Embed Size (px)
Citation preview

Monica Olson, Lilach O. Lerman and Amir LermanJoerg Herrmann, Ardan M. Saguner, Daniele Versari, Timothy E. Peterson, Alejandro Chade,
Chronic Proteasome Inhibition Contributes to Coronary Atherosclerosis
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2007 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.107.1529592007;101:865-874; originally published online September 6, 2007;Circ Res.
http://circres.ahajournals.org/content/101/9/865World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2007/09/06/CIRCRESAHA.107.152959.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from

Chronic Proteasome Inhibition Contributes toCoronary Atherosclerosis
Joerg Herrmann, Ardan M. Saguner, Daniele Versari, Timothy E. Peterson, Alejandro Chade,Monica Olson, Lilach O. Lerman, Amir Lerman
Abstract—The proteasome is responsible for the degradation of oxidized proteins, and proteasome inhibition has beenshown to generate oxidative stress in vitro. Atherosclerosis is thought to be initiated as a consequence of increasedendogenous oxidative stress. The current study was designed to assess whether chronic proteasome inhibition isassociated with early coronary atherosclerosis. Female pigs, 3 months of age, were randomized to a normal (N) orhigh-cholesterol (HC) diet (2% cholesterol, 15% lard) without or with twice weekly subcutaneous injections of theproteasome inhibitor (PSI) MLN-273 (0.08 mg/kg, N�PSI and HC�PSI) for a period of 12 weeks (n�5 per group).Coronary vasorelaxation to bradykinin (10�10.5 to 10�6.5 mol/L) and sodium nitroprusside (10�9 to 10�5 mol/L) wasassessed by in vitro organ chamber experiments, intima–media ratio by morphometric analysis of Elastica–vanGieson–stained slides, and intima superoxide production by dihydroethidium fluorescence. Vasorelaxation to 10�6.5
mol/L bradykinin was reduced in HC compared with N (69�7 versus 90�2%, P�0.05) and further reduced in N�PSIand HC�PSI (57�6 and 48�13%, P�0.05 versus N and HC for each). Compared with N (0.03�0.01), intima–mediaratio was higher in N�PSI (0.09�0.04, P�0.01) and HC�PSI (0.15�0.06, P�0.05). Compared with N (0.6�0.9% ofintima area), dihydroethidium fluorescence was higher in HC, N�PSI, and HC�PSI (8.9�1.6, 6.0�3.5, and 7.2�3.9%of intima area, P�0.05 for all). Thus, chronic proteasome inhibition is associated with increased coronary arteryoxidative stress and early atherosclerosis. These findings support the significance of the proteasome and related proteinquality control for vascular biology and pathology. (Circ Res. 2007;101:865-874.)
Key Words: atherosclerosis � endothelial dysfunction � oxidative stress � proteasome � ubiquitin
Atherosclerotic cardiovascular disease (ASCVD) is con-sidered to be initiated as a response to an injurious
stimulus, or clinically, a cardiovascular risk factor.1 A com-mon denominator of the pathophysiological mode of action ofmany cardiovascular risk factors is an increase in the gener-ation of reactive oxygen species, especially superoxide an-ions, surpassing antioxidant capabilities and resulting inoxidative stress.2,3 Superoxide anions rapidly react with NO,generating, for instance, the highly cytotoxic product per-oxynitrite and its footprint nitrotyrosine.4 In addition, modi-fication of signaling pathways leads to the alteration of theactivity and expression of transcription factors and growthfactors. The functional and structural consequences of thesemolecular changes include impairment of endothelium-dependent vasorelaxation and intimal thickening, constitutingthe early stage of atherosclerosis.4–6
Eighty to 90% of all intracellular proteins are degraded viathe 20S proteasome, a barrel-shaped complex formed by 2outer (�) rings and 2 inner (�) rings, each composed of 7subunits.7 The �1, �2, and �5 subunits harbor caspase-like,trypsin-like, and chymotrypsin-like activities, respectively,
with the latter being of utmost importance for overall protea-some function. The 20S proteasome is self-sufficient for thedegradation of oxidized proteins.8 It also operates in conjunc-tion with the ubiquitin system, which catalyzes the binding ofubiquitin molecules to target proteins, allowing their recog-nition by the 26S proteasome, ie, by the 19S subunits to eitherside of the 20S proteolytic complex.9 Possibly related to theimpairment of the function of the proteolytic core, accumu-lation of ubiquitin/ubiquitinated proteins can be seen inhuman atherosclerotic plaques and particularly in more activelesions, characterized by increased oxidative stress.10,11 In-deed, there is in vitro evidence that high levels of oxidativestress can impair proteasome function with cytotoxic conse-quences.12 In turn, impairment in proteasome function canincrease intracellular oxidative stress.13 Whereas these stud-ies support a detrimental effect of impaired proteasomefunction, other reports suggest that proteasome inhibitionmay be of benefit for atherosclerotic plaque progressionand complication.14,15 Hence, there has remained uncer-tainty with regard to the pathophysiological role of theproteasome in ASCVD.
Original received March 27, 2007; revision received August 16, 2007; accepted August 20, 2007.From the Divisions of Cardiovascular Diseases (J.H., A.M.S., D.V., T.E.P., M.O., A.L.) and Nephrology and Hypertension (A.C., L.O.L.), Department
of Internal Medicine, Mayo Clinic and College of Medicine, Rochester, Minn.Correspondence to Amir Lerman, MD, Division of Cardiology, Mayo Clinic Rochester, 200 First St SW, Rochester, MN 55905. E-mail
[email protected]© 2007 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.107.152959
865
Molecular Medicine
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from

Potent and specific pharmacological inhibitors of the pro-teasome have been tested extensively over the past years,such as the boronate-type inhibitor MLN-273.9 As a result,PS-341 (bortezomib [Velcade]), a proteasome inhibitor (PSI)very closely related to MLN-273, was approved for thetreatment of therapy-refractory multiple myeloma in 2003.16
Beyond therapeutic applications, the in vivo use of thesespecific inhibitors is an attractive means to gain pathophysi-ological insight. Thus, in the current study, chronic adminis-tration of MLN-273 was used to define the pathophysiolog-ical significance of the proteasome for ASCVD. Ourhypothesis was that by virtue of the reported stimulatingeffect on oxidative stress, chronic proteasome inhibitionwould impair the function and structure of coronary arteriesin normal animals and would aggravate the vascular changesof early atherosclerosis in hypercholesterolemic animals.
Materials and Methods
AnimalsThe current study was approved by the Mayo Foundation Institu-tional Animal Care and Use Committee, and all procedures were inaccordance with NIH guidelines. Three-month-old female domesticpigs (Pork Partners, Stewartsville, Mn, and Larson Products,Sargeant, Mn) were randomized to a normal chow diet (n�5) or ahigh-cholesterol diet (HC) (2% cholesterol, 15% lard; TD 93296,Harlan Teklad, Madison, Wisconsin; n�5) without or with twiceweekly subcutaneous injections of the boronate-type PSI MLN-273(0.08 mg/kg; Millennium Pharmaceuticals, Cambridge, Mass; nor-mal diet [N]�PSI [PSI] and HC�PSI, n�5 each). Similar to PS-341,this drug specifically inhibits the chymotrypsin-like activities of theproteasome, and in vivo analysis of chymotrypsin-like proteasomefunction was performed in treated animals to ensure a maximumdegree of 60% to 80% inhibition at 1 hour after drug administra-tion.17 After 11 weeks, mean arterial blood pressure and heart ratewere measured by an invasive signal transducer. After 12 weeks,blood was obtained for plasma analyses. Subsequently, animals wereeuthanized and the hearts were immediately harvested for coronaryvasoreactivity testing and tissue fixation, either in formalin forsubsequent paraffin embedding, or by snap freezing in liquidnitrogen for storage at �80°C.
Determination of Plasma Lipid Profile andOxidized Low-Density Lipoprotein LevelsAs described previously, plasma concentrations of triglycerides andtotal, HDL, and LDL cholesterol were determined with a commercialagent.18 Circulating levels of oxidized LDL were measured inplasma using a spectrophotometric enzyme immunoassay kit (Mer-codia, Uppsala, Sweden).19
Analysis of Proteasome ActivityProteasome activity in peripheral mononuclear cells was performedas described previously.17 Coronary artery proteasome activity wasquantified as outlined previously but with substantial modifications(see the expanded Materials and Methods section in the online datasupplement at http://circres.ahajournals.org).11,18
In Vitro Analysis of Vascular ReactivityOrgan chamber experiments were performed according to an estab-lished protocol.20–22
TUNEL StainingTerminal deoxynucleotidyl transferase end-labeling (TUNEL) stain-ing was performed by use of the ApopTag In Situ ApoptosisDetection Kit (Intergen Co, Purchase, NY), as described previously(see the online data supplement).10
Histologic and Morphometric AnalysesHematoxylin/eosin and Elastica–van Gieson staining was performedas previously reported.22,23 Using a digital image system, morpho-metric analyses on Elastica–van Gieson–stained slides were per-formed at a magnification of 10x (see the online data supplement).
Oil Red O StainingUnfixed frozen coronary artery segments were cut into 10-�m-thicksections, placed on a glass slide, and stored at �80°C until the dayof the experiment, which was performed as described previously.24,25
Immunostaining and ImmunoblottingImmunostaining and immunoblotting were performed as outlinedpreviously (see the online data supplement).10,18,21,23,25
Fluorescent MicroscopyDihydroethidium (DHE) (Sigma) was used to demonstrate in situlevels of superoxide production, as described previously.11,26,27 Thepercentage of intima area positive for immunofluorescence wasquantified as outlined for immunostaining.
