Embed Size (px)
Citation preview

Epidemiological studies of spectrin mutations relatedto hereditary elliptocytosis and spectrin polymorphismsin Benin
CLE MENT GL ELE-KAKAI,* MI CHEL GARBARZ, MARIE-CHRI STINE LECOMTE, SYLVIE LEBO RGNE, COLETTE GALAND,ODILE BOURNIER, ISAB ELLE DEVAUX, HUGUET TE GAUT ERO, ISIDORE ZOHOUN,* PATRIC K G. GAL LAGHER,†
BERNARD G. FORGET†
AND DIDIER DHE RMY INSERM U409, Faculte X. Bichat, Paris, France,*Centre National Hospitalier et Universitaire, Cotonou, Benin, and †Departments of Pediatrics,Internal Medicine and Genetics, Yale University School of Medicine, New Haven, Connecticut, U.S.A.
Received 12 February 1996; accepted for publication 21 June 1996
Summary. We studied an African population in Benin anddiscovered an unexpectedly high frequency (1.6%) ofhereditary elliptocytosis (HE) among the 1447 subjectsstudied. In approximately two-thirds of HE individuals weidentified molecular defects, primarily those in erythrocyte®-spectrin (dupL154, L260P and L207P mutations), as wellas a novel mutation of erythrocyte ¯-spectrin (¯-W2061Rmutation). We also identified the genetic basis of a previouslyidentified protein polymorphism of the ®III domain of
spectrin (R1331I mutation). The genetic background of HEin the African population was studied using a number ofpolymorphisms of the ®-spectrin gene, including the ®IIIdomain polymorphism. These studies suggest that the HEmutations appear to have originated from separate geneticbackgrounds in this population.
Keywords: spectrin, mutation, elliptocytosis, polymorphism.
Hereditary elliptocytosis (HE) is a heterogenous group ofdisorders characterized by the presence of elliptical red bloodcells. The clinical presentation of HE can range from thecommon asymptomatic condition with a variable number ofelliptical cells, to severe haemolytic anaemia with red cellfragmentation (poikilocytosis). Most of the abnormalitiesrecognized so far occur within two components of theerythrocyte skeleton, spectrin (Sp) and protein 4.1 (Dhermyet al, 1986; Lecomte et al, 1993; Lux & Palek, 1995). Sp iscomposed of two subunits, � and �, which are encodedby separate genes (Gallagher & Forget, 1993). The � and� chains intertwine in an antiparallel manner to form rod-like heterodimers which, in turn, self-associate head to headto form tetramers. Sp tetramers are secured to the membraneby an interaction with ankyrin, a protein that forms abridge to the transmembrane protein band 3. At theirends, the Sp tetramers are attached to the lattice junctions,which comprise short actin filaments, together with protein4.1.
Using partial trypsin digestion, Sp chains have been
dissected into domains (�I to �V, and �I to �IV domains)(Speicher & Marchesi, 1984) (Fig 1). The N-terminal �Idomain and the C-terminal �I domain participate in Spdimer self-association (Speicher et al, 1993). Almost all casesof HE related to a Sp molecular defect present a more or lesspronounced deficiency of the dimer self-association process.This functional defect has been related to structuralalterations of either �I, �II or �I tryptic domains. Thesestructural abnormalities were first revealed by mild trypticdigestion, and proteic variants were identified based on themolecular size of the additional tryptic peptide generatedinstead of the normal 80 kD �I peptide. Three of the eightSp�I variants described to date appear to be the mostfrequent: Sp�I/74; Sp�I/65 and Sp�I/46-50a (Dhermy et al,1986; Lecomte et al, 1993; Lux & Palek, 1995) and most ofthe genomic mutations responsible for these proteic pheno-types have been characterized (Lux & Palek, 1995; Delaunay& Dhermy, 1993) (see Fig 1).
We and others have observed that most HE subjects withabnormal Sp variants are of black extraction (Dhermy et al,1986; Lux & Palek, 1995). In 1988 we made a systematicsurvey of HE in West Africa (Benin, Burkina Faso, IvoryCoast and Togo) to confirm the possible prevalence of HE
British Journal of Haematology, 1996, 95, 57–66
57# 1996 Blackwell Science Ltd
Correspondence: Dr D. Dhermy, INSERM U409, Faculte X. Bichat,B.P. 416, 75870 Paris Cedex 18, France.

with abnormal Sp variants among black populations(Lecomte et al, 1988).
Since this first survey, genomic mutations related to the Spvariants have been identified. The Sp�I/65 variant that weobserved in Africa or in black patients who originated fromthe West Indies (Lecomte et al, 1985b; Garbarz et al, 1986)was related to a unique mutation: the duplication of leucine154 (dupL154) in the �-Sp chain (Lux & Palek, 1995; Sahret al, 1989, 1990a). The same mutation was also identifiedamong black Americans (Lawler et al, 1985), North Africans(Alloisio et al, 1986), as well as Southern Italian familieswith HE Sp�I/65 (Miraglia del Giudice et al, 1992; Qualtieriet al, 1995). The Sp�I/46-50a variant, recurrently found inSouthern of Benin and Togo among ethnic groups belongingto the Adja linguistic group (such as Fon, Adja or Eve peoplein Togo) (Lecomte et al, 1988), appears related to aLeu!Pro substitution at position 260 of the �-Sp chain(L260P) (Sahr et al, 1989, 1990a). However, the Sp�I/46-50a
variant is genetically heterogenous because another aminoacid substitution (L207P) was concurrently found in blackHE subjects originating from the West Indies or NorthAmerica (Gallagher et al, 1992) as well as in a Moroccanfamily (Venezia et al, 1993) (see Fig 1). This Sp variant wasretrospectively named SpSaint Louis (Gallagher & Forget,1994).
Several Sp polymorphisms have been characterized bymild tryptic digestion and 2-D electrophoresis (see Fig 1).Four polymorphisms of the �II domain (�II type 1–4) havebeen characterized (Knowles et al, 1984; Gallagher et al,1991; DiPaolo et al, 1993) and we have described apolymorphism of the �III domain (�III type 2) found in HEblack families originated from West Indies and bearing thevariant Sp�I/46-50a (Lecomte et al, 1985a).
Taking into account the data obtained from HE as well asfrom Sp polymorphisms since our first epidemiological studyin Africa, we undertook a new cytological screening ofelliptocytosis associated with genomic detection of Spmutations, related either to HE or to �II and �III poly-morphisms of the �-Sp chain in Benin. For this purpose, wehave identified the genomic mutation responsible for the �IIIpolymorphism type 2. Our results suggest that HE mutationsappear to have originated in separate genetic backgrounds inthis population and the relevance of a putative relationshipbetween Sp mutations and resistance to malaria is discussed.
