Embed Size (px)
Citation preview

Journal of Controlled Release 74 (2001) 213–224www.elsevier.com/ locate / jconrel
Drug targeting using thermally responsive polymers and localhyperthermia
a a a b a ,*D.E. Meyer , B.C. Shin , G.A. Kong , M.W. Dewhirst , A. ChilkotiaDepartment of Biomedical Engineering, Duke University, Durham, NC 27708, USA
bDepartment of Radiation Oncology, Duke University Medical Center, Durham, NC 27710, USA
Abstract
We report a new thermal targeting method in which a thermally responsive drug carrier selectively accumulates in a solidtumor that is maintained above physiological temperature by externally applied, focused hyperthermia. We synthesized twothermally responsive polymers that were designed to exhibit a lower critical solution temperature (LCST) transition slightlyabove physiological temperature: (1) a genetically engineered elastin-like polypeptide (ELP) and (2) a copolymer ofN-isopropylacrylamide (NIPAAm) and acrylamide (AAm). The delivery of systemically injected polymer–rhodamineconjugates to solid tumors was investigated by in vivo fluorescence video microscopy of ovarian tumors implanted in dorsalskin fold window chambers in nude mice, with and without local hyperthermia. When tumors were heated to 428C, theaccumulation of a thermally responsive ELP with a LCST of 408C was approximately twofold greater than the concentrationof the same polymer in tumors that were not heated. Similar results were also obtained for a thermally responsivepoly(NIPAAM–co-AAm), though the enhanced accumulation of this carrier in heated tumors was lower than that observedfor the thermally responsive ELP. These results suggest that enhanced delivery of drugs to solid tumors can be achieved byconjugation to thermally responsive polymers combined with local heating of tumors. 2001 Elsevier Science B.V. Allrights reserved.
Keywords: Drug delivery; Tumor; Hyperthermia; Thermally responsive polymer; Elastin-like polypeptide; Poly(N-isopropylacrylamide);Lower critical solution temperature; Inverse transition
1. Introduction systems such as low-molecular-weight prodrugs,liposomes, and micro- and nano-particles have been
New drug delivery modalities for cancer therapy developed [1–5], but many of these delivery vehiclesare urgently needed because of the currently limited have shown limited therapeutic efficacy in the clinictherapeutic activity and insolubility of administered [1]. The limitations of current therapy provide aanti-cancer drugs, problems of accessibility and compelling rationale for the development of alter-heterogeneity of the tumor, and toxicity / immuno- native modalities for the targeted delivery of thera-genicity of the delivery agent itself. Many different peutics to solid tumors.
The present work is motivated by two observa-tions from the literature. First, soluble polymeric*Corresponding author. Tel.: 11-919-660-5373; fax: 11-919-drug carriers are attractive because they improve660-5362.
E-mail address: [email protected] (A. Chilkoti). drug pharmacokinetics, and lead to increased tumor
0168-3659/01/$ – see front matter 2001 Elsevier Science B.V. All rights reserved.PI I : S0168-3659( 01 )00319-4

214 D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224
accumulation over free drug due to passive targeting obtained for a thermally responsive poly(NIPAAm–(also called the enhanced permeability and retention co-AAm) with an LCST of 398C, though the en-(EPR) effect) [6]. Polymeric carriers do not, however hanced accumulation of this carrier in heated tumorsintrinsically target a specified physiological site. was lower than that observed for the thermallySecond, hyperthermia has been clinically used in the responsive ELP. These results suggest that thermalmanagement of solid tumors because it can syner- targeting of therapeutics by conjugation to thermallygistically enhance tumor cytotoxicity when combined responsive polymers is a promising method forwith chemo- or radio-therapy [7–9]. Furthermore, targeted delivery of anti-cancer therapeutics to solidhyperthermia preferentially increases the permeabili- tumors.ty of tumor vasculature compared to normal vascula- Although thermally responsive polymers suchture, which can further enhance the delivery of drugs poly(NIPAAM) and its copolymers have been usedto tumors [10–12]. Combined, these facts suggested for drug delivery [17–19], these previous studiesthat thermal targeting of polymeric drug carriers may focused on crosslinked hydrogels [20,21], and moreoffer synergistic advantages over either treatment recently on polymeric micelles [22,23] and micro /modality used individually. nanoparticles [24], which are designed to release
We hypothesized that polymeric carriers that entrapped drugs in response to the LCST transition.undergo a lower critical solution temperature In contrast, this is the first study, to our knowledge,(LCST) phase transition (also known as an inverse that investigates the feasibility of using soluble,transition), could be designed to be systemically thermally responsive polymer conjugates for targetedsoluble when injected in vivo, but to become insolu- delivery to solid tumors in combination with focusedble and accumulate in locally heated regions. This hyperthermia of tumors. Furthermore, these studiescould be achieved if their transition temperature have been carried out in an in vivo model. This(LCST¯408C) was greater than physiologic body aspect of the study is of critical significance, becausetemperature (T ¯378C), but less than the tempera- significant transport barriers exist in solid tumors thatb
ture in a region of local hyperthermia (T ¯428C). limit the delivery of drugs [25]. Hence, investigatingh
Two polymer systems that exhibit a LCST phase a new drug delivery modality in a clinically relevanttransition are poly(N-isopropylacrylamide) (poly- animal model is critical to evaluating its feasibility,(NIPAAm)) [13,14] and elastin-like polypeptides as reported in this paper.(ELPs) [15,16], which are biopolymers of the penta-peptide repeat Val–Pro–Gly–Xaa–Gly (where Xaa,termed the ‘guest residue’, is any amino acid except 2. Materials and methodsPro). By exploiting the phase transition of thesepolymers, this method combines thermal targeting of 2.1. ELP synthesisthe polymer–drug conjugate with the establishedadvantages of polymeric carriers (e.g. increased Our approach to the design and synthesis of ELPsplasma half-life and a high loading capacity) and has been described elsewhere [26]. Briefly, geneshyperthermia (e.g. increased cytotoxicity and macro- encoding two different ELP sequences were syn-molecular extravasation). thesized: one with guest residues Val, Gly, and Ala in
The delivery of systemically injected polymer– a 5:3:2 ratio, respectively, and a target LCST of 408Crhodamine conjugates to solid tumors was investi- (termed ELP1); and a second, control ELP, in whichgated by in vivo fluorescence video microscopy of this ratio was 1:7:8 and the target LCST was 558Chuman ovarian tumors implanted in dorsal skin fold (ELP2). These guest residue ratios were selectedwindow chambers in nude mice, with and without based upon the previous results of Urry et al. whohyperthermia. When tumors were heated to 428C, the characterized the effect of substitution of differentaccumulation of a thermally responsive ELP with a amino acids at the fourth, ‘guest residue’ positionLCST of 408C was approximately twofold greater upon the LCST of the ELP [27]. Short gene seg-than the concentration of the same polymer in tumors ments (encoding 10 pentapeptides for ELP1, 16 forthat were not heated. Similar results were also ELP2) were constructed from synthetic oligonucleo-