Lucigenin ChemiluminescenceSuperoxide production was measured from aortic samples usinglucigenin-enhanced chemiluminescence (5 �mol/L), as describedpreviously, and validated previously.28,29
Statistical AnalysisContinuous data were expressed as means�SEM. Multiple groupcomparisons were made by 1-way ANOVA with Student–Newman–Keuls post hoc analysis. Two group comparisons were made byStudent’s t test. Statistical significance was accepted at P�0.05.
ResultsHemodynamic and Laboratory ParametersAnimals treated with MLN-273 had similar body weight andmean arterial blood pressure but slower heart rate compared
Table 1. Characteristics of the Experimental Groups
Characteristics N HC N�PSI HC�PSI
Body weight, kg 51�3 64�6 58�3 53�5
Mean arterial blood pressure, mm Hg 113�6 117�5 121�6 122�6
Heart rate, beats per minute 64�4 62�6 60�8 56�3†
Serum triglyceride, mg/dL 34�6 62�12† 88�30† 83�25†
Serum total cholesterol, mg/dL 72�5 479�41* 87�11 471�14*
Serum HDL, mg/dL 39�3 84�10* 28�5 99�4*
Serum LDL, mg/dL 26�4 382�34* 42�9 356�12*
Oxidized LDL, U/mL 12.5�1.6 19.1�1.8* 11.1�0.8 27.9�5.3*‡
Values are means�SEM. *P�0.05 vs N and N�PSI, †P�0.1 vs N, ‡P�0.05 vs HC.
866 Circulation Research October 26, 2007
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from
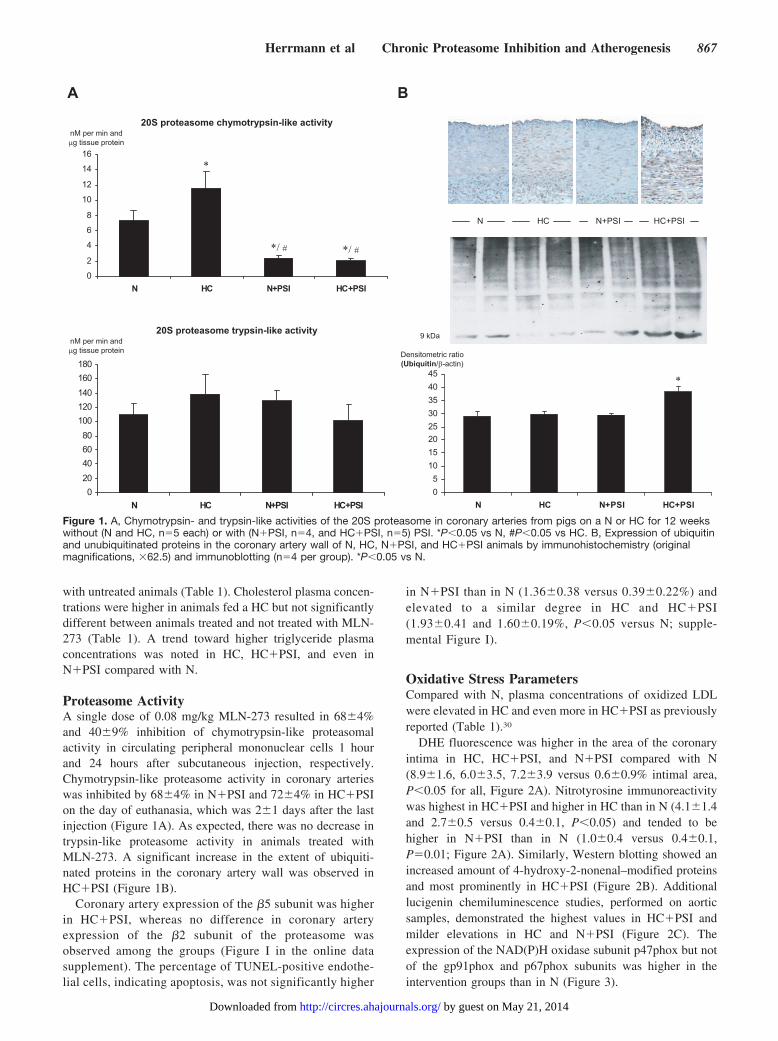
with untreated animals (Table 1). Cholesterol plasma concen-trations were higher in animals fed a HC but not significantlydifferent between animals treated and not treated with MLN-273 (Table 1). A trend toward higher triglyceride plasmaconcentrations was noted in HC, HC�PSI, and even inN�PSI compared with N.
Proteasome ActivityA single dose of 0.08 mg/kg MLN-273 resulted in 68�4%and 40�9% inhibition of chymotrypsin-like proteasomalactivity in circulating peripheral mononuclear cells 1 hourand 24 hours after subcutaneous injection, respectively.Chymotrypsin-like proteasome activity in coronary arterieswas inhibited by 68�4% in N�PSI and 72�4% in HC�PSIon the day of euthanasia, which was 2�1 days after the lastinjection (Figure 1A). As expected, there was no decrease intrypsin-like proteasome activity in animals treated withMLN-273. A significant increase in the extent of ubiquiti-nated proteins in the coronary artery wall was observed inHC�PSI (Figure 1B).
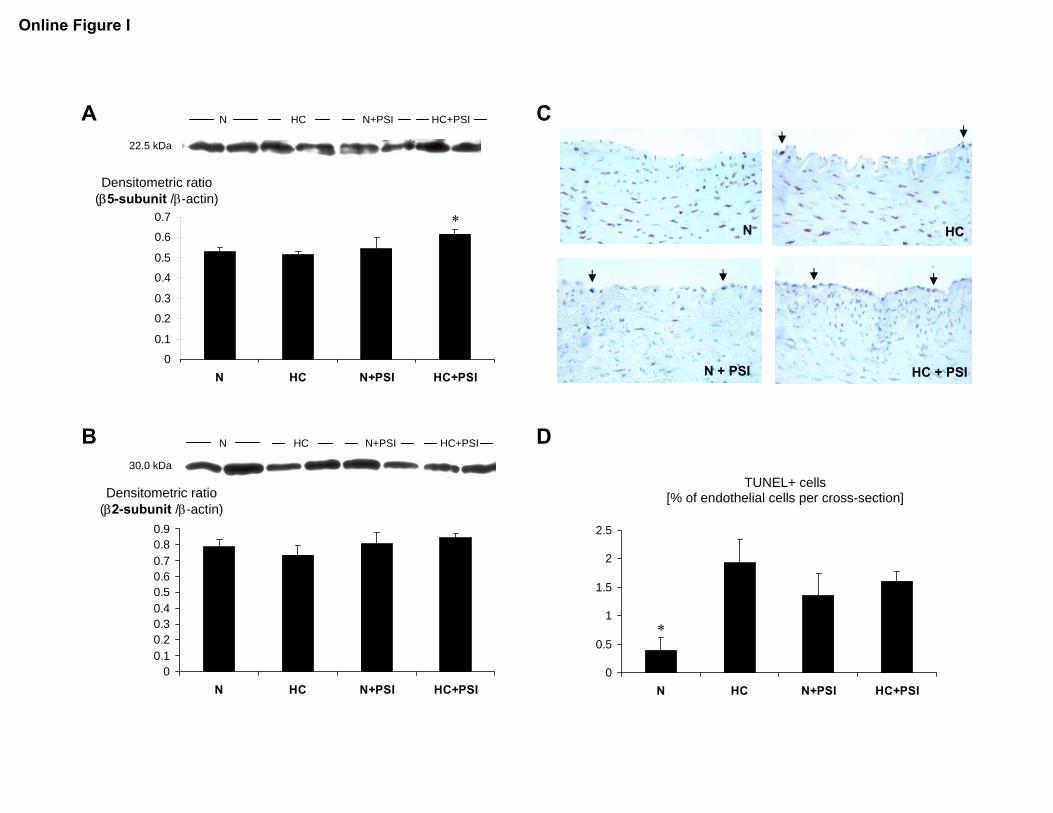
Coronary artery expression of the �5 subunit was higherin HC�PSI, whereas no difference in coronary arteryexpression of the �2 subunit of the proteasome wasobserved among the groups (Figure I in the online datasupplement). The percentage of TUNEL-positive endothe-lial cells, indicating apoptosis, was not significantly higher
in N�PSI than in N (1.36�0.38 versus 0.39�0.22%) andelevated to a similar degree in HC and HC�PSI(1.93�0.41 and 1.60�0.19%, P�0.05 versus N; supple-mental Figure I).
