Embed Size (px)
Citation preview

308 Virology 376 (2008) 308–322
Contents lists available at ScienceDirect
Virology
j ourna l homepage: www.e lsev ie r.com/ locate /yv i ro
Effect of transcription peptide inhibitors on HIV-1 replication
Rachel Van Duyne a, Jessica Cardenas a, Rebecca Easley a, Weilin Wu a, Kylene Kehn-Hall a, Zak Klase a,Susana Mendez b, Chen Zeng c, Hao Chen c, Mohammed Saifuddin d, Fatah Kashanchi a,e,f,⁎a The George Washington University Medical Center, Department of Microbiology, Immunology, and Tropical Medicine, Washington, DC 20037, USAb Cornell University, The College of Veterinary Medicine, Ithaca, NY 14853, USAc The George Washington University, Department of Physics, Washington, DC, 20037, USAd CONRAD, Eastern Virginia Medical School, 1611 North Kent Street, Suite 806, Arlington, Virginia 22209, USAe The Institute for Genomic Research, Rockville, MD 20850, USAf W.M. Keck Institute for Proteomics Technology and Applications, USA
a r t i c l e i n f o
⁎ Corresponding author. The George Washington U2300 Eye Street, NW, Ross Hall, Room 552, Washington, D1780.
E-mail addresses: [email protected] (R. Van Duyn(J. Cardenas), [email protected] (R. Easley), [email protected] (K. Kehn-Hall), [email protected]@cornell.edu (S. Mendez), [email protected] (C. [email protected] (M. Saifuddin), bcmfxk@gwumc
0042-6822/$ – see front matter. Published by Elsevier Idoi:10.1016/j.virol.2008.02.036
a b s t r a c t
Article history:Received 21 November 2007Returned to author for revision21 December 2007Accepted 27 February 2008Available online 2 May 2008
HIV-1 manipulates cellular machineries such as cyclin dependent kinases (cdks) and their cyclin elements, tostimulate virus production and maintain latent infection. Specifically, the HIV-1 viral protein Tat increasesviral transcription by binding to the TAR promoter element. This binding event is mediated by thephosphorylation of Pol II by complexes such as cdk9/Cyclin T and cdk2/Cyclin E. Recent studies have shownthat a Tat 41/44 peptide derivative prevents the loading of cdk2 onto the HIV-1 promoter, inhibiting geneexpression and replication. Here we show that Tat peptide analogs computationally designed to dock at thecyclin binding site of cdk2 have the ability to bind to cdk2 and inhibit the association of cdk2 with the HIVpromoter. Specifically, the peptide LAALS dissociated the complex and decreased kinase activity in vitro. Wealso describe our novel small animal model which utilizes humanized Rag2−/−γc
−/− mice. This small peptideinhibitor induces a decrease in HIV-1 viral transcription in vitro and minimizes viral loads in vivo.
Published by Elsevier Inc.
Keywords:HIVCyclin-dependent kinaseTatPeptide inhibitorTranscriptionCell cycleComputer modelingPBMCSmall animal modelStem cells
Introduction
Human immunodeficiency virus (HIV-1) is the etiological agent ofacquired immunodeficiency syndrome (AIDS). Approximately 40 mil-lion people are currently infected with HIV-1 and 4 million new caseswill be identified this year alone. When administered properly, thecurrently available HIV-1 medications are capable of controlling in-fection, however they do not represent a cure. Highly active antire-troviral therapy (HAART) is currently themosteffective strategy inHIV-1treatment and reduction in AIDS-related mortality. HAART is a multi-drug therapywhichhas proven to beeffective in loweringviral loads andimproving the host immune response to HIV-1 infection. While thedrugs merely prevent infection of new cells, latently infected cells
niversity, School of Medicine,C 20037, USA. Fax: +1 202 994
e), [email protected]@gwumc.edu (W. Wu),du (Z. Klase),g), [email protected] (H. Chen),.edu (F. Kashanchi).
nc.
continue to produce varying levels of wild type and mutant viruses.Additionally, the currently available drugs specifically target viralproteins, such that small polymorphisms largely affect the efficiency ofdrug action. The high rate ofmutation caused by the viral RTenzyme, forexample, and the large numbers of new virus produced during eachround of infection allow the virus to select variants with mutations thatmake them resistant to currently available therapies.
Current therapies in HIV-1 are insufficient due to their inability tocure the disease and the proficiency of the virus to become resistant tothe treatmentover time. New therapiesmust be developedwhich targetnovel mechanisms important to the viral life cycle. HIV encodes onlynine genes; a fact which forces the virus to interact with and subvertcellular processes for its own benefit. Identifying these interactions andtheir effect on both the virus and the cell will reveal a new range oftargets for future therapeutics. Alternative preventative measures suchas topical microbicides and vaccines are in development, however therate at which HIV-1 can replicate and mutate forces the need for an indepth understanding of the viral/host cell relationship.
Tat is the HIV-1 viral transactivator protein responsible for tran-scriptional activation and elongation. Tat has been known to not onlystimulate the HIV long terminal repeat (LTR) promoter, but also to
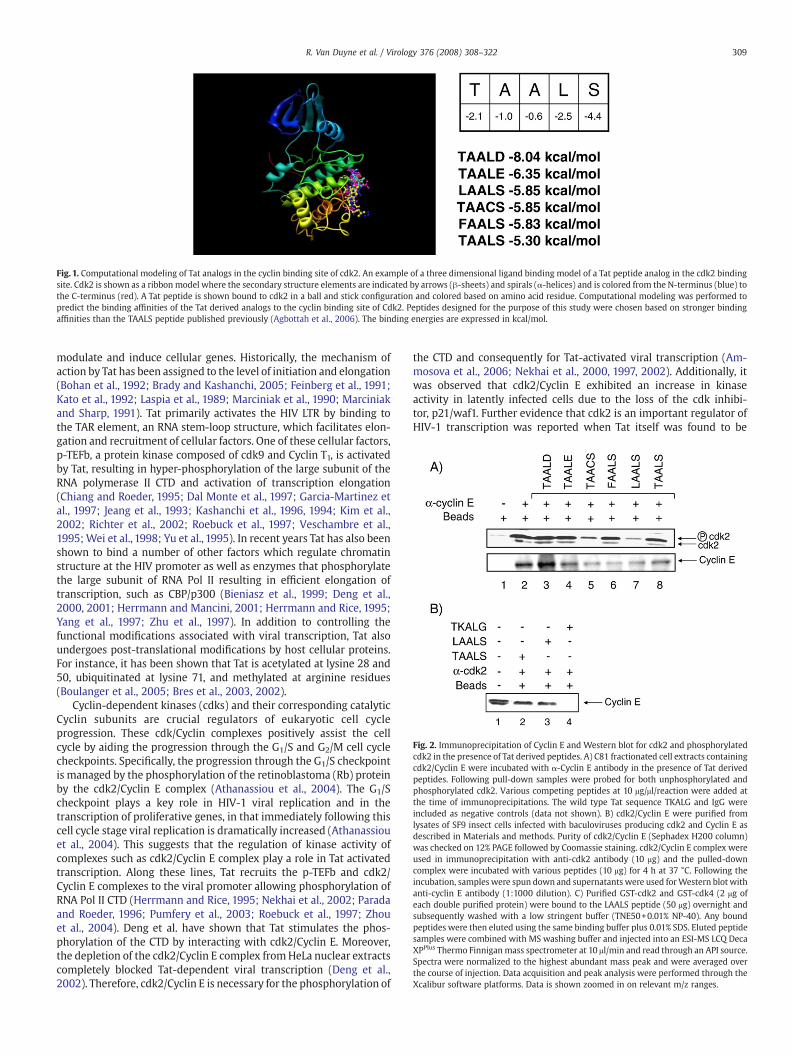
Fig. 1. Computational modeling of Tat analogs in the cyclin binding site of cdk2. An example of a three dimensional ligand binding model of a Tat peptide analog in the cdk2 bindingsite. Cdk2 is shown as a ribbon model where the secondary structure elements are indicated by arrows (β-sheets) and spirals (α-helices) and is colored from the N-terminus (blue) tothe C-terminus (red). A Tat peptide is shown bound to cdk2 in a ball and stick configuration and colored based on amino acid residue. Computational modeling was performed topredict the binding affinities of the Tat derived analogs to the cyclin binding site of Cdk2. Peptides designed for the purpose of this study were chosen based on stronger bindingaffinities than the TAALS peptide published previously (Agbottah et al., 2006). The binding energies are expressed in kcal/mol.
Fig. 2. Immunoprecipitation of Cyclin E and Western blot for cdk2 and phosphorylatedcdk2 in the presence of Tat derived peptides. A) C81 fractionated cell extracts containingcdk2/Cyclin E were incubated with α-Cyclin E antibody in the presence of Tat derivedpeptides. Following pull-down samples were probed for both unphosphorylated andphosphorylated cdk2. Various competing peptides at 10 μg/μl/reaction were added atthe time of immunoprecipitations. The wild type Tat sequence TKALG and IgG wereincluded as negative controls (data not shown). B) cdk2/Cyclin E were purified fromlysates of SF9 insect cells infected with baculoviruses producing cdk2 and Cyclin E asdescribed in Materials and methods. Purity of cdk2/Cyclin E (Sephadex H200 column)was checked on 12% PAGE followed by Coomassie staining. cdk2/Cyclin E complex wereused in immunoprecipitation with anti-cdk2 antibody (10 μg) and the pulled-downcomplex were incubated with various peptides (10 μg) for 4 h at 37 °C. Following theincubation, samples were spun down and supernatants were used forWestern blot withanti-cyclin E antibody (1:1000 dilution). C) Purified GST-cdk2 and GST-cdk4 (2 μg ofeach double purified protein) were bound to the LAALS peptide (50 μg) overnight andsubsequently washed with a low stringent buffer (TNE50+0.01% NP-40). Any boundpeptides were then eluted using the same binding buffer plus 0.01% SDS. Eluted peptidesamples were combined with MS washing buffer and injected into an ESI-MS LCQ DecaXPPlus Thermo Finnigan mass spectrometer at 10 μl/min and read through an API source.Spectra were normalized to the highest abundant mass peak and were averaged overthe course of injection. Data acquisition and peak analysis were performed through theXcalibur software platforms. Data is shown zoomed in on relevant m/z ranges.
309R. Van Duyne et al. / Virology 376 (2008) 308–322
modulate and induce cellular genes. Historically, the mechanism ofaction by Tat has been assigned to the level of initiation and elongation(Bohan et al., 1992; Brady and Kashanchi, 2005; Feinberg et al., 1991;Kato et al., 1992; Laspia et al., 1989; Marciniak et al., 1990; Marciniakand Sharp, 1991). Tat primarily activates the HIV LTR by binding tothe TAR element, an RNA stem-loop structure, which facilitates elon-gation and recruitment of cellular factors. One of these cellular factors,p-TEFb, a protein kinase composed of cdk9 and Cyclin T1, is activatedby Tat, resulting in hyper-phosphorylation of the large subunit of theRNA polymerase II CTD and activation of transcription elongation(Chiang and Roeder, 1995; Dal Monte et al., 1997; Garcia-Martinez etal., 1997; Jeang et al., 1993; Kashanchi et al., 1996, 1994; Kim et al.,2002; Richter et al., 2002; Roebuck et al., 1997; Veschambre et al.,1995;Wei et al., 1998; Yu et al., 1995). In recent years Tat has also beenshown to bind a number of other factors which regulate chromatinstructure at the HIV promoter as well as enzymes that phosphorylatethe large subunit of RNA Pol II resulting in efficient elongation oftranscription, such as CBP/p300 (Bieniasz et al., 1999; Deng et al.,2000, 2001; Herrmann and Mancini, 2001; Herrmann and Rice, 1995;Yang et al., 1997; Zhu et al., 1997). In addition to controlling thefunctional modifications associated with viral transcription, Tat alsoundergoes post-translational modifications by host cellular proteins.For instance, it has been shown that Tat is acetylated at lysine 28 and50, ubiquitinated at lysine 71, and methylated at arginine residues(Boulanger et al., 2005; Bres et al., 2003, 2002).
