Embed Size (px)
Citation preview

Describing Polytopal Rearrangements of Fluxional Molecules withCurvilinear Coordinates Derived from Normal Vibrational Modes AConceptual Extension of CremerminusPople Puckering CoordinatesWenli Zoupara Yunwen Taopara and Elfi Kraka
Cite This J Chem Theory Comput 2020 16 3162minus3193 Read Online
ACCESS Metrics amp More Article Recommendations sı Supporting Information
ABSTRACT In this work a new curvilinear coordinate system ispresented for the comprehensive description of polytopal rearrangementsof N-coordinate compounds (N = 4minus7) and systems containing an N-coordinate subunit It is based on normal vibrational modes and a naturalextension of the CremerminusPople puckering coordinates (J Am Chem Soc1975 97 1354) together with the ZouminusIzotovminusCremer deformationcoordinates (J Phys Chem A 2011 115 8731) for ring structures to N-coordinate systems We demonstrate that the new curvilinear coordinatesare ideal reaction coordinates describing fluxional rearrangement pathwaysby revisiting the Berry pseudorotation and the lever mechanism in sulfurtetrafluoride the Berry pseudorotation and two Muettertiesrsquo mechanismsin pentavalent compounds the chimeric pseudorotation in iodinepentafluoride Bailar and Ray-Dutt twists in hexacoordinate tris-chelates as well as the Bartell mechanism in iodine heptafluorideThe results of our study reveal that this dedicated curvilinear coordinate system can be applied to most coordination compoundsopening new ways for the systematic modeling of fluxional processes
1 INTRODUCTION
Fluxional processes in particular polytopal processes haverecently attracted attention caused by their potential to probesolventminussolute interactions1minus3 and to support self-healingprocesses4
The term polytopal rearrangement describes a dynamic processthat alters the positions of the vertices defined by the ligands in acoordination polyhedron ie it ldquointerconverts different orequivalent spatial arrangements of ligands about a central atomrdquoas stated in the IUPAC Gold Book5minus8 A well-known examplefor polytopal rearrangement found in most inorganic chemistrytextbooks is the Berry pseudorotation of the PF5 molecule910
Polytopal rearrangements being specific to coordinationcompounds are a subset of the broader class of f luxionality11minus13
Many studies use ldquofluxionalityrdquo14minus16 ldquotopomerizationrdquo17
ldquostereomutationenatiomerizationrdquo131718 ldquopseudorota-tionrdquo19minus21 ldquopermutational isomerizationrdquo1822 ldquopolyhedralrearrangementinterconversionrdquo2324 or ldquonon-dissociative li-gand exchangerdquo25 as a synonym for polytopal rearrangementsAlthough a polytopal rearrrangement does not involve any
ligand elimination andor addition (ie no bond breaking andor bond forming) it is of significant importance for chemists tounderstand the dynamics of such a process due to two reasons(i) Fluxional flexibility can lead to more than one thermallyaccessible structure or to molecules with a dynamic structurerecently coined as molecules with a quasistructure26 (ii)Polytopal rearrangements affect the stereoselectivity and the
reaction mechanism therefore they are critical for the reactivityand catalytic activity for many metal complexes27minus34
The tool extensively used by experimental chemists todetermine molecular fluxionality is dynamic NMR spectrosco-py1335minus38 On the basis of whether a polytopal rearrangement iseither fast or slow compared to the NMR time scale39 its rateconstant can be obtained by the Eyring equation1240
However NMR spectroscopy is not able to provide us withinformation about the actual path that the atomic nuclei aretracing during a polytopal rearrangement Therefore we have toresort to quantum chemical calculations to model this path andverify the proposed mechanism(s) by calculating the activationenergy4142 On the one hand often theory is guiding theexperiment1725 on the other hand numerous mechanismsderived from experimental results await theoretical validation34
One major obstacle in modeling polytopal rearrangements incoordination compounds is the lack of a generally applicable setof reaction coordinates tailored exclusively to distinctive N-coordinate geometries Although some tricks (eg combiningseveral bond angles18 or dihedral angles43) could be employed
Received December 20 2019Published March 25 2020
ArticlepubsacsorgJCTC
copy 2020 American Chemical Society3162
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
Dow
nloa
ded
via
SOU
TH
ER
N M
ET
HO
DIS
T U
NIV
on
May
27
202
0 at
14
391
7 (U
TC
)Se
e ht
tps
pub
sac
sor
gsh
arin
ggui
delin
es f
or o
ptio
ns o
n ho
w to
legi
timat
ely
shar
e pu
blis
hed
artic
les
when driving the reaction complex into a desired reactionchannel these workarounds cannot achieve a systematicexploration of the configuration space of the N-coordinatemotifs However it is of great necessity to determine all possiblefluxional mechanisms for a specific geometryNoteworthy is the work related to polytopal rearrangement by
Avnir Alvarez and co-workers who developed continuoussymmetry measure (CSM) and continuous shape measure(CShM)232444minus46 Their approach quantifies to what extent thegeometry of a coordination compound matches the geometry ofa reference compound with high symmetry or that of a regularpolyhedron By defining the ldquominimal distortion pathsrdquo forseveral polytopal rearrangement mechanisms one can position agiven coordination geometry onto the predefined minimaldistortion paths23 After enumerating all relevant X-ray crystalstructures with this approach they observed a concentrateddistribution of the geometries along the minimal distortion pathwhich they visualized as a ldquoshape maprdquo These resultsconvincingly proved the existence of a polytopal rearrangementmechanism with numerous discrete structures CSM and CShMmeasures offer a simplified way to describe and categorize thegeometries of coordination compounds However one caveat isappropriate the structural descriptors based on CSMCShMwill generally not span the complete configuration space of theN-coordinate motif and as such cannot serve as reactioncoordinates used for practical calculations modeling polytopalrearrangement pathwaysTherefore in this work we adopted the conceptual approach
of CremerminusPople ring puckering coordinates47 aiming at thedevelopment of a curvilinear coordinate system that providesappropriate reaction coordinates to (i) facilitate modelingpolytopal rearrangements in N-coordinate molecules and (ii)present a simplified but comprehensive description of theconformational changes taking place during these rearrange-mentsThe paper is structured in the following way First the ring
puckering and deformation coordinates are briefly reviewed interms of their underlying rationale as the basis to define the N-coordinate reference system ABn its symmetry configurationalspace and molecular vibrations Then the curvilinearcoordinate parameters for any N-coordinate geometry (N =4minus7) are derived from normal vibrational modes of thecorresponding reference system After the ComputationalDetails section nine different polytopal rearrangement mech-anisms ranging from tetracoordinated to heptacoordinatedcompounds are revisited using the curvilinear coordinatesderived in this work to demonstrate the general applicability ofour new approach The conclusions along with perspectives onfuture work are given in the last section
2 METHODOLOGY21 Retrospective on CremerminusPople Ring Puckering
and Deformation Coordinates The CremerminusPople ringpuckering coordinates proposed in 1975 have become a well-accepted standard tool to describe the out-of-plane conforma-tional flexibility of ring structures in cyclic molecules47minus53 It isbased on the concept of partitioning the (3N minus 6)-dimensionalconfiguration space of anN-membered ring structure (ldquoN-ringrdquo)into two subspaces where ring puckering and deformationmotions take place respectivelyAny N-ring has an (N minus 3)-dimensional puckering space
spanned by N minus 3 basis conformations whose linearcombinations are specified by ring puckering coordinates The
z-component of the Cartesian coordinates for each ring atom zj(j = 1 2 N) depicting the puckering conformation relative toan ideal planar form (mean ring plane) is connected to thepuckering amplitude qm and pseudorotation phase angle ϕm viathe following formulas depending on the parity of N4748
sum ϕπ
= +minus
=
minus Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildez
Nq
m jN
N2
cos2 ( 1)
( odd)jm
N
m m2
( 1)2
(1)
sum ϕπ
= +minus
+ minus
=
minus
minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildez
Nq
m jN
Nq N
2cos
2 ( 1)
1( 1) ( even)
jm
N
m m
Nj
2
( 2)2
21
(2)
where a pseudorotation cycle only starts from the five-membered ring (N minus 3 = 2) and the puckering coordinatepair (q2 ϕ2) is used to describe the motion in the puckeringsubspace Puckering starts from the four-membered ring (Nminus 3= 1) with the puckering amplitude q2The in-plane deformations of theN-ring take place in a (2Nminus
3)-dimensional subspace complementing the (N minus 3) out-of-plane motions to (3N minus 6) coordinates In recent work wederived the ring deformation coordinates (RDC) as thecomplement to CremerminusPople puckering coordinates renderinga whole picture of the ring configuration with curvilinearcoordinates54minus56 The definition of RDCs relies on furtherpartitioning of the (2N minus 3)-dimensional deformation spaceinto (1) a one-dimensional ring breathing mode subspacedetermined by the breathing radius R and (2) N minus 2 two-dimensional pseudorotational subspaces spanned by pairs ofamplitudes tn and phase angles τn (n = 1 2 N minus 2) After theN-ring is positioned into standard orientation the Cartesiancoordinates of the ring atoms are linked to the deformationcoordinate parameters R and (tn τn) with the followingformulas54
sum
π
τπ
= minusminus
+ minus+ minus
=
minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
x RjN
tn j
N
cos2 ( 1)
cos2 ( 1)( 1)
j
n
N
n n1
2
(3)
sum
π
τπ
=minus
+ minus+ minus
=
minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
y RjN
tn j
N
sin2 ( 1)
sin2 ( 1)( 1)
j
n
N
n n1
2
(4)
where all zj components as the out-of-plane deviations arerelated to puckering coordinates in eqs 1 and 2With the CremerminusPople ring puckering coordinates and the
recently developed ring deformation coordinates one obtains acomprehensive quantitative and intuitive description of ringconformations of any N-membered ring4754 Furthermore thepotential-energy surface (PES) or free-energy surface (FES) in3N minus 6 dimensions of theN-ring system can be reduced to two-or three-dimensional contour plots with deeper physical insightand higher interpretability53minus67
The success of these two curvilinear coordinate systems indescribing ring conformations can be attributed to theirgenerally applicable mathematical form for all N-rings more
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3163
importantly they turn out to be the natural choice incharacterizing ring conformations and their changes becauseboth the CremerminusPople puckering coordinates and the ringdeformation coordinates were inspired and derived from thenormal vibrational modes5154 Either the out-of-plane puckeringor in-plane deformationrearrangement of the N-ring isinitialized by a molecular vibration or the mixing of severaldegenerate vibrations68minus70 acting as the leading parameter71minus73
By adopting this conceptual approach we aimed in this work atdeveloping new curvilinear coordinates for more distinctivegeometries besides ring structures The novel curvilinearcoordinates derived from molecular vibrations are expected tobe especially useful in describing conformational changes andstructural rearrangements involving multiple atoms simulta-neouslyOne may question the extension of the underlying rationale in
ring coordinates (ie a specific vibration in a ring initiatespuckering or deformation motion) to other molecular geo-metries in general However it has been widely recognized thatmolecular vibrations can drive chemical reactions by exciting aspecific vibrational mode of the reactant along the reactioncoordinate and in this way overcoming the energy barrier towardthe product7475 Crim Zare and others have demonstrated thepossibilities of experimentally controlling the course of chemicalreactions in gas phase and on metal surfaces by selectivelyexciting certain vibrational modes with a laser76minus91 Thereforethe conceptual extension of ring coordinates to other geometriesbased on molecular vibrations has its physical foundation22 Vibration-Based Curvilinear Coordinates for N-
Coordinate Geometry Unlike for ring puckeringdeforma-tion coordinates there is no direct analytic mathematicalsolution to connect each atomrsquos Cartesian coordinates to thecurvilinear coordinates describing N-coordinate systems due tothe following reasons
bull The z-space can be separated from the xy-space whendefining the puckering47 and deformation54 coordinatesfor rings respectively However the conformationalchanges in N-coordinate geometries involve atomicmovement in the three-dimensional (3D) space whichis more complex
bull The mathematical expression of either CremerminusPoplepuckering or ring deformation coordinates has alreadyencompassed the normal vibrational modes of thecorresponding reference model systems (ie planar ringand regular polygon) implicitly However in the case ofan N-coordinate geometry we need to explicitly definethe normal vibrational modes as the basis vectorsspanning the configurational space of an N-coordinategeometry beforehand
In the following we elaborate how the curvilinear coordinatesderived from normal vibrational modes for N-coordinategeometries are defined Depending on the value of N thegeometry can be tricoordinate tetracoordinate pentacoordi-nate hexacoordinate heptacoordinate (eg IF7) or octacoor-dinate (eg XeF8
2minus)92 Within the same type of N-coordinategeometry the relative spatial arrangement of the ligating atomsbonded to the central atom may vary This complication can beresolved by utilizing point group information for the differ-entiation For example both Fe(CO)5 and ClF5 have apentacoordinate geometry however the former is trigonalbipyramidal and the latter is square pyramidal or expressed in
the point group language the former has D3h and the latter hasC4v symmetryAs shown in Figure 1 we define in this work the curvilinear
coordinates with respect to a tetracoordinate geometry with Td
symmetry pentacoordinate geometries with D3h and C4vsymmetry hexacoordinate geometries with Oh and C5vsymmetry along with heptacoordinate geometries with D5hand C6v symmetry to cover the vast majority of coordinationcompounds with fluxionalityThe pentacoordinate geometry in C4v symmetry is taken as an
example in the following to stepwise illustrate how thecurvilinear coordinate system derived from normal modes foran N-coordinate geometry is defined The same procedureequally applies to other geometries with various point groupsmentioned above
221 Reference System for a Given Symmetry As in thecase of defining ring puckering coordinates with regard to themean plane68minus70 we first need to stipulate a reference systemABn for the given symmetry egC4v where A is the central atomand n atoms of B are bonded to A as the first coordinationsphereThe definition of the reference system in Cartesian
coordinates has to fulfill the following requirementsbull The central atom A denoted as the first atom is placed at
the origin of the 3D Cartesian coordinate systembull All AminusB distances are set to be the same and assigned a
value of r1 This is equivalent to the situation that all atomsB are positioned on the surface of a sphere with radius r1and the center of sphere is placed at atom A
bull The relative positions of atoms B are adjusted under asymmetry constraint to guarantee that the geometricalcenter of ABn is located at the position of atom A
In addition the following two conditions are optional
bull The z-axis is chosen to be the highest n-fold rotation axisof ABn
bull Atoms B are grouped according to their z-values from topto bottom If the first group of B has more than one atom
Figure 1 Schematic diagram illustrating the definition of curvilinearcoordinates derived from normal vibrational modes for N-coordinategeometries
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3164
the first atom within this group is placed right above the+x-axis (y = 0) and the remaining atoms are numberedcounterclockwise when looking into the direction fromatom 2 to atom 1 If the first group of B has only one atomthen this atom is positioned on the z-axis Meanwhile thefirst atom of the second group is positioned in the +xdirection (y = 0)
For example the reference system AB5 with the C4v pointgroup (shown in Figure 2) has 18 Cartesian coordinates
collected in matrix R0 with dimension 6 times 3 shown in eq 5 Thethree columns correspond to x y and z components and the sixrows correspond to the six atoms of the reference systemordered according to the above description The first rowspecifies the position of A at the origin point
=minus
minus
minus minus
minus minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
r
r r
r r
r r
r r
R
0 0 00 0
0
0
0
0
0
1
1 1
1 1
1 1
1 1 (5)
where = 15 4 = 14 and r1 is the radius depending onthe real system which will be clarified belowA reference system for a given symmetry operates like a
template with fixed shape which can be scaled by r1Furthermore it serves as the basis for deriving the normalvibrational modes which are independent of r1222 Normal Vibrational Modes of Reference System In
order to span the complete configuration space of the referencesystem ABn with a given symmetry we need to derive one set ofnormal vibrational mode vectors acting as the basis We employfor this purpose the following two-step procedureFirst a model Hamiltonian accounting only for the pairwise
nuclear charge repulsion is used to describe the interatomicforces within the reference system geometry (with r1 temporarilytaking an arbitrary value between 1 and 2 Aring) leading to a modelHessian matrix which is calculated analytically With theassumption that all atoms in ABn have identical unit atomicmasses (1 amu) the normal-mode analysis is carried out leadingto an initial set of 3Mminus 6 normal vibrational modes whereM = n
+ 1 Besides three rotational normal modes calculated withmassless Eckart conditions9394 are obtained as column vectorsr1 r2 and r3 (Please note that r1 r2 and r3 must not be confusedwith r1 r2 and r3 representing amplitudes)Second this initial set of 3M minus 6 normal modes is fine-tuned
within the internal vibrational space by pairwise rotationaccording to the following rules
(1) Two nondegenerate normal vibrations with the sameirreducible representation can be mixed via a 2 times 2rotation matrix leading to another two normal vibrationswith the same symmetry
(2) The first vibrational mode should always be the totallysymmetric breathing mode of the present point groupThe amplitude for all n atoms B in this vibrational modevector must be identical while the central atom A has nocontribution
(3) The atom A can be distorted only in the last three normalmodes which correspond to the relative translationbetween atomA and the remaining n atoms B in x y and zdirections
(4) Doubly degenerate modes (denoted as E and Edagger) or triplydegenerate modes (TTdagger andTDagger) can be pairwise rotatedamong themselves to fulfill the following requirementsthat(a) for the E or T mode the distortions in the z
direction andor y direction are zero if possible(b) for the Tdagger mode the distortions in the y direction
andor x direction are zero if possible and(c) the Edagger or TDagger mode can be obtained by orthonormal
condition
Pairwise rotation as described between any two normalvibrational modes is a unitary transformation that keeps theorthonormality of 3M minus 6 normal modes and greatly simplifiesthe basis vibration vectors for later useFigure 3 shows all 12 normal vibrational modes of the AB5
reference system in the C4v point group The normal modevectors li (i = 1 2 3M minus 6) shown as (M times 3)-dimensionalmatrices are orthonormal to each other The collection ofnormal vibrational modes we derived for the other referencesystems can be found in Appendices A2minusA5
223 Real System in Standard Orientation For a realmolecular compound with N-coordinate geometry as its corethe central N-coordinate part (ldquoreal systemrdquo) deviates more orless from the reference geometry with a given symmetry duringconformational changes or reaction processes It would be idealto express such distortions in terms of the normal vibrationalmodes that we derived for selected reference systems In order todo so we have to order the atomic labels of the real systemaccording to those of the reference system for consistency andthen place the real system into a particular position where itsinternal coordinate space matches exactly the internal vibra-tional space of the reference system without any contaminationfrom the translational or rotational space of the referencesystem9596 This requires the following standard orientation tobe imposed on the real system geometry
bull The initial Cartesian coordinates of the real system withN-coordinate geometry are recorded as X which is an Mtimes 3 matrix
bull The real system needs to be translated so that itsgeometrical center is positioned at the origin of Cartesiancoordinate system current Cartesian coordinates of thereal system are collected in Xc
Figure 2 Ball-and-stick representation for the reference system AB5with C4v symmetry in Cartesian coordinates The schematic Cartesianaxes are shown for illustrating directions while the true origin is at thefirst atom A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3165
bull The real system is then rotated in the 3D space so that itsCartesian coordinates denoted Xr are orthogonal to therotational modes r1 r2 r3 of the reference system Thisfinal set of Cartesian coordinates Xr is named standardorientation for the real system
The rotation of the real system by a rotation matrix u can beexpressed by the product of three 3 times 3 elemental rotationmatrices
=u u u uz y x (6)
where ux uy and uz define the rotation around x- y- and z-axesrespectively
θ θ
θ θ
θ θ
θ θ
θ θ
θ θ
= minus =minus
=minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
u u
u
1 0 00 cos sin
0 sin cos
cos 0 sin
0 1 0sin 0 cos
cos sin 0
sin cos 0
0 0 1
x x x
x x
y
y y
y y
z
z z
z z
(7)
This leads to
=X X ur c (8)
Figure 3Twelve normal vibrational modes for the AB5 reference systemwithC4v symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3166
We rearrange the Cartesian coordinates Xc and Xr from matrixform into the (1 times 3M)-dimensional row vector form as Xc and
X r then a (3M times 3M)-dimensional block diagonal matrix U isconstructed with rotation matrix u as diagonal blocks and 03times3for nondiagonal blocksBy definition the standard orientation requires that
= =
= =
= =
l
mooooooo
n
ooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 c 1
r 2 c 2
r 3 c 3 (9)
where Xc r1 r2 and r3 are already known so that only threerotation angles θx θy and θz remain as the unknowns containedin U which can be determined by iteratively solving this systemof nonlinear equationsHowever practically solving the above system of equations
with numerical methods could lead to an undesired solution asmultiple sets of solutions could exist This issue arises when thestarting geometry in Cartesian coordinates Xc is close to anotherundesired standard orientation As a remedy the arbitrariness ofrotation matrix u may be eliminated by minimizing the rootmean-square deviation (RMSD) between the Cartesiancoordinates Xc and the reference system which has been provenby Kudin and Dymarsky97 to be closely related to the well-known Eckart axis conditions In this work we adopt thequaternion-based algorithm of Kearsley98 to first rotate the realsystem Noteworthy is that before calculating the best-fit
alignment between real system and reference system the AminusBdistance r1 in the reference system is temporarily set to be theaveraged AminusB distance of the real system With the real systemrotated its Cartesian coordinates change from Xc to Xf and thestandard orientation can be unambiguously obtained by solvingthe system of equations in eq 10
= =
= =
= =
l
mooooooo
nooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 f 1
r 2 f 2
r 3 f 3 (10)
where X r is guaranteed to be the desired standard orientationcalculated by numerical equation solver because Xf is close to Xr
and θx = θy = θz = 0 can be used as the initial guess Usually thecalculation of eq 10 converges after one iteration
224 Calculation of Curvilinear CoordinatesOnce the realsystem is positioned in the appropriate standard orientation itsgeometry Xr can be completely described by the set of 3M minus 6normal vibrational modes of the corresponding reference systemABnThe determination of the real system geometry along the
directions of normal vibrations is calculated by the projection ofCartesian coordinates Xr onto each normal mode vector li (i = 12 3M minus 6) using the dot product di
= d X li irT
(11)
Figure 4 r5 ϕ5 pseudorotational cycle specifying the (t2 τ2) deformation of the B4 ring within the reference system AB5 in C4v symmetry (see Table1) In the center of the cycle is the reference geometry for AB5 with r1 = 15 Aring and surrounding the cycle are the representative geometries for r5 = 035Aring and different ϕ5 values starting from 0deg with the interval of 225deg while all other rm values (except r1) are 0 The representative geometries are shownin the perspective of looking downward from atom 2 (red) to atom 1 (green) highlighting the in-plane deformation of the B4 ring consisting of atoms3minus6
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3167
where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

