Embed Size (px)
Citation preview

Journal of PathologyJ Pathol 2003; 200: 298–307.Published online 17 March 2003 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/path.1366
Original Paper
Discriminating expression of differentiation markersevolves in transplants of benign and malignant humanskin keratinocytes through stromal interactions‡‡
Pascal Tomakidi,1† Hans-Juergen Stark,1 Christel Herold-Mende,2 Franz Xaver Bosch,2 Heinrich Steinbauer,1
Norbert E Fusenig1 and Dirk Breitkreutz1*1Division of Differentiation and Carcinogenesis, German Cancer Research Centre (DKFZ), Heidelberg, Germany2Ear, Nose and Throat Hospital, Laboratory of Molecular Cell Biology, University of Heidelberg, Heidelberg, Germany†Present address: Department of Orthodontics and Dentofacial Orthopedics, Dental School, University of Heidelberg, Heidelberg, Germany
*Correspondence to:Dr Dirk Breitkreutz, Division ofDifferentiation andCarcinogenesis, B0600 GermanCancer Research Centre (DKFZ)Im Neuenheimer feld 280,D-69120 Heidelberg, Germany.E-mail: [email protected]
‡ This article is dedicated toProfessor Harald zur Hausen onthe occasion of his retirement asHead of the German CancerResearch Center (DeutschesKrebsforschungszentrum) inHeidelberg with gratitude andappreciation for 20 yearsleadership.
Received: 11 March 2002Revised: 29 October 2002Accepted: 28 January 2003
AbstractAccumulating evidence indicates a decisive role for the adjacent stroma in tumour growthand dissemination. However, it is not clear how far altered differentiation such as expressionof aberrant keratins and vimentin, common in invasive human carcinomas, may reflectintrinsic cell properties or a response to the tumour environment. We have addressed thisby transplanting benign and malignant human HaCaT-ras keratinocytes, seeded on collagenmatrix, onto nude mice. Initially, epithelia derived from benign and malignant cells, beingseparated from host stroma by collagen, were poorly organized and exhibited the samedifferentiation markers, as identified by immunofluorescence and in situ hybridization.Epidermal basal and suprabasal keratins were expressed persistently even upon contactwith newly formed stroma and malignant cell invasion. In contrast, non-epidermal keratins(K4/K13, K8/18, K19), which were similarly synthesized by benign and malignant cellsin culture and in early transplants, were differentially regulated with increasing stromalvicinity. While both proteins and mRNAs were downregulated in benign epithelia, themalignant, invasive tumour cells continuously expressed these non-epidermal keratinsthroughout (K19), suprabasally (K4/13) or at invasive sites (K8/18). Furthermore, themesenchymal protein vimentin was expressed de novo in invasive areas confronting tumourstroma. Thus, atypical tissue markers, similarly synthesized in isolated cells in vitro, aredownregulated in benign but maintained and upregulated in malignant epithelia. This ispresumably caused by the neighbouring stroma being permanently activated by malignantepithelia. Copyright 2003 John Wiley & Sons, Ltd.
Keywords: malignant human keratinocytes; mouse grafts; stromal reaction; atypicalkeratins; vimentin
Introduction
Over the past two decades, cancer research has beenlargely focused on events within cancer cells includ-ing genetic alterations of oncogenes and suppressorgenes [1,2]. However, tumours harbour a consider-able portion of stroma, consisting of connective tissue,fibroblasts, and endothelial and inflammatory cells,which plays an important role in tumour growth, inva-sion, and metastasis [3–5]. Apparently, the reciprocalexchange of molecular signals gives rise to alteredstromal cells which can enhance or establish the ulti-mate malignant phenotype of tumorigenic epithelialcells [6]. Authentic squamous cell carcinomas (SCCs)as well as transitional cell carcinomas (TCCs) derivedfrom different anatomical sites such as skin, oralmucosa, or other internal epithelia frequently exhibitalterations in keratin expression related to tumourgrade [7–9]. Thus, the simple epithelial keratin pairK8/K18 is frequently expressed in SCCs of the oral
cavity and epidermis, frequently at the invasive front,and also in some pre-malignant lesions [7,9]. Prefer-ential de novo synthesis of vimentin, an intermedi-ate filament protein common to cells of mesenchy-mal origin such as fibroblasts, has also been observedin that location [9]. Furthermore, downregulation ofdifferentiation-specific keratins closely correlated withtumour grade, while even loss of the basal cell-specificK5/K14 has been reported in undifferentiated tumours[7,9].
Assessment of alterations in these tissue markersin correlation to tumour growth can only mirror theactual, often advanced, tumour stage of squamous cellcarcinomas. Experimentally, the significance of thesealterations for tumour growth and development andin turn their modulation by the tumour environmentcannot be elucidated in conventional cell cultures, pre-dominantly because they lack stromal elements. Ingeneral, isolated epithelial cells undergo only limiteddifferentiation in vitro for that reason [10,11]. As
Copyright 2003 John Wiley & Sons, Ltd.