Cyclin-dependent kinases (cdks) and their corresponding catalyticCyclin subunits are crucial regulators of eukaryotic cell cycleprogression. These cdk/Cyclin complexes positively assist the cellcycle by aiding the progression through the G1/S and G2/M cell cyclecheckpoints. Specifically, the progression through the G1/S checkpointis managed by the phosphorylation of the retinoblastoma (Rb) proteinby the cdk2/Cyclin E complex (Athanassiou et al., 2004). The G1/Scheckpoint plays a key role in HIV-1 viral replication and in thetranscription of proliferative genes, in that immediately following thiscell cycle stage viral replication is dramatically increased (Athanassiouet al., 2004). This suggests that the regulation of kinase activity ofcomplexes such as cdk2/Cyclin E complex play a role in Tat activatedtranscription. Along these lines, Tat recruits the p-TEFb and cdk2/Cyclin E complexes to the viral promoter allowing phosphorylation ofRNA Pol II CTD (Herrmann and Rice, 1995; Nekhai et al., 2002; Paradaand Roeder, 1996; Pumfery et al., 2003; Roebuck et al., 1997; Zhouet al., 2004). Deng et al. have shown that Tat stimulates the phos-phorylation of the CTD by interacting with cdk2/Cyclin E. Moreover,the depletion of the cdk2/Cyclin E complex fromHeLa nuclear extractscompletely blocked Tat-dependent viral transcription (Deng et al.,2002). Therefore, cdk2/Cyclin E is necessary for the phosphorylation of
the CTD and consequently for Tat-activated viral transcription (Am-mosova et al., 2006; Nekhai et al., 2000, 1997, 2002). Additionally, itwas observed that cdk2/Cyclin E exhibited an increase in kinaseactivity in latently infected cells due to the loss of the cdk inhibi-tor, p21/waf1. Further evidence that cdk2 is an important regulator ofHIV-1 transcription was reported when Tat itself was found to be

Fig.
2(con
tinu
ed).
310 R. Van Duyne et al. / Virology 376 (2008) 308–322

Fig. 3. Loss of kinase activity of cdk2/Cyclin E in the presence of competing peptides.Immunoprecipitation of Cyclin E in the presence of Tat peptides was performed asdescribed in Materials and methods. Immunoprecipitated Cyclin E samples in thepresence of Tat peptides were assessed for kinase activity by washing the beads withkinase buffer to equilibrate the reaction. Histone H1 (5 μg/reaction) was added to eachreaction tube along with 2 μl of (γ-32P) ATP (3000 Ci/mmol). Reactions were incubatedat 37 °C for 30 min and stopped by the addition Laemmli buffer. The samples wereseparated on a 4–20% Tris–Glycine gel. Top panel represents samples ran on gel, driedand exposed to a PhosphorImager cassette and analyzed utilizing Molecular Dynamic'sImageQuant Software. Bottom panel represents samples that were stained withCoomassie blue, destained, and then dried for 2 h. Lane 2 served as a positive control(100%) activity without any competing peptides.
311R. Van Duyne et al. / Virology 376 (2008) 308–322
phosphorylated by cdk2 in vitro (Deng et al., 2002; Nekhai et al.,2000). siRNA against cdk2 was also found to inhibit Tat-induced HIV-1transcription and viral replication indicating that cdk2 is crucial forviral progression (Ammosova et al., 2005). Finally, cdk2 was found tobe non-essential for normal host cell proliferation as evident by theviability of cdk2 knock-out mice (Malumbres et al., 2003). Therefore,the inhibition of the cdk2/Cyclin E complex is an appealing target forHIV-1 anti-viral therapies.
Previously, we have shown that a Tat peptide (41/44) from theHIV-1 core domain can inhibit HIV-1 gene expression and act as areplication inhibitor (Agbottah et al., 2006). This Tat peptidewas synthesized by truncation of the sequences from both the N-and C-terminus of Tat and included specific double mutations. Thiscondensed analog retained the specificity necessary to inhibit HIV-1viral transactivation, and not other viral promoters from CMV orHTLV-1. Furthermore this peptide was capable of inhibiting thekinase activity of cdk2/Cyclin E in vitro (Agbottah et al., 2006; Denget al., 2002). Here we show a further refinement of these peptidesusing computational docking studies with the cdk2 structure,therefore characterizing the cdk2/Cyclin E binding interface andlocalizing potential dissociation sites both in vitro and in vivo. Weidentified a new set of peptides that have better inhibitory effect onactivated transcription. These peptides also inhibit virus replicationin a new humanized stem cell animal model in vivo.
Results
Predicted binding energies of Tat peptide analogs
To better design peptides that could possibly bind or compete forthe Cyclin E and cdk2 interface, we first utilized computationaldocking models. Ligand candidates based on the Tat 41/44 mutantpeptide from the previous study were computationally designed asindicated (Agbottah et al., 2006). Ligand candidates were prepared assingle mutations of TAALS, therefore, not straying too far from theoriginal Tat 41/44 sequence. Fig. 1 shows an example of a threedimensional computational docking model of a small peptide andcdk2. Here the structure of cdk2 is depicted in “ribbon” view such thatthe secondary structure elements α-helices and β-sheets are depictedas spirals and parallel arrows respectively. The cdk2 sequence is alsocolored from blue to red to correspond to the N- and the C-terminirespectively. The small ligand in Fig. 1 is docked to the Cyclin bindingsite of cdk2 and is represented in a “ball and stick” view. From energyminimization studies, the affinities of each ligand to the Cyclin bindingsite of cdk2 were also determined. Each amino acid substitutionprovides different bonding opportunities where each binding energyis different. All of the newly proposed peptides have a higherpredicted binding affinity than the Tat 41/44 peptide previouslydescribed (Agbottah et al., 2006) with TAALD having the highest. Thissuggests that these peptides may provide a higher percentage ofdissociation of the cdk2/Cyclin E complex.
Specificity of Tat peptide in possibly dissociating cdk2/Cyclin E complex
In order to detect any possible dissociation of the cdk2/Cyclin Ecomplex in the presence of the Tat peptides, immunoprecipitations ofCyclin E were performed. Cell extracts were first separated through asize-exclusion column into various fractions (total of 80). A repre-sentative amount of fractions were probed for cdk2 in order todetermine a size range in which the complex was likely to elute (datanot shown). This chromatographic step also minimized the chancethat non-specific binding interactions would occur with cdk/Cyclincomplexes. Also, in the previous Agbottah 2006 paper, we usedimmunoprecipitations from HIV-infected whole cell extracts, wherethe extracts contained two distinct populations of cdk2 complexes.When using a sizing column, one population that is fairly large comes
out at 440 kDa size and a second population elutes from 67 kDa up to150 kDa. The smaller size complexes (67 kDa–150 kDa) are active cdk2complexes whereas the large 440 kDa complex is largely inactive inkinase activity and contains as yet unpublished proteins and RNAbound molecules (Kashanchi and Nekhai, unpublished data; Denget al., 2002). Therefore, the smaller cdk2/Cyclin E containing fractions(29–32) were pooled together and the extract (250 μg) was used inimmunoprecipitations with antibodies against Cyclin E in thepresence or absence of the Tat peptides. Immunoprecipitates (IPs)were washed andWestern blotted for the presence of cdk2 and CyclinE. Results in Fig. 2A show the addition of six Tat derived peptides to theIP reaction. Lanes 1 and 2 serve as controls for non-specific bindingand control IP reaction, respectively. There is a varying degree ofdissociation amongst the various Tat peptide competitors. For instance,the LAALS peptide in lane 7 appears to have the highest level ofdissociation followed by TAACS. Based on the computational bindingenergy prediction, it would have been assumed that TAALD would havethe highest efficiency of dissociation due to the strong binding affinity(lane 3), but the actual biochemical test (IP followed by competition)showed a different result. Interestingly, the levels of detectablephosphorylated cdk2 in these IP/competition experiments varydepending on which peptide is used. For instance, LAALS and TAACSpeptides appear to have lower recovery levels of phosphorylated cdk2,while the other peptides appear to have no effect on the basal recoverylevels of phosphorylated cdk2. Also, the lack of efficiency of competitionof the other peptides for cdk2 indicates that both unphosphorylatedand phosphorylated cdk2 can equally bind to Cyclin E as seen in theimmunoprecipitates.
We next asked if the new peptide derivatives could indeeddissociate the cdk2/Cyclin E complex in vitro. We therefore used apurified system where Baculovirus expressed cdk2/Cyclin E complexes(Ammosova et al., 2006) were used in immunoprecipitation with anti-cdk2 antibody and the pulled-down complexes were incubated withvarious peptides for 4 h at 37 °C. Following the incubation, sampleswere spun down and supernatants were used for Western blots withanti-Cyclin E antibody. Results in Fig. 2B show that two peptides (TAALSand LAALS), but not the wild type peptide (TKALG), were able todissociate the Cyclin E away from the cdk2 by about ~50% (compareinput level to lanes 2 and 3). These results further indicate that the newLAALS peptide is able to at least partially dissociate the cdk2/Cyclin Ecomplex. Finally, we asked if we could indeed detect binding of theLAALS peptide to the cdk2 protein in the absence of its cyclin partner.
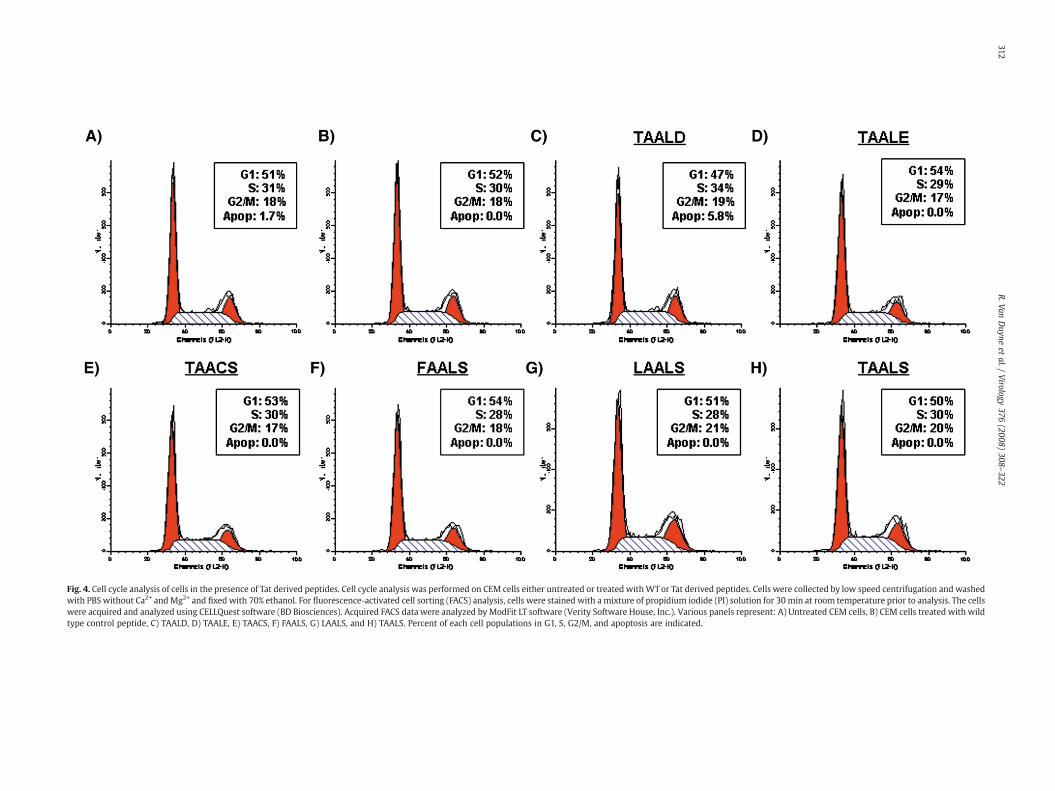
Fig. 4. Cell cycle analysis of cells in the presence of Tat derived peptides. Cell cycle analysis was performed on CEM cells either untreated or treated withWTor Tat derived peptides. Cells were collected by low speed centrifugation and washedwith PBS without Ca2+ andMg2+ and fixed with 70% ethanol. For fluorescence-activated cell sorting (FACS) analysis, cells were stained with a mixture of propidium iodide (PI) solution for 30 min at room temperature prior to analysis. The cellswere acquired and analyzed using CELLQuest software (BD Biosciences). Acquired FACS data were analyzed byModFit LT software (Verity Software House, Inc.). Various panels represent: A) Untreated CEM cells, B) CEM cells treated with wildtype control peptide, C) TAALD, D) TAALE, E) TAACS, F) FAALS, G) LAALS, and H) TAALS. Percent of each cell populations in G1, S, G2/M, and apoptosis are indicated.
