Embed Size (px)
Citation preview

Development and characterisation of novel fentanyl-deltaopioid receptor antagonist based bivalent ligandsM F. Bird1, R. S. Vardanyan2, V J. Hruby2, G. Calo3, R. Guerrini4, S. Salvadori4, C. Trapella4, J. McDonald1,D. J. Rowbotham1 and D. G. Lambert1*
1 Department of Cardiovascular Sciences, Division of Anaesthesia, Critical Care and Pain Management, University of Leicester, Robert KilpatrickClinical Sciences Building, Leicester Royal Infirmary, Leicester LE2 7LX, UK2 Department of Chemistry and Biochemistry, University of Arizona, Tucson, AZ 85721, USA3 Department of Medical Sciences, Section of Pharmacology, University of Ferrara and Italian Institute of Neuroscience, Ferrara, Italy4 Department of Chemical and Pharmaceutical Sciences and LTTA (Laboratorio per le Tecnologie delle Terapie Avanzate), University of Ferrara,Ferrara, Italy
* Corresponding author. E-mail: [email protected]
Editor’s key points
† There is considerable needfor opioids with reducedtolerance.
† A series of bifunctionalopioid receptor ligandswas tested for receptorbinding and function.
† Addition of a linker did notimpair receptor binding,but reduced m opioidreceptor functionalactivity, indicating theneed for alternateapproaches to reducingtolerance.
Background. Opioid tolerance is a limiting factor in chronic pain. Delta opioid peptide (DOP)(d)receptor antagonism has been shown to reduce tolerance. Here, the common clinical muopioid peptide (MOP)(m) receptor agonist fentanyl has been linked to the DOP antagonistDmt-Tic (2′,6′-dimethyl-L-tyrosyl-1,2,3,4-tetrahydrisoquinoline-3-carboxylic acid) to createnew bivalent compounds.
Methods. Binding affinities of bivalents(#9, #10, #11, #12 and #13) were measured in Chinesehamster ovary (CHO) cells expressing recombinant human MOP, DOP, Kappa opioidpeptide (KOP)(k) and nociceptin/orphanin FQ opioid peptide (NOP) receptors. Functional studies,measuring GTPg[35S] or b-arrestin recruitment, were performed in membranes or whole cellsrespectively expressing MOP and DOP.
Results.Thenewbivalentsbound toMOP(pKi : #9:7.31;#10:7.58;#11:7.91;#12:7.94;#13:8.03)andDOP (#9:8.03; #10:8.16; #11:8.17; #12:9.67; #13:9.71). In GTPg[35S] functional assays, compounds#9(pEC50:6.74; intrinsic activity:0.05) #10(7.13;0.34) and #11(7.52;0.27) showed weak partialagonist activity at MOP. Compounds #12 and #13, with longer linkers, showed no functionalactivity at MOP. In antagonist assays at MOP, compounds #9 (pKb:6.87), #10(7.55) #11(7.81)#12(6.91) and #13(7.05) all reversed the effects of fentanyl. At DOP, all compounds showedantagonist affinity (#9:6.85; #10:8.06; #11:8.11; #12:9.42; #13:9.00), reversing the effects ofDPDPE ([D-Pen2,5]enkephalin). In b-arrestin assays, compared with fentanyl (with response atmaximum concentration (RMC):13.62), all compounds showed reduced ability to activate b-arrestin (#9 RMC:1.58; #10:2.72; #11:2.40; #12:1.29; #13:1.58). Compared with fentanyl, theintrinsic activity was: #9:0.12; #10:0.20; #11:0.18; #12:0.09 and #13:0.12.
Conclusions. The addition of a linker between fentanyl and Dmt-Tic did not alter the ability to bindtoMOPandDOP,howeverasubstantial loss inMOPfunctionalactivitywasapparent.Thishighlightsthe difficulty in multifunctional opioid development.
Keywords: delta receptors; fentanyl; mu receptors; opioid; pharmacology - analgesics opioid
Accepted for publication: 8 October 2014
Opioid use in the pain clinic is hampered by development oftolerance, providing a dilemma in the treatment of chronicpain. The underlying causes of chronic pain are complex, theorigins of which include a wide range of diseases and/or injuries,making effective treatment difficult.1 For instance, the treat-ment of chronic pain in cancer has been shown to be ineffectivein up to �50% of patients in the last year of their lives.2 3 Mostclinically available opioids act solely at the mu opioid peptide(MOP) (m) receptor; a member of the Gi/o-protein coupledopioid receptor family of receptors, which also includes the
delta opioid peptide (DOP) (d), kappa opioid peptide (KOP) (k)and nociceptin/orphanin FQ opioid peptide (NOP) receptors.Substantial evidence has implicated the DOP receptor1 4 –6 inthe development of tolerance, hastening the development ofmultifunctional opioids.