MATERIAL AND METHODS
Clinical materials. Blood sampling was performed in theBlood Bank of Cotonou, Benin. Blood samples from 1447unrelated healthy blood donor volunteers (age range 18–45years) were drawn into heparinized capillary tubes from afinger puncture. 20–50 �l of blood were immediately dilutedand fixed in 1 ml of 5 mM phosphate buffer, 150 mM NaCl,pH 7.4, containing 0.3% glutaraldehyde. Cytological studieswere done using light phase-contrast microscopy. Simulta-neously, 50–100 �l of blood were blotted onto filter papersand DNA was microextracted ( Jinks et al, 1989). When ACDanticoagulated blood samples were available, DNA wasprepared by standard methods (Sambrook et al, 1989).
Spectrin biochemical analysis. Limited tryptic digestion ofSp, followed by one-dimensional SDS-PAGE and by two-dimensional electrophoresis, were performed as previouslyreported (Parquet et al, 1994). Microsequencings of Sptryptic domains was performed as described (Lecomte et al,1989).
Screening of Sp �-gene mutations. Four previously determined
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
58 Clement Glele-Kakai et al
Fig 1. Localization of the mutations responsible for HE and polymorphisms in spectrin � and � chains. The molecular model of spectrin isrestricted to the C terminus of the � chain and to the first three tryptic domains (�I to �III) of the � chain. The complementary anti-parallelchains are omitted. Each � or � conformational �106 aminoacid repeat is composed of three � helices. The point mutations responsible for HEtryptic phenotypes and polymorphisms cited in this study are indicated by open circles. The ankyrin-binding site is also indicated. The segment�10 corresponds to the SH3 domain.

59Epidemiological Studies of Spectrin Mutations in Benin
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
point mutations of the Sp �-gene were screened usingpolyacrylamide gel electrophoresis (PAGE) or restrictionenzyme digestion of PCR-amplified DNA fragments aspreviously described for DupL154 and L260P mutations(Boulanger et al, 1992), for L207P mutation (Gallagher et al,1992) and for mutations at codon 28 of �-Sp gene (Coetzeret al, 1991; Floyd et al, 1991).
Direct sequencing on PCR-amplified genomic DNA,corresponding to either exon 30 of the �-Sp gene or exon2 of the �-Sp gene, was performed automatically on anautomated fluorescent DNA sequencer (ALFTM DNA sequen-cer, Pharmacia Biotech) using the Autoread Sequencing Kit(Pharmacia Biotech) and sets of primers previously reported(Parquet et al, 1994).
Probing of exon 2 of the �-Sp gene for SSCP was performedas previously described (Parquet et al, 1994).
Identification of the DNA mutations associated with the �IIItype 2 polymorphism: synthesis and amplification of reticulocyte�Sp cDNA. RNA from reticulocytes was prepared fromperipheral blood as described (Kan et al, 1975). Total RNAwas transcribed into single-stranded cDNA and cDNA wasthen amplified by PCR as described (Garbarz et al, 1992).
Four series of primers (A/B, C/D, E/F and G/H) covering theentire �III domain cDNA coding sequence were used (Table I).Primers sense (A, C, E and G) were biotinylated in the 50 end.
Direct sequencing of amplified �-Sp cDNA. Amplified DNAproducts were analysed by electrophoresis in 2% agarose gelsand purified using the Geneclean II kit (Bio 101, La Jolla,Calif., U.S.A.) according to the manufacturer’s instructions.Single-stranded DNA was purified using streptavidin-coupledmagnetic beads (Dynabeads M280 streptavidin; Dynal Oslo,Norway) as described (Parquet et al, 1994). Sequencingreactions were performed using internal sequencing primers
(BI, DI, FI and HI in Table I) and commercially availablesequencing reagents. DNA sequencing was carried outmanually using the T7 DNA sequencing kit (PharmaciaBiotech) or automatically on an automated fluorescent DNAsequencer (ALFTM DNA sequencer, Pharmacia Biotech)using the Autoread Sequencing Kit (Pharmacia Biotech).
Detection of � spectrin gene mutations R1333I and R1496Wby mismatched PCR. For mutation R1333I an oligomerprimer was designed in the antisense orientation with aC substituted for a T at position -3 from the 30 end: 50-CTTTTGTAGTTTTACCTGACGT-30. After PCR with theupstream primer (nt 4088–4107) 50-TGCAGAACTGGAT-CAGTAG-30, when the nucleotide G (4175) is changed to Tproducing the R1333I mutation, a GACGT�C AatII site isabolished. In contrast, the normal G produces an AatII site.For mutations R1496W an oligomer primer was designatedin the sense orientation with a C substituted for an A atposition -2 from the 30 end: 50-AAGCACAACTGATTGAT-GCG-30. After PCR with the downstream antisense primer (nt4721–4702) 50-AGCTCCTCAAGGTCTCGGTA-30, when thenucleotide C(4663) is changed to T producing the R1496Wmutation, a GCG�C HhaI site is abolished. In contrast, thenormal C produces a HhaI site. For both R1333I andR1496W mutations, digestion of amplification productswith AatII and HhaI, respectively, followed by gel electro-phoresis enabled determination of presence or absence of therespective mutant allele.
Probing of other Sp �-gene polymorphisms. DNA mutationsresponsible for polymorphism of the �II domain were screenedas described by Gallagher et al (1991). Three couples of primers(I/J, K/L, M/N) were designed for PCR amplifications of exons16, 17 and 18 of the �-Sp gene, respectively.
DNA mutation responsible for the �V41 polymorphism
(L1857) was screened as previously described (Gallagheret al, 1993).