when driving the reaction complex into a desired reactionchannel these workarounds cannot achieve a systematicexploration of the configuration space of the N-coordinatemotifs However it is of great necessity to determine all possiblefluxional mechanisms for a specific geometryNoteworthy is the work related to polytopal rearrangement by
Avnir Alvarez and co-workers who developed continuoussymmetry measure (CSM) and continuous shape measure(CShM)232444minus46 Their approach quantifies to what extent thegeometry of a coordination compound matches the geometry ofa reference compound with high symmetry or that of a regularpolyhedron By defining the ldquominimal distortion pathsrdquo forseveral polytopal rearrangement mechanisms one can position agiven coordination geometry onto the predefined minimaldistortion paths23 After enumerating all relevant X-ray crystalstructures with this approach they observed a concentrateddistribution of the geometries along the minimal distortion pathwhich they visualized as a ldquoshape maprdquo These resultsconvincingly proved the existence of a polytopal rearrangementmechanism with numerous discrete structures CSM and CShMmeasures offer a simplified way to describe and categorize thegeometries of coordination compounds However one caveat isappropriate the structural descriptors based on CSMCShMwill generally not span the complete configuration space of theN-coordinate motif and as such cannot serve as reactioncoordinates used for practical calculations modeling polytopalrearrangement pathwaysTherefore in this work we adopted the conceptual approach
of CremerminusPople ring puckering coordinates47 aiming at thedevelopment of a curvilinear coordinate system that providesappropriate reaction coordinates to (i) facilitate modelingpolytopal rearrangements in N-coordinate molecules and (ii)present a simplified but comprehensive description of theconformational changes taking place during these rearrange-mentsThe paper is structured in the following way First the ring
puckering and deformation coordinates are briefly reviewed interms of their underlying rationale as the basis to define the N-coordinate reference system ABn its symmetry configurationalspace and molecular vibrations Then the curvilinearcoordinate parameters for any N-coordinate geometry (N =4minus7) are derived from normal vibrational modes of thecorresponding reference system After the ComputationalDetails section nine different polytopal rearrangement mech-anisms ranging from tetracoordinated to heptacoordinatedcompounds are revisited using the curvilinear coordinatesderived in this work to demonstrate the general applicability ofour new approach The conclusions along with perspectives onfuture work are given in the last section
2 METHODOLOGY21 Retrospective on CremerminusPople Ring Puckering
and Deformation Coordinates The CremerminusPople ringpuckering coordinates proposed in 1975 have become a well-accepted standard tool to describe the out-of-plane conforma-tional flexibility of ring structures in cyclic molecules47minus53 It isbased on the concept of partitioning the (3N minus 6)-dimensionalconfiguration space of anN-membered ring structure (ldquoN-ringrdquo)into two subspaces where ring puckering and deformationmotions take place respectivelyAny N-ring has an (N minus 3)-dimensional puckering space
spanned by N minus 3 basis conformations whose linearcombinations are specified by ring puckering coordinates The
z-component of the Cartesian coordinates for each ring atom zj(j = 1 2 N) depicting the puckering conformation relative toan ideal planar form (mean ring plane) is connected to thepuckering amplitude qm and pseudorotation phase angle ϕm viathe following formulas depending on the parity of N4748
sum ϕπ
= +minus
=
minus Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildez
Nq
m jN
N2
cos2 ( 1)
( odd)jm
N
m m2
( 1)2
(1)
sum ϕπ
= +minus
+ minus
=
minus
minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildez
Nq
m jN
Nq N
2cos
2 ( 1)
1( 1) ( even)
jm
N
m m
Nj
2
( 2)2
21
(2)
where a pseudorotation cycle only starts from the five-membered ring (N minus 3 = 2) and the puckering coordinatepair (q2 ϕ2) is used to describe the motion in the puckeringsubspace Puckering starts from the four-membered ring (Nminus 3= 1) with the puckering amplitude q2The in-plane deformations of theN-ring take place in a (2Nminus
3)-dimensional subspace complementing the (N minus 3) out-of-plane motions to (3N minus 6) coordinates In recent work wederived the ring deformation coordinates (RDC) as thecomplement to CremerminusPople puckering coordinates renderinga whole picture of the ring configuration with curvilinearcoordinates54minus56 The definition of RDCs relies on furtherpartitioning of the (2N minus 3)-dimensional deformation spaceinto (1) a one-dimensional ring breathing mode subspacedetermined by the breathing radius R and (2) N minus 2 two-dimensional pseudorotational subspaces spanned by pairs ofamplitudes tn and phase angles τn (n = 1 2 N minus 2) After theN-ring is positioned into standard orientation the Cartesiancoordinates of the ring atoms are linked to the deformationcoordinate parameters R and (tn τn) with the followingformulas54
sum
π
τπ
= minusminus
+ minus+ minus
=
minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
x RjN
tn j
N
cos2 ( 1)
cos2 ( 1)( 1)
j
n
N
n n1
2
(3)
sum
π
τπ
=minus
+ minus+ minus
=
minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
y RjN
tn j
N
sin2 ( 1)
sin2 ( 1)( 1)
j
n
N
n n1
2
(4)
where all zj components as the out-of-plane deviations arerelated to puckering coordinates in eqs 1 and 2With the CremerminusPople ring puckering coordinates and the
recently developed ring deformation coordinates one obtains acomprehensive quantitative and intuitive description of ringconformations of any N-membered ring4754 Furthermore thepotential-energy surface (PES) or free-energy surface (FES) in3N minus 6 dimensions of theN-ring system can be reduced to two-or three-dimensional contour plots with deeper physical insightand higher interpretability53minus67
The success of these two curvilinear coordinate systems indescribing ring conformations can be attributed to theirgenerally applicable mathematical form for all N-rings more
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3163
importantly they turn out to be the natural choice incharacterizing ring conformations and their changes becauseboth the CremerminusPople puckering coordinates and the ringdeformation coordinates were inspired and derived from thenormal vibrational modes5154 Either the out-of-plane puckeringor in-plane deformationrearrangement of the N-ring isinitialized by a molecular vibration or the mixing of severaldegenerate vibrations68minus70 acting as the leading parameter71minus73
By adopting this conceptual approach we aimed in this work atdeveloping new curvilinear coordinates for more distinctivegeometries besides ring structures The novel curvilinearcoordinates derived from molecular vibrations are expected tobe especially useful in describing conformational changes andstructural rearrangements involving multiple atoms simulta-neouslyOne may question the extension of the underlying rationale in
ring coordinates (ie a specific vibration in a ring initiatespuckering or deformation motion) to other molecular geo-metries in general However it has been widely recognized thatmolecular vibrations can drive chemical reactions by exciting aspecific vibrational mode of the reactant along the reactioncoordinate and in this way overcoming the energy barrier towardthe product7475 Crim Zare and others have demonstrated thepossibilities of experimentally controlling the course of chemicalreactions in gas phase and on metal surfaces by selectivelyexciting certain vibrational modes with a laser76minus91 Thereforethe conceptual extension of ring coordinates to other geometriesbased on molecular vibrations has its physical foundation22 Vibration-Based Curvilinear Coordinates for N-
Coordinate Geometry Unlike for ring puckeringdeforma-tion coordinates there is no direct analytic mathematicalsolution to connect each atomrsquos Cartesian coordinates to thecurvilinear coordinates describing N-coordinate systems due tothe following reasons
bull The z-space can be separated from the xy-space whendefining the puckering47 and deformation54 coordinatesfor rings respectively However the conformationalchanges in N-coordinate geometries involve atomicmovement in the three-dimensional (3D) space whichis more complex
bull The mathematical expression of either CremerminusPoplepuckering or ring deformation coordinates has alreadyencompassed the normal vibrational modes of thecorresponding reference model systems (ie planar ringand regular polygon) implicitly However in the case ofan N-coordinate geometry we need to explicitly definethe normal vibrational modes as the basis vectorsspanning the configurational space of an N-coordinategeometry beforehand
In the following we elaborate how the curvilinear coordinatesderived from normal vibrational modes for N-coordinategeometries are defined Depending on the value of N thegeometry can be tricoordinate tetracoordinate pentacoordi-nate hexacoordinate heptacoordinate (eg IF7) or octacoor-dinate (eg XeF8
2minus)92 Within the same type of N-coordinategeometry the relative spatial arrangement of the ligating atomsbonded to the central atom may vary This complication can beresolved by utilizing point group information for the differ-entiation For example both Fe(CO)5 and ClF5 have apentacoordinate geometry however the former is trigonalbipyramidal and the latter is square pyramidal or expressed in
the point group language the former has D3h and the latter hasC4v symmetryAs shown in Figure 1 we define in this work the curvilinear
coordinates with respect to a tetracoordinate geometry with Td
symmetry pentacoordinate geometries with D3h and C4vsymmetry hexacoordinate geometries with Oh and C5vsymmetry along with heptacoordinate geometries with D5hand C6v symmetry to cover the vast majority of coordinationcompounds with fluxionalityThe pentacoordinate geometry in C4v symmetry is taken as an
example in the following to stepwise illustrate how thecurvilinear coordinate system derived from normal modes foran N-coordinate geometry is defined The same procedureequally applies to other geometries with various point groupsmentioned above
221 Reference System for a Given Symmetry As in thecase of defining ring puckering coordinates with regard to themean plane68minus70 we first need to stipulate a reference systemABn for the given symmetry egC4v where A is the central atomand n atoms of B are bonded to A as the first coordinationsphereThe definition of the reference system in Cartesian
coordinates has to fulfill the following requirementsbull The central atom A denoted as the first atom is placed at
the origin of the 3D Cartesian coordinate systembull All AminusB distances are set to be the same and assigned a
value of r1 This is equivalent to the situation that all atomsB are positioned on the surface of a sphere with radius r1and the center of sphere is placed at atom A
bull The relative positions of atoms B are adjusted under asymmetry constraint to guarantee that the geometricalcenter of ABn is located at the position of atom A
In addition the following two conditions are optional
bull The z-axis is chosen to be the highest n-fold rotation axisof ABn
bull Atoms B are grouped according to their z-values from topto bottom If the first group of B has more than one atom
Figure 1 Schematic diagram illustrating the definition of curvilinearcoordinates derived from normal vibrational modes for N-coordinategeometries
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3164
the first atom within this group is placed right above the+x-axis (y = 0) and the remaining atoms are numberedcounterclockwise when looking into the direction fromatom 2 to atom 1 If the first group of B has only one atomthen this atom is positioned on the z-axis Meanwhile thefirst atom of the second group is positioned in the +xdirection (y = 0)
For example the reference system AB5 with the C4v pointgroup (shown in Figure 2) has 18 Cartesian coordinates
collected in matrix R0 with dimension 6 times 3 shown in eq 5 Thethree columns correspond to x y and z components and the sixrows correspond to the six atoms of the reference systemordered according to the above description The first rowspecifies the position of A at the origin point
=minus
minus
minus minus
minus minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
r
r r
r r
r r
r r
R
0 0 00 0
0
0
0
0
0
1
1 1
1 1
1 1
1 1 (5)
where = 15 4 = 14 and r1 is the radius depending onthe real system which will be clarified belowA reference system for a given symmetry operates like a
template with fixed shape which can be scaled by r1Furthermore it serves as the basis for deriving the normalvibrational modes which are independent of r1222 Normal Vibrational Modes of Reference System In
order to span the complete configuration space of the referencesystem ABn with a given symmetry we need to derive one set ofnormal vibrational mode vectors acting as the basis We employfor this purpose the following two-step procedureFirst a model Hamiltonian accounting only for the pairwise
nuclear charge repulsion is used to describe the interatomicforces within the reference system geometry (with r1 temporarilytaking an arbitrary value between 1 and 2 Aring) leading to a modelHessian matrix which is calculated analytically With theassumption that all atoms in ABn have identical unit atomicmasses (1 amu) the normal-mode analysis is carried out leadingto an initial set of 3Mminus 6 normal vibrational modes whereM = n
+ 1 Besides three rotational normal modes calculated withmassless Eckart conditions9394 are obtained as column vectorsr1 r2 and r3 (Please note that r1 r2 and r3 must not be confusedwith r1 r2 and r3 representing amplitudes)Second this initial set of 3M minus 6 normal modes is fine-tuned
within the internal vibrational space by pairwise rotationaccording to the following rules
(1) Two nondegenerate normal vibrations with the sameirreducible representation can be mixed via a 2 times 2rotation matrix leading to another two normal vibrationswith the same symmetry
(2) The first vibrational mode should always be the totallysymmetric breathing mode of the present point groupThe amplitude for all n atoms B in this vibrational modevector must be identical while the central atom A has nocontribution
(3) The atom A can be distorted only in the last three normalmodes which correspond to the relative translationbetween atomA and the remaining n atoms B in x y and zdirections
(4) Doubly degenerate modes (denoted as E and Edagger) or triplydegenerate modes (TTdagger andTDagger) can be pairwise rotatedamong themselves to fulfill the following requirementsthat(a) for the E or T mode the distortions in the z
direction andor y direction are zero if possible(b) for the Tdagger mode the distortions in the y direction
andor x direction are zero if possible and(c) the Edagger or TDagger mode can be obtained by orthonormal
condition
Pairwise rotation as described between any two normalvibrational modes is a unitary transformation that keeps theorthonormality of 3M minus 6 normal modes and greatly simplifiesthe basis vibration vectors for later useFigure 3 shows all 12 normal vibrational modes of the AB5
reference system in the C4v point group The normal modevectors li (i = 1 2 3M minus 6) shown as (M times 3)-dimensionalmatrices are orthonormal to each other The collection ofnormal vibrational modes we derived for the other referencesystems can be found in Appendices A2minusA5
223 Real System in Standard Orientation For a realmolecular compound with N-coordinate geometry as its corethe central N-coordinate part (ldquoreal systemrdquo) deviates more orless from the reference geometry with a given symmetry duringconformational changes or reaction processes It would be idealto express such distortions in terms of the normal vibrationalmodes that we derived for selected reference systems In order todo so we have to order the atomic labels of the real systemaccording to those of the reference system for consistency andthen place the real system into a particular position where itsinternal coordinate space matches exactly the internal vibra-tional space of the reference system without any contaminationfrom the translational or rotational space of the referencesystem9596 This requires the following standard orientation tobe imposed on the real system geometry
bull The initial Cartesian coordinates of the real system withN-coordinate geometry are recorded as X which is an Mtimes 3 matrix
bull The real system needs to be translated so that itsgeometrical center is positioned at the origin of Cartesiancoordinate system current Cartesian coordinates of thereal system are collected in Xc
Figure 2 Ball-and-stick representation for the reference system AB5with C4v symmetry in Cartesian coordinates The schematic Cartesianaxes are shown for illustrating directions while the true origin is at thefirst atom A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3165
bull The real system is then rotated in the 3D space so that itsCartesian coordinates denoted Xr are orthogonal to therotational modes r1 r2 r3 of the reference system Thisfinal set of Cartesian coordinates Xr is named standardorientation for the real system
The rotation of the real system by a rotation matrix u can beexpressed by the product of three 3 times 3 elemental rotationmatrices
=u u u uz y x (6)
where ux uy and uz define the rotation around x- y- and z-axesrespectively
θ θ
θ θ
θ θ
θ θ
θ θ
θ θ
= minus =minus
=minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
u u
u
1 0 00 cos sin
0 sin cos
cos 0 sin
0 1 0sin 0 cos
cos sin 0
sin cos 0
0 0 1
x x x
x x
y
y y
y y
z
z z
z z
(7)
This leads to
=X X ur c (8)
Figure 3Twelve normal vibrational modes for the AB5 reference systemwithC4v symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3166
We rearrange the Cartesian coordinates Xc and Xr from matrixform into the (1 times 3M)-dimensional row vector form as Xc and
X r then a (3M times 3M)-dimensional block diagonal matrix U isconstructed with rotation matrix u as diagonal blocks and 03times3for nondiagonal blocksBy definition the standard orientation requires that
= =
= =
= =
l
mooooooo
n
ooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 c 1
r 2 c 2
r 3 c 3 (9)
where Xc r1 r2 and r3 are already known so that only threerotation angles θx θy and θz remain as the unknowns containedin U which can be determined by iteratively solving this systemof nonlinear equationsHowever practically solving the above system of equations
with numerical methods could lead to an undesired solution asmultiple sets of solutions could exist This issue arises when thestarting geometry in Cartesian coordinates Xc is close to anotherundesired standard orientation As a remedy the arbitrariness ofrotation matrix u may be eliminated by minimizing the rootmean-square deviation (RMSD) between the Cartesiancoordinates Xc and the reference system which has been provenby Kudin and Dymarsky97 to be closely related to the well-known Eckart axis conditions In this work we adopt thequaternion-based algorithm of Kearsley98 to first rotate the realsystem Noteworthy is that before calculating the best-fit
alignment between real system and reference system the AminusBdistance r1 in the reference system is temporarily set to be theaveraged AminusB distance of the real system With the real systemrotated its Cartesian coordinates change from Xc to Xf and thestandard orientation can be unambiguously obtained by solvingthe system of equations in eq 10
= =
= =
= =
l
mooooooo
nooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 f 1
r 2 f 2
r 3 f 3 (10)
where X r is guaranteed to be the desired standard orientationcalculated by numerical equation solver because Xf is close to Xr
and θx = θy = θz = 0 can be used as the initial guess Usually thecalculation of eq 10 converges after one iteration
224 Calculation of Curvilinear CoordinatesOnce the realsystem is positioned in the appropriate standard orientation itsgeometry Xr can be completely described by the set of 3M minus 6normal vibrational modes of the corresponding reference systemABnThe determination of the real system geometry along the
directions of normal vibrations is calculated by the projection ofCartesian coordinates Xr onto each normal mode vector li (i = 12 3M minus 6) using the dot product di
= d X li irT
(11)
Figure 4 r5 ϕ5 pseudorotational cycle specifying the (t2 τ2) deformation of the B4 ring within the reference system AB5 in C4v symmetry (see Table1) In the center of the cycle is the reference geometry for AB5 with r1 = 15 Aring and surrounding the cycle are the representative geometries for r5 = 035Aring and different ϕ5 values starting from 0deg with the interval of 225deg while all other rm values (except r1) are 0 The representative geometries are shownin the perspective of looking downward from atom 2 (red) to atom 1 (green) highlighting the in-plane deformation of the B4 ring consisting of atoms3minus6
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3167
where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

importantly they turn out to be the natural choice incharacterizing ring conformations and their changes becauseboth the CremerminusPople puckering coordinates and the ringdeformation coordinates were inspired and derived from thenormal vibrational modes5154 Either the out-of-plane puckeringor in-plane deformationrearrangement of the N-ring isinitialized by a molecular vibration or the mixing of severaldegenerate vibrations68minus70 acting as the leading parameter71minus73
By adopting this conceptual approach we aimed in this work atdeveloping new curvilinear coordinates for more distinctivegeometries besides ring structures The novel curvilinearcoordinates derived from molecular vibrations are expected tobe especially useful in describing conformational changes andstructural rearrangements involving multiple atoms simulta-neouslyOne may question the extension of the underlying rationale in
ring coordinates (ie a specific vibration in a ring initiatespuckering or deformation motion) to other molecular geo-metries in general However it has been widely recognized thatmolecular vibrations can drive chemical reactions by exciting aspecific vibrational mode of the reactant along the reactioncoordinate and in this way overcoming the energy barrier towardthe product7475 Crim Zare and others have demonstrated thepossibilities of experimentally controlling the course of chemicalreactions in gas phase and on metal surfaces by selectivelyexciting certain vibrational modes with a laser76minus91 Thereforethe conceptual extension of ring coordinates to other geometriesbased on molecular vibrations has its physical foundation22 Vibration-Based Curvilinear Coordinates for N-
Coordinate Geometry Unlike for ring puckeringdeforma-tion coordinates there is no direct analytic mathematicalsolution to connect each atomrsquos Cartesian coordinates to thecurvilinear coordinates describing N-coordinate systems due tothe following reasons
bull The z-space can be separated from the xy-space whendefining the puckering47 and deformation54 coordinatesfor rings respectively However the conformationalchanges in N-coordinate geometries involve atomicmovement in the three-dimensional (3D) space whichis more complex
bull The mathematical expression of either CremerminusPoplepuckering or ring deformation coordinates has alreadyencompassed the normal vibrational modes of thecorresponding reference model systems (ie planar ringand regular polygon) implicitly However in the case ofan N-coordinate geometry we need to explicitly definethe normal vibrational modes as the basis vectorsspanning the configurational space of an N-coordinategeometry beforehand
In the following we elaborate how the curvilinear coordinatesderived from normal vibrational modes for N-coordinategeometries are defined Depending on the value of N thegeometry can be tricoordinate tetracoordinate pentacoordi-nate hexacoordinate heptacoordinate (eg IF7) or octacoor-dinate (eg XeF8
2minus)92 Within the same type of N-coordinategeometry the relative spatial arrangement of the ligating atomsbonded to the central atom may vary This complication can beresolved by utilizing point group information for the differ-entiation For example both Fe(CO)5 and ClF5 have apentacoordinate geometry however the former is trigonalbipyramidal and the latter is square pyramidal or expressed in
the point group language the former has D3h and the latter hasC4v symmetryAs shown in Figure 1 we define in this work the curvilinear
coordinates with respect to a tetracoordinate geometry with Td
symmetry pentacoordinate geometries with D3h and C4vsymmetry hexacoordinate geometries with Oh and C5vsymmetry along with heptacoordinate geometries with D5hand C6v symmetry to cover the vast majority of coordinationcompounds with fluxionalityThe pentacoordinate geometry in C4v symmetry is taken as an
example in the following to stepwise illustrate how thecurvilinear coordinate system derived from normal modes foran N-coordinate geometry is defined The same procedureequally applies to other geometries with various point groupsmentioned above
221 Reference System for a Given Symmetry As in thecase of defining ring puckering coordinates with regard to themean plane68minus70 we first need to stipulate a reference systemABn for the given symmetry egC4v where A is the central atomand n atoms of B are bonded to A as the first coordinationsphereThe definition of the reference system in Cartesian
coordinates has to fulfill the following requirementsbull The central atom A denoted as the first atom is placed at
the origin of the 3D Cartesian coordinate systembull All AminusB distances are set to be the same and assigned a
value of r1 This is equivalent to the situation that all atomsB are positioned on the surface of a sphere with radius r1and the center of sphere is placed at atom A
bull The relative positions of atoms B are adjusted under asymmetry constraint to guarantee that the geometricalcenter of ABn is located at the position of atom A
In addition the following two conditions are optional
bull The z-axis is chosen to be the highest n-fold rotation axisof ABn
bull Atoms B are grouped according to their z-values from topto bottom If the first group of B has more than one atom
Figure 1 Schematic diagram illustrating the definition of curvilinearcoordinates derived from normal vibrational modes for N-coordinategeometries
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3164
the first atom within this group is placed right above the+x-axis (y = 0) and the remaining atoms are numberedcounterclockwise when looking into the direction fromatom 2 to atom 1 If the first group of B has only one atomthen this atom is positioned on the z-axis Meanwhile thefirst atom of the second group is positioned in the +xdirection (y = 0)
For example the reference system AB5 with the C4v pointgroup (shown in Figure 2) has 18 Cartesian coordinates
collected in matrix R0 with dimension 6 times 3 shown in eq 5 Thethree columns correspond to x y and z components and the sixrows correspond to the six atoms of the reference systemordered according to the above description The first rowspecifies the position of A at the origin point
=minus
minus
minus minus
minus minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
r
r r
r r
r r
r r
R
0 0 00 0
0
0
0
0
0
1
1 1
1 1
1 1
1 1 (5)
where = 15 4 = 14 and r1 is the radius depending onthe real system which will be clarified belowA reference system for a given symmetry operates like a
template with fixed shape which can be scaled by r1Furthermore it serves as the basis for deriving the normalvibrational modes which are independent of r1222 Normal Vibrational Modes of Reference System In
order to span the complete configuration space of the referencesystem ABn with a given symmetry we need to derive one set ofnormal vibrational mode vectors acting as the basis We employfor this purpose the following two-step procedureFirst a model Hamiltonian accounting only for the pairwise
nuclear charge repulsion is used to describe the interatomicforces within the reference system geometry (with r1 temporarilytaking an arbitrary value between 1 and 2 Aring) leading to a modelHessian matrix which is calculated analytically With theassumption that all atoms in ABn have identical unit atomicmasses (1 amu) the normal-mode analysis is carried out leadingto an initial set of 3Mminus 6 normal vibrational modes whereM = n
+ 1 Besides three rotational normal modes calculated withmassless Eckart conditions9394 are obtained as column vectorsr1 r2 and r3 (Please note that r1 r2 and r3 must not be confusedwith r1 r2 and r3 representing amplitudes)Second this initial set of 3M minus 6 normal modes is fine-tuned
within the internal vibrational space by pairwise rotationaccording to the following rules
(1) Two nondegenerate normal vibrations with the sameirreducible representation can be mixed via a 2 times 2rotation matrix leading to another two normal vibrationswith the same symmetry
(2) The first vibrational mode should always be the totallysymmetric breathing mode of the present point groupThe amplitude for all n atoms B in this vibrational modevector must be identical while the central atom A has nocontribution
(3) The atom A can be distorted only in the last three normalmodes which correspond to the relative translationbetween atomA and the remaining n atoms B in x y and zdirections
(4) Doubly degenerate modes (denoted as E and Edagger) or triplydegenerate modes (TTdagger andTDagger) can be pairwise rotatedamong themselves to fulfill the following requirementsthat(a) for the E or T mode the distortions in the z
direction andor y direction are zero if possible(b) for the Tdagger mode the distortions in the y direction
andor x direction are zero if possible and(c) the Edagger or TDagger mode can be obtained by orthonormal
condition
Pairwise rotation as described between any two normalvibrational modes is a unitary transformation that keeps theorthonormality of 3M minus 6 normal modes and greatly simplifiesthe basis vibration vectors for later useFigure 3 shows all 12 normal vibrational modes of the AB5
reference system in the C4v point group The normal modevectors li (i = 1 2 3M minus 6) shown as (M times 3)-dimensionalmatrices are orthonormal to each other The collection ofnormal vibrational modes we derived for the other referencesystems can be found in Appendices A2minusA5
223 Real System in Standard Orientation For a realmolecular compound with N-coordinate geometry as its corethe central N-coordinate part (ldquoreal systemrdquo) deviates more orless from the reference geometry with a given symmetry duringconformational changes or reaction processes It would be idealto express such distortions in terms of the normal vibrationalmodes that we derived for selected reference systems In order todo so we have to order the atomic labels of the real systemaccording to those of the reference system for consistency andthen place the real system into a particular position where itsinternal coordinate space matches exactly the internal vibra-tional space of the reference system without any contaminationfrom the translational or rotational space of the referencesystem9596 This requires the following standard orientation tobe imposed on the real system geometry
bull The initial Cartesian coordinates of the real system withN-coordinate geometry are recorded as X which is an Mtimes 3 matrix
bull The real system needs to be translated so that itsgeometrical center is positioned at the origin of Cartesiancoordinate system current Cartesian coordinates of thereal system are collected in Xc
Figure 2 Ball-and-stick representation for the reference system AB5with C4v symmetry in Cartesian coordinates The schematic Cartesianaxes are shown for illustrating directions while the true origin is at thefirst atom A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3165
bull The real system is then rotated in the 3D space so that itsCartesian coordinates denoted Xr are orthogonal to therotational modes r1 r2 r3 of the reference system Thisfinal set of Cartesian coordinates Xr is named standardorientation for the real system
The rotation of the real system by a rotation matrix u can beexpressed by the product of three 3 times 3 elemental rotationmatrices
=u u u uz y x (6)
where ux uy and uz define the rotation around x- y- and z-axesrespectively
θ θ
θ θ
θ θ
θ θ
θ θ
θ θ
= minus =minus
=minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
u u
u
1 0 00 cos sin
0 sin cos
cos 0 sin
0 1 0sin 0 cos
cos sin 0
sin cos 0
0 0 1
x x x
x x
y
y y
y y
z
z z
z z
(7)
This leads to
=X X ur c (8)
Figure 3Twelve normal vibrational modes for the AB5 reference systemwithC4v symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3166
We rearrange the Cartesian coordinates Xc and Xr from matrixform into the (1 times 3M)-dimensional row vector form as Xc and
X r then a (3M times 3M)-dimensional block diagonal matrix U isconstructed with rotation matrix u as diagonal blocks and 03times3for nondiagonal blocksBy definition the standard orientation requires that
= =
= =
= =
l
mooooooo
n
ooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 c 1
r 2 c 2
r 3 c 3 (9)
where Xc r1 r2 and r3 are already known so that only threerotation angles θx θy and θz remain as the unknowns containedin U which can be determined by iteratively solving this systemof nonlinear equationsHowever practically solving the above system of equations
with numerical methods could lead to an undesired solution asmultiple sets of solutions could exist This issue arises when thestarting geometry in Cartesian coordinates Xc is close to anotherundesired standard orientation As a remedy the arbitrariness ofrotation matrix u may be eliminated by minimizing the rootmean-square deviation (RMSD) between the Cartesiancoordinates Xc and the reference system which has been provenby Kudin and Dymarsky97 to be closely related to the well-known Eckart axis conditions In this work we adopt thequaternion-based algorithm of Kearsley98 to first rotate the realsystem Noteworthy is that before calculating the best-fit
alignment between real system and reference system the AminusBdistance r1 in the reference system is temporarily set to be theaveraged AminusB distance of the real system With the real systemrotated its Cartesian coordinates change from Xc to Xf and thestandard orientation can be unambiguously obtained by solvingthe system of equations in eq 10
= =
= =
= =
l
mooooooo
nooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 f 1
r 2 f 2
r 3 f 3 (10)
where X r is guaranteed to be the desired standard orientationcalculated by numerical equation solver because Xf is close to Xr
and θx = θy = θz = 0 can be used as the initial guess Usually thecalculation of eq 10 converges after one iteration
224 Calculation of Curvilinear CoordinatesOnce the realsystem is positioned in the appropriate standard orientation itsgeometry Xr can be completely described by the set of 3M minus 6normal vibrational modes of the corresponding reference systemABnThe determination of the real system geometry along the
directions of normal vibrations is calculated by the projection ofCartesian coordinates Xr onto each normal mode vector li (i = 12 3M minus 6) using the dot product di
= d X li irT
(11)
Figure 4 r5 ϕ5 pseudorotational cycle specifying the (t2 τ2) deformation of the B4 ring within the reference system AB5 in C4v symmetry (see Table1) In the center of the cycle is the reference geometry for AB5 with r1 = 15 Aring and surrounding the cycle are the representative geometries for r5 = 035Aring and different ϕ5 values starting from 0deg with the interval of 225deg while all other rm values (except r1) are 0 The representative geometries are shownin the perspective of looking downward from atom 2 (red) to atom 1 (green) highlighting the in-plane deformation of the B4 ring consisting of atoms3minus6
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3167
where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

