Embed Size (px)
Citation preview

Ep
RTa
b
c
a
ARRAA
KBPLMSE
1
mhceoan
P
PC
2Ph
Journal of Asian Ceramic Societies 2 (2014) 268–274
Contents lists available at ScienceDirect
Journal of Asian Ceramic Societies
j ourna l ho me pa ge: www.elsev ier .com/ locate / jascer
thylene glycol assisted low-temperature synthesis of boron carbideowder from borate citrate precursors
afi-ud-dina,c,∗, G.H. Zahida, Z. Asghara, Muhammad Maqboolb, Ejaz Ahmada,anvir Azhara, Tayyab Subhanib, M. Shahzada
Materials Division, PINSTECH, Nilore, Islamabad, PakistanDepartment of Materials Science and Engineering, Institute of Space Technology, Islamabad, PakistanState Key Laboratory for Advanced Metals and Materials, School of Materials Science and Engineering, USTB, Beijing 100083, China
r t i c l e i n f o
rticle history:eceived 1 April 2014eceived in revised form 17 May 2014ccepted 22 May 2014vailable online 23 June 2014
eywords:
a b s t r a c t
B4C powders were synthesized by carbothermal reduction of ethylene glycol (EG) added borate citrateprecursors, and effects of EG additions (0–50 mol% based on citric acid) on the morphologies and yields ofsynthesized B4C powders were investigated. The conditions most suitable for the preparation of precursorwere optimized and optimum temperature for precursor formation was 650 ◦C. EG additions facilitatedlow-temperature synthesis of B4C at 1350 ◦C, which was around 100–300 ◦C lower temperature comparedto that without EG additions. The lowering of synthesis temperature was ascribed to the enlargement of
oron carbide (B4C)recursorsow-temperature synthesisicrostructure
pectroscopythylene glycol
interfacial area caused by superior homogeneity and dispersibility of precursors enabling the diffusionof reacting species facile. The 20% EG addition was optimal with free residual carbon lowered to 4%.For smaller EG additions, the polyhedral and rod-like particles of synthesized product co-existed. Withhigher EG additions, the morphology of synthesized product was transformed into needle and blade-likestructure.
© 2014 The Ceramic Society of Japan and the Korean Ceramic Society. Production and hosting by
. Introduction
Boron carbide (B4C) has a leading role in numerous high perfor-ance applications owing to its extreme hardness, low density,
igh melting point, high Young’s modulus, great resistance tohemical agents, excellent thermoelastic and thermoelectric prop-rties and high corrosion and oxidation resistance. The combinationf these properties renders B4C a promising material for numerouspplications including body and vehicle armor, abrasive powder,uclear applications and aerospace applications [1–4].
∗ Corresponding author at: Materials Division, PINSTECH, Nilore, Islamabad,akistan. Tel.: +92 51 9248848; fax: +92 51 9248808.
E-mail addresses: rafi [email protected], rafi [email protected] ( Rafi-ud-din).eer review under responsibility of The Ceramic Society of Japan and the Koreaneramic Society.
187-0764 © 2014 The Ceramic Society of Japan and the Korean Ceramic Society.roduction and hosting by Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.jascer.2014.05.011
Elsevier B.V. All rights reserved.
B4C is generally produced by carbothermal reduction of boronoxide at high temperatures according to reaction (1) and magne-siothermic reaction of boron oxide [5–10].
2B2O3 + 7C → B4C + 6CO (1)
However, this process has many associated problems, including,considerable amount of free carbon residue in the final product dueto substantial loss of boron by volatilization of its oxides; difficultyand associated greater cost to grind the product into fine particlesize for densification; requiring a high temperature furnace opera-tion; contamination in the final product; and time consumption.
An alternative to above processes is the utilization of polymerprecursors, e.g. sucrose and glucose, glycerin, polyols, and pheno-lic resin, as low-temperature synthetic route to B4C facilitated byimproving the homogeneity and dispersibility of B2O3 and carbon[11–22]. Nevertheless, the disadvantage of using organic precur-sors is the presence of residual carbon in the products synthesizedat lower temperatures. Citric acid (CA) has been employed to pro-duce B4C with significant residual carbon at 1500–1900 ◦C [15–18].Recently, it is indicated that the addition of tartaric acid to boric acid(BA)–glycerin ameliorates the dispersion of B2O3/C [21]. It is con-cluded that the hydroxyl group can be easily condensed with BA,enhancing the esterification (B O C) and yielding the precursors
with fine homogeneous dispersion.Ethylene glycol (containing hydroxyl groups) is extensively uti-lized to synthesize a variety of compounds by esterification withmetal citrate [23,24]. It is expected that EG addition to condensed
n Ceramic Societies 2 (2014) 268–274 269
breBopus
2
g9(wfEhpgc5c
m(m(wdwBwBasusbQAtia
3
saittd–taBihbaEa
Rafi-ud-din et al. / Journal of Asia
orate citrate will modify the thermodynamics of carbothermaleaction by promoting the esterification reaction. In this work, theffects of EG additions on the synthesis of B4C from condensedA-CA are investigated. The optimum starting compositions andptimum conditions for pyrolysis of precursor and preparation ofroducts have been determined. FTIR, XRD, and SEM analyses aretilized to investigate the yield and morphology changes of precur-ors and synthesized B4C.