The mechanisms underlying the development of toleranceare varied, involving a multitude of cellular functions,receptor properties and signalling processes, such as receptorinternalisation andb-arrestin recruitment.7 A large bodyof evi-dence, including various molecular studies4 8 9 and work in
& The Author 2015. Published by Oxford University Press on behalf of the British Journal of Anaesthesia. All rights reserved.For Permissions, please email: [email protected]
British Journal of Anaesthesia 114 (4): 646–56 (2015)Advance Access publication 13 February 2015 . doi:10.1093/bja/aeu454
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

mouse models,10 11 has implicated other members of theopioid receptor family, specifically the DOP receptor, as poten-tial mediators and/or instigators in the development of toler-ance. The involvement of two, or more, opioid receptors inthe progression of tolerance has provided a target for the de-velopment of drugs with multiple pharmacophores, i.e. bi-valent or bifunctional ligands. Bivalent and bifunctionalligands could present more predictable pharmacokinetic andpharmacodynamic traits than two separately acting ligands.Synergy between two conjoined pharmacophores can alsolead to improved potency, as well as a potential reduction inside-effects.6
Previous attempts at developing bivalent ligands havefocused on adaptations or derivatives of semi-synthetic orpeptide structures.12 13 We studied chemical adaptations offentanyl (N-(1-(2-phenylethyl)-4-piperidinyl)-N-phenylpropa-namide), a clinically available synthetic opioid, to acceptlinker molecules and a second pharmacophore (Dmt-Tic (2′,6′-dimethyl-L-tyrosyl-1,2,3,4-tetrahydrisoquinoline-3-carboxylic acid)). Previous studies have reported the synthesisand chemical adaptation of these carboxy-fentanyl moleculesthrough conjugation with DOP agonists (enkephalins) andneurokinin-1 (NK-1) antagonists.14 15 Fentanyl is a potentMOP agonist (10 fold greater than morphine) used in the treat-ment of pain in a clinical setting. Moreover, fentanyl is hydropho-bic allowing greater access to the central nervous system than,for instance, morphine.16 The potent analgesic activity andhigh bioavailability of fentanyl make it an ideal model on whichto base a mixed opioid. The second pharmacophore chosen forthe development of these new bivalent compounds is the DOPantagonist Dmt-Tic. Dmt-Tic has a long history of use in thedevelopment of multi-pharmacophoric compounds, and it hasbeen demonstrated that C-terminal chemical modifications ofDmt-Tic are tolerated, thus making it a good candidate for thedevelopmentof a[MOPagonist]-[DOPantagonist]complex.17–20
We characterised the binding and functional activity of fivenewlysynthesized fentanyl (MOP)/Dmt-Tic (DOP) bivalent com-pounds. As shown in previous studies, linker length can affectthe affinity and efficacy of either pharmacophore; with this inmind we have examined spacer length between the MOP andDOP pharmacophores.13 21 Finally we explored whether thesenovel bivalent molecules produce changes in signalling inG-protein activation andb-arrestin recruitment assays.5 9 22 – 24
MethodsMaterials
The reference molecules, Dmt-Tic and Nociceptin/OrphaninFQ (N/OFQ) were synthesised in house (Department ofChemical and Pharmaceutical Sciences, University of Ferrara).Tritiated UFP-101 ([3H]-UFP-101) was synthesized as des-cribed.25 Tritiated diprenorphine ([3H]-DPN) was purchasedfrom Perkin Elmer (UK). Morphine and naloxone was purchasedfrom Sigma-Aldrich Co. (Dorset, UK). Fentanyl, norbinaltorphi-mine (KOPantagonist, norBNI) and [D-Pen2, D-Pen5]-enkephalin(DOP agonist, DPDPE) were purchased from Tocris (Abingdon,UK). Tissue culture media and supplements were obtained
from Invitrogen (Paisley, UK). Nomenclature and structureare shown in Table 1. Synthesis of the novel bivalent ligandsis described in detail in the Supplementary material.
Cell culture
Chinese hamster ovary (CHO) cells expressing recombinanthuman opioid receptors were grown in either Hams F12 (forCHOhMOP, CHOhDOP and CHOhKOP cells) or DMEM/Hams F121:1 (for CHOhNOP cells). The media contained 10% fetalbovine serum, 100 IU ml21 penicillin, 100 mg ml21 streptomy-cin and 2.5mg ml21 fungizone. Stock cultures were maintainedthrough the addition of 200 mg ml21 G418 (for CHOhMOP,CHOhDOP and CHOhKOP cells). CHOhNOP cells were maintainedwith G418 (200 mg ml21) and hygromycin B (200 mg ml21). Cellcultures were maintained at 378C in 5% CO2/humidified air.Cells were used for experimentation when confluent.
Membrane preparation
Cells were harvested, homogenised, and membrane frag-ments were resuspended in either a wash buffer consisting
Table 1 Chemical structure and corresponding compoundabbreviation numbers
Structure Compound #
RRC2
RRC3
RRO
#9
#10
#11
#12
#13
MOP And DOP bivalent ligands BJA
647
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

of 50 mM Tris-HCl pH to 7.4 with KOH, for CHOhMOP, CHOhDOPand CHOhKOP; or additional 5 mM MgSO4 for CHOhNOP cellsfor saturation and displacement binding assays, or a hom-ogenisation buffer (50 mM Tris and 0.2 mM EGTA pH 7.4with NaOH) in GTPg[35S] functional assays. Membrane sus-pensions were centrifuged at 20374 × g at 48C for 10 min,and this process was repeated thrice. The resulting pelletwas resuspended in an appropriate volume of the desiredbuffer and protein concentration measured using the Lowryassay.26
Ligand selectivity and binding affinity- displacementbinding assays
Membrane protein (20–40 mg) was incubated in 0.5 ml of 50mM Tris, 0.5% bovine serum albumin (BSA) and �0.8nM[3H]-DPN (for CHOhMOP, CHOhDOP and CHOhKOP) or �0.8nM[3H]-UFP-101 (for CHOhNOPcells), as well as varying concentra-tions (1 pM-10 mM) of control ligands and test compounds.Nonspecific binding was determined in the presence of 10 mMnaloxone for CHOhMOP/DOP/KOP or 1 mM of N/OFQ forCHOhNOP cells. Samples were incubated for 1 h at roomtemperature, following which reactions were terminated byvacuum filtration onto PEI-soaked Whatman GF/B filters, usinga Brandel harvester (Semat, Germany).
Ligand functional activity- GTPg[35S] assays
Membrane protein (20–40mg) was incubated in 0.5 ml volumeof 50 mM Tris, 0.2 mM EGTA, 1 mM MgCl2, 100 mM NaCl, 0.1%BSA, 0.15 mM bacitracin; pH 7.4, GDP (33 mM for classical; 100mm for NOP), and �150 pM GTPg[35S]. A range of concentra-tions of both control (fentanyl, DPDPE) and the test compounds(1 pM – 10 mM) was added prior to incubation. Nonspecificbinding was determined in the presence of unlabeled GTPgS(10 mM).27 Samples were incubated for 1 h at 308C withgentle agitation. Reactions were terminated by vacuum filtra-tion through dry Whatman GF/B filters.
Beta-Arrestin assay
Assays were prepared as described in the DiscoveRx (UK)PathHunterw Assay protocol. Cells were incubated for 24 h(human OPRM1, MOP) or 48 h (human OPRD1, DOP) as sug-gested in 96 well plates provided. Following the desired incuba-tion period, desired concentrations of compounds were addedto the wells, and incubated at 378C for 90 min. For morphineand fentanyl, at MOP, and DPDPE, at DOP, a range of concentra-tions was used (1 nM–10mM). At MOP, only maximum concen-trations (10mM) compounds #9–#13 were used. In antagoniststudies at DOP, 10 nM of Dmt-Tic was incubated in a range ofconcentrations of DPDPE (1 nM–10 mM). High concentrationsof DPDPE (1 mM) were incubated with 100 nM (�EC80) of com-pounds #9–#13 to determine antagonist activity. The finaldevelopment reagent was added and plates were incubatedat room temperature for 1 h. Plates were read using a DynexMLX luminometer, Dynex Technologies, UK, set at 1 sec/wellto measure relative light units (RLU).
