Embed Size (px)
Citation preview

1
Polyelectrolyte capsules as growth factor releasing centers in tissue engineering
scaffolds
De Cock L.J.1, De Wever O.
2, Van Vlierberghe S.
3, Vanderleyden E.
3, Dubruel P.
3, De Vos F.
4, Vervaet C.
1, Remon
J.P.1
and De Geest B.G.1,*
1 Laboratory of Pharmaceutical Technology, Department of Pharmaceutics, Ghent University, Harelbekestraat 72, 9000
Ghent, Belgium
2 Laboratory of Experimental Cancer Research, Department of Radiotherapy and Nuclear Medicine, Ghent University
Hospital, Belgium
3 Polymer Chemistry and Biomaterials Research Group, Ghent University, Krijgslaan 281, 9000 Ghent, Belgium
4 Laboratory of Radiopharmacy, Department of Pharmaceutical analysis, Ghent University, Harelbekestraat 72, 9000 Ghent,
Belgium
* Corresponding author: Dr. Bruno De Geest
ABSTRACT
Spatial and temporal controlled delivery of growth factors which contribute to efficient repair upon
tissue injury or failure remains a major challenge in tissue engineering. Here we evaluate polyelectrolyte
multilayer microcapsules as growth factor carriers in order to allow spatial and temporal control over growth
factor release. Transforming growth factor-β1 (TGF-β1) was used as model growth factor and bio-specific
engineering of the microcapsules allowed to bind relative high amount of TGF-β1. Subsequently we investigated
the interactions between TGF-β1 and respectively microcapsules and gelatin based tissue engineering scaffolds.
By monitoring TGF-β1 induced transdifferentiation of fibroblasts into myofibroblasts we were able to
demonstrate that TGF-β1 encapsulation into polyelectrolyte multilayer microcapsules and subsequent
incorporation into gelatin based hydrogels did not alter the bioactivity of the growth factor.

2
1. INTRODUCTION
The field of regenerative medicine faces the challenge to treat damaged or diseased tissue in an efficient
and minimally invasive way. Tissue regeneration is a complex biological process requiring concerted action of
cell proliferation, differentiation and migration in addition to the generation of extracellular matrix components.
This process can be efficiently induced by appropriately combining cells, growth factors and scaffolds. The
scaffold which supports cell adhesion should degrade as soon as sufficient tissue regeneration has occurred and
this regeneration is enhanced by stimulating cells with growth factors.1 Growth factors are polypeptides which
transmit signals to control cellular activities such as proliferation, differentiation, migration, adhesion and
protein expression.2 Under normal circumstances, growth factors are produced by endogenous cells however, in
case of tissue repair, they must be provided exogenously.1 Optimal tissue regeneration requires the endogenous
profile of growth factors secreted during natural tissue regeneration to be closely mimicked.3
Delivery of growth factors to the site of tissue regeneration is challenging since these proteins have
short half-lives, high molecular weight and slow tissue penetration. Moreover, bolus injection should often be
avoided due to potential toxicity or undesirable side effects in the tissue surrounding the injection site. Growth
factors can be incorporated into polymeric scaffolds to stabilize the growth factor and to control their release
kinetics.4-6
Another promising strategy to enhance in vivo efficacy of growth factors is the use of growth factor
loaded drug delivery systems which release their payload in a controlled fashion. In this perspective, several
studies have focused on the encapsulation of growth factors into microsphere carriers based on PLGA7,8
,
gelatin9-11
, alginate12
, micelles13
, silk7, calcium carbonate
14 or polyelectrolytes
15,16. Incorporation of these
microcarriers into synthetic matrices 17-20
might be performed during scaffold preparation by mixing the
microcarriers with a building component of the synthetic scaffold 21,22
or after scaffold preparation through e.g.
microcapsule seeding 23
or injection 24
.
Fig. 1. Schematic overview of the design of growth factor binding polyelectrolyte multilayer microcapsules. (A) Synthesis of
heparin (orange) loaded CaCO3 microparticles by co-precipitating CaCl2 and Na2CO3 in the presence of heparin. (B) Layer-
by-layer coating of CaCO3 microparticles with heparin (polyanion; green) and poly-L-arginine (polycation; brown). (C) Hollow
microcapsules are obtained after dissolving the CaCO3 core templates with EDTA. (D) Growth factors (purple) are
postloaded into the capsules in slightly acidic buffer at low ionic strength.

3
In this paper we evaluate polyelectrolyte multilayer microcapsules25
as growth factor releasing centers
in tissue engineering scaffolds. These capsules are fabricated by layer-by-layer (LbL) coating of a sacrificial
template with alternately charged polyelectrolytes using electrostatic interaction as driving force, followed by
the decomposition of this template yielding hollow capsules.26-28
These capsules can be either preloaded or
postloaded with bio-active molecules.29
A preloading approach involves the therapeutic molecules of interest to
be embedded within the sacrificial template and subsequently to remain encapsulated within the hollow
capsules formed by LbL coating and decomposition of the core templates. This is an interesting strategy for the
encapsulation of rather high doses of larger proteins such as vaccine antigens.30,31
However, low dosed
expensive growth factors with mostly a molecular weight below 40 kDa, will be prone to premature release
upon dissolution of the core templates.29
Therefore, in this paper we introduce a postloading approach by
e gi eeri g the apsules i su h a way that they a t as growth fa tor i di g i ro-spo ges . This approa h is
schematically shown in Fig. 1 and comprises in a first step the synthesis of porous calcium carbonate (CaCO3)
microparticles filled with heparin in their pores by co-precipitating CaCl2 and Na2CO3 in the presence of heparin.
Subsequently, these microparticles are coated with 2 bilayers of again heparin as polyanion and poly-L-arginine
as polycation, followed by decomposition of the CaCO3 core in an aqueous EDTA solution. In this way, hollow
capsules are obtained with heparin both as membrane component as well as being suspended in their hollow
void. Heparin is a highly sulfated (thus bearing a net negative charge at physiological conditions)
glycosaminoglycan and has a high affinity for several growth factors.32-34
Therefore we hypothesize that
engineering the microcapsules with a high content of heparin will enhance their growth factor binding capacity.
Postloading with growth factor is then performed by incubating the microcapsules with growth factor in slightly
acidic buffer at low ionic strength to maximize the electrostatic interaction between growth factor (being more
protonated and thus bearing a higher net positive charge at lower pH) and heparin-engineered microcapsules.
Release of growth factor from the microcapsules is then expected to occur upon incubation in physiological
conditions through ionic competition and concentration gradient.
As model growth factor in this paper we encapsulate transforming growth factor-β1 (TGF-β1), a growth
factor known to induce transdifferentiation of fibroblasts into myofibroblasts, into heparin engineered
microcapsules. We demonstrate that TGF-β1 can be loaded and released without affecting its biological activity
and further demonstrate that these capsules can be incorporated within a gelatin tissue engineering cryogel
scaffold without affecting the properties of this scaffold.