. Experimental
Materials used were boric acid [(BA), Wako, 99.5%], ethylenelycol [(EG), Merck, 99.5%], and citric acid [(CA), Riedel-de Haen,9.5%]. BA/CA of 2:1 was used and EG additions were 0–50 mol%P0–P50) based on CA. The BA solution (2.5 M) in distilled wateras prepared at 80 ◦C. CA was added slowly to the solution and held
or 20 min until a yellowish solution was obtained. Subsequently,G was added to the stirred solution. The solution was then slowlyeated to 130 ◦C and maintained at this temperature to facilitate theolyesterification reaction. The condensed products changed fromolden yellow to transparent light yellow with EG additions. Theondensed products were pyrolyzed over the temperature range of50–800 ◦C in air for 2 h. The obtained dark gray precursors wereompacted and heated at 1050–1400 ◦C for 0–4 h in an Ar flow.
The B2O3 contents were determined by titration ofannitol–boric acid complex with standard NaOH solution
0.1 M). The residual carbon in the final products was deter-ined by the method used in [9,22]. Fourier transform infrared
FTIR) spectra of starting materials and condensed productsere recorded on Shimadzu Prestige 21 spectrometer. X-rayiffraction (XRD) patterns of pyrolyzed powder and productsere obtained using powder X-ray diffractometer (Rigaku). The
4C peak intensity ratio (IB4C/IB4C + IC + IB2O3 ) of the productsas estimated from the main peak intensities of each of the
4C, carbon, and B2O3. The morphology of pyrolyzed powdernd products was examined using a scanning electron micro-cope (LEO 440). A Cilas 1064 laser particle size analyzer wastilized for determining the average particle size and particleize distribution. The BET specific surface area, pore size distri-ution by pore diameter, and pore volume were determined byuanta chrome Autosorb 1C BET Surface Area & Pore Volumenalyzer. The surface areas and pore volumes (or pore size dis-
ribution) were determined from nitrogen adsorption–desorptionsotherms by using the Brunauer–Emmet–Teller (BET) equationnd Barret–Joyner–Halenda (BJH) methods.
. Results and discussion
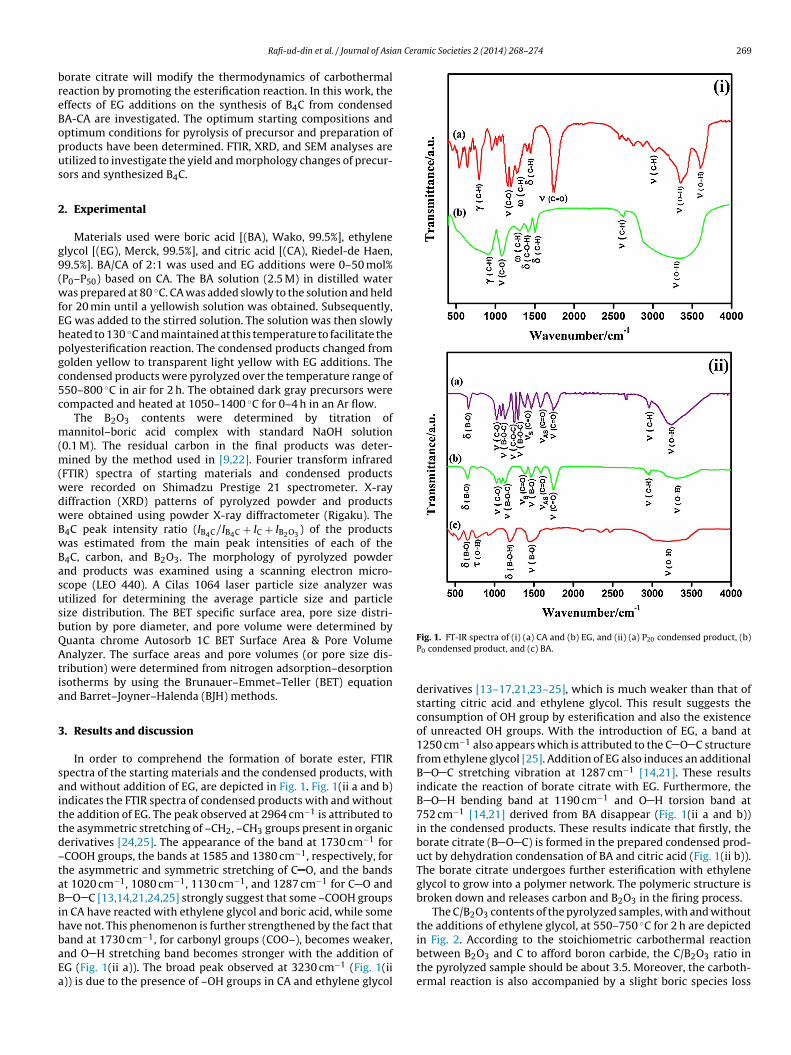
In order to comprehend the formation of borate ester, FTIRpectra of the starting materials and the condensed products, withnd without addition of EG, are depicted in Fig. 1. Fig. 1(ii a and b)ndicates the FTIR spectra of condensed products with and withouthe addition of EG. The peak observed at 2964 cm−1 is attributed tohe asymmetric stretching of –CH2, –CH3 groups present in organicerivatives [24,25]. The appearance of the band at 1730 cm−1 forCOOH groups, the bands at 1585 and 1380 cm−1, respectively, forhe asymmetric and symmetric stretching of C O, and the bandst 1020 cm−1, 1080 cm−1, 1130 cm−1, and 1287 cm−1 for C O and
O C [13,14,21,24,25] strongly suggest that some –COOH groupsn CA have reacted with ethylene glycol and boric acid, while someave not. This phenomenon is further strengthened by the fact that
and at 1730 cm−1, for carbonyl groups (COO–), becomes weaker,nd O H stretching band becomes stronger with the addition ofG (Fig. 1(ii a)). The broad peak observed at 3230 cm−1 (Fig. 1(ii)) is due to the presence of –OH groups in CA and ethylene glycolFig. 1. FT-IR spectra of (i) (a) CA and (b) EG, and (ii) (a) P20 condensed product, (b)P0 condensed product, and (c) BA.