Differentiation and stromal interactions in malignant cell transplants 299
an alternative tissue model tumorigenic keratinocyteclones, derived from the human keratinocyte lineHaCaT after transfection with the cellular Ha-rasoncogene (HaCaT- ras cells), were grown as surfacetransplants on nude mice. These cell grafts developbenign or malignant epithelia that allow the study ofthe early and intermediate stages of squamous epithe-lial malignancy [12–14]. Thus, we were able first todistinguish the stages of tumour morphogenesis andsecond to analyse molecular alterations and growthcharacteristics at early and late stages of the develop-ment of the malignant phenotype [15,16]. Previously,we had assessed in this model general defects in dif-ferentiation, alterations of the basement membrane(BM) zone, and specific host responses includingstroma formation and angiogenesis [12,14,17]. Hereinwe have investigated (i) whether and to what extentthe different dynamics of growth characteristics inbenign and malignant HaCaT-ras grafts are reflectedby alterations in keratin expression and acquisition ofvimentin expression and (ii) how this correlates withconversion to either transiently or permanently acti-vated stromal tissue. The finding that atypical keratinsgradually disappeared in benign grafts while these ker-atins persisted, with de novo expression of vimentin,in malignant grafts, strongly suggests differential func-tions of their respective stromal tissues.
Materials and methods
Cell culture and transplantation
Animal experiments were performed according tothe guidelines of the Ethics Commission of theGerman Cancer Research Centre, Heidelberg. Offi-cial approval was obtained from the State Govern-ment, the ‘Regierungspraesidium Karlsruhe’ in Baden-Wuerttemberg, given to Professor Norbert E. Fusenig,valid until 15 April 2003 (notification dated 22 March2002/KI; Dr Reichert, ref no 35-9185.81/34/99). Thehuman cell lines had been established previously.
The benign (I-7) and malignant HaCaT-ras clones(II-4) were derived from the human keratinocyteline HaCaT after transfection with mutated c-Ha-ras (EJ) in a neo-vector [15]. Cultures were grownin enriched medium (4 × MEM [12]) containing 5%fetal bovine serum and 400 µg/ml geneticin (G-418,GIBCO, Berlin, Germany) to maintain selection pres-sure. For transplantation, cells were cultured on typeI collagen matrix (rat tail tendon), grafted onto thedorsal muscle fascia of nude mice (BALB/c NU, IFACREDO, Lyon, France), and harvested at the timesindicated [14,17].
Immunohistochemistry (IHC)
Cryostat sections (6–8 µm) were mounted on adhe-sive slides (Histobond, Marienfeld, Germany), fixed in80% methanol and in acetone (5 min each, 4 ◦C), andincubated with primary antibodies overnight at 4 ◦C
following protocols described previously [14]. Mousemonoclonal antibodies (mabs) against keratins K1/10(Ks 8.60), K4, K8, K13, K18, and K19 were pur-chased from Dianova (Hamburg, Germany) and Pro-gen (Heidelberg, Germany), while mab LH8, detectingK14 only in basal cells, was donated by Irene Leigh(London, UK [18]). For indirect immunofluorescence(IIF) samples were washed in phosphate-bufferedsaline (PBS), incubated with secondary fluorochrome-conjugated antibodies (from Dianova and Sigma,Deisenhofen, Germany) for 1 h at room temperature,mounted in Vectashield (Vector, Wertheim, Germany),and photographed using a Zeiss ICM-405 invertedmicroscope (Oberkochen, Germany).
In situ hybridization (ISH)
mRNAs were detected on frozen sections by ISH usingα35S-CTP-labelled riboprobes according to Boschet al. [19]. Plasmids containing the cDNAs for K1,K5, K8, K14, K18, and vimentin were kindly pro-vided by W W Franke and R Leube (German CancerResearch Center, Heidelberg, Germany) [20]. Sectionswere fixed in 4% paraformaldehyde, digested withproteinase K (10 µg/ml), and hybridized at 53 ◦C in50% formamide overnight. Specimens were washed insaline sodium citrate (SSC) buffer containing 50% for-mamide, treated with RNase A (10 µg/ml), dehydratedin ethanol, and covered with NTB2 emulsion (Kodak,New Haven, USA). After exposure, slides were devel-oped, counterstained with haematoxylin and eosin,and mounted in Elvanol (Hoechst/Aventis, Frankfurt,Germany).
Results
Tumour morphology is determined by stromalinteractions
In the transplants of both benign (I-7) and malig-nant (II-4) HaCaT-ras cells the collagen matrix ini-tially prevented stromal contacts, but allowed forma-tion of multilayered, though poorly organized, epithe-lia within 1 week (Figure 1a, b). Concomitantly, accu-mulation of host stromal cells was visible beneath thegrafts particularly of the malignant cells, leading topenetration of host cells and blood vessels into thecollagen matrix (Figure 1b; arrows, advancing cells).Around week 2, benign cells had formed hyperplasticbut differentiated epithelia, still separated from hosttissue by a thin collagen layer (Figure 1c). Althoughmalignant cells formed differentiated epithelia, tis-sue polarity was disturbed and collagen was mostlyreplaced by host stroma. This contact with the newlyformed stroma, in particular the close vicinity of smallblood vessels, strongly correlated with focal inva-sion (Figure 1d, inset [14,17]). While benign epitheliaremained well structured and non-invasive upon stro-mal contact, and became highly keratinized (Figure 1e,3 weeks), malignant epithelia were heavily infiltrated
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

300 P Tomakidi et al
Figure 1. Histology of xenografts of benign I-7 (a, c, e) and malignant II-4 (b, d, f) HaCaT-ras cells on nude mice. Stratified butpoorly organized epithelia at week 1 (a, b) with preserved collagen lattices. Note the more irregular growth of malignant cellsand the accumulation of blood vessels at the collagen under-surface (b): arrows mark the advance of infiltrating cells from theconnective tissue. After 2 weeks, the epithelium of benign cells is slightly dysplastic but the collagen is largely intact (c) whereas theonset of epithelial invasion in the malignant graft is confronted by activated stroma replacing the collagen (d). The inset in (d) showsa group of micro-vessels in close vicinity to the epithelium. At week 3, there is a structured and differentiated non-invadingbenign epithelium with stromal contact (e) which contrasts with the formation of squamous cell carcinoma by malignant cells withpronounced epithelial invaginations by stromal strands (f). Bars 50 µm (a–e) and 100 µm (f)
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

Differentiation and stromal interactions in malignant cell transplants 301
by stromal strands containing numerous capillaries.Progressive growth eventually resulted in large, inva-sive tumour masses (Figure 1f) which resembled well-differentiated SCCs and displaced or destroyed hosttissue. In areas, dermal structures such as nerve, mus-cle and vascular tissue from the host dermis seemedto be engulfed by these malignant epithelia.