RESULTS
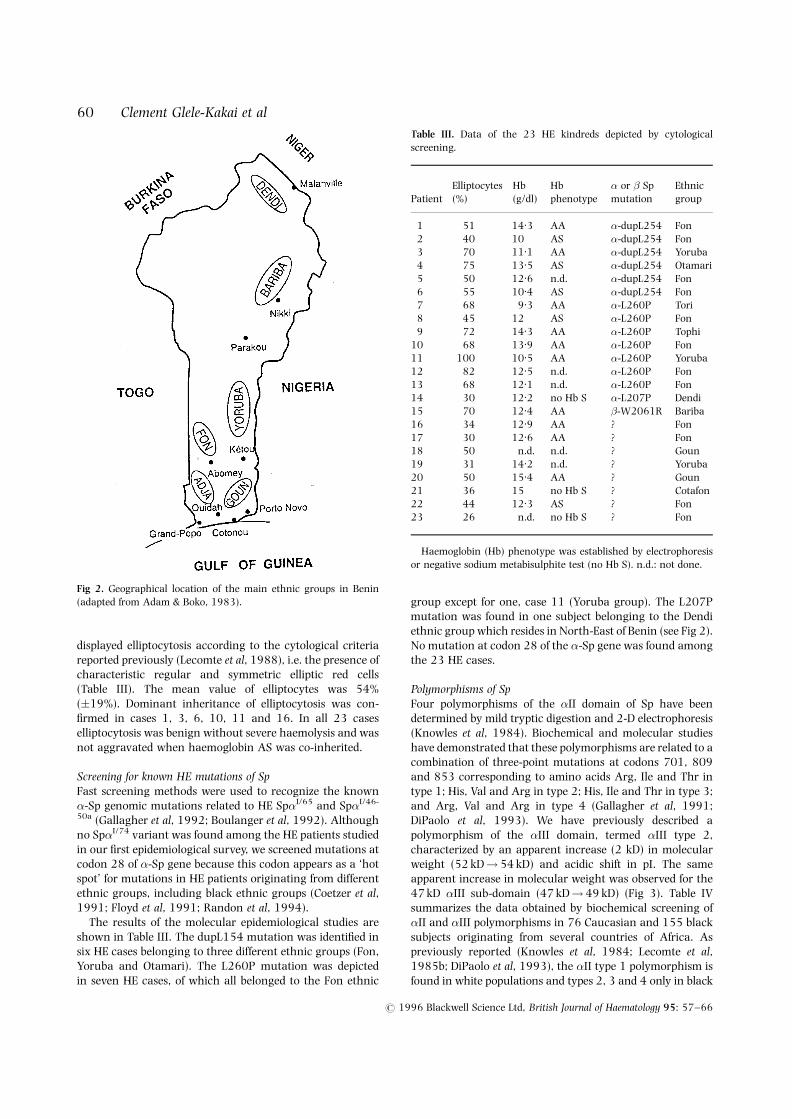
Half of the ethnic groups living in Benin (Adam & Boko,1983) were represented among the 1447 studied subjects(Table II). However, the Fon and Adja ethnic groups werepredominant (83.7%) because our study was made in apopulation residing in the area of Cotonou (Fig 2).
Among the 1447 subjects studied, 23 unrelated individuals
Table I. Oligonucleotide primers used in PCR andnucleotide sequencing. In primer M, the substitutionA!C* create a DdeI restriction site in conjunctionwith the mutation of the codon 853 (ACA!AGA)(Gallagher et al, 1991).
A: 50-GAGTTCCCGATGCTCCCACAGCG-30
B: 50-CAGCTGCCGCTGTTCATCTG-30
BI: 50-CAGCGGGCATTCAATTCCTG-30
C: 50-CAGAAGGAGCTCAAATCCGG-30
D: 50-GGTAGAATTTCTGGGCCTCA-30
DI: 50-TTACGATCCTTTGTACGCCC-30
E: 50-ATGAGGCCTGGGAAGACCT-30
F: 50-CTAGGATCTTCTTGCGTTTT-30
FI: 50-TCTCTCTCTAGCTTGACAGC-30
G: 50-TGATTTGGAGAAGGCTTGGG-30
H: 50-CTTCATGTGCAAAGGTCTGG-30
HI: 50-AGGTCCTCAAGGTCTCGGTA-30
I: 50-GGACCCAGTTGCATGAGGCC-30
J: 50-GCAGCCACAGCCGACTCCAG-30
K: 50-CTCCTATTACTTCCTACTAAATGATGG-30
L: 50-AGCAAGCTTTCTAGACTCCAACT-30
M: 50-GAACCACGCATTCAAGAGC*TAA-30
N: 50-TAGTACAAACTTCTTCCCAGAGG-30
Table II. Division of the ethnic groups of the 1447subjects studied in Benin.
Ethnic groups Percentage
Fon and related(Cotafon, Goun, Tophi, Tori) 67.5
Adja and related 16.2Yoruba and related 12.4Bariba and related 1.9Dendi and related 1.6Other groups 0.4
displayed elliptocytosis according to the cytological criteriareported previously (Lecomte et al, 1988), i.e. the presence ofcharacteristic regular and symmetric elliptic red cells(Table III). The mean value of elliptocytes was 54%(�19%). Dominant inheritance of elliptocytosis was con-firmed in cases 1, 3, 6, 10, 11 and 16. In all 23 caseselliptocytosis was benign without severe haemolysis and wasnot aggravated when haemoglobin AS was co-inherited.
Screening for known HE mutations of SpFast screening methods were used to recognize the known�-Sp genomic mutations related to HE Sp�I/65 and Sp�I/46-
50a (Gallagher et al, 1992; Boulanger et al, 1992). Althoughno Sp�I/74 variant was found among the HE patients studiedin our first epidemiological survey, we screened mutations atcodon 28 of �-Sp gene because this codon appears as a ‘hotspot’ for mutations in HE patients originating from differentethnic groups, including black ethnic groups (Coetzer et al,1991; Floyd et al, 1991; Randon et al, 1994).
The results of the molecular epidemiological studies areshown in Table III. The dupL154 mutation was identified insix HE cases belonging to three different ethnic groups (Fon,Yoruba and Otamari). The L260P mutation was depictedin seven HE cases, of which all belonged to the Fon ethnic
group except for one, case 11 (Yoruba group). The L207Pmutation was found in one subject belonging to the Dendiethnic group which resides in North-East of Benin (see Fig 2).No mutation at codon 28 of the �-Sp gene was found amongthe 23 HE cases.
