Embed Size (px)
Citation preview

Journal of Biomaterials Science 20 (2009) 1031–1047www.brill.nl/jbs
Delivery of Cisplatin from Pluronic Co-polymer Systems:Liposome Inclusion and Alginate Coupling
Jia-You Fang a,∗, Shu-Hui Hsu b, Yann-Lii Leu c and Jiuan-Wen Hu a
a Pharmaceutics Laboratory, Graduate Institute of Natural Products, Chang Gung University,259 Wen-Hwa 1st Road, Kweishan, Taoyuan, Taiwan
b Department of Pharmacy, Chia Nan University of Pharmacy and Science, Tainan County, Taiwanc Natural Products Laboratory, Graduate Institute of Natural Products, Chang Gung University,
Kweishan, Taoyuan, Taiwan
Received 29 November 2007; accepted 22 May 2008
AbstractThe aim of this work was to evaluate the use of Pluronic F127 (PF) hydrogels incorporating liposomesand/or grafted with alginate (AP) for the parenteral delivery of cisplatin. The physicochemical propertiessuch as scanning electron microscopy (SEM), polarity and sol–gel transition temperature, as well as in vitrodrug release of developed hydrogels were examined. The sol–gel temperature of PF, PF with liposomes andAP was 26.4, 19.7 and 26.6◦C, respectively. A hydrogel with stronger strength was obtained by alginatecoupling (AP) according to SEM images and viscosity kinetics as compared to PF. Both liposomes andhydrogels could sustain the release of cisplatin to a certain level. The drug release from the vehicles de-creased in the order of PF > liposomes � AP � PF/liposomes > AP/liposomes. The burst release effect ofcisplatin was inhibited when using the AP/liposomes composite system as the vehicle. Formulations wereadministered intratumorally in melanoma-bearing mice. The respective liposomes or hydrogels did not in-crease cisplatin accumulation in melanomas compared to the control (aqueous solution). PF/liposomes andAP/liposomes, respectively, increased cisplatin deposition by 2.5- and 4.4-fold. To our best of knowledge,the present work is the first report to investigate the drug delivery from PF–alginate conjugates.© Koninklijke Brill NV, Leiden, 2009
KeywordsCisplatin, liposomes, Pluronic F127, alginate, melanoma
1. Introduction
Cisplatin has long been widely used because of its broad spectrum of cytolyticactivity against solid tumors [1]. Because of its notable renal and auditory toxi-city, however, cisplatin was almost withdrawn from clinical trials [2]. Problems
* To whom correspondence should be addressed. Tel.: (886-3) 211-8800, ext. 5521; Fax: (886-3) 211-8236;e-mail: [email protected]
© Koninklijke Brill NV, Leiden, 2009 DOI:10.1163/156856209X444493

1032 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
associated with the parenteral administration of platinum drugs include systemicside-effects, low drug concentrations at the cancer site and short half-lives [3].Polymeric hydrogels have been extensively explored as drug-delivery vehicles toachieve higher concentrations of drugs in tumor tissues and controlled-release pro-files for extended time periods. Much interest has been focused on polymer systemsthat show a phase transition in response to temperature since they form a solid arti-ficial barrier and a prolonged drug release depot after injection into the body cavity[4, 5].
Pluronic F127 (PF) (also called Poloxamer 407) is a non-ionic polyoxyethylene–polyoxypropylene–polyoxyethylene (PEO–PPO–PEO) tri-block co-polymer whichforms micelles at low concentrations and clear thermo-reversible gels at concen-trations above 20% [6]. It can be a biocompatible drug-delivery system with theaim of controlled release. Hence, PF might be useful for prolonging cisplatin re-lease. Because of the relatively small molecular size of cisplatin, a stronger barrieris needed to prevent its rapid release [4]. In order to further achieve sustained re-lease, the drugs can be loaded into liposomes which are then incorporated intothermo-reversible formulations [7]. Liposomal systems are thought to be useful,since liposomes are essentially non-toxic and biodegradable, and they can delivercisplatin to specific sites and prolong the half-life of the drug [1]. Another ap-proach used for further improving drug delivery from PF hydrogels is chemicalmodification using synthetic methods [8, 9]. Chemically modified gels show highermechanical strengths and, thus, their residence times in the human body are pro-longed.
Previous studies have grafted PF onto hyaluronic acid, poly(lactic acid) orpoly(acrylic acid) to form hydrogels for controlled drug delivery [8, 9]. Grassi et al.[10] have first developed the blending of PF and alginate to examine its rheologicalproperties. However, the PF–alginate conjugates by chemical grafting are not yet in-vestigated. The aim of this study was to develop injectable thermo-sensitive systemsbased on PF with the inclusion of liposomes and/or chemical grafting with alginatefor delivering cisplatin. Alginate is a natural polysaccharide extracted from brownsea algae. It is highly hydrophilic, biocompatible and relatively economical [11].The drug release and physicochemical characteristics of the developed hydrogels,such as the polarity, electron microscopic images and viscosity, were determined inthis report. The in vivo drug accumulation in melanomas after intratumor adminis-tration of the hydrogels was also evaluated.
2. Materials and Methods
2.1. Materials
Cisplatin, phosphatidylethanolamine (PEA) from sheep brain, cholesterol, deoxy-cholic acid (DA), 4-nitrophenyl chloroformate, triethylamine, diaminoethylene,Nile red, Pluronic F127 (PF) and sodium alginate (1.2 × 105 Da) were pur-chased from Sigma (St. Louis, MO, USA). 1-Ethyl-3-(3-dimethylaminopropyl)-