Data analysis
Data are expressed as mean(SEM (n)) experiments. Resultswere analysed and graphs were fitted using GraphPadPRISM V6.02 (San Diego, USA). In displacement bindingstudies, the concentration of competitor which produced 50%displacement (IC50) was corrected for competing mass ofradiolabel according to Cheng and Prusoff28 (using Kd valuesfor [3H]-DPN of 125 pM (MOP); 323 pM (DOP); 134 pM (KOP) andfor [3H]-UFP-101 107 pM (NOP).29 30 GTPg [35S] binding dataare expressed as a stimulation factor (agonist stimulatedspecific binding/basal specific binding).27 Antagonist affinitieswere calculated using the Gaddum equation.31 b-arrestin re-cruitment is expressed as an activation factor (activated (RLU)/basal RLU). Data were analysed using t-tests and/or ANOVA, withBonferroni corrections as appropriate with P,0.05 consideredsignificant.
ResultsDisplacement binding assays-Fentanyl analoguesRRC2, RRC3 and RRO
RRC2; RRC3; RRO failed to displace [3H]-DPN in CHO cell mem-branes expressingMOP, DOPorKOPreceptors. Thesecompoundsalso failed to displace [3H]-UFP-101 in CHO cell membranesexpressing NOP receptors (Table 2).
Displacement binding assays-Final compounds#9, #10 and #11
At the MOP receptor, #9, #10, #11 displaced binding of [3H]-DPNin a concentration dependent and saturable manner. Bindingaffinities (pKi) were: #9(7.31), #10(7.58) and #11(7.91)(Table 2). Binding affinities of test compounds #10 and #11were not statistically different to that of the reference ligandfentanyl (8.13). Compound #9 was significantly different, dis-playing a lower binding affinity for MOP. At the DOP receptor,all ligands had nanomolar affinity: #9(8.03), #10(8.16) and#11(8.17) (Table 2). Of the test compounds, only #9 was signifi-cantly different compared to Dmt-Tic-OH (8.95). Compounds#9, #10, #11 displaced [3H]-DPN binding to CHOhKOP mem-branes [NorBNI (10.16); #9(7.29); #10(7.02); #11(7.13)].[3H]-UFP-101 was also displaced by the test compounds inCHOhNOP membranes [N/OFQ (10.69) #9(7.28); #10(6.96);#11(7.48)] (Table 2).
GTPg[35S] functional assays-Final compounds#9, #10 and #11
GTPg[35S] assays were used as a functional activity screen.Compounds #9, #10 and #11 stimulated the binding ofGTPg[35S] in a concentration dependent and saturable mannerat the MOP receptor (Fig 1A). pEC50 values of #10(7.13) and#11(7.52) were not significantly different to that of fentanyl(7.31) at MOP. The pEC50 for #9 (6.74) was significantly differentto fentanyl. Emax values, compared with fentanyl (3.88), forcompounds #9:1.18; #10:2.19; #11:1.98 were significantly dif-ferent. Relative intrinsic activity (a-Emax) and potency (pEC50)values are summarised in Table 3.
BJA Bird et al.
648
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

Since compounds #9, #10 and #11 showed reduced efficacyat CHOhMOP, these were screened in an antagonist assay(Fig. 1B; Table 2). A fixed concentration (1 mM) of the bivalentcompounds was added to varying concentrations of fentanylin CHOhMOP cells; there was a rightward shift in the fentanylconcentration response curve. Both #10 and #11 displayedEmax values similar to fentanyl (Table 2). Emax in the presenceof #9 was significantly different to fentanyl. The pKb valueswere: #9 (6.87), #10 (7.55) and #11 (7.81).
In CHOhDOP membranes DPDPE (D-Pen2, D-Pen5 Enkephalin)stimulatedthebindingof GTPg[35S] (pEC50:7.70;Emax:2.76).Com-pounds #9, #10 and #11 did not stimulate binding of GTPg[35S](Fig. 2A). When compounds #9, #10 and #11 were co-incubatedwith the DOP agonist DPDPE, all compounds caused a rightwardshift in the concentration- response curves (Fig. 2B). Compounds,#10 and #11, produced pKb values of 8.06 and 8.1, respectively.Compound #9 had a pKb of 6.85 (Table 3).
Functional assays were performed in cell membranes ex-pressing the KOP and NOP receptor; bivalents #9–#11showed no agonist or antagonist activity at these receptors(Fig. 3A and B).
With respect to reference compounds RRC2, RRC3 and RRO;compounds #9–#11 restore MOP binding. The substantial lossin MOPefficacy compared with fentanyl indicated an alterationin the interaction between the fentanyl pharmacophore andthe MOP receptor.
Displacement binding assays-Final compounds#12 and #13
Compounds #12 and #13 displaced [3H]-DPN at the MOP,DOP and KOP receptors in a concentration dependent and sat-urable manner, but failed to displace [3H]-UFP-101 (Table 2).At the MOP receptor, #12 (pKi:7.94) and #13(8.03) were
Table 2 Radioligand binding data for the [fentanyl]–[Dmt-Tic] bivalent ligands. Tritiated diprenorphine was used to determine binding affinity atCHOhMOP/DOP/KOP, while [3H]-UFP-101 was used to determine binding affinity at CHOhNOP. Data are mean (SEM) of five experiments for theCHOhMOP cell line and of three experiments for the remaining cell lines. Reference ligands: fentanyl (MOP), morphine (MOP), Dmt-Tic (DOP),norbinaltorphimine (Nor-BNI) (KOP), Nociception/Orphanin FQ (N/OFQ) (NOP). If a significant difference (ANOVA) was detected, post-hoc testingusing Bonferroni multiple comparisons was employed *P,0.05; ***P,0.0005; ****P,0.0001 compared with control
pKi (SEM)
CHOhMOP CHOhDOP CHOhKOP CHOhNOP
Control 8.13(0.04) 8.95(+0.04) 10.16(+0.02) 10.69(+0.10)
RRC2 N/A N/A N/A N/A
RRC3 N/A N/A N/A N/A
RRO N/A N/A N/A N/A
#9 7.31(0.06)* 8.03(0.28)
* 7.29(0.04)**** 7.28(0.07)
****
#10 7.58(0.10) 8.16(0.17) 7.02(0.08)**** 6.96(0.12)
****
#11 7.91(0.23) 8.17(0.22) 7.13(0.04)**** 7.48(0.05)
****
#12 7.94(0.04) 9.67(0.04)*** 8.14(0.03)
**** N/A
#13 8.03(0.04) 9.71(0.01)*** 7.35(0.04)
**** 6.63(0.26)****
Analysis of Variance significant significant Significant significant
5
4
3
2
1
0–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Ligand] (M)
Stim
ulat
ion
fact
or
Fentanyl#9#10#11
4
3
2
1
0–12 –11 –10 –9 –8 –7 –6 –5 –4 –3
Log [Fentanyl] (M)
Stim
ulat
ion
fact
or
Control+1mM #9
+1mM #10
+1mM #11
BA
Fig 1 (A) Ligand stimulated GTPg[35S] binding for fentanyl and compounds #9, #10 and #11 in CHOhMOP cell membranes. (B) Bivalent compoundswere co-incubated, at a concentration of 1 mM, with fentanyl. Data are shown as mean (SEM) for 5 experiments.
