Embed Size (px)
Citation preview

©2007 L
ANDES BIOSCI
ENCE.
DO NOT DIST
RIBUTE.
Report
Different Thresholds of MPF Inactivation Are Responsible for Controlling Different Mitotic Events in Mammalian Cell Division
[Cell Cycle 6:13, 1639-1645, 1 July 2007]; ©2007 Landes Bioscience
Naihan Xu1
Donald C. Chang1,2,*1Department of Biology; The Hong Kong University of Science and Technology; Clear Water Bay, Hong Kong, China
2Marine Biological Laboratory; Woods Hole, Massachusetts USA
*Correspondence to: Donald C. Chang; Department of Biology; Hong Kong University of Science and Technology; Clear Water Bay, Kowloon; Hong Kong, China; Tel.: +852.23587326; Fax: +852.23581559; Email: [email protected]
Original manuscript submitted: 05/04/07Manuscript accepted: 05/04/07
Previously published online as a Cell Cycle E-publication:http://www.landesbioscience.com/journals/cc/abstract.php?id=4385
Key worDs
MPF, Cdk1, cyclin B, metaphase/anaphase transition, chromosome separation, cytoki-nesis, nuclear reassembly, mitosis
ACKNowleDgemeNts
We thank Dr. Tony Hunter for providing the human cyclin B1 gene and Drs. Marc Kirschner and O. Stemmann for the separase gene. This work was supported by the Research Grants Council of Hong Kong (HKUST6466/05M and N_HKUST616/05).
Note
Supplemental material can be found at: www.landesbioscience.com/supplement/xuCC6-13-sup.pdf
AbstrACtWe present evidence for a paradigm that, during cell division, the decreasing activity
of MPF acts as a master signal, which utilizes different thresholds to control the initiation of different mitotic events. The key temporal control here is the degradation of cyclin B1. Using single cell analysis, we measured the kinetics of cyclin B1 degradation and deter‑mined quantitatively the thresholds of cyclin B1 level for different mitotic events within a HeLa cell. These observed thresholds were: 1.36 ± 0.49 mM (for chromosome separa‑tion), 0.75 ± 0.08 mM (for cytokinesis) and 0.54 ± 0.16 mM (for nuclear reassembly). By comparison, the average concentration of endogenous cyclin B1 within a prometaphase cell was found to be 2.92 ± 1.7 mM. We suggest that the decreasing order of these thresholds plays an important role in triggering the initiation of successive mitotic events in cell division.
INtroDuCtIoNIt is well known that MPF, an activated complex of Cdk1/cyclin B, is a key regulator of
mitosis. Up to now, it was widely thought that MPF acts basically like an ON/OFF switch for mitotic control.1-3 While a large amount of experimental data had supported this thinking, there were also conflicting observations. For example, there were diverse views on whether inactivation of MPF is required for anaphase onset or mainly for nuclear reas-sembly.4-10 More than a decade ago, in vitro studies using Xenopus egg extract suggested that degradation of cyclin B was required only for M/G1 transition but not anaphase onset.5 Similar conclusions were also reported in studies of budding or fission yeast.9,10 It was thought that the metaphase/ anaphase (M/A) transition is controlled mainly by the destruction of securin through the APC (anaphase promoting complex), which triggers the activation of separase to cleave the cohesin complex that holds the sister chromatids together.11-14 Destruction of cyclin B or inactivation of MPF was not required for the anaphase onset but required for later transition to G1.5-8
Recently, new evidence was discovered suggesting that this conclusion may not apply for mitosis in mammalian cells. First, we and Pines’ group observed that GFP-labeled cyclin B in HeLa and PtK2 cells was degraded mainly before the anaphase onset.15,16 We found the degradation kinetics was very similar between securin and cyclin B1.15 Second, when we overexpressed a nondegradable form of cyclin B in mammalian cells, cells were arrested mainly before anaphase onset.15 Third, this arrest was not due to the failure of activation of APC, since securin was degraded in the presence of nondegradable cyclin B.15 These findings suggested that, unlike yeast, the regulation of M/A transition in mammalian cells may involve inactivation of MPF. In a separated study by Stemmann et al, who reexamined the earlier in vitro study of Xenopus egg extract, also found that when a large amount of nondegradable cyclin B was added to the extract, anaphase onset could be blocked.17 They attributed this blockage to an inhibitory phosphorylation of separase either caused by a high activity of MPF or MAPK (ERK2).
In order to resolve the difference between our findings in mammalian cells and those in yeasts and Xenopus egg extract, we came to realize that the thinking of MPF as an ON/OFF switch could be a problem in explaining the controlling mechanism of mitosis. In fact, this reexamination touches on a more fundamental question on what type of mechanisms really controls the temporal order of the mitotic events. In general, there are two major ways to control the temporal order of a series of events: (1) The sequential (or domino) model, in which event A triggers event B, event B then triggers event C, etc. (2) The threshold model, in which the quantitative level of a decreasing inhibitory signal
www.landesbioscience.com Cell Cycle 1639

Thresholds of MPF Inactivation Controls Different Mitotic Events
(or an increasing activating signal) determines the initiation of a new event; if the threshold of event A is above that of event B, A will occur ahead of B (see Fig. 1A).