312R.Van
Duyne
etal./
Virology
376(2008)
308–322
313R. Van Duyne et al. / Virology 376 (2008) 308–322
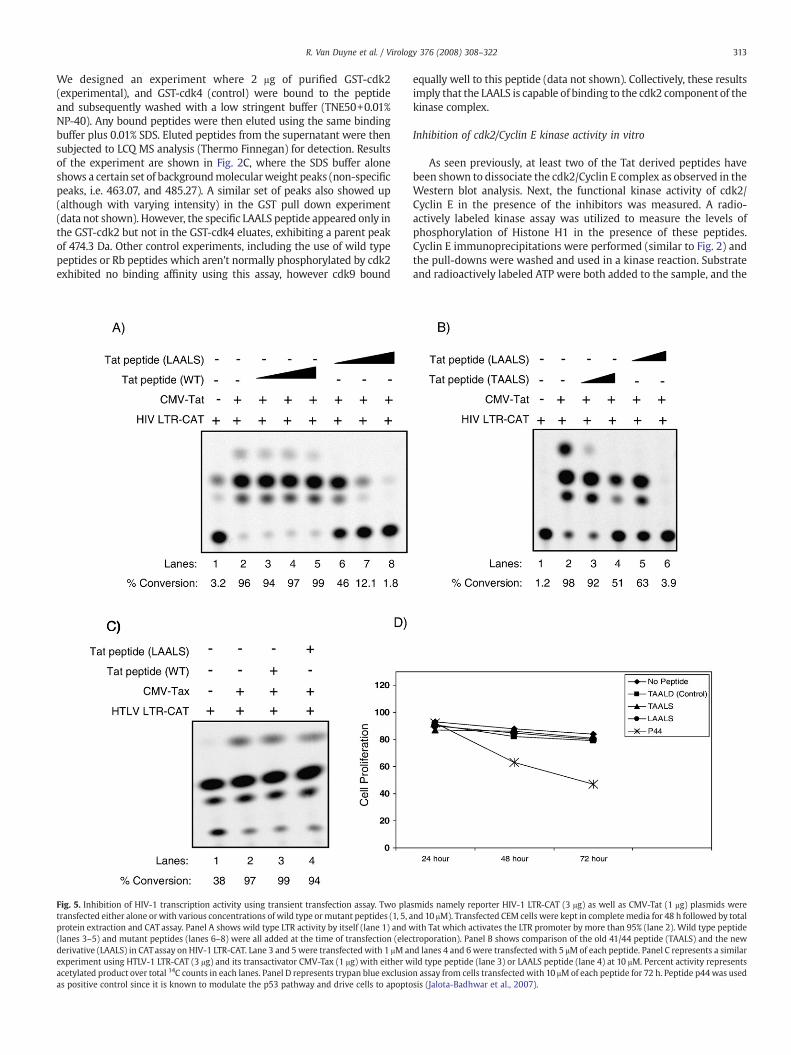
We designed an experiment where 2 μg of purified GST-cdk2(experimental), and GST-cdk4 (control) were bound to the peptideand subsequently washed with a low stringent buffer (TNE50+0.01%NP-40). Any bound peptides were then eluted using the same bindingbuffer plus 0.01% SDS. Eluted peptides from the supernatant were thensubjected to LCQ MS analysis (Thermo Finnegan) for detection. Resultsof the experiment are shown in Fig. 2C, where the SDS buffer aloneshows a certain set of backgroundmolecularweight peaks (non-specificpeaks, i.e. 463.07, and 485.27). A similar set of peaks also showed up(although with varying intensity) in the GST pull down experiment(data not shown). However, the specific LAALS peptide appeared only inthe GST-cdk2 but not in the GST-cdk4 eluates, exhibiting a parent peakof 474.3 Da. Other control experiments, including the use of wild typepeptides or Rb peptides which aren't normally phosphorylated by cdk2exhibited no binding affinity using this assay, however cdk9 bound
Fig. 5. Inhibition of HIV-1 transcription activity using transient transfection assay. Two platransfected either alone orwith various concentrations of wild type ormutant peptides (1, 5, aprotein extraction and CAT assay. Panel A shows wild type LTR activity by itself (lane 1) and w(lanes 3–5) and mutant peptides (lanes 6–8) were all added at the time of transfection (elecderivative (LAALS) in CATassay on HIV-1 LTR-CAT. Lane 3 and 5 were transfected with 1 μM anexperiment using HTLV-1 LTR-CAT (3 μg) and its transactivator CMV-Tax (1 μg) with either wacetylated product over total 14C counts in each lanes. Panel D represents trypan blue exclusioas positive control since it is known to modulate the p53 pathway and drive cells to apopto
equally well to this peptide (data not shown). Collectively, these resultsimply that the LAALS is capable of binding to the cdk2 component of thekinase complex.
Inhibition of cdk2/Cyclin E kinase activity in vitro
As seen previously, at least two of the Tat derived peptides havebeen shown to dissociate the cdk2/Cyclin E complex as observed in theWestern blot analysis. Next, the functional kinase activity of cdk2/Cyclin E in the presence of the inhibitors was measured. A radio-actively labeled kinase assay was utilized to measure the levels ofphosphorylation of Histone H1 in the presence of these peptides.Cyclin E immunoprecipitations were performed (similar to Fig. 2) andthe pull-downs were washed and used in a kinase reaction. Substrateand radioactively labeled ATP were both added to the sample, and the
smids namely reporter HIV-1 LTR-CAT (3 μg) as well as CMV-Tat (1 μg) plasmids werend 10 μM). Transfected CEM cells were kept in completemedia for 48 h followed by totalith Tat which activates the LTR promoter by more than 95% (lane 2). Wild type peptidetroporation). Panel B shows comparison of the old 41/44 peptide (TAALS) and the newd lanes 4 and 6were transfected with 5 μMof each peptide. Panel C represents a similarild type peptide (lane 3) or LAALS peptide (lane 4) at 10 μM. Percent activity representsn assay from cells transfected with 10 μMof each peptide for 72 h. Peptide p44was usedsis (Jalota-Badhwar et al., 2007).
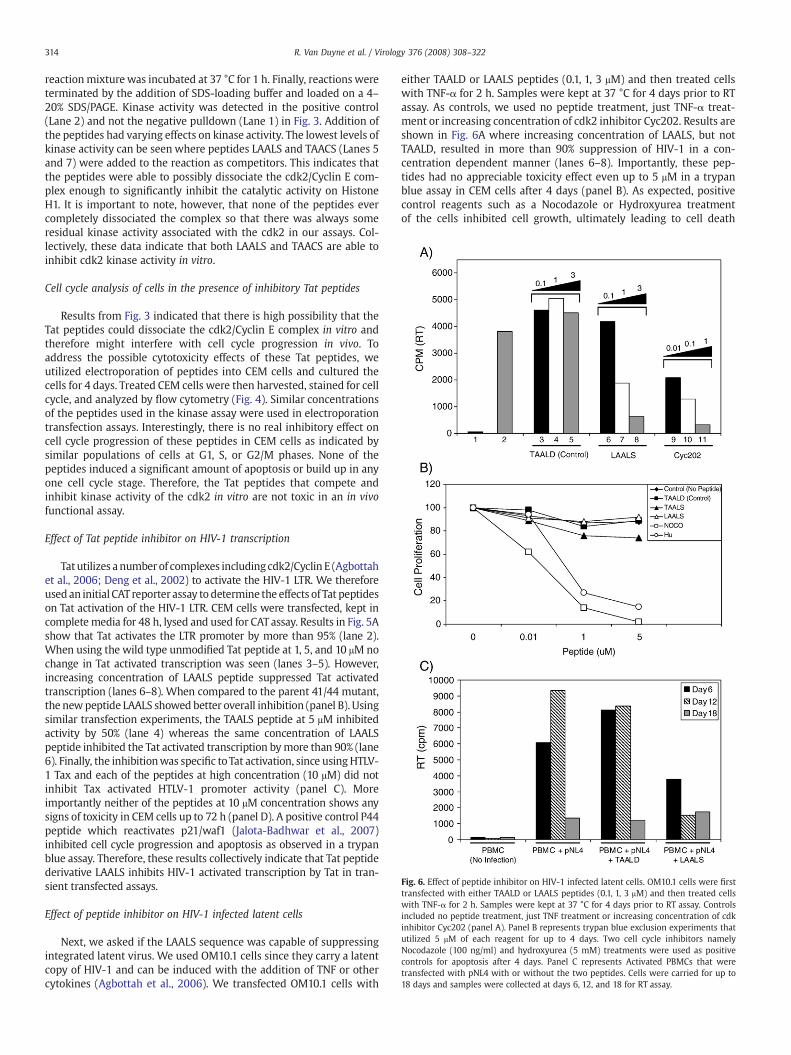
Fig. 6. Effect of peptide inhibitor on HIV-1 infected latent cells. OM10.1 cells were firsttransfected with either TAALD or LAALS peptides (0.1, 1, 3 μM) and then treated cellswith TNF-α for 2 h. Samples were kept at 37 °C for 4 days prior to RT assay. Controlsincluded no peptide treatment, just TNF treatment or increasing concentration of cdkinhibitor Cyc202 (panel A). Panel B represents trypan blue exclusion experiments thatutilized 5 μM of each reagent for up to 4 days. Two cell cycle inhibitors namelyNocodazole (100 ng/ml) and hydroxyurea (5 mM) treatments were used as positivecontrols for apoptosis after 4 days. Panel C represents Activated PBMCs that weretransfected with pNL4 with or without the two peptides. Cells were carried for up to18 days and samples were collected at days 6, 12, and 18 for RT assay.
314 R. Van Duyne et al. / Virology 376 (2008) 308–322
reactionmixture was incubated at 37 °C for 1 h. Finally, reactions wereterminated by the addition of SDS-loading buffer and loaded on a 4–20% SDS/PAGE. Kinase activity was detected in the positive control(Lane 2) and not the negative pulldown (Lane 1) in Fig. 3. Addition ofthe peptides had varying effects on kinase activity. The lowest levels ofkinase activity can be seen where peptides LAALS and TAACS (Lanes 5and 7) were added to the reaction as competitors. This indicates thatthe peptides were able to possibly dissociate the cdk2/Cyclin E com-plex enough to significantly inhibit the catalytic activity on HistoneH1. It is important to note, however, that none of the peptides evercompletely dissociated the complex so that there was always someresidual kinase activity associated with the cdk2 in our assays. Col-lectively, these data indicate that both LAALS and TAACS are able toinhibit cdk2 kinase activity in vitro.
Cell cycle analysis of cells in the presence of inhibitory Tat peptides
Results from Fig. 3 indicated that there is high possibility that theTat peptides could dissociate the cdk2/Cyclin E complex in vitro andtherefore might interfere with cell cycle progression in vivo. Toaddress the possible cytotoxicity effects of these Tat peptides, weutilized electroporation of peptides into CEM cells and cultured thecells for 4 days. Treated CEM cells were then harvested, stained for cellcycle, and analyzed by flow cytometry (Fig. 4). Similar concentrationsof the peptides used in the kinase assay were used in electroporationtransfection assays. Interestingly, there is no real inhibitory effect oncell cycle progression of these peptides in CEM cells as indicated bysimilar populations of cells at G1, S, or G2/M phases. None of thepeptides induced a significant amount of apoptosis or build up in anyone cell cycle stage. Therefore, the Tat peptides that compete andinhibit kinase activity of the cdk2 in vitro are not toxic in an in vivofunctional assay.
Effect of Tat peptide inhibitor on HIV-1 transcription
Tat utilizes anumberof complexes including cdk2/Cyclin E (Agbottahet al., 2006; Deng et al., 2002) to activate the HIV-1 LTR. We thereforeused an initial CAT reporter assay to determine the effects of Tat peptideson Tat activation of the HIV-1 LTR. CEM cells were transfected, kept incomplete media for 48 h, lysed and used for CATassay. Results in Fig. 5Ashow that Tat activates the LTR promoter by more than 95% (lane 2).When using the wild type unmodified Tat peptide at 1, 5, and 10 μM nochange in Tat activated transcription was seen (lanes 3–5). However,increasing concentration of LAALS peptide suppressed Tat activatedtranscription (lanes 6–8). When compared to the parent 41/44 mutant,the newpeptide LAALS showed better overall inhibition (panel B). Usingsimilar transfection experiments, the TAALS peptide at 5 μM inhibitedactivity by 50% (lane 4) whereas the same concentration of LAALSpeptide inhibited the Tat activated transcription bymore than 90% (lane6). Finally, the inhibitionwas specific toTat activation, since usingHTLV-1 Tax and each of the peptides at high concentration (10 μM) did notinhibit Tax activated HTLV-1 promoter activity (panel C). Moreimportantly neither of the peptides at 10 μM concentration shows anysigns of toxicity in CEM cells up to 72 h (panel D). A positive control P44peptide which reactivates p21/waf1 (Jalota-Badhwar et al., 2007)inhibited cell cycle progression and apoptosis as observed in a trypanblue assay. Therefore, these results collectively indicate that Tat peptidederivative LAALS inhibits HIV-1 activated transcription by Tat in tran-sient transfected assays.