the first atom within this group is placed right above the+x-axis (y = 0) and the remaining atoms are numberedcounterclockwise when looking into the direction fromatom 2 to atom 1 If the first group of B has only one atomthen this atom is positioned on the z-axis Meanwhile thefirst atom of the second group is positioned in the +xdirection (y = 0)
For example the reference system AB5 with the C4v pointgroup (shown in Figure 2) has 18 Cartesian coordinates
collected in matrix R0 with dimension 6 times 3 shown in eq 5 Thethree columns correspond to x y and z components and the sixrows correspond to the six atoms of the reference systemordered according to the above description The first rowspecifies the position of A at the origin point
=minus
minus
minus minus
minus minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
r
r r
r r
r r
r r
R
0 0 00 0
0
0
0
0
0
1
1 1
1 1
1 1
1 1 (5)
where = 15 4 = 14 and r1 is the radius depending onthe real system which will be clarified belowA reference system for a given symmetry operates like a
template with fixed shape which can be scaled by r1Furthermore it serves as the basis for deriving the normalvibrational modes which are independent of r1222 Normal Vibrational Modes of Reference System In
order to span the complete configuration space of the referencesystem ABn with a given symmetry we need to derive one set ofnormal vibrational mode vectors acting as the basis We employfor this purpose the following two-step procedureFirst a model Hamiltonian accounting only for the pairwise
nuclear charge repulsion is used to describe the interatomicforces within the reference system geometry (with r1 temporarilytaking an arbitrary value between 1 and 2 Aring) leading to a modelHessian matrix which is calculated analytically With theassumption that all atoms in ABn have identical unit atomicmasses (1 amu) the normal-mode analysis is carried out leadingto an initial set of 3Mminus 6 normal vibrational modes whereM = n
+ 1 Besides three rotational normal modes calculated withmassless Eckart conditions9394 are obtained as column vectorsr1 r2 and r3 (Please note that r1 r2 and r3 must not be confusedwith r1 r2 and r3 representing amplitudes)Second this initial set of 3M minus 6 normal modes is fine-tuned
within the internal vibrational space by pairwise rotationaccording to the following rules
(1) Two nondegenerate normal vibrations with the sameirreducible representation can be mixed via a 2 times 2rotation matrix leading to another two normal vibrationswith the same symmetry
(2) The first vibrational mode should always be the totallysymmetric breathing mode of the present point groupThe amplitude for all n atoms B in this vibrational modevector must be identical while the central atom A has nocontribution
(3) The atom A can be distorted only in the last three normalmodes which correspond to the relative translationbetween atomA and the remaining n atoms B in x y and zdirections
(4) Doubly degenerate modes (denoted as E and Edagger) or triplydegenerate modes (TTdagger andTDagger) can be pairwise rotatedamong themselves to fulfill the following requirementsthat(a) for the E or T mode the distortions in the z
direction andor y direction are zero if possible(b) for the Tdagger mode the distortions in the y direction
andor x direction are zero if possible and(c) the Edagger or TDagger mode can be obtained by orthonormal
condition
Pairwise rotation as described between any two normalvibrational modes is a unitary transformation that keeps theorthonormality of 3M minus 6 normal modes and greatly simplifiesthe basis vibration vectors for later useFigure 3 shows all 12 normal vibrational modes of the AB5
reference system in the C4v point group The normal modevectors li (i = 1 2 3M minus 6) shown as (M times 3)-dimensionalmatrices are orthonormal to each other The collection ofnormal vibrational modes we derived for the other referencesystems can be found in Appendices A2minusA5
223 Real System in Standard Orientation For a realmolecular compound with N-coordinate geometry as its corethe central N-coordinate part (ldquoreal systemrdquo) deviates more orless from the reference geometry with a given symmetry duringconformational changes or reaction processes It would be idealto express such distortions in terms of the normal vibrationalmodes that we derived for selected reference systems In order todo so we have to order the atomic labels of the real systemaccording to those of the reference system for consistency andthen place the real system into a particular position where itsinternal coordinate space matches exactly the internal vibra-tional space of the reference system without any contaminationfrom the translational or rotational space of the referencesystem9596 This requires the following standard orientation tobe imposed on the real system geometry
bull The initial Cartesian coordinates of the real system withN-coordinate geometry are recorded as X which is an Mtimes 3 matrix
bull The real system needs to be translated so that itsgeometrical center is positioned at the origin of Cartesiancoordinate system current Cartesian coordinates of thereal system are collected in Xc
Figure 2 Ball-and-stick representation for the reference system AB5with C4v symmetry in Cartesian coordinates The schematic Cartesianaxes are shown for illustrating directions while the true origin is at thefirst atom A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3165
bull The real system is then rotated in the 3D space so that itsCartesian coordinates denoted Xr are orthogonal to therotational modes r1 r2 r3 of the reference system Thisfinal set of Cartesian coordinates Xr is named standardorientation for the real system
The rotation of the real system by a rotation matrix u can beexpressed by the product of three 3 times 3 elemental rotationmatrices
=u u u uz y x (6)
where ux uy and uz define the rotation around x- y- and z-axesrespectively
θ θ
θ θ
θ θ
θ θ
θ θ
θ θ
= minus =minus
=minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
u u
u
1 0 00 cos sin
0 sin cos
cos 0 sin
0 1 0sin 0 cos
cos sin 0
sin cos 0
0 0 1
x x x
x x
y
y y
y y
z
z z
z z
(7)
This leads to
=X X ur c (8)
Figure 3Twelve normal vibrational modes for the AB5 reference systemwithC4v symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3166
We rearrange the Cartesian coordinates Xc and Xr from matrixform into the (1 times 3M)-dimensional row vector form as Xc and
X r then a (3M times 3M)-dimensional block diagonal matrix U isconstructed with rotation matrix u as diagonal blocks and 03times3for nondiagonal blocksBy definition the standard orientation requires that
= =
= =
= =
l
mooooooo
n
ooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 c 1
r 2 c 2
r 3 c 3 (9)
where Xc r1 r2 and r3 are already known so that only threerotation angles θx θy and θz remain as the unknowns containedin U which can be determined by iteratively solving this systemof nonlinear equationsHowever practically solving the above system of equations
with numerical methods could lead to an undesired solution asmultiple sets of solutions could exist This issue arises when thestarting geometry in Cartesian coordinates Xc is close to anotherundesired standard orientation As a remedy the arbitrariness ofrotation matrix u may be eliminated by minimizing the rootmean-square deviation (RMSD) between the Cartesiancoordinates Xc and the reference system which has been provenby Kudin and Dymarsky97 to be closely related to the well-known Eckart axis conditions In this work we adopt thequaternion-based algorithm of Kearsley98 to first rotate the realsystem Noteworthy is that before calculating the best-fit
alignment between real system and reference system the AminusBdistance r1 in the reference system is temporarily set to be theaveraged AminusB distance of the real system With the real systemrotated its Cartesian coordinates change from Xc to Xf and thestandard orientation can be unambiguously obtained by solvingthe system of equations in eq 10
= =
= =
= =
l
mooooooo
nooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 f 1
r 2 f 2
r 3 f 3 (10)
where X r is guaranteed to be the desired standard orientationcalculated by numerical equation solver because Xf is close to Xr
and θx = θy = θz = 0 can be used as the initial guess Usually thecalculation of eq 10 converges after one iteration
224 Calculation of Curvilinear CoordinatesOnce the realsystem is positioned in the appropriate standard orientation itsgeometry Xr can be completely described by the set of 3M minus 6normal vibrational modes of the corresponding reference systemABnThe determination of the real system geometry along the
directions of normal vibrations is calculated by the projection ofCartesian coordinates Xr onto each normal mode vector li (i = 12 3M minus 6) using the dot product di
= d X li irT
(11)
Figure 4 r5 ϕ5 pseudorotational cycle specifying the (t2 τ2) deformation of the B4 ring within the reference system AB5 in C4v symmetry (see Table1) In the center of the cycle is the reference geometry for AB5 with r1 = 15 Aring and surrounding the cycle are the representative geometries for r5 = 035Aring and different ϕ5 values starting from 0deg with the interval of 225deg while all other rm values (except r1) are 0 The representative geometries are shownin the perspective of looking downward from atom 2 (red) to atom 1 (green) highlighting the in-plane deformation of the B4 ring consisting of atoms3minus6
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3167
where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

bull The real system is then rotated in the 3D space so that itsCartesian coordinates denoted Xr are orthogonal to therotational modes r1 r2 r3 of the reference system Thisfinal set of Cartesian coordinates Xr is named standardorientation for the real system
The rotation of the real system by a rotation matrix u can beexpressed by the product of three 3 times 3 elemental rotationmatrices
=u u u uz y x (6)
where ux uy and uz define the rotation around x- y- and z-axesrespectively
θ θ
θ θ
θ θ
θ θ
θ θ
θ θ
= minus =minus
=minus
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
Auml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeAuml
Ccedil
AringAringAringAringAringAringAringAringAringAringAringAringAringAringAringAring
Eacute
Ouml
NtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtildeNtilde
u u
u
1 0 00 cos sin
0 sin cos
cos 0 sin
0 1 0sin 0 cos
cos sin 0
sin cos 0
0 0 1
x x x
x x
y
y y
y y
z
z z
z z
(7)
This leads to
=X X ur c (8)
Figure 3Twelve normal vibrational modes for the AB5 reference systemwithC4v symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3166
We rearrange the Cartesian coordinates Xc and Xr from matrixform into the (1 times 3M)-dimensional row vector form as Xc and
X r then a (3M times 3M)-dimensional block diagonal matrix U isconstructed with rotation matrix u as diagonal blocks and 03times3for nondiagonal blocksBy definition the standard orientation requires that
= =
= =
= =
l
mooooooo
n
ooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 c 1
r 2 c 2
r 3 c 3 (9)
where Xc r1 r2 and r3 are already known so that only threerotation angles θx θy and θz remain as the unknowns containedin U which can be determined by iteratively solving this systemof nonlinear equationsHowever practically solving the above system of equations
with numerical methods could lead to an undesired solution asmultiple sets of solutions could exist This issue arises when thestarting geometry in Cartesian coordinates Xc is close to anotherundesired standard orientation As a remedy the arbitrariness ofrotation matrix u may be eliminated by minimizing the rootmean-square deviation (RMSD) between the Cartesiancoordinates Xc and the reference system which has been provenby Kudin and Dymarsky97 to be closely related to the well-known Eckart axis conditions In this work we adopt thequaternion-based algorithm of Kearsley98 to first rotate the realsystem Noteworthy is that before calculating the best-fit
alignment between real system and reference system the AminusBdistance r1 in the reference system is temporarily set to be theaveraged AminusB distance of the real system With the real systemrotated its Cartesian coordinates change from Xc to Xf and thestandard orientation can be unambiguously obtained by solvingthe system of equations in eq 10
= =
= =
= =
l
mooooooo
nooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 f 1
r 2 f 2
r 3 f 3 (10)
where X r is guaranteed to be the desired standard orientationcalculated by numerical equation solver because Xf is close to Xr
and θx = θy = θz = 0 can be used as the initial guess Usually thecalculation of eq 10 converges after one iteration
224 Calculation of Curvilinear CoordinatesOnce the realsystem is positioned in the appropriate standard orientation itsgeometry Xr can be completely described by the set of 3M minus 6normal vibrational modes of the corresponding reference systemABnThe determination of the real system geometry along the
directions of normal vibrations is calculated by the projection ofCartesian coordinates Xr onto each normal mode vector li (i = 12 3M minus 6) using the dot product di
= d X li irT
(11)
Figure 4 r5 ϕ5 pseudorotational cycle specifying the (t2 τ2) deformation of the B4 ring within the reference system AB5 in C4v symmetry (see Table1) In the center of the cycle is the reference geometry for AB5 with r1 = 15 Aring and surrounding the cycle are the representative geometries for r5 = 035Aring and different ϕ5 values starting from 0deg with the interval of 225deg while all other rm values (except r1) are 0 The representative geometries are shownin the perspective of looking downward from atom 2 (red) to atom 1 (green) highlighting the in-plane deformation of the B4 ring consisting of atoms3minus6
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3167
where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

We rearrange the Cartesian coordinates Xc and Xr from matrixform into the (1 times 3M)-dimensional row vector form as Xc and
X r then a (3M times 3M)-dimensional block diagonal matrix U isconstructed with rotation matrix u as diagonal blocks and 03times3for nondiagonal blocksBy definition the standard orientation requires that
= =
= =
= =
l
mooooooo
n
ooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 c 1
r 2 c 2
r 3 c 3 (9)
where Xc r1 r2 and r3 are already known so that only threerotation angles θx θy and θz remain as the unknowns containedin U which can be determined by iteratively solving this systemof nonlinear equationsHowever practically solving the above system of equations
with numerical methods could lead to an undesired solution asmultiple sets of solutions could exist This issue arises when thestarting geometry in Cartesian coordinates Xc is close to anotherundesired standard orientation As a remedy the arbitrariness ofrotation matrix u may be eliminated by minimizing the rootmean-square deviation (RMSD) between the Cartesiancoordinates Xc and the reference system which has been provenby Kudin and Dymarsky97 to be closely related to the well-known Eckart axis conditions In this work we adopt thequaternion-based algorithm of Kearsley98 to first rotate the realsystem Noteworthy is that before calculating the best-fit
alignment between real system and reference system the AminusBdistance r1 in the reference system is temporarily set to be theaveraged AminusB distance of the real system With the real systemrotated its Cartesian coordinates change from Xc to Xf and thestandard orientation can be unambiguously obtained by solvingthe system of equations in eq 10
= =
= =
= =
l
mooooooo
nooooooo
X r X Ur
X r X Ur
X r X Ur
0
0
0
r 1 f 1
r 2 f 2
r 3 f 3 (10)
where X r is guaranteed to be the desired standard orientationcalculated by numerical equation solver because Xf is close to Xr
and θx = θy = θz = 0 can be used as the initial guess Usually thecalculation of eq 10 converges after one iteration
224 Calculation of Curvilinear CoordinatesOnce the realsystem is positioned in the appropriate standard orientation itsgeometry Xr can be completely described by the set of 3M minus 6normal vibrational modes of the corresponding reference systemABnThe determination of the real system geometry along the
directions of normal vibrations is calculated by the projection ofCartesian coordinates Xr onto each normal mode vector li (i = 12 3M minus 6) using the dot product di
= d X li irT
(11)
Figure 4 r5 ϕ5 pseudorotational cycle specifying the (t2 τ2) deformation of the B4 ring within the reference system AB5 in C4v symmetry (see Table1) In the center of the cycle is the reference geometry for AB5 with r1 = 15 Aring and surrounding the cycle are the representative geometries for r5 = 035Aring and different ϕ5 values starting from 0deg with the interval of 225deg while all other rm values (except r1) are 0 The representative geometries are shownin the perspective of looking downward from atom 2 (red) to atom 1 (green) highlighting the in-plane deformation of the B4 ring consisting of atoms3minus6
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3167
where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

where X r and li take the row vector form of matrices Xr and lirespectively and superscript T indicates the transposeUp to this point we are able to describe the geometry of the
real system merely by the set of 3Mminus 6 projection values di aslong as corresponding normal vibrational modes for thereference system have been determined beforehand Besidesfor those doubly or triply degenerate vibrations we derived forthe reference systems it would be more useful if thesedegenerate vibrations are combined using pseudorotationalcycles in polar coordinates or using the conformational globe incylindricalspherical coordinates as for ring puckering coor-dinates Even for nondegenerate vibrational modes they canalso be grouped together for convenienceAlong these lines we define three types of curvilinear
coordinate sets for an N-coordinate geometry(i) nondegenerate curvilinear coordinate rm
=r dm m (12)
where is the largest atomic amplitude within the normal modevector lm associated with the projection value dm For examplewithin the first breathing mode of the reference system AB5 inC4v symmetry all four atoms B have the largest amplitude as
α β+ = 122 2 Therefore takes the value of 12 for thisparticular vibrational modeWith as a scaling factor rm has the same fundamental unit
of distance as dm generally expressed in Angstrom units rm canbe either positive or negative(ii) doubly degenerate curvilinear coordinates rm ϕm
ϕ
ϕ
=
= prime
lmooonooo
r d
r d
cos
sin
m m m
m m m (13)
where dm and dmprime are the projections from real system geometryXr onto a pair of doubly degenerate (E and Edagger) vibrationalmodes and the curvilinear coordinates rmϕm are calculated bysolving above system of equationsThis curvilinear coordinate parameter set specifies a polar
coordinate system where we expect that
ϕ⩾ deg ⩽ lt degr 0 0 360m m (14)
During a relaxed PES scan or reaction pathway rm is allowedto take a negative value with the following equivalence relation
ϕ ϕ equiv minus plusmn degr r 180m m m m (15)
For a fixed amplitude rm a pseudorotational cycle arises whenthe phase angle ϕm gradually changes as shown in Figure 4When ϕm equals 0deg180deg the second degenerate vibration hasno contribution and when ϕm equals 90deg270deg thecontribution from the first degenerate vibration vanishesOtherwise both vibrations contribute to determine thegeometry of the real systemiii triply degenerate curvilinear coordinates rm θm ϕm
θ
θ ϕ
θ ϕ
=
= prime
= Prime
l
moooooo
noooooo
r d
r d
r d
cos
sin sin
sin cos
m m m
m m m m
m m m m (16)
where dm dmprime and dmPrime are the projections from Xr onto a triad oftriply degenerate (T Tdagger and TDagger) vibrational modes and rm θmϕm determines a spherical coordinate system where we expectthat
θ ϕ⩾ deg ⩽ ⩽ deg deg ⩽ lt degr 0 0 180 0 360m m m (17)
Also rm can take a negative value with the following equivalencerelation
θ ϕ θ ϕ
θ ϕ
equiv minus deg minus plusmn deg
equiv minus plusmn deg
r r
r
180 180
180
m m m m m m
m m m (18)
This set of curvilinear coordinate parameters controls thecontribution from three vibrations to the real system geometryin a similar fashion as the doubly degenerate set For a givenvalue of rm we adopt the idea of conformational globe from ringpuckering coordinates for describing the conformationalvariation in N-coordinate geometries as shown in Figure 6Consequently the geometry of the real system in the standard
orientation with regard to the selected reference system ABnhaving M atoms can be accurately determined by 3M minus 6curvilinear coordinate parameters of the above three typesTable 1 summarizes seven sets of curvilinear coordinate
parameters we constructed for the AB5 system with C4v
symmetry The geometry of the reference system is determinedonce r1 has been calculated with eq 12Note that we have combined the sixth and seventh normal
modes into a pair of doubly degenerate curvilinear coordinatesr5 ϕ5 although these two modes have different symmetriesThis is because these two vibrational modes are the basis vectorsfor the (t2 τ2) deformation of the B4 ring
54 and they are similarin their mathematical form with identical valuesFigure 4 shows a r5 ϕ5 pseudorotational cycle where a
single curvilinear coordinate parameter ϕ5 determines thegeometrical variation in AB5 Figure 5 illustrates a two-dimensional (2D) PES by relaxed scanning r5 and ϕ5simultaneously for the PF5 molecule as a showcase example todemonstrate one possibility of applying the curvilinearcoordinates developed in this work to model polytopalrearrangements Besides we have combined the pair ofdegenerate vibrations (10th and 11th) with the 12th vibrationalmode into a triply degenerate set of curvilinear coordinates r7θ7 ϕ7 because these three vibrational modes are similar in theirmathematical form and all of them describe the relativetranslations of the central atom
225 Conversion from Curvilinear Coordinates intoCartesian Coordinates During a quantum chemical modelingof an N-coordinate compound with its geometry described withthe curvilinear coordinates the back-conversion from curvi-linear coordinates to Cartesian coordinates is needed
Table 1 Summary of 12 Curvilinear Coordinate Parametersin 7 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in C4v Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB5 breathing2 r2 A1 antibreathing3 r3 B2 q2 puckering of B4 ring4 5 r4 ϕ4 E (t1 τ1) deformation of B4
ring6 7 r5 ϕ5 B2 + B1 (t2 τ2) deformation of B4
ring8 9 r6 ϕ6 E bending10 11 12 r7 θ7 ϕ7 E + A1 translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3168
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
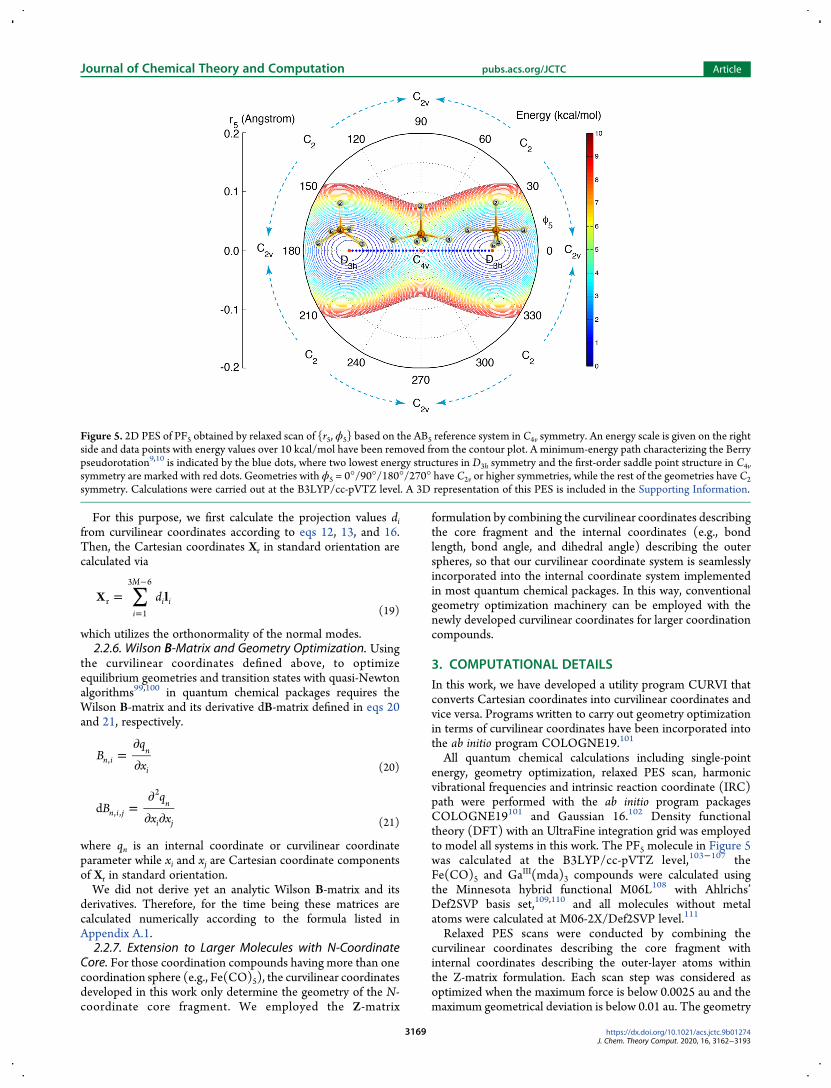
For this purpose we first calculate the projection values difrom curvilinear coordinates according to eqs 12 13 and 16Then the Cartesian coordinates Xr in standard orientation arecalculated via
sum==
minus
dX li
M
i ir1
3 6
(19)
which utilizes the orthonormality of the normal modes226 Wilson B-Matrix and Geometry Optimization Using
the curvilinear coordinates defined above to optimizeequilibrium geometries and transition states with quasi-Newtonalgorithms99100 in quantum chemical packages requires theWilson B-matrix and its derivative dB-matrix defined in eqs 20and 21 respectively
=partpart
Bq
xn in
i
(20)
=part
part partB
q
x xd n i j
n
i j
2
(21)
where qn is an internal coordinate or curvilinear coordinateparameter while xi and xj are Cartesian coordinate componentsof Xr in standard orientationWe did not derive yet an analytic Wilson B-matrix and its
derivatives Therefore for the time being these matrices arecalculated numerically according to the formula listed inAppendix A1227 Extension to Larger Molecules with N-Coordinate
Core For those coordination compounds having more than onecoordination sphere (eg Fe(CO)5) the curvilinear coordinatesdeveloped in this work only determine the geometry of the N-coordinate core fragment We employed the Z-matrix
formulation by combining the curvilinear coordinates describingthe core fragment and the internal coordinates (eg bondlength bond angle and dihedral angle) describing the outerspheres so that our curvilinear coordinate system is seamlesslyincorporated into the internal coordinate system implementedin most quantum chemical packages In this way conventionalgeometry optimization machinery can be employed with thenewly developed curvilinear coordinates for larger coordinationcompounds
3 COMPUTATIONAL DETAILSIn this work we have developed a utility program CURVI thatconverts Cartesian coordinates into curvilinear coordinates andvice versa Programs written to carry out geometry optimizationin terms of curvilinear coordinates have been incorporated intothe ab initio program COLOGNE19101
All quantum chemical calculations including single-pointenergy geometry optimization relaxed PES scan harmonicvibrational frequencies and intrinsic reaction coordinate (IRC)path were performed with the ab initio program packagesCOLOGNE19101 and Gaussian 16102 Density functionaltheory (DFT) with an UltraFine integration grid was employedto model all systems in this work The PF5 molecule in Figure 5was calculated at the B3LYPcc-pVTZ level103minus107 theFe(CO)5 and GaIII(mda)3 compounds were calculated usingthe Minnesota hybrid functional M06L108 with AhlrichsrsquoDef2SVP basis set109110 and all molecules without metalatoms were calculated at M06-2XDef2SVP level111
Relaxed PES scans were conducted by combining thecurvilinear coordinates describing the core fragment withinternal coordinates describing the outer-layer atoms withinthe Z-matrix formulation Each scan step was considered asoptimized when the maximum force is below 00025 au and themaximum geometrical deviation is below 001 au The geometry
Figure 5 2D PES of PF5 obtained by relaxed scan of r5 ϕ5 based on the AB5 reference system in C4v symmetry An energy scale is given on the rightside and data points with energy values over 10 kcalmol have been removed from the contour plot A minimum-energy path characterizing the Berrypseudorotation910 is indicated by the blue dots where two lowest energy structures in D3h symmetry and the first-order saddle point structure in C4vsymmetry are marked with red dots Geometries with ϕ5 = 0deg90deg180deg270deg have C2v or higher symmetries while the rest of the geometries have C2symmetry Calculations were carried out at the B3LYPcc-pVTZ level A 3D representation of this PES is included in the Supporting Information
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3169
optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