At present, most biologists tended to think along the sequential model in viewing the progression of mitotic events. This appears to be well justified for mitotic progression before metaphase. For example, DNA condensation (prophase) and spindle formation (prometaphase) must be ahead of the line-up of paired chromatids (metaphase). But is it also true for mitotic events following the metaphase? Is there any necessary temporal relationship between chromosome separation, cytokinesis and nuclear reassembly?
Here, we hypothesized that the mitotic events following meta-phase are controlled by the threshold model rather than by the sequential model. The master signal in this case is the inactivation of MPF, which is mainly regulated by the degradation of cyclin B1 during mitosis.1,6,7,18 Thus, inactivation of MPF should be a gradual event and it should act as an analog signal instead of an “ON/OFF” switch. This study is aimed to experimentally test this paradigm by examining the following questions: (1) At what point does the endogenous cyclin B1 within a cell start to degrade? (2) What is the time profile of cyclin B1 degradation? Is the degradation of cyclin B1 all or none? (3) Do different mitotic events triggered by different threshold levels of cyclin B1? (4) If the temporal gradient of MPF inactivation is destroyed, will the normal sequence of mitotic events be disrupted?
mAterIAls AND methoDsCell culture and synchronization. HeLa cells were cultured as a
monolayer at 37˚C in MEM supplemented with 10% fetal bovine serum and 100 IU/ml penicillin and streptomycin. To synchronize cells, cells were treated with 50 ng/ml nocodazole and cultured in 5% CO2 incubator at 37˚C for 6-8 hrs or 25 ng/ml nocodazole for 10-12 hrs. Mitotic cells were collected by shaking them off and washed three times with PBS by centrifugation (2000 rpm, 2 min). The cell pellet was resuspended into fresh medium for experiment.
Plasmid construction and fusion gene transfection. The human cyclin B1 gene was cloned between the HindIII and BamHI sites in a pEGFP-N1 vector. Cyclin B1-D85 was cut out from the full-length cyclin B1 (with a deletion of the first eighty-five amino acids in the N-terminus) and cloned behind the GFP gene between the KpnI and BamHI sites in a pEGFP-C3 vector. The fusion genes were intro-duced into HeLa cells by either electroporation or LipoFectamine TM 2000 reagent following standard protocols.1 Cells were usually cultured for at least 24 hrs for expressing the introduced genes.
siRNA treatment of HeLa cells. pSilencerTM 2.1-U6 hygro vector (Ambion Ltd. UK) was used for vector-based siRNA design. Specific siRNAs were generated for targeting human separase gene (KIAA 0165) corresponds to positions of 3505–3523 of the sepa-rase open reading frame (5'-ACAAACTCCTCCCCAGTCT- 3'), human Cdc20 gene corresponds to positions 541–559 of the Cdc20 open reading frame (5'-ATCCGAAATGACTATTACC-3'), and human Cdh1 gene corresponds to positions 201–219 of the Cdh1 open reading frame (5'-TGAGAAGTCTCCCAGT CAG-3'). The annealed hairpin siRNA insert was cloned into the pSilencer vector. The insertion was confirmed by DNA sequencing. HeLa cells were transfected with pSilencer-siRNA using LipoFectamineTM 2000 reagent. Cells were cultured for at least 24 hrs for expressing siRNA.
Immunofluorescence. HeLa cells released from nocodazole arrest were fixed with 4% paraformaldehyde plus 0.1% glutaraldehyde
and stained with anti-cyclin B1 monoclonal antibody (GNS-11, PharMingen). They were then stained with rhodamine-conjugated goat anti-mouse IgG (Calbiochem) antibody. Autofluorescence of glutaraldehyde was quenched with a freshly prepared solution of 0.1% NaBH4 (in phosphate-buffered saline) for 10 min. Nuclear DNA was stained with Hoechst 33342 (100 ng/ml) at 37˚C for 10 min.
Immunoblotting. HeLa cells transfected with GFP-cyclin B1-D85 were treated with 50 ng/ml nocodazole for 10 hrs. Mitotic cells were collected by shaking off and were released from nocodazole block. Cells were collected at 0, 1, 2, 3 and 4 hrs after nocodazole release. Cells were washed in ice-cold PBS by centrifugation and subse-quently lysed in lysis buffer (50 mM Tris, pH8.0, 150 mM NaCl, 1% NP-40, 50 mM NaF, 100 mg/ml PMSF, 20 mg/ml aprotinin, 20 mg/ml leupeptin, 2 mM EDTA, 10 mg/ml pepstatin A, 1 mM benzamidine, 0.5 mM okadaic acid). Equal amounts of protein deter-mined by Bradford assay (Bio-Rad) were separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blocked in 5% nonfat milk in Tris-buffered Saline containing 0.2% Tween 20. For immunoblotting analysis, blots were incubated with anti-phospho-Histone H1 (Upstate), anti-Cdc2 (Santa Cruz), anti-cyclin B1 (PharMingen) antibodies.
