Embed Size (px)
Citation preview
Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/43129723
Escherichiacoli-ClonedCFTRLociRelevantforHumanArtificialChromosomeTherapy
ArticleinHumangenetherapy·April2010
DOI:10.1089/hum.2009.225·Source:PubMed
CITATIONS
12
READS
63
11authors,including:
AnabelaSRamalho
UniversityofLisbon
34PUBLICATIONS463CITATIONS
SEEPROFILE
FiorentinaAscenzioni
SapienzaUniversityofRome
74PUBLICATIONS568CITATIONS
SEEPROFILE
MargaridaDAmaral
UniversityofLisbon-FacutyofSciences
209PUBLICATIONS3,002CITATIONS
SEEPROFILE
DirkSchindelhauer
ChromosomeMedicineProcurement
33PUBLICATIONS959CITATIONS
SEEPROFILE
AllcontentfollowingthispagewasuploadedbyMargaridaDAmaralon02December2016.
Theuserhasrequestedenhancementofthedownloadedfile.Allin-textreferencesunderlinedinbluearelinkedtopublicationsonResearchGate,lettingyouaccessandreadthemimmediately.

Escherichia coli-Cloned CFTR Loci Relevant for HumanArtificial Chromosome Therapy
Lucia Rocchi,1,2,* Carla Braz,1,3,4,* Sonja Cattani,5 Anabela Ramalho,4 Sulith Christan,1,5 Marlene Edlinger,1
Fiorentina Ascenzioni,2 Andreas Laner,5 Simone Kraner,1 Margarida Amaral,3,4 and Dirk Schindelhauer1,5,6
Abstract
Classical gene therapy for cystic fibrosis has had limited success because of immune response against viralvectors and short-term expression of cDNA-based transgenes. These limitations could be overcome by deliveringthe complete genomic CFTR gene on nonintegrating human artificial chromosomes (HACs). Here, we reportreconstruction of the genomic CFTR locus and analyze incorporation into HACs of three P1 phage-based and Ffactor bacteria-based artificial chromosomes (PACs/BACs) of various sizes: (1) 5A, a large, nonselectable BACcontaining the entire wild-type CFTR locus extending into both adjacent genes (296.8-kb insert, from kb �58.4 toþ51.4) containing all regulators; (2) CGT21, a small, selectable, telomerized PAC (134.7 kb, from kb �60.7 toþ 2)containing a synthetic last exon joining exon 10, EGFP, exon 24, and the 30 untranslated region; and (3) CF225, amidsized, nonselectable PAC (225.3 kb, from kb �60.7 to þ9.8) ligated from two PACs with optimized codonsand a silent XmaI restriction variant to discriminate transgene from endogenous expression. Cotransfection withtelomerized, blasticidin-S-selectable, centromere-proficient a-satellite constructs into HT1080 cells revealed aworkable HAC formation rate of 1 per ~25 lines when using CGT21 or 5A. CF225 was not incorporated into ade novo HAC in 122 lines analyzed, but integrants were expressed. Stability analyses suggest the feasibility ofprefabricating a large, tagged CFTR transgene that stably replicates in the proximity of a functional centromere.Although definite conclusions about HAC-proficient construct configurations cannot be drawn at this stage,important transfer resources were generated and characterized, demonstrating the promise of de novo HACs aspotentially ideal gene therapy vector systems.
Introduction
Cystic fibrosis (CF) is the most frequent, lethal geneticdisease associated with a single gene in the white popu-
lation, estimated to affect 1 per 2500–4000 newborns (WorldHealth Organization, 2004). CF results from mutations in theCF transmembrane conductance regulator (CFTR) gene,which encodes an ATP binding cassette (ABC) transporter thatfunctions as a phosphorylation-dependent and nucleotide-regulated chloride (Cl�) channel located in the apical mem-brane of epithelial cells (Riordan et al., 1989; Sheppard andWelsh, 1999) of many organs including the lungs, the intestine,the pancreas, the bladder, and the reproductive tracts.
Because CF is a recessive disorder, a single copy of thenormal CFTR gene is sufficient to achieve functional CFTRlevels that avoid CF, making CF an attractive candidatedisease for gene therapy. The airway epithelium is themajor target organ, as lung disease contributes mainly tomorbidity and mortality in CF patients. Moreover, thetherapeutic gene should target the cells that normally ex-press CFTR, preferably at physiological levels. It is expectedthat such physiological and stable CFTR expression levelsinside defective cells, or correction of even a fraction ofepithelial cells, could alter pathological epithelial physi-ology, thus being of clinical benefit (Dorin et al., 1996;Ramalho et al., 2002).
1Livestock Biotechnology, Life Sciences Center Weihenstephan, Technical University Munich (TUM), 85354 Freising, Germany.2Dipartimento Biologia Cellulare e dello Sviluppo, University of Rome La Sapienza, 00185 Rome, Italy.3Center for Biodiversity, Functional and Integrative Genomics (BioFIG), Faculty of Sciences, University of Lisbon, 1749-016 Lisbon,
Portugal.4Department of Human Genetics, National Institutes of Health, 1649-016 Lisbon, Portugal.5Department of Medical Genetics, Children’s Hospital, Ludwig Maximilians University, 80336 Munich, Germany.6Institute of Human Genetics, Technical University of Munich, 81675 Munich, Germany.*L.R. and C.B. contributed equally to this study.
HUMAN GENE THERAPY 21:1–16 (September 2010)ª Mary Ann Liebert, Inc.DOI: 10.1089/hum.2009.225
1

CF gene therapy, however, has failed until now because ofmultiple technical problems, among them gene silencing ofcDNA constructs and lack of stability. Thus, for successfulgene therapy, with persistent, tissue-specific expression ofthe transgene, delivery of the complete genomic DNA locusincluding native regulatory and promoter elements shouldbe achieved. This in turn also involves other technical diffi-culties, such as construct size. The primary transcript of theCFTR gene is ~189 kb long (Rommens et al., 1989) andcomprises 27 exons. The adjacent genes, GASZ (germ line-specific protein with ankyrin repeats, sterile a motif, basicleucine zipper) and CORTBP2 (widely expressed cortactinbinding protein-2), show different nuclear localization inhuman cells, depending on their differently regulated ex-pression (Zink et al., 2004; Sadoni et al., 2008), and thereforesequences of these genes are unlikely to belong to the chro-matin domain that regulates CFTR. Because the distancebetween the genomic regions of the primary transcripts ofthese adjacent genes is 283 kb, the size of the functional CFTRlocus should be between 189 and 283 kb. The gene order andexon structure around the CFTR locus is highly conserved invertebrates (Sadoni et al., 2008), bringing into question whythis order was maintained for more than 500 million years inthe absence of gene regulatory constraints. It is presently notknown whether all intronic and extragenic sequences of theCFTR locus are required for normal gene expression or playa role in locus stability.
To avoid random integration into the host chromosomesand allow stable inheritance during somatic cell divisions,our goal here is to achieve incorporation of the completeCFTR genomic locus into a human artificial chromosome(HAC). This requires additional genetic elements, the mostimportant being a functional centromere. HACs based oneither centromeric a-satellite DNA (i.e., long arrays of tan-dem repeats >80 kb) as the only human component in acircular P1 phage-based artificial chromosome (PAC), or onlinear, telomerized a-satellite DNA have been shown tofaithfully replicate and segregate during mitosis for manycell divisions in the absence of selection (Ebersole et al., 2000;Grimes et al., 2001).
In addition to a functional centromere, the vectors to betransfected into CFTR-expressing epithelial cells should alsocarry telomeres and should show correct splicing from thede novo formed HAC for correct expression of the transgene.P1- and F factor-based bacterial artificial chromosomes (PACs/BACs) show high stability, have a relatively large insertcapacity, and yield a large amount of DNA per culture vol-ume, making them ideal for cloning large genomic sequences(Shizuya et al., 1992; Ioannou et al., 1994). With the currentsequence information available for most BAC and PACclones, these artificial chromosomes have become a popularresource to generate artificial chromosome-based transgenes.
The de novo formation of HACs after transfer of nakedDNA molecules is a poorly understood process of DNA as-sembly, concatemerization, and chromatinization leading tonovel, individual chromosomes in the recipient cells. Co-transfections of gene loci and centromeres are usually inef-ficient, but in combination with intact DNA preparations canrepresent a workable strategy to characterize the function ofa cloned DNA fragment and determine its suitability for thefurther prefabrication of an HAC construct. Some of thede novo assembled structures may contain all transferred
sequences in a suitable composition to constitute a functionalHAC. This should include open chromatin regions for theregulated expression as well as a specialized open chromatindomain and replicating portions of heterochromatin forfaithful centromere formation (Nakano et al., 2008). Activecentromeres are marked by the histone H3 variant CENP-A,and need to be protected from adjacent gene expression.
We have previously reported the genomic PAC constructCGT21 containing about one-half of the CFTR gene locus anda tagged last exon, and demonstrated that it was stablypropagated in lung sarcoma cells, where it was expressedand correctly spliced (Laner et al., 2005). Thus, the next log-ical step was to generate a tagged genomic CFTR constructcarrying all 27 exons and flanking regulatory sequences forincorporation into an HAC. We describe herein ligation ofsuch a PAC construct, termed CF225, using previouslycharacterized DNA preparations of two resource PACs. Athird resource to study incorporation of a large version of aCFTR locus on an HAC was BAC clone 5A containing theentire wild-type (wt) sequence of a previously described,310 kb-sized insert of yeast artificial chromosome (YAC)37AB12 (Anand et al., 1991). This was shown to generate fulllevels of copy number-dependent expression and to correctthe detected features of the phenotype in CF null mice, andhas been recombined into a preexisting minichromosomeand expressed in Chinese hamster ovary (CHO) cells(Manson et al., 1997; Vassaux et al., 1997; Auriche et al., 2002).
To achieve incorporation of the CFTR locus into HACs, wecotransfected these three cloned CFTR loci with centromere-proficient a-satellite constructs and analyzed their expres-sion. To our knowledge, this is the first report of thesuccessful de novo formation of an HAC containing the entireCFTR locus.
Materials and Methods
PAC/BAC clones covering the CFTR locus
Details of the isolation of clones carrying the CFTR locusfrom PAC library RPCIP704 (Ioannou et al., 1994), thesequencing procedure, and primers are available online(Ramalho et al., 2004). The ends of PAC CF1 were mapped bypulsed-field gel electrophoresis (PFGE) and amplified withprimers In9F and T7 (30) or directly sequenced with PAC endprimer SP6 (50). The ends of PAC CF6 were mapped withprimers P77 and CF12R (50) and primers aMF and P86 in along PCR (30). For construction of the intermediate constructCF1-Met, primers MetF and MetR (Table 1) were used for theV470M exchange by site-directed mutagenesis in a plasmidcontaining an intron 9 and exon 10 portion during con-struction of CGT21, and primers E10XcF and CF10NMR(Table 1) for the introduction of the silent XmaI variant andintronic NotI site for PAC ligation. Introduced primersand PCR products were sequenced in the resulting clones.Construct CGT21 (EMBL/GenBank accession numberBN000167), and the centromere construct B2T8, containing a190 kb-sized a-satellite array of chromosome 7 in vectorpTAT-BS (17 kb), or TTE1 containing a 116 kb-sized a-satellite array of chromosome 5 in vector pTT (26 kb), aredescribed elsewhere (Laner et al., 2005). BAC 5A was gen-erated from a circularized YAC stuffed with a BAC vector inyeast cells. Intact BAC DNA was purified from yeast cells inagarose plugs and electroporated into DH10B (recA-deficient
2 ROCCHI ET AL.

Escherichia coli strain of large insert BACs/PACs) cells, re-sulting in the master culture EC100 cCFTR 5A. Insert endswere mapped in two single cell-derived sublines 5A a and b,using primers contYAC and 50CF (50) and primers 30CF andprim2. PCR products were sequenced and compared to thehuman genome sequence (build 37.1) using blastn at NCBI.The expected size of the insert was confirmed by pulsed-fieldgel analysis in both subclones.
Escherichia coli growth and agarose plug preparation
Escherichia coli DH10B strain [F mcrA D(mrr-hsdRMS-mcrBC) (F80dlacZDM15) DlacX74 deoR recA1araD139 D(ara-leu)7697 galU galK rpsL (SmR) endA1 l nupG] was grown inLB broth or agar medium. PAC clones were selected withkanamycin (30mg/ml). Telomerized PAC clones B2T8 andTTE1 were selected with both kanamycin (30 mg/ml) andampicillin (50 mg/ml), and CGT21 with both plus tetracycline(4 mg/ml). BAC 5A was selected with chloramphenicol(12.5 mg/ml).