D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224 215
tides (Integrated DNA Technologies, Coralville, IA, termined by gel-permeation chromatography usingUSA) and initially cloned into pUC19 (New England poly(ethylene glycol) calibration standards. The frac-Biolabs, Beverly, MA, USA). The genes were oligo- tion of each polymer (pNIPAAm1 and pNIPAAm2)merized by recursive directional ligation (resulting in with a M of |20 000 Da was dialyzed againstw
genes encoding 150 ELP pentapeptides for ELP1 and regularly freshened distilled water for a week using a160 pentapeptides for ELP2) and then cloned into a cellulose dialysis membrane (MWCO 10 000; Spec-modified pET25b (Novagen, Madison, WI, USA) tra /Por). The dialyzed polymer solution was heatedexpression vector. The expression vector contained to 508C to induce the LCST transition of the polymertranslation initiation and termination codons, and and the aggregated polymer was recovered by cen-codons for short leading (Ser–Lys–Gly–Pro–Gly) trifugation (16 000 g for 10 min at |458C). Theand trailing (Trp–Pro) sequences. The ELPs were polymer was dried in a vacuum oven, and storedexpressed in the BLR(DE3) E. coli strain (Novagen). under air at 48C. These polymers were furtherThe ELPs were purified from other E. coli proteins characterized by elemental analysis (Galbraith Lab-in the soluble fraction of the cell lysate by inverse oratories, Knoxville, TN, USA): pNIPAAm1: C,transition cycling [26,28], and stored frozen at 61.1%, 62% (calculated, experimental); H, 9.3%,2808C. The ELPs were characterized for purity and 10%; N, 13.3%, 12.6%; S, 0.25%, 0.23%;molecular weight by SDS–PAGE and visualized by pNIPAAm2: C, 61.4%, 61.0%; H, 9.2%, 9.2%; N,copper staining. The molecular weight of ELP1 was 13.6%, 13.6%; S, 0.25%, 0.24%. The number and59.2 kDa and that of ELP2 was 61.1 kDa. weight average molecular weights (Mn, Mw), and
polydispersity (Dp) of pNIPAAm1 and pNIPAAm22.2. Poly(NIPAAm–co-AAm) synthesis are summarized in Table 1.
Amine-terminated copolymers of NIPAAm and 2.3. Characterization of the LCST transitionAAm were synthesized by free radical polymeri-zation using a chain transfer agent. N-iso- The LCST transition of the polymers were char-propylacrylamide (NIPAAm) and 2,29-azobis- acterized by monitoring the optical density at 350 nmbutyronitrile (AIBN) were obtained from Aldrich as a function of temperature on a UV–visible(Milwaukee, WI, USA) and recrystallized in hexane– spectrophotometer equipped with a multicell ther-benzene (70:30, v /v) (Aldrich) and methanol, respec- moelectric temperature controller (Cary 300 Bio;tively. 2-Aminoethanethiol hydrochloride (AET) (Al- Varian, Melbourne, Australia). Reversibility of thedrich), acrylamide (Polyscience, Warrington, PA, LCST transition was examined by heating and thenUSA), and all other reagents were used without cooling a polymer solution at a rate of 18C/min. Thefurther purification. Synthesis of two polymers with LCST was defined as the temperature at 5% maxi-NIPAAm:AAm ratios of 84:16 mol /mol mum turbidity upon heating an aqueous solution of(pNIPAAm1) and 68:32 mol /mol (pNIPAAm2) was the ELP or poly(NIPAAm–co-AAm).carried out as follows: the monomers (100 mmoltotal) were dissolved in 40 ml of methanol con- 2.4. Labelingtaining AIBN as initiator (1 mmol) and AET as achain transfer agent (1 mmol). Dried nitrogen was Poly(NIPAAm–co-AAm) contained a single aminebubbled through the monomer solution for 20 min
Table 1prior to polymerization. After polymerization at 608CPhysical properties of the amine terminated poly(NIPAAm–co-for 20 h, the copolymers were obtained by precipi-AAm) copolymerstation of the reaction solution into diethyl ether. The
pNIPAAm1 pNIPAAm2precipitated copolymers were dissolved again inmethanol. This procedure was repeated twice, and AAm (mol%) 18 32
M (Da) 14 000 13 000the polymers were then separated by size using n
M (Da) 24 000 23 000wsolvent fractionation [29]. The weight average mo-Polydispersity (D ) 1.71 1.77plecular weight (M ) of the copolymers were de-w

216 D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224
end group, while the ELP contained two primary jected into a cannulated tail vein. Six groups of fiveamines (the N-terminal amine and a lysine residue). animals each were studied: ELP1 at 428C, ELP2 atRhodamine Red-X succinimidyl ester was conju- 428C, ELP1 at 348C, pNIPAAm1 at 428C,gated to the primary amines using succinimidyl ester pNIPAAm2 at 428C, and pNIPAAm1 at 348C. Im-chemistry (Molecular Probes, Eugene, OR, USA) for ages were recorded continuously for 40 s afterin vivo fluorescence microscopy. In a typical conju- injection and then for 10 s every 2 min for 60 min.gation reaction, polymer was dissolved in 100 mM The images were digitized and the average pixelsodium bicarbonate (pH 8.4). A 10-mg/ml solution intensity for the window (approximately 4003400of Rhodamine Red-X succinimidyl ester in dimethyl- mm) was determined using image analysis softwareformamide was slowly added to a stirred polymer (Scion Image, Scion, Frederick, MD, USA). Eachsolution, to a final molar excess of 4 for the time point was normalized to the initial intensity atconjugation of rhodamine-to-ELP, and 2 for 30 s post-injection, after subtraction of the pre-rhodamine-to-poly(NIPAAm–co-AAm). The reac- injection background. A more detailed analysis oftants were incubated with continuous stirring for 2 h the interstitial and vascular distribution of the poly-at room temperature. Insoluble matter was removed mers as a function of time will be reported elsewhereby centrifugation at 48C. The polymer was then [35].purified by inverse transition cycling [28] and gel
filtration using a PD-10 Sephadex G-25 column(Amersham Pharmacia Biotech). The labeling yield 3. Resultsof rhodamine was calculated for the ELP conjugatesusing UV–visible spectrophotometry. Typically, the The primary objective in the design of a thermallyrhodamine to ELP molar ratio was |0.5, and the responsive polymer for use as a thermally targetedtypical rhodamine to poly(NIPAAm–co-AAm) molar drug carrier is that it should undergo its LCSTratio was |0.3 in the purified conjugate. transition in vivo above normothermic temperature
(T ) but below the hyperthermia temperature (T )b h
2.5. In vivo fluorescence video microscopy within the heated tumor. We hypothesized thatsystemically delivered polymer–drug conjugates
Athymic mice were implanted with dorsal skinfold would be soluble under physiological conditions, butwindow chambers containing a small piece of tumor would selectively accumulate in a solid tumor main-
3tissue (|0.1 mm human ovarian carcinoma; SKOV- tained at |428C by externally induced local hy-3) placed near the center of the window [30–32]. perthermia because of the LCST-induced desolvationThe preparations were used 7–8 days after implanta- and precipitation of the polymer. We thereforetion, when the tumors had grown to 2–3 mm in targeted an LCST of 408C for the thermally respon-diameter. The mice were anesthetized with sodium sive polymer carriers because it is intermediatepentobarbital (80 mg/kg i.p.) and positioned laterally between the subcutaneous temperature of 348C andrecumbent on a microscope stage (Carl Zeiss, Thorn- the maximum in vivo T of 428C, which is easily andh