4
2. MATERIALS AND METHODS
2.1. Materials
Calcium chloride (CaCl2), sodium carbonate (Na2CO3), ethylenediaminetetraacetic acid (EDTA),
fluorescein isothiocyanate (FITC), rhodamine isothiocyanate (RITC), Hoechst 33258, potassium persulfate (KPS),
N,N,N ,N -tetramethylethylenediamine (TEMED), p-nitrophenyl phosphate (pNPP), poly-L-arginine (Mw > 70
kDa), anti-α-SMA (clone 1A4), anti-tubulin (clone B-5-1-2) and anti-N-cadherin were purchased from Sigma-
Aldrich. Anti-tenascin-C (anti-TNC; clone BC-8) was provided by L. Zardi. Anti-fibronectin was obtained from
Abcam. Heparin was purchased from Diosynth Biotechnology. Pronase was purchased from Roche. Human
dermal fibroblasts and fibroblast growth medium were obtained from Promocell. LysoTracker Red, CellTracker
Red and the FITC-conjugated secondary antibody were purchased from Invitrogen. TGF- 1 was purchased from
R&D systems. The DC protein assay was obtained from Biorad. Hybond membranes were purchased from
Amersham. Methacrylated gelatin (degree of substitution of 60 %) was synthesized according to Van Den Bulcke
et al.35
Glass bottom dishes were purchased from Mattek. Irgacure 2959 was obtained from Ciba. Tissue Tek was
purchased from Sakura. FlexiPERM rings were purchased from Vivascience.
2.2. Fabrication and degradation of polyelectrolyte capsules
Calcium carbonate (CaCO3) microparticles doped with heparin were fabricated by adding 625 µl CaCl2
and 625 µl Na2CO3 (both 1M in deionized water) to 5 ml of an aqueous (deionized water) 0.2 mg/ml heparin
solution (1 mg/ml; 0.5 M NaCl). The heparin doped CaCO3 microparticles were centrifuged and subsequently
incubated for 5 min. in a heparin solution. After two centrifugation and washing steps with deionized water to
remove non-adsorbed polyelectrolytes, the microparticles were incubated for 5 min. with poly-L-arginine (1
mg/ml; 0.5 M NaCl). This cycle was repeated twice to deposit 2 heparin/poly-L-arginine bilayers followed by
dissolving the CaCO3 core templates in an aqueous EDTA (0.2 M) solution, yielding hollow microcapsules.
Fluorescent labeling of the microcapsules was performed using FITC or RITC labeled poly-L-arginine.36
Microcapsule degradability was assessed by suspending the microcapsules in a pronase solution (0.5
mg/ml in Tris buffer containing 0.5% sodium lauryl sulfate) followed by monitoring the turbidity of the
suspension using an automated turbidity reader (Bioscreen-C). Microcapsules suspended in water were used as
control. All experiments were run in triplicate.

5
2.3. Microscopy
Fluorescent labeled samples were visualized on a Nikon EZ-C1 confocal microscope. Scanning electron
microscopy images of dry samples being sputtered with gold were recorded with a Quanta 200 FEG FEI scanning
electron microscope operating at an acceleration voltage of 5 kV.
2.4. Interaction between capsules and fibroblasts
Normal human dermal fibroblasts were cultured in fibroblast growth medium containing bFGF (1 ng/ml),
insulin (5 µg/ml) and antibiotics (penicillin and streptomycin). Cells were grown in a humidified atmosphere
containing 5 % CO2.
Fibroblasts were exposed to green fluorescently labeled microcapsules for 12 hours and subsequently
visualized by confocal microscopy. Lysosomal vesicels were stained red fluorescent using LysoTracker Red
(1/5000 Invitrogen). Cell nuclei were stained blue fluorescent with Hoechst.
Cytotoxicitiy of the microcapsules was assessed according to De Koker et al.37
Human dermal fibroblasts
were seeded in a 96-well plate at a density of 2500 cells per well and incubated for 6 hours with several
amounts of microcapsules (with and without dissolution of the CaCO3 core templates). After an additional
cultivation period of 48 hours, cell viability was evaluated using a p-nitrophenyl phosphate (pNPP) cell viability
assay as previously described.36
All experiments were run in triplicate.
2.5. Encapsulation and release of TGF-β1
Hollow microcapsules were postloaded with TGF-β1 by 1 h incubation in 1 µg/ml TGF-β1 acetate buffer
(pH 5; 50 mM; 0.1 % Tween 20). After the incubation period, the growth factor loaded microcapsules were
washed twice with acetate buffer and subsequently suspended in phosphate buffered saline (PBS; pH 7.4; 150
mm NaCl; 0.1 % Tween 20) for further experiments.
To quantify encapsulation and release, TGF-β1 was radioactive labeled with 131
I by incubating 10 μg TGF-
β1 with 131
I in a vial coated with 70 μg iodogen in PBS containing 0.1 % Tween 20 during 20 min. at room
temperature. Subsequently, residual 131
I was separated from 131
I-TGF-β1 on a PD-10 column by size exclusion
chromatography using acetate buffer (pH 5; 50 mM; 0.1 % Tween 20) as eluent. The concentration of the
radioactive growth factor was calculated via the percentage of nonspecific adsorption of TGF-β1 on the PD-10
column and the original amount of growth factor, and the concentrated 131
I-TGF-β1 solution was further diluted
in acetate buffer (pH 5; 50 mM; 0.1 % Tween 20). 131
I-TGF-β1 was postloaded into the microcpasules as

