Embed Size (px)
Citation preview

Neurogenetics ( . ) : 1–20DOI 10.1007/s10048-005-0218-3
REVIEW ARTICLE
Sara E. Mole . Ruth E. Williams . Hans H. Goebel
Correlations between genotype, ultrastructural morphologyand clinical phenotype in the neuronal ceroid lipofuscinoses
Received: 14 October 2004 / Accepted: 3 February 2005# Springer-Verlag
Abstract The neuronal ceroid lipofuscinoses (NCLs) are agroup of severe neurodegenerative diseases with onsetusually in childhood and characterised by the intracellularaccumulation of autofluorescent storage material. Withinthe last decade, mutations that cause NCL have been foundin six human genes (CLN1, CLN2, CLN3, CLN5, CLN6and CLN8). Mutations in two additional genes causedisease in animal models that share features with NCL-CTSD in sheep and mice and PPT2 in mice. Approxi-mately 160 NCL disease-causing mutations have now beendescribed (listed and fully cited in the NCL MutationDatabase, http://www.ucl.ac.uk/ncl/). Most mutations re-sult in a classic morphology and disease phenotype, butsome mutations are associated with disease that is of lateronset, less severe or protracted in its course, or withatypical morphology. Seven common mutations exist,some having a worldwide distribution and others asso-ciated with families originating from specific geographicalregions. This review attempts to correlate the gene, disease-causing mutation, morphology and clinical phenotype foreach type of NCL.
Keywords Neuronal ceroid lipofuscinoses . Battendisease . Genotype morphology . Phenotype
Introduction
The neuronal ceroid lipofuscinoses (NCLs) are a geneti-cally heterogeneous group of inherited progressive neuro-degenerative diseases of children and, occasionally, ofadults. They are marked clinically by visual impairment,gait abnormalities, seizures and dementia [1, 2]. Morpho-logically, they are identified by loss of neurons, predomi-nantly in the cerebral and cerebellar cortices and near-ubiquitous accumulation of NCL-specific lipopigments(Fig. 1) [2]. They were originally classified according tothe clinical onset of symptoms as infantile, late-infantile,juvenile and adult forms, but now several so-called variantforms are recognised [2, 3]. Molecular and biochemicaladvances have since enhanced this classification with agenetic perspective, based on mapping and identification ofthe genetic loci [4]. Currently, seven different childhoodforms and one generic form of adult NCL are known, i.e.genes CLN1 to CLN8 (Table 1). Neuropathologists havealways considered the NCLs as lysosomal diseases becauseof the intracellular accumulation of lysosomal lipopigmentresidual bodies. Biochemical and biological studies nowalso support the lysosomal classification of the NCLs.
Molecular advances
Classical lysosomal diseases have preserved a nosologybased on age of onset—infantile, late-infantile, juvenileand adult forms. The genetic elucidation of these disorders,such as the sphingolipidoses and mucopolysaccharidoses,followed their morphological and then biochemical clari-fication [5], examples of forward genetics, facilitated bythe principle of one-substrate one-(lysosomal)-enzymedeficiency. Such a course was not possible for the NCLsbecause the morphological hallmark of the lysosomalpathology, the lipopigments, were resistant to complete
S. E. Mole (*)MRC Laboratory for Molecular Cell Biology and Departmentof Paediatrics and Child Health, University College London,Gower Street,London, WC1E 6BT, UKe-mail: [email protected].: +44-207-6797257Fax: +44-207-6797805
R. E. WilliamsDepartment of Paediatric Neurology, Guy's Hospital,St Thomas Street,London, England, UK
H. H. GoebelDepartment of Neuropathology, Johannes GutenbergUniversity,Mainz, Germany
H. H. GoebelMedical Center,Langenbeckstrasse 1,55131 Mainz, Germany
18/05/05 - PROOF PAGEMAKEUP 10048_218 1
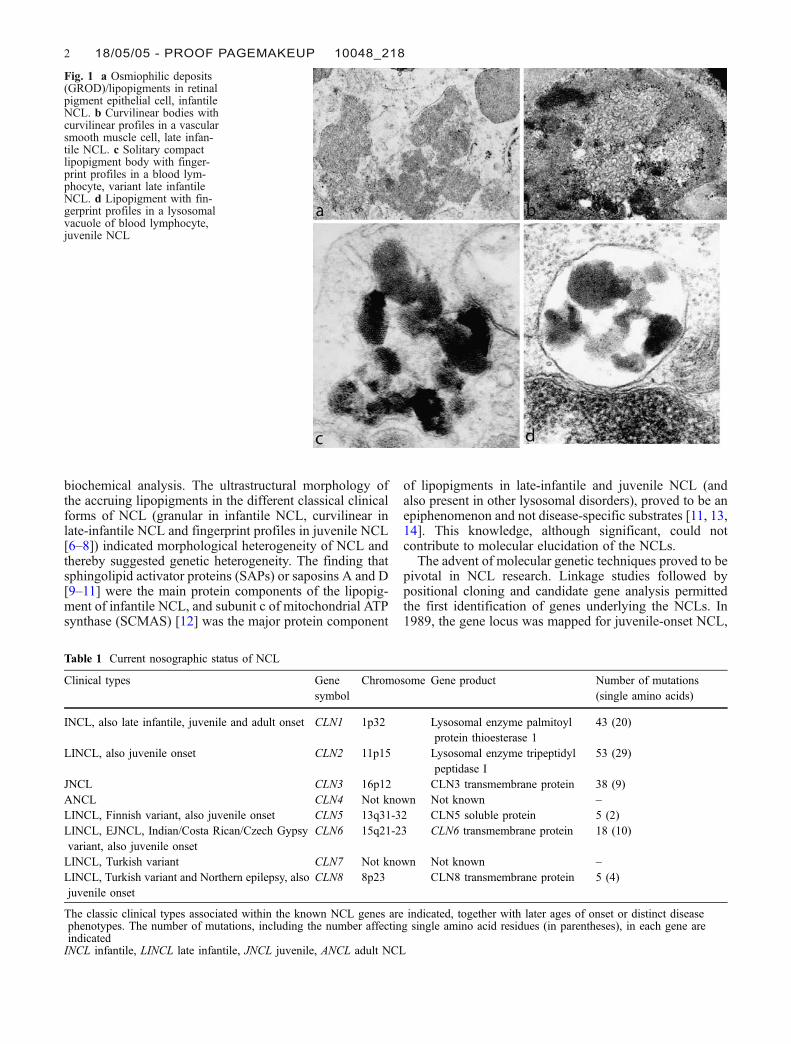
biochemical analysis. The ultrastructural morphology ofthe accruing lipopigments in the different classical clinicalforms of NCL (granular in infantile NCL, curvilinear inlate-infantile NCL and fingerprint profiles in juvenile NCL[6–8]) indicated morphological heterogeneity of NCL andthereby suggested genetic heterogeneity. The finding thatsphingolipid activator proteins (SAPs) or saposins A and D[9–11] were the main protein components of the lipopig-ment of infantile NCL, and subunit c of mitochondrial ATPsynthase (SCMAS) [12] was the major protein component
of lipopigments in late-infantile and juvenile NCL (andalso present in other lysosomal disorders), proved to be anepiphenomenon and not disease-specific substrates [11, 13,14]. This knowledge, although significant, could notcontribute to molecular elucidation of the NCLs.
The advent of molecular genetic techniques proved to bepivotal in NCL research. Linkage studies followed bypositional cloning and candidate gene analysis permittedthe first identification of genes underlying the NCLs. In1989, the gene locus was mapped for juvenile-onset NCL,
Fig. 1 a Osmiophilic deposits(GROD)/lipopigments in retinalpigment epithelial cell, infantileNCL. b Curvilinear bodies withcurvilinear profiles in a vascularsmooth muscle cell, late infan-tile NCL. c Solitary compactlipopigment body with finger-print profiles in a blood lym-phocyte, variant late infantileNCL. d Lipopigment with fin-gerprint profiles in a lysosomalvacuole of blood lymphocyte,juvenile NCL
Table 1 Current nosographic status of NCL
Clinical types Genesymbol
Chromosome Gene product Number of mutations(single amino acids)
INCL, also late infantile, juvenile and adult onset CLN1 1p32 Lysosomal enzyme palmitoylprotein thioesterase 1
43 (20)
LINCL, also juvenile onset CLN2 11p15 Lysosomal enzyme tripeptidylpeptidase I
53 (29)
JNCL CLN3 16p12 CLN3 transmembrane protein 38 (9)ANCL CLN4 Not known Not known –LINCL, Finnish variant, also juvenile onset CLN5 13q31-32 CLN5 soluble protein 5 (2)LINCL, EJNCL, Indian/Costa Rican/Czech Gypsyvariant, also juvenile onset
CLN6 15q21-23 CLN6 transmembrane protein 18 (10)
LINCL, Turkish variant CLN7 Not known Not known –LINCL, Turkish variant and Northern epilepsy, alsojuvenile onset
CLN8 8p23 CLN8 transmembrane protein 5 (4)
The classic clinical types associated within the known NCL genes are indicated, together with later ages of onset or distinct diseasephenotypes. The number of mutations, including the number affecting single amino acid residues (in parentheses), in each gene areindicatedINCL infantile, LINCL late infantile, JNCL juvenile, ANCL adult NCL
2 18/05/05 - PROOF PAGEMAKEUP 10048_218

the most frequent clinical type, to chromosome 16 [15, 16].Consequently, the principle of reverse genetics turned thestandstill in NCL nosology into forward progress and led toa new pathogenetic principle by demonstrating thatmutations in transmembrane proteins (CLN3, CLN6,CLN8) as well as soluble lysosomal proteins (CLN1,CLN5 and CLN2, identified using a biochemical approach)cause NCL disease. Such progress and an increasingknowledge of NCLs required a revision of the traditionalsystem of classification to incorporate the increasingnumber of NCL-related genes and their mutations [2, 7,17]. Within slightly more than one decade, delineation ofmutations in seven genes (including CTSD) have blurredthe formerly rather sharp borders between the individualclinical subtypes previously corroborated by clinicalsubtype-specific ultrastructural features. These featuresnow require reorientation within the entire nosologicalspectrum of NCL.
The NCLs can be described in terms of genotype andphenotype, both morphological and clinical. This realign-ment and correlation of the underlying genetic defect withthe clinical course and the ultrastructural features arenecessary as our knowledge of the NCLs expands. As theNCLs are a genetically heterogeneous group affectingdifferent genes (but only one gene is involved in each NCLtype), the correct correlation is not gene–phenotype, butrather mutation–phenotype. Loss of nerve cells andaccumulation of NCL-specific lipopigments are morpho-logical hallmarks of NCL, but for correlative genotype–phenotype purposes the ultrastructure of the lipopigmentsis of particular importance. In this extensive and detailedreview, each NCL gene that causes human disease will beconsidered separately and the mutations correlated withclinical phenotype and morphology of deposited material.
CLN1
CLN1 encodes the enzyme palmitoyl protein thioesterase 1(PPT1), which removes palmitate residues from proteins.Its in vivo substrates are not known. In non-neuronal cellsit is transported to the lysosome via the mannose-6-receptor-mediated pathway and is also secreted [18, 19]. Inmature neurons PPT1 does not appear to localise tolysosomes but is preferentially targeted to axons, inparticular, axonal varicosities and presynaptic terminals[20], where it may serve a function specific to neuronalcells. CLN1 expression is ubiquitous [21, 22] and is notparticularly elevated in any human tissue type [23].However, it may be important for the development ofneurons since its expression increases in the embryonicbrain from the beginning of cortical neurogenesis throughcortical development [24].
The crystal structure of the mature bovine enzyme,which is 95% identical to the human PPT1, has beendetermined [25]. PPT1 is a monomeric, globular enzymewith an α/β-hydrolase fold and glycosylated at three sites(Asn197, Asn212 and Asn232) [25]. It has a catalytic triadconsisting of conserved residues Ser115, Asp233 and
His289. Other conserved residues include some in thecatalytic groove and those donating hydrogen bonds, suchas Met41 and Gln116.
Mutational spectrum
Forty-three mutations have now been described in PPT1/CLN1. These are distributed throughout the gene andinclude 20 missense, 9 nonsense, 10 small deletions orinsertions and 4 mutations affecting splice sites. All casesof NCL caused by mutations in PPT1/CLN1 are associatedwith a granular appearance of the stored material. Areliable enzyme diagnostic test to determine the levels ofPPT activity in peripheral leukocytes or fibroblasts, andalso using saliva, is available [26–28].
Most mutations change amino acid residues that areconserved in diverse species (H39Q, G42E, C45ins, D79G,C96Y, G108R, G118D, R122W, C152Y, Q177E, V181L,V181M, L219Q, L222P; T75P, F85del, Y109P, E184K,F225S, Y247H, G250V). The geometry of Ser115 and itsactivation by His289 is critical to the enzymatic activity ofPPT1. Nonsense mutations or mutations causing frame-shifts that result in premature truncation of PPT1 arepredicted to lead to total loss of enzymatic activity [29]through deletion of His289, which is close to the Cterminus of the protein. Some mutations are predicted todisrupt the palmitate-binding pocket (Q177E, V181M,E184K, G250V) or to affect the geometry of the active siteor the conformation of PPT1 (H39D, G42Q, T75P, D79G,F85del, Y109D, G118D, R122W, C152Y, L219Q, F225S,Y247H, Q291X, W296X) [25, 30, 31]. Some mutationsthat cause incorrect folding cause retention of mutant PPT1in the ER or Golgi followed by degradation (R122W,H39Q, C45ins, T75P, D79G, F85del, G250V, Q177E, M1Iand Y109D) [18, 32–34]. Mutations R151X and L10X areassociated with an absence of mRNA, probably due to theproximity of the nonsense mutations to 3′ splice junctionsresulting in mRNA instability and nonsense-mediatedRNA decay [35]. F85del results from deletion of CTTfrom the repetitive sequence CTTCTTCTT, probably fromDNA slippage and mispairing during DNA replication.
Patients from certain countries share common mutationsthat facilitate DNA-based mutation and carrier testing.Patients of Scottish or Irish origin are enriched formutations T75P and L10X [29, 36]. Common mutationR151X is found in numerous patients of different ethnicorigins, suggesting it is an older mutation. Missensemutation, R122W, is enriched in Finland [34].
Clinical spectrum
The clinical spectrum of disease due to mutations in theCLN1 gene is extremely wide, with age at onset varyingfrom infancy to adulthood. The majority of patients arediagnosed within early childhood and in most the firstsymptoms are seen within the first 2 years of life. Thisinfantile-onset NCL (INCL) is seen predominantly in
18/05/05 - PROOF PAGEMAKEUP 10048_218 3

