Embed Size (px)
Citation preview

© Geneviève Marcoux, 2021
Étude du potentiel inflammatoire et immunitaire des vésicules extracellulaires dérivées des plaquettes
Thèse
Geneviève Marcoux
Doctorat en microbiologie-immunologie
Philosophiæ doctor (Ph. D.)
Québec, Canada

ii
Résumé
Les plaquettes sont très abondantes dans le sang où elles ont un rôle dans
l'hémostase, l’inflammation et l’immunité. Activées, elles vont subir un changement
de conformation qui permet la libération de nombreuses molécules effectrices ainsi
que la production de vésicules extracellulaires (EV). Les EV sont formées par le
bourgeonnement de la plaquette et emportent une partie de son contenu, incluant
des acides nucléiques, des protéines de surface et des organelles. Hétérogène, le
contenu diversifié des EV de plaquettes suggère qu’elles peuvent exercer de
nombreuses fonctions.
Dans le cadre de cette thèse, nous nous sommes d’abord intéressés à l’utilisation
de l’algorithme SPADE couplé à la cytométrie en flux pour améliorer la détection
d’EV et mieux apprécier leur hétérogénéité dans des concentrés plaquettaires (PC)
servant à la transfusion. Nous avons aussi évalué l’utilisation de cette approche pour
le développement de biomarqueurs dans le cadre de l’analyse d’EV dans le liquide
synovial de patients arthritiques. Cette étude a révélé que l’utilisation d’algorithmes
couplés à la cytométrie en flux tels que SPADE pourrait faciliter la compréhension
des fonctions des EV et le développement de leur étude comme biomarqueurs.
Nous nous sommes ensuite intéressés à une première sous-catégorie d’EV de
plaquettes, celles qui contiennent des mitochondries (mito+EV). Nous avons émis
l'hypothèse que ces mito+EV représentent un réservoir d'ADN mitochondrial, signal
de danger (motifs moléculaires associés aux dommages, DAMP) reconnu et présent
dans de nombreuses conditions inflammatoires, et qu’elles pourraient être
impliquées dans les réactions transfusionnelles. Nous avons observé que les
mito+EV étaient plus abondantes dans les PC impliqués dans des réactions
transfusionnelles et qu’elles corrèlent significativement avec l’ADN mitochondrial.
Comme la majorité de l’ADN mitochondrial est encapsulée dans des EV, cette étude
suggère que les EV peuvent être un biomarqueur utile pour la prédiction du risque
potentiel de réactions transfusionnelles, bien que des investigations soient

iii
nécessaires pour déterminer s'il existe un rôle pathogène causal du DAMP
mitochondrial encapsulé dans les EV par opposition à l'ADN mitochondrial en
solution.
Nous nous sommes enfin intéressés à une seconde sous-catégorie d’EV de
plaquettes, celles qui contiennent du protéasome. Les plaquettes contiennent un
protéasome actif et présentent des antigènes par l'intermédiaire du complexe majeur
d’histocompatibilité de classe I (CMH I) ce qui leur confère une fonction dans
l’immunité adaptative. Étant donné qu’il n’a jamais été examiné si le protéasome est
également transféré dans les EV de plaquettes auparavant, nous avons émis
l'hypothèse qu'une machinerie fonctionnelle pour l'apprêtement et la présentation de
l'antigène est transférée aux EV de plaquettes. En utilisant une combinaison
d'analyses moléculaires et fonctionnelles, nous avons montré la présence d'un
protéasome 20S actif dans diverses conditions où les plaquettes sont activées, ainsi
que du CMH I et des molécules de coactivation des lymphocytes. L’incubation d’EV
de plaquettes avec l’ovalbumine (OVA) a mis en évidence, par l'activation et la
prolifération des lymphocytes T CD8+ spécifiques de l'antigène OVA, qu’elles
peuvent apprêter et présenter efficacement l’antigène. Ces résultats suggèrent que
les EV de plaquettes contribuent à l'immunité adaptative.
Dans l’ensemble, nous avons montré que les EV de plaquettes ont un rôle dans
l’inflammation et l’immunité avec leur contenu en mitochondries et en protéasome.
Elles sont hétérogènes et peuvent être utilisées comme biomarqueurs dans
différents contextes.

iv
Abstract
Platelets are very abundant in the blood where they play a role in hemostasis,
inflammation and immunity. Activated, platelets will undergo a change of
conformation which allows the release of numerous effector molecules as well as
the production of extracellular vesicles (EVs). EVs are formed by the budding of the
platelet and bring some of its contents, including nucleic acids, surface proteins and
organelles. Heterogeneous, the diverse content of platelet EVs suggests that they
can perform many functions. As part of this thesis, we first looked at the use of the
SPADE algorithm coupled with flow cytometry to improve the detection of EVs and
allow a better appreciation of their heterogeneity in platelet concentrates (PC) used
for transfusion. We also evaluated the use of this approach for the development of
biomarkers in the analysis of EVs in the synovial fluid of arthritis patients. This study
revealed that the use of algorithms such as SPADE coupled with flow cytometry
could facilitate the understanding of the functions of EVs and the development of
their studies as biomarkers. We then looked at a first subcategory of platelet EV,
those that contain mitochondria (mito+EVs). We hypothesized that these mito+EVs
represent a reservoir of mitochondrial DNA, a damage-associated molecular pattern
(DAMP) recognized and present in many inflammatory conditions, and that they
could be involved in transfusion reactions. We observed that mito+EVs were more
abundant in PCs involved in transfusion reactions and that they correlate significantly
with mitochondrial DNA. As the majority of mitochondrial DNA is encapsulated in
EVs, this study suggests that EVs may be a useful biomarker for predicting the
potential risk of transfusion reactions, although further investigation is needed to
determine if there is a pathogenic role of mitochondrial DAMP encapsulated in EVs
as opposed to mitochondrial DNA in solution.
We finally focused on a second subcategory of platelet EVs, those that contain
proteasome. Platelets contain an active proteasome and present antigens through
the major histocompatibility complex I (MHC I), which gives them an important
function in adaptive immunity. Whether the proteasome is also transferred into the

v
platelet EVs has never been examined. We hypothesized that a functioning
machinery for the processing and presentation of the antigen is transferred to the
platelet EVs. Using a combination of molecular and functional assays, we have
demonstrated the presence of an active 20 S proteasome under various conditions
where platelets are activated, as well as MHC I and lymphocyte coactivation
molecules. Demonstrated by activation and proliferation of ovalbumin (OVA) antigen
specific CD8 + T lymphocytes, EVs from platelets incubated with OVA can efficiently
prepare and present antigen. These results suggest that platelet EVs contribute to
adaptive immunity.
Overall, we have shown that platelet EVs have a role in inflammation and immunity
with their mitochondria and proteasome content. They are heterogeneous and can
be used as biomarkers in different contexts.

vi
Table des matières
Résumé ........................................................................................................................................ ii
Abstract ...................................................................................................................................... iv
Table des matières ................................................................................................................. vi
Liste des figures ..................................................................................................................... ix
Liste des tableaux .................................................................................................................. xi
Liste des abréviations ......................................................................................................... xii
Remerciements ..................................................................................................................... xvi
Avant-propos ........................................................................................................................ xvii
Introduction ............................................................................................................................... 1 1.1 Les plaquettes ................................................................................................................................. 1
1.1.1 Description générale ............................................................................................................................1 1.1.2 À l’origine des plaquettes : les mégacaryocytes ...................................................................3 1.1.3 Rôles des plaquettes dans l’hémostase ....................................................................................5
1.1.3.1 Coagulation ........................................................................................................................................................... 5 1.1.3.2 Transfusion de plaquettes .............................................................................................................................. 7 1.1.3.3 Transfusion de plaquettes : les risques associés .................................................................................. 8 1.1.3.4 Les risques infectieux ....................................................................................................................................... 8 1.1.3.5 Les risques non infectieux .............................................................................................................................. 9 1.1.3.6 Facteurs connus à ce jour ............................................................................................................................ 10
1.1.4 Rôles des plaquettes et des mégacaryocytes dans l’immunité .................................. 11 1.1.4.1 Notions d’immunologie ................................................................................................................................ 12 1.1.4.2 Les plaquettes dans l’inflammation et l’immunité innée .............................................................. 20 1.1.4.3 Les mégakaryocytes dans l’immunité innée ....................................................................................... 24 1.1.4.4 Support des cellules de l’immunité adaptative par les plaquettes ............................................ 25 1.1.4.5 Apprêtement et présentation de l'antigène par les plaquettes .................................................. 27 1.1.4.6 Apprêtement et présentation de l'antigène par les mégacaryocytes....................................... 31
1.2 Les vésicules extracellulaires ................................................................................................ 34 1.2.1 Description générale et mécanisme de formation ............................................................. 34 1.2.2 Isolation et détection des vésicules extracellulaires. ....................................................... 37 1.2.3 Diversité des vésicules extracellulaires .................................................................................. 38 1.2.4 Organelles contenues dans les vésicules extracellulaires ............................................ 39
1.2.4.1 La mitochondrie ............................................................................................................................................... 39 1.2.4.2 Le protéasome .................................................................................................................................................. 41
1.2.5 Les vésicules extracellulaires de plaquettes ........................................................................ 41 1.3 Objectifs ........................................................................................................................................... 43
Chapitre 1: Revealing the diversity of extracellular vesicles using high-dimensional flow cytometry analyses. ........................................................................ 45
2.1 Résumé ............................................................................................................................................ 45 2.2 Abstract ............................................................................................................................................ 47 2.3 Introduction .................................................................................................................................... 48 2.4 Methods ........................................................................................................................................... 51 2.5 Results .............................................................................................................................................. 55 2.6 Discussion ...................................................................................................................................... 63

vii
2.7 References ...................................................................................................................................... 66 2.8 Figures and legends ................................................................................................................... 72 2.8 Supplementary figures and legends ................................................................................... 79 2.9 Supplementary tables ................................................................................................................ 83
Chapitre 2: Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions. .................................................................................................................................. 85
3.1 Résumé ............................................................................................................................................ 85 3.2 Abstract ........................................................................................................................................... 87 3.3 Introduction .................................................................................................................................... 88 3.4 Methods ........................................................................................................................................... 92 3.5 Results ............................................................................................................................................. 96 3.6 Discussion ................................................................................................................................... 100 3.7 References ................................................................................................................................... 104 3.8 Figures and legends ................................................................................................................ 111 3.9 Tables ............................................................................................................................................ 116 3.10 Supplementary methods ..................................................................................................... 120 3.11 Supplementary tables .......................................................................................................... 122 3.12 Supplementary references ................................................................................................. 129
Chapitre 3: Platelet-derived extracellular vesicles contain an active proteasome involved in protein processing for antigen presentation via class I major histocompatibility molecules ........................................................................ 130
4.1 Résumé ......................................................................................................................................... 130 4.2 Abstract ........................................................................................................................................ 133 4.3 Introduction ................................................................................................................................. 134 4.4 Methods ........................................................................................................................................ 136 4.5 Results .......................................................................................................................................... 138 4.6 Discussion ................................................................................................................................... 145 4.7 References ................................................................................................................................... 149 4.8 Figures and legends ................................................................................................................ 155 4.9 Supplementary methods ....................................................................................................... 166 4.10 Supplementary figures ........................................................................................................ 173 4.11 Supplementary references ................................................................................................. 175
Discussion ............................................................................................................................ 176 5.1 Mise en contexte ....................................................................................................................... 176 5.2 Résumé des découvertes et discussion ........................................................................ 177
Conclusion ............................................................................................................................ 185
Bibliographie ........................................................................................................................ 186
Annexe I: Mitochondrial damage-associated molecular patterns in blood transfusion products ........................................................................................................ 212
Abstract ................................................................................................................................................ 213 Mitochondria and inflammation ................................................................................................. 214 Mitochondrial release..................................................................................................................... 216 Extracellular mitochondria and adverse reactions ........................................................... 218 Extracellular mitochondria as a biomarker .......................................................................... 220 Summary ............................................................................................................................................. 221

viii
Acknowledgements ......................................................................................................................... 222 References .......................................................................................................................................... 223
Annexe II: Role of platelets and megakaryocytes in adaptive immunity .... 226 Abstract ................................................................................................................................................ 227 Introduction ........................................................................................................................................ 228 Platelet-leukocyte Interactions and Implications for Adaptive Immunity ................ 230 Antigen Processing and Presentation by Platelets........................................................... 232 Antigen Processing and Presentation by Megakaryocytes........................................... 236 Platelets Response to Antibodies ............................................................................................ 238 Conclusion and Perspectives ..................................................................................................... 245 References .......................................................................................................................................... 247
Annexe III: Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration ................................................... 256
Abstract ................................................................................................................................................ 258 Introduction ........................................................................................................................................ 259 Results .................................................................................................................................................. 261 Discussion .......................................................................................................................................... 278 Materials and Methods ................................................................................................................... 283 Acknowledgments ........................................................................................................................... 287 References .......................................................................................................................................... 288
Annexe IV: Autoantibodies in Systemic Lupus Erythematosus Target Mitochondrial RNA ............................................................................................................. 291
Abstract ................................................................................................................................................ 292 Introduction ........................................................................................................................................ 293 Materials and Methods ................................................................................................................... 297 Results .................................................................................................................................................. 303 Discussion .......................................................................................................................................... 313 References .......................................................................................................................................... 318
Annexe V: Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations ............. 323
Abstract ................................................................................................................................................ 324 Introduction ........................................................................................................................................ 325 Results .................................................................................................................................................. 330 Discussion .......................................................................................................................................... 341 Material and Methods ..................................................................................................................... 345 Acknowledgements ......................................................................................................................... 357 References .......................................................................................................................................... 358

ix
Liste des figures
Introduction Figure 1: Cascade de coagulation .......................................................................... 6 Figure 2: Vue générale de la cascade du complément ........................................ 13 Figure 3: Les sites de liaison du CMH I au CD8 et du CMH II au CD4 ................. 15 Figure 4: Structure du protéasome et de l’immunoprotéasome ............................ 17 Figure 5: Apprêtement du peptide et présentation par le CMH I .......................... 19 Figure 6: Les plaquettes soutiennent les cellules de l’immunité adaptative et présentent l’antigène ............................................................................................. 31 Figure 7: Les mégacaryocytes sont des cellules immunitaires ............................. 33 Figure 8: Échelle de taille des vésicules extracellulaire ........................................ 35 Figure 9: DAMPs dérivés de la mitochondrie ....................................................... 40 Chapitre 1 Figure 1. Characterization of extracellular vesicles .............................................. 72 Figure 2. Detection of platelet EV subpopulations by hs-FCM ............................. 73 Figure 3. SPADE analysis of human blood cells and EVs .................................... 75 Figure 4. SPADE tree to assess EVs in cellular perturbations ............................. 76 Figure 5. SPADE overlapping panels for EV diversity in plasma .......................... 77 Figure 6. SPADE and EV-based biomarkers in disease ....................................... 78 Supplementary Figure 1. Detection of red blood cell subpopulations by hs-FCM ... .............................................................................................................................. 79 Supplementary Figure 2. Protein content in cells and EVs from plasma, platelets and RBCs .............................................................................................................. 81 Supplementary Figure 3. Initial gating before performing SPADE analysis ........ 82 Chapitre 2 Figure 1. Assessment of platelet EV subpopulation by high-sensitivity flow cytometry............................................................................................................. 111 Figure 2. Platelet EV subpopulation levels are higher in platelet concentrates involved in adverse transfusion reaction ............................................................. 113 Figure 3. The majority of the mitochondrial DNA in platelet concentrates is present in EVs .................................................................................................................. 114 Figure 4. Levels of soluble CD62P, but not soluble CD40L, correlate with mitochondria containing platelet EVs .................................................................. 115

x
Chapitre 3 Figure 1. Platelets and PEVs contain proteasome .............................................. 155 Figure 2. Identification of proteasome-containing PEVs under physiological and pathological conditions ........................................................................................ 157 Figure 3. Platelets and PEVs load and process OVA onto MHC-I ...................... 158 Figure 4. PEVs in blood circulation can reach lymphoid organs and circulate in lymph ................................................................................................................... 159 Figure 5. Platelet and PEVs with loaded OVA peptide express activation and co-stimulatory molecules .......................................................................................... 161 Figure 6. PEVs can induce antigen-specific T cell activation and cytokine production through antigen presentation ............................................................................... 163 Figure 7. PEVs loaded with native OVA process and present OVA peptide to induce antigen-specific T cell lymphoproliferation ........................................................... 164 Supplementary Figure 1. Gate design for EV analysis ............................................ ............................................................................................................................ 173 Supplementary Figure 2. Characterization of proteasome PEV diversity using a fluorescent proteasomal activity-based probe ..................................................... 174

xi
Liste des tableaux
Chapitre 1 Supplementary Table 1. Staining panels for the different SPADE tree ............... 83 Supplementary Table 2. Demographic and clinical characteristics of RA patients .............................................................................................................................. 84 Chapitre 2 Table 1. Optimal cut-off values for EV subtypes as determined by Youden’s method ............................................................................................................................ 116 Table 2. EV subtype associations with soluble CD40L (n=92) and soluble CD62P (n=96) in platelet concentrates ............................................................................ 117 Table 3. Quantification of mitochondrial DNA, soluble CD40L and soluble CD62P in platelet concentrates ........................................................................................... 118 Table 4. Optimal cut-off values for sCD40L and sCD62P as determined by Youden’s method (Healthy donors: n=50, reaction n=48) ................................................... 119 Supplementary Table 1. Adverse transfusion reactions associated platelet concentrates........................................................................................................ 122 Supplementary Table 2. Qualitative information of the transfusion product ...... 123 Supplementary Table 3. Quantitative information on the donors and on the platelet concentrates........................................................................................................ 124 Supplementary Table 4. Quantification of EV subtypes in platelet concentrates ............................................................................................................................ 125 Supplementary Table 5. Cubic effect of EVs on adverse reaction .................... 126 Supplementary Table 6. Predictive values of EV subtypes adjusted for the different adverse reactions ................................................................................................ 127 Supplementary Table 7. EV subtype associations with mitochondrial DNA in platelet concentrates (n= 35) ............................................................................... 128

xii
Liste des abréviations
ACD (Acid-citrate-dextrose)
ADN Acide désoxyribonucléique
ADP Adénosine diphosphate
AHTR Réaction hémolytique aiguë (Acute Hemolytic Transfusion Reaction)
APC Cellules présentatrice d’antigène (Antigen presenting cells)
ARN Acide ribonucléique
ATP Adénosine triphosphate
β2-MG Microglobuline β2
BAL (Bronchoalveolar lavages)
BC Couche leucoplaquettaire (Buffy-Coat)
BCR Récepteur des cellules B (B cell receptor)
CAP (Carboxy(alkylpyrrole) protein adducts)
CD40L CD40 Ligand
CD62P Sélectine P
CLR Récepteurs lectine membranaire de type C (C-type Lectin Receptor)
CMFDA (Chloromethylfluorescein diacetate)
CMH Complexe majeur d’histocompatibilité
CPS-I Carbamoyl-phosphate synthétase I
CRP-XL (Cross-linked collagen-related peptide)
DAMPs Motifs moléculaires associés aux dommages (Damage-Associated
Molecular Patterns)
DC Cellules dendritiques (Dendritic cells)
DHTR Réaction hémolytique retardée (Delayed Hemolytic Transfusion
Reaction)
ERAAP (Endoplasmic reticulum amino peptidase)
ESCRT Complexe de tri endosomal nécessaire au transport (Endosomal
Sorting Complex Required for Transport)
EV Vésicule extracellulaire (Extracellular Vesicle)
FCM (Flow cytometry)

xiii
FNHTR Réaction fébrile non-hémolytique (Febrile Non-Hemolytic Transfusion
Reaction)
GPVI Glycoprotéine VI
HBSS (Hanks' Balanced Salt Solution)
HLA Antigènes leucocytaires humains (Human Leukocyte Antigen)
HMGB1 (High-mobility group box 1)
hs-FCM (high-sensitivity FCM)
HSP 70 (Heat shock protein 70)
IFN Interféron
Ig Immunoglobuline
IL Interleukine
ITP Thrombocytopénie immunitaire (Immune thrombocytopenia)
LPS Lipopolysaccharide
LBP Protéine liant le LPS (LPS binding protein)
Mac-1 lntégrine CD11b/CD18 (Macrophage-1 antigen)
MD-2 (Myeloid differentiation factor 2)
MIP (Macrophage Inflammatory Protein)
miRNA micro ARN
MK Mégacaryocytes (Megakaryocytes)
mRNA ARN messager
mtDNA ADN mitochondrial
MV Microvésicule
NET (Neutrophil extracellular traps)
NK Cellules tueuses naturelles (Natural killers)
NOD Nucleotide-binding Oligomerization Domain
OCS Système canaliculaire ouvert (Open Canalicular System)
ODN Oligodésoxyribonucléotides
OVA Ovalbumine
PAMP Motifs moléculaires associés aux pathogènes (Pathogen-associated
molecular patterns)
PBS (Phosphate buffered saline)
xiv
PC Concentrés plaquettaires (Platelet Concentrates)
PDGF (Platelet Derived Growth Factor)
PEV Vésicules extracellulaires de plaquettes (platelet-derived EV)
PF4 Facteur plaquettaire 4 (Platelet factor 4)
PLC Complexe chargeant le peptide (Peptide Loading Complex)
PMA (Phorbol myristate acetate)
PRP Plasma riche en plaquettes
PS Phosphatidylsérine
PSGL1 Ligand-1 de la glycoprotéine CD62P
PVDF (Polyvinylidene difluoride)
RANTES (Regulated on Activation, Normal T Cell Expressed and Secreted)
ROS Espèces réactives d’oxygène (Reactive oxygen species)
sCD40L CD40 Ligand soluble
SLC44A2 (Solute carrier family 44 member 2)
SLE Lupus érythémateux disséminé (Systemic Lupus Erythematosus)
SPADE (Spanning-tree Progression Analysis of Density-normalized Events)
TACO Surcharge circulatoire liée à la transfusion (Transfusion Associated
Circulatory Overload)
TA-GVHD Réaction du greffon contre l’hôte (Transfusion-Associated Graft-
Versus-Host Disease)
TAP Protéines de transport associées à l’antigène (Transporters
associated with Antigen Processing)
TCR Récepteur des cellules T (T cell receptor)
TEM (Transmission Electron Microscopy)
TF Facteur tissulaire (Tissue Factor)
TGF (Transforming growth factor)
TLR Récepteurs de type Toll (Toll-like receptors)
TRALI Syndrome respiratoire aigu post-transfusionnel (Transfusion-Related
Acute Lung Injury)
Tregs Lymphocytes T régulateurs
TNF Facteur de nécrose tumorale (Tumor necrosis factor)

xv
VIH Virus de l’immunodéficience humaine
VHB Virus de l’hépatite B
VHC Virus de l’hépatite C
vWf Facteur von Willebrand

xvi
Remerciements
Je tiens d'abord à remercier tout particulièrement le Dr Éric Boilard pour son
encadrement lors des sept dernières années. Merci de m’avoir encouragée à faire
une thèse à la suite de ma maitrise sur un sujet qui me passionne, pour la confiance
témoignée tout au long de mes études et pour m’avoir donné la liberté et l’autonomie
dont j’avais besoin. Merci aussi pour les multiples occasions de formations, congrès
et activités auxquelles j’ai pu assister sous sa supervision, ainsi que pour les
nombreuses activités d’équipe organisées sous son initiative.
Je remercie aussi le Dr Benoit Vingert et Marie Tamagne de m’avoir accueillie dans
leur laboratoire et de m’avoir fait une place dans leur famille. Les quelques mois
passés avec eux à Paris ont été exceptionnels et m’ont permis d’évoluer au sein
d’une dynamique de laboratoire complètement différente et de bénéficier de leur
expérience sur de nouvelles techniques et de nouvelles idées. J'aimerais également
remercier Tania Lévesque, une personne et une collègue exceptionnelle qui est
maintenant devenue une amie. J’aimerais aussi souligner Stephan, Anne, Yann,
Audrée, Julien, Andréa, Étienne, Jonathan, Anne-Claire et Vanessa sans qui la vie
au laboratoire n’aurait pas été aussi agréable. J'aimerais exprimer toute ma
reconnaissance à Isabelle Allaeys, Nathalie Cloutier et Emmanuelle Rollet-Labelle
pour leur encadrement et leur aide, ainsi que tous les autres membres de l’équipe
Boilard. Ils ont été une source importante de conseils et d'informations et des gens
avec qui il est agréable de travailler. J’inclus aussi tous les chercheurs, assistants
de recherche et étudiants de l’étage, ainsi que les membres du comité de
programme. Je remercie également la Société Canadienne du Sang, Mitacs et les
autres organismes ayant financé le projet.
Enfin, je souhaite exprimer toute ma gratitude à mes proches pour leur appui et pour
m’avoir toujours encouragé dans la poursuite de mes études. Merci aussi à mon
petit chat. Ton amour, ta confiance en moi et ton soutien m'ont permis de persévérer
et de surpasser mes objectifs personnels et professionnels.

xvii
Avant-propos
Le Dr Éric Boilard a conçu et dirigé le projet de recherche. Il a participé à l’analyse
des données et corrigé les articles des chapitres 1, 2 et 3. Le Dr Benoit Vingert a
codirigé une partie du projet mentionné au chapitre 3 de cette thèse.
J’ai conçu et réalisé les expériences, analysé et interprété les données, réalisé les
analyses statistiques et écrit les manuscrits des articles présentés aux chapitre 2 et
4 de ce document en collaboration avec les auteurs mentionnés ci-dessous.
Concernant l’article présent au chapitre 3, j’ai participé principalement à la
conception, l’analyse et l’interprétation des expériences pour les figures 1 et 2 de
l’article et pour la rédaction du manuscrit.
L’article qui constitue le chapitre 1, intitulé Revealing the diversity of extracellular
vesicles using high-dimensional flow cytometry analyses a été publié dans le
journal Scientific Reports en 2016 :
Marcoux, G., Duchez, A. C., Cloutier, N., Provost, P., Nigrovic, P. A., Boilard, E. (2016). Revealing the diversity of extracellular vesicles using high-dimensional flow cytometry analyses. Scientific Reports. 6:35928.
L’article qui constitue le chapitre 2, intitulé Platelet-derived extracellular vesicles
convey mitochondrial DAMPs in platelet concentrates and their levels are
associated with adverse reactions a été publié dans le journal Transfusion en
2019 :
Marcoux G, Magron A, Sut C, Laroche A, Laradi S, Hamzeh-Cognasse H, Allaeys I, Cabon O, Julien AS, Garraud O, Cognasse F, Boilard E. (2019). Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions. Transfusion. Apr 11. doi: 10.1111/trf.15300.

xviii
L’article qui constitue le chapitre 3, intitulé Platelet-derived extracellular vesicles
contain an active proteasome involved in protein processing for antigen
presentation via class I major histocompatibility molecules, est présentement
en révision pour le journal Blood (BLD-2020-009957):
Marcoux, G. Laroche, A., Hasse, S., Tamagne, M., Zufferey, A., Lévesque,T., Allaeys, I., Rebetz,J. Karakeussian-Rimbaud, A., Turgeon, J., Bourgoin,S.G., Hamzeh-Cognasse, H., Cognasse, F., Kapur, R., Semple, J.W., Hébert, M.J., Pirenne, F., Overkleeft, H.S., Florea, B.I., Dieude, M., Vingert, B. and Boilard, E. (2021) Platelet-derived extracellular vesicles contain an active proteasome involved in protein processing for antigen presentation via class I major histocompatibility molecules. Blood (BLD-2020-009957)
Au cours de mon doctorat, j’ai collaboré à la rédaction de deux revues de littérature.
La première qui est intitulée Mitochondrial damage-associated molecular
patterns in blood transfusion products est présente en Annexe I :
Marcoux, G. and Boilard, E. (2017). Mitochondrial damage-associated molecular patterns in blood transfusion products. ISBT Science Series. 12(4): 501-505. La seconde, intitulée Role of platelets and megakaryocytes in adaptive immunity, a été écrite en parallèle avec l’introduction de cette thèse, est présente en Annexe II et vient d’être publiée dans le journal Platelets : Marcoux, G., Laroche, A., Romero, J.E. and Boilard, E. (2020) Role of platelets and megakaryocytes in adaptive immunity. Platelets 2020: p. 1-12. doi: 10.1080/09537104.2020.1786043
J’ai aussi collaboré significativement à différents projets qui n’apparaissent pas dans
cette thèse. Parmi ceux qui ont été publiés, j’ai notamment contribué pour le modèle
LPS et l’imagerie en microscopie deux photons dans cet article en Annexe III qui
s’est mérité le prix PNAS Cozzarelli Prize 2018:
Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, Levesque T, Becker Y, Tessandier N, Melki I, Zhi H, Poirier G, Rondina MT, Italiano JE, Flamand L, McKenzie SE, Cote F, Nieswandt B, Khan WI, Flick MJ, Newman, PJ, Lacroix S, Fortin P, Boilard E. (2018). Platelets release pathogenic serotonin and return to

xix
circulation after immune complex-mediated sequestration. Proc Natl Acad Sci USA.115(7): 1550-1559.
J’ai aussi participé au projet sur l’étude des anticorps antimitochondriaux qui a mené
jusqu’à ce jour à la publication de deux articles en Annexes IV et V :
Becker Y, Marcoux G, Allaeys I, Julien A-S, Loignon R-C, Benk-Fortin H, Rollet-Labelle E, Rauch J, Fortin PR and Boilard E (2019). Autoantibodies in Systemic Lupus Erythematosus Target Mitochondrial RNA. Front. Immunol. 10:1026. doi: 10.3389/fimmu.2019.01026
Becker Y, Loignon RC, Julien AS, Marcoux G, Allaeys I, Lévesque T, Rollet-Labelle E, Benk-Fortin H, Cloutier N, Melki I, Eder L, Wagner É, Pelletier M, Hajj HE, Tremblay MÈ, Belleannée C, Hébert MJ, Dieudé M, Rauch J, Fortin PR, Boilard E. (2019). Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep. Mar 14;9(1):4530.
Enfin, j’ai aussi contribué à ces articles publiés depuis mon arrivée dans le
laboratoire du Dr Boilard :
Boudreau,L.H., Duchez,A.C., Cloutier,N., Soulet,D., Martin,N. Bollinger,J., Pare,A., Rousseau,M., Naika,G.S., Lévesque,T., Laflamme,C., Marcoux, G., Lambeau,G., Farndale,R.W., Pouliot,M., Hamzeh-Cognasse,H., Cognasse,F., Garraud,O., Nigrovic,P.A., Guderley,H., Lacroix,S., Thibault,L., Semple,J.W., Gelb,M.H. and Boilard,E (2014) Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation, Blood : 2014 Oct 2;124(14):2173-83. doi: 10.1182/blood-2014-05-573543. Marcoux, G., Duchez,A.C.,Rousseau,M., Lévesque,T., Boudreau,L.H., Thibault, L. andBoilard,E. (2016): Microparticle and mitochondrial release during extended storage of different types of platelet concentrates, Platelets, DOI: 10.1080/09537104.2016.1218455 Boudreau, L.H., G. Marcoux and E. Boilard, Platelet microparticles in transfusion
(2015). Vox Sanguinis, ISBT Science Series, 10: 305–308. doi: 10.1111/voxs.12142


1
Introduction
1.1 Les plaquettes
Giulio Bizzozero a décrit en 1882 un nouvel élément du sang ayant un rôle important
pour la thrombose et la coagulation. Maintenant connues sous le nom de
plaquettes[1], ces dernières circulent dans le sang pour une période d’environ 8 à
10 jours afin de détecter les vaisseaux endommagés[2, 3]. Il s’est écoulé près de
trente ans avant que James Homer Wright fasse la démonstration en 1910 que les
plaquettes sont dérivées des mégacaryocytes (MK-Megakaryocytes) de la moelle
osseuse[4, 5].
1.1.1 Description générale
Chez les mammifères, ces fragments anucléés de petite taille (environ 2 µm de
diamètre) sont rarement reconnus comme étant des cellules. Ne possédant pas de
noyau ni d’ADN génomique, les plaquettes héritent toutefois de l’ARN messager, de
microARN, de fragments de Golgi et de la machinerie nécessaire pour la traduction
de leurs précurseurs, ce qui leur confère la possibilité de synthétiser des
protéines[6]. De plus, le cytoplasme et des organelles telles que les granules ou les
mitochondries qui sont contenus dans la plaquette proviennent aussi des MK[6].
La membrane plasmique de la plaquette est constituée d’une bicouche de
phospholipides et possède un glycocalix qui est recouvert de glycolipides et de
glycoprotéines permettant l’interaction des plaquettes entre elles, avec les autres
cellules ou avec le milieu qui les entourent[7, 8]. Les plaquettes non activées sont
généralement lisses, mais possèdent des invaginations qui déterminent les entrées
du système canaliculaire ouvert (OCS-Open Canalicular System)[2]. Ce système
constitue une source importante de membrane mobilisable lors du changement de
conformation des plaquettes après leur activation[9] et permet l’importation ou la
sécrétion de protéines et d’autres molécules[2].

2
Le cytoplasme de plaquettes contient différents types de granules sécrétoires, dont
les granules alpha, les granules T, les granules denses, les peroxysomes et les
lysosomes. Les molécules contenues dans ces granules dérivent des MK, ou bien
sont incorporées par endocytose à partir du plasma[10]. La libération de granules
par les plaquettes est centrale pour assurer leurs diverses fonctions, que ce soit
pour l’adhésion, l’activation, l’agrégation ou la sécrétion de molécules anti-
inflammatoires et antimicrobiennes[11, 12].
Les granules alpha sont les plus abondantes[13] comptant en moyenne 50 à 80
granules par plaquettes[14]. D’une taille de 200 à 500 nm, ces granules
correspondent environ à 10% du volume de la plaquette. La surface membranaire
qui entoure les granules est équivalente à celle rendue disponible par l’OCS, ce qui
en fait aussi un réservoir important[14]. Combinées, la membrane plasmique des
granules alpha et celle de l’OCS permettent à la plaquette de multiplier de 2 à 4 fois
sa surface, lorsqu’activée[10]. Les granules alpha renferment plus de 300 protéines
dont des chimiokines (interleukine (IL)-8/CXCL8, Platelet Factor 4-PF4/CXCL4,
Regulated on Activation, Normal T Cell Expressed and Secreted-RANTES/CCL5),
des précurseurs du complément, des facteurs de coagulation, des protéines
d’adhésion et des intégrines (incluant la glycoprotéine VI (GPVI), αIIb, β3 et la
sélectine P (CD62P)), des facteurs de croissance et le complexe majeur
d’histocompatibilité (CMH) de classe I[10]. Des études ont montré que le cargo des
granules alpha est hétérogène, indiquant l’existence de sous-populations[15] dont
la libération pourrait être affectée par la force et la nature des agonistes utilisés pour
stimuler les plaquettes[16]. Les granules denses contiennent de petites molécules
comme le calcium, l’ADP (adénosine diphosphate) et l’ATP (adénosine
triphosphate), qui confèrent une apparence opaque en microscopie[6] ainsi que des
molécules telles que la sérotonine et l’histamine[11]. Des peroxysomes et des
lysosomes sont aussi présents dans le cytoplasme des plaquettes[17]. Ces derniers
libèrent des hydrolases acides dans l’environnement plaquettaire qui participent au
remodelage des thrombus et à la réponse antimicrobienne[16]. Les granules T,

3
identifiées récemment, ne contiennent pas de marqueurs typiques des autres types
de granules, mais contiennent des protéines disulfures isomérases qui catalysent le
repliement correct des peptides et le récepteur de type Toll (TLR/Toll-Like
Receptors) 9[18].
Le cytosquelette de la plaquette est constitué de trois composantes majeures, soit
la spectrine, l’actine et les microtubules, nécessaires pour le remodelage de la
plaquette et la libération de son contenu. Lorsque la plaquette est activée, elle subit
un changement de conformation important[19]. Ce processus est initié par un influx
de calcium[2] et implique le cytosquelette de la plaquette[19]. La contribution
spécifique de ces composantes a été démontrée en utilisant des molécules telles
que les cytochalasines B, D, E et le latrunculin A qui inhibent la polymérisation de
l’actine ou bien le nocodazole qui dépolymérise les microtubules[20, 21].
Même si les plaquettes ne sont pas considérées comme des cellules, elles partagent
de nombreuses caractéristiques communes. Elles possèdent des récepteurs
diversifiés lui permettant de répondre à de nombreux stimuli. Elles ont un
cytosquelette capable de se modifier rapidement lors de l’activation et des granules
sécrétoires contenant un grand éventail de molécules pouvant affecter leur
environnement. Bien qu’elles soient principalement reconnues pour leur rôle
physiologique primaire, soit l’initiation de la coagulation et la formation de thrombus
pour la prévention des saignements[22], la contribution des plaquettes dans
l’immunité innée et adaptative est de plus en plus reconnue. En effet, elles peuvent
reconnaitre et distinguer différents types de dangers (soi, non-soi, pathogènes) et
ajuster leurs réponses de façon appropriée[23].
1.1.2 À l’origine des plaquettes : les mégacaryocytes
Environ 100 milliards de plaquettes doivent être générées de façon quotidienne, par
le processus de thrombocytopoiëse afin de maintenir un compte plaquettaire normal
(150-400 x 109/litre de sang). Cette production peut être augmentée en cas de
baisse subite du compte plaquettaire, ce qui suggère une homéostasie réactive[24].
Un MK peut à lui seul libérer des milliers de plaquettes[25, 26].

4
Les MK sont de grandes cellules hétérogènes (50 à 100 µm) avec un noyau pouvant
contenir plusieurs copies de chaque chromosome (jusqu'à 128N)[27]. Leur fonction
principale est de produire et de libérer des plaquettes dans la circulation[28]. La
mégacaryocytopoïèse, qui est la production de MK dans la moelle osseuse, est
contrôlée par un processus complexe régulé principalement par la
thrombopoïétine[29]. Cette hormone est produite par le foie[30, 31] et son niveau
est inversement proportionnel au taux de plaquettes produites[32]. Sous l’action de
la thrombopoïétine et diverses autres cytokines, les cellules souches
hématopoïétiques vont se différentier lors de différents stades de maturation, soit le
progéniteur commun myéloïde, puis le progéniteur mégacaryocyte/érythrocyte pour
devenir finalement progéniteur mégacaryocytaire. C’est lors de ces étapes de
maturation que le MK va, par le processus d’endomitose[33], devenir une cellule
polyploïde ce qui permet l’augmentation de la synthèse protéique[34, 35]. La
formation d’un réseau de membranes de démarcation, l’augmentation du volume du
cytoplasme, la production de granules sécrétoires et l’expression des protéines
plaquettaires font aussi partie du processus de maturation et permettront aux
précurseurs mégacaryocytaires d’effectuer la formation des proplaquettes[36].
Un réarrangement du cytosquelette et du cytoplasme de façon à former une
succession d’extensions reliées par de ponts cytoplasmiques permet la formation
des proplaquettes[37, 38]. Ces proplaquettes, ressemblant à un collier de perles,
vont être produites jusqu’à l’utilisation complète du cytoplasme et des membranes
du MK. C’est aussi lors de ce processus que le contenu des MK incluant les
organelles est transféré aux proplaquettes[39-41]. Insérées dans les jonctions de la
paroi des vaisseaux sanguins, ces proplaquettes sont ensuite fragmentées et les
plaquettes individuelles sont libérées directement dans le courant sanguin où elles
pourront exercer leurs fonctions dans l’hémostase et dans l’immunité[42, 43].

5
1.1.3 Rôles des plaquettes dans l’hémostase
1.1.3.1 Coagulation
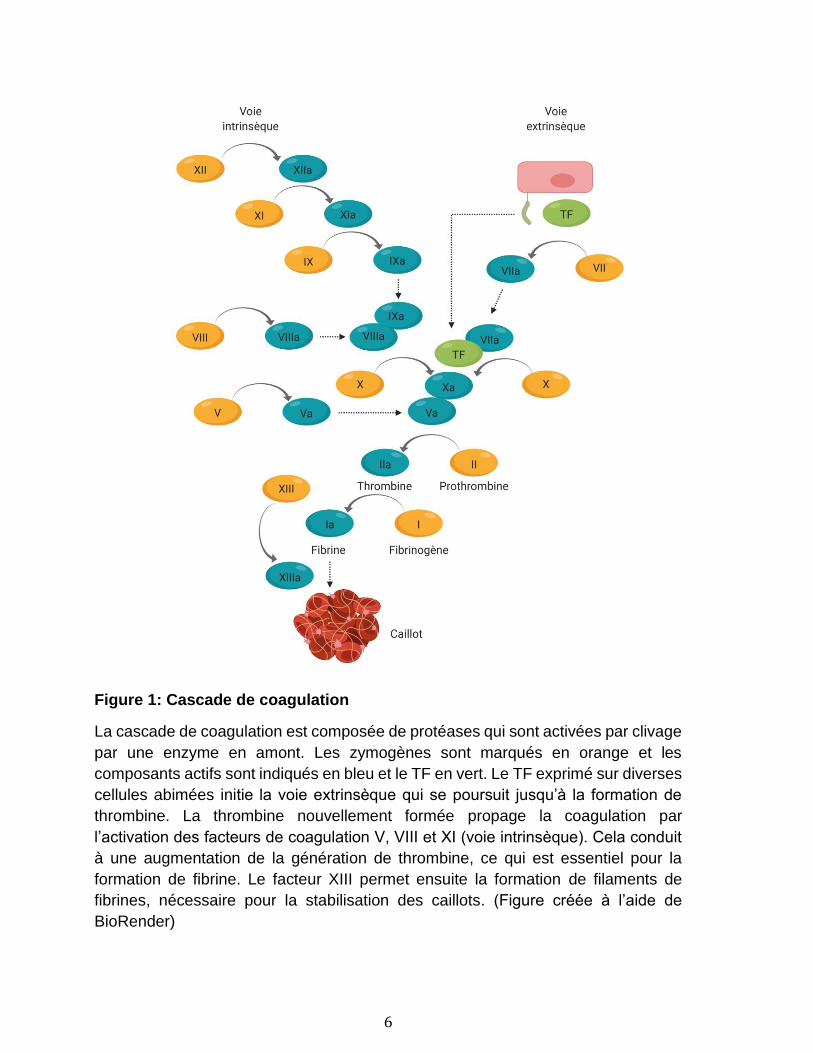
L’hémostase se définit comme l’arrêt de l'écoulement du sang, spontané ou
provoqué par différents moyens médicaux ou chirurgicaux. Les plaquettes et les
facteurs de coagulation permettent la formation spontanée d’un caillot et ont donc
un rôle majeur dans l’hémostase (Figure 1). À l’aide de leurs récepteurs de surface
pour le collagène sous-endothélial et le facteur von Willebrand (vWf) lié au
collagène[44], les plaquettes vont pouvoir s’activer, adhérer et agréger au site de
lésion vasculaire pour former un caillot. Étant donné la perte de l’asymétrie
membranaire, ces plaquettes activées exposent alors la phosphatidylsérine (PS) à
leur surface qui fournit un échafaudage aux complexes enzymatiques de
coagulation[45]. En parallèle, les cellules endothéliales abimées vont exposer à leur
surface le facteur tissulaire (TF-tissue factor)[44]. Le TF est ciblé par le facteur de
coagulation VII circulant dans le plasma, qui sera converti à sa forme activée
VIIa[44]. La liaison du facteur de coagulation VIIa au TF induit une cascade de
réactions impliquant les facteurs de coagulation présents dans le sang, ce qui
conduit à la génération de thrombine (voie extrinsèque)[44]. Les facteurs de
coagulation sont des protéines présentes dans le plasma sous forme de proenzymes
(zymogènes) qui doivent être clivées pour activer à leur tour la proenzyme suivante.
La thrombine va ensuite activer des plaquettes supplémentaires et permettre la
production de fibrine, tout en jouant un rôle crucial dans les phases d'amplification
et de propagation de la coagulation par l’activation des facteurs de coagulation V,
VIII et XI (voie intrinsèque), créant une boucle d’autoamplification de la génération
de thrombine. Le facteur XIII permet ensuite la formation d’un réseau de fibres de
fibrines, nécessaire pour la stabilisation des caillots[46].
6
Figure 1: Cascade de coagulation
La cascade de coagulation est composée de protéases qui sont activées par clivage
par une enzyme en amont. Les zymogènes sont marqués en orange et les
composants actifs sont indiqués en bleu et le TF en vert. Le TF exprimé sur diverses
cellules abimées initie la voie extrinsèque qui se poursuit jusqu’à la formation de
thrombine. La thrombine nouvellement formée propage la coagulation par
l’activation des facteurs de coagulation V, VIII et XI (voie intrinsèque). Cela conduit
à une augmentation de la génération de thrombine, ce qui est essentiel pour la
formation de fibrine. Le facteur XIII permet ensuite la formation de filaments de
fibrines, nécessaire pour la stabilisation des caillots. (Figure créée à l’aide de
BioRender)

7
1.1.3.2 Transfusion de plaquettes
La transfusion est une procédure médicale courante qui a pour but de substituer les
éléments du sang manquants à la suite d’une hémorragie, d’une maladie ou d’un
traitement[47-49]. La transfusion de plaquettes est utilisée plus spécifiquement pour
traiter ou prévenir les saignements en cas de thrombocytopénie ou de plaquettes
défectueuses[50]. La seule source de plaquettes viables, avant le début des années
1970, provenait de sang total frais[51]. Avec l’avancement de différentes
technologies, il est maintenant possible d’isoler les plaquettes sous forme de
concentrés plaquettaires (PC-platelet concentrate), facilitant ainsi la thérapie
transfusionnelle[52]. Ceux-ci sont préparés à partir du sang total par centrifugation
selon deux techniques distinctes par les vitesses de centrifugation, qui permettent
d’obtenir soit le plasma riche en plaquettes (PRP) ou bien la couche
leucoplaquettaire (BC-Buffy-Coat)[52-54]. Les leucocytes et les érythrocytes
résiduels sont éliminés par filtration ou centrifugation et les plaquettes sont remises
en suspension dans du plasma. Les PC peuvent aussi être préparés par aphérèse.
C’est le processus lors duquel le sang d’un donneur est mis en circulation en
système fermé à l’aide d’un cathéter dans l’appareil d’aphérèse. Les plaquettes sont
séparées des autres cellules par centrifugation afin d’être conservées, alors que les
autres composants du sang retournent au donneur par un second cathéter[55, 56].
L’aphérèse permet d’obtenir de plus grande quantité de plaquettes qu’avec un don
de sang total. La réduction du nombre de donneurs nécessaire avec la technique
d’aphérèse a l’avantage de diminuer le risque d’allo-immunisation et de transmission
de maladies au patient en comparaison des techniques de dons de sang total[57].
Outre le procédé de fabrication, de nombreux facteurs affectent la qualité des PC.
Le respect d’une période de repos avant de resuspendre les PC est important afin
de diminuer le niveau d’activation des plaquettes et d’améliorer leur morphologie[58-
62]. Les conditions d’entreposage, incluant la température, la durée et le type
d’agitation utilisé[63-71], la nature du dispositif contenant le PC[72-74] ainsi que
l’utilisation ou non d’une solution d’entreposage peuvent affecter grandement les
plaquettes[75-77].

8
Même sous des conditions optimales d’entreposage, des changements
morphologiques, structuraux et fonctionnels des plaquettes sont inévitables. Ces
altérations sont nommées lésions d’entreposage des plaquettes et sont la cause
d’une diminution progressive de la survie post-transfusion et des fonctions
hémostatiques des plaquettes[78]. Ces altérations incluent le changement des
protéines de surface des plaquettes[79-81], et la perte de la forme discoïde à la suite
d’un réarrangement du cytosquelette[82]. L’activation des plaquettes et leur
dégranulation lors de l’entreposage entrainent aussi la présence à leur surface de
CD62P et de PS[83] et l’accumulation de molécules bioactives[84]. L’entreposage
des PC entraine aussi la libération de vésicules extracellulaires (EV-extracellular
vesicle). Une section y sera consacrée.
1.1.3.3 Transfusion de plaquettes : les risques associés
Comme toute procédure médicale, les transfusions de produits sanguins présentent
certains risques pour les patients. En effet, cette procédure médicale peut mener à
la transmission de maladies infectieuses ou à des réactions transfusionnelles graves
pour le receveur. Les transfusions de plaquettes sont moins fréquentes que la
transfusion d’autres produits sanguins, mais présentent un risque supérieur[85].
1.1.3.4 Les risques infectieux
Que ce soit parce que le sang du donneur est contaminé, qu’un élément de la flore
bactérienne est introduit dans le composant sanguin lors du prélèvement ou qu’une
contamination survient lors de la préparation[86], la transfusion présente encore
aujourd’hui un risque infectieux. Le scandale du sang contaminé dans les années
1980, où la principale préoccupation concernait la transmission d’infections
virales[87], notamment du virus de l’immunodéficience humaine (VIH), de l’hépatite
B ou C (VHB ou VHC) a forcé l’instauration de mesures visant à prévenir la
transmission de maladies. L’application stricte de critères pour la détermination
d’éligibilité au don, incluant l’exclusion des pratiques sexuelles à risques, ont permis
une diminution nette de la transmission du VIH et de l’hépatite[88, 89]. Les critères
adressant les voyages à l’étranger ont aussi permis de limiter les risques de
contracter la malaria ou la maladie de Creutzfeldt-Jacob et sa variante. Une

9
sélection restrictive des donneurs de sang et l’utilisation de donneurs non rémunérés
permettent aussi de réduire les risques infectieux[90-92].
Depuis les années 1990, les efforts se sont davantage concentrés sur la réduction
de l’incidence de la contamination microbienne des produits sanguins,
principalement pour les PC qui sont conservés à température ambiante[87]. Des
procédures simples, comme la désinfection efficace de la peau au site de
prélèvement, permettent une réduction des bactéries de la flore normale jusqu’à
99%[93]. De plus, la méthode de diversion des premiers millilitres de sang a été
instaurée afin d’éviter la contamination causée par la carotte de peau pouvant se
retrouver dans 65% des cas dans l’aiguille lors de la phlébotomie[94]. Ces premiers
millilitres de sang, utilisés pour les tests de dépistage, se retrouvent maintenant dans
une poche annexe pour prévenir la contamination par la flore du donneur[95], ce qui
s’est avéré efficace pour réduire la contamination des produits sanguins[96-98].
L’introduction de nouvelles technologies pour détecter, réduire ou prévenir la
contamination qui sont maintenant appliquées couramment ont aussi permis la
réduction de ces risques infectieux[99-104].
1.1.3.5 Les risques non infectieux
L’implantation de ces nombreuses mesures préventives a mis de l’avant
l’importance des risques transfusionnels non infectieux.[105] Ceux-ci incluent la
réaction hémolytique aiguë (AHTR-Acute Hemolytic Transfusion Reaction) ou
retardée (DHTR-Delayed Hemolytic Transfusion Reaction) qui, en résultat d’une
incompatibilité entre les anticorps du donneur et les globules rouges du receveur,
entraine une destruction des globules rouges. Elle est considérée aiguë (AHTR)
quand les symptômes surviennent en moins de 24 heures suivant la transfusion ou
retardée (DHTR) s’ils surviennent entre 24 heures et trois semaines post-
transfusion[106]. Des réactions allergiques mineures, telles qu’une réaction
cutanée, ou majeures (anaphylactique) peuvent aussi être observées post-
transfusion[106]. Le mécanisme mis en cause dans ce type de réaction est une
hypersensibilité de type I due à la transfusion d’immunoglobulines (Ig) E présentes
dans le plasma[107].

10
Une surcharge circulatoire liée à la transfusion (TACO-Transfusion Associated
Circulatory Overload) peut se produire dans le cas de transfusion massive. Le cœur
étant incapable de pomper adéquatement un grand volume de produits sanguins, il
en résultera une insuffisance cardiaque et un oedème pulmonaire aigu[108]. La
thrombocytopénie immunitaire (ITP-Immune thrombocytopenia) est caractérisée par
une destruction des plaquettes du donneur et du receveur menant à une
thrombocytopénie sévère qui survient de 5 à 10 jours après la transfusion et entraine
des hémorragies pouvant être mortelles[106, 109]. Le mécanisme de destruction
des plaquettes reste inconnu, mais des antigènes plaquettaires absents des
plaquettes du receveur et présents chez le donneur semblent être en cause[109,
110]. Une réaction fébrile non hémolytique (FNHTR-Febrile Non-Hemolytic
Transfusion Reaction) est caractérisée par une élévation de température égale ou
supérieure à 1°C qui ne peut être reliée à la condition du patient ou à un autre type
de réaction transfusionnelle[106]. Une FNHTR peut être causée par plusieurs
facteurs et il existe une corrélation linéaire entre les niveaux de cytokines, la quantité
de leucocytes et la durée d’entreposage des PC conservés à 22°C[111, 112].
La réaction du greffon contre l’hôte (TA-GVHD-Transfusion-Associated Graft-
Versus-Host Disease), qui s’avère fatale dans 90% des cas, est causée par une
destruction des cellules du receveur par les lymphocytes du donneur, en cas de
différence des antigènes leucocytaires[106, 113]. Enfin, le syndrome respiratoire
aigu post-transfusionnel (TRALI-Transfusion-Related Acute Lung Injury) est
caractérisé par une détresse respiratoire aiguë, un œdème pulmonaire bilatéral et
une hypoxémie se produisant généralement dans les 2 heures suivant la
transfusion[106, 113]. Cette réaction, peu fréquente et encore mal comprise,
s’expliquerait par l’hypothèse du « Two-hit », où l’implication d’un anticorps ou d’un
médiateur soluble combiné à la condition clinique du receveur sont nécessaires pour
son développement[114, 115].
1.1.3.6 Facteurs connus à ce jour
Les réactions allergiques sont causées par des protéines plasmatiques comme l’IgA,

11
l’IgE et l’haptoglobine ainsi que certains allergènes chimiques ou alimentaires
présents dans le produit sanguin[85, 107, 116, 117]. Différents lipides bioactifs
relargués par les macrophages, les basophiles et les plaquettes, tels que certaines
lysophosphatidylcholines augmentent lors de l’entreposage des PC[118] et peuvent
activer les neutrophiles[119, 120]. Il en est de même pour de nombreuses cytokines
incluant l’IL-1α, l’IL-1β, l’IL-2, l’IL-6, IL-8/CXCL8, le Tumor Necrosis Factor (TNF),
l’interféron (IFN)-α, l’IFN-γ, le PF4/CXCL4, la β-thromboglobuline (β-TG),
RANTES/CCL5, le Macrophage Inflammatory Protein (MIP)-1α/CCL3, le
Transforming Growth Factor (TGF)-β et le Platelet Derived Growth Factor
(PDGF)[84, 111, 112, 121-129]. Le CD40 ligand (CD40L), aussi connu sous le nom
de CD154, est un médiateur pro-inflammatoire qui attire particulièrement l’attention.
Se trouvant sous forme soluble (sCD40L) ou associé aux cellules, il est entreposé
dans les granules alpha des plaquettes jusqu’à leur activation et s’accumule dans
les PC lors de l’entreposage[130-132]. Son effet activateur sur les macrophages
induit la libération de nombreuses cytokines associées aux réactions
transfusionnelles[133, 134]. Enfin, il peut y avoir production d’alloanticorps anti-
leucocytaires dirigés contre les antigènes leucocytaires humains de classes I et II
qui sont fréquemment retrouvés chez les femmes avec antécédent de grossesse ou
chez les donneurs ayant déjà été transfusés[135-137]. L’exclusion de ces derniers
aux dons a permis de réduire les risques de réactions transfusionnelles[138-140].
1.1.4 Rôles des plaquettes et des mégacaryocytes dans
l’immunité
Compte tenu du grand nombre de plaquettes dans le sang (40:1 par rapport aux
cellules immunitaires) et de leur vaste éventail de récepteurs immunitaires, la
capacité des plaquettes à soutenir les cellules immunitaires, promouvoir et participer
activement à l'immunité a fait l'objet de nombreuses investigations. Il est suggéré
que les MK et les plaquettes jouent un rôle dans la réponse immunitaire innée (révisé
en détail [2, 27, 141-144]) et dans l’immunité adaptative[145].

12
1.1.4.1 Notions d’immunologie
La capacité d'un organisme à se défendre contre des pathogènes ou des
substances étrangères nécessite la participation de deux composants du système
immunitaire : l'immunité innée et l’immunité adaptative. Les acteurs cellulaires
classiques de l’immunité innée sont les monocytes (forme immature et circulant dans
le sang), les macrophages (forme mature ayant migré dans les tissus), les
granulocytes (neutrophiles, éosinophiles et basophiles), les mastocytes et les
cellules dendritiques (DC-Dendritic Cells)[146]. L’immunité innée est la première à
se déclencher, prenant de quelques minutes à quelques heures pour détecter,
phagocyter et éliminer les micro-organismes tout en contribuant aux quatre points
cardinaux de l’inflammation (douleur, chaleur, rougeur, gonflement)[147].
Cette détection par le système immunitaire inné implique la reconnaissance de
motifs moléculaires connus sous le nom de motifs moléculaires liés aux pathogènes
ou associés aux dommages (respectivement PAMP ou DAMP)[148]. Les PAMP
sont des composants exogènes conservés parmi un large spectre de micro-
organismes et d'allergènes, tandis que les DAMP sont des signaux d'alarme
endogènes produits pendant le stress ou la mort cellulaire qui conduisent à des
réponses inflammatoires stériles[149, 150]. Les PAMP et les DAMP sont reconnus
par divers récepteurs reconnaissant des motifs structuraux répétés et comprennent
les récepteurs lectine membranaire de type C (CLR-C-type Lectin Receptor), les
TLR et les récepteurs de type NOD (Nucleotide-binding Oligomerization
Domain)[151]. La stimulation de ces récepteurs permet la sécrétion de cytokines et
de chimiokines pro-inflammatoires importantes pour le recrutement de cellules
immunitaires et l’activation du complément, pour activer directement la phagocytose
ou permettre la sécrétion de molécules nécessaires pour l’opsonisation, c’est-à-dire
la liaison de protéines au pathogène pour faciliter sa phagocytose[146, 148].
Le complément est un système de protéines plasmatiques qui entraine une réaction
en chaîne, tout comme la cascade de coagulation. Cette cascade peut être activée
par trois voies, la voie classique, la voie des lectines et la voie alternative. Ces

13
étapes précoces convergent toutes vers la formation de C3 convertase qui clive le
C3 en C3a et C3b et génère une succession de réactions de clivage menant au
recrutement des cellules inflammatoires et immunitaires, à l’opsonisation et à la
destruction des pathogènes et des cellules mortes, endommagées ou apoptotiques
(Figure 2)[152].
Figure 2: Vue générale de la cascade du complément
Elle peut être activée par trois voies, la voie classique, la voie des lectines et la voie
alternative. La voie classique est activée lorsque C1q se lie à la surface du
pathogène soit de manière directe, soit en liant la protéine C réactive ou bien en liant
un complexe antigène-anticorps. La voie des lectines est activée lors de la liaison
de lectines ou de ficolines présentes chez les phagocytes à des glucides présents
chez les pathogènes. Enfin, la voie alternative est déclenchée lors de la liaison sur
le pathogène du composant C3 activé spontanément dans le plasma. Ces étapes
précoces convergent toutes vers la formation de C3 convertase et mène au
recrutement des phagocytes, à la formation du complexe d’attaque membranaire, à
l’opsonisation et la destruction des pathogènes, des complexes immuns et des
cellules mortes, endommagées ou apoptotiques. (Figure créée à l’aide de
BioRender)
Bien qu’efficace pour éliminer la plupart des pathogènes, les réponses immunitaires
innées ne sont pas spécifiques et ne confèrent pas une immunité durable. En

14
revanche, le système immunitaire adaptatif confère une mémoire
immunologique[146]. Elle implique un processus appelé présentation antigénique
qui se produit pendant la réponse du système immunitaire inné et nécessite environ
deux semaines afin d’établir une réponse immunitaire plus forte, plus spécifique et
plus rapide à une rencontre subséquente[153].
Le système immunitaire adaptatif nécessite des acteurs supplémentaires, soit les
lymphocytes T impliqués dans la réponse immunitaire à médiation cellulaire et les
lymphocytes B, impliqués dans la réponse immunitaire humorale. À l’aide de leur
récepteur d’antigènes hautement spécifiques, respectivement le BCR (B cell
receptor) et le TCR (T cell receptor)[154], ils pourront détecter les antigènes
présentés à la surface des cellules présentatrices d’antigènes (APC-Antigen
presenting cells) qui font le pont entre l’immunité innée et adaptative[155].
Pour qu’un antigène soit reconnu par un lymphocyte T, il doit être présenté à la
surface de la cellule par les molécules du CMH, aussi connu chez l’humain sous le
nom d’antigène leucocytaire humain (HLA-Human Leukocytes Antigen), afin d’être
reconnu par le TCR[146]. Il existe deux catégories de molécules du CMH, soit les
molécules de CMH de classe I (HLA-A, -B et -C) et de classe II (HLA-DR, -DP, -DQ)
qui diffèrent par leur structure et leur distribution sur les différents types
cellulaires[156]. Le CMH I comprend une chaîne lourde liée de façon non covalente
à une chaîne légère microglobuline β2 (β2-MG) et est exprimé constitutivement par
toutes les cellules nucléées[157]. Le CMH II est composé d’une chaîne alpha et une
chaîne bêta et est exprimé sur les APC[158]. Ces protéines membranaires
possèdent un sillon extracellulaire qui permet l’insertion d’un fragment peptidique
dérivé de l’antigène et qui stabilise le complexe formé du peptide et d’une molécule
de CMH. Le CMH I a un sillon plus restrictif et lie généralement des peptides d’une
longueur de 8-10 acides aminés[159]. Le CMH II étant plus ouvert, la longueur des
peptides qu’il peut lier n’est pas restreinte, mais est généralement supérieure à 13
acides aminés[156]. En plus de la liaison entre le TCR et le CMH chargé d’un
peptide, une interaction supplémentaire avec les corécepteurs CD4 ou CD8
présents sur les lymphocytes est requise pour stabiliser l’interaction entre les deux
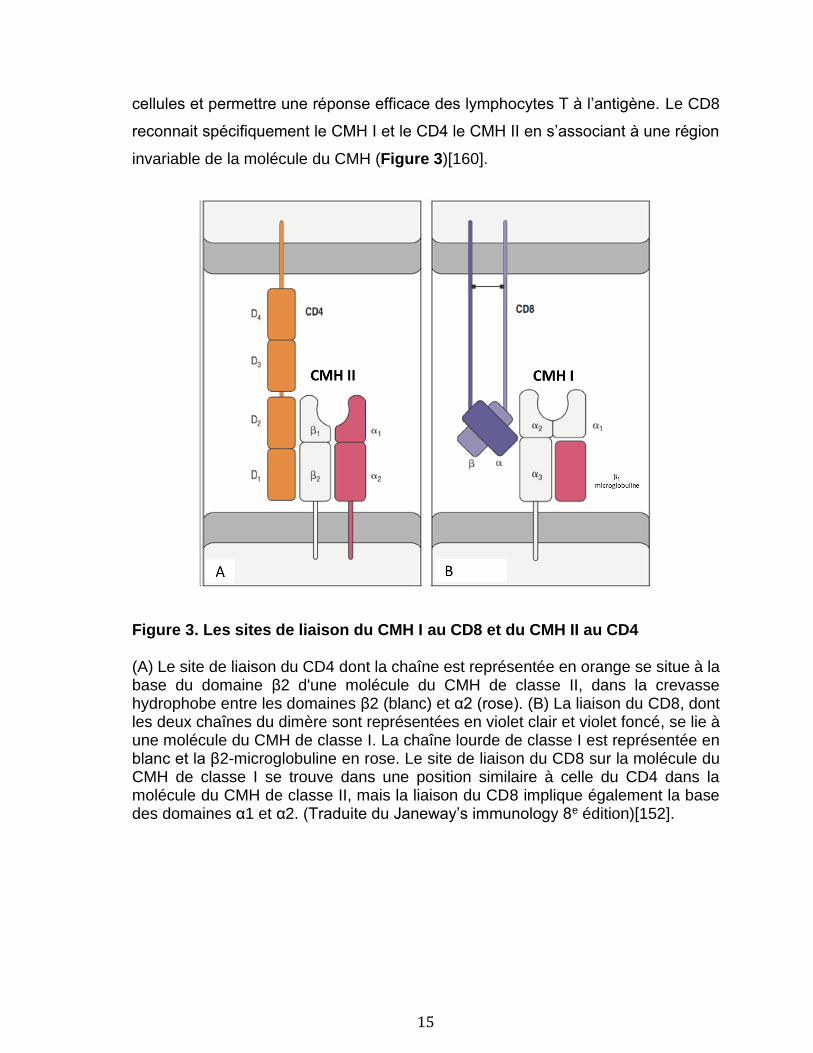
15
cellules et permettre une réponse efficace des lymphocytes T à l’antigène. Le CD8
reconnait spécifiquement le CMH I et le CD4 le CMH II en s’associant à une région
invariable de la molécule du CMH (Figure 3)[160].
Figure 3. Les sites de liaison du CMH I au CD8 et du CMH II au CD4
(A) Le site de liaison du CD4 dont la chaîne est représentée en orange se situe à la base du domaine β2 d'une molécule du CMH de classe II, dans la crevasse hydrophobe entre les domaines β2 (blanc) et α2 (rose). (B) La liaison du CD8, dont les deux chaînes du dimère sont représentées en violet clair et violet foncé, se lie à une molécule du CMH de classe I. La chaîne lourde de classe I est représentée en blanc et la β2-microglobuline en rose. Le site de liaison du CD8 sur la molécule du CMH de classe I se trouve dans une position similaire à celle du CD4 dans la molécule du CMH de classe II, mais la liaison du CD8 implique également la base des domaines α1 et α2. (Traduite du Janeway’s immunology 8e édition)[152].