6
described above and subsequently suspended in PBS. Encapsulation and release at several time points was
quantified by measuring the radioactivity in supernatant of centrifuged samples by a Packard cobra automated
γ-counter. All experiments were run in triplicate.
2.6. Biological activity of released TGF-β1
2.6.1. Immunocytochemistry
Fibroblasts were treated with cell medium, cell medium containing TGF-β1 (10 ng/ml), empty
microcapsules and TGF-β1 loaded microcapsules during 7 days. After treatment, the cells were fixed and
permeabilized using ice-cold methanol. Nonspecific binding was blocked by incubating the cells with Tris-
buffered saline (TBS) containing 5 % bovine serum albumin (BSA) for 30 minutes at room temperature. Alpha-
smooth muscle actin (α-SMA) was stained with a primary antibody (1/100; clone 1A4) followed by a FITC-
conjugated secondary antibody (1/1000). The cell nuclei were counterstained with Hoechst (1/5000).
2.6.2. Western blotting
Fibroblasts were treated with cell medium, cell medium containing TGF-β1 (10 ng/ml), empty capsules
and TGF-β1 loaded microcapsules during 7 days. Cells were lysed using Laemmli buffer and protein
concentrations were determined using a Lowry-based assay (DC protein assay). Samples were heated at 100°C
followed by loading on a 8 % SDS polyacrylamide gel. After transferring the separated proteins to a Hybond
membrane, nonspecific binding was blocked using PBS containing 5 % (w/v) nonfat milk powder and 0.5 % (w/v)
Tween 20 during 1 hour at room temperature. The separated proteins were then incubated with primary
antibodies against α-SMA (1/1000; clone 1A4), tubulin (1/4000; clone B-5-1-2), N-cadherin (1/1000), TNC
(1/1000; clone BC-8) and fibronectin (1/1000) during 1 hour at room temperature. After extensive washing in
PBS containing 5 % (w/v) nonfat milk powder and 0.5 % (w/v) Tween 20, membranes were incubated with HRP-
conjugated secondary antibodies. The intensity of the bands on the membrane was analyzed using Image J
software.
2.6.3. Collagen contraction assay
Fibroblast treated with cell medium, cell medium containing TGF-β1 (10 ng/ml), empty capsules and
TGF-β1 loaded microcapsules during 7 days were incorporated in a collagen gel. This was performed by
suspending the pretreated cells in a mixture of collagen type I, CMF-HBSS, MEM, NaHCO3 (0.25 M) and NaOH. A
final concentration of 250 000 cells/ml was obtained. 200 µl of the suspension was pipetted in a 24-well plate

7
followed by a 1 hour incubation period in a humidified atmosphere containing 5 % CO2 to allow gelification. The
fabricated gels were released and contraction of the gel was evaluated by measuring the diameter of the gel
after 24 hours. The procentual contraction was calculated according to the formula: [(diameter t0 – mean
diameter t24)/diameter t0)] were t0 represents the time point of collagen gel release and t24 represents the time
point 24 hours after collagen release.
2.6.4. Biological activity of TGF-β1 released from polyelectrolyte microcapsules incorporated within a gelatin
hydrogel film
TGF-β1 loaded microcapsules were suspended in 500 µl of a 10 % (w/v) methacrylated gelatin solution.
After adding 18 µl KPS (50 mg/ml) and 10 µl TEMED (neutralized with 4N HCl) to initiate radical polymerization
of the methacrylate moieties, the suspension was pipetted in a glass bottom dish. After 2 h reaction the
obtained hydrogel film was extensively rinsed with PBS to remove KPS and TEMED. Subsequently, fibroblasts
were seeded on the hydrogel film and after 7 days culturing, immunocytochemistry was performed to visualize
-SMA expression. Therefore, the cells were fixed and permeabilized using PBS containing 0.1 % Triton x-100.
Nonspecific binding was blocked by incubating the cells with Tris-buffered saline (TBS) containing 5 % bovine
BSA for 30 min. at roo te perature. α-SMA was stained with a primary antibody (1/100; clone 1A4) followed
by counterstaining with a FITC-conjugated secondary antibody (1/1000). Cell nuclei were stained with Hoechst
(1/5000).
2.7. Surface plasmon resonance (SPR)
Interaction between TGF-β1 and gelatin was evaluated using surface plasmon resonance (SPR; Biacore-
X). Gold sensor chips were spin coated with a 5 % (w/v) gelatin solution containing Irgacure 2959 (2 mol %) as
photoinitiator and exposed to UV light (276 nm; 10 mW/cm2; 5 min.) to crosslink the methacrylate moieties.
38
After inserting the chip in the SPR instrument, 50 µl TGF- 1 solution (Tris buffer; pH 7.4; 0.1 % Tween 20) was
injected (5 µl/min flow rate) and the surface plasmon resonance signal was monitored. Three concentrations
(i.e. 0.5, 1 and 2 µg/ml TGF-β1) were evaluated.
2.8. Incorporation of polyelectrolyte capsules in gelatin cryogels
1 g methacrylated gelatin was dissolved in 10 ml deionized water at 40 °C followed by the addition of
microcapsules and Irgacure 2959 (2 mol %) as photoinitiator. The mixture was injected into the mold of a cryo-
unit and was allowed to gel for 1 h at room temperature. Subsequently, crosslinking of the methacrylate
moieties was performed by exposing the hydrogel to UV light (276 nm; 10 mW/cm2) for 2 hours and pores were

8
formed using a freezing step (cryo-unit). Dry porous gelatin scaffolds were obtained through lyophilization of the
frozen hydrogels. Cryosections of Tissue Tek embedded cryogels were cut at 10 µm thickness.
The cryogels were mechanically tested using a TA 500 Texture Analyser equipped with a 10 N load cell.
An indentation of 2 mm was performed on a hydrated cryogel sample having a size of 1 mm x 1 mm x 0.7 mm.
Stiffness was calculated as slope of the load extension curve. For each data point, three samples were
measured.
2.9. Population of the synthetic scaffold with fibroblasts
Cell seeding was performed according to the method described by Dubruel et al.39
Briefly, hydrated
cryogels having a size of 15 mm x 4 mm x 3 mm were seeded with 160 000 cells using the drop-seeding method.
Seeded hydrogels were fixed under flexiPERM rings in a 6-well plate to prevent them from floating in the cell
medium. After 24 hours incubation, the flexiPERM rings were removed and the hydrogels were flipped. Cells
were stained red fluorescent using CellTracker Red (1/5000) while the gelatin was stained green fluorescent
through electrostatic adsorption of FITC-labeled poly-L-arginine.