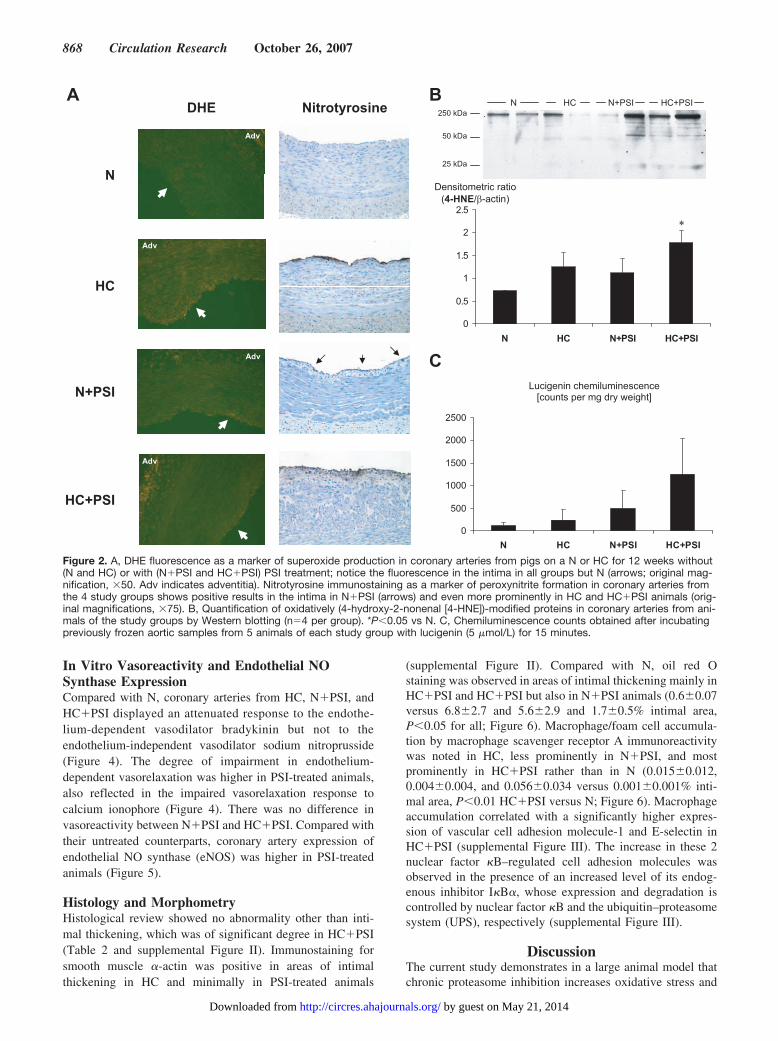
Oxidative Stress ParametersCompared with N, plasma concentrations of oxidized LDLwere elevated in HC and even more in HC�PSI as previouslyreported (Table 1).30
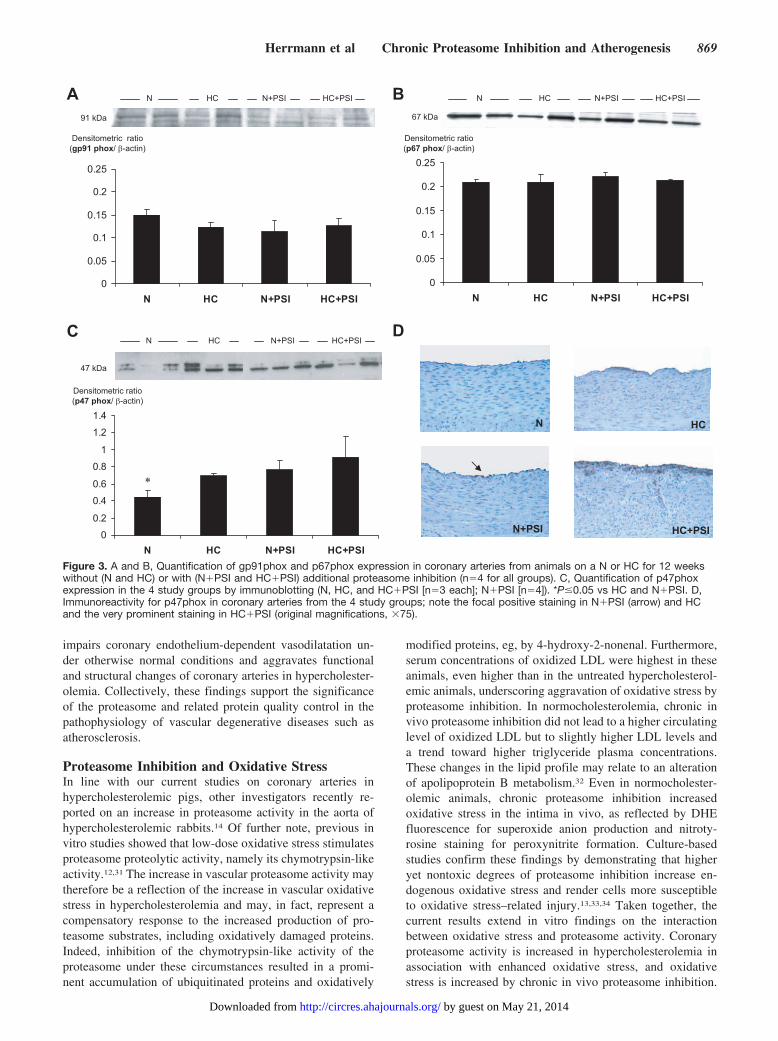
DHE fluorescence was higher in the area of the coronaryintima in HC, HC�PSI, and N�PSI compared with N(8.9�1.6, 6.0�3.5, 7.2�3.9 versus 0.6�0.9% intimal area,P�0.05 for all, Figure 2A). Nitrotyrosine immunoreactivitywas highest in HC�PSI and higher in HC than in N (4.1�1.4and 2.7�0.5 versus 0.4�0.1, P�0.05) and tended to behigher in N�PSI than in N (1.0�0.4 versus 0.4�0.1,P�0.01; Figure 2A). Similarly, Western blotting showed anincreased amount of 4-hydroxy-2-nonenal–modified proteinsand most prominently in HC�PSI (Figure 2B). Additionallucigenin chemiluminescence studies, performed on aorticsamples, demonstrated the highest values in HC�PSI andmilder elevations in HC and N�PSI (Figure 2C). Theexpression of the NAD(P)H oxidase subunit p47phox but notof the gp91phox and p67phox subunits was higher in theintervention groups than in N (Figure 3).
Figure 1. A, Chymotrypsin- and trypsin-like activities of the 20S proteasome in coronary arteries from pigs on a N or HC for 12 weekswithout (N and HC, n�5 each) or with (N�PSI, n�4, and HC�PSI, n�5) PSI. *P�0.05 vs N, #P�0.05 vs HC. B, Expression of ubiquitinand unubiquitinated proteins in the coronary artery wall of N, HC, N�PSI, and HC�PSI animals by immunohistochemistry (originalmagnifications, �62.5) and immunoblotting (n�4 per group). *P�0.05 vs N.
Herrmann et al Chronic Proteasome Inhibition and Atherogenesis 867
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from
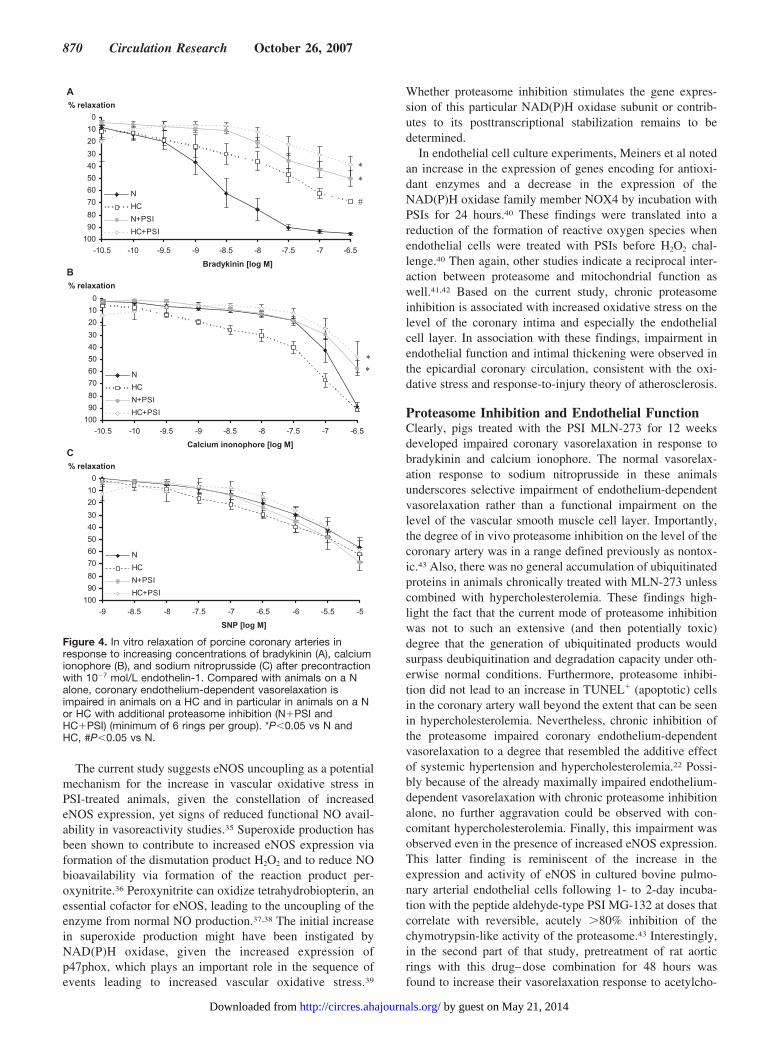
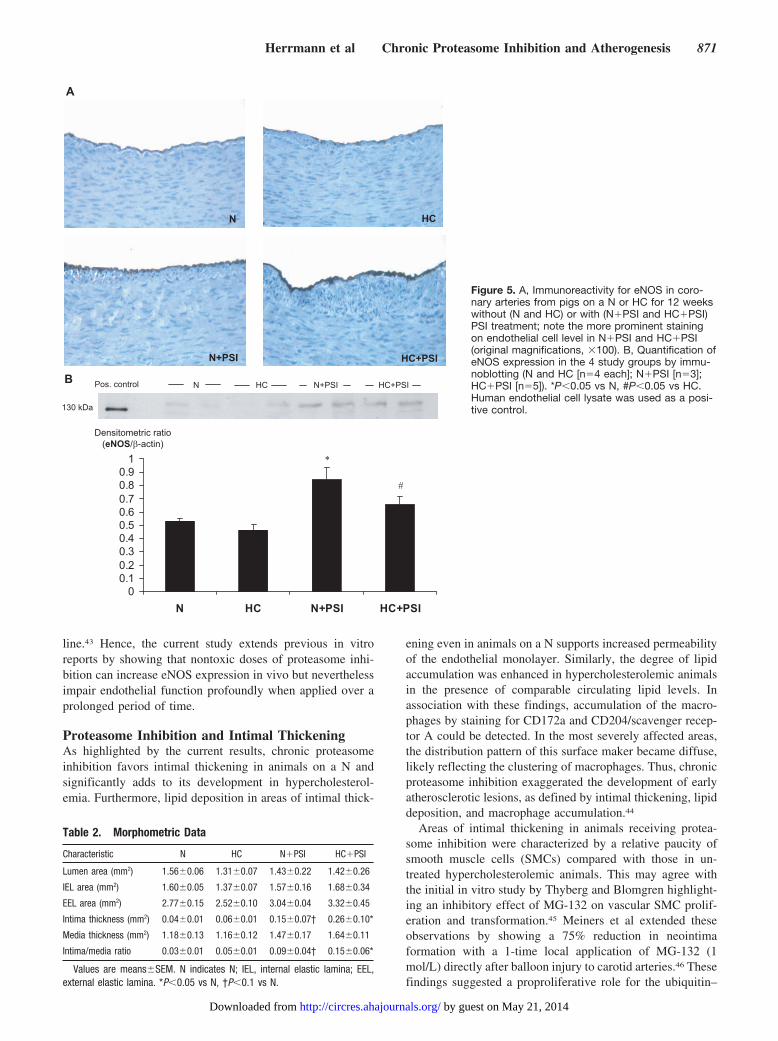
In Vitro Vasoreactivity and Endothelial NOSynthase ExpressionCompared with N, coronary arteries from HC, N�PSI, andHC�PSI displayed an attenuated response to the endothe-lium-dependent vasodilator bradykinin but not to theendothelium-independent vasodilator sodium nitroprusside(Figure 4). The degree of impairment in endothelium-dependent vasorelaxation was higher in PSI-treated animals,also reflected in the impaired vasorelaxation response tocalcium ionophore (Figure 4). There was no difference invasoreactivity between N�PSI and HC�PSI. Compared withtheir untreated counterparts, coronary artery expression ofendothelial NO synthase (eNOS) was higher in PSI-treatedanimals (Figure 5).