For large-scale growth simulation, single-cell subclones 2,3, and 5 of the master culture of CF225 were established onday 1, grown in 50-ml LB cultures at 378C, and tested forsequence-tagged site (STS) content (see Results) with follow-ups during subsequent growth phases, indicating full sta-bility. For the prolonged growth periods, the subclones weregrown in 1 liter of rich, buffered LB medium at 308C, fromwhich agarose plugs containing ~1015 cells were prepared onday 5. Subsequently, 1 ml of subclones 2 and 3 was trans-ferred to 1 liter of fresh medium every other day, resulting ina theoretical number of ~1021 and ~1027 bacteria on days 9and 13, respectively, when agarose plugs were prepared.
Preparation of intact DNA
Long DNA preparations in agarose plugs were carried outaccording to the protocol of Smith and colleagues (1988)supplemented by purification steps to either remove linearyeast chromosomes or E. coli fragments and damaged DNAfractions from the circular DNA preparations as described(Schindelhauer and Cooke, 1997).
Construction of CF225
In-gel restriction digestion and dephosphorylation. Forpartial digestion of CF1-Met PAC clones, 1 ml of NotI bufferwith 150 U of enzyme (New England BioLabs, Ipswich, MA)was added to 7 agarose plugs (~20 mg) whereas for total di-gestion of CF6, 600 U of enzyme in 500 ml of reaction bufferwith 1� bovine serum albumin (BSA) was added directlyonto a slice containing ~20 plug equivalents (~100 mg in2 ml). The plugs were left overnight at 08C. The next day,another 200 U of NotI was added directly on the CF6 slice.Digestions were carried out for 1 hr at 378C for the CF1-Metplugs and for 4 hr at 378C in a wet chamber for the CF6plugs. The partial digestion of the CF1-Met PACs wasstopped on ice by adding 10 vol% of 0.5 M EDTA, pH 7.9,and later NDS buffer (0.5 M EDTA, pH 9; 1% N-laur-oylsarcosine) containing proteinase K (10 mg/ml), incubatedat 558C overnight, whereas CF6 was immediately depho-sphorylated as follows. After removal of NotI buffer, the CF6plug slice was washed with bidistilled water, placed on ice,and incubated with 460 ml of 5� calf intestine phosphatase
(CIP) buffer containing 20 ml of CIP (New England BioLabs),which was directly added onto the plug slice and incubatedfor 2 hr. Incubation for 30 min at 378C was stopped on ice,using 250ml of 0.5 M EDTA, pH 7.9. CIP was inactivatedovernight with NDS buffer containing proteinase K (10 mg/ml) at 558C in a humid chamber. After equilibrating in 0.5�TAE, the slice was loaded on a 1% agarose pulsed-field gel.
PFGE separation and DNA isolation. After a 20-hr run at6 V/cm with a switch time of 1–30 sec in 0.5� TAE at 128C(CHEF DRII; Bio-Rad, Hercules, CA), the PAC DNA bandswere cut from the gel without UV exposure, and kept at 08C.DNA was recovered from gel slices by electroelution with aBioTrap BT1000 (Biometrics, Schleicher & Schuell, Dassel,Germany) placed in a CHEF DRII or III PFGE chamber in0.5�TAE.
Ligation and electroporation. The ligation reaction wascarried out with 200ml of eluate, 1� T4 DNA ligase buffer,and 20 ml of T4 DNA ligase (New England BioLabs), at 128C,overnight. Reaction mixtures without ligase were used ascontrols. Four microliters of the ligation reaction was addedto 5 ml of electrocompetent E. coli DH10B cells (ElectroMAXDH10B cells; Invitrogen, Carlsbad, CA) in a volume of 80 mlof bidistilled water and electroporated (Gene Pulser II;Bio-Rad) at 1.2–1.4 kV, 100O, and 25 mF using 0.1-cm gapcuvettes (Bio-Rad) chilled on ice. Warm SOC medium(500ml) was immediately added to the cuvette and the con-tent was transferred to sterile 10-ml white cap tubescontaining 5 ml of LB medium for 1 hr of growth at 378C withmoderate shaking. Spun bacteria were spread on LB agarcontaining kanamycin at 30mg/ml (Sigma, Munich, Germany),and incubated for a minimum of 24 hr at 378C.
Escherichia coli screening. Individual colonies werepicked into 30 ml of TTE buffer (0.01% Triton X-100, 20 mMTris-HCl [pH 8], 2 mM EDTA [pH 8]), heated for 1 min at958C, and spun for 15 min at 10,000 rpm. Two microliters ofsupernatant was used in a final volume of 50ml for a PCR of30 cycles with an annealing temperature of 558C with primerpair CFi10fus/R7 (Table 1). Two microliters of ligation re-action was used for the positive control. Aliquots of the PCRproduct were run on a 1% standard plus 1% low meltingpoint agarose gel.
Very long PCR. Agarose plug sections with purified,intact PAC/BAC template of ~10 ml were added to a totalreaction volume of 50 ml containing tuning buffer (1� Ep-pendorf ), 0.5 mM dNTPs, 0.5 ml Mg(OAc)2, 0.2 mM primers,2 U of Taq polymerase, and 2 U of TripleMaster enzyme mix(Eppendorf ). The program has an initial denaturation at928C for 30 sec, 6–9 cycles of denaturation for 12 sec, an-nealing at 60–648C for 1 min, and elongation at 668C for 15–45 min (20–60 kb), and a final extension at 668C for 5 min. Tento 20ml of the gellike product (0.2% LMP agarose) is mixedwith 10ml of loading buffer (Ficoll) and analyzed on apulsed-field gel.
Generation of stable HT1080 lines. Linearized DNA wasisolated with appropriate restriction enzymes (New EnglandBioLabs). NotI was used for TTE1 and CGT21, I-SceI for B2T8,and SalI for BAC 5A and CF225 inserts. PFGE separation,
CFTR-HAC 3

Ta
bl
e1.
Ol
ig
od
eo
xy
nu
cl
eo
tid
es
Use
din
St
ud
y
Pri
mer
Seq
uen
ce(50 -
30)
Loc
atio
nR
efer
ence
Use
dfo
r:
P77
-BG
GT
CG
AG
CT
TG
AC
AT
TG
TA
GG
pC
YP
AC
2N(r
ever
se)
Th
isw
ork
PC
R,
end
Seq
uen
cin
gP
86p
CY
PA
C2N
Th
isw
ork
En
dm
app
ing
T7
TA
AT
AC
GA
CT
CA
CT
AT
AG
GG
pC
YP
AC
2NS
chin
del
hau
eran
dS
chw
arz,
2002
;L
aner
etal
.,20
05P
CR
R7
CC
TC
TC
CC
TA
TA
GT
GA
GT
CG
pC
YP
AC
2N(r
ever
seco
mp
lem
ent
of
T7)
Th
isw
ork
PC
R
SP
6A
TT
TA
GG
TG
AC
AC
TA
TA
Gp
CY
PA
C2N
An
and
etal
.,19
91L
R-P
CR
En
dse
qu
enci
ng
Co
ntY
AC
TC
TC
GG
TA
GC
CA
AG
TT
GG
TT
YA
Cle
ftar
mT
his
wo
rkE
nd
map
pin
g/
seq
uen
cin
gL
R-P
CR
Pri
mer
2C
CG
GA
AT
TC
CG
GA
CC
GC
CG
CC
GC
AA
GG
AA
TG
GY
AC
rig
ht
arm
Th
isw
ork
En
dm
app
ing
/se
qu
enci
ng
LR
-PC
RC
F1-
5RG
TT
CC
AA
TT
CT
AT
AA
GA
TT
AT
CA
GC
FT
R50
reg
ion
of
CF
1T
his
wo
rkL
R-P
CR
En
dm
app
ing
/se
qu
enci
ng
50C
FC
CC
AT
GG
TG
GA
AT
AA
AG
TA
CC
FT
R50
reg
ion
of
5AT
his
wo
rkE
nd
map
pin
g/
seq
uen
cin
gL
R-P
CR
CF
-37F
CG
TG
TT
AG
GC
TG
AT
TT
TG
CA
GC
CF
TR
50re
gio
nT
his
wo
rkL
R-P
CR
CF
-37R
CG
AC
CA
GC
AC
AT
AA
CA
AC
TC
AG
CC
FT
R50
reg
ion
Th
isw
ork
LR
-PC
RC
F-1
8FG
AT
GT
CC
TG
CA
AC
TG
GC
AG
AG
CF
TR
50re
gio
nT
his
wo
rkL
R-P
CR
CF
-18R
CA
GA
GA
TC
TA
CA
TG
TG
AG
GG
CC
FT
R50
reg
ion
Th
isw
ork
LR
-PC
RC
F-1
FC
TC
AG
AG
AG
TT
GA
AG
AT
GG
CG
CF
TR
50re
gio
nT
his
wo
rkL
R-P
CR
CF
-1R
GT
GA
GT
GA
AC
TC
CA
AG
GG
TG
GC
FT
R50
reg
ion
Th
isw
ork
LR
-PC
RA
1RC
GA
GA
GA
CC
AT
GC
AG
AG
GT
CC
FT
Rex
on
1T
his
wo
rk;
Lan
eret
al.,
2005
RT
-PC
R
CF
3FC
TT
GG
GT
TA
AT
CT
CC
TT
GG
AC
FT
Rin
tro
n2
Th
isw
ork
;Z
iele
nsk
iet
al.,
1991
LR
-PC
R
CF
3RA
TT
CA
CC
AG
AT
TT
CG
TA
GT
CC
FT
Rin
tro
n3
Th
isw
ork
;Z
iele
nsk
iet
al.,
1991
LR
-PC
R
CF
c3F
GG
GA
TA
GA
GA
GC
TG
GC
TT
CC
FT
Rex
on
3T
his
wo
rkR
T-P
CR
Seq
uen
cin
gC
F4F
TC
AC
AT
AT
GG
TA
TG
AC
CC
TC
CF
TR
intr
on
3T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
4RT
TG
TA
CC
AG
CT
CA
CT
AC
CT
AC
FT
Rin
tro
n4
Th
isw
ork
;Z
iele
nsk
iet
al.,
1991
PC
R
CF
7bF
AG
AC
CA
TG
CT
CA
GA
TC
TT
CC
AT
CF
TR
intr
on
6T
his
wo
rk;
Zie
len
ski
etal
.,19
91L
R-P
CR
CF
7bR
GC
AA
AG
TT
CA
TT
AG
AA
CT
GA
TC
CF
TR
intr
on
7T
his
wo
rk;
Zie
len
ski
etal
.,19
91L
R-P
CR
B3F
AA
TG
TA
AC
AG
CC
TT
CT
GG
GA
GC
FT
Rex
on
8R
amal
ho
etal
.,20
02R
T-P
CR
Seq
uen
cin
g
4

In9F
CA
AG
TA
GC
AG
GT
GA
AG
CA
AG
TG
CC
FT
Rin
tro
n9
Th
isw
ork
PC
R Seq
uen
cin
gG
FP
1-A
LG
AA
CT
TC
AG
GG
TC
AG
CT
TG
CG
T21
,C
FT
R/
EG
FP
fusi
on
exo
n10
Lan
eret
al.,
2005
RT
-PC
RS
equ
enci
ng
CF
10F
GC
AG
AG
TA
CC
TG
AA
AC
AG
GA
CF
TR
intr
on
9/ex
on
10T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
10R
CA
TT
CA
CA
GT
AG
CT
TA
CC
CA
CF
TR
intr
on
10T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
LR
-PC
R
Met
Fa
GA
CT
TC
AC
TT
CT
AA
TG
AT
GA
TT
AT
GG
GA
GA
AC
TG
GC
FT
Rin
tro
n10
Th
isw
ork
PC
R,
V47
0Mex
chan
ge
Met
Ra
CC
AG
TT
CT
CC
CA
TA
AT
CA
TC
AT
TA
GA
AG
TG
AA
GT
CC
FT
Rex
on
10T
his
wo
rkP
CR
,V
470M
exch
ang
eE
10X
cFa
GG
AT
TA
TG
CC
CG
GG
AC
CA
TT
AA
AG
AA
AA
TA
TC
AT
CT
TT
GG
syn
thet
ic/
CF
TR
,ex
on
10T
his
wo
rkP
CR
,X
maI
site
CF
10N
MR
aC
GG
TG
GT
AC
GC
GT
GC
GG
CC
GC
CC
TA
AC
AT
TT
AC
AG
CA
AT
AA
syn
thet
ic/
CF
TR
,ex
on
10T
his
wo
rkP
CR
,N
otI/
Mlu
Isi
teC
16D
GT
TG
GC
AT
GC
TT
TG
AT
GA
CG
CT
TC
CF
TR
exo
n10
(rev
erse
)R
amal
ho
etal
.,20