Polymorphisms of SpFour polymorphisms of the �II domain of Sp have beendetermined by mild tryptic digestion and 2-D electrophoresis(Knowles et al, 1984). Biochemical and molecular studieshave demonstrated that these polymorphisms are related to acombination of three-point mutations at codons 701, 809and 853 corresponding to amino acids Arg, Ile and Thr intype 1; His, Val and Arg in type 2; His, Ile and Thr in type 3;and Arg, Val and Arg in type 4 (Gallagher et al, 1991;DiPaolo et al, 1993). We have previously described apolymorphism of the �III domain, termed �III type 2,characterized by an apparent increase (2 kD) in molecularweight (52 kD!54 kD) and acidic shift in pI. The sameapparent increase in molecular weight was observed for the47 kD �III sub-domain (47 kD!49 kD) (Fig 3). Table IVsummarizes the data obtained by biochemical screening of�II and �III polymorphisms in 76 Caucasian and 155 blacksubjects originating from several countries of Africa. Aspreviously reported (Knowles et al, 1984; Lecomte et al,1985b; DiPaolo et al, 1993), the �II type 1 polymorphism isfound in white populations and types 2, 3 and 4 only in black
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
60 Clement Glele-Kakai et alTable III. Data of the 23 HE kindreds depicted by cytologicalscreening.
Elliptocytes Hb Hb � or � Sp EthnicPatient (%) (g/dl) phenotype mutation group
1 51 14.3 AA �-dupL254 Fon2 40 10 AS �-dupL254 Fon3 70 11.1 AA �-dupL254 Yoruba4 75 13.5 AS �-dupL254 Otamari5 50 12.6 n.d. �-dupL254 Fon6 55 10.4 AS �-dupL254 Fon7 68 9.3 AA �-L260P Tori8 45 12 AS �-L260P Fon9 72 14.3 AA �-L260P Tophi
10 68 13.9 AA �-L260P Fon11 100 10.5 AA �-L260P Yoruba12 82 12.5 n.d. �-L260P Fon13 68 12.1 n.d. �-L260P Fon14 30 12.2 no Hb S �-L207P Dendi15 70 12.4 AA �-W2061R Bariba16 34 12.9 AA ? Fon17 30 12.6 AA ? Fon18 50 n.d. n.d. ? Goun19 31 14.2 n.d. ? Yoruba20 50 15.4 AA ? Goun21 36 15 no Hb S ? Cotafon22 44 12.3 AS ? Fon23 26 n.d. no Hb S ? Fon
Haemoglobin (Hb) phenotype was established by electrophoresisor negative sodium metabisulphite test (no Hb S). n.d.: not done.
Fig 2. Geographical location of the main ethnic groups in Benin(adapted from Adam & Boko, 1983).
61Epidemiological Studies of Spectrin Mutations in Benin
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
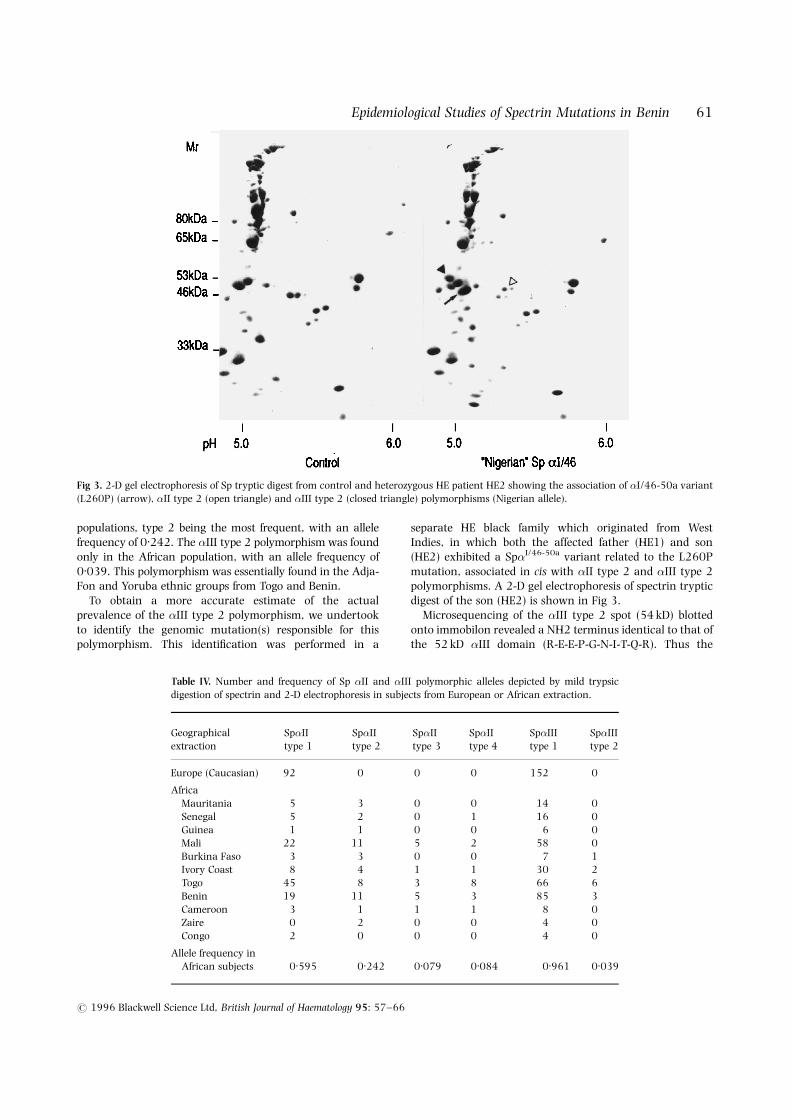
populations, type 2 being the most frequent, with an allelefrequency of 0.242. The �III type 2 polymorphism was foundonly in the African population, with an allele frequency of0.039. This polymorphism was essentially found in the Adja-Fon and Yoruba ethnic groups from Togo and Benin.
To obtain a more accurate estimate of the actualprevalence of the �III type 2 polymorphism, we undertookto identify the genomic mutation(s) responsible for thispolymorphism. This identification was performed in a
separate HE black family which originated from WestIndies, in which both the affected father (HE1) and son(HE2) exhibited a Sp�I/46-50a variant related to the L260Pmutation, associated in cis with �II type 2 and �III type 2polymorphisms. A 2-D gel electrophoresis of spectrin trypticdigest of the son (HE2) is shown in Fig 3.
Microsequencing of the �III type 2 spot (54 kD) blottedonto immobilon revealed a NH2 terminus identical to that ofthe 52 kD �III domain (R-E-E-P-G-N-I-T-Q-R). Thus the
Fig 3. 2-D gel electrophoresis of Sp tryptic digest from control and heterozygous HE patient HE2 showing the association of �I/46-50a variant(L260P) (arrow), �II type 2 (open triangle) and �III type 2 (closed triangle) polymorphisms (Nigerian allele).
Table IV. Number and frequency of Sp �II and �III polymorphic alleles depicted by mild trypsicdigestion of spectrin and 2-D electrophoresis in subjects from European or African extraction.