Effect of peptide inhibitor on HIV-1 infected latent cells
Next, we asked if the LAALS sequence was capable of suppressingintegrated latent virus. We used OM10.1 cells since they carry a latentcopy of HIV-1 and can be induced with the addition of TNF or othercytokines (Agbottah et al., 2006). We transfected OM10.1 cells with
either TAALD or LAALS peptides (0.1, 1, 3 μM) and then treated cellswith TNF-α for 2 h. Samples were kept at 37 °C for 4 days prior to RTassay. As controls, we used no peptide treatment, just TNF-α treat-ment or increasing concentration of cdk2 inhibitor Cyc202. Results areshown in Fig. 6A where increasing concentration of LAALS, but notTAALD, resulted in more than 90% suppression of HIV-1 in a con-centration dependent manner (lanes 6–8). Importantly, these pep-tides had no appreciable toxicity effect even up to 5 μM in a trypanblue assay in CEM cells after 4 days (panel B). As expected, positivecontrol reagents such as a Nocodazole or Hydroxyurea treatmentof the cells inhibited cell growth, ultimately leading to cell death
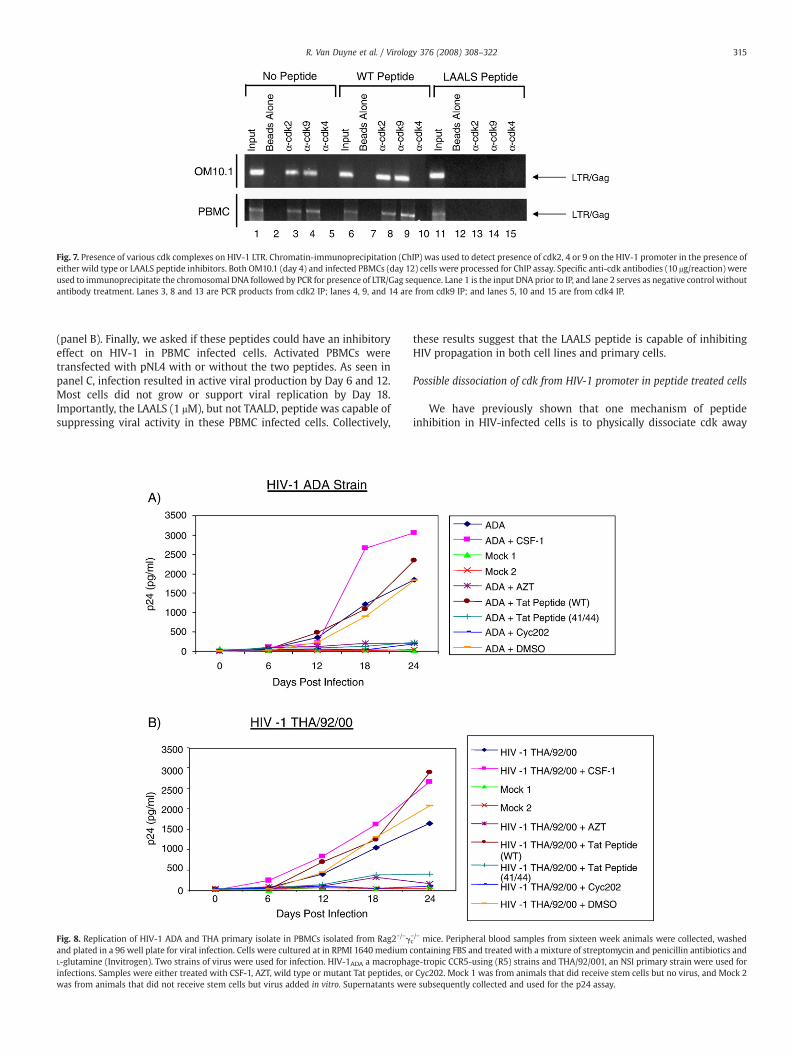
Fig. 7. Presence of various cdk complexes on HIV-1 LTR. Chromatin-immunoprecipitation (ChIP) was used to detect presence of cdk2, 4 or 9 on the HIV-1 promoter in the presence ofeither wild type or LAALS peptide inhibitors. Both OM10.1 (day 4) and infected PBMCs (day 12) cells were processed for ChIP assay. Specific anti-cdk antibodies (10 μg/reaction) wereused to immunoprecipitate the chromosomal DNA followed by PCR for presence of LTR/Gag sequence. Lane 1 is the input DNA prior to IP, and lane 2 serves as negative control withoutantibody treatment. Lanes 3, 8 and 13 are PCR products from cdk2 IP; lanes 4, 9, and 14 are from cdk9 IP; and lanes 5, 10 and 15 are from cdk4 IP.
315R. Van Duyne et al. / Virology 376 (2008) 308–322
(panel B). Finally, we asked if these peptides could have an inhibitoryeffect on HIV-1 in PBMC infected cells. Activated PBMCs weretransfected with pNL4 with or without the two peptides. As seen inpanel C, infection resulted in active viral production by Day 6 and 12.Most cells did not grow or support viral replication by Day 18.Importantly, the LAALS (1 μM), but not TAALD, peptide was capable ofsuppressing viral activity in these PBMC infected cells. Collectively,
Fig. 8. Replication of HIV-1 ADA and THA primary isolate in PBMCs isolated from Rag2−/−γand plated in a 96 well plate for viral infection. Cells were cultured at in RPMI 1640 medium cL-glutamine (Invitrogen). Two strains of virus were used for infection. HIV-1ADA a macrophainfections. Samples were either treated with CSF-1, AZT, wild type or mutant Tat peptides, owas from animals that did not receive stem cells but virus added in vitro. Supernatants wer
these results suggest that the LAALS peptide is capable of inhibitingHIV propagation in both cell lines and primary cells.
Possible dissociation of cdk from HIV-1 promoter in peptide treated cells
We have previously shown that one mechanism of peptideinhibition in HIV-infected cells is to physically dissociate cdk away
c−/− mice. Peripheral blood samples from sixteen week animals were collected, washedontaining FBS and treated with a mixture of streptomycin and penicillin antibiotics andge-tropic CCR5-using (R5) strains and THA/92/001, an NSI primary strain were used forr Cyc202. Mock 1 was from animals that did receive stem cells but no virus, and Mock 2e subsequently collected and used for the p24 assay.

Table 1Effect of HIV inhibitors on in vivo replication of HIV-1 ADA in mice
MouseID no.
HIV-infected(ADA)
Treatment Viral RNAcopies
Viral DNAcopies
3⁎ + – b100 b1004⁎ + – 1,237,925 855,1095⁎ + – 836,676 401,6646⁎ + – 574,730 988,1117⁎ + AZT 14,379 168,4508^ + AZT 26,892 190,3479^ + AZT 80,587 177,42010⁎ + Peptide (WT) 935,250 804,51011⁎ + Peptide (WT) 743,099 938,55312^ + Peptide (WT) 1,359,688 1,209,55313^ + Peptide (WT) 1,690,319 1,857,33614⁎ + Peptide (41/44) 58,477 640,65315⁎ + Peptide (41/44) 28,105 750,77216^ + Peptide (41/44) 103,266 1,800,84117^ + Peptide (41/44) 87,392 1,736,21718⁎ + Peptide (LAALS) 36,921 1,548,77019⁎ + Peptide (LAALS) 48,750 1,254,92920⁎ – – b100 b10021^ – – b100 b100
Animal #3 did not receive any human stem cells, and animals #16 and 17 did notreceive HIV, but RPMI media as mock. (⁎) denotes Rag/γc animals and (^) denotesNOD/SCID animals.
316 R. Van Duyne et al. / Virology 376 (2008) 308–322
from the LTR promoter (Agbottah et al., 2006). We used chromatinimmunoprecipitation (ChIP) assays to define molecules that arepresent or absent on the HIV promoter after peptide treatment. Wetherefore decided to perform similar experiments in the presence ofthe newly modified peptide. Both OM10.1 (Day 4) and infected PBMCs(Day 12) were cross linked and used for ChIP with anti-cdk2, anti-cdk9, and anti-cdk4 (control) antibodies. Following the ChIP assays,the recovered DNA was used in PCR with primers that span the LTR/Gag region (Agbottah et al., 2006). Results in Fig. 7 indicate that whencells were treated with no peptides or the control peptide (0.1 μM),both cdk2 and cdk9 were present on the HIV promoter region in bothcell line and PBMC infected cells (lanes 3, 4, 8 and 9). Importantly, bothcdk2 and cdk9 were dissociated from the HIV promoter when usingLAALS (0.1 μM) peptide (lanes 13 and 14). Negative control cdk4 wasnot present on the HIV promoter (lanes 5, 10 and 15). Collectively,these results point to dissociation of the cdk2 and cdk9 away from theHIV promoter in peptide treated cells.
Effect of peptide inhibitors on HIV-1 RNA production in a newlydeveloped mouse model
We have developed a mouse model where human cord blood stemcells are implanted into immunodeficient mice and allowed to re-constitute an immune system in vivo. The double-mutant mice withdeleted recombinase activating gene 2 (RAG)-2 and common cytokinereceptor gamma chain (Rag2−/−γc−/−) has been seen by us and others tobe a viable option in reconstitution experiments. These Rag2−/−γc−/−
mice exhibit an alymphoid phenotype with no spontaneous tumorformation and can be implanted with hematopoietic stem cells thatcan be further enhanced by administration of exogenous humancytokines (Kosco-Vilbois, 2004). A number of recent publications usingthis strain as well as reconstitution in NOD/SCID mice with humanstem cells show a very promising path for a feasible HIV/AIDS animalmodel (Baenziger et al., 2006; Berges et al., 2006; Gorantla et al., 2007;Watanabe et al., 2007a,b; Zhang et al., 2007). In our preliminaryexperiments, we found that mice tolerated a sub-lethal dose of 3.5 Gy(to destroy any residual endogenous immune system) and despiteinfertility, transplanted and non-transplanted animals showed noobvious defects compared with non-irradiated controls. We firstreconstituted newborn Rag2−/−γc−/− mice intrahepatically (i.h.) withCD34+ cord blood cells and subsequently analyzed between four andtwenty-six weeks of age for presence of human CD45+ hematopoieticcells. An increase in splenic and thymic cellularity was detectable, andas expected all mice beyond eight weeks developedmesenteric lymphnodes (Traggiai et al., 2004). The spleen, lymph node, and blood CD19+
cells expressed the IgM antigens. Mice beyond eight weeks developedmesenteric lymph nodes, and showed enlarged spleens whereaslymph nodes and enlarged spleens were never detected in non-transplanted controls (data not shown).
We next asked whether cells from the transplanted animals couldindeed allow replication of HIV-1 in vitro. PBMCs from implantedanimals were used for in vitro infection of HIV-1 using the lab adaptedmacrophage-tropic strain ADA and a primary isolate called THA/92/00.This particular primary strain has previously been used by us forinfection of human PBMCs in vitro (Agbottah et al., 2005). Sixteenweekperipheral blood sampleswere initially collected, processed to lyse redcells and stored in liquid N2. After confirmation that these animalscontained human hematopoietic cells, frozen mononuclear cells(5×105) were thawed, washed and plated in a 96 well plate for viralinfection. Cells were cultured in RPMI medium containing FBS andtreated with a mixture of streptomycin and penicillin antibiotics andL-glutamine. Infected cells were washed and subsequently treatedwith either wild type or mutant Tat peptides (experimental), AZT, orCyc202 (controls). Supernatant samples were collected for the presenceof Gag p24 for up to 24 days. As a control, cells were allowed to dif-ferentiate into mature macrophages in the presence of CSF-1 (M-CSF).
Figs. 8A and B shows that ADA could effectively replicate in these cellsand that CSF-1 can slightly increase the viral titers in cells obtainedfrom animals. Also, these three different inhibitors including Tatpeptide (our initial 41/44 peptide), AZT and Cyc202 inhibited virusreplicationwhen infectingwith the ADA or the THA strain. The effectsof Tat peptide inhibitors and Cyc202 have previously been shown byus (Agbottah et al., 2005). Collectively, these results imply thattransplantation of human CD34+ cells into Rag2−/−γc−/− mice allowsdifferentiation of cells into competent human cells necessary for HIV-1 infection in vitro and the virus can be inhibited with Tat peptides ordrugs that target pre- or post-integration events.