derivatives [13–17,21,23–25], which is much weaker than that ofstarting citric acid and ethylene glycol. This result suggests theconsumption of OH group by esterification and also the existenceof unreacted OH groups. With the introduction of EG, a band at1250 cm−1 also appears which is attributed to the C O C structurefrom ethylene glycol [25]. Addition of EG also induces an additionalB O C stretching vibration at 1287 cm−1 [14,21]. These resultsindicate the reaction of borate citrate with EG. Furthermore, theB O H bending band at 1190 cm−1 and O H torsion band at752 cm−1 [14,21] derived from BA disappear (Fig. 1(ii a and b))in the condensed products. These results indicate that firstly, theborate citrate (B O C) is formed in the prepared condensed prod-uct by dehydration condensation of BA and citric acid (Fig. 1(ii b)).The borate citrate undergoes further esterification with ethyleneglycol to grow into a polymer network. The polymeric structure isbroken down and releases carbon and B2O3 in the firing process.
The C/B2O3 contents of the pyrolyzed samples, with and withoutthe additions of ethylene glycol, at 550–750 ◦C for 2 h are depictedin Fig. 2. According to the stoichiometric carbothermal reaction
between B2O3 and C to afford boron carbide, the C/B2O3 ratio inthe pyrolyzed sample should be about 3.5. Moreover, the carboth-ermal reaction is also accompanied by a slight boric species loss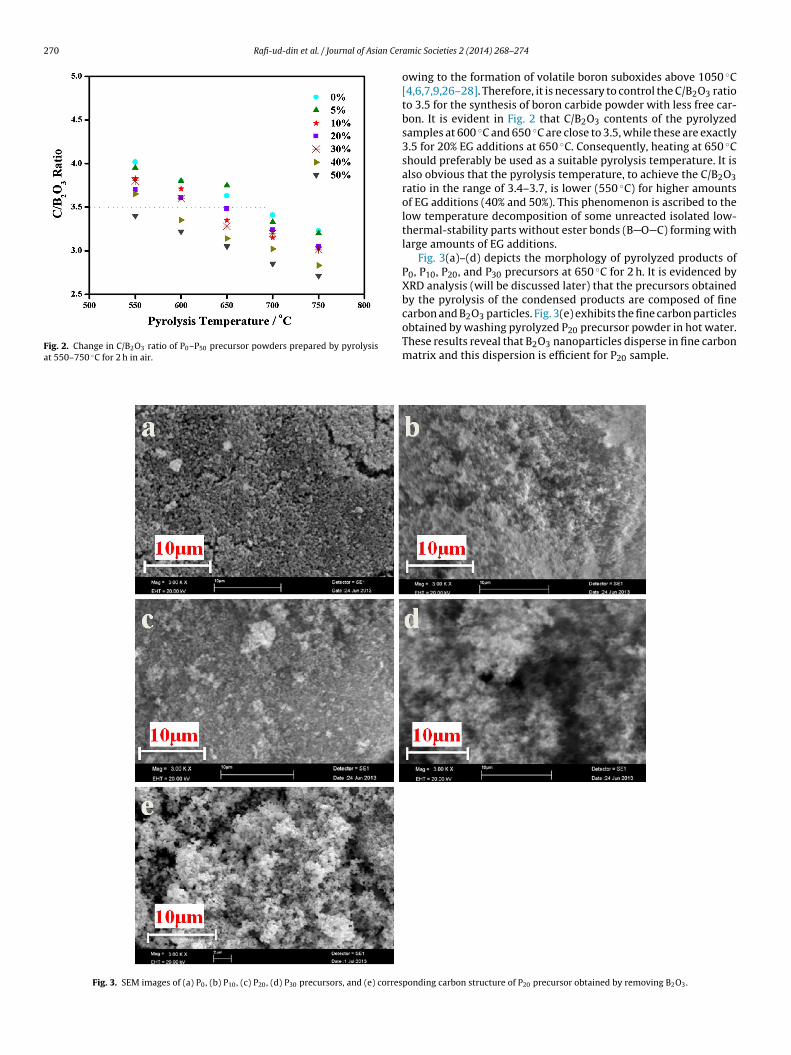
270 Rafi-ud-din et al. / Journal of Asian Cer
Fig. 2. Change in C/B2O3 ratio of P0–P50 precursor powders prepared by pyrolysisat 550–750 ◦C for 2 h in air.
Fig. 3. SEM images of (a) P0, (b) P10, (c) P20, (d) P30 precursors, and (e) corres
amic Societies 2 (2014) 268–274
owing to the formation of volatile boron suboxides above 1050 ◦C[4,6,7,9,26–28]. Therefore, it is necessary to control the C/B2O3 ratioto 3.5 for the synthesis of boron carbide powder with less free car-bon. It is evident in Fig. 2 that C/B2O3 contents of the pyrolyzedsamples at 600 ◦C and 650 ◦C are close to 3.5, while these are exactly3.5 for 20% EG additions at 650 ◦C. Consequently, heating at 650 ◦Cshould preferably be used as a suitable pyrolysis temperature. It isalso obvious that the pyrolysis temperature, to achieve the C/B2O3ratio in the range of 3.4–3.7, is lower (550 ◦C) for higher amountsof EG additions (40% and 50%). This phenomenon is ascribed to thelow temperature decomposition of some unreacted isolated low-thermal-stability parts without ester bonds (B O C) forming withlarge amounts of EG additions.