9
3. RESULTS AND DISCUSSION
3.1. Synthesis and characterization of (hep/pARG)2 capsules
As stated in the introduction and schematically represented in Fig. 1, we aim to engineer polyelectrolyte
microcapsules in such a way that they exhibit an enhanced affinity to bind growth factors. This is done by
initially co-precipitating heparin into calcium carbonate microparticles followed by the deposition of 2
polyelectrolyte bilayers composed of heparin and poly-L-arginine (further on denoted as (hep/pARG)2) and
subsequent dissolution of the calcium carbonate core templates in an aqueous EDTA solution. Fig. 2 shows
scanning electron microscopy (SEM) images of the calcium carbonate microparticles (A) before and (B) after LbL
coating. The hollow capsules obtained after EDTA treatment are visualized by SEM in Fig. 2C and confocal
microscopy in Fig. 2D. Note that the collapsed structure in Fig. 2C is due to the drying of the sample and a proof
of their hollow structure. The capsules are fairly monodisperse and have mean diameter of 3.7 µm as measured
by laser diffraction.
Fig. 2 Scanning electron microscopy images of (A) CaCO3 microparticles and (B) polyelectrolyte microparticles composed
of 2 bilayers heparin and poly-L-arginine before and (C) after dissolution of the CaCO3 cores. (D) Confocal microscopy image
of hollow (hep/pARG)2 microcapsules which are red fluorescent due to the use of RITC-labeled heparin in the LbL film.
To be incorporated into tissue engineering scaffolds that should degrade over time during tissue
regeneration, capsule degradability and biocompatibility is undoubtly an important issue. Capsule degradability
was investigated by incubating (hep/pARG)2 microcapsules in a pronase, a non-specific protease mixture,
solution while monitoring the turbidity of the suspension. Fig. 3A shows a decrease in turbidity during the first 6
hours of incubation followed by a constant turbidity which might be caused by reduction of the enzyme activity.
As tissue engineering scaffolds should degrade over weeks to months, with a continuous renewal of extracellular
proteases to which the capsule will be exposed, it is reasonable to conclude from the short term (i.e. 15 h) data
shown in Fig. 3A that progressive capsule degradation is likely to occur.
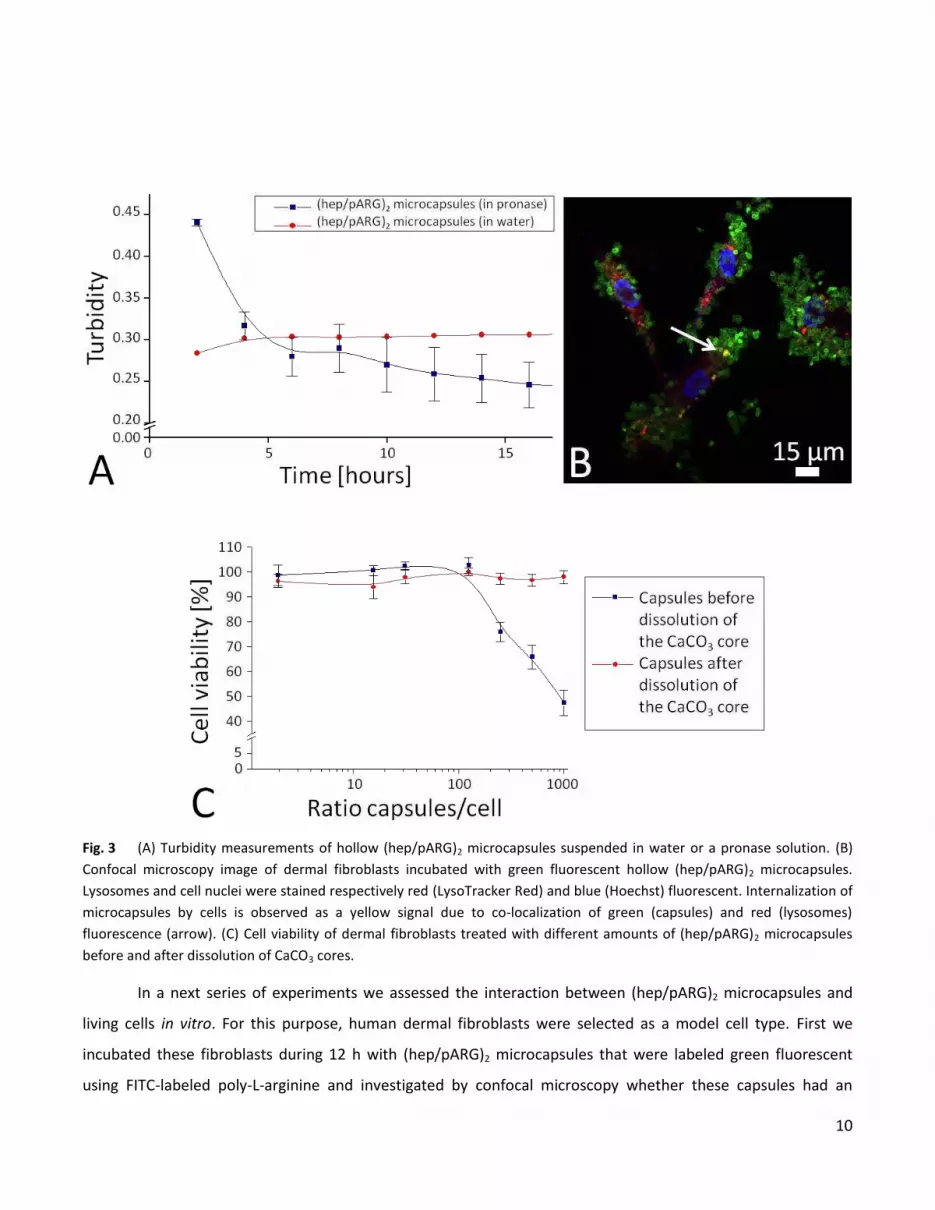
10
Fig. 3 (A) Turbidity measurements of hollow (hep/pARG)2 microcapsules suspended in water or a pronase solution. (B)
Confocal microscopy image of dermal fibroblasts incubated with green fluorescent hollow (hep/pARG)2 microcapsules.
Lysosomes and cell nuclei were stained respectively red (LysoTracker Red) and blue (Hoechst) fluorescent. Internalization of
microcapsules by cells is observed as a yellow signal due to co-localization of green (capsules) and red (lysosomes)
fluorescence (arrow). (C) Cell viability of dermal fibroblasts treated with different amounts of (hep/pARG)2 microcapsules
before and after dissolution of CaCO3 cores.
In a next series of experiments we assessed the interaction between (hep/pARG)2 microcapsules and
living cells in vitro. For this purpose, human dermal fibroblasts were selected as a model cell type. First we
incubated these fibroblasts during 12 h with (hep/pARG)2 microcapsules that were labeled green fluorescent
using FITC-labeled poly-L-arginine and investigated by confocal microscopy whether these capsules had an