Living cell imaging and confocal microscopy. HeLa cells expressing cyclin B1-GFP were synchronized at early mitosis by nocodazole treatment and mitotic cells were collected by shake-off. The cells were cultured on a 25-mm-diameter round coverslip, which was placed in a thermally regulated chamber (Medical System Corp.) mounted on the stage of a Leica TCS SP2 confocal microscope. To measure the dynamic changes of cyclin B1-GFP in living mitotic cells, fluorescent images and phase contrast images of cells expressing the fusion gene were recorded continuously for several hours using a laser line of 488 nm. We used the MetaMorph v6.0 software (Universal Image, West Chester, PA) to analyze fluorescent images and calculate the average fluorescence intensities of cyclin B1-GFP.
Histone H1 kinase assay. Cells were lysed in lysis buffer (20 mM Hepes, pH 7.35, 12.5 mM b-glycerophosphate, 150 mM NaCl, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 0.5 % Triton X-100, 2 mM EDTA, 1 mM NaVO3, 1 mM PMSF, 20 mg/ml aprotinin) and then centrifuged at 14,000 rpm for 15 min. The supernatant was mixed with 5 mg of histone H1 peptide, 100 uM ATP, 10 mM MgCl2, 20 mM MOPS and 1 mCi [g-32P]ATP in a final volume of 30 ml and incubated for 20 min at 30˚C. The reactions were termi-nated by the addition of 50% TCA, 32P incorporation into histone H1 was quantified by scintillation counting.
Determining the concentration of endogenous cyclin B1 in a single mitotic HeLa cell. GFP-cyclin B1-D85 was transfected into HeLa cells using LipoFectamineTM 2000 reagent. After fusion gene expressed for 16-18 hrs, cells were synchronized in early M phase by treating with nocodazole (50 ng/ml) for 8 hrs. Mitotic cells were collected by shake-off. The total cell number was determined using a hemacytometer before we lysed cells. Then, using a spectrofluorim-eter, we measured the fluorescence intensities of the cell lysate and a purified GFP protein solution of known concentration. The total amount of GFP-cyclin B1-D85 fusion protein in the cell lysate was determined based on the ratio of the fluorescence intensities between the lysate and the GFP solution. Next, we conducted a western blotting assay using an anti-cyclin B1 antibody to determine the ratio of endogenous cyclin B1 and GFP-cyclin B1-D85 in the cell lysate (see Fig. 2D). From this ratio and the total number of cells, we deter-mined the amount of endogenous cyclin B1 protein in a single cell.
1640 Cell Cycle 2007; Vol. 6 Issue 13

Thresholds of MPF Inactivation Controls Different Mitotic Events
Assuming the diameter of a mitotic HeLa cell is 15 mm, the concen-tration of cyclin B1 within a mitotic HeLa cell was calculated.
Measuring the concentration of GFP‑cyclin B1‑D85 in a single mitotic HeLa cell. HeLa cells were transfected with GFP-cyclin B1-D85 fusion gene construct by electroporation.1 Cells were treated with nocodazole (50 ng/ml) for 8 hrs after gene expression for 16-18 hrs. Mitotic cells were collected by shake-off and then released from nocodazole arrest by incubating them in fresh culture medium. After release for 2.5 hr, the fluorescence intensity (F) of GFP-cyclin B1-D85 in each individual cell was measured using a confocal
microscope (TCS SP2 by Leica). To convert F into protein concen-tration (PC), we determined the PC/F ratio by using the identical confocal condition to image droplets of purified GFP solution suspended in vegetable oil (see Fig. 2C). These droplets were gener-ated by vertexing a small amount of GFP protein solution in oil. Based on measurements of many droplets in diameters ranging from 5–30 mm, we determined the experimental value of the PC/F ratio. The protein concentration of GFP-cyclin B1-D85 in each individual mitotic cell was then determined from its fluorescence intensity based on this ratio.
Figure 1. Measuring degradation kinetics of endogenous and GFP‑labeled cyclin B1. (A) Two alternative models for controlling the timing of initiating a series of events. (B) Western blot showing that the endogenous cyclin B1 decreased after release from nocodazole arrest. The level of Cdc2 was used as a loading control. (C) Average endogenous cyclin B1 protein level following release from nocodazole arrest (based on five Western blotting experiments) and the percentage of cells after M/A transition (based on three experiments). (D) Western blot showing that Cdk1 activity, as indicated by the cellular phospho‑histone H1 level, decreased after cells were released from nocodazole arrest. (E) Average Cdk1 activity in synchronized HeLa cells determined from an in vitro Histone H1 kinase assay using [g‑32P]ATP following release from nocodazole block. (F) Immunostaining of cyclin B1 in HeLa cells at prometaphase (a), metaphase (b), anaphase (c), telophase (d) and early G1 (e). Scale bar, 10 mm. (G) Imaging records showing the degradation of cyclin B1‑GFP in a living HeLa cell following nocodazole release. (H) The relative endogenous cyclin B1 protein level (determined by immunostaining) in prometaphase (PM), metaphase (M), anaphase (A), telophase (T) and G1 phase cells. (I) Average degradation kinetics of cyclin B1‑GFP based on single living cell measure‑ments in nine different HeLa cells. Time zero represents the midpoint at which half of the cyclin B1‑GFP within the cell was degraded. The arrow shows the average time of anaphase onset. (J) A diagram comparing the percentages of cyclin B reduction at M/A transition with or without overexpressing cyclin B1‑GFP (see text).
