MS, Goldstein JL. Proc Natl Acad Sci USA 1999;96:11041–8.
[139] Brown MS, Rawson RB, Goldstein JL. Cell 2000;100:391–8.
[140] Simons K, Ikonen E. Science 2000;290:1721–6.
[141] Mann RK, Beachy PA. Biochim Biophys Acta 2000;1529:188–202.
390 W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391

[142] Incardona JP, Eaton S. Curr Opin Cell Biol 2000;12:193–203.
[143] Fielding CJ. Curr Opin Lipidol 2001;12:281–7.
[144] Dehart DB, Lanoue L, Tint GS, Sulik KK. Am J Med Genet 1997;68:328–37.
[145] Lanoue L, Dehart DB, Hinsdale ME, Maeda N, Tint GS, Sulik KK. Am J Med Genet 1997;73:24–31.
[146] Compagnone NA, Mellon SH. Front Neuroendocrinol 2000;21:1–56.
[147] Hunt MC, Solaas K, Kase BF, Alexson SHE. J Biol Chem 2002;277:1128–38.
W.J. Kovacs et al. / Progress in Lipid Research 41 (2002) 369–391 391





