wood, NY, USA). The window chamber, which was safely achieved in clinical hyperthermia withoutconnected to a temperature-controlled water bath inducing deleterious side effects in surrounding[33], was maintained either at 348C, the physiologic healthy tissue (e.g. edema and necrosis) [36,37].subcutaneous temperature in mice [34], or at 428C. A There are several variables that affect the LCST ofregion of the tumor containing clearly visible tumor the ELP and poly(NIPAAm) polymers: (1) polymermicrovasculature was selected under transillumina- composition, namely the mole fraction of thetion with a 203 objective. Then, under epi-illumina- alkylacrylamide that is copolymerized with NIPAAmtion using a dichroic filter set for rhodamine, images [38–40] and the identity and composition of thewere acquired by a SIT camera (Hamamatsu C2400- fourth guest residue (X) in the VPGXG pentapeptide08) and recorded in S-VHS format (Mitsubishi BV- repeat [27]; (2) cosolutes, such as salt and serum1000). In a typical experiment, 200 ml of rhodamine- proteins [41–43]; (3) conjugation of drug or reporterlabeled polymer in PBS (|1 mg/mouse) was in- groups; and (4) polymer concentration [44].

D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224 217
We sequentially present the effect of each of thesevariables on the LCST of these two polymerssystems in order to illustrate the strategy involved inthe design and synthesis of the thermally responsivepolymers, and selection of the appropriate ex-perimental parameters to achieve the target LCST of408C for in vivo delivery. We note that parameters(1) (3) and (4) are design variables, in that they canbe specified to modulate the LCST. In contrast, (2) islargely constrained by the experimental system: theeffect of salt and serum proteins on the LCST has tobe accounted for in the experimental design. Parame-ters (1) and (4) however, provide sufficient flexibili-
Fig. 1. Turbidity (optical density at 350 nm) as a function ofty to enable selection of the polymer compositiontemperature for the ELP (circles) and poly(NIPAAm–co-Am)
and the injected dose to achieve the target in vivo (squares) carriers. The turbidity profiles were obtained by heatingLCST of 408C. a 25 mM solution of each polymer at 18C/min in PBS. The LCST
transition was engineered to occur at |408C for the thermallyresponsive carriers (filled symbols). Thermally unresponsive con-3.1. Design of polymer carrierstrol carriers (open symbols) where designed with temperatureshigher than 408C such that they would remain soluble in heated
Both polymer carriers were designed to have a tumors.LCST of 408C, which was chosen because it ishigher than normal body temperature but lower thantemperatures that are routinely used in clinical from the turbidity profile obtained in the upwardhyperthermia [45]. We controlled the LCST of the thermal ramp. The LCST was 428C for ELP1, 418Cpolymers by adjusting their relative hydrophobicity. pNIPAAm1, 678C for ELP2, and 508C forFor ELP1, the LCST was increased to |408C from pNIPAAm2 (25 mM polymer solution in PBS).278C (LCST of the natural elastin repeat Val–Pro–Gly–Val–Gly) by substituting Ala and Gly for themore hydrophobic Val at the fourth position of the 3.2. Effect of conjugation of reporterpentapeptide [27]. Similarly, for pNIPAAm1, theLCST was increased to |408C from 328C [the LCST Conjugation of rhodamine reduced the ELP LCSTof poly(NIPAAm)] by the incorporation of the more by |48C. This effect was also observed forhydrophilic acrylamide co-monomer [poly- rhodamine-labeled poly(NIPAAm–co-AAm), al-(NIPAAm–co-AAm)] [38–41]. Control polymers though the shift was |18C (results not shown). This(ELP2 and pNIPAAm2) were similarly designed to is because the bulky, non-polar labels increase thebe more hydrophilic than ELP1 or pNIPAAm1, so as hydrophobicity of the polymer chain, and therebyto have a LCST greater than T to ensure that they decrease the LCST of the polymer. The decrease inh
would not undergo their phase transition in the LCST after conjugation has important implicationsheated tumors. for the loading of these polymers as therapeutic
The LCST transition of each polymer was char- carriers, particularly for drugs with significant hydro-acterized by monitoring solution turbidity as a phobic character. The decrease of LCST upon conju-function of temperature. Fig. 1 shows representative gation observed here does not require any compensa-heating turbidity profiles for the ELP and pNIPAAm tion other than adjustment of the polymer plasmacarriers. Below its LCST, each polymer solution concentration for in vivo delivery (discussed below).remained clear, but upon further heating the solution However, for drugs or reporters that are hydro-became turbid because of polymer aggregation over phobic, and especially for higher drug-to-carriera narrow temperature range of |28C. We defined the loading ratios, the carrier has to be designed to haveLCST as the temperature at 5% maximal turbidity a higher LCST than 408C in anticipation of a

218 D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224
substantial decrease in its LCST caused by conjuga- dence of the LCST is not unduly restrictive, how-tion of the drug. ever, because Fig. 2 illustrates that a large (|3-fold)
deviation from the target plasma concentration is3.3. Effect of cosolutes necessary to shift the LCST out of the therapeutic
window (e.g. |38–428C). For the in vivo experi-The LCST also depends on co-solutes in the ments, we selected a target plasma concentration to
solution, and is known to vary with ionic strength of result in a LCST of |408C in plasma from the resultssalts [42] and with the presence of other solutes (e.g. in Fig. 2, after correction for the decrease in LCSTglucose, protein) [43]. We hypothesized that the due to conjugation of rhodamine to the polymers. Wepresence of serum proteins in vivo would therefore then calculated the dose required to achieve thisdepress the LCST. Indeed, we found that murine concentration using an estimated mouse plasmaplasma reduced the LCST by 48C for the ELP and by volume of |1 ml [48,49].18C for poly(NIPAAm–co-AAm) when compared toPBS (results not shown). We found that the effect of 3.5. In vivo targetingmurine serum upon the LCST of both polymerscould be simulated by the addition of 0.9 mM BSA Temperature-controlled in vivo fluorescence vid-to PBS, and we typically determine the LCST in this eomicroscopy of systemically injected, polymer–mock serum to estimate the in vivo LCST. rhodamine conjugates enabled real-time visualization
of their accumulation within the tumor. The ther-3.4. Polymer concentration mally responsive polymers (ELP1 or pNIPAAm1)
were injected intravenously into mice with theThe LCST of the ELP and pNIPAAm carriers window chamber heated to 428C. Heated (thermally
decreases as a logarithmic function of polymer unresponsive ELP2 or pNIPAAm2 carriers at 428C)concentration (Fig. 2) [46,47]. Selection of the in and unheated (ELP1 or pNIPAAm1 at 348C) controlvivo concentration of the polymer carrier, therefore, groups were also studied. Image analysis of the totalis an important variable that can be utilized to ensure fluorescence intensity in the window allowed quan-that an LCST between T and T is attained in vivo titative comparison between the three groups becauseb h
after dilution in plasma. The concentration depen- the measured fluorescence intensity is linearly relatedto fluorophore concentration for our experimentalconditions [31]. We assumed that immediately afterinjection the carrier is present only in the vascula-ture, and we normalized to the initial fluorescenceintensity at 30 s post-injection to correct for varia-tions in injected dose or in absolute fluorescenceintensity between the polymers (e.g. due to differ-ences in rhodamine conjugation efficiency).