Finland and in families of Scandinavian or Finnish origin.Fewer numbers of cases are seen with onset of symptoms inlater childhood and adolescence (late infantile and juvenileonset) and these children are from various countries oforigin.
Infantile-onset CLN1 disease Classic INCL was firstdescribed in the 1970s in Finland and is also known asHaltia–Santavuori disease. Children have an uneventfulfirst few months of life and there are often no health ordevelopmental concerns at all in the first 6 months.Towards the end of the first year, development may slowand this particularly affects fine motor skills. Infants maybecome irritable and difficult to comfort. On examination,they may have some central hypotonia and there may beevidence of decelerating head growth. Development slowsfurther in the second year. Children may develop seizuresand jerks although myoclonus is less of a feature than seenin CLN2 disease. Sleep disturbance and irritability areseen in most children. Often children have characteristichand movements rather like those seen in Rett syndrome.Mobility and language skills are lost and vision deterio-rates. Spasticity of the limbs worsens later in the illness.Children come to be totally dependent on carers for alltheir needs and death usually occurs between 8 and 13years. Brain magnetic resonance imaging (MRI) showshypointensity of the thalami early and high signal intensityof the white matter later on T-weighted imaging. There isprogressive cerebral atrophy, and a recent study suggestedthat the combination of serial conventional and protonMRI could be useful for monitoring disease progressionand therapeutic interventions [37]. Neurophysiologicalfindings include early loss of changes on eye closure andsleep spindles together with extinction of the electroret-inogram and somatosensory potentials [38].
Scottish children studied before mutation analysisbecame available had developmental regression withinthe first 18 months of life. Visual impairment wasrecognised towards the end of the second year and theage at first seizure varied between 16 and 23 months [39].An Italian child is described, homozygous for a frameshiftmutation in exon 2, who developed normally to 12 monthsbefore starting to lose motor skills. Other skills were lostand seizures developed over the next few months. At 27months, MRI showed periventricular white matter andthalamic changes [29, 36, 39–41].
Late infantile onset CLN1 disease Wisniewski et al.reported five cases in 1998 with a late infantile onsetclinical phenotype and PPT deficiency. All had theultrastructural appearance of GROD. In these childrenthe onset of symptoms was a little earlier (between 1.5 and3 years) but the clinical features were otherwise not clearlydifferent from children presenting with classical lateinfantile onset NCL caused by CLN2 mutations [29, 42].An Italian child was homozygous for an exon 7 mutationresulting in an amino acid substitution. She developeddevelopmental regression, behaviour problems and visualimpairment at 4 years of age. She was ataxic with brisk
deep tendon reflexes in all four limbs and had apigmentary retinopathy. Diagnosis was made as a resultof ultrastructural examination of a skin biopsy [43].
Juvenile-onset CLN1 disease A number of children havenow been described with an atypical or variant juvenile-onset clinical phenotype, but granular osmiophilic deposits(GROD) on ultrastructural analysis and PPT deficiency ormutations in CLN1. Most present with initial visualimpairment but others have learning difficulties at thestart of their illness. Vacuolated lymphocytes are not foundin the peripheral blood and this is a very importantdiagnostic feature which, in addition to the ultrastructuralfindings, distinguishes these cases from those with CLN3mutations [29, 36, 39, 41].
Adult-onset CLN1 disease Two sisters have been describedpresenting in the fourth decade with depressive symptoms.There was a progressive deterioration of cognitive skills;then motor skills and vision became severely impaired.Neurological examination revealed cerebellar ataxia, ex-trapyramidal movement disorder, and a general cognitiveimpairment. They had optic atrophy and retinal pigmen-tation, which is not usually found in the rare adult-onsetNCL, Kufs disease. Imaging revealed generalised atrophyand neurophysiology was non-specific. PPT deficiencywas documented and CLN1 sequencing in the youngersister showed that she was a compound heterozygoteR151X/G108R [44].
Congenital NCL disease A few papers [45, 46] havesuggested a non-familial congenital NCL. These wereautopsy reports on children who had died shortly afterbirth and in whom lipopigment accretion of the granulartype in cerebral neurons was described, suggesting veryearly onset, although no molecular analyses have beenpublished.
Morphology of lipopigments
In disease caused by mutations in CLN1, lipopigmentshave a uniformly granular appearance forming lysosomalresidual bodies, also called granular osmiophilic deposits.These may take a globular appearance and coalesce,forming larger complexes. The granularity is described asfine in cerebral neurons and somewhat coarser inextraneuronal cells. Whereas densely packed in cerebralneurons, the granular lipopigments in extraneuronal cells,especially outside of the central nervous system (CNS),may appear more loosely structured. Occasionally, at veryhigh magnification, lamellar profiles may be encountered.Lipid droplets as a component of granular lipopigments arerare, allowing good diagnostic differentiation from regularlipofuscin, the age or ‘wear-and-tear’ lipopigment. Thedistribution of cell types carrying granular lipopigments isalmost ubiquitous.
For diagnostic purposes, blood lymphocytes are wellsuited. The skin, too, is a good biopsy target because
4 18/05/05 - PROOF PAGEMAKEUP 10048_218

numerous dermal cell types, e.g. secretory eccrine sweatgland epithelia, smooth muscle cells, mural vascular cells,Schwann cells and fibroblasts, harbour granular lipopig-ments, whereas mast cells and epidermal cells do not.However, if the disease commences after infancy, granularlipopigments may need to be differentiated from physio-logically accruing lipopigments in secretory cells of eccrinesweat glands. Here, again, lipid droplets present within thelipopigments are most likely due to non-specific lipopig-ments and not NCL-specific lipopigments. In mural cells ofchorionic vessels, granular lipopigments may appear afterthe 8th week of gestation [47]. Whether amniotic fluid cellsmay also carry granular lipopigments has not beenunequivocally determined. The gestation at which granularlipopigments first appear in non-chorionic fetal tissues, forinstance, in skin or skeletal muscle, is also currentlyunclear. Taking biochemical PPT1 deficiency as evidenceof CLN1, lymphocytes of certain patients have shownpeculiar mixed granular–curvilinear or granular–finger-print patterns [29].
In congenital NCL, still considered a separate form ofNCL and not yet assigned to any genetic NCL type, theultrastructure of the accruing lipopigments is also granular,suggesting an early-infantile or even ‘pre-infantile’ form ofCLN1, but no molecular or biochemical data have beenreported in the few published cases studied only postmor-tem [48]. In juvenile-onset NCL with GRODs, nowconsidered juvenile CLN1, the granular lipopigments ofblood lymphocytes are not located within membrane-bound vacuoles, unlike the fingerprint complexes inclassical juvenile NCL caused by mutations in CLN3.
Genotype, clinico-phenotype correlation
The clinical phenotype caused by mutations in CLN1/PPT1is the most variable of the NCLs, although within familieswith more than one affected child the clinical presentationis consistent [29]. In very general terms, the severity of theclinical disease can be predicted from the kind of mutation.Mutations predicted to result in loss of protein or a severelytruncated protein tend to result in earlier onset disease andmore rapid disease progression. Milder mutations result inlater onset of symptoms and a less severe clinicalphenotype with a slower rate of neurodegeneration.
Whatever the pattern of symptoms, all cases with provenmutations in CLN1 have reduced PPT activity in whitecells, and granular osmiophilic deposits are seen onultrastructural examination of nerve and other cell types.Thus, this finding on biopsy material should lead theclinician to confirm the diagnosis using enzyme assay andmutation analysis regardless of the clinical picture.
Most mutations (around half of cases in the USA [29])cause classical infantile NCL. In Finland, almost allchildren with infantile-onset CLN1 disease are homozy-gous for the R122W missense mutation, which isassociated with severely reduced PPT activity. Childrenfrom other more genetically heterogeneous populationshave slightly different clinical phenotypes and a variety of
mutations. The most common is R151X, which is alsoassociated with an infantile disease onset. Some mutations,however, are associated with later onset. This can be in thelate infantile (2–4 years) or juvenile age range (>5 years) orin adulthood. Mutations M1I, T75P, D79G, C96Y, Q177E,V181L, L219Q, L222P, Y247H. G250V and W296X haveall been associated with onset in late infancy or the juvenileage range [29, 36, 43]. Those that are predicted to affect theconformation of PPT1 presumably do so less dramaticallythan those that cause onset in infancy. Certainly some ofthese mutations are associated with retention of residualenzyme activity [30, 33]. Missense mutation G108R wasassociated with onset in adulthood in two siblings [44].However, PPT1 activity was still severely reduced in thesepatients, although in the upper part of the range observed inINCL patients.
Decreased enzyme stability is probably more importantin determining the disease course than decreased catalyticactivity [30]. Whether the level of residual enzymaticactivity or localisation of mutant PPT1 polypeptides inexperimental cell systems correlates with the severity ofdisease phenotype is unclear, although it has beensuggested that the extent of trafficking and migration ofthe mutant polypeptide within neuronal cells does correlate[29, 33]. Mutation M1I results from a mutated initiationcodon (ATG>ATA). Since this mutation is associated withlater disease onset then there must be sufficient translationto allow significant enzyme activity in patient brain cells[30].
Genotype–morphotype correlation
In disease caused by mutations in CLN1, irrespective of theclinical subtype and which now encompass infantile, late-infantile, juvenile and adult forms [44], the ultrastructuralpattern of CLN1-specific lipopigments is granular in anytype of cell and disease specific.
CLN1 granular lipopigments need to be distinguishedfrom granular lipofuscin, accumulating with age, and fromthe finely granular lipopigment accruing in vitamin Edeficiency [49]. Distinguishing between the ultrastructuralappearance of deposits in disease caused by mutations inCLN1 and other NCL genes is normally straightforwardexcept when curvilinear bodies or mixed fingerprintinclusions in non-CLN1 cases are heavily condensed andobscure the nature of the curvilinear and fingerprintprofiles, respectively. The observation by Das et al. [29]that certain patients with PPT1 deficiency had mixedgranular–curvilinear and granular–fingerprint patterns inlipopigments of blood lymphocytes remains to be furtherexplained because such ‘mixed’ ultrastructure of lipopig-ments in CLN1 is not common experience. Whether suchmixed ultrastructural patterns occur in other cell types ofPPT1-deficient patients is also unknown.
18/05/05 - PROOF PAGEMAKEUP 10048_218 5

CLN2
CLN2 encodes a lysosomal enzyme tripeptidyl peptidase(TPP1), a member of a recently defined family of serine-carboxyl proteinases [50], that removes tripeptides fromthe N terminus of small proteins such as subunit c ofmitochondrial ATP synthase that accumulates in mostforms of NCL [51–53]. It is also implicated in thedegradation of certain neuronal transmitters and hormones[52, 54, 55]. TPPI is produced as a glycoslyated precursorpolypeptide that is processed to the mature enzyme in thelysosome [56]. The N terminus of the mature form beginsat residue L196 and it has an apparent molecular weight of46 kDa [57]. TPPI is reported to interact with CLN5 protein[58]. The crystal structure of the related enzyme PSCP hasbeen resolved allowing a structural model for TPPI to becreated. The catalytic triad in PSCP was identified asGlu80, Asp84 and Ser287, with Asp170 also crucial to thecatalytic activity of the enzyme [50, 59]. The equivalentresidues in human TPPI are Glu272, Asp276 and Ser495for the catalytic triad and Asp360 as a crucial residue.CLN2 is widely expressed and not particularly elevated inany human tissue type [23, 60]. Enzyme activity is at adultlevels by the age of 2 years in the cerebral cortex [61]. TPPIactivity can be measured by simple enzyme assay enablingaccurate diagnosis of NCL caused by mutations in CLN2[28, 62, 63].
Mutational spectrum
Fifty-two mutations and 22 polymorphisms have beenreported in CLN2. The spectrum of mutations encompasses7 small deletions, 2 small insertions, 29 missensemutations, 5 nonsense mutations and 9 mutations affectingsplice sites or intronic sequence. There are two commonmutations: IVS5-1G>C, which affects a splice site in intron5, and nonsense mutation R208X. Some mutations(Q509X in Italy [64] and G284V in Canadian patientsfrom Newfoundland [65]) are more common in particularcountries, raising the possibility of DNA-based testing forcarriers.
Clinical spectrum
Mutations in CLN2 usually cause a disease with onset inthe late infantile age range, which is characterised byseizures, myoclonic jerks and developmental regression inthe early stages. Visual failure occurs later. A small numberof mutations are associated with a later disease onset andslower symptom progression.
Classical late-infantile NCLThis is one of the commonestinherited neurodegenerative disorders in Europe. Claussenet al. calculated an incidence of 0.46 in 100,000 live birthsin West Germany in 1992 [66] and the British PaediatricSurveillance Unit reports approximately six new casesdiagnosed per year in the UK (BPSU, 17th Annual Report,
2002–2003, Royal College of Paediatrics and ChildHealth). The two most common mutations in the NorthernEuropean and US populations are the splice site mutationIV5-1G>C and the nonsense R208X [67]. Patients aredescribed from all over the world [65, 68–71].
Whichever of the common mutations are present, theclinical phenotype is broadly similar. However, inter- andintrafamilial variation in disease severity can be consider-able. Children are healthy and developmentally normal inthe first year of life. During the second year, developmentmay slow when compared in retrospect with healthysiblings, and this particularly affects speech. Children maybe referred for speech and language therapy beforediagnosis. Developmental progress may, however, fallwell within the normal range. The onset of seizures, whichmay be of any type, occurs between 2 and 4 years of age.An earlier or later onset is suggestive of a variant late-infantile NCL caused by mutations in CLN6 or other NCLgenes. As seizures become more frequent and often notcompletely responsive to medication, developmental skillsare lost—gross and fine motor, play and independence,cognitive skills and vision. Sometimes disease progressionis rapid over 6–8 months with loss of independentmobility and language at around the age of 4–5 years.Some children become extremely irritable and distressed.Limb spasticity may become prominent with truncalhypotonia and loss of head control. Gastrostomy feedingis usually necessary by 6 years. Death occurs at any timeup to middle teenage. A very clear description of theneurological examination findings is given by Lavrov etal. [70].
From early in the disease, the neurophysiologicalfindings are very characteristic and should enable promptdiagnosis. An occipital spike response to slow flash (1–2 Hz) stimulation may be present before the onset ofseizures. This becomes more exaggerated as symptomsdevelop. The ERG is diminished, often before parentsnotice any visual impairment. Brain imaging is usuallyperformed as part of the diagnostic process but thefindings are non-specific with cerebellar and later cerebralatrophy. Vacuolated lymphocytes in the peripheral bloodare not present. Diagnosis is made on enzyme analysis forTPP1 activity and confirmed with CLN2 mutation anal-ysis. Biopsy is no longer required unless enzyme levels arenormal.
Atypical phenotypes Occasionally, children known to haveTPP1 deficiency and/or mutations in CLN2 show adelayed disease onset and slower progression. Sleat et al.[72] described two compound heterozygotes with diseaseonset at 8 years and deaths in the fourth and fifth decades.These cases each had one of the common mutations andR447H, which is a missense mutation, on the other allele.Other authors have reported similarly late onset disease incompound heterozygotes, but published clinical details arelimited [73–75]. Surprisingly one child is described,homozygous for the R208X mutation, with early speechdelay, learning disabilities diagnosed at age 6 years but noseizures until the age of 8 years. At the age of 10 years,
6 18/05/05 - PROOF PAGEMAKEUP 10048_218