Geographical Sp�II Sp�II Sp�II Sp�II Sp�III Sp�IIIextraction type 1 type 2 type 3 type 4 type 1 type 2
Europe (Caucasian) 92 0 0 0 152 0
AfricaMauritania 5 3 0 0 14 0Senegal 5 2 0 1 16 0Guinea 1 1 0 0 6 0Mali 22 11 5 2 58 0Burkina Faso 3 3 0 0 7 1Ivory Coast 8 4 1 1 30 2Togo 45 8 3 8 66 6Benin 19 11 5 3 85 3Cameroon 3 1 1 1 8 0Zaire 0 2 0 0 4 0Congo 2 0 0 0 4 0
Allele frequency inAfrican subjects 0.595 0.242 0.079 0.084 0.961 0.039

63Epidemiological Studies of Spectrin Mutations in Benin
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
Cotonou to Paris within 48 h. In patient 15 the pattern of Sptryptic digestion revealed the presence of a Sp�I/74 variant(data not shown). As previously demonstrated (Lecomte et al,1990), this Sp variant can be related either to �- or to �-Spmutations, lying in the self-association site formed by theassociation of the incomplete repeating segment �17(Winkelmann et al, 1990), encoded by the exon 30 of �-Spgene, with a complementary solitary �-helix at the NH2terminus of the � chain, encoded by the exon 2 of thecorresponding gene (Speicher et al, 1993; Tse et al, 1990)(see Fig 1). Direct sequencing of exon 30 of the �-Sp gene inpatient 15 revealed a new mutation of the �-Sp gene atposition 2061 (TGG!AGG) causing the replacement oftryptophan by arginine. Sequencing of exon 2 of the �-Spgene was normal. This new variant was termed SpCotonou.In patient 16 the pattern of Sp tryptic digestion was normal,except for the presence of homozygous �III type 2 poly-morphism. In patient 16 and in the seven HE cases whereblood samples were not available for additional biochemicalstudies, exon 30 of �-Sp was sequenced and exon 2 of the�-Sp gene was probed for SSCP. No DNA abnormality couldbe detected using these techniques.
Since molecular diagnosis of HE has been available, familystudies have confirmed the reality of ‘silent carriers’ of HEwho do not exhibit apparent elliptocytosis (Lux & Palek,1995). This was observed particularly in patients bearingthe SpSaint Louis variant (L207P) (Venezia et al, 1993). Thepossibility that our cytological screening might have under-estimated the frequency of HE related to Sp mutations wastested by studying 100 additional non-elliptocytic subjects,randomly chosen among the 1447 studied subjects. DNAscreening of the �-Sp genomic mutations dupL154, L260Pand L207P and mutations modifying the codon 28 of �-Spgene did not show any abnormality.
DISCUSSION
Our present prospective cytological study revealed that theprevalence of elliptocytosis in Benin was 3 times higher(1.6%) than that estimated from our first study (0.6%)(Lecomte et al, 1988), which reported the results from fourcountries of West Africa (including the percentage of 0.5%from Benin). This increased prevalence could be explained byan improvement in our morphological studies. Moreover,this last study was made in a large area around Cotonouwhere the ethnic groups reside in which several HE caseswere identified in our first study. Two �-Sp mutations appearto be responsible for most of the HE depicted: the duplicationof leucine 154 (dupL154) and the replacement of leucine byproline at position 260 (L260P).
We confirmed that dupL154 mutation was diffusedamong black populations, because it was identified in threedifferent ethnic groups of Benin (Fon, Yoruba and Otamari).So far, we have observed 56 HE cases with the Sp�I/65
variant, who originated either from Africa or the WestIndies. In 22 informative families studied we have found thatthe dupL154 mutation is always associated in cis with the�II type 1 polymorphism and that all subjects heterozygousfor the dupL154 mutation have at least one allele bearing
the �II type 1 polymorphism. Analysis of �II polymorphismsin the six HE patients bearing the dupL154 mutation are inagreement with our previous data (see Table VI). These dataare consistent with a monocentric origin of the mutation(Guetarni et al, 1990; Delaunay & Dhermy, 1993).
In contrast, the L260P mutation was found in a restrictedarea (Southern Benin) among two ethnic groups: Fon andYoruba. In fact, Iarocci et al (1988) reported HE Sp�I/46-50a
in a black boy from the Yoruba tribe, Kwara Sate, in Nigeria.Historical, linguistic and customary studies have shown thatboth Fon and Yoruba ethnic groups are highly related (Adam& Boko, 1983; Cornevin, 1981; Felix Idoko, personalcommunication). Whether Fon are descendants of theYoruba people, the main ethnic group of the SouthernNigeria, or Fon and Yoruba share a common origin has notyet been established. Moreover, the L260P mutation appearsto be more often associated with both �II type 2 and �III type2 polymorphisms. Including the data reported in Table VI,we observed 21 non-related black HE cases bearing theL260P mutation (Sp�I/46-50a variant) and in 19 cases HEpatients were also heterozygous for both �II type 2 and �IIItype 2 polymorphisms. Family studies were available in fourcases and demonstrated that these two polymorphisms werein cis to the L260P mutation. In 10/19 both �R1331I and�R1496W mutations were identified. We propose to termthe allele bearing together the L260P HE mutation, the �IItype 2 polymorphism, the �III type 2 polymorphism(R1331I mutation) and the R1496W mutation as theNigerian allele.
For the first time, we identified SpSaint Louis (L207Pmutation) in a black African patient, originating from theNorth of Benin and belonging to the Dendi ethnic group.Customary data reported that the Dendi group had migratedfrom Mali (area of Gao and Tira), along the left bank of Nigerriver (Cornevin, 1981). As in six other HE patients bearingthe L207P mutation observed previously, the L207P mutationfound in the present study is in cis of both the �LELY allele andthe �II type 1 polymorphism (Gallagher et al, 1991).
As pointed out by Gallagher et al (1991), all these resultssuggest that the two different mutations responsible for theSp�I/46a-50 variant (L260P and L207P) arose on separatechromosomal backgrounds, implying a founder effect foreach mutation.
After the initial screening for known �-Sp mutations, nomolecular diagnosis could be made in nine HE patients. Inpatient 15 the discovery of a Sp�I/74 variant led us to identifya new �-Sp gene mutation: �-W2061R (SpCotonou). Accord-ing to the three-helix model established by Yan et al (1993),this mutation lies in the middle of the last �-helix B of theSp-� repeat 17 and it is the C-terminal mutation mostdescribed not associated with a truncated �-Sp chain. Itreplaces an uncharged hydrophobic amino acid with acharged hydrophilic amino acid that may disrupt hydro-phobic packing (i.e. it may disrupt interchain and intrachaininteractions). It is not found at, near, or adjacent to, aconserved amino acid residue of �-Sp chain.