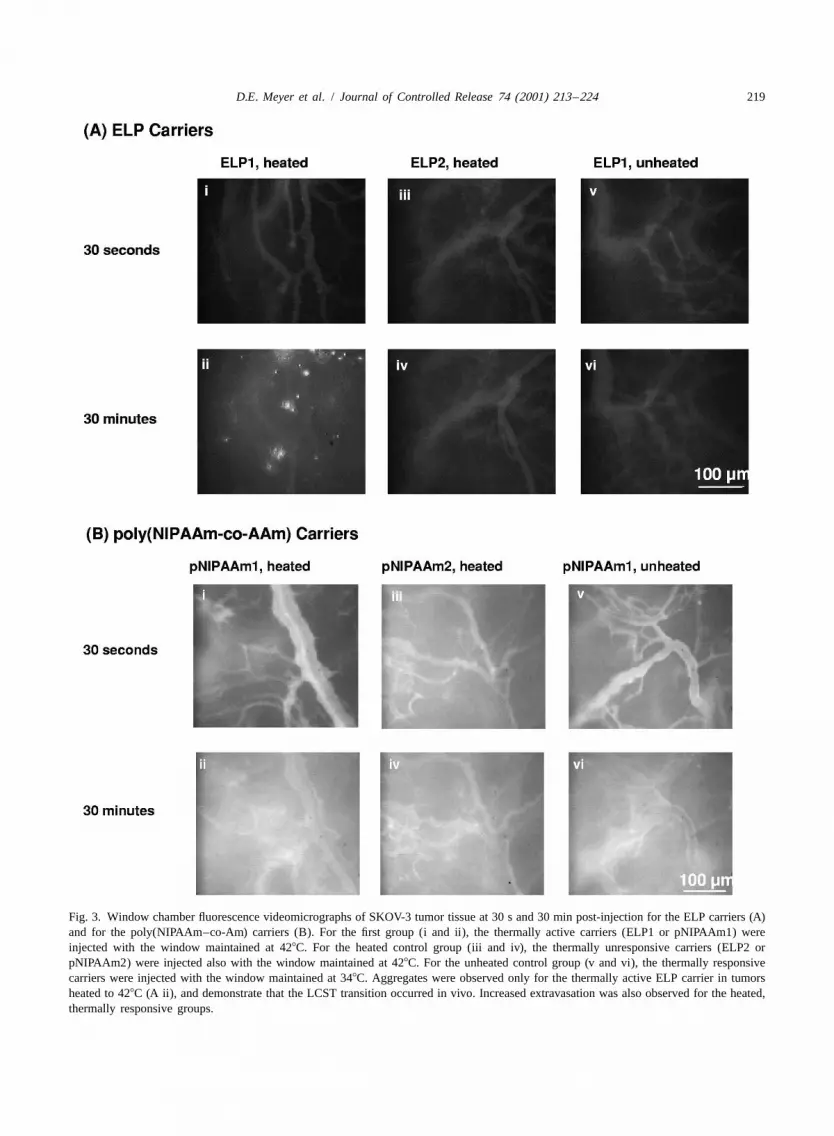
Fig. 3A and B shows representative videomicrog-raphs acquired at 30 s and 30 min after injection ofthe ELP and poly(NIPAAm–co-AAm) carriers, re-spectively. Initially at 30 s, the labeled carrier wasconfined to the vasculature, and the three groupswere indistinguishable for both the ELP and the
Fig. 2. LCST (defined as the temperature at the onset of turbidity) poly(NIPAAm–co-AAm) carriers. Within one to twois shown as a function of polymer concentration. For both ELP1 min after the thermally responsive ELP1 was in-and pNIPAAm1, a logarithmic relationship (solid lines) was jected into mice with heated window chambers,observed between LCST and concentration. The experiments were
fluorescent particles were observed in the vasculatureperformed in PBS supplemented with 0.9 mM BSA (mock serum).(Fig. 3A, ii). Similar particles were also observed inFrom these data, a dose was selected to achieve a desired target in
vivo LCST after dilution in plasma. some of the animals injected with pNIPAAm1. No
D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224 219
Fig. 3. Window chamber fluorescence videomicrographs of SKOV-3 tumor tissue at 30 s and 30 min post-injection for the ELP carriers (A)and for the poly(NIPAAm–co-Am) carriers (B). For the first group (i and ii), the thermally active carriers (ELP1 or pNIPAAm1) wereinjected with the window maintained at 428C. For the heated control group (iii and iv), the thermally unresponsive carriers (ELP2 orpNIPAAm2) were injected also with the window maintained at 428C. For the unheated control group (v and vi), the thermally responsivecarriers were injected with the window maintained at 348C. Aggregates were observed only for the thermally active ELP carrier in tumorsheated to 428C (A ii), and demonstrate that the LCST transition occurred in vivo. Increased extravasation was also observed for the heated,thermally responsive groups.