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1033
carbodiimide (EDC) and N-hydroxylsuccinimide (NHS) were obtained from AcrosOrganics (Geel, Belgium). The melanoma cell line (B16-F0) was supplied by Amer-ican Type Culture Collection (Rockville, MD, USA).
2.2. Preparation of Liposomes
PEA (160 mg) and cholesterol (40 mg) were dissolved in a 5-ml volume of a chlo-roform/methanol (2:1) solution to form a liposomal formulation (L1). DA (10 mg)was further added as an anionic surfactant in another formulation (L2). The or-ganic solvent was evaporated in a rotary evaporator at 50◦C, and solvent traceswere removed by maintaining the lipid film under a vacuum for 6 h. The films werehydrated with double-distilled water (ddH2O) containing cisplatin using a probe-type sonicator (VCX 600, Sonics and Materials, Newtown, CT, USA) at 25 W for30 min. The total volume of the final formulations was 10 ml with a drug concen-tration at 2.6 mM.
2.3. Size and Zeta Potential of Liposomes
The mean vesicle size and zeta potential of the liposomes were measured by alaser scattering method (Nano ZS® 90, Malvern, UK). Liposomal suspensions werediluted 100-fold with ddH2O before the measurement. The determination was re-peated three times for each batch. The mean values were measured from threebatches per liposomal system.
2.4. Drug Encapsulation Percentage in Liposomes
The un-entrapped drug was removed by size exclusion chromatography on aSephadex G-15 column using ddH2O as the eluent. The encapsulation efficiencywas determined by dissolving 500 µl of the liposomes obtained from gel chro-matography in 500 µl Triton X-100 (1%) and diluting this with ddH2O (200×),followed by measurement of the absorbance by graphite furnace (flameless) atomicabsorption spectrophotometry (AAS; Z-5000, Hitachi, Tokyo, Japan).
2.5. PF–Alginate Composite (AP) Synthesis
Graft co-polymers were prepared by coupling mono amine-terminated PF (MATP)with an alginate backbone using EDC and NHS as coupling agents. MATP wasfirst prepared by a two-step reaction modified from Cho et al. [12]. In the first step,PF (5 g) was reacted with 4-nitrophenyl chloroformate (80 mg) dissolved in 50 mlmethylene chloride in the presence of triethylamine (0.15 ml) at room temperaturefor 4 h to yield an intermediate. This intermediate was recovered by extraction threetimes using petroleum ether. In the second step, the intermediate was reacted withdiaminoethylene (1 ml) dissolved in methylene chloride (50 ml) at room tempera-ture for 12 h. After reacting, the mixture was extracted three times with petroleumether, then dialyzed against ddH2O using a membrane with a MW cut-off of 3500for 3 days, and then was freeze-dried to obtain the resulting product.
MATP and alginate were dissolved in 50 ml ddH2O. EDC and NHS were addedto the above solution for 24 h. After reacting, the mixture was dialyzed for 3 days

1034 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
using a membrane with a MW cut-off of (12–14) × 103, and then freeze-dried toobtain the AP co-polymer.
2.6. Fourier-Transformed Infrared (FT-IR) Absorption Spectroscopy
The chemical structure of the AP co-polymer was verified by FT-IR. Tablets of thegrafted co-polymer and alginate were obtained using KBr pellets. The measurementwas carried out on a Jasco FT/IR-410 spectrometer (Tokyo, Japan).
2.7. Polarity of Co-polymers
The hydrophobic fluorescent marker Nile red (2.5 × 10−5%, w/w) was used as themodel solute, and the molecular environment (polarity) was elucidated by fluoro-metric spectroscopy due to the solvatochromism of Nile red. Emission fluorescencespectra were determined with a Hitachi F-2500 fluorescence spectrophotometer.The spectra of the aqueous solutions of polymers loaded with Nile red wererecorded at room temperature with both slit widths set to 10 nm. The excitationwavelength was fixed at 546 nm, and emission spectra were recorded from 550 to700 nm at a scanning speed of 300 nm/min.
2.8. Preparation of Hydrogels
The PF or AP co-polymer was mixed with ddH2O with or without liposomes(20 mg/ml lipids in the final formulations) to a concentration of 20% (w/w). The re-sulting solution was left overnight in a refrigerator at 4◦C. A homogenous solutionwith a clear appearance was obtained. The mixtures were allowed to reach roomtemperature (approx. 25◦C) before the experiments.
2.9. Scanning Electron Microscopy (SEM)
Hydrogels were frozen at −80◦C and then lyophilized by a freeze-drying method[13]. Samples were fractured in liquid nitrogen and sputter-coated with gold. Theresulting dried samples were examined using a Hitachi S3000N SEM.
2.10. Sol–Gel Transition Temperature
Viscosity measurements were used to assess the gelation behavior of the hydro-gels. The viscosities of these systems were determined using a Brookfield RVDV-IIviscometer (Middleboro, MA, USA). The spindle (s29, 40 mm) was rotated in thesamples, and the viscosity was determined from 15 to 38◦C.
2.11. In Vitro Release of Cisplatin from the Hydrogels
Drug release from the hydrogels was measured using a Franz diffusion cell. A cellu-lose membrane (Spectrapor® 3, with a MW cut-off of 3500, Spectrum Laboratories,Fort Lauderdale, FL, USA) was mounted between the donor and receptor com-partments. The donor medium consisted of 0.5 ml of a hydrogel formulation. Thereceptor medium consisted of 5.5 ml of citrate–phosphate buffer at pH 7.4. Theavailable diffusion area between cells was 1.13 cm2. The stirring rate and temper-ature were maintained at 600 rpm and 37◦C, respectively. At appropriate intervals,