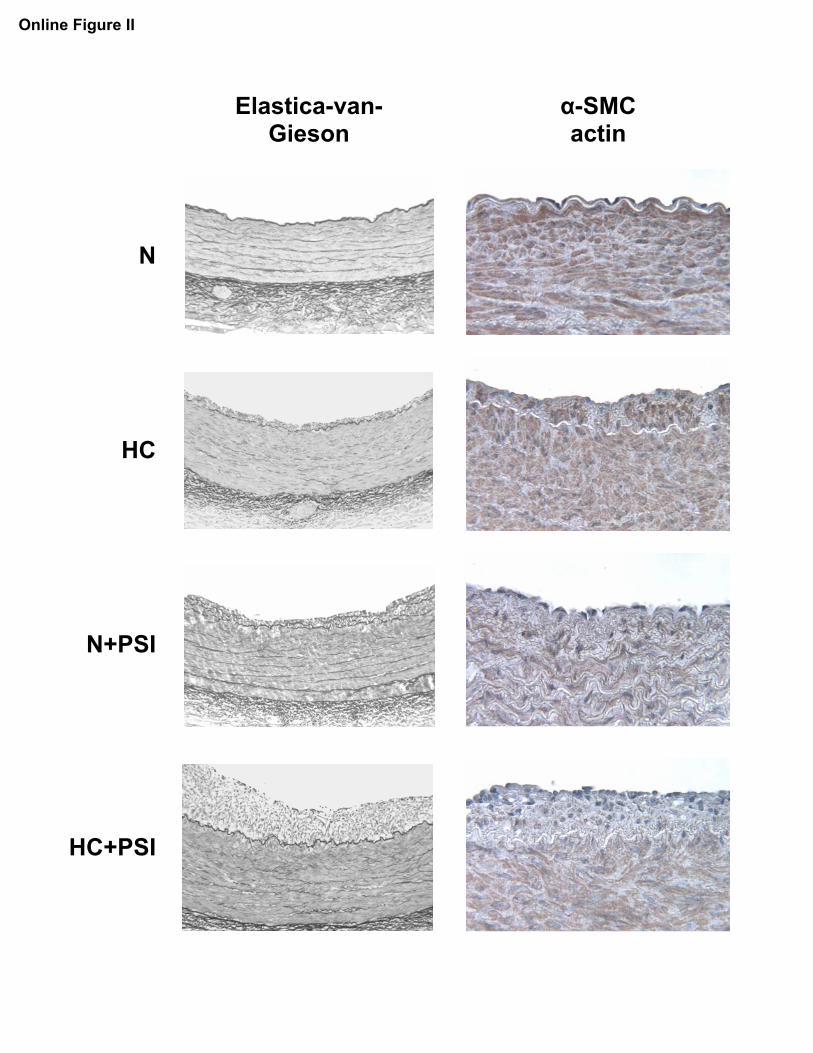
Histology and MorphometryHistological review showed no abnormality other than inti-mal thickening, which was of significant degree in HC�PSI(Table 2 and supplemental Figure II). Immunostaining forsmooth muscle �-actin was positive in areas of intimalthickening in HC and minimally in PSI-treated animals
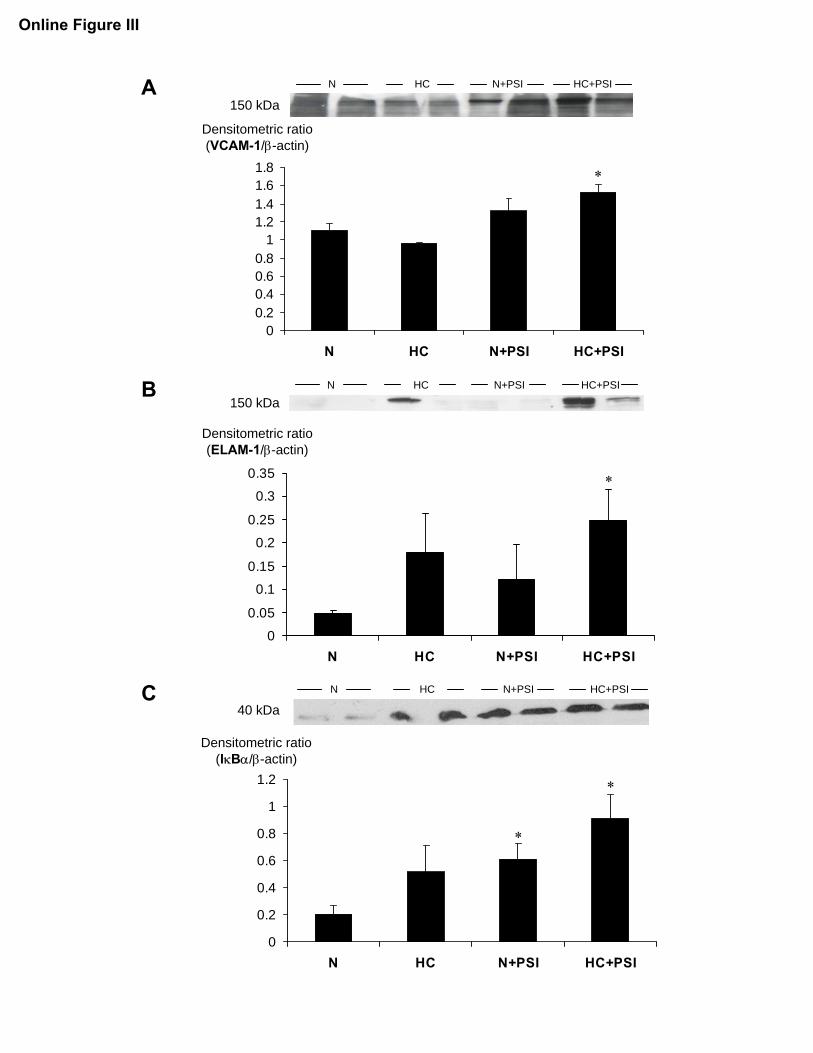
(supplemental Figure II). Compared with N, oil red Ostaining was observed in areas of intimal thickening mainly inHC�PSI and HC�PSI but also in N�PSI animals (0.6�0.07versus 6.8�2.7 and 5.6�2.9 and 1.7�0.5% intimal area,P�0.05 for all; Figure 6). Macrophage/foam cell accumula-tion by macrophage scavenger receptor A immunoreactivitywas noted in HC, less prominently in N�PSI, and mostprominently in HC�PSI rather than in N (0.015�0.012,0.004�0.004, and 0.056�0.034 versus 0.001�0.001% inti-mal area, P�0.01 HC�PSI versus N; Figure 6). Macrophageaccumulation correlated with a significantly higher expres-sion of vascular cell adhesion molecule-1 and E-selectin inHC�PSI (supplemental Figure III). The increase in these 2nuclear factor �B–regulated cell adhesion molecules wasobserved in the presence of an increased level of its endog-enous inhibitor I�B�, whose expression and degradation iscontrolled by nuclear factor �B and the ubiquitin–proteasomesystem (UPS), respectively (supplemental Figure III).