220 D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224
such particles were observed in any of the heated or selected for the ELP and poly(NIPAAm–co-AAm)unheated control animals for either carrier. The carriers to illustrate the enhancement of tumoraggregates often attached to the vessel wall, and accumulation that can be achieved by each system.grew in intensity and size over time. In some At 44 min post-injection, the total window intensityinstances, the particles were sheared from the vessel increased by |250% for heated ELP1 relative to itswalls and carried away by the blood flow. Because initial window intensity, indicating significant ac-the LCST transition is completely reversible, any cumulation of the carrier occurred over time. Afterparticles washed from the heated tissue would be 26 min, pNIPAAm1 had similarly increased 180%expected to rapidly disaggregate and resolubilize. relative to its initial window intensity. Total accumu-The presence of these aggregates only in the heated lation for the thermally responsive carriers was |2-window chamber provides clear evidence that the fold greater than that for the heated and unheatedLCST can be engineered to occur at a specific LCST control groups (P,0.05 for heated pNIPAAm1even in the complex solution environment within the versus both control groups and for ELP1 versus thebody. Accumulation of the thermally responsive unheated control group; P,0.1 for heated ELP1carriers (ELP1 and pNIPAAm1) in heated tumors versus the heated ELP2 control group). The vid-increased with time, and by 30 min, extravascular eomicroscopy results (Fig. 3A and B) suggest thatareas of the window often became so bright that they the observed increases in total window intensity inwere indistinguishable from vessels (Fig. 3A and B). heated tumors of the thermally responsive carriers
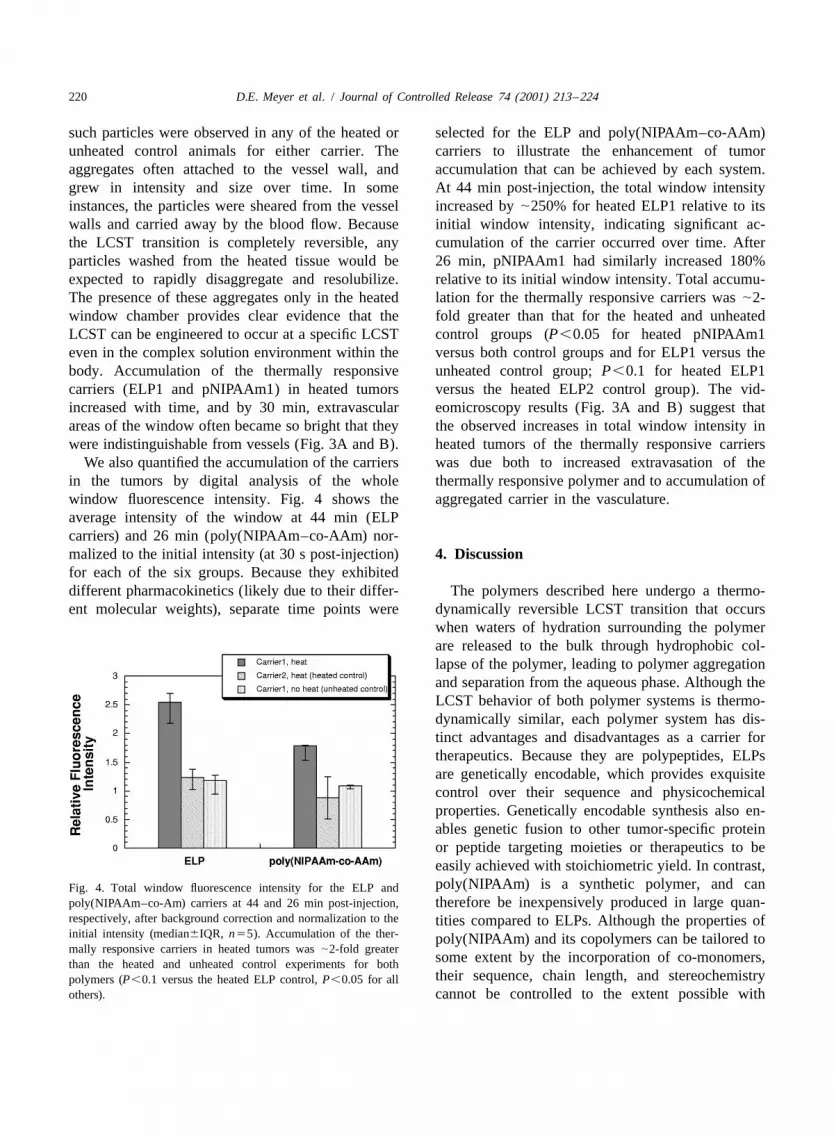
We also quantified the accumulation of the carriers was due both to increased extravasation of thein the tumors by digital analysis of the whole thermally responsive polymer and to accumulation ofwindow fluorescence intensity. Fig. 4 shows the aggregated carrier in the vasculature.average intensity of the window at 44 min (ELPcarriers) and 26 min (poly(NIPAAm–co-AAm) nor-malized to the initial intensity (at 30 s post-injection) 4. Discussionfor each of the six groups. Because they exhibiteddifferent pharmacokinetics (likely due to their differ- The polymers described here undergo a thermo-ent molecular weights), separate time points were dynamically reversible LCST transition that occurs
when waters of hydration surrounding the polymerare released to the bulk through hydrophobic col-lapse of the polymer, leading to polymer aggregationand separation from the aqueous phase. Although theLCST behavior of both polymer systems is thermo-dynamically similar, each polymer system has dis-tinct advantages and disadvantages as a carrier fortherapeutics. Because they are polypeptides, ELPsare genetically encodable, which provides exquisitecontrol over their sequence and physicochemicalproperties. Genetically encodable synthesis also en-ables genetic fusion to other tumor-specific proteinor peptide targeting moieties or therapeutics to beeasily achieved with stoichiometric yield. In contrast,poly(NIPAAm) is a synthetic polymer, and canFig. 4. Total window fluorescence intensity for the ELP and
poly(NIPAAm–co-Am) carriers at 44 and 26 min post-injection, therefore be inexpensively produced in large quan-respectively, after background correction and normalization to the tities compared to ELPs. Although the properties ofinitial intensity (median6IQR, n55). Accumulation of the ther- poly(NIPAAm) and its copolymers can be tailored tomally responsive carriers in heated tumors was |2-fold greater
some extent by the incorporation of co-monomers,than the heated and unheated control experiments for boththeir sequence, chain length, and stereochemistrypolymers (P,0.1 versus the heated ELP control, P,0.05 for all
others). cannot be controlled to the extent possible with

D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224 221
genetically encoded ELPs. Control of the yield and responsive soluble polymers for systemic delivery ofstoichiometry of poly(NIPAAm) conjugates of thera- anticancer therapeutics. The first and unique advan-peutic or targeting moieties is also considerably more tage of these polymers over other thermally sensitivedifficult to achieve. carriers, such as temperature sensitive liposomes
Both polymer carriers were designed to have a [50], is that accumulation of the drug in the targetLCST of 408C, which was chosen because it is tissue occurs through the LCST transition of thehigher than normal body temperature but lower than carrier rather than through triggered release of thetemperatures that are routinely used in clinical drug. A concentration gradient is therefore nothyperthermia [45]. We controlled the LCST of the required to drive accumulation of thermally respon-polymers by adjusting their relative hydrophobicity. sive polymers in the heated tumor, unlike otherFor ELP1, the LCST was increased to |408C from delivery systems. Thermally responsive polymers278C (LCST of the natural elastin repeat Val–Pro– will continue to accumulate because of aggregationGly–Val–Gly) by substituting Ala and Gly for the in a heated tumor even when their blood concen-more hydrophobic Val at the fourth position of the tration is less than the concentration in the tumor.pentapeptide [27]. Similarly, for pNIPAAm1, the This is a significant feature because it enables theLCST was increased to |408C from 328C (the LCST polymer–drug conjugate to be injected at low con-of the poly(NIPAAm) homopolymer) by the incorpo- centration systemically, while still achieving a higherration of the more hydrophilic acrylamide co-mono- concentration in the tumor.mer [40–42]. Control polymers (ELP2 and Second, thermal targeting is attractive because it ispNIPAAm2) were similarly designed to have a not specific to a particular cell type, and thereforeLCST greater than T such that they would not any organ or tissue can be targeted, independent ofh