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1035
300-µl aliquots of the receptor medium were withdrawn and immediately replacedwith an equal volume of fresh buffer. The amount of drugs was determined by mea-suring the absorbance by AAS.
2.12. In Vivo Intratumor Administration
Melanoma cells were seeded and incubated in medium (10% fetal bovine serum,89% DMEM and 1% penicillin–streptomycin). The solid tumors were obtainedby a subcutaneous injection of 106 melanoma cells into the back of female nudemice (ICR-Foxn/nu strain, 8 weeks old). All animal experiments were reviewedand approved by the Institutional Animal Care Committee at Chang Gung Univer-sity. On day 7 when the size of the melanomas had reached approx. 250 mm3, 50 µlof hydrogels containing cisplatin was intratumorally injected into the tumor tissuewith a 29-gauge needle. Tumor volume was determined by direct measurement withcalipers. The tumor volume was calculated using the formula w2 × l × π/6, wherethe length (l) is the longest dimension and the width (w) is the dimension perpen-dicular to the length. At 24 h after administration, tumor sites were excised andweighed. The tissues were minced with scissors, positioned in a glass homogenizercontaining 1 ml of 4.31% sulfosalicylic acid, and ground for 5 min at 300 rpm. Theresulting solution was centrifuged for 10 min at 104 rpm and then filtered througha PVDF membrane (with a pore size of 0.45 µm, Millipore, Billerica, MA, USA).The drug amount in the supernatant was determined by AAS.
2.13. Statistical Analysis
Statistical analyses of differences between the various treatments were performedusing the unpaired Student’s t-test. A 0.05 level of probability (P < 0.05) wastaken as the level of significance. An analysis of variance (ANOVA) test was alsoused.
3. Results
3.1. Preparation of Liposomes
As shown in previous studies [1, 14], conventional liposomes prepared for cisplatinoffer low encapsulation efficacy. We previously developed liposomes composedof PEA, which showed greater cisplatin encapsulation compared to conventionalliposomes composed of phosphatidylcholine [15]. The PEA liposomes (L1) couldefficiently entrap cisplatin into the vesicles based on the high encapsulation percent-age of approx. 63%, as shown in Table 1. DA as an anionic surfactant can reducethe rigidity of phospholipids bilayers [16, 17]. DA was also incorporated into lipo-somes to examine the effect of bilayer rigidity on physicochemical characteristics.The incorporation of DA into the liposomes (L2) showed comparable encapsula-tion ability (no significant difference) with the system without DA. The sizes andzeta potentials of the prepared liposomes are summarized in Table 1. The liposomes

1036 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
Table 1.The cisplatin encapsulation percentage, size and zeta potential of liposomes in the absence or presenceof Pluronic F127 co-polymer in a solution type
L1 L2
Encapsulation (%) 63.7 ± 9.4 70.0 ± 6.9Size (nm) 97.1 ± 5.1 127.5 ± 3.5Size (nm) in the presence of PF 146.0 ± 2.2 169.0 ± 1.4Zeta potential (mV) −59.7 ± 7.0 −56.4 ± 0.8Zeta potential (mV) in the presence of PF −55.2 ± 1.8 −60.0 ± 3.9
L1, liposomal formulation consisting of phosphatidylethanolamine and cholesterol (4:1); L2, li-posomal formulation consisting of phosphatidylethanolamine, cholesterol and deoxycholic acid(4:1:0.25). Each value represents the mean ± SD (n = 3).
made from PEA and cholesterol (L1) had a relatively small size of 97 nm. The fur-ther addition of DA (L2) significantly increased (P < 0.05) the size of the vesicles.
As shown in Table 1, the inclusion of liposomes into PF hydrogels significantlyincreased (P < 0.05) the vesicle size by approx. 1.5-fold. The surface charge of theliposomes remained unchanged (no significant difference) after hydrogel combin-ing.
3.2. AP Composite Synthesis
The grafted co-polymer was prepared by coupling PF with alginate by theEDC/NHS method. Amide bonds were formed through the reaction between theamino groups of MATP (PF with mono amine terminal) and carboxyl ones of algi-nate. Figure 1 shows the FT-IR spectra of alginate and the grafted AP co-polymer.The alginate spectrum shows the characteristics peaks at 3381 (OH stretching),1606 (COOH stretching) and 1035 cm−1 (C–O–C stretching). In the FT-IR spec-trum of the grafted co-polymer, the carbonyl absorption band of carboxylate sodiumsalt in alginate disappeared, and a new characteristic amide I band appearedat 1644 cm−1. This result suggests that amide bonds formed between alginateand PF.
The efficiency of grafting (%) was 90.9% which was calculated by the equation:((WAP − Walginate)/WMATP) × 100%, where WAP is the weight of the freeze-driedgraft co-polymer, and Walginate and WMATP are the weights of alginate and MATP inthe feed, respectively [18]. The results are summarized in Table 2. The synthesis ofAP attained a satisfied yield to a level of 91.3%. The estimated molecular mass ofAP was approx. 32.9 × 105 Da, as determined by the end-group analysis [19]. Thezeta potential of these co-polymers in ddH2O (20%, w/w) was determined to exam-ine the electrical charge before and after conjugation. The ionizable amino groupsin MATP were protonated (+15.4 mV). The anionic characteristics of alginate ledto the high negative charge of the AP co-polymer (−47.5 mV). This suggests thatmost of the amino groups in MATP were conjugated with alginate.