ERG and brain imaging were reported to be normal. At 12years, the child had evidence of significant decline incognitive skills but at 14, still had good vision [76].
Morphology of lipopigments
The basic ultrastructural feature is curvilinear profiles thataggregate in membrane-bound lysosomal residual bodies,i.e. curvilinear bodies. The pure curvilinear body, largelywithout a clear lipid droplet, is the hallmark of diseasecaused by mutations in CLN2 and is a reliable diagnostictarget. It can be observed within the nervous system andextraneural tissues including blood lymphocytes. Exami-nation of somatic fetal cells, skin and lymphocytes mayalso show curvilinear bodies in affected pregnancies [77].Curvilinear bodies accrue in amniotic fluid cells but have,so far, not been demonstrated in chorionic tissue, perhapsbecause investigators and diagnosticians have alwaysconcentrated on amniotic fluid cells. It would be surprisingif—during an affected pregnancy—chorionic tissue did notaccumulate curvilinear bodies, even late or at term.However, it would not be of any diagnostic value, asscant fingerprint profiles may be encountered rarely withina curvilinear body in disease caused by mutations in CLN2[78].
When mutations in CLN2 cause NCL of later onset,curvilinear bodies may not be pure and may also containfingerprint profiles and granular osmiophilic deposits aswell [79]. In addition, curvilinear profiles may be observedas part of a mixed morphology of lipopigments in othergenetic NCL types (see CLN3, CLN5, CLN6, CLN7 andCLN8).
Another differential diagnostic aspect is the reportedformation of curvilinear profiles and curvilinear bodiesafter therapy and intoxication by chloroquine where theseinclusions also contain SCMAS [80], similar to thepresence of SCMAS as a major part of NCL-specificcurvilinear bodies [81]. This demonstrates the use ofincorrect terminology because the term curvilinear body orcurvilinear profile should be limited to NCL. Similarly,another misleading misnomer is the description of lysoso-mal residual bodies in Farber disease or ceramidosiscontaining curved profiles, though somewhat different tocurvilinear profiles, as curvilinear bodies. However, thisnomenclatorial confusion only underscores our ignoranceof the precise biochemical constituents of the respectivecurvilinear or curved profiles in NCL, chloroquine intox-ication, or ceramidosis.
Genotype, clinico-phenotype correlation
Mutations in CLN2 abolish detectable levels of TPP1activity. Patients homozygous for the common splicemutation or for mutation R208X have undetectable proteinby Western analysis [56]. Missense mutations may directlyaffect the catalytic activity of TPP1 or the structuralintegrity of the enzyme leading to abolition of enzymatic
activity. In a patient heterozygous for a missense mutationaffecting residue S475 (S475L) of the active site, TPP1enzyme was processed normally but enzymatic activitywas undetectable [56, 57]. At least three mutations areassociated with later age of onset or a more protracteddisease course. These include mutations R447H [57, 72],S153P [82], R127Q [75] and also a mutation in which thereis an insertion into intronic sequence [83]. Mutations, suchas G77R, R127Q and S153P, which affect residues withinthe N terminus of the immature precursor protein, mayaffect processing or stability of the protein preventing gainof enzymatic activity. However, G77R and R127Q alterinvariant residues at splice junctions and so may in fact beaffecting the splicing of CLN2 rather than the stability ofimmature TPPI. Mutation Q422H may also affect splicingsince an invariant residue at a splice junction is changed.Q389E and R447H may affect the structural stability ofmature TPP1 and, certainly, no residual enzyme activitywas detected in expression studies of mutant proteincontaining R447H [57]. Mutation N286S affects one of thefive glycolysation sites utilized in TPP1, and in vitrosignificantly reduced enzyme activity [84] by affectingprotein folding [85], although it is associated with a slightlymore protracted disease progression [75] probably becausethe mutant protein is not retained in the ER but is secretedand exerts residual activity if taken up and transported tothe lysosome where it is further processed [85]. Occasion-ally patients carrying common mutations can have a moreprotracted disease course with onset in the juvenile agerange [73]. In one case, severe caudate atrophy anddystonia presenting at 4 months of age was associated withhomozygosity for R208X [86], the unusual disease courseperhaps caused by additional genetic or environmentalfactors.
Genotype–morphotype correlation
In classic late-infantile NCL or CLN2, lipopigments have apure curvilinear pattern in lymphocytes, skin and brain, butthe entire morphological spectrum in visceral cell types hasnot completely been explored. In juvenile CLN2 geneti-cally characterized by compound heterozygosity of R447Hand R208X, compound curvilinear bodies with fingerprintprofiles and granular matrix were also observed [79].
CLN3
Mutations in CLN3 cause most cases of NCL of juvenileonset. The CLN3 gene encodes a 438-amino-acid glyco-sylated membrane protein (size up to 60 kDa depending ontissue) almost certainly located in the lysosome and inadditional locations in neurones [87–91]. CLN3 ispredicted to be a transmembrane protein, although itsexact topology is not certain [87, 92–94], and it may beassociated with lipid rafts [95]. Its expression is highest ingastrointestinal and glandular/secretory tissues [23]. CLN3is thought to perform a fundamental but still unknown
18/05/05 - PROOF PAGEMAKEUP 10048_218 7

cellular function since it is a conserved protein withorthologous genes present in all eukaryotes down to yeasts.Using yeast as a model system, it has been implicated invacuolar homeostasis [96, 97] and has also been reported tobind the protein CLN5 in mammalian cells [58].
Mutational spectrum
Thirty-eight mutations have now been defined in CLN3.Five polymorphisms have been reported. The spectrum ofmutations in CLN3 includes 5 small deletions, 5 smallinsertions, 4 large deletions, 9 missense mutations, 8nonsense mutations, 5 mutations affecting splice sites and 1intron change. A 1-kb deletion that is present onapproximately 85% of disease chromosomes is the mostcommon mutation [98, 99]. A few mutations affect splicingof mRNA [100, 101], and it is possible that some mis-spliced transcripts encode protein with partial function. Allmissense mutations affect residues conserved betweendiverse species. Some change residues that are predicted tolie on the same topological face of the CLN3 protein(L170, G187, V330, R334), possibly interfering with itsbinding to a cellular partner, and some affect residues thatare predicted to lie within a membrane (L101 and E295),possibly affecting CLN3 topology. Certain mutations areknown to prevent CLN3 from reaching the correctintracellular location and others are known not to affecttrafficking of CLN3, thereby being directly implicated inthe function of the CLN3 protein, as discussed below.Novel mutations still remain to be defined in some patients,so mutations that affect the production or stability of themRNA species may therefore exist. Patients with mutationsin CLN3 have been found all over the world. The sharingof a common 1-kb deletion by the majority of CLN3patients indicates their European ancestry [98, 99].
Clinical spectrum
Classical juvenile-onset NCL caused by mutations inCLN3 usually presents with progressive visual failure fromthe early school years. Most cases in Northern Europe andelsewhere are homozygous for the common 1-kb deletion.This group is therefore genetically homogeneous and hasbeen most extensively studied in Finland. The remainingyoung people are mostly compound heterozygotes for the1-kb deletion and one of the many other mutations so fardescribed. In some patients, the second mutation has notyet been identified. The clinical phenotype shows greatervariation within this compound heterozygous groupcompared with the homozygous group.
Homozygous 1-kb deletion Visual failure is the initialsymptom, progressing rapidly over 2–3 years to anappreciation of light and dark only. There is a pigmentaryretinopathy and children may be diagnosed with retinitispigmentosa or a cone dystrophy. In most, deterioration incognitive skills, speech and mobility occurs in the early
teenage years together with the onset of seizures. Seizuresmay be generalised tonic–clonic or partial seizures and areoften reasonably well controlled on valproate or lamo-trigene. Behaviour may become problematic at this time.Aggressiveness, psychosis, mood disturbance and anxietyare well recognised. Speech becomes slightly dysarthricand dysfluent with echolalia. Mobility becomes character-istically slow and shuffling with a slightly stooped posture.As the disease progresses myoclonic jerks and parkinso-nian features become prominent. Communication, mobil-ity and self-help skills are lost.
The EEG is abnormal but has no diagnostic or specificfeatures. The ERG is reduced very early on and thesomatosensory-evoked potentials may be enhanced (butnot to the same degree as is commonly seen in the CLN2form). MRI brain imaging is normal in the early stages ofdisease. There may be abnormalities of periventricularwhite matter, thalami and basal ganglia later.
Interestingly, Järvelä et al. [102] found considerableinter- and intrafamilial variation in a group of homozygouscases, suggesting that environmental factors and modify-ing genes must influence the clinical phenotype.
Compound heterozygotes Lauronen et al. [103] described aseries of ten compound heterozygotes. In six children ofthree families, the second mutation was a deletion of exons10–13. In these cases the rate of disease progression wasslower than is usually seen in Finnish 1-kb homozygotes.The disease presented with progressive visual impairment,but motor and cognitive deterioration occurred slightlylater and was slower. In the same study, two young peopleaged 32 and 16 years, with missense mutations E295K orR334H resulting from the second disease allele, had visualimpairment but no motor or cognitive signs of disease.These patients resemble those previously described asprotracted JNCL [104]. Intrafamilial variability is againrecognised [100, 102].
Morphology of lipopigments
The ultrastructural hallmark of CLN3 is fingerprintprofiles. They can occur with three different appearances:(1) pure within a lysosomal residual body, as notinfrequently seen in cerebral and extracerebral neurons;(2) in conjunction with curvilinear profiles or rectilinearprofiles; (3) as a small component within rather largemembrane-bound lysosomal vacuoles [105–107]. In muralcells of vessels, both endothelial cells and smooth musclecells, mixed fingerprint bodies may particularly beencountered. The combination of fingerprint complexeswithin lysosomal vacuoles is a regular feature of bloodlymphocytes and an occasional feature in neuronal peri-karya and secretory eccrine sweat gland epithelia. Thesignificance and the biochemical composition of thevacuolar component in these fingerprint-bearing lysosomalvacuoles are unknown but may reflect a role for CLN3protein in the metabolism of components in addition to
8 18/05/05 - PROOF PAGEMAKEUP 10048_218

lipopigment. This electron-lucent or vacuolar structure is aCLN3-typical ultrastructural feature.
Whereas fingerprint profiles, pure or mixed, may aboundin diverse types of cells in organs, skeletal muscle fibresharbour lipopigments devoid of fingerprint profiles, butinstead consisting of curved or curvilinear and straight orrectilinear profiles. These lipopigments within striatedmuscle cells are apparently not distinct from similarlipopigments in other NCL forms, except CLN1, but mayrepresent a cell-specific uniform appearance amonggenetically different NCL forms [108]. Furthermore,lipopigments or inclusions considered to be lipopigmentsand typical of NCL have been reported in biopsiedchorionic tissue, where they have a lamellar appearance,distinct from the ultrastructural features of lipopigments inpostnatal disease [109, 110]. However, whether theserather few prenatal observations encompass the entireultrastructural spectrum of lipopigments in chorionic tissueis not known because very few samples have beeninvestigated. Current practice emphasizes the molecularstudies of biopsied chorionic tissue antenatally; thus, theultrastructural spectrum of lipopigments in CLN3 chorionictissues may remain unclear. However, the few but signif-icant observations on lamellar lipopigments in fetal cells,both endothelial cells of chorionic stromal vessels and inspinal cord, would seem to suggest a developmentalchange in ultrastructure of lipopigments in CLN3 [106].
Rarely, lamellar inclusions resembling membraneouscytoplasmic bodies have been observed in extracerebralCLN3 neurons [106, 111], probably not representinglipopigments, but a ganglioside-type storing material. Inthis context, it is curious to note that another lysosomaldisorder, mucolipidosis IV, characterized by a mutanttransmembrane protein, the product of the MCOLN1 gene,may also be associated with lysosomal lamellar inclusionsresembling membraneous cytoplasmic bodies [112] andlipopigments of the curvilinear and fingerprint types [113].
Finally, a protracted form of juvenile NCL has beendescribed showing fingerprint profiles as fine structure ofthe accruing lipopigments [103, 104, 114, 115]. Theultrastructural appearances do not differ in variants ofdisease caused by mutations in CLN3, such as late infantileand protracted juvenile, from those in classical juvenile-onset disease.
Genotype, clinico-phenotype correlation
There is some evidence of correlation between CLN3genotype and clinical disease phenotype. Compoundheterozygotes for the 1-kb deletion and another disease-causing allele exhibit a spectrum of disease severity thatcan be roughly predicted from the kind of mutation presenton the second allele. For examples, mutations that severelyaffect CLN3 structure (such as nonsense mutations) oraffect critical resides (some missense mutations) cause aclassic JNCL phenotype, whereas other missense muta-tions, presumably affecting less important resides, result in
later onset of cognitive and motor decline and slowersymptom progression.
In all cases of NCL caused by mutations in CLN3, visualfailure has occurred by 10 years, suggesting the importanceof CLN3 function in the retina. However, in some instancesseizures, motor dysfunction, or mental regression can besignificantly delayed. Four missense mutations are asso-ciated with an atypical disease course. In three patients,E295K is associated with markedly protracted JNCL [100,103, 116] and one patient with R334H had a moreprotracted disease course [100, 103]. Patients with L101Pand L170P also had atypical disease courses [100].Occasional atypical JNCL cases are seen including a caseof granular deposits in some tissue in a patient homozygousfor the 1-kb deletion [117] and onset at 5 months of age in apatient heterozygous for the 1-kb deletion [118].
The 1-kb deletion encodes a truncated CLN3 protein ofapproximately 24 kDa that is retained in the endoplasmicreticulum, either because it is recognised as an aberrantprotein or because it lacks some critical trafficking motif[90]. Missense mutations L101P, L170P, E295K, V330Fand R334H do not abolish trafficking of the mutant CLN3protein to the endosome–lysosome system [88], nor, in thecase of mutation E295K, to vesicles containing thepresynaptic marker synaptic vesicle protein 2 in neurons[90]. Mutations L170P and E295K, when expressed as partof the orthologous Saccharomyces cerevisiae protein donot abolish protein function, whereas mutations L101P,V330F, R334H and C435S permit only partial CLN3function [88]. Mutation G187A, which is associated withclassic JNCL, when expressed as part of the orthologous S.pombe protein retarded trafficking of CLN3 in the ER (S.Codlin and S. Mole, unpublished data).
Genotype–morphotype correlation
A genotype–morphotype correlation for disease caused bymutations in CLN3 has hardly ever been attempted becausehistorically a considerable temporal gap existed betweenthe morphological diagnosis and subsequent molecularanalysis in individual patients and families. Non-vacuolarmixed fingerprint–curvilinear lipopigments in circulatinglymphocytes [17] has been associated with protracteddisease caused by compound heterozygosity for CLN3mutations [103, 116].
CLN4
Currently, the gene symbol CLN4 represents a heteroge-neous and rare group of recessively inherited adult forms ofNCL not yet linked to a genetic locus. Two adult siblingswere found to have reduced PPT1 enzyme levels [44] andnow carry the diagnosis of adult CLN1 rather than CLN4.Autosomal dominant inheritance of ANCL has also beenreported [119–123]. It remains to be determined whethercases of adult onset are caused by mutations in novel genes
18/05/05 - PROOF PAGEMAKEUP 10048_218 9