Fig. 3(a)–(d) depicts the morphology of pyrolyzed products ofP0, P10, P20, and P30 precursors at 650 ◦C for 2 h. It is evidenced byXRD analysis (will be discussed later) that the precursors obtainedby the pyrolysis of the condensed products are composed of finecarbon and B2O3 particles. Fig. 3(e) exhibits the fine carbon particles
obtained by washing pyrolyzed P20 precursor powder in hot water.These results reveal that B2O3 nanoparticles disperse in fine carbonmatrix and this dispersion is efficient for P20 sample.ponding carbon structure of P20 precursor obtained by removing B2O3.
n Cer
oiXBttwtwiit5itBws
Fa
Fm
Rafi-ud-din et al. / Journal of Asia
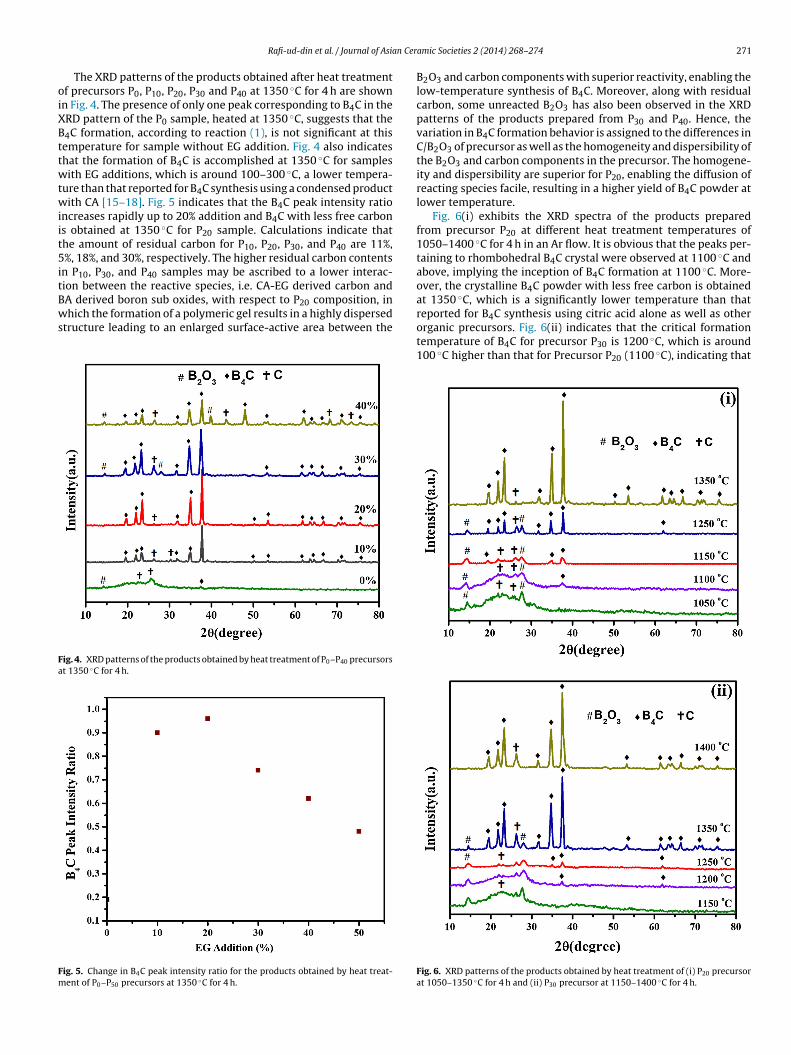
The XRD patterns of the products obtained after heat treatmentf precursors P0, P10, P20, P30 and P40 at 1350 ◦C for 4 h are shownn Fig. 4. The presence of only one peak corresponding to B4C in theRD pattern of the P0 sample, heated at 1350 ◦C, suggests that the4C formation, according to reaction (1), is not significant at thisemperature for sample without EG addition. Fig. 4 also indicateshat the formation of B4C is accomplished at 1350 ◦C for samplesith EG additions, which is around 100–300 ◦C, a lower tempera-
ure than that reported for B4C synthesis using a condensed productith CA [15–18]. Fig. 5 indicates that the B4C peak intensity ratio
ncreases rapidly up to 20% addition and B4C with less free carbons obtained at 1350 ◦C for P20 sample. Calculations indicate thathe amount of residual carbon for P10, P20, P30, and P40 are 11%,%, 18%, and 30%, respectively. The higher residual carbon contents
n P10, P30, and P40 samples may be ascribed to a lower interac-ion between the reactive species, i.e. CA-EG derived carbon and
A derived boron sub oxides, with respect to P20 composition, inhich the formation of a polymeric gel results in a highly dispersedtructure leading to an enlarged surface-active area between the
ig. 4. XRD patterns of the products obtained by heat treatment of P0–P40 precursorst 1350 ◦C for 4 h.
ig. 5. Change in B4C peak intensity ratio for the products obtained by heat treat-ent of P0–P50 precursors at 1350 ◦C for 4 h.
amic Societies 2 (2014) 268–274 271
B2O3 and carbon components with superior reactivity, enabling thelow-temperature synthesis of B4C. Moreover, along with residualcarbon, some unreacted B2O3 has also been observed in the XRDpatterns of the products prepared from P30 and P40. Hence, thevariation in B4C formation behavior is assigned to the differences inC/B2O3 of precursor as well as the homogeneity and dispersibility ofthe B2O3 and carbon components in the precursor. The homogene-ity and dispersibility are superior for P20, enabling the diffusion ofreacting species facile, resulting in a higher yield of B4C powder atlower temperature.