MOP And DOP bivalent ligands BJA
649
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

not significantly different from fentanyl (8.13). At the DOPreceptor, #12(9.67) and #13(9.71) both showed increa-sed affinity for the DOP receptor, when compared with both#11(8.37) and the parent compound Dmt-Tic-OH (8.95).Differences in binding affinity between the two Gly-linkerextended molecules were evident at the KOP receptor; #12(8.14) displaying nanomolar affinity. While there was no sig-nificant difference in the binding affinities of #11(7.13) and#13(7.35), #12 showed statistical differences from both of
these compounds. The Gly extended compounds showedeither weak (#13; 6.63), or no affinity (#12; inactive) for theNOP receptor (Table 2).
GTPg[35S] functional assays-Final compounds#12 and #13
The Gly-extended compounds #12 and #13 failed to stimul-ate the binding of GTPg[35S] with no efficacy at the MOP
Table 3 Agonist and antagonist activity of the [fentanyl]–[Dmt-Tic] bivalent pharmacophores at CHOhMOP and CHOhDOP. Antagonist affinity wasdetermined against Fentanyl (MOP) and DPDPE [D-Pen2, D-Pen5 Enkephalin] (DOP). Relative intrinsic activity was determined by removal of basalactivity and as a ratio of full agonist Emax. All experiments are represented as the mean (SEM) with n≥3. If a significant difference was detected(ANOVA), post-hoc testing using Bonferroni multiple comparisons was employed *P,0.05; **P,0.005; ****P,0.0001 compared with fentanyl for MOPand Dmt-Tic for DOP
Agonist Activity Antagonist Activity
(pEC50) Relative Intrinsic activity pKb
CHOhMOP
Fentanyl 7.32(0.12) 1 Inactive
#9 6.74(1.02)* 0.05 6.87(0.61)
#10 7.13(0.29) 0.34 7.55(0.28)
#11 7.52(0.27) 0.27 7.81(0.18)
#12 Inactive Inactive 6.91(0.08)
#13 Inactive Inactive 7.05(0.11)
Analysis of Variance Significant N/A N/A
CHOhDOP
Dmt-Tic Inactive Inactive 8.77(0.11)
#9 Inactive Inactive 6.85(0.19)****
#10 Inactive Inactive 8.06(0.09)*
#11 Inactive Inactive 8.11(0.16)
#12 Inactive Inactive 9.42(0.14)**
#13 Inactive Inactive 9.00(0.15)
Analysis of Variance N/A N/A significant
–13 –12 –11 –10 –9 –8 –7 –6 –5 –40
1
2
3
4 DPDPE
#11
#9
#10
Log[Ligand] (M)
Stim
ulat
ion
Fac
tor
Stim
ulat
ion
Fac
tor
–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
1
2
3
4 Control
+100nM # 11
Log[DPDPE] (M)
+100nM # 9
+100nM # 10
A B
Fig 2 (A) DPDPE stimulated GTPg[35S] binding. Compounds (#9, #10, and #11) showed no activity in CHOhDOP cell membranes. (B) DPDPE stimulatedbinding in the absence and presence of 100 nM of bivalent compounds (#9, #10, and #11). All data are mean (SEM) of 5 experiments. Referenceligand, DPDPE; [D-Pen2, D-Pen5 Enkephalin].
BJA Bird et al.
650
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

receptor (Fig. 4A; Table 3). At 300 nM, compounds #12 and #13behaved as weak antagonists with pKb values of 6.91and 7.05,respectively (Fig. 4B).
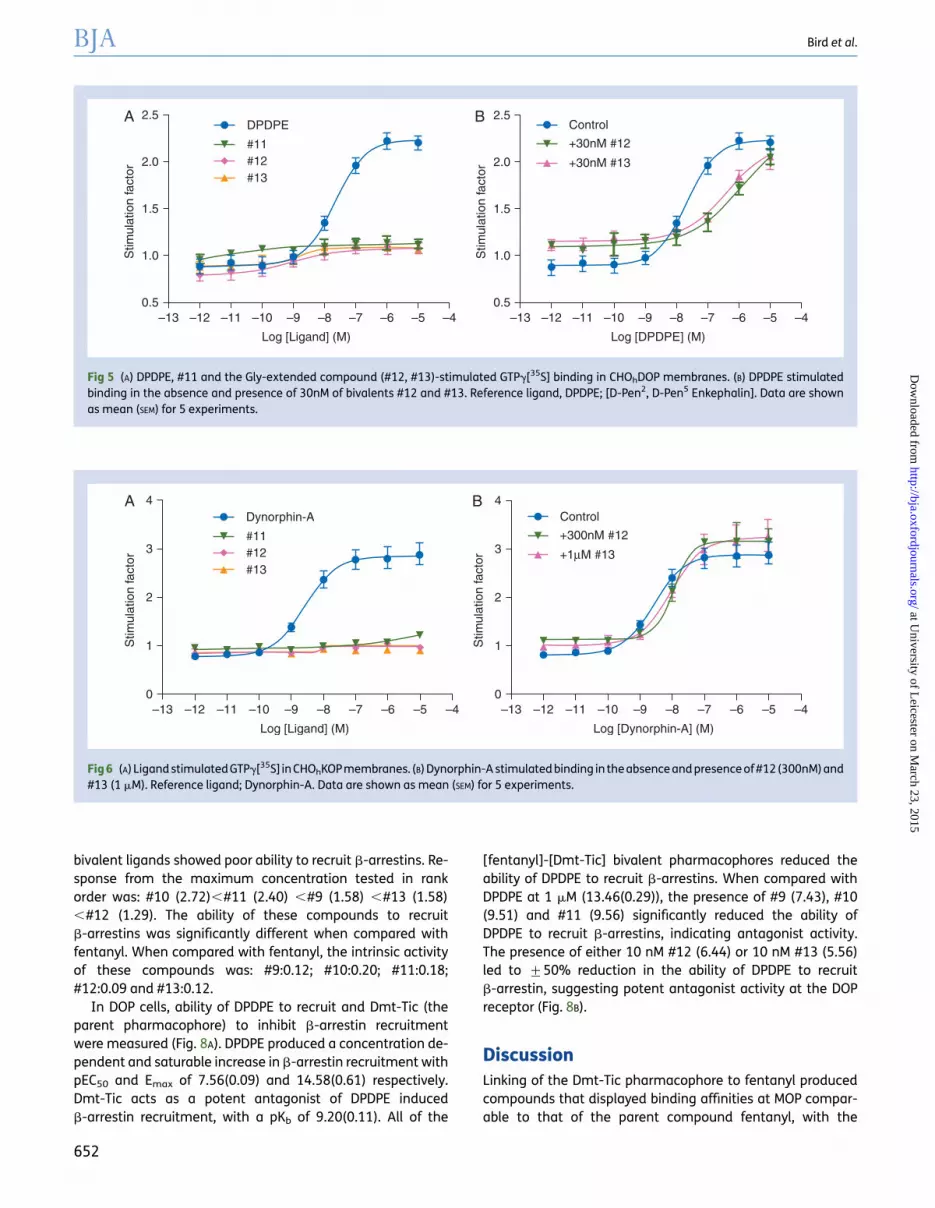
Compounds #12 and #13 showed no activity at the DOPreceptor (Fig. 5A, Table 3). However, when co-incubated withthe DOPagonist DPDPE, both #12 (pKb:9.42) and #13 (9.00) pro-duced a rightward shift in the agonist concentration responsecurve (Fig. 5B, Table 3).