or some represent milder mutations in genes that also causechildhood onset NCL.
Clinical spectrum
Since the seminal review by Berkovic and colleagues[124], two major clinical forms of adult NCL have beendelineated and accepted: type A, marked by myoclonusepilepsy, dementia and ataxia and type B, marked bybehavioural changes and dementia as well as peculiar facialdyskinesia. Retinal vision is not impaired in either type andis an important clinical (and electrophysiological) featureto distinguish adult NCL from protracted juvenile NCL,particularly for those patients considered to have adultNCL but with clinical onset of their disease in adolescence[125]. There is some visual impairment in adult CLN1 [44],but no electroretinographic abnormalities. Age of onset andduration of the disease are not strikingly different betweentypes A and B.
The two major types A and B of adult NCL weredelineated by including those previously classed as Kufsdisease or adult amaurotic familial idiocy but excludingcertain earlier reports [124]. A similar recent approachsuggests that the complete clinical spectrum of adult NCLmight be variably defined by different clinicians since anoverlap between types A and B, which sometimes defyprecise separation, was noted [125]. Late adult-onset NCLhas also been reported [126] that has a rather long durationover several decades [125].
Morphology of lipopigments
Two aspects of lipopigment accretion are important in adultNCL—the ultrastructural pattern and the distribution ofabnormally accumulated lipopigments. Ultrastructuralpatterns comprise all of the previously described features:granular, curvilinear, or fingerprint in different cell typesand organs of the same patient, but a combination ofpatterns, such as granular and fingerprint, have also beenfound within the same cell.
In terms of the distribution of NCL-specific lipopig-ments, some patients displayed accretion of lipopigmentsin the CNS only, in spite of a diligent search inextracerebral tissues [125], whereas others showed accu-mulation in both cerebral and extracerebral cells [127–129]. Storage may occur in extracerebral neurons, usuallyin the rectum and visceral organs [125]. So far, nobody hasreported blood lymphocytes carrying NCL-specific lipo-pigments in adult NCL. This carries very importantdiagnostic implications, as a negative extracranial tissuebiopsy cannot therefore exclude the diagnosis of adult-onset NCL and a brain biopsy may be necessary.
Genotype–phenotype correlation
As currently no specific genes are known to be involved inadult NCL—apart from adult CLN1—such correlation canonly be confined to the two major hereditary formsobserved in adult NCL, the autosomal-recessive form,which is largely based on sibling studies, and theautosomal-dominant form, also called Parry disease, bothof which overlap with the clincal types A and B describedabove. Families with autosomal-dominant NCL are rare[119, 121] and ultrastructurally, the lipopigments havebeen reported as being granular and confined to the CNS.Ultrastructural diversity and involvement of both cerebraland extracerebal including visceral tissues have only beendocumented in siblings with autosomal recessive adultNCL. Thus, at present it is not known whether adult NCLcases are caused by mutations in the same gene or inseveral genes.
CLN5
CLN5 encodes a 407-amino-acid glycosylated protein withan apparent molecular weight of 60 kDa. It appears to beconfined to vertebrates. Its expression increases duringcortical neurogenesis [24]. CLN5 is synthesised as fourprecursor forms, all of which are targeted to the lysosome.Most forms are thought to be soluble, but the longestspecies may be associated with the lysosome membrane[58, 130]. CLN5 is reported to interact with CLN2/TPP1and CLN3 proteins [58].
Mutational spectrum
Five mutations and one polymorphism are now known inCLN5. Two mutations are nonsense mutations (W75X,Y392X) predicted to result in truncated proteins. W75X iscaused by a single base change and W392X by a 2-bpdeletion. One mutation (c.669insC) is predicted to result ina truncated protein caused by a translation frameshift. Onemissense mutation (D279N) changes a conserved andcharged aspartate residue to a polar but unchargedasparagine residue. Residue D279 is located in a hydro-phobic region [131]. Most cases of NCL caused bymutations in CLN5 are from Finland. Patients from Swedenshare mutations found in Finland and one patient from theNetherlands carries a unique mutation [132]. MutationY392X is particularly common in Finland. All thesemutations are associated with the classic disease course.Recently missense mutation R112H has been described inan extended family from Columbia [133]. This mutationwas associated with onset in the juvenile age range and anatypical protracted NCL. There may be other patients withso-called ‘milder’ mutations in CLN5 that are not recog-nised as vLINCL.
10 18/05/05 - PROOF PAGEMAKEUP 10048_218

Clinical spectrum
Mutations in CLN5 [131] were shown to underlie a Finnishvariant of late infantile NCL that was first described in1991 [134]. The clinical phenotype and diagnosticinvestigations of the Finnish cases have been describedin detail [132, 134–136]. The initial symptoms are usuallyclumsiness and/or difficulties in concentration noticed inthe preschool and early school years. Developmentalregression, visual impairment, ataxia, myoclonus andepilepsy follow. The neurophysiological findings aresimilar to those seen in classical late infantile NCL(CLN2 form) but may occur later in the course of theillness: giant VEPs, exaggerated somatosensory potentialsand occipital spikes in response to slow photic stimulation.There are no vacuolated lymphocytes in the peripheralblood, distinguishing cases from CLN3 juvenile-onsetdisease. Imaging shows early-onset cerebellar atrophy andhypointensity of periventricular white matter on MRI.Diagnosis is confirmed by ultrastructural analysis andmutation testing.
A multiply consanguineous family from Columbia hasbeen described recently [133]. Two siblings and a cousinpresented with a similar clinical phenotype with what wasclinically considered to be juvenile-onset NCL. Visualfailure at 9 years was the initial symptom. The visualimpairment progressed to blindness within a year and wascomplicated by ataxia, jerks, seizures and later a progres-sive loss of motor and cognitive skills. Death occurred intwo of the children at 14 years. Brain imaging revealedcortical and cerebellar atrophy. Ultrastructural examinationof a skin biopsy showed a mixed pattern suggestive ofvariant NCL. A novel mutation, R112H, was found in thisfamily. CLN5 must now therefore be considered acandidate gene for NCL disease with an age of onsetfrom late infancy onwards where the genetic defect has notbeen identified.
Morphology of lipopigments
The ultrastructural spectrum of lipopigments accruing bothwithin and outside of the CNS, with an extracerebraldistribution similar to that in other childhood forms ofNCL, encompasses classical fingerprint and curvilinearprofiles, but in addition also curvilinear profiles andsometimes even lamellar inclusions [137, 138] andcondensed fingerprint profiles occasionally associatedwith lipid droplets [133]. Mural cells of chorionic stromalvessels contain lamellar lipopigments similar to those seenin chorionic tissue in disease caused by mutations in CLN3,so far reported as an unpublished observation [38].
Genotype, clinico-phenotype correlation
The clinical phenotype of most patients carrying somecombination of the first known CLN5 mutations aresimilar, suggesting that these mutations severely disturb
the function of CLN5 [132]. Therefore, missense mutationD279N must have a deleterious effect on the function ofCLN5 as the truncating mutations. In one study, it wasreported that mutation Y392X interfered with lysosomaltargeting of overexpressed CLN5 [130]; however, inanother study, mutations Y392X and D279N did notprevent targeting of at least some overexpressed CLN5polypeptide to the lysosome [58], although Y392X was nolonger associated with the lysosome membrane. The effectof R112H on CLN5 has not been studied but it is associatedwith a later age of onset [133]. The disease phenotype maybe a result of mutant CLN5 not reaching the lysosome or alack of function in this location. It is not known whateffects mutations in CLN5 have on its location in neurons.In non-neuronal cells at least three of the known mutationsare reported to interfere with its binding to TPP1 [58].
Genotype–morphotype correlation
Both the ultrastructure of lipopigments and distribution oflipopigments are similar to those in the other variant late-infantile forms. Although originally denied, involvementof circulating lymphocytes has later been reported [139]upon re-examination of earlier lymphocyte specimens.Unlike the ultrastructure of lipopigments in both CLN2 andCLN3 circulating lymphocytes, lipopigments in CLN5lymphocytes appeared as non-vacuolar complex lipopig-ments containing mixed curvilinear and fingerprint pro-files, which are not distinguishable from respectivelipopigments in CLN6 and CLN7 lymphocytes.
CLN6
Mutations in the gene CLN6 cause variant LINCL. Thegene is predicted to encode a 311-amino-acid membraneprotein [140, 141] located in the endoplasmic reticulum[142, 143]. CLN6 has no homology with known proteins orfunctional domains and is confined to vertebrates. Itsfunction is unknown. It exists as a dimer and its absenceaffects the degradation of the endocytosed index proteinarylsulfatase A [143].
Mutational spectrum
Eighteen mutations have been reported in CLN6. Tenmutations change or delete conserved amino acid residues.Six mutations including one nonsense mutation arepredicted to result in truncated proteins caused by aframeshift in translation. Two mutations occur at splicesites and are predicted to cause aberrant splicing. Onepolymorphism has been reported. One mutation(c.316dupC), an insertion of a single cytosine in a run ofsix cytosine residues, is identical to that in nclf, thenaturally occurring mouse model for CLN6. No majorfounder effect for CLN6 is observed, although patientsfrom the same country may share the same mutation [144].
18/05/05 - PROOF PAGEMAKEUP 10048_218 11

Diagnostic tests that detect specific mutations in CLN6may be worthwhile for certain populations (e.g. Costa Rica,Pakistan, Portugal).
Clinical spectrum
Mutations in CLN6 are now known to cause NCL withonset in late infancy that is distinct from that caused bymutations in CLN2. Originally, children outside Finlandwith later onset of symptoms (between 5 and 6 years), aslower rate of disease progression, and with a mixedpattern of curvilinear bodies and fingerprint profiles onultrastructural analysis were described as having ‘early-juvenile’ NCL [145–147]. As the genetic studies of the lateinfantile onset NCLs progressed in the 1990s, it was soonevident that these children could be given a diagnosis ofclassical (onset of seizures and developmental delaybetween 2 and 4 years, together with the ultrastructuralappearances of curvilinear bodies in lymphocytes, neuronsand other cell types) or variant (earlier or later diseaseonset, mixed curvilinear and fingerprint profiles) lateinfantile NCL based on their clinical and morphologicaldisease phenotype. Almost all those with classical lateinfantile NCL were shown to have TPPI deficiency andmutations in CLN2. A subgroup of variant late infantileNCL families was shown to have genetic linkage to a locuson chromosome 15 and, subsequently, mutations in CLN6[140, 141]. Other families did not appear to be linked tothis locus, suggesting that other NCL gene loci remain tobe found [148].
The CLN6 form is seen in a number of differentgeographical populations including South America and theIndian subcontinent. A large number of mutations havenow been identified, some more common in certainpopulations, but some apparently unique. The pattern ofdisease corresponding to these mutations also showsvariability in age of onset and rate of symptom progression.The order in which symptoms develop is quite consistent,seizures and motor difficulties presenting early with visualimpairment later. Several case reports and series have beenpublished with a clinical phenotype and/or linkage datasuggestive of CLN6, but without mutation data.
Children from Costa Rica have onset of symptomsbetween 5 and 7 years and death (in four cases only) wasbetween 4.5 and 11.8 years, which tends to be later than inchildren from other populations. The great majority arehomozygous for E72X in exon 4 [140, 144]. One child hasbeen described in detail. She presented after the onset ofseizures at just before 3 years of age. At 4 years, she hadmyoclonic jerks and frequent falls. She had lost develop-mental skills, was ataxic with pyramidal signs and opticatrophy. Neurophysiological studies revealed high-ampli-tude discharges posteriorly in response to intermittentphotic stimulation. T2-weighted brain MRI revealed high-intensity periventricular white matter and decreased signalintensity in the thalami and putamen with some cerebellaratrophy. Skin biopsy examination showed curvilinearbodies and genetic studies demonstrated linkage to the
chromosome 15 locus. At 5 years of age, she was blind andunable to walk independently. Brain imaging showed aprogressive generalised cerebral atrophy. At 9 years, shehad no useful vision, frequent myoclonic jerks andgeneralized seizures [149].
A series of NCL cases was reported from the CzechRepublic that included 27 children from 23 families (9were of Romany origin). The age of onset of variantLINCL ranged from 18 months to 8 years with the majoritybetween 3 and 5.3 years. Gait and speech disturbance,seizures and developmental delay were early features. Thedisease progressed rapidly with death occurring between18 and 32 years. EEG showed an early photic response (2–4 years) that was lost later [150]. Several of these Czechfamilies show good evidence, supporting linkage to CLN6,but mutations have so far not been identified [144].
A series of 26 Portuguese cases included 11 showing avariant LINCL phenotype. Clinical information from 8cases gave age at onset of symptoms between 3 and 8years, blindness between 4 and 9 years, cognitive declinebetween 4 and 10 years, and loss of independent walkingbetween 4 and 10 years. Thus, again there appears to besubstantial clinical variation [151]. The majority of thesefamilies were subsequently found to be homozygous for anamino acid deletion (I154del). A small number werecompound heterozygotes for this mutation and deletion/insertion in exon 7, and in 3 cases with a similar clinicalphenotype, no mutations in CLN6 could be identified at all[141, 152].
Nardocci [153] described 11 atypical late infantile casesseen in Italy. Onset of symptoms was between 2.5 and 5.5years with seizures or ataxia the leading symptoms. TheERG was normal in two children aged 3.4 years and 4.3years but later became extinguished in both. The EEGresponse to intermittent photic stimulation was present[154–156]. The ultrastructural appearance consisted of amixed pattern of curvilinear bodies and fingerpint profilesor fingerprint profiles only. Imaging findings in one caseare described by Iannetti [157]. Genetic studies of Italianvariant LINCL families have confirmed linkage to CLN6but to date a mutation in only one Italian family has beendescribed [144].
Morphology of lipopigments
Distribution of lipopigments in cerebral and extracerebralcell types mimics that seen in other childhood forms ofNCL. The ultrastructure of the lipopigments comprisesfingerprint and curvilinear profiles as well as the rectilinearcomplex [12, 105, 145, 147, 158].
Genotype, clinico-phenotype correlation
Most of the known mutations are associated with a similardisease course with variations as detailed above [144]. Onepatient with a more protracted disease progression carriedY221S on one chromosome and an as yet undefined
12 18/05/05 - PROOF PAGEMAKEUP 10048_218