optimization for stationary points was carried out with a tightconvergence criterionIRC calculations were performed in Cartesian coordinates
without mass-weighting in order to map out the minimum-energy path (MEP) Most MEPs in this work were obtainedusing Hessian-based predictor-corrector (HPC) integra-tor112minus114 with a stepsize of 005 bohr and analytic Hessianevery five steps For larger compounds discussed in sections 42and 43 either the GS2115116 or local quadratic approximation(LQA)117118 algorithm was employed as the path integratorwith the stepsize of 020minus050 bohr
4 RESULTS AND DISCUSSIONThe Berry pseudorotation of PF5molecule910 has been shown inFigure 5 as an example where the multidimensional PES isreduced to a 2D PES spanned by the curvilinear coordinate pairr5 ϕ5 and the reaction path connecting the stationary points isthen straightforwardly determined We need to note that thechoice of the reference system AB5 in C4v symmetry for this
example is based on its known mechanism ie the transitionstate takes a square pyramidal (C4v) geometry However wheninvestigating fluxional molecules with uncertain mechanisms wehave to start from the local minimum structures (reactantsproducts) on the PES Besides the scanned PES with twodegrees of freedom can be further simplified into a 1D PES if thereaction coordinate is properly chosen
Figure 6Conformational globe spanned by the parameter set r7 θ7 ϕ7 specifying the translation of central atom A within the AB5 reference system(r1 = 15 Aring) in C4v symmetry (see Table 1) Any point on the globe surface has r7 = 03 Aring while all other rm values (except r1) are 0 Some distinctlongitudes and latitudes are shown including the equator (θ = 90deg) in red and the Tropic of Cancer (θ = 665deg) and the Tropic of Capricorn (θ =1135deg) in magenta Among five representative structures (ϕ7 = 0deg θ7 = 0deg665deg90deg1135deg180deg) bond 1minus2 first shortens and then elongates(controlled by sin θ7) bond 1minus3 elongates and bond 1minus5 shortens (controlled by cos θ7) continuously from north pole to south pole Among sevenrepresentative structures on the equator (θ7 = 90degϕ7 = 180deg210deg240deg270deg300deg330deg360deg) bond 1minus2 shortens (controlled by cosϕ7) from leftto right bond 1minus6 first shortens and then elongates and bond 1minus4 changes in the opposite way (controlled by sin ϕ7)
Table 2 Curvilinear Coordinate Parameters of SF4 at ItsEquilibriumGeometry Based on the AB4 Reference System inTd Symmetrya
m rm θm ϕm
1 141862 04673 27003 02157 00 004 04493 900 00b
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively bϕ4 = 0deg is equivalent to ϕ4 = 360deg
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3170
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
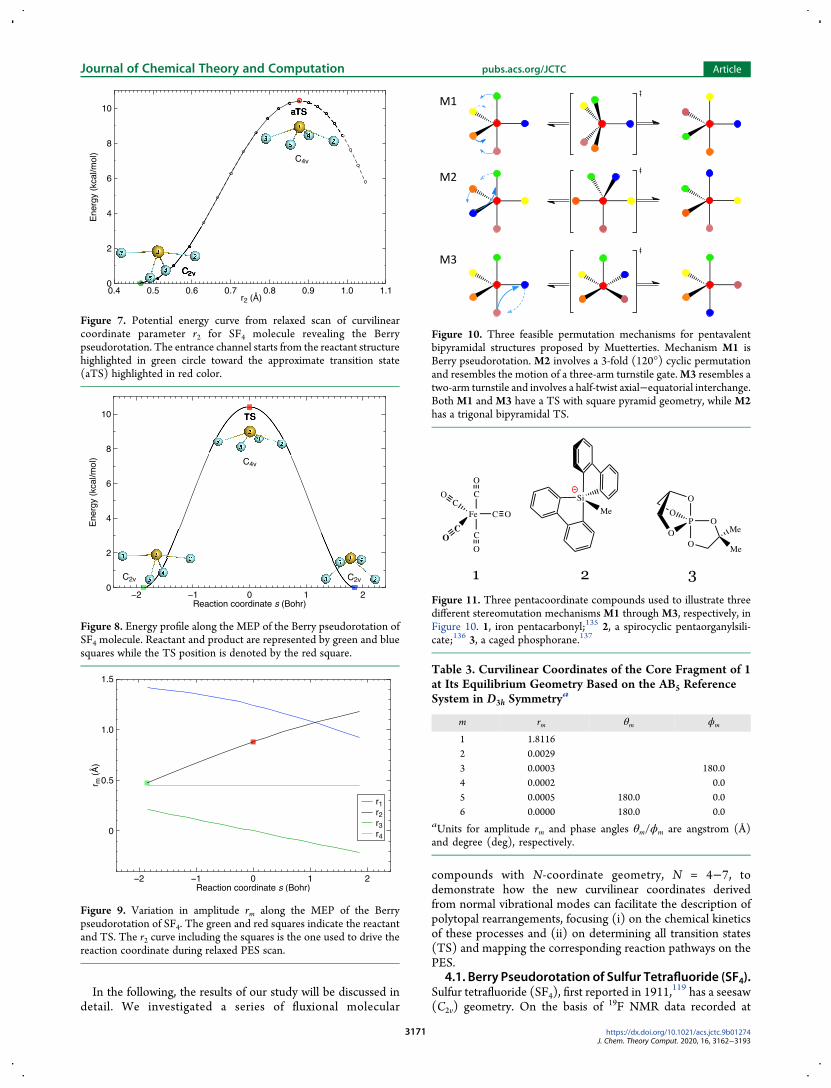
In the following the results of our study will be discussed indetail We investigated a series of fluxional molecular
compounds with N-coordinate geometry N = 4minus7 todemonstrate how the new curvilinear coordinates derivedfrom normal vibrational modes can facilitate the description ofpolytopal rearrangements focusing (i) on the chemical kineticsof these processes and (ii) on determining all transition states(TS) and mapping the corresponding reaction pathways on thePES
41 Berry Pseudorotation of Sulfur Tetrafluoride (SF4)Sulfur tetrafluoride (SF4) first reported in 1911
119 has a seesaw(C2v) geometry On the basis of 19F NMR data recorded at
Figure 7 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r2 for SF4 molecule revealing the Berrypseudorotation The entrance channel starts from the reactant structurehighlighted in green circle toward the approximate transition state(aTS) highlighted in red color
Figure 8 Energy profile along the MEP of the Berry pseudorotation ofSF4 molecule Reactant and product are represented by green and bluesquares while the TS position is denoted by the red square
Figure 9 Variation in amplitude rm along the MEP of the Berrypseudorotation of SF4 The green and red squares indicate the reactantand TS The r2 curve including the squares is the one used to drive thereaction coordinate during relaxed PES scan
Figure 10 Three feasible permutation mechanisms for pentavalentbipyramidal structures proposed by Muetterties Mechanism M1 isBerry pseudorotation M2 involves a 3-fold (120deg) cyclic permutationand resembles the motion of a three-arm turnstile gateM3 resembles atwo-arm turnstile and involves a half-twist axialminusequatorial interchangeBoth M1 and M3 have a TS with square pyramid geometry while M2has a trigonal bipyramidal TS
Figure 11 Three pentacoordinate compounds used to illustrate threedifferent stereomutation mechanisms M1 through M3 respectively inFigure 10 1 iron pentacarbonyl135 2 a spirocyclic pentaorganylsili-cate136 3 a caged phosphorane137
Table 3 Curvilinear Coordinates of the Core Fragment of 1at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 181162 000293 00003 18004 00002 005 00005 1800 006 00000 1800 00
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3171
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
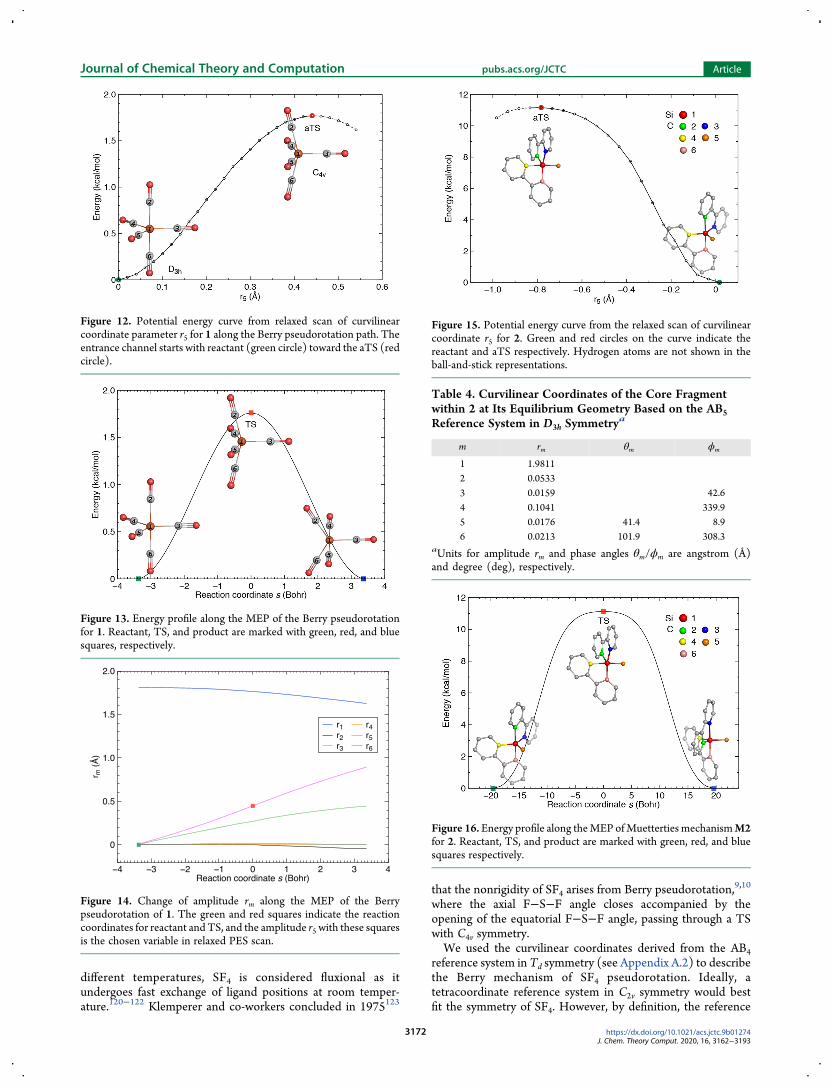
different temperatures SF4 is considered fluxional as itundergoes fast exchange of ligand positions at room temper-ature120minus122 Klemperer and co-workers concluded in 1975123
that the nonrigidity of SF4 arises from Berry pseudorotation910
where the axial FminusSminusF angle closes accompanied by theopening of the equatorial FminusSminusF angle passing through a TSwith C4v symmetryWe used the curvilinear coordinates derived from the AB4
reference system inTd symmetry (see Appendix A2) to describethe Berry mechanism of SF4 pseudorotation Ideally atetracoordinate reference system in C2v symmetry would bestfit the symmetry of SF4 However by definition the reference
Figure 12 Potential energy curve from relaxed scan of curvilinearcoordinate parameter r5 for 1 along the Berry pseudorotation path Theentrance channel starts with reactant (green circle) toward the aTS (redcircle)
Figure 13 Energy profile along the MEP of the Berry pseudorotationfor 1 Reactant TS and product are marked with green red and bluesquares respectively
Figure 14 Change of amplitude rm along the MEP of the Berrypseudorotation of 1 The green and red squares indicate the reactioncoordinates for reactant and TS and the amplitude r5 with these squaresis the chosen variable in relaxed PES scan
Figure 15 Potential energy curve from the relaxed scan of curvilinearcoordinate r5 for 2 Green and red circles on the curve indicate thereactant and aTS respectively Hydrogen atoms are not shown in theball-and-stick representations
Table 4 Curvilinear Coordinates of the Core Fragmentwithin 2 at Its Equilibrium Geometry Based on the AB5Reference System in D3h Symmetrya
m rm θm ϕm
1 198112 005333 00159 4264 01041 33995 00176 414 896 00213 1019 3083
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 16 Energy profile along theMEP ofMuetterties mechanismM2for 2 Reactant TS and product are marked with green red and bluesquares respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3172
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
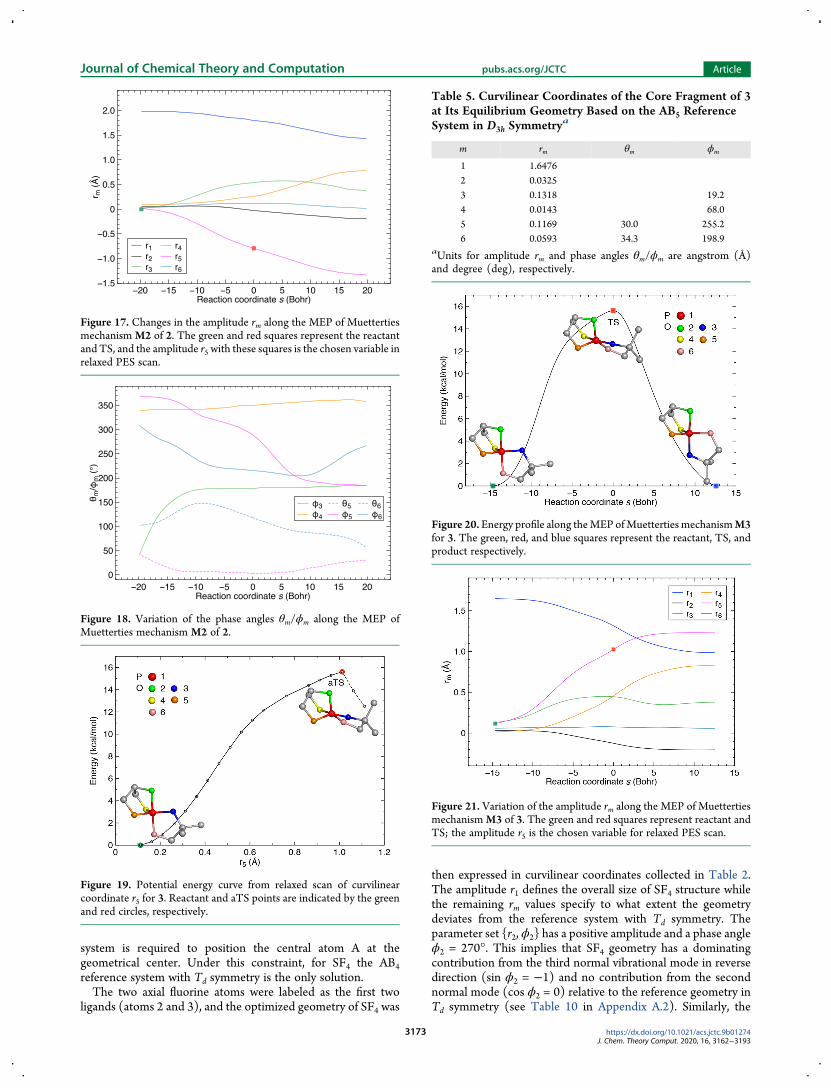
system is required to position the central atom A at thegeometrical center Under this constraint for SF4 the AB4reference system with Td symmetry is the only solutionThe two axial fluorine atoms were labeled as the first two
ligands (atoms 2 and 3) and the optimized geometry of SF4 was
then expressed in curvilinear coordinates collected in Table 2The amplitude r1 defines the overall size of SF4 structure whilethe remaining rm values specify to what extent the geometrydeviates from the reference system with Td symmetry Theparameter set r2 ϕ2 has a positive amplitude and a phase angleϕ2 = 270deg This implies that SF4 geometry has a dominatingcontribution from the third normal vibrational mode in reversedirection (sin ϕ2 = minus1) and no contribution from the secondnormal mode (cos ϕ2 = 0) relative to the reference geometry inTd symmetry (see Table 10 in Appendix A2) Similarly the
Figure 17 Changes in the amplitude rm along the MEP of MuettertiesmechanismM2 of 2 The green and red squares represent the reactantand TS and the amplitude r5 with these squares is the chosen variable inrelaxed PES scan
Figure 18 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M2 of 2
Figure 19 Potential energy curve from relaxed scan of curvilinearcoordinate r5 for 3 Reactant and aTS points are indicated by the greenand red circles respectively
Table 5 Curvilinear Coordinates of the Core Fragment of 3at Its Equilibrium Geometry Based on the AB5 ReferenceSystem in D3h Symmetrya
m rm θm ϕm
1 164762 003253 01318 1924 00143 6805 01169 300 25526 00593 343 1989
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 20 Energy profile along theMEP ofMuetterties mechanismM3for 3 The green red and blue squares represent the reactant TS andproduct respectively
Figure 21 Variation of the amplitude rm along the MEP of MuettertiesmechanismM3 of 3 The green and red squares represent reactant andTS the amplitude r5 is the chosen variable for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3173
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
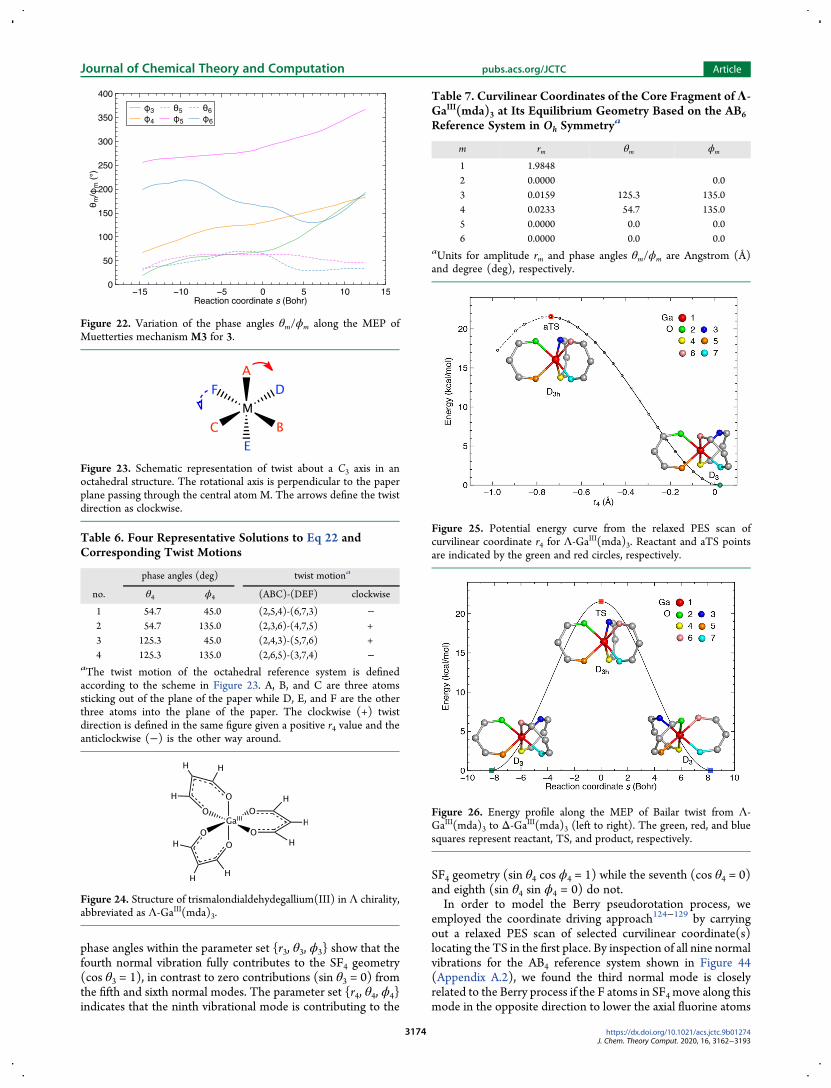
phase angles within the parameter set r3 θ3 ϕ3 show that thefourth normal vibration fully contributes to the SF4 geometry(cos θ3 = 1) in contrast to zero contributions (sin θ3 = 0) fromthe fifth and sixth normal modes The parameter set r4 θ4 ϕ4indicates that the ninth vibrational mode is contributing to the
SF4 geometry (sin θ4 cos ϕ4 = 1) while the seventh (cos θ4 = 0)and eighth (sin θ4 sin ϕ4 = 0) do notIn order to model the Berry pseudorotation process we
employed the coordinate driving approach124minus129 by carryingout a relaxed PES scan of selected curvilinear coordinate(s)locating the TS in the first place By inspection of all nine normalvibrations for the AB4 reference system shown in Figure 44(Appendix A2) we found the third normal mode is closelyrelated to the Berry process if the F atoms in SF4 move along thismode in the opposite direction to lower the axial fluorine atoms
Figure 22 Variation of the phase angles θmϕm along the MEP ofMuetterties mechanism M3 for 3
Figure 23 Schematic representation of twist about a C3 axis in anoctahedral structure The rotational axis is perpendicular to the paperplane passing through the central atom M The arrows define the twistdirection as clockwise
Table 6 Four Representative Solutions to Eq 22 andCorresponding Twist Motions
phase angles (deg) twist motiona
no θ4 ϕ4 (ABC)-(DEF) clockwise
1 547 450 (254)-(673) minus2 547 1350 (236)-(475) +3 1253 450 (243)-(576) +4 1253 1350 (265)-(374) minus
aThe twist motion of the octahedral reference system is definedaccording to the scheme in Figure 23 A B and C are three atomssticking out of the plane of the paper while D E and F are the otherthree atoms into the plane of the paper The clockwise (+) twistdirection is defined in the same figure given a positive r4 value and theanticlockwise (minus) is the other way around
Figure 24 Structure of trismalondialdehydegallium(III) in Λ chiralityabbreviated as Λ-GaIII(mda)3
Table 7 Curvilinear Coordinates of the Core Fragment ofΛ-GaIII(mda)3 at Its Equilibrium Geometry Based on the AB6Reference System in Oh Symmetrya
m rm θm ϕm
1 198482 00000 003 00159 1253 13504 00233 547 13505 00000 00 006 00000 00 00
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Figure 25 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 for Λ-GaIII(mda)3 Reactant and aTS pointsare indicated by the green and red circles respectively
Figure 26 Energy profile along the MEP of Bailar twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent reactant TS and product respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3174
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
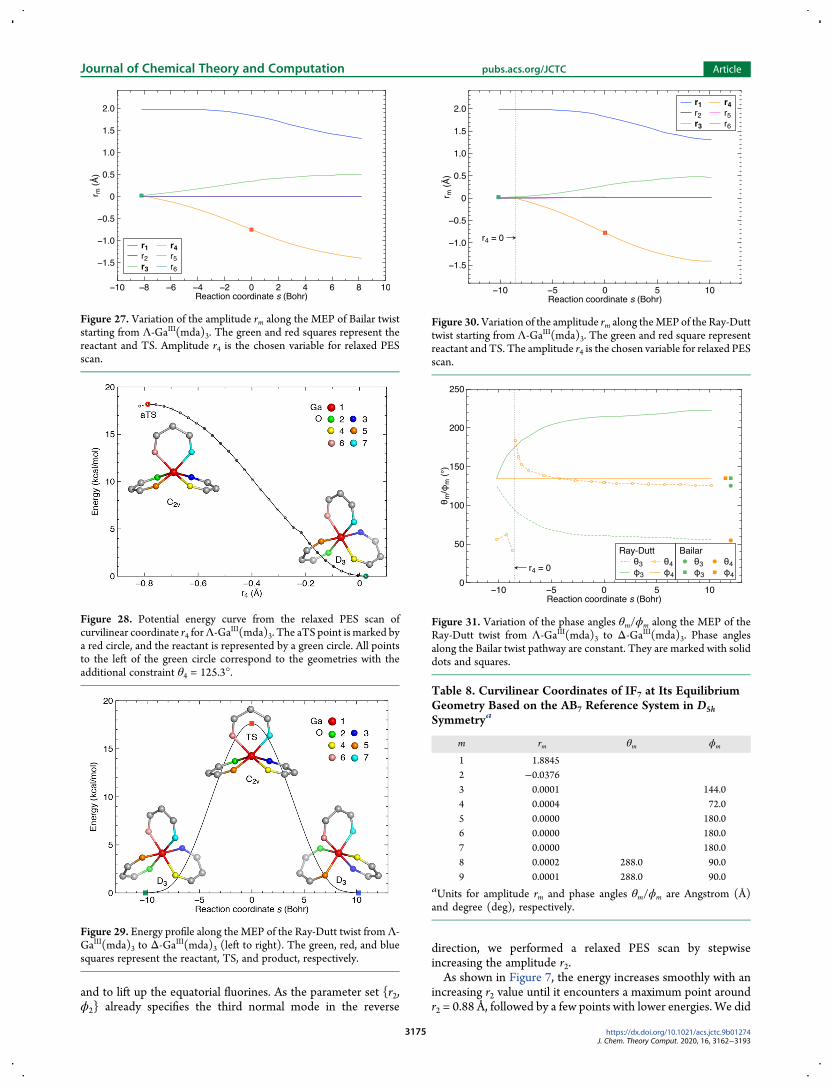
and to lift up the equatorial fluorines As the parameter set r2ϕ2 already specifies the third normal mode in the reverse
direction we performed a relaxed PES scan by stepwiseincreasing the amplitude r2As shown in Figure 7 the energy increases smoothly with an
increasing r2 value until it encounters a maximum point aroundr2 = 088 Aring followed by a few points with lower energies We did
Figure 27 Variation of the amplitude rm along the MEP of Bailar twiststarting from Λ-GaIII(mda)3 The green and red squares represent thereactant and TS Amplitude r4 is the chosen variable for relaxed PESscan
Figure 28 Potential energy curve from the relaxed PES scan ofcurvilinear coordinate r4 forΛ-GaIII(mda)3 The aTS point is marked bya red circle and the reactant is represented by a green circle All pointsto the left of the green circle correspond to the geometries with theadditional constraint θ4 = 1253deg
Figure 29 Energy profile along the MEP of the Ray-Dutt twist fromΛ-GaIII(mda)3 to Δ-GaIII(mda)3 (left to right) The green red and bluesquares represent the reactant TS and product respectively
Figure 30Variation of the amplitude rm along theMEP of the Ray-Dutttwist starting from Λ-GaIII(mda)3 The green and red square representreactant and TS The amplitude r4 is the chosen variable for relaxed PESscan
Figure 31 Variation of the phase angles θmϕm along the MEP of theRay-Dutt twist from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 Phase anglesalong the Bailar twist pathway are constant They are marked with soliddots and squares
Table 8 Curvilinear Coordinates of IF7 at Its EquilibriumGeometry Based on the AB7 Reference System in D5hSymmetrya
m rm θm ϕm
1 188452 minus003763 00001 14404 00004 7205 00000 18006 00000 18007 00000 18008 00002 2880 9009 00001 2880 900
aUnits for amplitude rm and phase angles θmϕm are Angstrom (Aring)and degree (deg) respectively
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3175
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
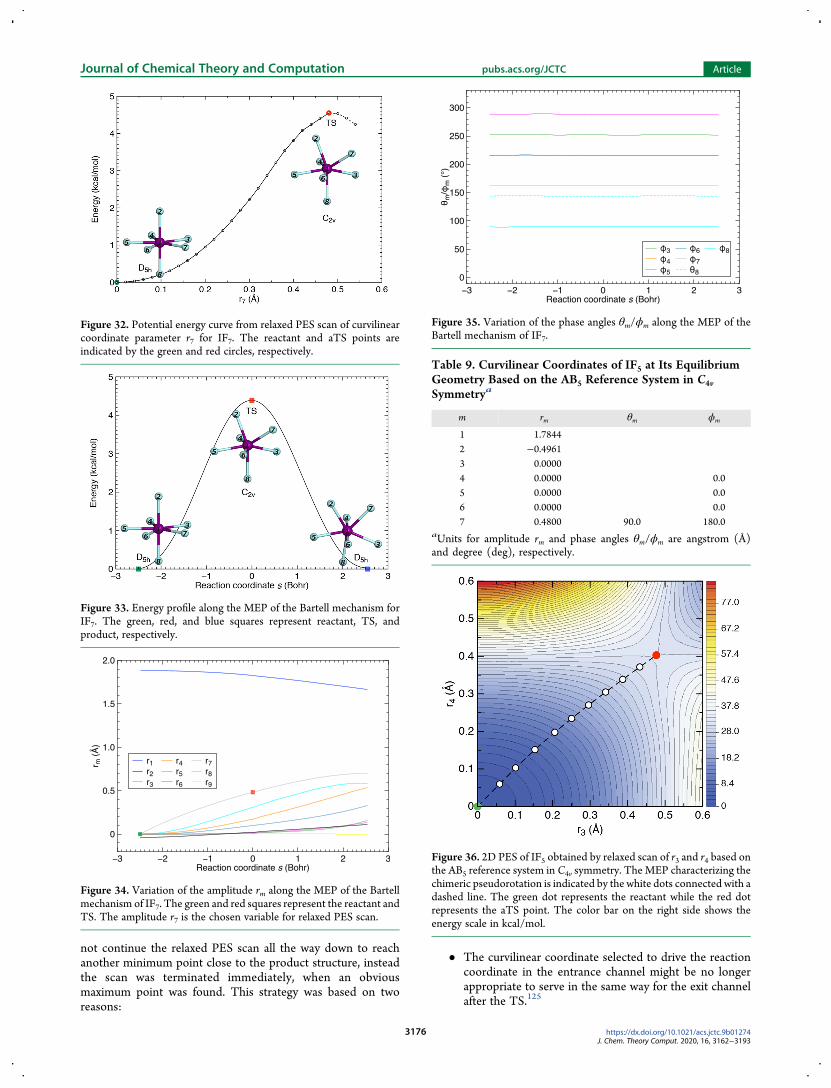
not continue the relaxed PES scan all the way down to reachanother minimum point close to the product structure insteadthe scan was terminated immediately when an obviousmaximum point was found This strategy was based on tworeasons
bull The curvilinear coordinate selected to drive the reactioncoordinate in the entrance channel might be no longerappropriate to serve in the same way for the exit channelafter the TS125
Figure 32 Potential energy curve from relaxed PES scan of curvilinearcoordinate parameter r7 for IF7 The reactant and aTS points areindicated by the green and red circles respectively
Figure 33 Energy profile along the MEP of the Bartell mechanism forIF7 The green red and blue squares represent reactant TS andproduct respectively
Figure 34 Variation of the amplitude rm along the MEP of the Bartellmechanism of IF7 The green and red squares represent the reactant andTS The amplitude r7 is the chosen variable for relaxed PES scan
Figure 35 Variation of the phase angles θmϕm along the MEP of theBartell mechanism of IF7
Table 9 Curvilinear Coordinates of IF5 at Its EquilibriumGeometry Based on the AB5 Reference System in C4vSymmetrya
m rm θm ϕm
1 178442 minus049613 000004 00000 005 00000 006 00000 007 04800 900 1800
aUnits for amplitude rm and phase angles θmϕm are angstrom (Aring)and degree (deg) respectively
Figure 36 2D PES of IF5 obtained by relaxed scan of r3 and r4 based onthe AB5 reference system in C4v symmetry TheMEP characterizing thechimeric pseudorotation is indicated by the white dots connected with adashed line The green dot represents the reactant while the red dotrepresents the aTS point The color bar on the right side shows theenergy scale in kcalmol
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3176
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
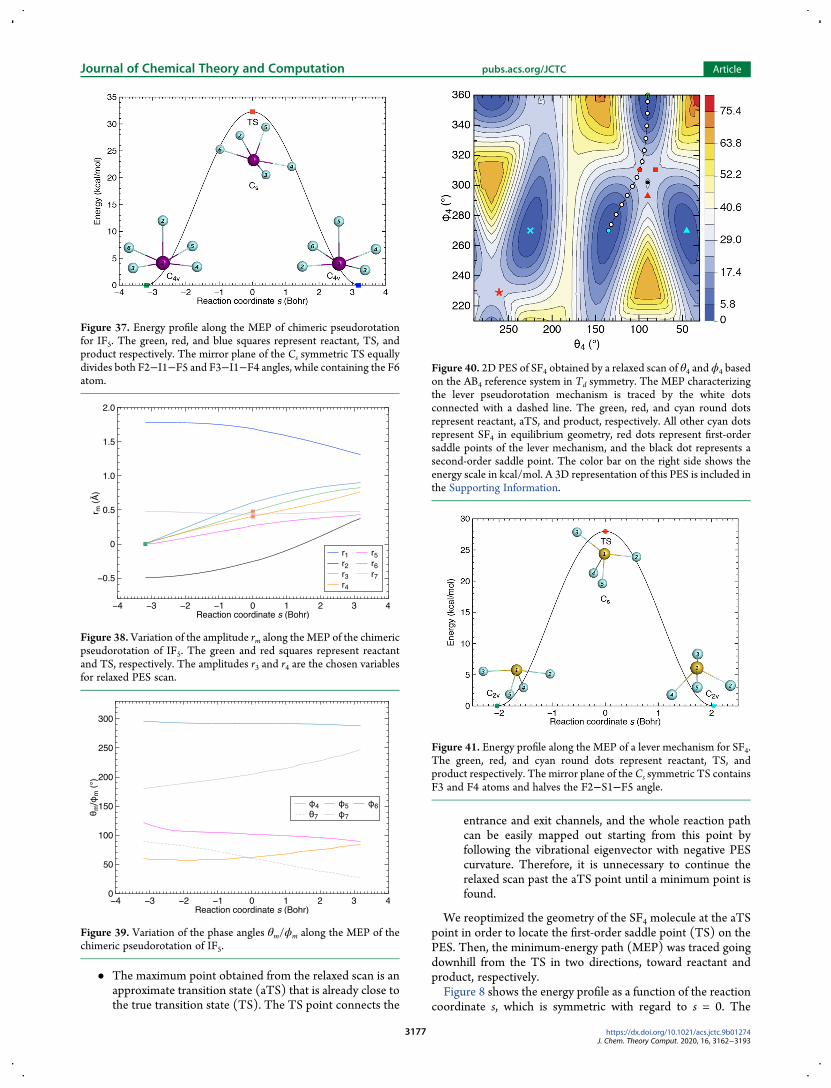
bull The maximum point obtained from the relaxed scan is anapproximate transition state (aTS) that is already close tothe true transition state (TS) The TS point connects the
entrance and exit channels and the whole reaction pathcan be easily mapped out starting from this point byfollowing the vibrational eigenvector with negative PEScurvature Therefore it is unnecessary to continue therelaxed scan past the aTS point until a minimum point isfound
We reoptimized the geometry of the SF4 molecule at the aTSpoint in order to locate the first-order saddle point (TS) on thePES Then the minimum-energy path (MEP) was traced goingdownhill from the TS in two directions toward reactant andproduct respectivelyFigure 8 shows the energy profile as a function of the reaction
coordinate s which is symmetric with regard to s = 0 The
Figure 37 Energy profile along the MEP of chimeric pseudorotationfor IF5 The green red and blue squares represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS equallydivides both F2minusI1minusF5 and F3minusI1minusF4 angles while containing the F6atom
Figure 38 Variation of the amplitude rm along theMEP of the chimericpseudorotation of IF5 The green and red squares represent reactantand TS respectively The amplitudes r3 and r4 are the chosen variablesfor relaxed PES scan
Figure 39 Variation of the phase angles θmϕm along the MEP of thechimeric pseudorotation of IF5
Figure 40 2D PES of SF4 obtained by a relaxed scan of θ4 and ϕ4 basedon the AB4 reference system in Td symmetry The MEP characterizingthe lever pseudorotation mechanism is traced by the white dotsconnected with a dashed line The green red and cyan round dotsrepresent reactant aTS and product respectively All other cyan dotsrepresent SF4 in equilibrium geometry red dots represent first-ordersaddle points of the lever mechanism and the black dot represents asecond-order saddle point The color bar on the right side shows theenergy scale in kcalmol A 3D representation of this PES is included inthe Supporting Information
Figure 41 Energy profile along the MEP of a lever mechanism for SF4The green red and cyan round dots represent reactant TS andproduct respectively The mirror plane of the Cs symmetric TS containsF3 and F4 atoms and halves the F2minusS1minusF5 angle
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3177
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
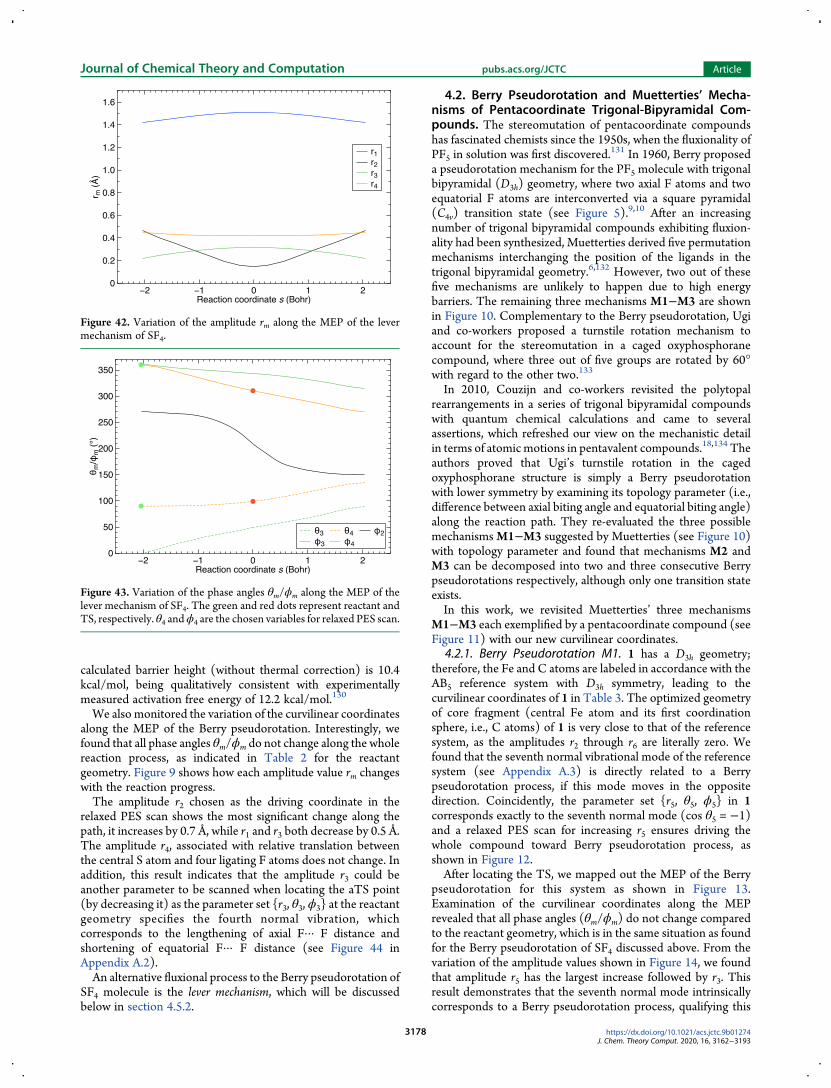
calculated barrier height (without thermal correction) is 104kcalmol being qualitatively consistent with experimentallymeasured activation free energy of 122 kcalmol130
We also monitored the variation of the curvilinear coordinatesalong the MEP of the Berry pseudorotation Interestingly wefound that all phase angles θmϕm do not change along the wholereaction process as indicated in Table 2 for the reactantgeometry Figure 9 shows how each amplitude value rm changeswith the reaction progressThe amplitude r2 chosen as the driving coordinate in the
relaxed PES scan shows the most significant change along thepath it increases by 07 Aring while r1 and r3 both decrease by 05 AringThe amplitude r4 associated with relative translation betweenthe central S atom and four ligating F atoms does not change Inaddition this result indicates that the amplitude r3 could beanother parameter to be scanned when locating the aTS point(by decreasing it) as the parameter set r3 θ3 ϕ3 at the reactantgeometry specifies the fourth normal vibration whichcorresponds to the lengthening of axial Fmiddotmiddotmiddot F distance andshortening of equatorial Fmiddotmiddotmiddot F distance (see Figure 44 inAppendix A2)An alternative fluxional process to the Berry pseudorotation of
SF4 molecule is the lever mechanism which will be discussedbelow in section 452
42 Berry Pseudorotation and Muettertiesrsquo Mecha-nisms of Pentacoordinate Trigonal-Bipyramidal Com-pounds The stereomutation of pentacoordinate compoundshas fascinated chemists since the 1950s when the fluxionality ofPF5 in solution was first discovered
131 In 1960 Berry proposeda pseudorotation mechanism for the PF5 molecule with trigonalbipyramidal (D3h) geometry where two axial F atoms and twoequatorial F atoms are interconverted via a square pyramidal(C4v) transition state (see Figure 5)910 After an increasingnumber of trigonal bipyramidal compounds exhibiting fluxion-ality had been synthesized Muetterties derived five permutationmechanisms interchanging the position of the ligands in thetrigonal bipyramidal geometry6132 However two out of thesefive mechanisms are unlikely to happen due to high energybarriers The remaining three mechanisms M1minusM3 are shownin Figure 10 Complementary to the Berry pseudorotation Ugiand co-workers proposed a turnstile rotation mechanism toaccount for the stereomutation in a caged oxyphosphoranecompound where three out of five groups are rotated by 60degwith regard to the other two133
In 2010 Couzijn and co-workers revisited the polytopalrearrangements in a series of trigonal bipyramidal compoundswith quantum chemical calculations and came to severalassertions which refreshed our view on the mechanistic detailin terms of atomicmotions in pentavalent compounds18134 Theauthors proved that Ugirsquos turnstile rotation in the cagedoxyphosphorane structure is simply a Berry pseudorotationwith lower symmetry by examining its topology parameter (iedifference between axial biting angle and equatorial biting angle)along the reaction path They re-evaluated the three possiblemechanismsM1minusM3 suggested by Muetterties (see Figure 10)with topology parameter and found that mechanisms M2 andM3 can be decomposed into two and three consecutive Berrypseudorotations respectively although only one transition stateexistsIn this work we revisited Muettertiesrsquo three mechanisms
M1minusM3 each exemplified by a pentacoordinate compound (seeFigure 11) with our new curvilinear coordinates
421 Berry Pseudorotation M1 1 has a D3h geometrytherefore the Fe and C atoms are labeled in accordance with theAB5 reference system with D3h symmetry leading to thecurvilinear coordinates of 1 in Table 3 The optimized geometryof core fragment (central Fe atom and its first coordinationsphere ie C atoms) of 1 is very close to that of the referencesystem as the amplitudes r2 through r6 are literally zero Wefound that the seventh normal vibrational mode of the referencesystem (see Appendix A3) is directly related to a Berrypseudorotation process if this mode moves in the oppositedirection Coincidently the parameter set r5 θ5 ϕ5 in 1corresponds exactly to the seventh normal mode (cos θ5 = minus1)and a relaxed PES scan for increasing r5 ensures driving thewhole compound toward Berry pseudorotation process asshown in Figure 12After locating the TS we mapped out the MEP of the Berry
pseudorotation for this system as shown in Figure 13Examination of the curvilinear coordinates along the MEPrevealed that all phase angles (θmϕm) do not change comparedto the reactant geometry which is in the same situation as foundfor the Berry pseudorotation of SF4 discussed above From thevariation of the amplitude values shown in Figure 14 we foundthat amplitude r5 has the largest increase followed by r3 Thisresult demonstrates that the seventh normal mode intrinsicallycorresponds to a Berry pseudorotation process qualifying this
Figure 42 Variation of the amplitude rm along the MEP of the levermechanism of SF4
Figure 43 Variation of the phase angles θmϕm along the MEP of thelever mechanism of SF4 The green and red dots represent reactant andTS respectively θ4 andϕ4 are the chosen variables for relaxed PES scan
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3178
mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