Fig. 6(i) exhibits the XRD spectra of the products preparedfrom precursor P20 at different heat treatment temperatures of1050–1400 ◦C for 4 h in an Ar flow. It is obvious that the peaks per-taining to rhombohedral B4C crystal were observed at 1100 ◦C andabove, implying the inception of B4C formation at 1100 ◦C. More-over, the crystalline B4C powder with less free carbon is obtainedat 1350 ◦C, which is a significantly lower temperature than that
reported for B4C synthesis using citric acid alone as well as otherorganic precursors. Fig. 6(ii) indicates that the critical formationtemperature of B4C for precursor P30 is 1200 ◦C, which is around100 ◦C higher than that for Precursor P20 (1100 ◦C), indicating thatFig. 6. XRD patterns of the products obtained by heat treatment of (i) P20 precursorat 1050–1350 ◦C for 4 h and (ii) P30 precursor at 1150–1400 ◦C for 4 h.
2 an Cer
tecftEiarancSfsa
fac
Fm
F1
72 Rafi-ud-din et al. / Journal of Asi
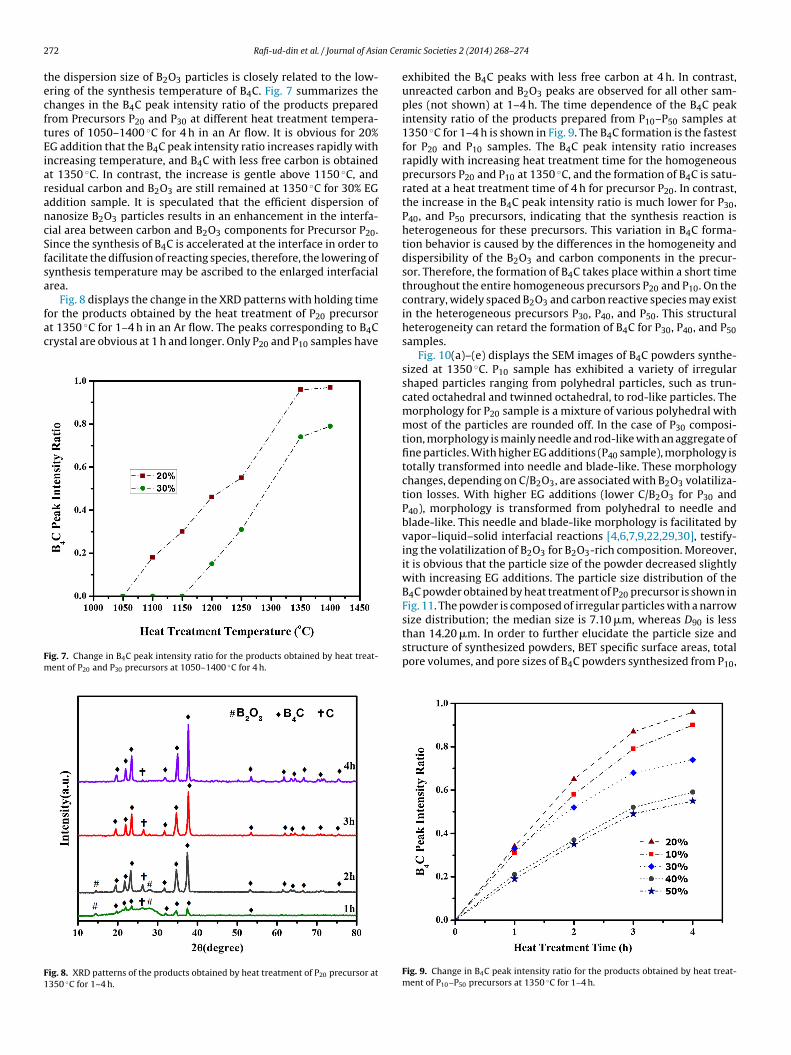
he dispersion size of B2O3 particles is closely related to the low-ring of the synthesis temperature of B4C. Fig. 7 summarizes thehanges in the B4C peak intensity ratio of the products preparedrom Precursors P20 and P30 at different heat treatment tempera-ures of 1050–1400 ◦C for 4 h in an Ar flow. It is obvious for 20%G addition that the B4C peak intensity ratio increases rapidly withncreasing temperature, and B4C with less free carbon is obtainedt 1350 ◦C. In contrast, the increase is gentle above 1150 ◦C, andesidual carbon and B2O3 are still remained at 1350 ◦C for 30% EGddition sample. It is speculated that the efficient dispersion ofanosize B2O3 particles results in an enhancement in the interfa-ial area between carbon and B2O3 components for Precursor P20.ince the synthesis of B4C is accelerated at the interface in order toacilitate the diffusion of reacting species, therefore, the lowering ofynthesis temperature may be ascribed to the enlarged interfacialrea.