11
influence on cell morphology and whether they were phagocyted. We did not observe an effect of the capsules
on cell spreading and morphology, indicating that these capsules are well tolerated by living cells. Furthermore,
by counterstaining the cellular lysosomes red fluorescent with LysoTracker Red 40-42
we only sparsely observed
co-localization of green and red fluorescence (yielding a yellow/orange signal), indicating that capsule
internalization by primary fibroblasts is unlike to occur (Fig. 3B). The observation that these capsules were well
tolerated was further confirmed by quantifying cell viability according to the method described by De Koker et
al.37
Fibroblasts were incubated for 6 hours with (hep/pARG)2 microcapsules (with and without the CaCO3 cores
being dissolved) followed by an additional cultivation period of 48 hours in cell medium followed by measuring
cell viability. As shown in Fig. 3C, hollow microcapsules did not affect cell viability up to an amount of 1000
microcapsules per cell. This result is in agreement with earlier published data showing the relatively inert
character of hollow polyelectrolyte microcapsules on cell viability.37
In contrast, incubation of fibroblasts with
microcapsules that still contained their CaCO3 core, did have a negative influence on cell viability starting from
250 microcapsules per cell or more. The higher mass of these CaCO3 containing microcapsules, thus exerting a
higher mechanical pressure onto the cells is assumed to be responsible for this.
3.2. Encapsulation, realease and biological activity of TGF-β1
Postloading polyelectrolyte multilayer microcapsules with proteins results mostly in protein
accumulation within the capsule membrane.29
Therefore we engineered our capsules in such a way that they are
strongly anionic by virtue of heparin both in their shell as well as in their hollow void. TGF-β1 abundantly
possesses basic residues (the isoelectric point of TGF-β1 is 9.5) which bind through electrostatic interaction with
heparin.32
Furthermore, we loaded the capsules in acetate buffer at pH 5 to enhance the electrostatic
i tera tio etwee the hepari s a io i sulfate groups a d the a i o groups of TGF-β1 which should be
extensively protonated at pH 5. We also kept the ionic strength of the loading buffer low, i.e. 50 mM, to
minimize charge shielding by the buffer ions. Radioactive labeling of TGF-β1 and subsequent determination of
radioactivity within the capsules after loading and washing allowed us to measure an amount of 59 ± 3 ng TGF-
β1 per 106 capsules. Related to the amount of TGF-β1 in the postloading solution, this is an encapsulation
efficiency of 35 ± 2 %. When these TGF-β1 loaded capsules are incubated in phosphate buffered saline at pH 7.4,
thus mimicking physiological conditions, an initial burst release of TGF-β1 is observed followed by a sustained
release over several days (Fig. 4).

12
0 20 40 60 80 100 120 140 160 1800
10
20
30
TG
F-
1 r
ele
ase
[%
]
Time [hours]
Fig. 4 Cummulative release profile of radioactive labeled TGF-β1 from (hep/pARG)2 microcapsules.
TGF-β1 is a homodimer of two 12.5 kDa peptides joined by a sulphydryl bond forming a 25 kDa molecule
and is involved in wound healing by affecting cellular processes as growth inhibition, differentiation,
angiogenesis and deposition of extracellular matrix components.43-45
After injury, blood platelets release a TGF-
β1 bolus to recruit inflammatory cells to the site of injury. Moreover, TGF-β1 stimulates transdifferentiation of
fibroblasts into myofibroblasts and stimulates (myo)fibroblasts to produce collagen and fibronectin in addition
to contract the newly formed connective tissue to bring together the edges of the wound in order to reduce the
wound size.46-51
Transdifferentiation of fibroblasts into myofibroblasts is expressed by an upregulation of several
proteins including tenascin (TNC), fibronectin, N-cadherin (N-cadh) and alpha-s ooth us le a ti (α-SMA). To
evaluate whether TGF-β1 retained its biological activity after being loaded and released from the microcapsules,
we exposed fibroblasts for 7 days to TGF-β1 loaded microcapsules. Subsequently, cell lysates were analyzed by
western blotting to determine several differentiation and functional markers of myofibroblasts such as tenascin,
fibronectin, N-cadherin and α-SMA. Tubulin was used as control to verify that equal amounts of proteins were
loaded in each lane as tubulin should exhibit the same relative intensity for each sample. The western blots in
Fig. 5A show increased expression of tenascin, fibronectin, N-cadherin and α-SMA after fibroblast treatment
with TGF-β1 both in solution (10 ng/ml) as well as released from the microcapsules, compared to fibroblasts
treated with empty capsules or cell medium without growth factor. Immunocytochemistry revealed that α-SMA
was organized in stress fibers (Fig. 5B1-5B2) after fibroblast treatment with both native TGF-β1 (10 ng/ml) as
well as TGF-β1 that was released from the microcapsules. Since contractile activity of (myo)fibroblasts is
enhanced when de novo expressed alpha-s ooth us le a ti (α-SMA) is incorporated in stress fibers,
fu tio ality of α-SMA incorporated in stress fibers was evaluated by incorporating TGF-β1 treated fibroblasts in

