Embed Size (px)
Citation preview

DnaA protein interacts with RNA polymerase and partiallyprotects it from the effect of rifampicin
Ingvild Flåtten, Morigen and Kirsten Skarstad*Department of Cell Biology, Institute for CancerResearch, The Norwegian Radium Hospital,Rikshospitalet, University of Oslo, 0310 Oslo, Norway.
Summary
The Escherichia coli DnaA protein forms an oligomerat the origin and initiates chromosome replicationwith the aid of architectural elements and transcrip-tion by RNA polymerase. Rifampicin inhibits initiationof transcription by RNA polymerase and thus alsoinitiation of replication. Here, we report that wild-typecells undergo rifampicin-resistant initiation of replica-tion during slow growth in acetate medium. Therifampicin-resistant initiation was prevented byreducing the availability of DnaA. In vitro experimentsshowed that the DnaA protein interacted with RNApolymerase and that it afforded a partial protectionfrom the negative effect of rifampicin. It is possiblethat rifampicin-resistant rounds of replication occurwhen a surplus of DnaA is available at the origin. Inrich medium wild-type cells do not exhibit rifampicin-resistant rounds of replication, possibly indicatingthat there is no surplus DnaA, and that DnaA activityis the factor limiting the process of initiation. Duringgrowth in acetate medium, on the contrary, DnaAactivity is not limiting in the same way because aninitiation potential is present and can be turned intoextra rounds of replication when rifampicin is added.The result suggests that regulation of replication ini-tiation may differ at different growth rates.
Introduction
Escherichia coli has a single circular chromosome that isreplicated bidirectionally from the origin, oriC. In rapidlygrowing E. coli cells, the duration of replication and seg-regation is longer than one generation, and replicationcycles overlap (Cooper and Helmstetter, 1968; Skarstadet al., 1986), whereas in slowly growing cells a round ofreplication is confined within the same generation. The
DnaA protein is the main actor in initiation of replication. Itbinds to specific DnaA boxes within oriC (Fuller et al.,1984; McGarry et al., 2004; Speck and Messer, 2001),unwinds the DNA of an AT-rich region (Sekimizu et al.,1987; Bramhill and Kornberg, 1988) and forms an opencomplex aided by integration host factor (IHF), otherarchitectural elements (Dixon and Kornberg, 1984; Grim-wade et al., 2000) and transcriptional activation by RNApolymerase (Lark, 1972; Messer, 1972; Baker and Korn-berg, 1988; Skarstad et al., 1990). Finally, it recruits theDnaB helicase to the single-stranded region (Marszalekand Kaguni, 1994).
The DnaA protein binds both ATP and ADP with highaffinity (Sekimizu et al., 1987). Both forms of the proteinbind equally well to the 9-mer DnaA boxes in oriC(Sekimizu et al., 1987; Schaper and Messer, 1995). Inaddition the ATP-form of DnaA binds specifically to lowaffinity 6-mer sequences called ATP–DnaA boxes (Yungand Kornberg, 1989; Speck et al., 1999) and 9-mersequences termed I-sites (McGarry et al., 2004). Duringinitiation, binding of ATP–DnaA to these boxes is neces-sary for formation of the open complex (Speck andMesser, 2001; McGarry et al., 2004).
After initiation the new origins become sequestered bythe SeqA protein and immediate re-initiation is thus pre-vented (Russell and Zinder, 1987; Campbell and Kleck-ner, 1990; Lu et al., 1994). In addition, the initiationpotential is reduced by a decrease in the level of activeATP–DnaA by hydrolysis of the bound ATP by regulatoryinactivation of DnaA (RIDA). In this process the DnaAprotein’s intrinsic ATPase activity is stimulated by Hda andthe b-clamp of the DNA polymerase III (Katayama et al.,1998; Kato and Katayama, 2001). The level of free DnaAin the cell is also reduced by titration of the DnaA to thehigh-affinity datA site (Hansen et al., 1991a; Kitagawaet al., 1998; Morigen et al., 2001), and by autoregulationand sequestration of the dnaA gene (Atlung et al., 1985;Braun et al., 1985; Kücherer et al., 1986; Campbell andKleckner, 1990; Bogan and Helmstetter, 1997).
It has previously been shown that both deletion of thehda gene and deletion of the datA region lead to anasynchrony phenotype when analysed by flow cytometry(Kitagawa et al., 1998; Camara et al., 2003). In bothcases it was concluded that the asynchrony was a resultof re-initiation of replication, presumably due to increased
Accepted 16 December, 2008. *For correspondence. E-mail [email protected]; Tel. (+47) 22 93 42 55; Fax (+47) 22 9345 80.
Molecular Microbiology (2009) 71(4), 1018–1030 � doi:10.1111/j.1365-2958.2008.06585.xFirst published online 13 January 2009
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd

proportions of ATP–DnaA and unbound DnaA re-spectively. However, when analysed further it was shownthat the asynchrony in the datA-deleted strain was a resultof rifampicin-resistant initiations rather than (rifampicin-sensitive) re-initiation of replication (Morigen et al., 2005).
The rifampicin drug binds to the b-subunit of RNA poly-merase and inhibits transcription and thereby initiation ofreplication (Hartmann et al., 1967; Wehrli et al., 1968).Mutations in the rpoB gene, encoding the b-subunit of theRNA polymerase, can make the polymerase resistant tothe rifampicin drug (Rabussay and Zillig, 1969; Ovchinni-kov et al., 1983; Jin and Gross, 1988; Severinov et al.,1993). Certain of these mutations can also suppress dnaA(ts) mutations in an allele-specific manner, suggesting aninteraction between the DnaA protein and the RNA poly-merase (Bagdasarian et al., 1977; Atlung, 1984). Experi-ments investigating replication of l and l-plasmids inE. coli has also suggested an interaction between theDnaA protein and the b-subunit of the RNA polymerase(Szalewska-Palasz et al., 1998).
In this work we have investigated the occurrence ofrifampicin-resistant rounds of replication in wild-type cells,and have found evidence in vitro for an interactionbetween DnaA and RNA polymerase.
Results
Rifampicin-resistant initiations occur in wild-type cellsgrown in acetate medium
We noticed that growth in poor medium gave similarresults with respect to rifampicin-resistant rounds of rep-lication in wild-type cells as found by von Freieslebenet al. in cells without IHF (von Freiesleben et al., 2000)and by Morigen et al. in cells without the datA site(Morigen et al., 2005). Here, we present studies of growthin minimal medium with acetate as the carbon source. Inthis medium the cells grew with doubling times of about5 h, with the replication period confined within onegeneration. Such cells contain one, between one and two,or two chromosomes, being either not replicating, in theprocess of replicating or have completed replication, butnot yet divided. A common way to estimate the origindistribution in exponentially growing cultures is to addrifampicin and cephalexin (Skarstad et al., 1986; Boyeand Lobner-Olesen, 1991). This inhibits new initiationsand blocks cell division, but allows ongoing rounds ofreplication to finish. When analysed by flow cytometry,cells that were replicating at the time of drug addition willbe found in the 2-chromosome peak in the rifampicinrun-out sample together with the other 2-chromosomecells (see Fig. 1). The number of 2-chromosome cells inthe rifampicin run-out histogram thus should reflect thenumber of cells with two origins at the time of drug
addition. The cells with one chromosome at the time ofdrug addition should not undergo further initiations, andso the percentage of these cells should be the same in thehistogram of exponential cells and the drug-treated cells.
Slowly growing wild-type CM735 cells were analysed byflow cytometry, but did not follow the expected patternafter treatment with rifampicin and cephalexin. Onaverage the fraction of cells containing one chromosomedecreased from 31% in the untreated cells (Fig. 1A) to11% in the rifampicin/cephalexin-treated cells (Fig. 1B).This means that 20% of the cells in the population hadinitiated replication after the addition of rifampicin.
Earlier work has shown that in cells without the datA sitethe extent of replication after rifampicin addition could bereduced by increasing the concentration of rifampicin(Morigen et al., 2005). Treatment of slowly growing wild-
Fig. 1. Rifampicin-resistant initiations in slowly growing wild-typecells. Cells grown in acetate medium were analysed with flowcytometry before (A) and after (B) treatment with rifampicin andcephalexin (see Experimental procedures). The percentage of cellswith one chromosome in the exponential sample (A) and therifampicin/cephalexin-treated sample (B) was calculated in WinMDI2.8. The percentages shown in the figure are the average of sevenexperiments.