02P
CR
Seq
uen
cin
gR
T-P
CR
CF
i10f
us
GT
GA
CT
GC
AA
TT
CT
TT
GA
TG
CC
FT
Rin
tro
n10
Th
isw
ork
PC
RS
equ
enci
ng
CF
11F
CA
AC
TG
TG
GT
TA
AA
GC
AA
TA
GT
GT
CF
TR
intr
on
10T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
11R
GC
AC
AG
AT
TC
TG
AG
TA
AC
CA
TA
AT
CF
TR
intr
on
11T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
Seq
uen
cin
g
CF
12F
GT
GA
AT
CG
AT
GT
GG
TG
AC
CA
CF
TR
intr
on
11T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
12R
CT
GG
TT
TA
GC
AT
GA
GG
CG
GT
CF
TR
intr
on
12T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
14cR
GT
AT
GT
GT
TC
CA
TG
TA
GT
CA
CT
GC
CF
TR
exo
n14
Th
isw
ork
LR
-PC
RC
F14
iFT
TC
TA
TG
GA
TC
AT
GA
GC
AG
CC
AC
CF
TR
intr
on
14T
his
wo
rkL
R-P
CR
CF
17b
FT
TC
AA
AG
AA
TG
GC
AC
CA
GT
GT
CF
TR
intr
on
17a
Th
isw
ork
;Z
iele
nsk
iet
al.,
1991
PC
R
CF
17b
RA
TA
AC
CT
AT
AG
AA
TG
CA
GC
AC
FT
Rin
tro
n17
bT
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
19cF
GC
AA
AA
TA
CA
CA
GA
AG
GT
GG
AA
AT
GC
CC
FT
Rex
on
19T
his
wo
rkL
R-P
CR
CF
21F
AA
TG
TT
CA
CA
AG
GG
AC
TC
CA
CF
TR
intr
on
20T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
21R
CA
AA
AG
TA
CC
TG
TT
GC
TC
CA
CF
TR
intr
on
21T
his
wo
rk;
Zie
len
ski
etal
.,19
91P
CR
CF
aFC
TA
GG
GT
GA
TA
TT
AA
CC
AG
GG
CF
TR
po
ly(A
)re
gio
nT
his
wo
rkP
CR
CF
aRG
AG
GC
TT
GA
AG
AC
AT
TA
TG
CT
AG
CF
TR
po
ly(A
)re
gio
nT
his
wo
rkP
CR
aM2R
GT
TG
CA
GC
GC
TA
TG
AG
GC
TT
GA
AG
AC
AT
TA
TG
CT
AG
syn
thet
ic/
CF
TR
,þ
2k
b(s
top
)T
his
wo
rkC
GT
21co
nst
ruct
ion
LR
-PC
RC
F6-
4FC
TC
TG
TG
AA
GG
AG
GT
TC
TA
AG
AA
CC
FT
Rþ
9.5
kb
(sto
p)
Th
isw
ork
LR
-PC
RE
nd
seq
uen
cin
gC
Fþ
23F
TG
TC
TA
CT
GA
GC
AG
AG
GT
AG
CC
FT
Rþ
23k
b(s
top
)T
his
wo
rkL
R-P
CR
CFþ
23R
GT
AG
CA
GA
GT
CT
AG
AA
TT
AG
TC
CC
FT
Rþ
23k
b(s
top
)T
his
wo
rkL
R-P
CR
30C
FC
TC
TG
TG
AT
TC
AC
AC
TG
AA
AG
CF
TRþ
51k
b(s
top
)T
his
wo
rkE
nd
map
pin
g/
seq
uen
cin
gL
R-P
CR
AcF
GC
AC
TC
TT
CC
AG
CC
TT
CC
b-A
ctin
Th
isw
ork
;L
aner
etal
.,20
05R
T-P
CR
AcR
AG
AA
AG
GG
TG
TA
AC
GC
AA
CT
AA
Gb-
Act
inT
his
wo
rkR
T-P
CR
aM
od
ified
pri
mer
s.
5

excision without UV illumination, and electroelution was asdescribed (Laner et al., 2005) with the modification of placingthe BioTrap elution chamber in a CHEF DRII apparatus.
Cell culture. HT1080 cells (pseudodiploid, epitheloid fi-brosarcoma of the lung) were grown in Dulbecco’s modifiedEagle’s medium (DMEM; PAA Laboratories, Pasching,Austria) supplemented with 10% fetal calf serum (FCS; PAALaboratories), 1% Glutamine 100� (GIBCO, Karlsruhe,Germany), and 2% (v/v) penicillin–streptomycin (GIBCO) at378C and 5% CO2.
Lipofection. Ten-centimeter tissue culture plates con-taining ~50% confluent HT1080 cells were washed withphosphate-buffered saline (PBS; PAA Laboratories). For eachplate, 6ml of Lipofectamine 2000 reagent (Invitrogen) and294 ml of Opti-MEM I (GIBCO) were mixed and incubated for5 min at room temperature.
Variable volumes of DNA eluate were gently mixed withOpti-MEM, resulting in a volume of 300ml, which was gentlyadded to the Lipofectamine/Opti-MEM mixture. The tubewas turned twice and incubated for 20 min at room tem-perature. The solution was directly added onto PBS-rinsedcells, using wide-bore plastic Pasteur pipettes to avoid DNAshearing. Plates were incubated at 378C for 12 hr and the cellswere washed with medium and incubated for 1 day withoutselection. Medium supplemented with blasticidin S (BS,4 mg/ml; InvivoGen, Toulouse, France) was added on day 3and changed every other day.
Clone expansion and isolation. Transfected plates werescreened by bright-field and fluorescence microscopy withan Axiovert 10 (Zeiss, Oberkochen, Germany) in a dark roomequipped with a 100-W Hg lamp and filters for blue excita-tion and green detection of enhanced green fluorescentprotein (EGFP). Individual BS-resistant cell clones wereisolated with cloning rings, and expanded in 12-well dishes,25- and 75-cm2 flasks for PCR screening, growth on and offselection, DNA extraction, RNA extraction, freezing, andfluorescence in situ hybridization (FISH) analyses. DNA forPCR screening was extracted with lysis buffer (0.1 M Tris[pH 8.3], 5 mM EDTA, 0.2 M NaCl, 0.2% sodium dodecylsulfate [SDS], proteinase K [10mg/ml]) and ethanol precipi-tation.
Expression analysis and sequencing
RT-PCR. Total RNA was isolated from confluent BS-resistant cells grown in T25 flasks, using TRIzol reagent(Invitrogen) according to the manufacturer’s protocol, withadditional DNase digestion (Ambion, Austin, TX). The re-verse transcription (RT) reaction was also performed ac-cording to the Invitrogen protocol, using a SuperScript IIIone-step RT-PCR kit. Primer sequences used for RT-PCR(polymerase chain reaction with a reverse-transcribed cDNAtemplate) are shown in Table 1. The efficiency of RNA ex-traction and the RNA levels in the various samples werecontrolled by RT-PCR with 1ml of each RNA sample toamplify a b-actin fragment of 385 bp, using primers AcF andAcR (Table 1). Primers A1R/C16D were used to amplify a1584-bp product of wt CFTR and primers A1R/GFP1-AL fora 1668-bp product specific for CGT21. Primers B3F/C16D
were used to amplify a 391-bp product indicating correctsplicing, and a minor product of 208 bp indicating skippingof exon 9, or primers B3F/GFP1-AL resulting in a 474-bpproduct specific for CGT21, also showing a minor band of291 bp indicating exon 9 skipping. B3F/C16D were also usedfor analyzing a silent XmaI variant present in transgeneCF225 as follows. To distinguish CF225 and endogenousCFTR transcripts in the respective lines, 15ml of each RT-PCRwas digested in a 25-ml reaction with 20 U of XmaI, resultingin 310- and 81-bp fragments derived of the transgene (and areduction of the minor 208-bp band to 121 bp from exon 9skipping of the transgene). Fragments were analyzed on 1%standard plus 1% low melting point agarose gels and imageswere registered on an UV Gene Genius bioimaging systemdigital analyzer (Syngene, Cambridge, UK). RT-PCR prod-ucts corresponding to the main transcript (without exon 9skipping) were cut from agarose gels, purified, and se-quenced.
FISH and immuno-FISH analyses
Fluorescence in situ hybridization (FISH) was carried outas described (Laner et al., 2004) after 30 days on and off BSselection. After initial screening, a minimum of 20 (mostly>30)metaphases were analyzed for each growth phase. Theprobes used were rsf for the vector sequence including the BSmarker (Laner et al., 2005) labeled by PCR with biotin-16-dUTP (Roche, Mannheim, Germany) and Cy3.5 avidin, nick-translated PAC inserts CF1 (biotin-16-dUTP, Cy3.5) and CF6(digoxigenin-11-dUTP, fluorescein isothiocyanate [FITC]),nick-translated 2.6 kb EcoRI repeats excised of PAC B2 con-taining the cen17 a-satellite array (digoxigenin-11-dUTP,FITC), and a PCR-generated, nick-translated probe E1 ofvector TTE1 (diethylaminocoumarin [DEAC]), hybridizing tocen5 and a-satellite arrays on chromosomes 1 and 19. Signalswere visualized with an Axiovert 200 microscope (Zeiss,Oberkochen, Germany) and digitally captured. Triple-colorFISH and immuno-FISH analyses were carried out as de-scribed. Briefly, for immuno-FISH, CENP-A antibodies(Valdivia et al., 1998) were bound to unfixed, cytospunchromosomes and stained with Cy3.5 (red) before fixation,followed by fixation and FISH analysis with probes E1(DEAC, pink) and the CFTR region of PAC CF6 (DIG,green).
Results
Construction of the fusion PAC CF225from characterized resource clones
Here, we aimed at constructing a tagged version of theentire genomic CFTR gene cloned in a P1 phage- or F factor-based artificial chromosome suitable for large-scale, high-quality DNA preparation. In addition to the previouslydescribed construct CGT21 carrying a tagged half-locus(Laner et al., 2005) and BAC 5A with the YAC-derived wtinsert (Anand et al., 1991), we constructed PAC CF225 car-rying the human CFTR gene with all exons and introns plusregulatory sequences by ligating two PACs, CF1-Met (i.e.,with M at the M470V locus) and CF6, each containingroughly half of the CFTR gene and flanking regions (Fig. 1).PAC CF1-Met carries an insert running from �60.7 kb up-stream from the start of translation in exon 1 to intron 10,
6 ROCCHI ET AL.

and PAC CF6 carries an insert running from intron 10 toþ9.7 kb (relative to the end of translation) of downstreamDNA. Because of the choice of the resource PACs, both in-trons 9 and 10 are substantially shortened by 5.1 and 27.1 kb,respectively (Fig. 1A). As a consequence, the describedDNase I-hypersensitive sites (DHS) in intron 10 (McCarthyand Harris, 2005) are excluded from locus CF225.