www.landesbioscience.com Cell Cycle 1641

Thresholds of MPF Inactivation Controls Different Mitotic Events
resultsIn previous studies, overexpressed GFP-labeled cyclin B1 was
found to degrade mainly before anaphase onset in mammalian cells.15-16 But, does it truly represent the degradation kinetics of the endogenous cyclin B? Thus, we compared the degradation kinetics of endogenous and overexpressed cyclin B1 in synchronized HeLa cells. Using Western blot analysis, we found the majority of endogenous cyclin B was degraded in the first hour following nocodazole release (Fig. 1B). This timing coincided with the period when most cells were undergoing M/A transition (Fig. 1C). The kinetics of cyclin B1 degradation was parallel with the kinetics of the inactivation of Cdk1, as measured either by Western blot analysis using phospho-Histone H1 antibody (Fig. 1D) or based on in vitro kinase assay using radioactive 32P-ATP (Fig. 1E).
These results suggest that degradation kinetics of cyclin B1 is not very sharp. However, these average data from synchronized cells may not necessarily reflect the degradation kinetics within an individual cell. To obtain the true degradation kinetics of cyclin B1, one must use single cell measurements. Thus, we measured the endogenous cyclin B1 protein levels by immunostaining cells fixed at different times after nocodazole release (Fig. 1F). For purpose of comparison, degradation of exogenous cyclin B1-GFP following nocodazole release was also measured using a living cell imaging technique (Fig. 1G). Figure 1H shows the average results obtained from three independent experiments on 719 cells. We found that while most endog-enous cyclin B1 was degraded before the M/A transition, about 30% of endogenous cyclin B1 still remained un-degraded when cells entered anaphase. This was significantly different from results of single living cell measurements on cyclin B1-GFP (Fig. 1I), which indicated that the amount of un-degraded cyclin B1 at anaphase onset was less than 10%.
Why was there a discrepancy between the two measurements? After a careful analysis, we found that this seemingly discrepancy was not real. The situation is explained in (Fig. 1J). Let us suppose that the endogenous cyclin B1 must decrease to about 30% of its prometaphase value before M/A transition can take place. When cyclin B1-GFP was overexpressed, the total amount of cyclin B1 increased substantially. For example, if the overexpressed cyclin B1-GFP is 2 fold of the endogenous cyclin B1, the total amount of cyclin B1 in the prometaphase cell would now become 300% of the endogenous cyclin B1. But in order to trigger the M/A transition, the cell must wait until the total cyclin B1 decreased to a level equal to 30% of the endogenous cyclin B1 at prometaphase. This means that 90% of the total cyclin B1 must be degraded before the M/A transition. This prediction is consistent with the results shown in Figure 1I.
In order to directly determine the threshold levels of cyclin B1 between different mitotic events, we conducted a series of experiments to measure the cyclin B1 levels using single cell analysis. HeLa cells were transfected with nondegradable GFP-cyclin B1-D85 and then were synchronized by nocodazole. At 2.5 hr after nocodazole release, the endogenous cyclin B1 was completely degraded.15 Then, we used a confocal microscope to record the fluo-rescent images of cells arrested in different mitotic stages, including prometaphase (PM), metaphase (Meta), anaphase (Ana), cytokinesis and G1 (Fig. 2A-B). Then, under the identical confocal conditions, we measured the fluorescent intensities of purified GFP droplets (with known concentration) in oil bath (Fig. 2C). (For details, see Materials and Methods). By comparing the fluorescent intensities of cells with that of GFP droplets, we were able to determine the concentration of GFP-cyclin B1-D85 in each individual cell.
Figure 2. Determination of the level of cyclin B1 within a single cell at various mitotic stages. (A) Sample confocal imaging records of HeLa cells expressing nondegradable GFP‑cyclin B1‑D85 that were arrested in different mitotic stages. Chromosomes were visualized by RFP‑labeled histone H4. Scale bar, 10 mm. (B) A diagram summarized the detected levels of GFP‑cyclin B1‑D85 in cells arrested in different mitotic stages based on confocal measurements. Each dot represents the result of one cell. The blue arrows (in PM and Meta) indicate the range of the mid 90% of data; the green arrows represent the Mean ± SD of the data. (C) Confocal images of purified GFP protein droplets suspended in vegetable oil. These droplets were used to convert fluorescent signals into protein concentrations. Scale bar, 10 mm. (D) Western blot analysis using anti‑cyclin B1 antibody to determine the ratio of endogenous cyclin B1 and GFP‑cyclin B1‑D85 in the cell lysate. (E) Western blot results showing the Cdk1 activity in HeLa cells collected at different times following nocodazole release under three treatments: (1) control, (2) treated with 50 mM roscovitine, and (3) 15 mM purvalanol A. Drugs were applied at 0.5 hr after nocodazole release. (F) The dosage‑dependent effects of purvalanol A (PA) on overcoming the mitotic blocks induced by cyclin B1‑D85. HeLa cells expressing GFP‑cyclin B1‑D85 were synchronized by nocodazole treatment. PA was added at 0.5 hr after nocodazole release. Cells were fixed after 3 hr and their distribution in different mitotic stages was measured.