16
Les lymphocytes T exprimant le CD4 (effecteurs) reconnaissent des antigènes
extracellulaires liés aux molécules du CMH II et activent d’autres cellules
immunitaires incluant les lymphocytes B[161]. Les lymphocytes B vont ensuite
entrainer la production d’anticorps nécessaire pour la neutralisation ou
l’opsonisation de l’antigène et l’activation du complément. Les lymphocytes T
exprimant le CD8 (cytotoxiques) vont reconnaitre des APC présentant des antigènes
du cytosol (pathogènes intracellulaires et tumeurs) liés aux molécules du CMH I et
mener à la destruction de la cellule infectée ou cancéreuse[161]. Certaines APC
peuvent aussi présenter des antigènes extracellulaires aux lymphocytes T CD8+
(restreints au CMH I) lors d’un processus appelé présentation croisée ou l’antigène
est capturé par endocytose ou phagocytose puis dégradé par des protéases dans
une vacuole[161].
Les peptides présentés par le CMH I ne pourraient l’être sans avoir été
préalablement apprêtés par le protéasome, l’une des deux voies principales de
dégradation des protéines intracellulaires qui existent chez les eucaryotes avec la
voie vacuolaire qui est impliquée dans la présentation croisée[160, 162-164]. Le
protéasome est une protéase multicatalytique de haut poids moléculaire
principalement connu pour sa fonction dans le maintien de l'hémostase des
protéines cellulaires et l’élimination des protéines mal repliées ou marquées
d’ubiquitine[165, 166]. Il est aussi impliqué dans d’autres fonctions cellulaires, telles
que la différenciation cellulaire, la progression du cycle cellulaire, la transcription, la
réponse au stress oxydatif et l'apoptose[162, 164]. Le protéasome est délimité du
reste de la cellule par sa structure fermée qui forme un espace compartimenté
analogue à la lumière d'une organelle[167]. Étant une structure spécialisée contenue
dans le cytoplasme qui a une fonction spécifique, nous allons considérer le
protéasome comme une organelle.
Le complexe central, nommé protéasome 20S, est formé de quatre anneaux
superposés comprenant chacune 7 sous-unités différentes pour un total de 28 sous-
unités distinctes (Figure 4)[168]. Les anneaux aux extrémités sont composés de

17
sous-unités α et les anneaux du centre sont composés des sous-unités β incluant
les sous-unités catalytiques β1 (activité de type caspase), β2 (type trypsine) et β5
(type chymotrypsine)[168, 169]. Ces différentes activités permettent au protéasome
20S de dégrader n’importe quel type de protéines en peptides d’une longueur de 3
à 15 acides aminés[163, 170]. Ce complexe central peut être associé avec le
régulateur 19S, qui contient la sous-unité ATPase permettant l’ouverture des
anneaux α, pour former le protéasome 26S[168]. Le 19S est important pour la
reconnaissance des protéines ubiquitinylées et permet leur linéarisation pour
l’insertion dans le complexe central[163]. Le protéasome 20S peut aussi s’associer
au régulateur 11S pour former l’immunoprotéasome, dans lequel les sous-unités
catalytiques β1, β2 et β5 sont remplacées par leurs homologues β1i, β2i et β5i[168].
Il est impliqué dans la génération de peptides améliorés pour la présentation
antigénique du CMH I[171].
Figure 4. Structure du protéasome et de l’immunoprotéasome
Le cœur du protéasome standard 20S est composé de 28 sous-unités non identiques qui sont disposées en quatre anneaux; deux composés de sept sous-unités α et deux composés de sept sous-unités β. Les sous-unités (β1, β2 et β5) sont catalytiquement actives (centre). Le protéasome 20S peut s’associer à deux régulateurs 19S pour former le protéasome 26S (à gauche). Le protéasome peut aussi s’associer au régulateur 11S pour former l’immunoprotéasome, qui contient trois sous-unités catalytiquement actives différentes (β1i, β2i et β5i). (Tirée de Klockenbusch et al[168].)

18
En effet, l’immunoprotéasome présente une activité de type caspase réduite et une
activité de type trypsine et chymotrypsine renforcée en comparaison du protéasome
standard, ce qui génère des peptides de nature hydrophobe ou basique ayant une
plus grande affinité avec la poche de liaison aux peptides du CMH I[163, 172-175].
Exprimé constitutivement dans les cellules immunitaires des tissus lymphoïdes tels
que la rate, le thymus et les ganglions lymphatiques, l’immunoprotéasome peut
aussi être induit dans les autres cellules en présence d’IFN γ et de TNF[162, 176].
En effet, la libération de cytokines proinflammatoires libérées par les cellules de
l’immunité innée contribue à la substitution du protéasome standard par
l’immunoprotéasome dans les tissus inflammés, en plus d’induire la surexpression
d’autres protéines liées à la présentation antigénique dont le CMH I et les protéines
de transport associées à l’antigène (TAP-Transporters associated with Antigen
Processing)[177]. Les sous-unités catalytiques de l’immunoprotéasome influencent
aussi la sélection des lymphocytes T en modulant le répertoire de TCR; les peptides
apprêtés en absence de l’une ou l’autre des sous-unités vont avoir des séquences
différentes qui peuvent mener à une réponse moindre ou même absente des
lymphocytes T cytotoxiques[178-180]. L’utilisation d’inhibiteurs sélectifs de
l’immunoprotéasome dans les dernières années à aussi mis en évidence sa
contribution dans les maladies auto-immunes, dans divers syndromes auto-
inflammatoires, dans le cancer et dans le rejet de greffe[181-184].
Les peptides, une fois clivés par le protéasome, sont ensuite transférés par le
transporteur TAP (composé des protéines TAP1 et TAP2) dans la lumière du
réticulum endoplasmique[185]. C’est à cet endroit qu’ils seront apprêtés davantage
au besoin par les aminopeptidases ERAAP (Endoplasmic reticulum amino
peptidase )1 et 2 et qu’ils vont s’associer au CMH I à l’aide du complexe chargeant
le peptide (PLC-peptide loading complex)[164]. Le PLC est composé des
chaperones tapasine, calnexine, calréticuline et la protéine disulfure isomérase
ERp57 qui aident au chargement et à la stabilisation du peptide dans le sillon du
CMH I en plus de vérifier la qualité du complexe formé[185]. Enfin le complexe CMH

19
I-peptide est exporté à travers le Golgi puis exposé à la surface de la cellule pour
être détecté par les lymphocytes T CD8+[164]. (Figure 5)
Figure 5: Apprêtement du peptide et présentation par le CMH I
Une chaine de CMH I nouvellement synthétisée se lie temporairement à la calnexine
dans le réticulum endoplasmique (RE) jusqu’à ce que ce complexe se lie à la β2-
microglobuline (β2-MG). Une fois stabilisé, le complexe partiellement replié se
dissocie ensuite de la calnexine, pour se lier à la tapasine, au transporteur TAP et
aux chaperonnes ERp57 et la calréticuline pour former le complexe de chargement
peptidique. Le protéasome, situé dans le cytosol, génère des peptides qui sont
transportés dans la lumière du RE par le transporteur TAP. La liaison d'un peptide
au CMH I achève le repliement de la molécule du CMH I et lui permet de se libérer
du complexe de chargement peptidique et de quitter le RE pour être exposé à la
surface cellulaire. L’interaction avec les lymphocytes T CD8+ pour la présentation
antigénique pourra alors se produire. (Figure créée à l’aide de BioRender)

20
1.1.4.2 Les plaquettes dans l’inflammation et l’immunité innée
Le rôle des plaquettes n'est pas limité à la réponse hémostatique. Les plaquettes
sont capables de phagocytose, mécanisme inné conservé des thrombocytes
(l’équivalent nucléé des plaquettes chez les vertébrés inférieurs), et cette activité
permettant de capturer des pathogènes s’effectue grâce à l’OCS[186-189]. Parmi
les premières cellules recrutées à l’endothélium inflammé ou endommagé, les
plaquettes expriment également un ensemble de récepteurs qui leur permettent de
détecter et d’éliminer les pathogènes directement ou en activant d’autres cellules
immunitaires[2, 143, 190].
Les plaquettes expriment de nombreux membres de la famille des TLR. Les TLR 1
à 10 ainsi que leurs voies de signalisation, molécules adaptatrices et facteurs de
transcription ont été identifiés chez l’humain et sont tous exprimés par les
plaquettes[191]. Ces TLR permettent à la plaquette de jouer un rôle critique dans
l’immunité innée par la reconnaissance de PAMP et de DAMP. Fait intéressant,
l’expression des différents TLR est plus élevée chez les plaquettes des femmes et
est associée avec l’expression de CD62P[191]. Les TLR 1-2-4-5-6 et 10 sont
présents à la surface de la cellule et les TLR 3-7-8 et 9 sont localisés dans les
endosomes et dans les granules T pour le TLR 9[18, 192]. En plus de leur
localisation cellulaire, les TLR peuvent aussi être classés en fonction des
pathogènes qu’ils ciblent. Le TLR 10 n’a pas encore de fonction connue et ne sera
pas décrit davantage. Les TLR 1 à 6 reconnaissent principalement des molécules
composant les bactéries, les mycètes (les TLR 2,4 et 6), les parasites et parfois des
composants structuraux des virus (TLR 2 et 4)[192]. Les acides nucléiques des
pathogènes sont aussi reconnus par des TLR, soit le TLR 3 pour l’ARN double brins,
les TLR 7 et 8 pour l’ARN simple brin et le TLR 9 pour l’ADN double brins[192]. Étant
donné qu’un modèle murin d’injection de lipopolysaccharide (LPS), ligand majeur du
TLR 4, a été utilisé lors de ma thèse, et que l’ADN mitochondrial (mtDNA-
mitochondrial DNA), ligand du TLR 9, est un sujet récurrent dans les prochains
chapitres, ces deux TLR seront décrits plus en détail.

21
Le TLR 4 reconnaît le LPS, un composant majeur de la paroi des bactéries Gram-
négatives[193]. La liaison du LPS par le TLR 4 n’est pas directe, elle implique deux
protéines plasmatiques accessoires, la protéine liant le LPS (LBP-LPS binding
protein), et le CD14. Le CD14 est présent à la surface des cellules, mais pas à la
surface des plaquettes, c’est donc sa forme soluble (sCD14) présente dans le sang
qui permet l’interaction avec deux complexes TLR 4-MD-2 (myeloid differentiation
factor 2) et qui entraine leur dimérisation[193, 194]. Les plaquettes humaines
peuvent discerner diverses isoformes de LPS bactérien via le TLR 4, entrainant des
profils de sécrétion de cytokines distincts et une réponse modulée en fonction des
différentes espèces de pathogènes[195, 196]. L’effet du LPS sur les plaquettes est
controversé et semble être affecté par le mode d’isolation et de préparation des
plaquettes[197]. Il pourrait augmenter l’activation des plaquettes par divers
agonistes ou promouvoir directement leur activation et entrainer la production
d’EV[196, 198-201]. Les plaquettes, en contact avec le LPS, seraient importantes
dans la production du TNF-α[202], de TF[203] et de sCD40L[204] en plus
d’augmenter la phagocytose des plaquettes qui sont liées à des auto-anticorps[205].
La stimulation du TLR 4 peut entrainer une thrombocytopénie ainsi que
l’accumulation de plaquettes aux poumons[198]. Les plaquettes, une fois leurs TLR
4 activés, se lient aussi aux neutrophiles pour augmenter la sécrétion de peptides
antimicrobiens et la production de NET (neutrophil extracellular traps) par ces
derniers, ce qui permet de piéger les pathogènes[206, 207]. En plus du LPS,
d’autres ligands incluant des motifs viraux et des DAMPs sont reconnus par le TLR
4, tels que les histones[208], l’hyaluronane soluble, la bêta-défensine 2, la protéine
HMGB1 (high-mobility group box 1)[209], HSP 70 (heat shock protein 70)[210],
S100A8/A9[211] et les pentaglucosides de Candida albicans[212].
Le TLR 9, exprimé par les plaquettes humaines et murines, reconnaît les motifs
d'ADN 2´-désoxyribo (cytidine-phosphate guanosine) non méthylés, communément
appelés motifs CpG. Ces motifs sont retrouvés dans l'ADN bactérien et viral, mais
aussi dans l’ADN mitochondrial[213]. L’ARNm du TLR 9 est exprimé chez les MK
matures et est régulé à la hausse durant la production de proplaquettes[18]. Les

22
plaquettes non activées expriment une quantité basale importante de TLR 9 à leur
surface. Celle-ci peut être augmentée chez l’humain en cas de stimulation avec de
la trombine, du collagène de type IV, de l’ADP, du PMA (phorbol myristate acetate)
et du CRP-XL (cross-linked collagen-related peptide)[18]. Ceci suggère d’une part
que le TLR 9 existe également dans les compartiments intracellulaires, mais d’autre
part que son expression est régulée différentiellement en fonction des espèces
puisque ce n’est pas le cas chez la souris[202]. Des oligodéoxyribonucléotides
(ODN) synthétiques contenant des motifs CpG nonméthylés sont généralement
utilisés comme agonistes pour activer le TLR 9. Lorsque les plaquettes sont
stimulées avec des ODN, elles séquestrent ces derniers ce qui favorise
l’augmentation de TLR 9 et de CD62P à leur surface[18]. Il a été montré que
l’activation de la voie TLR 9 augmente le risque de thromboses et d’accidents
cardiovasculaires[214]. En plus de reconnaitre les motifs CpG, le TLR 9 détecte
aussi des ligands endogènes altérés lors de conditions physiopathologiques
associées au stress oxydatif ou aux infections[215]. Ces protéines générées lors du
stress oxydatif sont groupées sous le terme CAP (carboxy(alkylpyrrole) protein
adducts) et sont fréquemment retrouvées dans les tumeurs, les plaies cicatrisantes
ou des maladies comme le diabète ou l’athérosclérose[215].
En plus d’exprimer différents TLR, les plaquettes humaines expriment des
récepteurs Fc pour les Ig A (IgA), E (IgE) et G (IgG), soit respectivement le
FcαRI[216], le FcεRI et FcεRII[217-219], et le FcγRIIA[220]. Les récepteurs FcαRI
et FcγRIIA sont absents chez la souris[221, 222], mais un modèle de souris
transgénique exprimant le FcγRIIA humain a été développé pour étudier le rôle de
ce récepteur[222]. L’activation du récepteur FcαRI des plaquettes par des IgA,
immunoglobulines participantes à l’immunité des muqueuses, entraine la production
de cytokines proinflammatoires (incluant l’IL-1β) et de TF qui sont critiques pour la
pathogenèse de plusieurs maladies inflammatoires et pour la thrombose[216]. La
stimulation des plaquettes via les récepteurs FcεRI et FcεRII, induit la libération de
sérotonine et de RANTES/CCL5, appuyant le rôle des plaquettes dans
l’inflammation allergique et la défense contre les parasites[217-219]. Le FcγRIIA est

23
lui aussi impliqué dans la thrombose et dans la pathogenèse de certaines maladies
auto-immunes[223], car il est important pour l’activation des plaquettes par le
vWf[224] et l'élimination des complexes immuns contenant des IgG circulants[225].
L’activation du FcγRIIA plaquettaire induit l’activation d’intégrines dont αIIbβ3 (le
complexe GPIIb-IIIa dont l’expression est unique aux plaquettes et induit leur
aggrégation), l’aggrégation des plaquettes, la sécrétion du contenu des granules, la
génération d’espèces réactives d’oxygène (ROS-reactive oxygen species), et
l’augmentation d’expression de surface de CD62P[226-232]. Cette cascade
d’évènements peut mener à un choc systémique tel qu’observé lors d’injection de
complexes immuns dans les souris transgéniques[233-236]. La présence du
FcγRIIA sur la plaquette permet aussi l’internalisation de bactéries opsonisées ou
leur élimination par la libération de molécules antimicrobiennes comme le
PF4/CXCL4[228, 237-239]. Il est aussi important pour le rôle des plaquettes dans la
lutte contre les infections virales, parasitaires et fongiques[240-249].
Évidemment l’ensemble des récepteurs de plaquette est vaste et ne se limite pas
uniquement aux TLR et aux récepteurs Fc. Elles expriment notamment six intégrines
différentes (α2β1, α5β1, α6β1, αLβ2, αIIbβ3, et ανβ3) importantes pour l’adhésion
cellulaire et leur activation. Elles possèdent aussi les récepteurs pour de nombreux
agonistes dont la thrombine[250], l’ADP[251], des prostaglandines[252-256], des
lipides[257-259] et des chimokines[260]. Une fois activées, les plaquettes vont
libérer de nombreuses molécules pouvant permettre l’interaction avec d’autres
cellules immunitaires, induire l’inflammation ou sa résolution, l’angiogenèse,
l’apoptose et/ou détruire les pathogènes[261].
Différentes molécules participent aux interactions entre les plaquettes et les cellules
immunitaires. Les plaquettes interagissent via CD62P ou la glycoprotéine Ib, avec
le ligand-1 de la glycoprotéine CD62P (PSGL-1) et l'intégrine CD11b/CD18 (Mac-1)
trouvés sur les leucocytes[262, 263]. Ces interactions déclenchent l’activation des
leucocytes et l’expression d’intégrines nécessaire pour l’adhésion aux parois des
vaisseaux sanguins[264]. Le dépôt de RANTES/CCL5 et de PF4/CXCL4 par les

24
plaquettes sur les cellules endothéliales aux sites d’inflammation favorise aussi
l’adhésion ferme et la transmigration des leucocytes vers les tissus inflammés,
endommagés et/ou infectés[265]. La forme activée du récepteur plaquettaire au
fibrinogène, le récepteur αIIbβ3, peut aussi se lier à SLC44A2 (solute carrier family
44 member 2) ainsi que Mac-1 sur le neutrophile. Cette interaction entraine la
production de cytokines pro-inflammatoires dont l’IL-1b, l’IL-8/CXCL8 et la libération
de sérotonine, ce qui a pour effet d’augmenter la perméabilité des cellules
endothéliales, de causer une vasodilatation et des fuites vasculaires[266, 267]. Les
différents médiateurs relargués par les plaquettes favorisent aussi l’activation des
neutrophiles et leur activité bactéricide, notamment en promouvant la production de
ROS[207, 268, 269], en augmentant l’activité phagocytaire et leur capacité à
produire des NET, soit des filets formés de chromatine décondensée chargée
d'enzymes protéolytiques et d'autres molécules antibactériennes expulsés des
neutrophiles activés[270, 271]. Il a été démontré que les NET favorisent l’activation,
la dégranulation et l’aggrégation des plaquettes, créant ainsi une boucle
d’amplification[272, 273]. En plus d’avoir un rôle dans la défense antimicrobienne,
les NET sont aussi générés lors d’une réponse à un stimulus non
conventionnel[274]. La formation non sollicitée ou un défaut dans l’élimination des
NET peut entrainer de l’inflammation et une surexposition à des auto-antigènes, ce
qui entraine des dommages tissulaires et facilite le développement de l’auto-
immunité[275-278].
1.1.4.3 Les mégakaryocytes dans l’immunité innée
Les mégakaryocytes expriment de nombreux récepteurs immunitaires y compris les
TLR 1 à 6 (l'ARNm du TLR 5 a été identifié dans les mégakaryocytes pulmonaires)
et le TLR 9 lors de la production de proplaquettes[18]. Le TLR 4 est notamment
impliqué dans la régulation de la production de plaquettes. Elles expriment aussi les
récepteurs d’immunoglobulines (FcRγIIA et FcεRI pour les mégakaryocytes
humains et FcγRI sur une sous-population de mégakaryocytes murins) et des
molécules co-stimulatrices, telles que le CD40L[27]. Les MK de la niche médullaire
sont en contact avec des cellules souches hémopoïétiques. Par sécrétion de

25
plusieurs molécules comme la thrombopoïétine, PF4/CXCL4 ou TGF-β, ils sont ainsi
capables de réguler la quiescence ou la prolifération des cellules souches
hématopoïétiques[242, 243]. Non seulement ils communiquent avec d'autres
cellules via ces molécules, mais les mégakaryocytes sont également capables
d'empéripolèse, un phénomène dynamique où elles engloutissent les neutrophiles
et où il se produit un échange membranaire[279].
1.1.4.4 Support des cellules de l’immunité adaptative par les plaquettes
Pendant l'immunité adaptative, les plaquettes peuvent soutenir les APC classiques
à travers plusieurs molécules (résumé en Figure 6, panneau de gauche). Par
exemple, le PF4/CXCL4, une chimiokine produite en abondance par les plaquettes
activées, inhibe la prolifération des lymphocytes T humains activés et la libération
des cytokines qu’ils produisent[280]. Le PF4/CXCL4 peut également influencer la
fonction et la différenciation des DC, conduisant à des DC différenciées qui sont
capables d'internaliser l'antigène, mais qui ne peuvent l’apprêter
adéquatement[281]. La protéine CD62P est emmagasinée dans des granules α et
libérée lors de l'activation plaquettaire. Cette protéine joue un rôle dans le
recrutement des lymphocytes aux sites d'inflammation[282, 283]. La contribution de
CD62P dans l'immunité adaptative implique également une présentation croisée
augmentée par les DC. En effet, les plaquettes modulent la maturation des DC
dérivées de monocytes par des interactions directes entre CD62P et PSGL1. En
effet, les DC générées par la co-incubation avec CD62P sont plus efficaces que les
DC dérivées à partir de cytokines pour générer une immunité tumeur-spécifique des
lymphocytes T[284]. La sérotonine n’est pas synthétisée par les plaquettes, mais
capturée et emmagasinée via leur transporteur de sérotonine dans les granules
denses plaquettaires[285]. Cette molécule est capable de favoriser le recrutement
des lymphocytes T CD4+ et CD8+ [286, 287] et d’affecter la différenciation des DC.
Les DC traitées à la sérotonine montrent une expression réduite des molécules co-
stimulatrices et une production accrue d'IL-10, diminuant leur capacité d’activer les
lymphocytes T[288].

26
Alors que le CD40L est localisé de manière intracellulaire dans les plaquettes
inactivées, les plaquettes activées arborent le CD40L à leur surface, ce qui leur
permet d'interagir avec les cellules porteuses de CD40 telles que les autres
plaquettes ou les cellules endothéliales, en plus des lymphocytes et des DC. Il a été
démontré que le CD40L dérivé des plaquettes induit la différenciation des
monocytes en DC et leur maturation, en plus de réguler positivement l’expression
de leurs molécules costimulatrices[289-291]. Cette fonction du CD40L plaquettaire
peut être très pertinente pour le lupus érythémateux systémique (SLE-Systemic
Lupus Erythematosus), maladie auto-immune dans laquelle les plaquettes se sont
révélées capables d'induire la différenciation des DC et la libération d'IFN, favorisant
ainsi la sécrétion d'anticorps par les lymphocytes B[292]. Une description plus
détaillée des interactions plaquettaires via le CD40L dans l'immunité adaptative a
déjà été étudiée en détail dans plusieurs revues[293-296]. Les plaquettes exprimant
le CD40L ont également été identifiées dans différentes pathologies où elles activent
directement l'endothélium[297] ou bien elles contribuent au recrutement de
neutrophiles et de lymphocytes T à l'endothélium endommagé, plus particulièrement
dans l'intima et dans les plaques dans l'athérosclérose[298]. Ainsi, conformément
aux investigations in vivo et in vitro qui ont révélé un rôle du CD40L plaquettaire
dans la commutation isotypique des lymphocytes B et dans l'augmentation de la
fonction des lymphocytes T CD8+ durant l'infection, ces multiples études suggèrent
que les plaquettes, via le CD40L, peuvent avoir un impact sur les lymphocytes et les
DC dans les étapes clés de l'immunité adaptative[299].
Les plaquettes sont également importantes dans l'induction de la tolérance
immunitaire, car leur déplétion réduit la fréquence des lymphocytes T régulateurs
(Tregs) Foxp3+ dans la peau inflammée et dans les ganglions lymphatiques
drainants[300]. Elles sont considérées comme protectives, car elles augmentent
l'activation des lymphocytes T régulateurs CD4+ et elles produisent aussi du TGF-β,
un régulateur reconnu de la fonction des lymphocytes T CD8+[301, 302].

27
1.1.4.5 Apprêtement et présentation de l'antigène par les plaquettes
Le protéasome a été purifié de la fraction cytosolique des plaquettes humaines pour
la première fois au début des années 90[303, 304]. Depuis, la présence de toutes
les sous-unités catalytiquement actives du protéasome 20S dans les plaquettes
humaines a été confirmée, incluant les sous-unités composant
l'immunoprotéasome[168]. Toutes ces composantes peuvent s'assembler pour
obtenir un immunoprotéasome constitutivement actif dans les plaquettes, même en
l’absence de stimulation par l’IFN[168]. De plus, il a été montré que l’activité du
protéasome des plaquettes est régulée positivement lors de septicémie
bactérienne[305].
La pertinence de cet organite dans les plaquettes n'est pas encore entièrement
comprise étant donné la faible production de protéines par les plaquettes, mais
certaines fonctions clés ont été identifiées et examinées récemment[306]. Le
protéasome est nécessaire pour la production de plaquettes par les MK[307, 308],
pour limiter la durée de vie des plaquettes[309], pour leur activation[310, 311]
(contesté par Koessler et al.[312]), et pour la libération d’EV[313]. Ces résultats sont
d'une importance clinique, car le bortézomib, un inhibiteur du protéasome utilisé
pour traiter les patients atteints de lymphome multiple, induirait une
thrombocytopénie[307, 308].
Une autre fonction du protéasome est d'hydrolyser les protéines en petits peptides.
Ce processus est d'une importance critique pour le chargement des peptides dans
le CMH de classe I et pour la présentation antigénique aux cellules T CD8+ dans
l'immunité adaptative. On trouve des molécules de CMH I sur les plaquettes murines
et humaines[314, 315], mais une large proportion (70 à 80%) s'est avérée provenir
de l’adsorption de CMH I circulant dans le plasma[316]. Non seulement les
plaquettes peuvent l’adsorber, elles sont également capables de transférer leur
CMH de classe I à la surface des cellules tumorales, ce qui leur confère un
phénotype normal[317]. En enrobant les cellules tumorales, les plaquettes
interfèrent avec la capacité des cellules tueuses naturelles (NK-Natural Killers) à

28
reconnaitre les cellules nocives auxquelles il manque le CMH I. Les tumeurs
cachées échappent ainsi à la reconnaissance des cellules NK, empêchant la
cytotoxicité et la production d'IFN γ[317]. Ce rôle insidieux du CMH I plaquettaire
permet la croissance des tumeurs et l'établissement du cancer.
En plus du CMH I et de la β2-MG, une chaperone moléculaire pour le complexe du
CMH I, la spectrométrie de masse a permis de caractériser le protéome des granules
α des plaquettes et d’identifier 45 protéines liées à la voie « d’apprêtement et
présentation de l'antigène »[318]. Parmi ces protéines, les auteurs ont identifié les
protéines TAP1 et TAP2 (antigen peptide transporter 1 and 2), qui permettent la
translocation du peptide traité dans le réticulum endoplasmique plaquettaire avant
leur chargement sur le CMH I[318]. Les composants du PLC (ERp57, calréticuline,
calnexine et tapasine), ainsi que les molécules costimulatrices des cellules T, dont
le CD40, ICOSL et CD86 (cette dernière chez l'homme uniquement)[318, 319] se
trouvent dans les plaquettes, ce qui suggère que les plaquettes contiennent toute la
machinerie pour l’apprêtement et la présentation des antigènes aux lymphocytes T.
Alors que la plupart des molécules ont été identifiées dans les compartiments
intracellulaires, leur présence à la surface des plaquettes est nécessaire à la
formation de synapses immunologiques avec les lymphocytes T. Une analyse par
microscopie confocale a révélé que les protéines CMH I, β2-MG, TAP1 et TAP2
pouvaient être localisées à la surface des plaquettes activées par la thrombine,
indiquant leur sécrétion lors de l'activation[318]. De plus, les nouvelles plaquettes
présentent des quantités plus grandes de CMH I sur leur membrane plasmique que
les plaquettes plus anciennes chez les souris, ce qui suggère que l'expression de
surface du CMH I est modulée tout au long de la durée de vie des plaquettes[320].
L'activation des plaquettes lors d’une infection peut également conduire à
l'expression du CMH I sur la membrane plasmique. En effet, chez les plaquettes
murines le CMH I est augmenté en cas d'infection par Plasmodium berghei et le
virus de la dengue[319, 321].

29
Les travaux par Chapman et al. ont montré la capacité des plaquettes à apprêter et
à présenter des antigènes aux lymphocytes T[319]. Les auteurs, en chargeant les
plaquettes avec la protéine ovalbumine (OVA) ont confirmés qu’elles possédaient la
machinerie nécessaire pour transformer efficacement l'OVA en peptides, y compris
la séquence peptidique antigénique SIINFEKL. Cette séquence, si elle est présentée
par les molécules du CMH I, peut être reconnue par le TCR exprimé par toutes les
cellules T chez les souris transgéniques OT-1. En utilisant des approches in vitro et
in vivo, ils ont montré que les plaquettes pouvaient en effet présenter l'antigène de
l'OVA via le CMH I aux lymphocytes T et, étant donné l'expression de molécules
costimulatrices par les plaquettes, pouvaient stimuler l’activation et la production
d'IL-2 des lymphocytes T[319]. Ces résultats indiquent une utilisation potentielle des
plaquettes dans un vaccin à base de cellules. Cette hypothèse a été testée avec un
véritable pathogène, Plasmodium berghei, parasite responsable du paludisme. En
utilisant le parasite Plasmodium berghei transgénique pour les acides aminés C-
terminaux 150-368 d'OVA (Pba-OVA), il a été montré que les plaquettes, via leur
CMH I, sont capables de charger et de présenter des antigènes dérivés de PbA-
OVA pour générer des lymphocytes T protecteurs[319]. L'ensemble de ces
expériences montrent que les plaquettes sont capables d’apprêter et de présenter
l'antigène.
Bien que les plaquettes puissent internaliser d'autres agents pathogènes que
Plasmodium berghei, tels que le virus de la dengue, le virus de la grippe et le VIH
par phagocytose ou endocytose[240, 243, 322, 323], elles augmentent également
l’élimination bactérienne chez les souris septicémiques[208]. Non seulement elles
sont capables de tuer directement des agents pathogènes comme C. albicans[324],
les plaquettes favorisent aussi l'interaction entre des agents pathogènes comme le
virus de l'hépatite B[325], L. monocytogenes ou d'autres bactéries Gram positifs
avec des cellules présentatrices d’antigènes professionnelles[326], soit en se liant
au pathogène et en le conduisant aux DCs, soit en recrutant les cellules
immunitaires au site d'infection et en modulant leur réponse. Un rôle pour le CD40L
plaquettaire a également été mis en évidence dans le contexte d’infections

30
bactériennes et virales. L'infection par le virus de la dengue chez l'homme augmente
l'expression de CD40L à la membrane plaquettaire et induit la sécrétion de
sCD40L[321]. Le CD40L dérivé des plaquettes améliore également l'absorption et
la destruction de Staphylococcus aureus par les DC grâce à l’augmentation de
l’expression du CD80 et la production accrue du TNF-α et des cytokines pro-
inflammatoires IL-12 et IL-6[327].
Un autre groupe de recherche a montré que le CD40L dérivé des plaquettes
améliorait la réponse des lymphocytes T CD8+ spécifiques de l'OVA et augmentait
l’élimination d'une Listeria monocytogenes transgénique pour le peptide OVA[328].
Le rôle des plaquettes dans la présentation antigénique a été montré depuis peu
(voir Figure 6, panneau de droite). Il reste cependant à déterminer si les antigènes
microbiens transformés peuvent être présentés par les molécules du CMH I des
plaquettes activées pour jouer un rôle dans l'immunité adaptative contre ces agents
pathogènes.
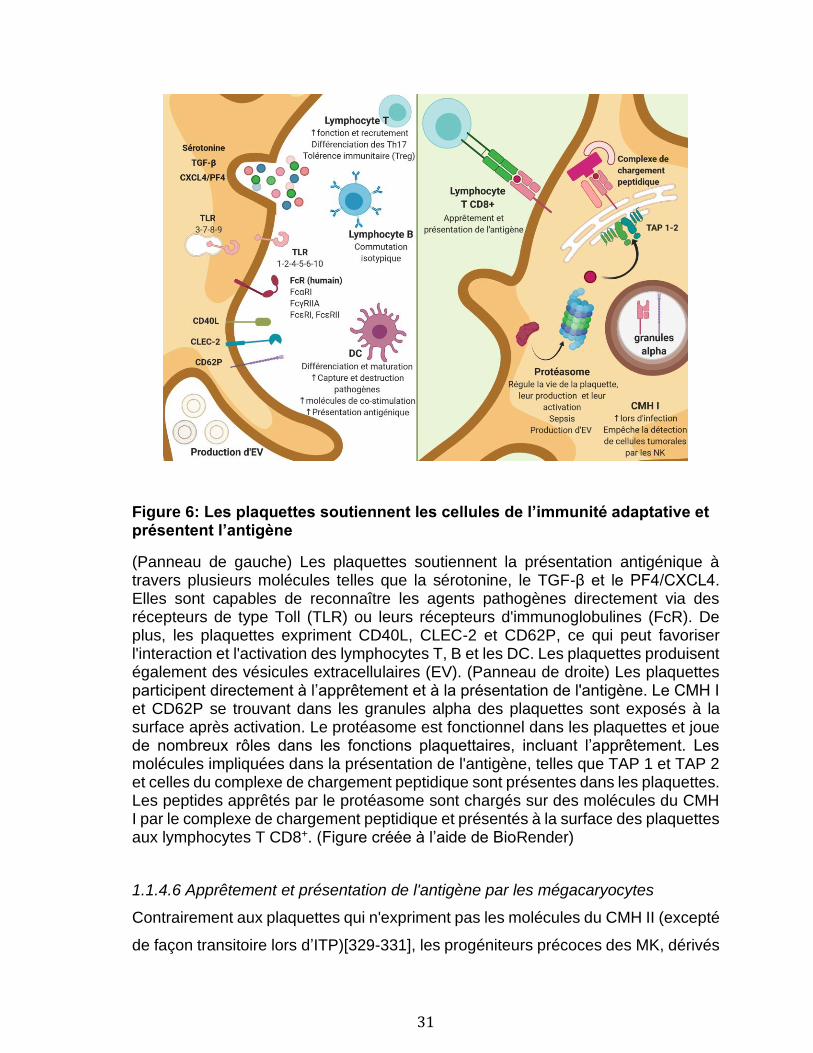
31
Figure 6: Les plaquettes soutiennent les cellules de l’immunité adaptative et présentent l’antigène
(Panneau de gauche) Les plaquettes soutiennent la présentation antigénique à travers plusieurs molécules telles que la sérotonine, le TGF-β et le PF4/CXCL4. Elles sont capables de reconnaître les agents pathogènes directement via des récepteurs de type Toll (TLR) ou leurs récepteurs d'immunoglobulines (FcR). De plus, les plaquettes expriment CD40L, CLEC-2 et CD62P, ce qui peut favoriser l'interaction et l'activation des lymphocytes T, B et les DC. Les plaquettes produisent également des vésicules extracellulaires (EV). (Panneau de droite) Les plaquettes participent directement à l’apprêtement et à la présentation de l'antigène. Le CMH I et CD62P se trouvant dans les granules alpha des plaquettes sont exposés à la surface après activation. Le protéasome est fonctionnel dans les plaquettes et joue de nombreux rôles dans les fonctions plaquettaires, incluant l’apprêtement. Les molécules impliquées dans la présentation de l'antigène, telles que TAP 1 et TAP 2 et celles du complexe de chargement peptidique sont présentes dans les plaquettes. Les peptides apprêtés par le protéasome sont chargés sur des molécules du CMH I par le complexe de chargement peptidique et présentés à la surface des plaquettes aux lymphocytes T CD8+. (Figure créée à l’aide de BioRender)
1.1.4.6 Apprêtement et présentation de l'antigène par les mégacaryocytes
Contrairement aux plaquettes qui n'expriment pas les molécules du CMH II (excepté
de façon transitoire lors d’ITP)[329-331], les progéniteurs précoces des MK, dérivés

32
des cellules souches hématopoïétiques humaines, expriment le CMH II. Ces MK
jouent des rôles qui rappellent celui des APC. Il a été montré que ces MK soutiennent
l'activation des lymphocytes T et augmentent la prolifération des Th17, des Th1 et
des cellules doubles positives Th17/Th1 lorsqu'ils sont incubés en présence de
lymphocytes de sujets normaux ou de patients atteints de SLE[332]. Les
progéniteurs de MK sont aussi efficaces pour monter une réponse immunitaire
contre le pathogène opportuniste Candida albicans, suggérant ainsi que la
mobilisation des cellules souches hématopoïétiques peut conduire à la génération
d'une population de MK avec des fonctions immunitaires[333]. Classiquement, les
molécules du CMH II présentent des peptides dérivés de pathogènes
extracellulaires via les endosomes, qui est un site d'entrée pour les virus. Il est connu
que le virus de la dengue peut se développer dans les progéniteurs de MK et les MK
matures peuvent générer une immunité antivirale qui implique des molécules d'IFN
en réponse au virus de la dengue ainsi qu'au virus de l’influenza[334]. Il reste à
établir, cependant, si les MK peuvent traiter et présenter des peptides viraux dans
le contexte d'une infection virale[335]. Les MK peuvent aussi être localisés dans les
poumons et, selon des études de transcriptome, ces derniers présentent un
phénotype de cellules immunitaires (cytokines et voies de signalisation activées)
contrairement aux MK de la moelle osseuse[336]. Comme les MK pulmonaires ont
un accès privilégié aux pathogènes de l'air tels que le virus de l’influenza ainsi qu'aux
allergènes, ils pourraient être en mesure de contribuer à la présentation antigénique
dans cet organe. Non seulement les mégacaryocytes des poumons ont une
expression de gènes similaire aux APC, mais ce phénotype est plastique et dépend
de l’environnement tissulaire comme il a été montré lors du transfert de
mégacaryocytes de la moelle osseuse dans les poumons[337]. Ces derniers
acquièrent une expression augmentée du CMH II dans l’environnement pulmonaire,
tout comme lorsqu’ils sont stimulés avec des PAMP tels que le LPS[337]. Cet article,
publié tout récemment, a aussi mis en évidence que les mégacaryocytes des
poumons peuvent internaliser, apprêter et présenter l’antigène via le CMH II aux
lymphocytes T CD4+[337].
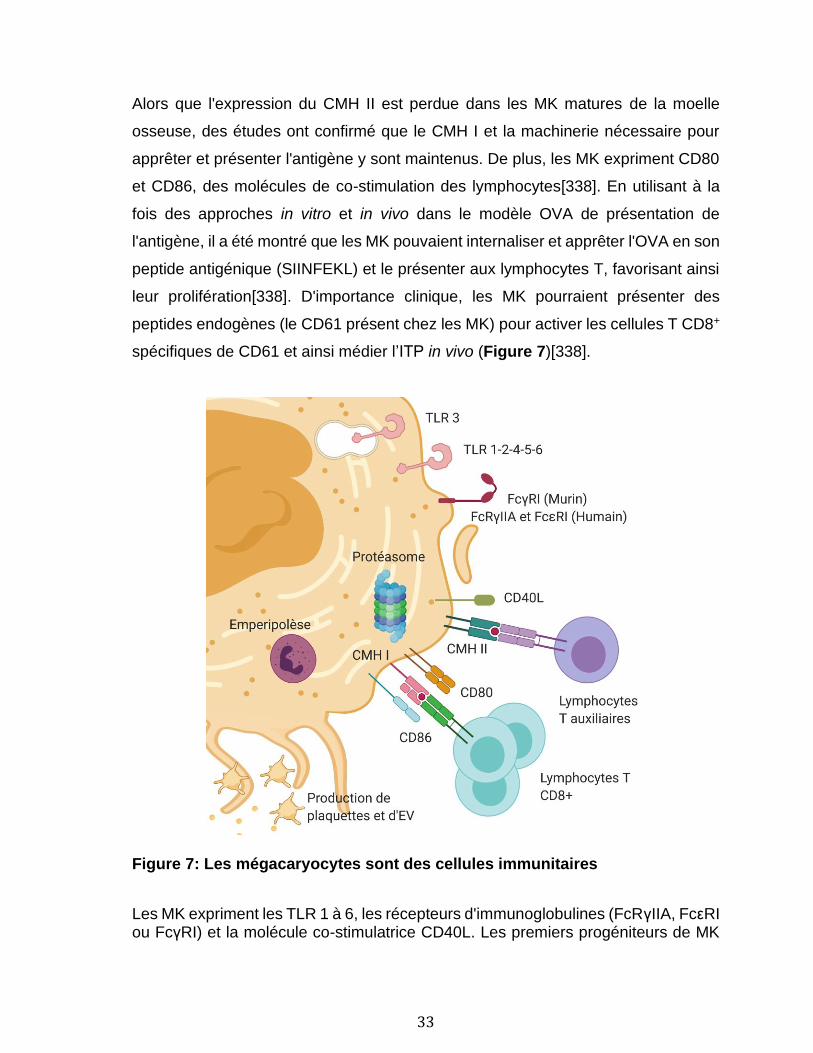
33
Alors que l'expression du CMH II est perdue dans les MK matures de la moelle
osseuse, des études ont confirmé que le CMH I et la machinerie nécessaire pour
apprêter et présenter l'antigène y sont maintenus. De plus, les MK expriment CD80
et CD86, des molécules de co-stimulation des lymphocytes[338]. En utilisant à la
fois des approches in vitro et in vivo dans le modèle OVA de présentation de
l'antigène, il a été montré que les MK pouvaient internaliser et apprêter l'OVA en son
peptide antigénique (SIINFEKL) et le présenter aux lymphocytes T, favorisant ainsi
leur prolifération[338]. D'importance clinique, les MK pourraient présenter des
peptides endogènes (le CD61 présent chez les MK) pour activer les cellules T CD8+
spécifiques de CD61 et ainsi médier l’ITP in vivo (Figure 7)[338].
Figure 7: Les mégacaryocytes sont des cellules immunitaires
Les MK expriment les TLR 1 à 6, les récepteurs d'immunoglobulines (FcRγIIA, FcεRI ou FcγRI) et la molécule co-stimulatrice CD40L. Les premiers progéniteurs de MK

34
expriment également le CMH II qui est important pour soutenir l'activation des cellules T et l'expansion des cellules T auxiliaires. Les MK contiennent du protéasome, expriment le CMH I et les molécules co-stimulatrices CD80 et CD86. Ces dernières leur permettent d’apprêter et de présenter l'antigène et de déclencher l'activation des lymphocytes T CD8+ et leur lymphoprolifération. Les MK sont capables d'emperipolèse et produisent les plaquettes et des vésicules extracellulaires (EV). (Figure créée à l’aide de BioRender) En raison de leur abondance dans le sang et les poumons, les plaquettes et les MK
sont idéalement positionnés pour réagir rapidement à un antigène et le présenter
aux lymphocytes. Un résumé de leurs rôles dans l’immunité innée et adaptative est
présenté en Figures 6 et 7. Toutefois, les plaquettes et les MK ne peuvent pas
atteindre les organes lymphoïdes en circulant à travers la lymphe comme le ferait
un leucocyte. Les plaquettes peuvent interagir directement avec les lymphocytes de
la rate ou les organes lymphoïdes secondaires et tertiaires par le biais des veinules
postcapillaires. Les plaquettes activées libèrent aussi des EV qui apportent avec
elles du contenu de la plaquette et qui pourraient être impliquées dans cette
interaction.
1.2 Les vésicules extracellulaires
1.2.1 Description générale et mécanisme de formation
Les EV sont des éléments sphériques de petite taille, composées de cytosol et d’une
membrane lipidique dont le contenu est enrichi en cholestérol, sphingomyélines, PS
et glycosphingolipides[339]. Les EV sont produites lors de l’activation ou de
l’apoptose de tout type cellulaire confondu, incluant les cellules procaryotes et
eucaryotes d’organismes unicellulaires ou pluricellulaires[340], et diffèrent en
fonction de leur taille, de leur contenu, de leur composition et de leur processus de
formation. Elles sont retrouvées dans tous les liquides biologiques ainsi que dans le
surnageant de culture cellulaire. Les EV ont de nombreux rôles, notamment pour
l’élimination de déchets cellulaires, la communication intracellulaire, l’hémostase, le
développement et l’inflammation[341-345].

35
Certaines EV sont associées à des pathologies et peuvent servir de biomarqueur
comme dans les fluides de patients atteints de maladies auto-immunes[346, 347].
Leur implication dans la progression de certaines maladies est également étudiée.
Par exemple, les EV de plaquettes et de cellules endothéliales sont plus abondantes
chez les patients atteints du syndrome anti-phospholipides[348]. Les EV de
plaquettes jouent aussi un rôle dans l’arthrite, où elles sont présentes abondamment
dans le liquide synovial des patients, et chez les patients atteints de SLE[349, 350].
En plus de présenter un intérêt comme source de biopsie liquide, les EV offrent aussi
un potentiel thérapeutique. Des études récentes s’intéressent à l’utilisation d’EV
pour effectuer du transport de médicament à des sites spécifiques dans l’organisme
ou bien à leur utilisation pour le développement de vaccins dans des maladies auto-
immunes[351-353].
Classées en trois groupes, soit les exosomes, les microvésicules (MV) et les corps
apoptotiques, les EVs ont des caractéristiques et des fonctions distinctes (Figure
8)[345].
Figure 8: Échelle de taille des vésicules extracellulaires Tirée de György, B. et al[354]. Taille respective des cellules et des différentes vésicules extracellulaires en comparaison avec d’autres éléments connus.

36
Issus de la fragmentation de cellules en apoptose, les corps apoptotiques ont une
taille comprise entre 1 et 5 μm, chevauchant celle des plaquettes[354]. Les
exosomes, qui sont formés par l’exocytose des corps multivésiculaires (MVB). Le
mécanisme de formation des MVB est dirigé par le complexe de tri endosomal
nécessaire au transport (ESCRT-endosomal sorting complex required for transport)
et requiert la participation d’une trentaine de protéines[355]. Les MVB ainsi formés
seront libérés dans le milieu extracellulaire lors de leur fusion avec la membrane
plasmique[356]. Les exosomes ont une taille comprise entre 50 et 100 nm de
diamètre qui est similaire aux virus[354]. De façon intéressante, de nombreux virus
enveloppés, principalement ceux à ARN, sont générés en utilisant la voie
ESCRT[357]. Les exosomes peuvent aussi être formés de façon ESCRT-
indépendante[358]. Les MV, également connues sous le nom d’ectosomes ou de
microparticules, sont des structures dont la taille comprise entre 100 nm et 1 µm
chevauche celle des complexes immuns et des bactéries (Figure 8)[354, 359]. Bien
que le système ESCRT semble aussi être impliqué dans la production de MV[360,
361], une combinaison d’autres facteurs sont importants. Ceci inclut le remodelage
de la membrane et la redistribution des phospholipides dont l’externalisation de la
PS via l’activité de flippases, floppases et de scramblases[362-364]. Une
augmentation du calcium intracellulaire est aussi nécessaire pour le remodelage du
cytosquelette qui permet le bourgeonnement et la libération des MV[365]. Les EV
principalement étudiées lors de cette thèse entrent dans la catégorie des MV et
seront décrites plus en détail.
Étant donné l’augmentation rapide de la recherche sur les EV dans les dernières
années, une société nommée ISEV (International Society for Extracellular Vesicles)
qui regroupe des scientifiques ayant une expertise dans le domaine a été fondée en
2011 dans le but d’améliorer, d’uniformiser et d’encadrer la recherche sur les EV.
Trois ans plus tard, l’ISEV a publié un article de référence indiquant les exigences
expérimentales minimales pour la définition des EV et de leurs fonctions[366]. Ces
exigences ont été révisées en 2018[367], et des recommandations entourant le
prélèvement, le transport et l’entreposage des EV ont été décrites en 2019[368]. Des

37
bases de données telles que ExoCarta (http://www.exocarta.org), EVpedia
(http://www.evpedia.info) et Vesiclepedia (http://www.microvesicles.org) concernant
les exosomes ou les EV ont aussi été créées[369-371].
1.2.2 Isolation et détection des vésicules extracellulaires.
L’isolation des EV pour leur caractérisation peut s’avérer complexe et présente de
nombreux obstacles. Non seulement, le milieu duquel sont extraites les EV contient
de nombreux contaminants tels que des acides nucléiques, des lipoprotéines, des
agrégats protéiques et des complexes immuns[354, 366, 372], mais une séparation
stricte entre les exosomes et les MV basée sur la taille ou par leur mécanisme de
formation n’est pas possible à ce jour et il n’existe pas de consensus sur les
marqueurs spécifiques à utiliser pour discriminer les deux populations[373]. Les
méthodes actuellement utilisées pour l’isolation incluent la centrifugation,
l’ultracentrifugation par gradient de densité, la chromatographie d’exclusion de
taille[372] et l’utilisation de trousses commerciales qui combinent plusieurs
approches[366].
Les EV peuvent être étudiées et caractérisées de nombreuses façons, notamment
par l’utilisation de marqueurs pour différentier les EV entre elles pour l’origine
cellulaire par exemple. Elles peuvent aussi être analysées par mesure de la diffusion
dynamique de la lumière (mouvement brownien), par analyse de suivi des
nanoparticules, par la mesure d’impédance, par immunobavardage de type
Western, ou des analyses protéomiques globales utilisant la spectrométrie de
masse[366, 374]. D’autres techniques qui permettent d’étudier les EV de façon
individuelle et qualitative sont la microscopie à fluorescence et la vidéomicroscopie
(pour les EV de grande tailles)[375, 376], ainsi que la microscopie électronique, la
microscopie à force atomique et la cytométrie en flux[377]. Étant donné la complexité
et le temps nécessaire pour l’analyse par microscopie, la cytométrie en flux est
l'approche la plus largement utilisée (70-75% des cas) pour la détection des EV et
présente l’avantage d’être quantitative en plus de permettre l’utilisation de plusieurs
marqueurs[378, 379]. Les cytomètres conventionnels, initialement développés pour

38
l’analyse de cellules offrent une faible résolution pour les EV de taille inférieure à 0,5
µm[380]. Cette différence de taille a un impact majeur pour l’analyse par cytométrie
si l’on considère qu’une cellule fait en moyenne 100 fois le diamètre d’une EV et
donc 10 000 fois sa surface[381]. Heureusement des cytomètres de haute sensibilité
ont été développés et offrent une résolution suffisante pour différentier les sous-
types de EV, incluant ceux qui contiennent des organelles[382, 383]. Cette approche
sera la principale utilisée pour l’analyse des EV de plaquettes.
De nombreux critères doivent être respectés pour une bonne caractérisation des EV
par cytométrie en flux afin de respecter les recommandations de l’ISEV.[367] Les
informations concernant la collection, l’isolation et l’entreposage des EV doivent être
présente dans le manuscrit avec toutes les étapes du marquage. Une description
détaillée des réactifs utilisés et du cytomètre (type de cytomètre, lasers utilisés,
seuils (treshold) et intensité, débit, etc.) est aussi exigé. Des contrôles de marquage
incluant l’analyse du tampon, des échantillons non marqués, des simples
marquages et l’utilisation d’isotype sont demandés. La nature membranaire des EV
doit être confirmée par un traitement à l’aide d’un détergent tel que le Triton X-100.
Des dilutions en série doivent être faites afin de démontrer l’absence de coïncidence
lors de l’analyse et de la quantification des EV. Enfin des détails sur la méthodologie
utilisée pour estimer la taille des EVs doivent aussi être fournis.
1.2.3 Diversité des vésicules extracellulaires
Le développement de techniques pour l’isolation et la caractérisation des EV a
permis de mieux apprécier leur diversité. En effet, les EV circulant dans le sang
peuvent provenir de différentes cellules vasculaires (plaquettes, monocytes, cellules
endothéliales, globules rouges et granulocytes). Ces EV expriment des marqueurs
de surface telles que des intégrines et transportent des acides nucléiques et des
protéines qui proviennent de la cellule mère, suggérant que ces EV pourraient avoir
une fonction distincte selon la nature de la cellule de laquelle elles proviennent et de
l’état (activé ou non)[378]. Les EV ont un rôle important pour l’hémostase et la

39
défense de l’hôte[384]. Elles ont toutefois un rôle majeur dans le développement et
la progression de nombreuses maladies comme le cancer, les maladies
cardiovasculaires, la prééclampsie, le diabète et les maladies
neurodégénératives[342, 384-387].
1.2.4 Organelles contenues dans les vésicules extracellulaires
1.2.4.1 La mitochondrie
La mitochondrie possède une membrane interne, site de la respiration cellulaire, qui
est repliée de façon à former de nombreuses invaginations et d’une membrane
externe. Elle possède son propre génome, l’ADN mitochondrial qui s’apparente a
celui de Rickettsia prowazekii[388]. Cette molécule circulaire, double-brin, est
formée de 16 569 paires de bases qui codent pour 2 ARN ribosomaux, 22 ARN de
transfert et 13 protéines[389], et qui possède des îlots CpG non méthylés[213]. La
mitochondrie joue de nombreux rôles au sein de la cellule. Elle est essentielle pour
la production d’énergie (ATP)[390], pour la synthèse d’hormones[391], pour
l’homéostasie du calcium[392, 393], et pour la régulation de la mort cellulaire
(apoptose)[394, 395].
Possédant de nombreuses caractéristiques communes avec les procaryotes, la
mitochondrie est une source importante de DAMPs (Figure 9). La mtDNA peut, via
le TLR 9, induire une réponse immunitaire dans différentes pathologies telles que le
cancer et les maladies auto-immunes[396, 397]. La présence de mtDNA est aussi
observée chez les patients gravement blessés, lors de sepsis ou de dommages aux
organes et est liée à l’évolution clinique[398-405]. Les produits sanguins, incluant
les PC, contiennent aussi de la mtDNA, qui est d’ailleurs impliquée dans le
développement de certaines réactions transfusionnelles comme le TRALI[406].
Les peptides formylés provenant de la mitochondrie s’accumulent aux sites de
dommages tissulaires. Ces derniers activent les récepteurs de peptides formylés, ce
qui induit l’accumulation de cellules pro-inflammatoires telles que les
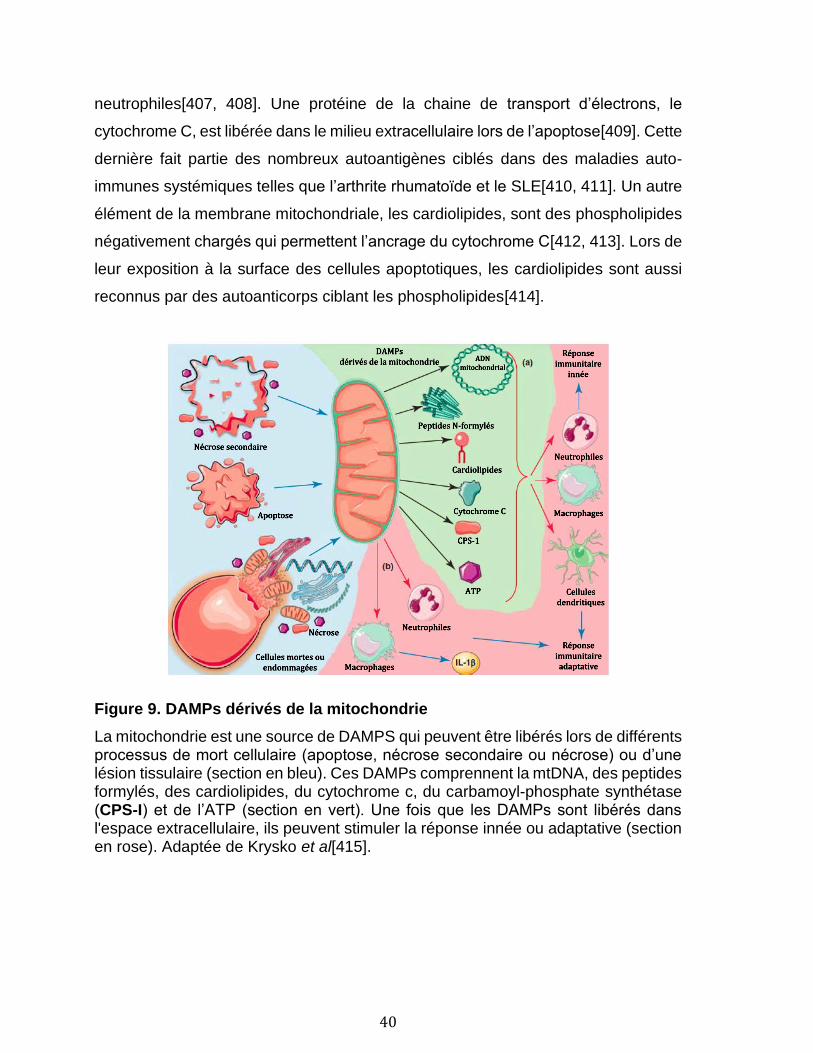
40
neutrophiles[407, 408]. Une protéine de la chaine de transport d’électrons, le
cytochrome C, est libérée dans le milieu extracellulaire lors de l’apoptose[409]. Cette
dernière fait partie des nombreux autoantigènes ciblés dans des maladies auto-
immunes systémiques telles que l’arthrite rhumatoïde et le SLE[410, 411]. Un autre
élément de la membrane mitochondriale, les cardiolipides, sont des phospholipides
négativement chargés qui permettent l’ancrage du cytochrome C[412, 413]. Lors de
leur exposition à la surface des cellules apoptotiques, les cardiolipides sont aussi
reconnus par des autoanticorps ciblant les phospholipides[414].
Figure 9. DAMPs dérivés de la mitochondrie
La mitochondrie est une source de DAMPS qui peuvent être libérés lors de différents processus de mort cellulaire (apoptose, nécrose secondaire ou nécrose) ou d’une lésion tissulaire (section en bleu). Ces DAMPs comprennent la mtDNA, des peptides formylés, des cardiolipides, du cytochrome c, du carbamoyl-phosphate synthétase (CPS-I) et de l’ATP (section en vert). Une fois que les DAMPs sont libérés dans l'espace extracellulaire, ils peuvent stimuler la réponse innée ou adaptative (section en rose). Adaptée de Krysko et al[415].