13
a 3D collagen gel. Contraction of the collagen matrix was evaluated 24 hours after fabrication of the collagen gel
(Fig. 5C-5D1-5D2). Fibroblasts exposed to TGF-β1 in solution (10 ng/ml) or to TGF-β1 released from microcapsules
showed enhanced contraction of the collagen gel compared to fibroblasts exposed to cell medium without
growth factor or blanco microcapsules. Taken together, these results clearly demonstrate that TGF-β1 loaded
and released from (hep/pARG)2 polyelectrolyte microcapsules is still bioactive and can stimulate fibroblast
transdifferentiation into myofibroblasts as potent as a native TGF-β1 solution.
Fig. 5 Fibroblast transdifferentiation into myofibroblasts by exposure to cell medium without growth factor, TGF-β1
solution (10 ng/ml), blanco microcapsules and TGF-β1 loaded microcapsules for 7 days. (A) Western blot analysis of
cellysates. Co fo al i ros opy i ages of α-SMA and nuclei staining of fibroblasts exposed to (B1) TGF-β1 loaded

14
microcapsules and (B2) empty microcapsules as control. α-SMA was stai ed gree fluores e t with α-SMA antibody and
FITC-labeled secondary antibody while the nuclei were stained blue fluorescent with Hoechst. (C) Contraction of collagen
gels incorporating fibroblasts exposed to cell medium without growth factor, TGF-β1 solution (10 ng/ml), empty
microcapsules and TGF-β1 loaded microcapsules for 7 days. Digital images of collagen gels incorporating fibroblasts exposed
during 7 days to (D1) TGF-β1 loaded microcapsules and (D2) blanco microcapsules after a 24 h incubation period.
3.3. Incorporation and interaction of TGF-β1 in tissue engineering scaffolds
The final aim of this paper is to incorporate growth factor loaded microcapsules into tissue engineering
scaffolds and to evaluate their interaction with these scaffolds. Dubruel et al. previously demonstrated the
potential of gelatin based cryogels as tissue engineering scaffolds. Gelatin is a natural polymer derived from
collagen and allows excellent cellular adhesion and growth. To obtain gels which are stable at physiological
temperature, gelatin was grafted with methacrylate moieties which can be crosslinked by radical
polymerization.35
Cryogel scaffolds are 3D structures with interconnected macropores (size of 100 µm and more)
which are formed by a cryogenic treatment (i.e. controlled cooling cyclus) of an aqueous gelatin solution which
induces phase separation of the solution into ice crystals, which will form the pores after freeze-drying, and a
gelatin rich phase which is then further stabilized by crosslinking the methacrylate moieties.39,52
Here we used
these gelatin cryogels as a model scaffold to incorporate TGF-β1 loaded hollow polyelectrolyte microcapsules. In
order to assess whether TGF-β1 released from the microcapsules would bind to gelatin and whether this would
preserve its bioactivity we first investigated the interaction between TGF-β1 and planar gelatin films. Surface
plasmon resonance (SPR) was used to specifically investigate the adsorption behavior of TGF-β1 onto gelatin.
Therefore, gold sensor chips were spin coated with methacrylated gelatin followed by UV irradiation, in the
presence of Irgacure 2959 as photoinitiator, to form a stable crosslinked hydrogel film. Subsequently, the chips
were placed in the flow cell of the SPR apparatus and TGF-β1 solutions were injected. At the start of the injection
phase (t = 500 s) TGF-β1 bound to the gelatin film which resulted in an increase of the response units and, as
shown in Fig. 6A, this binding was concentration dependent. The driving force for adsorption is electrostatic
interaction since TGF-β1 (IEP = 9.5) and gelatin (IEP = 5) are oppositely charged at physiological conditions. At the
end of the injection phase (t = 1100 s) loosely bound TGF-β1 dissociates from the gelatin film resulting in a
limited decrease of the response units. At t = 1700 s an extra washing step was performed however this did not
evoke a further decrease of the response units (data not shown) demonstrating that TGF-β1 was stably adsorbed
onto the gelatin.
To investigate whether TGF-β1 retains its biological activity upon encapsulation in (hep/pARG)2
microcapsules and subsequent incorporation within gelatin hydrogels we incorporated TGF-β1 loaded
microcapsules in a gelatin hydrogel film by mixing the TGF-β1 loaded microcapsules with a gelatin solution. After

15
adding the agents to crosslink the gelati s pe di g etha rylates, the mixture was quickly transferred into a
glass bottom dish to allow the formation of a stable hydrogel film. Next, fibroblasts were cultured for 7 days
onto this gelatin film followed by immunocytochemical staining of -SMA expression. As shown in Fig. 6B,
fibroblasts cultured on a gelatin film containing TGF-β1 loaded microcapsules did express -SMA which was
organized in stress fibers. By contrast, -SMA expression was absent in fibroblasts cultured on a gelatin film
containing empty microcapsules. This demonstrates that upon encapsulation in polyelectrolyte multilayer
capsules and subsequent incorporation into gelatin hydrogels, TGF-β1 is released from the microcapsules and
can reach cells cultured on the surface of the gelatin hydrogels while retaining its biological activity.
Fig. 6 (A) Surface plasmon resonance sensorgram showing the interaction between TGF-β1 and gelatin. (B) -SMA
expression by fibroblasts cultured on a gelatin film containing TGF-β1 loaded microcapsules. α-SMA was stained green
fluorescent with a primary antibody against α-SMA and FITC-labeled secondary antibody while the nuclei were stained blue