DnaA protects RNA polymerase from rifampicin 1019
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030
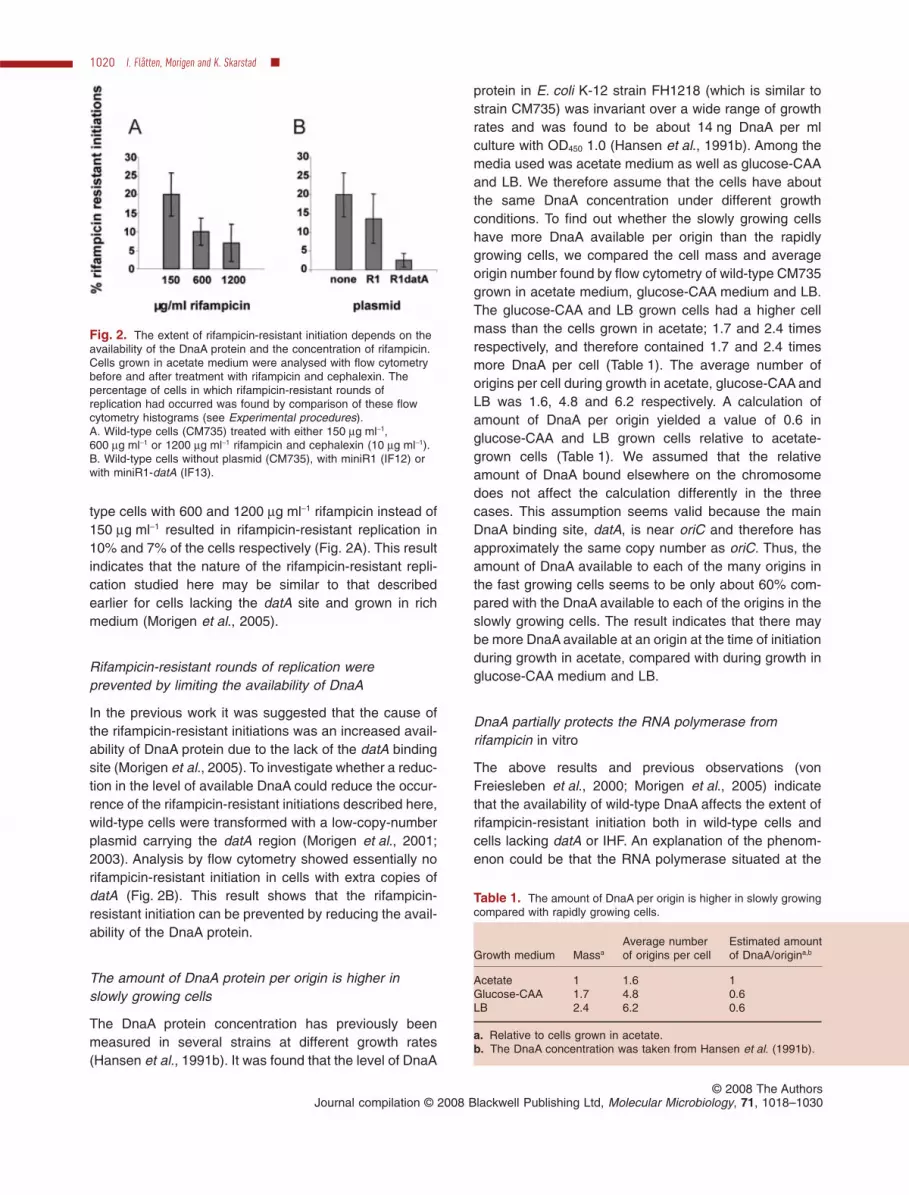
type cells with 600 and 1200 mg ml-1 rifampicin instead of150 mg ml-1 resulted in rifampicin-resistant replication in10% and 7% of the cells respectively (Fig. 2A). This resultindicates that the nature of the rifampicin-resistant repli-cation studied here may be similar to that describedearlier for cells lacking the datA site and grown in richmedium (Morigen et al., 2005).
Rifampicin-resistant rounds of replication wereprevented by limiting the availability of DnaA
In the previous work it was suggested that the cause ofthe rifampicin-resistant initiations was an increased avail-ability of DnaA protein due to the lack of the datA bindingsite (Morigen et al., 2005). To investigate whether a reduc-tion in the level of available DnaA could reduce the occur-rence of the rifampicin-resistant initiations described here,wild-type cells were transformed with a low-copy-numberplasmid carrying the datA region (Morigen et al., 2001;2003). Analysis by flow cytometry showed essentially norifampicin-resistant initiation in cells with extra copies ofdatA (Fig. 2B). This result shows that the rifampicin-resistant initiation can be prevented by reducing the avail-ability of the DnaA protein.
The amount of DnaA protein per origin is higher inslowly growing cells
The DnaA protein concentration has previously beenmeasured in several strains at different growth rates(Hansen et al., 1991b). It was found that the level of DnaA
protein in E. coli K-12 strain FH1218 (which is similar tostrain CM735) was invariant over a wide range of growthrates and was found to be about 14 ng DnaA per mlculture with OD450 1.0 (Hansen et al., 1991b). Among themedia used was acetate medium as well as glucose-CAAand LB. We therefore assume that the cells have aboutthe same DnaA concentration under different growthconditions. To find out whether the slowly growing cellshave more DnaA available per origin than the rapidlygrowing cells, we compared the cell mass and averageorigin number found by flow cytometry of wild-type CM735grown in acetate medium, glucose-CAA medium and LB.The glucose-CAA and LB grown cells had a higher cellmass than the cells grown in acetate; 1.7 and 2.4 timesrespectively, and therefore contained 1.7 and 2.4 timesmore DnaA per cell (Table 1). The average number oforigins per cell during growth in acetate, glucose-CAA andLB was 1.6, 4.8 and 6.2 respectively. A calculation ofamount of DnaA per origin yielded a value of 0.6 inglucose-CAA and LB grown cells relative to acetate-grown cells (Table 1). We assumed that the relativeamount of DnaA bound elsewhere on the chromosomedoes not affect the calculation differently in the threecases. This assumption seems valid because the mainDnaA binding site, datA, is near oriC and therefore hasapproximately the same copy number as oriC. Thus, theamount of DnaA available to each of the many origins inthe fast growing cells seems to be only about 60% com-pared with the DnaA available to each of the origins in theslowly growing cells. The result indicates that there maybe more DnaA available at an origin at the time of initiationduring growth in acetate, compared with during growth inglucose-CAA medium and LB.
DnaA partially protects the RNA polymerase fromrifampicin in vitro
The above results and previous observations (vonFreiesleben et al., 2000; Morigen et al., 2005) indicatethat the availability of wild-type DnaA affects the extent ofrifampicin-resistant initiation both in wild-type cells andcells lacking datA or IHF. An explanation of the phenom-enon could be that the RNA polymerase situated at the
Fig. 2. The extent of rifampicin-resistant initiation depends on theavailability of the DnaA protein and the concentration of rifampicin.Cells grown in acetate medium were analysed with flow cytometrybefore and after treatment with rifampicin and cephalexin. Thepercentage of cells in which rifampicin-resistant rounds ofreplication had occurred was found by comparison of these flowcytometry histograms (see Experimental procedures).A. Wild-type cells (CM735) treated with either 150 mg ml-1,600 mg ml-1 or 1200 mg ml-1 rifampicin and cephalexin (10 mg ml-1).B. Wild-type cells without plasmid (CM735), with miniR1 (IF12) orwith miniR1-datA (IF13).
Table 1. The amount of DnaA per origin is higher in slowly growingcompared with rapidly growing cells.
Growth medium MassaAverage numberof origins per cell
Estimated amountof DnaA/origina,b
Acetate 1 1.6 1Glucose-CAA 1.7 4.8 0.6LB 2.4 6.2 0.6
a. Relative to cells grown in acetate.b. The DnaA concentration was taken from Hansen et al. (1991b).