mode as suitable parameter for a relaxed PES scan The reasonwhy r3 has a significant change is because the third normal modein the opposite direction (cos ϕ3 = minus1) corresponds to theopening of the equatorial CminusFeminusC bite angle (see Figure 45 inAppendix A3) which is also observed in the Berrypseudorotation The changes in the amplitude values of r2 r4and r6 are only marginal422 Muetterties Mechanism M2 According to the work of
Couzijn Lammertsma and co-workers18 the seeminglycomplicated 3-fold cyclic permutation mechanism M2 consistsof two consecutive Berry pseudorotation steps Following thisidea we first identified in 2 the pivot atom which does notparticipate the Berry pseudorotation involving the other fourligating atoms As shown in Figure 15 we chose one equatorialcarbon atom of a biphenyl-22prime-diyl group as the pivot atom (no3) and the other atoms were labeled according to the referencesystem Then the geometry of the core fragment in 2 is expressedin curvilinear coordinates shown in Table 4As revealed from the previous example the seventh normal
mode of the AB5 reference system with D3h symmetry is closelyrelated to a Berry pseudorotation The curvilinear coordinates
can ideally describe the Berry mechanism if the phase angle θ5 isexpected to be close to 180degwhile r5 is positiveWe found that inthe reactant geometry of 2 (see Table 4) the amplitude r5 ispositive cos θ5 = 075 reveals the dominance of the seventhnormal vibration over the eighth (sin θ5 sin ϕ5 = 010) and ninth(sin θ5 cos ϕ5 = 065) vibrations because 075 is closer to 1However the phase angle θ5 specifies a reverse direction towardthe desired Berry pseudorotation path A simple solution tomitigate this issue is to push the amplitude r5 into the negativedirection Therefore we carried out a relaxed PES scan for 2 bydecreasing the amplitude r5 as shown in Figure 15 and the aTSpoint pertaining to mechanism M2 was successfully locatedAfter the refinement of the TS point the complete reaction
path shown in Figure 16 was mapped out Figures 17 and 18show how the curvilinear coordinates (ie amplitudes and phaseangles) change along the MEPAmplitude r5 shows the largest variation with a steady decline
along the whole path r3 starts to increase in the middle of theentrance channel while the increase of r4 becomes obvious in theexit channelWe were particularly interested in the parameter setr5 θ5 ϕ5 monitoring its contribution from the seventh normal
Figure 44 List of 9 normal vibrational modes for AB4 reference system with Td symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3179
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
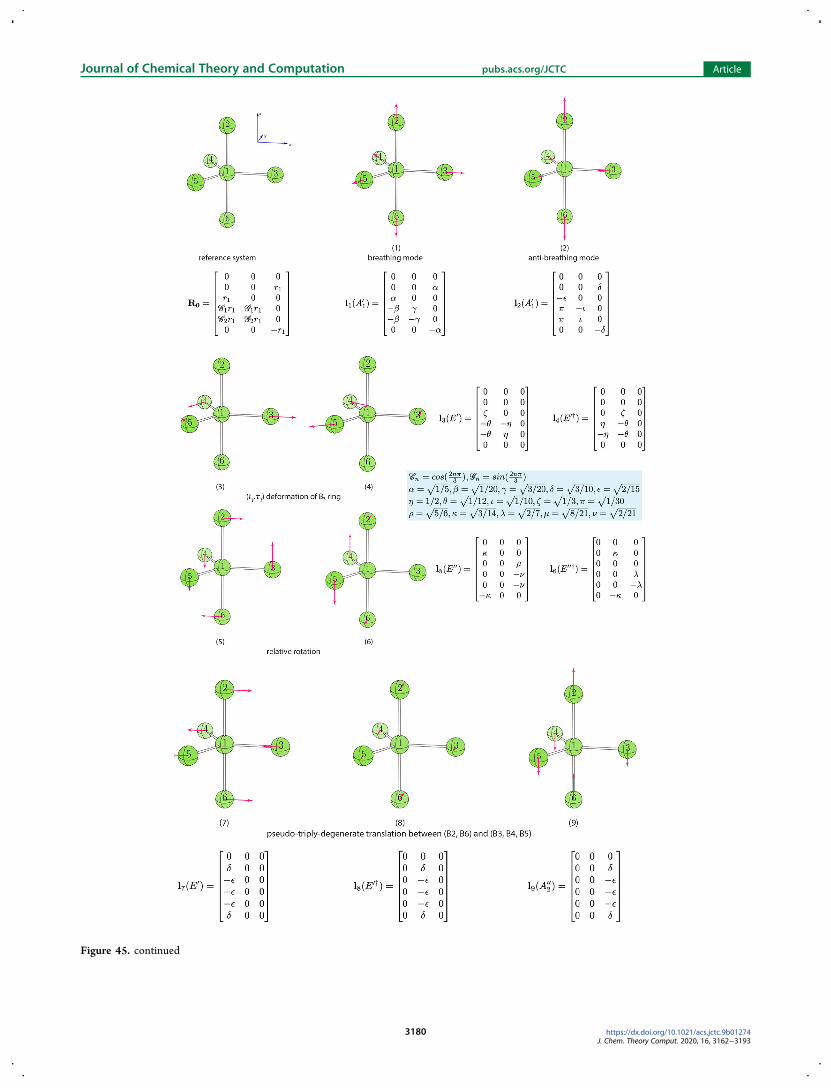
Figure 45 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3180
vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