1642 Cell Cycle 2007; Vol. 6 Issue 13
Thresholds of MPF Inactivation Controls Different Mitotic Events
Results of these measurements on 287 cells are summarized in (Fig. 2B), which showed that the nondegradable cyclin B1 levels differ significantly between cells arrested in different mitotic stages. From these results, the threshold for the M/A transition can be directly determined; it is between the lower limit of cyclin B1-D85 level in metaphase cells ( [cyclin_low]meta) and the upper limit of cyclin B1-D85 level in anaphase cells ( [cyclin_hi]ana). The latter can be taken as the “Mean+Standard Deviation (SD)” value of GFP-cyclin B1-D85 in anaphase cells (i.e., 1.26+0.59=1.85 mM). The value of [cyclin_low]meta, however, cannot be taken as the “Mean-SD” value in metaphase cells, since the mean level of nondegradable cyclin B1 in the metaphase arrested cells was dependent solely on the overexpressing efficiency of the transfected gene and thus had no physiological significance. Hence, we used a different criterion, i.e., we defined [cyclin_low]meta as the level of nondegradable cyclin B1 that was lower than 95% of cells arrested in metaphase (i.e., 0.86 mM). Based on these two limits, we determined that the threshold of cyclin B1 level for the M/A transition was 1.36±0.49 mM.
To measure the thresholds for cytokinesis and nuclear reassembly, we used a similar approach, i.e., they were determined from the lower limit (Mean-SD) and the upper limit (Mean+SD) of nondegrad-able cyclin B1 levels between two successive mitotic stages. From (Fig. 2B), the thresholds of cyclin B1 level for cytokinesis and nuclear reassembly were estimated to be 0.75±0.08 and 0.54±0.16 mM, respectively.
Finally, we determined the level of endogenous cyclin B1 at prometaphase using the following method. First, we transfected cells with GFP-cyclin B1-D85 and synchronized them at prometaphase by nocodazole. Then, using a spectrofluorimeter, we determined the concentration of GFP-cyclin B1-D85 in the cell extract by comparing its fluorescence with
a GFP solution of known concentration. Finally, using Western blot analysis with an anti-cyclin B1 antibody, we measured the ratio between the endogenous cyclin B1 and overexpressed GFP-cyclin B1-D85 (Fig. 2D). (For details, see Materials and Methods). Using this method, we determined that the average concentration of endog-enous cyclin B1 within a prometaphase cell was 2.92±1.7 mM (from four independent experiments).
To exclude the possibility that the mitotic arrest caused by overex-pressing GFP-cyclin B1-D85 could be due to factors other than the failure of MPF inactivation, we tested if applying Cdk1 inhibitors could rescue the mitotic arrest. Indeed, we found applying 50 mM roscovitine or 15 mM purvalanol A, which can effectively abolish the Cdk1 activity in mitotic cells overexpressing nondegradable GFP-cyclin B1-D85 (Fig. 2E), can also overcome their mitotic arrest (Fig. S1). Furthermore, we found that only inhibitors of Cdk1, but not inhibitors of other kinases, including ERK2, PI3K/Akt and CaMKII, could rescue the metaphase arrest by cyclin B1-D85 (Fig. S1B and C). These results suggest that Cdk1 must be specifi-cally inactivated to allow M/A transition in mammalian cells.
To further test the threshold model, we examined the dosage- dependent effects of Cdk1 inhibitor on overcoming the mitotic blocks induced by nondegradable cyclin B1. In this experiment, cells expressing GFP-cyclin B1-D85 were synchronized by nocodazole treatment. Half an hour after nocodazole release, we treated cells with different amount of purvalanol A. Three hours later, we measured the
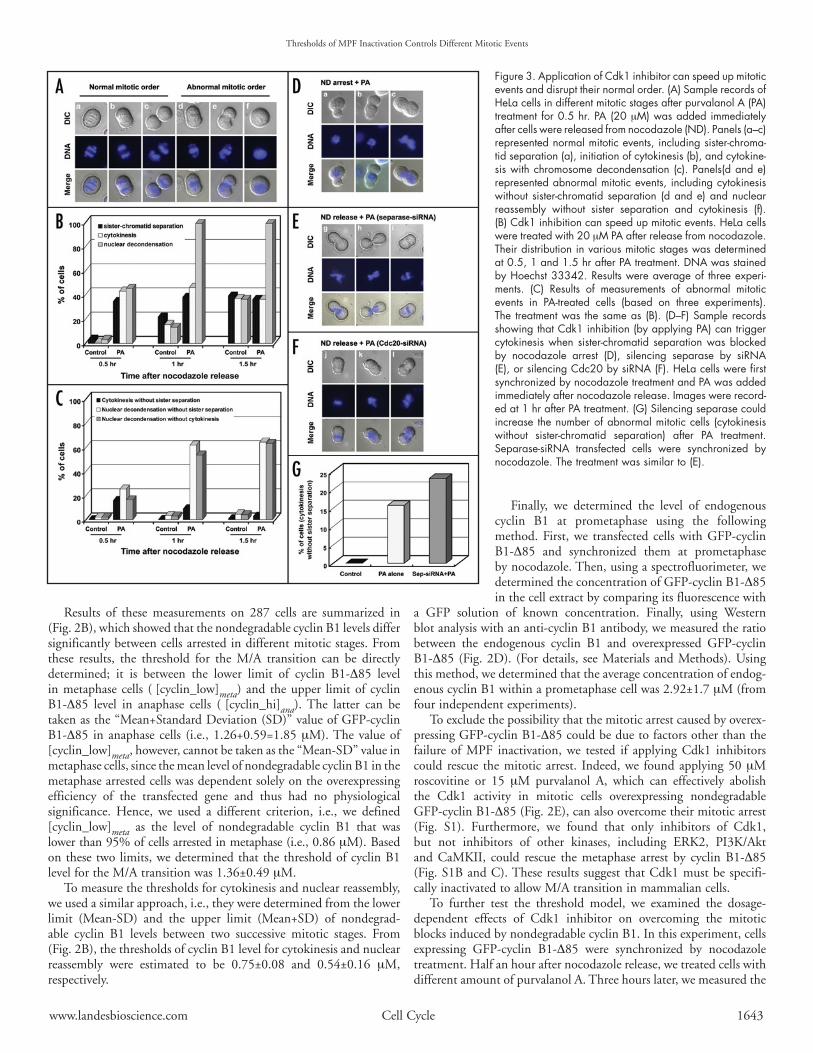