Maintained expression of epidermal keratins in celltransplants
In spite of differences in growth behaviour and tissuemorphology, benign and malignant cells continued to
express the epidermal keratins K5/K14 and K1/K10,though with slight irregularities in localization. Thiswas independent of stromal influence and irrespectiveof the final tumour phenotype, reflecting the differenti-ation potential in culture (Table 1 [10,22]). During theinitial growth phase K5 and K14 mRNA and proteinwere present throughout benign and malignant grafts(not shown). These keratins finally became restrictedto basal cells, as in the epidermis [11,21], in trans-plants of normal or HaCaT. This also occurred par-tially in benign, but did not occur at all in malignant,HaCaT-ras grafts, several suprabasal layers remain-ing positive (Figure 2a, b). Herein K14 was detected
Figure 2. (a, b) Maintenance of basal cell-specific K14 (IIF) in I-7 (a) and II-4 (b) transplants after a grafting period of 4 weeks,appearing largely normal in (a), but extending up the epithelium in (b). (c–h) Changes in expression (ISH, 35S-labelled antisenseprobe; bright-field) and protein localization (IIF) of the differentiation markers K1 and K10 in benign I-7 (g) and malignant II-4epithelia (c-f, h). Analysis of the expression of K1 mRNA (c, e), and localization of the keratin pair K1/10 (d, f, g, h) 3 days (c, d),7 days (e, f), and 4 weeks (g, h) after grafting shows persisting normalization only in the benign graft (g). In (h) type IV collagenstaining is superimposed for clarity. This marks a marginal basement membrane zone (arrows) and approaching blood vessels inlongitudinal and cross-section. Arrows indicate epithelial–matrix interface; bars 50 µm (a, b, g) and 100 µm (c-f, h)
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

302 P Tomakidi et al
Table 1. Keratin patterns in HaCaT/HaCaT-rasculturesa
KeratinbHaCaTc
(n-t)I-7
(ben)II-3
(mal)
K1 +++ + +/−K4 + + +K5 +++ +++ +++K8 + ++ ++K10 ++ + +/−K13 ++ ++ ++K14 +++ +++ +++K18 + + +K19 + ++ ++a Arbitrary values (+/− to +++) modified fromprevious data [10,22], 2D gel electrophoresis(NEPHGE/SDS-PAGE) of total or cytoskeletal proteinextracts from confluent cultures.b Other keratins present (K6, 7, 15, 16, 17), not listedfor simplicity.c HaCaT cells (non-tumorigenic), ras clones I-7(benign), and II-3 (malignant; similar to II-4).
with the conformation-dependent antibody LH8 whichexclusively stains basal cells in normal epidermis, butmore cell layers in hyperproliferative processes such ashealing wounds or psoriasis [18]. For the keratin pairK5/K14 the patterns at the mRNA and protein levelcorresponded to one another (as in Figure 2a, b) butwere mutually exclusive to K1/K10. Thus, with pro-ceeding differentiation K1 mRNA and K1/K10 proteinexpanded within the suprabasal compartment. Thisis shown for malignant transplants by comparing 3(Figure 2c, d) and 7 days (Figure 2e, f). While up tothis stage these patterns developed similarly towards
normality in benign and malignant grafts, the K1/K10distribution became largely regular in benign epithe-lia thereafter, starting mostly in the first suprabasallayer (Figure 2g, 4 weeks; also [11,14,21]). In con-trast, malignant epithelia showed a general delay in theexpression of K1/K10 retracting to upper suprabasalpositions at later stages, although this was still seenin large invasive tumour masses (Figure 2h, 4 weeks).For better visibility, labelling for type IV collagen issuperimposed showing remnants of the epithelial base-ment membrane and adjacent blood vessels (for detailssee [17,21]).
Aberrant keratins are downregulated in benign butnot in malignant grafts
Generally, parental HaCaT, benign, and malignantcells expressed keratins typical for regenerating epi-dermis (K6/K16), mucosa (K4/K13), simple (K8/K18),and transitional epithelia (K19) in addition to the epi-dermal keratins (K5/K14, K1/K10) in culture. Sincekeratin expression was similarly modulated in all celllines, virtually independent of transformation stage(Table 1, [10,22]), benign and malignant cells couldnot be discriminated on that basis. Although all ker-atins expressed in vitro were initially present in benignand malignant transplants, too, increasing interac-tions with host stroma disclosed differential regulation.Thus, a progressive downregulation of aberrant ker-atins was observed in benign epithelia with approach-ing stroma. This shift was most striking for K19which was strongly expressed throughout after 1 week(Figure 3a), but only faintly seen in the uppermost
Figure 3. Loss of keratin K19 in benign I-7 (a, c) and constitutive abundance in malignant II-4 transplants (b, d). K19 is distributedhomogeneously in I-7 (a) and II-4 (b) epithelia at week 1. However, it is restricted to the shedding superficial layers in I-7 grafts atweek 3 (c) but persists in malignant epithelium at the onset of invasion (d, 2 weeks). Arrows indicate epithelial–matrix interface;bars 50 µm (a, b) and 100 µm (c, d)
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.