DiscussionThe current study demonstrates in a large animal model thatchronic proteasome inhibition increases oxidative stress and
Figure 2. A, DHE fluorescence as a marker of superoxide production in coronary arteries from pigs on a N or HC for 12 weeks without(N and HC) or with (N�PSI and HC�PSI) PSI treatment; notice the fluorescence in the intima in all groups but N (arrows; original mag-nification, �50. Adv indicates adventitia). Nitrotyrosine immunostaining as a marker of peroxynitrite formation in coronary arteries fromthe 4 study groups shows positive results in the intima in N�PSI (arrows) and even more prominently in HC and HC�PSI animals (orig-inal magnifications, �75). B, Quantification of oxidatively (4-hydroxy-2-nonenal [4-HNE])-modified proteins in coronary arteries from ani-mals of the study groups by Western blotting (n�4 per group). *P�0.05 vs N. C, Chemiluminescence counts obtained after incubatingpreviously frozen aortic samples from 5 animals of each study group with lucigenin (5 �mol/L) for 15 minutes.
868 Circulation Research October 26, 2007
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from
impairs coronary endothelium-dependent vasodilatation un-der otherwise normal conditions and aggravates functionaland structural changes of coronary arteries in hypercholester-olemia. Collectively, these findings support the significanceof the proteasome and related protein quality control in thepathophysiology of vascular degenerative diseases such asatherosclerosis.
Proteasome Inhibition and Oxidative StressIn line with our current studies on coronary arteries inhypercholesterolemic pigs, other investigators recently re-ported on an increase in proteasome activity in the aorta ofhypercholesterolemic rabbits.14 Of further note, previous invitro studies showed that low-dose oxidative stress stimulatesproteasome proteolytic activity, namely its chymotrypsin-likeactivity.12,31 The increase in vascular proteasome activity maytherefore be a reflection of the increase in vascular oxidativestress in hypercholesterolemia and may, in fact, represent acompensatory response to the increased production of pro-teasome substrates, including oxidatively damaged proteins.Indeed, inhibition of the chymotrypsin-like activity of theproteasome under these circumstances resulted in a promi-nent accumulation of ubiquitinated proteins and oxidatively
modified proteins, eg, by 4-hydroxy-2-nonenal. Furthermore,serum concentrations of oxidized LDL were highest in theseanimals, even higher than in the untreated hypercholesterol-emic animals, underscoring aggravation of oxidative stress byproteasome inhibition. In normocholesterolemia, chronic invivo proteasome inhibition did not lead to a higher circulatinglevel of oxidized LDL but to slightly higher LDL levels anda trend toward higher triglyceride plasma concentrations.These changes in the lipid profile may relate to an alterationof apolipoprotein B metabolism.32 Even in normocholester-olemic animals, chronic proteasome inhibition increasedoxidative stress in the intima in vivo, as reflected by DHEfluorescence for superoxide anion production and nitroty-rosine staining for peroxynitrite formation. Culture-basedstudies confirm these findings by demonstrating that higheryet nontoxic degrees of proteasome inhibition increase en-dogenous oxidative stress and render cells more susceptibleto oxidative stress–related injury.13,33,34 Taken together, thecurrent results extend in vitro findings on the interactionbetween oxidative stress and proteasome activity. Coronaryproteasome activity is increased in hypercholesterolemia inassociation with enhanced oxidative stress, and oxidativestress is increased by chronic in vivo proteasome inhibition.
Figure 3. A and B, Quantification of gp91phox and p67phox expression in coronary arteries from animals on a N or HC for 12 weekswithout (N and HC) or with (N�PSI and HC�PSI) additional proteasome inhibition (n�4 for all groups). C, Quantification of p47phoxexpression in the 4 study groups by immunoblotting (N, HC, and HC�PSI [n�3 each]; N�PSI [n�4]). *P�0.05 vs HC and N�PSI. D,Immunoreactivity for p47phox in coronary arteries from the 4 study groups; note the focal positive staining in N�PSI (arrow) and HCand the very prominent staining in HC�PSI (original magnifications, �75).
Herrmann et al Chronic Proteasome Inhibition and Atherogenesis 869
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from
The current study suggests eNOS uncoupling as a potentialmechanism for the increase in vascular oxidative stress inPSI-treated animals, given the constellation of increasedeNOS expression, yet signs of reduced functional NO avail-ability in vasoreactivity studies.35 Superoxide production hasbeen shown to contribute to increased eNOS expression viaformation of the dismutation product H2O2 and to reduce NObioavailability via formation of the reaction product per-oxynitrite.36 Peroxynitrite can oxidize tetrahydrobiopterin, anessential cofactor for eNOS, leading to the uncoupling of theenzyme from normal NO production.37,38 The initial increasein superoxide production might have been instigated byNAD(P)H oxidase, given the increased expression ofp47phox, which plays an important role in the sequence ofevents leading to increased vascular oxidative stress.39
Whether proteasome inhibition stimulates the gene expres-sion of this particular NAD(P)H oxidase subunit or contrib-utes to its posttranscriptional stabilization remains to bedetermined.
In endothelial cell culture experiments, Meiners et al notedan increase in the expression of genes encoding for antioxi-dant enzymes and a decrease in the expression of theNAD(P)H oxidase family member NOX4 by incubation withPSIs for 24 hours.40 These findings were translated into areduction of the formation of reactive oxygen species whenendothelial cells were treated with PSIs before H2O2 chal-lenge.40 Then again, other studies indicate a reciprocal inter-action between proteasome and mitochondrial function aswell.41,42 Based on the current study, chronic proteasomeinhibition is associated with increased oxidative stress on thelevel of the coronary intima and especially the endothelialcell layer. In association with these findings, impairment inendothelial function and intimal thickening were observed inthe epicardial coronary circulation, consistent with the oxi-dative stress and response-to-injury theory of atherosclerosis.
Proteasome Inhibition and Endothelial FunctionClearly, pigs treated with the PSI MLN-273 for 12 weeksdeveloped impaired coronary vasorelaxation in response tobradykinin and calcium ionophore. The normal vasorelax-ation response to sodium nitroprusside in these animalsunderscores selective impairment of endothelium-dependentvasorelaxation rather than a functional impairment on thelevel of the vascular smooth muscle cell layer. Importantly,the degree of in vivo proteasome inhibition on the level of thecoronary artery was in a range defined previously as nontox-ic.43 Also, there was no general accumulation of ubiquitinatedproteins in animals chronically treated with MLN-273 unlesscombined with hypercholesterolemia. These findings high-light the fact that the current mode of proteasome inhibitionwas not to such an extensive (and then potentially toxic)degree that the generation of ubiquitinated products wouldsurpass deubiquitination and degradation capacity under oth-erwise normal conditions. Furthermore, proteasome inhibi-tion did not lead to an increase in TUNEL� (apoptotic) cellsin the coronary artery wall beyond the extent that can be seenin hypercholesterolemia. Nevertheless, chronic inhibition ofthe proteasome impaired coronary endothelium-dependentvasorelaxation to a degree that resembled the additive effectof systemic hypertension and hypercholesterolemia.22 Possi-bly because of the already maximally impaired endothelium-dependent vasorelaxation with chronic proteasome inhibitionalone, no further aggravation could be observed with con-comitant hypercholesterolemia. Finally, this impairment wasobserved even in the presence of increased eNOS expression.This latter finding is reminiscent of the increase in theexpression and activity of eNOS in cultured bovine pulmo-nary arterial endothelial cells following 1- to 2-day incuba-tion with the peptide aldehyde-type PSI MG-132 at doses thatcorrelate with reversible, acutely �80% inhibition of thechymotrypsin-like activity of the proteasome.43 Interestingly,in the second part of that study, pretreatment of rat aorticrings with this drug–dose combination for 48 hours wasfound to increase their vasorelaxation response to acetylcho-
Figure 4. In vitro relaxation of porcine coronary arteries inresponse to increasing concentrations of bradykinin (A), calciumionophore (B), and sodium nitroprusside (C) after precontractionwith 10�7 mol/L endothelin-1. Compared with animals on a Nalone, coronary endothelium-dependent vasorelaxation isimpaired in animals on a HC and in particular in animals on a Nor HC with additional proteasome inhibition (N�PSI andHC�PSI) (minimum of 6 rings per group). *P�0.05 vs N andHC, #P�0.05 vs N.
870 Circulation Research October 26, 2007
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from
line.43 Hence, the current study extends previous in vitroreports by showing that nontoxic doses of proteasome inhi-bition can increase eNOS expression in vivo but neverthelessimpair endothelial function profoundly when applied over aprolonged period of time.