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1037
Figure 1. FT-IR spectra of alginate and the Pluronic F127–alginate conjugate.
Table 2.The efficiency of grafting, estimated molecular mass and yield (%)of PF–alginate (AP) co-polymer
Value
Grafting efficiency (%) 90.9Estimated molecular mass (×10−5 Da) 32.9Yield (%) 91.3
The efficiency of grafting (%) = ((WAP − Walginate)/
WMATP) × 100, where WAP is the weight of the freeze-dried graftco-polymer, and Walginate and WMATP are the weights of alginateand MATP in the feed, respectively.
3.3. Polarity of Co-polymers
Nile red is a dye whose absorption bands vary in shape, position and intensitywith the nature of the environment. The fluorescence of Nile red is quenched in amore-hydrophilic environment [20]. The emission spectra of Nile red in co-polymersolutions are shown in Fig. 2. Grafting of alginate to PF showed a reduction in thefluorescence intensity at 604 nm (2726–2475 a.u.). This indicates an increase inenvironmental polarity after conjugation.
3.4. SEM Examination
The material structures of PF and AP hydrogels (20%, w/w) in cross-sections wereexamined by SEM, as shown in Fig. 3. The PF nanostructure in hydrogels assemblesinto a transient polymeric network as observed in Fig. 3A. The PF hydrogel struc-

1038 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
Figure 2. Fluorescence emission spectra of Nile red (2.5 × 10−5%, w/w) in Pluronic F127 (PF) andPF–alginate (AP) conjugate systems.
ture in the presence of liposomes (PL1) is shown in Fig. 3B. Although liposomalvesicles could not be observed in the image because of the manipulation for SEMexamination, it can be seen that the incorporation of liposomes did not change thebackbone structure of the PF hydrogel. The SEM image of the AP hydrogel shows aregularly interconnected texture with pores (Fig. 3C). One remarkable characteris-tic of the AP hydrogel was the deep pores. The pore size of the AP hydrogel rangedbetween 10 and 15 µm. The nanostructure of AP hydrogel was not changed by SEMobservation after the liposome inclusion (APL1, Fig. 3D).
3.5. Sol–Gel Transition Temperature
Gel formation was traced through the viscosity of the formulations, which signif-icantly increased during gelation. Figure 4 illustrates the temperature-dependentsol–gel transitions of various systems. The gelation kinetics were dependent onthe types of co-polymer and the inclusion of liposomes (L1). The PF system(20%, w/w) began to gel at 26.4◦C. The sol–gel temperature of the PF systemdecreased to 19.7◦C after inclusion of liposomes (PL1). An abrupt increase in vis-cosity at approx. 26.6◦C marks the onset of the gelation process. Similar to theresults of the PF system, the critical gel temperature of the AP system with lipo-somes (APL1) was considerably lower than that of the parent AP hydrogel.
3.6. In Vitro Release of Cisplatin from Hydrogels
The levels of in vitro cisplatin release from PF hydrogels with or without liposomes(L1 and L2) are given in Fig. 5. The amount of cisplatin released was plotted asa function of time. The aqueous solution of ddH2O was used as the control. Thedrug release of the control was virtually complete by 6 h. The encapsulation of cis-platin in the liposomes showed drug release percentages from 77% to 32–40% aftera 12-h application. Incorporation of liposomes into PF hydrogels (PL1 and PL2)
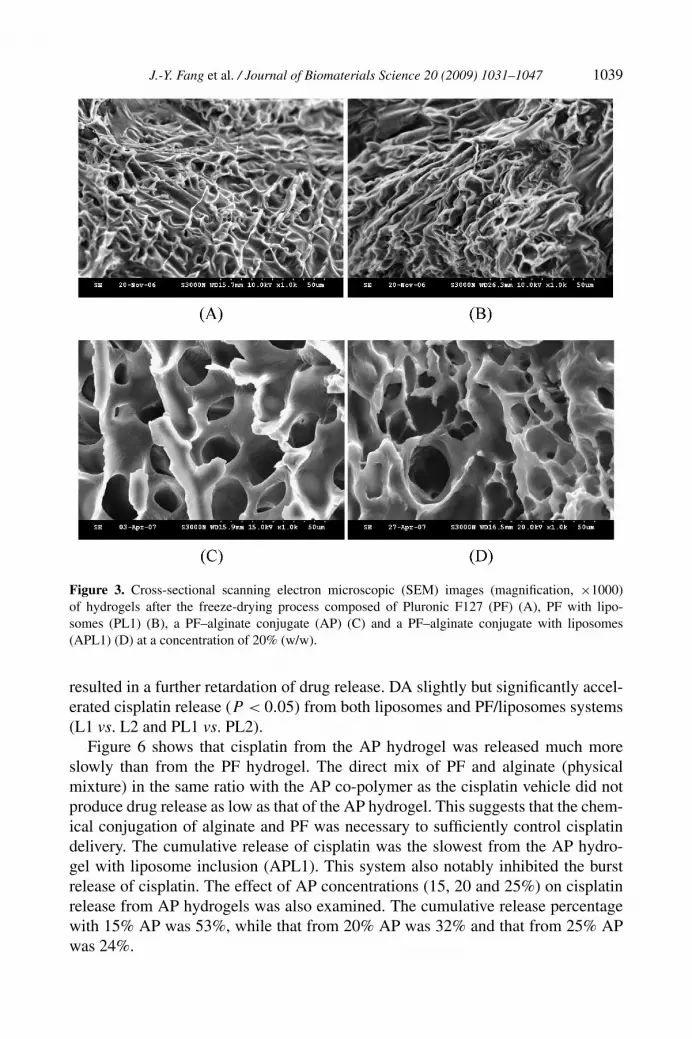
J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1039
Figure 3. Cross-sectional scanning electron microscopic (SEM) images (magnification, ×1000)of hydrogels after the freeze-drying process composed of Pluronic F127 (PF) (A), PF with lipo-somes (PL1) (B), a PF–alginate conjugate (AP) (C) and a PF–alginate conjugate with liposomes(APL1) (D) at a concentration of 20% (w/w).
resulted in a further retardation of drug release. DA slightly but significantly accel-erated cisplatin release (P < 0.05) from both liposomes and PF/liposomes systems(L1 vs. L2 and PL1 vs. PL2).
Figure 6 shows that cisplatin from the AP hydrogel was released much moreslowly than from the PF hydrogel. The direct mix of PF and alginate (physicalmixture) in the same ratio with the AP co-polymer as the cisplatin vehicle did notproduce drug release as low as that of the AP hydrogel. This suggests that the chem-ical conjugation of alginate and PF was necessary to sufficiently control cisplatindelivery. The cumulative release of cisplatin was the slowest from the AP hydro-gel with liposome inclusion (APL1). This system also notably inhibited the burstrelease of cisplatin. The effect of AP concentrations (15, 20 and 25%) on cisplatinrelease from AP hydrogels was also examined. The cumulative release percentagewith 15% AP was 53%, while that from 20% AP was 32% and that from 25% APwas 24%.