Differentiation and stromal interactions in malignant cell transplants 303
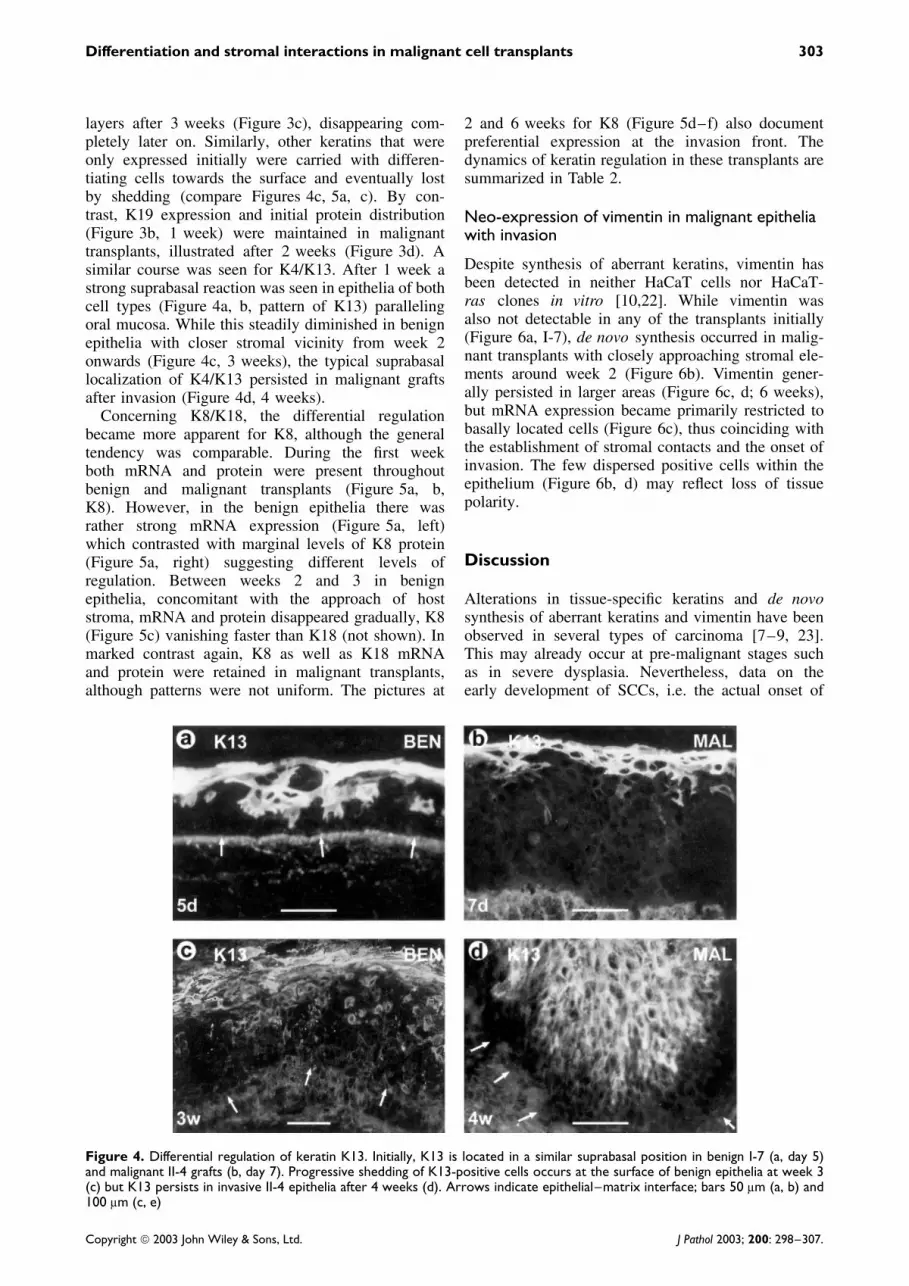
layers after 3 weeks (Figure 3c), disappearing com-pletely later on. Similarly, other keratins that wereonly expressed initially were carried with differen-tiating cells towards the surface and eventually lostby shedding (compare Figures 4c, 5a, c). By con-trast, K19 expression and initial protein distribution(Figure 3b, 1 week) were maintained in malignanttransplants, illustrated after 2 weeks (Figure 3d). Asimilar course was seen for K4/K13. After 1 week astrong suprabasal reaction was seen in epithelia of bothcell types (Figure 4a, b, pattern of K13) parallelingoral mucosa. While this steadily diminished in benignepithelia with closer stromal vicinity from week 2onwards (Figure 4c, 3 weeks), the typical suprabasallocalization of K4/K13 persisted in malignant graftsafter invasion (Figure 4d, 4 weeks).