16
fluorescent with Hoechst. (C) Scanning electron microscopy image of a gelatin cryogel. (D) Confocal microscopy image of a
microtomed section of a cryogel (labeled green fluorescent through adsorption of FITC-labeled pARG) loaded with red
fluorescence (due to RITC-labeled poly-L-arginine) microcapsules. (E) Overview and (F) zoomed confocal microscopy image
of a cryogel (green fluorescent) populated with fibroblasts (red fluorescent). The green fluorescence is due to adsorption of
FITC-labeled pARG to the gelatin. The red fluorescence is due to staining of the fibroblasts with CellTracker Red.
In a next series of experiments we incorporated the microcapsules in a 3D gelatin cryogel by mixing the
microcapsules with methacrylated gelatin in solution, followed by a freezing and thawing cycle to induce
porosity through phase separation of ice crystals (i.e. the pores) and a gelatin rich phase. UV irradiation of the
matrix resulted in crosslinking of the methacrylate moieties to covalently crosslink the cryogel. Fig. 6C shows a
scanning electron microscopy image of a cryogel, clearly demonstrating its 3D structure. Confocal microscopy
image (Fig. 6D) of a microtomed section shows that the microcapsules (labeled red fluorescent) are
homogeneously distributed throughout the cryogel (green fluorescent). To assess whether the incorporation of
microcapsules had an impact on the mechanical properties of the cryogels the stiffness of the scaffolds without
and with different amount of capsules (2.8*107, 7*10
7 and 14*10
7) was measured. These measurements
indicated that, independent on the amount of incorporated capsules, the stiffness remained around 0.3 N/mm.
This demonstrates that one is able to incorporate high amounts of microcapsules within the scaffold without
affecting its mechanical properties. This is beneficial for the design of scaffolds with specific mechanical
properties which are independent on the presence of growth factor releasing centers.
Finally we aimed to assess whether gelatin cryogels containing microcapsules could still be populated
with living cells in vitro. Therefore, dermal fibroblasts were drop-seeded on cryogels. This drop-seeding
procedure is a static cell seeding method involving the deposition of a drop of cells suspended in culture
medium on top of the cryogels as described by Dubruel et al. After 7 days culturing, confocal microscopy was
used to visualize the distribution of the cells throughout the gelatin cryogels. For this purpose the cells were
stained red fluorescent with CellTracker Red and the cryogels were stained green fluorescent through
electrostatic adsorption of FITC-labeled poly-L-arginine. As shown in Fig. 6E and 6F complete and dense cellular
population within the cryogels was observed.
4. CONCLUSIONS
In this paper, we evaluated the potential of hollow (hep/pARG)2 microcapsules as growth factor
releasing centers for tissue engineering applications. First we optimized the microcapsule design and the loading
conditions to optimally bind TGF- 1 which was used as model growth factor in this work. Incubation of these
capsules in physiological buffer induced an initial burst release followed by a sustained release of TGF- 1,

17
without affecting its biological activity. Subsequently we demonstrated the incorporation of these growth factor
loaded microcapsules within a gelatin cryogel tissue engineering scaffold which did not alter the cryogel
morphology, the mechanical properties and the bioactivity of released TGF- 1.
5. ACKNOWLEDGEMENTS
LJDC wishes to express her gratitude to the Institute for the Promotion of Innovation by Science and
Technology in Flanders (IWT-Flanders) for their financial support. BGDG acknowledges the FWO-Flanders for a
postdoctoral scholarship.
6. REFERENCES
1. R. Vasita and D. S. Katti, Expert Rev Med Devices, 2006, 3, 29-47.
2. J. E. Babensee, L. V. McIntire and A. G. Mikos, Pharm Res, 2000, 17, 497-504.
3. M. J. Whitaker, R. A. Quirk, S. M. Howdle and K. M. Shakesheff, J Pharm Pharmacol, 2001, 53, 1427-
1437.
4. K. Y. Lee, M. C. Peters, K. W. Anderson and D. J. Mooney, Nature, 2000, 408, 998-1000.
5. M. Ozeki, T. Ishii, Y. Hirano and Y. Tabata, J Drug Target, 2001, 9, 461-471.
6. M. Matsusaki, H. Sakaguchi, T. Serizawa and M. Akashi, Journal of Biomaterials Science-Polymer Edition,
2007, 18, 775-783.
7. X. Q. Wang, E. Wenk, X. H. Zhang, L. Meinel, G. Vunjak-Novakovic and D. L. Kaplan, Journal of Controlled
Release, 2009, 134, 81-90.
8. A. Perets, Y. Baruch, F. Weisbuch, G. Shoshany, G. Neufeld and S. Cohen, Journal of Biomedical Materials
Research Part A, 2003, 65A, 489-497.
9. X. H. Zhu, Y. Tabata, C. H. Wang and Y. W. Tong, Tissue Eng Part A, 2008, 14, 1939-1947.
10. Z. S. Patel, H. Ueda, M. Yamamoto, Y. Tabata and A. G. Mikos, Pharm Res, 2008, 25, 2370-2378.
11. T. A. Holland, Y. Tabata and A. G. Mikos, J Control Release, 2003, 91, 299-313.
12. S. M. Jay and W. M. Saltzman, J Control Release, 2009, 134, 26-34.
13. J. S. Lee, J. W. Bae, Y. K. Joung, S. J. Lee, D. K. Han and K. D. Park, International Journal of Pharmaceutics,
2008, 346, 57-63.
14. D. Green, D. Walsh, X. B. Yang, S. Mann and R. O. C. Oreffo, Journal of Materials Chemistry, 2004, 14,
2206-2212.
15. S. Facca, C. Cortez, C. Mendoza-Palomares, N. Messadeq, A. Dierich, A. P. R. Johnston, D. Mainard, J. C.
Voegel, F. Caruso and N. Benkirane-Jessel, Proc. Natl. Acad. Sci. U. S. A., 107, 3406-3411.
16. Y. Itoh, M. Matsusaki, T. Kida and M. Akashi, Biomacromolecules, 2008, 9, 2202-2206.
17. T. A. Holland, Y. Tabata and A. G. Mikos, Journal of Controlled Release, 2005, 101, 111-125.
18. S. Young, Z. S. Patel, J. D. Kretlow, M. B. Murphy, P. M. Mountziaris, L. S. Baggett, H. Ueda, Y. Tabata, J.
A. Jansen, M. Wong and A. G. Mikos, Tissue Engineering Part A, 2009, 15, 2347-2362.
19. Z. S. Patel, S. Young, Y. Tabata, J. A. Jansen, M. E. K. Wong and A. G. Mikos, Bone, 2008, 43, 931-940.
20. E. Wenk, A. J. Meinel, S. Wildy, H. P. Merkle and L. Meinel, Biomaterials, 2009, 30, 2571-2581.
21. D. Banerjee I Fau - Mishra, T. K. Mishra D Fau - Maiti and T. K. Maiti.