Since #12 and #13 displayed binding affinity for KOP,GTPg[35S] functional assays were performed. Both compoundswere inactive (Fig. 6A). In antagonist experiments, 300 nM and1mM for #12 and #13, respectively, produced a weak but meas-urable rightward shift in the concentration response curvewhen co-incubated with dynorphin-A. Compound #12 and#13 produced pKb values of 6.96 and 6.45, respectively
(Fig. 6B). In view of the low affinity at NOP for #12 and #13 func-tional assays were not performed.
Beta-Arrestin assays
b-arrestin recruitment in MOP cells was determined for fen-tanyl and morphine in CHOhMOP cells lines (Fig. 7A). Maximallyeffective concentrations of bivalent compounds were used tocompare against fentanyl and morphine (Fig. 7B). Fentanyl(Emax of 13.62(0.45)) produced a greaterb-arrestin recruitmentthan morphine (Emax: 10.80(0.38)). Fentanyl (pEC50: 7.45(0.07)) also has increased potency compared with morphine(6.88(0.12)). The potency of fentanyl forb-arrestin recruitmentwas similar to its potency of GTPgS activation (Table 2). Whencompared with the maximum response of fentanyl, the
2.0
1.5
1.0
0.5–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Ligand] (M)
Stim
ulat
ion
fact
orDynorphin A#9#10#11
8
10
6
4
2
0–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Ligand] (M)
Stim
ulat
ion
fact
or
N/OFQ#9
#10
#11
BA
Fig 3 (A) Dynorphin-A stimulated GTPg[35S] binding. Compounds (#9, #10, and #11) showed no activity in CHOhKOP cell membranes. (B) N/OFQ sti-mulated GTPg[35S] binding. Bivalent compounds (#9, #10, and #11) showed no activity in CHOhNOP cell membranes. Data are shown as the mean(SEM) for 3 experiments.
6
5
1
2
3
4
0–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Ligand] (M)
Stim
ulat
ion
fact
or
Fentanyl
#11#12#13
3
4
2
1
0–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Fentanyl] (M)
Stim
ulat
ion
fact
or
Control
+300nM #12
+300nM #13
BA
Fig 4 (A) Fentanyl stimulated GTPg[35S] binding for #11 and the Gly-extended compounds (#12, #13), is shown in CHOhMOP cell membranes. (B)Fentanyl stimulated binding in the absence and presence of 300 nM of bivalents #12 and #13. Reference ligand, fentanyl. Data are shown asmean (SEM) for 5 experiments.
MOP And DOP bivalent ligands BJA
651
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from
bivalent ligands showed poor ability to recruit b-arrestins. Re-sponse from the maximum concentration tested in rankorder was: #10 (2.72),#11 (2.40) ,#9 (1.58) ,#13 (1.58),#12 (1.29). The ability of these compounds to recruitb-arrestins was significantly different when compared withfentanyl. When compared with fentanyl, the intrinsic activityof these compounds was: #9:0.12; #10:0.20; #11:0.18;#12:0.09 and #13:0.12.
In DOP cells, ability of DPDPE to recruit and Dmt-Tic (theparent pharmacophore) to inhibit b-arrestin recruitmentwere measured (Fig. 8A). DPDPE produced a concentration de-pendent and saturable increase in b-arrestin recruitment withpEC50 and Emax of 7.56(0.09) and 14.58(0.61) respectively.Dmt-Tic acts as a potent antagonist of DPDPE inducedb-arrestin recruitment, with a pKb of 9.20(0.11). All of the
[fentanyl]-[Dmt-Tic] bivalent pharmacophores reduced theability of DPDPE to recruit b-arrestins. When compared withDPDPE at 1 mM (13.46(0.29)), the presence of #9 (7.43), #10(9.51) and #11 (9.56) significantly reduced the ability ofDPDPE to recruit b-arrestins, indicating antagonist activity.The presence of either 10 nM #12 (6.44) or 10 nM #13 (5.56)led to +50% reduction in the ability of DPDPE to recruitb-arrestin, suggesting potent antagonist activity at the DOPreceptor (Fig. 8B).
DiscussionLinking of the Dmt-Tic pharmacophore to fentanyl producedcompounds that displayed binding affinities at MOP compar-able to that of the parent compound fentanyl, with the
2.5
2.0
1.0
1.5
0.5–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Ligand] (M)
Stim
ulat
ion
fact
orDPDPE
#11#12#13
2.0
2.5
1.5
1.0
0.5–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [DPDPE] (M)
Stim
ulat
ion
fact
or
Control
+30nM #12
+30nM #13
BA
Fig 5 (A) DPDPE, #11 and the Gly-extended compound (#12, #13)-stimulated GTPg[35S] binding in CHOhDOP membranes. (B) DPDPE stimulatedbinding in the absence and presence of 30nM of bivalents #12 and #13. Reference ligand, DPDPE; [D-Pen2, D-Pen5 Enkephalin]. Data are shownas mean (SEM) for 5 experiments.