mutation on the second [144]. It is highly likely that so farunidentified mutations in CLN6 could cause a milderdisease that is not clinically diagnosed as vLINCL.
Recently it was shown that some CLN6 patients had nodetectable CLN6 protein [142] and the mitochondrialenzyme MnSOD (manganese-dependent superoxide dis-mutase) was increased in several fibroblast cell lines fromCLN6 patients [159], perhaps laying the foundations forCLN6-specific diagnostic tests.
Genotype–morphotype correlation
Whereas distribution and ultrastructure of lipopigments indisease caused by mutation in CLN6 does not differ fromrespective features in disease caused by mutations inCLN5, it had been claimed that subunit c of mitochondrialATP synthase is not present in hepatic, adrenal andendocrine pancreatic cells [12], which is different fromthe distribution of SCMAS in CLN2. Another featuredistinguishing CLN6 disease from CLN2 disease is thepresence of lipopigment-carrying ‘histiocytes’ in intestinalsmooth muscle layers in the former [158]. Bloodlymphocytes are affected by the formation of compactnon-vacuolar fingerprint-containing lipopigments as inCLN5 and CLN7.
CLN7
The gene CLN7was assigned to a group of consanguineousfamilies from Turkey with variant late infantile NCL [148,160]. A subset of these Turkish vLINCL families wasrecently shown to contain mutations in the CLN8 gene[161] and see below. It is not yet known whether theremaining Turkish families with vLINCL represent a novellocus (CLN7) or are allelic to other genetically knownLINCL forms.
Clinical spectrum
At present the clinical phenotype is considered to be thesame as Turkish patients now known to carry mutations inCLN8.
Morphology of lipopigments
No data on neuronal loss and brain atrophy are easilyaccessible. In Turkish patients diagnosed with variant lateinfantile NCL (and which therefore may carry mutations inCLN8 or a novel gene such as CLN7), the distribution andultrastructure of lipopigments are similar to those seen inCLN5 and CLN6, encompassing fingerprint and curvilinearprofiles in a mixed pattern as well as rectilinear complexes.Circulating lymphocytes are non-vacuolar, containingfingerprint profiles and amorphous, somewhat granularmaterial [162].
CLN8
CLN8 is a predicted membrane protein containing 286amino acids [163] with an apparent molecular weight of33 kDa [164]. It is located in the endoplasmic reticulum(ER) and also shuttles between the ER and ER–Golgiintermediate complex (ERGIC) [164]. An additionallocation outside of the ER has been suggested in polarizedcells including neurones [165]. The exact function ofCLN8 is unknown. It is a member of a large protein family(TLC; TRAM-LAG1-CLN8) that may contain lipid-sensing domains [166], raising the possibility that thisNCL is caused by defects in lipid biosynthesis or transport.A mouse model, mnd, that has a one base pair insertion inthe orthologous mouse Cln8 gene predicted to result in atruncated CLN8 protein, exists [163].
Mutational spectrum
Five mutations have been identified in the CLN8 gene thatcause two distinct disease phenotypes. Three mutations aremissense changes (R24G, R204C, W263C) and anotherdisease allele exists containing two different amino acidchanges (L16M;T170M). The other mutation is the resultof the deletion of a single base pair (c.88delG) and ispredicted to result in a truncated CLN8 protein due to aframeshift after residue 29. Two polymorphisms have beenreported. Two mutations cause a residue change to cysteine(R204C, W263C), possibly resulting in inappropriatedisulphide bridge formation in the mutant polypeptide.The R204C change affects an arginine residue that isabsolutely conserved in the TLC family. The linkedchanges (L16M:T170M) could result in a methionine thatcan function as an alternative translation initiation codon.A mutation changing a conserved residue (L164P) hasrecently been found to cause NCL in the English Setter dogmodel [167].
Clinical spectrum
Mutations in CLN8 cause two distinct diseases: Northernepilepsy in Finland, also known as progresive epilepsywith mental retardation (EPMR), and a form of variant lateinfantile onset NCL so far described in families of Turkishorigin only.
Northern epilepsy was initially described in 1994 [168]and to date has not been reported outside of Finland. Allaffected families (22 cases) were found to be related to eachother and homozygous for the same missense mutationR24G [163]. It is not known whether other slowlyprogressive epilepsy syndromes seen outside Finland arecaused by mutations in this or other NCL genes. Northernepilepsy is characterised by the onset of generalised tonic–clonic seizures usually between 5 and 10 years of agetogether with complex partial seizures in some children.Developmental progress is normal up until the onset of
18/05/05 - PROOF PAGEMAKEUP 10048_218 13

seizures. Seizures are drug resistant, and there is aprogressive decline of cognitive skills from 2 to 5 yearsafter the onset of seizures. In adulthood the seizuresbecome less frequent but slow deterioration of cognitiveskills continues. Behavioural difficulties are described andmay require medication. Unsteadiness and poor fine motorskills may become evident in adulthood. Myoclonus is notseen and vision is preserved until late. Of the small numberof cases described, a number have survived into the fifthand sixth decades. Neurophysiological studies show pro-gressive slowing of the background EEG activity and thedisappearance of normal features of sleep. The detailedstudy of brain tissue from autopsy samples revealedabnormal neuronal storage material with autofluorescenceon light microscopy.
All types of NCL are seen in Turkey where there is ahigh rate of consanguinity. The most common form is,however, a variant of late infantile-onset disease. Seizuresand deteriorating motor skills are often the leadingsymptoms between 2 and 7 years of age. There are thetypical neurophysiological findings commonly seen inother late infantile NCLs (occipital discharges with slowphotic stimulation). Disease progression is often rapid withmyoclonus, visual impairment and loss of cognitive skillsover 2 years from the time of diagnosis. Mutations in theCLN8 gene have been identified in 9 of 17 families studied[161]. In two families only one disease-causing allele wasidentified, suggesting that other mutations remain to bediscovered in this gene. Data from other Turkish variantlate infantile onset families suggest that another geneticlocus may be responsible for a proportion of variant lateinfantile NCL [160]. These families are assumed to havemutations in the as yet unidentified CLN7 gene.
Morphology of lipopigments
The morphological features of disease caused by mutationsin CLN8 have been identified only recently so data are stillsparse. Ultrastructural analysis of lymphocytes in Turkishpatients shows abnormal storage resembling fingerprintprofiles or a mixed pattern of fingerprint profiles andcurvilinear bodies [169]. In Northern epilepsy, no grossabnormalities were noted and only mild neuronal loss wasencountered in the CA2 section of the hippocampus. It wasthe enlargement of the neuronal perikarya by lipopigmentsthat drew a major interest. Lipopigments had not onlyaccrued within cortical and subcortical neuronal cells buthave also been expressed extracerebrally in numeroustissues including the heart, liver and kidney [170].Vacuoles could not be ascertained within circulatinglymphocytes but detailed ultrastructural studies have notyet been reported. The fine structure of lipopigments hasbeen described as curvilinear and granular [171], contain-ing high proportions of subunit c of mitochondrial ATPsynthase, thus classifying this epilepsy syndrome withinthe NCLs [169].
Genotype, clinico-phenotype correlations
Four mutations are associated with a typical vLINCLdisease course [161]. W263C is associated with a slightlymilder disease than R204C, L16M;T170M and c.88delGsince onset of visual failure and seizures is slightly later infamilies homozygous or heterozygous for W263C [161].Three children with variant late infantile onset NCL andhomozygous for the R204C mutation are described. Theyall had onset of seizures with loss of skills and visualimpairment at around 3.5–4 years. In three childrendescribed with homozygosity for W263C the onset ofseizures and other symptoms was from around 6–8 years.However, one missense mutation in CLN8 (R24G) causes avery different and protracted disease course, Northernepilepsy. R24G can therefore be classed as a particularlymild mutation that causes a protracted and atypical NCLdisease, whereas the four other known mutations affectCLN8 protein function more severely. Four mutations inCLN8 that cause vLINCL are located in a region of theCLN8 protein homologous to the TLC family of proteins,suggesting that mutations at these residues affect the TLC-specific function, whereas R24G that underlies Northernepilepsy is located prior to the TLC region of homology[166] in a region of the protein with sequence unique toCLN8. T170M and R204C are located within predictedtransmembrane domains and W263C is located at the TLCC-terminal boundary. Polymorphism 155V/A is locatedwithin a predicted transmembrane domain of the TLCregion but the residue is not conserved across the TLCfamily. Three mutations (R204C, W263C and R24G) didnot affect the intracellular location of CLN8 in non-neuronal or neuronal cells [165].
Conclusion and outlook
The neuronal ceroid lipofuscinoses are the most commongroup of neurodegenerative diseases in children. JuvenileNCL was the first recognised NCL form and was describedin Norway (1826) [172], possibly as a result of its higherincidence in Scandinavian countries. Since then, variousnosographic stages have been passed through, from theearly stage of ‘amaurotic family idiocy’ defined by carefulclinical and morphological observations; through morerefined diagnostic procedures using fluorescence lightmicroscopy and electron microscopy, which distinguishedNCL from gangliosidoses and resulted in the more preciseclinicopathological classification of several NCL clinicalsubtypes [5, 173, 174], and finally to the current stage ofmolecular classification and genotype–phenotype correla-tions [3, 8]. Remarkably, the morphology, i.e. loss ofneurons and almost universal accretion of lipopigmentswith distinct ultrastructure, and the clinical similaritybetween the different genetic types are in puzzling contrastto the diversity of gene loci and gene products, i.e. enzymesas well as membrane proteins, with locations in differentsubcellular compartments. Recent progress has improvedthe genetic classification; for example, a set of Turkish
14 18/05/05 - PROOF PAGEMAKEUP 10048_218