vibration compared with that from the eighth and ninth normalmodes Figure 18 shows that the phase angle θ5 dropsimmediately toward 5deg as the reaction starts This ensures thedominance of the seventh normal mode as its cosine value of0996 is very close to 1 Although the phase angle θ5 starts toincrease in the middle of exit channel its maximum value at theend is around 30deg which still claims the dominant role of theseventh normal mode Given the significant change in r5throughout the whole path we conclude that the reactionpath of the Muetterties mechanism M2 of 2 is dictated by thecontinuous displacement of the seventh normal vibration of thecorresponding reference systemIt seems that our findings are inconsistent with Couzijnrsquos
claim18 that this mechanism consists of two consecutive Berryprocesses However one needs to be aware that their analysiswas based on a so-called topology parameter that relies on thedefinition of an equatorial pivot atom in order to describe theBerry process The well-suited topology parameter describingthe entrance (exit) channel of M2 mechanism is no longersuitable to describe the geometrical changes in the exit(entrance) channel Couzijn and co-workers thus defined forthis reaction two separate topology parameters whose valuesmeet at the TS pointDespite the simplicity introduced into the Muetterties
mechanism M2 topology parameters are by nature discontin-uous along the complete reaction path In contrast ourcurvilinear coordinate system provides a comprehensive andcontinuous description of the geometry along reaction path interms of distortions from a reference geometry423 Muetterties Mechanism M3 Although Couzijnrsquos
work18 suggests the location of an aTS point of the MuettertiesmechanismM3 for 3 via a relaxed PES scan following the Berryprocess led by the seventh normal vibrational mode as in the lastexample the vibration library data (see Appendix A3) suggestthat the eighth normal mode is the most important motiondescribing this mechanism One also has to consider that theseventh and eighth vibrations form a doubly degenerate pairTherefore we first reordered the atoms in the core fragment of 3as shown in Figure 19 to ensure that the bending of O2minusP1minusO6angle in the minusy direction could optimally correspond to the
target mechanism The geometry of the core fragment expressedin curvilinear coordinates is collected in Table 5Compared with the AB5 reference geometry inD3h symmetry
the relative contributions of the seventh eighth and ninthnormal modes to the geometry of 3 are determined by the phaseangles θ5 and ϕ5 as this set of pseudotriply degenerate normalmodes is controlled by the parameter set r5 θ5 ϕ5Noteworthy is that the contribution from the seventh vibration(cos θ5 = 087) to the reactant geometry is still dominating andthe eighth vibration (sin θ5 sin ϕ5 = minus048) is the seconddominant vibration Current phase angles for the eighthvibration correctly specify the desired motion responsible forthe reaction mechanism M3 The relaxed PES scan was carriedout by gradually increasing the amplitude r5As shown in Figure 19 we located a maximum point at r5 =
102 Aring followed by a sudden decrease in energy leading to a cuspHowever the subsequent refinement showed that the geometryof this maximum point is already very close to the real TS pointfor mechanismM3 which means the approximate TS point wassuccessfully identified through relaxed PES scan of r5 On thebasis of the refined TS point a smooth MEP was mapped out asshown in Figure 20 The corresponding variation of thecurvilinear coordinates along the MEP is shown in Figures 21and 22The amplitude r5 shows themost significant change before the
TS by increasing from 01 to 12 Aring In the exit channel changesof all amplitude values are only marginal especially after s = 50bohr As amplitude r5 is the major driving coordinate inMuetterties mechanismM3 in the entrance channel monitoringof the phase angles θ5 and ϕ5 is of great importance for theunderstanding of this mechanism θ5 gradually increases from30deg to 60deg at the TS and ϕ5 increases from 255deg to 290deg ie theeighth normal mode (sin θ5 sin ϕ5 =minus048rarrminus081) takes overthe role as the dominant vibration after s = minus125 bohrcompared to the seventh normal mode (cos θ5 = 087rarr 050)The contribution from the ninth vibration remains low (sin θ5cos ϕ5 = minus013 rarr minus030)Amplitude r4 shows also a significant change in the entrance
channel by increasing from 00 to 05 Aring The associated phaseangle ϕ4 increases from 68deg to 130deg at the TS point indicatingthe prevailing contribution from the sixth normal mode over the
Figure 45 List of 12 normal vibrational modes for AB5 reference systemwithD3h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3181
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
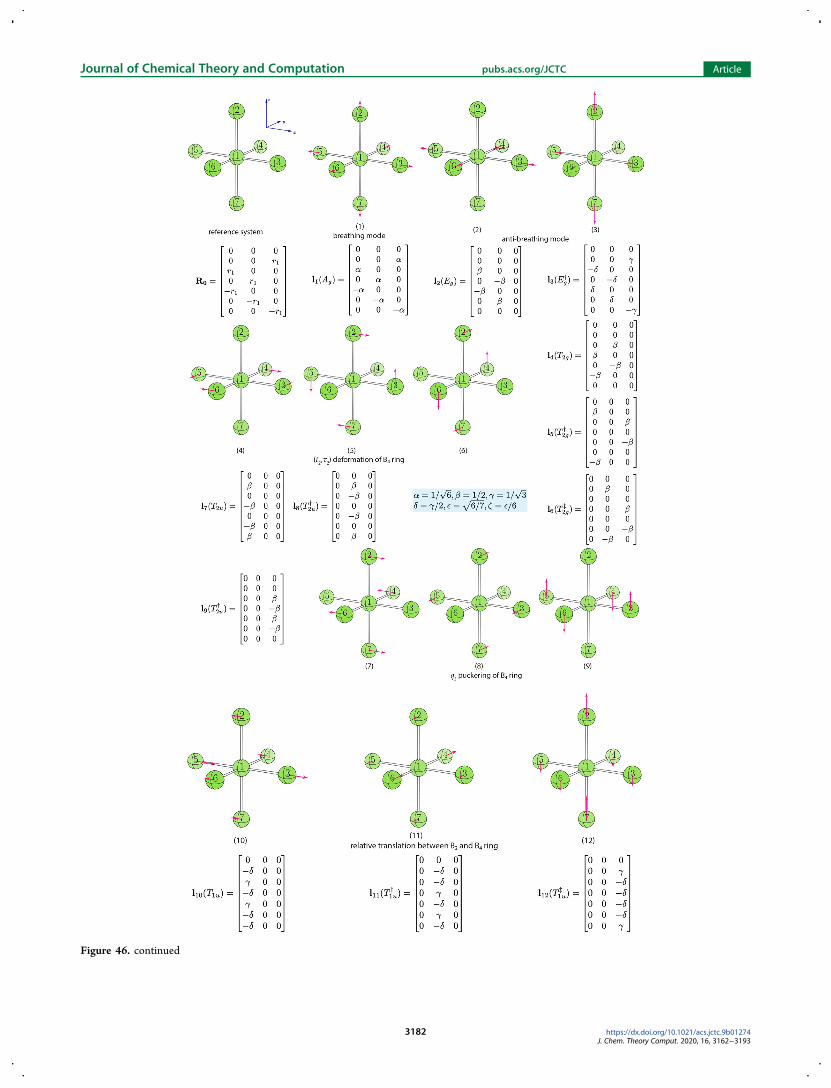
Figure 46 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3182
fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

fifth While the eighth normal mode brings the O2minusP1minusO6angle into the minusy direction the sixth normal mode pulls the O6atom into the minusy direction but the O2 atom is pushed to +ydirection The combined effects of these two modes result in theO6 atom being lifted up in the minusy direction while the motionsfor O2 atom are canceled out leading to the square pyramidalTSThe above analysis on mechanismM3 is based on the present
atomic ordering for the core fragment shown in Figure 19 Wealso could have chosen another more straightforward set ofatomic ordering (ie pick the yellow C atom as the pivot) inorder to utilize Couzijnrsquos conclusion18 that mechanism M3consists of three consecutive Berry processes expecting adominating contribution from the seventh normal modethroughout the whole path Important to note is that accordingto our findings the initial assignment of reference to real system(ie ordering of atoms within the core fragment) is not strictlylimited to only one possibility This allows a certain flexibilitywhich makes our curvilinear coordinate system robust43 Bailar and RayminusDutt Twists of Tris-chelate Metal
Complexes Tris-chelate metal complexes containing threechelating ligands typically adopt an octahedral (hexacoordinate)geometry in the center and they can undergo nondissociativeligand permutation processes (Figure 23) altering their helicalchirality (ie from Δ to Λ form and vice versa)29138 Two well-known mechanisms for racemization of chiral tris-chelates are(1) the Bailar twist139 a trigonal twist around the realC3 axis of ametal tris-chelate and (2) the Ray-Dutt twist140 a rhombic twistaround a pseudo-C3 axis The different pathways describingthese two conformational changes originate in the differentrotation axes leading to D3h and C2v TSs for the Bailar and theRayminusDutt twists respectively Twists converting an octahedralstructure to a trigonal prismatic one at the TS are oftenassociated with spin-state changes of the central metal atomenabled by spinminusorbit coupling between different spinstates141142 However in this work we focus on the aspect ofgeometrical changes during the twisting motionIn order to derive the appropriate curvilinear coordinates
describing the two twist mechanisms we first analyzed 15normal modes for the AB6 reference system with Oh symmetry(see Figure 47 in Appendix A4) to search for one vibration thatcan best describe the twist motion about the C3 axis in anoctahedral geometry However we could not identify a single
vibration serving for this purpose The only solution to obtainsuch a twist vibration turned out to equally mix the seventheighth and ninth vibrations which are triply degenerate normalmodes describing q2 puckering of four ligating atoms in a planewithin the AB6 reference system The phase angles of parameterset r4 θ4 ϕ4 describing the twisting motions have thefollowing relationship
θ θ ϕ θ ϕ| | = | | = | |cos sin sin sin cosm m m m m (22)
There are in total eight sets of solutions to eq 22 in thespherical coordinate system Four of them are listed in Table 6The other four solutions are equivalent if the amplitude r4 ismultiplied by minus1To illustrate how our curvilinear coordinates can be applied to
study Bailar and Ray-Dutt twists we selected a gallium(III)complex coordinated with three malondialdehyde ligands (seeFigure 24) as an example which was taken from a DFT study byRzepa and Cass143 We pick this tris-chelate complex with GaIII
as the central atom instead of other transition metals (eg ScIII
and TiIII) to avoid the complication from possible crossing of thePESs for two different spin states during the twist process43
These spin-crossings may be the reason why Rzepa and Cass143
were unable the locate the TS for Bailar and Ray-Dutt twists ofCoIII(mda)3 on the singlet PES described with DFTThe starting geometry of the core fragment of Λ-GaIII(mda)3
expressed in curvilinear coordinates is shown in Table 7431 Bailar Twist For Λ-GaIII(mda)3 the Bailar twist
corresponds to the second solution (ie θ4 = 547degϕ4 = 1350deg)of eq 22 see Table 6 However the twist direction has to beanticlockwise to initiate this conformational change From thedata in Table 7 we learn that the curvilinear coordinates θ4 andϕ4 of the reactant geometry are identical with those of thesecond solution ie we can search for the aTS point of theBailar twist simply via a relaxed PES scan for decreasingamplitude r4 as shown in Figure 25The MEP starting from the refined TS structure shown in
Figure 26 shows the conformational changes from Λ-GaIII(mda)3 to Δ-GaIII(mda)3 via a Bailar twistConcerning the geometrical changes in terms of curvilinear
coordinates for the above Bailar twist process one interestingobservation is that the values of all phase angles (θmϕm) stayconstant during the whole reaction path as in the reactant
Figure 46 List of 15 normal vibrational modes for AB6 reference system withOh symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3183
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
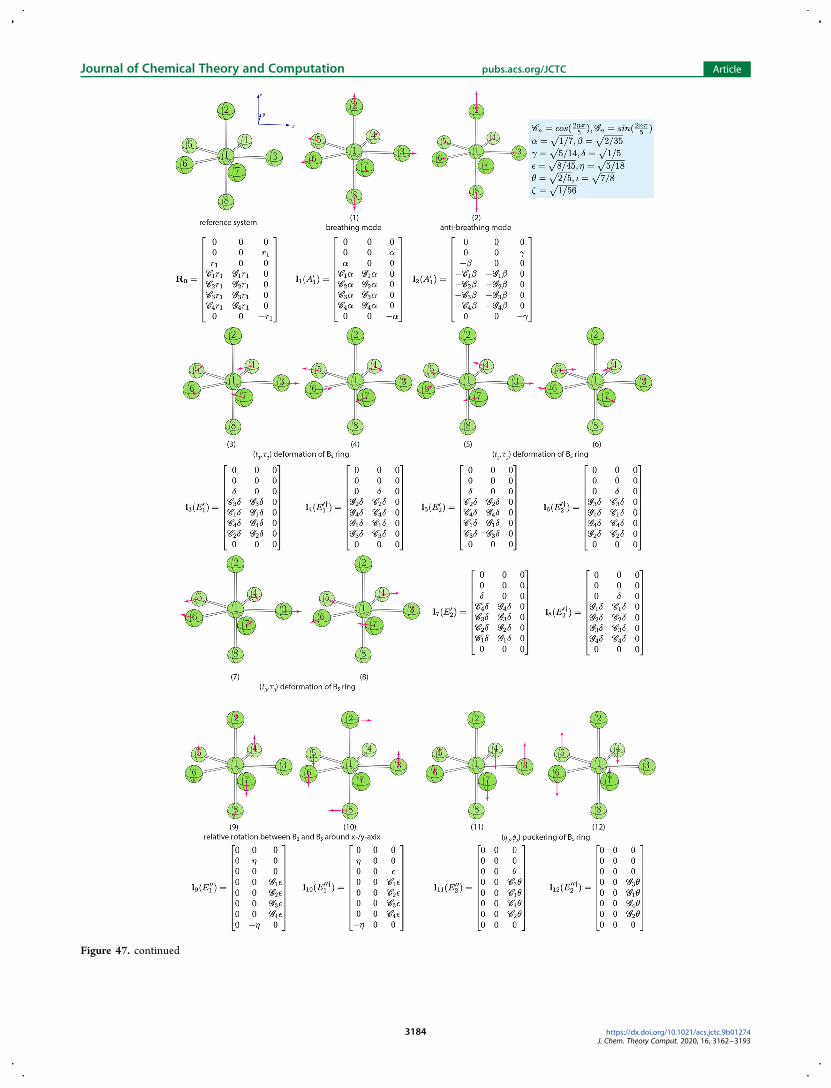
Figure 47 continued
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3184
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193
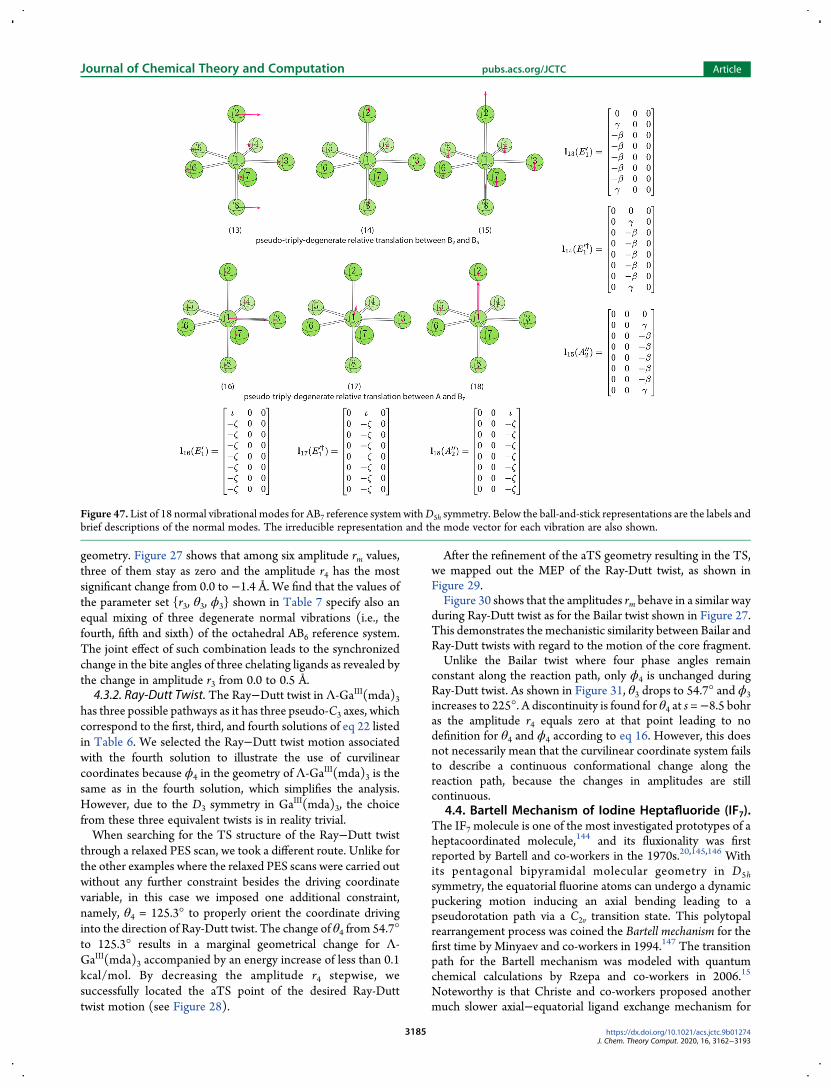
geometry Figure 27 shows that among six amplitude rm valuesthree of them stay as zero and the amplitude r4 has the mostsignificant change from 00 to minus14 Aring We find that the values ofthe parameter set r3 θ3 ϕ3 shown in Table 7 specify also anequal mixing of three degenerate normal vibrations (ie thefourth fifth and sixth) of the octahedral AB6 reference systemThe joint effect of such combination leads to the synchronizedchange in the bite angles of three chelating ligands as revealed bythe change in amplitude r3 from 00 to 05 Aring432 Ray-Dutt Twist The RayminusDutt twist inΛ-GaIII(mda)3
has three possible pathways as it has three pseudo-C3 axes whichcorrespond to the first third and fourth solutions of eq 22 listedin Table 6 We selected the RayminusDutt twist motion associatedwith the fourth solution to illustrate the use of curvilinearcoordinates because ϕ4 in the geometry of Λ-GaIII(mda)3 is thesame as in the fourth solution which simplifies the analysisHowever due to the D3 symmetry in GaIII(mda)3 the choicefrom these three equivalent twists is in reality trivialWhen searching for the TS structure of the RayminusDutt twist
through a relaxed PES scan we took a different route Unlike forthe other examples where the relaxed PES scans were carried outwithout any further constraint besides the driving coordinatevariable in this case we imposed one additional constraintnamely θ4 = 1253deg to properly orient the coordinate drivinginto the direction of Ray-Dutt twist The change of θ4 from 547degto 1253deg results in a marginal geometrical change for Λ-GaIII(mda)3 accompanied by an energy increase of less than 01kcalmol By decreasing the amplitude r4 stepwise wesuccessfully located the aTS point of the desired Ray-Dutttwist motion (see Figure 28)
After the refinement of the aTS geometry resulting in the TSwe mapped out the MEP of the Ray-Dutt twist as shown inFigure 29Figure 30 shows that the amplitudes rm behave in a similar way
during Ray-Dutt twist as for the Bailar twist shown in Figure 27This demonstrates themechanistic similarity between Bailar andRay-Dutt twists with regard to the motion of the core fragmentUnlike the Bailar twist where four phase angles remain
constant along the reaction path only ϕ4 is unchanged duringRay-Dutt twist As shown in Figure 31 θ3 drops to 547deg and ϕ3increases to 225deg A discontinuity is found for θ4 at s =minus85 bohras the amplitude r4 equals zero at that point leading to nodefinition for θ4 and ϕ4 according to eq 16 However this doesnot necessarily mean that the curvilinear coordinate system failsto describe a continuous conformational change along thereaction path because the changes in amplitudes are stillcontinuous
44 Bartell Mechanism of Iodine Heptafluoride (IF7)The IF7 molecule is one of the most investigated prototypes of aheptacoordinated molecule144 and its fluxionality was firstreported by Bartell and co-workers in the 1970s20145146 Withits pentagonal bipyramidal molecular geometry in D5hsymmetry the equatorial fluorine atoms can undergo a dynamicpuckering motion inducing an axial bending leading to apseudorotation path via a C2v transition state This polytopalrearrangement process was coined the Bartell mechanism for thefirst time by Minyaev and co-workers in 1994147 The transitionpath for the Bartell mechanism was modeled with quantumchemical calculations by Rzepa and co-workers in 200615
Noteworthy is that Christe and co-workers proposed anothermuch slower axialminusequatorial ligand exchange mechanism for
Figure 47 List of 18 normal vibrational modes for AB7 reference systemwithD5h symmetry Below the ball-and-stick representations are the labels andbrief descriptions of the normal modes The irreducible representation and the mode vector for each vibration are also shown
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3185
IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