undergo their phase transition in the heated tissues. the availability of specific ligands or antibodies. ForIn addition to the effect of polymer composition the delivery of radiotherapeutics, regional targeting
upon the LCST of the polymer, accounting for the to the tumor (as opposed to tumor cells) is sufficienteffect of concentration upon the LCST is a critical for therapy, and studies in progress are thereforeconsideration in the design of a thermally responsive focused on their delivery to solid tumors.polymeric drug carrier. Because the injected dose is Third, in addition to radionuclides, thermal target-rapidly diluted in the plasma volume, the LCST of ing is also applicable to tumor-specific delivery ofthe diluted polymer will increase upon injection, other therapeutic moieties that are conjugated to thewhich must be taken into account when selecting the polymer. To be effective, however, most other drugs,injected dose of the polymer–drug conjugate. On the such as small molecule chemotherapeutics, andother hand, the dilution effect can also be exploited biomolecular therapeutics (proteins, DNA, RNA,in designing an administration protocol, because it carbohydrates or their mimics) require extravasationprovides flexibility in controlling the LCST of the of the drug from the tumor microvasculature andthermally responsive polymer in vivo by selecting a transport to, and uptake by tumor cells [25]. Whendose that will yield a LCST of |408C upon dilution these drugs are conjugated to a polymer carrier,in plasma. This increase in LCST with dilution is extravasation and transport might require release ofespecially useful in experimental design, because it the drug from the polymer carrier via the incorpora-compensates for the decrease in LCST that occurs tion of proteolytically or hydrolytically labile linkersupon conjugation of drugs / reporters and due to between the drug and the polymer [1]. Conjugationinteraction of the polymer with serum proteins. of this class of drugs to a thermally responsiveSelection of an appropriate in vivo concentration of polymer is advantageous, however, because it willthe polymer carrier, therefore, is an important vari- lead to significant improvement in the first step ofable that can be utilized to specify the in vivo LCST systemic targeting of the polymer–drug conjugate toof the polymer–drug conjugate. tumors. For these applications, thermal targeting
In addition to the increased accumulation of the could also be synergistically combined with a tumorthermally responsive carriers in heated tumors, there cell-specific affinity targeting moiety by gene-levelare several other attractive features of thermally fusion or chemical conjugation of the targeting

222 D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224
moiety [51–53]. The thermally sensitive polymer lar weight, the injected dose, and the mode ofwould then serve as a first stage, regional targeting administration (e.g. bolus versus steady infusion).method to rapidly increase the concentration of the Finally, synergistic thermal and affinity targetingpolymer-drug conjugate in the tumor, followed by schemes offer the possibility of further improving therelease of the drug through a labile linker, which efficiency of targeted delivery. We conclude thatcould also incorporate a secondary, affinity targeting enhanced delivery of therapeutics to solid tumors canmoiety specific to tumor cells. be achieved by conjugation to thermally responsive
Finally, the ability to maintain focused hyperther- synthetic polymers, in concert with targeted hyper-mia in tumors is greatly facilitated both by the thermia, by precisely specifying the LCST of theabnormal physiology of tumors and the availability thermally responsive polymer.of hyperthermia applicators for clinical use. Uponapplication of external hyperthermia to normal tis-sues, vasodilation occurs in the vascular bed, with a Acknowledgementssubsequent increase in blood perfusion rate, whichregulates tissue temperature [54–57]. Tumors, how-
This work was supported by a Whitaker Founda-ever, are unable to efficiently vasodilate upon heat-
tion research grant (A.C.), NSF CAREER awarding. Thus, as the tumor is heated, vasodilation does
BES-97-33009 (A.C.), a grant from the Targon Corp.not occur and the tumor temperature continues to rise
through the Comprehensive Cancer Center at Dukebecause of a lack of increased blood perfusion. In
University Medical Center (A.C.), and the Nationaladdition, heat transfer out of tumors is impeded by
Institutes of Health, grant CA42745 (MW.D). Wethe absence of functional artery–vein pairs, which
also thank the Whitaker Foundation for support ofallow efficient heat transfer out of normal tissues
D.E.M. as a graduate fellow.[58]. The design of hyperthermia applicators is also amature technology; several different hyperthermiaapplicators based on microwave arrays [59,60] and
Referencesultrasound transducers [61,62] have been developedfor clinical use.
[1] R. Duncan, Drug–polymer conjugates: potential for im-proved chemotherapy, Anticancer Drugs 3 (1992) 175–210.
[2] M. Jones, J. Leroux, Polymeric micelles — a new generation5. Conclusionsof colloidal drug carriers, Eur. J. Pharm. Biopharm. 48(1999) 101–111.