Figure 3. Application of Cdk1 inhibitor can speed up mitotic events and disrupt their normal order. (A) Sample records of HeLa cells in different mitotic stages after purvalanol A (PA) treatment for 0.5 hr. PA (20 mM) was added immediately after cells were released from nocodazole (ND). Panels (a–c) represented normal mitotic events, including sister‑chroma‑tid separation (a), initiation of cytokinesis (b), and cytokine‑sis with chromosome decondensation (c). Panels(d and e) represented abnormal mitotic events, including cytokinesis without sister‑chromatid separation (d and e) and nuclear reassembly without sister separation and cytokinesis (f). (B) Cdk1 inhibition can speed up mitotic events. HeLa cells were treated with 20 mM PA after release from nocodazole. Their distribution in various mitotic stages was determined at 0.5, 1 and 1.5 hr after PA treatment. DNA was stained by Hoechst 33342. Results were average of three experi‑ments. (C) Results of measurements of abnormal mitotic events in PA‑treated cells (based on three experiments). The treatment was the same as (B). (D–F) Sample records showing that Cdk1 inhibition (by applying PA) can trigger cytokinesis when sister‑chromatid separation was blocked by nocodazole arrest (D), silencing separase by siRNA (E), or silencing Cdc20 by siRNA (F). HeLa cells were first synchronized by nocodazole treatment and PA was added immediately after nocodazole release. Images were record‑ed at 1 hr after PA treatment. (G) Silencing separase could increase the number of abnormal mitotic cells (cytokinesis without sister‑chromatid separation) after PA treatment. Separase‑siRNA transfected cells were synchronized by nocodazole. The treatment was similar to (E).
www.landesbioscience.com Cell Cycle 1643

Thresholds of MPF Inactivation Controls Different Mitotic Events
distribution of cells arrested in various mitotic stages. We found that, in the control case, overexpressing nondegradable cyclin B arrested cells mainly before M/A transition. When increasing amounts of purvalanol A were added, cells were driven toward later mitotic stages (Fig. 2F). These findings again demonstrated that an increasing inhibition of the MPF activity could trigger the cell cycle to progress from M/A transition to cytokinesis and then nuclear reassembly.
If the threshold model is correct, i.e., the order of mitotic events should depend on the temporal gradient of the MPF inactivation, one can make several direct predictions: (1) Premature inactivation of MPF should trigger the various post-metaphase mitotic events at an earlier time. (2) If the temporal gradient of MPF inactivation is destroyed by quickly inhibiting Cdk1, the normal order of mitotic events could be disrupted. (3) Following metaphase, one should be able to trigger any mitotic event simply by inactivating MPF to a sufficiently low level, regardless other mitotic events normally before it were blocked or not.
We conducted a series of experiments to test these predictions. First, we used living cell imaging technique to examine if applying purvalanol A to cells released from nocodazole-arrest can speed up the various post-metaphase mitotic events. The results clearly supported
the predictions of the threshold model (Fig. 3A–C). For example, at 0.5 hr after nocodazole release, control cells normally have not yet entered anaphase or cytokinesis; but under the purvalanol A treatment, about 30–40% of cells had entered those mitotic stages (Fig. 3B). So, premature inhibition of Cdk1 can definitely speed up the initiation of these post-metaphase mitotic events. Furthermore, under the purvalanol A treatment, the order of mitotic events was altered in many cells, which entered cytokinesis or nuclear reassembly without undergoing chromosome separation first (Fig. 3A and C). This result demonstrated that an abrupt inactivation of MPF could disrupt the normal order of mitotic progression. We also observed that under the purvalanol A treatment, some dividing cells could later reverse their cytokinesis and fused back (see Fig. S2). Apparently, Cdk1 inactivation can mainly trigger the initiation of cytokinesis but its completion will require additional signals.