families with variant late-infantile NCL were shown tohave mutations in CLN8, bringing this disease phenotypeinto the same genetic group as Northern epilepsy/EPMRpatients [161].
As a result of these diagnostic advances, future researchtrends are already apparent: (1) to complete and fullydescribe the genetic spectrum of human NCL by searchingfor additional gene loci in those families that have beenexcluded from known NCL genes by genetic linkage andmutational analysis; (2) to add the genetic spectrum ofanimal models (natural and engineered) to the increasingnumber of NCL genes since some of these will representvery rare human NCL disease types—CTSD in sheep [175]and mice [176], PPT2 in mice [177, 178] are examples ofNCL disease caused by mutations in known genes, andmany other animal models exist [179] that may representadditional novel genes[180]; (3) to understand the biologyof the NCL gene products. The function of NCL proteins,especially the membrane forms, are still little understood,both in their normal physiological role as well as their roleas mutated proteins, thus severely hampering any rationaltherapeutic approach; (4) to expand the use of simplersystems to further elucidate the role of physiological andmutant NCL proteins, such as in vitro tissue culture studies,studies on the budding and fission yeasts (S. cerevisiae andS. pombe) the nematode worm Caenorhabditis elegans andthe fruitfly Drosophila melanogaster as well as the mouseand larger animal models.
In summary, the wealth of data and information accruedover the past decade of the NCLs and the CLN genes hasbroadened the field of research and opened a newnosographic era in the NCL.
Acknowledgements We are grateful to Dr. Hannah Mitchison forcritical reading of the manuscript and Ms. Astrid Wöber for editorialassistance.
References
1. Santavuori P (1988) Neuronal ceroid lipofuscinosis in child-hood. Brain Dev 10:80–83
2. Goebel HH, Mole SE, Lake BD (eds) (1999) The neuronalceroid lipofuscinoses (Batten disease). Biomedical and healthresearch. IOS, Amsterdam, pp 211
3. Mole SE (2004) The genetic spectrum of human neuronalceroid-lipofuscinoses. Brain Pathol 14:70–76
4. Mole S (2004) Neuronal ceroid lipofuscinoses (NCL). Eur JPaediatr Neurol 8:101–103
5. Zeman W (1970) Historical development of the nosologicalconcept of amaurotic familial idiocy. In: Vinken PJ, Bruyn GW(eds) Handbook of clinical neurology, vol. 10. North-Holland,Amsterdam, pp 212–232
6. Goebel HH (1995) The neuronal ceroid-lipofuscinoses. J ChildNeurol 10:424–437
7. Goebel HH (2000) The new nosography of the neuronal ceroid-lipofuscinoses. Ann Pathol 20:479–491
8. Goebel HH, Wisniewski KE (2004) Current state of clinical andmorphological features in human NCL. Brain Pathol 14:61–69
9. Tyynelä J, Baumann M, Henseler M, Sandhoff K, Haltia M(1995) Sphingolipid activator proteins in the neuronal ceroid-lipofuscinoses: an immunological study. Acta Neuropathol89:391–398
10. Tyynelä J, Suopanki J, Baumann M, Haltia M (1997)Sphingolipid activator proteins (SAPs) in neuronal ceroid-lipofuscinoses (NCL). Neuropediatrics 28:49–52
11. Tyynelä J, Palmer DN, Baumann M, Haltia M (1993) Storageof saposins A and D in infantile neuronal ceroid-lipofuscinosis.FEBS Lett 330:8–12
12. Elleder M, Sokolova J, Hrebicek M (1997) Follow-up study ofsubunit c of mitochondrial ATP synthase (ASCMAS) in Battendisease and in unrelated lysosomal disorders. Acta Neuropathol93:379–390
13. Palmer DN, Martinus RD, Cooper SM, Midwinter GG, ReidJC, Jolly RD (1989) Ovine ceroid lipofuscinosis. The majorlipopigment protein and the lipid-binding subunit of mitochon-drial ATP synthase have the same NH2-terminal sequence. JBiol Chem 264:5736–5740
14. Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, WolfeLS, Haltia M, Martinus RD, Jolly RD (1992) MitochondrialATP synthase subunit c storage in the ceroid-lipofuscinoses(Batten disease). Am J Med Genet 42:561–567
15. Eiberg H, Gardiner RM, Mohr J (1989) Batten disease(Spielmeyer–Sjögren disease) and haptoglobins (HP): indica-tion of linkage and assignment to chromosome 16. Clin Genet36:217–218
16. Zeman W, Rider JA (eds) (1975) The dissection of adegenerative disease. Elsevier, New York
17. Wisniewski KE, Kida E, Golabek A, Kaczmarski W, Connell F,Zhong N (2001) Neuronal ceroid lipofuscinoses: classificationand diagnosis. In: Wisniewski KE, Zhong N (eds) Battendisease: diagnosis, treatment, and research. Academic, SanDiego, pp 1–34
18. Hellsten E, Vesa J, Olkkonen VM, Jalanko A, Peltonen L(1996) Human palmitoyl protein thioesterase: evidence forlysosomal targeting of the enzyme and disturbed cellularrouting in infantile neuronal ceroid lipofuscinosis. EMBO J15:5240–5245
19. Verkruyse LA, Hofmann SL (1996) Lysosomal targeting ofpalmitoyl-protein thioesterase. J Biol Chem 271:15831–15836
20. Ahtiainen L, Van Diggelen OP, Jalanko A, Kopra O (2003)Palmitoyl protein thioesterase 1 is targeted to the axons inneurons. J Comp Neurol 455:368–377
21. Camp LA, Hofmann SL (1993) Purification and properties of apalmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J Biol Chem 268:22566–22574
22. Camp LA, Verkruyse LA, Afendis SJ, Slaughter CA, HofmannSL (1994) Molecular cloning and expression of palmitoyl-protein thioesterase. J Biol Chem 269:23212–23219
23. Chattopadhyay S, Pearce DA (2000) Neural and extraneuralexpression of the neuronal ceroid lipofuscinoses genes CLN1,CLN2, and CLN3: functional implications for CLN3. MolGenet Metab 71:207–211
24. Heinonen O, Salonen T, Jalanko A, Peltonen L, Copp A (2000)CLN-1 and CLN-5, genes for infantile and variant late infantileneuronal ceroid lipofuscinoses, are expressed in the embryonichuman brain. J Comp Neurol 426:406–412
25. Bellizzi JJ III, Widom J, Kemp C, Lu JY, Das AK, HofmannSL, Clardy J (2000) The crystal structure of palmitoyl proteinthioesterase 1 and the molecular basis of infantile neuronalceroid lipofuscinosis. Proc Natl Acad Sci U S A 97:4573–4578
26. van Diggelen OP, Keulemans JL, Winchester B, Hofman IL,Vanhanen SL, Santavuori P, Voznyi YV (1999) A rapidfluorogenic palmitoyl-protein thioesterase assay: pre- andpostnatal diagnosis of INCL. Mol Genet Metab 66:240–244
27. Voznyi YV, Keulemans JL, Mancini GM, Catsman-BerrevoetsCE, Young E, Winchester B, Kleijer WJ, van Diggelen OP(1999) A new simple enzyme assay for pre- and postnataldiagnosis of infantile neuronal ceroid lipofuscinosis (INCL)and its variants. J Med Genet 36:471–474
18/05/05 - PROOF PAGEMAKEUP 10048_218 15

28. Halac IN, Kramer RD, Kohan R, Tapia V, Guelbert N, Oller-Ramírez AM, Ghio A, Cismondi IA, Depetris-Boldini C,Paschini-Capra A, Giner-Ayala A (2004) Evaluation of thescreening of PPT1 and TPP-I in saliva in the infantile (INCL)and late infantile (LINCL) neuronal ceroid lipofuscinoses. InProceedings of ‘Neuronal ceroid lipofuscinoses and othergenetic lysosomal storage disorders: from the clinical to themolecular aspects’, Córdoba, Argentina, 18–20 September2003. Rev Neurol 38:292–298
29. Das AK, Becerra CHR, Yi W, Lu J-Y, Siakotos AN,Wisniewski KE, Hofmann SL (1998) Molecular genetics ofpalmitoyl-protein thioesterase deficiency in the U.S. J ClinInvest 102:361–370
30. Das AK, Lu JY, Hofmann SL (2001) Biochemical analysis ofmutations in palmitoyl-protein thioesterase causing infantileand late-onset forms of neuronal ceroid lipofuscinosis. HumMol Genet 10:1431–1439
31. Bach G (2001) Mucolipidosis type IV. Mol Genet Metab73:197–203
32. Lehtovirta M, Kyttälä A, Eskelinen E-L, Hess M, Heinonen O,Jalanko A (2001) Palmitoyl protein thioesterase (PPT) localizesinto synaptosomes and synaptic vesicles in neurons: implica-tions for infantile neuronal ceroid lipofuscinosis (INCL). HumMol Genet 10:69–75
33. Salonen T, Heinonen-Kopra O, Vesa J, Jalanko A (2001)Neuronal trafficking of palmitoyl protein thioesterase providesan excellent model to study the effects of different mutationswhich cause infantile neuronal ceroid lipofuscinocis. Mol CellNeurosci 18:131–140
34. Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J,Santavuori P, Hofmann SL, Peltonen L (1995) Mutations in thepalmitoyl protein thioesterase gene causing infantile neuronalceroid lipofuscinosis. Nature 376:584–587
35. Hentze MW, Kulozik AE (1999) A perfect message: RNAsurveillance and nonsense-mediated decay. Cell 96:307–310
36. Mitchison HM, Hofmann SL, Becerra CHR, Munroe PB, LakeBD, Crow YJ, Stephenson JBP, Williams RE, Hofman IL,Taschner PEM, Martin J-J, Philippart M, Andermann E,Andermann F, Mole SE, Gardiner RM, O'Rawe AM (1998)Mutations in the palmitoyl-protein thioesterase gene (PPT;CLN1) causing juvenile neuronal ceroid lipofuscinosis withgranular osmiophilic deposits. Hum Mol Genet 7:291–297
37. Vanhanen SL, Puranen J, Autti T, Raininko R, Liewendahl K,Nikkinen P, Santavuori P, Suominen P, Vuori K, Hakkinen AM(2004) Neuroradiological findings (MRS, MRI, SPECT) ininfantile neuronal ceroid-lipofuscinosis (infantile CLN1) atdifferent stages of the disease. Neuropediatrics 35:27–35
38. Santavuori P, Gottlob I, Haltia M, Rapola J, Lake BD, TyynelaJ, Peltonen L (1999) CLN1—infantile and other types of NCLwith GROD. In: Goebel HH, Mole SE, Lake BD (eds) Theneuronal ceroid lipofuscinoses (Batten disease). IOS, Amster-dam, pp 16–36
39. Crow YJ, Tolmie JL, Howatson AG, Patrick WJA, StephensonJBP (1997) Batten disease in the West of Scotland 1974–1995including five cases of the juvenile form with granularosmiophilic deposits. Neuropediatrics 28:140–144
40. Santorelli FM, Bertini E, Petruzzella V, Di Capua M, CalvieriS, Gasparini P, Zeviani M (1998) A novel insertion mutation(A169i) in the CLN1 gene is associated with infantile neuronalceroid lipofuscinosis in an Italian patient. Biochem BiophysRes Commun 245:519–522
41. Stephenson JB, Greene ND, Leung KY, Munroe PB, Mole SE,Gardiner RM, Taschner PE, O'Regan M, Naismith K, Crow YJ,Mitchison HM (1999) The molecular basis of GROD-storingneuronal ceroid lipofuscinoses in Scotland. Mol Genet Metab66:245–247
42. Wisniewski KE, Connell F, Kaczmarski W, Kaczmarski A,Siakotos A, Becerra CR, Hofmann SL (1998) Palmitoyl-proteinthioesterase deficiency in a novel granular variant of LINCL.Pediatr Neurol 18:119–123
43. Mazzei R, Conforti FL, Magariello A, Bravaccio C, MiliterniR, Gabriele AL, Sampaolo S, Patitucci A, Di Iorio G, MugliaM, Quattrone A (2002) A novel mutation in the CLN1 gene in apatient with juvenile neuronal ceroid lipofuscinosis. J Neurol249:1398–1400
44. van Diggelen OP, Thobois S, Tilikete C, Zabot MT, KeulemansJL, van Bunderen PA, Taschner PE, Losekoot M, Voznyi YV(2001) Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of achildhood disease. Ann Neurol 50:269–272
45. Humphreys S, Lake BD, Scholtz CL (1985) Congenitalamaurotic idiocy—a pathological, histochemical, biochemicaland ultrastructural study. Neuropathol Appl Neurobiol 11:475–484
46. Garborg I, Torvik A, Hals J, Tangsrud SE, Lindemann R (1987)Congenital neuronal ceroid lipofuscinosis. A case report. ActaPathol Microbiol Immunol Scand A 95:119–125
47. Rapola J, Salonen R, Ammala P, Santavuori P (1990) Prenataldiagnosis of the infantile type of neuronal ceroid lipofuscinosisby electron microscopic investigation of human chorionic villi.Prenat Diagn 10:553–559
48. Kohlschütter A, Lake BD (1999) NCL variants without geneticassignment. In: Goebel HH, Mole SE, Lake BD (eds) Theneuronal ceroid lipofuscinoses (Batten disease). IOS, Amster-dam, pp 125–127
49. Burck U, Goebel HH, Kuhlendahl HD, Meier C, Goebel KM(1981) Neuromyopathy and vitamin E deficiency in man.Neuropediatrics 12:267–278
50. Wlodawer A, Li M, Dauter Z, Gustchina A, Uchida K, OyamaH, Dunn BM, Oda K (2001) Carboxyl proteinase fromPseudomonas defines a novel family of subtilisin-like enzymes.Nat Struct Biol 8:442–446
51. Ezaki J, Takeda-Ezaki M, Kominami E (2000) Tripeptidylpeptidase I, the late infantile neuronal ceroid lipofuscinosisgene product, initiates the lysosomal degradation of subunit c ofATP synthase. J Biochem (Tokyo) 128:509–516
52. Junaid MA, Wu G, Pullarkat RK (2000) Purification andcharacterization of bovine brain lysosomal pepstatin-insensitiveproteinase, the gene product deficient in the human late-infantile neuronal ceroid lipofuscinosis. J Neurochem 74:287–294
53. Warburton MJ, Bernardini F (2001) The specificity of lysoso-mal tripeptidyl peptidase-I determined by its action on angio-tensin-II analogues. FEBS Lett 500:145–148
54. Bernardini F, Warburton MJ (2002) The lysosomal degradationof cholecystokinin-(29–33)-amide in mouse brain is dependenton tripeptidyl peptidase: I. Implications for the degradation andstorage of peptides in classical late infantile neuronal ceroidlipofuscinosis. Biochem J 366:521–529
55. Kopan S, Sivasubramaniam U, Warburton MJ (2004) Thelysosomal degradation of neuromedin B is dependent ontripeptidyl peptidase: I. Evidence for the impairment ofneuropeptide degradation in late-infantile neuronal ceroidlipofuscinosis. Biochem Biophys Res Commun 319:58–65
56. Golabek AA, Kida E, Walus M, Wujek P, Mehta P, WisniewskiKE (2003) Biosynthesis, glycosylation, and enzymatic proces-sing in vivo of human tripeptidyl-peptidase I. J Biol Chem278:7135–7145
57. Lin L, Sohar I, Lackland H, Lobel P (2001) The human CLN2protein/tripeptidyl-peptidase I is a serine protease that auto-activates at acidic pH. J Biol Chem 276:2249–2255
58. Vesa J, Chin MH, Oelgeschlager K, Isosomppi J, DellAngelicaEC, Jalanko A, Peltonen L (2002) Neuronal ceroid lipofusci-noses are connected at molecular level: interaction of CLN5protein with CLN2 and CLN3. Mol Biol Cell 13:2410–2420
59. Wlodawer A, Durell SR, Li M, Oyama H, Oda K, Dunn BM(2003) A model of tripeptidyl-peptidase I (CLN2), a ubiquitousand highly conserved member of the sedolisin family of serine-carboxyl peptidases. BMC Struct Biol 3:8
16 18/05/05 - PROOF PAGEMAKEUP 10048_218