In the eight HE cases in which no molecular diagnosiscould be made, we attempted to sequence exon 30 of the�-Sp gene and to screen exon 2 of the �-Sp for SSCP. No

DNA abnormality was found using this strategy, but domi-nant HE can be also related to rare �-Sp gene mutationslocated outside the tetramerization site (Delaunay & Dhermy,1993). Elliptocytosis could be secondary to another patho-logical process, such as iron deficiency, which is frequent inAfrica and can lead to marked elliptocytosis and even tosplenomegaly. However, such clinical features imply severeiron deficiency but no anaemia and hypochromia wereobserved in the eight HE kindreds. Elliptocytes can be alsofound in thalassaemia but the cytological features suggestiveof this diagnosis such as hypochromia, microcytosis andtarget cells were not observed.
Cytological screening of HE in Benin and complementarymolecular epidemiologic studies, indicated that about 1% ofHE can be related to Sp genetic defects which can impair thedimer self-association process. These defects have a 20 timeshigher incidence than that reported previously for HE in theCaucasian population (Bannerman & Renwick, 1962;Bernard et al, 1953) and are observed in populationswhich are (or have been) exposed to malaria. So, thequestion has to be raised whether these Sp mutationsprovide the heterozygotes with some protection againstmalaria. However, none of the above Sp mutations reachessuch a high frequency as, for instance, the protein band 3mutation accounting for ovalocytosis in Southern Asia( Jarolim et al, 1991; Schofield et al, 1992) or sickle celldisease in Africa (Nagel & Roth, 1989). We therefore testedthe possibility that our cytological screening might haveunderestimated the frequency of HE related to Sp mutations.No HE silent carrier was found in a cohort of 100 additionalnon-elliptocytic subjects in which we found concomitantly20% of subjects carrying HbS (as previously reported inBenin (Beauvais, 1994)). The putative relationship betweenHE related to Sp mutations and resistance to malaria cannotbe assessed in this molecular epidemiological study. However,in vitro studies (Lecomte et al, 1991; Schulman et al, 1990)on the malarial infection of erythrocytes carrying geneticallydefined membrane abnormalities have shown the necessityof a qualitatively and quantitatively normal membraneskeleton for an efficient invasion and a complete maturationof the parasite. Studies of extracellular development of theerythrocyte cycle of Plasmodium falciparum have also shownthat constituents of the erythrocyte membrane favour thedevelopment of rings into trophozoites and schizonts(Williams et al, 1995).
If in vitro studies have suggested that some elliptocytogenicalleles of Sp could be additional genetic factors to malarialresisitance (such as dupL154 and L207P), it has not yet beenproved that this potential resistance is expressed in vivo.Moreover, Sp polymorphisms have to be also discussed in thetopic of malaria resistance. As pointed out by DiPaolo et al(1993), the high frequency of the Sp �II type 2 polymorph-ism, which is a compound of types 3 and 4, suggests aselective advantage of this polymorphism since separatemutations represented by type 3 and type 4 occur at lowfrequency.
It now becomes obvious that several genes may beinvolved in the innate resistance to malaria and that theirrespective part in this complex process will be difficult to
recognize. One of the most promising developments con-cerning the role of the erythrocyte skeleton is the discovery ofdirect and relevant interactions between host skeletalproteins and parasite proteins such as MSP-1, RESA andMESA (Gratzer & Dluzewski, 1993).
ACKNOWLEDGMENTS
We acknowledge the contribution of L. Denoroy (CNRS,service central d’analyse, Vernaison, France) who performedpartial amino acid sequencing.
This work was supported in part by grants from theAssociation Francaise contre les Myopathies and from theUSPHS National Institutes of Health.
REFERENCES
Adam, K.S. & Boko, M. (1983) Le Benin (ed. by K. S. Adam and M.Boko).
Alloisio, N., Guetarni, D., Morle, L., Pothier, B., Ducluzeau, M.T.,Soun, A., Colonna, P., Clerc, M., Philippe, N. & Delaunay, J. (1986)Sp�I/65 hereditary elliptocytosis in North Africa. American Journalof Hematology, 23, 113–122.
Bannerman, R.M. & Renwick, J.H. (1962) The hereditary ellipto-cytoses: clinical and linkage date. Annals of Human Genetics, 26,23–38.
Beauvais, P. (1994) La drepanocytose. Revue de Pediatrie, 7, 4–19.Bernard, J., Dugas, M. & Cotlenko, M. (1953) L’elliptocytose
genotypique; a propos de dix observations personnelles. Annalesde Medecine, 54, 652–678.
Boulanger, L., Lecomte, M.C., Dhermy, D. & Garbarz, M. (1992) Fastscreening methods to detect mutations of spectrin in subjects withhereditary elliptocytosis. American Journal of Human Genetics, 51,440–442.
Coetzer, T.L., Sahr, K., Prchal, J., Blacklock, H., Peterson, L.A.,Koler, R., Doyle, J., Manaster, J. & Palek, J. (1991) Four differentmutations in codon 28 of � spectrin are associated withstructurally and functionally abnormal spectrin �I/74 in heredi-tary elliptocytosis. Journal of Clinical Investigation, 88, 743–749.
Cornevin, R. (1981) La republique populaire du Benin des originesDahomeennes a nos jours. G. P. Maisonneuve & Larose, Paris.
Delaunay, J. & Dhermy, D. (1993) Mutations involving the spectrinheterodimer contact site: clinical expression and alterations inspecific function. Seminars in Hematology, 30, 21–33.
Dhermy, D., Garbarz, M., Lecomte, M., Feo, C., Bournier, O.,Chaveroche, I., Gautero, H., Galand, C. & Boivin, P. (1986)Hereditary elliptocytosis: clinical, morphological and biochemicalstudies of 38 cases. Nouvelle Revue Francaise d’Hematologie, 28,129–140.
DiPaolo, B.R., Speicher, K.D. & Speicher, D.W. (1993) Identification ofthe amino acid mutations associated with human erythrocytespectrin �II domain polymorphisms. Blood, 82, 284–291.
Floyd, P.B., Gallagher, P.G., Valentino, L.A., Davis, M., Marchesi, S.L.& Forget, B.G. (1991) Heterogeneity of the molecular basis ofhereditary pyropoikilocytosis and hereditary elliptocytosis associ-ated with increased levels of the spectrin �I/74-kilodalton trypticpeptide. Blood, 78, 1364–1372.
Gallagher, P.G. & Forget, B.G. (1993) Spectrin genes in health anddisease. Seminars in Hematology, 30, 4–21.