1020 I. Flåtten, Morigen and K. Skarstad �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

origin, providing the transcription necessary for initiationof replication, is protected from the action of rifampicin byorigin-bound DnaA. To investigate whether DnaA canprotect RNA polymerase from the rifampicin drug in vitro,we performed transcription assays with two differenttemplates. First, we used the dnaA promoter region whichcontains only one strong and a few weak DnaA bindingsites, in order to obtain one, or a small number, of repro-ducible multimer complexes of DnaA. In these experi-ments a 665 bp PCR fragment containing the two dnaApromoters, dnaAp1 and dnaAp2, was used (Fig. 3A). TheDnaA protein (0, 10 or 50 nM) was incubated with thetemplate for 10 min at 37°C, and then RNA polymerase(66 nM) and the four ribonucleotides were added. Thetranscription products were separated on an 8% sequenc-ing gel and quantified in ImageQuant (see Experimentalprocedures). The DnaA protein is known to be an autore-pressor (Atlung et al., 1985; Braun et al., 1985; Küchereret al., 1986; Speck et al., 1999). As expected, and inaccordance with previous results (Speck et al., 1999), thetranscription from both dnaA promoters was inhibited byDnaA. The transcription from dnaAp1 (which in our handsgave more transcription than dnaAp2) was reduced byabout 10% and 35% in the presence of 10 nM and 50 nMDnaA respectively (Fig. 3B, black bars). We also foundan expected reduction in transcription when rifampicin(0.15 mg ml-1) was added to the reaction (Fig. 3B, 0 nMDnaA, streaked bar).
At the low concentration of DnaA (10 nM) an alleviationof the rifampicin inhibition was found. This result indicatesthat the DnaA protein is capable of protecting the RNA
polymerase from the activity of rifampicin in vitro (Fig. 3B,10 nM DnaA). At the higher DnaA concentration the alle-viation could not be clearly seen, presumably because theDnaA-mediated repression was quite strong at this DnaAconcentration (Fig. 3B, 50 nM DnaA).
Fig. 3. DnaA partially protects the RNA polymerase fromrifampicin in vitro.A. A schematic representation of the 665 bp dnaA promoterfragment used in the in vitro transcription assay and cross-linkingexperiment. Arrows indicate the two promoters, black rectangleindicates the consensus (strong) DnaA box, dark grey rectanglesindicate 9-mer DnaA boxes with mismatches (weak boxes) andlight grey rectangles indicate ATP–DnaA boxes (Speck et al., 1999;Hansen et al., 2007). At the site of ATP–DnaA box a there is also apartially overlapping weak 9-mer DnaA box which is not indicatedhere (Hansen et al., 2007).B. DnaA protein (0, 10 or 50 nM) was incubated with the dnaApromoter fragment (6 nM) for 10 min. Then RNA polymerase(74 nM) and rifampicin 0.15 mg ml-1 ( ) or 0 mg ml-1 ( ) wereadded, followed by another 10 min incubation. The reaction wasstarted by adding ribonucleotides, including radioactively labelledUTP, and incubated for 10 min. The transcription products were runon 8% sequencing gel, dried and exposed in a phosphor screencassette and scanned on a Storm 840. The intensity of the bandswas quantified in ImageQuant.C. A schematic representation of the 600 bp oriC fragment used inthe in vitro transcription assay and cross-linking experiment.Symbols are as in A (Speck and Messer, 2001; McGarry et al.,2004; Hansen et al., 2007).D. DnaA protein (0, 50, 100 or 200 nM) was incubated with the oriCfragment (6 nM) for 10 min. Then RNA polymerase (74 nM) andrifampicin, 0.05 mg ml-1 ( ) or 0 mg ml-1 ( ), were added. Theincubation and conditions otherwise were as described for B.
DnaA protects RNA polymerase from rifampicin 1021
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

We also performed the in vitro transcription assay usingan oriC fragment as a template. This fragment containsthe gidA promoter. In addition, two weak promoters, oriLand oriR1, which have been detected in oriC are presenton the fragment (Fig. 3C). The gidA promoter is the stron-gest of these three promoters and a band correspondingto the size of a transcript from this promoter was repro-ducibly detected in the assays and identified as the gidAtranscript by Northern blot (data not shown). The amountof this transcript increased with increasing amounts of theDnaA protein (Fig. 3D, black bars). The result suggeststhat DnaA acts as a transcriptional activator at the gidApromoter. This assay was also done with rifampicinpresent in the reaction (Fig. 3D, streaked bars). This leadsto a reduction in transcription of 46% when no DnaAprotein was present in the reaction. When 50 or 200 nMDnaA was present in the reactions, a lower reduction intranscription was observed, 34% and 36% respectively,indicating that DnaA may have the ability to protect theRNA polymerase against rifampicin also at this promoter.However, at lower concentrations of DnaA than 50 nM,only small effects were seen, and at 100 nM DnaA, theinhibition by rifampicin was reproducibly found to beincreased by DnaA, to 57% on average. The results indi-cate that more DnaA is required to bind to the oriC frag-ment compared with the dnaA fragment in order to seeeffects at the promoters. The main effect of DnaA at thegidA promoter was a stimulation of transcription. A pos-sible protection against rifampicin afforded by DnaA wasonly seen at certain concentrations, and may indicate thatseveral kinds of multimeric complexes of DnaA form onthe oriC fragment depending on protein concentration,and that an interaction between RNA polymerase andDnaA is dependent on which complex is formed.
Protection of RNA polymerase with both ATP- andADP-form DnaA at the dnaA promoter
To investigate whether the ADP-form of DnaA behaveddifferently from the ATP-form with respect to the protectionof RNA polymerase against rifampicin at the dnaA pro-moter, DnaA protein purified with a denaturation step thatremoves the nucleotide was used (Sekimizu et al., 1988).This DnaA preparation was incubated with either ATP orADP before it was used in the transcription assaydescribed above. When no rifampicin was added to thereaction, ATP–DnaA inhibited transcription from thepromoter. At a concentration of 60 nM DnaA the level oftranscription was reduced with about 50% (Fig. 4A, blackbars). In contrast, ADP–DnaA did not inhibit, but stimu-lated transcription at low concentrations (15 and 30 nM).At higher concentrations transcription was inhibited,although not to the same extent as with the ATP-form ofthe DnaA protein (Fig. 4A, grey bars). This result suggests
Fig. 4. In vitro transcription assay with ADP-form and ATP-form ofDnaA. The in vitro transcription assay was performed using theDnaA promoter fragment as described in legend to Fig. 3B, but withnucleotide-free DnaA protein (0–240 nM), incubated with 5 mM ATPor ADP for 20 min on ice prior to use in the transcription assay.A. Comparison of the ATP–DnaA ( ) and ADP–DnaA ( ) in the invitro transcription assay.B. In vitro transcription with ATP–DnaA, without ( ) or with ( )rifampicin (0.15 mg ml-1) present in the reaction.C. In vitro transcription with ADP–DnaA without ( ) or with ( )rifampicin (0.15 mg ml-1) present in the reaction.
1022 I. Flåtten, Morigen and K. Skarstad �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

that ADP–DnaA acts as an activator as well as a repressorat dnaAp1, depending on the concentration.
With ATP–DnaA, an alleviation of the rifampicin inhibi-tion of transcription was seen at the lower concentrationsof DnaA (30 nM) (Fig. 4B). In the case of ADP–DnaA therewas also an increase in the level of transcription whenboth rifampicin and DnaA were added to the reactioncompared with the reaction without DnaA (Fig. 4C). But atthe lowest concentrations (15 and 30 nM) this increasedid not exceed the increase in the transcription that wasseen when DnaA alone was added to the reaction.However, at 60 nM ADP–DnaA a clear alleviation ofrifampicin inhibition was seen. This shows that also theADP-form of the DnaA protein can protect the RNA poly-merase from the effect of rifampicin at the dnaA promoterin vitro, but that a higher concentration of ADP–DnaA maybe required compared with that of ATP–DnaA.
The ADP-form of DnaA was also included in the tran-scription assay with the oriC fragment. No stimulation oftranscription from the gidA promoter was detected and noprotection of the RNA polymerase against rifampicin, atconcentrations of ADP–DnaA of 200 nM or below (datanot shown).
Cross-linking of DnaA and RNA polymerase
To investigate whether the protection and the stimulationof the RNA polymerase by the DnaA protein were due toa direct interaction between the two proteins, a cross-linking experiment was performed. The two DNA frag-ments described above were incubated with DnaA andRNA polymerase before adding Disuccinimidyl suberate(DSS) cross-linker. The cross-linked products were sepa-rated in a polyacrylamide-agarose gel designed for sepa-ration of large complexes (Lavoie et al., 1991), blottedand analysed with antibody against DnaA (Fig. 5).