The nine PAC clones, CF1–CF10 (Fig. 1C), covering thehuman CFTR gene locus (a resource from the Human Gen-ome Project) were analyzed here for exon content by PCR.Because approximately 1 in 25 white individuals carries aCFTR mutation, and functionally relevant polymorphismsexist, we sequenced all exons and splice junctions of the re-source PACs before construction. PCR primers and se-quencing data are summarized online (Ramalho et al., 2004)and sequences are available under EMBL/GenBank acces-sion numbers AJ574939–AJ575055. We found that PACsCF1–CF5 contained the common splice variant (TG)11T7
described to result in a small proportion of an alternativelyspliced product lacking exon 9 (Cuppens et al., 1998;Ramalho et al., 2002), but mostly producing a normal tran-script. Exon 10 of PACs CF3–CF5 was found to encode valineat amino acid position 470, which we exchanged to methio-nine during construction of CF1-Met PAC, giving rise to a1.7-fold higher chloride conductance activity (Cuppens et al.,1998). Construction of CF1-Met PAC required a number ofengineering steps. In short, we used here again a stored DNApreparation of a cloning intermediate based on PAC CF1,which was used for the construction of CGT21, a synthetichalf-CFTR locus that was shown to be functional and thatexpressed the intended M470 variant and tag sequences(Laner et al., 2005). Using the silent XmaI restriction variantintroduced into exon 10 downstream of M470, the EGFP/exon 24 portion of CGT21 was replaced by the missing exon10 and flanking intronic sequences by ligating a PCR frag-ment of this region from PAC CF3, resulting in PAC
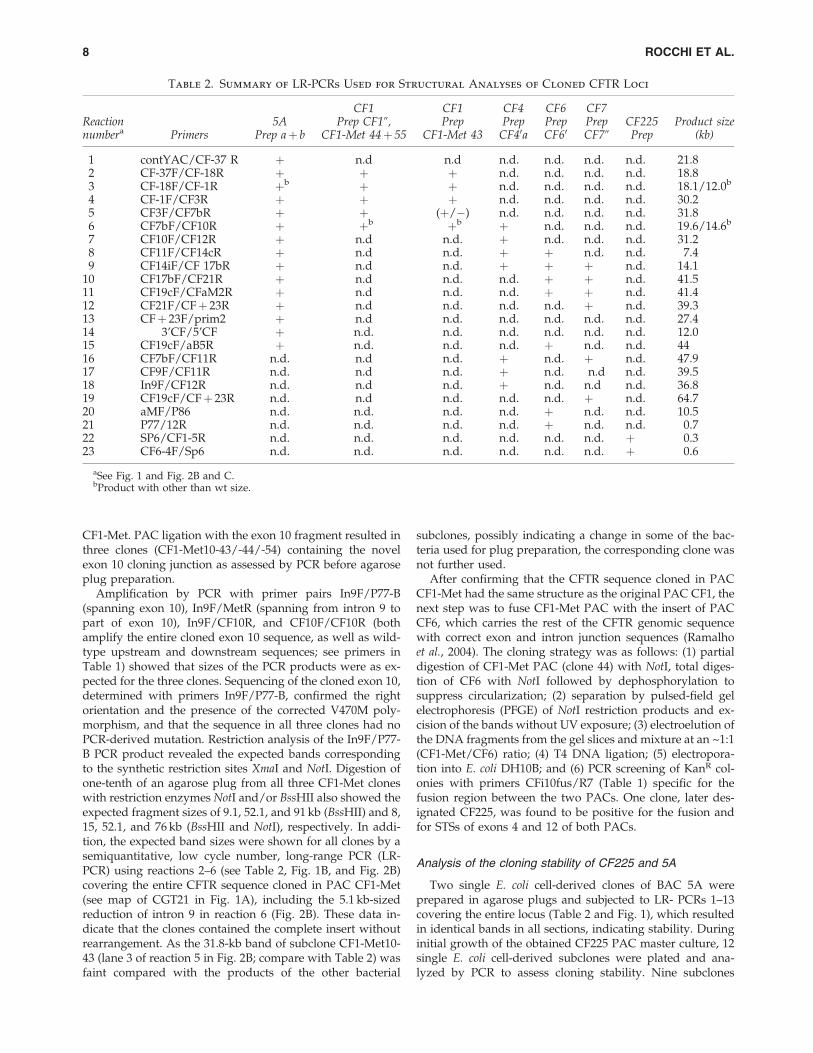
FIG. 1. Schematic of theCFTR locus, constructs andresource clones drawn toscale. (A) The bar shows theposition on the chromosome7 sequence, according tobuild 37.1 of the human ge-nome sequence. The primarytranscripts of the CFTR geneand the differently regulated,adjacent genes GASZ andCORTBP2 are shown withtheir intron and exon struc-ture and transcript direction.The three construct inserts(boldface lines) used in thisstudy are shown and theCFTR exon/intron contentand adjacent regions are gi-ven. CGT21 contains one-halfof the CFTR locus and isbased on PAC CF1, whichwas joined to an engineeredexon 10 (open box) consistingof partial exon 10, EGFP-coding, and partial exon 24sequences, including the stopcodon of CFTR and 0.5-kb 30
region. Regions not coveredby the construct are joined bythin lines. Intron 9 of con-struct CGT21 lacks 5.1 kb,which was known not toimpair expression and correct splicing (Laner et al., 2005). Construct CF225 is based on a modified PAC CF1 (CF1-Metrequired during construction of CGT21) and CF6, which were joined with an engineered exon 10 sequence including splicejunctions. Intron 9 lacks a 5.1-kb region and intron 10 lacks a 27.1-kb region not present in the chosen resource PACs. BothCGT21 and CF225 contain an optimized sequence in exon 10 (asterisk) encoding the 470M variant and contain the silent XmaIvariant introduced for the unambiguous transcript detection compared with endogenous human CFTR genes. Construct 5Arepresents the entire wild-type (wt) CFTR locus and extends into adjacent genes, suggesting the presence of all regulatoryregions of the CFTR-related chromatin domain. (B) Shown are 13 long-range PCRs covering the entire CFTR locus, except fora 3-kb gap between reactions 8 and 9 in intron 14. LR-PCR-based fine mapping was used to compare the structure of theCFTR locus cloned in the various PAC and BAC resources (Table 2). All products were obtained from the various sources. Forcomparison, positions of BssHII restriction sites are given. (C) Characterized resource PACs with published exon/intronjunction sequences (Ramalho et al., 2004) covering the CFTR locus.
CFTR-HAC 7
CF1-Met. PAC ligation with the exon 10 fragment resulted inthree clones (CF1-Met10-43/-44/-54) containing the novelexon 10 cloning junction as assessed by PCR before agaroseplug preparation.
Amplification by PCR with primer pairs In9F/P77-B(spanning exon 10), In9F/MetR (spanning from intron 9 topart of exon 10), In9F/CF10R, and CF10F/CF10R (bothamplify the entire cloned exon 10 sequence, as well as wild-type upstream and downstream sequences; see primers inTable 1) showed that sizes of the PCR products were as ex-pected for the three clones. Sequencing of the cloned exon 10,determined with primers In9F/P77-B, confirmed the rightorientation and the presence of the corrected V470M poly-morphism, and that the sequence in all three clones had noPCR-derived mutation. Restriction analysis of the In9F/P77-B PCR product revealed the expected bands correspondingto the synthetic restriction sites XmaI and NotI. Digestion ofone-tenth of an agarose plug from all three CF1-Met cloneswith restriction enzymes NotI and/or BssHII also showed theexpected fragment sizes of 9.1, 52.1, and 91 kb (BssHII) and 8,15, 52.1, and 76 kb (BssHII and NotI), respectively. In addi-tion, the expected band sizes were shown for all clones by asemiquantitative, low cycle number, long-range PCR (LR-PCR) using reactions 2–6 (see Table 2, Fig. 1B, and Fig. 2B)covering the entire CFTR sequence cloned in PAC CF1-Met(see map of CGT21 in Fig. 1A), including the 5.1 kb-sizedreduction of intron 9 in reaction 6 (Fig. 2B). These data in-dicate that the clones contained the complete insert withoutrearrangement. As the 31.8-kb band of subclone CF1-Met10-43 (lane 3 of reaction 5 in Fig. 2B; compare with Table 2) wasfaint compared with the products of the other bacterial
subclones, possibly indicating a change in some of the bac-teria used for plug preparation, the corresponding clone wasnot further used.
After confirming that the CFTR sequence cloned in PACCF1-Met had the same structure as the original PAC CF1, thenext step was to fuse CF1-Met PAC with the insert of PACCF6, which carries the rest of the CFTR genomic sequencewith correct exon and intron junction sequences (Ramalhoet al., 2004). The cloning strategy was as follows: (1) partialdigestion of CF1-Met PAC (clone 44) with NotI, total diges-tion of CF6 with NotI followed by dephosphorylation tosuppress circularization; (2) separation by pulsed-field gelelectrophoresis (PFGE) of NotI restriction products and ex-cision of the bands without UV exposure; (3) electroelution ofthe DNA fragments from the gel slices and mixture at an ~1:1(CF1-Met/CF6) ratio; (4) T4 DNA ligation; (5) electropora-tion into E. coli DH10B; and (6) PCR screening of KanR col-onies with primers CFi10fus/R7 (Table 1) specific for thefusion region between the two PACs. One clone, later des-ignated CF225, was found to be positive for the fusion andfor STSs of exons 4 and 12 of both PACs.
Analysis of the cloning stability of CF225 and 5A
Two single E. coli cell-derived clones of BAC 5A wereprepared in agarose plugs and subjected to LR- PCRs 1–13covering the entire locus (Table 2 and Fig. 1), which resultedin identical bands in all sections, indicating stability. Duringinitial growth of the obtained CF225 PAC master culture, 12single E. coli cell-derived subclones were plated and ana-lyzed by PCR to assess cloning stability. Nine subclones
Table 2. Summary of LR-PCRs Used for Structural Analyses of Cloned CFTR Loci
5ACF1 CF1 CF4 CF6 CF7
CF225 Product sizeReactionnumbera Primers Prep aþ b
Prep CF1@,CF1-Met 44þ 55
PrepCF1-Met 43
PrepCF40a
PrepCF60
PrepCF7@ Prep (kb)
1 contYAC/CF-37 R þ n.d n.d n.d. n.d. n.d. n.d. 21.82 CF-37F/CF-18R þ þ þ n.d. n.d. n.d. n.d. 18.83 CF-18F/CF-1R þb þ þ n.d. n.d. n.d. n.d. 18.1/12.0b
4 CF-1F/CF3R þ þ þ n.d. n.d. n.d. n.d. 30.25 CF3F/CF7bR þ þ (þ/�) n.d. n.d. n.d. n.d. 31.86 CF7bF/CF10R þ þb þb þ n.d. n.d. n.d. 19.6/14.6b
7 CF10F/CF12R þ n.d n.d. þ n.d. n.d. n.d. 31.28 CF11F/CF14cR þ n.d n.d. þ þ n.d. n.d. 7.49 CF14iF/CF 17bR þ n.d n.d. þ þ þ n.d. 14.1
10 CF17bF/CF21R þ n.d n.d. n.d. þ þ n.d. 41.511 CF19cF/CFaM2R þ n.d n.d. n.d. þ þ n.d. 41.412 CF21F/CFþ 23R þ n.d n.d. n.d. n.d. þ n.d. 39.313 CFþ 23F/prim2 þ n.d n.d. n.d. n.d. n.d. n.d. 27.414 3’CF/5’CF þ n.d. n.d. n.d. n.d. n.d. n.d. 12.015 CF19cF/aB5R þ n.d. n.d. n.d. þ n.d. n.d. 4416 CF7bF/CF11R n.d. n.d n.d. þ n.d. þ n.d. 47.917 CF9F/CF11R n.d. n.d n.d. þ n.d. n.d n.d. 39.518 In9F/CF12R n.d. n.d n.d. þ n.d. n.d n.d. 36.819 CF19cF/CFþ 23R n.d. n.d n.d. n.d. n.d. þ n.d. 64.720 aMF/P86 n.d. n.d. n.d. n.d. þ n.d. n.d. 10.521 P77/12R n.d. n.d. n.d. n.d. þ n.d. n.d. 0.722 SP6/CF1-5R n.d. n.d. n.d. n.d. n.d. n.d. þ 0.323 CF6-4F/Sp6 n.d. n.d. n.d. n.d. n.d. n.d. þ 0.6
aSee Fig. 1 and Fig. 2B and C.bProduct with other than wt size.
8 ROCCHI ET AL.
contained the eight tested PCR STSs covering the locus atpositions �18 kb (CF-18F/R), �1 kb (CF-1F/R), exon 3(CF3F/R), exon 10 corrected/fusion (CFi10fus/R7), exon 11(CF11F/R), exon 17 (CF17bF/R), exon 21 (CF21F/R), and thepoly(A) region (CFaF/R; primer sequences are described inTable 1). To further analyze cloning stability, three singlecell-derived cultures of PAC CF225 were grown for varioustime periods simulating a potential final yield of 1015, 1021,and 1027 E. coli cells, each containing up to ~10 completelyreplicated copies of the construct (Ioannou et al. 1994). ADNA yield of a 1015 cell preparation represents a normallaboratory scale required for functional testing. The 1021 scalemay represent the upper range required for extensive testingin a multicenter gene therapy trial. Larger cell numbers couldperhaps represent the range of a continuous production of aDNA-based drug for the clinical setting.