41
1.2.4.2 Le protéasome
Du protéasome extracellulaire a été observé dans le sang pour la première fois il y
a bientôt 30 ans[416]. Connu sous le nom de protéasome circulant, sa présence est
augmentée chez les patients souffrant de maladies auto-immunes, de cancers, de
trauma et de sepsis[417]. Il n’est toutefois pas exclu que ce protéasome puisse être
relargué par les cellules. D’ailleurs, il a récemment été observé que les lymphocytes
T et les cellules souches mésenchymateuses relarguent des EV contenant du
protéasome[418, 419]. De plus, il a été démontré que le protéasome 20S est présent
et actif dans les EV dérivées de cellules endothéliales apoptotiques[169]. Non
seulement ces EV régulent la formation des structures lymphoïdes tertiaires, elles
sont aussi importantes dans la production d'auto-anticorps et le rejet de greffe après
la transplantation[169, 420]. Le transfert du protéasome dans les EV de plaquettes
est plausible étant donné sa présence dans les plaquettes et les MK, mais n’a pas
été vérifié à ce jour.
1.2.5 Les vésicules extracellulaires de plaquettes
Les EV de plaquettes représentent environ 80% des EV circulantes dans le
sang[421, 422]. Anciennement appelées poussière de plaquettes[423], elles sont
formées lors de l’activation des plaquettes par divers agonistes comme la thrombine,
le collagène et le complément[424, 425]. Elles peuvent aussi être formées en
l’absence d’activation sous contrainte de cisaillement et lors de l’entreposage dans
les PC[426-428].
Les EV de plaquettes, lors de leur genèse, peuvent apporter avec elles des
mitochondries situées à proximité de la membrane plasmique[382]. Ces EV,
présentes dans les liquides biologiques et dans les PC forment une population
hétérogène qui peut être divisée en trois catégories[382], soit les EV qui ne
contiennent pas de mitochondries, les EV qui contiennent une ou des mitochondries.
Il est aussi possible d’observer des mitochondries extracellulaires, qui ne sont pas
entourées d’une membrane de plaquette (freeMitos). L’ADN mitochondrial est aussi

42
retrouvé dans les EV[429, 430].
Il est suggéré que les EV de plaquettes jouent de nombreux rôles dans la régulation
des fonctions physiologiques et pathologiques[431]. Elles ont une activité
procoagulante en exposant la PS et elles accélèrent la génération de thrombine[426,
432-434]. Elles expriment aussi du TF qui permet l’initiation de la cascade de
coagulation in vivo[435-437]. Les EV de plaquettes ont aussi un rôle dans la
régulation de l’immunité (stimulation et suppression), où elles peuvent interagir avec
les lymphocytes T et réguler leur différenciation et leur activité régulatrices[438, 439].
De plus, elles peuvent favoriser la formation de centres germinatifs et la production
d'IgG par les lymphocytes B en fonction de leur contenu en CD40L[440, 441].
Comme les EV de plaquettes peuvent circuler dans la lymphe[442, 443], elles ont la
possibilité de transporter des molécules de l'immunité adaptative vers les organes
lymphoïdes.
Le cargo transporté par les EV de plaquettes (facteurs de croissance, protéines,
acides nucléiques, organelles, etc) leur permet aussi de jouer un rôle dans la
communication intracellulaire[354, 444]. Par l’échange d’acides nucléiques (mRNA,
miRNA et autres types d’ARN), elles peuvent réguler les cellules à des niveaux post-
transcriptionnels[445-447]. Le cargo des EV de plaquettes leur donne aussi rôle
dans l’inflammation en libérant de nombreux médiateurs pro-inflammatoires.
L’emphase de cette thèse sera sur deux organelles contenues dans les EV, soit la
mitochondrie et le protéasome.

43
1.3 Objectifs
Les plaquettes favorisent l'hémostase en jouant un rôle important dans la
coagulation, mais elles ont aussi un rôle dans l’immunité. Lors de leur activation,
elles produisent des EV dont le cargo est variable. Le but de cette étude vise d’une
part à approfondir les connaissances fondamentales sur les EV de plaquettes et
leurs rôles dans l’inflammation et l’immunité en s’attardant plus spécifiquement aux
organelles pouvant être contenues dans les EV de plaquettes. Notre hypothèse
générale est que le cargo des EV de plaquettes, plus précisément les organelles,
leur confèrent un rôle dans l’inflammation et l’immunité.
Dans le cadre de cette thèse, nous avions comme premier objectif d’évaluer
l’utilisation d’un algorithme (SPADE) couplé à la cytométrie en flux pour améliorer la
détection d’EV. Notre hypothèse est que l’utilisation de SPADE peut permettre de
mieux apprécier l’hétérogénéité des EV dans des concentrés plaquettaires (CP)
servant à la transfusion.
Un second objectif était d’évaluer si les EV de plaquettes contenant des
mitochondries pouvaient représenter un réservoir d'ADN mitochondrial, DAMP
reconnu et présent dans de nombreuses conditions inflammatoires. Notre hypothèse
est que les EV de plaquettes contenant des mitochondries peuvent être un réservoir
d’ADN mitochondrial et que cet ADN mitochondrial peut être utilisé pour évaluer le
risque d’incident transfusionnel des PC.
Un troisième objectif était de vérifier si le protéasome, ainsi qu’une machinerie
fonctionnelle pour l'apprêtement et la présentation de l'antigène sont transférés dans
les EV de plaquettes. Notre hypothèse est que les EV de plaquettes contiennent un
protéasome fonctionnel et que le protéasome confère aux EV de plaquettes un rôle
dans l’immunité.

44
Dans l’ensemble, les objectifs ont été atteints et nous avons démontré que les EV
de plaquettes ont un rôle dans l’inflammation et l’immunité avec leur contenu en
mitochondries et en protéasome.

45
Chapitre 1: Revealing the diversity of extracellular
vesicles using high-dimensional flow cytometry
analyses.
2.1 Résumé
Les vésicules extracellulaires (EV) produites par les cellules activées ou
apoptotiques sont hétérogènes et cette diversité pourrait permettre leur utilisation
comme biomarqueur. Malgré les progrès concernant l’amélioration des techniques
d’isolation et de caractérisation des EV, leur analyse par cytométrie en flux est
encore limitée concernant leur séparation du bruit de fond de l’appareil et l’analyse
de plusieurs paramètres conjoints. Nous avons appliqué l’algorithme SPADE à
l’analyse de nos EV par cytométrie en flux à haute sensibilité. Celui-ci nous a permis
d’organiser les sous-populations d’EV de plaquettes et de globules rouges et
d’apprécier leur hétérogénéité en plus de mettre en évidence des sous-types de
patients arthritiques selon la composition des EV présentes dans leur liquide
synovial. Notre étude a révélé que l’utilisation d’algorithmes couplés à la cytométrie
en flux tels que SPADE pourrait faciliter la compréhension des fonctions des EV et
le développement de leur étude comme biomarqueurs.

46
Revealing the diversity of extracellular vesicles using high-dimensional flow
cytometry analyses
Geneviève Marcoux1, Anne-Claire Duchez1, Nathalie Cloutier1, Patrick Provost1,
Peter A. Nigrovic2 & Eric Boilard1
1 Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de
Médecine de l’Université Laval, Département de microbiologie et immunologie,
Québec, QC, Canada, G1V 4G2
2 Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de
Médecine de l’Université Laval, Département de Psychiatrie et Neurosciences,
1Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de
Médecine de l’Université Laval, Département de microbiologie et immunologie,
Québec, QC, Canada.
2Department of Medicine, Division of Rheumatology Immunology and Allergy,
Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA.
Corresponding author:
Eric Boilard, PhD
Centre de Recherche en Rhumatologie et Immunologie
Centre de Recherche du Centre Hospitalier Universitaire de Québec
Faculté de Médecine de l’Université Laval
2705 Laurier Blvd., room T1-49
Québec, QC
Canada G1V 4G2
E-mail: [email protected]

47
2.2 Abstract
Extracellular vesicles (EV) are small membrane vesicles produced by cells upon
activation and apoptosis. EVs are heterogeneous according to their origin, mode of
release, membrane composition, organelle and biochemical content, and other
factors. Whereas it is apparent that EVs are implicated in intercellular
communication, they can also be used as biomarkers. Continuous improvements in
pre-analytical parameters and flow cytometry permit more efficient assessment of
EVs; however, methods to more objectively distinguish EVs from cells and
background, and to interpret multiple single-EV parameters are lacking. We used
spanning-tree progression analysis of density-normalized events (SPADE) as a
computational approach for the organization of EV subpopulations released by
platelets and erythrocytes. SPADE distinguished EVs, and logically organized EVs
detected by highsensitivity flow cytofluorometry based on size estimation,
granularity, mitochondrial content, and phosphatidylserine and protein receptor
surface expression. Plasma EVs were organized by hierarchy, permitting
appreciation of their heterogeneity. Furthermore, SPADE was used to analyze EVs
present in the synovial fluid of patients with inflammatory arthritis. Its algorithm
efficiently revealed subtypes of arthritic patients based on EV heterogeneity
patterns. Our study reveals that computational algorithms are useful for the analysis
of high-dimensional single EV data, thereby facilitating comprehension of EV
functions and biomarker development.

48
2.3 Introduction
Extracellular vesicles (EV) are small membrane vesicles released by cells into the
extracellular milieu. They are subdivided into three major groups, primarily based on
the process underlying their release. Exosomes are produced by exocytosis of
multivesicular bodies and have a diameter that ranges between approximately 50-
150 nm. Microvesicles (MV), also termed microparticles or ectosomes, are vesicles
of approximately 100–1000 nm in diameter generated by cytoplasmic membrane
budding and fission. Apoptotic bodies are released by apoptotic cells and generally
possess dimensions larger than 1000 nm1–3. However, it should be noted that these
definitions are still subject to change, as larger exosomes (up to 250 nm) have been
described, and apoptotic cells also release exosome-like vesicles3,4. Hence, the term
EV is increasingly utilized, as it more liberally encompasses all vesicle types
released by cells.
As EVs carrying components from donor cells can be internalized by cellular
recipients, they are implicated in intercellular communication5–7. Eukaryotic, and
prokaryotic cells produce EVs, suggesting that this mechanism is well-conserved
throughout evolution, which points to its significance. Furthermore, EVs are induced
by cell activation, and EV levels in biological fluids are altered in different
pathologies, such as cancer, rheumatic diseases, neurodegenerative disorders,
organ damage, and infectious diseases2,3,8. Hence, EVs appear to be potent
biomarkers, and their detection in blood and other biological fluids is improving with
the recent establishment of pre-analytical conditions, standardization of
quantification approaches, and development of more efficient means of detection9–
15.
EVs (more specifically MVs) have been described in the blood of healthy individuals.
They mainly comprise MVs derived from platelets and red blood cells (RBC)16–19.
Cell surface markers allow the cellular origin of MVs to be distinguished as CD41a
and CD235a are detected on the surface of platelets and RBC MVs, respectively.

49
Studies show that megakaryocytes (MK) can also generate CD41a+-MVs, and that
MVs in blood derived from MKs can be distinguished from those of platelets by
surface expression of P-selectin (CD62P), lysosomal-associated membrane protein
1 (LAMP-I) and immunoreceptor-based activation motif (ITAM) receptors16,20,21.
Thus, cell lineage markers provide useful information for determining the cellular
source of EVs.
EV heterogeneity is further complicated by the presence or absence of other
components. Phosphatidylserine (PS) exposure by EVs, which permits identification
of the latter with probes conjugated to PS-binding proteins such as annexin V and
lactadherin, can in fact appear undetectable on a substantial proportion of EVs16,22–
24. EV subpopulations may contain active proteasome, organelles (e.g.
mitochondria), and may undergo post-translational modification in disease, such as
citrullination in inflammatory arthritis4,16,23,25–27. Furthermore, EVs are decorated with
autoantibodies in autoimmune diseases such as systemic lupus erythematosus
(SLE) and rheumatoid arthritis (RA)16,23,28, and different platelet stimuli were shown
to trigger release of different EV subpopulations18,24. In summary, EVs are highly
heterogeneous, and the recognition of EV subtypes is necessary for the
comprehension of EV functions and consistent design of biomarkers.
Seminal research has established optimal pre-analytical conditions for the
preparation of samples for EV analyses12,29,30. Flow cytometry (FCM) remains the
most convenient methodology for assessment of EVs in biological fluids, especially
when a large sample number is analyzed, and when further characterization is
required using multiple markers on hundreds of thousands of EVs simultaneously.
Whereas traditional flow cytometers were not designed for the detection of such
small vesicles, the recent development of high-sensitivity FCM (hs-FCM) has greatly
improved investigators’ capabilities of analyzing EVs and developing biomarkers
based on EV heterogeneity10,12,13,15,23.
However, even with up-to-date approaches for the detection of EVs, investigators

50
are left with tremendous amounts of high-dimensional datasets that are impossible
to interpret using traditional gating on bivariate dot plots. Furthermore, with more
powerful methods of detection, distinguishing potentially relevant, but rare,
subpopulations of EVs from the generally important non-specific background is a
recognized issue12. There is currently no study that reports a means of facilitating
interpretation of flow cytofluorometric analyses of EVs. Thus, with the rapidly growing
improvements in detection technologies, and recognition of the vast heterogeneity
of EVs, the development of methods to objectively interpret multiple single-EV
parameters is urgent. Scientists implicated in the study of EVs are, of course, not
the first to face this sort of challenge. For instance, in immunology, an understanding
of the functions of discrete immune cells has evolved greatly since the discovery of
the different immune cell subtypes. It is now well-accepted that T lymphocytes are
heterogeneous and plastic, with CD4 and CD8 expression, and the identification of
several subtypes such as Th1, Th2, Th17, regulatory T cells and follicular-helper T
cells31. Moreover, dendritic cells32,33, monocytes, macrophages34 and
neutrophils35,36 are subdivided into different classes based on surface receptor
expression and density. From this perspective, traditional methods to analyze
multidimensional single-cell data are inadequate, as they are subjective and
restricted to our actual knowledge of the cellular phenotypes.
Spanning-tree progression analysis of density-normalized events (SPADE) is a
versatile computational approach that can be applied to mass and FCM data37.
SPADE does not require manual gating, which is considered more subjective37,38,
and allows the visualization of rare cell types, which would be lost or simply excluded
if they were unexpected. Also, it provides a global overview of cellular heterogeneity
by showing a continuity of phenotype, instead of clustering events in an independent
and strictly-defined population. Those features were successfully used to
characterize cell populations in different contexts37,39–43. In this present study, we
successfully used SPADE algorithms to build an EV hierarchy for the appreciation
of EV diversity in biological fluids.

51
2.4 Methods
2.4.1 Ethics
The study was approved by the medical ethics committee of CHU de Québec and
Université Laval. Methods were carried out in accordance with the approved
guidelines. Platelets and RBC were obtained from the citrated blood of healthy
human volunteers under informed consent according to a protocol approved by an
Institutional Review Board (Centre de Recherche du Centre Hospitalier Universitaire
de Québec). Synovial fluid (SF) was obtained from volunteers with rheumatoid
arthritis (RA) with approval of the ethic committee (Brigham and Women’s Hospital)
and under informed consent.
2.4.2 Platelets
Platelets were isolated after centrifugation of blood (282 x g for 10 min) at room
temperature (RT). Platelet-rich plasma (PRP) was obtained after an additional
centrifugation of the supernatant at 600 x g for 3 min at RT. The supernatant was
then centrifuged at 1,300g for 5 min at RT and the platelet-containing pellet was
resuspended in Tyrode’s buffer (pH 7.4). Platelets were counted (Cellometer
AutoM10; Nexcelom Bioscience) and adjusted to a density of 100 x 106 cells/ml in
Tyrode’s buffer (pH 7.4). For platelet EVs, platelets were stimulated with thrombin
(0.5 U/ml, Sigma) for 1 h at 37 °C after addition of 5 mM of calcium. Platelet activation
was stopped by addition of 20 mM of EDTA, and remnant platelets were removed
by centrifugation at 1,300 x g for 5 min at RT, performed twice.
2.4.3 Platelet-free plasma (PFP)
PRP from healthy volunteers was prepared as above without any stimulation and
was centrifuged at 2,500 x g for 20 min at RT. Then, the supernatant was centrifuged
twice at 3,200 x g for 5 min when platelet-free plasma (PFP) was required.
2.4.4 Red blood cells (RBCs)
Red blood cells were isolated after centrifugation of blood (282 x g for 10 min at RT).

52
Platelet-rich plasma and buffy coat fractions were eliminated to conserve the RBC
pellet. Red blood cells were counted (Cellometer AutoM10), adjusted to a density of
100 x 106 cells/mL in Tyrode’s buffer (pH 7.4) and contained less than 0.004% (n =
3) contaminating platelets. For RBC EVs, 200 μL of this preparation was added to
50 ml of distilled water (filtered through a 0.22-μm membrane) for 5 min, and 5 ml of
PBS 10X (filtered through a 0.22-μm membrane) was then added to stop the
hypotonic reaction. Remnant RBC were removed by centrifugation at 1,300 x g for
5 min at RT13.
2.4.5 Characterization of EVs
Hs-FCM approach. All the analyses were performed on a BD Canto II Special Order
Research Product (BD Biosciences) equipped with a small particle option, as
described previously13,23. The forward scatter (FSC) on this dedicated equipment is
coupled to a photomultiplier tube (PMT) with a 488 nm solid state, 100 mW output
blue laser (rather than the conventional 20 mW), and includes a 633 nm HeNe, 20
mW output red laser and a 405 nm solid state diode, 50 mW output violet laser. The
hs-FCM includes an FSC-PMT and a Fourier optical transformation unit, which
reduces the background/noise and increases the angle of diffusion, thereby
enhancing the detection of small-diameter particles.
FCM performance tracking was performed daily before all analyses using the BD
cytometer setup and tracking beads (BD Biosciences, San Jose, CA, USA). The
assigned voltage for FSC-PMT was 300 volts (V). For side scatter (SSC), the
assigned voltage was 460 V and the threshold was 200. Voltage was set to 360 V
for FITC, 450 V for PE-Cy7, 500 V for Deep Red (or APC), 500 V for PE, 500 V for
Alexa Fluor 700 (used only for the counting beads) and 305 V for V450. Acquisition
was performed at low speed (~10 μL/min) and, to remain quantitative, a known
quantity of (fluorescent) polystyrene microsphere (15-μm diameter: Polysciences,
PA, USA) was added to each tube, and a constant number of beads detected on the
basis of (auto) fluorescence was acquired for each sample throughout all the study.
Silica particles (Kisker Biotech GmbH & Co. Steinfurt, Germany) of known

53
dimensions (100 nm, 500 nm and 1 μm in diameter) were used for instrument set-
up standardization13,23. Fluorophores implicating distinct lasers were intentionally
chosen to minimise compensation. For every performed experiment, the only
necessary compensation was 5.00% for APC-V450.
Detection of platelets, RBCs and EVs by flow cytometry. Platelets and EVs were
prepared as above and 5 μL per sample was labeled in a total reaction volume of
100 μL (as indicated in Table S1) at 37 °C for 30 min. Then, the sample was diluted
by adding 400 μL of the labeling buffer prior analysis by hs-FCM. RBC-derived
samples were labeled as described above with the exception of MitoTracker, which
was omitted given the known absence of mitochondria in RBCs. For Triton or EDTA
treatment, samples (5 μL) were incubated 30 min at RT with 0.05% Triton X-100 or
50 μM EDTA (with PBS instead of Annexin V buffer for EDTA) before labeling. For
the ultracentrifugation treatment, samples were centrifuge at 100 000 g x 1 h at 20
°C to pellet EVs, and the supernatant was labeled as presented above.
Plasma EV diversity with overlapping staining panels. EVs contained in platelet-
free plasma (5 μL for 100 μL reaction) were labeled as indicated in Table S1 with the
four different cocktails (5a, b, c and d). The reaction was stopped by adding 400 μL
of the labeling buffer.
EV diversity in RA synovial fluid. Freshly obtained SF, collected without
anticoagulant, was centrifuged at 1,900 x g for 30 min at 4 °C to remove leukocytes
and then stored at − 80 °C. EVs present within the SF (5 μL for 100 μL reaction)
were labeled as indicated in Table S1 and the reaction was stopped by adding 400
μL of the labeling buffer.
Immunoblotting. Cells were pelleted at 1,300 x g for 5 min: EVs derived from
platelets and RBC and those present in plasma were centrifuged at 18,000 x g for
90 min5 and processed in lysis buffer (20 mM Tris HCl pH 7.8, 1.25 mM EDTA, 0.5%
Triton X-100, 0.5% NP-40, 120 mM NaCl, 2 mM phenylmethylsulfonyl fluoride.

54
Lysate protein content was measured using a bicinchoninic acid protein assay kit
(Fisher). Proteins (10 μg) were separated by 10% SDS-PAGE, transferred to a
polyvinylidene difluoride membrane and the candidate proteins were detected using
antibodies against platelet MV marker CD41a (Abcam used at 1/1,000), an enzyme
reportedly present in platelet MVs; 12-lipoxygenase (12LO) (Santa Cruz, used at 1
μg/ml); cytoskeleton marker β-actin (Sigma, clone AC-15 used at 1/15,000); the EV
markers, tumor susceptibility 101 (TSG101)(Abcam, clone 4A10 used at 0.1 mg/ml)
and ALG-interacting protein X (ALIX) (Santa Cruz, clone 3A9 used at 1 μg/ml); the
RBC marker CD235a (Santa Cruz, clone YTH89.1 used at 1 μg/mL); and the
mitochondrial markers, voltage-dependent anion channel (VDAC) (Cell Signaling,
used at 11.6 μg/ml) and translocase of outer mitochondrial membrane TOMM-22
(Abcam, used at 2.5 μg/ml)5,44. The PVDF membranes were incubated with
peroxidase-conjugated antibodies recognizing the primary antibodies (Jackson
ImmunoResearch, used at 0.08 μ g/ml).
Size analysis. Silica beads were diluted 1/100 in PBS 1X (filtered through a 0.22-
μm membrane) and analyzed by Zetasizer Nano S (Malvern Instruments, Ltd.,
Malvern, UK).
2.4.6 Statistical analyses
The results are presented as mean ± SEM, and were analyzed with Prism 6
(GraphPad Software, CA, USA).
2.4.7 SPADE analyses
The pre-compiled standalone version of SPADE-3 for Mac without Matlab was
downloaded at http://pengqiu.gatech.edu/software/SPADE/. FCS files were
exported from BD FACSDiva™ software (BD Bioscience) in FCS 3.0 format and
analyzed using FlowJo (FlowJo, LLC, OR, USA) software to exclude counting beads
and events with dimensions smaller than 100-nm silica beads. Specific details for
each SPADE analysis are provided within the results section.

55
2.5 Results
2.5.1 Optimization and validation of high sensitivity flow
cytofluorometric methods for the detection of EVs.
In the first set of experiments, we validated that platelets, RBC, and their daughter
EVs, were efficiently resolved by hs-FCM. EVs were not pelleted prior to hs-FCM
analyses, given the reported deleterious impact of this procedure on EV integrity45.
Furthermore, as one goal of this study was to appreciate EV diversity, we chose to
maintain the complexity of our EV preparations by avoiding exosome and MV
enrichment. Therefore, EV preparations comprised a mixture of exosomes and MVs
derived from platelets and RBC, and the hs-FCM conditions were optimally designed
to detect EVs larger than 100 nm silica beads.
Given that size is a factor of interest in these analyses, we used microspheres of
known dimensions to standardize the instrument setup. Whereas polystyrene
microspheres are frequently utilized for the determination of size, their refraction
index (1.59) differs considerably from that of membrane vesicles (1.39)13,46,47.
Hence, while size, shape, surface roughness, granularity and the angle of collection
impact light scattering, the intensity of the scattered light greatly depends on the
refraction index for particles with dimensions smaller than the wavelength of light (in
this case 488 nm). Thus, for our analyses, we used silica beads, which have a
refraction index (1.42)13,46,47 closer to that of membrane vesicles, to establish the
lower limit of the EV gate, and we included intact platelets and RBC to ensure that
they were efficiently distinguished from their respective daughter EVs.
We validated that silica microspheres ranging from 100–1000 nm in diameter (Fig.
1A) were efficiently resolved by hs-FCM (Fig. 1B). Resting platelets were as
expected larger than 1000 nm silica beads (Fig. 1C)48. To generate platelet EVs,
platelets were triggered by thrombin and remnant platelets were removed by
centrifugation. In these preparations, platelets were undetectable (Fig. 1D), and

56
platelet EVs were detected by hs-FCM (Fig. 1E), and were clearly distinguishable
from intact platelets (compared to Fig. 1C). As expected, RBC (mean diameter
between 7–8 μ m) appeared much larger than platelets in our hs-FCM analyses (Fig.
1F), and were triggered to release EVs by osmotic shock. RBCs were largely absent
from EV preparations (Fig. 1H). RBC EVs were efficiently distinguished from intact
RBC (Fig. 1H), although they displayed apparent larger dimensions than platelet
EVs in hs-FCM (compared to Fig. 1E). This observation will not be investigated
further in the present study. Thus, small EVs are detected in our hs-FCM analyses.
Fluorochrome-conjugated probes and antibodies can form submicron aggregates in
solution, which can be mistakenly interpreted as EVs by hs-FCM49. Furthermore,
multiple EVs can be detected simultaneously if present at a too elevated
concentration or analyzed at high acquisition speed, a process called coincidence
or swarm that compromises the interpretation of EV multicolor labeling47. To ensure
that genuine EVs were detected, and that no signals arose from aggregated
fluorochromes, we used an established detergent assay49,50. Under these
conditions, the membrane moiety of the EVs is dissolved by Triton X-100 treatment
while protein aggregates are left intact13,23,49,50. In addition, the specificity of PS
recognition by annexin V-conjugated fluorochromes, which is a calcium-dependent
event, was confirmed by calcium chelation using EDTA13,22. As EVs can be pelleted
by centrifugation, we also verified that no EVs were detected in fluids after
ultracentrifugation.
Platelet EVs may contain mitochondria, and can express surface CD41a and PS5,25.
Thus, platelet EVs, detected by a combination of mitochondrial dye MitoTracker,
anti-CD41a antibody and annexin V (Fig. 2A), were treated with detergent (Fig. 2B),
or EDTA (Fig. 2C). Furthermore, all the fluorescent probes were incubated with fluids
that underwent ultracentrifugation (Fig. 2D). Under these conditions, the vast
majority of EVs positive for CD41a (Fig. 2B,D,E), MitoTracker (Fig. 2B,D,F), and
annexin V (Fig. 2G), were eliminated by detergent and centrifugation. Conversely,
EDTA primarily affected annexin V labeling (Fig. 2G), with only a modest impact on

57
CD41a (Fig. 2C,E) and MitoTracker signals (Fig. 2C,F), thereby validating the
specificity of our multicolor labeling of platelet EVs and further confirming the
heterogeneity of EVs.
We next verified the absence of coincidence in our hs-FCM conditions and validated
our quantitative strategies. In the absence of coincidence, the concentration of EVs
should be reduced according to dilution factors, while the mean fluorescence
intensity should remain constant13. We confirmed the lack of coincidence, as the
concentrations of EVs positive for CD41a (Fig. 2H), MitoTracker (Fig. 2K) and
annexin V (data not shown for platelet EVs) were consistently reduced without any
impact on the mean and median fluorescence intensity (Fig. 2I,J,L,M). Using anti-
CD235a and annexin V-conjugated probes, we also confirmed the specificity of our
signals and the absence of coincidence in our flow cytofluorometric acquisitions of
RBC EVs (Figure S1).
Depending on their mechanism of release, EVs may contain distinct sets of proteins
and organelles44. Of note is that immunoblotting confirmed that our EV preparations
contained proteins reportedly present in EVs5,25,44. As expected, CD235a was
absent in EVs derived from platelets, whereas the surface protein CD41a, the
cytosolic platelet 12-LO, the cytoskeleton protein actin, the EV proteins TSG101 and
ALIX, and the mitochondrial proteins VDAC and TOMM-22 were detected in platelet
EVs. CD41a, platelet 12-LO, and mitochondrial markers were absent in RBC EVs,
whereas cytoskeleton and EV markers were detected (Figure S2). Together, these
observations confirm that our strategies are optimal for the establishment of optimal
high-dimensional dataset analyses of EVs.
2.5.2 Analysis of RBCs, platelets and their EVs using SPADE
Contrary to traditional gating analysis, where gates must be manually designed,
SPADE uses topological methods to reveal distinct populations of cells from high-
dimensional data sets37,38,43, and also equally represents rare and abundant cell
types (and potentially EVs). This is important, because rare, but biologically relevant

58
EVs, might be masked if outnumbered by background or noise; a particularly
frequent issue in FCM analyses of EVs. Events (e.g. cells, or potentially EVs here)
that share similitudes on the basis of marker expression are clustered within the
same node. Each node can be colored according to their median intensity for a given
marker expression (low to high; blue to red, respectively) and the size of the node
reflects the number of events that it contains43. Nodes that belong to the same
branch on the tree are more likely to be related to each other than nodes found on
different branches, and the length of the branches is determined automatically by
the program43. Thus, using multiple fluorescent markers, in addition to lightscatter
(FSC-PMT and SSC), it might become possible to identify groups of EVs that are
similar with respect to each measured parameter.
RBCs, platelets, and their respective EVs generated in vitro, as above (n = 3 blood
donors), were detected by hs-FCM on the basis of expression of CD41a, CD235a,
PS exposure, mitochondrial content, size (FSC-PMT) and inner complexity (SSC).
FCS files were pre-analyzed to exclude counting beads (Fig S3A) and events smaller
than 100 nm silica beads (Figs S3B and 1B). The files were used to build the SPADE
tree with the following markers: FSC-H (for cells), FSC-PMT-H, SSC-H, MitoTracker-
H, CD41a-H, CD235a-H and annexin V-H. An inverse hyperbolic sine transformation
with cofactor 150 was applied in order to scale the data, and the maximum allowable
cells/EVs in the pooled down-sampled data was set to 50,000. The outlier was set
to the 1st percentile of local densities and the target density was set such that a fixed
number of 20,000 cells would remain. The number of desired clusters was 200, as
a high EV heterogeneity was expected, and the K-means algorithm was chosen as
the clustering parameter.
Using the semi-automated annotation tool (button “Auto Suggest Annotation”), which
relies on all markers used to build the tree, a tree was automatically generated (Fig.
3A), distinguishing 10 sub-populations (namely 1–10). The first autosuggestion
revealed a strong difference in CD235a expression, size and inner complexity, and
isolated the CD235a (RBCs) high branch (1–3) from the rest of the tree. A second

59
autosuggestion highlighted a subpopulation (6) presenting high expression of
CD41a, MitoTracker, SSC, FSC and FSC-PMT, which correspond to platelets. The
three subsequent autosuggestions revealed PS-expressing EVs produced from
platelets (9–10) and from RBCs (3), which were also smaller than their mother cells.
As (1–2) subpopulations showed a homogeneous distribution for every marker
except for CD235a expression, the software suggested division of this branch into
two. Autosuggestions also distinguished mitochondria-containing EVs that did not
present RBC (CD235a−) or platelet (CD41a−) markers (subpopulation 8),and allowed
the partition of subpopulations (9–10) according to CD41a, MitoTracker and PS
expression. It is important to note that these populations were objectively identified
without any gating or prior knowledge. Only subpopulations (4) and (7) were drawn
manually, mainly because of their bright intensities in distinctive markers.
The (1–10) subpopulations were then annotated, and the SPADE tree was
interpreted. Subpopulation (1) includes cells (high intensity for FSC-PMT-H, FSC-H
and SSC-H) that were not RBCs (low expression of CD235a) or platelets (low
expression of CD41a), potentially representing a low number of contaminating
leukocytes or RBC ghosts generated by RBC activation. Subpopulation (2) contains
RBCs, which show high intensity for FSC-PMT, FSC and SSC and also high
expression of CD235a markers, but low expression of CD41a. Subpopulation (3)
contains RBC EVs with intermediate intensity for FSC-PMT, FSC and SSC, high
expression of CD235a and low expression of CD41a. All RBC EVs detected in those
samples exposed PS (high intensity for annexin V expression). With a low
expression of all 7 markers, the (5) subpopulation was annotated as background,
although it might also contain EVs left unidentified using this set of markers.
Subpopulation (6) contains platelets, which show relatively high light scatter for FSC-
PMT, FSC and SSC and expression of CD41a and MitoTracker. Subpopulation (8)
showed low expression of all markers except for MitoTracker, suggesting that they
might be naked mitochondria or mitochondria encapsulated in EVs lacking
expression of CD41a. Subpopulations (7, 9 and 10) represent platelet EVs
(intermediate CD41a expression and SSC, low CD235a expression and low light

60
scatter for FSC-PMT and FSC). Subpopulation (4) includes EVs with variable
expression levels of CD41a (low to high), with those presenting the brightest CD41a
intensity representing 1,15+/-1,99% of this subpopulation (data not shown).
Subpopulation (10) includes EVs containing mitochondria (high MitoTracker
expression), with variable exposure of PS (intermediate to high intensity annexin V
binding). More than 40 classical analyses with bivariate plots were necessary to
interpret the data (Fig. 3B–E). These observations confirm that upon treatment by
SPADE analyses, homogeneous EV subpopulations were identified, and further
highlight the complexity of analyzing high-dimensional flow cytometry data without
appropriate computerized tools.
2.5.3 Analysis of platelet response to thrombin stimulation using
SPADE
EV production is evidence of cellular activation or apoptosis. Thus, the SPADE
analysis as above was used to appreciate the platelet response to thrombin
stimulation (Fig. 4). Both resting and activated platelets were portrayed in the
constructed tree. The fold-change in subpopulation frequencies varied upon
activation (i.e. red, increase; blue, decrease), indicating that platelet EV
subpopulations (9–10), and extracellular mitochondria (8), were produced, while
platelets (6) lost their dominance. Background/debris (5) varied following the
stimulation, pointing to the generation of debris following platelet activation or the
presence of unidentified EVs using this set of markers. These observations confirm
the ability of SPADE to distinguish cell and EV populations, and to appreciate cellular
plasticity and EV biogenesis in response to stimuli.
2.5.4 Generating overlapping panels in plasma EV analyses.
SPADE also permits the integration of multiple staining using overlapping marker
panels (cocktail 5a-d in Table S1). For these experiments, we evaluated endogenous
EVs present in healthy human platelet-free plasma samples and generated a new
tree. The overlapping markers CD41a, MitoTracker, FSC-PMT and SSC, were
present in every condition, and we also included annexin V, CD62P, GPVI and

61
CLEC-2 as interchangeable markers within the tree (Fig. 5)16,20,21. FCS files were
exported and analyzed as above. The SPADE parameters were the same except for
the target densities that were fixed to 10,000 cells/EVs. Platelet-derived EVs (high
CD41a expression) were mostly located at the bottom of the tree (the lower branches
1–3), some of them expressing GPVI and CLEC-2 (branches 1,2). Of note was that
theupper part of the tree (CD41a−) also revealed high expression of GPVI and CLEC-
2 on EVs (branches 7–9). Thus, the SPADE algorithm provides analyses of high-
dimensional data that is scalable with an increasing number of markers useful for
EV analysis. Furthermore, these data demonstrate that SPADE can identify
subpopulations, like the presence of three subpopulations of platelet-derived EVs
that could have been overlooked with classical dot plot analyses.
2.5.5 SPADE for the appreciation of EVs as biomarker in disease.
Different cellular lineages contribute to EV accumulation in the synovial fluid (SF) of
RA patients. Platelet-derived EVs have been identified in RA SF5,13,25,51,52, and
present with heterogeneous dimensions and mitochondrial content (Fig. 6A). We
quantitatively identified EVs in the SF of 20 RA patients on the unique basis of 4
markers (i.e. CD41a, MitoTracker, FSC-PMT and SSC) to generate a new SPADE
tree (Fig. 6A). SPADE parameters were the same as in the first tree (Figs 3 and 4)
with the exception that 150 clusters were used instead of 200 given the reduced
number of parameters measured. Using the autosuggestion tool to objectively
identify EV subpopulations, two major subpopulations were revealed: i.e. CD41+,
MitoTracker− and CD41+, MitoTracker+ EVs. SPADE tree highlighted the great
variability between RA patients, as some EV subtypes appeared to be completely
absent (empty nodes in white) in some patients (Fig. 6B,C). These differences in EV
expression patterns did not seem to correlate with rheumatoid factor (RF) and anti-
citrullinated protein auto-antibody (anti-CCP) levels (Table S2). These data suggest
that EV pattern recognition using the objective analysis tool SPADE can highlight
differences in EV content in disease, providing a tool for the determination of potent
biomarkers in disease.


63
2.6 Discussion
The emergence of EVs as important players in intercellular communication has
opened the way to intensive research on this topic. EV levels are modulated in
certain pathologies, and studies have established the vast diversity of EVs produced
by cells, suggesting that EVs might be used as biomarkers2,7. Groups of scientists
have established the most appropriate pre-analytical conditions for the study of EVs
and for the design of modern methodologies for their fine characterization3,9–
11,13,14,17,18,26,27,29,30,48–50. Furthermore, efforts have been made in recent years to
institute a coherent nomenclature for EVs44. Not withstanding these major
improvements, it remains an obvious challenge to objectively interpret the large
quantity of high-dimensional data in EV analyses. For instance, although
fluorescence, rather than light scatter, used as trigger greatly improves EV detection,
the distinction of EVs from background in FCM is still an obstacle in complex
fluids12,13,17. Furthermore, platelets are abundant in blood and represent an important
source of EVs; however, they are frequently misinterpreted as EVs given their
relatively small dimensions11. The absence of such analytical tools for the
comprehension of EV functions prompted our study.
SPADE offers the advantage of using an encompassing panel of markers to cluster
the data, which allows the identification of rare cell types and facilitates new,
unanticipated, biological discoveries37,38. Our study shows that SPADE is a versatile
computerized tool to objectively handle hundreds of thousands of hs-FCM EV data
and to reveal unpredicted EV subtypes. Most importantly, SPADE can be utilized for
the analysis of EV data obtained with any flow cytometer, assuming that EVs are
detected correctly. Whereas there exist other algorithms (other than SPADE)
available for the interpretation of FCM data53, SPADE is among the most appropriate
to reveal rare subpopulations of events by flow cytometry37,38,43. Investigators,
however, need to compare trees from one condition to another to identify changes
in nodes between conditions, which can be challenging if FCM based biomarkers
are examined in multiple patients, for example. Future improvements to these

64
applications by engineers in the field might include high throughput tree comparison.
SPADE permitted the establishment of a tree that portrays EVs from platelets and
RBC, the two main sources of EVs reported in blood. Of interest is that SPADE
successfully recognized platelet activation based on platelet-derived EV production,
suggesting that it might be used as a tool to assess EV-based cellular perturbations.
Unanticipated subpopulations of EVs present in plasma were revealed by a second
SPADE analysis with overlapping markers. Prior studies revealed that the majority
of the platelet-derived EVs in blood in fact originate from MKs, the cells from which
platelets are produced16,20. The immunoreceptor-based activation motif (ITAM)
receptors GPVI and CLEC-2 were reported absent on the surface of platelet-derived
EVs, but were found on the surface of (MK)-derived EVs, which also express
CD41a16,20,21. Whereas SPADE might have revealed MK-derived EVs in plasma
(CD41+ GPVI+ CLEC-2+ EVs), it unexpectedly shed light on CD41−-EVs expressing
GPVI and CLEC-2. Although the functions of these EVs remain to be established,
we hypothesize that these EVs might also originate from MKs. As for any data
interpretation implicating flow cytometry, complementary approaches, such as
electron microscopy, functional assays and biochemistry, should be utilized to
confirm the actual presence of novel populations of EVs and to verify their biological
significance.
The analysis of EVs contained in bio-specimens from disease patients permits the
identification of biomarkers. With a rather simple analysis that included assessment
of CD41a and mitochondrial content in EVs from 20 patients, a third SPADE analysis
highlighted the heterogeneity that prevails in RA. The possibility to extract the
number of events in each node for all patients, similarly to the exportation of statistics
in traditional gating, allows a relatively easy comparison between patients. RA
patients with higher EV diversity were usually those with higher platelet EV
concentrations, pointing to a more important platelet contribution to the pathology in
those patients. As it is suggested that platelet EVs invade the synovial space due to
enhanced joint vascular permeability54, it might also provide information on the

65
integrity of the vasculature in these patients. RA patients have enhanced risks of
death due to cardiovascular disorders. Future studies might determine if increased
levels of EV subtypes are an indication of certain comorbidities (other than those
that were presented in Table S2) or impact response to different treatment in RA.
Furthermore, we suggest that the combination of these approaches might be used
for a qualitative and a quantitative stratification of patients suffering from
heterogeneous diseases, such as RA and potentially other rheumatic diseases. In
this study, SPADE was applied to the analysis of high-dimensional data based on
EV detection. We suggest that this approach may be utilized for the assessment of
more numerous EV markers by FCM or mass cytometry. An in-depth understanding
of EV subtypes modelled as high-dimensional point clouds will accelerate the
implementation of EV subtypes as biomarkers and will facilitate the understanding
of their role(s) in different contexts such as coagulation, inflammation, cancer, and
immunity.

66
2.7 References
1 Gyorgy, B. et al. Membrane vesicles, current state-of-the-art: emerging role of
extracellular vesicles. Cell Mol Life Sci 68, 2667–2688, doi: 10.1007/s00018-
011-0689-3 (2011).
2 S, E. L. A., Mager, I., Breakefield, X. O. & Wood, M. J. Extracellular vesicles:
biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12, 347–
357, doi: 10.1038/nrd3978 (2013).
3 van der Pol, E., Boing, A. N., Gool, E. L. & Nieuwland, R. Recent developments
in the nomenclature, presence, isolation, detection and clinical impact of
extracellular vesicles. J Thromb Haemost 14, 48–56, doi: 10.1111/jth.13190
(2016).
4 Dieude, M. et al. The 20S proteasome core, active within apoptotic exosome-
like vesicles, induces autoantibody production and accelerates rejection. Sci
Transl Med 7, 318ra200, doi: 10.1126/scitranslmed.aac9816 (2015).
5 Duchez, A. C. et al. Platelet microparticles are internalized in neutrophils via the
concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc
Natl Acad Sci USA 112, E3564–E3573, doi: 10.1073/pnas.1507905112 (2015).
6 Laffont, B. et al. Activated platelets can deliver mRNA regulatory
Ago2*microRNA complexes to endothelial cells via microparticles. Blood 122,
253–261, doi: 10.1182/blood-2013-03-492801 (2013).
7 Thery, C., Ostrowski, M. & Segura, E. Membrane vesicles as conveyors of
immune responses. Nat Rev Immunol 9, 581–593, doi: 10.1038/nri2567 (2009).
8 Malda, J., Boere, J., van de Lest, C. H., van Weeren, P. & Wauben, M. H.
Extracellular vesicles-new tool for joint repair and regeneration. Nat Rev
Rheumatol 12, 243–249, doi: 10.1038/nrrheum.2015.170 (2016).
9 Burnouf, T., Goubran, H. A., Chou, M. L., Devos, D. & Radosevic, M. Platelet
microparticles: detection and assessment of their paradoxical functional roles in
disease and regenerative medicine. Blood Rev 28, 155–166, doi:
10.1016/j.blre.2014.04.002 (2014).
10 Robert, S. et al. High-sensitivity flow cytometry provides access to standardized

67
measurement of small-size microparticles–brief report. Arterioscler Thromb
Vasc Biol 32, 1054–1058, doi: 10.1161/ATVBAHA.111.244616 (2012).
11 Chandler, W. L. Measurement of microvesicle levels in human blood using flow
cytometry. Cytometry B Clin Cytom 90, 326–336, doi: 10.1002/cyto.b.21343
(2016).
12 Poncelet, P. et al. Tips and tricks for flow cytometry-based analysis and counting
of microparticles. Transfus Apher Sci 53, 110–126, doi:
10.1016/j.transci.2015.10.008 (2015).
13 Rousseau, M. et al. Detection and quantification of microparticles from different
cellular lineages using flow cytometry. Evaluation of the impact of secreted
phospholipase A2 on microparticle assessment. PLoS One 10, e0116812, doi:
doi: 10.1371/journal.pone.0116812 (2015).
14 Stoner, S. A. et al. High sensitivity flow cytometry of membrane vesicles.
Cytometry A 89, 196–206, doi: 10.1002/cyto.a.22787 (2016).
www.nature.com/scientificreports/Scientific Reports | 6:35928 | DOI:
10.1038/srep35928 12
15 van der Vlist, E. J., Nolte-‘t Hoen, E. N., Stoorvogel, W., Arkesteijn, G. J. &
Wauben, M. H. Fluorescent labeling of nano-sized vesicles released by cells
and subsequent quantitative and qualitative analysis by high-resolution flow
cytometry. Nat Protoc 7, 1311–1326, doi: 10.1038/nprot.2012.065 (2012).
16 Boilard, E., Duchez, A. C. & Brisson, A. The diversity of platelet microparticles.
Curr Opin Hematol 22, 437–444, doi: 10.1097/MOH.0000000000000166
(2015).
17 Arraud, N., Gounou, C., Turpin, D. & Brisson, A. R. Fluorescence triggering: A
general strategy for enumerating and phenotyping extracellular vesicles by flow
cytometry. Cytometry A 89, 184–195, doi: 10.1002/cyto.a.22669 (2016).
18 Aatonen, M. T. et al. Isolation and characterization of platelet-derived
extracellular vesicles. J Extracell Vesicles 3, doi: 10.3402/jev.v3.24692 (2014).
19 Heijnen, H. F., Schiel, A. E., Fijnheer, R., Geuze, H. J. & Sixma, J. J. Activated
platelets release two types of membrane vesicles: microvesicles by surface
shedding and exosomes derived from exocytosis of multivesicular bodies and

68
alpha-granules. Blood 94,3791–3799 (1999).
20 Flaumenhaft, R. et al. Megakaryocyte-derived microparticles: direct
visualization and distinction from platelet-derived microparticles. Blood 113,
1112–1121, doi: 10.1182/blood-2008-06-163832 (2009).
21 Gitz, E. et al. CLEC-2 expression is maintained on activated platelets and on
platelet microparticles. Blood 124, 2262–2270, doi: 10.1182/blood-2014-05-
572818 (2014).
22 Arraud, N. et al. Extracellular vesicles from blood plasma: determination of their
morphology, size, phenotype and concentration. J Thromb Haemost 12, 614–
627, doi: 10.1111/jth.12554 (2014).
23 Cloutier, N. et al. The exposure of autoantigens by microparticles underlies the
formation of potent inflammatory components: the microparticle-associated
immune complexes. EMBO Mol Med 5, 235–249, doi:
10.1002/emmm.201201846 (2013).
24 Perez-Pujol, S., Marker, P. H. & Key, N. S. Platelet microparticles are
heterogeneous and highly dependent on the activation mechanism: studies
using a new digital flow cytometer. Cytometry A 71, 38–45, doi:
10.1002/cyto.a.20354 (2007).
25 Boudreau, L. H. et al. Platelets release mitochondria serving as substrate for
bactericidal group IIA-secreted phospholipase A2 to promote inflammation.
Blood 124, 2173–2183, doi: 10.1182/blood-2014-05-573543 (2014).
26 Tamir, A. et al. The macromolecular architecture of platelet-derived
microparticles. J Struct Biol 193, 181–187, doi: 10.1016/j.jsb.2015.12.013
(2016).
27 Gupta, N., Li, W., Willard, B., Silverstein, R. L. & McIntyre, T. M. Proteasome
proteolysis supports stimulated platelet function and thrombosis. Arterioscler
Thromb Vasc Biol 34, 160–168, doi: 10.1161/ATVBAHA.113.302116 (2014).
28 Fortin, P. R. et al. Distinct Subtypes of Microparticle-containing Immune
Complexes Are Associated with Disease Activity, Damage, and Carotid Intima-
media Thickness in Systemic Lupus Erythematosus. J Rheumatol, doi:
10.3899/jrheum.160050 (2016).

69
29 Lacroix, R. et al. Standardization of pre-analytical variables in plasma
microparticle determination: results of the International Society on Thrombosis
and Haemostasis SSC Collaborative workshop. J Thromb Haemost, doi:
10.1111/jth.12207 (2013).
30 Fendl, B., Weiss, R., Fischer, M. B., Spittler, A. & Weber, V. Characterization of
extracellular vesicles in whole blood: Influence of pre-analytical parameters and
visualization of vesicle-cell interactions using imaging flow cytometry. Biochem
Biophys Res Commun, doi: 10.1016/j.bbrc.2016.07.073 (2016).
31 Zhu, J. & Paul, W. E. Heterogeneity and plasticity of T helper cells. Cell Res 20,
4–12, doi: 10.1038/cr.2009.138 (2010).
32 Belz, G. T. & Nutt, S. L. Transcriptional programming of the dendritic cell
network. Nat Rev Immunol 12, 101–113, doi: 10.1038/nri3149 (2012).
33 Watchmaker, P. B. et al. Comparative transcriptional and functional profiling
defines conserved programs of intestinal DC differentiation in humans and mice.
Nat Immunol 15, 98–108, doi: 10.1038/ni.2768 (2014).
34 Gordon, S. & Taylor, P. R. Monocyte and macrophage heterogeneity. Nat Rev
Immunol 5, 953–964, doi: 10.1038/nri1733 (2005).
35 Sagiv, J. Y. et al. Phenotypic diversity and plasticity in circulating neutrophil
subpopulations in cancer. Cell Rep 10, 562–573, doi:
10.1016/j.celrep.2014.12.039 (2015).
36 Scapini, P. & Cassatella, M. A. Social networking of human neutrophils within
the immune system. Blood 124, 710–719, doi: 10.1182/blood-2014-03-453217
(2014).
37 Qiu, P. et al. Extracting a cellular hierarchy from high-dimensional cytometry
data with SPADE. Nat Biotechnol 29, 886–891, doi: 10.1038/nbt.1991 (2011).
38 Mair, F. et al. The end of gating? An introduction to automated analysis of high
dimensional cytometry data. Eur J Immunol 46, 34–43, doi:
10.1002/eji.201545774 (2016).
39 Bendall, S. C. et al. Single-cell mass cytometry of differential immune and drug
responses across a human hematopoietic continuum. Science 332, 687–696,
doi: 10.1126/science.1198704 (2011).

70
40 Diggins, K. E., Ferrell, P. B. Jr. & Irish, J. M. Methods for discovery and
characterization of cell subsets in high dimensional mass cytometry data.
Methods 82, 55–63, doi: 10.1016/j.ymeth.2015.05.008 (2015).
41 Sen, N., Mukherjee, G. & Arvin, A. M. Single cell mass cytometry reveals
remodeling of human T cell phenotypes by varicella zoster virus. Methods, doi:
10.1016/j.ymeth.2015.07.008 (2015).
42 Jobin, C., Cloutier, M., Simard, C. & Neron, S. Heterogeneity of in vitro-cultured
CD34+ cells isolated from peripheral blood. Cytotherapy 17, 1472–1484, doi:
doi: 10.1016/j.jcyt.2015.05.006 (2015).
43 Anchang, B. et al. Visualization and cellular hierarchy inference of single-cell
data using SPADE. Nat Protoc 11, 1264–1279, doi: 10.1038/nprot.2016.066
(2016).
44 Lotvall, J. et al. Minimal experimental requirements for definition of extracellular
vesicles and their functions: a position statement from the International Society
for Extracellular Vesicles. J Extracell Vesicles 3, 26913, doi:
10.3402/jev.v3.26913 (2014).
45 Linares, R., Tan, S., Gounou, C., Arraud, N. & Brisson, A. R. High-speed
centrifugation induces aggregation of extracellular vesicles. J Extracell Vesicles
4, 29509, doi: 10.3402/jev.v4.29509 (2015).
46 Chandler, W. L., Yeung, W. & Tait, J. F. A new microparticle size calibration
standard for use in measuring smaller microparticles using a new flow
cytometer. J Thromb Haemost 9, 1216–1224, doi: 10.1111/j.1538-
7836.2011.04283.x (2011).
47 van der Pol, E., van Gemert, M. J., Sturk, A., Nieuwland, R. & van Leeuwen, T.
G. Single vs. swarm detection of microparticles and exosomes by flow
cytometry. J Thromb Haemost 10, 919–930, doi: 10.1111/j.1538-
7836.2012.04683.x (2012).
48 Parida, B. K., Garrastazu, H., Aden, J. K., Cap, A. P. & McFaul, S. J. Silica
microspheres are superior to polystyrene for microvesicle analysis by flow
cytometry. Thromb Res 135, 1000–1006, doi: 10.1016/j.thromres.2015.02.011
(2015).

71
49 Gyorgy, B. et al. Detection and isolation of cell-derived microparticles are
compromised by protein complexes resulting from shared biophysical
parameters. Blood 117, e39–e48, doi: 10.1182/blood-2010-09-307595 (2011).
50 Inglis, H. C. et al. Techniques to improve detection and analysis of extracellular
vesicles using flow cytometry. Cytometry A 87, 1052–1063, doi:
10.1002/cyto.a.22649 (2015).
51 Gyorgy, B. et al. Improved flow cytometric assessment reveals distinct
microvesicle (cell-derived microparticle) signatures in joint diseases. PLoS One
7, e49726, doi: 10.1371/journal.pone.0049726 (2012).
52 Boilard, E. et al. Platelets amplify inflammation in arthritis via collagen-
dependent microparticle production. Science 327, 580–583, doi:
10.1126/science.1181928 (2010).
53 Aghaeepour, N. et al. Critical assessment of automated flow cytometry data
analysis techniques. Nat Methods 10, 228–238, doi: 10.1038/nmeth.2365
(2013).
54 Cloutier, N. et al. Platelets can enhance vascular permeability. Blood 120,
1334–1343, doi: 10.1182/blood-2012-02-413047 (2012).
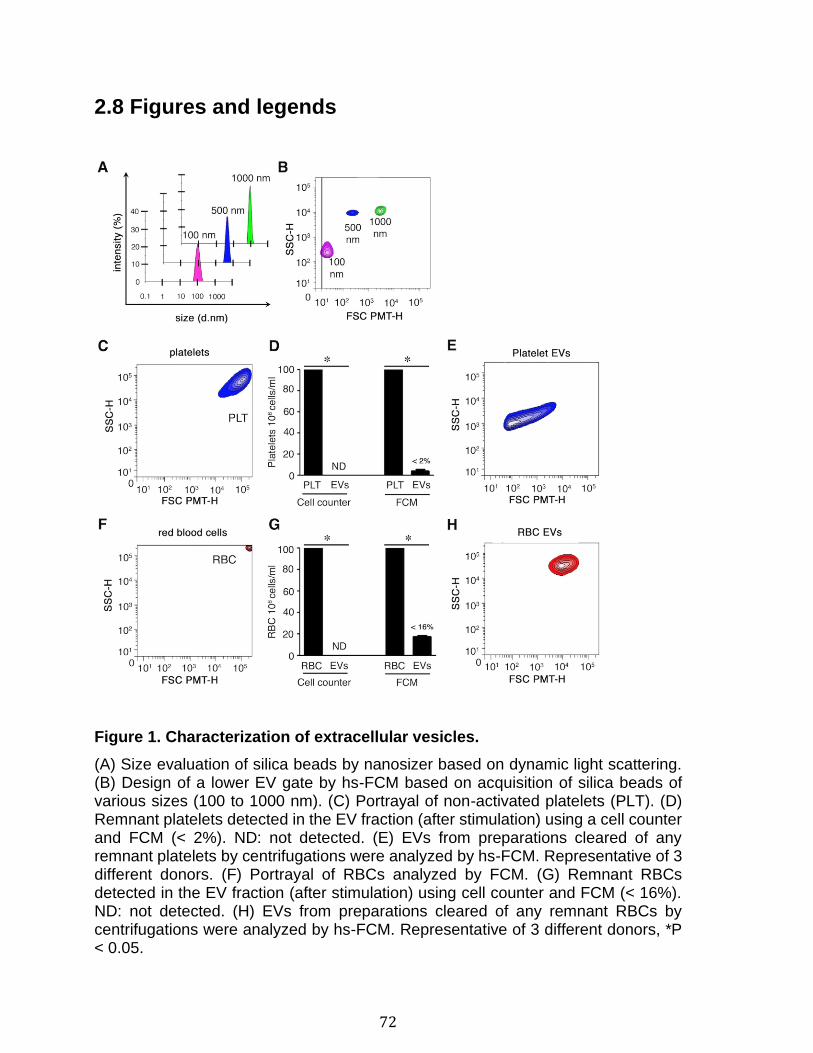
72
2.8 Figures and legends
Figure 1. Characterization of extracellular vesicles.
(A) Size evaluation of silica beads by nanosizer based on dynamic light scattering. (B) Design of a lower EV gate by hs-FCM based on acquisition of silica beads of various sizes (100 to 1000 nm). (C) Portrayal of non-activated platelets (PLT). (D) Remnant platelets detected in the EV fraction (after stimulation) using a cell counter and FCM (< 2%). ND: not detected. (E) EVs from preparations cleared of any remnant platelets by centrifugations were analyzed by hs-FCM. Representative of 3 different donors. (F) Portrayal of RBCs analyzed by FCM. (G) Remnant RBCs detected in the EV fraction (after stimulation) using cell counter and FCM (< 16%). ND: not detected. (H) EVs from preparations cleared of any remnant RBCs by centrifugations were analyzed by hs-FCM. Representative of 3 different donors, *P < 0.05.
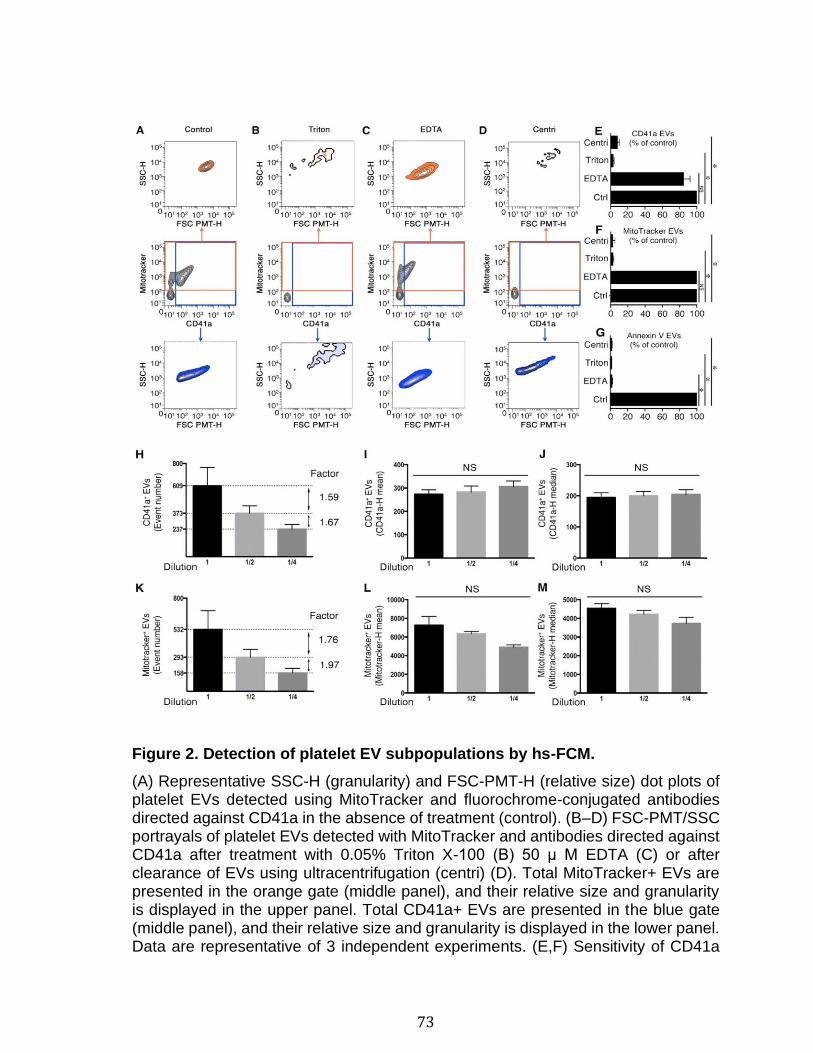
73
Figure 2. Detection of platelet EV subpopulations by hs-FCM.
(A) Representative SSC-H (granularity) and FSC-PMT-H (relative size) dot plots of platelet EVs detected using MitoTracker and fluorochrome-conjugated antibodies directed against CD41a in the absence of treatment (control). (B–D) FSC-PMT/SSC portrayals of platelet EVs detected with MitoTracker and antibodies directed against CD41a after treatment with 0.05% Triton X-100 (B) 50 μ M EDTA (C) or after clearance of EVs using ultracentrifugation (centri) (D). Total MitoTracker+ EVs are presented in the orange gate (middle panel), and their relative size and granularity is displayed in the upper panel. Total CD41a+ EVs are presented in the blue gate (middle panel), and their relative size and granularity is displayed in the lower panel. Data are representative of 3 independent experiments. (E,F) Sensitivity of CD41a

74
(E) MitoTracker (F) and annexin V (G) EVs to clearance by ultracentrifugation (centri), Triton and EDTA, presented as % of untreated (Ctrl). (H–M) CD41a+ and MitoTracker+ EVs were serially diluted twice (2-fold dilution) and quantitatively analyzed by hs-FCM using counting microspheres. Their concentration (H,K), the mean of fluorescence (I,L) and the median of fluorescence (J,M), are presented. Data are presented as mean ± SEM of 3 independent experiments, *P < 0.05 compared with the control (Ctrl); NS: Non-significant; Wilcoxon test.
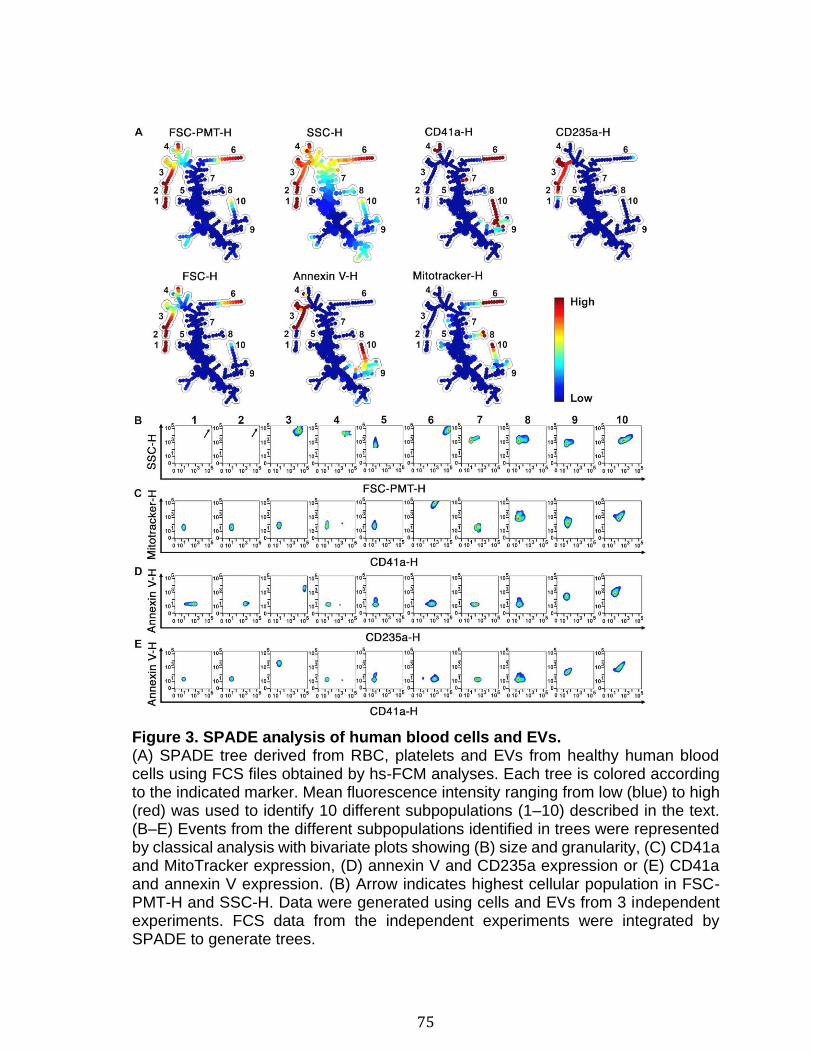
75
Figure 3. SPADE analysis of human blood cells and EVs. (A) SPADE tree derived from RBC, platelets and EVs from healthy human blood cells using FCS files obtained by hs-FCM analyses. Each tree is colored according to the indicated marker. Mean fluorescence intensity ranging from low (blue) to high (red) was used to identify 10 different subpopulations (1–10) described in the text. (B–E) Events from the different subpopulations identified in trees were represented by classical analysis with bivariate plots showing (B) size and granularity, (C) CD41a and MitoTracker expression, (D) annexin V and CD235a expression or (E) CD41a and annexin V expression. (B) Arrow indicates highest cellular population in FSC-PMT-H and SSC-H. Data were generated using cells and EVs from 3 independent experiments. FCS data from the independent experiments were integrated by SPADE to generate trees.