1040 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
Figure 4. Effect of temperature on the viscosities of hydrogels with PF or the PF–alginate conjugate(20%, w/w) determined using a Brookfield RVDV-II viscometer.
3.7. In Vivo Intratumor Administration
Cisplatin in formulations was intratumorally injected into melanomas on the backsof nude mice to examine drug deposition. Figure 7 shows the retention of cis-platin in melanomas at 24 h after the injection. Cisplatin accumulation within themelanomas was low in the free drug form (control). A similar result (no signifi-cant difference) was detected for the formulations of PF and AP hydrogels, as wellas liposomes (L1). The enhanced cisplatin deposition in melanomas was only ob-served in the systems combining co-polymers and liposomes (PL1 and APL1). Thetumor cisplatin level following treatment with the PF and liposome combinationwas more than 2.5-fold higher than the control. Among all the tested groups, thehydrogel incorporating AP co-polymer and liposomes showed the most superioreffect on cisplatin residence in the melanomas. The cisplatin accumulation in thetumor by the system of AP/liposomes was 4.4-fold higher compared to the con-trol.
4. Discussion
4.1. Physicochemical Characteristics of Liposomes
Cisplatin is a platinum-chelated complex with four ligands: two ammonias and twochlorides. The two chloride ligands are gradually substituted with H2O moleculesin water [21]. Both Pt(NH3)2Cl(OH2)
+ and Pt(NH3)2Cl(OH2)22+ can be formed

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1041
Figure 5. Release percentage (%)–time profiles of cisplatin across a cellulose membrane from the freeform (control), a Pluronic F127 hydrogel (PF), liposomes (L1 and L2) and the PF–liposome composite(PL1 and PL2). Each value represents the mean ± SD (n = 4).
by the aqueous complex. It is easy for the aqueous complex to form a chelate withthe lone paired electrons in PEA [15], resulting in the high cisplatin entrapment inPEA liposomes. However, the encapsulation efficiency was not increased by addingDA although the vesicle size was enlarged. This can be due to the drug leakage fromDA-containing liposomes as a result of the elastic bilayer properties [16, 17].
The charge of PEA is negative at a pH of near 7.0 [15]. This pH approximates thevalue of PEA liposomes (pH 6.7). This may have contributed to the highly negativecharge of PEA liposomes. PF is an amphiphilic thermo-gelling compound. PF mayintercalate into the PEA bilayers of liposomes in the presence of the PF co-polymer,resulting in destabilization of the liposomes and subsequent aggregation [7]. Thisleads to enlargement of the vesicle size after incorporation of liposomes into the PFsystem.
4.2. Physicochemical Characteristics of Hydrogels
PF is a PEO–PPO–PEO block co-polymer. Both PEO and PPO homo-polymershave a lower critical solution temperature in water and the solubility of PEO andPPO decreases with increasing temperature. PF is surface active and forms micellesand liquid lyotropic crystalline phases. At higher temperatures (above 25◦C), themicelles become ordered into a lattice [5]. Thermal gelation occurs by micellar de-solvation and swelling to form a pseudo-cross-linkage among the co-polymer struc-tures [22], which was confirmed by the SEM images (Fig. 2A). PF hydrogels are,thus, considered to be systems with nanostructured domains that possess thermo-reversible gelation property. The structure of the AP hydrogel greatly differed from