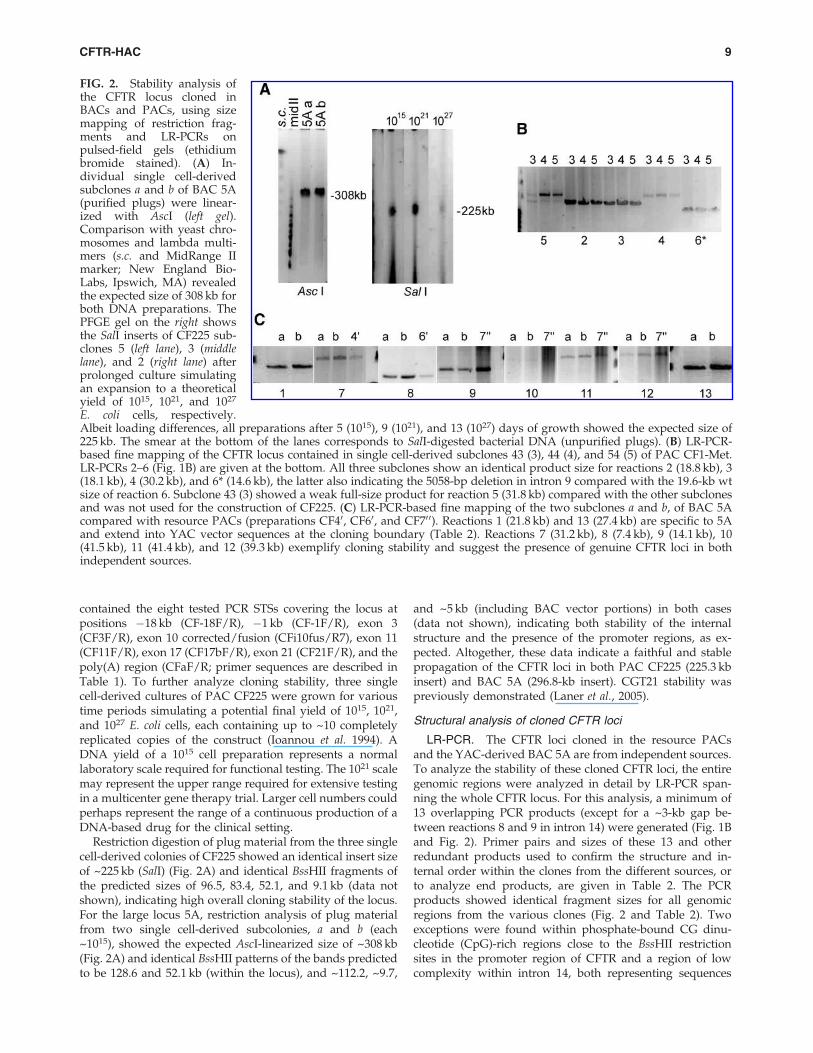
Restriction digestion of plug material from the three singlecell-derived colonies of CF225 showed an identical insert sizeof ~225 kb (SalI) (Fig. 2A) and identical BssHII fragments ofthe predicted sizes of 96.5, 83.4, 52.1, and 9.1 kb (data notshown), indicating high overall cloning stability of the locus.For the large locus 5A, restriction analysis of plug materialfrom two single cell-derived subcolonies, a and b (each~1015), showed the expected AscI-linearized size of ~308 kb(Fig. 2A) and identical BssHII patterns of the bands predictedto be 128.6 and 52.1 kb (within the locus), and ~112.2, ~9.7,
and ~5 kb (including BAC vector portions) in both cases(data not shown), indicating both stability of the internalstructure and the presence of the promoter regions, as ex-pected. Altogether, these data indicate a faithful and stablepropagation of the CFTR loci in both PAC CF225 (225.3 kbinsert) and BAC 5A (296.8-kb insert). CGT21 stability waspreviously demonstrated (Laner et al., 2005).
Structural analysis of cloned CFTR loci
LR-PCR. The CFTR loci cloned in the resource PACsand the YAC-derived BAC 5A are from independent sources.To analyze the stability of these cloned CFTR loci, the entiregenomic regions were analyzed in detail by LR-PCR span-ning the whole CFTR locus. For this analysis, a minimum of13 overlapping PCR products (except for a ~3-kb gap be-tween reactions 8 and 9 in intron 14) were generated (Fig. 1Band Fig. 2). Primer pairs and sizes of these 13 and otherredundant products used to confirm the structure and in-ternal order within the clones from the different sources, orto analyze end products, are given in Table 2. The PCRproducts showed identical fragment sizes for all genomicregions from the various clones (Fig. 2 and Table 2). Twoexceptions were found within phosphate-bound CG dinu-cleotide (CpG)-rich regions close to the BssHII restrictionsites in the promoter region of CFTR and a region of lowcomplexity within intron 14, both representing sequences
FIG. 2. Stability analysis ofthe CFTR locus cloned inBACs and PACs, using sizemapping of restriction frag-ments and LR-PCRs onpulsed-field gels (ethidiumbromide stained). (A) In-dividual single cell-derivedsubclones a and b of BAC 5A(purified plugs) were linear-ized with AscI (left gel).Comparison with yeast chro-mosomes and lambda multi-mers (s.c. and MidRange IImarker; New England Bio-Labs, Ipswich, MA) revealedthe expected size of 308 kb forboth DNA preparations. ThePFGE gel on the right showsthe SalI inserts of CF225 sub-clones 5 (left lane), 3 (middlelane), and 2 (right lane) afterprolonged culture simulatingan expansion to a theoreticalyield of 1015, 1021, and 1027
E. coli cells, respectively.Albeit loading differences, all preparations after 5 (1015), 9 (1021), and 13 (1027) days of growth showed the expected size of225 kb. The smear at the bottom of the lanes corresponds to SalI-digested bacterial DNA (unpurified plugs). (B) LR-PCR-based fine mapping of the CFTR locus contained in single cell-derived subclones 43 (3), 44 (4), and 54 (5) of PAC CF1-Met.LR-PCRs 2–6 (Fig. 1B) are given at the bottom. All three subclones show an identical product size for reactions 2 (18.8 kb), 3(18.1 kb), 4 (30.2 kb), and 6* (14.6 kb), the latter also indicating the 5058-bp deletion in intron 9 compared with the 19.6-kb wtsize of reaction 6. Subclone 43 (3) showed a weak full-size product for reaction 5 (31.8 kb) compared with the other subclonesand was not used for the construction of CF225. (C) LR-PCR-based fine mapping of the two subclones a and b, of BAC 5Acompared with resource PACs (preparations CF40, CF60, and CF70 0). Reactions 1 (21.8 kb) and 13 (27.4 kb) are specific to 5Aand extend into YAC vector sequences at the cloning boundary (Table 2). Reactions 7 (31.2 kb), 8 (7.4 kb), 9 (14.1 kb), 10(41.5 kb), 11 (41.4 kb), and 12 (39.3 kb) exemplify cloning stability and suggest the presence of genuine CFTR loci in bothindependent sources.
CFTR-HAC 9
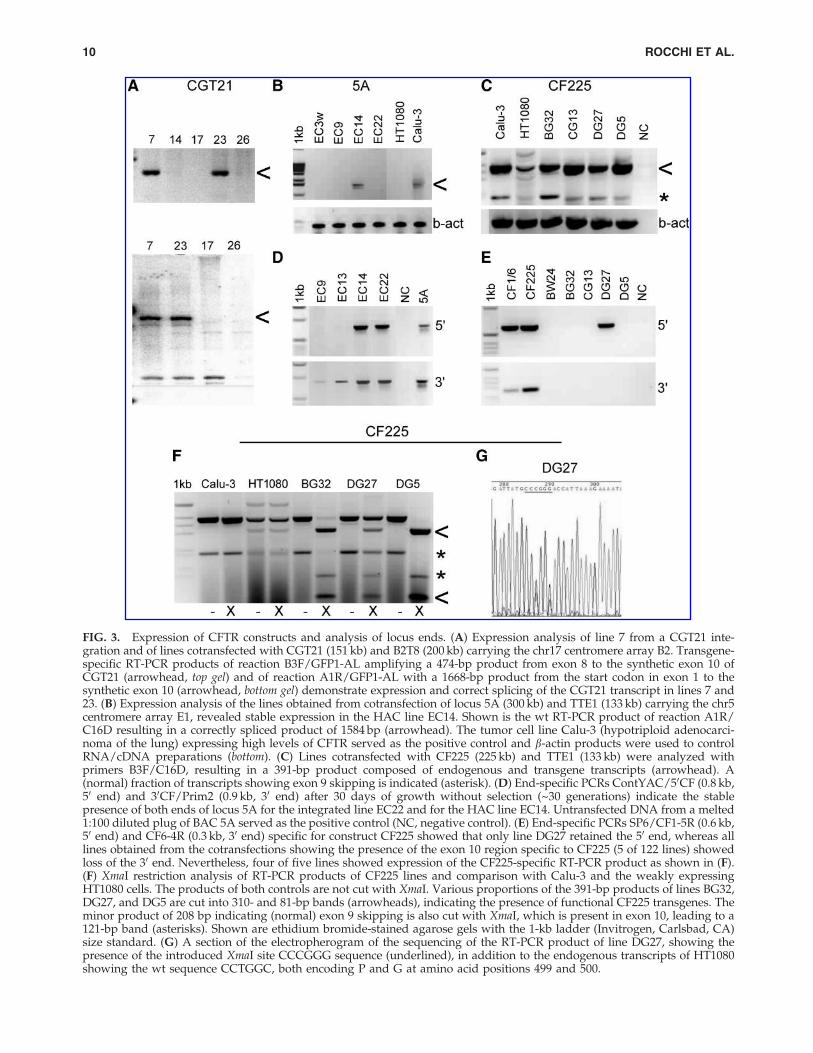
FIG. 3. Expression of CFTR constructs and analysis of locus ends. (A) Expression analysis of line 7 from a CGT21 inte-gration and of lines cotransfected with CGT21 (151 kb) and B2T8 (200 kb) carrying the chr17 centromere array B2. Transgene-specific RT-PCR products of reaction B3F/GFP1-AL amplifying a 474-bp product from exon 8 to the synthetic exon 10 ofCGT21 (arrowhead, top gel) and of reaction A1R/GFP1-AL with a 1668-bp product from the start codon in exon 1 to thesynthetic exon 10 (arrowhead, bottom gel) demonstrate expression and correct splicing of the CGT21 transcript in lines 7 and23. (B) Expression analysis of the lines obtained from cotransfection of locus 5A (300 kb) and TTE1 (133 kb) carrying the chr5centromere array E1, revealed stable expression in the HAC line EC14. Shown is the wt RT-PCR product of reaction A1R/C16D resulting in a correctly spliced product of 1584 bp (arrowhead). The tumor cell line Calu-3 (hypotriploid adenocarci-noma of the lung) expressing high levels of CFTR served as the positive control and b-actin products were used to controlRNA/cDNA preparations (bottom). (C) Lines cotransfected with CF225 (225 kb) and TTE1 (133 kb) were analyzed withprimers B3F/C16D, resulting in a 391-bp product composed of endogenous and transgene transcripts (arrowhead). A(normal) fraction of transcripts showing exon 9 skipping is indicated (asterisk). (D) End-specific PCRs ContYAC/50CF (0.8 kb,50 end) and 30CF/Prim2 (0.9 kb, 30 end) after 30 days of growth without selection (~30 generations) indicate the stablepresence of both ends of locus 5A for the integrated line EC22 and for the HAC line EC14. Untransfected DNA from a melted1:100 diluted plug of BAC 5A served as the positive control (NC, negative control). (E) End-specific PCRs SP6/CF1-5R (0.6 kb,50 end) and CF6-4R (0.3 kb, 30 end) specific for construct CF225 showed that only line DG27 retained the 50 end, whereas alllines obtained from the cotransfections showing the presence of the exon 10 region specific to CF225 (5 of 122 lines) showedloss of the 30 end. Nevertheless, four of five lines showed expression of the CF225-specific RT-PCR product as shown in (F).(F) XmaI restriction analysis of RT-PCR products of CF225 lines and comparison with Calu-3 and the weakly expressingHT1080 cells. The products of both controls are not cut with XmaI. Various proportions of the 391-bp products of lines BG32,DG27, and DG5 are cut into 310- and 81-bp bands (arrowheads), indicating the presence of functional CF225 transgenes. Theminor product of 208 bp indicating (normal) exon 9 skipping is also cut with XmaI, which is present in exon 10, leading to a121-bp band (asterisks). Shown are ethidium bromide-stained agarose gels with the 1-kb ladder (Invitrogen, Carlsbad, CA)size standard. (G) A section of the electropherogram of the sequencing of the RT-PCR product of line DG27, showing thepresence of the introduced XmaI site CCCGGG sequence (underlined), in addition to the endogenous transcripts of HT1080showing the wt sequence CCTGGC, both encoding P and G at amino acid positions 499 and 500.