Next, we used three different methods to block sister chromatid separation and tested if cytokinesis or nuclear reassembly can still be initiated by inactivating MPF. These blocking methods included: (a) Using nocodazole to prevent the formation of mitotic spindle, so that APC cannot be activated due to the spindle checkpoint. (b) Using siRNA to silence Cdc20 to prevent the activation of APC. (c) Using siRNA to silence the protease separase. We first synchronized cells in prometaphase using nocodazole. In (a), we fixed cells at 1.5 hours after applying purvalanol A (20 mM) to inactivate MPF without washing out nocodazole. We observed that, although chromosome separation was blocked, some cells could still undergo cytokinesis when purvalanol A was applied (Fig. 3D). The blockage of sister chromatid separation under such a treatment was further verified by conducting a chromosome spread experiment (Fig. S3).
In (b) or (c), we used siRNA to silence either Cdc20 or sepa-rase before synchronizing cells with nocodazole (See Materials and Methods). After nocodazole release, we applied purvalanol A to inhibit Cdk1. Again, we observed a significant fraction of these cells undergoing cytokinesis or nuclear reassembly without sister chromatid separation (Fig. 3E–G). These results strongly suggested that inactivation of MPF alone is capable to trigger various mitotic events; they do not need to go through a sequential order. In fact, besides our own experiments, evidence supporting the predictions of the threshold model can also be found in recent literatures. It had been observed that under a sudden inhibition of Cdk1, cytokinesis or nuclear reassembly could be triggered without chromosome sepa-ration.19-21 Particularly, Potapova et al. recently reported that, under the presence of nondegradable cyclin B, inhibiting Cdk1 activity using flavopiridol could trigger cytokinesis in almost 40% of cells without prior separation of sister chromatids.21 They further demon-strated that cytokinesis was reversible when flavopiridol was washed out. Their observations can fit very well with our model.
DIsCussIoNOne may wonder why MPF inactivation can independently
trigger different mitotic events. Based on findings reported in the literature and our own studies, we proposed a detailed mechanism as shown in Figure 4A. First, the triggering of M/A transition is mainly due to the removal of Cdk1 inhibition on separase. Using Xenopus extract, Stemmann et al showed earlier that high activity of MPF can inhibit separase by phosphorylating its Ser1126 site.17 Previously, it was not clear if the required MPF activity for such inhibition was physiologically meaningful or not.15,17,22 We found in mammalian cells that one third of endogenous MPF was sufficient to inhibit
Figure 4. Diagrams showing our proposed paradigm and the underlying molecular mechanisms. (A) Outline of the molecular mechanisms that allow MPF to independently control the initiation of various mitotic events, including separation of sister chromatids, cytokinesis and nuclear reassembly. The phosphatases (PPT 1–4) responsible for counter‑acting Cdk1 on the different substrates are still under active investigation. (For details, please see supple‑mental materials). (B) A proposed model showing that different thresholds of MPF activity can determine the timing of initiating different post‑metaphase mitotic events.
1644 Cell Cycle 2007; Vol. 6 Issue 13

M/A transition.15 We also showed that mutating (or deleting) the inhibitory site Ser1126 in separase could prevent MPF to inhibit the M/A transition in HeLa cells (see Fig. S4). Second, the triggering of cytokinesis by MPF inactivation is mainly through MLC (myosin light chain). It had been suggested that MPF could phosphorylate the inhibitory sites (Ser1 and Ser2) of MLC and thus block the formation of the contractile ring.23 Alternatively, Cdk1 could inhibit the RhoA pathway through the phosphorylation of ETC2.20 Hence, MPF must be inactivated before cytokinesis can begin. Finally, the requirement of MPF inactivation for nuclear reassembly is known to be related to the dephosphorylation of condensin, histone H1 and nuclear lamin.24-28 (For details, see supplemental material).
These pathways also explain why the MPF thresholds for trig-gering different mitotic events are different. As can be seen in Figure 4A, the substrates of Cdk1 in controlling different mitotic events are not the same. Furthermore, the phosphatases responsible for counter-acting Cdk1 on these substrates can also be different. Thus, it is not surprising that their thresholds for removing the inhibitory effects of Cdk1 are different.
In conclusion, we believe that MPF serves as a master signal for controlling the temporal order of different mitotic events (Fig. 4B). At the G2/M transition, MPF is rapidly activated to allow the cell to enter M phase (due to a positive feedback mechanism). In this situation, MPF acts almost like an “ON” switch. MPF remains at a high level until the spindle checkpoint is turned off during meta-phase. Then, MPF is gradually inactivated due to the destruction of cyclin B1 by the Cdc20/APC. (We have shown that cyclin B1 was degraded by Cdc20-activated APC instead of Cdh1-activated APC, see Fig. S5). In this situation, MPF acts almost like an “analog” signal (as used in most electronic device). Different levels of MPF inactiva-tion would trigger the initiation of different mitotic events, and their temporal order is determined by their thresholds (Fig. 4B).