4
3
1
2
0–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Ligand] (M)
Stim
ulat
ion
fact
or
Dynorphin-A
#11#12#13
3
4
2
1
0–13 –12 –11 –10 –9 –8 –7 –6 –5 –4
Log [Dynorphin-A] (M)
Stim
ulat
ion
fact
or
Control
+300nM #12
+1mM #13
BA
Fig 6 (A) Ligand stimulated GTPg[35S] in CHOhKOP membranes. (B) Dynorphin-A stimulated binding in the absence and presence of #12 (300nM) and#13 (1 mM). Reference ligand; Dynorphin-A. Data are shown as mean (SEM) for 5 experiments.
BJA Bird et al.
652
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

exception of #9. Compound #9 showed a decrease in bindingaffinity for MOP. Final compounds #9–13 displayed affinityfor the DOP receptor consistent with the presence of aDmt-Tic pharmacophore. Compounds #10 and #11 displayedsimilar binding affinity to that of Dmt-Tic, whilst compound#9 again showed a decrease in binding affinity. Compoundswith glycine-extended linkers (#12 and #13) showed a relativeincrease in binding at DOP. In functional studies, all compoundsshowed either weak partial agonist activity or no functionalactivity at MOP relative to fentanyl, with compounds #9, #10and #11 acting as weak partial agonists, while the extendedlinker compounds (#12 and #13) showed no functional activity
at this receptor. In antagonist assays at MOP, compounds #10and #11 antagonised fentanyl, producing pKb values similar toboth their pKi and pEC50 values, inherent qualities of a partialagonist. Compounds #12 and #13 displayed low antagonist af-finity at MOP, but displayed increased antagonist affinity, whencompared with Dmt-Tic, at DOP. The antagonist affinity dis-played at DOP by #12 and #13 matched their pKi values forDOP. The weak intrinsic activity, regarding GTPg[35S] binding,was mirrored in their ability to recruit b-arrestins. Compounds#10 and #11 produced the highest recruitment; however thiswas approximately 5-fold lower than that of the parent com-pound fentanyl.
–9 –8 –7 –6 –5
Log [Ligand] (M)
Act
ivat
ion
fact
or (
RLU
)20
15
10
5
0
Fentanyl
Morphine
Act
ivat
ion
fact
or (
RLU
)
20
15
10
5
0
Fent
anyl
Mor
phine #9 #1
0#1
1#1
2#1
3
*
** *
* *
A B
Fig 7 (A) Fentanyl and morphine concentration response curves for b-arrestin recruitment. (B) shows the recruitment of b-arrestin in the presenceof 10mM of fentanyl, morphine and the various bivalent ligands in CHOhDOP cells. The activation factor is a ratio relative to basalb-arrestin activity.Data were significant (ANOVA) and *P,0.0001 compared with fentanyl post hoc Bonferroni test. Data are shown as mean (SEM) for n≥5.
–9 –8 –7 –6 –5
Log [Ligand] (M)
Act
ivat
ion
fact
or (
RLU
)
20
15
10
5
0
DPDPE
DPDPE vs Dmt-Tic
Act
ivat
ion
fact
or (
RLU
)
15
10
5
0
DPDPE
DPDPE + #
9
DPDPE + #
10
DPDPE + #
11
DPDPE + #
12
DPDPE + #
13
**
* *
****
A B
Fig 8 (A) Depicts a concentration response curve in the absence, and presence, of 10 nM Dmt-Tic. (B) Shows the inhibition caused by selected con-centrations of the various bivalent ligands in CHOhDOP cells. Data were significant (ANOVA) and *P,0.0005; **P,0.0001 compared with DPDPE posthoc Bonferroni test. Data are shown as mean (SEM) for n≥5.
MOP And DOP bivalent ligands BJA
653
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

Prior to the development of the [fentanyl]-[Dmt-Tic]ligands, the carboxy fentanyl series had been conjugatedwith both DOP agonists (enkephalins)15 and NK-1 antago-nists.14 In the case of [fentanyl]-[NK-1 antagonist], a seriesof compounds where tested using radioligand binding andex vivo tissue functional studies. In agreement with thepresent study, functionalised fentanyl derivatives showed aloss of functional ability at MOP. Following conjugation withan NK1 antagonist, most compounds showed a reduction inbinding affinity for MOP and functional activity in guinea pigileum contraction assays.14 Further studies, using the RRC3fentanyl derivative demonstrated significant analgesia inwarm water tail flick tests, following spinal administration,after conjugation with enkephalins. This compound producedantiallodynic and thermal antihypersensitive effects in nerveinjured animals.15
The involvement of DOP receptors in MOP receptor function-al activity, trafficking and endocytosis has been demonstratedin a large number of studies. Initially, experiments involvingantagonism of the DOP receptor during morphine treatmentdemonstrated an attenuation of tolerance.1 The role of DOPin tolerance was further demonstrated in studies involvingeither DOP knockout or ppENK knockout mice, wherebyremoval of either the receptor or endogenous agonist led to areduction in morphine tolerance. Furthermore, DOP receptorexpression and trafficking is also up-regulated during long-term morphine treatment.4 5 7 10 32 33 Co-expression of bothMOP and DOP in neurones and cells in the pain pathway hasled to the hypothesis of a heterodimer, with unique signallingproperties that influence opioid tolerance. The involvementof the DOP receptor in this heterodimer is believed to includedifferential phosphorylation and recruitment of b-arrestins,such that selectivity for the ubiquitination pathway is favouredafter endocytosis.34 35 This makes it an ideal target for a drugwith dual selectivity. Due to the more predictable pharmaco-kinetics and pharmacodynamics of a single drug, previouswork has either focused on novel bifunctional drugs (UFP-505) or bivalent pharmacophores (MDAN21).6 13 In the devel-opment of drugs with dual agonist targets, it is important thatthe individual affinities produced by the drug for differentreceptors are similar. This would remove any favourabilityfor a particular receptor, thereby increasing the possibility ofthe drug acting at both target sites. In the case of both ourcompounds and UFP-505, their affinities for DOP are higherthan their affinities for MOP. However, since both have antag-onist activity at DOP, this could be beneficial as a higher affin-ity for DOP ensures that DOP is fully blocked when MOP isactivated.