60. Sleat DE, Donnelly RJ, Lackland H, Liu C-G, Sohar I,Pullarkat RK, Lobel P (1997) Association of mutations in alysosomal protein with classical late-infantile neuronal ceroidlipofuscinosis. Science 277:1802–1805
61. Kurachi Y, Oka A, Itoh M, Mizuguchi M, Hayashi M,Takashima S (2001) Distribution and development of CLN2protein, the late-infantile neuronal ceroid lipofuscinosis geneproduct. Acta Neuropathol (Berl) 102:20–26
62. Young EP, Worthington VC, Jackson M, Winchester BG (2001)Pre- and postnatal diagnosis of patients with CLN1 and CLN2by assay of palmitoyl-protein thioesterase and tripeptidyl-peptidase I activities. Eur J Paediatr Neurol 5(Suppl A):193–196
63. Lukacs Z, Santavuori P, Keil A, Steinfeld R, Kohlschutter A(2003) Rapid and simple assay for the determination oftripeptidyl peptidase and palmitoyl protein thioesterase activ-ities in dried blood spots. Clin Chem 49:509–511
64. Tessa A, Simonati A, Tavoni A, Bertini E, Santorelli FM (2000)A novel nonsense mutation (Q509X) in three Italian late-infantile neuronal ceroid-lipofuscinosis children. Hum Mutat(online) 15:577
65. Ju W, Zhong R, Moore S, Moroziewicz D, Currie JR, Parfrey P,Brown WT, Zhong N (2002) Identification of novel CLN2mutations shows Canadian specific NCL2 alleles. J Med Genet39:822–825
66. Claussen M, Heim P, Knispel J, Goebel HH, Kohlschutter A(1992) Incidence of neuronal ceroid lipofuscinosis in WestGermany: variation of a method for studying autosomalrecessive disorders. Am J Med Genet 42:536–538
67. Zhong N, Wisniewski KE, Hartikainen J, Ju W, MoroziewiczDN, McLendon L, Brooks SS, Brown WT (1998) Twocommon mutations in the CLN2 gene underlie late infantileneuronal ceroid lipofuscinosis. Clin Genet 54:234–238
68. Ko CH, Kong CK, Chow TC, Lee KC (2001) Classic lateinfantile neuronal ceroid lipofuscinosis in a Chinese patient.Hong Kong Med J 7:93–96
69. Lam CW, Poon PM, Tong SF, Ko CH (2001) Two novel CLN2gene mutations in a Chinese patient with classical late-infantileneuronal ceroid lipofuscinosis. Am J Med Genet 99:161–163
70. Lavrov AY, Ilyna ES, Zakharova EY, Boukina AM, TishkaninaSV (2002) The first three Russian cases of classical, late-infantile, neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol6:161–164
71. Barisic N, Logan P, Pikija S, Skarpa D, Blau N (2003) R208Xmutation in CLN2 gene associated with reduced cerebrospinalfluid pterins in a girl with classic late infantile neuronal ceroidlipofuscinosis. Croat Med J 44:489–493
72. Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower-Brooks S,Pullarkat RK, Palmer DN, Lerner TJ, Boustany R-M, Uldall P,Siakotos AN, Donnelly RJ, Lobel P (1999) Mutational analysisof the defective protease in classical late-infantile neuronalceroid lipofuscinosis, a neurodegenerative lysosomal storagedisorder. Am J Hum Genet 64:1511–1523
73. Zhong N, Moroziewicz D, Jurkiewicz A, Ju W, Johnston L,Wisniewski KE, Brown WT (2000) Heterogeneity of lateinfantile neuronal ceroid lipofuscinoses. Genet Med 2:312–318
74. Lin L, Lobel P (2001) Expression and analysis of CLN2variants in CHO cells: Q100R represents a polymorphism, andG389E and R447H represent loss-of-function mutations. HumMutat 18:165
75. Steinfeld R, Heim P, Von Gregory H, Meyer K, Ullrich K,Goebel HH, Kohlschutter A (2002) Late infantile neuronalceroid lipofuscinosis: quantitative description of the clinicalcourse in patients with CLN2 mutations. Am J Med Genet112:347–354
76. Wisniewski KE, Kida E, Connell F, Zhong N (2000) Neuronalceroid lipofuscinoses: research update. Neurol Sci 21:S49–S56
77. Chow CW, Borg J, Billson VR, Lake BD (1993) Fetal tissueinvolvement in the late infantile type of neuronal ceroidlipofuscinosis. Prenat Diagn 13:833–841
78. Williams RE, Gottlob I, Lake BD, Goebel HH, Winchester BG,Wheeler RB (1999) CLN2—classic late infantile NCL. In:Goebel HH, Mole SE, Lake BD (eds) The neuronal ceroidlipofuscinoses (Batten disease). IOS, Amsterdam, pp 37–54
79. Wisniewski KE, Kaczmarski A, Kida E, Connell F, KaczmarskiW, Michalewski MP, Moroziewicz DN, Zhong N, Das AM,Jolly RD, Kohlschutter A (1999) Reevaluation of neuronalceroid lipofuscinoses: atypical juvenile onset may be the resultof CLN2 mutations. Mol Genet Metab 66:248–252
80. Goebel HH, Kominami E, Neuen-Jacob E, Wheeler RB (2001)Morphological studies on CLN2. Eur J Paediatr Neurol 5(SupplA):203–207
81. Rowan SA, Lake BD (1995) Tissue and cellular distribution ofsubunit c of ATP synthase in Batten disease (neuronal ceroid-lipofuscinosis). Am J Med Genet 57:172–176
82. Caillaud C, Manicom J, Puech JP, Lobel P, Poenaru L (1999)Enzymatic and molecular pre natal and postnatal diagnosis ofceroid lipofuscinoses. Am J Hum Genet 65(Suppl):A232
83. Hartikainen JM, Ju W, Wisniewski KE, Moroziewicz DN,Kaczmarski AL, McLendon L, Zhong D, Suarez CT, BrownWT, Zhong N (1999) Late infantile neuronal ceroid lipofusci-nosis is due to splicing mutations in the CLN2 gene. Mol GenetMetab 67:162–168
84. Tsiakas K, Steinfeld R, Storch S, Ezaki J, Lukacs Z, KominamiE, Kohlschutter A, Ullrich K, Braulke T (2004) Mutation of theglycosylated asparagine residue 286 in human CLN2 proteinresults in loss of enzymatic activity. Glycobiology 14:1C–5C
85. Wujek P, Kida E, Walus M, Wisniewski KE, Golabek AA(2004) N-Glycosylation is crucial for folding, trafficking, andstability of human tripeptidyl-peptidase I. J Biol Chem279:12827–12839
86. Simonati A, Santorum E, Tessa A, Polo A, Simonetti F,Bernardina BD, Santorelli FM, Rizzuto N (2000) A CLN2 genenonsense mutation is associated with severe caudate atrophyand dystonia in LINCL. Neuropediatrics 31:199–201
87. Ezaki J, Takeda-Ezaki M, Koike M, Ohsawa Y, Taka H, MinekiR, Murayama K, Uchiyama Y, Ueno T, Kominami E (2003)Characterization of Cln3p, the gene product responsible forjuvenile neuronal ceroid lipofuscinosis, as a lysosomal integralmembrane glycoprotein. J Neurochem 87:1296–1308
88. Haskell RE, Carr CJ, Pearce DA, Bennett MJ, Davidson BL(2000) Batten disease: evaluation of CLN3 mutations on proteinlocalization and function. Hum Mol Genet 9:735–744
89. Järvelä I, Sainio M, Rantamaki T, Olkkonen VM, Carpén O,Peltonen L, Jalanko A (1998) Biosynthesis and intracellulartargeting of the CLN3 protein defective in Batten disease. HumMol Genet 7:85–90
90. Järvelä I, Lehtovirta M, Tikkanen R, Kyttälä A, Jalanko A(1999) Defective intracellular transport of CLN3 is themolecular basis of Batten disease (JNCL). Hum Mol Genet8:1091–1098
91. Luiro K, Kopra O, Lehtovirta M, Jalanko A (2001) CLN3protein is targeted to neuronal synapses but excluded fromsynaptic vesicles: new clues to Batten disease. Hum Mol Genet10:2123–2131
92. Janes RW, Munroe PB, Mitchsion HM, Gardiner RM, Mole SE,Wallace BA (1996) A model for Batten disease protein CLN3:functional implications from homology and mutations. FEBSLett 399:75–77
93. Mao Q, Foster BJ, Xia H, Davidson BL (2003) Membranetopology of CLN3, the protein underlying Batten disease.FEBS Lett 541:40–46
94. Kyttälä A, Ihrke G, Vesa J, Schell MJ, Luzio JP (2003) Twomotifs target Batten disease protein CLN3 to lysosomes intransfected non-neuronal and neuronal cells. Mol Biol Cell
95. Rakheja D, Narayan SB, Pastor JV, Bennett MJ (2004) CLN3P,the Batten disease protein, localizes to membrane lipid rafts(detergent-resistant membranes). Biochem Biophys Res Com-mun 317:988–991
96. Pearce DA, Ferea T, Nosel SA, Das B, Sherman F (1999)Action of BTN1, the yeast orthologue of the gene mutated inBatten disease. Nat Genet 22:55–58
18/05/05 - PROOF PAGEMAKEUP 10048_218 17

97. Chattopadhyay S, Muzaffar NE, Sherman F, Pearce DA (2000)The yeast model for Batten disease: mutations in BTN1, BTN2,and HSP30 alter pH homeostasis. J Bacteriol 182:6418–6423
98. The International Batten Disease Consortium (1995) Isolationof a novel gene underlying Batten disease, CLN3. Cell 82:949–957
99. Mitchison HM, O'Rawe AM, Taschner PEM, Santavuori P, deVos N, Breuning MH, Mole SE, Gardiner RM, Järvelä IE(1995) Batten disease (CLN3): linkage disequilibrium mappingin the Finnish population and analysis of European haplotypes.Am J Hum Genet 56:654–662
100. Munroe PB, Mitchison HM, O'Rawe AM, Anderson JW,Boustany R-M, Lerner TJ, Taschner PEM, de Vos N, BreuningMH, Gardiner RM, Mole SE (1997) Spectrum of mutations inthe Batten disease gene, CLN3. Am J Hum Genet 61:310–316
101. Munroe PB, O'Rawe AM, Mitchison HM, Järvelä IE,Santavuouri P, Lerner TJ, Taschner PEM, Gardiner RM,Mole SE (1997) Strategy for mutation detection in CLN3:characterisation of two Finnish mutations. Neuropediatrics28:15–17
102. Järvelä I, Autti T, Lamminranta S, Åberg L, Raininko R,Santavuori P (1997) Clinical and magnetic resonance imagingfindings in Batten disease: analysis of the major mutation(1.02-kb deletion). Ann Neurol 42:799–802
103. Lauronen L, Munroe PB, Järvelä I, Autti T, Mitchison HM,O'Rawe AM, Gardiner RM, Mole SE, Puranen J, Häkkinen A-M, Kirveskari E, Santavuori P (1999) Delayed classic andprotracted phenotypes of compound heterozygous juvenileneuronal ceroid lipofuscinosis. Neurology 52:360–365
104. Goebel HH, Pilz H, Gullotta F (1976) The protracted form ofjuvenile neuronal ceroid-lipofuscinosis. Acta Neuropathol(Berl) 36:393–396
105. Elleder M, Lake BD, Goebel HH, Rapola J, Haltia M,Carpenter S (1999) Definitions of the ultrastructural patternsfound in NCL. In: Goebel HH, Mole SE, Lake BD (eds) Theneuronal ceroid lipofuscinoses (Batten disease). IOS, Amster-dam, pp 5–15
106. Hofman I, Kohlschütter A, Santavuori P, Gottlob I, GoebelHH, Lake BD, Schutgens RBH, Greene NDE, Leung K-Y,Mitchison HM, Munroe PB, Taschner PEM (1999) CLN3—juvenile NCL. In: Goebel HH, Mole SE, Lake BD (eds) Theneuronal ceroid lipofuscinoses (Batten disease). IOS, Amster-dam, pp 55–70
107. Goebel HH (2004) Introduction. Symposium: the neuronalceroid lipofuscinopses (NCL)—a group of lysosomal storagediseases come of age. Brain Pathol 14:59–60
108. Goebel HH, Zeman W, Pilz H (1975) Significance of musclebiopsies in neuronal ceroid-lipofuscinoses. J Neurol NeurosurgPsychiatry 38:985–993
109. Munroe PB, Rapola J, Mitchison HM, Mole SE, Gardiner RM,Järvelä IE (1996) Prenatal diagnosis of Batten disease. Lancet347:1014–1015
110. Lake BD, Young EP, Winchester BG (1998) Prenatal diagnosisof lysosomal storage diseases. Brain Pathol 8:133–149
111. Goebel HH, Zeman W, Patel VK, Pullarkat RK, Lenard HG(1979) On the ultrastructural diversity and essence of residualbodies in neuronal ceroid-lipofuscinosis. Mech Ageing Dev10:53–70
112. Goebel HH, Kohlschutter A, Lenard HG (1982) Morphologicand chemical biopsy findings in mucolipidosis IV. ClinNeuropathol 1:73–82
113. Bargal R, Goebel HH, Latta E, Bach G (2002) MucolipidosisIV: novel mutation and diverse ultrastructural spectrum in theskin. Neuropediatrics 33:199–202
114. Goebel HH (1993) Protracted juvenile neuronal ceroid-lipofuscinosis. J Inherit Metab Dis 16:233–236
115. Wisniewski KE, Zhong N, Kaczmarski W, Kaczmarski A,Sklower-Brooks S, Brown WT (1998) Studies of atypicalJNCL suggest overlapping with other NCL forms. PediatrNeurol 18:36–40
116. Wisniewski KE, Zhong N, Kaczmarski W, Kaczmarski A,Kida E, Brown WT, Schwarz KO, Lazzarini AM, Rubin AJ,Stenroos ES, Johnson WG, Wisniewski TM (1998) Compoundheterozygous genotype is associated with protracted juvenileneuronal ceroid lipofuscinosis. Ann Neurol 43:106–110
117. Åberg L, Järvelä I, Rapola J, Autti T, Kirveskari E, Lappi M,Sipilä L, Santavuori P (1998) Atypical juvenile neuronalceroid lipofuscinosis with granular osmiophilic deposit-likeinclusions in the autonomic nerve cells of the gut wall. ActaNeuropathol 95:306–312
118. de los Reyes E, Dyken PR, Phillips P, Brodsky M, Bates S,Glasier C, Mrak RE (2004) Profound infantile neuroretinaldysfunction in a heterozygote for the CLN3 genetic defect. JChild Neurol 19:42–46
119. Boehme DH, Cottrell JC, Leonberg SC, Zeman W (1971) Adominant form of neuronal ceroid-lipofuscinosis. Brain94:745–760
120. Nijssen PC, Ceuterick C, van Diggelen OP, Elleder M, MartinJJ, Teepen JL, Tyynela J, Roos RA (2003) Autosomaldominant adult neuronal ceroid lipofuscinosis: a novel formof NCL with granular osmiophilic deposits without palmitoylprotein thioesterase 1 deficiency. Brain Pathol 13:574–581
121. Ferrer I, Arbizu T, Pena J, Serra JP (1980) A Golgi andultrastructural study of a dominant form of Kufs' disease. JNeurol 222:183–190
122. Dyken P, Wisniewski K (1995) Classification of the neuronalceroid-lipofuscinoses: expansion of the atypical forms. Am JMed Genet 57:150–154
123. Josephson SA, Schmidt RE, Millsap P, McManus DQ, MorrisJC (2001) Autosomal dominant Kufs' disease: a cause of earlyonset dementia. J Neurol Sci 188:51–60
124. Berkovic SF, Carpenter S, Andermann F, Andermann E, WolfeLS (1988) Kufs' disease: a critical reappraisal. Brain 111:27–62
125. Sadzot B, Reznik M, Arrese-Estrada JE, Franck G (2000)Familial Kufs' disease presenting as a progressive myoclonicepilepsy. J Neurol 247:447–454
126. Constantinidis J, Wisniewski KE, Wisniewski TM (1992) Theadult and a new late adult forms of neuronal ceroid lipofus-cinosis. Acta Neuropathol (Berl) 83:461–468
127. Dom R, Brucher JM, Ceuterick C, Carton H, Martin JJ (1979)Adult ceroid-lipofuscinosis (Kufs' disease) in two brothers.Retinal and visceral storage in one; diagnostic muscle biopsyin the other. Acta Neuropathol (Berl) 45:67–72
128. Vercruyssen A, Martin JJ, Ceuterick C, Jacobs K, Swerts L(1982) Adult ceroid-lipofuscinosis: diagnostic value ofbiopsies and of neurophysiological investigations. J NeurolNeurosurg Psychiatry 45:1056–1059
129. Martin JJ, Libert J, Ceuterick C (1987) Ultrastructure of brainand retina in Kufs' disease (adult type-ceroid-lipofuscinosis).Clin Neuropathol 6:231–235
130. Isosomppi J, Vesa J, Jalanko A, Peltonen L (2002) Lysosomallocalization of the neuronal ceroid lipofuscinosis CLN5protein. Hum Mol Genet 11:885–891
131. Savukoski M, Klockars T, Holmberg V, Santavuori P, LanderES, Peltonen L (1998) CLN5, a novel gene encoding a putativetransmembrane protein mutated in Finnish variant late infantileneuronal ceroid lipofuscinosis. Nat Genet 19:286–288
132. Holmberg V, Lauronen L, Autti T, Santavuori P, Savukoski M,Uvebrant P, Hofman I, Peltonen L, Järvelä I (2000) Pheno-type–genotype correlation in eight patients with Finnishvariant late infantile NCL (CLN5). Neurology 55:579–581
133. Pineda-Trujillo N, Cornejo W, Wheeler RB, Múnera S,Valencia A, Agudelo-Arango J, Cogollo A, Anderson G,Bedoya G, Mole SE, Ruíz-Linares A (2004) A novel mutationin CLN5 underlying juvenile onset neuronal ceroid lipofusci-nosis in South America. Neurology (in press)
134. Santavuori P, Rapola J, Nuutila A, Raininko R, Lappi M,Launes J, Herva R, Saino K (1991) The spectrum of Jansky–Bielschowsky disease. Neuropediatrics 22:92–96
18 18/05/05 - PROOF PAGEMAKEUP 10048_218