This paradigm is not only consistent with our findings reported here, it can also reconcile with a large amount of earlier findings which previously appeared as contradicting. For example, from works on various biological models systems, there were conflicting reports on whether inactivation of MPF was required for exit of M phase or anaphase onset.4-10 With the threshold model, one can explain all these findings. Since MPF acts like an analog signal that can control multiple events, these earlier reports may not necessarily be mutually exclusive.
References1. Murray AW, Solomon MJ, Kirschner MW. The role of cyclin synthesis and degradation in
the control of maturation promoting factor activity. Nature 1989; 339:280-6.2. Ghiara JB, Richardson HE, Sugimoto K, Henze M, Lew DJ, Wittenberg C, Reed SI. A
cyclin B homolog in S. cerevisiae: chronic activation of the Cdc28 protein kinase by cyclin prevents exit from mitosis. Cell 1991; 65:163-74.
3. Luca FC, Shibuya EK, Dohrmann CE, Ruderman JV. Both cyclin A delta 60 and B delta 97 are stable and arrest cells in M- phase, but only cyclin B delta 97 turns on cyclin destruction. EMBO J 1991; 10:4311-20.
4. Gallant P, Nigg EA. Cyclin B2 undergoes cell cycle-dependent nuclear translocation and, when expressed as a non-destructible mutant, causes mitotic arrest in HeLa cells. J Cell Biol 1992; 117:213-24.
5. Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell 1993; 73:1393-402.
6. King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science 1996; 274:1652-9.
7. Morgan DO. Regulation of the APC and the exit from mitosis. Nat Cell Biol 1999; 1:E47-53.
8. Farr KA, Cohen-Fix O. The metaphase to anaphase transition: a case of productive destruc-tion. Eur J Biochem 1999; 263:14-9.
9. Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J 1993; 12:1969-78.
10. Yamano H, Gannon J, Hunt T. The role of proteolysis in cell cycle progression in Schizosaccharomyces pombe. EMBO J 1996; 15:5268-79.
11. Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999; 400:37-42.
12. Nasmyth K, Peters JM, Uhlmann F. Splitting the chromosome: Cutting the ties that bind sister chromatids. Science 2000; 288:1379-85.
13. Hauf S, Waizenegger IC, Peters JM. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science 2001; 293:1320-3.
14. Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 2000; 103:399-410.
15. Chang DC, Xu N, Luo KQ. Degradation of cyclin B is required for the onset of anaphase in Mammalian cells. J Biol Chem 2003; 278:37865-73.
16. Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol 1999; 1:82-7.
17. Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW. Dual inhibition of sister chromatid separation at metaphase. Cell 2001; 107:715-26.
18. Hunt T, Luca FC, Ruderman JV. The requirements for protein synthesis and degradation, and the control of destruction of cyclins A and B in the meiotic and mitotic cell cycles of the clam embryo. J Cell Biol 1992; 116:707-24.
19. Canman JC, Cameron LA, Maddox PS, Straight A, Tirnauer JS, Mitchison TJ, Fang G, Kapoor TM, Salmon ED. Determining the position of the cell division plane. Nature 2003; 424:1074-8.
20. Niiya F, Xie X, Lee KS, Inoue H, Miki T. Inhibition of cyclin-dependent kinase 1 induces cytokinesis without chromosome segregation in an ECT2 and MgcRacGAP-dependent manner. J Biol Chem 2005; 280:36502-9.
21. Potapova TA, Daum JR, Pittman BD, Hudson JR, Jones TN, Satinover DL, Stukenberg PT, Gorbsky GJ. The reversibility of mitotic exit in vertebrate cells. Nature 2006; 440:954-8.
22. Hagting A, Den Elzen N, Vodermaier HC, Waizenegger IC, Peters JM, Pines J. Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J Cell Biol 2002; 157:1125-37.
23. Satterwhite LL, Pollard TD. Cytokinesis. Curr Opin Cell Biol 1992; 4:43-52.24. Shimada A, Ohsumi K, Kishimoto T. An indirect role for cyclin B-Cdc2 in inducing
chromosome condensation in Xenopus egg extracts. Biol Cell 1998; 90:519-30.25. Roberge M, Th’ng J, Hamaguchi J, Bradbury EM. The topoisomerase II inhibitor VM-26
induces marked changes in histone H1 kinase activity, histones H1 and H3 phosphoryla-tion, and chromosome condensation in G2 phase and mitotic BHK cells. J Cell Biol 1990; 111:1753-62.
26. Kimura K, Hirano M, Kobayashi R, Hirano T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 1998; 282:487-90.
27. Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 1990; 61: 591-602.
28. Luscher B, Brizuela L, Beach D, Eisenman RN. A role for the p34cdc2 kinase and phospha-tases in the regulation of phosphorylation and disassembly of lamin B2 during the cell cycle. EMBO J 1991; 10:865-75.
Thresholds of MPF Inactivation Controls Different Mitotic Events
www.landesbioscience.com Cell Cycle 1645





