Due to the differences in functional activity seen with com-pounds #9, #10 and #11 compared with fentanyl, it was initiallyhypothesised that the distance between the pharmacophoresaffected their ability to interact with MOP. The importance ofspacer molecules (size, distance, charge) has been previouslydemonstrated in a number of models, none more so effectivelythan in the development of the MDAN series of compounds.13 Inorder to validate this, we extended the linker molecules, usingthe amino acid glycine, of the most efficacious compound,
#11. Either a single glycine molecule (#12) or two glycine mole-cules (#13) were added to the linker structure. The increase inspacer length between the MOP and DOP pharmacophorescompletely prevented MOP receptor activation. In contrast,DOP antagonist activity was retained. These results inferredthat the changes of efficacy determined here, most likely layin a combination of adaptation of the chemical structure ofthe fentanyl conjugates and linker length. It should be empha-sized that simple functionalization of fentanyl with an acidicmoiety as in RRC2, RRC3 and RRO produced loss of MOP binding.
b-arrestin recruitment has been implicated in the internal-isation and recycling or ubqiuitination of opioid receptors.36
In this study, bivalent compounds showed poor efficacy ingeneral and substantially reduced ability to recruit b-arrestinsto the MOP receptor, collectively suggesting a limited ability topromote receptor internalisation. Interestingly, the bivalentligand with the shortest linker length (#9) displayed weakerinteractions with MOP. The compound displayed weak partialagonist activity, potency and antagonist affinity which wasnot similar to that of its binding affinity. The similarity of thebinding affinity of compound #9 to fentanyl suggests thatthe initial interactions between drug and receptor are notaltered. However, due to the marked decrease in potency andantagonist binding affinity, when compared with the pKi, wehypothesise that the close proximity of the two pharmaco-phores negatively affects the ability of the compound toengage with the binding pocket of the MOP receptor toproduce a functional response. Similar results have been re-cently obtained with MOP/DOP bivalent ligands composed ofmorphine and Dmt-Tic pharmacophores.37 Moreover, in thiscase the molecules lose MOP functional activity, behaving asMOP partial or antagonist ligands. In contrast, both fentanyland morphine Dmt-Tic bivalent ligands maintain potent DOPantagonist properties corroborating the evidence that C-terminalmodifications of Dmt-Tic pharmacophore are tolerated.38 39
Limitations
While the compounds demonstrated the desired antagonistaffinity at DOP, they demonstrated poor, or no, efficacy atMOP in our high expression system. Coupled with a modest an-tagonist affinity at the MOP receptor, these compounds wouldbe predicted to have absent or ultra-low analgesic action. Inthe clinical setting the goal would be to design a high efficacyMOP agonist (retain the activity of fentanyl) coupled with DOPantagonism. The main limitation of our study is the lack ofin vivo testing. However, we would argue that as the MOPagonist activity is lost there would be little point in undertakingantinociceptive testing in vivo.
In conclusion, the development of clinically available opioidswith reduced tolerance profile is of high importance; MOPagonist - DOP antagonist ligands are possible candidates. Inthis study, the potent clinical MOP opioid fentanyl was linkedto the DOP antagonist, Dmt-Tic to produce a new series of biva-lents. This series of compounds was unable to retain the potentfunctional activity of fentanyl at MOP whilst retaining potentantagonist activity at DOP.
BJA Bird et al.
654
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

Supplementary materialSupplementary Material is available at British Journal ofAnaesthesia online.
Authors’ contributionsM.F.B., and J. M.: collected experimental data, wrote paper.R.S.V., and V.J.H.: synthesis of functionalised fentanyls, wrotechemistry and reviewed drafts. G.C., R.G., S.S., and C.T.: synthe-sis of bivalent ligands, wrote chemistry and reviewed drafts.D.J.R., and D.G.L.: conceived project, obtained funding (forMFB), supervised all functional testing, wrote paper.
AcknowledgementMark F. Bird is a British Journal of Anaesthesia/Royal College ofAnaesthetists funded PhD student.
Declaration of interestD.G.L. and D.J.R. are board members and directors of BritishJournal of Anaesthesia. D.G.L. is also Administration Directorand Company Secretary for British Journal of Anaesthesia.
FundingFunding from BJA/ROCA to D.G.Land D.J.R. Funding from ItalianMinistry of University grant FIRB 2010 Future in ricercuCRBFR109 SBM to C.T.
References1 Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham DJ, Lambert DG.
Simultaneous targeting of multiple opioid receptors: a strategy toimprove side-effect profile. Br J Anaesth 2009; 103: 38–49
2 Addington-Hall J, McCarthy M. Dying from cancer: results of a na-tional population-based investigation. Palliat Med 1995; 9:295–305
3 Colvin LA, Lambert DG. Pain medicine: advances in basic sciencesand clinical practice. Br J Anaesth 2008; 101: 1–4
4 Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selectiveblockage of delta opioid receptors prevents the development ofmorphine tolerance and dependence in mice. J Pharmacol ExpTher 1991; 258: 299–303
5 Hasbi A, Nguyen T, Fan T, et al. Trafficking of Preassembled Opioidm2d Heterooligomer2Gz Signaling Complexes to the PlasmaMembrane: Coregulation by Agonists†. Biochemistry 2007; 46:12997–3009
6 Schiller PW. Bi- or multifunctional opioid peptide drugs. Life Sci2010; 86: 598–603
7 ZuoZ.TheRoleofOpioidReceptorInternalizationandb-Arrestins intheDevelopment of Opioid Tolerance. Anesth Analge 2005; 101: 728–34
8 He L, Fong J, von Zastrow M, Whistler JL. Regulation of Opioid Recep-tor Trafficking and Morphine Tolerance by Receptor Oligomeriza-tion. Cell 2002; 108: 271–82