1042 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
Figure 6. Release percentage (%)–time profiles of cisplatin (measured as platinum by atomic ab-sorbance) across a cellulose membrane from the free form (control), a Pluronic F127 hydrogel (PF),an PF–alginate conjugate hydrogel (AP), a physical mixture of PF and alginate, and an AP–liposomecomposite (APL1). Each value represents the mean ± SD (n = 4).
Figure 7. In vivo tumor uptake of cisplatin (measured as platinum by atomic absorbance) at 24 hfrom an aqueous solution (control) and various hydrogel systems after an intratumor injection intomelanomas on the backs of nude mice. Each value represents the mean ± SD (n = 6). ∗P < 0.05 ascompared to the control group.
that of the PF hydrogel. The AP system showed a reticular, sponge-like structure(Fig. 2C). A stronger gel was formed by AP based on this three-dimensional net-work. The incorporation of liposomes in AP hydrogel did not change the original

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1043
structure of the hydrogel. Both PEA liposomes and AP co-polymers showed neg-ative charges according to the zeta potential determination. The repulsive forcesbetween liposomes and the co-polymer minimize possible interactions betweenthem, thus reducing the influence of liposomes on the hydrogel backbone.
Packing of micelles and micellar entanglements is a possible mechanism of PFsolution gelation with an increase in temperature [23]. The onset of gelation inthe AP system occurred approximately at the sol–gel temperature of the parent PF.However, as observed in Fig. 4, the attainment of complete gelling (>16 000 cP)after initiation of gelation was slow for PF (26.4–38◦C). The viscosity of the APsystem sharply increased, and it eventually became a non-flowing mass at ap-prox. 27◦C. This result suggests that the hydrophilic property of alginate interferedwith the hydrophobic interactions of PF. A stronger gel structure was formed due toalginate coupling compared to the parent PF co-polymer. The slow gelation processis time-consuming and limiting with regard to the drug loading level. Moreover, in-complete gelation can be observed in vivo, resulting in high initial drug release andlocal or systemic toxicity [24]. Therefore, a delivery formulation where gelationand drug loading can quickly and simultaneously be achieved in an aqueous envi-ronment would be attractive [25]. AP may be applicable to achieve this purpose.
The presence of liposomes in the vicinity of the PF formulation greatly reducedthe sol–gel temperature from 26.4 to 19.7◦C. A reduction in the transition tempera-ture for PF was previously reported by the addition of salts because of their abilityto reduce water activity of the system, which effectively increases the aqueous poly-mer concentration by a salting-out effect [5, 26]. The anionic PEA may also producea similar effect as salts in decreasing the critical micelle concentration of PF and,thus, the gelation temperature. Another mechanism for lowering the transition tem-perature is the presence of hydrophobic or amphiphilic molecules which can lowerthe temperature required to achieve the desired liquid crystalline structure [24, 27].The low transition temperature of the PF/liposomes system makes this formulationunsuitable for in vivo administration. This phenomenon can be improved by usingthe novel AP co-polymer. The inclusion of liposomes into the AP hydrogel (APL1)led to a limited reduction in the transition temperature from 26.6 to 24.4◦C.
4.3. In Vitro Release of Cisplatin from Hydrogels
Compared to the free control, entrapment in liposomes resulted in a prolongationof cisplatin release. Figure 5 shows that the initial phase of cisplatin release wasfaster than the terminal phase. The burst release was likely to occur by the rapiddiffusion of unencapsulated cisplatin (approx. 30%) in the liposomal system. Theremaining approx. 70% cisplatin may be stably retained in the vesicles for a deter-mined duration, followed by its slow release into the external phase. The releasestudy showed that inclusion of an anionic species, DA (L2), produced faster releaseof cisplatin. It is possible that DA in liposomes further disrupted the rigidity of thebilayers, resulting in cisplatin being more easily transferred across the membrane.The results of combining PF and liposomes (PL1 and PL2) suggest that both PF and