Proteasome Inhibition and Intimal ThickeningAs highlighted by the current results, chronic proteasomeinhibition favors intimal thickening in animals on a N andsignificantly adds to its development in hypercholesterol-emia. Furthermore, lipid deposition in areas of intimal thick-
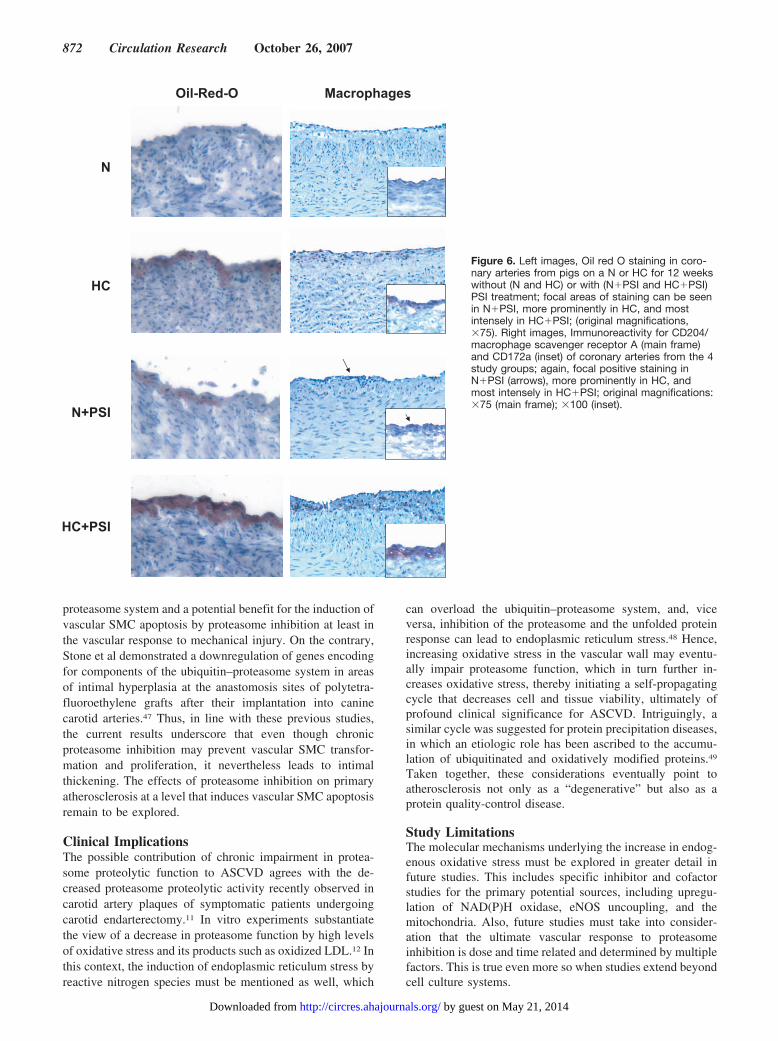
ening even in animals on a N supports increased permeabilityof the endothelial monolayer. Similarly, the degree of lipidaccumulation was enhanced in hypercholesterolemic animalsin the presence of comparable circulating lipid levels. Inassociation with these findings, accumulation of the macro-phages by staining for CD172a and CD204/scavenger recep-tor A could be detected. In the most severely affected areas,the distribution pattern of this surface maker became diffuse,likely reflecting the clustering of macrophages. Thus, chronicproteasome inhibition exaggerated the development of earlyatherosclerotic lesions, as defined by intimal thickening, lipiddeposition, and macrophage accumulation.44
Areas of intimal thickening in animals receiving protea-some inhibition were characterized by a relative paucity ofsmooth muscle cells (SMCs) compared with those in un-treated hypercholesterolemic animals. This may agree withthe initial in vitro study by Thyberg and Blomgren highlight-ing an inhibitory effect of MG-132 on vascular SMC prolif-eration and transformation.45 Meiners et al extended theseobservations by showing a 75% reduction in neointimaformation with a 1-time local application of MG-132 (1mol/L) directly after balloon injury to carotid arteries.46 Thesefindings suggested a proproliferative role for the ubiquitin–
Figure 5. A, Immunoreactivity for eNOS in coro-nary arteries from pigs on a N or HC for 12 weekswithout (N and HC) or with (N�PSI and HC�PSI)PSI treatment; note the more prominent stainingon endothelial cell level in N�PSI and HC�PSI(original magnifications, �100). B, Quantification ofeNOS expression in the 4 study groups by immu-noblotting (N and HC [n�4 each]; N�PSI [n�3];HC�PSI [n�5]). *P�0.05 vs N, #P�0.05 vs HC.Human endothelial cell lysate was used as a posi-tive control.
Table 2. Morphometric Data
Characteristic N HC N�PSI HC�PSI
Lumen area (mm2) 1.56�0.06 1.31�0.07 1.43�0.22 1.42�0.26
IEL area (mm2) 1.60�0.05 1.37�0.07 1.57�0.16 1.68�0.34
EEL area (mm2) 2.77�0.15 2.52�0.10 3.04�0.04 3.32�0.45
Intima thickness (mm2) 0.04�0.01 0.06�0.01 0.15�0.07† 0.26�0.10*
Media thickness (mm2) 1.18�0.13 1.16�0.12 1.47�0.17 1.64�0.11
Intima/media ratio 0.03�0.01 0.05�0.01 0.09�0.04† 0.15�0.06*
Values are means�SEM. N indicates N; IEL, internal elastic lamina; EEL,external elastic lamina. *P�0.05 vs N, †P�0.1 vs N.
Herrmann et al Chronic Proteasome Inhibition and Atherogenesis 871
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from
proteasome system and a potential benefit for the induction ofvascular SMC apoptosis by proteasome inhibition at least inthe vascular response to mechanical injury. On the contrary,Stone et al demonstrated a downregulation of genes encodingfor components of the ubiquitin–proteasome system in areasof intimal hyperplasia at the anastomosis sites of polytetra-fluoroethylene grafts after their implantation into caninecarotid arteries.47 Thus, in line with these previous studies,the current results underscore that even though chronicproteasome inhibition may prevent vascular SMC transfor-mation and proliferation, it nevertheless leads to intimalthickening. The effects of proteasome inhibition on primaryatherosclerosis at a level that induces vascular SMC apoptosisremain to be explored.
Clinical ImplicationsThe possible contribution of chronic impairment in protea-some proteolytic function to ASCVD agrees with the de-creased proteasome proteolytic activity recently observed incarotid artery plaques of symptomatic patients undergoingcarotid endarterectomy.11 In vitro experiments substantiatethe view of a decrease in proteasome function by high levelsof oxidative stress and its products such as oxidized LDL.12 Inthis context, the induction of endoplasmic reticulum stress byreactive nitrogen species must be mentioned as well, which
can overload the ubiquitin–proteasome system, and, viceversa, inhibition of the proteasome and the unfolded proteinresponse can lead to endoplasmic reticulum stress.48 Hence,increasing oxidative stress in the vascular wall may eventu-ally impair proteasome function, which in turn further in-creases oxidative stress, thereby initiating a self-propagatingcycle that decreases cell and tissue viability, ultimately ofprofound clinical significance for ASCVD. Intriguingly, asimilar cycle was suggested for protein precipitation diseases,in which an etiologic role has been ascribed to the accumu-lation of ubiquitinated and oxidatively modified proteins.49
Taken together, these considerations eventually point toatherosclerosis not only as a “degenerative” but also as aprotein quality-control disease.
Study LimitationsThe molecular mechanisms underlying the increase in endog-enous oxidative stress must be explored in greater detail infuture studies. This includes specific inhibitor and cofactorstudies for the primary potential sources, including upregu-lation of NAD(P)H oxidase, eNOS uncoupling, and themitochondria. Also, future studies must take into consider-ation that the ultimate vascular response to proteasomeinhibition is dose and time related and determined by multiplefactors. This is true even more so when studies extend beyondcell culture systems.
Figure 6. Left images, Oil red O staining in coro-nary arteries from pigs on a N or HC for 12 weekswithout (N and HC) or with (N�PSI and HC�PSI)PSI treatment; focal areas of staining can be seenin N�PSI, more prominently in HC, and mostintensely in HC�PSI; (original magnifications,�75). Right images, Immunoreactivity for CD204/macrophage scavenger receptor A (main frame)and CD172a (inset) of coronary arteries from the 4study groups; again, focal positive staining inN�PSI (arrows), more prominently in HC, andmost intensely in HC�PSI; original magnifications:�75 (main frame); �100 (inset).
872 Circulation Research October 26, 2007
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from

In contrast to our previous study, we performed more rapidtissue homogenization with a glass grinder and simultaneous,plate reader–based measurements of all probes and usedepoxomicin, the most selective PSI currently available, tocalculate the proteasome-attributable degradation of the flu-orogenic substrate.18 It is very likely, for this reason, that thecurrent results differ from our previous findings in theidentification of an increase in chymotrypsin-like activity ofthe proteasome in coronary arteries of hypercholesterolemicanimals. Indeed, as indicated by others, the outcome ofproteasome activity studies is subject to and hence limited bythe methodology.50
ConclusionsChronic proteasome inhibition is associated with increasedoxidative stress, impairment in coronary endothelium-dependent vasorelaxation and intimal thickening, resemblingand aggravating the vascular effects of traditional cardiovas-cular risk factors such as hypercholesterolemia. These find-ings support the significance of the proteasome and relatedprotein quality-control mechanisms for vascular biology andpathology.
AcknowledgmentsWe are very grateful to Millenium Pharmaceutics forproviding MLN-273.
Sources of FundingThis work was supported by NIH grants R01 HL63911-04 and K24HL69840-01 (to A.L.) and RO1 HL77131 and RO1 DK73608 (toL.O.L.), the Mayo Foundation, and the Mayo Stiftung. A.L. is anEstablished Investigator of the American Heart Association.
DisclosuresNone.
References1. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;
340:115–126.2. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases:
the role of oxidant stress. Circ Res. 2000;87:840–844.3. Sorescu D, Szocs K, Griendling KK. NAD(P)H oxidases and their rel-
evance to atherosclerosis. Trends Cardiovasc Med. 2001;11:124–131.4. Herrmann J, Lerman A. The endothelium: dysfunction and beyond.
J Nucl Cardiol. 2001;8:197–206.5. De Nigris F, Lerman LO, Condorelli M, Lerman A, Napoli C. Oxidation-
sensitive transcription factors and molecular mechanisms in the arterialwall. Antioxid Redox Signal. 2001;3:1119–1130.
6. Napoli C, de Nigris F, Palinski W. Multiple role of reactive oxygenspecies in the arterial wall. J Cell Biochem. 2001;82:674–682.
7. Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and26S proteasomes. Annu Rev Biochem. 1996;65:801–847.
8. Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugationis not required for the degradation of oxidized proteins by proteasome.J Biol Chem. 2003;278:311–318.
9. Herrmann J, Ciechanover A, Lerman LO, Lerman A. The ubiquitin-proteasome system in cardiovascular diseases-a hypothesis extended.Cardiovasc Res. 2004;61:11–21.
10. Herrmann J, Edwards WD, Holmes DR Jr, Shogren KL, Lerman LO,Ciechanover A, Lerman A. Increased ubiquitin immunoreactivity inunstable atherosclerotic plaques associated with acute coronary syn-dromes. J Am Coll Cardiol. 2002;40:1919–1927.
11. Versari D, Herrmann J, Gossl M, Mannheim D, Sattler K, Meyer FB,Lerman LO, Lerman A. Dysregulation of the ubiquitin-proteasomesystem in human carotid atherosclerosis. Arterioscler Thromb Vasc Biol.2006;26:2132–2139.
12. Vieira O, Escargueil-Blanc I, Jurgens G, Borner C, Almeida L, SalvayreR, Negre-Salvayre A. Oxidized LDLs alter the activity of the ubiquitin-proteasome pathway: potential role in oxidized LDL-induced apoptosis.FASEB J. 2000;14:532–542.
13. Lee MH, Hyun DH, Jenner P, Halliwell B. Effect of proteasome inhi-bition on cellular oxidative damage, antioxidant defenses and nitric oxideproduction. J Neurochem. 2001;78:32–41.
14. Tan C, Li Y, Tan X, Pan H, Huang W. Inhibition of the ubiquitin-proteasome system: a new avenue for atherosclerosis. Clin Chem LabMed. 2006;44:1218–1225.
15. Marfella R, D’Amico M, Di Filippo C, Baldi A, Siniscalchi M, Sasso FC,Portoghese M, Carbonara O, Crescenzi B, Sangiuolo P, Nicoletti GF,Rossiello R, Ferraraccio F, Cacciapuoti F, Verza M, Coppola L, Rossi F,Paolisso G. Increased activity of the ubiquitin-proteasome system inpatients with symptomatic carotid disease is associated with enhancedinflammation and may destabilize the atherosclerotic plaque: effects ofrosiglitazone treatment. J Am Coll Cardiol. 2006;47:2444–2455.
16. Richardson PG. A review of the proteasome inhibitor bortezomib inmultiple myeloma. Expert Opin Pharmacother. 2004;5:1321–1331.
17. Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott PJ.Proteasome inhibition measurements: clinical application. Clin Chem.2000;46:673–683.
18. Hermann J, Gulati R, Napoli C, Woodrum JE, Lerman LO, Rodriguez-Porcel M, Sica V, Simari RD, Ciechanover A, Lerman A. Oxidativestress-related increase in ubiquitination in early coronary atherogenesis.FASEB J. 2003;17:1730–1732.
19. Chade AR, Mushin OP, Zhu X, Rodriguez-Porcel M, Grande JP, TextorSC, Lerman A, Lerman LO. Pathways of renal fibrosis and modulation ofmatrix turnover in experimental hypercholesterolemia. Hypertension.2005;46:772–779.
20. Herrmann J, Lerman LO, Rodriguez-Porcel M, Holmes DR Jr, RichardsonDM, Ritman EL, Lerman A. Coronary vasa vasorum neovascularizationprecedes epicardial endothelial dysfunction in experimental hypercholester-olemia. Cardiovasc Res. 2001;51:762–766.
21. Rodriguez-Porcel M, Lerman LO, Holmes DR Jr, Richardson D, NapoliC, Lerman A. Chronic antioxidant supplementation attenuates nuclearfactor-kappa B activation and preserves endothelial function in hypercho-lesterolemic pigs. Cardiovasc Res. 2002;53:1010–1018.
22. Rodriguez-Porcel M, Lerman LO, Herrmann J, Sawamura T, Napoli C,Lerman A. Hypercholesterolemia and hypertension have synergistic del-eterious effects on coronary endothelial function. Arterioscler ThrombVasc Biol. 2003;23:885–891.
23. Herrmann J, Samee S, Chade A, Rodriguez Porcel M, Lerman LO,Lerman A. Differential effect of experimental hypertension and hyper-cholesterolemia on adventitial remodeling. Arterioscler Thromb VascBiol. 2005;25:447–453.
24. de Nigris F, Lerman LO, Rodriguez-Porcel M, De Montis MP, Lerman A,Napoli C. c-myc activation in early coronary lesions in experimentalhypercholesterolemia. Biochem Biophys Res Commun. 2001;281:945–950.
25. Wilson SH, Caplice NM, Simari RD, Holmes DR Jr, Carlson PJ, LermanA. Activated nuclear factor-kappaB is present in the coronary vasculaturein experimental hypercholesterolemia. Atherosclerosis. 2000;148:23–30.
26. Chade AR, Krier JD, Rodriguez-Porcel M, Breen JF, McKusick MA,Lerman A, Lerman LO. Comparison of acute and chronic antioxidantinterventions in experimental renovascular disease. Am J Physiol RenalPhysiol. 2004;286:F1079–F1086.
27. Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL,Lerman A, Lerman LO. Cortical microvascular remodeling in the stenotickidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol.2004;24:1854–1859.
28. Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increasesendothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551.
29. Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA,Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotionin apoE-deficient mice: implications for interactions between per-oxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288.
30. Chade AR, Herrmann J, Zhu X, Krier JD, Lerman A, Lerman LO. Effectsof proteasome inhibition on the kidney in experimental hypercholester-olemia. J Am Soc Nephrol. 2005;16:1005–1012.
31. Gomes-Marcondes MC, Tisdale MJ. Induction of protein catabolism andthe ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett.2002;180:69–74.
Herrmann et al Chronic Proteasome Inhibition and Atherogenesis 873
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from

32. Liang JS, Distler O, Cooper DA, Jamil H, Deckelbaum RJ, Ginsberg HN,Sturley SL. HIV protease inhibitors protect apolipoprotein B from deg-radation by the proteasome: a potential mechanism for protease inhibi-tor-induced hyperlipidemia. Nat Med. 2001;7:1327–1331.
33. Lee CS, Tee LY, Warmke T, Vinjamoori A, Cai A, Fagan AM, Snider BJ.A proteasomal stress response: pre-treatment with proteasome inhibitorsincreases proteasome activity and reduces neuronal vulnerability to oxi-dative injury. J Neurochem. 2004;91:996–1006.
34. Lev N, Melamed E, Offen D. Proteasomal inhibition hypersensitizesdifferentiated neuroblastoma cells to oxidative damage. Neurosci Lett.2006;399:27–32.
35. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vasculardisease: from marvel to menace. Circulation. 2006;113:1708–1714.
36. Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Tran-scriptional and posttranscriptional regulation of endothelial nitric oxidesynthase expression by hydrogen peroxide. Circ Res. 2000;86:347–354.
37. Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite:implications for vascular endothelial function. Biochem Biophys ResCommun. 1999;263:681–684.
38. Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions ofperoxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implicationsfor uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–22554.
39. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM,Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads touncoupling of endothelial cell nitric oxide synthase in hypertension.J Clin Invest. 2003;111:1201–1209.
40. Meiners S, Ludwig A, Lorenz M, Dreger H, Baumann G, Stangl V, Stangl K.Nontoxic proteasome inhibition activates a protective antioxidant defenseresponse in endothelial cells. Free Radic Biol Med. 2006;40:2232–2241.
41. Dhanasekaran A, Kotamraju S, Karunakaran C, Kalivendi SV, Thomas S,Joseph J, Kalyanaraman B. Mitochondria superoxide dismutase mimeticinhibits peroxide-induced oxidative damage and apoptosis: role of mito-chondrial superoxide. Free Radic Biol Med. 2005;39:567–583.
42. Ding Q, Dimayuga E, Keller JN. Proteasome regulation of oxidativestress in aging and age-related diseases of the CNS. Antioxid RedoxSignal. 2006;8:163–172.
43. Stangl V, Lorenz M, Meiners S, Ludwig A, Bartsch C, Moobed M,Vietzke A, Kinkel HT, Baumann G, Stangl K. Long-term up-regulation ofeNOS and improvement of endothelial function by inhibition of theubiquitin-proteasome pathway. FASEB J. 2004;18:272–279.
44. Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr, RosenfeldME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definitionof initial, fatty streak, and intermediate lesions of atherosclerosis. A reportfrom the Committee on Vascular Lesions of the Council on Arterioscle-rosis, American Heart Association. Circulation. 1994;89:2462–2478.
45. Thyberg J, Blomgren K. Effects of proteasome and calpain inhibitors onthe structural reorganization and proliferation of vascular smooth musclecells in primary culture. Lab Invest. 1999;79:1077–1088.
46. Meiners S, Laule M, Rother W, Guenther C, Prauka I, Muschick P,Baumann G, Kloetzel PM, Stangl K. Ubiquitin-proteasome pathway as anew target for the prevention of restenosis. Circulation. 2002;105:483–489.
47. Stone DH, Sivamurthy N, Contreras MA, Fitzgerald L, LoGerfo FW,Quist WC. Altered ubiquitin/proteasome expression in anastomoticintimal hyperplasia. J Vasc Surg. 2001;34:1016–1022.
48. Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, Austin RC.Peroxynitrite causes endoplasmic reticulum stress and apoptosis in humanvascular endothelium: implications in atherogenesis. Arterioscler ThrombVasc Biol. 2005;25:2623–2629.