18
22. S. Huang, T. Z. Deng, H. Wu, F. M. Chen and Y. Jin, Journal of Microencapsulation, 2006, 23, 277-290.
23. G. B. Wei, Q. M. Jin, W. V. Giannobile and P. X. Ma, Biomaterials, 2007, 28, 2087-2096.
24. K. Kawai, S. Suzuki, Y. Tabata, Y. Ikada and Y. Nishimura, Biomaterials, 2000, 21, 489-499.
25. L. J. De Cock, S. De Koker, B. G. De Geest, J. Grooten, C. Vervaet, J. P. Remon, G. B. Sukhorukov and M. N.
Antipina, Angew Chem Int Ed Engl.
26. D. V. Volodkin, N. I. Larionova and G. B. Sukhorukov, Biomacromolecules, 2004, 5, 1962-1972.
27. A. I. Petrov, D. V. Volodkin and G. B. Sukhorukov, Biotechnology Progress, 2005, 21, 918-925.
28. D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398-3406.
29. Z. She, M. N. Antipina, J. Li and G. B. Sukhorukov, Biomacromolecules, 2010, 11, 1241-1247.
30. S. De Koker, T. Naessens, B. G. De Geest, P. Bogaert, J. Demeester, S. De Smedt and J. Grooten, J
Immunol, 2010, 184, 203-211.
31. S. De Koker, B. G. De Geest, S. K. Singh, R. De Rycke, T. Naessens, Y. Van Kooyk, J. Demeester, S. C. De
Smedt and J. Grooten, Angewandte Chemie-International Edition, 2009, 48, 8485-8489.
32. M. Lyon, G. Rushton and J. T. Gallagher, Journal of Biological Chemistry, 1997, 272, 18000-18006.
33. S. Faham, R. E. Hileman, J. R. Fromm, R. J. Linhardt and D. C. Rees, Science, 1996, 271, 1116-1120.
34. C. H. Heldin and B. Westermark, Physiol. Rev., 1999, 79, 1283-1316.
35. A. I. Van Den Bulcke, B. Bogdanov, N. De Rooze, E. H. Schacht, M. Cornelissen and H. Berghmans,
Biomacromolecules, 2000, 1, 31-38.
36. L. J. De Cock, S. De Koker, F. De Vos, C. Vervaet, J. P. Remon and B. G. De Geest, Biomacromolecules, 11,
1002-1008.
37. S. De Koker, B. G. De Geest, C. Cuvelier, L. Ferdinande, W. Deckers, W. E. Hennink, S. De Smedt and N.
Mertens, Advanced Functional Materials, 2007, 17, 3754-3763.
38. S. Van Vlierberghe, E. Vanderleyden, P. Dubruel, F. De Vos and E. Schacht, Macromol Biosci, 2009, 9,
1105-1115.
39. P. Dubruel, R. Unger, S. Van Vlierberghe, V. Cnudde, P. J. S. Jacobs, E. Schacht and C. J. Kirkpatrick,
Biomacromolecules, 2007, 8, 338-344.
40. A. M. Javier, O. Kreft, M. Semmling, S. Kempter, A. G. Skirtach, O. T. Bruns, P. del Pino, M. F. Bedard, J.
Raedler, J. Kaes, C. Plank, G. B. Sukhorukov and W. J. Parak, Advanced Materials, 2008, 20, 4281-4287.
41. B. G. De Geest, R. E. Vandenbroucke, A. M. Guenther, G. B. Sukhorukov, W. E. Hennink, N. N. Sanders, J.
Demeester and S. C. De Smedt, Advanced Materials, 2006, 18, 1005-+.
42. A. Szarpak, D. Cui, F. Dubreuil, B. G. De Geest, L. J. De Cock, C. Picart and R. Auzely-Velty,
Biomacromolecules, 11, 713-720.
43. D. A. Clark and R. Coker, International Journal of Biochemistry & Cell Biology, 1998, 30, 293-298.
44. H. B. Fan, Y. Y. Hu, L. Qin, X. S. Li, H. Wu and R. Lv, Journal of Biomedical Materials Research Part A, 2006,
77A, 785-794.
45. I. Y. Kim, M. M. Kim and S. J. Kim, Journal of Biochemistry and Molecular Biology, 2005, 38, 1-8.
46. R. Montesano and L. Orci, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 4894-4897.
47. P. Lijnen, V. Petrov and R. Fagard, Journal of the Renin-Angiotensin-Aldosterone System, 2003, 4, 113-
118.
48. B. Hinz, G. Celetta, J. J. Tomasek, G. Gabbiani and C. Chaponnier, Molecular Biology of the Cell, 2001, 12,
2730-2741.
49. H. Denys, L. Derycke, A. Hendrix, W. Westbroek, A. Gheldof, K. Narine, P. Pauwels, C. Gespach, M.
Bracke and O. De Wever, Cancer Letters, 2008, 266, 263-274.
50. A. Desmouliere, A. Geinoz, F. Gabbiani and G. Gabbiani, Journal of Cell Biology, 1993, 122, 103-111.
51. K. Narine, O. De Wever, D. Van Valckenborgh, K. Francois, M. Bracke, S. DeSmet, M. Mareel and G. Van
Nooten, Tissue Eng, 2006, 12, 2707-2716.
52. S. Van Vlierberghe, V. Cnudde, P. Dubruel, B. Masschaele, A. Cosijns, I. De Paepe, P. J. S. Jacobs, L. Van
Hoorebeke, J. P. Remon and E. Schacht, Biomacromolecules, 2007, 8, 331-337.

19





