1044 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
liposomes played roles in controlling cisplatin delivery. The drug can be released intwo phases with barrier functions, phospholipid bilayers, and PF structures, whichled to a slower release rate. The increase in the microviscosity within the aqueousregion of the PF hydrogel by the addition of phospholipids is another mechanismproducing reduced release. A similar phenomenon was observed with the additionof salts to a PF hydrogel [5, 26].
Cisplatin in hydrogel or liposomes sustained the release amount to a 6-h period,as shown in Figs 5 and 6. Previous studies have indicated a prolonged release ofcisplatin over a period of 30 days from the nanoparticles [27–29]. The dialysis bagmethod is used in these studies. The quick release in our study may have beendue to the use of Franz cell. Since a drug is released to the definitive space ofreceptor (5.5 ml) and diffusion area (1.13 cm2), the drug loading in the receptor islimited. There is no longer a concentration gradient between the donor and receptorcompartments. Nevertheless, this method is still useful for differentiating the releaseof various formulations, especially the free control.
When the cross-linking structure was formed by grafting alginate and PF, cis-platin release was further reduced. A considerable decrease in the diffusion ofcisplatin is believed to have occurred when the strong cross-linkage was formed[30]. According to the release results of hydrogels with different AP concentra-tions, the amount of cisplatin released was a function of the amount of AP usedin the hydrogels. The entanglement was more marked at a higher AP concentra-tion, producing an increase in gel strength. This indicates the possible importanceof strength on cisplatin release from hydrogels. There are two possible interactionsbetween cisplatin and AP which may have contributed to the slow drug delivery. Ithas been reported that cisplatin is coordinately associated with carboxyl groups ofpolymer materials [31, 32]. Alginate has many carboxyl groups in its molecules. Asa result, firm interactions between cisplatin and the AP co-polymer would reducediffusion. Ionic interactions between the cisplatin aqueous complex and the anionicAP backbone are another possible mechanism reducing cisplatin release. Both in-teractions could not have occurred with PF because of the lack of carbonyl groupsand negative charges.
The burst release observed in PF and AP hydrogels was likely to occur by rapiddiffusion of the surface-located and loosely entrapped cisplatin in the hydrogel ma-trices upon incubation [33]. The burst effect was minimized by combining AP andliposomes (APL1). The vehicle with AP and liposomes was best able to retain cis-platin. PF can permeabilize bilayers of liposomes and trigger the rapid leakageof liposomal contents [7, 34]. This phenomenon might not be observed with APco-polymers, resulting in the sufficient control of cisplatin from AP/liposome com-posites.
4.4. In Vivo Intratumor Administration
Tumors have large interstitial spaces composed predominantly of a collagen andelastic fiber network. The vascular structure inside tumors is very leaky [35]. A rel-

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1045
atively low accumulation of cisplatin in melanomas was achieved with the free form(control). Since a low-MW drug like cisplatin is rapidly passed into the blood cir-culation and the time period of retention in the tumor is very short, no significantlyhigh and prolonged antitumor effects were expected [3].
Sustained drug release from liposomes and thermo-sensitive hydrogels loadedwith cisplatin in vitro was observed in this report. Providing that similar releasecharacteristics occur in vivo, this result could be potentially important for develop-ing a sustained-release formulation. The use of thermo-sensitive dispersions suchas chitosan/glycerophosphate and PF as intratumor preparations can constitute agreat depot of paclitaxel within tumors [24, 36, 37]. However, the enhanced depo-sition could not be detected with the PF and AP systems containing cisplatin in ourcase. Melanomas show an abundant vasculature around the tumors [38], contribut-ing to a higher percentage of capillary volume to the entire tumor. This results indifficulty maintaining cisplatin within melanomas. The poor cisplatin retention byhydrogels was improved by incorporating liposome-encapsulated cisplatin. The ra-tionale behind these formulations is to maintain the liposomes at the delivery siteand to avoid clearance from the system. The preferential accumulation of nano-sized drug carriers in tumors is explained by the so-called ‘enhanced permeabilityand retention (EPR)’ effect, which is caused by the disorganized vascularizationand defective vascular architecture of tumors [39]. The entrapment of cisplatin inliposomes can improve its entry into tumor cells by a endocytosis process [40]. Li-posomes also enhanced the drug stability in the targeted site. A hydrogel scaffold,of course, also provides an important cisplatin reservoir for drug protection andslow release. Another observation is that AP possessed a greater ability to retaincisplatin accumulation in melanomas as compared to PF. AP was highly negativelyionized. The negatively charged glycocalyx in the vascular walls may have repelledthe AP, thus retaining the structures within the tumor interstitium.
In conclusion, the injectable thermo-sensitive hydrogels were fabricated bychemically grafting alginate and PF. A porous network of the cross-section of APhydrogel was obtained. The complete gel-forming process could be shortened by al-ginate conjugation. This system showed higher mechanical strength; therefore, thecisplatin release was more sustained. The further incorporation of PE liposomes inhydrogels could greatly sustain cisplatin delivery, resulting in higher drug accumu-lation in tumors after in vivo intratumor injection. These results can be potentiallyimportant for future development of cisplatin carrier systems for cancer treatment.
References
1. C. Xiao, X. Qi, Y. Maitani and T. Nagai, J. Pharm. Sci. 93, 1718 (2004).
2. K. Avgoustakis, A. Beletsi, Z. Panagi, P. Klepetsanis, A. G. Karydas and D. S. Ithakissios, J. Con-trol. Rel. 79, 123 (2002).
3. M. Konishi, Y. Tabata, M. Kariya, A. Suzuki, M. Mandai, K. Nanbu, K. Takakura and S. Fujii,J. Control. Rel. 92, 301 (2003).