10 ROCCHI ET AL.

that amplify inconsistently. Analysis of these regions re-vealed either short side products, which were identical inboth BAC subclones studied (Table 2), or resulted in failureof amplification in all resource clones (3-kb gap in intron 14).In addition, both regions do not differ from the expectedfragment sizes in a BssHII restriction analysis. Thus, the longPCR and pulsed-field restriction analysis demonstrates thepresence of the expected structure and a lack of internal re-arrangements in clones of both resources. These data indicatethat the entire CFTR gene can be stably cloned in yeast andin E. coli. Moreover, recloning of the YAC insert into the BACvector via yeast recombination did not introduce internalrearrangements or deletions. On occasion, deletions havebeen observed under prorecombinatorial conditions in E. coliexpressing RecE and RecT in the PAC host DH10B, which isRecA negative for the stable propagation of large inserts(Shizuya et al., 1992; Ioannou et al., 1994), as reported for arepetitive region 50 kb 30 to the b-globin locus (Imam et al.,2000). In addition, electroporation of an intact DNA prepa-ration of BAC 5A apparently did not result in a deletion, ashas frequently been observed with circularized YAC/BACs>250 kb (Cocchia et al., 2000). Overall, these structuralanalyses indicate high cloning stability of the CFTR locusin E. coli, suggesting that a genomic DNA of clinical usecan be produced in sufficient quantity and quality.
End sequencing of loci CF225 and 5A. To determine theends of CF225, LR-PCRs were carried out with primer pairsSP6/CF1-5R (50 end) and CF6-4F/SP6 (30 end) (Tables 1 and2). These were found to amplify 605- and 265-bp products(Fig. 3E), respectively. SP6 hybridizes to a sequence presenton both sides of the ligated PAC vector backbone of CF225.The amplified fragments were sequenced and compared tothe human genome build 37.1 using blastn at the NationalCenter for Biotechnology Information (NCBI). The re-constructed CFTR locus runs from nucleotide position�60651 relative to the start of translation to nucleotide po-sition þ9767 relative to the end of translation. Both endscoincide with a Sau3AI site, in agreement with the partiallydigested genomic DNA cloned in the BamHI site of the PACvector pCYPAC2N in library RPCIP704 (Ioannou et al., 1994).
As a result of the cloning procedure, CF225 has two de-letions within introns 9 and 10, representing regions thatwere not covered by the genuine PACs CF1 (intron 9) andCF6 (intron 10), and were omitted by reconstructing exon 10and its flanking intron sequences. The resulting 5.1-kb dele-tion in intron 9 was already known from the functionalanalysis of CGT21 (Laner et al., 2005) not to affect expressionand correct splicing in this region. To precisely locate anddetermine the extent of those deletions, PCRs were carriedout with primer pairs In9F/C16D (intron 9/exon 10) andCFi10fus/CF11R (intron 10/intron 11) (Table 1), whichgenerate DNA fragments of 735 and 773 bp, respectively. ThePCR products were sequenced and compared against pub-lished human BAC sequences using blastn at NCBI. In9Fhybridizes to nucleotide positions 73794–73816, and C16D tonucleotide positions 79552–79529 on the CFTR genomic se-quence (relative to the start of translation). The deletion inintron 9 is located from nucleotide position 74148 to 79204and is 5058 bp long. CFi10fus hybridizes to nucleotide posi-tions 80085–80105, and CF11R to nucleotide positions107929–107952. The deletion in intron 10 runs from nucleo-
tide positions 80281 to 107408 and is 27,128 bp long. DNAfrom HT1080 cells served as a control for the absence ofamplification from wt CFTR loci (data not shown).
To determine the ends of the large locus of BAC 5A, PCRswere carried out with primer pairs ContYAC/50CF (50 end)and 30CF/Prim2 (30 end) (Table 1), which were found toamplify ~0.8- and ~0.9-kb products, including 735 and558 bp of the insert, respectively (Fig. 3D). The amplifiedfragments were sequenced and blasted against human ge-nome build 37.1. The YAC-derived CFTR locus in BAC 5Aruns from nucleotide position �58351 relative to the start oftranslation to nucleotide position þ51381 relative to the endof translation. Both ends coincide with an EcoRI site, inagreement with the partially digested genomic DNA ligatedto the arms of the YAC vector pYAC4 (Anand et al., 1991).
Cotransfection experiments
To analyze the functional incorporation of the obtainedCFTR loci in a de novo formed human artificial chromosome(HAC), we employed simple cotransfection experimentswith linearized DNA components. HAC formation by co-transfection is not efficient, but has successfully been used toincorporate an HPRT gene into a de novo formed HAC(Grimes et al., 2001). In addition, it is advantageous not toprefabricate a fixed composition regarding the size of thelocus, the orientation with respect to the centromere, and thetype of centromere included. Indeed, cotransfection ofthe HAC components separately may to some extent increasethe flexibility of the assembly of an HAC, which may adoptsome rules of how stable structures need to be composed. Onthe other hand, the ongoing repair and recombination pro-cesses required to generate a stable genetic entity may bychance alter the input DNA. Thus, cotransfections representa workable tool to initialize HAC formation studies.
Construct CGT21 (Fig. 1) was cotransfected with constructB2T8 (for both see Laner et al., 2005) carrying a centromere-competent a-satellite array of 190 kb derived from chr17,which was cloned in the telomerized PAC vector pTAT-BS(17 kb) (Ebersole et al., 2000) and transfected as an ~200-kbconstruct. Cell lines 1–7 were transfected with the telomer-ized, ~151 kb-sized construct CGT21 alone, which contains aBS-selectable marker, resulting in integration. Cell line 7showed expression of the tagged transgene by RT-PCR (Fig.3A), and sequencing of the entire coding region demon-strated correct expression and splicing, and the presence ofthe improved 470M variant (the analyzed cDNA sequence isavailable at EMBL/GenBank under accession numberAY299332; Laner et al., 2005). From 24 cotransfected lines,numbered 8–32, 13 lines were screened by FISH. Cen17-based HACs lacking CFTR sequences were observed in fourlines, and three lines showed integration of the vector probersf only. Four lines did not allow a consistent detection of thetransferred constructs, and two lines showed HACs withCFTR and cen17 sequences. Only HAC line 23 showed ex-pression of the CGT21 transgene and persistence still after 30and 50 generations without selection. This was demonstratedby RT-PCR analyses specific for the transgene, using primersB3F and GFP1-AL, amplifying a 474-bp product betweenexons 8 and 10 (synthetic portion) (Fig. 3A, top), or primersA1R and GFP1-AL, generating a 1668-bp product from exon1 to the synthetic exon 10 (Fig. 3A, bottom). Despite correct
CFTR-HAC 11

sequences and expression, the EGFP tag did not result invisible green fluorescence under the microscope, as previ-ously described for integrated lines (Laner et al., 2005), sug-gesting that a functional CFTR/EGFP fusion protein is notsufficiently accumulating in HT1080 cells. FISH analyses ofline 23 after 30 days of growth with BS selection, and after 30and 50 days without selection (approximating 80 generationsin total), revealed that the HACs contained a stable structureconsisting of cen17 and CF1 sequences (Fig. 4A). However, atlater culture stages, an integration of HAC material in a hostchromosome was detected. This integrated material showedtruncation products with separated CFTR and cen17 signalswith both probes, in addition to an HAC. Attempts to selectout a single cell-derived subline only containing an HACwith CFTR and cen17 sequences were not successful afterexpanding four sublines, two isolated and expanded on BS,and two off selection.
The ~300-kb insert of BAC 5A was cotransfected with the116 kb-sized, centromere-proficient a-satellite array of chro-mosome 5, termed ‘‘E1’’ (Laner et al., 2004, 2005), whichwas cloned in the tetratelomeric PAC vector pTT (26 kb),
resulting in construct TTE1 (142 kb). Construct TTE1 con-sistently results in about two-thirds green clones, when usinga stored preparation with two-thirds of the molecules havingan EGFP marker (data not shown). The EGFP marker isprone to deletion from the vector because of its location be-tween two canonical telomere repeats, and the lack of anantibiotic marker gene within this fragment of the vectorprecluding its selection against deletion. Nevertheless, once agood DNA preparation has been characterized, a high rate ofgreen fluorescent clones can be obtained. Cotransfection ofthe 133 kb-sized TTE1 fragment containing a duplicated BS-selectable marker gene and the EGFP marker, with the un-selected, ~300-kb fragment of BAC 5A resulted in 24 greenclones. Three independent green lines (EC9, EC14, and EC22)were found to be positive for both ends of the 5A locus in aPCR screening (data not shown). After expansion for anadditional 30 days off BS selection, lines EC14 and EC22stably contained both ends (Fig. 3D). In a FISH screen, EC9and EC22 showed either signals for cointegration of bothvector and CFTR, or of vector, centromere, and CFTR in anendogenous chromosome. Cell line EC14 showed a free HAC
FIG. 4. FISH-based HAC detection andanalysis of centromere chromatin, usingimmuno-FISH. (A) Dual-color FISH analysisof line 23 containing a CGT21 (151 kb)- andB2T8 (200 kb)-cotransfected HAC (arrow-head) off selection. The HAC carries signalsfor the cen17 a-satellite probe B2 (green) andprobe CF1 (red). Chromosomes are stainedwith diamidinophenylindole (DAPI) (lightblue). (B) Triple-color FISH analysis of lineDG27 containing the CF225 (225 kb)- andTTE1 (133 kb)-cotransfected material inte-grated into chromosome 7 close to the en-dogenous CFTR locus (arrow). Probe CF1(red), probe CF6 (green), and traces of probeE1 (pink) staining the centromere 5 a-satelliteDNA and related centromeres of chromo-somes 1 and 19 are present at the site ofintegration. A rare case (1 in 130 meta-phases) of fragility at the site of integrationsuggests the presence of multiple copies(right inset). Vector probe rsf spanning the BSmarker (red) is present at the integrated lo-cus marked by the CF6 probe (green) andthe cen5 probe E1 (pink) (left inset). (C)Triple-color FISH of line EC14 showing anHAC from a cotransfection of 5A (300 kb)and TTE1 (133 kb). The free HAC (arrow-head) and the HAC material integrated intochromosome 1 (indicated) both carry signalsfor the centromere 5 a-satellite probe E1(pink), probe CF1 for the left half of theCFTR locus (red), and probe CF6 for theright half of the CFTR locus (green)(appearing yellow when merged). Chromo-somes are stained with DAPI (blue). Single-
color channels of a detailed HAC section were captured with a reduced exposure time to reduce blooming (bottom inset). (D)Triple-color immuno-FISH analysis of line EC14, demonstrating that the cen5 (probe E1, pink)-based HAC contains afunctional centromere in proximity to the entire CFTR wt locus. The centromere-binding protein CENP-A, indicative of activecentromeres, is stained with anti-CENP-A antibodies (red). CENP-A is present on the free HAC (arrowhead), but absent onthe integrated HAC material (arrow). CFTR sequences are hybridized with probe CF6 (green), and chromosomes are stainedwith DAPI (blue). Single channels of a section containing the free HAC are shown (bottom inset).
12 ROCCHI ET AL.

in addition to an integration of the HAC material intochromosome 1, which was present on and after 30 days offselection (Fig. 4C). The integrated centromere at a distance of~20 Mb from the endogenous chr1 centromere was in-activated and did not bind CENP-A. In contrast, the free,de novo formed HAC showed CENP-A in a combined im-muno-FISH analysis (Fig. 4D), indicating the formation of afunctional centromere in the vicinity of the CFTR locus onthe free, de novo formed HAC. Completeness of the CFTRlocus on the HACs is suggested by the presence of the ends,as confirmed by PCR and by RT-PCR analyses using primersA1R and C16D, which amplify a 1584-bp product from exon1 to exon 10 (Fig. 3B). Although these showed various levelsat different culture phases and between experiments, in-creased amounts of wt sequences were present in line EC14,compared with the weaker expression of the endogenous lociin the HT1080 cells. Expression appeared still increased after30 days of culture (approximating 30 generations) in theabsence of selection. Expression of the transgene was furthersupported by detection of the 470V polymorphism of theBAC by sequencing the RT-PCR product of line EC14. Incontrast, sequencing of a genomic PCR product of exon 10 ofthe endogenous loci in an HT1080 control line, using primersCF10F/CF10R, revealed that it was homozygous for 470M(data not shown). The presence of multiple complete loci isalso suggested from the FISH signals of both locus halves onthe HACs, which appear to be organized into multiple do-mains arranged around the comparably tiny centromericportion, like the leaves of a flower, each domain containingCF1 and CF6 sequences in a triple-color FISH analysis (seeFig. 4D inset, captured under low-exposure conditions toavoid blooming).