Concerning K8/K18, the differential regulationbecame more apparent for K8, although the generaltendency was comparable. During the first weekboth mRNA and protein were present throughoutbenign and malignant transplants (Figure 5a, b,K8). However, in the benign epithelia there wasrather strong mRNA expression (Figure 5a, left)which contrasted with marginal levels of K8 protein(Figure 5a, right) suggesting different levels ofregulation. Between weeks 2 and 3 in benignepithelia, concomitant with the approach of hoststroma, mRNA and protein disappeared gradually, K8(Figure 5c) vanishing faster than K18 (not shown). Inmarked contrast again, K8 as well as K18 mRNAand protein were retained in malignant transplants,although patterns were not uniform. The pictures at
2 and 6 weeks for K8 (Figure 5d–f) also documentpreferential expression at the invasion front. Thedynamics of keratin regulation in these transplants aresummarized in Table 2.
Neo-expression of vimentin in malignant epitheliawith invasion
Despite synthesis of aberrant keratins, vimentin hasbeen detected in neither HaCaT cells nor HaCaT-ras clones in vitro [10,22]. While vimentin wasalso not detectable in any of the transplants initially(Figure 6a, I-7), de novo synthesis occurred in malig-nant transplants with closely approaching stromal ele-ments around week 2 (Figure 6b). Vimentin gener-ally persisted in larger areas (Figure 6c, d; 6 weeks),but mRNA expression became primarily restricted tobasally located cells (Figure 6c), thus coinciding withthe establishment of stromal contacts and the onset ofinvasion. The few dispersed positive cells within theepithelium (Figure 6b, d) may reflect loss of tissuepolarity.
Discussion
Alterations in tissue-specific keratins and de novosynthesis of aberrant keratins and vimentin have beenobserved in several types of carcinoma [7–9, 23].This may already occur at pre-malignant stages suchas in severe dysplasia. Nevertheless, data on theearly development of SCCs, i.e. the actual onset of
Figure 4. Differential regulation of keratin K13. Initially, K13 is located in a similar suprabasal position in benign I-7 (a, day 5)and malignant II-4 grafts (b, day 7). Progressive shedding of K13-positive cells occurs at the surface of benign epithelia at week 3(c) but K13 persists in invasive II-4 epithelia after 4 weeks (d). Arrows indicate epithelial–matrix interface; bars 50 µm (a, b) and100 µm (c, e)
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

304 P Tomakidi et al
Figure 5. Transient expression of keratin K8 in benign I-7 (a, c) and continuity in malignant II-4 epithelia (b, d–f). While K8 mRNA(a, on the left, ISH, dark-field; also e) and protein are detectable in I-7 (a) and II-4 grafts (b) at week 1, both are disappearing in I-7epithelia from week 2 onwards (c), but remain in II-4 grafts (d–f) which show expression of mRNA after 6 (e) and K8 protein after2 (d) and 6 weeks (f). Small arrows indicate epithelial–matrix interface; large arrows indicate epithelial invaginations by protrudingstroma; bars 50 µm (a, b) and 100 µm (c–f)
Table 2. Keratins and vimentin in I-7 and II-4 transplants
Filament type I-7 (ben) II-4 (mal)
Keratins Transplant (weeks) Transplant (weeks)Category 1 2 3 & 4 1 2 4 & 6
K5, 14 +++ ++ ++ +++ ++ ++Epid.–basalK1, 10 ++ +++ +++ ++ ++ ++SuprabasalK19 ++ + − ++ ++ ++TransitionalK4∗ , 13∗∗ ++ −∗/+∗∗ − ++ ++ ++InternalK8 + − − + + +K18 ++ + − ++ ++ +Simple
Vimentin − − − − + ++
Relative quantity of keratins and vimentin (arbitrary scale):+++, strong; ++, intermediate variable; +, low or irregular reaction;−, complete loss or only exceptional expression.
invasion, are scarce. As an experimental alternative,we were able to reproduce the sequence of events
from early to late stages, representing a fully malignantphenotype, in transplants of human HaCaT-ras cloneswith different neoplastic potential [12,15].
Divergent epithelial development upon exposureto stromal tissue
Because cells were grafted as epithelial sheets oncollagen matrix, early events in both the epithe-lial and host connective tissue compartments wereclearly distinguishable. The early epithelial phenotypein grafts of benign and malignant cells was similar, aswas their keratin profile. Besides epidermal keratins,this included keratins present in stratified internal,transitional, and simple epithelia, in this regard resem-bling profiles from expanding cultures (Table 1). Note-worthy in this context is that high density and externalstimuli that promoted differentiation suppressed theaberrant keratins equally in both benign and malig-nant cell cultures [10]. While vicinity to host stromaled to stable, differentiated epithelia with downregula-tion of non-epidermal keratins in the benign grafts,
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

Differentiation and stromal interactions in malignant cell transplants 305
Figure 6. Synthesis of vimentin in malignant II-4 (b–d), but not in benign I-7 epithelia (a, 1 week). There is partial overlap ofvimentin mRNA (c, ISH, dark-field) and the more abundant protein (b, d) largely at the epithelial periphery, shown 2 (b) and6 weeks after grafting (c, d). Some scattered positive cells usually remain, as observed for other markers in malignant epithelia.Arrows indicate epithelial–matrix interface; bars 50 µm (a, d) and 100 µm (b, c)
aberrant keratins remained expressed upon stromalconfrontation in malignant epithelia. Although stillcircumstantial evidence, this strongly suggests dis-tinct regulatory influences by the respective micro-environment when taken together with the in vitrodata. Intimate contact with a bona fide tumour stroma(see below) may actually be responsible for increas-ingly polarized expression of K8/K18 and vimentintowards the stromal interface. Overall levels of the epi-dermal markers K5/K14 and K1/K10 were fairly wellmaintained but, in the malignant epithelia, differentia-tion specific K1/K10 expression became delayed withthe expanding proliferative compartment [14]. Never-theless, other pathological conditions may alter expres-sion patterns as well. Thus, K19 is newly induced inkeratinized oral gingiva by inflammation [24]. Simi-larly, K1/K10 is induced in the buccal mucosa, whichis not normally keratinized, of smokers [25], whereasK1/K10 is decreased in psoriatic epidermis [18].