49. Dudek EJ, Shang F, Valverde P, Liu Q, Hobbs M, Taylor A. Selectivityof the ubiquitin pathway for oxidatively modified proteins: relevance toprotein precipitation diseases. FASEB J. 2005;19:1707–1709.
50. Powell SR, Davies KJ, Divald A. Optimal determination of heart tissue26S-proteasome activity requires maximal stimulating ATP concen-trations. J Mol Cell Cardiol. 2007;42:265–269.
874 Circulation Research October 26, 2007
by guest on May 21, 2014http://circres.ahajournals.org/Downloaded from

Online Materials and Methods
Analysis of coronary proteasome activity
Frozen coronary arteries were homogenized by a motorized tissue grinder at 550 RPM
while keeping the samples at ice-cold temperatures and using a lysis buffer of the
following composition: 50 nM HEPES (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1% Triton
X-100. Subsequently, the homogenate was centrifuged at 10,000x g at 40 C for 15
minutes. The supernatant was obtained, its protein content was determined by Bradford
assay, and 30 µg were used for the individual reaction set-up. The reaction buffer was of
the following composition: 25 mM HEPES (pH 7.5), 5 mM EDTA, 0.5% NP-40 with and
without 0.01% SDS and the substrates 25 µM LLVY-AMC and 50 µM VGA-AMC
(BioMol, Plymouth Meeting, PA) for the determination of chymotrypsin-like and trypsin-
like activity, respectively. The final reaction volume in the 96-well plate set-up was 100
µL. Increase in the concentration of the fluorogenic compound 7-amino-4-methyl-
coumarin (AMC) after an incubation period of 60 minutes at 370 C was measured by use
of a 380/460 nm filtered fluorometric plate reader (SpectraMax Gemini XPS, Global
Medical Instrumentation Inc., Ramsey, MN). Measurements were performed in
duplicates and in the presence and absence of epoxomicin (25 µM, A.G. Scientific Inc.,
San Diego, CA). Individual proteasome proteolytic activity was calculated as the
difference between non-inhibited and inhibited activity and expressed as nanomoles per
minute and mg tissue protein.

Histologic and morphometric analyses
Intima thickness was calculated as IEL area minus lumen area and media thickness as
EEL area minus IEL area, allowing calculation of the intima-media ratio (IMR). IMRs
were not different between proximal and distal segments (0.071±0.045 vs. 0.072±0.048)
and between two different coronary arteries of the same pig (0.084±0.12 vs. 0.085±0.10)
with all values falling within two standard deviations of the mean difference on Bland-
Altman plots. Hence, one cross-section was taken per animal for the morphometric
analyses in this study.
TUNEL staining
Apoptotic cells were defined as TUNEL positive cells with condensed, pyknotic nuclei.
Female rodent mammary gland tissue, 3-5 days after weaning, was used as a positive
control with a rate of apoptotic cells of 4-7%, underscoring a high sensitivity assay setup
(standard sensitivity 1-2%). Absence of staining was confirmed on slides prepared with
omission of the TdT enzyme from the labeling (negative control).
Immunostaining
Porcine coronary artery slides were deparaffinized and rehydrated, followed by antigen
retrieval with steaming in citrate acid (1M, pH 6.0) and quenching of endogenous tissue
peroxidase activity by incubation with 3% H202 solution. For CD172a/macrophage
staining, 10 µm frozen slides were fixed in acetone at 4ºC for 10 minutes prior to the
incubation with 3% H202 solution. Primary antibodies, including anti-ubiquitin (Covance,
Berkeley, CA, dilution 1:200), anti-nitrotyrosine (Zymed, San Francisco, dilution 1:500),

anti-p47phox (Santa Cruz Biotechnology, Santa Cruz, CA, dilution 1:50), anti-eNOS
(Assay Designs, Ann Arbor, MI, dilution 1:100), anti-alpha smooth muscle cell (α-SMC)
actin (Dako Corp., Carpinteria, CA, dilution 1:1000), anti-CD172a/macrophage (VMRD
Inc., Pullman, WA, 1:2000) and anti-CD204/macrophage scavenger receptor-A (Cosmo
Bio, Carlsbad, CA, dilution 1:25), were applied at 40C overnight and were detected with
the EnVision kit (Dako) in peroxidase-labeling technique with 3,3-diaminobenzidine
tetra-hydrochloride or NovaRed as chromogens (Vector Laboratories, Inc., Burlingame,
CA). Incubations with unspecific isotype antibodies served as specificity controls. All
sections were counterstained with Gill No. 2 hematoxylin. Immunoreactivity was
quantified by use of a computer-aided image analysis program (MetaMorph 4.6,
Molecular Devices Corp., Sunnyvale, CA).
Immunoblotting
Protein content in coronary homogenates was analyzed by a Bradford assay (Bio-Rad,
Hercules, CA), and under reducing conditions (unless indicated otherwise) equal amounts
of protein (at least 30 µg) from coronary artery homogenates were dissolved in SDS-
polyacrylamide gels and electrophoretically transferred on polyvinylidene difluoride or
nitrocellulose membranes. These membranes were blocked in 1xPBS/3% non-fat milk
overnight and incubated for 1 hours at room temperature with the following primary
antibodies: anti-ubiquitin (Covance, Berkeley, CA, dilution 1:1000), anti-proteasome
subunit β5 and β2 (BioMol, dilution 1:500), anti-eNOS (Transduction Laboratories,
Lexington, KY, dilution 1:200), anti-vascular cell adhesion molecule-1 (VCAM-1, Santa
Cruz Biotechnology, dilution 1:100), anti-E-selectin (Abcam, Cambridge, MA, dilution

1:100, non-reducing conditions), anti-4-hydroxynonenal (4-HNE, Abcam, dilution
1:1000, non-reducing conditions), anti-gp91phox, anti-p67phox and anti-p47phox (Santa
Cruz Biotechnology, dilution 1:100, 1:100, and 1:200, respectively). Primary antibody-
labelled membranes were then rinsed with 1xPBS/Tween20, incubated with horseradish-
linked secondary antibodies for 1 hour, and subjected to enhanced chemiluminescence
(Pierce, Rockford, IL). Following membrane exposure to an X-ray film (Kodak,
Rochester, NY), optical density of immunoblots was determined by use of NIH Image
and expressed relative to the expression of β-actin (Sigma, dilution 1:2000). Human
endothelial cell lysate (Transduction Laboratories, 1 mg/mL) was used as a positive
control for eNOS immunoblotting.

Online Figure Legends
Online Figure I.
Panels A and B: Quantification of the expression of the β5- and β2-subunits of the
proteasome in coronary arteries from animals on a normal or high-cholesterol diet for 12
weeks without (N and HC) or with additional proteasome inhibition (N+PSI and
HC+PSI, n=4 for all groups); *p<0.05 vs. N.
Panel C: TUNEL staining of coronary arteries from pigs on a normal or highcholesterol
diet for 12 weeks without (N and HC) or with proteasome inhibitor treatment (N+PSI and
HC+PSI). Original magnification: 100x.
Panel D: Bar graphs illustrating the percentage of TUNEL-positive endothelial cells in
the four study groups (n=5 for N and HC+PSI, n=4 for HC and N+PSI); * p<0.05 vs. HC
and N+PSI.
Online Figure II.
Left panels: Elastica-van-Gieson (EvG) staining of coronary arteries from pigs on a
normal or high-cholesterol diet for 12 weeks without (N and HC) or with proteasome
inhibitor treatment (N+PSI and HC+PSI) with black-white display to enhance contrast;
intimal thickening can be seen of minor degree in HC and N+PSI and of major degree in
HC+PSI; original magifications 50x.
Right panels: alpha-smooth muscle cell (α-SMC) actin immunostaining of coronary
arteries from the four study groups; notice the relative paucity of α-SMC actin-positive
cells in the area of intimal thickening in proteasome inhibitor-treated animals; original
magnifications 150x.

Online Figure III.
Panels A-C: Quantification of the expression of VCAM-1, ELAM-1 (E-selectin), and
IκBα in coronary arteries from animals on a normal or high-cholesterol diet for 12 weeks
without (N and HC) or with additional proteasome inhibition (N+PSI and HC+PSI, n=4
for all groups); *p<0.05 vs. N.
HC
HC + PSI
0
0.5
1
1.5
2
2.5
N HC N+PSI HC+PSI
*
TUNEL+ cells [% of endothelial cells per cross-section]
N + PSI
N
C
D
00.10.20.30.40.50.60.70.80.9
N HC N+PSI HC+PSI
Densitometric ratio (β5-subunit /β-actin)
0
0.1
0.2
0.3
0.4
0.5
0.6* 0.7
N HC N+PSI HC+PSI
N HC N+PSI HC+PSI
N HC N+PSI HC+PSI
Densitometric ratio (β2-subunit /β-actin)
22.5 kDa
30.0 kDa
Online Figure I
A
B
Online Figure II
Elastica-van-Gieson
α-SMC actin
N HC N+PSI HC+PSI
Online Figure III
A N HC N+PSI HC+PSI
150 kDa Densitometric ratio
(VCAM-1/β-actin)
00.20.40.60.8
11.21.41.61.8
N HC N+PSI HC+PSI
* B N HC N+PSI HC+PSI 150 kDa Densitometric ratio
(ELAM-1/β-actin)
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
N HC N+PSI HC+PSI
*
C N HC N+PSI HC+PSI
40 kDa
0
0.2
0.4
0.6
0.8
1
1.2
N HC N+PSI HC+PSI
*
*
Densitometric ratio (IκBα/β-actin)





