9 Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role forheterodimerization of m and d opiate receptors in enhancing mor-phine analgesia. Proc Natl Acad Sci U S A 2004; 101: 5135–9
10 Zhu Y, King MA, Schuller AGP, et al. Retention of Supraspinal Delta-like Analgesia and Loss of Morphine Tolerance in d Opioid ReceptorKnockout Mice. Neuron 1999; 24: 243–52
11 Nitsche JF, Schuller AGP, King MA, Zengh M, Pasternak GW, Pintar JE.Genetic Dissociation of Opiate Tolerance and Physical Dependencein d-Opioid Receptor-1 and Preproenkephalin Knock-Out Mice. JNeurosci 2002; 22: 10906–13
12 Lipkowski AW, Konecka AM, Sroczynska I, Przewlocki R, Stala L,Tam SW. Bivalent opioid peptide analogues with reduced distancesbetween pharmacophores. Life Sci 1987; 40: 2283–8
13 Daniels DJ, Lenard NR, Etienne CL, Law P-Y, Roerig SC, Portoghese PS.Opioid-induced tolerance and dependence in mice is modulated bythe distance between pharmacophores in a bivalent ligand series.Proc Natl Acad Sci U S A 2005; 102: 19208–13
14 Vardanyan R, Kumirov VK, Nichol GS, et al. Synthesis and biologicalevaluation of new opioid agonist and neurokinin-1 antagonistbivalent ligands. Bioorg Med Chem 2011; 19: 6135–42
15 Podolsky AT, Sandweiss A, Hu J, et al. Novel fentanyl-based dualmu/delta-opioid agonists for the treatment of acute and chronicpain. Life Sci 2013; 93: 1010–6
16 Mayes S, Ferrone M. Fentanyl HCl Patient-Controlled IontophoreticTransdermal System for the Management of Acute PostoperativePain. Ann Pharmacother 2006; 40: 2178–86
17 Balboni G, Guerrini R, Salvadori S, et al. Evaluation of the Dmt2TicPharmacophore: Conversion of a Potent d-Opioid Receptor Antag-onist into a Potent d Agonist and Ligands with Mixed Properties.J Med Chem 2002; 45: 713–20
18 Balboni G, Guerrini R, Salvadori S, et al. Conversion of thePotent d-Opioid Agonist H-Dmt-Tic-NH-CH2-Bid into d-OpioidAntagonists by N1-Benzimidazole Alkylation1. J Med Chem 2005;48: 8112–4
19 Fichna J, do-Rego J-C, Chung NN, et al. Synthesis andCharacterization of Potent and Selective m-Opioid ReceptorAntagonists, [Dmt1, d-2-Nal4]endomorphin-1 (Antanal-1) and[Dmt1, d-2-Nal4]endomorphin-2 (Antanal-2). J Med Chem 2007;50: 512–20
20 Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Dmt andopioid peptides: A potent alliance. Pept Sci 2003; 71: 86–102
21 Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Synthesis and Prelimin-ary In vitro Investigation of Bivalent Ligands Containing Homo- andHeterodimeric Pharmacophores at m, d, and k Opioid Receptors.J Med Chem 2005; 49: 256–62
22 Gupta A, Mulder J, Gomes I, et al. Increased Abundance of OpioidReceptor Heteromers After Chronic Morphine Administration. SciSignal 2010; 3: ra54-
23 Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switchin signaling: b-arrestin2-mediated ERK activation by m-d opioidreceptor heterodimers. FASEB J 2007; 21: 2455–65
24 Garzon J, Rodrıguez-Munoz M, Sanchez-Blazquez P. Morphine altersthe selective association between mu-opioid receptors and specificRGS proteins in mouse periaqueductal gray matter. Neuropharma-cology 2005; 48: 853–68
25 Ibba M, Kitayama M, McDonald J, et al. Binding of the novel radioli-gand [(3)H]UFP-101 to recombinant human and native rat nocicep-tin/orphanin FQ receptors. Naunyn Schmiedebergs Arch Pharmacol2008; 378: 553–61
26 Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measure-ment with the folin reagent. J Biol Chem 1951; 193: 265–75
27 McDonald J, Calo G, Guerrini R, Lambert DG. UFP-101, a high affinityantagonist for the nociceptin/orphanin FQ receptor: radioligandand GTPg35S binding studies. Naunyn Schmiedebergs Arch Phar-macol 2003; 367: 183–7
28 Cheng Y, Prusoff WH. Relationship between the inhibition constant(K1) and the concentration of inhibitor which causes 50 per centinhibition (I50) of an enzymatic reaction. Biochem Pharmacol1973; 22: 3099–108
MOP And DOP bivalent ligands BJA
655
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from

29 Hashiba E, Harrison C, Calo G, et al. Characterisation and comparisonof novel ligands for the nociceptin/orphanin FQ receptor. NaunynSchmiedebergs Arch Pharmacol 2001; 363: 28–33
30 Dietis N, McDonald J, Molinari S, et al. Pharmacological character-ization of the bifunctional opioid ligand H-Dmt-Tic-Gly-NH-Bzl(UFP-505). Br J Anaesth 2012; 108: 262–70
31 Gaddum JH. Theories of drug antagonism. Pharmacol Rev 1957; 9:211–8
32 Ballesta JJ, Cremades J, Rodrıguez-Munoz M, Garzon J, Faura CC.Sensitivity to m-opioid receptor-mediated anti-nociception isdetermined by cross-regulation betweenm- and d-opioid receptorsat supraspinal level. Br J Pharmacol 2012; 166: 309–26
33 Ong E, Cahill C. Molecular Perspectives for mu/delta Opioid ReceptorHeteromers as Distinct, Functional Receptors. Cells 2014; 3: 152–79
34 Liggett SB. Phosphorylation Barcoding as a Mechanism of DirectingGPCR Signaling. Sci Signal 2011; 4: pe36-
35 Nobles KN, Xiao K, Ahn S, et al. Distinct Phosphorylation Sites on the{beta}2-Adrenergic Receptor Establish a Barcode That Encodes Dif-ferential Functions of {beta}-Arrestin. Sci Signal 2011; 4: ra51-
36 Williams JT, Ingram SL, Henderson G, et al. Regulation of mu-opioidreceptors: desensitization, phosphorylation, internalization, andtolerance. Pharmacol Rev 2013; 65: 223–54
37 Balboni G, Salvadori S, Marczak ED, et al. Opioid bifunctional ligandsfrom morphine and the opioid pharmacophore Dmt-Tic. European JMed Chem 2011; 46: 799–803
38 Snyder KR, Story SC, Heidt ME, Murray TF, DeLander GE, Aldrich JV.Effect of modification of the basic residues of dynorphin A-(1–13)amide on opioid receptor selectivity and opioid activity. J MedChem 1992; 35: 4330–3
39 Story SC, Murray TF, Delander GE, Aldrich JV. Synthesis and opioidactivity of 2-substituted dynorphin A-(1–13) amide analogues.Int J Pept Protein Res 1992; 40: 89–96
Handling editor: H. C. Hemmings
BJA Bird et al.
656
at University of L
eicester on March 23, 2015
http://bja.oxfordjournals.org/D
ownloaded from





