Fig. 8 displays the change in the XRD patterns with holding time
or the products obtained by the heat treatment of P20 precursort 1350 ◦C for 1–4 h in an Ar flow. The peaks corresponding to B4Crystal are obvious at 1 h and longer. Only P20 and P10 samples haveig. 7. Change in B4C peak intensity ratio for the products obtained by heat treat-ent of P20 and P30 precursors at 1050–1400 ◦C for 4 h.
ig. 8. XRD patterns of the products obtained by heat treatment of P20 precursor at350 ◦C for 1–4 h.
amic Societies 2 (2014) 268–274
exhibited the B4C peaks with less free carbon at 4 h. In contrast,unreacted carbon and B2O3 peaks are observed for all other sam-ples (not shown) at 1–4 h. The time dependence of the B4C peakintensity ratio of the products prepared from P10–P50 samples at1350 ◦C for 1–4 h is shown in Fig. 9. The B4C formation is the fastestfor P20 and P10 samples. The B4C peak intensity ratio increasesrapidly with increasing heat treatment time for the homogeneousprecursors P20 and P10 at 1350 ◦C, and the formation of B4C is satu-rated at a heat treatment time of 4 h for precursor P20. In contrast,the increase in the B4C peak intensity ratio is much lower for P30,P40, and P50 precursors, indicating that the synthesis reaction isheterogeneous for these precursors. This variation in B4C forma-tion behavior is caused by the differences in the homogeneity anddispersibility of the B2O3 and carbon components in the precur-sor. Therefore, the formation of B4C takes place within a short timethroughout the entire homogeneous precursors P20 and P10. On thecontrary, widely spaced B2O3 and carbon reactive species may existin the heterogeneous precursors P30, P40, and P50. This structuralheterogeneity can retard the formation of B4C for P30, P40, and P50samples.
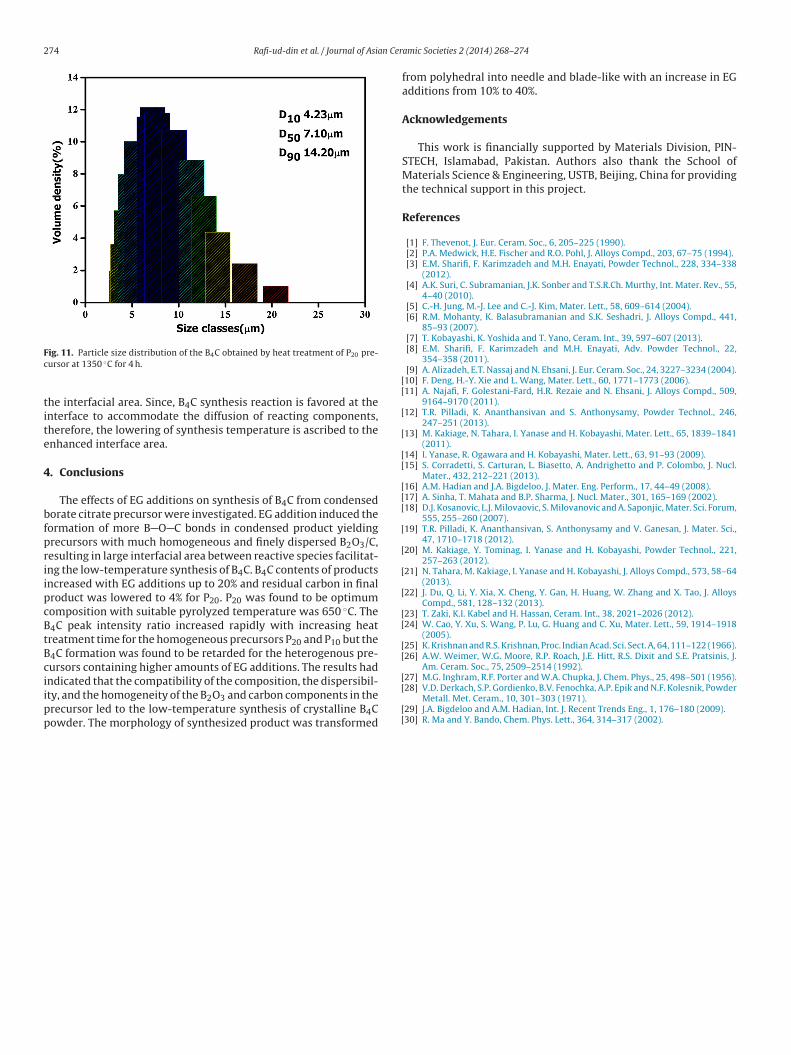
Fig. 10(a)–(e) displays the SEM images of B4C powders synthe-sized at 1350 ◦C. P10 sample has exhibited a variety of irregularshaped particles ranging from polyhedral particles, such as trun-cated octahedral and twinned octahedral, to rod-like particles. Themorphology for P20 sample is a mixture of various polyhedral withmost of the particles are rounded off. In the case of P30 composi-tion, morphology is mainly needle and rod-like with an aggregate offine particles. With higher EG additions (P40 sample), morphology istotally transformed into needle and blade-like. These morphologychanges, depending on C/B2O3, are associated with B2O3 volatiliza-tion losses. With higher EG additions (lower C/B2O3 for P30 andP40), morphology is transformed from polyhedral to needle andblade-like. This needle and blade-like morphology is facilitated byvapor–liquid–solid interfacial reactions [4,6,7,9,22,29,30], testify-ing the volatilization of B2O3 for B2O3-rich composition. Moreover,it is obvious that the particle size of the powder decreased slightlywith increasing EG additions. The particle size distribution of theB4C powder obtained by heat treatment of P20 precursor is shown inFig. 11. The powder is composed of irregular particles with a narrowsize distribution; the median size is 7.10 �m, whereas D90 is less
than 14.20 �m. In order to further elucidate the particle size andstructure of synthesized powders, BET specific surface areas, totalpore volumes, and pore sizes of B4C powders synthesized from P10,Fig. 9. Change in B4C peak intensity ratio for the products obtained by heat treat-ment of P10–P50 precursors at 1350 ◦C for 1–4 h.