76
Figure 4. SPADE tree to assess EVs in cellular perturbations.
After stimulation of platelets with thrombin, cell frequency decrease (blue) in platelets (6) and increase (red) in platelet EVs (9–10). Red blood cell markers (CD235a) (2,3), other cells (1), and noise/background (5) were mostly unchanged. Platelet CD41a+ EVs (9,10) and extracellular mitochondria lacking CD41a+ (8, naked mitochondria) were induced (red). Data are representative of 3 independent experiments.

77
Figure 5. SPADE overlapping panels for EV diversity in plasma.
EVs present in plasma were detected by hs-FCM, and FCS files were used to build a SPADE tree. Each tree is colored according to the indicated marker intensity. Mean fluorescence intensity ranging from low (blue) to high (red) for size (FSC-PMT-H), granularity (SSC-H), fluorescent-conjugated antibodies (CD41a−H, CD62P-H, GPVI-H, CLEC-2-H), MitoTracker and annexin V. Nine subpopulations were highlighted by manual identification based on the expression of the different markers, starting from the bottom of the tree: (1) Platelet EVs, positive for every marker (high intensity shown in red); (2) Platelet EVs positive for every marker except annexin V and CLEC-2 (low intensity shown in blue); (3) platelet EVs (CD41a high) with weak expression (shown in yellow) of MitoTracker and GPVI, and negative for annexin V, CD62P and CLEC-2; (4) EVs negative for CD41a, but positive for MitoTracker, annexin V and GPVI; (5) EVs presenting high expression of CD41a with low light scattering in FSC and SSC; (6) EVs with a weak expression of CD41a, negative for all other markers; (7) EVs negative for CD41a, and positive for all other markers, including activation markers (annexin V and CD62P); (8) EVs positive for every marker and presenting higher inner complexity (SSC-H) and (9) EVs negative for CD41a and presenting higher intensity in FSC-PMT. Data are representative of 3 independent experiments.
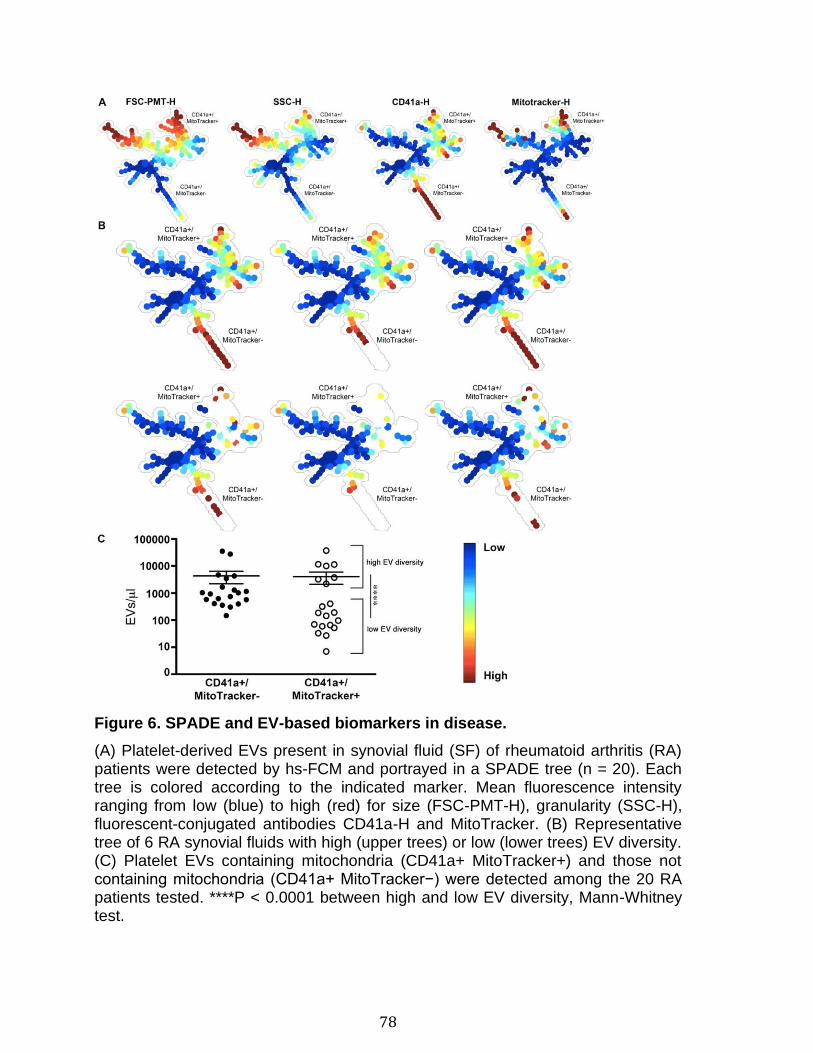
78
Figure 6. SPADE and EV-based biomarkers in disease.
(A) Platelet-derived EVs present in synovial fluid (SF) of rheumatoid arthritis (RA) patients were detected by hs-FCM and portrayed in a SPADE tree (n = 20). Each tree is colored according to the indicated marker. Mean fluorescence intensity ranging from low (blue) to high (red) for size (FSC-PMT-H), granularity (SSC-H), fluorescent-conjugated antibodies CD41a-H and MitoTracker. (B) Representative tree of 6 RA synovial fluids with high (upper trees) or low (lower trees) EV diversity. (C) Platelet EVs containing mitochondria (CD41a+ MitoTracker+) and those not containing mitochondria (CD41a+ MitoTracker−) were detected among the 20 RA patients tested. ****P < 0.0001 between high and low EV diversity, Mann-Whitney test.
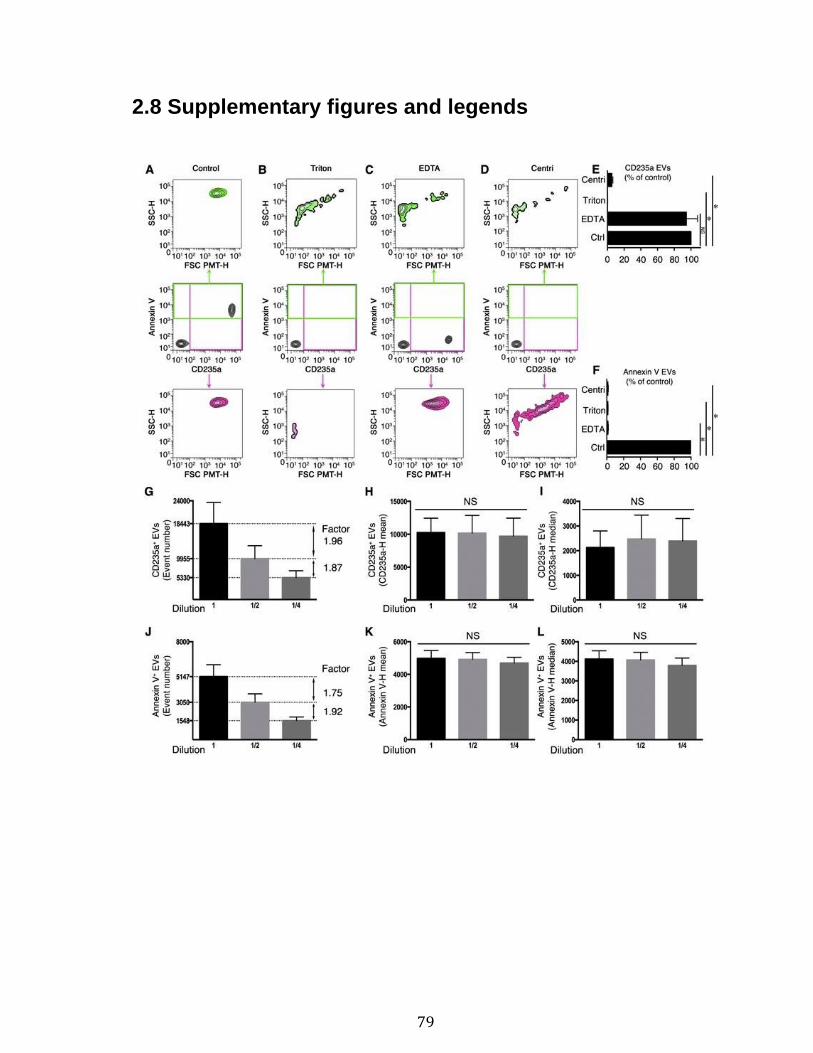
79
2.8 Supplementary figures and legends

80
Supplementary Figure 1. Detection of RBC subpopulations by hs-FCM. (A) Representative SSC-H (granularity) and FSC-PMT-H (relative size) dot plots of RBC EVs detected using annexin V and fluorochrome-conjugated antibodies directed against CD235a in the absence of treatment (control). (b–d) FSC-PMT/SSC portrayals of RBC EVs detected with fluorochrome-conjugated annexin V and fluorochrome-conjugated antibodies directed against CD235a and treated with 0.05% Triton X-100, (B) 50 μM EDTA, (C) or after clearance of EVs using ultracentrifugation (centri)(D). Total annexin V+ events are presented in the green gate (middle panel) and their relative size and granularity is displayed in the upper panel. Total CD235a+ events are presented in the blue gate (middle panel) and their relative size and granularity is displayed in the lower panel. Data are representative of 3 independent experiments. (E–F) Sensitivity of CD235a (E) and annexin V (F) EVs to clearance by ultracentrifugation (centri), Triton and EDTA, presented as % of untreated (Ctrl). (G–L) CD235a+ and annexin V+ EVs were serially diluted twice (2-fold dilution) and quantitatively analyzed by hs-FCM using counting microspheres. Their concentration (G and J), the mean of fluorescence (H and K) and the median of fluorescence (I and L), are presented. Data are presented as the mean ± SEM of 3 independent experiments, *P < 0.05 compared with the control (Ctrl); NS : Non significant ; Wilcoxon test.
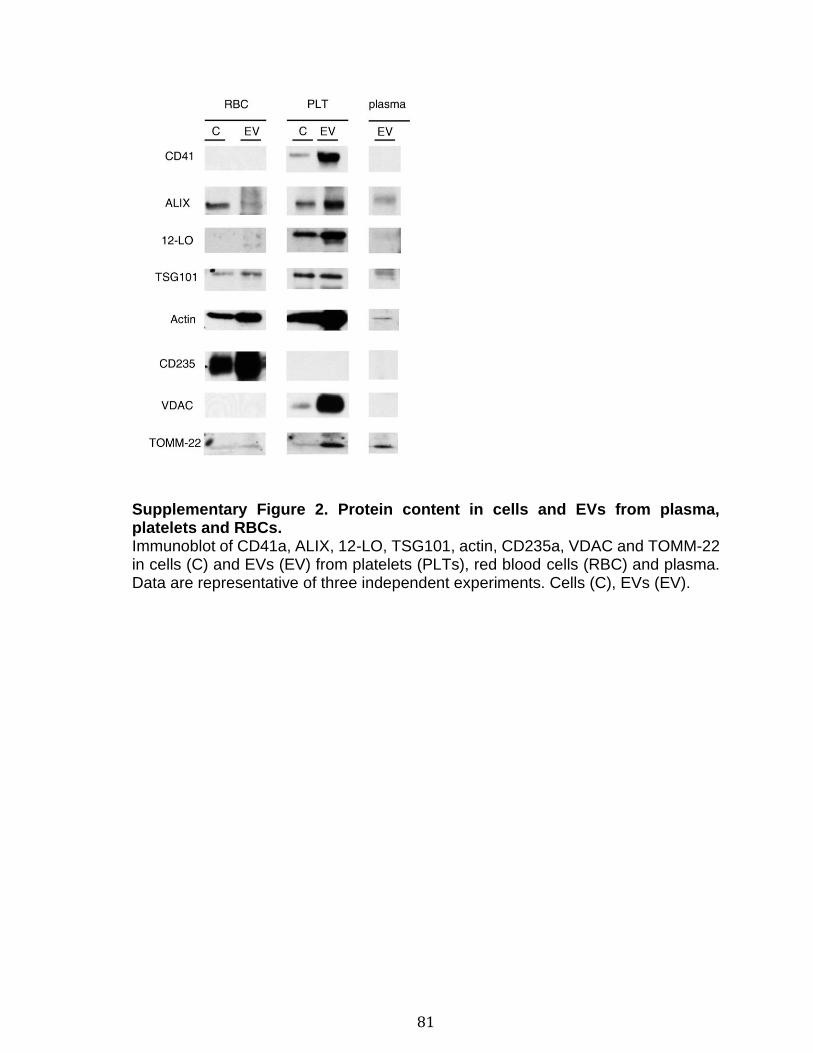
81
Supplementary Figure 2. Protein content in cells and EVs from plasma, platelets and RBCs. Immunoblot of CD41a, ALIX, 12-LO, TSG101, actin, CD235a, VDAC and TOMM-22 in cells (C) and EVs (EV) from platelets (PLTs), red blood cells (RBC) and plasma. Data are representative of three independent experiments. Cells (C), EVs (EV).
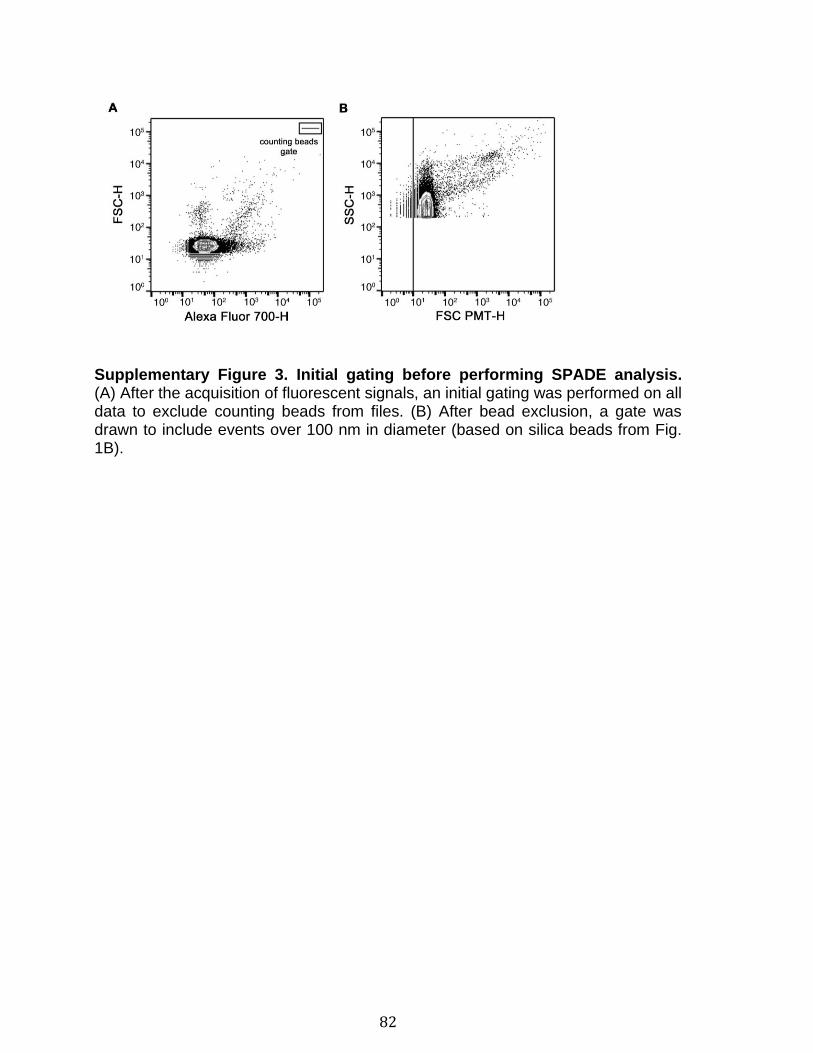
82
Supplementary Figure 3. Initial gating before performing SPADE analysis. (A) After the acquisition of fluorescent signals, an initial gating was performed on all data to exclude counting beads from files. (B) After bead exclusion, a gate was drawn to include events over 100 nm in diameter (based on silica beads from Fig. 1B).
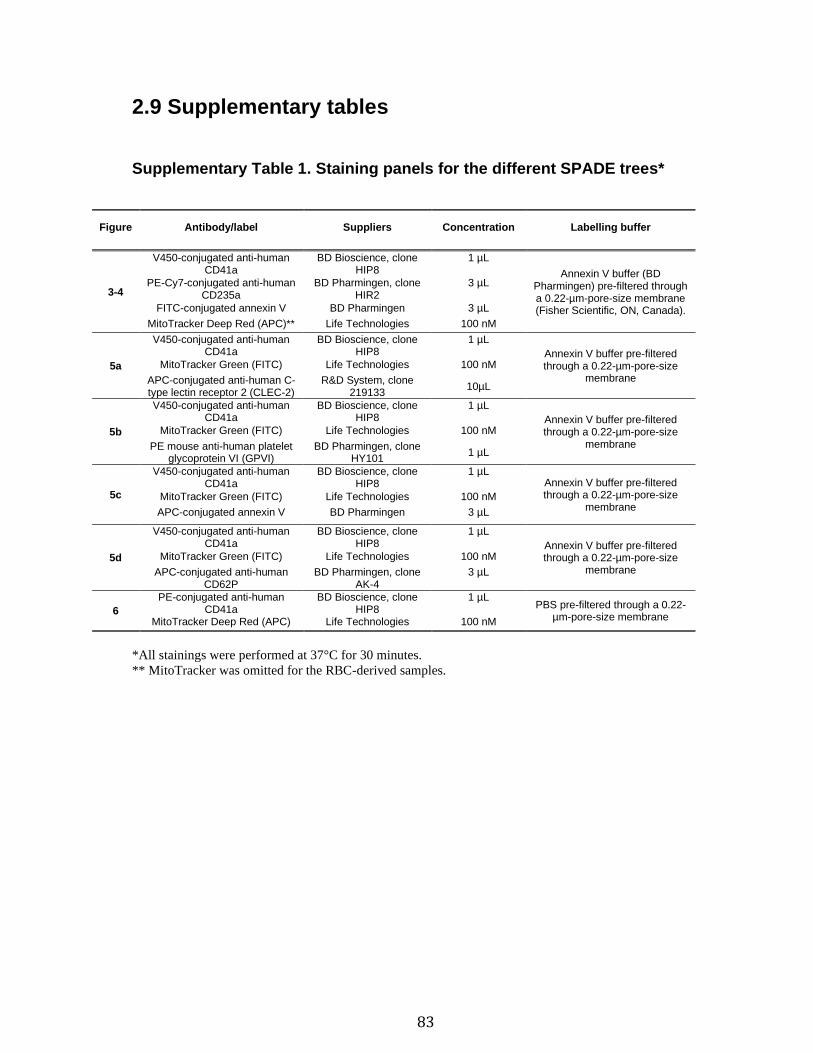
83
2.9 Supplementary tables
Supplementary Table 1. Staining panels for the different SPADE trees*
Figure Antibody/label Suppliers Concentration Labelling buffer
3-4
V450-conjugated anti-human CD41a
BD Bioscience, clone HIP8
1 µL
Annexin V buffer (BD Pharmingen) pre-filtered through a 0.22-µm-pore-size membrane (Fisher Scientific, ON, Canada).
PE-Cy7-conjugated anti-human CD235a
BD Pharmingen, clone HIR2
3 µL
FITC-conjugated annexin V BD Pharmingen 3 µL
MitoTracker Deep Red (APC)** Life Technologies 100 nM
5a
V450-conjugated anti-human CD41a
BD Bioscience, clone HIP8
1 µL
Annexin V buffer pre-filtered through a 0.22-µm-pore-size
membrane
MitoTracker Green (FITC) Life Technologies 100 nM
APC-conjugated anti-human C-type lectin receptor 2 (CLEC-2)
R&D System, clone 219133
10µL
5b
V450-conjugated anti-human CD41a
BD Bioscience, clone HIP8
1 µL
Annexin V buffer pre-filtered through a 0.22-µm-pore-size
membrane
MitoTracker Green (FITC) Life Technologies 100 nM
PE mouse anti-human platelet glycoprotein VI (GPVI)
BD Pharmingen, clone HY101
1 µL
5c
V450-conjugated anti-human CD41a
BD Bioscience, clone HIP8
1 µL Annexin V buffer pre-filtered through a 0.22-µm-pore-size
membrane MitoTracker Green (FITC) Life Technologies 100 nM
APC-conjugated annexin V BD Pharmingen 3 µL
5d
V450-conjugated anti-human CD41a
BD Bioscience, clone HIP8
1 µL
Annexin V buffer pre-filtered through a 0.22-µm-pore-size
membrane
MitoTracker Green (FITC) Life Technologies 100 nM
APC-conjugated anti-human CD62P
BD Pharmingen, clone AK-4
3 µL
6
PE-conjugated anti-human CD41a
BD Bioscience, clone HIP8
1 µL PBS pre-filtered through a 0.22-
µm-pore-size membrane MitoTracker Deep Red (APC) Life Technologies 100 nM
*All stainings were performed at 37°C for 30 minutes.
** MitoTracker was omitted for the RBC-derived samples.
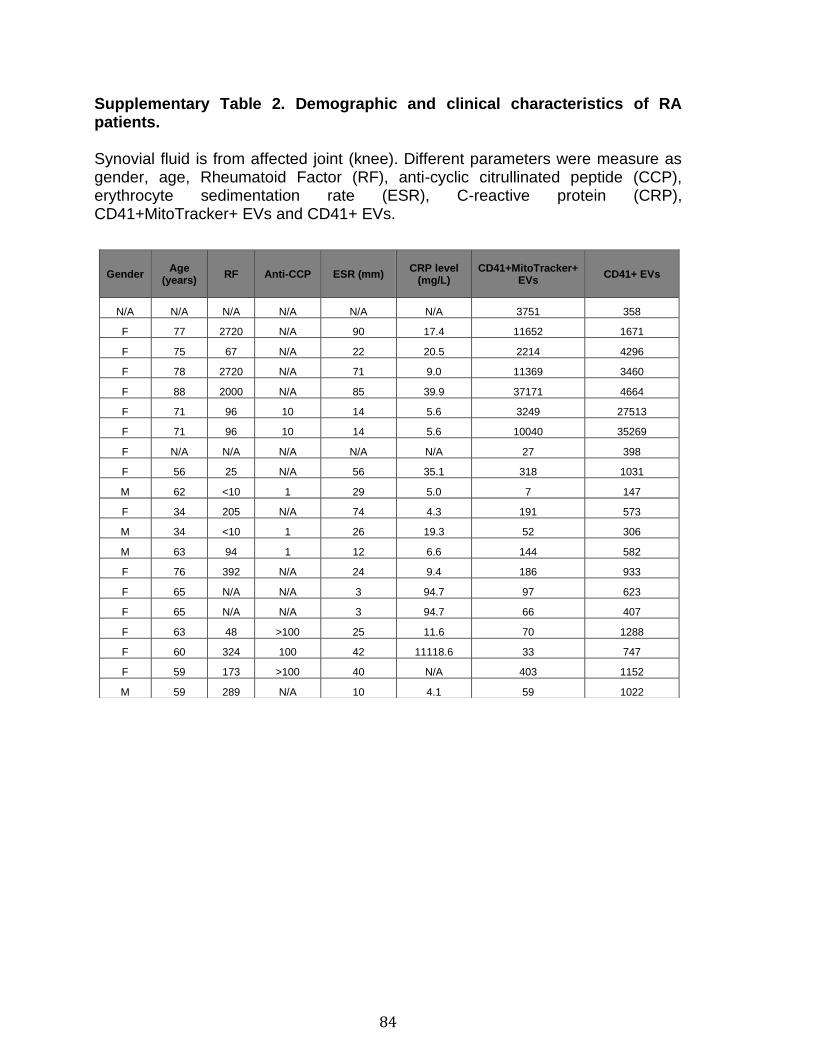
84
Supplementary Table 2. Demographic and clinical characteristics of RA patients. Synovial fluid is from affected joint (knee). Different parameters were measure as gender, age, Rheumatoid Factor (RF), anti-cyclic citrullinated peptide (CCP), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), CD41+MitoTracker+ EVs and CD41+ EVs.
Gender Age
(years) RF Anti-CCP ESR (mm)
CRP level (mg/L)
CD41+MitoTracker+ EVs
CD41+ EVs
N/A N/A N/A N/A N/A N/A 3751 358
F 77 2720 N/A 90 17.4 11652 1671
F 75 67 N/A 22 20.5 2214 4296
F 78 2720 N/A 71 9.0 11369 3460
F 88 2000 N/A 85 39.9 37171 4664
F 71 96 10 14 5.6 3249 27513
F 71 96 10 14 5.6 10040 35269
F N/A N/A N/A N/A N/A 27 398
F 56 25 N/A 56 35.1 318 1031
M 62 <10 1 29 5.0 7 147
F 34 205 N/A 74 4.3 191 573
M 34 <10 1 26 19.3 52 306
M 63 94 1 12 6.6 144 582
F 76 392 N/A 24 9.4 186 933
F 65 N/A N/A 3 94.7 97 623
F 65 N/A N/A 3 94.7 66 407
F 63 48 >100 25 11.6 70 1288
F 60 324 100 42 11118.6 33 747
F 59 173 >100 40 N/A 403 1152
M 59 289 N/A 10 4.1 59 1022

85
Chapitre 2: Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions.
3.1 Résumé Les plaquettes activées libèrent des vésicules extracellulaires (EV) pouvant contenir
des mitochondries (mito+EV). Ces EV contiennent des motif moléculaires associés
aux dommages (DAMP) comme l’ADN mitochondrial, détecté en conditions
inflammatoires et dans les concentrés plaquettaires (CP) destinés à la transfusion.
Nous avons émis l'hypothèse que les mito+EV représentent un réservoir d'ADN
mitochondrial et avons exploré ce lien dans les CP qui ont induit ou non des
réactions transfusionnelles. Non seulement les mito+EV étaient plus abondantes
dans les CP impliqués dans des réactions transfusionnelles, mais elles corrèlent
significativement avec l’ADN mitochondrial dont la majorité est encapsulée dans des
EV. Bien que notre travail implique que des investigations sont nécessaires pour
déterminer s'il existe un rôle pathogène causal du DAMP mitochondrial encapsulé
dans les EV par opposition à l'ADN mitochondrial en solution, cette étude suggère
que les EV peuvent être un biomarqueur utile pour la prédiction du risque potentiel
de réactions transfusionnelles.

86
Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions Genevieve Marcoux,1,† Audrey Magron,1,† Caroline Sut,2,3 Audree Laroche,1 Sandrine Laradi,2,3 Hind Hamzeh-Cognasse,2 Isabelle Allaeys,1 Ophelie Cabon,1 Anne-Sophie Julien,4 Olivier Garraud,2 Fabrice Cognasse,2,3,† and Eric Boilard1,5,† From the 1Department of Infectious Diseases and Immunity, Centre de Recherche du CHU de Québec - Université Laval, the 4Department of Mathematics and Statistic, Université Laval, Quebec City, Québec, and the 5Canadian National Transplantation Research Program, Edmonton, Alberta, Canada; the 2Université de Lyon, UJM-Saint-Etienne, GIMAP, EA 3064 and 3Département Scientifique, Établissement Français du Sang Auvergne- Rhône-Alpes, Saint-Étienne, France. Address reprint requests to: Eric Boilard, PhD, Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de Médecine de l’Université Laval, 2705 Laurier Boulevard, Room T1-49, Québec, QC, Canada G1V 4G2; e-mail: eric.boilard@ crchudequebec.ulaval.ca; or Fabrice Cognasse, PhD, Etablissement Français du Sang Auvergne-Rhône-Alpes, Département Scientifique, 25 Boulevard Pasteur, 42100 Saint-Etienne, France; e-mail: fabrice. [email protected]. † Equally contributed. This work was supported by “Amis de Rémi Association” to (FC), the Etablissement Français du Sang (EFS) to (FC and EB), and in part by a foundation grant from the Canadian Institutes of Health Research (CIHR) to EB. EB is the recipient of an investigator award from CIHR. GM has an award from the Canadian Blood Services, and AM is recipient of an award from MITACS. Received for publication January 9, 2019; revision received March 6, 2019, and accepted March 10, 2019. doi:10.1111/trf.15300 © 2019 AABB TRANSFUSION 2019;00;1–12

87
3.2 Abstract
BACKGROUND: Whereas platelet transfusion is a common medical procedure,
inflammation still occurs in a fraction of transfused individuals despite the absence
of any apparent infectious agents. Platelets can shed membrane vesicles, called
extracellular vesicles (EVs), some of which contain mitochondria (mito+EV). With its
content of damage-associated molecular pattern (DAMP), the mitochondrion can
stimulate the innate immune system. Mitochondrial DNA (mtDNA) is a recognized
DAMP detected in the extracellular milieu in numerous inflammatory conditions and
in platelet concentrates. We hypothesized that platelet-derived mitochondria
encapsulated in EVs may represent a reservoir of mtDNA.
STUDY DESIGN AND METHODS: Herein, we explored the implication of mito+EVs
in the occurrence of mtDNA quantified in platelet concentrate supernatants that
induced or did not induce transfusion adverse reactions.
RESULTS: We observed that EVs were abundant in platelet concentrates, and
platelet-derived mito+EVs were more abundant in platelet concentrates that induced
adverse reactions. A significant correlation (rs = 0.73; p < 0.0001) between platelet-
derived mito+EV levels and mtDNA concentrations was found. However, there was
a non-significant correlation between the levels of EVs without mitochondria and
mtDNA concentrations (rs =−0.11; p = 0.5112). The majority of the mtDNA was
encapsulated into EVs.
CONCLUSION: This study suggests that platelet derived EVs, such as those that
convey mitochondrial DAMPs, may be a useful biomarker for the prediction of
potential risk of adverse transfusion reactions. Moreover, our work implies that
investigations are necessary to determine whether there is a causal pathogenic role
of mitochondrial DAMP encapsulated in EVs as opposed to mtDNA in solution.

88
3.3 Introduction
Platelet transfusion is a common medical procedure necessary for individuals with
low platelet counts or defective platelets.1 It requires the collection of platelets as
platelet concentrates and storage, usually up to 5 or 7 days depending on the country
and blood bank involved. Whereas transfusion is a lifesaving operation, it
can also generate unwanted reactions in a proportion of patients.1,2
The establishment of strict procedures to avoid the transfusion of microbial
components has greatly reduced the transmission of infections in recipients.
However, sterile inflammation and organ injury in transfused recipients still occur in
the absence of any apparent infectious agents.3 Transfusion-related acute lung
injury is the leading cause of transfusion-related death and is initiated by soluble
mediators in plasma, such as antibodies against the human leukocyte antigen on
WBCs of the recipient or lysophospholipids, and may be modulated by regulatory T
cells or the recipient gastrointestinal microbiota.4–7 Febrile nonhemolytic transfusion
reactions (FNHTRs) are more frequent; they affect approximately 1% to 5% of total
platelet transfusions and are experienced by 30% of multitransfused patients.1,8 As
transfusion of the plasma fraction (without cells or platelets) is sufficient to trigger
these reactions,9 it is suggested that mediators released by cells or platelets during
the platelet preparation or storage (commonly referred to as “storage lesion”) are
involved in adverse reactions.10
Notwithstanding the various improvements that may preserve platelet integrity
during the preparation of platelet concentrates and storage, the occurrence of
platelet activation or storage lesions may lead to release of immunomodulatory
molecules in the platelet concentrates capable of mediating leukocyte activation.1
Lipid mediators (e.g., prostaglandins, lysophospholipids), cytokines and chemokines
(e.g., IL1β, CD40L, RANTES [CCL5], platelet factor 4 [PF4]), all may play a role in
immunologic reactions.1 For example, the release of soluble CD40L (sCD40L) has

89
been described during platelet storage, and its elevated concentration is associated
with a higher occurrence of adverse reactions.11–13
Activated or apoptotic cells and platelets can also shed small (approx. 100-1000 nm
in diameter) membrane vesicles, called extracellular vesicles (EVs) that are
generally identified as microparticles in transfusion research.14,15 They harbor CD41
and CD61 markers, and a proportion of them can express and P-selectin (CD62P).
During plasma membrane budding and shedding, there is engulfment of platelet
cytoplasmic molecules into certain EVs. Molecules potentially implicated in adverse
reactions following platelet transfusion have been described in EVs.14,15 For
instance, CD40L carried by EVs from apheresis platelet concentrates activate
respiratory burst in neutrophils.16,17 Moreover, phosphatidylserine (PS), a
phospholipid implicated in the prothrombotic role of platelet-derived EVs and their
clearance,18–20 is harbored by only a fraction of them.21–23
The mitochondrion is the organelle that mediates the oxidative phosphorylation
necessary for adenosine triphosphate production.24 It also plays an important role in
cell division and apoptosis.24,25 Intriguingly, mitochondria share several features with
bacteria, such as the presence of a double membrane comprising cardiolipin,
formylated peptides, and a circular genome containing hypomethylated CpG DNA
motifs.26–29 Mitochondrial damage-associated molecular patterns (DAMPs), such as
mitochondrial DNA (mtDNA) and formylated peptides, activate toll-like receptors
(TLRs; particularly TLR9) and formyl peptide receptors, respectively.30–36
Mitochondrial DAMPs have been described in a multitude of inflammatory
conditions.37–44 Thus, if released from a damaged cell, mitochondrial DAMPs are
suggested to contribute to inflammation.45
Each platelet contains approximately four mitochondria in the cytoplasm, which have
the potential of being wrapped into EVs, thereby forming mito+EVs.21,38,46 Early
proteomic analyses have already identified certain mitochondrial proteins in platelet-
derived EVs,47 thus pointing to an EV mitochondrial content. The study by Boudreau

90
et al.38 established the presence of actual respiratory-competent mitochondria,
inside a subpopulation of EVs generated by stimulating platelets using various
stimuli or from platelet concentrates. Confirmatory studies by multiple laboratories
have further contributed to unequivocally identifying extracellular mitochondria in
platelet concentrates.46,48–50 How mitochondria are released by platelets in platelet
concentrates is not completely understood, but factors such as the methodology
used to prepare platelet concentrates (e.g., platelet-rich plasma, apheresis, and
buffy coat) and the utilization of pathogen inactivation systems have been
suggested.45,46,48–51
Quantitative polymerase chain reaction (qPCR) to assess mtDNA is a powerful
quantitative approach to indirectly measure mitochondria in the extracellular milieu.
It was used by different laboratories to determine the presence of mtDNA in platelet
concentrates, plasma or stored red blood cells used for transfusion.38,46,48,52–55 It was
determined that higher levels of mtDNA are present in platelet concentrates that
induced FNHTR in transfused individuals than in those that were transfused without
complications.38,50,52,54,55 As the experimental conditions (qPCR) to quantify mtDNA
in the extracellular milieu do not allow distinction of mtDNA in solution from the
mtDNA encapsulated in EVs, none of these studies actually provide insights into the
source of mtDNA or the potential pathogenic mechanism. Hence, whether mtDNA
plays a pathogenic role in transfusion cannot be adequately mechanistically
assessed unless it is initially determined whether it is in solution or encapsulated in
mito+EVs.
Although a contribution by EVs in adverse reactions following transfusion has been
proposed, experimental evidence in human studies supporting this concept is
scarce.1 Blood was collected from patients with side effects (urticaria, fever,
erythema, dyspnea, and hypotension) attributed to platelet transfusion, and
significantly more platelet-derived EVs were detected in the circulation of transfused
patients undergoing hematologic disease compared to nonhematologic disease.56
Others have examined platelet-derived EVs present in platelet concentrates and an

91
association with adverse reactions; levels of platelet-derived EVs did not correlate
with the occurrence of transfusion-related acute lung injury or posttransfusion
reaction with dyspnea.57
In the present study, we examined platelet-derived EVs in platelet concentrates as
a potential biomarker predictive of adverse reaction, taking into account the diversity
of EVs and their content in mitochondrial DAMP. We monitored EVs based on their
mitochondrial content and expression of CD62P and PS using flow cytometry.
Moreover, we quantified mtDNA in the same platelet concentrates, and we
determined whether there was an association between the levels of EV subtypes
and mtDNA and whether they were associated with a higher risk of FNHTR in
transfused recipients.

92
3.4 Methods
3.4.1 Preparation and storage of platelet concentrates
Single-donor apheresis platelets were collected from regular anonymized blood
donors (Regional Blood Bank, EFS Auvergne-Rhône-Alpes;
http://www.dondusang.net)13,50 who volunteered to provide blood for research
purposes and signed a consent form, approved by the ethical committees of
Etablissement Français du Sang. Platelet concentrates were identified with
barcodes, were leukoreduced to less than 106/bag and included 35% native plasma
and 65% platelet additive solution Intersol (Fenwal) or SSP+ (Macopharma). Platelet
concentrates were stored at 22°C with gentle rotation and shaking (60 rpm) for a
maximum of 5 days before being used. Platelet transfusions were conducted as part
of routine care in the university-affiliated hospital. If the transfusion induced adverse
reactions, the incident/accident declaration, strictly conformed to national protocols,
was reported to hemovigilance departments describing symptoms and forwarding
the platelet concentrate identification number. Immediately after the identification of
adverse reactions, the incriminated platelet concentrate bags were shipped back to
blood establishment for immediate processing. The platelet concentrate bag tubing
(controls and adverse reactions) was then stripped and sufficient volume samples
were recovered for the analyses.58 To generate supernatants for the EV analysis,
platelet concentrate samples were centrifuged at 402 x g, during 10 minutes at 22°C.
Supernatants were kept frozen (−80°C) until testing. Samples obtained from platelet
concentrate not associated with adverse transfusion reaction were processed in the
same conditions and were used as controls. All the data relative to the transfused
recipients were provided by the physician in charge and were identified using
barcodes to protect their anonymity, according to the French regulation. Any
information regarding the transfused patient, other than the occurrence of adverse
reaction and type of adverse reaction, are not accessible to the study given the
absence of consent from the patients and in agreement with the French regulation.
For this study, we investigated 48 platelet concentrates that were associated with
transfusion reaction (e.g., allergic reaction, FNHTR, hemodynamic trouble) for which
we had 50 matched controls according to storage duration. All the available

93
information related to platelet concentrates and transfusions are presented in Tables
S1through S3, available as supporting information in the online version of this paper.
3.4.2 Measures of soluble CD62P and CD40L in platelet concentrate
supernatants
Control platelet concentrates and platelet concentrates that resulted in adverse
reactions were shipped almost immediately to the blood establishment.
Supernatants were collected and frozen (−80°C) until assay, with a maximum of 12
hours between the occurrence of a reaction and the end of the processing. Levels
of CD40L and soluble CD62P (sCD62P) in PC supernatants were measured using
Luminex technology, according to the manufacturer’s instructions. Data were
expressed in ng/mL and adjusted to 109 platelets.
3.4.3 EV depletion using centrifugation and magnetic microspheres
Platelet concentrate supernatants were diluted five times in 0.2-μm filtered PBS. Fifty
microliters of each diluted sample was used for the following treatments.
Ultracentrifugation
Samples were centrifuged at 100,000 x g for 1 hour at 18°C to pellet all EVs, and
supernatants were used to extract nucleic acids.
CD41 depletion
Twenty-five microliters of protein G magnetic beads (EMD Millipore) were washed
and incubated for 2 hours at room temperature with 10 μg of antihuman CD41
antibody in 100 μL PBS (clone M148, NovusBio) or 10 μg of irrelevant antibody in
100 μL phosphate-buffered saline (antihuman NFATc1, clone 7A6, Santa Cruz
Biotechnology) as control. After 4 washes, beads were incubated with 50 μL of
diluted PC’s supernatant, for 2 hours at room temperature. A magnet was used to
retain magnetic beads, and the remaining samples were used to extract nucleic
acids.

94
Benzonase digestion
Samples were digested or not for 30 minutes at 37°C with a genetically engineered
endonuclease (Benzonase, Sigma- Aldrich), 100 U/mL in 40mM of Tris, 10 mM of
NaCl, 6 mM of MgCl2, 1 mM of CaCl2, pH 7.9); nuclease activity was stopped by
adding 20mM of ethylenediaminetetraacetic acid and samples were then used to
extract nucleic acids. Benzonase activity on free mtDNA was evaluated by adding
purified human mtDNA in platelet concentrate supernatant. In all conditions, mtDNA
was further quantified by real-time qPCR.
3.4.4 Statistical analyses
Descriptive statistics are presented as frequency with percentage for categorical
variables, while interquartile ranges are presented for continuous variables. Graphic
results are presented as mean±standard error of the mean. The concentrations of
EVs and soluble factors in platelet concentrates were compared between the control
and reaction groups using Mann–Whitney test. The statistical significance for
comparisons between groups for the settings of optimal flow cytometry conditions
and for the benzonase nuclease assays were determined using one-way repeated
measures-analysis of variance or paired Student’s t test. Correlations between the
variables were assessed using Spearman correlation coefficient.
Associations between the diverse types of EVs and occurrence of adverse reaction
on transfusion were studied by univariable and multivariable logistic regressions.
The latter were adjusted for sex of donor, storage duration, and the concentration of
platelets in the platelet concentrates.
Receiver operating characteristic curves were generated to assess the predictive
ability of EVs on reactions, and their area under the curve was calculated. Results
were considered positive for the different EV subtypes when their value was above
the cutoff value identified after maximizing Youden’s index. A 95% confidence
interval was obtained for the cutoff using 10,000 bootstrap samples. Performance
measures of these values (sensitivity, specificity, negative and positive predictive

95
value, accuracy, and area under the curve) are presented. Results were considered
statistically significant when the p value was less than 0.05. All statistical analyses
were done using computer software (Prism, version 6, GraphPad Software; and SAS
version 9.4, SAS Institute Inc.).
More details on detection of EV and mitochondria by flow cytometry and
quantification of mtDNA are in Appendix S1, available as supporting information in
the online version of this paper.

96
3.5 Results
In this retrospective case control study, 9206 consecutive platelet concentrates were
collected, for which 140 were associated with adverse reactions. We examined 48
single donor apheresis platelet concentrates that were associated with transfusion
reaction (e.g., allergic reaction, FNHTR, hemodynamic trouble) and for which we
had 50 controls, matched on method of platelet preparation and storage duration.
None of these platelet concentrates had been analyzed previously for their mtDNA
nor the EV contents. All the transfused patients had received a single platelet
transfusion with the exclusion of other components, with the exception of four who
had also received RBC transfusion more than 16 hours before the platelet
transfusion (Tables S1 and S2). The respective information on transfused products
and the blood donors is presented in Table S3.
Flow cytometry was used to quantify the different subtypes of EVs. We first ensured
the validity of our approach and confirmed that we could detect particles as small as
100-nm silica beads (EV gate, Fig. 1A), which are also smaller than resting platelets
(Fig. 1B). We activated washed platelets in vitro using thrombin to generate EVs
(Fig. 1C) and verified that the different EV subtypes were quantified with high degree
of specificity. Events in the EV gate (Fig. 1D, upper left) were displayed to assess
the presence of mitotracker and CD41 expression in reactive platelet concentrates
(Fig. 1D, lower left). Among CD41+EVs containing mitochondria (red gate) or not
(blue gate), PS, as determined by the binding of annexin V (Fig. 1D, upper right), or
CD62P expression (Fig. 1D, lower right) were also determined for all platelet
concentrates. To assess the specificity of our EV detection method, we used several
controls schematized in Fig. 1E: Given their membrane moiety, EVs were quantified
in samples that were either treated or left untreated with detergent Triton-X100. We
confirmed that in the presence of detergent, CD41+EVs, CD41+CD62P+EVs,
CD41+PS+EVs and CD41+mito+EVs were dissolved and thereby not detected (Fig.
1E-F). Furthermore, given that EVs can be eliminated from supernatants by high-
speed centrifugation, all EV types were significantly decreased in biospecimens that
underwent ultracentrifugation (Fig. 1E-F). PS, reportedly present on the surface of a

97
subpopulation of EVs, was assessed using the PS-binding probe annexin V. As
annexin V binding to PS requires calcium ions, we confirmed the absence of
CD41+PS+EVs in the presence of the calcium chelator ethylenediaminetetraacetic
acid (Fig. 1E-G). Serial dilution of the samples led to consistent dilution of the EVs
without affecting the median and mean CD41 fluorescence intensity, further
confirming our quantitative approach (Fig. 1H and I).
The various subtypes of EVs were quantified in platelet concentrate supernatants by
a blinded operator. Our study confirms the abundance of CD41+EVs in platelet
concentrates and indicates that their levels are on average significantly higher in
samples that induced adverse reactions compared to controls (Fig. 2A). Moreover,
when the mitochondrial content and/or other markers (PS, CD62P) were taken into
account, all the EV subtypes were significantly more elevated in adverse reactions
than in controls (Fig. 2B-F and Table S4, available as supporting information in the
online version of this paper.). Cutoff values were calculated by Youden’s index for
all subtypes of CD41+EVs. We found that total number of CD41+EVs gave the best
predictive ability; hence, a concentration of 40.87 x106 CD41+EV/mL could predict
90% of the true adverse reactions (Table 1).
Despite the efforts to identify perfectly matched platelet concentrates in each group
(no reaction and reaction), small but significant differences were noticed in variables
such as storage duration and platelet levels in circulation in the blood donors, and a
near significant difference in platelet concentration present in platelet concentrates
(Table S3). Logistic regressions to predict adverse reactions showed a cubic relation
for total CD41+EVs, as well as CD41+mito+EVs and CD41+mito−EVs. When adjusting
for sex of the transfused product, storage duration of the platelet concentrates, and
the platelet concentration in platelet concentrates, the previous relationships were
still highly significant (Table S5, available as supporting information in the online
version of this paper.), further proving the robustness of the EV markers.

98
Moreover, we verified the predictive ability of EV subtypes on particular adverse
reactions using receiver operating characteristics and their area under the curve
(Table S6, available as supporting information in the online version of this paper.).
We observed that total CD41+EVs, CD41+mito−EVs and CD41+mito−PS+ EVs were
promising predictors of all adverse reactions. Mitochondria containing EVs were
poorly predictive of allergic reaction, whereas CD41+mito−CD62P+ EVs could predict
allergic and hemodynamic trouble but not the FNHTRs. Although adverse reaction
types are not necessarily exclusive, the number of incidents that implicated two
reactions types was insufficient to make further prediction analyses.
To determine whether CD41+mito+EVs could account for the mtDNA present in
platelet concentrates, the concentration of mtDNA in the same samples was
determined using a qPCR approach, and correlations with CD41+mito+EV levels
were examined. We found a strong and significant correlation between the two
markers (rs = 0.73; p < 0.0001), whereas no significant correlation was found
between CD41+mito−EVs and mtDNA (Fig. 3A-B and Table S7, available as
supporting information in the online version of this paper.). To further confirm the
involvement of EVs with mtDNA levels, EVs (of all types) were eliminated from
supernatants by ultracentrifugation. We found a near complete (98%) reduction in
mtDNA levels in EV-depleted samples (Fig. 3C). A 50% reduction in mtDNA was
determined when CD41+EVs were specifically depleted using magnetic microbeads,
which confirmed the presence of mtDNA associated with CD41+EVs (Fig. 3C). The
efficient depletion of CD41+mito+EVs with ultracentrifugation and magnetic beads
was validated by flow cytometry (Fig. 3D). Moreover, while mtDNA in platelet
concentrates was protected from DNAse, suggesting that it is encapsulated within
mito+EVs (Fig. 3E), mtDNA isolated from hepatocytes and spiked into plasma was
indeed sensitive to DNAse treatment (in those conditions, 95.9±1.9% [n = 3] of the
exogenous mtDNA was hydrolyzed) (Fig. 3F). Thus, CD41+EVs, including those
containing mitochondria (CD41+mito+EVs), are more abundant in platelet
concentrates that have induced adverse reactions and their presence explains, at
least in part, the occurrence of mtDNA. Higher levels of sCD40L have been reported

99
in adverse reactions.11–13 However, whether CD40L release occurs concomitantly
with the production of mito+CD41+EVs in platelet concentrates is as yet unknown.
We measured sCD40L in platelet concentrate supernatants and verified the
correlation with CD41+mito+EVs. We found a good correlation between the two
measurements, suggesting that they are released concomitantly (Fig. 4A and Table
2). In the same way, the occurrence of CD41+mito+EVs correlated strongly with
sCD62P, suggesting that the liberation of CD41+mito+EV and α-granule content may
occur jointly (Fig. 4B and Table 2). We also observed that both sCD40L and sCD62P
levels were higher in platelet concentrates that have induced adverse reactions (Fig.
4C-D and Table 3). Cutoff values were calculated by Youden’s index for sCD40L
and sCD62P. We found that sCD62P has a good predictive ability; however, sCD40L
measurement shows a poor sensitivity (Table 4).

100
3.6 Discussion
Although platelet-derived EVs have long been incriminated as a potential player in
adverse reactions, no study has formally verified the presence of platelet-derived
EVs and their subtypes in platelet concentrates confirmed as having triggered
adverse reactions. Furthermore, the fact that platelet EVs transport mitochondria,
and the multiple studies from different investigators that indicated higher levels of
mtDNA in adverse reactions,38,46,48,52–55 prompted our examination of platelet-
derived EVs as the actual source of mtDNA in platelet concentrates. Our study
highlights the broad diversity of platelet-derived EVs and shows that platelet EVs
convey mitochondria in platelet concentrates.
Extracellular vesicles encompass exosomes, small vesicles stored in intracellular
compartments, and microparticles or microvesicles, produced by plasma membrane
budding and shedding.14 Platelets can release both exosomes and microparticles59
and in the absence of consistent markers to distinguish both types of vesicles, it is
difficult to unequivocally determine the cellular compartment from which the
microparticles have emerged.60 However, given that smaller EVs may have been
overlooked by the flow cytometry approach used herein, and given that exosomes
are not expected to contain organelles,14,60 the EVs described in the present study
mainly include microparticles shed from the plasma membrane. The exact
mechanism of mitochondria release from platelets is unclear, but rearrangement of
the cytoskeleton is implied and may involve proteins in the apoptosis cascade.38
If replicated in a larger cohort of samples, our study suggests that the quantification
of CD41+EV may be a useful approach to identify platelet concentrates at risk of
provoking adverse reactions. Moreover, we found that extracellular mitochondria,
assessed using mtDNA or mito+EVs, were also significantly elevated in adverse
reactions. Although the quantification of platelet EVs requires only a few microliters
of fluids (5 μL were used herein), which is useful in the design of potent biomarker,
it also implicates experienced investigators and sophisticated flow cytometry
equipment. Given that the levels of mtDNA and mito+EVs showed a strong

101
correlation, it suggests that the measurement of mtDNA, using a quantitative
approach such as qPCR, may more conveniently permit to indirectly assess the
number of platelet-derived EVs in platelet concentrates.
The observation that DNAse treatment did not impact mtDNA levels suggests that it
is protected within EVs. Interpretation of the data could be complicated by the
presence of PF4 in platelet concentrates, as the former also protects DNA from
hydrolysis by DNAse.61 However, the fact that mtDNA was completely eliminated
from platelet concentrate supernatants by centrifugation provided further
confirmation that most, if not all, mtDNA in platelet concentrates is in fact
encapsulated in EVs. The targeted depletion of CD41+EVs using magnetic
microspheres led to consistent but partial reduction of mtDNA in the extracellular
milieu. We hypothesize that the absence or the too low expression of CD41 on a
certain proportion of platelet-derived mito+EVs, the presence of free mitochondrial
organelles in the extracellular milieu, or the liberation of mito+EV by leukocytes
through the blood processing could explain how mtDNA was not completely
eliminated by this approach.
While the release of CD40L is suggested to occur with longer storage duration,62,63
the preparation method of the transfusion product is suggested to be the main cause
of EV and mitochondrial release.46,49 We found that mitochondrial release correlates
with sCD40L and sCD62P, suggesting that similar pathways may have led to their
liberation. Given that technical approaches (e.g., enzyme-linked immunosorbent
assay, multiplex) used to quantify sCD40L and sCD62P cannot distinguish whether
these molecules are in solution or associated with EVs, we further speculate that
similarly to mtDNA, CD40L and CD62P molecules in platelet concentrates are in fact
conveyed by platelet-derived EVs.
While associations between various EV subtypes and adverse reactions were
examined, the study design did not address causality. Whether platelet-derived EVs
play an actual role in adverse reactions remains to be established experimentally,
but they may contribute through their broad repertoire of inflammatory

102
molecules,14,15 such as sCD40L and CD62P and their interaction with their
respective counterreceptors on leukocytes, namely, CD40 or integrin β3 and
platelet-selectin glycoprotein ligand-1.56,64 As PS is also exposed on a proportion of
the mito+EVs, they may also contribute to adverse reaction through the support of
the coagulation cascade and thrombosis. With the identification of mtDNA in platelet
concentrates, prior investigations have logically focused on verifying the impact of
purified mtDNA on various cellular lineages both in vitro and in vivo.54 While these
studies are crucial for the understanding of potential mechanisms related to
transfusion-induced adverse reactions, they may not fully recapitulate the actual
pathogenesis if mtDNA is in fact typically encapsulated into EVs and not free in
solution. It is frequently suggested that mitochondrial DAMPs activate TLRs (e.g.,
TLR9) in endosomes. Thus, it remains to be established whether mitochondrial
DAMPs inside EVs actually reach endosomes to activate TLRs. As platelet EVs,
including mito+EVs, were previously found internalized by neutrophils,65 this pathway
of activation remains a promising avenue. Moreover, mtDNA has been assessed in
a multitude of inflammatory conditions, including trauma,37,44 burn injury,43 hepatitis,
cancer,66 rheumatoid arthritis,38,39 and systemic lupus erythematosus.40,41
Considering that the induction of platelet EVs is frequently observed in inflammation,
it would be interesting to consider whether their presence explains the occurrence
of mtDNA in all of these conditions.
To summarize, we demonstrated the potential of EV based biomarker research with
an appreciation of EV diversity. Transfusion is viewed as a triad of intricate
parameters: 1) the donor (physiologic and genetic characteristic, environmental—
e.g., stress, pollution, self-prescribed drugs, food intake), 2) the labile blood
component (collection, preparation/processing, storage, delivery condition, etc.),
and 3) the recipient (physiologic and genetic characteristics, pathology, etc.). All
these variables must be considered to improve outcomes. The identification of
biomarker or the identification of storage lesion and transfusion outcomes may
contribute to the improvement of storage conditions and personalized transfusion.
The EV cargo is vast, and the development of additional tools may permit the

103
distinction of a larger spectrum of EV heterogeneity for the identification of novel
potent biomarkers and an understanding of the roles of EVs in inflammation.

104
3.7 References
1. Sahler J, Grimshaw K, Spinelli SL, et al. Platelet storage and transfusions: new
concerns associated with an old therapy. Drug Discov Today Dis Mech
2011;8:e9-e14.
2. Refaai MA, Phipps RP, Spinelli SL, et al. Platelet transfusions: impact on
hemostasis, thrombosis, inflammation and clinical outcomes. Thromb Res
2011;127:287-91.
3. Garraud O, Sut C, Haddad A, et al. Transfusion-associated hazards: a revisit of
their presentation. Transfus Clin Biol 2018; 25:118-35.
4. Semple JW, McVey MJ, Kim M, et al. Targeting transfusion related acute lung
injury: the journey from basic science to novel therapies. Crit Care Med
2018;46:e452-e8.
5. Silliman CC, Curtis BR, Kopko PM, et al. Donor antibodies to HNA-3a implicated
in TRALI reactions prime neutrophils and cause PMN-mediated damage to
human pulmonary microvascular endothelial cells in a two-event in vitro
model. Blood 2007;109:1752-5.
6. Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect
against transfusion-related acute lung injury via IL-10. Blood 2017;129:2557-
69.
7. Kapur R, Kim M, Rebetz J, et al. Gastrointestinal microbiota contributes to the
development of murine transfusion-related acute lung injury. Blood Adv
2018;2:1651-63.
8. Heddle NM. Febrile nonhemolytic transfusion reactions to platelets. Curr Opin
Hematol 1995;2:478-83.
9. Heddle NM, Klama L, Singer J, et al. The role of the plasma from platelet
concentrates in transfusion reactions. N Engl J Med 1994;331:625-8.
10. Sut C, Tariket S, Aubron C, et al. The non-hemostatic aspects of transfused
platelets. Front Med (Lausanne) 2018;5:42.

105
11. Blumberg N, Gettings KF, Turner C, et al. An association of soluble CD40 ligand
(CD154) with adverse reactions to platelet transfusions. Transfusion
2006;46:1813-21.
12. Sahler J, Spinelli S, Phipps R, et al. CD40 ligand (CD154) involvement in platelet
transfusion reactions. Transfus Clin Biol 2012;19:98-103.
13. Cognasse F, Sut C, Fromont E, et al. Platelet soluble CD40-ligand level is
associated with transfusion adverse reactions in a mixed threshold-and-hit
model. Blood 2017;130: 1380-3.
14. Melki I, Tessandier N, Zufferey A, et al. Platelet microvesicles in health and
disease. Platelets 2017;28:214-21.
15. Burnouf T, Goubran HA, Chou ML, et al. Platelet microparticles: detection and
assessment of their paradoxical functional roles in disease and regenerative
medicine. Blood Rev 2014;28:155-66.
16. Xie RF, Hu P, Li W, et al. The effect of platelet-derived microparticles in stored
apheresis platelet concentrates on polymorphonuclear leucocyte respiratory
burst. Vox Sang 2014;106: 234-41.
17. Xie RF, Hu P, Wang ZC, et al. Platelet-derived microparticles induce
polymorphonuclear leukocyte-mediated damage of human pulmonary
microvascular endothelial cells. Transfusion 2015;55:1051-7.
18. Dasgupta SK, Le A, Chavakis T, et al. Developmental endothelial locus-1 (Del-
1) mediates clearance of platelet microparticles by the endothelium.
Circulation 2012;125:1664-72.
19. Fujii T, Sakata A, Nishimura S, et al. TMEM16F is required for phosphatidylserine
exposure and microparticle release in activated mouse platelets. Proc Natl
Acad Sci U S A 2015;112: 12800-5.
20. Happonen KE, Tran S, Morgelin M, et al. The Gas6-Axl interaction mediates
endothelial uptake of platelet microparticles. J Biol Chem 2016;291:10586-
601.
21. Boilard E, Duchez AC, Brisson A. The diversity of platelet microparticles. Curr
Opin Hematol 2015;22:437-44.

106
22. Cloutier N, Tan S, Boudreau LH, et al. The exposure of autoantigens by
microparticles underlies the formation of potent inflammatory components:
the microparticle-associated immune complexes. EMBO Mol Med
2013;5:235-49.
23. Arraud N, Linares R, Tan S, et al. Extracellular vesicles from blood plasma:
determination of their morphology, size, phenotype and concentration. J
Thromb Haemost 2014;12:614-27.
24. Andersen JL, Kornbluth S. The tangled circuitry of metabolism and apoptosis.
Mol Cell 2013;49:399-410.
25. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet
2009;43:95-118.
26. Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science 1999;283:1476-
81.
27. Arechaga I. Membrane invaginations in bacteria and mitochondria: common
features and evolutionary scenarios. J Mol Microbiol Biotechnol 2013;23:13-
23.
28. Lang BF, Burger G, O’Kelly CJ, et al. An ancestral mitochondrial DNA resembling
a eubacterial genome in miniature. Nature 1997;387:493-7.
29. Friedman JR, Nunnari J. Mitochondrial form and function. Nature 2014;505:335-
43.
30. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause
inflammatory responses to injury. Nature 2010; 464:104-7.
31. McDonald B, Pittman K, Menezes GB, et al. Intravascular danger signals guide
neutrophils to sites of sterile inflammation. Science 2010;330:362-6.
32. Sandhir R, Halder A, Sunkaria A. Mitochondria as a centrally positioned hub in
the innate immune response. Biochim Biophys Acta 1863;2017:1090-7.
33. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity.
Nat Immunol 2017;18:488-98.
34. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate
and adaptive immunity. Immunity 2015;42:406-17.