Four rounds of colipofection of the 225-kb insert of PACCF225, with the characterized preparation of constructTTE1 (133 kb fragment), resulted in 185 (white and green)cell clones, 122 of which were expanded and screened byPCR with primers CFi10fus/R7 (Table 1), specific forCF225. Five individual cell clones, BW24, BG32, CG13,DG27, and DG5, were positive for the exon 10 junction re-gion, indicating that only 1 in ~25 cell clones was co-transfected with both CF225 and TTE1 DNA. Although theratio of intact molecules per liposome preparation was notfurther assessed in the four individual transfer experimentsresulting in the 122 analyzed clones, there is no obviousexplanation for this low cotransfection efficiency. Othercotransfections >100 kb regularly approached efficiencies of1 in ~3–10 clones when equimolar DNA preparations wereused under similar conditions, regardless of whether one orboth components carried the BS marker. Successful co-transfections leading to HAC formation were possible athigher efficiency even if one component lacked telomericrepeats, as was the case here for the 5A and CF225 inserts.Nevertheless, the cotransfections of CF225 still representeda workable means of selecting out stable clones, allowing aninitial functional assessment of the locus not carrying aselectable marker.
RT-PCRs were carried out with primers B3F and C16D(Table 1), generating a spliced product of 391 bp betweenexons 8 and 10, which represents a mixture of productsfrom endogenous CFTR genes of the HT1080 cell line andthe transgene loci. RNA/cDNA preparations were con-trolled with b-actin primers (Fig. 3C). All lines showed
various levels of CFTR expression after 30 days off selec-tion. To distinguish between endogenous and transgeneexpression, the RT-PCR products were digested with XmaIcutting the engineered exon 10 from CF225 into two frag-ments of 310 and 81 bp (Fig. 3F). In four cell lines, variousproportions of the CFTR transcript resulted from thetransgene, which demonstrated expression levels of thetransgene above wt background in most cases, and showedcorrect splicing (Fig. 3F). Cell line BW24 did not express thetransgene (data not shown). The expressing cell lines andparental HT1080 cells were further analyzed by sequencingof the RT-PCR products with the same primers and primerCFc3F (Table 1), demonstrating that all lines contained boththe 470M polymorphism and the synthetic XmaI variant inexon 10, confirming transgene origin. Figure 3G shows asection of the electropherogram from sequencing of lineDG27, evidencing both the XmaI site (CCCGGG) expressedfrom CF225 and the wt sequence (CCTGGC) from theHT1080 loci, both encoding wt amino acids 499P and 500G(corresponding to silent exchanges).
To check the integrity of construct CF225 in the clonal celllines, PCRs were carried out with primer pairs SP6/CF1-5Rfor the left vector/CFTR junction of 605 bp, and CF-4F/SP6for the right CFTR/vector junction of 265 bp (Table 1). Of thefive cell lines that were positive for the exon 10 junction PCR,line DG27 has kept the left end of CF225, confirmed by se-quencing the PCR product. All five lines were negative forthe right end junction PCR (Fig. 3E) and for two other LR-PCRs extending approximately 2 and 3 kb from the right endinto the CFTR locus (data not shown), suggesting that 30
DNA of CF225 was lost in all four expressing lines. Loss oflocus ends may have occurred during integration or subse-quent propagation.
Triple-color FISH analyses of these cell lines after 30 daysof growth on and 30 days off BS selection revealed eitherintegration of the CF225 locus into host chromosomes, orintegration and truncation in all five clonal cell lines. NoHACs carrying the CFTR locus were observed. Clonal lineDG27 showed a stable cointegration close to the endoge-nous CFTR gene on chr7 (arrow in Fig. 4B), which waspositive for CF1, CF6, and rsf signals on and off selection.Weak signals for E1 were regularly visible at the site ofintegration. Line BG32 revealed a distal/telomeric integra-tion into a chromosome (non-7), which was positive forCF1, CF6, and rsf, but not for E1 signals, indicating that thea-satellite DNA was either not cotransfected or lost. Cellline DG5 showed integration of CF1, CF6, and E1 signals ina distal position of chr19q, and line CG13 showed integra-tion of CF1 and CF6 portions in the p arm of a metacentricchromosome (non-7) accompanied by truncation. Thetruncated portion containing the endogenous centromereand signals for CF1 and CF6 was stably maintainedwhereas the p arm portion positive for signals E1 and CF6,but not CF1, was frequently lost. In cell line BW24 only CF6sequences were detected on a small truncated chromosome.Overall, we conclude that CF225 and the E1 centromere didnot efficiently form a stably replicating structure together.Instead, rare stable clones were selected out that containedat least the BS marker and various portions of locus CF225lacking the very 30 end, which nevertheless showed ex-pression of the tagged exon 10 sequence in four of five linesobtained.
CFTR-HAC 13

Discussion
CF gene therapy, with delivery of the correct CFTR geneto cells, promises a great benefit for all CF patients, irre-spective of the type of mutation. However, classical genetherapy to cystic fibrosis has had limited success due toimmune response or short-term transgene expression ofcDNA delivered by viral vectors or cationic lipids due tosilencing. These limitations could be overcome by deliveringthe complete CFTR gene with relevant regulatory regionswithin its genomic context on nonintegrating human artifi-cial chromosomes. Thus, for gene therapy success, a stableand functional CFTR locus containing all exons, introns, andregulatory elements should be inserted into a human artifi-cial chromosome (HAC) vector that can be delivered to cells.
CGT21, a genomic construct containing about one-half ofthe CFTR locus from which expression and correct splicingwas demonstrated (Laner et al., 2005), is a functional genewith 10 exons encoding a nonfunctioning CFTR protein de-rivative. Because of its reduced size and the tagged exon,such a construct can ease certain transfer studies. Anotherversion, CGT20, with opposite orientation with respect to theBS marker, and basic PAC version CG2 are also available(our unpublished results).
Here, we aimed at producing and analyzing the stableincorporation of the entire CFTR locus (including all exonsand introns, and regulators) on a de novo HAC, which couldbe used for gene therapy. The de novo approach based on thetransfection of ‘‘naked’’ DNA molecules to the cells of thepatient offers a technically feasible strategy toward long-term gene therapy, provided sufficient numbers of largeconstructs can be produced in a functional form and deliv-ered.
We describe the successful generation of a large constructcontaining the CFTR locus with a reduced size (PAC CF225),constructed from characterized CFTR PAC resources, with afunctionally optimized polymorphism and a silent restrictionvariant in exon 10, and also used the 296.8 kb-sized BAC 5Acovering the entire wt CFTR locus with all regulators (seebelow). For reconstructing the tagged CFTR locus, weadopted a technically simple approach based on a ‘‘rarecutter’’ (NotI) restriction site present at the PAC vectorboundary, thus allowing ‘‘conventional’’ cloning on a geno-mic scale, followed by electroporation of the entire CFTRfusion PAC into E. coli DH10B cells. To our knowledge,cloning of PAC fragments of this size, which is based onintact DNA preparations, represents a novel method bywhich to assemble genomic loci.
To define the CFTR locus, we used our previous data,describing that the human CFTR gene and its adjacent geneslocalize individually and differentially to distinct nuclearregions, according to their transcriptional activity. Thissuggests that the principal CFTR regulatory information iscontained within the limits of the two adjacent genes, that is,<283 kb (Zink et al., 2004; Sadoni et al., 2008). In addition todevelopmentally regulated alternative 50 exons (Mouchelet al., 2003) numerous DNase I-hypersensitive sites (DHSs)have been described and analyzed within this region(McCarthy and Harris, 2005; Ott et al., 2009). Most of theseare included in CF225, namely DHSs that lie at �20.9 kb (tothe start of translation); in introns 1, 2, 3, 11, 16, 17a, 18, 20,and 21; and 30 to the gene at þ5.4, þ6.8, þ7, and þ7.4 kb
relative to the end of translation. DHSs not present in CF225likely belong to the adjacent genes, or are in the reducedregion of intron 10, including the DHS that has been shownto be active in intestinal cells (Ott et al., 2009), and DHSs atþ15.6, þ17.2, and þ20.1 kb (to the end of translation). Thelatter are placed in the vicinity of the start position of an ~17-kb transcript mapped in opposite direction of the CFTR gene(source: human genome build 37.1 at the NCBI), running infrom the intergenic region between CFTR and CORTBP2toward the end of the CFTR transcript.
Similar transcripts in the opposite strand can be found inthe 30 region of many genes, for example, the genes FOXP2,MDFIC, CAV2, CAV1, MET, and CAPZA2, to name just afew in the vicinity of the CFTR locus (our unpublished ob-servation). The common presence of such 30 transcriptssuggests an important genetic function, for example, in generegulation, formation of a chromatin domain, or stability.
By cotransfecting CF225 and TTE1, we obtained five BS-resistant cell lines that, however, did not show de novo for-mation of HACs. All obtained integrated cell lines have lostthe 30 end of CF225 DNA, which could include loss of DHS atþ5.4, þ6.8, þ7, and þ7.4 kb. Although these DHSs are po-tentially absent and the ones at þ15.6 kb and downstreamare not in the construct, it was possible to detect the XmaI-specific, correctly spliced transcript in RT-PCR productsfrom four of five cell lines, albeit at various levels relativelyto the endogenous CFTR genes of HT1080.
Because of the poor cotransfection efficiency observedwith PAC CF225, it remains unknown whether the CF225locus is complete and replication-competent (autonomously)when ligated to a centromere. It seems possible that addi-tional 30 sequences, such as an origin of replication, orchromatin barriers separating gene expression from centro-mere function, could be required on a prefabricated HAC.PAC CF225 contains three NotI sites at both locus ends andin intron 10, which is 27 kb reduced in size compared withthe wt locus. Thus, the possibly critically short 30 end, or thelacking DHS in intron 10 can easily be altered by cloningsuspected regulatory regions into the NotI sites, representingan interesting system to verify expression regulation andstability on an HAC in suitable cell or animal models.
To study incorporation of the three distinct versions of theE. coli-cloned CFTR locus into de novo HACs, colipofectionexperiments were carried out in HT1080 cells. A workablenumber of 24 and 25 clones, respectively, revealed de novoformation of a stable HAC when using either the short, BS-selected construct CGT21 (151 kb, linearized with I-SceI) andthe BS-selected cen17 construct B2T8 (~200 kb, linearizedwith I-SceI), or when using the long, nonselected wt locus 5A(~300-kb SalI insert) and the BS-selected cen5 construct TTE1(133 kb, linearized with NotI). In contrast, the more elaborateanalysis of 122 clones from the cotransfection of the mid-sized, nonselected locus CF225 (225 kb, SalI insert) with theBS-selected cen5 construct TTE1 (133 kb, linearized withNotI) did not reveal HAC formation. Only five integratedlines positive for CF225 were obtained. Although the num-bers are not sufficient to conclude that CF225 is less HACcompetent than 5A, it is tempting to speculate that CF225 isprone to instability. Although the larger 5A construct haskept both locus ends in 2 individual cell lines (of 25 analyzedlines, which represent fewer than 25 individual lines be-cause double-picked lines were excluded when they have
14 ROCCHI ET AL.

been identified), including an integrated and the HAC line,all 5 integrated lines of the midsized locus CF225 (of 122lines) have lost the 30 end of the locus. Nevertheless, thespecific RT-PCR analysis demonstrated that CF225 is ex-pressed in four of five cell lines, and that the introducedXmaI variant in exon 10 can be used to distinguish betweentransgene and endogenous transcripts. It is possible thatCF225 underwent a 30 trimming process to generate a stablestructure, which may have reduced the cotransfection effi-ciency and thus HAC incorporation.