Inhibition of HIV-1 RNA production with Tat peptide derivative intransplanted Rag2−/−γc
−/− mice in vivo
To address whether HIV-1 could indeed replicate in these animals,we used the ADA viral stock with ~100,000 TCID50/ml for our initialinfections into animals. Animals received virus alone and/or ~1000 µg/ml of AZT at the time of injection. A volume of 0.3 ml was injected intothe intra-peritoneal region of anesthetized Rag2−/−γc−/− mice with ablunt-tipped 30-gauge needle. Animals were infected twelve weekspost-transplantation. Animals that received HIV-1 and stem cellsalone (Table 1; # 4, 5 and 6), showed a presence of viral RNA andintegrated DNA. Animals treated with inhibitors including AZT and Tatpeptide 41/44 showed a dramatic drop in viral RNA (Table 1; #7, 14and 15). Both animals #3 and #20 served as negative controls andshowed no HIV-1 RNA or DNA in these animals. We also observedsimilar inhibitory effects using LAALS peptide (animals #18 and 19). Itis important to note that, although we were not able to successfullydetect significant p24 levels in the serum of these animals, we co-cultivated (1:10 ratio) the infected PBMCs with human monocytes(U937 and THP) and human T-cells (CEM, H9, Jurkat) cells in vitro andfound varying ng/ml quantities of HIV-1 Gag p24 in the supernatantsof both monocytes and T-cells (data not shown).
To further confirm that the human stem cell transplantation couldindeed support HIV-1 ADA growth and inhibition in vivo, and thatthese results were not an artifact of using this particular animal strain,we decided to use another strain of mice for similar subsequentstudies. NOD/SCID mice (generous gift of Dr. Leonard Schultz, theJackson Lab) were irradiated three weeks after birth and transplanted

317R. Van Duyne et al. / Virology 376 (2008) 308–322
with stem cells and treated either with AZT or peptide inhibitors.Results of these studies showed a consistent pattern of viral inhibitionin AZT and Tat peptide treated animals (Table 1, #16 and 17). In-terestingly, even though the transplantation efficiency at the time ofcell and serum harvest was fairly high (27–61%) for these animals, stillno reproducible levels of Gag p24 antigen could be detected in theserum of these animals after twoweeks. However, both cell associatedHIV-1 DNA and RNA was detectable in these animals (Table 1).
Discussion
The development of alternative antiretroviral therapies is crucial tothe progression of HIV-1 treatment. Currently, this therapy involvesthe use of agents from distinct classes of antivirals: protease in-hibitors, transcriptase inhibitors, fusion inhibitors, and integraseinhibitors. Recently, compounds interacting with Tat/TAR (or siRNAagainst Tat) have also been studied which inhibit HIV replication in alow micromolar range (Ammosova et al., 2006; Christensen et al.,2007; Mukerjee et al., 2007). However, most current antiretroviraltherapies result in more drug resistant viral strains, poor inhibition,and low efficiency.
Pharmacological cdk inhibitors (PCIs) are an attractive concept forthe next phase of antiretroviral therapies. Drugs that would targetvarious cdk/Cyclin complexes which are vital in controlling viral tran-scription, replication, and progeny formation may allow for theinhibition of virally-induced cell cycle progression and result in theinhibition of viral transcription and replication. Artificial peptideinhibitors have been developed, and have shown selectivity for cdksthrough binding near the active site, however, in depth knowledge ofboth substrate and kinase is needed in order to be effective.
Previously, we have shown success in mimicking the core domainof the HIV-1 viral protein Tat sequence, and making simple alterationsin order to impart an inhibitory effect on HIV-1 gene expression(Agbottah et al., 2006). The 41/44 peptide analog was found to actupon the cdk2/Cyclin E interface, which through a series ofbiochemical assays, was determined to be present on the HIV-1promoter. In previous studies, a model for the interaction of cdk2/Cyclin E on the viral promoter was established where in the presenceof TAR, Tat regulates this complex and is essential in phosphorylatingtranscriptional machinery, including the RNAPII CTD. A model for thepeptide analog induced inhibition was also established, where thebinding of the peptide to cdk2 causes it to dissociate from the Cyclinsubunit, therefore inactivating the kinase and preventing activation oftranscription factors through phosphorylation. Here, we continuedthese studies including additional peptides with varying efficacies forcdk2 inhibition and evaluate the inhibitory effects into an in vivomodel.
In this study, the initial Tat 41/44 double-mutant, TAALS, wasmodified to include one additional point mutation to generate thepeptides indicated in Fig.1. Given the computationally derived bindingaffinities, the initial inhibition assay performed was meant to test thein vitro inhibitory effects of each peptide. The cdk2/Cyclin E IPWesterndata in Fig. 2 indicated that peptides TAACS and LAALS showed thehighest degree of possible dissociation, and subsequently the greatestinhibition of the kinase activity (Fig. 3). This in vitro data suggests thatnot only are there varying degrees of specificity of each peptide forbinding to cdk2, as predicted, but the dissociation seen is alsoindicative of loss of kinase activity. Interestingly, the two peptidesshown to dissociate the complex most successfully did not have thehighest predicted binding affinity energy as compared to all thepredicted ligands. It is also important to mention that the two pep-tides specifically bound to the unphosphorylated form of cdk2,implying that the phosphorylated subunit has an induced conforma-tional change, therefore closing the ligand binding site. These in vitrostudies were carried out using immunoprecipitated complexes, andit is important to note that we have never observed a complete dis-
sociation of the complex when using these Tat peptides. We trans-fected these inhibitory peptides into proliferating cells to monitor thelevel of apoptosis induced by inhibition of the cdk2/Cyclin E complex.Complementary concentrations of inhibitors were used in the cellcycle analysis as in the in vitro activity studies, and consequentlyproved that the amounts of peptides needed to inhibit the complex forHIV-1 transcription were not toxic to the cells.
The inhibitory peptide LAALS has thus far been the most adept atdissociating and inhibiting cdk2/Cyclin E as well as conferring littletoxic effect on non-infected cells. We tested LAALS inhibitory effectson a simulated viral promoter. The effectiveness of LAALS in sup-pressing transcription of the HIV-1 LTR was monitored in Fig. 5.Increasing concentrations of LAALS in the presence of Tat resulted in adramatic decrease in the LTR-CAT reporter activity as compared to thewild type peptide. Fig. 5C indicates that the activity of LAALS is specificfor the HIV-1 promoter and not for HTLV (in the presence of Tax).
We tested the efficacy of LAALS in inhibiting viral replication in alatently infected cell line. Results in OM10.1 cells showed a decrease inviral replication correlating with an increase in concentration ofLAALS treatment. This treatment exhibited an effect comparable to thewell known cdk inhibitor drug Cyc202, and more importantly, thecontrol peptide TAALD did not result in reduced viral replication.Again, cell proliferationwas still not affected by this treatment. Fig. 6Cfurther confirmed the drop in viral load by looking at the treatment ofinfected PBMCs with LAALS and a control peptide. The PBMCs treatedwith LAALS showed an overall decrease in viral load as compared tountreated and control treated cells.
The data thus far has indicated and implied a change in protein–protein interactions resulting in a lower amount of viral transcriptionin the presence of the peptide LAALS. This has been inferred by activitylevels at the viral LTR as well as monitoring the levels of virus present.
It is well known that key cellular factors (i.e., NF-kB, Sp1 and cdk9)interact with the viral LTR resulting in Tat activated transcriptionwhich can be detected using ChIP assays. Fig. 7 shows a ChIP assaywhere latently infected cells and PBMC infected cells were eitheruntreated, treatedwith control or LAALS peptides and screened for thepresence of cdk2, cdk9, and cdk4 at the LTR. No changes were seenwhen cells were treated with the wild type peptide, however, in thepresence of LAALS, there was no pulldown of cdk2 or cdk9 in eitherlatent cells or PBMCs. Loss of cdk9 from the LTR in the presence ofLAALS is very interesting andmay prove to be significant. It is possiblethat the inhibition of cdk2/Cyclin E prevents a phosphorylationevent which is otherwise necessary for the recruitment/modificationof p-TEFb or that the peptide has some homology to the cdk9/CyclinT interface site. Along these lines we have recently found that cdk9/Cyclin T is a substrate for cdk2/Cyclin E complex and may in factenhance the auto-phosphorylation of cdk9 that is normally seen priorto Tat activated transcription (S. Nekhai and F. Kashanchi, unpublisheddata).
Finally, the current work utilizes a novel animal model that usesreconstitution of the human immune system when implantingirradiated mice with human CD34+ stem cells. A previous pioneeringreport by the Manz group has indicated that the human T-cells wereobserved in mouse secondary lymphoid organs which underwent anumber of post-thymic cell divisions as measured by TCR-rearrange-ment excision circles (TRECs) (Traggiai et al., 2004). Inspired by theseresults, and more recent similar publications (Baenziger et al., 2006;Berges et al., 2006; Gorantla et al., 2007; Watanabe et al., 2007a,b;Zhang et al., 2007) we injected CD34+ cord blood cells into sub-lethallyirradiated newborn Rag2−/−γc−/− mice and observed a significant num-ber of human CD45+ hematopoietic cells when using FACS analysis.Our attempts to infect these cells ex-vivo and in vivo were successfulusing either T- or lab adapted macrophage-tropic viruses. When usingthe ADA and THA/92/00 viruses, we observed a healthy infectionin vitro and were able to follow virus replication up to 24 days.These results were encouraging as they indicated that the in vivo

318 R. Van Duyne et al. / Virology 376 (2008) 308–322
differentiated human cells could in fact be infected through normalHIV receptor/co-receptor pathway. However, to date we have notexpanded our virus repertoire and do not know if virus replication isunique to these particular strains or can they be infected by a broadrange of HIV-1 clades. Future studies will determine the efficiency ofinfection, level of pathogenesis, and whether certain viruses canreplicate better in the ex-vivo system as compared to the in vivomodel.
We were also encouraged by the inhibitor studies using AZT,Cyc202, and Tat peptide inhibitors as these compounds reduceddetectable virus in the mouse model. Our in vivo infection studies inthe transplanted animals are the most exciting results. Our initialinjection of HIV orHIV+AZTutilized an i.p. route and seems to facilitateviral entry into cells. As such, we were able to observe HIV infection inthese animals and a marked decrease of Gag DNA and RNA in AZTtreated cells, indicating that the virusmay be able to enter the cells andgo through the RT process followed by integration and subsequenttranscription. However, many factors, including better titration ofvirus, peptides, and drugs, will have to be performed to obtain validviral kinetic data in vivo. Finally, wewere not able to successfully detectp24 antigen levels in the serum of these animals; however upon co-cultivation of infected cells, we were able to observe ng/ml quantitiesof HIV-1 Gag in a tissue culture setting (data not shown). We believethat this may be due to many reasons including possible low viralproduction in vivo, rapid clearance of the virus within 24–48 h ofrelease, possible accumulation of virus in specific organs as opposed toPBMCs, few rounds of viral replication, possible depletion of human T-and monocytic cells, or differing viral replication kinetics betweentissue culture isolates and primary virus isolated from patients.Ongoing future experiments will clearly define the limits of thisanimal model and will determine the extent that the model could beused to study peptide inhibitors, drugs, siRNA, or vaccine candidates.
Materials and methods
Computational modeling
Computational docking studies were carried out as previouslydescribed by Agbottah et al. (2006). We took advantage of the existingcrystal structure of cdk2 to mimic binding of each of the Tat peptideanalogs to the cdk/Cyclin binding site. The cdk2 structure used wasobtained from the Protein Data Bank ID 1FIN (resolution 1.85 A°).Docking was performed using the software AutoDock 3.0 with thesimulated annealing search algorithm. The receptor protein (cdk2)was taken as rigid and the ligands (thewild type andmutant peptides)as flexible. All torsion angles were allowed to change (except thoseabout the peptide bond), giving 19 rotatable bonds for the wild typepeptide and 16 for the mutant TAALS. The annealing temperature wasreduced with a geometric scheme, and each docking run took about2 h on a 2.8 GHz processor. A Linux computer cluster with 64 nodeswas employed for our simulation. For each docked ligand conforma-tion, computer scripts were written to breakdown the total ligand–receptor interaction energy by ligand atoms (this assists in ligandoptimization). To determine the binding site of the wild type andmutant peptides on cdk2, binding energy per residue was calculatedfor the low-energy docked conformations, and each cdk2 residue wasranked by average energy for these conformations. To further exploitthis mechanism of cdk inhibition, we screened for other peptides thatcan potentially target this pocket more effectively with strongerbinding affinities. To this end, we performed molecular docking simu-lations for all 95 possible singlemutants of TAALS while restricting theallowed configurational space of these peptides to the local regionaround this new binding pocket of cdk2 by TAALS. The simulationsresulted in 15 mutant variants of TAALS that have stronger bindingaffinities than that of TAALS, the top five of which are listed in Fig. 1and are selected for further experimental studies.