107
35. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat
Rev Immunol 2011;11:389-402.
36. Krysko DV, Agostinis P, Krysko O, et al. Emerging role of damage-associated
molecular patterns derived from mitochondria in inflammation. Trends
Immunol 2011;32:157-64.
37. Zhao Z, Wang M, Tian Y, et al. Cardiolipin-mediated procoagulant activity of
mitochondria contributes to traumatic brain injury-associated coagulopathy in
mice. Blood 2016;127: 2763-72.
38. Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria
serving as substrate for bactericidal group IIA-secreted phospholipase A2 to
promote inflammation. Blood 2014;124:2173-83.
39. Hajizadeh S, DeGroot J, TeKoppele JM, et al. Extracellular mitochondrial DNA
and oxidatively damaged DNA in synovial fluid of patients with rheumatoid
arthritis. Arthritis Res Ther 2003;5:R234-40.
40. Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched
in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like
disease. Nat Med 2016; 22:146-53.
41. Caielli S, Athale S, Domic B, et al. Oxidized mitochondrial nucleoids released by
neutrophils drive type I interferon production in human lupus. J Exp Med
2016;213:697-713.
42. Garcia-Martinez I, Santoro N, Chen Y, et al. Hepatocyte mitochondrial DNA
drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest
2016;126:859-64.
43. Simmons JD, Lee YL, Mulekar S, et al. Elevated levels of plasma mitochondrial
DNA DAMPs are linked to clinical outcome in severely injured human
subjects. Ann Surg 2013;258: 591-6 discussion 6-8.
44. Nakahira K, Kyung SY, Rogers AJ, et al. Circulating mitochondrial DNA in
patients in the ICU as a marker of mortality: derivation and validation. PLoS
Med 2013;10:e1001577; discussion e.
45. Marcoux G, Boilard E. Mitochondrial damage-associated molecular patterns in
blood transfusion products. ISBT Sci Ser 2017;12:501-5.

108
46. Marcoux G, Duchez AC, Rousseau M, et al. Microparticle and mitochondrial
release during extended storage of different types of platelet concentrates.
Platelets 2017;28:272-80.
47. Dean WL, Lee MJ, Cummins TD, et al. Proteomic and functional characterisation
of platelet microparticle size classes. Thromb Haemost 2009;102:711-8.
48. Chen Z, Schubert P, Bakkour S, et al. p38 mitogen-activated protein kinase
regulates mitochondrial function and microvesicle release in riboflavin- and
ultraviolet light-treated apheresis platelet concentrates. Transfusion
2017;57:1199-207.
49. Bakkour S, Acker JP, Chafets DM, et al. Manufacturing method affects
mitochondrial DNA release and extracellular vesicle composition in stored red
blood cells. Vox Sang 2016;111: 22-32.
50. Cognasse F, Aloui C, Anh Nguyen K, et al. Platelet components associated with
adverse reactions: predictive value of mitochondrial DNA relative to biological
response modifiers. Transfusion 2016;56:497-504.
51. Magron A, Laugier J, Provost P, et al. Pathogen reduction technologies: the pros
and cons for platelet transfusion. Platelets 2018;29: 2-8.
52. Simmons JD, Lee YL, Pastukh VM, et al. Potential contribution of mitochondrial
DNA damage associated molecular patterns in transfusion products to the
development of acute respiratory distress syndrome after multiple
transfusions. J Trauma Acute Care Surg 2017;82:1023-9.
53. Bakkour S, Chafets DM, Wen L, et al. Development of a mitochondrial DNA real-
time polymerase chain reaction assay for quality control of pathogen
reduction with riboflavin and ultraviolet light. Vox Sang 2014;107:351-9.
54. Yasui K, Matsuyama N, Kuroishi A, et al. Mitochondrial damage associated
molecular patterns as potential proinflammatory mediators in post-platelet
transfusion adverse effects. Transfusion 2016;56:1201–12.
55. Lee YL, King MB, Gonzalez RP, et al. Blood transfusion products contain
mitochondrial DNA damage-associated molecular patterns: a potential
effector of transfusion-related acute lung injury. J Surg Res 2014;191:286-9.

109
56. Nomura S, Okamae F, Abe M, et al. Platelets expressing Pselectin and platelet-
derived microparticles in stored platelet concentrates bind to PSGL-1 on
filtrated leukocytes. Clin Appl Thromb Hemost 2000;6:213-21.
57. Maslanka K, Uhrynowska M, Lopacz P, et al. Analysis of leucocyte antibodies,
cytokines, lysophospholipids and cell microparticles in blood components
implicated in post transfusion reactions with dyspnoea. Vox Sang 2015;108:
27-36.
58. Millar D, Murphy L, Labrie A, et al. Routine screening method for microparticles
in platelet transfusions. J Vis Exp 2018;131: e56893.
https://doi.org/10.3791/56893.
59. Heijnen HF, Schiel AE, Fijnheer R, et al. Activated platelets release two types of
membrane vesicles: microvesicles by surface shedding and exosomes
derived from exocytosis of multivesicular bodies and alpha-granules. Blood
1999;94:3791-9.
60. Lotvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for
definition of extracellular vesicles and their functions: a position statement
from the International Society for Extracellular Vesicles. J Extracell Vesicles
2014;3:26913.
61. Kandace Gollomp IJ, Kim M, Zhai L, et al. Platelet factor 4 (PF4)-mediated
neutrophil extracellular trap compaction limits endothelial injury and promotes
survival following lipopolysaccharide challenge. Blood 2017;130(Suppl
1):997.
62. Khan SY, Kelher MR, Heal JM, et al. Soluble CD40 ligand accumulates in stored
blood components, primes neutrophils through CD40, and is a potential
cofactor in the development of transfusion-related acute lung injury. Blood
2006;108:2455-62.
63. Phipps RP, Kaufman J, Blumberg N. Platelet derived CD154 (CD40 ligand) and
febrile responses to transfusion. Lancet 2001;357:2023-4.
64. Andre P, Prasad KS, Denis CV, et al. CD40L stabilizes arterial thrombi by a
beta3 integrin–dependent mechanism. Nat Med 2002;8:247-52.

110
65. Duchez AC, Boudreau LH, Bollinger J, et al. Platelet microparticles are
internalized in neutrophils via the concerted activity of 12-lipoxygenase and
secreted phospholipase A2-IIA. Proc Natl Acad Sci U S A 2015;112:E3564-
73.
66. Kohler C, Radpour R, Barekati Z, et al. Levels of plasma circulating cell free
nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol
Cancer 2009;8:105.
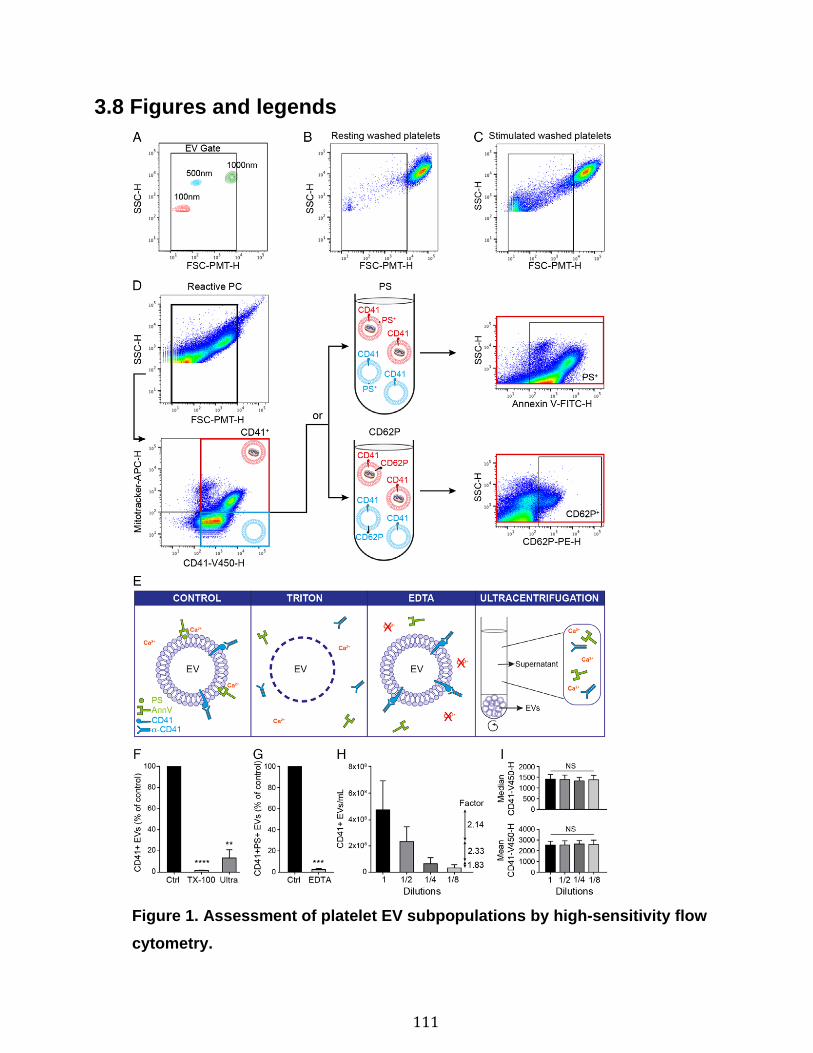
111
3.8 Figures and legends
Figure 1. Assessment of platelet EV subpopulations by high-sensitivity flow
cytometry.

112
Fig. 1. (A) Acquisition by flow cytometry (BD Canto II Special Order Research Product) of silica beads of various size (100, 500, and 1000 nm) displayed in side scatter (SSC)-H (granularity) and forward scatter (FSC)-photomultiplier tube (PMT)-H (relative size) to design the lower (100 nm) and upper (1000 nm) limits of the EV gate. (B) SSC-H and FSC-PMT-H portrayal of nonactivated platelets. (C) Remnant platelets (outside of the EV gate) and EVs (inside the gate) generated after stimulation of platelets with thrombin. (D) Representative SSC-H and FSC-PMT-H dot plots of platelet EVs from a PC supernatant implicated in transfusion reaction (upper left). EVs detected from this sample in the EV gate (in black) are displayed (lower left) according to their content in mitochondria using 100-nm MitoTracker Deep Red (MitoTracker-APC-H) and their expression of V450 fluorochrome-conjugated antibodies directed against CD41a (CD41-V450-H). All EVs expressing CD41 (with mitochondria in the red section, and no mitochondria in blue) were also analyzed for PS expression using fluorescein isothiocyanate– conjugated annexin V (upper right) or activation using phycoerythrin-conjugated CD62P (lower right). (E) Several controls were done to assess the specificity of our EV detection. (F) Sensitivity of CD41a+ EVs to clearance by 0.05% Triton X-100 (TX-100) and ultracentrifugation (Ultra), presented as % of untreated (Ctrl). Data are presented as mean±standard error of the mean (SEM) of 3 independent experiments, paired t test (**p < 0.01 and ****p < 0.0001) compared with the control (Ctrl); (G) Sensitivity of CD41a+PS+ EVs to clearance by ethylenediaminetetraacetic acid treatment presented as percentage of untreated (Ctrl). Data are presented as mean±SEM of four independent experiments, paired t test (***p < 0.001) compared with the control (Ctrl). (H, I) CD41a+EVs were serially diluted (twofold dilution) and quantitatively analyzed by flow cytometry using counting microspheres. Their concentration and the calculated dilution factor (H), the median of fluorescence (I, upper) and the mean of fluorescence (I, lower) are presented. Data are presented as mean±SEM of four independent experiments, Repeated Measures one-way ANOVA; NS, non-significant. [Color figure can be viewed at wileyonlinelibrary.com] as the mean ± SEM. Differences between day 1 and 7 were verified using t-test, *p<0.05.
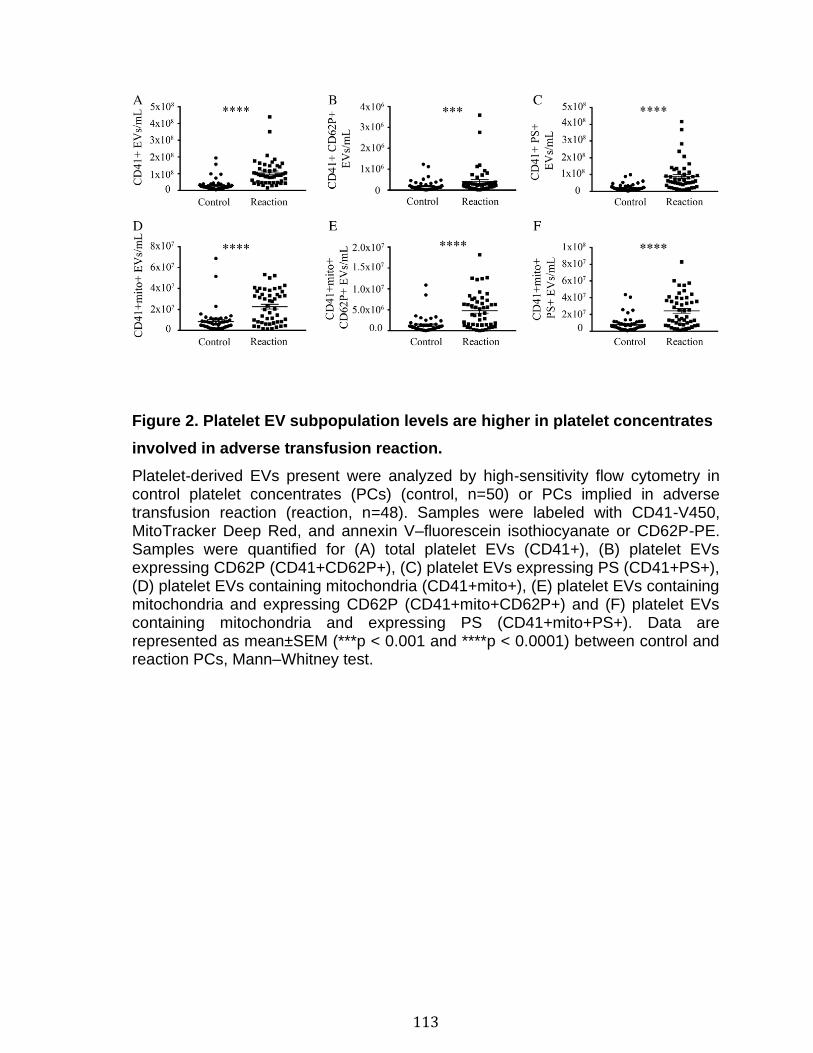
113
Figure 2. Platelet EV subpopulation levels are higher in platelet concentrates
involved in adverse transfusion reaction.
Platelet-derived EVs present were analyzed by high-sensitivity flow cytometry in control platelet concentrates (PCs) (control, n=50) or PCs implied in adverse transfusion reaction (reaction, n=48). Samples were labeled with CD41-V450, MitoTracker Deep Red, and annexin V–fluorescein isothiocyanate or CD62P-PE. Samples were quantified for (A) total platelet EVs (CD41+), (B) platelet EVs expressing CD62P (CD41+CD62P+), (C) platelet EVs expressing PS (CD41+PS+), (D) platelet EVs containing mitochondria (CD41+mito+), (E) platelet EVs containing mitochondria and expressing CD62P (CD41+mito+CD62P+) and (F) platelet EVs containing mitochondria and expressing PS (CD41+mito+PS+). Data are represented as mean±SEM (***p < 0.001 and ****p < 0.0001) between control and reaction PCs, Mann–Whitney test.
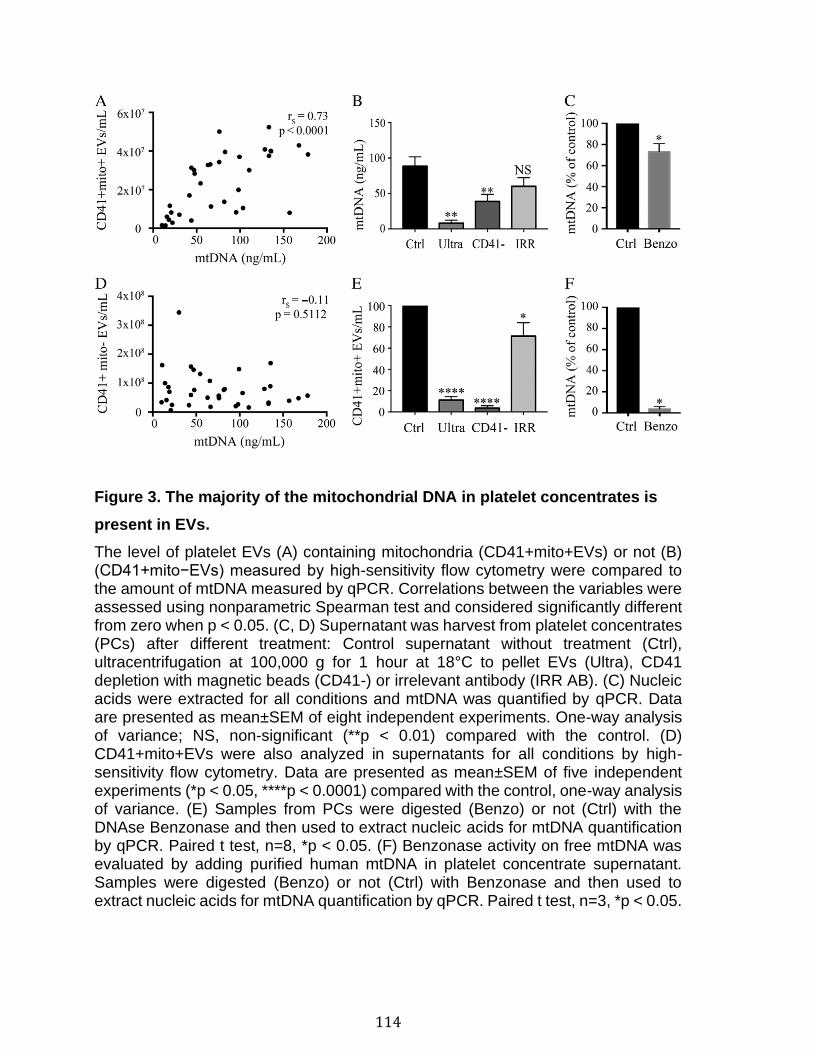
114
Figure 3. The majority of the mitochondrial DNA in platelet concentrates is
present in EVs.
The level of platelet EVs (A) containing mitochondria (CD41+mito+EVs) or not (B) (CD41+mito−EVs) measured by high-sensitivity flow cytometry were compared to the amount of mtDNA measured by qPCR. Correlations between the variables were assessed using nonparametric Spearman test and considered significantly different from zero when p < 0.05. (C, D) Supernatant was harvest from platelet concentrates (PCs) after different treatment: Control supernatant without treatment (Ctrl), ultracentrifugation at 100,000 g for 1 hour at 18°C to pellet EVs (Ultra), CD41 depletion with magnetic beads (CD41-) or irrelevant antibody (IRR AB). (C) Nucleic acids were extracted for all conditions and mtDNA was quantified by qPCR. Data are presented as mean±SEM of eight independent experiments. One-way analysis of variance; NS, non-significant (**p < 0.01) compared with the control. (D) CD41+mito+EVs were also analyzed in supernatants for all conditions by high-sensitivity flow cytometry. Data are presented as mean±SEM of five independent experiments (*p < 0.05, ****p < 0.0001) compared with the control, one-way analysis of variance. (E) Samples from PCs were digested (Benzo) or not (Ctrl) with the DNAse Benzonase and then used to extract nucleic acids for mtDNA quantification by qPCR. Paired t test, n=8, *p < 0.05. (F) Benzonase activity on free mtDNA was evaluated by adding purified human mtDNA in platelet concentrate supernatant. Samples were digested (Benzo) or not (Ctrl) with Benzonase and then used to extract nucleic acids for mtDNA quantification by qPCR. Paired t test, n=3, *p < 0.05.
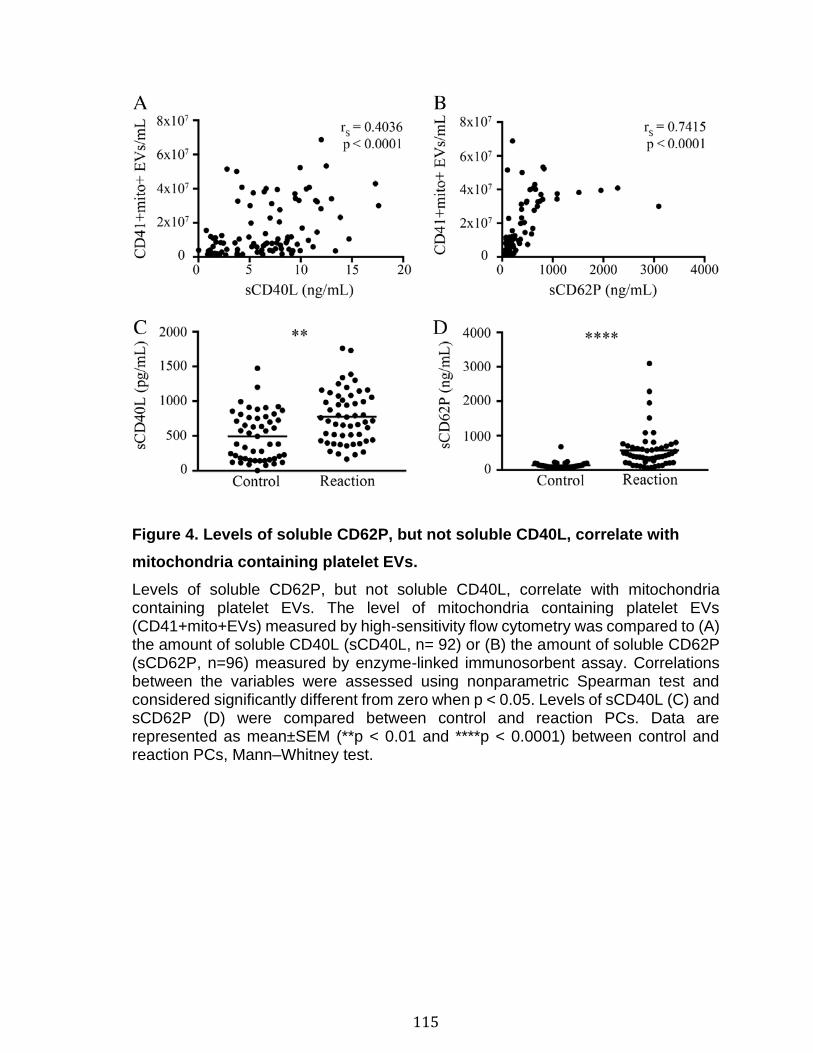
115
Figure 4. Levels of soluble CD62P, but not soluble CD40L, correlate with
mitochondria containing platelet EVs.
Levels of soluble CD62P, but not soluble CD40L, correlate with mitochondria containing platelet EVs. The level of mitochondria containing platelet EVs (CD41+mito+EVs) measured by high-sensitivity flow cytometry was compared to (A) the amount of soluble CD40L (sCD40L, n= 92) or (B) the amount of soluble CD62P (sCD62P, n=96) measured by enzyme-linked immunosorbent assay. Correlations between the variables were assessed using nonparametric Spearman test and considered significantly different from zero when p < 0.05. Levels of sCD40L (C) and sCD62P (D) were compared between control and reaction PCs. Data are represented as mean±SEM (**p < 0.01 and ****p < 0.0001) between control and reaction PCs, Mann–Whitney test.
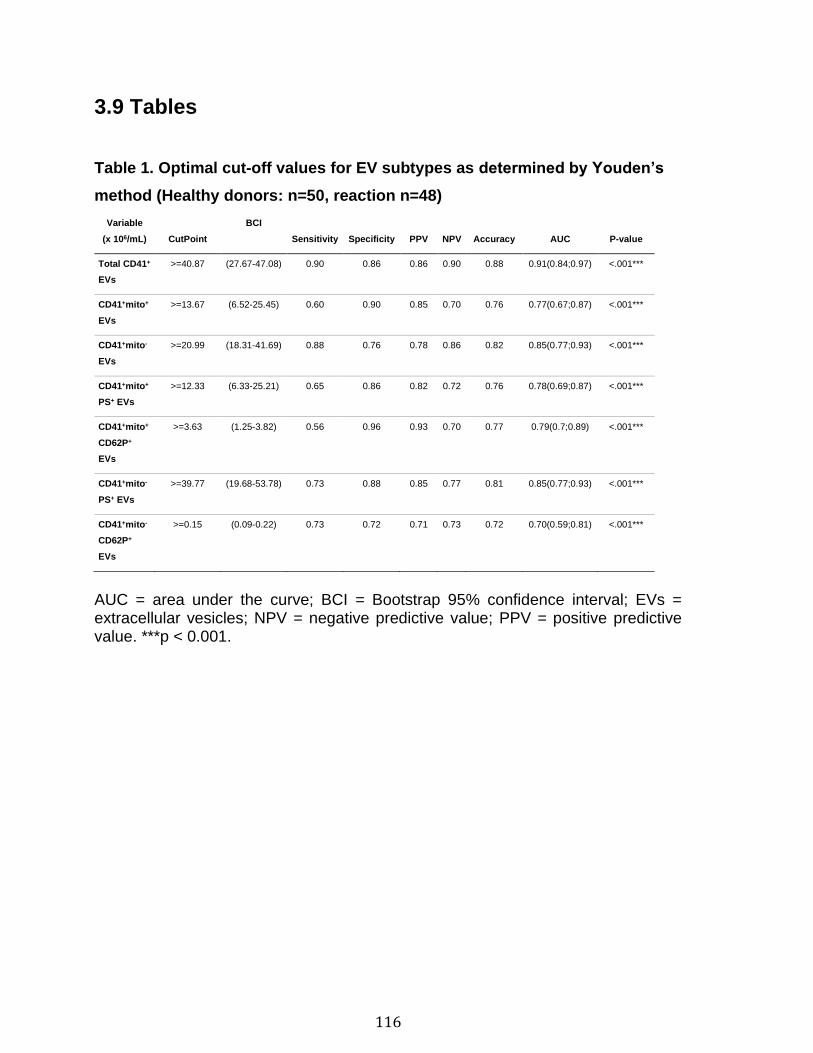
116
3.9 Tables
Table 1. Optimal cut-off values for EV subtypes as determined by Youden’s
method (Healthy donors: n=50, reaction n=48)
Variable
(x 106/mL) CutPoint
BCI
Sensitivity Specificity PPV NPV Accuracy AUC P-value
Total CD41+
EVs
>=40.87 (27.67-47.08) 0.90 0.86 0.86 0.90 0.88 0.91(0.84;0.97) <.001***
CD41+mito+
EVs
>=13.67 (6.52-25.45) 0.60 0.90 0.85 0.70 0.76 0.77(0.67;0.87) <.001***
CD41+mito-
EVs
>=20.99 (18.31-41.69) 0.88 0.76 0.78 0.86 0.82 0.85(0.77;0.93) <.001***
CD41+mito+
PS+ EVs
>=12.33 (6.33-25.21) 0.65 0.86 0.82 0.72 0.76 0.78(0.69;0.87) <.001***
CD41+mito+
CD62P+
EVs
>=3.63 (1.25-3.82) 0.56 0.96 0.93 0.70 0.77 0.79(0.7;0.89) <.001***
CD41+mito-
PS+ EVs
>=39.77 (19.68-53.78) 0.73 0.88 0.85 0.77 0.81 0.85(0.77;0.93) <.001***
CD41+mito-
CD62P+
EVs
>=0.15 (0.09-0.22) 0.73 0.72 0.71 0.73 0.72 0.70(0.59;0.81) <.001***
AUC = area under the curve; BCI = Bootstrap 95% confidence interval; EVs = extracellular vesicles; NPV = negative predictive value; PPV = positive predictive value. ***p < 0.001.

117
Table 2. EV subtype associations with soluble CD40L (n=92) and soluble
CD62P (n=96) in platelet concentrates
Total
CD41+ EVs
CD41+mito
+ EVs
CD41+mito
- EVs
CD41+mito
+ PS+ EVs
CD41+mito+
CD62P+ EVs
CD41+mito
- PS+ EVs
CD41+mito-
CD62P+ EVs
sCD40L rs 0.45 0.40 0.35 0.44 0.38 0.34 0.24
p <.0001 <.0001 0.0006 <.0001 0.0002 0.0009 0.0189
sCD62P rs 0.65 0.74 0.51 0.72 0.75 0.49 0.33
p <.0001 <.0001 <.0001 <.0001 <.0001 <.0001 0.0011
Values are presented as Spearman correlation coefficient (rs) and p-value (p). sCD40L: soluble CD40L. sCD62P: soluble CD62P.
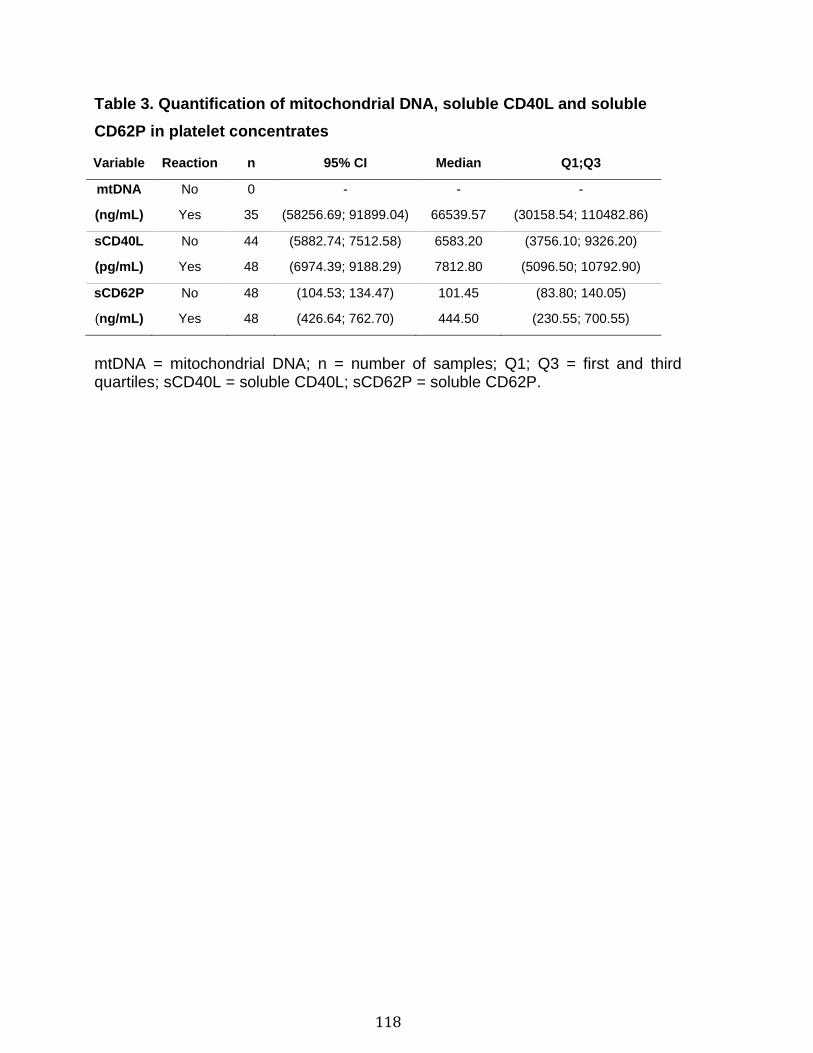
118
Table 3. Quantification of mitochondrial DNA, soluble CD40L and soluble
CD62P in platelet concentrates
Variable Reaction n 95% CI Median Q1;Q3
mtDNA No 0 - - -
(ng/mL) Yes 35 (58256.69; 91899.04) 66539.57 (30158.54; 110482.86)
sCD40L No 44 (5882.74; 7512.58) 6583.20 (3756.10; 9326.20)
(pg/mL) Yes 48 (6974.39; 9188.29) 7812.80 (5096.50; 10792.90)
sCD62P No 48 (104.53; 134.47) 101.45 (83.80; 140.05)
(ng/mL) Yes 48 (426.64; 762.70) 444.50 (230.55; 700.55)
mtDNA = mitochondrial DNA; n = number of samples; Q1; Q3 = first and third quartiles; sCD40L = soluble CD40L; sCD62P = soluble CD62P.
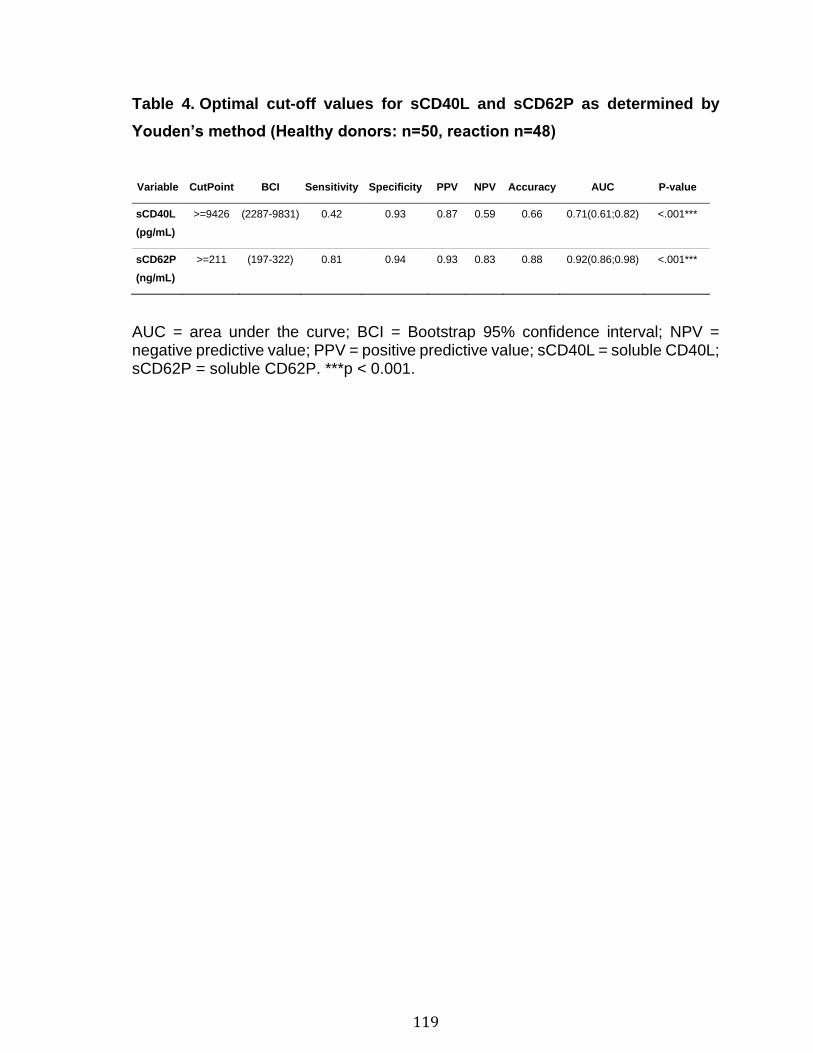
119
Table 4. Optimal cut-off values for sCD40L and sCD62P as determined by
Youden’s method (Healthy donors: n=50, reaction n=48)
Variable CutPoint BCI Sensitivity Specificity PPV NPV Accuracy AUC P-value
sCD40L
(pg/mL)
>=9426 (2287-9831) 0.42 0.93 0.87 0.59 0.66 0.71(0.61;0.82) <.001***
sCD62P
(ng/mL)
>=211 (197-322) 0.81 0.94 0.93 0.83 0.88 0.92(0.86;0.98) <.001***
AUC = area under the curve; BCI = Bootstrap 95% confidence interval; NPV = negative predictive value; PPV = positive predictive value; sCD40L = soluble CD40L; sCD62P = soluble CD62P. ***p < 0.001.

120
3.10 Supplementary methods
Detection of platelet EVs and mitochondria
Samples were labeled with 100nM MitoTracker Deep Red (ThermoFisher Scientific),
which accumulates in activated mitochondria, V450 anti-human CD41a (BD
Biosciences) which targets glycoprotein IIb on platelets and platelet-derived EVs and
PE anti-CD62P (Beckman Coulter, CA, USA), which targets the activated platelets
and platelet-derived EVs or FITC-annexinV (BD Biosciences) which targets
phosphatidylserine (PS). After a 30 minutes incubation period at 37ºC, samples were
diluted with PBS pre-filtered through a 0.22µm pore size membrane (Fisher
Scientific, ON, Canada) or 1x AnnexinV-binding buffer. EVs were then analyzed by
Flow cytometry with a BD Canto II Special Order Research Product (BD
Biosciences), as described previously.[378, 448] Briefly, a forward scatter (FSC)
coupled to photomultiplier tube (PMT) was mounted on the flow cytometer and
parameters were set to allow optimal detection of silica microspheres from 100 to
1000nm. Flow cytometer performance tracking was performed daily before all
analyses. For the FSC-PMT, the assigned voltage was 300V, 450 for FITC, 435 for
PE, 500 for APC and 500 for V450. For SSC, the assigned voltage was 400V and
the threshold was 200. EV acquisition was performed at low speed (~10µl/min) and
for quantification, a known quantity of fluorescents beads (3µm diameter:
Polysciences inc., Warrington, PA, USA) was added to each tube and a constant
number of beads was acquired for each sample. Silica particles of known dimension
(100nm, 500nm and 1µm in diameter (Kisker Biotech GmbH &Co., Steinfurt,
Germany) were used to standardize FSC-PMT parameters, and EVs were defined
as being smaller than 1µm silica particles and platelets. The results were analyzed
using the software FlowJo V10.1r5 in order to identify different EV subpopulations
on the chosen parameters; namely CD41a, MitoTracker, CD62P, PS, relative size
(FSC-PMT) and granularity (SSC-H).
To assess the detection specificity, we made several controls: For Triton treatment,
samples were incubated 30 minutes at room temperature (RT) with 0.05% Triton X-
100 before labeling. For EDTA, samples were treated for 30 minutes at RT with

121
50μM EDTA (using PBS instead of Annexin V buffer as diluent). For
ultracentrifugation treatment, samples were centrifuge at 100 000g for 1 hour at 20°C
to pellet EVs, and supernatants were labeled. When washed platelets were
stimulated to produce EVs, 0.5 U/mL of thrombin (Sigma) was added for 1 hours at
37°C after addition of 5 mM of calcium.
Quantification of mtDNA
For each sample that resulted in adverse reactions, nucleic acids were extracted
from 50 μl of PC’s supernatant with the QIAamp DNA Micro extraction kit (QIAGEN,
Toronto, ON, Canada) and mtDNA was quantified by real-time quantitative PCR as
described previously.[382, 449]
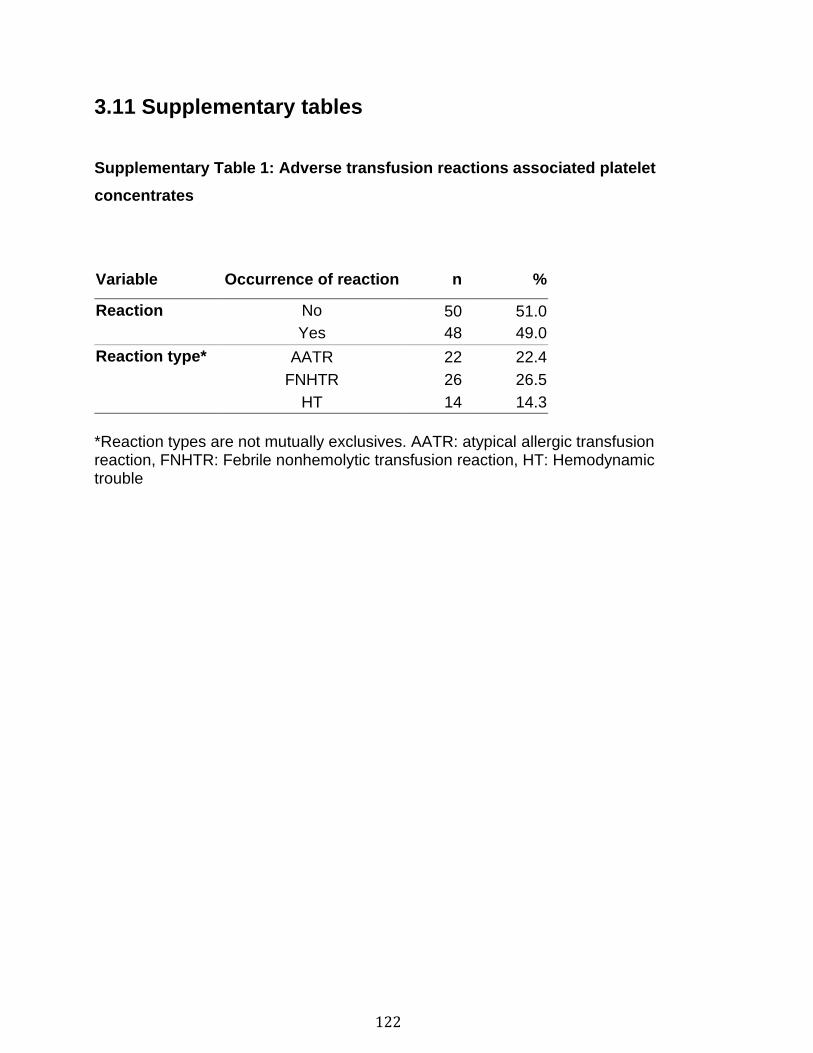
122
3.11 Supplementary tables
Supplementary Table 1: Adverse transfusion reactions associated platelet
concentrates
Variable Occurrence of reaction n %
Reaction No 50 51.0
Yes 48 49.0
Reaction type* AATR 22 22.4
FNHTR 26 26.5
HT 14 14.3
*Reaction types are not mutually exclusives. AATR: atypical allergic transfusion reaction, FNHTR: Febrile nonhemolytic transfusion reaction, HT: Hemodynamic trouble

123
Supplementary Table 2: Qualitative information of the transfusion product
Variable Level n %
Transfusion alone No 54 55.1 Yes 44 44.9
Transfusion with RBCs No 94 95.9 Yes 4 4.1
Transfusion with FFP No 98 100
Sex of the donor Female 36 36.7
Male 62 63.3
RBCs: red blood cells, FFP: fresh frozen plasma
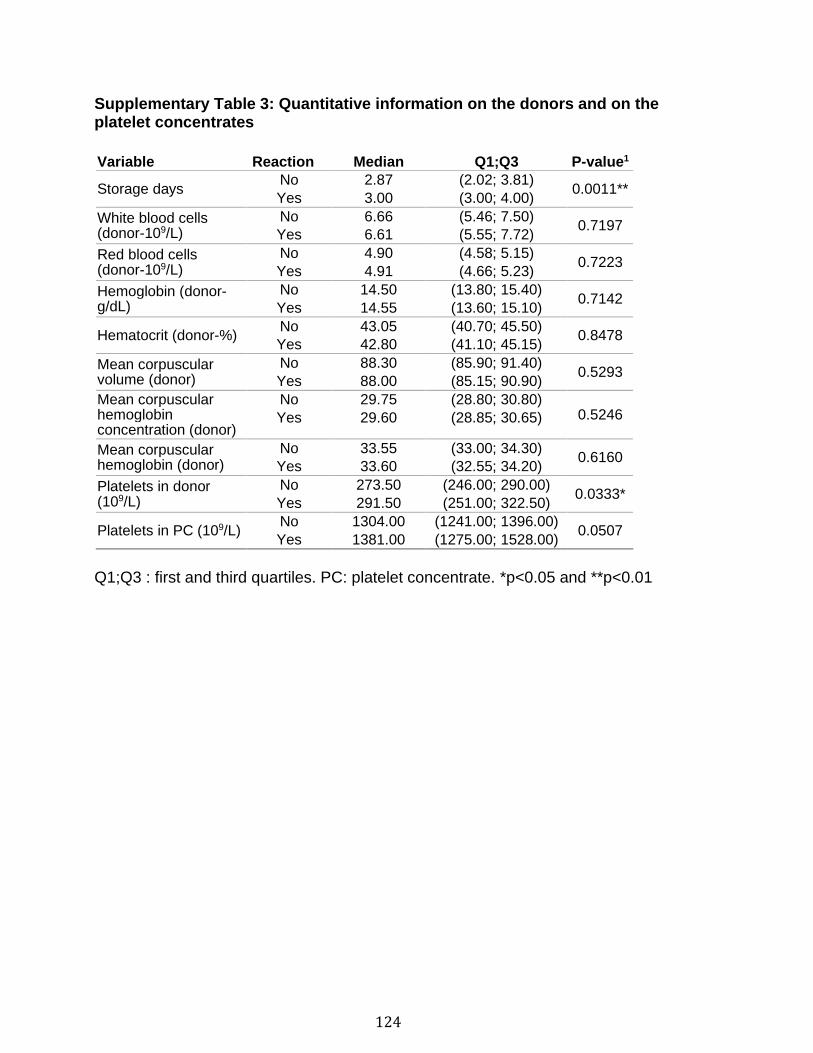
124
Supplementary Table 3: Quantitative information on the donors and on the platelet concentrates
Variable Reaction Median Q1;Q3 P-value1
Storage days No 2.87 (2.02; 3.81)
0.0011** Yes 3.00 (3.00; 4.00)
White blood cells (donor-109/L)
No 6.66 (5.46; 7.50) 0.7197
Yes 6.61 (5.55; 7.72)
Red blood cells (donor-109/L)
No 4.90 (4.58; 5.15) 0.7223
Yes 4.91 (4.66; 5.23)
Hemoglobin (donor-g/dL)
No 14.50 (13.80; 15.40) 0.7142
Yes 14.55 (13.60; 15.10)
Hematocrit (donor-%) No 43.05 (40.70; 45.50)
0.8478 Yes 42.80 (41.10; 45.15)
Mean corpuscular volume (donor)
No 88.30 (85.90; 91.40) 0.5293
Yes 88.00 (85.15; 90.90)
Mean corpuscular hemoglobin concentration (donor)
No 29.75 (28.80; 30.80) 0.5246 Yes 29.60 (28.85; 30.65)
Mean corpuscular hemoglobin (donor)
No 33.55 (33.00; 34.30) 0.6160
Yes 33.60 (32.55; 34.20)
Platelets in donor (109/L)
No 273.50 (246.00; 290.00) 0.0333*
Yes 291.50 (251.00; 322.50)
Platelets in PC (109/L) No 1304.00 (1241.00; 1396.00)
0.0507 Yes 1381.00 (1275.00; 1528.00)
Q1;Q3 : first and third quartiles. PC: platelet concentrate. *p<0.05 and **p<0.01
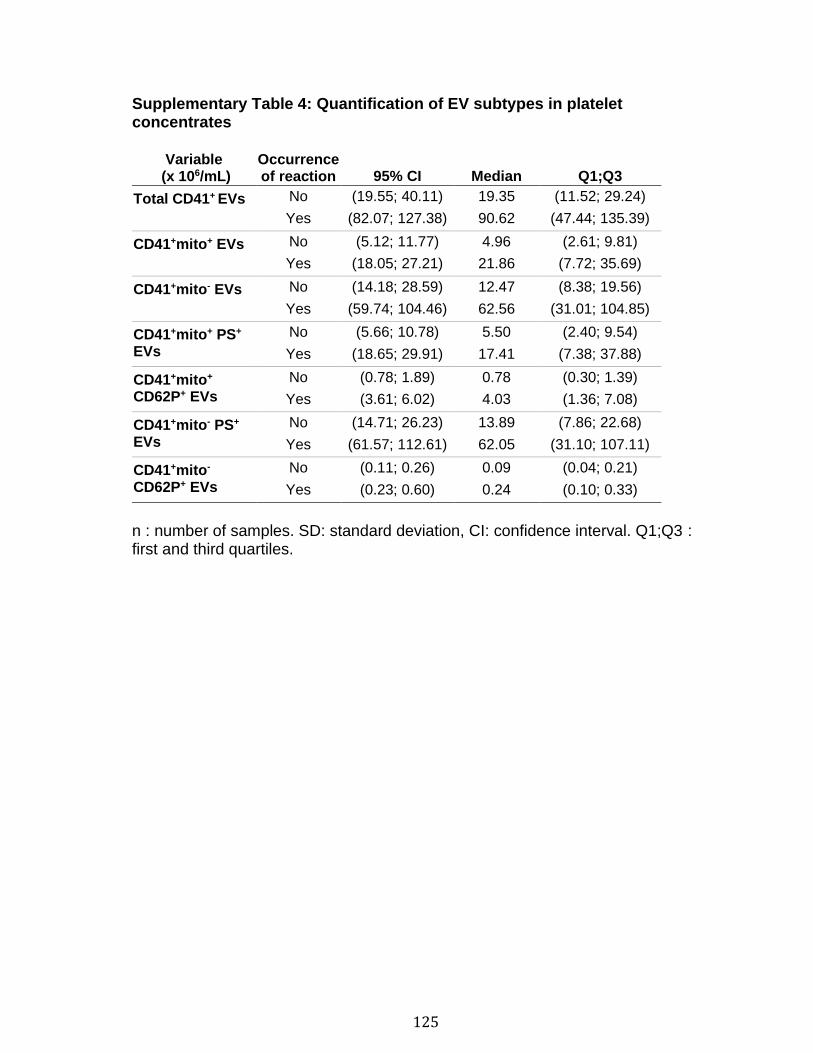
125
Supplementary Table 4: Quantification of EV subtypes in platelet concentrates
Variable (x 106/mL)
Occurrence of reaction 95% CI Median Q1;Q3
Total CD41+ EVs No (19.55; 40.11) 19.35 (11.52; 29.24)
Yes (82.07; 127.38) 90.62 (47.44; 135.39)
CD41+mito+ EVs No (5.12; 11.77) 4.96 (2.61; 9.81)
Yes (18.05; 27.21) 21.86 (7.72; 35.69)
CD41+mito- EVs No (14.18; 28.59) 12.47 (8.38; 19.56)
Yes (59.74; 104.46) 62.56 (31.01; 104.85)
CD41+mito+ PS+ EVs
No (5.66; 10.78) 5.50 (2.40; 9.54)
Yes (18.65; 29.91) 17.41 (7.38; 37.88)
CD41+mito+ CD62P+ EVs
No (0.78; 1.89) 0.78 (0.30; 1.39)
Yes (3.61; 6.02) 4.03 (1.36; 7.08)
CD41+mito- PS+ EVs
No (14.71; 26.23) 13.89 (7.86; 22.68)
Yes (61.57; 112.61) 62.05 (31.10; 107.11)
CD41+mito- CD62P+ EVs
No (0.11; 0.26) 0.09 (0.04; 0.21)
Yes (0.23; 0.60) 0.24 (0.10; 0.33)
n : number of samples. SD: standard deviation, CI: confidence interval. Q1;Q3 : first and third quartiles.

126
Supplementary Table 5: Cubic effect of EVs on adverse reaction
Variable Univariable model Multivariable model
Total CD41+ EVs 29.31 (<0.0001) 23.15 (<0.0001)
CD41+mito+ EVs 15.90 (0.0012) 18.28 (0.0004)
CD41+mito- EVs 27.31 (<0.0001) 24.62 (<0.0001)
Results are shown as Chi-Square statistic (p-value) for the cubic effect of each variable. Multivariable model also includes sex of the donor, storage duration and concentration of platelets.

127
Supplementary Table 6: Predictive values of EV subtypes adjusted for the different adverse reactions
Variable
AUC (95% CI)
AATR FNHTR HT
Total CD41+ EVs 0.79(0.70;0.88)*** 0.74(0.65;0.84) *** 0.85(0.77;0.93)***
CD41+mito+ EVs 0.61(0.48;0.74) 0.75(0.63;0.86)*** 0.7(0.55;0.86)*
CD41+mito- EVs 0.78(0.69;0.87)*** 0.68(0.56;0.80)** 0.84(0.75;0.93)***
CD41+mito+ PS+ EVs 0.62(0.49;0.76) 0.75(0.64;0.86)*** 0.71(0.56;0.86)**
CD41+mito+ CD62P+ EVs 0.64(0.51;0.77)* 0.74(0.62;0.86)*** 0.68(0.54;0.83)*
CD41+mito- PS+ EVs 0.79(0.70;0.89)*** 0.68(0.56;0.80)** 0.81(0.72;0.90)***
CD41+mito- CD62P+ EVs 0.66(0.55;0.78)** 0.41(0.28;0.54) 0.73(0.61;0.84)***
AATR: atypical allergic transfusion reaction, FNHTR: Febrile nonhemolytic transfusion reactions, HT: Hemodynamic trouble, AUC: area under the curve. * p < 0.05, ** p < 0.01, *** p <0.001, where p comes from the test H0: AUC = 50% vs H1: AUC ≠ 50%

128
Supplementary Table 7: EV subtype associations with mitochondrial DNA in platelet concentrates (n= 35)
Total
CD41+ EVs
CD41+mito+
EVs
CD41+mito-
EVs
CD41+mito+
PS+ EVs
CD41+mito+
CD62P+ EVs
CD41+mito-
PS+ EVs
CD41+mito-
CD62P+ EVs
rs 0.07 0.73 -0.11 0.59 0.74 -0.07 -0.14
p 0.7012 <.0001 0.5112 0.0002 <.0001 0.6706 0.4214
Values are presented as Spearman correlation coefficient (rs) and p-value (p).

129
3.12 Supplementary references
1 Rousseau, M. et al. Detection and quantification of microparticles from
different cellular lineages using flow cytometry. Evaluation of the impact of
secreted phospholipase A2 on microparticle assessment. PLoS One 10,
e0116812, doi:10.1371/journal.pone.0116812 (2015).
2 Marcoux, G. et al. Revealing the diversity of extracellular vesicles using high-
dimensional flow cytometry analyses. Sci Rep 6, 35928,
doi:10.1038/srep35928 (2016).
3 Marcoux, G. et al. Microparticle and mitochondrial release during extended
storage of different types of platelet concentrates. Platelets 28, 272-280,
doi:10.1080/09537104.2016.1218455 (2017).
4 Boudreau, L. H. et al. Platelets release mitochondria serving as substrate for
bactericidal group IIA-secreted phospholipase A2 to promote inflammation.
Blood 124, 2173-2183, doi:10.1182/blood-2014-05-573543 (2014).

130
Chapitre 3: Platelet-derived extracellular vesicles
contain an active proteasome involved in protein
processing for antigen presentation via class I
major histocompatibility molecules
4.1 Résumé
Les plaquettes sont maintenant reconnues pour leur contribution à l'immunité. Elles
contiennent un protéasome actif et présentent des antigènes par l'intermédiaire du
CMH I. Une fois activées, elles libèrent des vésicules extracellulaires (PEV) qui sont
hétérogènes, ce qui suggère qu'elles jouent diverses fonctions. Il n’a jamais été
examiné si le protéasome est emporté dans les EV de plaquettes lors de leur
formation. Nous avons émis l'hypothèse qu'une machinerie fonctionnelle
d’apprêtement et de présentation de l'antigène se retrouve dans les EV de
plaquettes lors de leur activation. En utilisant une combinaison d'analyses
moléculaires et fonctionnelles, nous avons montré la présence d'un protéasome 20S
actif dans diverses conditions ou les plaquettes sont activées, ainsi que du CMH I et
des molécules de coactivation des lymphocytes. Démontré par l'activation et la
prolifération des lymphocytes T CD8+ spécifiques de l'ovalbumine (OVA), les PEV
incubées avec l’OVA peuvent apprêter et présenter l’antigène. Ces résultats
suggèrent que les PEV contribuent à l'immunité adaptative.

131
Platelet-derived extracellular vesicles contain an active proteasome involved
in protein processing for antigen presentation via class I major
histocompatibility molecules
Genevieve Marcoux1, Audrée Laroche1, Stephan Hasse1, Marie Tamagne2,3,4, Anne
Zufferey1, Tania Lévesque1, Isabelle Allaeys1, Johan Rebetz5, Annie Karakeussian-
Rimbaud6,7, Julie Turgeon6,7, Sylvain G. Bourgoin1, Hind Hamzeh-Cognasse8,
Fabrice Cognasse8,9, Rick Kapur10, John W. Semple5, Marie-Josée Hébert6,7, France
Pirenne2,3,4, Herman S. Overkleeft11, Bogdan I. Florea11, Mélanie Dieude6,7,12, Benoit
Vingert2,3,4, Eric Boilard1,7.
1 Centre de recherche du CHU de Québec- Université Laval, Département de
microbiologie-infectiologie et d’immunologie, Centre ARThrite de l’Université Laval,
Québec, QC, Canada.
2 Univ Paris Est Creteil, INSERM, IMRB, F-94010 Creteil, France
3 Etablissement Français du Sang, Ivry sur Seine, F-94200, France
4 Laboratory of Excellence GR-Ex, Paris, France
5 Division of Hematology and Transfusion Medicine, Lund University, Lund, Sweden,
Departments of Pharmacology and Medicine, University of Toronto, Toronto,
Canada
6 Research Centre, Centre hospitalier de l'Université de Montréal (CRCHUM),
Montréal, Québec, Canada.
7 Canadian Donation and Transplantation Research Program, Edmonton, Alberta,
Canada
8 Université de Lyon, Université Jean Monnet, INSERM U1059, Saint-Etienne,
France
9 Établissement Français du Sang Auvergne-Rhône-Alpes, Saint-Etienne, France.

132
10 Sanquin Research, Department of Experimental Immunohematology, Amsterdam
and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam,
Amsterdam, the Netherlands.
11 Gorlaeus Laboratories, Leiden Institute of Chemistry and Netherlands Proteomics
Centre, Leiden, The Netherlands.
12 Département Microbiologie, Infectiologie et Immunologie, Faculté de Médecine,
Université de Montréal, Montréal, Québec, Canada.
Correspondence should be sent to:
Eric Boilard, PhD
Centre de Recherche du Centre Hospitalier Universitaire de Québec
Faculté de Médecine de l’Université Laval
2705 Laurier Blvd, room T1-49, Québec, QC, Canada G1V 4G2
Phone: +1 418-525-4444, extension 46175, Fax: +1 418-654-2765
Or
Benoît Vingert, PhD
Établissement Français du Sang
Institut Mondor de Recherche Biomédicale-INSERM U955,
5 rue Gustave Eiffel, 94017 Créteil, France
Phone: +33 1 56 72 17 40, Fax: +33 1 56 72 17 47

133
4.2 Abstract
In addition to their hemostatic role, platelets play a significant role in immunity. Once
activated, platelets release extracellular vesicles (EVs) formed by budding of their
cytoplasmic membranes. Because of their heterogeneity, platelet EVs (PEVs) are
thought to perform diverse functions. It is unknown, however, whether the
proteasome is transferred from platelets to PEVs or whether proteasomal protein
processing function is retained. We hypothesized that functional protein processing
and antigen presentation machinery is bearing by PEVs derived from activated
platelets. Using a combination of molecular and functional assays, we showed the
presence of an active 20S proteasome in PEVs. Moreover, MHC-I and lymphocyte
costimulatory molecules (CD40L and OX40L) were found in proteasome-containing
PEVs. Proteasome-containing PEVs were identified in healthy donor blood, in
platelet concentrates used for transfusion, in inflammatory conditions and in the
murine lymphatic system. Injection of PEVs into mice revealed that they could reach
lymphoid organs. The PEV proteasome could process exogenous ovalbumin (OVA)
and efficiently load its antigenic peptide onto MHC-I molecules which promoted
OVA-specific CD8+ T lymphocyte proliferation. These results suggest that PEVs
contribute to adaptive immunity through cross-presentation of antigens and have
privileged access to immune cells through the lymphatic system, a tissue location
that is inaccessible to platelets.

134
4.3 Introduction
After erythrocytes, platelets are the second most abundant lineage in blood and are
best known for their role in hemostasis1. Platelets are small fragments produced by
the large multinucleated megakaryocytes in the bone marrow. Although
approximately 100 billion platelets are produced in humans each day, most are
eliminated through the mononuclear phagocytic system in the spleen and liver
without having encountered injured endothelium.
Platelets bear receptors that permit the recruitment of immune cells and carry an
extensive set of immune and inflammatory molecules (e.g. cytokines/chemokines,
lipid mediators, hormones) that are stored in their granules or cytoplasm, or
synthesized and upregulated by mRNA translation following platelet activation.
Thus, while platelets may mount an innate immune response to injury, which is
critical to combat pathogen invasion, organ and tissue damages may also favor
platelet activation and inflammation in chronic inflammatory diseases2-8.
In contrast to innate immunity, adaptive immunity encompasses the mechanisms
underlying the specific response to pathogens. This involves lymphocytes, antigen-
presenting cells (APCs) and the production of effector cells and antibodies9-31. The
differentiation and activation of APCs are enhanced by platelet-derived molecules,
such as platelet factor 4 (PF4) and soluble CD40L, while platelet adhesion receptors
can also promote the recruitment of lymphocytes11,12,15-17.
Albeit anucleate, the platelet cytoplasm includes numerous molecules comprising
the proteasome, which are transferred from megakaryocytes to their progeny. The
proteasome is a high molecular weight cylindrical protein complex through which
unwanted or damaged proteins are degraded32,33. The central complex part, called
the 20S proteasome, is made up of twenty-eight distinct subunits34, comprising the
three catalytic subunits necessary for the degradation of proteins into peptides of
three to fifteen amino acids in length34,35. Proteasome activity in megakaryocytes is
required for platelet production36,37 and in platelets, the proteasome regulates

135
platelet lifespan38, activation39-41 and the release of PEVs42,43. The platelet
proteasome can hydrolyze proteins into smaller peptides34,44,45, thereby enabling
peptide loading onto the platelet major histocompatibility complex (MHC) class I
molecules (MHC-1)46-48. Components of the peptide loading complex are also
expressed in platelets and are found in close proximity with MHC-I during platelet
activation49,50. As platelets can efficiently form an immunological synapse with T-
lymphocytes to activate lymphocyte proliferation49,51,52, they are known to fulfill roles
in cross-presentation of antigens in adaptive immunity. In a similar manner,
megakaryocytes cross-present antigens to CD8 T-lymphocytes, thereby suggesting
that they may also play a dual role in innate and adaptive immunity48,53-55.
Extracellular vesicles, produced in abundance by platelets, are small (up to 1 µm in
diameter) membrane-bound vesicles released from the plasma membrane or
endosomal compartments of activated cells. Platelet EVs are heterogeneous in
terms of surface molecules and content (e.g. nucleic acids, lipids, transcription
factors, enzymes, mitochondria) and as such may play diverse functions beyond
hemostasis56-58. For instance, PEVs convey mitochondrial components that are
associated with inflammation and adverse transfusion reactions (ATRs)59-61. Despite
the fact that platelets are restricted to the blood circulation, PEVs can cross tissue
barriers and enter synovial fluid62,63, lymph64,65 and bone marrow66 where they can
deliver platelet-derived molecules and modulate target cells57. For instance, PEVs
promote the formation of germinal centers and the production of IgG by B-cells67,68.
They also interact with and modulate regulatory T cell differentiation and activity69,70.
Thus, PEVs may be able to transport platelet-derived molecules relevant to adaptive
immunity into lymphoid organs. However, it is unknown whether the proteasome and
the molecules necessary for antigen presentation are also transferred during the
budding of PEVs. In this study, we evaluated whether a functional protein processing
and antigen presentation machinery is transferred to PEVs by activated platelets.