Gallagher, P.G. & Forget, B.G. (1994) Spectrin St Louis and the�LELY allele. Blood, 84, 1684–1686.
Gallagher, P.G., Kotula, L., DiPaulo, B., Curtis, P., Speicher, D. &
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
64 Clement Glele-Kakai et al

65Epidemiological Studies of Spectrin Mutations in Benin
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
Forget, B.G. (1991) Polymorphisms of the �II domain of human �
spectrin and of the 30 untranslated region of � spectrin mRNA.Blood, 78, 364a
Gallagher, P.G., Roberts, W.E., Benoit, L., Speicher, D.W.,Marchesi, S.L. & Forget, B.G. (1993) Poikilocytic hereditaryelliptocytosis associated with spectrin Alexandria: an �I/50b Kdvariant that is caused by a single amino acid deletion. Blood, 82,2210–2215.
Gallagher, P.G., Tse, W.T., Coetzer, T., Lecomte, M.C., Garbarz, M.,Zarkowsky, H.S., Baruchel, A., Ballas, S.K., Dhermy, D., Palek, J. &Forget, B.G. (1992) A common type of the spectrin �I 46–50a-kDpeptide abnormality in hereditary elliptocytosis and pyropoikilo-cytosis is associated with a mutation distant from the proteolyticcleavage site: evidence for the functional importance of the triplehelical model of spectrin. Journal of Clinical Investigation, 89, 892–898.
Garbarz, M., Boulanger, L., Pedroni, S., Lecomte, M.C., Gautero, H.,Galand, C., Boivin, P., Feldman, L. & Dhermy, D. (1992)Spectrin beta Tandil, a novel shortened beta-chain variantassociated with hereditary elliptocytosis is due to a deletionalframeshift mutation in the beta-spectrin gene. Blood, 80, 1066–1073.
Garbarz, M., Lecomte, M.C., Dhermy, D., Feo, C., Chaveroche, I.,Gautero, H., Bournier, O., Picat, C., Goepp, A. & Boivin, P.(1986) Double inheritance of an alpha I/65 spectrin variantin a child with homozygous elliptocytosis. Blood, 67, 1661–1667.
Gratzer, W.B. & Dluzewski, A.R. (1993) The red blood cell andmalaria parasite invasion. Seminars in Hematology, 30, 232–247.
Guetarni, D., Roux, A.F., Alloisio, N., Morle, F., Ducluzeau, M.T.,Forget, B.G., Colonna, P., Delaunay, J. & Godet, J. (1990) Evidencethat expression of Sp�I/65 hereditary elliptocytosis is com-pounded by a genetic factor that is linked to the homologous�-spectrin allele. Human Genetics, 85, 627–630.
Iarocci, T.A., Wagner, G.M., Mohandas, N., Lane, P.A. &Mentzer, W.C. (1988) Hereditary poikilocytic anemia associatedwith the co-inheritance of two alpha spectrin abnormalities.Blood, 71, 1390–1396.
Jarolim, P., Palek, J., Amato, D., Hassan, K., Sapak, P., Nurse, G.T.,Rubin, H.L., Zhai, S., Sahr, K.E. & Liu, S. (1991) Deletion inerythrocyte band 3 gene in malaria-resistant Southeast Asianovalocytosis. Proceedings of the National Academy of Sciences of theUnited States of America, 88, 11022–11026.
Jinks, D.C., Minter, M., Tarver, D.A., Vanderford, M., Hejtmanick, J.F.& McCabe, R.B. (1989) Molecular genetic diagnosis of sickle celldisease using dried blood specimens in blotters used for newbornscreening. Human Genetics, 81, 363–366.
Kan, Y.W., Holland, J.P., Dozy, A.M. & Varmus, H.E. (1975)Demonstration of non-functional �-globin mRNA on homozygous��-thalassemia. Proceedings of the National Academy of Sciences of
the United States of America, 72, 5140–5144.Knowles, W.J., Bologna, M.L., Chasis, J.A., Marchesi, S.L. &
Marchesi, V.T. (1984) Common structural polymorphisms inhuman erythrocyte spectrin. Journal of Clinical Investigation, 73,973–979.
Lawler, J., Coetzer, T.L., Palek, J., Jacob, H.S. & Luban, N. (1985)Spectrin �I/65: a new variant of the � subunit of spectrinhereditary elliptocytosis. Blood, 66, 706–709.
Lecomte, M.C., Barrault, C., Deguercy, A., Boivin, P., Schrevel, J. &Dhermy, D. (1991) Inhibition of P. falciparum growth in elliptocytesbearing spectrin �I-domain mutations. (Abstract). Nouvelle RevueFrancaise d’Hematologie, 33, 133.
Lecomte, M.C., Dhermy, D., Garbarz, M., Feo, C., Gautero, H.,Bournier, O., Picat, C., Chaveroche, I., Ester, A., Galand, C. &
Boivin, P. (1985a) Pathologic and nonpathologic variants of thespectrin molecule in two black families with hereditary ellipto-cytosis. Human Genetics, 71, 351–357.
Lecomte, M.C., Dhermy, D., Gautero, H., Bournier, O., Galand, C. &Boivin, P. (1988) L’elliptocytose hereditaire en Afrique de l’Ouest:frequence et repartition des variants de la spectrine. ComptesRendus de l’Academie des Sciences, 306, 43–46.
Lecomte, M.C., Dhermy, D., Solis, C., Ester, A., Feo, C., Gautero, H.,Bournier, O. & Boivin, P. (1985b) A new abnormal variant ofspectrin in black patients with hereditary elliptocytosis. Blood, 65,1208–1217.
Lecomte, M.C., Garbarz, M., Gautero, H., Bournier, O., Galand, C.,Boivin, P. & Dhermy, D. (1993) Molecular basis of clinical andmorphological heterogeneity in hereditary elliptocytosis (HE)with spectrin �I variants. British Journal of Haematology, 85,584–595.
Lecomte, M.C., Garbarz, M., Grandchamp, B., Feo, C., Gautero, H.,Devaux, I., Bournier, O., Galand, C., d’Auriol, L., Galibert, F.,Sahr, K.E., Forget, B.G., Boivin, P. & Dhermy, D. (1989) Sp�I/78: amutation of the �I spectrin domain in a white kindred with HEand HPP phenotypes. Blood, 74, 1126–1133.
Lecomte, M.C., Gautero, H., Garbarz, M., Boivin, P. & Dhermy, D.(1990) Abnormal tryptic peptide from the spectrin �-chainresulting from �- or �-chain mutations: two genetically distinctforms of the Sp �I/74 variant. British Journal of Haematology, 76,406–413.