In the experiment with the dnaA promoter fragment allthree reactions contained the same amounts ofATP–DnaA(64 nM), one of them included RNA polymerase (74 nM)(Fig. 5A, lane 3) and two included DSS (Fig. 5A, lanes 2and 3). In the reaction without DSS (Fig. 5A, lane 1), a bandrepresenting the monomer form of the DnaA protein(52 kDa) is seen. In lane 2, one can see bands represent-ing dimer, trimer and possibly tetramer of DnaA. Whencomparing this lane with lane 3 (containing the reactionincluding RNA polymerase) it can be seen that a bandrepresenting a size larger than 250 kDa has appeared. Theexperiment was performed with bothATP–DnaAandADP–DnaA. Both forms of DnaA gave similar results, as didexperiments were RNA polymerase was added to thereaction first (data not shown). The size of this complex(indicated by an arrow in Fig. 5A) corresponds to approxi-mately the size of an RNA polymerase holoenzyme(449 kDa) plus one to four DnaA proteins (52–208 kDa).
The experiment was also done using an antibodyagainst the b-subunit of the RNA polymerase. In this blotit was not possible to distinguish the band containing theDnaA–RNA polymerase complex from the smear of otherbands containing different combinations of the RNA poly-merase subunits (data not shown). A protein that does notinteract with DnaA, bovine serum albumin (BSA), wastested under the same conditions as the RNApolymerase. No cross-linking occurred between these twoproteins (data not shown).
The cross-linking experiment was also performed withthe oriC fragment. This fragment contains higher numbersof both strong and weak DnaA boxes than the dnaApromoter fragment (Fig. 3C). The experiment was per-formed in the same way as described above (Fig. 5B).Also shown is one reaction performed with addition of the
Fig. 5. DnaA interacts with the RNA polymerase at the dnaApromoter and at oriC. DnaA protein (64 nM) was incubated withDNA fragment (4 nM), RNA polymerase (74 nM) and DSScross-linker and separated on an agarose-polyacrylamide gel. Afterblotting, detection of DnaA protein was carried out usingDnaA-antibody and an ECF fluorescence kit.A. ATP–DnaA protein and dnaA promoter fragment, without (lanes1 and 2) and with RNA polymerase (lane 3), and without (lane 1)and with the DSS cross-linker (lanes 2 and 3).B. ATP–DnaA protein and oriC fragment without (lanes 1 and 2)and with RNA polymerase (lanes 3 and 4), and without (lane 1) andwith the DSS cross-linker (lanes 2, 3 and 4). In lane 3 the DnaAprotein was added to the reaction before the RNA polymerase andin lane 4 the RNA polymerase was added before the DnaA protein.The positions of the molecular weight standard and of DnaAmonomer and multimer forms are indicated. Arrows indicatemultimer forms found in the presence of RNA polymerase.
DnaA protects RNA polymerase from rifampicin 1023
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

RNA polymerase before DnaA protein (Fig. 5B, lane 4).The reaction without RNA polymerase exhibited a largenumber of higher multimeric forms of the DnaA protein(Fig. 5B, lane 2). This difference between the complexesin lane 2 in Fig. 5A and lane 2 in Fig. 5B probably reflectsthe difference in numbers of binding sites on the respec-tive DNA fragments. The oriC region contains a highernumber, and more a complex organization, of DnaA boxescompared with the dnaA promoter. In the reactions includ-ing RNA polymerase (Fig. 5B, lanes 3 and 4), two slowermigrating bands can be seen, of which one (top arrow inFig. 5B) is at a position that is higher than the smear of thebands containing only DnaA seen in lane 2. The resultsuggests that an interaction between the DnaA proteinand RNA polymerase occurs also in the presence of anoriC fragment.
DnaA interacts directly with RNA polymerase
To rule out the possibility that the interaction between theDnaA protein and RNA polymerase seen in the cross-linking experiment was just a result of the two proteinsbeing localized near each other on the DNA fragment, anenzyme-linked immunosorbent assay (ELISA) wasperformed. This assay has previously been used to dem-onstrate specific interaction between the HU a-subunitand DnaA (Chodavarapu et al., 2008a) and betweenDnaA and DnaB (Marszalek and Kaguni, 1994). Here, weused this assay to confirm the interaction between theDnaA protein and RNA polymerase. For a positive controlwe used the previously shown interaction between DnaAand DnaB (Marszalek and Kaguni, 1994). To check thatthe antibodies did not exhibit unspecific binding, whichcould give false positives, ParE, a protein that does notinteract with any of the proteins, was used as a negativecontrol. The first protein (DnaA or ParE) was immobilizedto individual wells of a microtiter plate. The unboundprotein was removed and the wells washed with buffercontaining BSA and milk to quench the reactive surfacesof the plate. Increasing amounts of the second protein(RNA polymerase or DnaB) were then added and theplates incubated for 1 h at room temperature. Afterremoval of unbound proteins, a mouse antibody againstthe RNA polymerase b-subunit and a rabbit antibodyagainst DnaB were added to the respective reactions andincubated for 1 h. After further incubation with secondaryanti-mouse/anti-rabbit antibody linked to horseradish per-oxidase, O-phenylenediamine and hydrogen peroxidewere added as substrate for the peroxidase, and absor-bance at 492 nm was measured (Fig. 6). An elevatedabsorbance relative to the controls indicates an interac-tion between the proteins in question. As expected thereaction containing DnaA and DnaB gave an increase inthe absorbance at 492 nM (Fig. 6A). In the wells contain-
ing the DnaA protein and RNA polymerase a significantincrease in absorbance was also observed (Fig. 6B), sug-gesting an interaction between the proteins present in thereaction. None of the reactions containing ParE gave asignificant increase in absorbance, confirming that theantibodies against DnaB and RNAP do not exhibit unspe-cific binding. ParE is one of two subunits of the enzymeTopoisomerase IV and when ParE was tested with theother subunit of this enzyme, ParC, this gave about sev-
Fig. 6. DnaA interacts with RNA polymerase independently ofDNA binding. An ELISA assay was performed as described inExperimental procedures to detect interaction between DnaA andRNA polymerase and as a positive control, between DnaA andDnaB. Both experiments were also performed with ParE as anegative control. Immune complexes were detected colorimetricallyat 492 nm (y-axis). The plot represents an average of threeexperiments and the vertical lines represent the standard error ofthe experiments.A. Different amounts of DnaB protein (0–40 nM) were incubatedwith immobilized DnaA (filled squares) or immobilized ParE (filledtriangles).B. Different amounts of RNA polymerase (0–40 nM) were incubatedwith immobilized DnaA (filled squares) or immobilized ParE (filledtriangles).
1024 I. Flåtten, Morigen and K. Skarstad �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

enfold increase in absorption at 492 nm (data not shown).This result confirmed that the preparation of ParE wasactive and that the results with RNA polymerase andDnaB were not false negatives.
These results indicate that the DnaA protein interactsspecifically with RNA polymerase. As there is no DNApresent in the reaction, this also shows that the interactionis not dependent on the proteins being bound to DNA.
Discussion
The mechanism of regulation of replication initiationmay differ at different growth rates
In this work we found that extra rounds of replication occurin the presence of rifampicin in cells growing slowly inacetate medium. A reduction in the availability of the DnaAprotein, by introduction of extra copies of the datA region,prevented most of the rifampicin-resistant initiations. Asthe rifampicin-resistant initiations occur, there must be aninitiation potential present which is not released duringexponential growth (before rifampicin is added). That is,there must be an unknown limitation or inhibition that isalleviated after incubation in the presence of rifampicin. Itis thus possible that an initiation complex is assembledsome time before initiation of replication in acetate growncells, but this does not immediately lead to initiation ofreplication. In contrast, during rapid growth in glucose-CAA medium or LB, the assembly of an initiation complexmay be the limiting step.
As cells growing rapidly do not exhibit rifampicin-resistant replication (Morigen et al., 2005), we investi-gated whether there could be a difference between theavailability of DnaA in rapidly growing cells and the avail-ability of DnaA in slowly growing cells. The result suggeststhat a surplus of DnaA is present in acetate-grown cells(relative to glucose-CAA- and LB-grown cells) at the timeof initiation, and may explain why only these, and notthe glucose-CAA- and LB-grown cells, exhibit rifampicin-resistant initiation.