In summary, thermally responsive polymeric car- [3] V.P. Torchilin, Polymer-coated long-circulating microparticu-late pharmaceuticals, J. Microencapsul. 15 (1998) 1–19.riers in combination with hyperthermia achieve a
[4] T.M. Allen, Liposomal drug formulations — Rationale forsignificant increase in delivery to solid tumorsdevelopment and what we can expect in the future, Drugs 56compared to the same polymers without hyperther-(1998) 747–756.
mia and compared to thermally unresponsive poly- [5] R. Langer, Drug delivery and targeting, Nature 392 (Suppl.)mers of similar molecular weight and composition (1998) 5–10.
[6] Y. Matsamura, H. Maeda, A new concept for macromolecu-with hyperthermia. The window chamber studieslar therapeutics in cancer chemotherapy: mechanisms ofreveal that much of the increased accumulation oftumoritropic accumulation of protein and the antitumor agentthe thermally responsive polymers (ELP1 andSMANCS, Cancer Res. 46 (1986) 6387–6392.
pNIPAAm1) in heated tumors is due to aggregation [7] S.B. Field, J.W. Hand, An Introduction To the Practicalof the polymer (for ELP1) and increased extravasa- Aspects of Clinical Hyperthermia, Taylor and Francis,
London, 1990.tion of both classes of thermally responsive carriers[8] K. Engin, D.B. Leeper, L. Tupchong, F.M. Waterman,within the tumor upon undergoing their LCST
Thermoradiotherapy in the management of superficial malig-transition. This thermal targeting system has not yetnant tumors, Clin. Cancer Res. 1 (1995) 1139–1145.
been optimized and we believe that accumulation of [9] M.W. Dewhirst, L. Prosnitz, D. Thrall, D. Prescott, S. Clegg,the thermally responsive carriers in tumors can be C. Charles, J. MacFall, G. Rosner, T. Samulski, E. Gillette,further increased by optimizing the polymer molecu- S. LaRue, Hyperthermic treatment of malignant diseases:

D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224 223
current status and a view toward the future, Semin. Oncol. 24 inverse temperature transition depends on mean residue(1997) 616–625. hydrophobicity, J. Am. Chem. Soc. 113 (1991) 4346–4348.
[10] K. Engin, Biological rationale and clinical experience with [28] D.T. McPherson, J. Xu, D.W. Urry, Product purification byhyperthermia, Control. Clin. Trials 17 (1996) 316–342. reversible phase transition following Escherichia coli expres-
sion of genes encoding up to 251 repeats of the elastomeric[11] T. Feyerabend, R. Steeves, G.J. Wiedemann, E. Richter, H.I.pentapeptide GVGVP, Protein Exp. Purif. 7 (1996) 51–57.Robins, Rationale and clinical status of local hyperthermia,
radiation, and chemotherapy in locally advanced malignan- [29] L.H. Tung, in: Fractionation of Synthetic Polymers, Marcelcies, Anticancer Res. 17 (1997) 2895–2897. Dekker, New York, 1977, p. 103.
[12] R. Issels, Hyperthermia combined with chemotherapy — [30] M.W. Dewhirst, C.Y. Tso, R. Oliver, C.S. Gustafson, T.W.biological rationale, clinical application, and treatment re- Secomb, J.F. Gross, Morphologic and hemodynamic com-sults, Onkologie 22 (1999) 374–381. parison of tumor and healing normal tissue microvasculature,
Int. J. Radiat. Oncol. Biol. Phys. 17 (1989) 91–99.[13] M. Heskins, J.E. Guillet, Solution properties of poly(N-isopropylacrylamide), J. Macromol. Sci. Chem. A 2 (1968) [31] N.Z. Wu, D. Da, T.L. Rudoll, D. Needham, A.R. Whorton,1441. M.W. Dewhirst, Increased microvascular permeability contri-
butes to preferential accumulation of stealth liposomes in[14] H. Schild, Poly(NIPAAm): Experiment, theory, and applica-tumor tissue, Cancer Res. 53 (1993) 3765–3770.tions, Prog. Polym. Sci. 17 (1992) 163–249.
[32] N.Z. Wu, B. Klitzman, G. Rosner, D. Needham, M.W.[15] D.W. Urry, Free energy transduction in polypeptides andDewhirst, Measurement of material extravasation in mi-proteins based on inverse temperature transitions, Prog.crovascular networks using fluorescence video-microscopy,Biophys. Mol. Biol. 57 (1992) 23–57.Microvasc. Res. 46 (1993) 231–253.[16] D.W. Urry, Physical chemistry of biological free energy
[33] M.H. Gaber, N.Z. Wu, K. Hong, S.K. Huang, M.W. Dewhirst,transduction as demonstrated by elastic protein-based poly-D. Papahadjopoulos, Thermosensitive liposomes: extravasa-mers, J. Phys. Chem. B 101 (1997) 11007–11028.tion and release of contents in tumor microvascular net-[17] A.S. Hoffman, Intelligent polymers in medicine and bio-works, Int. J. Radiat. Oncol. Biol. Phys. 36 (1996) 1177–technology, Macromol. Sym. 98 (1995) 645–664.1187.[18] R. Yoshida, K. Sakai, T. Okano, Y. Sakurai, Pulsatile drug
[34] J.F. Gross, R. Roemer, M.W. Dewhirst, M. Meyer, A uniformdelivery systems using hydrogels, Adv. Drug Deliv. Rev. 11thermal field in a hyperthermia chamber for microvascular(1993) 85–108.studies, Int. J. Heat Mass Transfer. 25 (1982) 1313–1320.[19] I.Y. Galaev, B. Mattiasson, ‘Smart’ polymers and what they
could do in biotechnology and medicine, Trends. Biotechnol. [35] D.E. Meyer, G.H. Kong, M.W. Dewhirst, M. Zalutsky, A.17 (1999) 335–339. Chilkoti, Targeting a genetically engineered elastin-like
polypeptide to solid tumors by local hyperthermia, Cancer[20] S.H. Yuk, Y.H. Bae, Phase-transition polymers for drugRes. 61 (2001) 1548–1554.delivery, Crit. Rev. Ther. Drug Carrier Syst. 16 (1999)
385–423. [36] M.W. Dewhirst, Future directions in hyperthermia biology,Int. J. Hypertherm. 10 (1994) 339–345.[21] C.S. Brazel, N.A. Peppas, Pulsatile local delivery of throm-
bolytic and antithrombotic agents using poly(N-iso- [37] S.F. Badylak, C.F. Babbs, T.M. Skojac, W.D. Voorhees, R.C.propylacrylamide–co-methacrylic acid) hydrogels, J. Con- Richardson, Hyperthermia-induced vascular injury in normaltrol. Release 39 (1996) 57–64. and neoplastic tissue, Cancer 56 (1985) 991–1000.