1046 J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047
4. N. Kashyap, N. Kumar and M. N. V. Ravi Kumar, Crit. Rev. Ther. Drug Carrier Syst. 22, 107(2005).
5. J. J. Escobar-Chávez, M. López-Cervantes, A. Naïk, Y. N. Kalia, D. Quintanar-Guerrero andA. Ganem-Quintanar, J. Pharm. Pharm. Sci. 9, 339 (2006).
6. T. Moore, S. Croy, S. Mallapragada and N. Pandit, J. Control. Rel. 67, 191 (2000).7. E. Ruel-Gariépy, G. Leclair, P. Hildgen, A. Gupta and J. C. Leroux, J. Control. Rel. 82, 373 (2002).8. X. Y. Xiong, K. C. Tam and L. H. Gan, J. Nanosci. Nanotechnol. 6, 2638 (2006).9. L. Bromberg, J. Phys. Chem. B 102, 1956 (1998).
10. G. Grassi, A. Crevatin, R. Farra, G. Guarnieri, A. Pascotto, B. Rehimers, R. Lapasin and M. Grassi,J. Colloid Interface Sci. 301, 282 (2006).
11. O. Séchoy, G. Tissié, C. Sébastian, F. Maaurin, J.-Y. Driot and C. C. Trinquand, Int. J. Pharm.207, 109 (2000).
12. K. Y. Cho, T. W. Chung, B. C. Kim, M. K. Kim, J. H. Lee, W. R. Wee and C. S. Cho, Int. J. Pharm.260, 83 (2003).
13. N. Bhattari, H. R. Ramay, J. Gunn, F. A. Matsen and M. Zhang, J. Control. Rel. 103, 609 (2005).14. M. S. Newman, G. T. Colbern, P. K. Working, C. Engbers and M. A. Amantea, Cancer Chemother.
Pharmacol. 43, 1 (1999).15. T. L. Hwang, W. R. Lee, S. C. Hua and J. Y. Fang, J. Dermatol. Sci. 46, 11 (2007).16. J. Y. Fang, C. F. Hung, T. L. Hwang and Y. L. Huang, J. Drug Target. 13, 19 (2005).17. J. Y. Fang, W. R. Lee, S. C. Shen and Y. L. Huang, J. Dermatol. Sci. 42, 101 (2006).18. J. P. Chen and T. H. Cheng, Macromol. Biosci. 6, 1026 (2006).19. M. P. Stevens (Ed.), in: Polymer Chemistry, Chapter 2, p. 4. Oxford University Press, New York,
NY (1990).20. K. Jores, A. Haberland, S. Wartewig, K. Mäder and W. Mehnert, Pharm. Res. 22, 1887 (2005).21. T. Yotsuyanagi, M. Usami, Y. Noda and M. Nagata, Int. J. Pharm. 246, 95 (2002).22. M. Guzmán, F. F. García, J. Molpeceres and M. R. Aberturas, Int. J. Pharm. 80, 119 (1992).23. A. Cabana, A. Ait-Kadi and J. Juhasz, J. Colloid Interface Sci. 190, 307 (1997).24. E. Ruel-Gariépy and J.-C. Leroux, Eur. J. Pharm. Biopharm. 58, 409 (2004).25. J. Li, X. Ni and K. W. Leong, J. Biomed. Mater. Res. A 65, 196 (2003).26. N. K. Pandit and J. Kisaka, Int. J. Pharm. 145, 129 (1996).27. V. DiTizio, C. Karlgard, L. Lilge, A. E. Khoury, M. W. Mittelman and F. DiCosmo, J. Biomed.
Mater. Res. 51, 96 (2000).28. T. Chandy, J. Biomater. Appl. 16, 275 (2002).29. S. Cafaggi, E. Russo, R. Stefani, R. Leardi, G. Caviglioli, B. Parodi, G. Bignardi, D. De Totero,
C. Aiello and M. Viale, J. Control. Rel. 121, 110 (2007).30. R. Narayani and K. P. Rao, Int. J. Pharm. 138, 121 (1996).31. J. S. Zhang, T. Imai, A. Suenaga and M. Odagiri, Int. J. Pharm. 240, 23 (2002).32. M. Konishi, Y. Tabata, M. Kariya, H. Hosseinkhani, A. Suzuki, K. Fukuhara, M. Mandai,
K. Takakura and S. Fujii, J. Control. Rel. 103, 7 (2005).33. M. R. Kim and T. G. Park, J. Control. Rel. 80, 69 (2002).34. A. Bochot, E. Fattal, A. Gulik, G. Couarraze and P. Couvreur, Pharm. Res. 15, 1364 (1998).35. K. Maruyama, O. Ishida, T. Takizawa and K. Moribe, Adv. Drug Deliv. Rev. 40, 89 (1999).36. G. M. Zentner, R. Rathi, C. Shih, J. C. McRea, M. H. Seo, H. Oh, B. G. Rhee, J. J. Mestecky,
Z. Moldoveanu, M. Morgan and S. Weitman, J. Control. Rel. 72, 203 (2001).37. M. M. Amiji, P. K. Lai, D. B. Shenoy and M. Rao, Pharm. Dev. Technol. 7, 195 (2002).38. K. Arashiro, Plast. Reconstr. Surg. 110, 717 (2002).

J.-Y. Fang et al. / Journal of Biomaterials Science 20 (2009) 1031–1047 1047
39. J. H. Kim, Y. S. Kim, K. Park, S. Lee, H. Y. Nam, K. H. Min, H. G. Jo, J. H. Park, K. Choi,S. Y. Jeong, R. W. Park, I. S. Kim, K. Kim and I. C. Kwon, J. Control. Rel. 127, 41 (2008).
40. A. D. Carvalho Júnior, F. P. Vieira, V. J. De Melo, M. T. P. Lopes, J. N. Silveira, G. A. Ramaldes,A. Garnier-Suillerot, E. C. Pereira-Maia and M. C. De Oliveira, Braz. J. Med. Biol. Res. 40, 1149(2007).





