Aberrant cytoskeletal proteins in experimentaltumours
The abundance of aberrant keratins in our graftswas comparable to (i) tumours generated in the two-stage mouse carcinogenesis model which reveal strongK4/K13 expression [26] and (ii) a tumour modelin transgenic mice with epidermis-targeted HPV16expression activating K8, K13, and K19 [27]. Inanother grafting system using v-ras transfected mouse
keratinocyte lines, K8 was upregulated by TGF-αspecifically in malignant variants [28], showing asimilar localization of K8/K18 and vimentin at thetumour invasion front to that shown in our study.This restricted localization also explains why wecould not detect these proteins biochemically in corre-sponding specimens, despite metabolic labelling andhigh-resolution analysis [10,12]. Interestingly, tran-sient expression of vimentin correlating with motilityhas been observed in mouse keratinocyte lines [29,30],human keratinocyte cultures [31], and conventionalco-cultures with fibroblasts [32]. Similarly, K8/K18may correlate with migratory potential as postulatedfor embryonic development and intestinal epithelium.Immunoelectron microscopy of our malignant graftssuggests that the fine filament bundles in basal cellscontain other keratins than the regular K5/K14 pair,such as K8/K18 [33].
Impact of mutual epithelial–stromal interactions
Although ultimate mechanisms providing definiteproof remain elusive, the different dynamics of aber-rant keratin expression most likely reflect the roleof stroma in the development of the tumour pheno-type. One destabilizing effect that presumably pro-motes progression of tumour development is the vastbasement membrane destruction in malignant grafts
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

306 P Tomakidi et al
[12,14]. Loss of this negatively charged biochemi-cal barrier based on release of basement membrane-integrated proteoglycans, especially of perlecan [34],may enhance activating stimuli from neighbouringstroma and relieve constraints on cell movements.Such changes in the intimate matrix scaffold shouldprofoundly influence cell–matrix interactions. Accord-ingly, little or no basement membrane material wasobserved by immunofluorescence and ultrastructurerevealed close association of malignant cells with typeI collagen fibrils (Mirancea et al., in press) not seenin other transplants [11,14,21]. This also suggests anincreasing dominance of β1 integrins, namely α2β1 asthe main collagen receptor, over α6β4, which bindslaminins [34,35]; this is compatible with the alteredintegrin distribution and defective hemidesmosomes(14; Mirancea et al., in press).
Concerning the postulated stromal reaction, malig-nant tumour cells, unlike healing epithelia, seem toactivate surrounding connective tissue constitutively,giving rise to a typical cell- and matrix-rich tumourstroma [13,16,17]. The detection of vascular basementmembrane components (type IV collagen, laminins) orendothelial surface molecules (e.g. PECAM, α6 inte-grin, Ulex europeus lectin-binding) revealed intensevessel growth, preferentially directed towards malig-nant epithelia (not shown; examples in [17]). Othersurface markers are currently being applied in orderto characterize in more detail the host cell infil-trate at the graft site, with the major focus being onmyofibroblasts, smooth muscle cells, monocytes andmacrophages. While dermal tissue elements may beengulfed by tumour tissue, very recent ultrastructuralobservations (own unpublished observations) supportthe view that active, directed endothelial migration,with infiltration of vessels and intimate epithelial con-tacts, occurs. In turn, this stromal activation appar-ently not only supports expanding tumour growth, butalso modulates differentiation mediated by extracellu-lar matrix as in mammary gland development [35,36]or by stromal factors as demonstrated for fibrob-lasts in a skin model in vitro [37]. There are severallines of evidence indicating the essential regulatoryfunctions of activated stroma in the development ofthe tumour phenotype. (i) Interfering with angiogene-sis using VEGF-receptor neutralizing antibodies pre-vented epithelial invasion of II-4 cells in this transplantmodel, and also normalized gross morphology [17].(ii) By transfection-mediated constitutive expressionof PDGF, which generally induces granulation tissueand angiogenesis, the non-tumorigenic HaCaT cellsprovoked stromal activation similar to that in tumours.In turn, this enhanced epithelial growth and these cellsformed benign tumours upon subcutaneous injection[38]. (iii) As an additional hint, our I-7 and II-4 cellsgrowing readily on type I collagen in 3D cultures werefurther stimulated by the presence of fibroblasts. Thisinduced in epithelia derived from II-4 cells a con-siderable loss of tissue polarity marked by extended,
irregular integrin α6β4 patterns resembling the phe-notype in vivo (own unpublished observations). (iv) Ina similar model with ‘initiated’ prostate tumour cellsonly tumour stroma-derived, not normal fibroblasts,promoted a neoplastic phenotype [39]. (v) Along theselines, HPV16-transfected human keratinocytes formedmore regular or metaplastic epithelia in co-culture witheither 3T3 cells or skin fibroblasts respectively [40].