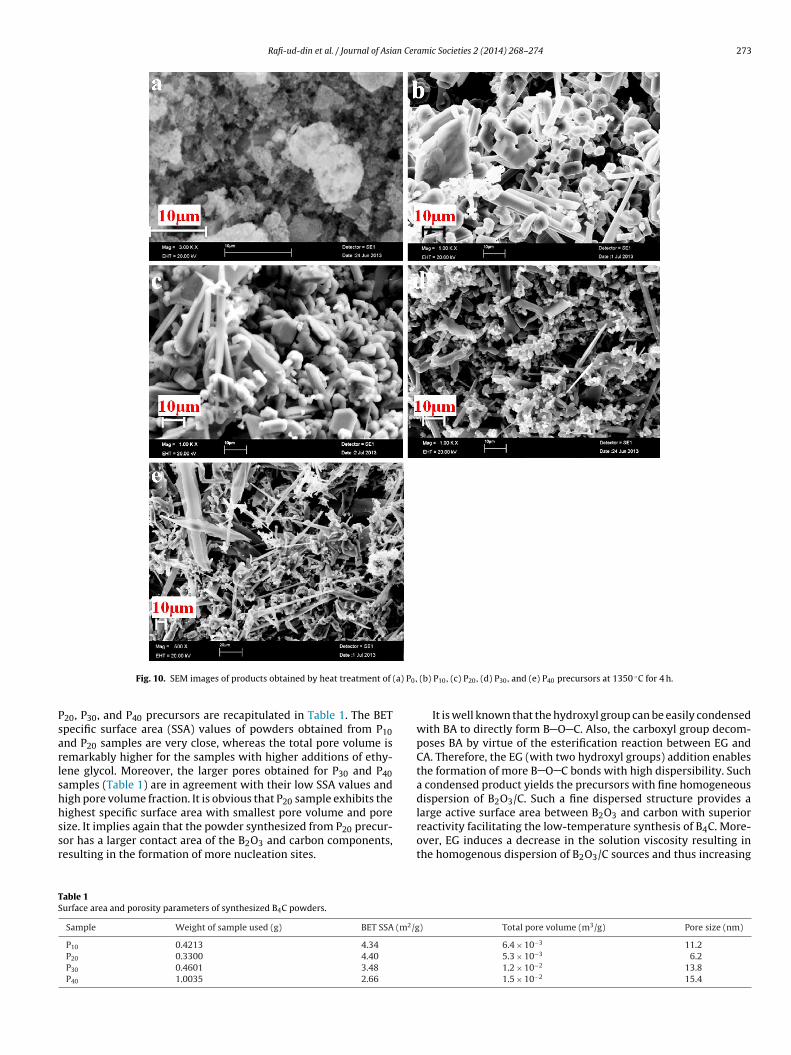
Rafi-ud-din et al. / Journal of Asian Ceramic Societies 2 (2014) 268–274 273
(a) P0,
Psarlshhssr
TS
Fig. 10. SEM images of products obtained by heat treatment of
20, P30, and P40 precursors are recapitulated in Table 1. The BETpecific surface area (SSA) values of powders obtained from P10nd P20 samples are very close, whereas the total pore volume isemarkably higher for the samples with higher additions of ethy-ene glycol. Moreover, the larger pores obtained for P30 and P40amples (Table 1) are in agreement with their low SSA values andigh pore volume fraction. It is obvious that P20 sample exhibits the
ighest specific surface area with smallest pore volume and poreize. It implies again that the powder synthesized from P20 precur-or has a larger contact area of the B2O3 and carbon components,esulting in the formation of more nucleation sites.able 1urface area and porosity parameters of synthesized B4C powders.
Sample Weight of sample used (g) BET SSA (m2/g
P10 0.4213 4.34
P20 0.3300 4.40
P30 0.4601 3.48
P40 1.0035 2.66
(b) P10, (c) P20, (d) P30, and (e) P40 precursors at 1350 ◦C for 4 h.
It is well known that the hydroxyl group can be easily condensedwith BA to directly form B O C. Also, the carboxyl group decom-poses BA by virtue of the esterification reaction between EG andCA. Therefore, the EG (with two hydroxyl groups) addition enablesthe formation of more B O C bonds with high dispersibility. Sucha condensed product yields the precursors with fine homogeneousdispersion of B2O3/C. Such a fine dispersed structure provides a
large active surface area between B2O3 and carbon with superiorreactivity facilitating the low-temperature synthesis of B4C. More-over, EG induces a decrease in the solution viscosity resulting inthe homogenous dispersion of B2O3/C sources and thus increasing) Total pore volume (m3/g) Pore size (nm)
6.4 × 10−3 11.25.3 × 10−3 6.21.2 × 10−2 13.81.5 × 10−2 15.4
274 Rafi-ud-din et al. / Journal of Asian Cer
Fc
tite
4
bfpriipcBtBciipp
[[
[
[
[[
[[[
[
[
[
[
[[
[[
ig. 11. Particle size distribution of the B4C obtained by heat treatment of P20 pre-ursor at 1350 ◦C for 4 h.
he interfacial area. Since, B4C synthesis reaction is favored at thenterface to accommodate the diffusion of reacting components,herefore, the lowering of synthesis temperature is ascribed to thenhanced interface area.