135. Autti T, Raininko R, Launes J, Nuutila A, Santavuori P (1992)Jansky–Bielschowsky variant disease: CT, MRI, and SPECTfindings. Paediatric Neurol 8:121–126
136. Lauronen L, Huttunen J, Kirveskari E, Wikstrom H, Sainio K,Autti T, Santavuori P (2002) Enlarged SI and SII somatosen-sory evoked responses in the CLN5 form of neuronal ceroidlipofuscinosis. Clin Neurophysiol 113:1491–1500
137. Santavuori P, Rapola J, Saino K, Raitta C (1982) A variant ofJansky–Bielschowsky disease. Neuropediatrics 13:135–141
138. Tyynelä J, Suopanki J, Santavuori P, Baumann M, Haltia M(1997) Variant late infantile neuronal ceroid-lipofuscinosis:pathology and biochemistry. J Neuropathol Exp Neurol56:369–375
139. Rapola J, Lake BD (2000) Lymphocyte inclusions in Finnish-variant late infantile neuronal ceroid lipofuscinosis (CLN5).Neuropediatrics 31:33–34
140. Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L,Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, BronsonRT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME(2002) Mutations in a novel CLN6-encoded transmembraneprotein cause variant neuronal ceroid lipofuscinosis in man andmouse. Am J Hum Genet 70:324–335
141. Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE,Mole SE (2002) The gene mutated in variant late infantileneuronal ceroid lipofuscinosis (CLN6) and nclf mutant miceencodes a novel predicted transmembrane protein. Am J HumGenet 70:537–542
142. Mole SE, Michaux G, Codlin S, Wheeler RB, Sharp JD, CutlerDF (2004) CLN6, which is associated with a lysosomal storagedisease, is an endoplasmic reticulum protein. Exp Cell Res298:399–406
143. Heine C, Koch B, Storch S, Kohlschutter A, Palmer DN,Braulke T (2004) Defective ER-resident membrane proteinCLN6 affects lysosomal degradation of endocytosed arylsul-fatase A. J Biol Chem 279:22347–22352
144. Sharp JD, Wheeler RB, Parker KA, Gardiner RM, WilliamsRE, Mole SE (2003) Spectrum of CLN6 mutations in variantlate infantile neuronal ceroid lipofuscinosis. Hum Mutat22:35–42
145. Lake BD, Cavanagh NPC (1978) Early-juvenile Batten'sdisease—a recognisable subgroup distinct from other formsof Batten's disease. J Neurol Sci 36:265–271
146. Kimura S, Goebel HH (1987) Electron microscopic studies onskin and lymphocytes in early juvenile neuronal ceroidlipofuscinosis. Brain Dev 9:576–580
147. Andermann E, Jacob JC, Andermann F, Carpenter S, Wolfe L,Berkovic SF (1988) The Newfoundland aggregate of neuronalceroid lipofuscinosis. Am J Med Genet Suppl 5:111–116
148. Wheeler RB, Sharp JD, Mitchell WA, Bate SL, Williams RE,Lake BD, Gardiner RM (1999) A new locus for variant lateinfantile neuronal ceroid lipofuscinosis (LINCL)—CLN7. MolGenet Metab 66:337–338
149. Peña JA, Cardozo JJ, Montiel CM, Molina OM, Boustany R(2001) Serial MRI findings in the Costa Rican variant ofneuronal ceroid-lipofuscinosis. Pediatr Neurol 25:78–80
150. Elleder M, Franc J, Kraus J, Nevsímalová S, Sixtová K,Zeman J (1997) Neuronal ceroid lipofuscinosis in the CzechRepublic: analysis of 57 cases. Report of the ‘Prague NCLGroup’. Eur J Paediatr Neurol 4:109–114
151. Teixeira C, Guimaraes A, Bessa C, Ferreira MJ, Lopes L, PintoE, Pinto R, Boustany RM, Sa Miranda MC, Ribeiro MG(2003) Clinicopathological and molecular characterization ofneuronal ceroid lipofuscinosis in the Portuguese population. JNeurol 250:661–667
152. Teixeira CA, Espinola JA, Huo L, Kohlschütter J, PersaudSawin D-A, Minassian B, Bessa CJP, Guimarães A, StephanAA, Clara Sà Miranda M, MacDonald ME, Gil Ribeiro M,Boustany R-M (2003) Novel mutations in the CLN6 genecausing a variant late infantile neuronal ceroid lipofuscinosis.Hum Mutat 21:502–508
153. Nardocci N, Morbin M, Bugiani M, Lamantea E, Bugiani O(2000) Neuronal ceroid lipofuscinosis: detection of atypicalforms. Neurol Sci 21:S57–S61
154. Binelli S, Canafoglia L, Panzica F, Pozzi A, Franceschetti S(2000) Electroencephalographic features in a series of patientswith neuronal ceroid lipofuscinoses. Neurol Sci 21:S83–S87
155. Scaioli V, Nardocci N (2000) A pathophysiological study ofneuronal ceroid lipofuscinoses in 17 patients: critical reviewand methodological proposal. Neurol Sci 21:S89–S92
156. Veneselli E, Biancheri R, Perrone MV, Buoni S, Fois A (2000)Neuronal ceroid lipofuscinoses: clinical and EEG findings in alarge study of Italian cases. Neurol Sci 21:S75–S81
157. Iannetti P, Messa C, Spalice A, Lucignani G, Fazio F (1994)Positron emission tomography in neuronal ceroid lipofuscino-sis (Jansky–Bielschowsky disease): a case report. Brain Dev16:459–462
158. Williams RE, Lake BD, Elleder M, Sharp J (1999) CLN6—variant late infantile/early juvenile NCL. In: Goebel HH, MoleSE, Lake BD (eds) The neuronal ceroid lipofuscinoses (Battendisease). IOS, Amsterdam, pp 102–113
159. Heine C, Tyynela J, Cooper JD, Palmer DN, Elleder M,Kohlschutter A, Braulke T (2003) Enhanced expression ofmanganese-dependent superoxide dismutase in human andsheep CLN6 tissues. Biochem J 376:369–376
160. Mitchell WA, Wheeler RB, Sharp JD, Bate SL, Gardiner RM,Ranta US, Lonka L, Williams RE, Lehesjoki AE, Mole SE(2001) Turkish variant late infantile neuronal ceroid lipofus-cinosis (CLN7) may be allelic to CLN8. Eur J Paediatr Neurol5(Suppl A):21–27
161. Ranta S, Topçu M, Tegelberg S, Tan H, Üstübütün A, Saatci I,Dufke A, Enders H, Pohl K, Alembik Y, Mitchell WA, MoleSE, Lehesjoki A-E (2004) Variant late infantile neuronal ceroidlipofuscinosis in a subset of Turkish patients is allelic toNorthern epilepsy. Hum Mutat 23:300–305
162. Williams RE, Topçu M, Lake BD, Mitchell WA, Mole SE(1999) CLN7—Turkish variant late infantile NCL. In: GoebelHH, Mole SE, Lake BD (eds) The neuronal ceroidlipofuscinoses (Batten disease). IOS, Amsterdam, pp 114–116
163. Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A,Sharp J, Wheeler R, Kusumi K, Mole S, Liu W, Soares MB, deFatima Bonaldo M, Hirvasniemi A, de la Chapelle A, GilliamTC, Lehesjoki AE (1999) The neuronal ceroid lipofuscinosesin human EPMR and mnd mutant mice are associated withmutations in CLN8. Nat Genet 23:233–236
164. Lonka L, Kyttälä A, Ranta S, Jalanko A, Lehesjoki AE (2000)The neuronal ceroid lipofuscinosis CLN8 membrane protein isa resident of the endoplasmic reticulum. Hum Mol Genet9:1691–1697
165. Lonka L, Salonen T, Siintola E, Kopra O, Lehesjoki AE,Jalanko A (2004) Localization of wild-type and mutantneuronal ceroid lipofuscinosis CLN8 proteins in non-neuronaland neuronal cells. J Neurosci Res 76:862–871
166. Winter E, Ponting CP (2002) TRAM, LAG1 and CLN8:members of a novel family of lipid-sensing domains? TrendsBiochem Sci 27:381–383
167. Katz ML, Khan S, Awano T, Shahid SA, Siakotos AN,Johnson GS (2005) A mutation in the CLN8 gene in Englishsetter dogs with neuronal ceroid-lipofuscinosis. BiochemBiophys Res Commun 327:541–547
168. Hirvasniemi A, Lang H, Lehesjoki AE, Leisti J (1994)Northern epilepsy syndrome: an inherited childhood onsetepilepsy with associated mental deterioration. J Med Genet31:177–182
169. Topçu M, Tan H, Yalnizoglu D, Usubutun A, Saatci I, AynaciM, Anlar B, Topaloglu H, Turanli G, Kose G, Aysun S (2004)Evaluation of 36 patients from Turkey with neuronal ceroidlipofuscinosis: clinical, neurophysiological, neuroradiologicaland histopathologic studies. Turk J Pediatr 46:1–10
170. Herva R, Tyynela J, Hirvasniemi A, Syrjakallio-Ylitalo M,Haltia M (2000) Northern epilepsy: a novel form of neuronalceroid-lipofuscinosis. Brain Pathol 10:215–222
18/05/05 - PROOF PAGEMAKEUP 10048_218 19

171. Haltia M, Tyynelä J, Hirvasniemi A, Herva R, Ranta US,Lehesjoki A-E (1999) CLN8—Northern epilepsy. In: GoebelHH, Mole SE, Lake BD (eds) The neuronal ceroidlipofuscinoses (Batten disease). IOS, Amsterdam, pp 117–124
172. Stengel OC (1826) Account of a singular illness among foursiblings in the vicinity of RØraas. Ceroid-lipofuscinosis(Batten's disease). Elsevier/North Holland Biomedical Press,1982, Amsterdam, pp 17–19
173. Zeman W, Dyken P (1969) Neuronal ceroid-lipofuscinosis(Batten's disease): relationship to amaurotic familial idiocy?Pediatrics 44:570–583
174. Zeman W (1971) Morphological approaches to the nosologyof nervous system defects. Birth Defects Orig Artic Ser 3:23–30
175. Tyynelä J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, BaumannM, Haltia M, Lobel P (2000) A mutation in the ovine cathepsinD gene causes a congenital lysosomal storage disease withprofound neurodegeneration. EMBO J 19:2786–2792
176. Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y,Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S,Shibata M, Yamamoto K, Kominami E, Peters C, von FiguraK, Uchiyama Y (2000) Cathepsin D deficiency induceslysosomal storage with ceroid lipofuscin in mouse CNSneurons. J Neurosci 20:6898–6906
177. Gupta P, Soyombo AA, Atashband A, Wisniewski KE, SheltonJM, Richardson JA, Hammer RE, Hofmann SL (2001)Disruption of PPT1 or PPT2 causes neuronal ceroid lipofus-cinosis in knockout mice. Proc Natl Acad Sci U S A98:13566–13571
178. Gupta P, Soyombo AA, Shelton JM, Wilkofsky IG,Wisniewski KE, Richardson JA, Hofmann SL (2003) Disrup-tion of PPT2 in mice causes an unusual lysosomal storagedisorder with neurovisceral features. Proc Natl Acad Sci U S A100:12325–12330
179. Jolly RD, Martinus RD, Palmer DN (1992) Sheep and otheranimals with ceroid lipofuscinoses: their relevance to Battendisease. Am J Med Genet 42:609–614
180. Lingaas F, Aarskaug T, Sletten M, Bjerkas I, Grimholt U, MoeL, Juneja RK, Wilton AN, Galibert F, Holmes NG, Dolf G(1998) Genetic markers linked to neuronal ceroid lipofusci-nosis in English setter dogs. Anim Genet 29:371–376
20 18/05/05 - PROOF PAGEMAKEUP 10048_218





