Peptide synthesis
All peptides used for this study were commercially synthesized(SynBioSci, Livermore, CA) with the following sequences:
NH2-T-A-A-L-D-OHNH2-T-A-A-L-E-OHNH2-L-A-A-L-S-OHNH2-T-A-A-C-S-OHNH2-F-A-A-L-S-OHNH2-T-A-A-L-S-OH
The purity of each peptide was analyzed by HPLC to greater than98%. Mass spectral analysis was also performed to confirm the identityof each peptide as compared to the theoretical mass (AppliedBiosystems Voyager System 1042). Peptides were resuspended indH2O to a concentration of 1 mg/ml and stored at −70C. Peptides wereonly thawed once prior to use for biochemical or in vivo experiments.
Cell culture
C81 is an HTLV-1-infected T-cell line that expresses Tax protein,and CEM (12D7) is an uninfected human T-cell line established frompatients with T-cell leukemia. C81 and CEM cells were cultured inRPMI 1640 containing 10% fetal bovine serum, 1% penicillin/strepto-mycin, and 1% L-glutamine (Quality Biological) and were incubated ina 5% CO2 incubator at 37 °C. Cells were cultured to confluency andpelleted at 4 °C for 15 min at 3,000 rpm. The cell pellets were washedtwice with 25 ml of phosphate-buffered saline (PBS) with Ca2+ andMg2+ (Quality Biological) and centrifuged once more. Cell pellets wereresuspended in lysis buffer (50 mM Tris–HCl, pH 7.5, 120 mM NaCl,5 mM EDTA, 0.5% NP-40, 50 mM NaF, 0.2 mM Na3VO4, 1 mM DTT, onecomplete protease cocktail tablet/50 ml) and incubated on ice for20 min, with a gentle vortexing every 5 min. Cell lysates were trans-ferred to Eppendorf tubes and were centrifuged at 10,000 rpm for10 min. Supernatants were transferred to a fresh tube where proteinconcentrations were determined using Bio-Rad protein assay (Bio-Rad, Hercules, CA).
Size-exclusion chromatography
C81 cell lysate (30 mg/ml) was fractionated on a Superose 6 HR 10/30 column (AmershamBiosciences, Piscataway, NJ) in Buffer D (20mMHEPES (pH 7.9), 0.05M KCl, 0.2 mM EDTA, 0.5 mM PMSF, 0.05 DTT, and20% Glycerol). Flow-through was collected at 0.5 ml for 50 fractions.Every 10th fractionwas analyzed by immunoblotting for cdk2 in orderto determine the elution location of the cdk2/Cyclin E complex.
Immunoprecipitation
Cdk2 containing chromatography fractions (28–32) were pooledtogether for immunoprecipitation. The pooled C81 extracts (250 μgeach) were combined with 10 μg/μl of each respective peptide.Cyclin E antibody (Santa Cruz, sc-198) was added to each reactiontube (10 μl, 2 μg), the reaction mixture was brought up to 500 μl withTNE50+0.1% NP-40 (100 mM Tris, pH 8.0; 50 mM NaCl; 1 mM EDTA,0.1% Nonidet P-40) and was allowed to incubate while rotatingovernight at 4 °C. α-IgG was added to extract as a negative control,and an IP was performed in the absence of competing peptide, actingas a positive control. The following day, 30 μl of a 30% Protein A & Gbead slurry (CalBioChem, La Jolla, CA) was added to each reactiontube and allowed to incubate while rotating for 2 h at 4 °C. Sampleswere spun and washed 2× with TNE300 + 0.1% NP-40 (100 mM Tris,pH 8.0; 300 mM NaCl; 1 mM EDTA, 0.1% Nonidet P-40) and 1× withTNE50+0.1% NP-40 to remove non-specifically bound proteins. 2×Laemmli buffer was added to each sample and heated at 95 °C for

319R. Van Duyne et al. / Virology 376 (2008) 308–322
3 min. Samples were loaded and run on a 4–20% Tris–Glycine SDS/PAGE gel to be used for both Western blots and kinase assays.
Cdk2 and Cyclin E were purified from lysates of SF9 insect cellsinfected with baculoviruses producing cdk2 and Cyclin E. Briefly, 1 mlof cells (from 250 ml of culture) was lysed with 16 ml of lysis buffer(50 mM Tris–HCl, pH 8.0, 10 mM 2-mercaptoethanol, 10% glycerol andPMSF), homogenized on ice and centrifuged at 45,000 ×g for 1 h at4 °C. Supernatant was loaded on Mono-Q 10/10 column (Amersham,USA). Two separate cell cultures, one infected with cdk2-expressingbaculovirus and the other one infected with Cyclin E-expressingbaculovirus were used for purification. The Mono-Q fractions contain-ing cdk2 or Cyclin E weremixed 1:1 and loaded onto Superdex column(Sephadex H200, Amersham, USA). Purity of cdk2/Cyclin E waschecked on 12% PAGE followed by Coomassie staining. We alsoanalyzed the Superdex fractions by immunoblotting with anti-cdk2antibodies and assayed their enzymatic activity using histone H1 andpurified Tat proteins as substrates. Fractions containing cdk2/Cyclin Ewere concentrated using Microcon tubes (Amicon, USA).
Western blots
Immunoprecipitated Cyclin E samples in the presence of Tatpeptides were probed for cdk2. IP samples separated on SDS/PAGEgels were transferred to a nitrocellulose membrane via a constantcurrent of 70mA overnight. Themembranewas blockedwith a 3% BSAsolution in PBS containing 0.1% Tween-20, rocking for 2 h at 4 °C. A1:1000 dilution of α-cdk2 antibody (Santa Cruz, sc-163) was added tothe blocking solution and incubated rocking overnight at 4 °C. Themembrane was washed with a fresh PBS+0.1% Tween-20 solution inorder to wash off any residual primary antibody solution. A 1:1000dilution of α-rabbit secondary antibody was added to a fresh 3% BSAsolution in PBS+0.1% Tween-20 and incubated with the membrane,rocking for 2 h at 4 °C. The membrane was washed 2× with PBS+0.1%Tween-20 and 1× with PBS to remove any residual antibody. Themembranewas exposed to chemiluminescence reagent (Pierce) in thedark for 5 min., and was developed using a Kodak Imager.
GST-cdk binding assay to peptide in vitro
GST-cdk2, cdk4, cdk9 and GST alone were expressed and purifiedtwice over Glutathione Sepharose beads. The elution and re-bindingdropped the non-specific background significantly. Peptides (50 μg)were allowed tobindvariouspurifiedbead-GSTproteins (2 μg) overnightat 4 °C. Next day theywerewashedwith TNE50+0.01%NP-40 and elutedwith 20 μl of elution buffer (TNE50+0.01% NP-40+0.01% SDS). Followingcentrifugation eluates were used for further analysis. Eluted peptidesamples were combined with MS washing buffer (50:50 Methanol:Water+0.1% TFA) and loaded onto a Hamilton Gastight 250 μl syringe.Samplesweremanually injected into an ESI-MS LCQDecaXPPlus ThermoFinniganmass spectrometer at 10 μl/min and read throughanAPI source.Spectra were normalized to the highest abundant mass peak and wereaveraged over the course of injection. Data acquisition and peak analysiswere performed through theXcalibur software platforms. Data is shownzoomed in on relevant m/z ranges.
Kinase assay
Immunoprecipitated Cyclin E samples in the presence of Tatpeptides were assessed for kinase activity. After the final TNE50+0.1%NP-40 wash, beads were washed with kinase buffer (50 mM HEPES,10 mM MgCl2, 5 mM MnCl2, 1 mM DTT, 50 mM NaF, 0.2 mM Na3VO4
and one complete tablet of protease cocktail inhibitor/50 ml buffer) toequilibrate the reaction. The indicated amount of Histone H1 wasadded to each reaction tube alongwith the indicated amount of (γ-32P)ATP (3000 Ci/mmol). Reactions were incubated at 37 °C for 30min andstopped by the addition of 15 μl Laemmli buffer. The samples were
separated by reducing SDS-PAGE on a 4-20% Tris–Glycine gel. Gelswere stained with Coomassie blue, destained, and then dried for 2 h.Following drying, the gels were exposed to a PhosphorImager cassetteand analyzed utilizing Molecular Dynamic's ImageQuant Software.
Flow cytometry
Cell cycle analysis was performed on CEM cells either untreated ortreated with WT or Tat derived peptides. Cells were collected by lowspeed centrifugation andwashed with PBS without Ca2+ andMg2+ andfixed with 70% ethanol. For fluorescence-activated cell sorting (FACS)analysis, cells were stained with a mixture of propidium iodide (PI)solution (PBS with Ca2+ and Mg2+, 10 µg/ml RNase A, 0.1% Nonidet P-40, and 50 µg/ml PI) for 30 min at room temperature prior to analysis.The cells were acquired and analyzed using CELLQuest software (BDBiosciences). Acquired FACS datawere analyzed byModFit LT software(Verity Software House, Inc.).
CAT assay
Wild type LTR constructs from both HIV and HTLV-1 were elec-troporated into CEM cells. Extracts were prepared for chlorampheni-col acetyltransferase (CAT) assay. The cells were harvested, washedonce with phosphate-buffered saline (PBS) without Ca2+ and Mg2+,pelleted, and resuspended in 150 µl of 0.25 M Tris (pH 7.8). The cellswere freeze-thawed three times with vortexing and then incubatedfor 5 min at 68 °C followed by centrifugation. The supernatants weretransferred to 1.5-ml Eppendorf tubes. After one final spin, the super-natant was again transferred to 1.5-ml Eppendorf tubes, and theprotein concentration was determined. CAT assays were performedwith 25 µg of protein as described in Kashanchi et al., 2000.
ChIP assays
Ten million OM10.1 cells in log phase were first electroporatedwith peptides and then incubated for 2 h with or 5 μg/ml TNF-α toinduce transcription of latent proviral DNA. After 4 days, cells werecross linked (1% formaldehyde, 10 min at 37 °C) and samples weresonicated to reduce DNA fragments to 200–800 nt lengths for ChIPassays. Infected PBMCs (Day 12) were also treated for ChIP analysis.Specific cdk complexes were immunoprecipitated with appropriateantibodies (10 μg/reaction). DNA sequences in the immunoprecipi-tates were detected by PCR using primers specific for the HIV-1 LTR/Gag (Agbottah et al., 2006).
Mice
A set of male and female Rag2−/−γc−/− mice were graciously donatedby Dr. Anton Berns (The Netherlands Cancer Institute). Subsequentexperiments utilized similar animals obtained from Dr. RameshAkkina (Colorado State University). Both sets of animal havepreviously been described and are identical to animals described inthe Traggiai et al. manuscript (Traggiai et al., 2004). A breeding colonywas successfully established at the GWUMC. All procedures andpractices associated with the use of Rag2−/−γc−/− mice was beenapproved by the GWUMC Committee on Human Research andCommittee on Animal Research (IACUCC #007-4,5A1 and animalassurance #A-3205-01). The GWUMC Committee on Animal Researchwas charged with carrying out the regulations of the federalgovernment's Animal Welfare Act governing the care and use ofanimals in research and instruction. The animals are housed in anAAALAC-approved vivarium GWUMC (BSL3 facility). Animal housingconsisted of a Techniplast caging system and all changes were com-pleted in a biosafety hood. Bedding, food and water were allautoclaved. Animals in the vivarium were monitored almost dailyand project protocols were reviewed and approved by the University

320 R. Van Duyne et al. / Virology 376 (2008) 308–322
Animal Use Care Committee. Data in Table 1 are also from Balb/c Rag2−/−γc
−/− animals.The NOD/SCID mice (NOD.Cg-Prkdc^scid Il2rg^tm1Wjl/SzJ; Stock
Number: 005557) were kindly donated by Dr. Leonard Schultz fromthe Jackson Laboratory Animal Resource Department. These micewere maintained in an AAALAC-approved vivarium housed withinGWUMC's BSL-3 suite at all times. Ten three week old NOD/SCID pupswere sub-lethally irradiated at a dose of 3.5 Gy using a CS-137irradiator. Twenty-four hours later, the mice were given a 100 μl intra-peritoneal (IP) injection of 100,000 human CD34+ cord blood stemcells. These stem cells were allowed to differentiate for three weeksfollowed by infection with HIV-1. Each animal was given an IPinjection of virus, virus plus drug, peptides or RPMI media. Twoweeksafter infection, the mice were sacrificed and blood was collectedthrough cardiac puncture for further DNA and RNA analysis.