Conclusions
Distinct key features of this model were (i) thepersistence of aberrant non-epidermal keratins andneo-expression of vimentin in malignant transplantsand (ii) the downregulation of aberrant keratinsin benign transplants. Altered expression of thesecytoskeletal proteins may reflect more general phe-nomena in carcinogenesis. Quite a few genes, normallysilent in the epidermis, may be either constitutively orfacultatively expressed upon appropriate environmen-tal stimulation. Some may serve merely as diagnosticmarkers such as K4/K13 or K19, perhaps without pro-found functional consequences (‘blind passengers’),while others such as K8/K18 and vimentin may pro-mote migratory activities, facilitating invasion. Thedifferent levels of regulation in cells or tissues presum-ably reflect the degree of transformation towards thefully malignant phenotype, progressing in our systemfrom pre-malignant HaCaT and benign to malignantHaCaT-ras cells. On the other hand, the discriminatingregulation of differentiation markers at distinct stagesof carcinogenesis is most likely determined by the stateof stromal activation induced by the epithelial tumourcells. Although not providing definitive proof, thesefindings contribute to the growing body of evidencefor a crucial role of mutual tumour–stroma interac-tions in carcinoma progression.
Acknowledgements
We are grateful to Drs Irene Leigh and Rudolf Leube for theirgenerosity in providing antibodies and probes. We would alsolike to thank Drs Petra Boukamp and Hans Smola for sup-port and valuable discussion, Elke Tomakidi and Antje Schu-mann for excellent technical assistance, the photo departmentat DKFZ for artwork, and Martina Kegel for expert typing.This work was supported by grants from the TumorzentrumHeidelberg-Mannheim (FB) and the Deutsche Forschungsge-meinschaft (DFG PT/KO 1531/1-2; DFG FU 91/5-1).
References
1. Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW,Vogelstein B. Evidence that genetic instability occurs at an earlystage of colorectal tumorigenesis. Cancer Res 2001; 61: 818–822.
2. McCormick F. Signalling networks that cause cancer. Trends CellBiol 1999; 9: M53–M56.
3. Fidler IJ. Critical factors in the biology of human cancermetastasis: twenty-eight award lecture. Cancer Res 1990; 50:6130–6138.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.

Differentiation and stromal interactions in malignant cell transplants 307
4. Mareel MM, Van Roy FM, Bracke ME. How and when do tumorcells metastasize? Crit Rev Oncogene 1993; 4: 559–594.
5. Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cellinteractions with the extracellular matrix during invasion andmetastasis. Annu Rev Cell Biol 1993; 9: 541–573.
6. Rinehart CA, Torti VR. Aging and cancer: the role of stromalinteractions with epithelial cells. Mol Carcinogenesis 1997; 18:187–192.
7. Markey AC, Lane EB, Churchill LJ, McDonald DM, Leigh IM.Expression of simple epithelial keratins 8 and 18 in epidermalneoplasia. J Invest Dermatol 1991; 97: 763–770.
8. Schaafsma HE, Van Der Velden LA, Manni JJ, et al. Increasedexpression of cytokeratins 8, 18 and vimentin in the invasion frontof mucosal squamous cell carcinoma. J Pathol 1993; 170: 77–86.
9. Morgan PR, Su L. Intermediate filaments in oral neoplasia. I. Oralcancer and epithelial dysplasia. Eur J Cancer 1994; 30B: 160–166.
10. Breitkreutz D, Stark HJ, Plein P, Baur M, Fusenig NE. Differ-ential modulation of epidermal keratinization in immortalized(HaCaT) and tumorigenic human skin keratinocytes (HaCaT-ras)by retinoic acid and extracellular Ca2+. Differentiation 1993; 54:201–217.
11. Breitkreutz D, Stark HJ, Mirancea N, Tomakidi P, Steinbauer H,Fusenig NE. Integrin and basement membrane normalization inmouse grafts of human keratinocytes: implications for epidermalhomeostasis. Differentiation 1997; 61: 195–209.
12. Breitkreutz D, Boukamp P, Ryle CM, Stark HJ, Roop DR,Fusenig NE. Epidermal morphogenesis and keratin expression inc-Ha-ras-transfected tumorigenic clones of the human HaCaT cellline. Cancer Res 1991; 51: 4402–4409.
13. Fusenig NE, Skobe M, Vosseler S, et al. Tissue models to studytumor–stroma interactions. In Proteases and their Inhibitorsin Cancer Metastasis, Muschel RJ, Foidart J-M (eds). Kluwer:Dordrecht (in press).
14. Tomakidi P, Mirancea N, Fusenig NE, Herold-Mende C,Bosch FX, Breitkreutz D. Defects of basement membrane andhemidesmosome structure correlate with malignant phenotypeand stromal interactions in HaCaT-ras xenografts. Differentiation1999; 64: 263–275.
15. Boukamp P, Stanbridge EJ, Yin Foo D, Cerutti PA, Fusenig NE.c-Ha-ras oncogene expression in immortalized human ker-atinocytes (HaCaT) alters growth potential in vivo but lacks cor-relation with malignancy. Cancer Res 1990; 50: 2840–2847.
16. Mueller MM, Peter W, Mappes M, et al. Tumor progression ofskin carcinoma cells in vivo promoted by clonal selection,mutagenesis, and autocrine growth regulation by granulocytecolony-stimulating factor and granulocyte–macrophage colony-stimulating factor. Am J Pathol 2001; 159: 1567–1579.
17. Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE.Halting angiogenesis suppresses carcinoma cell invasion. Nat Med1997; 11: 1222–1227.
18. Purkis PE, Steel JB, Mackenzie IC, Nathrath WB, Leigh IM,Lane EB. Antibody markers of basal cells in complex epithelia.J Cell Sci 1990; 97: 39–50.
19. Bosch FX, Udvarhelyi N, Venter E, et al. Expression of the his-tone H3 gene in benign, semi-malignant and malignant lesions ofthe head and neck: a reliable proliferation marker. Eur J Cancer1993; 10: 1454–1461.
20. Bosch FX, Leube RE, Achtstatter T, Moll R, Franke WW. Expres-sion of simple epithelial type cytokeratins in stratified epithelia asdetected by immunolocalization and hybridization in situ . J CellBiol 1988; 106: 1635–1648.
21. Breitkreutz D, Schoop VM, Mirancea N, Baur M, Stark HJ,Fusenig NE. Epidermal differentiation and basement membraneformation by HaCaT cells in surface transplants. Eur J Cell Biol1998; 75: 273–286.
22. Ryle CM, Breitkreutz D, Stark HJ, et al. Density-dependent mod-ulation of synthesis of keratins 1 and 10 in the human keratinocytecell line HaCaT and in ras-transfected clones. Differentiation 1989;40: 42–54.
23. Iyer PV, Leong AS. Poorly differentiated squamous cell carcino-mas of the skin express vimentin. J Cutan Pathol 1992; 19: 34–39.
24. Ouhayoun JP, Goffaux JC, Sawaf MH, Shabana AH, Collin C,Forest N. Changes in cytokeratin expression in gingiva duringinflammation. J Periodont Res 1990; 25: 283–292.
25. Bloor BK, Su L, Shirlaw PJ, Morgan PR. Gene expression ofdifferentiation-specific keratins (4/13 and 1/10) in normal humanbuccal mucosa. Lab Invest 1998; 78: 787–795.
26. Tennenbaum T, Weiner AK, Belanger AJ, Glick AB, Hennings H,Yuspa SH. The suprabasal expression of alpha 6 beta 4 integrinis associated with a high risk for malignant progression in mouseskin carcinogenesis. Cancer Res 1993; 15: 4803–4810.
27. Coussens LM, Hanahan D, Arbeit JM. Genetic predisposition andparameters of malignant progression in K14-HPV16 transgenicmice. Am J Pathol 1996; 149: 1899–1917.
28. Cheng C, Tennenbaum T, Dempsey PJ, Coffey RJ, Yuspa SH,Dlugosz AA. Epidermal growth factor receptor ligands regulatekeratin 8 expression in keratinocytes, and transforming growthfactor alpha mediates the induction of keratin 8 by the v-rasHaoncogene. Cell Growth Differ 1993; 4: 317–327.
29. Breitkreutz D, Boukamp P, Lueder M, Fusenig NE. Morpholog-ical and biochemical criteria for keratinization in primary andpermanent mouse epidermal cell cultures. Front Matrix Biol 1981;9: 57–82.
30. Ben-Ze’ev A. differential control of cytokeratins and vimentinsynthesis by cell–cell contact and cell spreading in culturedepithelial cells. J Cell Biol 1984; 99: 1424–1433.
31. Van Muijen GN, Warnaar SO, Ponec M. Differentiation-relatedchanges of cytokeratin expression in cultured keratinocytes andin fetal, newborn, and adult epidermis. Exp Cell Res 1987; 171:331–345.
32. Atula S, Grenman R, Syrjanen S. Fibroblasts can modulate thephenotype of malignant epithelial cells in vitro. Exp Cell Res 1997;235: 180–187.
33. Mirancea N, Schmidt C, Daum N, et al. Basement membranedefects in xeno-grafts of malignant human cells. In Proceedings ofthe Second International Conference on Tumor Microenvironment,Progression, Therapy and Prevention, Witz IP (ed.). MonduzziEditore: Bologna, 2002; 55–58.
34. Timpl R, Brown JC. Supramolecular assembly of basement mem-branes. Bioessays 1996; 18: 123–132.
35. Muschler J, Lochter A, Roskelley CD, Yurchenco P, Bissell MJ.Division of labor among the α6ß4 integrin, β1 integrins, and an E3laminin receptor to signal morphogenesis and β-casein expressionin mammary epithelial cells. Mol Biol Cell 1999; 10: 2817–2828.
36. Roskelley CD, Srebrow A, Bissell MJ. A hierarchy of ECM-mediated signaling regulates tissue-specific gene expression. CurrOpin Cell Biol 1995; 7: 736–747.
37. Szabowski A, Maas-Szabowski NM, Andrecht S, et al. c-Junand JunB antagonistically control cytokine-regulated mesenchy-mal–epidermal interaction in skin. Cell 2000; 103: 745–755.
38. Skobe M, Fusenig NE. Tumorigenic conversion of immortalhuman keratinocytes through stromal activation. Proc Natl AcadSci USA 1998; 95: 1050–1055.
39. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD,Cunha GR. Carcinoma-associated fibroblasts direct tumor progres-sion of initiated human prostatic epithelium. Cancer Res 1999; 59:5002–5011.
40. Kaur P, Carter WG. Integrin expression and differentiation intransformed human epidermal cells is regulated by fibroblasts. JCell Sci 1992; 103: 755–763.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 298–307.




