. Conclusions
The effects of EG additions on synthesis of B4C from condensedorate citrate precursor were investigated. EG addition induced theormation of more B O C bonds in condensed product yieldingrecursors with much homogeneous and finely dispersed B2O3/C,esulting in large interfacial area between reactive species facilitat-ng the low-temperature synthesis of B4C. B4C contents of productsncreased with EG additions up to 20% and residual carbon in finalroduct was lowered to 4% for P20. P20 was found to be optimumomposition with suitable pyrolyzed temperature was 650 ◦C. The4C peak intensity ratio increased rapidly with increasing heatreatment time for the homogeneous precursors P20 and P10 but the4C formation was found to be retarded for the heterogenous pre-ursors containing higher amounts of EG additions. The results had
ndicated that the compatibility of the composition, the dispersibil-ty, and the homogeneity of the B2O3 and carbon components in therecursor led to the low-temperature synthesis of crystalline B4Cowder. The morphology of synthesized product was transformed[[
[[
amic Societies 2 (2014) 268–274
from polyhedral into needle and blade-like with an increase in EGadditions from 10% to 40%.
Acknowledgements
This work is financially supported by Materials Division, PIN-STECH, Islamabad, Pakistan. Authors also thank the School ofMaterials Science & Engineering, USTB, Beijing, China for providingthe technical support in this project.
References
[1] F. Thevenot, J. Eur. Ceram. Soc., 6, 205–225 (1990).[2] P.A. Medwick, H.E. Fischer and R.O. Pohl, J. Alloys Compd., 203, 67–75 (1994).[3] E.M. Sharifi, F. Karimzadeh and M.H. Enayati, Powder Technol., 228, 334–338
(2012).[4] A.K. Suri, C. Subramanian, J.K. Sonber and T.S.R.Ch. Murthy, Int. Mater. Rev., 55,
4–40 (2010).[5] C.-H. Jung, M.-J. Lee and C.-J. Kim, Mater. Lett., 58, 609–614 (2004).[6] R.M. Mohanty, K. Balasubramanian and S.K. Seshadri, J. Alloys Compd., 441,
85–93 (2007).[7] T. Kobayashi, K. Yoshida and T. Yano, Ceram. Int., 39, 597–607 (2013).[8] E.M. Sharifi, F. Karimzadeh and M.H. Enayati, Adv. Powder Technol., 22,
354–358 (2011).[9] A. Alizadeh, E.T. Nassaj and N. Ehsani, J. Eur. Ceram. Soc., 24, 3227–3234 (2004).10] F. Deng, H.-Y. Xie and L. Wang, Mater. Lett., 60, 1771–1773 (2006).11] A. Najafi, F. Golestani-Fard, H.R. Rezaie and N. Ehsani, J. Alloys Compd., 509,
9164–9170 (2011).12] T.R. Pilladi, K. Ananthansivan and S. Anthonysamy, Powder Technol., 246,
247–251 (2013).13] M. Kakiage, N. Tahara, I. Yanase and H. Kobayashi, Mater. Lett., 65, 1839–1841
(2011).14] I. Yanase, R. Ogawara and H. Kobayashi, Mater. Lett., 63, 91–93 (2009).15] S. Corradetti, S. Carturan, L. Biasetto, A. Andrighetto and P. Colombo, J. Nucl.
Mater., 432, 212–221 (2013).16] A.M. Hadian and J.A. Bigdeloo, J. Mater. Eng. Perform., 17, 44–49 (2008).17] A. Sinha, T. Mahata and B.P. Sharma, J. Nucl. Mater., 301, 165–169 (2002).18] D.J. Kosanovic, L.J. Milovaovic, S. Milovanovic and A. Saponjic, Mater. Sci. Forum,
555, 255–260 (2007).19] T.R. Pilladi, K. Ananthansivan, S. Anthonysamy and V. Ganesan, J. Mater. Sci.,
47, 1710–1718 (2012).20] M. Kakiage, Y. Tominag, I. Yanase and H. Kobayashi, Powder Technol., 221,
257–263 (2012).21] N. Tahara, M. Kakiage, I. Yanase and H. Kobayashi, J. Alloys Compd., 573, 58–64
(2013).22] J. Du, Q. Li, Y. Xia, X. Cheng, Y. Gan, H. Huang, W. Zhang and X. Tao, J. Alloys
Compd., 581, 128–132 (2013).23] T. Zaki, K.I. Kabel and H. Hassan, Ceram. Int., 38, 2021–2026 (2012).24] W. Cao, Y. Xu, S. Wang, P. Lu, G. Huang and C. Xu, Mater. Lett., 59, 1914–1918
(2005).25] K. Krishnan and R.S. Krishnan, Proc. Indian Acad. Sci. Sect. A, 64, 111–122 (1966).26] A.W. Weimer, W.G. Moore, R.P. Roach, J.E. Hitt, R.S. Dixit and S.E. Pratsinis, J.
Am. Ceram. Soc., 75, 2509–2514 (1992).
27] M.G. Inghram, R.F. Porter and W.A. Chupka, J. Chem. Phys., 25, 498–501 (1956).28] V.D. Derkach, S.P. Gordienko, B.V. Fenochka, A.P. Epik and N.F. Kolesnik, PowderMetall. Met. Ceram., 10, 301–303 (1971).29] J.A. Bigdeloo and A.M. Hadian, Int. J. Recent Trends Eng., 1, 176–180 (2009).30] R. Ma and Y. Bando, Chem. Phys. Lett., 364, 314–317 (2002).





