Human Stem cells
CD34 cells were either obtained from Cambrex Bio ScienceWalkersville, Inc. (Gaithersburg, MD) or prepared by us using thefollowing method. Cells were separated using the CD34 MicroBead Kit(Miltenyi Biotec, Auburn, CA). This kit formerly termed Direct CD34Progenitor Cell Isolation Kit is a single-step labeling system whichallows for a fairly fast and easy isolation of CD34+ cells. By using directMicroBeads instead of an indirect labeling system, the washing stepduring the labeling procedure is abolished and resulting cell loss isavoided. CD34+ cells were isolated from pooled cord blood samples.The CD34 MicroBead Kit contained FcR blocking reagent andMicroBeads which were conjugated to the monoclonal mouse anti-human CD34 antibody, QBEND/10. The CD34 antigen is a singletransmembrane glycoprotein which is expressed on human hemato-poietic progenitor cells andmost endothelial cells. Fluorescent controlstaining of magnetically labeled cells requires a monoclonal CD34antibody recognizing an epitope other than QBEND/10, e.g. cloneAC136. All cells were washed with PBS prior to implantation.
FACS analysis for cells from animal studies
Samples (i.e, twenty-four weeks post-transplantation) werecollected from euthanized animals and blood, thymus, bone marrow,mesenteric lymph nodes and spleen were collected from bothtransplanted and non-transplanted animals. In most cases to obtainperipheral blood cells, mice were bled from retro orbital venous sinusunder anesthesia. After euthanasia, single cell suspensions wereprepared from various organs and red cells were lysed in cold ACKlysis buffer (NH4Cl 8.29 g/l, KHCO3 1.00 g/l, disodium EDTA 2H2O0.0372 g/l, membrane filtered, pH: 7.4±0.2, Osmolality: 290±5%mOsm/kg H2O). Remaining cells were then spun, washed once in 2 mlPBS and pelleted again. Samples were cross linked with 1 ml of 4%paraformaldehyde to fix cells and virus at room temperature for10 min. Subsequently they were washed in 1 ml PBS/BSA/10% NMS(normal mouse serum), spun, washed in 100 μl PBS/BSA/NMS andadded 20 μl of each antibody to cells. Samples were incubated in darkat RT, added 1 ml PBS/BSA/NMS, spun, and resuspended in 500 μl ofPBS/BSA/NMS.
Lymph nodes, spleens and thymuses were cut in small pieces andhomogenized on a 70 µ strainer with a syringe plunger. Cellhomogenates were washed once with 30 ml of RPMI 1640 supple-mented with 10% FCS, and once with RPMI 1640 alone, centrifugedand stained with antibodies for flow cytometric analysis. Thefollowing surface markers were used: CD45–FITC, CD3–FITC, CD19–FITC, CD34–FITC and CD14-APC and CD33-Cy5. Antibodies werepurchased from e-Bioscience except for CD34 (BD Bioscience, SanJose, CA). These antibodies were chosen to identify subsets ofhematopoetic cells that may have derived from the transplant. Cellswere analyzed using a FACScalibur (Becton Dickinson Immunocyto-
metry Systems). At least 200,000 cells were acquired from eachsample for FACS analysis.
In vitro infection of human cells
Sixteen weeks peripheral blood samples from animals werecollected, washed and platted for viral infection. After confirmationthat these animals contained human hematopoietic cells, frozenmononuclear cells (5×105) were thawed, washed and platted in a 96well plate for viral infection. Cells were cultured at 37 °C in RPMI 1640medium containing 10% fetal bovine serum (FBS) and treated with amixture of 1% streptomycin, penicillin antibiotics, and 1% L-glutamine(Invitrogen, Carlsbad, CA). Two strains of virus were used for infectionfor 2 h in vitro. HIV-1ADA is a macrophage-tropic CCR5-using (R5)strains (Jacque et al., 2002; Schmidtmayerova et al., 1997) and THA/92/001 is an NSI primary strain (Agbottah et al., 2005). ADA (2×105 cpmof RT activity) and THA/92/001 (Thailand strain, subtype E envelope,5 ng of p24 Gag antigen) were used for these initial infections. Bothviruses have previously been used by our group with successfulinfection rates. Washed cells were subsequently either treated with2 ng/ml CSF-1 (Sigma, St. Louis, MO), AZT (100 µg/ml), wild type ormutant Tat peptides (100 nM), or Cyc202 (0.36 µM). Mock 1 was foranimals that did receive stem cells but no virus, and Mock 2 was foranimals that did not receive stem cells but virus added in vitro.Supernatants were collected and used for detection of p24 antigens(Agbottah et al., 2005).
In vivo infection of HIV-1 in Rag2−/−γc−/− mice
Sixteen weeks post-transplantation animals were infected withADA strain (~100,000 TCID50/ml). Various animalswould receive virusalone, virus plus ~1000 µg/ml of AZT, or various peptides at 3 mg/kgusing intrapertenial administration. To detect viral DNA in theseanimals (data not shown) four weeks later, chromosomal DNA wasisolated from PBMCs and examined for presence of HIV-1 DNA. A totalof 0.1, 0.01, and 0.001 mg of total DNA were used in PCRs to amplifyboth HIV and human HLA-specific sequences in parallel. The primersused were SK38 (59-ATA ATC CAC CTA TCC CAG TAG GAG AAAT-39)and SK39 (59-TTT GGT CCT TGT CTT ATG TCC AGA ATG C-39),which amplifies a 115-bp region of the HIV-1 Gag gene, and GH26(59-GTGCTG CAG GTG TAA ACT TGT ACC AG-39) and GH27 (59-CACGGA TCCGGT AGC AGC GGT AGA GTT G-39), which amplifies a 242-bpregion of HLA-DQ-a. Mutant Gag primers included SK38 M (59-ATAATC CACGACTTTGAGTAGGAGAAAT-39) and SK39M (59-TTTGGTCCTCGATTTGAGTCC AGAATG C-39). Amplification conditionswere 1minof denaturation at 95 °C, 2 min of annealing at 55 °C, and 3 min ofextension at 72 °C for 35 cycles; followed by 7 min at 68 °C. FollowingPCR, sampleswere runon2% agarose gels and EtBr stained for presenceof DNA products.
For virological analysis from these animals, both viral RNA andDNA amplification was used as previously described (Watanabe et al.,2007a,b). Cell associated viral RNA (from 5×104) copy numbers weremeasured using a real-time quantification assay based on the TaqMansystem (Applied Biosystems, Foster City, CA). Cell associated viral RNAwas extracted and purified using a QIAamp Viral RNAMini Kit (Qiagen,Valencia, CA). This kit can be used to isolates plasma RNA, cell cultureRNA, and cellular RNA. The RNA was subjected to reverse-transcrip-tion and amplification using a TaqMan one-step RT-PCR Master MixReagents Kit (PE Biosystems) with HIV-1 Gag consensus primers(forward, 5′-GGACA TCAAGCAGCCATGCAA-3′, and reverse, 5′-TGCT-ATGTCACTTCCCCTTGG-3′) and an HIV-1 Gag consensus TaqMan probe(FAM-5′-ACCATCAATGAGGAAGCTGCAG AA-3′-TAMRA). Probed pro-ducts were quantitatively monitored by their fluorescence intensitywith the ABI7300 Real-Time PCR system (PE Biosystems). To obtaincontrol RNA for quantification, HIV-1 Gag RNA was synthesized usingT7 RNA polymerase and pKS460. Viral DNAwas extracted and purified

321R. Van Duyne et al. / Virology 376 (2008) 308–322
using a QIAamp DNA Mini Kit (Qiagen). Determination of HIV-1 DNAcopy numbers was performed by real-time PCR assay with TaqManMaster mixture (PE Biosystems). Primers (forward, 5′-GGCTAAC-TAGGGAACCCACTG-3′; reverse, 5′ CTGCTAGAGATTTTCC ACACT-3′)and probes (FAM-5′ TAGTGTGTGCCCGTCTG TTGTGTGAC-3′-TAMRA)were designed for targeting the HIV-1 long terminal repeat region, R/U5. The viral DNAwas quantified using LightCycler (Roche). Viral RNAand DNA were calculated based on the standard curve of control RNAand DNA. All assays were carried out in triplicate.
Acknowledgments
This work was supported by grants from the George WashingtonUniversity REF funds to FK, and Akos Vertes, and by an NIH grantAI071903-01 to FK, and by an NSF grant DMR0313129 to CZ. We aregrateful to both Dr. Anton Berns and Ramesh Akkina for providing uswith initial seed colony animals. This work was also supported bygrant from a subproject (MSA-06-437) provided by CONRAD, EasternVirginia Medical School under a Cooperative Agreement (HRN-A-00-98-00020-00) with the United States Agency for International De-velopment (USAID).
References
Agbottah, E., de la Fuente, C., Nekhai, S., Barnett, A., Gianella-Borradori, A., Pumfery, A.,Kashanchi, F., 2005. Antiviral activity of CYC202 in HIV-1-infected cells. J. Biol.Chem. 280 (4), 3029–3042.
Agbottah, E., Zhang, N., Dadgar, S., Pumfery, A., Wade, J.D., Zeng, C., Kashanchi, F., 2006.Inhibition of HIV-1 virus replication using small soluble Tat peptides. Virology 345 (2),373–389.
Ammosova, T., Berro, R., Kashanchi, F., Nekhai, S., 2005. RNA interference directed toCDK2 inhibits HIV-1 transcription. Virology 341 (2), 171–178.
Ammosova, T., Berro, R., Jerebtsova, M., Jackson, A., Charles, S., Klase, Z., Southerland,W.,Gordeuk, V.R., Kashanchi, F., Nekhai, S., 2006. Phosphorylation of HIV-1 Tat by CDK2in HIV-1 transcription. Retrovirology 3, 78.
Athanassiou, Z., Dias, R.L., Moehle, K., Dobson, N., Varani, G., Robinson, J.A., 2004.Structural mimicry of retroviral tat proteins by constrained beta-hairpin peptido-mimetics: ligands with high affinity and selectivity for viral TAR RNA regulatoryelements. J. Am. Chem. Soc. 126 (22), 6906–6913.
Baenziger, S., Tussiwand, R., Schlaepfer, E., Mazzucchelli, L., Heikenwalder,M., Kurrer, M.O.,Behnke, S., Frey, J., Oxenius, A., Joller, H., Aguzzi, A., Manz, M.G., Speck, R.F., 2006.Disseminated and sustained HIV infection in CD34+ cord blood cell-transplantedRag2−/−gamma c−/− mice. Proc. Natl. Acad. Sci. U. S. A. 103 (43), 15951–15956.
Berges, B.K., Wheat, W.H., Palmer, B.E., Connick, E., Akkina, R., 2006. HIV-1 infection andCD4 Tcell depletion in the humanized Rag2−/−gamma c−/− (RAG-hu) mouse model.Retrovirology 3, 76.
Bieniasz, P.D., Grdina, T.A., Bogerd, H.P., Cullen, B.R., 1999. Recruitment of cyclin T1/P-TEFbto anHIV type 1 long terminal repeat promoter proximal RNA target is both necessaryand sufficient for full activation of transcription. Proc. Natl. Acad. Sci. U. S. A. 96 (14),7791–7796.
Bohan, C.A., Kashanchi, F., Ensoli, B., Buonaguro, L., Boris-Lawrie, K.A., Brady, J.N., 1992.Analysis of Tat transactivation of human immunodeficiency virus transcription invitro. Gene Expr. 2 (4), 391–407.
Boulanger, M.C., Liang, C., Russell, R.S., Lin, R., Bedford, M.T., Wainberg, M.A., Richard, S.,2005. Methylation of Tat by PRMT6 regulates human immunodeficiency virus type1 gene expression. J. Virol. 79 (1), 124–131.
Brady, J., Kashanchi, F., 2005. Tat gets the “green” light on transcription initiation.Retrovirology 2, 69.