IF7 based on NMR evidence in 199314 However this processseemed to be too complicated and was never confirmedafterwardIn this work we focused on the Bartell mechanism being
described with our curvilinear coordinates The optimizedgeometry of IF7 molecule is described in Table 8 where threephase anlges ϕm associated with rm = 0 was intentionally set to180degAs described in Bartellrsquos original work this pseudorotation
path is dominated by E2Prime puckering20 Analyzing the vibrationlibrary data of the reference system (see Appendix A5) we foundthat the parameter set r7ϕ7 corresponds to doubly degenerate(q2 ϕ2) ring puckering vibrations By carrying out a relaxed PESscan with the increasing amplitude r7 we successfully located theaTS point for the Bartell mechanism as shown in Figure 32After refinement of the TS point the complete MEP was
mapped out as shown in Figure 33 revealing the Bartellmechanism The changes in amplitudes rm and phase angles θmϕm of IF7 along the MEP are shown in Figures 34 and 35Along the MEP amplitudes r2 through r8 are increased to
different extent The amplitude r9 adopts the zero valuethroughout the whole path and its associated phase angles arethus not shown Similar to several other dynamic fluxionalprocesses discussed above the phase angles also adopt constantvalues along the whole reaction path Amplitude r7 has thelargest increase from 00 to 07 Aring followed by the amplitude r8from 00 to 06 Aring Phase angle ϕ7 takes the value of 162degindicating a dominant contribution from the 11th normal mode(cos ϕ7 = minus095) compared with the 12th normal mode (sin ϕ7= 031) For the parameter set r8 θ8 ϕ8 θ8 is 145deg and ϕ8equals 90deg which specifies the mixing of the 13th and 14thvibrational modes without any contribution from the 15thnormal mode leading to an axial bending motion In summarythe conformational changes described in terms of curvilinearcoordinates are consistent with Bartellrsquos original description ofdistortion from D5h symmetry for IF7
20
45 Two Technically Complicated Fluxional Mecha-nisms In the above examples illustrating the usefulness of ournewly developed curvilinear coordinate system for thedescription of dynamic fluxional processes we have demon-strated that the search of the aTS point for the polytopalrearrangement processes can be done in a systematic way via arelaxed PES scan starting from equilibrium geometry using asingle amplitude rm from the complete set of curvilinearcoordinates as a variableIn the following we discuss two examples for which the single-
parameter PES scan protocol fails to locate the aTS point and wepresent a solution for successfully finding the aTS point in thesecases Rather than undermining the value of the curvilinearcoordinates developed in this work these two examples reflectrobustness and flexibility of our approach451 Chimeric Pseudorotation of Iodine Pentafluoride
(IF5) Similar to IF7 the iodine pentafluoride (IF5) is also ahypervalent halogen compound Its square pyramidal (C4v)geometry was first reported by Lord and co-workers in 1950148
Several years later in 1957 Muetterties and Phillips carried out19F NMR studies on IF5 and they observed IF5 being stable atroom temperature However the absence of FminusF couplingpattern in NMR spectra at elevated temperatures indicatedapical-basal fluorine exchange121 Therefore they concludedthat such apical-basal exchange has a relatively high energybarrier over 25 kcalmol and they hypothesized that this processwould proceed via a dimer structure During the following years
little progress was made with regard to a more detailedunderstanding of the fluxional mechanism of IF5 until in 2006when Rzepa and Cass suggested a nondissociative (ieintramolecular) mechanism based on quantum chemicalcalculations25 Their reaction path analysis led to the conclusionthat the fluxional mechanism of IF5 is composed of three distinctfluxional processes mixed together which they coined chimericpseudorotation Noteworthy is that the BrF5 can undergo thesame chimeric pseudorotation as IF5 via a Cs TS according toRzeparsquos calculations while the ClF5 adopts a different path via aC2v TS resembling a Berry pseudorotation which wasexemplified by PF5 SF4 and Fe(CO)5 as discussed above Inthis work we studied the chimeric pseudorotation of IF5 withcurvilinear coordinates to further explore Rzeparsquos findingsEquilibrium geometry data of IF5 are collected in Table 9In order to locate the aTS point for the chimeric
pseudorotation mechanism we performed a relaxed PES scanof a single amplitude parameter using r3 through r7 one at a timecombined with various phase angle possibilities but failed tocapture the desired aTS point After closer inspection of the IF5equilibrium geometry compared with the TS geometrycalculated by Rzepa25 we found that the geometrical changefrom reactant toward TS can be attributed to a q2 puckeringmotion of four basal fluorines combined with a (t1 τ1)deformation of the same four-membered ring According tothe vibration library for the AB5 reference system in C4vsymmetry (see Figure 3) the displacement of q2 puckeringand (t1 τ1) deformation for the B4 ring is controlled by r3 and r4ϕ4 respectively Therefore we performed next a 2D relaxedPES scan of r3 and r4 as shown in Figure 36 and the aTS pointcould be correctly identifiedAfter the refinement of the TS point the completeMEP of the
chimeric pseudorotation for IF5 was mapped out as shown inFigure 37The change in the amplitude values along the MEP shown in
Figure 38 is rather simple as r3 through r6 all increase from 00 Aringin the very beginning Noteworthy is that r3 and r4 stay close toeach other and progress in a synchronized fashion The phaseangle change in Figure 39 shows ϕ4 gradually increases from600deg to 850deg indicating more contribution from the fifthnormal mode than the fourthFor this showcase example one might suggest combining the
third normal mode from the vibrational library with the fourthand fifth modes forming a pseudotriply degenerate curvilinearcoordinate parameter set as these three vibrational modes arehighly similar and in this way allowing to locate the aTS point viaa single-parameter (ie amplitude) relaxed PES scan which issimpler than 2D scan It is feasible to implement the curvilinearcoordinates in that way However the grouping of the vibrationswith different symmetries into either pairs or triads is notarbitrary We rather prefer to group vibrations depending ontheir types of motion The third vibration is the puckeringmotion of the four-membered ring of the basal F atoms whilethe fourth and fifth vibrations are related to ring deformationSeparating these two well-defined motions is physically moremeaningful
452 Lever Mechanism of Sulfur Tetrafluoride (SF4) Inaddition to the Berry pseudorotation of SF4 discussed in section41 other mechanisms were proposed to enable completescrambling of its four fluorine atoms149 The most crediblealternative to Berry pseudorotation termed lever mechanism wasfirst proposed by Minyaev and co-workers147 and further
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3186
investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

investigated by Mauksch and Schleyer with theoreticalcalculations17
We failed to locate the aTS point pertaining to the levermechanism in SF4 with curvilinear coordinates via a relaxed PESscan of either one single amplitude or two amplitudes as we didfor IF5 The correct aTS point was successfully disclosed by arelaxed PES scan of the two phase angles of the parameter set r4θ4 ϕ4 which corresponds to relative translations of the centralatomAs shown in the right half of the energy contour plot in Figure
40 there are three reaction valleys interconnected with eachother by three first-order saddle points The green round dot inthe upper reaction valley corresponds to the reactant geometryfor SF4 described in Table 2 In the middle of three first-ordersaddle points there is a second-order saddle point correspond-ing to a C3v geometry17 with slightly higher energy (sim10 kcalmol) The left region of this contour map seems to be centrallysymmetric to the right partOur results are consistent with the qualitative energy
landscape of Mauksch and Schleyer which led them to theconclusion that three MEPs of lever mechanism encircle thesecond-order saddle point17 They named such an energysurface where three valleys meet on a hilltop having twoimaginary frequencies150151 as ldquoeffective monkey saddle pointrdquowhich has to be distinguished from an ideal ldquomonkey saddlerdquowhere doubly degenerate zero curvatures are required152153 Inthis sense we have now consolidated the work of Mauksch andSchleyer17 for the first time by presenting a detailed descriptionof the unusual PES of the SF4 lever mechanism with the help ofour new curvilinear coordinatesOn the basis of a refined TS point theMEPmarked out in the
above 2D PES contour plot was mapped out in Figure 41The conformational change of SF4 expressed in curvilinear
coordinates during the lever pseudorotation is shown in Figures42 and 43The amplitude values along the MEP are symmetric with
regard to s = 0 bohr Amplitude r4 has the least change althoughits associated phase angles θ4 andϕ4 play a central role in drivingthe reaction coordinatesAmong all phase angles θ4 and ϕ4 show a moderate change
along the reaction path Throughout the whole path these twophase angles change from 90deg and 360deg toward 135deg and 270degrespectively specifying the full contribution from the ninthnormal vibration (sin θ cos ϕ = 1) for the reactant geometry andequal contributions from the seventh and eighth normal modes(cos θ = sin θ sin ϕ = minus 2 2) for the product geometry Thismeans that the reactant geometry is basically a result of theupward translation of the central sulfur atom in the +z directionWith the progress of lever mechanism the contribution of thisupward translation decays (ie central sulfur moves downward)and the sulfur atom gradually moves into the minusx and minusydirectionA similar variation is also found for θ3 and ϕ3 On the basis of
this observation we performed a relaxed PES scan of these twophase angles in order to locate the aTS point However it turnedout that the only solution leading to the correct aTS point is viarelaxed scanning of θ4 and ϕ4
5 CONCLUSIONS
We developed in this work a novel curvilinear coordinate systemdescribing polytopal rearrangements in N-coordinate com-pounds whereN can take any number from four through seven
(1) The physical foundation of this curvilinear coordinatesystem is based on the fact that a chemical reaction can beinitiated and even accelerated via the excitation of vibrationalmodes of the reaction complex68minus707475 The polytopalrearrangements in N-coordinate molecules like conformationalchanges can be considered as unimolecular reactions which areamenable to the promotion by certain vibrations In this sensethe new curvilinear coordinate system is a conceptual extensionof the CremerminusPople ring puckering coordinates47 as well as therecently proposed ring deformation coordinates54
(2) The curvilinear coordinates developed in this work arederived from the normal vibrational modes of N-coordinatereference systems ABn for which 3Mminus 6 curvilinear coordinates(whereM =N + 1) are sufficient to describe the geometry of anyN-coordinate motif(3) A reference system ABn defines a geometrical template
(ie its size is scalable) with a highly symmetric N-coordinatestructure that fulfill several preconditions In this work we havestipulated reference systems for different N-coordinatestructures ranging from tetracoordinate to heptacoordinate indifferent point groups including AB4 in Td symmetry AB5 inD3hand C4v symmetries AB6 in Oh and C5v symmetries along withAB7 in D5h and C6v symmetries These reference systems coverthe vast majority of single-center coordination compounds(versus binuclear and polynuclear complexes)(4) In a coordination compound the geometry of its N-
coordinate core fragment (ldquoreal systemrdquo) can be unambiguouslyexpressed by the nonredundant set of 3M minus 6 normal vibrationsof corresponding reference system ABn if the internal coordinatespace of the real system matches exactly the internal vibrationspace of the reference system This is realized by translating androtating the real system into a standard orientation from whichthe geometrical information is encoded by the projection ofCartesian coordinates onto the 3M minus 6 normal vibrations of thereference system Depending on the degeneracy and type ofmotion of the 3Mminus 6 normal vibrational modes some of the 3Mminus 6 projections are grouped into pairs or triads leading to threekinds of curvilinear coordinate parameter sets (i) non-degenerate rm (ii) doubly degenerate rm ϕm and (iii)triply degenerate rm θmϕm The latter two parameter sets withphase angles (θmϕm) linearly combine two and three vibrationsrespectively(5) Any complete set of internal or Cartesian coordinates of
an N-coordinate structure can be transformed into 3M minus 6curvilinear coordinates In turn the curvilinear coordinates canbe used to derive the internal or Cartesian coordinates of the N-coordinate structure(6) The new curvilinear coordinates are well-suited reaction
coordinates for describing polytopal rearrangements in twoaspects First these curvilinear coordinates can directly be usedfor 1D2D relaxed PES scan starting from an equilibriumgeometry to locate the first-order saddle point (TS) In mostcases the equilibrium geometry of the reactant is close to that ofthe corresponding reference system with many amplitudeparameters rm being almost zero (except r1) A relaxed PESscan in which one or two amplitudes rm are steadily increasedsimulates the process of the vibration(s) (associated with thescanned amplitudes) being excited to obtain pronounced atomicdisplacements Noteworthy is that like using internalcoordinates for relaxed PES scans the choice of curvilinearcoordinations as the constraint variables requires them to beclosely related to the vibrations initiating a polytopal rearrange-ment process Even in the rare case that the molecular geometry
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3187
is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

is different from its best suited reference system in terms ofsymmetry (eg SF4 with C2v symmetry versus its referencesystem with Td symmtry) a relaxed PES scan with ourcurvilinear coordinate system can reveal important mechanisticdetails Second the polytopal rearrangement pathways(especially in the entrance channel before TS) can be betterunderstood in terms of one or two leading vibrational motionsSuch a simplification has been widely used in understanding theconformational change of proteins154
(7) Nine different polytopal rearrangement processes intetracoordinate through heptacoordinate compounds werestudied including the Berry pseudorotation and the levermechanism for SF4 the chimeric pseudorotation for IF5 theBerry pseudorotation and the Muettertiesrsquo mechanisms fortrigonal bipyramidal structures Bailar and Ray-Dutt twists fortris-chelates and the Bartell mechanism for IF7 These showcaseexamples endorse the general applicability of our curvilinearcoordinates for the description of polytopal rearrangements fordifferent coordination structuresNoteworthy is that in the case of several polytopal
rearrangement mechanisms studied in this work the phaseangles (θmϕm) are unchanged along the whole pathwayincluding the Berry pseudorotations for SF4 and Fe(CO)5 theBailar twist for tris-chelates and the Bartell mechanism for IF7This finding is closely related to the observation of AvnirAlvarez and co-workers that the Berry pseudorotation inpentacoordinate compounds and the Bailar twist possessminimal distortion pathways according to their CShManalysis23
On the basis of the curvilinear coordinate frameworkintroduced in this work work in progress covers the followingdirections
bull The curvilinear coordinates will be employed to exploreall possible polytopal rearrangement mechanisms fornewly discovered and intriguing coordination structures
bull Unconfirmed polytopal mechanisms reported in theliterature will be revisited like the chimeric pseudor-otation for IF5 rediscovered and studied by Rzepa andCass25
bull More reference systems and associated curvilinearcoordinates are to be designed for heptacoordinate andoctacoordinate compounds which are less exploredcompared with lower coordinated counterparts in thehope of discovering novel polytopal rearrangementmechanisms
bull The curvilinear coordinate system can be incorporatedinto automatic workflows for high-throughput screeningfor polypotal rearrangements as well as rational design oftransition-metal complexes Related work in this directionhas been pioneered by the Kulik group155minus158
bull Collective variables based on the curvilinear coordinatesdeveloped in this work will be implemented in order tosample free energy surfaces of polytopal rearrangementswith molecular dynamics simulations159
In conclusion this work introduces a dedicated curvilinearcoordinate system for the description of polytopal rearrange-ments in coordination compounds opening a new avenue for thesystematic study and comparison of these fluxional processes
APPENDIX A
A1 Formula for Wilson B- and dB-MatrixWe have derived the formulas to calculate the Wilson B-matrixand its derivative for the curvilinear coordinate systemdeveloped in this work using finite difference method(i) Nondegenerate curvilinear coordinate rm
partpart
=ΔΔ
rx
dx
m
i
m
i (23)
partpart part
=Δ
Δ Δr
x xd
x xm
i j
m
i j
2 2
(24)
(ii) Doubly degenerate curvilinear coordinates rm ϕm
ϕ ϕpartpart
=ΔΔ
+Δ primeΔ
ikjjjjj
yzzzzz
rx
dx
dx
cos sinm
im
m
im
m
i (25)
ϕϕ ϕ
partpart
= minusΔΔ
+Δ primeΔ
ikjjjjj
yzzzzzx r
dx
dx
sin cosm
i mm
m
im
m
i (26)
ϕ ϕ
ϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
rx x
cos sinm
i jm
m
i jm
m
i j
mm
i
m
j
2 2 2
(27)
ϕϕ ϕ
ϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
sin cos
1
m
i j mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
2 2 2
(28)
(iii) Triply degenerate curvilinear coordinates rm θm ϕm
θ θ ϕ
θ ϕ
partpart
=ΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
rx
dx
dx
dx
cos sin sin
sin cos
m
im
m
im m
m
i
m mm
i (29)
θθ θ ϕ
θ ϕ
partpart
= minusΔΔ
+Δ primeΔ
+Δ PrimeΔ
ikjjjjj
yzzzzz
x rdx
dx
dx
sin cos sin
cos cos
m
i mm
m
im m
m
i
m mm
i (30)
ϕθ
ϕ ϕpartpart
=Δ primeΔ
minusΔ PrimeΔ
ikjjjjj
yzzzzzx r
dx
dxsin
cos sinm
i m mm
m
im
m
i (31)
θ θ ϕ
θ ϕθ θ
θϕ ϕ
partpart part
=Δ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ+
ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
rx x
dx x
dx x
dx x
rx x
rx x
cos sin sin
sin cos
sin
m
i jm
m
i jm m
m
i j
m mm
i jm
m
i
m
j
m mm
i
m
j
2 2 2
2
2
(32)
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3188
θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

θθ θ ϕ
θ ϕ
θ θ
θ θϕ ϕ
partpart part
= minusΔ
Δ Δ+
Δ primeΔ Δ
+Δ Prime
Δ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
dx x
rrx x
rx x
x x
sin cos sin
cos cos
1
sin cos
m
i j mm
m
i jm m
m
i j
m mm
i j
m
m
i
m
j
m
j
m
i
m mm
i
m
j
2 2 2
2
(33)
ϕθ
ϕ ϕ
ϕ ϕ
θθ ϕ θ ϕ
partpart part
=Δ prime
Δ Δminus
Δ PrimeΔ Δ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
minusΔΔ
ΔΔ
+ΔΔ
ΔΔ
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
i
kjjjjjj
y
zzzzzz
x x rd
x xd
x x
rrx x
rx x
x x x x
sincos sin
1
1tan
m
i j m mm
m
i jm
m
i j
m
m
i
m
j
m
j
m
i
m
m
i
m
j
m
j
m
i
2 2 2
(34)
A2 Td Reference System and Its VibrationsThe geometry and the vibration library for AB4 reference systemin Td symmetry are shown in Figure 44 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 10A3 D3h Reference System and Its VibrationsThe geometry and the vibration library for AB5 reference systemin D3h symmetry are shown in Figure 45 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 11A4 Oh Reference System and Its VibrationsThe geometry and the vibration library for AB6 reference systemin Oh symmetry are shown in Figure 46 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 12A5 D5h Reference System and Its VibrationsThe geometry and the vibration library for AB7 reference systemin D5h symmetry are shown in Figure 47 The definition ofcurvilinear coordinate parameters associated with this referencesystem is collected in Table 13
ASSOCIATED CONTENTsı Supporting InformationThe Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjctc9b01274
3D PES of Berry pseudorotation for PF5 molecule 3DPES of lever mechanism for SF4 molecule and referencesystem and associated normal modes for AB6 in C5vsymmetry and AB7 in C6v symmetry (PDF)
AUTHOR INFORMATIONCorresponding Author
Elfi Kraka minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United Statesorcidorg0000-0002-9658-5626 Email ekraka
gmailcom
AuthorsWenli Zou minus Institute of Modern Physics Northwest Universityand Shaanxi Key Laboratory for Theoretical Physics FrontiersXirsquoan Shaanxi 710127 P R China Department of ChemistrySouthern Methodist University Dallas Texas 75275-0314United States orcidorg0000-0002-0747-2428
Yunwen Tao minus Department of Chemistry Southern MethodistUniversity Dallas Texas 75275-0314 United States
Complete contact information is available at
Table 10 Summary of 9 Curvilinear Coordinate Parametersin 4 Sets Based on 9 Normal Vibrational Modes for theReference System AB4 in Td Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1 AB4 breathing2 3 r2 ϕ2 E doubly degenerate
vibrations4 5 6 r3 θ3 ϕ3 T2 triply degenerate vibrations7 8 9 r4 θ4 ϕ4 T2 translation of A
Table 11 Summary of 12 Curvilinear Coordinate Parametersin 6 Sets Based on 12 Normal Vibrational Modes for theReference System AB5 in D3h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB5 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 Eprime (t1 τ1) deformation of B3 ring5 6 r4 ϕ4 EPrime relative rotation within B5
7 8 9 r5 θ5 ϕ5 Eprime + A2Prime relative translation within B5
10 11 12 r6 θ6 ϕ6 Eprime + A2Prime translation of A
Table 12 Summary of 15 Curvilinear Coordinate Parametersin 6 Sets Based on 15 Normal Vibrational Modes for theReference System AB6 in Oh Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 Ag AB6 breathing2 3 r2 ϕ2 Eg antibreathing4 5 6 r3 θ3 ϕ3 T2g (t2 τ2) deformation of B4 ring7 8 9 r4 θ4 ϕ4 T2u q2 puckering of B4 ring10 11 12 r5 θ5 ϕ5 T1u relative translation within B6
13 14 15 r6 θ6 ϕ6 T1u translation of A
Table 13 Summary of 18 Curvilinear Coordinate Parametersin 9 Sets Based on 18 Normal Vibrational Modes for theReference System AB7 in D5h Symmetry
normalmode
curvilinearcoordinates symmetry description
1 r1 A1prime AB7 breathing2 r2 A1prime antibreathing3 4 r3 ϕ3 E1prime (t1 τ1) deformation of B5 ring5 6 r4 ϕ4 E2prime (t2 τ2) deformation of B5 ring7 8 r5 ϕ5 E2prime (t3 τ3) deformation of B5 ring9 10 r6 ϕ6 E1Prime relative rotation within B7
11 12 r7 ϕ7 E2Prime (q2 ϕ2) puckering of B5 ring13 14 15 r8 θ8 ϕ8 E1prime + A2Prime relative translation within B7
16 17 18 r9 θ9 ϕ9 E1prime + A2Prime translation of A
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3189
httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

httpspubsacsorg101021acsjctc9b01274
Author ContributionsparaContributed equally to this workNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThis work was financially supported by the National ScienceFoundation (Grant CHE 1464906) We thank SMU forproviding generous computational resources WZ alsoacknowledges the financial support by National Natural ScienceFoundation of China (Grant No 21673175) and the DoubleFirst-Class University Construction Project of NorthwestUniversity
DEDICATIONDedicated to Dieter Cremer (1944minus2017)
REFERENCES(1) Portius P Buhl M George M W Grevels F-W Turner J JStructure and Dynamics of Iron Pentacarbonyl Organometallics 201938 4288minus4297(2) Schilter D Trading places Nat Rev Chem 2020 4 2(3) Cahoon J F Sawyer K R Schlegel J P Harris C BDetermining Transition-State Geometries in Liquids Using 2D-IRScience 2008 319 1820minus1823(4) Lai J-C Jia X-Y Wang D-P Deng Y-B Zheng P Li C-HZuo J-L Bao Z Thermodynamically Stable Whilst Kinetically LabileCoordination Bonds Lead to Strong and Tough Self-Healing PolymersNat Commun 2019 10 1164(5) Moss G P Basic Terminology of Stereochemistry (IUPACRecommendations 1996) Pure Appl Chem 1996 68 2193minus2222(6)Muetterties E L Topological Representation of StereoisomerismI Polytopal Rearrangements J Am Chem Soc 1969 91 1636minus1643(7) Muetterties E L Storr A T Topological Analysis of PolytopalRearrangements Sufficient Conditions for Closure J Am Chem Soc1969 91 3098minus3099(8) Asatryan R Ruckenstein E Hachmann J Revisiting thePolytopal Rearrangements in Penta-Coordinate d7-MetallocomplexesModified Berry Pseudorotation Octahedral Switch and ButterflyIsomerization Chem Sci 2017 8 5512minus5525(9) Berry R S Correlation of Rates of Intramolecular TunnelingProcesses with Application to Some Group V Compounds J ChemPhys 1960 32 933minus938(10) Berry R S Time-Dependent Measurements and MolecularStructure Ozone Rev Mod Phys 1960 32 447minus454(11)Muller P Glossary of Terms used in Physical Organic Chemistry(IUPAC Recommendations 1994) Pure Appl Chem 1994 66 1077minus1184(12) McKee M L Fluctional Molecules WIREs Comput Mol Sci2011 1 943minus951(13) Nikitin K OrsquoGara R Mechanisms and Beyond Elucidation ofFluxional Dynamics by Exchange NMR Spectroscopy Chem - Eur J2019 25 4551minus4589(14) Christe K O Curtis E C Dixon D A On the Problem ofHeptacoordination Vibrational Spectra Structure and Fluxionality ofIodine Heptafluoride J Am Chem Soc 1993 115 1520minus1526(15) Cass M E Hii K K Rzepa H S Mechanisms ThatInterchange Axial and Equatorial Atoms in Fluxional ProcessesIllustration of the Berry Pseudorotation the Turnstile and the LeverMechanisms via Animation of Transition State Normal VibrationalModes J Chem Educ 2006 83 336(16) Lonnon D G Ball G E Taylor I Craig D C Colbran S BFluxionality in a Paramagnetic Seven-Coordinate Iron(II) Complex AVariable-Temperature Two-Dimensional NMR and DFT Study InorgChem 2009 48 4863minus4872
(17) Mauksch M von Rague Schleyer P Effective Monkey SaddlePoints and Berry and Lever Mechanisms in the Topomerization of SF4and Related Tetracoordinated AX4 Species Inorg Chem 2001 401756minus1769(18) Couzijn E P A Slootweg J C Ehlers A W Lammertsma KStereomutation of Pentavalent Compounds Validating the BerryPseudorotation Redressing Ugirsquos Turnstile Rotation and Revealing theTwo- and Three-Arm Turnstiles J Am Chem Soc 2010 132 18127minus18140(19) Russegger P Brickmann J Pseudorotation of TrigonalBipyramidal Molecules Berry Rotation Contra ldquoTurnstilerdquo Rotationin PF5 Chem Phys Lett 1975 30 276minus278(20) Adams W J Thompson H B Bartell L S StructurePseudorotation and Vibrational Mode Coupling in IF7 An ElectronDiffraction Study J Chem Phys 1970 53 4040minus4046(21) Montgomery C D Mechanisms of Pentacoordinate Pseudor-otation A Molecular Modeling Study of PF5 J Chem Educ 2001 78844(22) Ramirez F Ugi I Turnstile Rearrangement and Pseudorotationin the Permutational Isomerization of Pentavalent PhosphorusCompounds In Adv Phys Org Chem Gold V Ed Elsevier 1971Vol 9 pp 25minus126(23) Casanova D Cirera J Llunell M Alemany P Avnir DAlvarez S Minimal Distortion Pathways in Polyhedral Rearrange-ments J Am Chem Soc 2004 126 1755minus1763(24) Alvarez S Alemany P Casanova D Cirera J Llunell MAvnir D Shape Maps and Polyhedral Interconversion Paths inTransitionMetal Chemistry Coord Chem Rev 2005 249 1693minus1708(25) Rzepa H S Cass M E A Computational Study of theNondissociative Mechanisms that Interchange Apical and EquatorialAtoms in Square Pyramidal Molecules Inorg Chem 2006 45 3958minus3963(26) Csaszar A G Fabri C Sarka J Quasistructural moleculesWiley Interdiscip Rev Comput Mol Sci 2020 10 No e1432(27) Cotton F A Fluxional Organometallic Molecules Acc ChemRes 1968 1 257minus265(28) Muetterties E L Stereochemically Nonrigid Structures AccChem Res 1970 3 266minus273(29) Wentworth R Trigonal Prismatic vs Octahedral Stereo-chemistry in Complexes Derived from Innocent Ligands CoordChem Rev 1972 9 171minus187(30) der Heyde T A Determination of Reaction Paths forPentacoordinate Metal Complexes with the Structure CorrelationMethod Angew Chem Int Ed Engl 1994 33 823minus839(31) Gielen M Willem R Wrackmeyer B Eds FluxionalOrganometallic and Coordination Compounds Wiley Online Library2004(32) Westmoreland T D Symmetry Control of Chemical ReactionsApplications to the Berry Pseudorotation of Five-CoordinateTransition Metal Complexes Inorg Chim Acta 2008 361 1187minus1191(33) Faller J W Stereochemical Nonrigidity of OrganometallicComplexes In Encyclopedia of Inorganic and Bioinorganic ChemistryScott R A Ed Wiley Online Library 2011 pp 1minus34(34) Villafane F Dynamic Behavior in Solution of Seven-Coordinated Transition Metal Complexes Coord Chem Rev 2014281 86minus99(35) Sandstrom JDynamic NMR Spectroscopy Academic Press 1982(36) Oki M Applications of Dynamic NMR Spectroscopy to OrganicChemistry VCH Publishers 1985(37) Perrin C L Dwyer T J Application of Two-Dimensional NMRto Kinetics of Chemical Exchange Chem Rev 1990 90 935minus967(38) Bain A D Chemical Exchange in NMR Prog Nucl Magn ResonSpectrosc 2003 43 63minus103(39) Bryant R G The NMR Time Scale J Chem Educ 1983 60933minus935(40) Eyring H The Activated Complex in Chemical Reactions JChem Phys 1935 3 107minus115(41) Houk K N Liu F Holy Grails for Computational OrganicChemistry and Biochemistry Acc Chem Res 2017 50 539minus543
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3190
(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