We have not identified the element that regulates initia-tion of replication during growth in acetate medium. Thereare several possibilities. The element could for instancebe (i) a nucleoid-associated protein; (ii) transcriptionalactivation; and (iii) a protein required downstream, suchas DnaB, primase or a polymerase subunit. In order toinvestigate whether recruitment of the DnaB helicasecould be a limiting factor, cells with 50% overproduction ofDnaB were analysed. These cells still exhibited the sameamount of rifampicin-resistant initiations, however, indicat-ing that the limiting step is not recruitment of DnaB (datanot shown). Bacillus subtilis primase has recently beenfound to be inhibited by the starvation-induced nucle-otides ppGpp and pppGpp (Wang et al., 2007). This
causes replication fork arrest in the bacterium after aminoacid starvation. Whether E. coli primase is sensitive to(p)ppGpp is not known, but RNA polymerase is regulatedby this starvation signal, which causes less transcriptionfrom stringently regulated promoters (Dennis et al., 2004;Vrentas et al., 2005). Thus, the transcriptional activationrequired for initiation of replication might be sensitive to(p)ppGpp. Finally, the composition of nucleoid-associatedproteins is expected to be different in rich and poor media.Among proteins that possibly are more abundant duringgrowth in acetate medium compared with glucose-CAAmedium or LB is the Dps protein which is the most abun-dant nucleoid-associated protein in stationary phase (AliAzam et al., 1999). In LB medium this protein is much lessabundant than Fis, Hfq and HU (Ali Azam et al., 1999).Recently Dps was found to inhibit strand opening by DnaAprotein in vitro (Chodavarapu et al., 2008b). If Dps inhibi-tion of DnaA activity also occurs in vivo, a possibility couldbe that the Dps protein is responsible for holding backinitiation of replication during growth in acetate medium.
The DnaA protein interacts with RNA polymerase
In this work we have shown, with two different types ofassay (ELISA and cross-linking), that the DnaA proteininteracts with RNA polymerase. We did not determinewhich of the RNA polymerase subunits is responsible forthe interaction. However, earlier experiments suggest thatit is the b-subunit that interacts with DnaA (Bagdasarianet al., 1977; Atlung, 1984; Szalewska-Palasz et al., 1998).Rifampicin is known to bind to the RNA polymeraseb-subunit (Ovchinnikov et al., 1983; Jin and Gross, 1988;Severinov et al., 1993), although more recent research hasshown that it might also bind to the s-subunit (Wegrzynet al., 1998; Artsimovitch et al., 2005). In this study it wasfound that binding of rifamycins to the b- or s-subunit of theRNA polymerase not only sterically block the RNA synthe-sis, but might induce an allosteric signal that leads tore-positioning of the Mg2+ in the active site. Thus, it is notquite clear whether DnaA interaction with RNA polymerasemay prevent simple binding of rifampicin or whether DnaAmay contribute to keeping RNA polymerase in an activeconformation with Mg2+ in the correct position.
Both the ATP-form and ADP-form of the DnaA proteinwere found to alleviate the inhibition of RNA polymeraseby rifampicin at the dnaA promoter, although a higherconcentration was necessary to achieve the effect whenthe ADP-form of the DnaA protein was used. Structuralstudies of the major part of Aquifex aeolicus DnaA (thewhole protein except the N-terminal domain) indicate thatATP–DnaA can form a helical multimer through head-to-tail interactions of domain III, whereas ADP–DnaA cannot(Erzberger et al., 2006). Studies of the N-terminal domainof DnaA indicate that the head-to-tail multimer also forms
DnaA protects RNA polymerase from rifampicin 1025
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

dimers through hydrophobic head-to-head interactions indomain I (Abe et al., 2007). Recent work has also uncov-ered that a small protein, DiaA, specifically promotes oli-gomerization of ATP–DnaA via this domain (Ishida et al.,2004; Keyamura et al., 2007). It is not known whetherATP–DnaA forms one type of multimer in vivo and ADP–DnaA does not, or whether several different types of mul-timer can be formed and some of these can be formed byboth ATP–DnaA and ADP–DnaA. It is also possible thatsingle or few molecules of DnaA are sufficient to promotestimulation and/or protection of RNA polymerase and thata large multimer is not necessary. Our in vitro resultsshowed cross-linking of two, three and four DnaA mol-ecules at the dnaA promoter both when ATP–DnaA andADP–DnaA was used, indicating that several moleculesinteract upon binding also to this DNA fragment whichonly has one strong binding site. In the presence of 30 nMADP–DnaA, transcription was stimulated 25%. Thus,ADP–DnaA functioned as an activator at low concentra-tions and a repressor at higher concentrations. In accor-dance with previous findings, a higher concentration ofADP–DnaA was required to achieve repressor functioncompared with ATP–DnaA (Speck et al., 1999). In light ofthis it was not surprising that the maximum protectionof RNA polymerase at the dnaA promoter was foundwith higher concentrations of ADP–DnaA compared withATP–DnaA.
At the gidA promoter the ATP-form of DnaA was foundto stimulate transcription, but not the ADP-form. This indi-cates that at this promoter the DnaA binding sites weresituated in such a way that only ATP–DnaA could interactwith the RNA polymerase. In the origin region, two mainpromoters act in opposite ways on initiation of replicationin vivo. Transcription from the gidA promoter is directedaway from the origin and thus stimulates initiation of rep-lication (Liu and Wang, 1987; Asai et al., 1990; 1992;Ogawa and Okazaki, 1991; 1994). Transcription from themioC promoter is directed towards the origin, thus inhib-iting strand separation (Su’etsugu et al., 2003). The tran-scription from the mioC promoter has been shown to beinhibited by DnaA (Nozaki et al., 1988), whereas we showhere that transcription from the gidA promoter is stimu-lated by ATP–DnaA. Thus, the active ATP-form of theDnaA protein acts in several ways to drive initiation ofreplication, first by inhibiting negatively acting transcrip-tion from the mioC promoter, second, by stimulating posi-tively acting transcription from the gidA promoter andthird, by forming the opened oriC complex.
Several weak promoters have also been found withinthe origin; oriR1, and oriR direct transcription away fromthe 13-mer region and oriL directs transcription towards the13-mer region (Messer and Weigel, 1996). Transcriptionfrom these promoters may also contribute to regulation ofinitiation for instance by altering the supercoiling in the
AT-rich region. It has been shown that transcription fromthe oriL promoter is repressed by the DnaA protein (Asaiet al., 1992), whereas transcription from the two otherpromoters is stimulated by DnaA (Asai et al., 1992). In ourin vitro experiment, transcription from the oriR1 and oriLpromoters could not be detected, probably because RNApolymerase preferred to bind to the stronger gidA pro-moter. (The oriR promoter was not present on the tem-plate.) In vivo, however, it is possible that if transcription isrequired during rifampicin-resistant initiation, it comes fromone of these weak promoters, because little and inconsis-tent protection of RNA polymerase was observed at thegidA promoter in our in vitro experiment. It is also possiblethat the rifampicin-resistant initiation involves the protec-tion of RNA polymerase situated at the gidA promoter, butthat it was not possible to build a correct DnaA complex onthe linear fragment in the in vitro experiment. We haveshown here that DnaA and RNA polymerase interact, butwe have not shown that this interaction directly causes aprotection of RNA polymerase from rifampicin. An effectmay also be indirect, for instance, through effects of DnaAon the DNA conformation.
It should be noted that because the element respon-sible for holding back initiation of replication during growthin acetate medium is unknown, details about its activity inrelation to transcriptional activation by RNA polymeraseare also unknown. Thus, it is not known whether protec-tion of origin-bound RNA polymerase from rifampicinoccurs in vivo during growth in acetate medium. If tran-scription by RNA polymerase is required, or is itself thelimiting element, it is reasonable to assume that protectionof origin bound RNA polymerase does occur duringrifampicin-resistant replication in vivo.
Experimental procedures
Bacterial strains and growth conditions
All strains used are E. coli K-12 CM735 (metE46, trp-3, his-4,thi-1, galK2, lacY1 or lacZ4, mtl-1, ara-9, tsx-3, ton-1, rps-8 orrps-9, supE44 l -) (Hansen and von Meyenburg, 1979). StrainIF12 and IF13 was made by transforming CM735 with miniR1plasmid and miniR1 plasmid containing the datA region(Morigen et al., 2001) respectively. Cells were grown in ABminimal medium (Clark and Maaloe, 1967) supplementedwith 10 mg ml-1 thiamine, 20 mg ml-1 methionine, tryptophanand histidine and 0.4% sodium acetate as a carbon source(acetate medium) at 30°C, AB minimal medium supple-mented with 10 mg ml-1 thiamine, 20 mg ml-1 tryptophan, 0.5%casamino acids and 0.2% glucose (glucose-CAA medium) orLB medium at 37°C. Ampicillin was added at 25 mg ml-1 whennecessary.