136
4.4 Methods
More details are presented in supplemental methods
Labeling of murine platelets, DCs and PEVs Platelets were isolated from
C57BL/6J mice by retro-orbital puncture in 200 µL ACD (acid-citrate-dextrose),
350 µL Tyrode’s buffer pH 6.5. Whole blood was centrifuged at 600xg for 3 min and
then at 400xg for 2 min to remove red blood cells. Supernatant was spun at 1,300xg
for 5 min and the platelet-containing pellet was gently resuspended in 600 µL
Tyrode’s buffer pH 7.4. Platelets were either left nonactivated or activated with
thrombin (0.1 U/mL) after addition of 5 mM of calcium for 90 min at RT. Platelet EVs
were obtained by two rounds of centrifugation of stimulated platelets at 1,300xg for
5 min at RT. Either activated platelets, EVs or DC were pulsed with 100 µg/mL OVA
protein (Sigma-Aldrich), 200 µg/mL of OVA peptide (SIINFEKL [Invivogen, San
Diego, CA]) or left unpulsed for 4 h at RT. These conditions were either left unlabeled
for lymphoproliferation or intracellular staining experiments or labelled for hs-FCM
experiments.
Five µL of PEVs or platelet suspensions were labeled with 250 nM LWA300
proteasome probe in a total volume of 100 μl for 90 min at 30°C. Samples were then
incubated with the following antibodies for 30 min at RT prior to dilution in Annexin
V binding buffer and analysis by hs-FCM: BUV395 anti-CD41, BV650 anti-CD62p,
BV650 anti-CD62p, BV711 Annexin V, BV421 anti-OX40L, BUV737 anti-CD154 (all
BD Biosciences), PeCy7 anti-CD40, PeCy7 anti-MHC-I (AF6-88.5) and PE anti-
MHC-I bound to OVA peptide (25D1.16) (all from Biolegend).
In vitro antigen-specific CD8+ T-cell activation In vitro antigen-specific CD8+ T-
cell activation was performed using purified OVA-specific OT-1 CD8+ T cells in
combination with C57BL/6J mice platelets, EVs and DCs. Purified splenocytes were
obtained as described above.

137
For proliferation, splenocytes were stained with 5-(and 6)-carboxyfluorescein
diacetate, succinimidyl ester (CFSE, Thermo Fisher Scientific) and 3 × 105 cells in
100 µL were then stimulated for five days at 37°C in 96-well plates in the presence
of CD41+ PEVs (or platelets or DCs) pulsed with either OVA protein, OVA peptide
(SIINFEKL) or left unpulsed (ratio of 1 CD41+ PEV for 1 OT-1 cell). Cultured OT-1
cells were washed twice by centrifugation at 1,400xg for 7 min in PBS and labeled
with BUV395 mouse anti-CD3 (BD Biosciences) and PerCP-Cy5.5 mouse anti-CD8
(Biolegend) for 30 min at 4°C. Cells were washed and fixed with 150 µL PFA 1% for
analysis by hs-FCM.
For intracellular staining, 3 × 105 splenocytes were cocultured (1:1 ratio) in the
presence of CD41+ EVs (or platelets or DC) pulsed with either OVA protein, OVA
peptide (SIINFEKL) or left unpulsed. After 1-h incubation at 37°C, brefeldin A and
monensin (0.25× final concentration, Thermo Fisher Scientific) were added in the
culture media to determine the production of cytokines and the coculture was
pursued overnight. Cells were washed twice and membrane proteins were labeled
with PE anti-CD40 (Thermo Fisher Scientific), BUV395 anti-CD3, PerCP-Cy5.5 anti-
CD8, BV421 anti-CD134 (Biolegend) and aqua live/dead (Invitrogen) for 30 min at
4°C. Cells were fixed and permeabilized with a commercial kit (Thermo Fisher
Scientific) for intramembranous staining and then stained with AF647 anti-IL2 (BD
Biosciences) and AF700 IFN-γ (BD Biosciences). Cells were washed and fixed with
150 µL PFA 1% for analysis by flow cytometry.

138
4.5 Results
4.5.1 PEVs contain functional proteasome
Immunoblotting was used to confirm that PEVs released from thrombin-activated
human platelets contained the proteasome 20S α subunit (Figure 1A). Using
platelets as a positive control, we assessed proteasome function in PEVs.
Proteasome-associated caspase-like activity was detectable in both platelets and
the PEV fraction, but was undetectable in the supernatant (Figure 1B), indicating
that the catalytically active proteasome was transferred to PEVs upon their release.
Visualization of immunogold-labeled proteasome 20S α subunit by transmission
electron microscopy confirmed the presence of proteasomes in PEVs (Figure 1C).
LWA300 is a conjugate between epoxomicin, a natural inhibitor of the 20S
proteasome, and BODIPY FL fluorophore and generates an activity-based,
fluorescent, and plasma membrane-permeable inhibitor that can identify the
proteasome in cells71,71. Using LWA300, we designed assays to detect and quantify
active proteasome-containing PEVs71,72. Confocal microscopic visualization of
platelets as positive controls, and PEVs from thrombin-activated platelets labeled
with LWA300 revealed that both platelets and a subpopulation of PEVs contained
active proteasome (Figure 1D). Moreover, hs-FCM confirmed PEV heterogeneity
following platelet activation by thrombin (Figure 1E). Approximately 16.66.5% of
the larger (i.e. 500–900 nm) PEV microvesicles73, contained proteasome whereas
smaller PEV (i.e. less than 500 nm) exosomes73 had no detectable proteasome
(Figure 1E and Supplementary figure 1). The detection specificity of proteasome-
containing PEVs by hs-FCM was confirmed using a combination of controls. We
confirmed efficient competition of the LWA300 probe by unlabeled epoxomicin, and
we determined the particulate nature and membrane moiety of proteasome-
containing PEVs, as they were respectively pelleted by ultracentrifugation and
sensitive to detergent treatment (Figure 1F-G).

139
Hs-FCM was further used to characterize proteasome-containing PEVs in terms of
surface markers and mitochondrial content. Approximately half of the proteasome-
containing PEVs expressed exposed phosphatidylserine while the vast majority
expressed surface P-selectin (Supplementary figure 2A). Furthermore, 68.37.8%
of the proteasome-containing PEVs also contained mitochondria (Supplementary
figure 2A). Investigation of the mechanisms underlying release of active
proteasome positive PEVs revealed that the total number of PEVs (with and without
proteasomes) and proteasome-containing PEVs were significantly reduced in the
presence of actin inhibitors (cytochalasin B, D, E and latrunculin A) but not by the
tubulin polymerization inhibitor nocodazole (Supplementary figure 2B).
Proteasome release in PEVs was not unique to thrombin stimulation as the platelet
agonists ADP, cross-linked collagen related peptide (CRP-XL) and heat-aggregated
IgG (HA-IgG) also triggered release of proteasome-containing PEVs
(Supplementary figure 2C).
4.5.2 Identification of proteasome-containing PEVs under
physiological and pathological conditions
The presence of proteasome-containing PEVs was assessed ex vivo and in vivo
under conditions conducive to platelet activation and PEV release. A mean of 1.82
× 106 (range:1.13 × 105 to 8.11 × 106, n = 6) proteasome-containing PEVs/mL were
detected by hs-FCM in the blood of healthy individuals, pointing to their constitutive
presence under physiological conditions.
PEVs were quantified in platelet concentrates known to have caused ATRs and
compared with control concentrates that did not induce ATRs. Given the reported
increase in mitochondria-containing PEVs in ATRs59,60, we also determined the
levels of mitochondria-containing PEVs. Documented ATRs included febrile
nonhemolytic reactions, skin manifestations (e.g. itching or skin rash) and
cardiovascular events such as hypotension or tachycardia. High levels of
proteasome-containing PEVs were found in all tested platelet concentrates
(Figure 2A) but the concentrations of proteasome-containing PEVs (with or without

140
mitochondria) were not significantly elevated in platelet concentrates that induced
ATR (Figure 2A). In contrast, compared with control platelet concentrates not
associated with ATR, the concentrations of mitochondria-containing PEVs were
increased in ATR-associated concentrates, consistent with prior findings59,60. These
data reveal that proteasome-containing PEVs are an expected component in platelet
concentrates and suggest that the presence of extracellular mitochondria in PEVs,
rather than the proteasome, may indicate increased risks of ATR.
Transfusion-related acute lung injury (TRALI) is a potentially lethal adverse reaction
that can result from transfusion of platelet concentrates74. Thus, we quantified
proteasome-containing PEVs in murine bronchoalveolar lavages in an inducible
TRALI model75,76. Proteasome-containing PEVs were detected in bronchoalveolar
lavages from both TRALI and control mice (Figure 2B), however, no significant
difference was observed between the two groups (Figure 2B). This suggests that
proteasome-containing PEVs are not increased during lung inflammation in this
model.
Our in vitro investigations pointed to the high potency of immune complexes (HA-
IgG) in generating proteasome-containing PEVs (Supplementary figure 2C).
Although mice lack FcγRIIA, this is the only Fcγ receptor expressed by human
platelets that is capable of responding to immune complexes77. Recent findings
indicate that circulating immune complexes stimulate the release of mitochondria-
containing PEVs in mice expressing the FcγRIIA transgene78,79. To determine
whether the proteasome is also released under these conditions, we injected
immune complexes into FcγRIIA-expressing mice and quantified proteasome-
containing PEVs79. Compared with diluent injected control mice, there were
significantly elevated levels of proteasome-containing PEVs in plasma of mice with
immune-complexes challenge (Figure 2C). These findings confirmed that
proteasome-containing PEVs are present under various physiological and
pathological conditions.

141
4.5.3 Protein processing by proteasome-containing PEVs
In order to study proteasome function in PEVs, we investigated its ability to process
proteins into smaller peptides by assessing their successful loading into the antigen-
binding groove of MHC-I molecules. We confirmed the expression of MHC-I on
resting and thrombin-activated murine platelets and verified whether MHC-I is
maintained on PEVs. We found that washed resting platelets did not express MHC-
I on their surface (Figure 3A), however, thrombin activation led to a significant
increase in surface MHC-I expression (Figure 3A), consistent with the reported
presence of this molecule in α-granules and its release upon activation49,50,80,81. A
small proportion (1.9 0.9%) of the spontaneously released PEVs expressed MHC-
I, but this proportion significantly increased upon platelet activation with thrombin
(means of 7.7 2.1%).
To determine whether PEV MHC-I can indeed load small peptides, we pulsed PEVs
with the ovalbumin (OVA) peptide SIINFEKL and monitored its association with
MHC-I molecules using the 25D1.16 monoclonal antibody, which specifically
recognizes MHC-I/SIINFEKL complexes82. Similarly with platelets, PEVs loaded the
SIINFEKL peptide onto their MHC-I molecules (Figure 3B). Native OVA was also
efficiently processed by platelets and the SIINFEKL peptide was loaded in MHC-I
(Figure 3C), consistent with prior work49. Of interest, incubation of native OVA with
PEVs resulted in proteolysis of the former and retrieval of the SIINFEKL peptide from
MHC-I molecules expressed by the PEVs. Taken together, the data show that PEVs
can process native proteins into smaller peptides thereby enabling antigen
presentation through MHC-I.
4.5.4 Proteasome-containing PEVs can reach lymphoid organs and
circulate through the lymphatic system
Intravenously injected PEVs have a limited circulation time in human blood, ranging
from 10 min to hours depending on studies83,84. It is unclear, however, whether they
can reach lymphoid organs. Hence, fluorescently labeled PEVs were intravenously
injected into mice and their presence in spleen and lymph nodes was monitored

142
60 min post injection. Platelet EVs were no longer detectable in blood at this time
(Figure 4A) but were observed in the tested lymphoid organs (Figure 4B). Platelet
EVs can circulate through the lymphatic system, which is the main circulatory system
permitting dissemination of antigens to lymphoid organs. Moreover, the levels of
PEVs in lymph are increased in mouse models of atherosclerosis and autoimmune
inflammatory arthritis57,64,65. Using the lymph from mice, we evaluated whether PEVs
were associated with proteasome and MHC-I molecules. We found that a fraction of
the PEVs in lymph expressed MHC-I (11.2 2.2%) and contained an active
proteasome (12.0 3.9%). Remarkably, a small, but detectable proportion (1.6
0.7%) of the lymph PEVs contained both proteasome and MHC-I molecules
(Figure 4C). This finding was significant given the substantial number of PEVs in
lymph (mean of 2.5 × 107/mL in mice64).
4.5.5 Proteasome-containing PEVs express lymphocyte co-
stimulatory molecules
Efficient stimulation of adaptive immunity requires both recognition of the antigen-
MHC-I complexes by the T cell receptor (TCR) and the activity of co-stimulatory
molecules. Therefore, we evaluated whether platelets or proteasome-containing
PEVs loaded with SIINFEKL expressed co-stimulatory molecules in addition to other
known PEV markers. Compared with PEVs that had undetectable SIINFEKL
loading, both platelets and PEVs loaded with SIINFEKL (25D1.16-positive)
expressed higher levels of proteasome (Figure 5). Moreover, in contrast to
thrombin-activated platelets, where phosphatidylserine expression is increased
when loaded with SIINFEKL, both PEVs bearing SIINFEKL and those negative for
SIINFEKL expressed similar levels of phosphatidylserine (Figure 5). Furthermore,
both platelets and SIINFEKL-bearing PEVs expressed higher levels of P-selectin,
and the co-stimulatory molecules CD40L, CD40 and OX40L (Figure 5). Thus,
among the different subtypes of PEVs, those with a higher density of antigen–MHC-
I complexes show more abundant expression of lymphocyte co-stimulatory
molecules and bear a higher content of active proteasome.

143
4.5.6 Proteasome-containing PEVs can support antigen-specific T
cell activation
T cells isolated from OT-1 mice100 were co-incubated for 18 hours with PEVs that
were either pulsed or not with the SIINFEKL peptide or native OVA. Dendritic cells
and platelets were treated similarly as positive controls and for comparison
(Figure 6A). The T cells (CD3+CD8+) were then washed and the expression of
CD40, OX40, IL-2 and IFN-γ was evaluated to assess T cell activation.
Compared with DCs and platelets, PEVs could induce a significant release of IFN-γ
when pulsed with the OVA peptide, whereas native OVA led to an increase in IFN-γ
but did not reach statistical significance (Figure 6B). Moreover, DCs, and to a lesser
extent platelets and PEVs, were only capable of inducing significant CD40
expression by T lymphocytes previously pulsed with the OVA peptide (Figure 6C).
In contrast, OX40 and IL-2 expression were not induced by DCs, platelets or PEVs
under these experimental conditions (Figure 6D-6E).
Whether PEVs could stimulate T cell proliferation, a hallmark response by the
lymphocyte antigen-MHC-I complex was evaluated. T cells from OT-1 mice were
labeled with CFSE to monitor cellular division and co-incubated for 5 days with either
DCs, activated platelets or PEVs, which were either pulsed or not with either
SIINFEKL or native OVA (Figure 7A). Lymphoproliferation would be represented by
a decrease in the mean fluorescence intensity histogram, i.e. a dilution of CFSE
fluorescence. (Figure 7B-C).
As expected, we found that the proportion of lymphoproliferative cells was
significantly higher when OT-1 T lymphocytes were incubated with peptide- or native
OVA-pulsed DCs or activated platelets (Figure 7B-C). Of particular note is that PEVs
also supported T cell proliferation when pulsed with either the SIINFEKL or native
OVA (Figure 7D). In addition, when PEVs were removed from pulsed conditions by
ultracentrifugation, no proliferation was observed, confirming that the pulsed proteins
alone cannot support proliferation (Figure 7E). Furthermore, inhibition of PEV

144
proteasome by epoxomicin before pulsing with native OVA inhibited the ability of
PEV to induce T cell proliferation (Figure 7F left panel). The effect was directed
toward PEV proteasome, as addition of epoxomicin prior to peptide pulsing at the
same concentration used on DCs did not inhibit proliferation (Figure 7F right panel).
In summary, PEVs are capable of processing native proteins into smaller peptides
through their active proteasome, thereby enabling peptide loading onto MHC-I.
Platelet EVs express co-stimulatory molecules, and their interaction with positively
selected T lymphocytes promotes lymphocyte cytokine production and proliferation.

145
4.6 Discussion
Megakaryocytes and platelets are emerging as active players in innate and adaptive
immunity7,53,54. The platelet role in immunity is mainly confined to the blood
circulation, while megakaryocytes are localized in bone marrow and lungs. The latter
location potentially provides the megakaryocyte with more direct access to airborne
pathogens and allergens55,85,86. In contrast, PEVs can disseminate into organs and
tissues that are inaccessible to megakaryocytes and platelets and this may be
possibly due to their small dimensions and the presence of unique surface
molecules. In this study, we found that the proteasome and the necessary machinery
to process and present antigens to CD8+ T cells are packaged into PEVs by
platelets. Thus, PEVs may extend the immune functions played by platelets and
megakaryocytes outside the confines of the blood.
Platelet EVs are heterogeneous in terms of surface molecules and their platelet-
derived content. The presence of mitochondria within PEVs is well
documented59,60,87, but it was unknown whether other organelles were also
transferred from the platelet. The proteasome is much more abundant than
mitochondria, at around 800,000 copies per cell88 in contrast to approximately 3–7
mitochondria per platelet60. Further investigation will be necessary to determine if
the presence of multiple organelles within a single vesicle is the result of a specific
sorting mechanism, or because those vesicles are larger and may have more
storage capacity. Nonetheless, as previously shown for mitochondria-containing
PEVs, we observed that the release of proteasome-containing PEVs requires
cytoskeleton remodeling via intact actin microfilament dynamics and that a broad
array of platelet agonists induce the release of proteasome-containing PEVs60.
The presence of an extracellular proteasome has already been documented in
normal human blood, and elevated levels have been found in patients suffering from
autoimmune diseases, sepsis or trauma89. Moreover, the 20S proteasome core is
present and active within EVs derived from apoptotic endothelial cells and regulates
tertiary lymphoid structures formation, autoantibody production and graft rejection

146
following transplantation35. While some evidence supports that a circulating
extracellular proteasome may be transported by EVs, we show that EVs of platelet
origin, among the most abundant EVs in blood, do contain the proteasome.
Consistent with this, mass spectrometry analysis of the human PEV proteome
identified numerous proteasomal subunits90-92. These include subunits of the 20S
catalytic core (PSMA1, PSMA2, PSMA3, PSMA4, PSMA5, PSMA6, PSMA7,
PSMB1, PSMB2, PSMB3, PSMB4, PSMB5, PSMB6), an immunoproteasome
subunit (PSMB8), subunits of the 19S regulator (PSMC2, PSMC4, PSMC5, PSMC6,
PSMD2, PSMD3, PSMD11, PSMD13) and the 11S regulator (PSME1 and
PSME2)90-92. Thus, while proteomic data points to proteasome proteins in PEVs, the
present work unequivocally demonstrates its presence and documents for the first
time that the extracellular proteasome in PEVs is functional and can contribute to
antigen processing.
We used complementary approaches and developed a hs-FCM-based assay to
detect active proteasome at the single EV level, thereby permitting the quantification
and assessment of other molecules expressed by proteasome-containing EVs. Of
particular note the proteasome-containing PEVs also expressed MHC-I and
lymphocyte co-stimulatory molecules, which enabled lymphocyte activation,
proliferation and cytokine generation. This process is well regulated, as it strictly
implicated protein processing by the proteasome, subsequent loading onto MHC-I
molecules for recognition by CD8+ T cells. These findings demonstrate a novel and
potentially important role for PEVs in adaptive immunity. While our work suggests
that PEVs may be involved in adaptive immunity through antigen presentation, it
does not necessarily exclude that other cells may release proteasome-containing
EVs capable of playing this role. Indeed, EVs derived from DCs, B and T
lymphocytes, macrophages and NK cells can perform cross-presentation,
suggesting that they also contain the necessary antigen processing machinery93-98.
Further studies will be necessary to determine the impact and the importance of
PEVs as antigen presenting elements.

147
We identified proteasome-containing PEVs in the blood of healthy donors. As most
PEVs in blood under healthy conditions are suggested to originate from
megakaryocytes99,100, the latter may also constitutively release proteasome-
containing EVs. Moreover, we found that numerous stimuli of human platelets, as
well as the in vivo stimulation of mouse platelets by immune complexes could induce
release of proteasomes in PEVs, suggesting that proteasome release is at least
conserved in humans and mice and takes place under conditions implicating platelet
activation. Platelet potency at actively generating immunity against the Plasmodium
berghei parasite has been demonstrated49. Moreover, megakaryocytes can be
infected by Dengue virus101, and they can also ingest and phagocytose E. coli55.
Given their small size, intact microorganisms may not necessarily be present inside
PEVs, but PEVs might process cytosolic microbial proteins derived from infected
cells, such as platelets or megakaryocytes that lack the ability to disseminate through
the lymphatic system. Thus, PEVs may be implicated in immune surveillance and
might contribute to presentation of microbial antigens. Furthermore, the presence of
proteasome-containing PEVs in platelet concentrates was not associated with
increased risks of ATR or TRALI in a mouse model. It remains to be verified whether
the presentation of platelet antigens (e.g. CD41 or CD61) by PEVs from platelet
concentrates might contribute to generation of anti-platelet immunity in transfused
recipients, although this has been shown with megakaryocytes56. It is also unknown
whether the proteasome in EVs (from platelets, megakaryocytes, or other cells)
displays other functions in addition to antigen processing. Future investigations
might reveal whether the proteasome impact protein half-life in EVs.
Activated platelets release PEVs containing both mitochondria and the proteasome.
Our findings in mouse lymph revealed that proteasome-containing PEVs can
circulate in the lymphatic system, potentially explaining their accumulation in
lymphoid organs following intravenous injection. It is intriguing that these findings
contrast with mitochondria-containing PEVs, which are essentially absent from
lymph64. This privileged access to the lymphatic system by proteasome-containing
PEVs may reveal a new immune route for PEVs to reach lymphoid organs or infected

148
tissues. Our study highlights the diversity of PEVs, and supports the concept that
different subtypes of PEVs may play different roles depending on their cargo and
tissue distribution.

149
4.7 References
1. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med.
2007;357(24):2482-2494.
2. Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of Innate Immune
Responses by Platelets. Front Immunol. 2019;10:1320.
3. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune
cell interactions in thrombo-inflammation. Res Pract Thromb Haemost. 2020;4(1):23-35.
4. Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat
Rev Immunol. 2011;11(4):264-274.
5. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets
as immune and inflammatory cells. Blood. 2014;123(18):2759-2767.
6. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served
with inflammation. J Immunol. 2015;194(12):5579-5587.
7. Cunin P, Nigrovic PA. Megakaryocytes as immune cells. J Leukoc Biol.
2019;105(6):1111-1121.
8. Garraud O, Cognasse F. Are Platelets Cells? And if Yes, are They Immune Cells?
Front Immunol. 2015;6:70.
9. Silva-Cardoso SC, Affandi AJ, Spel L, et al. CXCL4 Exposure Potentiates TLR-
Driven Polarization of Human Monocyte-Derived Dendritic Cells and Increases
Stimulation of T Cells. J Immunol. 2017;199(1):253-262.
10. Moore KL, Thompson LF. P-selectin (CD62) binds to subpopulations of human
memory T lymphocytes and natural killer cells. Biochem Biophys Res Commun.
1992;186(1):173-181.
11. Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to
postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets.
Hepatology. 2006;43(2):306-315.
12. Han P, Hanlon D, Arshad N, et al. Platelet P-selectin initiates cross-presentation and
dendritic cell differentiation in blood monocytes. Sci Adv. 2020;6(11):eaaz1580.
13. Laberge S, Cruikshank WW, Beer DJ, Center DM. Secretion of IL-16 (lymphocyte
chemoattractant factor) from serotonin-stimulated CD8+ T cells in vitro. J Immunol.
1996;156(1):310-315.
14. Lang PA, Contaldo C, Georgiev P, et al. Aggravation of viral hepatitis by platelet-
derived serotonin. Nat Med. 2008;14(7):756-761.
15. Martinson J, Bae J, Klingemann HG, Tam Y. Activated platelets rapidly up-regulate
CD40L expression and can effectively mature and activate autologous ex vivo
differentiated DC. Cytotherapy. 2004;6(5):487-497.
16. Kaneider NC, Kaser A, Tilg H, Ricevuti G, Wiedermann CJ. CD40 ligand-
dependent maturation of human monocyte-derived dendritic cells by activated platelets. Int
J Immunopathol Pharmacol. 2003;16(3):225-231.
17. Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to
antigen-presenting cells: a potential bridge between injury and immune activation. Exp
Hematol. 2004;32(2):135-139.
18. Katoh N, Soga F, Nara T, et al. Effect of serotonin on the differentiation of human
monocytes into dendritic cells. Clin Exp Immunol. 2006;146(2):354-361.

150
19. Xia CQ, Kao KJ. Effect of CXC chemokine platelet factor 4 on differentiation and
function of monocyte-derived dendritic cells. Int Immunol. 2003;15(8):1007-1015.
20. Duffau P, Seneschal J, Nicco C, et al. Platelet CD154 potentiates interferon-alpha
secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med.
2010;2(47):47ra63.
21. Sowa JM, Crist SA, Ratliff TL, Elzey BD. Platelet influence on T- and B-cell
responses. Arch Immunol Ther Exp (Warsz). 2009;57(4):235-241.
22. Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive
immunity. Cell Immunol. 2005;238(1):1-9.
23. Blumberg N, Spinelli SL, Francis CW, Taubman MB, Phipps RP. The platelet as an
immune cell-CD40 ligand and transfusion immunomodulation. Immunol Res. 2009;45(2-
3):251-260.
24. Sprague DL, Sowa JM, Elzey BD, Ratliff TL. The role of platelet CD154 in the
modulation in adaptive immunity. Immunol Res. 2007;39(1-3):185-193.
25. Elzey BD, Tian J, Jensen RJ, et al. Platelet-mediated modulation of adaptive
immunity. A communication link between innate and adaptive immune compartments.
Immunity. 2003;19(1):9-19.
26. Andre P, Prasad KS, Denis CV, et al. CD40L stabilizes arterial thrombi by a beta3
integrin--dependent mechanism. Nat Med. 2002;8(3):247-252.
27. Buchner K, Henn V, Grafe M, de Boer OJ, Becker AE, Kroczek RA. CD40 ligand
is selectively expressed on CD4+ T cells and platelets: implications for CD40-CD40L
signalling in atherosclerosis. J Pathol. 2003;201(2):288-295.
28. Fleischer J, Grage-Griebenow E, Kasper B, et al. Platelet factor 4 inhibits
proliferation and cytokine release of activated human T cells. J Immunol. 2002;169(2):770-
777.
29. Adnot S, Houssaini A, Abid S, Marcos E, Amsellem V. Serotonin transporter and
serotonin receptors. Handb Exp Pharmacol. 2013;218:365-380.
30. Mudd JC, Panigrahi S, Kyi B, et al. Inflammatory Function of CX3CR1+ CD8+ T
Cells in Treated HIV Infection Is Modulated by Platelet Interactions. J Infect Dis.
2016;214(12):1808-1816.
31. Shi G, Field DJ, Ko KA, et al. Platelet factor 4 limits Th17 differentiation and
cardiac allograft rejection. J Clin Invest. 2014;124(2):543-552.
32. Opperman CM, Sishi BJ. Tumor necrosis factor alpha stimulates p62 accumulation
and enhances proteasome activity independently of ROS. Cell Biol Toxicol. 2015;31(2):83-
94.
33. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser
B Phys Biol Sci. 2009;85(1):12-36.
34. Klockenbusch C, Walsh GM, Brown LM, et al. Global proteome analysis identifies
active immunoproteasome subunits in human platelets. Mol Cell Proteomics.
2014;13(12):3308-3319.
35. Dieude M, Bell C, Turgeon J, et al. The 20S proteasome core, active within
apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection.
Sci Transl Med. 2015;7(318):318ra200.
36. Shi DS, Smith MC, Campbell RA, et al. Proteasome function is required for platelet
production. J Clin Invest. 2014;124(9):3757-3766.

151
37. Murai K, Kowata S, Shimoyama T, et al. Bortezomib induces thrombocytopenia by
the inhibition of proplatelet formation of megakaryocytes. Eur J Haematol.
2014;93(4):290-296.
38. Nayak MK, Kulkarni PP, Dash D. Regulatory role of proteasome in determination
of platelet life span. J Biol Chem. 2013;288(10):6826-6834.
39. Grundler K, Rotter R, Tilley S, et al. The proteasome regulates collagen-induced
platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb Res.
2016;148:15-22.
40. Nayak MK, Kumar K, Dash D. Regulation of proteasome activity in activated
human platelets. Cell Calcium. 2011;49(4):226-232.
41. Grundler Groterhorst K, Mannell H, Pircher J, Kraemer BF. Platelet Proteasome
Activity and Metabolism Is Upregulated during Bacterial Sepsis. Int J Mol Sci.
2019;20(23).
42. Gupta N, Li W, Willard B, Silverstein RL, McIntyre TM. Proteasome proteolysis
supports stimulated platelet function and thrombosis. Arterioscler Thromb Vasc Biol.
2014;34(1):160-168.
43. Colberg L, Cammann C, Greinacher A, Seifert U. Structure and function of the
ubiquitin-proteasome system in platelets. J Thromb Haemost. 2020.
44. Yukawa M, Sakon M, Kambayashi J, et al. Purification and characterization of
endogenous protein activator of human platelet proteasome. J Biochem. 1993;114(3):317-
323.
45. Yukawa M, Sakon M, Kambayashi J, et al. Proteasome and its novel endogeneous
activator in human platelets. Biochem Biophys Res Commun. 1991;178(1):256-262.
46. Boegel S, Lower M, Bukur T, Sorn P, Castle JC, Sahin U. HLA and proteasome
expression body map. BMC Med Genomics. 2018;11(1):36.
47. Semple JW, Speck ER, Milev YP, Blanchette V, Freedman J. Indirect
allorecognition of platelets by T helper cells during platelet transfusions correlates with
anti-major histocompatibility complex antibody and cytotoxic T lymphocyte formation.
Blood. 1995;86(2):805-812.
48. Zufferey A, Speck ER, Machlus KR, et al. Mature murine megakaryocytes present
antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv.
2017;1(20):1773-1785.
49. Chapman LM, Aggrey AA, Field DJ, et al. Platelets present antigen in the context
of MHC class I. J Immunol. 2012;189(2):916-923.
50. Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization
of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J
Proteomics. 2014;101:130-140.
51. Iannacone M, Sitia G, Isogawa M, et al. Platelets mediate cytotoxic T lymphocyte-
induced liver damage. Nat Med. 2005;11(11):1167-1169.
52. Verschoor A, Neuenhahn M, Navarini AA, et al. A platelet-mediated system for
shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb
and complement C3. Nat Immunol. 2011;12(12):1194-1201.
53. Maouia A, Rebetz J, Kapur R, Semple JW. The Immune Nature of Platelets
Revisited. Transfus Med Rev. 2020.
54. Marcoux G, Laroche A, Espinoza Romero J, Boilard E. Role of platelets and
megakaryocytes in adaptive immunity. Platelets. 2020:1-12.

152
55. Pariser DN, Hilt ZT, Ture SK, et al. Lung megakaryocytes are immune modulatory
cells. J Clin Invest. 2020.
56. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and
disease. Platelets. 2017;28(3):214-221.
57. Puhm F, Boilard E, Machlus KR. Platelet Extracellular Vesicles: Beyond the Blood.
Arterioscler Thromb Vasc Biol. 2020:ATVBAHA120314644.
58. Siljander PR. Platelet-derived microparticles - an updated perspective. Thromb Res.
2011;127 Suppl 2:S30-33.
59. Marcoux G, Magron A, Sut C, et al. Platelet-derived extracellular vesicles convey
mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse
reactions. Transfusion. 2019;59(7):2403-2414.
60. Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria serving
as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation.
Blood. 2014;124(14):2173-2183.
61. Burnouf T, Chou ML, Goubran H, Cognasse F, Garraud O, Seghatchian J. An
overview of the role of microparticles/microvesicles in blood components: Are they
clinically beneficial or harmful? Transfus Apher Sci. 2015;53(2):137-145.
62. Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis
via collagen-dependent microparticle production. Science. 2010;327(5965):580-583.
63. Gyorgy B, Szabo TG, Turiak L, et al. Improved flow cytometric assessment reveals
distinct microvesicle (cell-derived microparticle) signatures in joint diseases. PLoS One.
2012;7(11):e49726.
64. Tessandier N, Melki I, Cloutier N, et al. Platelets Disseminate Extracellular
Vesicles in Lymph in Rheumatoid Arthritis. Arterioscler Thromb Vasc Biol.
2020:ATVBAHA119313698.
65. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular
vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell
Vesicles. 2016;5:31427.
66. French SL, Butov KR, Allaeys I, et al. Platelet-derived extracellular vesicles
infiltrate and modify the bone marrow during inflammation. Blood Adv. 2020;4(13):3011-
3023.
67. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-
mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-
derived membrane vesicles. Blood. 2008;111(10):5028-5036.
68. Yari F, Motefaker M, Nikougoftar M, Khayati Z. Interaction of Platelet-Derived
Microparticles with a Human B-Lymphoblast Cell Line: A Clue for the Immunologic
Function of the Microparticles. Transfus Med Hemother. 2018;45(1):55-61.
69. Sadallah S, Amicarella F, Eken C, Iezzi G, Schifferli JA. Ectosomes released by
platelets induce differentiation of CD4+T cells into T regulatory cells. Thromb Haemost.
2014;112(6):1219-1229.
70. Dinkla S, van Cranenbroek B, van der Heijden WA, et al. Platelet microparticles
inhibit IL-17 production by regulatory T cells through P-selectin. Blood.
2016;127(16):1976-1986.
71. Verdoes M, Florea BI, Menendez-Benito V, et al. A fluorescent broad-spectrum
proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol.
2006;13(11):1217-1226.

153
72. Raz V, Raz Y, Paniagua-Soriano G, et al. Proteasomal activity-based probes mark
protein homeostasis in muscles. J Cachexia Sarcopenia Muscle. 2017;8(5):798-807.
73. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release
two types of membrane vesicles: microvesicles by surface shedding and exosomes derived
from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94(11):3791-
3799.
74. Semple JW, Rebetz J, Kapur R. Transfusion-associated circulatory overload and
transfusion-related acute lung injury. Blood. 2019;133(17):1840-1853.
75. Kapur R, Kim M, Rebetz J, et al. Gastrointestinal microbiota contributes to the
development of murine transfusion-related acute lung injury. Blood Adv. 2018;2(13):1651-
1663.
76. Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect
against transfusion-related acute lung injury via IL-10. Blood. 2017;129(18):2557-2569.
77. McKenzie SE, Taylor SM, Malladi P, et al. The role of the human Fc receptor Fc
gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol.
1999;162(7):4311-4318.
78. Cloutier N, Allaeys I, Marcoux G, et al. Platelets release pathogenic serotonin and
return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S
A. 2018;115(7):E1550-E1559.
79. Melki I, Allaeys I, Tessandier N, et al. Platelets release
mitochondrial antigens in systemic lupus erythematosus: Science Translational
Medicine (in press); 2020.
80. Angenieux C, Dupuis A, Gachet C, de la Salle H, Maitre B. Cell surface expression
of HLA I molecules as a marker of young platelets. J Thromb Haemost. 2019;17(9):1511-
1521.
81. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME,
Corona-de la Pena NA, Salazar MI. Dengue Virus Induces the Release of sCD40L and
Changes in Levels of Membranal CD42b and CD40L Molecules in Human Platelets.
Viruses. 2018;10(7).
82. Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization,
quantitation, and in situ detection of specific peptide-MHC class I complexes using a
monoclonal antibody. Immunity. 1997;6(6):715-726.
83. Rand ML, Wang H, Bang KW, Packham MA, Freedman J. Rapid clearance of
procoagulant platelet-derived microparticles from the circulation of rabbits. J Thromb
Haemost. 2006;4(7):1621-1623.
84. Rank A, Nieuwland R, Crispin A, et al. Clearance of platelet microparticles in vivo.
Platelets. 2011;22(2):111-116.
85. Lefrancais E, Ortiz-Munoz G, Caudrillier A, et al. The lung is a site of platelet
biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105-
109.
86. Campbell RA, Schwertz H, Hottz ED, et al. Human megakaryocytes possess
intrinsic antiviral immunity through regulated induction of IFITM3. Blood.
2019;133(19):2013-2026.
87. Marcoux G, Duchez AC, Rousseau M, et al. Microparticle and mitochondrial
release during extended storage of different types of platelet concentrates. Platelets.
2017;28(3):272-280.

154
88. Princiotta MF, Finzi D, Qian SB, et al. Quantitating protein synthesis, degradation,
and endogenous antigen processing. Immunity. 2003;18(3):343-354.
89. Sixt SU, Dahlmann B. Extracellular, circulating proteasomes and ubiquitin -
incidence and relevance. Biochim Biophys Acta. 2008;1782(12):817-823.
90. Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet
microparticle proteome. J Proteome Res. 2005;4(5):1516-1521.
91. Dean WL, Lee MJ, Cummins TD, Schultz DJ, Powell DW. Proteomic and
functional characterisation of platelet microparticle size classes. Thromb Haemost.
2009;102(4):711-718.
92. Capriotti AL, Caruso G, Cavaliere C, Piovesana S, Samperi R, Laganà A.
Proteomic characterization of human platelet-derived microparticles. Anal Chim Acta.
2013;776:57-63.
93. Chen Z, Larregina AT, Morelli AE. Impact of extracellular vesicles on innate
immunity. Curr Opin Organ Transplant. 2019;24(6):670-678.
94. Lindenbergh MFS, Stoorvogel W. Antigen Presentation by Extracellular Vesicles
from Professional Antigen-Presenting Cells. Annu Rev Immunol. 2018;36:435-459.
95. Lindenbergh MFS, Wubbolts R, Borg EGF, van 't Veld EM, Boes M, Stoorvogel
W. Dendritic cells release exosomes together with phagocytosed pathogen; potential
implications for the role of exosomes in antigen presentation. J Extracell Vesicles.
2020;9(1):1798606.
96. Federici C, Shahaj E, Cecchetti S, et al. Natural-Killer-Derived Extracellular
Vesicles: Immune Sensors and Interactors. Front Immunol. 2020;11:262.
97. Zeng F, Morelli AE. Extracellular vesicle-mediated MHC cross-dressing in immune
homeostasis, transplantation, infectious diseases, and cancer. Semin Immunopathol.
2018;40(5):477-490.
98. Noulsri E. Effects of Cell-Derived Microparticles on Immune Cells and Potential
Implications in Clinical Medicine. Lab Med. 2020.
99. Flaumenhaft R, Dilks JR, Richardson J, et al. Megakaryocyte-derived
microparticles: direct visualization and distinction from platelet-derived microparticles.
Blood. 2009;113(5):1112-1121.
100. Gitz E, Pollitt AY, Gitz-Francois JJ, et al. CLEC-2 expression is maintained on
activated platelets and on platelet microparticles. Blood. 2014;124(14):2262-2270.
101. Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue viruses infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis. 2019;13(11):e0007837.
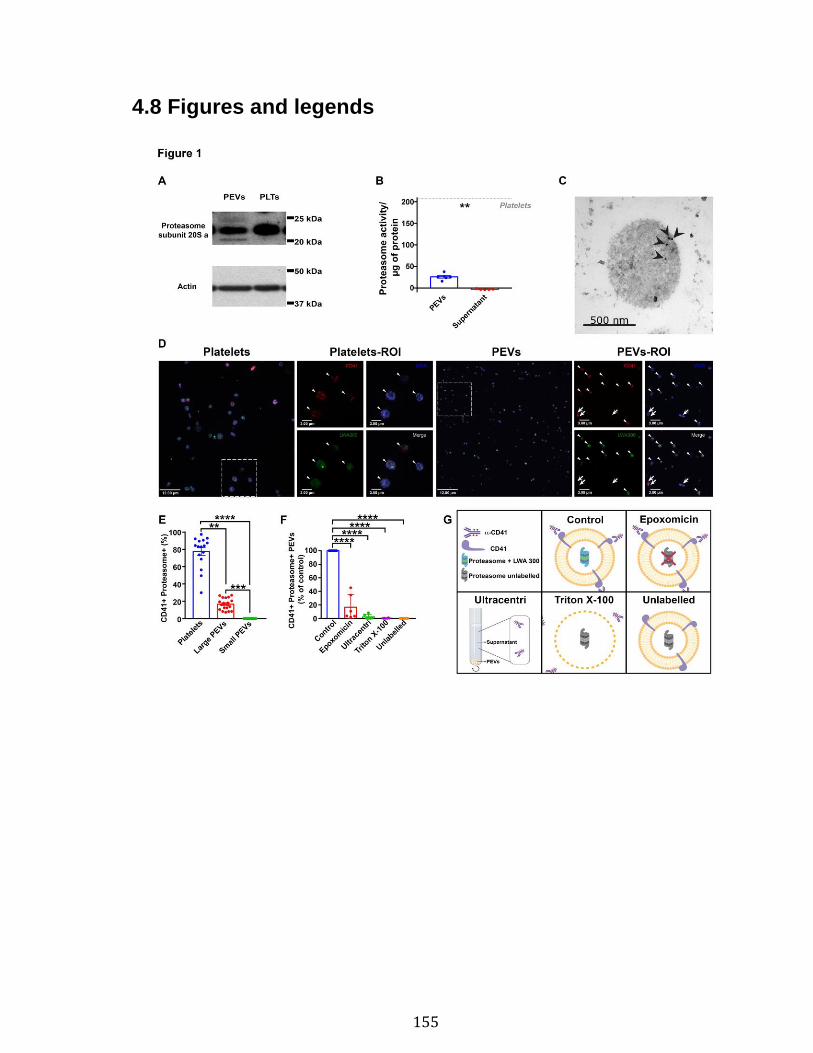
155
4.8 Figures and legends

156
Figure 1. Platelets and PEVs contain proteasome
(A) Proteasome 20S α subunit and actin in human platelet extracellular vesicles
(PEVs) and platelet (PLTs) preparations (25 μg protein per lane) were assessed by
immunoblotting. Results are representative of six distinct preparations. (B)
Proteasome function was assessed by measuring the caspase-like activity of PEVs
and platelet supernatants. Platelets were used as positive control (Gray dotted line)
using the Proteasome-Glo Caspase-like Cell-based Assay. Fifteen μg of proteins
were used per condition. Mean ± SEM, n = 7, ** P < 0.01, Mann-Whitney. (C)
Transmission Electron Microscopy (TEM) visualization of immunogold labelling of
proteasome 20S α subunit in PEVs released from thrombin-activated platelets. Data
are representative of three independent experiments. (D) Confocal microscopy
visualization of proteasome content associated with platelets (left panel) and PEVs
(right panel). Visualization of CD41, wheat germ agglutinin (WGA) to determine
plasma membrane surface, proteasome (LWA300) and merge is displayed in the
region of interest (ROI). Populations originating from dashed lines squares and
represented in ROI are triple positives (white arrowheads) or CD4- and WGA-
positive but proteasome-negative (white arrows). (E) High-sensitivity flow cytometry
(hs-FCM) analysis of resting platelets and thrombin-activated platelets. Two distinct
populations of PEVs, i.e. larger PEVs (approximately 17% of these PEVs contain
active proteasome) and smaller PEVs, not containing active proteasome. (n = 20
data are presented as mean ± SEM, ** P < 0.01, *** P < 0.001 and **** P < 0.0001,
Kruskal–Wallis). (F-G) Controls were performed to assess the specificity of PEV
detection using hs-FCM. Sensitivity of CD41+Proteasome+ PEVs to competition by
epoxomicin, ultracentrifugation (Ultracentri) or 0.05% Triton X-100 and unlabeled
samples are presented as % of untreated (Control). Data are presented as mean ±
SEM of 5 independent experiments, paired t-test ****P < 0.0001 compared with the
control.
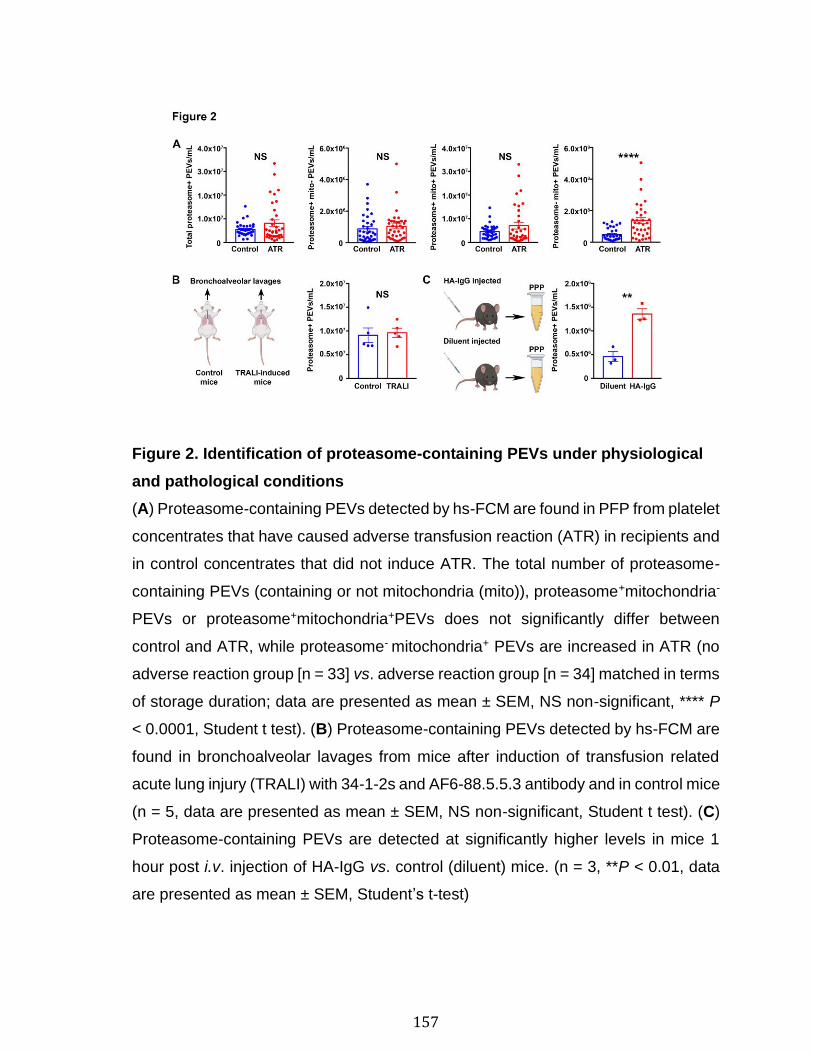
157
Figure 2. Identification of proteasome-containing PEVs under physiological
and pathological conditions
(A) Proteasome-containing PEVs detected by hs-FCM are found in PFP from platelet
concentrates that have caused adverse transfusion reaction (ATR) in recipients and
in control concentrates that did not induce ATR. The total number of proteasome-
containing PEVs (containing or not mitochondria (mito)), proteasome+mitochondria-
PEVs or proteasome+mitochondria+PEVs does not significantly differ between
control and ATR, while proteasome- mitochondria+ PEVs are increased in ATR (no
adverse reaction group [n = 33] vs. adverse reaction group [n = 34] matched in terms
of storage duration; data are presented as mean ± SEM, NS non-significant, **** P
< 0.0001, Student t test). (B) Proteasome-containing PEVs detected by hs-FCM are
found in bronchoalveolar lavages from mice after induction of transfusion related
acute lung injury (TRALI) with 34-1-2s and AF6-88.5.5.3 antibody and in control mice
(n = 5, data are presented as mean ± SEM, NS non-significant, Student t test). (C)
Proteasome-containing PEVs are detected at significantly higher levels in mice 1
hour post i.v. injection of HA-IgG vs. control (diluent) mice. (n = 3, **P < 0.01, data
are presented as mean ± SEM, Student’s t-test)
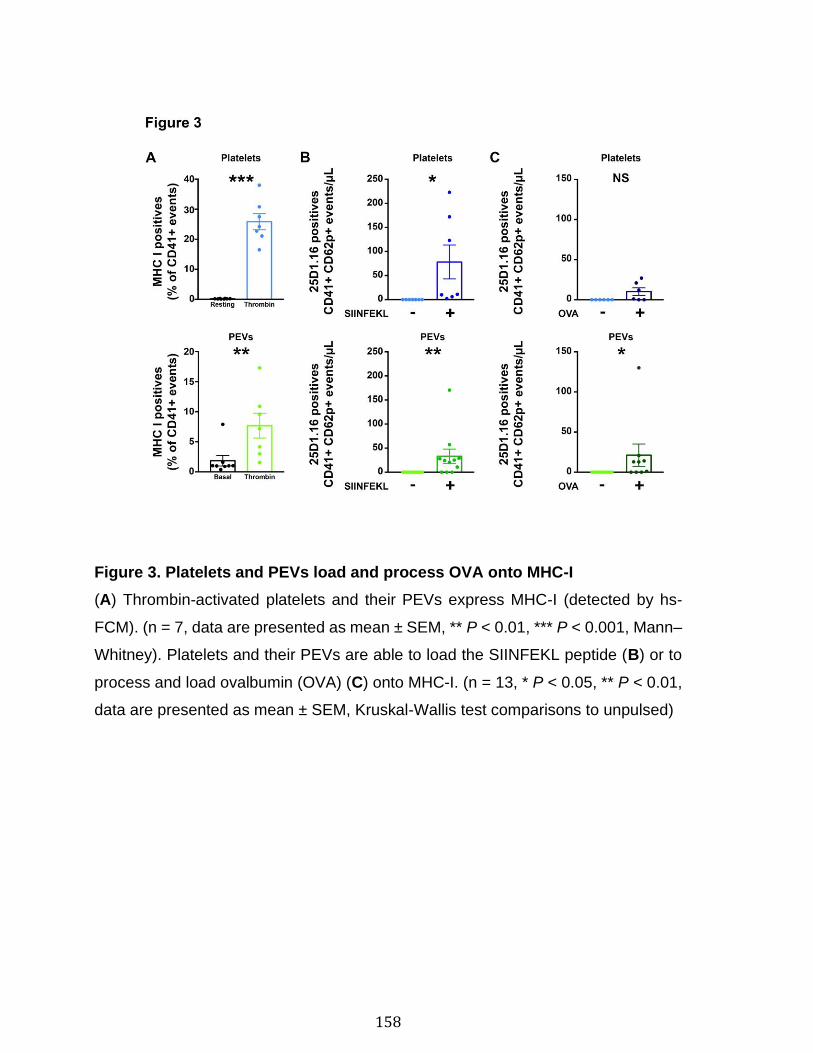
158
Figure 3. Platelets and PEVs load and process OVA onto MHC-I
(A) Thrombin-activated platelets and their PEVs express MHC-I (detected by hs-
FCM). (n = 7, data are presented as mean ± SEM, ** P < 0.01, *** P < 0.001, Mann–
Whitney). Platelets and their PEVs are able to load the SIINFEKL peptide (B) or to
process and load ovalbumin (OVA) (C) onto MHC-I. (n = 13, * P < 0.05, ** P < 0.01,
data are presented as mean ± SEM, Kruskal-Wallis test comparisons to unpulsed)
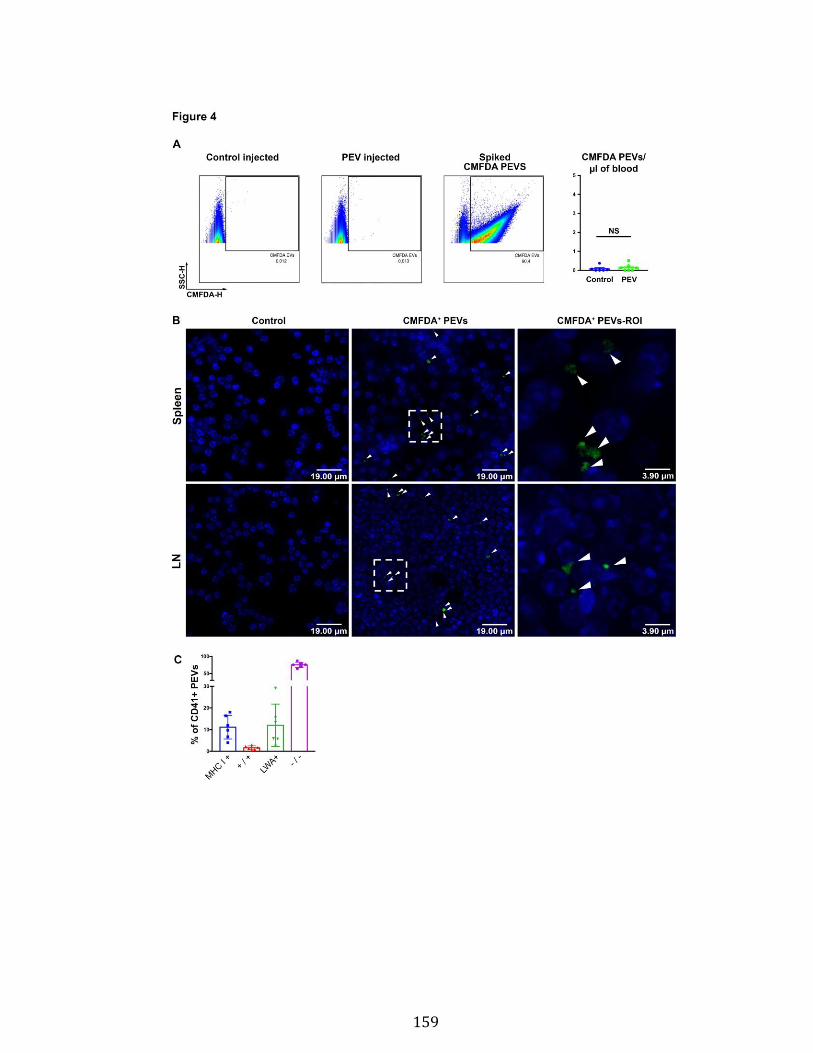
159

160
Figure 4. PEVs in blood circulation can reach lymphoid organs and circulate
in lymph
(A) One-hour post i.v. injection, CMFDA fluorescently labeled PEVs are
undetectable in blood circulation. CMFDA fluorescently labeled PEVs are easily
detected if spiked in blood as a positive control. The number of CMFDA PEVs per
µL of blood was quantified and no difference was observed between control (not
injected) and PEV-injected mice (lower left). (n = 7, data are presented as mean ±
SEM, NS non-significant, Mann–Whitney). (B) CMFDA-labeled PEVs localized in the
spleen and in lymph nodes (LN) are indicated by white arrowhead. ROI: region of
interest. Results are representative of observations made in seven mice per group.
(C) PEVs in lymph were detected by hs-FCM and expression of MHC-I and
proteasome (LWA300) was determined. +/+ double positive and −/− double negative
for MHC-I and proteasome. (n = 6, data are presented as mean ± SEM).
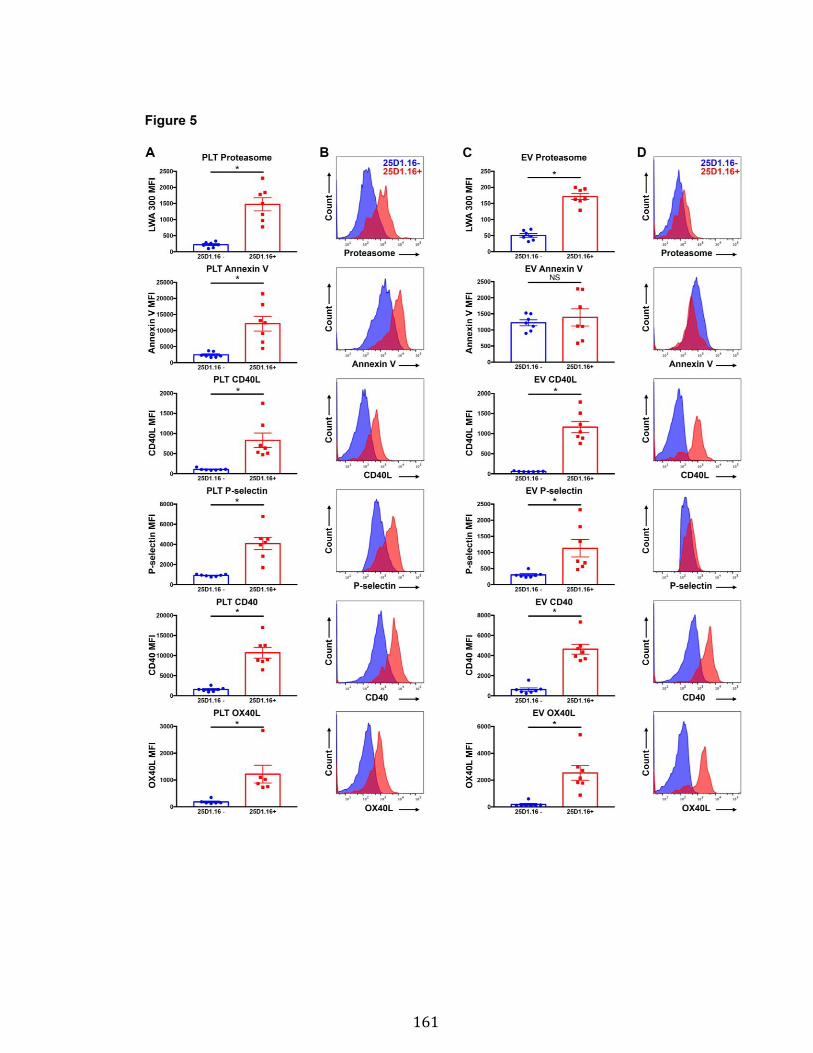
161

162
Figure 5. Platelets and PEVs with loaded OVA peptide express activation and
co-stimulatory molecules
(A-B) Platelets and (C-D) PEVs loaded with OVA peptide (25D1.16+) express higher
levels of proteasome, and activation and co-stimulatory molecules. (A,C) Mean
fluorescence intensity (MFI) of the different markers assessed by hs-FCM (n = 7,
data are mean ± SEM, NS non-significant, * P < 0.05, Student’s t-test). (B,D)
Representative MFI histogram of the 25D1.16 negative and positive populations for
each marker.
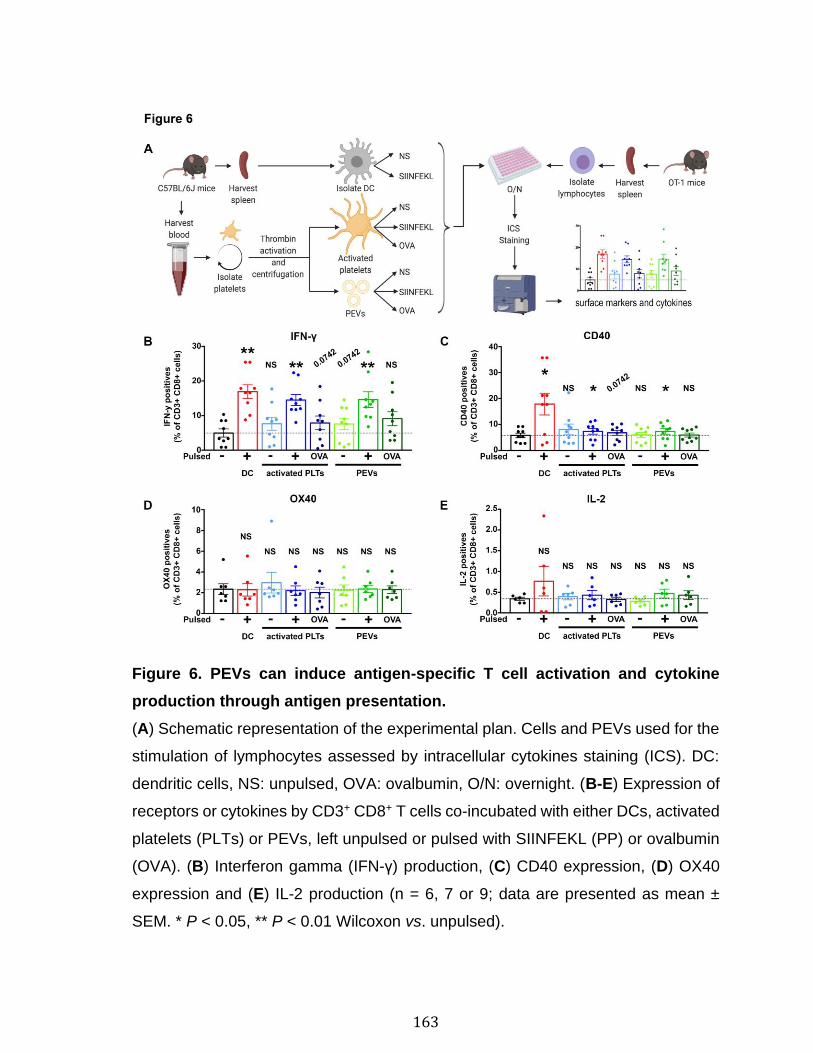
163
Figure 6. PEVs can induce antigen-specific T cell activation and cytokine
production through antigen presentation.
(A) Schematic representation of the experimental plan. Cells and PEVs used for the
stimulation of lymphocytes assessed by intracellular cytokines staining (ICS). DC:
dendritic cells, NS: unpulsed, OVA: ovalbumin, O/N: overnight. (B-E) Expression of
receptors or cytokines by CD3+ CD8+ T cells co-incubated with either DCs, activated
platelets (PLTs) or PEVs, left unpulsed or pulsed with SIINFEKL (PP) or ovalbumin
(OVA). (B) Interferon gamma (IFN-γ) production, (C) CD40 expression, (D) OX40
expression and (E) IL-2 production (n = 6, 7 or 9; data are presented as mean ±
SEM. * P < 0.05, ** P < 0.01 Wilcoxon vs. unpulsed).
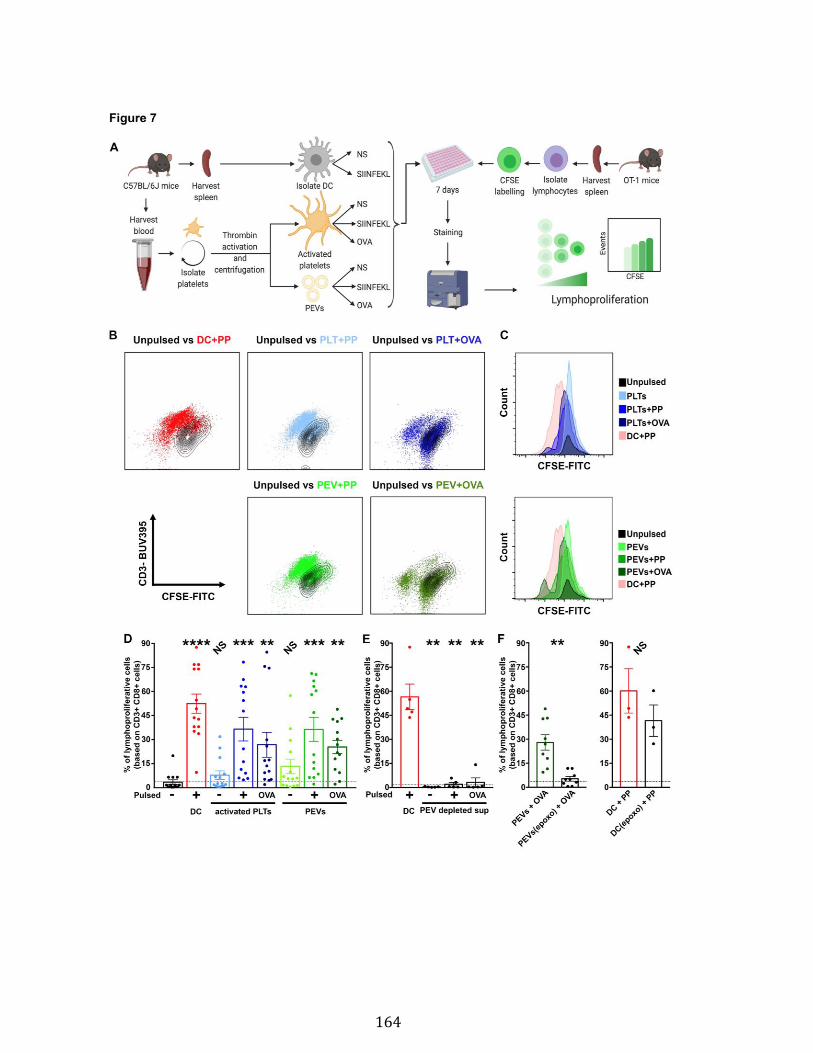
164

165
Figure 7. PEVs loaded with native OVA process and present OVA peptide to
induce antigen-specific T cell lymphoproliferation.
(A) Schematic representation of the experimental plan. DC: dendritic cells, NS:
unpulsed, OVA: ovalbumin, CFSE: Carboxyfluorescein succinimidyl ester. (B-C) Hs-
FCM dot plot (B) or histogram (C) showing CFSE fluorescence shift of CD3+ CD8+
T cells populations when co-incubated with either dendritic cells (DC), activated
platelets (PLTs) or PEVs left unpulsed or pulsed with SIINFEKL peptide (PP) or
ovalbumin (OVA) for 7 days. (D) Percentage of CD3+ CD8+ lymphoproliferative cells
after co-incubation with either DCs, PLTs or PEVs unpulsed or pulsed with PP or
OVA for 7 days. (n = 14; data are presented as mean ± SEM. NS non-significant, **
P < 0.01, *** P < 0.001, ****P < 0.0001, Friedman test followed by Dunn’s post-test
for multiple comparisons to unpulsed). (E) Percentage of CD3+ CD8+
lymphoproliferative cells after 7 days co-incubation with either PP-pulsed DCs or
supernatant (surn) depleted of PEVs by ultracentrifugation, left unpulsed or pulsed
with PP or OVA. (n=5; data are presented as mean ± SEM, ** P < 0.01, Mann-
Whitney vs. unpulsed). (F) Proportion of CD3+ CD8+ lymphoproliferative cells after 7
days co-incubation with OVA-pulsed PEVs treated or not with epoxomicin (epoxo)
for 2 hours and PP-pulsed DCs(DC + PP) treated or not with epoxomicin (epoxo). (n
= 9 for PEVs and n = 3 for DC; data are mean ± SEM, NS non-significant, ** P <
0.01, Wilcoxon).