[22] J. Taillefer, M.C. Jones, N. Brasseur, J.E. van Lier, J.C. [38] C.K. Chiklis, J.M. Grasshof, Swelling of thin films. I.Leroux, Preparation and characterization of pH-responsive Acrylamide–N-isopropylacrylamide copolymers in water, J.polymeric micelles for the delivery of photosensitizing Polym. Sci. 8 (Part A-2) (1970) 1617–1626.anticancer drugs, J. Pharm. Sci. 89 (2000) 52–62. [39] L.D. Taylor, L.D. Cerankowski, Preparation of films ex-
[23] J.E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. hibiting a balanced temperature dependence to permeation bySakurai, T. Okano, Thermo-responsive drug delivery from aqueous solutions — a study of lower cosolute behavior, J.polymeric micelles constructed using block copolymers of Polym. Sci. Polym. Chem. 13 (1975) 2551–2570.poly(N-isopropylacrylamide) and poly(butylmethacrylate), J. [40] J.H. Priest, S.L. Murray, R.J. Nelson, A.S. Hoffman, LowerControl. Release 62 (1999) 115–127. critical solution temperatures of aqueous copolymers of N-
[24] C. Ramkissoon-Ganorkar, F. Liu, M. Baudys, S.W. Kim, isopropylacrylamide and other N-substituted acrylamides,Modulating insulin release profile from release from pH/ ACS Sym. Ser. 350 (1987) 255–264.temperature sensitive polymeric beads through polymer [41] H. Feil, Y.H. Bae, J. Feijen, S.W. Kim, Effect of comonomermolecular weight, J. Control. Release 59 (1999) 287–298. hydrophilicity and ionization on the lower critical solution
[25] R.K. Jain, Barriers to drug delivery in solid tumors, Sci. Am. temperature of N-isopropylacrylamide copolymers, Macro-271 (1994) 58–65. molecules 26 (1993) 2497–2500.
[26] D.E. Meyer, A. Chilkoti, Purification of recombinant proteins [42] T. Baltes, F. Garret-Flaudy, R. Freitag, Investigation of theby fusion with thermally responsive polypeptides, Nature. LCST of polyacrylamides as a function of molecular parame-Biotechnol. 17 (1999) 1112–1115. ters and the solvent composition, J. Polym. Sci. A. Polym.
Chem. 37 (1999) 2977–2989.[27] D.W. Urry, C.H. Luan, T.M. Parker, D.C. Gowda, K.U.Prasad, M.C. Reid, A. Safavy, Temperature of polypeptide [43] Y.-H. Kim, I.C. Kwon, Y.H. Bae, S.W. Kim, Saccharide

224 D.E. Meyer et al. / Journal of Controlled Release 74 (2001) 213 –224
effect on the lower critical solution temperature of ther- [53] Z.R. Lu, P. Kopeckova, J. Kopecek, Polymerizable Fab9
mosensitive polymers, Macromolecules 28 (1995) 939–944. antibody fragments for targeting of anticancer drugs, Nature[44] H.G. Schild, M. Muthukumar, D.A. Tirrell, Cononsolvency Biotechnol. 279 (1999) 377–380.
in mixed aqueous solutions of poly(N-isopropylacrylamide), [54] P. Vaupel, F. Kallinoqwski, Physiological effects of hy-Macromolecules 24 (1991) 948–952. perthermia, Recent Results Cancer Res. 104 (1987) 71–109.
[45] M.W. Dewhirst, in: M.H. Seegenschmiedt, P. Fessenden, C.C. [55] C.W. Song, A. Lokshina, J.G. Rhee, M. Patten, S.H. Levitt,Vernon (Eds.), Principles and Practice of Ther- Implication of blood flow in hyperthermic treatment ofmoradiotherapy and Thermochemotherapy, Vol. I, Springer- tumors, IEEE Trans. Biomed. Eng. 31 (1984) 9–16.Verlag, Berlin, 1995, pp. 123–136. [56] R.B. Roemer, J.R. Oleson, T.C. Cetas, Oscillatory tempera-
[46] H.G. Schild, D.A. Tirrell, Microcalorimetric detection of ture response to constant power applied to canine muscle,lower critical solution temperatures in aqueous polymer Am. J. Physiol. 249 (1985) R153–R157.solutions, J. Phys. Chem. 94 (1990) 4352–4356. [57] H.S. Reinhold, B. Endrich, Tumor microcirculation as a
[47] D.W. Urry, T.L. Trapane, K.U. Prasad, Phase-structure target for hyperthermia: A review, Int. J. Hypertherm. 2transitions of the elastin polypentapeptide–water system (1986) 111–137.within the framework of composition–temperature studies, [58] M.N. Gaze, The current status of targeted radiotherapy inBiopolymer 24 (1985) 2345–2356. clinical practice, Phys. Med. Biol. 41 (1996) 1895–1903.
[48] R.W. Barbee, B.D. Perry, R.N. Re, J.P. Murgo, Microsphere [59] P.F. Turner, Mini-annular phased array for limb hyperther-and dilution techniques for the determination of blood flows mia, IEEE Trans. Microwave Theory Tech. 34 (1986) 508–and volumes in conscious mice, Am. J. Physiol. 263 (1992) 513.R728–R733. [60] T.V. Samulski, D.S. Kapp, P. Fessenden, A. Lohrbach,
[49] P.W. Durbin, N. Jeung, B. Kullgren, G.K. Clemons, Gross Heating deep seated eccentrically located tumors with ancomposition and plasma and extracellular water volumes of annular phased array system: A comparative clinical studytissues of a reference mouse, Health Phys. 63 (1992) 427– using two annular array operating configurations, Int. J.442. Radiat. Oncol. Biol. Phys. 14 (1987) 83–94.
[50] G.H. Kong, M.W. Dewhirst, Hyperthermia and liposomes, [61] P.F. Fessenden, E.R. Lee, T.L. Anderson, J.W. Strohbehn,Int. J. Hypertherm. 15 (1999) 345–370. J.L. Meyel, T.V. Samulski, J.B. Marmor, Experience with a
[51] W. Arap, R. Pasqualini, E. Ruoslahti, Cancer treatment by multitransducer ultrasound system for localized hyperthermiatargeted drug delivery to tumor vasculature in a mouse of deep tissues, IEEE Trans. Biomed. Eng. 31 (1984) 126–model, Science 279 (1998) 377–380. 135.
[52] R. Panchagnula, C.S. Dey, Monoclonal antibodies in drug [62] C.J. Diederich, K. Hynynen, Ultrasound technology fortargeting, J. Clin. Pharm. Ther. 22 (1997) 7–19. hyperthermia, Ultrasound Med. Biol. 25 (1999) 871–887.





