Lux, S.E. & Palek, J. (1995) Disorders of the red cell membrane.Blood: Principles and Practice of Hematology (ed. by R. I. Handin, S. E.Lux and T. P. Stossel), p. 1701. Lippincott, Philadelphia.
Miraglia del Giudice, E., Ducluzeau, M.T., Alloisio, N., Wilmotte, R.,Delaunay, J., Perrotta, S., Cutillo, S. & Iolascon, A. (1992) �I/65hereditary elliptocytosis in Southern Italy: evidence for an Africanorigin. Human Genetics, 89, 553–556.
Nagel, R.L. & Roth, E.F.J. (1989) Malaria and red cell genetic defects.Blood, 74, 1213–1221.
Parquet, N., Devaux, I., Boulanger, L., Galand, C., Boivin, P.,Lecomte, M.C., Dhermy, D. & Garbarz, M. (1994) Identification ofthree novel spectrin �I/74 mutations in hereditary elliptocytosis:further support for a triple-stranded folding unit model of thespectrin heterodimer contact site. Blood, 84, 303–308.
Qualtieri, A., Bisconte, M.G., Pasqua, A., Bria, M. & Brancati, C.(1995) Sp�I/65 hereditary elliptocytosis in Calabria (southernItaly). Human Genetics, 95, 359–362.
Randon, J., Boulanger, L., Marechal, J., Garbarz, M., Vallier, A.,Ribeiro, L., Tamagnini, G., Dhermy, D. & Delaunay, J. (1994)A variant of spectrin low expression allele alphaLELY carryinga hereditary elliptocytosis mutation in codon 28. British Journalof Haematology, 88, 534–540.
Sahr, K.E., Garbarz, M., Dhermy, D., Lecomte, M.C., Boivin, P.,Agre, P., Laughinghouse, K., Scarpa, A., Coetzer, T., Palek, J.,Marchesi, S.L. & Forget, B.G. (1990a) Use of the polymerase chainreaction for the detection and characterization of mutationscausing hereditary elliptocytosis. Cellular and Molecular Biology ofNormal and Abnormal Erythroid Membranes (ed. by C. M. Cohenand J. Palek), p. 201. Wiley-Liss, New York.
Sahr, K.E., Laurila, P., Kotula, L., Scarpa, A.L., Coupal, E., Leto, T.L.,Linnebach, A.J., Winkelmann, J.C., Speicher, D.W., Marchesi, V.T.,Curtis, P.J. & Forget, B.G. (1990b) The complete cDNA andpolypeptide sequences of human erythroid �-spectrin. Journal ofBiological Chemistry, 265, 4434–4443.
Sahr, K.E., Tobe, T., Scarpa, A., Laughinghouse, K., Marchesi, S.L.,Agre, P., Linnenbach, A.J., Marchesi, V.T. & Forget, B.G. (1989)Sequence and exon-intron organization of the DNA encoding the�I domain of human spectrin: application to the study of

mutations causing hereditary elliptocytosis. Journal of ClinicalInvestigation, 84, 1243–1252.
Sambrook, J., Fritch, E.F. & Maniatis, T. (1989) Molecular Cloning,2nd edn. Cold Spring Harbor Laboratory Press, New York.
Schofield, A.E., Tanner, M.J.A., Pinder, J.C., Clough, B., Bayley, P.M.,Nash, G.B., Dluzewski, A.R., Reardon, D.M., Cox, T.M.,Wilson, R.J.M. & Gratzer, W.B. (1992) Basis of unique red cellmembrane properties in hereditary ovalocytosis. Journal ofMolecular Biology, 223, 949–958.
Schulman, S., Roth, E.F.J., Cheng, B., Rybicki, A.C., Sussman, I.I.,Wong, M., Wang, W., Ranney, H.M., Nagel, R.L. & Schwartz, R.S.(1990) Growth of Plasmodium falciparum in human erythrocytescontaining abnormal membrane proteins. Proceedings of theNational Academy of Sciences of the United States of America, 87,7339–7343.
Speicher, D.W., DeSilva, T.M., Speicher, K.D., Ursitti, J.A., Hembach, P.& Weglarz, L. (1993) Location of the human red cell spectrintetramer binding site and detection of a related ‘closed’ hairpinloop dimer using proteolytic footprinting. Journal of BiologicalChemistry, 268, 4227–4235.
Speicher, D.W. & Marchesi, V.T. (1984) Erythrocyte spectrin iscomprised of many homologous triple helical segments. Nature,311, 177–180.
Tse, W.T., Lecomte, M.C., Costa, F.F., Garbarz, M., Feo, C., Boivin, P.,Dhermy, D. & Forget, B.G. (1990) A point mutation in the �
spectrin gene associated with �I/74 hereditary elliptocytosis:implications for the mechanism of spectrin dimer self-association.Journal of Clinical Investigation, 86, 909–916.
Venezia, N.D., Wilmotte, R., Morle, L., Forissier, A., Parquet, N.,Garbarz, M., Rousset, T., Dhermy, D., Alloisio, N. & Delaunay, J.(1993) An �-spectrin mutation responsible for hereditaryelliptocytosis associated in cis with the �V/41 polymorphism.Human Genetics, 90, 641–644.
Williams, J.H., Gill, G.S. & Trager, W. (1995) Effect of erythrocytemembrane on extracellular development of the erythrocytic cycleof Plasmodium falciparum. Proceedings of the National Academy ofSciences of the United States of America, 92, 566–568.
Wilmotte, R., Marechal, J., Morle, L., Baklouti, F., Philippe, N.,Kastally, R., Kotula, L., Delaunay, J. & Alloisio, N. (1993) Lowexpression allele �LELY of red cell spectrin is associated withmutations in exon 40 (�V/41 polymorphism) and intron 45 andwith partial skipping of exon 46. Journal of Clinical Investigation,91, 2091–2096.
Winkelmann, J.C., Chang, J.G., Tse, W.T., Scarpa, A.L., Marchesi, V.T.& Forget, B.G. (1990) Full-length sequence of the cDNA forhuman erythroid �-spectrin. Journal of Biological Chemistry, 265,11827–11832.
Yan, Y., Winograd, E., Viel, A., Harrison, S.C. & Branton, D. (1993)Crystal structure of the repetitive segments of spectrin. Science,262, 2027–2030.
# 1996 Blackwell Science Ltd, British Journal of Haematology 95: 57–66
66 Clement Glele-Kakai et al





