166
4.9 Supplementary methods
Ethics The study was approved by the medical ethics committee of CHU de Québec
and Université Laval. Methods were carried out in accordance with approved
guidelines. Platelets were obtained from the citrated blood of healthy human
volunteers under informed consent according to a protocol approved by an
Institutional Review Board (Centre de Recherche du Centre Hospitalier Universitaire
de Québec). With approval from the ethics committees of Etablissement Français du
Sang, single-donor apheresis platelet concentrates were collected from healthy
anonymized volunteer blood donors (Regional Blood Bank, EFS Auvergne-Rhône-
Alpes) who gave consent for research purposes1-3.
Human platelets and EVs Platelets were isolated from healthy volunteers after
centrifugation of citrated whole blood (282xg for 10 min) at room temperature (RT).
The supernatant was centrifuged at 600xg for 3 min at RT to obtain platelet-rich
plasma (PRP). The latter was then centrifuged at 1,300xg for 5 min at RT and the
platelet-containing pellet was resuspended in Tyrode’s buffer (pH 7.4). Platelets
were counted (Cellometer AutoM10; Nexcelom Bioscience Lawrence, MA, USA),
adjusted to a density of 100 x 106 cells/mL in Tyrode’s buffer (pH 7.4) and, unless
stated in the legend, were either left nonactivated or activated with thrombin for 3 h
to obtain PEVs. In order to obtain PEVs, 5 mM calcium was added to platelets, which
were then stimulated with either thrombin (0.5 U/mL, Sigma-Aldrich), crosslinked
collagen-related peptide (CRP-XL) (1 µg/mL), ADP (20 µM, Sigma-Aldrich) or heat-
aggregated immunoglobulin G (HA-IgG) (0.5 mg/mL MP Biomedicals), for the
duration described in the corresponding legend. Platelet activation was stopped by
addition of 20 mM EDTA, and remnant platelets were removed by two rounds of
centrifugation at 1,300xg for 5 min at RT.
Characterization of proteasome-containing platelets and PEVs
Western Blot Platelet EVs and platelets were freshly prepared and centrifuged at
18,000xg for 1 h 30 min at 18°C or 1,500xg for 10 min, respectively. Pellets were

167
stored in phosphate-buffered saline (PBS) buffer containing protease inhibitor
cocktail, at −80°C until analysis (completeTM mini EDTA-free Protease Inhibitor
Cocktail, Sigma-Aldrich). Protein content was determined by Pierce BCA Protein
Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s guidelines.
Protein samples were separated by 12.5% SDS-PAGE followed by transfer onto a
polyvinylidene difluoride (PVDF) membrane. After blocking in milk, proteins of
interest were detected by primary antibodies against Proteasome 20S alpha3
subunit (Novus Biologicals, Centennial, CO, USA) and actin (Abcam). The PVDF
membranes were then incubated with horseradish peroxidase-conjugated antibody
against the primary antibodies (Jackson ImmunoResearch Laboratories).
Proteasome function assay Proteasome function was assessed by measuring the
caspase-like activity of PEVs using the Proteasome-Glo Caspase-like Cell-based
Assay (Promega Corporation), according to the manufacturer’s instructions.
Platelets served as the positive control. Fifteen μg of proteins were used per
condition.
Transmission electron microscopy Freshly obtained PEVs were fixed in 2% PFA
for 30 min and stored at 4°C prior to embedding in LR White acrylic resin. Eighty-
nanometer sections were mounted on an electron microscopy (EM) grid. Colloidal
bead staining was conducted by incubating the different EM grids face down on a 15
μL-droplet on a Parafilm sheet in a humidity chamber at RT. Electron microscopy
grids were incubated in PBS-glycine 50 mM for 15 min followed by a 15-min blocking
step in PBS containing 1% OVA. The grids were labeled for 30 min with
proteasome 20S alpha 3 antibody (Novus Biological) at 1:50, followed by 20 min
with 18 nm colloidal Gold-AffiniPure Donkey anti-rabbit IgG (H+L) at 1:150 (Jackson
ImmunoResearch Laboratories). Between each incubation, the EM grids were
washed under a stream of PBS. Following labelling, the grids were stained with 3%
uranyl acetate in water for 3 min prior to visualization on a FEI Tecnai G2 Spirit
BioTWIN transmission electron microscope at 80kV.

168
Confocal microscopy Platelets or PEVs were labeled with 1 µM LWA300
(proteasome probe) for 1 h 30 min at 30°C. Labeled and unlabeled platelets or PEVs
were added to a 24-well plate at 4°C for 18 h to promote adhesion to round glass
coverslips precoated with poly-L-lysine (Sigma-Aldrich). Immunocytofluorescence
staining was performed on the coverslips after centrifugation at 2,000xg for 5 min.
After three washes with PBS, the coverslips coated with platelets or EVs were
removed from the 24-well plate processed for staining on a parafilm sheet (10 ×
20 cm) organized in a gridpattern for each step: Each coverslip was flipped over (cell
side down) in the appropriate solution droplets (100 µL) for each staining step. The
samples were first blocked with 5% FBS (fetal bovine serum), 5% horse serum,
0.05% saponin in PBS for 20 min at room temperature. The primary antibodies were
diluted in the blocking solution: Integrin alpha 2b/CD41 antibody (1:100, mouse,
Novus Biologicals) and wheat germ agglutinin (WGA) - Alexa Fluor® 594 conjugate
(1:200, Invitrogen). The coverslips were transferred into the primary antibody
droplets, protected from light and incubated overnight at 4°C. After three washes in
PBS droplets, the coverslips were incubated for 2 h at room temperature with the
secondary antibodies diluted in blocking buffer: AlexaFluor647 F(ab’)2 anti-mouse
(1:300, Jackson ImmunoResearch Laboratories). The coverslips were washed three
times and mounted onto microscope slides in DAKO fluorescent mounting medium
(Agilent Technologies). Images were acquired with a Quorum Wave FX Spinning
Disk Confocal Microscope (Borealis - Leica DMI 6000B) at 63× (LEICA HCX PL
APO 63× 1.30 glycerol objective).
High-sensitivity flow cytometry (hs-FCM) All analyses were performed on a BD
Canto II Special Order Research Product (BD Biosciences) equipped with a small
particle option, as described previously (Figures 1 to 5)4, or with 20 parameters
LSRFortessa with small particle option (BD Biosciences) (Figures 6 to 8). Hs-FCM
performance tracking was performed daily before all analyses for both cytometers
using the BD cytometer setup and tracking beads (BD Biosciences,) or BD
FACSDiva CS&T Research Beads (BD Biosciences). Flow cytometer performance
was confirmed before each assay. Acquisition was performed at low speed and

169
quantification was conducted using either fluorescent polystyrene microspheres (3
μm diameter: Polysciences) of known concentration added to each tube, or
TruCount tubes (BD Biosciences). Silica particles (Kisker Biotech GmbH & Co.
Steinfurt) of known dimensions (100 nm, 500 nm and 1 μm in diameter) were used
for instrument set-up standardization. Fluorophore compensation was optimized for
every antibody panel.
Labeling of human platelets and EVs Five µl of PEV or platelet suspension were
labeled with 250 nM LWA300 proteasome probe in a total volume of 100 μL for 1 h
30 min at 30°C. Samples were then incubated with V450 anti-human CD41a
(clone HIP8) (BD Biosciences) and/or APC anti-human CD62P (clone AK-4, BD
Biosciences), APC Annexin V for phosphatidylserine (BD Biosciences),
MitoTrackerTM Deep Red FM for mitochondria (Invitrogen) for 30 min at RT and then
diluted into 500 µL PBS and analyzed by hs-FCM. Extracellular vesicles were first
gated according to their size and inner complexity (FSC PMT-H vs. SSC-H) to
consider events between approximately 100 and 1000 nm. CD41 and proteasome
expression levels were then verified in EVs. The presence of EVs was confirmed by
several negative controls: 1) EVs were ultracentrifuged at 100,000xg for 1 h at 20°C
and the supernatant was labeled as described above; 2) EVs were lysed using
0.05% Triton X-100 for 30 min at RT, before proteasome labeling and hs-FCM
analysis. 3) In addition, functionality of the EV-associated proteasomes was
confirmed by specific inhibition by epoxomicin (2.5 µM). The latter is a non-
fluorescent competitive inhibitor of the LWA300 probe and was used prior to
LWA300 labeling. To induce EV release, platelets (after addition of 5 mM of calcium)
were treated with diluent (PBS), cytochalasin B (20 µM; Sigma-Aldrich), cytochalasin
D (1 µM; Sigma-Aldrich), cytochalasin E (4 µM; Cayman Chemical), latrunculin A (10
µM; Cayman Chemical), or nocodazole (5 µM; Cayman Chemical) for 15 min at RT
and then treated with thrombin (0.5 U/mL, 4 h at RT). Platelet activation was
terminated by addition of 20 mM EDTA, and remnant platelets were removed by two
rounds of centrifugation at 1,300xg for 5 min at RT.

170
Mice C57BL/6J, FcγRIIATGN hemizygous mice and OT-1 mice were obtained from
The Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed and bred in the
research center animal facility, under pathogen-free conditions. Guidelines of the
Canadian Council on Animal Care were followed in a protocol approved by the
Animal Welfare Committee at Laval University (#2017-122-2).
Antibody-mediated murine TRALI model TRALI in mice was induced as reported
previously5-6. In brief, 18 hours prior to TRALI induction, C57BL/6 mice were injected
intraperitoneally with the monoclonal antibody GK1.5 (4.5 mg/kg) to deplete CD4+ T
cells. Mice then received a single i.v. injection of a cocktail of 34-1-2S (45 mg/kg)
and AF6-88.5.5.3 (4.5 mg/kg) in a 600 µL volume on the day of experimentation.
After 90 min, mice were anesthetized with a mixture of Ketaminol and Rompun and
exsanguinated by cardiac puncture. Bronchoalveolar lavages (BAL) were collected
by flushing the lungs three times with PBS. BAL were spun at 600xg for 5 min at RT.
The supernatant was transferred in a clean tube and spun at 1,300xg for 5 min at
RT. Supernatants were stored at −80°C until hs-FCM analysis.
Immune complex injection model HA-IgG (600 μg and 750 μg in males and
females, respectively) were i.v. injected in mice as previously reported7. Mice were
sacrificed after 1 h and blood was collected by cardiac puncture in 200 µL ACD,
350 µL Tyrode’s buffer pH 6.5. Platelet-free plasma (PFP) was prepared by two
rounds of centrifugation at 2,500xg for 15 min at RT and stored at −80°C until hs-
FCM analysis.
Lymph Lymph was collected as previously described from 18-h-fasted C57BL/6J
mice8-9. For PEV analysis, lymph was centrifuged twice at 2,500xg for 15 min to
remove all residual cells. Freshly collected lymph was used for hs-FCM analysis and
frozen (at −80°C) lymph was used for immunoblotting. Platelet EVs in 1 µL lymph
were labeled with LWA300 (250 nM). The following antibodies were added for 30 min
at RT: BV421 anti-mouse CD41 (clone MWReg30, BD Biosciences), and MHC

171
Class I (H-2kb) monoclonal antibody (AF6-88.5.5.3) APC (Invitrogen). Samples were
then diluted with PBS and analyzed by hs-FCM.
Intravenous injection of PEVs PEVs were prepared from freshly isolated, washed
platelets adjusted to a density of 100 × 106 cells/mL, labeled with 1 μM 5-
chloromethylfluorescein diacetate (CMFDA, Invitrogen) for 10 min at RT and then
stimulated overnight at RT with collagen (0.5 μg/mL) after addition of
5 mM calcium10. Platelet activation was terminated by addition of 20 mM EDTA, and
remnant platelets were removed by centrifugation at 2,000xg for 10 min at RT.
Platelet EVs were then ultracentrifuged (18,000xg for 90 min at 18C), resuspended
in Tyrode’s Buffer 7.4 with 5 mM calcium and stored at −80°C. CMFDA-labelled EVs
were thawed just before injection and fluorescence and count were verified by hs-
FCM. Mice were injected i.v. with 5 × 108 CMFDA-labeled EVs in 100 µL or
equivalent volume of control (PBS / Tyrode’s buffer pH 7.4 with 5 mM calcium). After
1 h, mice were sacrificed and blood, lymph nodes (LNs) (popliteal, inguinal,
mesenteric and mandibular) and spleen were harvested and processed in the dark.
Platelet-free plasma (PFP) was prepared by centrifugation of whole blood as above
and 5 µL of PFP was fixed in 500 µL final volume of PBS 2% PFA for analysis by hs-
FCM. Spleen and pooled LNs were dissected into small pieces and incubated in
1 mL of collagenase type IV (0.4 mg/mL in HBSS 1×, Biochemical Corporation) for
90 min at 37°C in a 6-well plate. Organ disruption was completed by pipetting up and
down followed by passage through a 70 µm cell strainer with the addition of 1 mL
PBS. Cells were centrifuged at 500xg for 5 min at 4°C. Spleen pellets (but not LN
pellets) were resuspended in 1 mL of ultrapure H2O for 20 s to remove RBCs. Lysis
was stopped by the addition of 110 µL Hanks' Balanced Salt Solution (HBSS) 10×
and cells were centrifuged at 500xg for 5 min at 4°C. Spleen and LN pellets were
resuspended in PBS. One hundred µL of cell suspension were labeled with Hoechst
(Invitrogen) for 10 min at RT and fixed with 100 µL 4% PFA before cytospinning
(500xg for 3 min at RT). Cells were mounted with SlowFadeTM Gold Antifade
Mountant (Invitrogen) on microscope slides. Images were acquired with a Quorum
Wave FX Spinning Disk Confocal Microscope (Borealis - Leica DMI 6000B) at 63×

172
(LEICA HCX PL APO 63× 1.30 glycerol objective). Images were processed using
Volocity software.
Dendritic cell preparation Splenocytes were obtained from C57BL/6J mice by
treating the spleen with collagenase D (400 Mandl U/mL; Roche Diagnostics) and
DNAse (50 μg/mL; Roche Diagnostics) for 30 min at 37°C as previously described11-
12. Fetal bovine serum was added and the spleen was dilacerated between two
microscope slides. The cells were then filtered through 40 µm cell strainers in
complete medium: (RPMI 1640–glutamate, 50 U/mL penicillin-streptomycin, 10 mM
HEPES, 1 mM sodium pyruvate [Thermo Fisher Scientific], and 5% SVF). Cells were
washed twice in complete medium by centrifugation at 700xg for 10 min. Dendritic
cells were enriched from splenocytes with 250 µL of the Dendritic Cell Enrichment
Set (BD Biosciences) biotinylated antibodies for 15 min at 4°C. Cells were washed
and 250 µL streptavidin beads were added for 30 min at 4°C before magnetic
separation (10 min on the magnet). Dendritic cells were counted and used
immediately.
Data analysis Hs-FCM data were analyzed using FlowJo software V.10.1 (BD
Biosciences). Statistical analyses were performed using GraphPad Prism 7.0
software (GraphPad Software). Gaussian distribution of the datasets was tested
prior to performing the statistical analysis. The test performed for each comparison
is specified in the corresponding legend, i.e. Wilcoxon, Mann–Whitney or Student’s
t-tests for 2 group comparisons and Friedman test followed by Dunn’s post-test for
multiple comparison for multi-experimental comparisons or Kruskal-Wallis. Only
relevant statistical tests are reported in the figures. Results are expressed as mean
± standard error of mean (SEM). The p-values (P) <0.05 were considered statistically
significant.
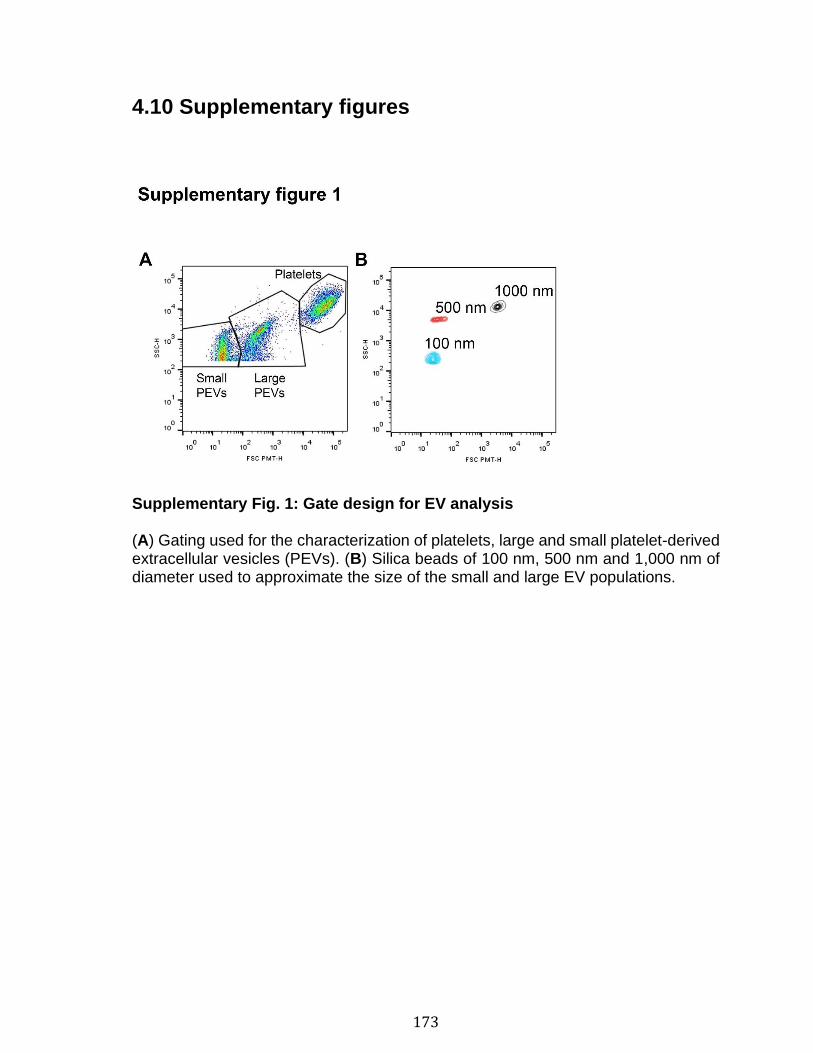
173
4.10 Supplementary figures
Supplementary Fig. 1: Gate design for EV analysis (A) Gating used for the characterization of platelets, large and small platelet-derived extracellular vesicles (PEVs). (B) Silica beads of 100 nm, 500 nm and 1,000 nm of diameter used to approximate the size of the small and large EV populations.
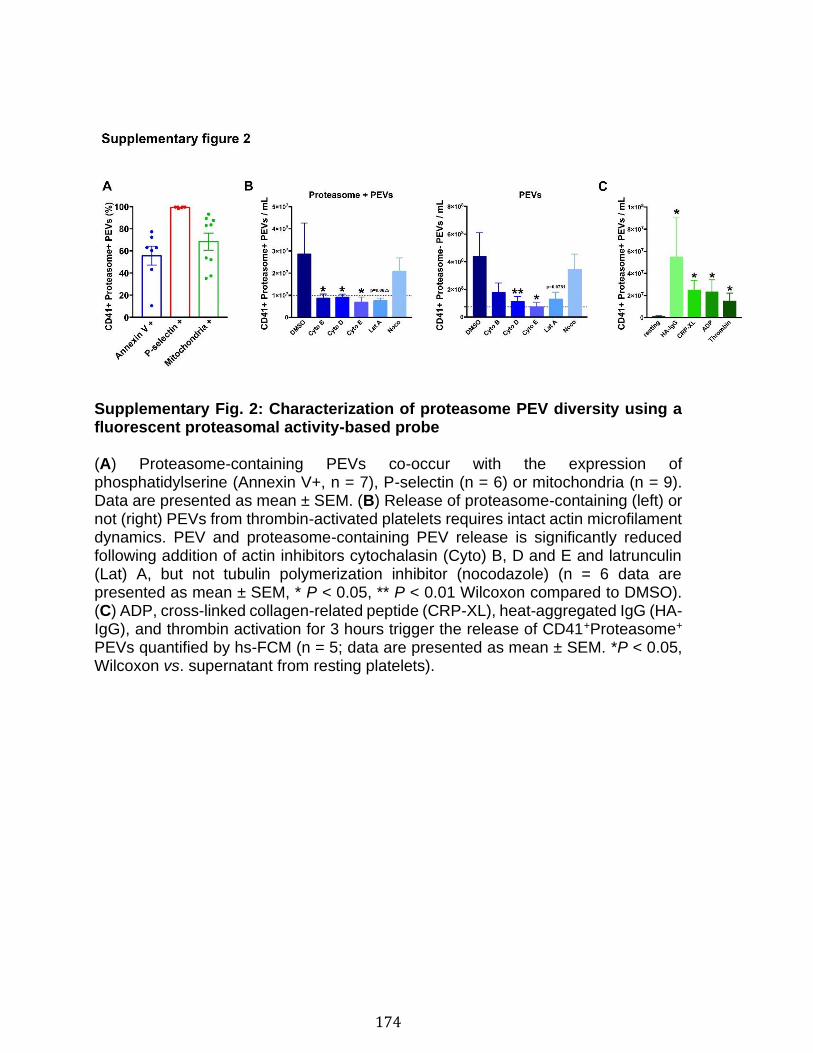
174
Supplementary Fig. 2: Characterization of proteasome PEV diversity using a fluorescent proteasomal activity-based probe (A) Proteasome-containing PEVs co-occur with the expression of phosphatidylserine (Annexin V+, n = 7), P-selectin (n = 6) or mitochondria (n = 9). Data are presented as mean ± SEM. (B) Release of proteasome-containing (left) or not (right) PEVs from thrombin-activated platelets requires intact actin microfilament dynamics. PEV and proteasome-containing PEV release is significantly reduced following addition of actin inhibitors cytochalasin (Cyto) B, D and E and latrunculin (Lat) A, but not tubulin polymerization inhibitor (nocodazole) (n = 6 data are presented as mean ± SEM, * P < 0.05, ** P < 0.01 Wilcoxon compared to DMSO). (C) ADP, cross-linked collagen-related peptide (CRP-XL), heat-aggregated IgG (HA-IgG), and thrombin activation for 3 hours trigger the release of CD41+Proteasome+ PEVs quantified by hs-FCM (n = 5; data are presented as mean ± SEM. *P < 0.05, Wilcoxon vs. supernatant from resting platelets).

175
4.11 Supplementary references
1. Cognasse F, Aloui C, Anh Nguyen K, et al. Platelet components associated with
adverse reactions: predictive value of mitochondrial DNA relative to biological response
modifiers. Transfusion. 2016;56(2):497-504.
2. Cognasse F, Sut C, Fromont E, Laradi S, Hamzeh-Cognasse H, Garraud O. Platelet
soluble CD40-ligand level is associated with transfusion adverse reactions in a mixed
threshold-and-hit model. Blood. 2017;130(11):1380-1383.
3. Marcoux G, Magron A, Sut C, et al. Platelet-derived extracellular vesicles convey
mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse
reactions. Transfusion (Paris). 2019;59(7):2403-2414.
4. Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria serving
as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation.
Blood. 2014;124(14):2173-2183.
5. Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect
against transfusion-related acute lung injury via IL-10. Blood. 2017;129(18):2557-2569.
6. Kapur R, Kim M, Rebetz J, et al. Gastrointestinal microbiota contributes to the
development of murine transfusion-related acute lung injury. Blood Adv. 2018;2(13):1651-
1663.
7. Cloutier N, Allaeys I, Marcoux G, et al. Platelets release pathogenic serotonin and
return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S
A. 2018;115(7):E1550-E1559.
8. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular
vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell
Vesicles. 2016;5:31427.
9. Tessandier N, Melki I, Cloutier N, et al. Platelets Disseminate Extracellular
Vesicles in Lymph in Rheumatoid Arthritis. Arterioscler Thromb Vasc Biol.
2020:ATVBAHA119313698.
10. Rousseau M, Belleannee C, Duchez AC, et al. Detection and quantification of
microparticles from different cellular lineages using flow cytometry. Evaluation of the
impact of secreted phospholipase A2 on microparticle assessment. PLoS One.
2015;10(1):e0116812.
11. Elayeb R, Tamagne M, Bierling P, Noizat-Pirenne F, Vingert B. Red blood cell
alloimmunization is influenced by the delay between Toll-like receptor agonist injection
and transfusion. Haematologica. 2016;101(2):209-218.
12. Elayeb R, Tamagne M, Pinheiro M, et al. Anti-CD20 Antibody Prevents Red Blood
Cell Alloimmunization in a Mouse Model. J Immunol. 2017;199(11):3771-3780.

176
Discussion
5.1 Mise en contexte
Les EV sont des vésicules membranaires de petite taille produites par tout type
cellulaire et libérées dans le milieu extracellulaire. L’émergence de leur importance
dans de nombreuses fonctions, principalement pour la communication intercellulaire
et la régulation de conditions physiologiques et pathologiques[341-343], a forcé la
mise en place et le développement de nombreux éléments afin d’améliorer la
recherche dans le domaine. Que ce soit via la création de l’ISEV et de bases de
données sur les EV ou bien via la mise en place de nombreux congrès, les
scientifiques ont contribué lors des dernières années à améliorer la qualité de la
recherche sur les EV[366, 367, 369-371]. En 2019, la dernière version des
exigences expérimentales minimales pour la définition des EV et de leurs fonctions
a été publiée[368]. Cette version est la troisième publiée depuis mon arrivée dans le
laboratoire (2013) et le début de ma thèse (fin 2015). Étant donné les efforts mis en
place dans les dernières années, notamment concernant l’institution d’une
nomenclature cohérente des EV[366], vous aurez probablement remarqué une
évolution entre les articles insérés dans ce manuscrit.
Dès le début de ma thèse, un effort constant a été déployé pour respecter les
exigences expérimentales minimales. Bien que la taille des EV que j’ai étudiées par
cytométrie en flux corresponde davantage aux microvésicules qu’aux exosomes,
nous avons toujours fait preuve de prudence dans notre nomenclature. Nous avons
évité le terme microvésicule/microparticule, n’ayant jamais visualisé la production
des vésicules par des techniques d’imagerie en temps réel ce qui est nécessaire
selon le MISEV pour confirmer leur origine[367]. Concernant la caractérisation des
EV par cytométrie en flux, les articles des trois chapitres respectent les exigences
minimales. En effet, nous avons toujours fait preuve de transparence dans notre
caractérisation des EV en donnant une description détaillée de la collection, de

177
l’isolation, de l’entreposage, des protocoles de marquages et des caractéristiques
du cytomètre utilisé. Chacun des articles présente, dans ses premières figures, tous
les contrôles nécessaires pour confirmer la spécificité des marquages et la nature
membranaire des EV étudiées ainsi que leur taille et leur quantité. Non seulement
le domaine a grandement évolué, mes connaissances se sont aussi approfondies.
Cette discussion aura donc pour but de faire un résumé des découvertes, mais aussi
de prendre du recul sur les résultats publiés au cours des dernières années et de
leur impact.
Mes travaux présentés dans cette thèse portent sur diversité des EV de
plaquettes, plus précisément en ce qui concerne leur contenu en
mitochondries et en protéasome et les différentes fonctions qu’elles peuvent
exercer dans l’inflammation et l’immunité
5.2 Résumé des découvertes et discussion
Un regard neuf sur la diversité des EV
Les EV sont hétérogènes et cette diversité pourrait permettre leur utilisation comme
biomarqueur[378, 383, 450, 451]. Bien que les techniques d’isolation et de
caractérisation des EV aient été améliorées dans les dernières années, l’utilisation
de la cytométrie en flux pour leur analyse n’est toujours pas optimale. La séparation
entre les EV et le bruit de fond de l’appareil est difficile, de même que la mise en
évidence de populations rares[451]. De plus, en cherchant à étudier la diversité et
l’hétérogénéité des EVs, il est nécessaire d’utiliser plusieurs marqueurs ce qui
complique l’analyse et celle-ci peut varier considérablement entre les utilisateurs.
Comme l’interprétation des résultats était un défi important et qu’à cette époque il
n’y avait aucune étude rapportant un moyen de faciliter l'interprétation des analyses
cytofluorométriques des EV, nous avons cherché une solution. Parmi les approches
déjà utilisées pour l’analyse des cellules, nous avons sélectionné l’algorithme

178
SPADE[452, 453]. Cet algorithme présentait l’avantage d’une analyse automatique,
donc non influencée par l’utilisateur[452, 454]. Il permettait également la
visualisation de populations peu abondantes et d’obtenir une vue d'ensemble de
l'hétérogénéité cellulaire en montrant une continuité du phénotype.
L’application de l’algorithme SPADE à l’analyse de nos EV par cytométrie en flux à
haute sensibilité nous a permis d’organiser les sous-populations d’EV de plaquettes
et de globules rouges et d’apprécier leur hétérogénéité. Nous avons ainsi pu
appliquer cette technique à l’analyse de la composition des EV présentes dans le
liquide synovial de patients arthritiques. Notre étude a révélé que l’utilisation
d’algorithmes couplés à la cytométrie en flux tels que SPADE pourrait faciliter la
compréhension des fonctions des EV et le développement de leur étude comme
biomarqueurs.
Avec du recul, cette combinaison entre l’utilisation d’un algorithme et l’analyse de la
diversité des EV par cytométrie en flux était innovante. Bien que l’article ait eu un
impact limité dans le domaine avec 19 citations[455-471], nous avons pu montrer
que l’hétérogénéité des EV ajoute un niveau de complexité supérieur pour l’analyse,
mais que celui-ci est nécessaire pour l’avancement des connaissances dans le
domaine. L’étude des sous-populations d’un même type d’EV, de plaquettes dans
le cas présent, a permis de stratifier des patients arthritiques. De plus, cette étude a
encouragé d’autres équipes de recherche à utiliser SPADE et des algorithmes
similaires pour étudier les EV[468, 469]. Alors qu’en 2015-2016, il était nécessaire
d’avoir des connaissances en bio-informatique pour pouvoir utiliser SPADE, cet
algorithme et d’autres sont maintenant intégrés directement dans les logiciels
d’analyse classiques de cytométrie en flux tels que FlowJo et FCS Express. Bien
que j’ai personnellement aimé apprendre des notions de bio-informatique, je crois
qu’il était nécessaire de rendre l’utilisation d’algorithmes plus accessible à tous pour
favoriser leur application. En conclusion, nous avons contribué à mettre l’emphase
sur l’importance de l’analyse de l’hétérogénéité des EV. Une meilleure
caractérisation de l’hétérogénéité des EV, basée non seulement sur leur source et

179
leur taille, mais aussi sur leur cargo pourra nous permettre une meilleure
compréhension de leur rôle critique dans la physiologie et les pathologies.
dans l’inflammation…
Dans la continuité de ce projet, je me suis intéressée à la diversité du cargo contenu
dans les EV de plaquettes, notamment concernant le contenu en organelles. Notre
laboratoire a déjà montré que les EV de plaquettes, lors de leur genèse, peuvent
apporter avec elles des mitochondries fonctionnelles situées à proximité de la
membrane plasmique[382]. Ces dernières forment une population hétérogène selon
la présence ou l’absence de mitochondries[382]. Des mitochondries extracellulaires,
qui ne sont pas entourées d’une membrane de plaquette (freeMitos), ont aussi été
observées dans les liquides biologiques et les PC servant pour la transfusion[382].
L’hydrolyse de la membrane mitochondriale par la phospholipase A2 IIA entraîne
non seulement la libération de médiateurs inflammatoires[382], mais de l’ADN
mitochondrial est aussi retrouvé dans les EV et est potentiellement
inflammatoire[429, 430].
Lors de mon projet de maitrise, je m’étais intéressée à la libération des sous-
populations d’EV de plaquettes et de leur contenu en mitochondries dans les PC
préparés selon trois techniques différentes, soit le plasma riche en plaquettes, la
couche leucoplaquettaire et l’aphérèse sur une période de 7 jours. Cette étude nous
a permis de conclure que le mode de préparation, plutôt que la durée de stockage,
a un impact sur la libération des EV contenant ou non des mitochondries dans les
PC[449]. Toutefois, même après 7 jours d’entreposage, les PC étudiés dans cette
étude avaient un niveau d’ADN mitochondrial bien inférieur à la quantité détectée
dans les PC impliqués dans des réactions transfusionnelles[382, 449]. Ainsi, bien
que les EV de plaquettes semblent être impliquées dans les réactions
transfusionnelles, des études supplémentaires adressant le mécanisme de
libération de l’ADN mitochondrial dans les PC et le mécanisme d’implication des EV
étaient nécessaires.

180
Comme aucune étude n'avait officiellement vérifié la présence de sous-populations
d’EV de plaquettes dans les PC impliqués dans des réactions transfusionnelles
indésirables, nous avons exploré ce lien dans les PC qui ont induit ou non des
réactions transfusionnelles. Nous avons donc émis l’hypothèse que les EV de
plaquettes contenant des mitochondries représentent un réservoir d'ADN
mitochondrial. Nous avons observé que non seulement les EV contenant des
mitochondries étaient plus abondantes dans les PC impliqués dans des réactions
transfusionnelles, mais qu’elles corrélaient significativement avec l’ADN
mitochondrial.[472] De plus, nous avons confirmé que la vaste majorité de l’ADN
mitochondrial détecté dans les PC est encapsulé dans des EV[472]. Cette étude
nécessite d’être répliquée dans une cohorte plus grande, mais elle suggère que la
quantification d’EV de plaquette pourrait être un biomarqueur potentiel pour la
prévention de réactions transfusionnelles.
La présence d’un cargo mitochondrial n’est pas unique aux vésicules de plaquettes
et sont utilisation en tant que biomarqueur est de plus en plus envisagée en dehors
du domaine transfusionnel. Par exemple, des EV contenant des DAMPS
mitochondriaux sont présentes en plus grande quantité chez les patients atteints de
la maladie de Parkinson[473, 474]. Ces EV contenant des mitochondries sont aussi
augmentées dans le plasma de patients atteints de mélanome, de cancer des
ovaires et de cancer du sein[475]. L’absence ou la diminution du cargo mitochondrial
peut aussi être indicateur de maladies. Par exemple, bien qu’une grande quantité
d’EV de plaquettes soit produite lors d’infection par le HIV, la proportion d’EV
contenant des mitochondries et leur densité est inférieure chez les malades que
chez les contrôles[476]. La diminution du contenu mitochondrial dans les EV est
aussi un marqueur potentiel pour le diagnostic des troubles du spectre autistique et
le syndrome du X fragile[477].
Bien qu’elle requiert un faible volume d’échantillon à analyser, cette approche serait
toutefois limitée étant donné qu’elle implique l’utilisation d’un cytomètre adapté à
l’analyse des EV. De plus, l’harmonisation de l’analyse des EV entre les différents

181
laboratoires est complexe et nécessite d’avoir des utilisateurs expérimentés. Étant
donné la forte corrélation entre la présence d’EV de plaquettes contenant des
mitochondries et la quantité d’ADN mitochondrial dans les PC, une approche plus
simple et plus accessible serait l’utilisation de la PCR quantitative ou d’une technique
similaire pour quantifier l’ADN mitochondrial.
Cette étude suggère que les EV et leur cargo en mitochondrie peuvent être un
biomarqueur utile pour la prédiction du risque potentiel de réactions
transfusionnelles. Il reste toutefois à déterminer si l’ADN mitochondrial encapsulé
dans les EV, par opposition à l'ADN mitochondrial libre dans le plasma, est aussi
inflammatoire. Considérant que l'induction des EV de plaquettes est observée
fréquemment en conditions inflammatoires, il serait pertinent de valider si leur
présence explique aussi la présence d’ADN mitochondrial. Il faudrait donc évaluer
si l’ADN mitochondrial encapsulé a le potentiel d’activer le TLR9 in vivo aussi
efficacement que l’ADN libre. De plus, il sera important de considérer l’effet du reste
du cargo inflammatoire de la mitochondrie, tel que les cardiolipides, les ROS, l’ARN
mitochondrial[478] ou l’ATP qui sont responsables de dommages aux poumons[405]
en plus du cargo inflammatoire des EV de plaquettes.
Un modèle in vivo pertinent de réaction transfusionnel pour répondre à ces questions
serait le modèle de TRALI murin. Ce modèle a déjà été utilisé pour montrer
l’implication de plaquettes conservées plusieurs jours et d’EV libérées lors de
l’entreposage dans le développement du TRALI[479, 480]. Une continuité de ce
projet serait de continuer à décortiquer l’hétérogénéité des EV de plaquettes dans
le modèle de TRALI murin et d’évaluer leur effet dans l’initiation du TRALI et dans
les dommages aux poumons.
Un article récent a montré que l'emballage sélectif des protéines mitochondriales
dans des vésicules extracellulaires empêche la libération de DAMP
mitochondriaux[481]. Puisque la membrane vésiculaire confère une protection
contre la dégradation lors du transport[472], et que les EV de plaquettes sont
retrouvées en grande quantité dans la circulation, il serait important de considérer

182
un aspect que nous avons négligé. Les EV de nombreuses sources cellulaires
peuvent être utilisées pour effectuer un transfert horizontal du contenu mitochondrial
dans le traitement de diverses maladies. Par exemple, les cellules souches
mésenchymateuses sécrètent des EV contenant des mitochondries
fonctionnelles[482]. Celles-ci peuvent transférer leurs mitochondries à d’autres
cellules telles que les cellules épithéliales du poumon, ce qui améliore leur capacité
à refermer des plaies[483], ou bien aux macrophages pour les polariser vers un
phénotype anti-inflammatoire[482, 484]. Des EV de cellules souches dérivées de
cardiomyocytes contenant des mitochondries ont aussi été utilisées pour améliorer
les fonctions cardiaques post ischémie et offrent une possibilité thérapeutique pour
les maladies cardiaques[485]. Les cellules souches neuronales peuvent aussi
transférer leurs mitochondries via la production d’EV aux phagocytes
mononucléaires dans un modèle murin de sclérose en plaques. Ce transfert à
permis de rétablir les dysfonctions mitochondriales présentes et d’améliorer l’état
clinique ce qui suggère un traitement potentiel non seulement pour la sclérose en
plaques, mais aussi pour les autres maladies neurodégénératives[486]. D’ailleurs,
les EV d’astrocytes contenant des mitochondries se sont avérées efficaces dans le
traitement de maladies telles que l’Alzheimer[487] et les accidents vasculaires
cérébraux[477]. Ces exemples sont la preuve que non seulement les EV de
plaquettes contenant des mitochondries ont un potentiel comme biomarqueur, elles
pourraient aussi être utilisées d’un point de vue thérapeutique.
…et dans l’immunité
Lors de ma thèse, j’ai eu la chance de collaborer sur un autre projet visant à mettre
en lumière la diversité du contenu en organelles des EV de plaquettes, non pas
uniquement dans un contexte d’inflammation, mais égalemment d’immunité. J’ai
initialement contribué à ce projet pour finalement l’amener à terme lors du départ de
la post-doctorante qui l’avait initié. Ceci m’a permis de m’éloigner du domaine de la
transfusion et de m’intéresser davantage au rôle immunitaire des plaquettes,
élargissant mon champ d’expertise développé pendant le doctorat.

183
Les plaquettes et les mégacaryocytes contribuent à l’immunité innée et
adaptative[488]. Elles contiennent un protéasome actif et présentent des antigènes
par l'intermédiaire du CMH I[143, 318, 319, 338]. Pour ce projet, nous avons émis
l'hypothèse qu'une machinerie fonctionnelle d’apprêtement et de présentation de
l'antigène est transférée aux EV de plaquettes lors de leur activation. Nous avons
non seulement démontré la présence d'un protéasome 20S actif dans les EV de
plaquettes dans diverses conditions où les plaquettes sont activées, mais aussi
démontré que les PEV incubées avec l’OVA peuvent apprêter et présenter
efficacement l’antigène.
Les PEV ne sont pas les seuls types d'EV qui contribuent à la présentation de
l'antigène. Il a déjà été prouvé que les EV dérivées de cellules immunitaires telles
que les DC, les lymphocytes B, les lymphocytes T, les macrophages et les cellules
NK ont un rôle dans la présentation antigénique [80-85]. Tout comme nous l’avons
montré pour les plaquettes, il a été mis en évidence que les EV dérivées de cellules
NK contiennent des composants du système protéasome-ubiquitine[489] et que les
lymphocytes T exportent du protéasome via leurs vésicules, ce qui explique une
grande partie du protéasome extracellulaire observé dans de nombreuses
conditions pathologiques[418, 467]. Les cellules endothéliales libèrent aussi des EV
contenant du protéasome et ces dernières induisent la production d’autoanticorps et
accélèrent le rejet de greffe[169]. Les EV dérivées de macrophages associées aux
tumeurs, de cellules souches mésenchymateuses et d’ostéoclastes contiennent
aussi du protéasome et participent au remodelage tissulaire[419, 490, 491]. Cette
fonction de remodelage est d’ailleurs utilisée par de nombreux pathogènes tels que
Plasmodium falciparum, Acanthamoeba castellanii, Toxoplasma gondii,
Echinococcus granulosus, Leishmania, Trichuris muris, Trichomonas vaginalis et
certains champignons pour altérer la membrane cellulaire de son hôte et faciliter
l’infection[492]. Ceci est un élément supplémentaire appuyant le rôle des EV, et par
extension des plaquettes, dans la régulation de l’immunité en conditions
physiologiques ou pathologiques[493, 494]. De plus, en plus de la présentation

184
antigénique, il n’est pas exclu que les EV de plaquettes puissent elle aussi participer
au rejet de greffe, à la production d’autoanticorps, à la réponse antitumorale et au
remodelage tissulaire comme c’est le cas pour les EV d’autres types cellulaires.
Il a récemment été démontré que les MK pulmonaires peuvent induire l’activation de
lymphocytes T CD4+ via le CMH II in vitro et in vivo[337]. Cet article a soulevé de
nombreuses questions. Premièrement, est-ce que les EV de plaquettes pouvant
effectuer la présentation antigénique proviennent de plaquettes strictement dérivées
des MK pulmonaires? Deuxièmement, est-ce que les plaquettes dérivées des MK
pulmonaires expriment aussi le CMH II et, par conséquent, est-ce que les EV de MK
pulmonaires et de plaquettes peuvent aussi effectuer la présentation aux
lymphocytes T CD4+ ? Mon étude s’est focalisée sur les EV de plaquettes, mais il
faudra aussi vérifier si les EV dérivées de MK sont capables d’effectuer la
présentation antigénique via le CMH I ou le CMH II en émettant l’hypothèse que la
présentation puisse même être augmentée dans les cas des EV dérivées des MK
pulmonaires.
Les EV de plaquettes ayant un accès privilégié au système lymphatique et aux
organes lymphoïdes, elles contribueraient à une fonction immunitaire
supplémentaire des plaquettes, cette fois en dehors de la circulation sanguine. Il
sera cependant nécessaire de déterminer l’impact et l’importance réelle des EV de
plaquettes en tant qu’éléments présentant l’antigène. Une autre limite de cette étude
est que le mécanisme exact d’apprêtement et de présentation de l’antigène n’est
pas connu. Ceci est d’ailleurs le cas pour la présentation antigénique par les
plaquettes et par les autres types d’EV. Un effort collectif sera nécessaire dans les
prochaines années pour mieux décortiquer et comprendre la contribution des EV
dans la présentation antigénique.

185
Conclusion
Les travaux présents dans cette thèse montrent que les plaquettes activées libèrent
à la fois des mitochondries et du protéasome. En étudiant la diversité du contenu en
organelles des EV de plaquettes en contexte inflammatoire et immunitaire, cette
étude a mis en évidence l’importance de s’intéresser à l’hétérogénéité des EV et
soutient le concept selon lequel différents sous-types de PEV peuvent jouer des
rôles différents en fonction de leur cargo.

186
Bibliographie
1. Ribatti, D. and E. Crivellato, Giulio Bizzozero and the discovery of platelets. Leuk
Res, 2007. 31(10): p. 1339-41.
2. Semple, J.W., J.E. Italiano, Jr., and J. Freedman, Platelets and the immune
continuum. Nat Rev Immunol, 2011. 11(4): p. 264-74.
3. Pietersz, R.N., Pooled platelet concentrates: an alternative to single donor
apheresis platelets? Transfus Apher Sci, 2009. 41(2): p. 115-9.
4. Wright, J.H., The histogenesis of the blood platelets. 1910, n. p.
5. Kuter, D.J., Milestones in understanding platelet production: a historical overview.
Br J Haematol, 2014. 165(2): p. 248-58.
6. King, S.M. and G.L. Reed, Development of platelet secretory granules. Semin Cell
Dev Biol, 2002. 13(4): p. 293-302.
7. Leung, R., et al., Persistence of procoagulant surface expression on activated
human platelets: involvement of apoptosis and aminophospholipid translocase
activity. J Thromb Haemost, 2007. 5(3): p. 560-70.
8. Kunicki, T.J., Platelet glycoprotein antigens and immune receptors. Prog Clin Biol
Res, 1988. 283: p. 87-123.
9. Choi, W., Z.A. Karim, and S.W. Whiteheart, Protein expression in platelets from
six species that differ in their open canalicular system. Platelets, 2010. 21(3): p.
167-75.
10. Blair, P. and R. Flaumenhaft, Platelet alpha-granules: basic biology and clinical
correlates. Blood Rev, 2009. 23(4): p. 177-89.
11. Sharda, A. and R. Flaumenhaft, The life cycle of platelet granules. F1000Res, 2018.
7: p. 236.
12. Chen, Y., Y. Yuan, and W. Li, Sorting machineries: how platelet-dense granules
differ from α-granules. Biosci Rep, 2018. 38(5).
13. Reed, G.L., Platelet secretory mechanisms. Semin Thromb Hemost, 2004. 30(4): p.
441-50.
14. Frojmovic, M.M. and J.G. Milton, Human platelet size, shape, and related functions
in health and disease. Physiol Rev, 1982. 62(1): p. 185-261.
15. Koseoglu, S. and R. Flaumenhaft, Advances in platelet granule biology. Curr Opin
Hematol, 2013. 20(5): p. 464-71.
16. Pagel, O., et al., Taking the stock of granule cargo: Platelet releasate proteomics.
Platelets, 2017. 28(2): p. 119-128.
17. White, J.G., Ultrastructural studies of the gray platelet syndrome. Am J Pathol,
1979. 95(2): p. 445-62.
18. Thon, J.N., et al., T granules in human platelets function in TLR9 organization and
signaling. J Cell Biol, 2012. 198(4): p. 561-74.
19. Hartwig, J.H., Mechanisms of actin rearrangements mediating platelet activation. J
Cell Biol, 1992. 118(6): p. 1421-42.
20. Cooper, J.A., Effects of cytochalasin and phalloidin on actin. J Cell Biol, 1987.
105(4): p. 1473-8.

187
21. Florian, S. and T.J. Mitchison, Anti-Microtubule Drugs. Methods Mol Biol, 2016.
1413: p. 403-21.
22. Davi, G. and C. Patrono, Platelet activation and atherothrombosis. N Engl J Med,
2007. 357(24): p. 2482-94.
23. Garraud, O. and F. Cognasse, Are Platelets Cells? And if Yes, are They Immune
Cells? Front Immunol, 2015. 6: p. 70.
24. Deutsch, V.R. and A. Tomer, Megakaryocyte development and platelet production.
Br J Haematol, 2006. 134(5): p. 453-66.
25. Patel, S.R., et al., Differential roles of microtubule assembly and sliding in
proplatelet formation by megakaryocytes. Blood, 2005. 106(13): p. 4076-85.
26. Thon, J.N. and J.E. Italiano, Platelet formation. Semin Hematol, 2010. 47(3): p.
220-6.
27. Cunin, P. and P.A. Nigrovic, Megakaryocytes as immune cells. J Leukoc Biol,
2019. 105(6): p. 1111-1121.
28. Thon, J.N. and J.E. Italiano, Platelets: production, morphology and ultrastructure.
Handb Exp Pharmacol, 2012(210): p. 3-22.
29. Kaushansky, K., The molecular mechanisms that control thrombopoiesis. J Clin
Invest, 2005. 115(12): p. 3339-47.
30. Peck-Radosavljevic, M., et al., Is inadequate thrombopoietin production a major
cause of thrombocytopenia in cirrhosis of the liver? J Hepatol, 1997. 27(1): p. 127-
31.
31. Peck-Radosavljevic, M., Thrombocytopenia in liver disease. Can J Gastroenterol,
2000. 14 Suppl D: p. 60D-66D.
32. Emmons, R.V., et al., Human thrombopoietin levels are high when
thrombocytopenia is due to megakaryocyte deficiency and low when due to
increased platelet destruction. Blood, 1996. 87(10): p. 4068-71.
33. Nagata, Y., Y. Muro, and K. Todokoro, Thrombopoietin-induced polyploidization
of bone marrow megakaryocytes is due to a unique regulatory mechanism in late
mitosis. J Cell Biol, 1997. 139(2): p. 449-57.
34. Raslova, H., et al., Megakaryocyte polyploidization is associated with a functional
gene amplification. Blood, 2003. 101(2): p. 541-4.
35. Raslova, H., et al., Interrelation between polyploidization and megakaryocyte
differentiation: a gene profiling approach. Blood, 2007. 109(8): p. 3225-34.
36. Patel, S.R., J.H. Hartwig, and J.E. Italiano, Jr., The biogenesis of platelets from
megakaryocyte proplatelets. J Clin Invest, 2005. 115(12): p. 3348-54.
37. Schulze, H. and R.A. Shivdasani, Molecular mechanisms of megakaryocyte
differentiation. Semin Thromb Hemost, 2004. 30(4): p. 389-98.
38. Cramer, E.M., et al., Ultrastructure of platelet formation by human megakaryocytes
cultured with the Mpl ligand. Blood, 1997. 89(7): p. 2336-46.
39. Landry, P., et al., Existence of a microRNA pathway in anucleate platelets. Nat
Struct Mol Biol, 2009. 16(9): p. 961-6.
40. Denis, M.M., et al., Escaping the nuclear confines: signal-dependent pre-mRNA
splicing in anucleate platelets. Cell, 2005. 122(3): p. 379-91.
41. Richardson, J.L., et al., Mechanisms of organelle transport and capture along
proplatelets during platelet production. Blood, 2005. 106(13): p. 4066-75.

188
42. Sabri, S., et al., Deficiency in the Wiskott-Aldrich protein induces premature
proplatelet formation and platelet production in the bone marrow compartment.
Blood, 2006. 108(1): p. 134-40.
43. Jiang, J., D.S. Woulfe, and E.T. Papoutsakis, Shear enhances thrombopoiesis and
formation of microparticles that induce megakaryocytic differentiation of stem cells.
Blood, 2014. 124(13): p. 2094-103.
44. Ito, T., PAMPs and DAMPs as triggers for DIC. J Intensive Care, 2014. 2(1): p. 67.
45. Furie, B. and B.C. Furie, Mechanisms of thrombus formation. N Engl J Med, 2008.
359(9): p. 938-49.
46. Jackson, S.P., W.S. Nesbitt, and S. Kulkarni, Signaling events underlying thrombus
formation. J Thromb Haemost, 2003. 1(7): p. 1602-12.
47. Khorana, A.A., et al., Blood transfusions, thrombosis, and mortality in hospitalized
patients with cancer. Arch Intern Med, 2008. 168(21): p. 2377-81.
48. Lozano, M. and J. Cid, Transfusion medicine as of 2014. F1000Prime Rep, 2014. 6:
p. 105.
49. Gill, R., Practical management of major blood loss. Anaesthesia, 2015. 70 Suppl 1:
p. 54-7, e19-20.
50. Parker, R.I., Transfusion in critically ill children: indications, risks, and challenges.
Crit Care Med, 2014. 42(3): p. 675-90.
51. Hersh, E.M., et al., Causes of Death in Acute Leukemia: A Ten-Year Study of 414
Patients from 1954-1963. JAMA, 1965. 193: p. 105-9.
52. Slichter, S.J. and L.A. Harker, Preparation and storage of platelet concentrates. I.
Factors influencing the harvest of viable platelets from whole blood. Br J Haematol,
1976. 34(3): p. 395-402.
53. Pietersz, R.N., J.A. Loos, and H.W. Reesink, Platelet concentrates stored in plasma
for 72 hours at 22 degrees C prepared from buffycoats of citrate-phosphate-
dextrose blood collected in a quadruple-bag saline-adenine-glucose-mannitol
system. Vox Sang, 1985. 49(2): p. 81-5.
54. Cardigan, R. and L.M. Williamson, The quality of platelets after storage for 7 days.
Transfus Med, 2003. 13(4): p. 173-87.
55. Hagberg, I.A., et al., Apheresis-induced platelet activation:comparison of three
types of cell separators. Transfusion, 2000. 40(2): p. 182-92.
56. Macher, S., et al., Function and activation state of platelets in vitro depend on
apheresis modality. Vox Sang, 2010. 99(4): p. 332-40.
57. Murphy, M.F. and A.H. Waters, Clinical aspects of platelet transfusions. Blood
Coagul Fibrinolysis, 1991. 2(2): p. 389-96.
58. American Association of Blood Banks, Preparation of platelets from whole blood. .
2008, Roback JD (ed). : Bethesda (MD): American Association of Blood Banks. p.
199.
59. Fijnheer, R., et al., Platelet activation during preparation of platelet concentrates: a
comparison of the platelet-rich plasma and the buffy coat methods. Transfusion,
1990. 30(7): p. 634-8.
60. Metcalfe, P., et al., Activation during preparation of therapeutic platelets affects
deterioration during storage: a comparative flow cytometric study of different
production methods. Br J Haematol, 1997. 98(1): p. 86-95.

189
61. Skripchenko, A., et al., Platelet products prepared by different methods of
sedimentation undergo platelet activation differently during storage. Transfusion,
2008. 48(7): p. 1469-77.
62. Wagner, S.J., S. Seetharaman, and J. Kurtz, A rest period does not affect in vitro
storage properties in apheresis platelets collected from the buffy coat layer.
Transfusion, 2012. 52(11): p. 2427-31.
63. Murphy, S. and F.H. Gardner, Effect of storage temperature on maintenance of
platelet viability--deleterious effect of refrigerated storage. N Engl J Med, 1969.
280(20): p. 1094-8.
64. Lelkens, C.C., et al., Experiences with frozen blood products in the Netherlands
military. Transfus Apher Sci, 2006. 34(3): p. 289-98.
65. Johnson, L., et al., In vitro comparison of cryopreserved and liquid platelets:
potential clinical implications. Transfusion, 2014.
66. Macdonald, N.E., S.F. O'Brien, and G. Delage, Transfusion and risk of infection in
Canada: Update 2012. Paediatr Child Health, 2012. 17(10): p. e102-6.
67. Stroncek, D.F. and P. Rebulla, Platelet transfusions. Lancet, 2007. 370(9585): p.
427-38.
68. AuBuchon, J.P., et al., Experience with universal bacterial culturing to detect
contamination of apheresis platelet units in a hospital transfusion service.
Transfusion, 2002. 42(7): p. 855-61.
69. Dumont, L.J., et al., Seven-day storage of single-donor platelets: recovery and
survival in an autologous transfusion study. Transfusion, 2002. 42(7): p. 847-54.
70. Ezuki, S., et al., Survival and recovery of apheresis platelets stored in a polyolefin
container with high oxygen permeability. Vox Sang, 2008. 94(4): p. 292-8.
71. van der Meer, P.F. and D. de Korte, Platelet preservation: agitation and containers.
Transfus Apher Sci, 2011. 44(3): p. 297-304.
72. Murphy, S. and F.H. Gardner, Platelet storage at 22 degrees C: role of gas
transport across plastic containers in maintenance of viability. Blood, 1975. 46(2):
p. 209-18.
73. Kilkson, H., S. Holme, and S. Murphy, Platelet metabolism during storage of
platelet concentrates at 22 degrees C. Blood, 1984. 64(2): p. 406-14.
74. Prowse, C.V., et al., Commercially available blood storage containers. Vox Sang,
2014. 106(1): p. 1-13.
75. Murphy, S., The efficacy of synthetic media in the storage of human platelets for
transfusion. Transfus Med Rev, 1999. 13(3): p. 153-63.
76. Seghatchian, J. and P. Krailadsiri, Current methods for the preparation of platelet
concentrates: laboratory and clinical aspects. Transfus Sci, 1997. 18(1): p. 27-32.
77. de Wildt-Eggen, J., et al., Reactions and platelet increments after transfusion of
platelet concentrates in plasma or an additive solution: a prospective, randomized
study. Transfusion, 2000. 40(4): p. 398-403.
78. Seghatchian, J. and P. Krailadsiri, The platelet storage lesion. Transfus Med Rev,
1997. 11(2): p. 130-44.
79. Bessos, H., et al., Glycoproteins Ib and IIb/IIIa in the quality assessment of platelet
concentrates during storage. Blood Coagul Fibrinolysis, 1992. 3(5): p. 633-6.
80. George, J.N., E.B. Pickett, and R. Heinz, Platelet membrane glycoprotein changes
during the preparation and storage of platelet concentrates. Transfusion, 1988.
28(2): p. 123-6.