(42) Harvey J N Himo F Maseras F Perrin L Scope andChallenge of Computational Methods for Studying Mechanism andReactivity in Homogeneous Catalysis ACS Catal 2019 9 6803minus6813(43) Ashley D C Jakubikova E Ray-Dutt and Bailar Twists inFe(II)-Tris(22prime-bipyridine) Spin States Sterics and Fe-N BondStrengths Inorg Chem 2018 57 5585minus5596(44) Zabrodsky H Peleg S Avnir D Continuous SymmetryMeasures J Am Chem Soc 1992 114 7843minus7851(45) Alvarez S Distortion Pathways of Transition Metal Coordina-tion Polyhedra Induced by Chelating Topology Chem Rev 2015 11513447minus13483(46) Alemany P Casanova D Alvarez S Dryzun C Avnir DContinuous Symmetry Measures A New Tool in Quantum ChemistryIn Reviews in Computational Chemistry Parrill A L Lipkowitz K BEds John Wiley amp Sons Inc 2017 Vol 30 Chapter 7 pp 289minus352(47) Cremer D Pople J A General Definition of Ring PuckeringCoordinates J Am Chem Soc 1975 97 1354minus1358(48) Cremer D RING - A Coordinate Transformation Program forEvaluating the Degree and Type of Puckering of a Ring CompoundQuantum Chemical Program Exchange 1975 288 1minus8(49) Cremer D On the Correct Usage of the CremerminusPoplePuckering Parameters as Quantitative Descriptors of Ring Shapes - AReply to Recent Criticism By Petit Dillen And Geise Acta CrystallogrSect B Struct Sci 1984 40 498minus500(50) Cremer D Calculation of Puckered Rings with AnalyticalGradients J Phys Chem 1990 94 5502minus5509(51) Kraka E Cremer D Dieter Cremerrsquos Contribution to the Fieldof Theoretical Chemistry Int J Quantum Chem 2019 119No e25849(52) Kraka E Preface Dieter Cremerrsquos Scientific JourneyMol Phys2019 117 1047minus1058(53) Lyu S Beiranvand N Freindorf M Kraka E Interplay of RingPuckering and Hydrogen Bonding in Deoxyribonucleosides J PhysChem A 2019 123 7087minus7103(54) Zou W Izotov D Cremer D New Way of Describing Staticand Dynamic Deformations of the Jahn-Teller Type in Ring MoleculesJ Phys Chem A 2011 115 8731minus8742(55) Zou W Filatov M Cremer D Bond Pseudorotation Jahn-Teller and Pseudo-Jahn-Teller Effects in the Cyclopentadienyl Cationand its Pentahalogeno Derivatives Int J Quantum Chem 2012 1123277minus3288(56) Zou W Cremer D Description of Bond Pseudorotation BondPseudolibration and Ring Pseudoinversion Processes Caused by thePseudo-Jahn-Teller Effect Fluoro Derivatives of the CyclopropaneRadical Cation Aust J Chem 2014 67 435minus443(57) Cremer D Theoretical Determination of Molecular Structureand Conformation III The Pseudorotation Surface of 123-Trioxolaneand 124-Trioxolane J Chem Phys 1979 70 1898minus1910(58) Cremer D Szabo K J Ab Initio Studies of Six-MemberedRings Present Status and Future Developments In ConformationalBehavior of Six-Membered Rings Juaristi E Ed Wiley-VCHWeinheim 1995 pp 59minus135(59) Biarnes X Ardevol A Planas A Rovira C Laio AParrinello M The Conformational Free Energy Landscape of β-D-Glucopyranose Implications for Substrate Preactivation in β-GlucosideHydrolases J Am Chem Soc 2007 129 10686minus10693(60) Sega M Autieri E Pederiva F On the Calculation ofPuckering Free Energy Surfaces J Chem Phys 2009 130 225102(61) Autieri E Sega M Pederiva F Guella G Puckering FreeEnergy of Pyranoses A NMR and Metadynamics-Umbrella SamplingInvestigation J Chem Phys 2010 133 095104(62) Sega M Autieri E Pederiva F Pickett Angles and CremerminusPople Coordinates as Collective Variables for the Enhanced Samplingof Six-Membered Ring ConformationsMol Phys 2011 109 141minus148(63) Huang M Giese T J Lee T-S York D M Improvement ofDNA and RNA Sugar Pucker Profiles from Semiempirical QuantumMethods J Chem Theory Comput 2014 10 1538minus1545(64) Iglesias-Fernandez J Raich L Ardevol A Rovira C TheComplete Conformational Free Energy Landscape of β-Xylose Reveals
a Two-Fold Catalytic Itinerary for β-Xylanases Chem Sci 2015 61167minus1177(65) Lindner J O Sultangaleeva K Rohr M I S Mitric RmetaFALCON A Program Package for Automatic Sampling of ConicalIntersection Seams Using Multistate Metadynamics J Chem TheoryComput 2019 15 3450minus3460(66) Paoloni L Rampino S Barone V Potential-Energy Surfacesfor Ring-Puckering Motions of Flexible Cyclic Molecules throughCremerminusPople Coordinates Computation Analysis and Fitting JChem Theory Comput 2019 15 4280minus4294(67) Alibay I Bryce R A Ring Puckering Landscapes ofGlycosaminoglycan-Related Monosaccharides from Molecular Dy-namics Simulations J Chem Inf Model 2019 59 4729minus4741(68) Cremer D Kraka E From Molecular Vibrations to BondingChemical Reactions and Reaction Mechanism Curr Org Chem 201014 1524minus1560(69) Kraka E Reaction PathHamiltonian and its Use for InvestigatingReaction Mechanism In Encyclopedia of Computational ChemistrySchleyer P Allinger N Clark T Gasteiger J Kollman P SchaeferH III Schreiner P Eds John Wiley amp Sons New York 1998 pp2437minus2463(70) Kraka E Reaction Path Hamiltonian and the Unified ReactionValley Approach WIREs Comput Mol Sci 2011 1 531minus556(71) Cremer D Larsson J A Kraka E New Developments in theAnalysis of Vibrational Spectra on the Use of Adiabatic InternalVibrational Modes In Theoretical and Computational ChemistryParkanyi C Ed Elsevier Amsterdam 1998 Vol 5 pp 259minus327(72) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra I Derivation of Adiabatic Internal Modes Int J QuantumChem 1998 67 1minus9(73) Konkoli Z Cremer D A New Way of Analyzing VibrationalSpectra III Characterization of Normal Vibrational Modes in terms ofInternal Vibrational Modes Int J Quantum Chem 1998 67 29minus40(74) Levine R Bernstein R Molecular Reaction Dynamics andChemical Reactivity Oxford University Press New York 1987(75) Brouard M Reaction Dynamics Oxford Chemistry PrimersOxford University Press New York 1998(76) Crim F F Vibrational State Control of Bimolecular ReactionsDiscovering and Directing the Chemistry Acc Chem Res 1999 32877minus884(77) Crim F F Chemical Dynamics of Vibrationally ExcitedMolecules Controlling Reactions in Gases and on Surfaces ProcNatl Acad Sci U S A 2008 105 12654minus12661(78) Zare R N Laser Control of Chemical Reactions Science 1998279 1875minus1879(79) Sinha A Hsiao M C Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water with HydrogenAtoms J Chem Phys 1991 94 4928minus4935(80) Sinha A Thoemke J D Crim F F Controlling BimolecularReactions Mode and Bond Selected Reaction of Water withTranslationally Excited Chlorine Atoms J Chem Phys 1992 96372minus376(81) Simpson W R Rakitzis T P Kandel S A Orr-Ewing A JZare R N Reaction of Cl with Vibrationally Excited CH4 and CHD3State-to-state Differential Cross Sections and Steric Effects for the HClProduct J Chem Phys 1995 103 7313minus7335(82) Yoon S Henton S Zivkovic A N Crim F F The RelativeReactivity of the Stretch-Bend Combination Vibrations of CH4 in theCl(2P32)+CH4 Reaction J Chem Phys 2002 116 10744minus10752(83) Bechtel H A Camden J P Brown D J A Zare R NComparing the Dynamical Effects of Symmetric and AntisymmetricStretch Excitation of Methane in the Cl+CH4 Reaction J Chem Phys2004 120 5096minus5103(84) Camden J P Bechtel H A Brown D J A Zare R N Effectsof C-H Stretch Excitation on theH+CH4 Reaction J Chem Phys 2005123 134301(85) Crim F F Vibrationally Mediated Photodissociation ExploringExcited-State Surfaces and Controlling Decomposition PathwaysAnnu Rev Phys Chem 1993 44 397minus428
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3191
(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

(86) Reid S A Reisler H Experimental Studies of Resonances inUnimolecular Decomposition Annu Rev Phys Chem 1996 47 495minus525(87) Chen J Zhou X Zhang Y Jiang B Vibrational Control ofSelective BondCleavage in Dissociative Chemisorption ofMethanol onCu(111) Nat Commun 2018 9 4039(88) Shirhatti P R Rahinov I Golibrzuch K Werdecker JGeweke J Altschaffel J Kumar S Auerbach D J Bartels CWodtke A M Observation of the Adsorption and Desorption ofVibrationally Excited Molecules on a Metal Surface Nat Chem 201810 592minus598(89) Stensitzki T Yang Y Kozich V Ahmed A A Kossl FKuhn O Heyne K Acceleration of a Ground-State Reaction bySelective Femtosecond-Infrared-Laser-Pulse Excitation Nat Chem2018 10 126minus131(90) Stei M Carrascosa E Dorfler A Meyer J Olasz B CzakoG Li A Guo H Wester R Stretching Vibration is a Spectator inNucleophilic Substitution Sci Adv 2018 4 No eaas9544(91) Heyne K Kuhn O Infrared Laser Excitation ControlledReaction Acceleration in the Electronic Ground State J Am Chem Soc2019 141 11730minus11738(92) Gillespie R Fifty Years of the VSEPR Model Coord Chem Rev2008 252 1315minus1327(93) Eckart C Some Studies Concerning Rotating Axes andPolyatomic Molecules Phys Rev 1935 47 552minus558(94) Sayvetz A The Kinetic Energy of PolyatomicMolecules J ChemPhys 1939 7 383minus389(95) Zou W Kalescky R Kraka E Cremer D Relating NormalVibrational Modes to Local Vibrational Modes with the Help of anAdiabatic Connection Scheme J Chem Phys 2012 137 084114(96) Tao Y Tian C Verma N Zou W Wang C Cremer DKraka E Recovering Intrinsic Fragmental Vibrations Using theGeneralized Subsystem Vibrational Analysis J Chem Theory Comput2018 14 2558minus2569(97) Kudin K N Dymarsky A Y Eckart Axis Conditions and theMinimization of the Root-Mean-Square Deviation Two CloselyRelated Problems J Chem Phys 2005 122 224105(98) Kearsley S K On the Orthogonal Transformation Used forStructural Comparisons Acta Crystallogr Sect A Found Crystallogr1989 A45 208minus210(99) Peng C Ayala P Y Schlegel H B Frisch M J UsingRedundant Internal Coordinates to Optimize Equilibrium Geometriesand Transition States J Comput Chem 1996 17 49minus56(100) Li X Frisch M J Energy-Represented Direct Inversion in theIterative Subspace within a Hybrid Geometry Optimization Method JChem Theory Comput 2006 2 835minus839(101) Kraka E Zou W Filatov M Tao Y Grafenstein J IzotovD Gauss J He Y Wu A Konkoli Z Polo V Olsson L He ZCremer D COLOGNE2019 SMU 2019 see httpwwwsmueducatco(102) Frisch M J Trucks G W Schlegel H B Scuseria G ERobb M A Cheeseman J R Scalmani G Barone V Petersson GA Nakatsuji H Li X Caricato M Marenich A V Bloino JJanesko B G Gomperts R Mennucci B Hratchian H P Ortiz JV Izmaylov A F Sonnenberg J L Williams-Young D Ding FLipparini F Egidi F Goings J Peng B Petrone A Henderson TRanasinghe D Zakrzewski V G Gao J Rega N Zheng G LiangW Hada M Ehara M Toyota K Fukuda R Hasegawa J IshidaM Nakajima T Honda Y Kitao O Nakai H Vreven TThrossell KMontgomery J A Jr Peralta J E Ogliaro F BearparkM J Heyd J J Brothers E N Kudin K N Staroverov V N KeithT A Kobayashi R Normand J Raghavachari K Rendell A PBurant J C Iyengar S S Tomasi J Cossi M Millam J M KleneM Adamo C Cammi R Ochterski J W Martin R L MorokumaK Farkas O Foresman J B Fox D J Gaussian 16 Revision B01Gaussian Inc Wallingford CT 2016(103) Becke A D Density-Functional Thermochemistry III TheRole of Exact Exchange J Chem Phys 1993 98 5648minus5652
(104) Lee C Yang W Parr R G Development of the Colle-SalvettiCorrelation-Energy Formula into A Functional of the Electron DensityPhys Rev B Condens Matter Mater Phys 1988 37 785minus789(105) Vosko S H Wilk L Nusair M Accurate Spin-DependentElectron Liquid Correlation Energies for Local Spin DensityCalculations A Critical Analysis Can J Phys 1980 58 1200minus1211(106) Stephens P J Devlin F J Chabalowski C F Frisch M J AbInitio Calculation of Vibrational Absorption and Circular DichroismSpectra Using Density Functional Force Fields J Phys Chem 1994 9811623minus11627(107) Dunning T H Gaussian Basis Sets for Use in CorrelatedMolecular Calculations I The Atoms Boron Through Neon andHydrogen J Chem Phys 1989 90 1007minus1023(108) Zhao Y Truhlar D G A New Local Density Functional forMain-Group Thermochemistry Transition Metal Bonding Thermo-chemical Kinetics and Noncovalent Interactions J Chem Phys 2006125 194101(109) Weigend F Ahlrichs R Balanced Basis Sets of Split ValenceTriple Zeta Valence and Quadruple Zeta Valence Quality for H to RnDesign and Assessment of Accuracy Phys Chem Chem Phys 2005 73297minus3305(110) Weigend F Accurate Coulomb-Fitting Basis Sets for H to RnPhys Chem Chem Phys 2006 8 1057minus1065(111) Zhao Y Truhlar D G The M06 Suite of Density Functionalsfor Main Group Thermochemistry Thermochemical Kinetics Non-covalent Interactions Excited States and Transition Elements TwoNew Functionals and Systematic Testing of Four M06-Class Func-tionals and 12 Other Functionals Theor Chem Acc 2008 120 215minus241(112) Hratchian H P Schlegel H B Accurate Reaction Paths usinga Hessian Based Predictor-Corrector Integrator J Chem Phys 2004120 9918minus9924(113) Hratchian H P Schlegel H B Using Hessian Updating ToIncrease the Efficiency of a Hessian Based Predictor-CorrectorReaction Path Following Method J Chem Theory Comput 2005 161minus69(114) Hratchian H P Kraka E Improved Predictor-CorrectorIntegrators For Evaluating Reaction Path Curvature J Chem TheoryComput 2013 9 1481minus1488(115) Gonzalez C Schlegel H B An Improved Algorithm forReaction Path Following J Chem Phys 1989 90 2154minus2161(116) Gonzalez C Schlegel H B Reaction Path Following in Mass-Weighted Internal Coordinates J Phys Chem 1990 94 5523minus5527(117) Page M McIver J W On Evaluating the Reaction PathHamiltonian J Chem Phys 1988 88 922minus935(118) Page M Doubleday C McIver J W Following SteepestDescent Reaction Paths The Use of Higher Energy DerivativesWith abinitio Electronic Structure Methods J Chem Phys 1990 93 5634minus5642(119) Ruff O Heinzelmann A Uber das Uranhexafluorid Z AnorgAllg Chem 1911 72 63minus84(120) Pople J Schneider W Bernstein H J High-ResolutionNuclear Magnetic Resonance McGraw-Hill 1959 p 223(121) Muetterties E L Phillips W D Structure of ClF3 andExchange Studies on Some Halogen Fluorides by Nuclear MagneticResonance J Am Chem Soc 1957 79 322minus326(122) Muetterties E L Phillips W D Structure and ExchangeProcesses in Some Inorganic Fluorides by Nuclear MagneticResonance J Am Chem Soc 1959 81 1084minus1088(123) Klemperer W G Krieger J K McCreary M D MuettertiesE L Traficante D D Whitesides G M Dynamic Nuclear MagneticResonance Study of Fluorine Exchange in Liquid Sulfur Tetrafluoride JAm Chem Soc 1975 97 7023minus7030(124) Rothman M J Lohr L L Analysis of an Energy MinimizationMethod for Locating Transition States on Potential Energy Hyper-surfaces Chem Phys Lett 1980 70 405minus409(125) Williams I H Maggiora G M Use and Abuse of theDistinguished-Coordinate Method for Transition-State StructureSearching J Mol Struct THEOCHEM 1982 89 365minus378
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3192
(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193

(126) Fadrna E Koc a J Single-Coordinate-Driving MethodCoupled with Simulated Annealing An Efficient Tool To SearchConformational Space J Phys Chem B 1997 101 7863minus7868(127) Yang M Zou J Wang G Li S Automatic Reaction PathwaySearch via Combined Molecular Dynamics and Coordinate DrivingMethod J Phys Chem A 2017 121 1351minus1361(128) Yang M Yang L Wang G Zhou Y Xie D Li SCombined Molecular Dynamics and Coordinate Driving Method forAutomatic Reaction Pathway Search of Reactions in Solution J ChemTheory Comput 2018 14 5787minus5796(129) Plessow P N Efficient Transition State Optimization ofPeriodic Structures through Automated Relaxed Potential EnergySurface Scans J Chem Theory Comput 2018 14 981minus990(130) Taha A N True N S LeMaster C B LeMaster C LNeugebauer-Crawford S M Gas-Phase Nuclear Magnetic ResonanceStudy of Berry Pseudorotation of SF4 Comparison of Experimental andCalculated Kinetic Parameters and Falloff Kinetics J Phys Chem A2000 104 3341minus3348(131) Gutowsky H S McCall D W Slichter C P NuclearMagnetic Resonance Multiplets in Liquids J Chem Phys 1953 21279minus292(132) Muetterties E L Topological Representation of Stereo-isomerism II The Five-Atom Family J Am Chem Soc 1969 914115minus4122(133) Ugi I Marquarding D Klusacek H Gillespie P Ramirez FBerry Pseudorotation and Turnstile Rotation Acc Chem Res 1971 4288minus296(134) Moberg C Stereomutation in Trigonal-Bipyramidal SystemsA Unified Picture Angew Chem Int Ed 2011 50 10290minus10292(135) Braga D Grepioni F Orpen A G Nickel Carbonyl(Ni(CO)4) and Iron Carbonyl (Fe(CO)5) Molecular Structures inthe Solid State Organometallics 1993 12 1481minus1483(136) Deerenberg S Schakel M de Keijzer A H J F KranenburgM Lutz M Spek A L Lammertsma K TetraalkylammoniumPentaorganosilicates The First Highly Stable Silicates with FiveHydrocarbon Ligands Chem Commun 2002 348minus349(137) Campbell B S DersquoAth N J Denney D B Denney D ZKipnis I S Min T B Some Caged Polycyclic Phosphoranes J AmChem Soc 1976 98 2924minus2927(138) Alvarez S Distortion Pathways of Transition MetalCoordination Polyhedra Induced by Chelating Topology Chem Rev2015 115 13447minus13483(139) Bailar J C Jr Some Problems in the Stereochemistry ofCoordination Compounds Introductory Lecture J Inorg Nucl Chem1958 8 165minus175(140) Ray P Dutt N Kinetics and Mechanism of Racemization ofOptically Active Cobaltic Trisbiguanide Complex J Indian Chem Soc1943 20 81minus92(141)Marian CM Spin-Orbit Coupling and IntersystemCrossing inMolecules WIREs Comput Mol Sci 2012 2 187minus203(142) Tao Y Pei Z Bellonzi N Mao Y Zou Z Liang W YangZ Shao Y Constructing Spin-Adiabatic States for the Modeling ofSpin-Crossing Reactions I A Shared-Orbital Implementation Int JQuantum Chem 2020 120 No e26123(143) Rzepa H S Cass M E In Search of the Bailar and Ray-DuttTwist Mechanisms That Racemize Chiral Trischelates A Computa-tional Study of ScIII TiIV CoIII ZnII GaIII and GeIV Complexes of aLigand Analogue of Acetylacetonate Inorg Chem 2007 46 8024minus8031(144) Claassen H H Gasner E L Selig H Vibrational Spectra ofIF7 and ReF7 J Chem Phys 1968 49 1803minus1807(145) Bartell L S Rothman M J Gavezzotti A PseudopotentialSCF-MO Studies of Hypervalent Compounds IV Structure Vibra-tional Assignments and Intramolecular Forces in IF7 J Chem Phys1982 76 4136minus4143(146) Kuczkowski R Lawrence S Bartell Biographical Notes JMol Struct 1999 485minus486 ximinusxxvii
(147) Minyaev R M Wales D J Transition Vector Symmetry andthe Internal Pseudo-Rotation and Inversion Paths of CIF4
+ J ChemSoc Faraday Trans 1994 90 1831minus1837(148) Lord R C Lynch M A Schumb W C Slowinski E J TheVibrational Spectra and Structures of Iodine Pentafluoride andHeptafluoride J Am Chem Soc 1950 72 522minus527(149) Musher J I Cowley A H Intramolecular Ligand Rearrange-ments in Four- and Five-Coordinate Sulfur Compounds Inorg Chem1975 14 2302minus2304(150) Heidrich D Quapp W Saddle Points of Index 2 on PotentialEnergy Surfaces and Their Role in Theoretical Reactivity Inves-tigations Theor Chem Acc 1986 70 89minus98(151) Li Y Houk K N The Dimerization of Cyclobutadiene An abInitio CASSCF Theoretical Study J Am Chem Soc 1996 118 880minus885(152)Mezey P G Potential Energy Hypersurfaces Elsevier New York1987 p 76(153) Sadasivam D V Theoretical and Experimental Studies of SomeUnusual Potential Energy Surfaces and Pseudopericyclic Reactions PhDthesis Texas Tech University 2006(154) Tama F Sanejouand Y-H Conformational Change ofProteins Arising fromNormal Mode Calculations Protein Eng Des Sel2001 14 1minus6(155) Ioannidis E I Gani T Z H Kulik H J molSimplify AToolkit for Automating Discovery in Inorganic Chemistry J ComputChem 2016 37 2106minus2117(156) Ioannidis E I Automated Structure Generation for First-Principles Transition-Metal Catalysis PhD thesis MassachusettsInstitute of Technology 2018(157) Foscato M Venkatraman V Jensen V R DENOPTIMSoftware for Computational de Novo Design of Organic and InorganicMolecules J Chem Inf Model 2019 59 4077minus4082(158) Kulik H J Making Machine Learning a Useful Tool in theAccelerated Discovery of Transition Metal Complexes WileyInterdiscip Rev Comput Mol Sci 2020 10 No e1439(159) Fiorin G Klein M L Henin J Using Collective Variables toDrive Molecular Dynamics Simulations Mol Phys 2013 111 3345minus3362
Journal of Chemical Theory and Computation pubsacsorgJCTC Article
httpsdxdoiorg101021acsjctc9b01274J Chem Theory Comput 2020 16 3162minus3193
3193





