Flow cytometry
Exponentially growing cells (OD450 = 0.15) were treatedwith 150 mg ml-1 rifampicin, unless otherwise stated, and
1026 I. Flåtten, Morigen and K. Skarstad �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

10 mg ml-1 cephalexin for 1.5–2 generations to allow ongoingrounds of replication to finish. Rifampicin inhibits transcrip-tion, which results in inhibition of initiation of replication,whereas cephalexin inhibits cell division. In the presence ofthese drugs cells end up with integral numbers ofchromosomes. 1.5 ml of exponentially growing or rifampicinand cephalexin-treated cells was harvested, washed andresuspended in 100 ml TE buffer and fixed by adding 1 ml77% ethanol. The cells were washed in 0.1 M phosphatebuffer, pH 9.0, and total protein content was stained overnightin 1.5 mg ml-1 fluorescein isothiocyanate (Sigma-Aldrich) inthe same buffer (Wold et al., 1994). The cells were washed in0.02 M Tris buffered saline, pH 7.5, and resuspended in thesame buffer. The DNA was stained for 30 min in 1.5 mg ml-1
Hoechst 33258 (Sigma-Aldrich) in the same buffer (Torheimet al., 2000). The samples were analysed on LSR II flowcytometer (BD Biosciences). The average cell mass wasdetermined by taking the average of the fluorescein isothio-cyanate fluorescence intensity distribution. Quantification ofthe number of origins in the fast growing cells (Table 1) wasdone by using the histogram of the rifampicin/cephalexin-treated cells, while in the slowly growing situation the histo-gram of the exponential sample was used.
Calculation of the fraction of cells in whichrifampicin-resistant initiation occurred
Quantification of the percentage of cells with one, one to two,two and more than two chromosome equivalents wasperformed in WinMDI 2.8. The fraction of cells in whichrifampicin-resistant initiation occurred was estimated by thedifference between the percentage of 1-chromosome cells inthe histograms of the exponential and the drug-treated cells.
In vitro transcription assay
The in vitro transcription assay was performed essentially asdescribed (Speck et al., 1999).
A 665 bp fragment containing the DnaA promoter region(containing 339 bp upstream and 326 bp downstream of the+1 transcription start site of dnaAp1) was amplified using theprimers 5′-GCCTGCAATGGGCGTATCC and 5′-GGCTTTATTGGATATCCGCCG. The oriC fragment including the gidApromoter was amplified using primers 5′-AATGCGTTATGCGCTACGACG and 5′-GGATCGTGGGTTAATTTACTCAAAT.This gave a fragment of 2269 bp. To make a shorter fragmentmore suitable for in vitro transcription, the PCR product wascut with ClaI, giving a fragment of 600 bp. In the experimentswhere the nucleotide-free DnaA was used, the DnaA proteinwas incubated with 5 mM ADP or ATP for 20 min on icebefore use. The DNA template (6 nM) was pre-incubated withvarious amounts of DnaA for 10 min at 37°C (unless statedotherwise) before adding RNA polymerase (Epicentre Bio-technologies) and 0.15 mg ml-1 rifampicin in the case of theDnaA promoter and 0.05 mg ml-1 in the case of the oriC frag-ment followed by another 10 min of incubation at 37°C. Tran-scription was initiated by addition of the four ribonucleotides,including [a-32P]-UTP (10 mCi, 3000 Ci mmol-1) (all from GEHealthcare) and incubated for 10 more minutes. The reactionwas stopped by addition of 25 mM EDTA and precipitated
with ethanol and yeast tRNA (Sigma-Aldrich) overnight at-20°C. The samples were resuspended in 10 ml of water,20 ml formamide with 0.05% xylene cyanol was added andthe samples were incubated for 5 min at 96°C prior to elec-trophoresis in an 8% sequencing gel at 450 V for 2 h. The gelwas dried and exposed in a phosphor screen cassette andscanned on a Storm 840 (Molecular Dynamics). The intensityof the bands was quantified using ImageQuant software(Molecular Dynamics).
Protein purification
The ParC and ParE proteins were purified as described(Peng and Marians, 1993). DnaA protein was purified asdescribed (Krause et al., 1997). In this purification, the DnaAprotein was kept in native form throughout the purificationand therefore probably retained a bound nucleotide. Thebound nucleotide is assumed to be predominantly ATPbecause the level of ATP in the cell is higher than the level ofADP, causing newly made DnaA to be bound by ATP(Bochner and Ames, 1982). As the DnaA was purified fromcells with overproduction of DnaA, relatively little of it waspresumed to be a target for RIDA. The preparation wasshown to be active in an origin unwinding assay supportingthis assumption (data not shown). DnaA protein withoutbound nucleotide was purified with a denaturing step asdescribed in Sekimizu et al. (1988).
Cross-linking with DSS
The cross-linking was performed in the presence of the dnaApromoter fragment or the oriC fragment described above(4 nM) in a total reaction volume of 30 ml. The DnaA proteinwas incubated with 5 mM ADP or ATP for 20 min on icebefore use. One hundred nanograms (64 nM) DnaA proteinwas then incubated with the DNA for 10 min at 37°C before1 mg (74 nM) RNA polymerase or 130 ng (65 nM) BSA wasadded. The reaction was further incubated for 10 min at 37°Cbefore 1.5 ml of 20 mg ml-1 DSS (2.7 mM) (Sigma-Aldrich)was added and the incubation continued at 37°C for 2.5 h.The reaction was quenched by adding 50 mM Tris-HCl,pH 7.5 and incubated for 30 min at room temperature. Thecross-linked products were loaded onto a 16 cm gel contain-ing 0.5% agarose, 2.5% polyacrylamide, 0.1% bis, 0.15%ammonium per sulphate and 0.05% TEMED in 1¥ TAE with0.1% SDS, and run at 50 mA for about 4 h (Lavoie et al.,1991). The proteins were then transferred to a PVDF mem-brane (Millipore) and detection of DnaA protein was carriedout using DnaA antibody and ECF fluorescence kit (GEHealthcare).
Enzyme-linked immunosorbent assay
The ELISA was performed essentially as described (Marsza-lek and Kaguni, 1994; Chodavarapu et al., 2008a). 40 nM ofDnaA or ParE was incubated in the wells of a 96 well micro-titerplate in 100 ml binding buffer (40 mM HEPES-KOHpH 7.6, 10 mM MgCl2, 50 mM NaCl, 4% glycerol and 2 mMDTT) overnight at 4°C. The next day unbound proteins wereremoved and the wells were washed for 10 min with washing
DnaA protects RNA polymerase from rifampicin 1027
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

buffer (binding buffer supplemented with 2 mg ml-1 BSA and2 mg ml-1 milk) at 4°C. This wash was repeated three timesand serves to quench any reactive surface in the wells. Then0–40 nM of the second protein (RNA polymerase or DnaB)was added in 100 ml binding buffer and the reaction incubatedat room temperature for 1 h. The second proteins wereremoved and the wells washed three times for 10 min withwashing buffer at room temperature. Then an antibodyagainst the second protein was added, mouse monoclonalantibody against the b-subunit of the RNA polymerase andrabbit polyclonal antibody against the DnaB protein. The anti-body was added in 100 ml of washing buffer and incubated for1 h at room temperature before the wells were washed threetimes as described above.
Anti-mouse/anti-rabbit antibody linked to horseradish per-oxidise was then added in 100 ml of washing buffer andincubated for 1 h at room temperature before it was removedand the wells were washed three times. As a substrate 100 mlof 50 mM sodium citrate, pH 4.0 with 0.4 mg ml-1O-phenylenediamine and 0.03% hydrogen peroxide was addedand the reactions were incubated until a yellow colourappeared. This was about 7 min for the reactions with DnaBand 3–4 min for the reactions with RNA polymerase. Thenegative controls were incubated the same time as theirpositive counterparts.
The reactions were stopped by adding 100 ml 4 N sulphuricacid and the absorbance measured at 492 nm with Lab-systems iEMS reader.