The HAC present in cell line EC14 was highly stable on andoff BS selection and contained the entire wt CFTR locus.Moreover, the cen5 a-satellite DNA present on the HACbound CENP-A, indicating formation of an active centromere.Accordingly, the HAC in line EC14 represents the first de novoformed human artificial chromosome carrying the entireCFTR locus. Cell line EC14 showed expression of the CFTRlocus above the endogenous background on and off selection,suggesting that the functional locus was transferred. Un-ambiguous proof of expression from the CFTR genomic con-struct that localizes in the vicinity of the active centromere isnot possible at present, because high copy numbers are likelyto be present on the HAC, resulting in a greater distance to thecentromere of some copies. Moreover, HAC material inte-grated into chr1 could also contribute to expression.
We previously demonstrated that PAC-cloned, >100 kb-sized human a-satellite arrays with ~99% identical 2-kb re-peats did not result in internal recombination or deletionduring a prolonged growth period of 2� ~400 E. coli gener-ations in a colony-replating experiment (Schindelhauer andSchwarz, 2002). Here, we provide extensive structural data,including a set of LR-PCRs covering the CFTR loci fromvarious cloning sources, that indicate stability. Moreover, wecould confirm cloning stability after prolonged growth pe-riods in fluid culture, which would be required to test andproduce an HAC-based DNA therapeutic product. Overall,the cumulative data presented here suggest that the con-struction of a prefabricated CFTR-HAC based on the definedresource clones is feasible. These results are thus promisingfor the development of HAC-based therapy.
Acknowledgments
The authors thank Sebastian Beck for RT-PCR advice andSonia Pedro and Ana Cardoso for RT-PCR productsequencing, Ulrich Zissler for PAC end sequencing, Steffi Si-mon for setting up large-scale growth simulations, MichaelSpeicher and Burgis Cleve for access and support with theFISH microscope, Manuel Valdivia for the kind gift of CENP-A antibodies, Christina Auriche for BAC handling, and An-gelika Schnieke for excellent working conditions, a supportiveatmosphere, and hospitality at her chair. This work was fi-nanced by WZW/TUM for student bench fees, two grants ofthe Forschungsgemeinschaft Mukoviszidose (D.S.), Vaincre laMucoviscidose grants (D.S. and A.L.), two grants of the Frie-drich Baur Stiftung (D.S.), Deutsche Forschungsgemeinschaft(D.S.), and multiannual funding from BioFIG (FCT, Portugaland FEDER/EU), the European Community (QLK3-CT-2002-02119) and partially Fondazione Italiana per la ricerca sullafibrosi cistica and Istituto Pasteur-Fondazione Cenci Bolog-netti, Sapienza University (F.A.). L.R. was a guest scientistemployed by La Sapienza University, Rome (Dottorato in
Biologia Cellulare e dello Sviluppo). C.B. is the recipient ofdoctoral fellowship SFRH/BD/17912/2004 (FCT, Portugal).
Author Disclosure Statement
No competing financial interests exist.
References
Anand, R., Ogilvie, D.J., Butler, R., Riley, J.H., Finniear, R.S.,Powell, S.J., Smith, J.C., and Markham, A.F. (1991). A yeastartificial chromosome contig encompassing the cystic fibrosislocus. Genomics 9, 124–130.
Auriche, C., Carpani, D., Conese, M., Caci, E., Zegarra-Moran,O., Donini, P., Ascenzioni, F. (2002). Functional human CFTRproduced by a stable minichromosome. EMBO Rep. 3, 862–868.
Cocchia, M., Kouprina, N., Kim, S.J., Larionov, V., Schlessinger,D., and Nagaraja, R. (2000). Recovery and potential utility ofYACs as circular YAC/BACs. Nucleic Acids Res. 28, E81.
Cuppens, H., Lin, W., Jaspers, M., Costes, B., Teng, H., Van-keerberghen, A., Jorissen, M., Droogmans, G., Reynaert, I.,Goossens, M., Nilius, B., and Cassiman, J.J. (1998). Polyvariantmutant cystic fibrosis transmembrane conductance regulatorgenes. The polymorphic (Tg)m locus explains the partialpenetrance of the T5 polymorphism as a disease mutation. J.Clin. Invest. 101, 487–496.
Dorin, J.R., Farley, R., Webb, S., Smith, S.N., Farini, E., Delaney,S.J., Wainwright, B.J., Alton, E.W., Porteous, D.J. (1996). Ademonstration using mouse models that successful genetherapy for cystic fibrosis requires only partial gene correction.Gene Ther. 3, 797–801.
Ebersole, T.A., Ross, A., Clark, E., McGill, N., Schindelhauer, D.,Cooke, H., Grimes, B. (2000). Mammalian artificial chromo-some formation from circular alphoid input DNA does notrequire telomere repeats. Hum. Mol. Genet. 9, 1623–1631.
Grimes, B.R., Schindelhauer, D., MGill, N.I., Ross, A., Ebersole,T.A., and Cooke, H.J. (2001). Stable gene expression from amammalian artificial chromosome. EMBO Rep. 2, 910–914.
Imam, A.M., Patrinos, G.P., De Krom, M., Bottardi, S., Janssens,R.J., Katsantoni, E., Wai, A.W., Sherratt, D.J., Grosveld, F.G.(2000). Modification of human b-globin locus PAC clones byhomologous recombination in Escherichia coli. Nucleic AcidsRes. 28, E65.
Ioannou, P.A., Amemiya, C.T., Garnes, J., Kroisel, P.M., Shizuya,H., Chen, C., Batzer, M.A., and De Jong, P.J. (1994). A newbacteriophage P1-derived vector for the propagation of largehuman DNA fragments. Nat. Genet. 6, 84–89.
Laner, A., Schwarz, T., Christan, S., and Schindelhauer, D.(2004). Suitability of a CMV/EGFP cassette to monitor stableexpression from human artificial chromosomes but not tran-sient transfer in the cells forming viable clones. Cytogenet.Genome Res. 107, 9–13.
Laner, A., Goussard, S., Ramalho, A.S., Schwarz, T., Amaral,M.D., Courvalin, P., Schindelhauer, D., and Grillot-Courvalin,C. (2005). Bacterial transfer of large functional genomic DNAinto human cells. Gene Ther. 12, 1559–1572.
Manson, A.L., Trezise, A.E., Macvinish, L.J., Kasschau, K.D.,Birchall, N., Episkopou, V., Vassaux, G., Evans, M.J., Colledge,W.H., Cuthbert, A.W., and Huxley, C. (1997). Complementa-tion of null CF mice with a human CFTR YAC transgene.EMBO J. 16, 4238–4249.
McCarthy, V.A., and Harris, A. (2005). The CFTR gene andregulation of its expression. Pediatr. Pulmonol. 40, 1–8.
Mouchel, N., Broackes-Carter, F., and Harris, A. (2003). Alter-native 50 exons of the CFTR gene show developmental regu-lation. Hum. Mol. Genet. 12, 759–769.
CFTR-HAC 15

Nakano, M., Cardinale, S., Noskov, V.N., Gassmann, R., Vag-narelli, P., Kandels-Lewis, S., Larionov, V., Earnshaw, W.C.,Masumoto, H. (2008). Inactivation of a human kinetochore byspecific targeting of chromatin modifiers. Dev. Cell 14, 507–522.
Ott, C.J., Blackledge, N.P., Kerschner, J.L., Leir, S.-H., Crawford,G.E., Cotton, C.U., Harris, A. (2009). Intronic enhancers co-ordinate epithelial-specific looping of the active CFTR locus.Proc. Natl. Acad. Sci. U.S.A. 106, 19934–19939.
Ramalho, A.S., Beck, S., Meyer, M., Penque, D., Cutting, G.R.,and Amaral, M.D. (2002). Five percent of normal cystic fibrosisconductance regulator mRNA ameliorates the severity ofpulmonary disease in cystic fibrosis. Am. J. Respir. Cell. Mol.Biol. 27, 619–627.
Ramalho, A.S., Beck, S., Schindelhauer, D., and Amaral, M.D.(2004). Sequence of CFTR exons present in clones from PAClibrary RPCIP704. Virtual Repository of the European Work-ing Group on CFTR Expression F.4.1. Available at http://central.igc.gulbenkian.pt/cftr/vr/f/ramalho_sequence_cftr_exons_clones_pac_library_rpcip704.pdf (accessed June 2010).
Riordan, J.R., Rommens, J.M., Kerem, B., Alon, N., Rozmahel, R.,Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N., Chou, J.L.,Drumm, M.L., Iannouzzi, M.C., Collins, F.S., and Tsui, L.-C.(1989). Identification of the cystic fibrosis gene: Cloning andcharacterization of complementary DNA. Science 245, 1066–1073.
Rommens, J.M., Iannuzzi, M.C., Kerem, B., Drumm, M.L.,Melmer, G., Dean, M., Rozmahel, R., Cole, J.L., Kennedy, D.,Hidaka, N., Zsiga, M., Buchwald, M., Riordan, J., Tsui, L.-C.,Collins, F.S. (1989). Identification of the cystic fibrosisgene: Chromosome walking and jumping. Science 245, 1050–1065.
Sadoni, N., Targosz, B.S., Englmann, A., Fesser, S., Koch, J.,Schindelhauer, D., Zink, D. (2008). Transcription-dependentspatial arrangements of CFTR and conserved adjacent loci arenot conserved in human and murine nuclei. Chromosoma 117,381–397.
Schindelhauer, D., and Cooke, H.J. (1997). Efficient combinationof large DNA in vitro: In gel site specific recombination(IGSSR) of PAC fragments containing a satellite DNA and thehuman HPRT gene locus. Nucleic Acids Res. 25, 2241–2243.
Schindelhauer, D., and Schwarz, T. (2002). Evidence for a fast,intrachromosomal conversion mechanism from mapping ofnucleotide variants within a homogeneous a-satellite DNAarray. Genome Res. 12, 1815–1826.
Sheppard, D.N., and Welsh, M.J. (1999). Structure and functionof the CFTR chloride channel. Physiol. Rev. 79, S23–S45.
Shizuya, H., Birren, B., Kim, U.J., Mancino, V., Slepak, T.,Tachiiri, Y., and Simon, M. (1992). Cloning and stable main-tenance of 300-kilobase-pair fragments of human DNA inEscherichia coli using an F-factor-based vector. Proc. Natl.Acad. Sci. U.S.A. 89, 8794–8797.
Smith, C.L., Klco, S.R., and Cantor, C.R. (1988). Pulsed field gelelectrophoresis and the technology of large DNA molecules.In: K.E. Davies, ed. Genome Analysis (IRL Oxford) pp. 47–71.
Valdivia, M.M., Figueroa, J., Iglesias, C., and Ortız, M. (1998). Anovel centromere monospecific serum to a human autoepitopeon the histone H3-like protein CENP-A. FEBS Lett. 422, 5–9.
Vassaux, G., Manson, A.L., Huxley, C. (1997). Copy number-dependent expression of a YAC-cloned human CFTR gene in ahuman epithelial cell line. Gene Ther. 4, 618–623.
World Health Organization (2004). The Molecular Genetic Epi-demiology of Cystic Fibrosis: Report of a Joint Meeting ofWHO/ECFTN/ICF(M)A/ECFS, Genoa, Italy, June 19, 2002.Available at http://www.cfww.org/who/article/203/The_molecular_genetic_epidemiology_of_cystic_fibrosis (accessedJune 2010).
Zielenski, J., Rozmahel, R., Bozon, D., Kerem, B., Grzelczak, Z.,Riordan, J.R., Rommens, J., and Tsui, L.C. (1991). GenomicDNA sequence of the cystic fibrosis transmembrane conduc-tance regulator (CFTR) gene. Genomics 10, 214–228.
Zink, D., Amaral, M.D., Englmann, A., Lang, S., Clarke, L.A.,Rudolph, C., Alt, F., Luther, K., Braz, C., Sadoni, N., Rose-necker, J., Schindelhauer, D. (2004). Transcription-dependentspatial arrangements of CFTR and adjacent genes in humancell nuclei. J. Cell Biol. 166, 815–825.
Address correspondence to:Dr. Dirk Schindelhauer
Chromosome Medicine ProcurementTherese-Studer-Str. 47
80797 Munich, Germany
E-mail: [email protected]
Received for publication December 23, 2009;accepted after revision April 12, 2010.
Published online: July 7, 2010.
16 ROCCHI ET AL.





