Bres, V., Tagami, H., Peloponese, J.M., Loret, E., Jeang, K.T., Nakatani, Y., Emiliani, S.,Benkirane, M., Kiernan, R.E., 2002. Differential acetylation of Tat coordinates itsinteraction with the co-activators cyclin T1 and PCAF. EMBO J. 21 (24), 6811–6819.
Bres, V., Kiernan, R.E., Linares, L.K., Chable-Bessia, C., Plechakova, O., Treand, C., Emiliani,S., Peloponese, J.M., Jeang, K.T., Coux, O., Scheffner, M., Benkirane, M., 2003. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter.Nat. Cell Biol. 5 (8), 754–761.
Chiang, C.M., Roeder, R.G., 1995. Cloning of an intrinsic human TFIID subunit thatinteracts with multiple transcriptional activators. Science 267 (5197), 531–536.
Christensen, H.S., Daher, A., Soye, K.J., Frankel, L.B., Alexander, M.R., Laine, S., Bannwarth,S., Ong, C.L., Chung, S.W., Campbell, S.M., Purcell, D.F., Gatignol, A., 2007. Smallinterfering RNAs against the TAR RNA binding protein, TRBP, a Dicer cofactor, inhibithuman immunodeficiency virus type 1 long terminal repeat expression and viralproduction. J. Virol. 81 (10), 5121–5131.
Dal Monte, P., Landini, M.P., Sinclair, J., Virelizier, J.L., Michelson, S., 1997. TAR and Sp1-independent transactivation of HIV long terminal repeat by the Tat protein in thepresence of human cytomegalovirus IE1/IE2. Aids 11 (3), 297–303.
Deng, L., de la Fuente, C., Fu, P., Wang, L., Donnelly, R., Wade, J.D., Lambert, P., Li, H., Lee,C.G., Kashanchi, F., 2000. Acetylation of HIV-1 Tat by CBP/P300 increasestranscription of integrated HIV-1 genome and enhances binding to core histones.Virology 277 (2), 278–295.
Deng, L., Wang, D., de la Fuente, C., Wang, L., Li, H., Lee, C.G., Donnelly, R., Wade, J.D.,Lambert, P., Kashanchi, F., 2001. Enhancement of the p300 HAT activity by HIV-1 Taton chromatin DNA. Virology 289 (2), 312–326.
Deng, L., Ammosova, T., Pumfery, A., Kashanchi, F., Nekhai, S., 2002. HIV-1 Tatinteraction with RNA polymerase II C-terminal domain (CTD) and a dynamicassociation with CDK2 induce CTD phosphorylation and transcription from HIV-1promoter. J. Biol. Chem. 277 (37), 33922–33929.
Feinberg, M.B., Baltimore, D., Frankel, A.D., 1991. The role of Tat in the humanimmunodeficiency virus life cycle indicates a primary effect on transcriptionalelongation. Proc. Natl. Acad. Sci. U. S. A. 88 (9), 4045–4049.
Garcia-Martinez, L.F., Ivanov, D., Gaynor, R.B.,1997. Association of Tatwith purifiedHIV-1and HIV-2 transcription preinitiation complexes. J. Biol. Chem. 272 (11), 6951–6958.
Gorantla, S., Sneller, H., Walters, L., Sharp, J.G., Pirruccello, S.J., West, J.T., Wood, C.,Dewhurst, S., Gendelman, H.E., Poluektova, L., 2007. Human immunodeficiencyvirus type 1 pathobiology studied in humanized BALB/c-Rag2−/−gammac−/− mice.J. Virol. 81 (6), 2700–2712.
Herrmann, C.H., Rice, A.P., 1995. Lentivirus Tat proteins specifically associate with acellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminaldomain of the large subunit of RNA polymerase II: candidate for a Tat cofactor.J. Virol. 69 (3), 1612–1620.
Herrmann, C.H.,Mancini,M.A., 2001. The Cdk9 and cyclinTsubunits of TAK/P-TEFb localizeto splicing factor-rich nuclear speckle regions. J. Cell Sci. 114 (Pt 8), 1491–1503.
Jacque, J.M., Triques, K., Stevenson, M., 2002. Modulation of HIV-1 replication by RNAinterference. Nature 418 (6896), 435–438.
Jalota-Badhwar, A., Kaul-Ghanekar, R., Mogare, D., Boppana, R., Paknikar, K.M.,Chattopadhyay, S., 2007. SMAR1-derived P44 peptide retains its tumor suppressorfunction through modulation of p53. J. Biol. Chem. 282 (13), 9902–9913.
Jeang, K.T., Chun, R., Lin, N.H., Gatignol, A., Glabe, C.G., Fan, H., 1993. In vitro and in vivobinding of human immunodeficiency virus type 1 Tat protein and Sp1 transcriptionfactor. J. Virol. 67 (10), 6224–6233.
Kashanchi, F., Agbottah, E.T., Pise-Masison, C.A., Mahieux, R., Duvall, J., Kumar, A., Brady,J.N., 2000. Cell cycle-regulated transcription by the human immunodeficiency virustype 1 Tat transactivator. J. Virol. 74 (2), 652–660.
Kashanchi, F., Piras, G., Radonovich, M.F., Duvall, J.F., Fattaey, A., Chiang, C.M., Roeder, R.G.,Brady, J.N., 1994. Direct interaction of human TFIID with the HIV-1 transactivator tat.Nature 367 (6460), 295–299.
Kashanchi, F., Khleif, S.N., Duvall, J.F., Sadaie, M.R., Radonovich, M.F., Cho, M., Martin, M.A.,Chen, S.Y., Weinmann, R., Brady, J.N., 1996. Interaction of human immunodeficiencyvirus type 1 Tatwith a unique site of TFIID inhibits negative cofactorDr1 and stabilizesthe TFIID–TFIIA complex. J. Virol. 70 (8), 5503–5510.
Kato, H., Sumimoto, H., Pognonec, P., Chen, C.H., Rosen, C.A., Roeder, R.G., 1992. HIV-1 Tatacts as a processivity factor in vitro in conjunction with cellular elongation factors.Genes Dev. 6 (4), 655–666.
Kim, Y.K., Bourgeois, C.F., Isel, C., Churcher, M.J., Karn, J., 2002. Phosphorylation of theRNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible forhuman immunodeficiency virus type 1 Tat-activated transcriptional elongation.Mol. Cell. Biol. 22 (13), 4622–4637.
Kosco-Vilbois, M.H., 2004. A mightier mouse with human adaptive immunity. Nat.Biotechnol. 22 (6), 684–685.
Laspia, M.F., Rice, A.P., Mathews, M.B., 1989. HIV-1 Tat protein increases transcriptionalinitiation and stabilizes elongation. Cell 59 (2), 283–292.
Malumbres, M., Hunt, S.L., Sotillo, R., Martin, J., Odajima, J., Martin, A., Dubus, P., Ortega,S., Barbacid, M., 2003. Driving the cell cycle to cancer. Adv. Exp. Med. Biol. 532, 1–11.
Marciniak, R.A., Calnan, B.J., Frankel, A.D., Sharp, P.A., 1990. HIV-1 Tat protein trans-activates transcription in vitro. Cell 63 (4), 791–802.
Marciniak, R.A., Sharp, P.A., 1991. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 10 (13), 4189–4196.
Mukerjee, R., Sawaya, B.E., Khalili, K., Amini, S., 2007. Associationofp65 andC/EBPbetawithHIV-1 LTR modulates transcription of the viral promoter. J. Cell. Biochem. 100 (5),1210–1216.
Nekhai, S., Shukla, R.R., Kumar, A., 1997. A human primary T-lymphocyte-derivedhuman immunodeficiency virus type 1 Tat-associated kinase phosphorylates the C-terminal domain of RNA polymerase II and induces CAK activity. J. Virol. 71 (10),7436–7441.
Nekhai, S., Shukla, R.R., Fernandez, A., Kumar, A., Lamb, N.J., 2000. Cell cycle-dependentstimulation of the HIV-1 promoter by Tat-associated CAK activator. Virology 266 (2),246–256.
Nekhai, S., Zhou, M., Fernandez, A., Lane, W.S., Lamb, N.J., Brady, J., Kumar, A., 2002. HIV-1 Tat-associated RNA polymerase C-terminal domain kinase, CDK2, phosphorylatesCDK7 and stimulates Tat-mediated transcription. Biochem. J. 364 (Pt 3), 649–657.
Parada, C.A., Roeder, R.G., 1996. Enhanced processivity of RNA polymerase II triggered byTat-induced phosphorylation of its carboxy-terminal domain. Nature 384 (6607),375–378.
Pumfery, A., Deng, L., Maddukuri, A., de la Fuente, C., Li, H., Wade, J.D., Lambert, P.,Kumar, A., Kashanchi, F., 2003. Chromatin remodeling andmodification during HIV-1 Tat-activated transcription. Curr. HIV Res. 1 (3), 343–362.
Richter, S., Ping, Y.H., Rana, T.M., 2002. TAR RNA loop: a scaffold for the assembly of aregulatory switch in HIV replication. Proc. Natl. Acad. Sci. U. S. A. 99 (12), 7928–7933.
Roebuck, K.A., Rabbi, M.F., Kagnoff, M.F., 1997. HIV-1 Tat protein can transactivate aheterologous TATAA element independent of viral promoter sequences and thetrans-activation response element. Aids 11 (2), 139–146.
Schmidtmayerova, H., Nuovo, G.J., Bukrinsky, M., 1997. Cell proliferation is not requiredfor productive HIV-1 infection of macrophages. Virology 232 (2), 379–384.
Traggiai, E., Chicha, L., Mazzucchelli, L., Bronz, L., Piffaretti, J.C., Lanzavecchia, A., Manz,M.G., 2004. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 304 (5667), 104–107.

322 R. Van Duyne et al. / Virology 376 (2008) 308–322
Veschambre, P., Simard, P., Jalinot, P., 1995. Evidence for functional interaction betweenthe HIV-1 Tat transactivator and the TATA box binding protein in vivo. J. Mol. Biol.250 (2), 169–180.
Watanabe, S., Ohta, S., Yajima, M., Terashima, K., Ito, M., Mugishima, H., Fujiwara, S.,Shimizu, K., Honda, M., Shimizu, N., Yamamoto, N., 2007a. Humanized NOD/SCID/IL2R{gamma}null mice transplanted with hematopoietic stem cells under non-myeloablative condition show prolonged lifespans and allow detailed analysis ofHIV-1 pathogenesis. J. Virol. 81 (23), 13259–13264.
Watanabe, S., Terashima, K., Ohta, S., Horibata, S., Yajima, M., Shiozawa, Y., Dewan, M.Z.,Yu, Z., Ito, M., Morio, T., Shimizu, N., Honda, M., Yamamoto, N., 2007b.Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develophuman lymphoid systems and induce long-lasting HIV-1 infection with specifichumoral immune responses. Blood 109 (1), 212–218.
Wei, P., Garber, M.E., Fang, S.M., Fischer, W.H., Jones, K.A., 1998. A novel CDK9-associatedC-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92 (4), 451–462.
Yang,X.,Gold,M.O., Tang,D.N., Lewis,D.E., Aguilar-Cordova, E., Rice, A.P.,Herrmann,C.H.,1997.TAK, anHIV Tat-associated kinase, is amember of the cyclin-dependent family of proteinkinases and is induced by activation of peripheral blood lymphocytes and differentiationof promonocytic cell lines. Proc. Natl. Acad. Sci. U. S. A. 94 (23), 12331–12336.
Yu, L., Loewenstein, P.M., Zhang, Z., Green, M., 1995. In vitro interaction of the humanimmunodeficiency virus type 1 Tat transactivator and the general transcriptionfactor TFIIB with the cellular protein TAP. J. Virol. 69 (5), 3017–3023.
Zhang, L., Kovalev, G.I., Su, L., 2007. HIV-1 infection and pathogenesis in a novelhumanized mouse model. Blood 109 (7), 2978–2981.
Zhou, M., Deng, L., Lacoste, V., Park, H.U., Pumfery, A., Kashanchi, F., Brady, J.N., Kumar,A., 2004. Coordination of transcription factor phosphorylation and histonemethylation by the P-TEFb kinase during human immunodeficiency virus type 1transcription. J. Virol. 78 (24), 13522–13533.
Zhu, Y., Pe'ery, T., Peng, J., Ramanathan, Y., Marshall, N., Marshall, T., Amendt, B.,Mathews, M.B., Price, D.H., 1997. Transcription elongation factor P-TEFb is requiredfor HIV-1 tat transactivation in vitro. Genes. Dev. 11 (20), 2622–2632.





