Acknowledgements
We are grateful to the Department of Radiation Biology, TheNorwegian Radium Hospital for sharing their flow cytometryfacility and to Tania Baker for suggesting the use of com-posite agarose-polyacrylamide gels. We also thank AnneWahl for excellent technical assistance, Elliott Crooke forthe kind gift of DnaA protein, Line Johnsen and IngvildOdsbu for critical reading of the manuscript and one anony-mous reviewer for thoughtful comments. This work wassupported by The Norwegian Research Council FUGEprogram.
References
Abe, Y., Jo, T., Matsuda, Y., Matsunaga, C., Katayama, T.,and Ueda, T. (2007) Structure and function of DnaAN-terminal domains: specific sites and mechanisms ininter-DnaA interaction and in DnaB helicase loading onoriC. J Biol Chem 282: 17816–17827.
Ali Azam, T., Iwata, A., Nishimura, A., Ueda, S., and Ishi-hama, A. (1999) Growth phase-dependent variation inprotein composition of the Escherichia coli nucleoid.J Bacteriol 181: 6361–6370.
Artsimovitch, I., Vassylyeva, M.N., Svetlov, D., Svetlov, V.,Perederina, A., Igarashi, N., et al. (2005) Allosteric modu-lation of the RNA polymerase catalytic reaction is anessential component of transcription control by rifamycins.Cell 122: 351–363.
Asai, T., Takanami, M., and Imai, M. (1990) The AT richnessand gid transcription determine the left border of the repli-
cation origin of the E. coli chromosome. EMBO J 9: 4065–4072.
Asai, T., Chen, C.P., Nagata, T., Takanami, M., and Imai, M.(1992) Transcription in vivo within the replication origin ofthe Escherichia coli chromosome: a mechanism for acti-vating initiation of replication. Mol Gen Genet 231: 169–178.
Atlung, T. (1984) Allele-specific suppression of dnaA(Ts)mutations by rpoB mutations in Escherichia coli. Mol GenGenet 197: 125–128.
Atlung, T., Clausen, E.S., and Hansen, F.G. (1985) Autoregu-lation of the dnaA gene of Escherichia coli K12. Mol GenGenet 200: 442–450.
Bagdasarian, M.M., Izakowska, M., and Bagdasarian, M.(1977) Suppression of the DnaA phenotype by mutations inthe rpoB cistron of ribonucleic acid polymerase in Salmo-nella typhimurium and Escherichia coli. J Bacteriol 130:577–582.
Baker, T.A., and Kornberg, A. (1988) Transcriptional activa-tion of initiation of replication from the E. coli chromosomalorigin: an RNA-DNA hybrid near oriC. Cell 55: 113–123.
Bochner, B.R., and Ames, B.N. (1982) Complete analysisof cellular nucleotides by two-dimensional thin layerchromatography. J Biol Chem 257: 9759–9769.
Bogan, J.A., and Helmstetter, C.E. (1997) DNA sequestrationand transcription in the oriC region of Escherichia coli. MolMicrobiol 26: 889–896.
Boye, E., and Lobner-Olesen, A. (1991) Bacterial growthcontrol studied by flow cytometry. Res Microbiol 142: 131–135.
Bramhill, D., and Kornberg, A. (1988) Duplex opening byDnaA protein at novel sequences in initiation of replicationat the origin of the E. coli chromosome. Cell 52: 743–755.
Braun, R.E., O’Day, K., and Wright, A. (1985) Autoregulationof the DNA replication gene dnaA in E. coli K-12. Cell 40:159–169.
Camara, J.E., Skarstad, K., and Crooke, E. (2003) Controlledinitiation of chromosomal replication in Escherichia colirequires functional Hda protein. J Bacteriol 185: 3244–3248.
Campbell, J.L., and Kleckner, N. (1990) E. coli oriC and thednaA gene promoter are sequestered from Dam methyl-transferase following the passage of the chromosomal rep-lication fork. Cell 62: 967–979.
Chodavarapu, S., Felczak, M.M., Yaniv, J.R., and Kaguni,J.M. (2008a) Escherichia coli DnaA interacts with HU ininitiation at the E. coli replication origin. Mol Microbiol 67:781–792.
Chodavarapu, S., Gomez, R., Vicente, M., and Kaguni, J.M.(2008b) Escherichia coli Dps interacts with DnaA protein toimpede initiation: a model of adaptive mutation. Mol Micro-biol 67: 1331–1346.
Clark, D.J., and Maaloe, O. (1967) DNA replication and thedivision cycle in Escherichia coli. J Mol Biol 23: 99–112.
Cooper, S., and Helmstetter, C.E. (1968) Chromosome rep-lication and the division cycle of Escherichia coli B/r. J MolBiol 31: 519–540.
Dennis, P.P., Ehrenberg, M., and Bremer, H. (2004) Controlof rRNA synthesis in Escherichia coli: a systems biologyapproach. Microbiol Mol Biol Rev 68: 639–668.
Dixon, N.E., and Kornberg, A. (1984) Protein HU in the enzy-
1028 I. Flåtten, Morigen and K. Skarstad �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

matic replication of the chromosomal origin of Escherichiacoli. Proc Natl Acad Sci USA 81: 424–428.
Erzberger, J.P., Mott, M.L., and Berger, J.M. (2006) Struc-tural basis for ATP-dependent DnaA assembly andreplication-origin remodeling. Nat Struct Mol Biol 13: 676–683.
von Freiesleben, U., Rasmussen, K.V., Atlung, T., andHansen, F.G. (2000) Rifampicin-resistant initiation of chro-mosome replication from oriC in ihf mutants. Mol Microbiol37: 1087–1093.
Fuller, R.S., Funnell, B.E., and Kornberg, A. (1984) TheDnaA protein complex with the E. coli chromosomal repli-cation origin (oriC) and other DNA sites. Cell 38: 889–900.
Grimwade, J.E., Ryan, V.T., and Leonard, A.C. (2000) IHFredistributes bound initiator protein, DnaA, on supercoiledoriC of Escherichia coli. Mol Microbiol 35: 835–844.
Hansen, F.G., and von Meyenburg, K. (1979) Characteriza-tion of the dnaA, gyrB and other genes in the dnaA regionof the Escherichia coli chromosome on specialized trans-ducing phages lambda tna. Mol Gen Genet 175: 135–144.
Hansen, F.G., Christensen, B.B., and Atlung, T. (1991a) Theinitiator titration model: computer simulation of chromo-some and minichromosome control. Res Microbiol 142:161–167.
Hansen, F.G., Atlung, T., Braun, R.E., Wright, A., Hughes, P.,and Kohiyama, M. (1991b) Initiator (DnaA) protein concen-tration as a function of growth rate in Escherichia coli andSalmonella typhimurium. J Bacteriol 173: 5194–5199.
Hansen, F.G., Christensen, B.B., and Atlung, T. (2007)Sequence characteristics required for cooperative bindingand efficient in vivo titration of the replication initiatorprotein DnaA in E. coli. J Mol Biol 367: 942–952.
Hartmann, G., Honikel, K.O., Knusel, F., and Nuesch, J.(1967) The specific inhibition of the DNA-directed RNAsynthesis by rifamycin. Biochim Biophys Acta 145: 843–844.
Ishida, T., Akimitsu, N., Kashioka, T., Hatano, M., Kubota, T.,Ogata, Y., et al. (2004) DiaA, a novel DnaA-bindingprotein, ensures the timely initiation of Escherichia colichromosome replication. J Biol Chem 279: 45546–45555.
Jin, D.J., and Gross, C.A. (1988) Mapping and sequencing ofmutations in the Escherichia coli rpoB gene that lead torifampicin resistance. J Mol Biol 202: 45–58.
Katayama, T., Kubota, T., Kurokawa, K., Crooke, E., andSekimizu, K. (1998) The initiator function of DnaA protein isnegatively regulated by the sliding clamp of the E. colichromosomal replicase. Cell 94: 61–71.
Kato, J., and Katayama, T. (2001) Hda, a novel DnaA-relatedprotein, regulates the replication cycle in Escherichia coli.EMBO J 20: 4253–4262.
Keyamura, K., Fujikawa, N., Ishida, T., Ozaki, S., Su’etsugu,M., Fujimitsu, K., et al. (2007) The interaction of DiaA andDnaA regulates the replication cycle in E. coli by directlypromoting ATP DnaA-specific initiation complexes. GenesDev 21: 2083–2099.
Kitagawa, R., Ozaki, T., Moriya, S., and Ogawa, T. (1998)Negative control of replication initiation by a novel chromo-somal locus exhibiting exceptional affinity for Escherichiacoli DnaA protein. Genes Dev 12: 3032–3043.
Krause, M., Rnckert, B., Lurz, R., and Messer, W. (1997)Complexes at the replication origin of Bacillus subtilis with
homologous and heterologous DnaA protein. J Mol Biol274: 365–380.
Kücherer, C., Lother, H., Kölling, R., Schauzu, M.A., andMesser, W. (1986) Regulation of transcription of the chro-mosomal dnaA gene of Escherichia coli. Mol Gen Genet205: 115–121.
Lark, K.G. (1972) Evidence for the direct involvement of RNAin the initiation of DNA replication in Escherichia coli 15T-.J Mol Biol 64: 47–60.
Lavoie, B.D., Chan, B.S., Allison, R.G., and Chaconas, G.(1991) Structural aspects of a higher order nucleoproteincomplex: induction of an altered DNA structure at theMu-host junction of the Mu type 1 transpososome. EMBOJ 10: 3051–3059.
Liu, L.F., and Wang, J.C. (1987) Supercoiling of the DNAtemplate during transcription. Proc Natl Acad Sci USA 84:7024–7027.
Lu, M., Campbell, J.L., Boye, E., and Kleckner, N. (1994)SeqA: a negative modulator of replication initiation inE. coli. Cell 77: 413–426.
McGarry, K.C., Ryan, V.T., Grimwade, J.E., and Leonard,A.C. (2004) Two discriminatory binding sites in the Escheri-chia coli replication origin are required for DNA strandopening by initiator DnaA-ATP. Proc Natl Acad Sci USA101: 2811–2816.
Marszalek, J., and Kaguni, J.M. (1994) DnaA protein directsthe binding of DnaB protein in initiation of DNA replicationin Escherichia coli. J Biol Chem 269: 4883–4890.
Messer, W. (1972) Initiation of deoxyribonucleic acid replica-tion in Escherichia coli B-r: chronology of events and tran-scriptional control of initiation. J Bacteriol 112: 7–12.
Messer, W., and Weigel, C. (1996) Initiation of chromosomereplication. In Escherichia coli and Salmonella, Cellular,Molecular Biology. Neidhardt, F.C., Curtiss, R., Ingraham,J.L., Lin, E.C.C., Low, K.B., Magasanik, B., et al. (eds).Washington, DC: American Society for Microbiology, pp.1579–1601.
Morigen, Boye, E., Skarstad, K., and Lobner-Olesen, A.(2001) Regulation of chromosomal replication by DnaAprotein availability in Escherichia coli: effects of the datAregion. Biochim Biophys Acta 1521: 73–80.
Morigen, Lobner-Olesen, A., and Skarstad, K. (2003) Titra-tion of the Escherichia coli DnaA protein to excess datAsites causes destabilization of replication forks, delayedreplication initiation and delayed cell division. Mol Microbiol50: 349–362.
Morigen, Molina, F., and Skarstad, K. (2005) Deletion of thedatA site does not affect once-per-cell-cycle timing butinduces rifampin-resistant replication. J Bacteriol 187:3913–3920.
Nozaki, N., Okazaki, T., and Ogawa, T. (1988) In vitro tran-scription of the origin region of replication of the Escheri-chia coli chromosome. J Biol Chem 263: 14176–14183.
Ogawa, T., and Okazaki, T. (1991) Concurrent transcriptionfrom the gid and mioC promoters activates replication of anEscherichia coli minichromosome. Mol Gen Genet 230:193–200.
Ogawa, T., and Okazaki, T. (1994) Cell cycle-dependenttranscription from the gid and mioC promoters of Escheri-chia coli. J Bacteriol 176: 1609–1615.
Ovchinnikov, Y.A., Monastyrskaya, G.S., Guriev, S.O.,
DnaA protects RNA polymerase from rifampicin 1029
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030

Kalinina, N.F., Sverdlov, E.D., Gragerov, A.I., et al. (1983)RNA polymerase rifampicin resistance mutations inEscherichia coli: sequence changes and dominance. MolGen Genet 190: 344–348.
Peng, H., and Marians, K.J. (1993) Escherichia coli topoi-somerase IV. Purification, characterization, subunit struc-ture, and subunit interactions. J Biol Chem 268: 24481–24490.
Rabussay, D., and Zillig, W. (1969) A rifampicin resistantRNA-polymerase from E. coli altered in the beta-subunit.FEBS Lett 5: 104–106.
Russell, D.W., and Zinder, N.D. (1987) Hemimethylation pre-vents DNA replication in E. coli. Cell 50: 1071–1079.
Schaper, S., and Messer, W. (1995) Interaction of the initiatorprotein DnaA of Escherichia coli with its DNA target. J BiolChem 270: 17622–17626.
Sekimizu, K., Bramhill, D., and Kornberg, A. (1987) ATPactivates DnaA protein in initiating replication of plasmidsbearing the origin of the E. coli chromosome. Cell 50: 259–265.
Sekimizu, K., Yung, B.Y., and Kornberg, A. (1988) The DnaAprotein of Escherichia coli. Abundance, improved purifica-tion, and membrane binding. J Biol Chem 263: 7136–7140.
Severinov, K., Soushko, M., Goldfarb, A., and Nikiforov, V.(1993) Rifampicin region revisited. New rifampicin-resistant and streptolydigin-resistant mutants in the betasubunit of Escherichia coli RNA polymerase. J Biol Chem268: 14820–14825.
Skarstad, K., Boye, E., and Steen, H.B. (1986) Timing ofinitiation of chromosome replication in individual Escheri-chia coli cells. EMBO J 5: 1711–1717.
Skarstad, K., Baker, T.A., and Kornberg, A. (1990) Strandseparation required for initiation of replication at the chro-mosomal origin of E. coli is facilitated by a distant RNA-DNA hybrid. EMBO J 9: 2341–2348.
Speck, C., and Messer, W. (2001) Mechanism of originunwinding: sequential binding of DnaA to double- andsingle-stranded DNA. EMBO J 20: 1469–1476.
Speck, C., Weigel, C., and Messer, W. (1999) ATP- andADP-DnaA protein, a molecular switch in gene regulation.EMBO J 18: 6169–6176.
Su’etsugu, M., Emoto, A., Fujimitsu, K., Keyamura, K., andKatayama, T. (2003) Transcriptional control for initiation ofchromosomal replication in Escherichia coli: fluctuation ofthe level of origin transcription ensures timely initiation.Genes Cells 8: 731–745.
Szalewska-Palasz, A., Wegrzyn, A., Blaszczak, A., Taylor, K.,and Wegrzyn, G. (1998) DnaA-stimulated transcriptionalactivation of orilambda: Escherichia coli RNA polymerasebeta subunit as a transcriptional activator contact site. ProcNatl Acad Sci USA 95: 4241–4246.
Torheim, N.K., Boye, E., Lobner-Olesen, A., Stokke, T., andSkarstad, K. (2000) The Escherichia coli SeqA proteindestabilizes mutant DnaA204 protein. Mol Microbiol 37:629–638.
Vrentas, C.E., Gaal, T., Ross, W., Ebright, R.H., and Gourse,R.L. (2005) Response of RNA polymerase to ppGpp:requirement for the omega subunit and relief of thisrequirement by DksA. Genes Dev 19: 2378–2387.
Wang, J.D., Sanders, G.M., and Grossman, A.D. (2007)Nutritional control of elongation of DNA replication by(p)ppGpp. Cell 128: 865–875.
Wegrzyn, A., Szalewska-Palasz, A., Blaszczak, A., Liberek,K., and Wegrzyn, G. (1998) Differential inhibition of tran-scription from s70- and s32-dependent promoters byrifampicin. FEBS Lett 440: 172–174.
Wehrli, W., Knusel, F., Schmid, K., and Staehelin, M. (1968)Interaction of rifamycin with bacterial RNA polymerase.Proc Natl Acad Sci USA 61: 667–673.
Wold, S., Skarstad, K., Steen, H.B., Stokke, T., and Boye, E.(1994) The initiation mass for DNA replication in Escheri-chia coli K-12 is dependent on growth rate. EMBO J 13:2097–2102.
Yung, B.Y., and Kornberg, A. (1989) The DnaA initiatorprotein binds separate domains in the replication origin ofEscherichia coli. J Biol Chem 264: 6146–6150.
1030 I. Flåtten, Morigen and K. Skarstad �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 71, 1018–1030





