190
81. Shattil, S.J., et al., Changes in the platelet membrane glycoprotein IIb.IIIa complex
during platelet activation. J Biol Chem, 1985. 260(20): p. 11107-14.
82. Klinger, M.H., The storage lesion of platelets: ultrastructural and functional
aspects. Ann Hematol, 1996. 73(3): p. 103-12.
83. Tzima, E., et al., Annexin V relocates to the periphery of activated platelets
following thrombin activation: an ultrastructural immunohistochemical approach.
Cell Biol Int, 1999. 23(9): p. 629-35.
84. Stack, G. and E.L. Snyder, Cytokine generation in stored platelet concentrates.
Transfusion, 1994. 34(1): p. 20-5.
85. Hirayama, F., Current understanding of allergic transfusion reactions: incidence,
pathogenesis, laboratory tests, prevention and treatment. Br J Haematol, 2013.
160(4): p. 434-44.
86. Blajchman, M.A. and M. Goldman, Bacterial contamination of platelet
concentrates: incidence, significance, and prevention. Semin Hematol, 2001. 38(4
Suppl 11): p. 20-6.
87. Blajchman, M.A., A.M. Ali, and H.L. Richardson, Bacterial contamination of
cellular blood components. Vox Sang, 1994. 67 Suppl 3: p. 25-33.
88. Busch, M.P., Transfusion-transmitted viral infections: building bridges to
transfusion medicine to reduce risks and understand epidemiology and
pathogenesis. Transfusion, 2006. 46(9): p. 1624-40.
89. Benjamin, R.J., et al., Deferral of males who had sex with other males. Vox Sang,
2011. 101(4): p. 339-67.
90. Webert, K.E., et al., Proceedings of a Consensus Conference: pathogen
inactivation-making decisions about new technologies. Transfus Med Rev, 2008.
22(1): p. 1-34.
91. Eastlund, T., Monetary blood donation incentives and the risk of transfusion-
transmitted infection. Transfusion, 1998. 38(9): p. 874-82.
92. Alter, H.J., et al., Posttransfusion hepatitis after exclusion of commercial and
hepatitis-B antigen-positive donors. Ann Intern Med, 1972. 77(5): p. 691-9.
93. McDonald, C.P., et al., Evaluation of donor arm disinfection techniques. Vox Sang,
2001. 80(3): p. 135-41.
94. Gibson, T. and W. Norris, Skin fragments removed by injection needles. Lancet,
1958. 2(7054): p. 983-5.
95. Satake, M., et al., Frequency of bacterial contamination of platelet concentrates
before and after introduction of diversion method in Japan. Transfusion, 2009.
49(10): p. 2152-7.
96. Wagner, S.J., et al., Diversion of initial blood flow to prevent whole-blood
contamination by skin surface bacteria: an in vitro model. Transfusion, 2000. 40(3):
p. 335-8.
97. Bruneau, C., et al., Efficacy of a new collection procedure for preventing bacterial
contamination of whole-blood donations. Transfusion, 2001. 41(1): p. 74-81.
98. Eder, A.F., et al., Limiting and detecting bacterial contamination of apheresis
platelets: inlet-line diversion and increased culture volume improve component
safety. Transfusion, 2009. 49(8): p. 1554-63.
99. Bashir, S., et al., Pathogen inactivation of platelets using ultraviolet C light: effect
on in vitro function and recovery and survival of platelets. Transfusion, 2013. 53(5):
p. 990-1000.

191
100. Slichter, S.J., et al., Extended storage of buffy coat platelet concentrates in plasma
or a platelet additive solution. Transfusion, 2014. 54(9): p. 2283-91.
101. Morel, P., et al., [Prevention of bacterial risk: pathogen inactivation/detection of
bacteria]. Transfus Clin Biol, 2013. 20(2): p. 109-14.
102. Palavecino, E.L., R.A. Yomtovian, and M.R. Jacobs, Bacterial contamination of
platelets. Transfus Apher Sci, 2010. 42(1): p. 71-82.
103. Ness, P.M., An incremental solution to bacterial contamination: taking the first
step. Transfusion, 2004. 44(3): p. 311-2.
104. Yomtovian, R., Bacterial contamination of blood: lessons from the past and road
map for the future. Transfusion, 2004. 44(3): p. 450-60.
105. Gilliss, B.M., M.R. Looney, and M.A. Gropper, Reducing noninfectious risks of
blood transfusion. Anesthesiology, 2011. 115(3): p. 635-49.
106. Kleinman, S., P. Chan, and P. Robillard, Risks associated with transfusion of
cellular blood components in Canada. Transfus Med Rev, 2003. 17(2): p. 120-62.
107. Johansson, S.G., et al., Passive IgE-sensitization by blood transfusion. Allergy,
2005. 60(9): p. 1192-9.
108. Alam, A., et al., The prevention of transfusion-associated circulatory overload.
Transfus Med Rev, 2013. 27(2): p. 105-12.
109. Hawkins, J., R.H. Aster, and B.R. Curtis, Post-Transfusion Purpura: Current
Perspectives. J Blood Med, 2019. 10: p. 405-415.
110. Taaning, E. and F. Tonnesen, Pan-reactive platelet antibodies in post-transfusion
purpura. Vox Sang, 1999. 76(2): p. 120-3.
111. Heddle, N.M., Pathophysiology of febrile nonhemolytic transfusion reactions. Curr
Opin Hematol, 1999. 6(6): p. 420-6.
112. Heddle, N.M., Febrile nonhemolytic transfusion reactions to platelets. Curr Opin
Hematol, 1995. 2(6): p. 478-83.
113. Brown, C.J. and C.V. Navarrete, Clinical relevance of the HLA system in blood
transfusion. Vox Sang, 2011. 101(2): p. 93-105.
114. Silliman, C.C., D.R. Ambruso, and L.K. Boshkov, Transfusion-related acute lung
injury. Blood, 2005. 105(6): p. 2266-73.
115. Fung, Y.L., et al., Recipient T lymphocytes modulate the severity of antibody-
mediated transfusion-related acute lung injury. Blood, 2010. 116(16): p. 3073-9.
116. Sandler, S.G., et al., IgA anaphylactic transfusion reactions. Transfus Med Rev,
1995. 9(1): p. 1-8.
117. Shimada, E., et al., Anaphylactic transfusion reactions in haptoglobin-deficient
patients with IgE and IgG haptoglobin antibodies. Transfusion, 2002. 42(6): p. 766-
73.
118. Silliman, C.C., et al., Analysis of the priming activity of lipids generated during
routine storage of platelet concentrates. Transfusion, 1996. 36(2): p. 133-9.
119. Harr, J.N., et al., Antiplatelet therapy is associated with decreased transfusion-
associated risk of lung dysfunction, multiple organ failure, and mortality in trauma
patients. Crit Care Med, 2013. 41(2): p. 399-404.
120. Vlaar, A.P., et al., Supernatant of stored platelets causes lung inflammation and
coagulopathy in a novel in vivo transfusion model. Blood, 2010. 116(8): p. 1360-8.
121. Kanter, J., et al., Oncogenic and angiogenic growth factors accumulate during
routine storage of apheresis platelet concentrates. Clin Cancer Res, 2008. 14(12):
p. 3942-7.

192
122. Muylle, L., et al., Increased tumor necrosis factor alpha (TNF alpha), interleukin 1,
and interleukin 6 (IL-6) levels in the plasma of stored platelet concentrates:
relationship between TNF alpha and IL-6 levels and febrile transfusion reactions.
Transfusion, 1993. 33(3): p. 195-9.
123. Bayraktaroglu, Z., et al., Platelet storage time and cytokine (IL-2R, IL-8, TNF-
alpha) levels. East Mediterr Health J, 2007. 13(1): p. 79-84.
124. Wakamoto, S., et al., Biologic activity of RANTES in apheresis PLT concentrates
and its involvement in nonhemolytic transfusion reactions. Transfusion, 2003.
43(8): p. 1038-46.
125. Heddle, N.M., et al., The role of the plasma from platelet concentrates in
transfusion reactions. N Engl J Med, 1994. 331(10): p. 625-8.
126. Palmer, D.S., et al., Prevention of cytokine accumulation in platelets obtained with
the COBE spectra apheresis system. Vox Sang, 1998. 75(2): p. 115-23.
127. Ferrer, F., et al., Evaluation of leukocyte-depleted platelet concentrates obtained by
in-line filtration. Vox Sang, 2000. 78(4): p. 235-41.
128. Ferrer, F., et al., Evaluation of pooled platelet concentrates using prestorage versus
poststorage WBC reduction: impact of filtration timing. Transfusion, 2000. 40(7): p.
781-8.
129. Cardigan, R., et al., The influence of platelet additive solutions on cytokine levels
and complement activation in platelet concentrates during storage. Vox Sang,
2003. 84(1): p. 28-35.
130. Inwald, D.P., et al., CD40 is constitutively expressed on platelets and provides a
novel mechanism for platelet activation. Circ Res, 2003. 92(9): p. 1041-8.
131. Charafeddine, A.H., et al., Platelet-derived CD154: ultrastructural localization and
clinical correlation in organ transplantation. Am J Transplant, 2012. 12(11): p.
3143-51.
132. Cognasse, F., et al., Release of potential immunomodulatory factors during platelet
storage. Transfusion, 2006. 46(7): p. 1184-9.
133. Phipps, R.P., J. Kaufman, and N. Blumberg, Platelet derived CD154 (CD40 ligand)
and febrile responses to transfusion. Lancet, 2001. 357(9273): p. 2023-4.
134. Blumberg, N., et al., An association of soluble CD40 ligand (CD154) with adverse
reactions to platelet transfusions. Transfusion, 2006. 46(10): p. 1813-21.
135. Bux, J., Transfusion-related acute lung injury (TRALI): a serious adverse event of
blood transfusion. Vox Sang, 2005. 89(1): p. 1-10.
136. Triulzi, D.J., et al., The effect of previous pregnancy and transfusion on HLA
alloimmunization in blood donors: implications for a transfusion-related acute lung
injury risk reduction strategy. Transfusion, 2009. 49(9): p. 1825-35.
137. Kakaiya, R.M., et al., Prevalence of HLA antibodies in remotely transfused or
alloexposed volunteer blood donors. Transfusion, 2010. 50(6): p. 1328-34.
138. Eder, A.F., et al., Effective reduction of transfusion-related acute lung injury risk
with male-predominant plasma strategy in the American Red Cross (2006-2008).
Transfusion, 2010. 50(8): p. 1732-42.
139. Nakazawa, H., et al., Impact of fresh-frozen plasma from male-only donors versus
mixed-sex donors on postoperative respiratory function in surgical patients: a
prospective case-controlled study. Transfusion, 2009. 49(11): p. 2434-41.

193
140. Powers, A., et al., Testing only donors with a prior history of pregnancy or
transfusion is a logical and cost-effective transfusion-related acute lung injury
prevention strategy. Transfusion, 2008. 48(12): p. 2549-58.
141. Ribeiro, L.S., L. Migliari Branco, and B.S. Franklin, Regulation of Innate Immune
Responses by Platelets. Front Immunol, 2019. 10: p. 1320.
142. Rayes, J., et al., The dual role of platelet-innate immune cell interactions in
thrombo-inflammation. Res Pract Thromb Haemost, 2020. 4(1): p. 23-35.
143. Morrell, C.N., et al., Emerging roles for platelets as immune and inflammatory
cells. Blood, 2014. 123(18): p. 2759-67.
144. Kapur, R., et al., Nouvelle cuisine: platelets served with inflammation. J Immunol,
2015. 194(12): p. 5579-87.
145. Marcoux, G., et al., Role of platelets and megakaryocytes in adaptive immunity.
Platelets, 2020: p. 1-12.
146. Nicholson, L.B., The immune system. Essays Biochem, 2016. 60(3): p. 275-301.
147. Netea, M.G., et al., A guiding map for inflammation. Nat Immunol, 2017. 18(8): p.
826-831.
148. Akira, S., S. Uematsu, and O. Takeuchi, Pathogen recognition and innate immunity.
Cell, 2006. 124(4): p. 783-801.
149. Minnicozzi, M., R.T. Sawyer, and M.J. Fenton, Innate immunity in allergic disease.
Immunol Rev, 2011. 242(1): p. 106-27.
150. Zindel, J. and P. Kubes, DAMPs, PAMPs, and LAMPs in Immunity and Sterile
Inflammation. Annu Rev Pathol, 2020. 15: p. 493-518.
151. Corridoni, D. and A. Simmons, Innate immune receptors for cross-presentation:
The expanding role of NLRs. Mol Immunol, 2019. 113: p. 6-10.
152. Murphy, K., et al., Janeway's Immunobiology. 8th ed. ed. 2012, New York.
153. Farber, D.L., et al., Immunological memory: lessons from the past and a look to the
future. Nat Rev Immunol, 2016. 16(2): p. 124-8.
154. Netea, M.G., et al., Innate and Adaptive Immune Memory: an Evolutionary
Continuum in the Host's Response to Pathogens. Cell Host Microbe, 2019. 25(1): p.
13-26.
155. Danilova, N., The evolution of adaptive immunity. Adv Exp Med Biol, 2012. 738: p.
218-35.
156. Wieczorek, M., et al., Major Histocompatibility Complex (MHC) Class I and MHC
Class II Proteins: Conformational Plasticity in Antigen Presentation. Front
Immunol, 2017. 8: p. 292.
157. Yewdell, J.W., D. Dersh, and R. Fåhraeus, Peptide Channeling: The Key to MHC
Class I Immunosurveillance? Trends Cell Biol, 2019. 29(12): p. 929-939.
158. Jurewicz, M.M. and L.J. Stern, Class II MHC antigen processing in immune
tolerance and inflammation. Immunogenetics, 2019. 71(3): p. 171-187.
159. Bjorkman, P.J., MHC restriction in three dimensions: a view of T cell
receptor/ligand interactions. Cell, 1997. 89(2): p. 167-70.
160. Embgenbroich, M. and S. Burgdorf, Current Concepts of Antigen Cross-
Presentation. Front Immunol, 2018. 9: p. 1643.
161. Van Kaer, L., et al., Role of autophagy in MHC class I-restricted antigen
presentation. Mol Immunol, 2019. 113: p. 2-5.

194
162. Kasahara, M. and M.F. Flajnik, Origin and evolution of the specialized forms of
proteasomes involved in antigen presentation. Immunogenetics, 2019. 71(3): p.
251-261.
163. Tanaka, K., The proteasome: overview of structure and functions. Proc Jpn Acad
Ser B Phys Biol Sci, 2009. 85(1): p. 12-36.
164. Vigneron, N. and B.J. Van den Eynde, Proteasome subtypes and regulators in the
processing of antigenic peptides presented by class I molecules of the major
histocompatibility complex. Biomolecules, 2014. 4(4): p. 994-1025.
165. Rock, K.L. and A.L. Goldberg, Degradation of cell proteins and the generation of
MHC class I-presented peptides. Annu Rev Immunol, 1999. 17: p. 739-79.
166. Tomko, R.J. and M. Hochstrasser, Molecular architecture and assembly of the
eukaryotic proteasome. Annu Rev Biochem, 2013. 82: p. 415-45.
167. Rubin, D.M. and D. Finley, Proteolysis. The proteasome: a protein-degrading
organelle? Curr Biol, 1995. 5(8): p. 854-8.
168. Klockenbusch, C., et al., Global proteome analysis identifies active
immunoproteasome subunits in human platelets. Mol Cell Proteomics, 2014.
13(12): p. 3308-19.
169. Dieude, M., et al., The 20S proteasome core, active within apoptotic exosome-like
vesicles, induces autoantibody production and accelerates rejection. Sci Transl
Med, 2015. 7(318): p. 318ra200.
170. Opperman, C.M. and B.J. Sishi, Tumor necrosis factor alpha stimulates p62
accumulation and enhances proteasome activity independently of ROS. Cell Biol
Toxicol, 2015. 31(2): p. 83-94.
171. Kloetzel, P.M. and F. Ossendorp, Proteasome and peptidase function in MHC-
class-I-mediated antigen presentation. Curr Opin Immunol, 2004. 16(1): p. 76-81.
172. Kisselev, A.F., et al., The sizes of peptides generated from protein by mammalian
26 and 20 S proteasomes. Implications for understanding the degradative
mechanism and antigen presentation. J Biol Chem, 1999. 274(6): p. 3363-71.
173. Gaczynska, M., K.L. Rock, and A.L. Goldberg, Gamma-interferon and expression
of MHC genes regulate peptide hydrolysis by proteasomes. Nature, 1993.
365(6443): p. 264-7.
174. Gaczynska, M., et al., Peptidase activities of proteasomes are differentially
regulated by the major histocompatibility complex-encoded genes for LMP2 and
LMP7. Proc Natl Acad Sci U S A, 1994. 91(20): p. 9213-7.
175. Driscoll, J., et al., MHC-linked LMP gene products specifically alter peptidase
activities of the proteasome. Nature, 1993. 365(6443): p. 262-4.
176. Stohwasser, R., et al., Molecular cloning of the mouse proteasome subunits MC14
and MECL-1: reciprocally regulated tissue expression of interferon-gamma-
modulated proteasome subunits. Eur J Immunol, 1997. 27(5): p. 1182-7.
177. Sijts, E.J. and P.M. Kloetzel, The role of the proteasome in the generation of MHC
class I ligands and immune responses. Cell Mol Life Sci, 2011. 68(9): p. 1491-502.
178. Chen, W., et al., Immunoproteasomes shape immunodominance hierarchies of
antiviral CD8(+) T cells at the levels of T cell repertoire and presentation of viral
antigens. J Exp Med, 2001. 193(11): p. 1319-26.
179. Pang, K.C., et al., Immunoproteasome subunit deficiencies impact differentially on
two immunodominant influenza virus-specific CD8+ T cell responses. J Immunol,
2006. 177(11): p. 7680-8.

195
180. Basler, M., et al., An altered T cell repertoire in MECL-1-deficient mice. J
Immunol, 2006. 176(11): p. 6665-72.
181. Basler, M., J. Li, and M. Groettrup, On the role of the immunoproteasome in
transplant rejection. Immunogenetics, 2019. 71(3): p. 263-271.
182. Feist, E., G.R. Burmester, and E. Krüger, The proteasome - victim or culprit in
autoimmunity. Clin Immunol, 2016. 172: p. 83-89.
183. Basler, M., et al., Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is
required to block autoimmunity. EMBO Rep, 2018. 19(12).
184. Yasutomo, K., Dysregulation of immunoproteasomes in autoinflammatory
syndromes. Int Immunol, 2019. 31(10): p. 631-637.
185. Praest, P., et al., New insights into the structure of the MHC class I peptide-loading
complex and mechanisms of TAP inhibition by viral immune evasion proteins. Mol
Immunol, 2019. 113: p. 103-114.
186. Lewis, J.C., J.E. Maldonado, and K.G. Mann, Phagocytosis in human platelets:
localization of acid phosphatase-positive phagosomes following latex uptake.
Blood, 1976. 47(5): p. 833-40.
187. Male, R., W.E. Vannier, and J.D. Baldeschwieler, Phagocytosis of liposomes by
human platelets. Proc Natl Acad Sci U S A, 1992. 89(19): p. 9191-5.
188. Nagasawa, T., et al., Phagocytosis by Thrombocytes is a Conserved Innate Immune
Mechanism in Lower Vertebrates. Front Immunol, 2014. 5: p. 445.
189. White, J.G., Platelets are covercytes, not phagocytes: uptake of bacteria involves
channels of the open canalicular system. Platelets, 2005. 16(2): p. 121-31.
190. Rick Kapur, A.Z., Eric Boilard, and John W. Semple, Nouvelle Cuisine: Platelets
Served with Inflammation. Journal of Immunology, 2015. 12(194).
191. Koupenova, M., et al., Sex differences in platelet toll-like receptors and their
association with cardiovascular risk factors. Arterioscler Thromb Vasc Biol, 2015.
35(4): p. 1030-7.
192. Kawai, T. and S. Akira, Toll-like receptors and their crosstalk with other innate
receptors in infection and immunity. Immunity, 2011. 34(5): p. 637-50.
193. Palsson-McDermott, E.M. and L.A. O'Neill, Signal transduction by the
lipopolysaccharide receptor, Toll-like receptor-4. Immunology, 2004. 113(2): p.
153-62.
194. Damien, P., et al., LPS stimulation of purified human platelets is partly dependent
on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-
Ligand. BMC Immunol, 2015. 16: p. 3.
195. Berthet, J., et al., Human platelets can discriminate between various bacterial LPS
isoforms via TLR4 signaling and differential cytokine secretion. Clin Immunol,
2012. 145(3): p. 189-200.
196. Zhang, G., et al., Lipopolysaccharide stimulates platelet secretion and potentiates
platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase
pathway. J Immunol, 2009. 182(12): p. 7997-8004.
197. Koessler, J., et al., The Role of Human Platelet Preparation for Toll-Like Receptors
2 and 4 Related Platelet Responsiveness. TH Open, 2019. 3(2): p. e94-e102.
198. Andonegui, G., et al., Platelets express functional Toll-like receptor-4. Blood, 2005.
106(7): p. 2417-23.

196
199. Rivadeneyra, L., et al., Regulation of platelet responses triggered by Toll-like
receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB.
Thromb Res, 2014. 133(2): p. 235-43.
200. Montrucchio, G., et al., Mechanisms of the priming effect of low doses of lipopoly-
saccharides on leukocyte-dependent platelet aggregation in whole blood. Thromb
Haemost, 2003. 90(5): p. 872-81.
201. Brown, G.T. and T.M. McIntyre, Lipopolysaccharide signaling without a nucleus:
kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich
microparticles. J Immunol, 2011. 186(9): p. 5489-96.
202. Aslam, R., et al., Platelet Toll-like receptor expression modulates
lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha
production in vivo. Blood, 2006. 107(2): p. 637-41.
203. Matus, V., et al., Human platelet interaction with E. coli O111 promotes tissue-
factor-dependent procoagulant activity, involving Toll like receptor 4. PLoS One,
2017. 12(9): p. e0185431.
204. Cognasse, F., et al., Lipopolysaccharide induces sCD40L release through human
platelets TLR4, but not TLR2 and TLR9. Intensive Care Med, 2007. 33(2): p. 382-4.
205. Semple, J.W., et al., Platelet-bound lipopolysaccharide enhances Fc receptor-
mediated phagocytosis of IgG-opsonized platelets. Blood, 2007. 109(11): p. 4803-5.
206. Schmid, W., et al., Platelets Toll-like receptor-4 in Crohns disease. Eur J Clin
Invest, 2017. 47(2): p. 109-116.
207. Clark, S.R., et al., Platelet TLR4 activates neutrophil extracellular traps to ensnare
bacteria in septic blood. Nat Med, 2007. 13(4): p. 463-9.
208. Carestia, A., et al., Platelets Promote Macrophage Polarization toward Pro-
inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep, 2019.
28(4): p. 896-908 e5.
209. Yang, X., et al., HMGB1: a novel protein that induced platelets active and
aggregation via Toll-like receptor-4, NF-κB and cGMP dependent mechanisms.
Diagn Pathol, 2015. 10: p. 134.
210. Rigg, R.A., et al., Heat shock protein 70 regulates platelet integrin activation,
granule secretion and aggregation. Am J Physiol Cell Physiol, 2016. 310(7): p.
C568-75.
211. Lood, C., et al., Platelet-Derived S100A8/A9 and Cardiovascular Disease in
Systemic Lupus Erythematosus. Arthritis Rheumatol, 2016. 68(8): p. 1970-80.
212. Molteni, M., S. Gemma, and C. Rossetti, The Role of Toll-Like Receptor 4 in
Infectious and Noninfectious Inflammation. Mediators Inflamm, 2016. 2016: p.
6978936.
213. Cardon, L.R., et al., Pervasive CpG suppression in animal mitochondrial genomes.
Proc Natl Acad Sci U S A, 1994. 91(9): p. 3799-803.
214. Hally, K.E., et al., Toll-like receptor 9 expression and activation in acute coronary
syndrome patients on dual anti-platelet therapy. Thromb Res, 2016. 148: p. 89-95.
215. Panigrahi, S., et al., Engagement of platelet toll-like receptor 9 by novel endogenous
ligands promotes platelet hyperreactivity and thrombosis. Circ Res, 2013. 112(1):
p. 103-12.
216. Qian, K., et al., Functional expression of IgA receptor FcalphaRI on human
platelets. J Leukoc Biol, 2008. 84(6): p. 1492-500.

197
217. Hasegawa, S., et al., Functional expression of the high affinity receptor for IgE
(FcepsilonRI) in human platelets and its' intracellular expression in human
megakaryocytes. Blood, 1999. 93(8): p. 2543-51.
218. Joseph, M., et al., Expression and functions of the high-affinity IgE receptor on
human platelets and megakaryocyte precursors. Eur J Immunol, 1997. 27(9): p.
2212-8.
219. Capron, M., et al., Functional study of a monoclonal antibody to IgE Fc receptor
(Fc epsilon R2) of eosinophils, platelets, and macrophages. J Exp Med, 1986.
164(1): p. 72-89.
220. King, M., P. McDermott, and A.D. Schreiber, Characterization of the Fc gamma
receptor on human platelets. Cell Immunol, 1990. 128(2): p. 462-79.
221. Maruoka, T., T. Nagata, and M. Kasahara, Identification of the rat IgA Fc receptor
encoded in the leukocyte receptor complex. Immunogenetics, 2004. 55(10): p. 712-
6.
222. McKenzie, S.E., et al., The role of the human Fc receptor Fc gamma RIIA in the
immune clearance of platelets: a transgenic mouse model. J Immunol, 1999.
162(7): p. 4311-8.
223. Nimmerjahn, F., Activating and inhibitory FcgammaRs in autoimmune disorders.
Springer Semin Immunopathol, 2006. 28(4): p. 305-19.
224. Canobbio, I., et al., Platelet activation by von Willebrand factor requires
coordinated signaling through thromboxane A2 and Fc gamma IIA receptor. J Biol
Chem, 2001. 276(28): p. 26022-9.
225. Huang, Z.Y., et al., Human platelet FcgammaRIIA and phagocytes in immune-
complex clearance. Mol Immunol, 2011. 48(4): p. 691-6.
226. Kerrigan, S.W., et al., Molecular basis for Staphylococcus aureus-mediated platelet
aggregate formation under arterial shear in vitro. Arterioscler Thromb Vasc Biol,
2008. 28(2): p. 335-40.
227. Mitrugno, A., et al., A novel and essential role for FcgammaRIIa in cancer cell-
induced platelet activation. Blood, 2014. 123(2): p. 249-60.
228. Arman, M., et al., Amplification of bacteria-induced platelet activation is triggered
by FcgammaRIIA, integrin alphaIIbbeta3, and platelet factor 4. Blood, 2014.
123(20): p. 3166-74.
229. Zhi, H., et al., Cooperative integrin/ITAM signaling in platelets enhances thrombus
formation in vitro and in vivo. Blood, 2013. 121(10): p. 1858-67.
230. Boulaftali, Y., et al., Platelet immunoreceptor tyrosine-based activation motif
(ITAM) signaling and vascular integrity. Circ Res, 2014. 114(7): p. 1174-84.
231. Arthur, J.F., et al., ITAM receptor-mediated generation of reactive oxygen species
in human platelets occurs via Syk-dependent and Syk-independent pathways. J
Thromb Haemost, 2012. 10(6): p. 1133-41.
232. Ilkan, Z., et al., P2X1 Receptors Amplify FcgammaRIIa-Induced Ca2+ Increases
and Functional Responses in Human Platelets. Thromb Haemost, 2018. 118(2): p.
369-380.
233. Meyer, T., et al., CD32a antibodies induce thrombocytopenia and type II
hypersensitivity reactions in FCGR2A mice. Blood, 2015. 126(19): p. 2230-8.
234. Jonsson, F., et al., Human FcgammaRIIA induces anaphylactic and allergic
reactions. Blood, 2012. 119(11): p. 2533-44.

198
235. Reilly, M.P., et al., Heparin-induced thrombocytopenia/thrombosis in a transgenic
mouse model requires human platelet factor 4 and platelet activation through
FcgammaRIIA. Blood, 2001. 98(8): p. 2442-7.
236. Cloutier, N., et al., Platelets release pathogenic serotonin and return to circulation
after immune complex-mediated sequestration. Proc Natl Acad Sci U S A, 2018.
115(7): p. E1550-E1559.
237. Krijgsveld, J., et al., Thrombocidins, microbicidal proteins from human blood
platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem, 2000.
275(27): p. 20374-81.
238. Yeaman, M.R., The role of platelets in antimicrobial host defense. Clin Infect Dis,
1997. 25(5): p. 951-68; quiz 969-70.
239. Riaz, A.H., et al., Human platelets efficiently kill IgG-opsonized E. coli. FEMS
Immunol Med Microbiol, 2012. 65(1): p. 78-83.
240. Boilard, E., et al., Influenza virus H1N1 activates platelets through FcgammaRIIA
signaling and thrombin generation. Blood, 2014. 123(18): p. 2854-63.
241. Hudson, J.B., L. Weinstein, and T.W. Chang, Thrombocytopenic purpura in
measles. J Pediatr, 1956. 48(1): p. 48-56.
242. Hirst, G.K., The Quantitative Determination of Influenza Virus and Antibodies by
Means of Red Cell Agglutination. J Exp Med, 1942. 75(1): p. 49-64.
243. Terada, H., et al., Interaction of influenza virus with blood platelets. Blood, 1966.
28(2): p. 213-28.
244. Chomette, G., M. Auriol, and F. Guilbert, [Pleomorphic adenoma. Diagnostic
difficulties. Value of ultrastructural and immunohistochemical studies. Apropos of 2
cases]. Rev Stomatol Chir Maxillofac, 1987. 88(5): p. 356-8.
245. Ojha, A., et al., Platelet activation determines the severity of thrombocytopenia in
dengue infection. Sci Rep, 2017. 7: p. 41697.
246. Kelton, J.G., et al., Immune-mediated thrombocytopenia of malaria. J Clin Invest,
1983. 71(4): p. 832-6.
247. Charkes, N.D., Purpuric chickenpox: report of a case, review of the literature, and
classification by clinical features. Ann Intern Med, 1961. 54: p. 745-59.
248. Hsieh, Y.C., et al., Influenza pandemics: past, present and future. J Formos Med
Assoc, 2006. 105(1): p. 1-6.
249. Ghuman, H., et al., Mucor circinelloides induces platelet aggregation through
integrin alphaIIbbeta3 and FcgammaRIIA. Platelets, 2019. 30(2): p. 256-263.
250. Xu, W.F., et al., Cloning and characterization of human protease-activated
receptor 4. Proc Natl Acad Sci U S A, 1998. 95(12): p. 6642-6.
251. Hollopeter, G., et al., Identification of the platelet ADP receptor targeted by
antithrombotic drugs. Nature, 2001. 409(6817): p. 202-7.
252. Hirata, M., et al., Cloning and expression of cDNA for a human thromboxane A2
receptor. Nature, 1991. 349(6310): p. 617-20.
253. Maeda, H., et al., Possible involvement of protein-tyrosine kinases such as p72syk in
the disc-sphere change response of porcine platelets. J Biochem, 1995. 117(6): p.
1201-8.
254. Katsuyama, M., et al., Cloning and expression of a cDNA for the human
prostacyclin receptor. FEBS Lett, 1994. 344(1): p. 74-8.
255. Boie, Y., et al., Molecular cloning and characterization of the human prostanoid
DP receptor. J Biol Chem, 1995. 270(32): p. 18910-6.

199
256. Fabre, J.E., et al., Activation of the murine EP3 receptor for PGE2 inhibits cAMP
production and promotes platelet aggregation. J Clin Invest, 2001. 107(5): p. 603-
10.
257. Burgers, J.A. and J.W. Akkerman, Regulation of the receptor for platelet-activating
factor on human platelets. Biochem J, 1993. 291 ( Pt 1): p. 157-61.
258. Bandoh, K., et al., Molecular cloning and characterization of a novel human G-
protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem, 1999.
274(39): p. 27776-85.
259. Motohashi, K., et al., Identification of lysophospholipid receptors in human
platelets: the relation of two agonists, lysophosphatidic acid and sphingosine 1-
phosphate. FEBS Lett, 2000. 468(2-3): p. 189-93.
260. Clemetson, K.J., et al., Functional expression of CCR1, CCR3, CCR4, and CXCR4
chemokine receptors on human platelets. Blood, 2000. 96(13): p. 4046-54.
261. Vieira-de-Abreu, A., et al., Platelets: versatile effector cells in hemostasis,
inflammation, and the immune continuum. Semin Immunopathol, 2012. 34(1): p. 5-
30.
262. Ehlers, R., et al., Targeting platelet-leukocyte interactions: identification of the
integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J
Exp Med, 2003. 198(7): p. 1077-88.
263. Furie, B. and B.C. Furie, The molecular basis of platelet and endothelial cell
interaction with neutrophils and monocytes: role of P-selectin and the P-selectin
ligand, PSGL-1. Thromb Haemost, 1995. 74(1): p. 224-7.
264. Martins, P.A.D.C., et al., Platelet binding to monocytes increases the adhesive
properties of monocytes by up-regulating the expression and functionality of β. J
Leukoc Biol, 2006. 79(3): p. 499-507.
265. Gaertner, F. and S. Massberg, Patrolling the vascular borders: platelets in immunity
to infection and cancer. Nat Rev Immunol, 2019. 19(12): p. 747-760.
266. Hottz, E.D., et al., Platelet activation and apoptosis modulate monocyte
inflammatory responses in dengue. J Immunol, 2014. 193(4): p. 1864-72.
267. Michels, M., et al., Platelet function alterations in dengue are associated with
plasma leakage. Thromb Haemost, 2014. 112(2): p. 352-62.
268. Suzuki, K., et al., Activated platelets in ulcerative colitis enhance the production of
reactive oxygen species by polymorphonuclear leukocytes. Scand J Gastroenterol,
2001. 36(12): p. 1301-6.
269. McDonald, B., et al., Intravascular neutrophil extracellular traps capture bacteria
from the bloodstream during sepsis. Cell Host Microbe, 2012. 12(3): p. 324-33.
270. Constantinescu-Bercu, A., et al., Activated alphaIIbbeta3 on platelets mediates
flow-dependent NETosis via SLC44A2. Elife, 2020. 9.
271. Flick, M.J., et al., Fibrin(ogen) exacerbates inflammatory joint disease through a
mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest, 2007.
117(11): p. 3224-35.
272. Elaskalani, O., N.B. Abdol Razak, and P. Metharom, Neutrophil extracellular traps
induce aggregation of washed human platelets independently of extracellular DNA
and histones. Cell Commun Signal, 2018. 16(1): p. 24.
273. Fuchs, T.A., et al., Extracellular DNA traps promote thrombosis. Proc Natl Acad
Sci U S A, 2010. 107(36): p. 15880-5.

200
274. Podolska, M.J., et al., Autoimmune, rheumatic, chronic inflammatory diseases:
Neutrophil extracellular traps on parade. Autoimmunity, 2018. 51(6): p. 281-287.
275. Sangaletti, S., et al., Neutrophil extracellular traps mediate transfer of cytoplasmic
neutrophil antigens to myeloid dendritic cells toward ANCA induction and
associated autoimmunity. Blood, 2012. 120(15): p. 3007-18.
276. Villanueva, E., et al., Netting neutrophils induce endothelial damage, infiltrate
tissues, and expose immunostimulatory molecules in systemic lupus erythematosus.
J Immunol, 2011. 187(1): p. 538-52.
277. Bruschi, M., et al., Neutrophil Extracellular Traps Profiles in Patients with Incident
Systemic Lupus Erythematosus and Lupus Nephritis. J Rheumatol, 2020. 47(3): p.
377-386.
278. Hsu, C.C., et al., Neutrophil Extracellular Traps Enhance Staphylococcus Aureus
Vegetation Formation through Interaction with Platelets in Infective Endocarditis.
Thromb Haemost, 2019. 119(5): p. 786-796.
279. Cunin, P., et al., Megakaryocyte emperipolesis mediates membrane transfer from
intracytoplasmic neutrophils to platelets. Elife, 2019. 8.
280. Fleischer, J., et al., Platelet factor 4 inhibits proliferation and cytokine release of
activated human T cells. J Immunol, 2002. 169(2): p. 770-7.
281. Silva-Cardoso, S.C., et al., CXCL4 Exposure Potentiates TLR-Driven Polarization
of Human Monocyte-Derived Dendritic Cells and Increases Stimulation of T Cells.
J Immunol, 2017. 199(1): p. 253-262.
282. Moore, K.L. and L.F. Thompson, P-selectin (CD62) binds to subpopulations of
human memory T lymphocytes and natural killer cells. Biochem Biophys Res
Commun, 1992. 186(1): p. 173-81.
283. Khandoga, A., et al., CD4+ T cells contribute to postischemic liver injury in mice
by interacting with sinusoidal endothelium and platelets. Hepatology, 2006. 43(2):
p. 306-15.
284. Han, P., et al., Platelet P-selectin initiates cross-presentation and dendritic cell
differentiation in blood monocytes. Sci Adv, 2020. 6(11): p. eaaz1580.
285. Adnot, S., et al., Serotonin transporter and serotonin receptors. Handb Exp
Pharmacol, 2013. 218: p. 365-80.
286. Laberge, S., et al., Secretion of IL-16 (lymphocyte chemoattractant factor) from
serotonin-stimulated CD8+ T cells in vitro. J Immunol, 1996. 156(1): p. 310-5.
287. Lang, P.A., et al., Aggravation of viral hepatitis by platelet-derived serotonin. Nat
Med, 2008. 14(7): p. 756-61.
288. Katoh, N., et al., Effect of serotonin on the differentiation of human monocytes into
dendritic cells. Clin Exp Immunol, 2006. 146(2): p. 354-61.
289. Martinson, J., et al., Activated platelets rapidly up-regulate CD40L expression and
can effectively mature and activate autologous ex vivo differentiated DC.
Cytotherapy, 2004. 6(5): p. 487-97.
290. Kaneider, N.C., et al., CD40 ligand-dependent maturation of human monocyte-
derived dendritic cells by activated platelets. Int J Immunopathol Pharmacol, 2003.
16(3): p. 225-31.
291. Czapiga, M., A.D. Kirk, and J. Lekstrom-Himes, Platelets deliver costimulatory
signals to antigen-presenting cells: a potential bridge between injury and immune
activation. Exp Hematol, 2004. 32(2): p. 135-9.

201
292. Duffau, P., et al., Platelet CD154 potentiates interferon-alpha secretion by
plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med, 2010.
2(47): p. 47ra63.
293. Sowa, J.M., et al., Platelet influence on T- and B-cell responses. Arch Immunol
Ther Exp (Warsz), 2009. 57(4): p. 235-41.
294. Elzey, B.D., D.L. Sprague, and T.L. Ratliff, The emerging role of platelets in
adaptive immunity. Cell Immunol, 2005. 238(1): p. 1-9.
295. Blumberg, N., et al., The platelet as an immune cell-CD40 ligand and transfusion
immunomodulation. Immunol Res, 2009. 45(2-3): p. 251-60.
296. Sprague, D.L., et al., The role of platelet CD154 in the modulation in adaptive
immunity. Immunol Res, 2007. 39(1-3): p. 185-93.
297. Andre, P., et al., CD40L stabilizes arterial thrombi by a beta3 integrin--dependent
mechanism. Nat Med, 2002. 8(3): p. 247-52.
298. Buchner, K., et al., CD40 ligand is selectively expressed on CD4+ T cells and
platelets: implications for CD40-CD40L signalling in atherosclerosis. J Pathol,
2003. 201(2): p. 288-95.
299. Elzey, B.D., et al., Platelet-mediated modulation of adaptive immunity. A
communication link between innate and adaptive immune compartments. Immunity,
2003. 19(1): p. 9-19.
300. Hotta, E., R. Tamagawa-Mineoka, and N. Katoh, Platelets are important for the
development of immune tolerance: Possible involvement of TGF-beta in the
mechanism. Exp Dermatol, 2019. 28(7): p. 801-808.
301. Bergmann, C.B., et al., Platelets modulate the immune response following trauma
by interaction with CD4+ T regulatory cells in a mouse model. Immunol Res, 2016.
64(2): p. 508-17.
302. Mudd, J.C., et al., Inflammatory Function of CX3CR1+ CD8+ T Cells in Treated
HIV Infection Is Modulated by Platelet Interactions. J Infect Dis, 2016. 214(12): p.
1808-1816.
303. Yukawa, M., et al., Purification and characterization of endogenous protein
activator of human platelet proteasome. J Biochem, 1993. 114(3): p. 317-23.
304. Yukawa, M., et al., Proteasome and its novel endogeneous activator in human
platelets. Biochem Biophys Res Commun, 1991. 178(1): p. 256-62.
305. Grundler Groterhorst, K., et al., Platelet Proteasome Activity and Metabolism Is
Upregulated during Bacterial Sepsis. Int J Mol Sci, 2019. 20(23).
306. Colberg, L., et al., Structure and function of the ubiquitin-proteasome system in
platelets. J Thromb Haemost, 2020.
307. Shi, D.S., et al., Proteasome function is required for platelet production. J Clin
Invest, 2014. 124(9): p. 3757-66.
308. Murai, K., et al., Bortezomib induces thrombocytopenia by the inhibition of
proplatelet formation of megakaryocytes. Eur J Haematol, 2014. 93(4): p. 290-6.
309. Nayak, M.K., P.P. Kulkarni, and D. Dash, Regulatory role of proteasome in
determination of platelet life span. J Biol Chem, 2013. 288(10): p. 6826-34.
310. Grundler, K., et al., The proteasome regulates collagen-induced platelet
aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb Res, 2016.
148: p. 15-22.
311. Nayak, M.K., K. Kumar, and D. Dash, Regulation of proteasome activity in
activated human platelets. Cell Calcium, 2011. 49(4): p. 226-32.

202
312. Koessler, J., et al., The role of proteasome activity for activating and inhibitory
signalling in human platelets. Cell Signal, 2019. 62: p. 109351.
313. Gupta, N., et al., Proteasome proteolysis supports stimulated platelet function and
thrombosis. Arterioscler Thromb Vasc Biol, 2014. 34(1): p. 160-8.
314. Boegel, S., et al., HLA and proteasome expression body map. BMC Med Genomics,
2018. 11(1): p. 36.
315. Semple, J.W., et al., Indirect allorecognition of platelets by T helper cells during
platelet transfusions correlates with anti-major histocompatibility complex antibody
and cytotoxic T lymphocyte formation. Blood, 1995. 86(2): p. 805-12.
316. Kao, K.J., D.J. Cook, and J.C. Scornik, Quantitative analysis of platelet surface
HLA by W6/32 anti-HLA monoclonal antibody. Blood, 1986. 68(3): p. 627-32.
317. Placke, T., et al., Platelet-derived MHC class I confers a pseudonormal phenotype
to cancer cells that subverts the antitumor reactivity of natural killer immune cells.
Cancer Res, 2012. 72(2): p. 440-8.
318. Zufferey, A., et al., Characterization of the platelet granule proteome: evidence of
the presence of MHC1 in alpha-granules. J Proteomics, 2014. 101: p. 130-40.
319. Chapman, L.M., et al., Platelets present antigen in the context of MHC class I. J
Immunol, 2012. 189(2): p. 916-23.
320. Angenieux, C., et al., Cell surface expression of HLA I molecules as a marker of
young platelets. J Thromb Haemost, 2019. 17(9): p. 1511-1521.
321. Nunez-Avellaneda, D., et al., Dengue Virus Induces the Release of sCD40L and
Changes in Levels of Membranal CD42b and CD40L Molecules in Human
Platelets. Viruses, 2018. 10(7).
322. Simon, A.Y., M.R. Sutherland, and E.L. Pryzdial, Dengue virus binding and
replication by platelets. Blood, 2015. 126(3): p. 378-85.
323. Koupenova, M., et al., The role of platelets in mediating a response to human
influenza infection. Nat Commun, 2019. 10(1): p. 1780.
324. Schultz, C.M., et al., Stepping up to the Plate(let) against C. albicans. Infect
Immun, 2020.
325. Iannacone, M., et al., Platelets mediate cytotoxic T lymphocyte-induced liver
damage. Nat Med, 2005. 11(11): p. 1167-9.
326. Verschoor, A., et al., A platelet-mediated system for shuttling blood-borne bacteria
to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3.
Nat Immunol, 2011. 12(12): p. 1194-201.
327. Nishat, S., L.M. Wuescher, and R.G. Worth, Platelets Enhance Dendritic Cell
Responses against Staphylococcus aureus through CD40-CD40L. Infect Immun,
2018. 86(9).
328. Elzey, B.D., et al., Platelet-derived CD154 enables T-cell priming and protection
against Listeria monocytogenes challenge. Blood, 2008. 111(7): p. 3684-91.
329. Boshkov, L.K., J.G. Kelton, and P.F. Halloran, HLA-DR expression by platelets in
acute idiopathic thrombocytopenic purpura. Br J Haematol, 1992. 81(4): p. 552-7.
330. Lazarus, A.H., et al., Comparison of platelet immunity in patients with SLE and with
ITP. Transfus Sci, 2000. 22(1-2): p. 19-27.
331. Semple, J.W., et al., Differences in serum cytokine levels in acute and chronic
autoimmune thrombocytopenic purpura: relationship to platelet phenotype and
antiplatelet T-cell reactivity. Blood, 1996. 87(10): p. 4245-54.

203
332. Kang, H.K., et al., Megakaryocyte progenitors are the main APCs inducing Th17
response to lupus autoantigens and foreign antigens. J Immunol, 2012. 188(12): p.
5970-80.
333. Finkielsztein, A., et al., Human megakaryocyte progenitors derived from
hematopoietic stem cells of normal individuals are MHC class II-expressing
professional APC that enhance Th17 and Th1/Th17 responses. Immunol Lett, 2015.
163(1): p. 84-95.
334. Clark, K.B., et al., Characterization of dengue virus 2 growth in megakaryocyte-
erythrocyte progenitor cells. Virology, 2016. 493: p. 162-72.
335. Campbell, R.A., et al., Human megakaryocytes possess intrinsic antiviral immunity
through regulated induction of IFITM3. Blood, 2019. 133(19): p. 2013-2026.
336. Lefrancais, E., et al., The lung is a site of platelet biogenesis and a reservoir for
haematopoietic progenitors. Nature, 2017. 544(7648): p. 105-109.
337. Pariser, D.N., et al., Lung megakaryocytes are immune modulatory cells. J Clin
Invest, 2020.
338. Zufferey, A., et al., Mature murine megakaryocytes present antigen-MHC class I
molecules to T cells and transfer them to platelets. Blood Adv, 2017. 1(20): p.
1773-1785.
339. Raposo, G. and W. Stoorvogel, Extracellular vesicles: exosomes, microvesicles, and
friends. J Cell Biol, 2013. 200(4): p. 373-83.
340. Colombo, M., G. Raposo, and C. Thery, Biogenesis, secretion, and intercellular
interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol,
2014. 30: p. 255-89.
341. Tetta, C., et al., Extracellular vesicles as an emerging mechanism of cell-to-cell
communication. Endocrine, 2013. 44(1): p. 11-9.
342. Zara, M., et al., Biology and Role of Extracellular Vesicles (EVs) in the
Pathogenesis of Thrombosis. Int J Mol Sci, 2019. 20(11).
343. Yanez-Mo, M., et al., Biological properties of extracellular vesicles and their
physiological functions. J Extracell Vesicles, 2015. 4: p. 27066.
344. Duchez, A.C., et al., Platelet microparticles are internalized in neutrophils via the
concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc Natl
Acad Sci U S A, 2015. 112(27): p. E3564-73.
345. Thery, C., M. Ostrowski, and E. Segura, Membrane vesicles as conveyors of
immune responses. Nat Rev Immunol, 2009. 9(8): p. 581-93.
346. van der Pol, E., et al., Recent developments in the nomenclature, presence,
isolation, detection and clinical impact of extracellular vesicles. J Thromb
Haemost, 2016. 14(1): p. 48-56.
347. Malda, J., et al., Extracellular vesicles - new tool for joint repair and regeneration.
Nat Rev Rheumatol, 2016. 12(4): p. 243-9.
348. Breen, K.A., et al., Endothelial and platelet microparticles in patients with
antiphospholipid antibodies. Thromb Res, 2015. 135(2): p. 368-74.
349. Boilard, E., et al., Platelets amplify inflammation in arthritis via collagen-dependent
microparticle production. Science, 2010. 327(5965): p. 580-3.
350. Pereira, J., et al., Circulating platelet-derived microparticles in systemic lupus
erythematosus. Association with increased thrombin generation and procoagulant
state. Thromb Haemost, 2006. 95(1): p. 94-9.

204
351. Słomka, A., et al., Large Extracellular Vesicles: Have We Found the Holy Grail of
Inflammation? Front Immunol, 2018. 9: p. 2723.
352. de Jong, O.G., et al., Drug Delivery with Extracellular Vesicles: From Imagination
to Innovation. Acc Chem Res, 2019. 52(7): p. 1761-1770.
353. Urbanelli, L., et al., The Role of Extracellular Vesicles in Viral Infection and
Transmission. Vaccines (Basel), 2019. 7(3).
354. Gyorgy, B., et al., Membrane vesicles, current state-of-the-art: emerging role of
extracellular vesicles. Cell Mol Life Sci, 2011. 68(16): p. 2667-88.
355. Hanson, P.I. and A. Cashikar, Multivesicular body morphogenesis. Annu Rev Cell
Dev Biol, 2012. 28: p. 337-62.
356. Record, M., et al., Extracellular vesicles: lipids as key components of their
biogenesis and functions. J Lipid Res, 2018. 59(8): p. 1316-1324.
357. Mathieu, M., et al., Specificities of secretion and uptake of exosomes and other
extracellular vesicles for cell-to-cell communication. Nat Cell Biol, 2019. 21(1): p.
9-17.
358. Carayon, K., et al., Proteolipidic composition of exosomes changes during
reticulocyte maturation. J Biol Chem, 2011. 286(39): p. 34426-39.
359. Gyorgy, B., et al., Detection and isolation of cell-derived microparticles are
compromised by protein complexes resulting from shared biophysical parameters.
Blood, 2011. 117(4): p. e39-48.
360. Hurley, J.H., ESCRTs are everywhere. EMBO J, 2015. 34(19): p. 2398-407.
361. Nabhan, J.F., et al., Formation and release of arrestin domain-containing protein 1-
mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101
protein. Proc Natl Acad Sci U S A, 2012. 109(11): p. 4146-51.
362. Beer, K.B., et al., Extracellular vesicle budding is inhibited by redundant regulators
of TAT-5 flippase localization and phospholipid asymmetry. Proc Natl Acad Sci U S
A, 2018. 115(6): p. E1127-E1136.
363. Fujii, T., et al., TMEM16F is required for phosphatidylserine exposure and
microparticle release in activated mouse platelets. Proc Natl Acad Sci U S A, 2015.
112(41): p. 12800-5.
364. Akers, J.C., et al., Biogenesis of extracellular vesicles (EV): exosomes,
microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol, 2013.
113(1): p. 1-11.
365. Morel, O., et al., Cellular mechanisms underlying the formation of circulating
microparticles. Arterioscler Thromb Vasc Biol, 2011. 31(1): p. 15-26.
366. Lotvall, J., et al., Minimal experimental requirements for definition of extracellular
vesicles and their functions: a position statement from the International Society for
Extracellular Vesicles. J Extracell Vesicles, 2014. 3: p. 26913.
367. Théry, C., et al., Minimal information for studies of extracellular vesicles 2018
(MISEV2018): a position statement of the International Society for Extracellular
Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles, 2018. 7(1):
p. 1535750.
368. Clayton, A., et al., Considerations towards a roadmap for collection, handling and
storage of blood extracellular vesicles. J Extracell Vesicles, 2019. 8(1): p. 1647027.
369. Kalra, H., et al., Vesiclepedia: a compendium for extracellular vesicles with
continuous community annotation. PLoS Biol, 2012. 10(12): p. e1001450.

205
370. Keerthikumar, S., et al., ExoCarta: A Web-Based Compendium of Exosomal Cargo.
J Mol Biol, 2016. 428(4): p. 688-692.
371. Kim, D.K., et al., EVpedia: A community web resource for prokaryotic and
eukaryotic extracellular vesicles research. Semin Cell Dev Biol, 2015. 40: p. 4-7.
372. Boing, A.N., et al., Single-step isolation of extracellular vesicles by size-exclusion
chromatography. J Extracell Vesicles, 2014. 3.
373. Witwer, K.W., et al., Standardization of sample collection, isolation and analysis
methods in extracellular vesicle research. J Extracell Vesicles, 2013. 2.
374. Chandler, W.L., Measurement of microvesicle levels in human blood using flow
cytometry. Cytometry B Clin Cytom, 2016. 90(4): p. 326-36.
375. Di Vizio, D., et al., Large oncosomes in human prostate cancer tissues and in the
circulation of mice with metastatic disease. Am J Pathol, 2012. 181(5): p. 1573-84.
376. Muralidharan-Chari, V., et al., ARF6-regulated shedding of tumor cell-derived
plasma membrane microvesicles. Curr Biol, 2009. 19(22): p. 1875-85.
377. Aatonen, M.T., et al., Isolation and characterization of platelet-derived
extracellular vesicles. J Extracell Vesicles, 2014. 3.
378. Rousseau, M., et al., Detection and quantification of microparticles from different
cellular lineages using flow cytometry. Evaluation of the impact of secreted
phospholipase A2 on microparticle assessment. PLoS One, 2015. 10(1): p.
e0116812.
379. Gardiner, C., et al., Techniques used for the isolation and characterization of
extracellular vesicles: results of a worldwide survey. J Extracell Vesicles, 2016. 5:
p. 32945.
380. Chandler, W.L., W. Yeung, and J.F. Tait, A new microparticle size calibration
standard for use in measuring smaller microparticles using a new flow cytometer. J
Thromb Haemost, 2011. 9(6): p. 1216-24.
381. Nolan, J.P. and J.C. Jones, Detection of platelet vesicles by flow cytometry.
Platelets, 2017. 28(3): p. 256-262.
382. Boudreau, L.H., et al., Platelets release mitochondria serving as substrate for
bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood,
2014. 124(14): p. 2173-83.
383. Cloutier, N., et al., The exposure of autoantigens by microparticles underlies the
formation of potent inflammatory components: the microparticle-associated
immune complexes. EMBO Mol Med, 2013. 5(2): p. 235-49.
384. Nieuwland, R., et al., Essentials of extracellular vesicles: posters on basic and
clinical aspects of extracellular vesicles. J Extracell Vesicles, 2018. 7(1): p.
1548234.
385. Sedgwick, A.E. and C. D'Souza-Schorey, The biology of extracellular
microvesicles. Traffic, 2018. 19(5): p. 319-327.
386. Kohli, S., et al., Maternal extracellular vesicles and platelets promote preeclampsia
via inflammasome activation in trophoblasts. Blood, 2016. 128(17): p. 2153-2164.
387. Ciardiello, C., et al., Large extracellular vesicles: Size matters in tumor
progression. Cytokine Growth Factor Rev, 2020. 51: p. 69-74.
388. Andersson, S.G., et al., The genome sequence of Rickettsia prowazekii and the
origin of mitochondria. Nature, 1998. 396(6707): p. 133-40.
389. Taanman, J.W., The mitochondrial genome: structure, transcription, translation
and replication. Biochim Biophys Acta, 1999. 1410(2): p. 103-23.

206
390. Ryan, M.T. and N.J. Hoogenraad, Mitochondrial-nuclear communications. Annu
Rev Biochem, 2007. 76: p. 701-22.
391. Ramalho-Santos, J. and S. Amaral, Mitochondria and mammalian reproduction.
Mol Cell Endocrinol, 2013. 379(1-2): p. 74-84.
392. Osellame, L.D., T.S. Blacker, and M.R. Duchen, Cellular and molecular
mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab,
2012. 26(6): p. 711-23.
393. Baughman, J.M., et al., Integrative genomics identifies MCU as an essential
component of the mitochondrial calcium uniporter. Nature, 2011. 476(7360): p.
341-5.
394. Tait, S.W. and D.R. Green, Mitochondrial regulation of cell death. Cold Spring
Harb Perspect Biol, 2013. 5(9).
395. Elkholi, R., et al., Putting the pieces together: How is the mitochondrial pathway of
apoptosis regulated in cancer and chemotherapy? Cancer Metab, 2014. 2: p. 16.
396. Kohler, C., et al., Levels of plasma circulating cell free nuclear and mitochondrial
DNA as potential biomarkers for breast tumors. Mol Cancer, 2009. 8: p. 105.
397. Collins, L.V., et al., Endogenously oxidized mitochondrial DNA induces in vivo and
in vitro inflammatory responses. J Leukoc Biol, 2004. 75(6): p. 995-1000.
398. Kung, C.T., et al., Plasma nuclear and mitochondrial DNA levels as predictors of
outcome in severe sepsis patients in the emergency room. J Transl Med, 2012. 10: p.
130.
399. Sjovall, F., et al., Cytokine and nitric oxide levels in patients with sepsis--temporal
evolvement and relation to platelet mitochondrial respiratory function. PLoS One,
2014. 9(7): p. e103756.
400. Oka, T., et al., Mitochondrial DNA that escapes from autophagy causes
inflammation and heart failure. Nature, 2012. 485(7397): p. 251-5.
401. Marques, P.E., et al., Chemokines and mitochondrial products activate neutrophils
to amplify organ injury during mouse acute liver failure. Hepatology, 2012. 56(5):
p. 1971-82.
402. Simmons, J.D., et al., Elevated levels of plasma mitochondrial DNA DAMPs are
linked to clinical outcome in severely injured human subjects. Ann Surg, 2013.
258(4): p. 591-6; discussion 596-8.
403. Yamanouchi, S., et al., Plasma mitochondrial DNA levels in patients with trauma
and severe sepsis: time course and the association with clinical status. J Crit Care,
2013. 28(6): p. 1027-31.
404. Nakahira, K., et al., Circulating mitochondrial DNA in patients in the ICU as a
marker of mortality: derivation and validation. PLoS Med, 2013. 10(12): p.
e1001577; discussion e1001577.
405. Zhang, Q., et al., Circulating mitochondrial DAMPs cause inflammatory responses
to injury. Nature, 2010. 464(7285): p. 104-7.
406. Lee, Y.L., et al., Blood transfusion products contain mitochondrial DNA damage-
associated molecular patterns: a potential effector of transfusion-related acute lung
injury. J Surg Res, 2014. 191(2): p. 286-9.
407. Wenceslau, C.F., et al., Mitochondrial-derived N-formyl peptides: novel links
between trauma, vascular collapse and sepsis. Med Hypotheses, 2013. 81(4): p.
532-5.

207
408. Carp, H., Mitochondrial N-formylmethionyl proteins as chemoattractants for
neutrophils. J Exp Med, 1982. 155(1): p. 264-75.
409. Jemmerson, R., B. LaPlante, and A. Treeful, Release of intact, monomeric
cytochrome c from apoptotic and necrotic cells. Cell Death Differ, 2002. 9(5): p.
538-48.
410. Kruse, K., et al., Inefficient clearance of dying cells in patients with SLE: anti-
dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis, 2010.
15(9): p. 1098-113.
411. Pullerits, R., et al., Extracellular cytochrome c, a mitochondrial apoptosis-related
protein, induces arthritis. Rheumatology (Oxford), 2005. 44(1): p. 32-9.
412. Schlame, M., D. Rua, and M.L. Greenberg, The biosynthesis and functional role of
cardiolipin. Prog Lipid Res, 2000. 39(3): p. 257-88.
413. Wenceslau, C.F., et al., Mitochondrial damage-associated molecular patterns and
vascular function. Eur Heart J, 2014. 35(18): p. 1172-7.
414. Sorice, M., et al., Cardiolipin on the surface of apoptotic cells as a possible trigger
for antiphospholipids antibodies. Clin Exp Immunol, 2000. 122(2): p. 277-84.
415. Krysko, D.V., et al., Emerging role of damage-associated molecular patterns
derived from mitochondria in inflammation. Trends Immunol, 2011. 32(4): p. 157-
64.
416. Wada, M., et al., Serum concentration and localization in tumor cells of
proteasomes in patients with hematologic malignancy and their pathophysiologic
significance. J Lab Clin Med, 1993. 121(2): p. 215-23.
417. Sixt, S.U. and B. Dahlmann, Extracellular, circulating proteasomes and ubiquitin -
incidence and relevance. Biochim Biophys Acta, 2008. 1782(12): p. 817-23.
418. Bochmann, I., et al., T lymphocytes export proteasomes by way of microparticles: a
possible mechanism for generation of extracellular proteasomes. J Cell Mol Med,
2014. 18(1): p. 59-68.
419. Lai, R.C., et al., Proteolytic Potential of the MSC Exosome Proteome: Implications
for an Exosome-Mediated Delivery of Therapeutic Proteasome. Int J Proteomics,
2012. 2012: p. 971907.
420. Dieudé, M., et al., Extracellular vesicles derived from injured vascular tissue
promote the formation of tertiary lymphoid structures in vascular allografts. Am J
Transplant, 2020. 20(3): p. 726-738.
421. Flaumenhaft, R., et al., Megakaryocyte-derived microparticles: direct visualization
and distinction from platelet-derived microparticles. Blood, 2009. 113(5): p. 1112-
21.
422. Benameur, T., et al., Molecular Mechanisms Underpinning Microparticle-Mediated
Cellular Injury in Cardiovascular Complications Associated with Diabetes. Oxid
Med Cell Longev, 2019. 2019: p. 6475187.
423. Wolf, P., The nature and significance of platelet products in human plasma. Br J
Haematol, 1967. 13(3): p. 269-88.
424. Sims, P.J., et al., Assembly of the platelet prothrombinase complex is linked to
vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an
isolated defect in platelet procoagulant activity. J Biol Chem, 1989. 264(29): p.
17049-57.

208
425. Sandberg, H., et al., Expression of coagulant activity in human platelets: release of
membranous vesicles providing platelet factor 1 and platelet factor 3. Thromb Res,
1985. 39(1): p. 63-79.
426. Keuren, J.F., et al., Effects of storage-induced platelet microparticles on the
initiation and propagation phase of blood coagulation. Br J Haematol, 2006.
134(3): p. 307-13.
427. Reininger, A.J., et al., Mechanism of platelet adhesion to von Willebrand factor and
microparticle formation under high shear stress. Blood, 2006. 107(9): p. 3537-45.
428. Holme, P.A., et al., Shear-induced platelet activation and platelet microparticle
formation at blood flow conditions as in arteries with a severe stenosis. Arterioscler
Thromb Vasc Biol, 1997. 17(4): p. 646-53.
429. Balaj, L., et al., Tumour microvesicles contain retrotransposon elements and
amplified oncogene sequences. Nat Commun, 2011. 2: p. 180.
430. Guescini, M., et al., Astrocytes and Glioblastoma cells release exosomes carrying
mtDNA. J Neural Transm (Vienna), 2010. 117(1): p. 1-4.
431. Melki, I., et al., Platelet microvesicles in health and disease. Platelets, 2017. 28(3):
p. 214-221.
432. Keuren, J.F., et al., Microparticles adhere to collagen type I, fibrinogen, von
Willebrand factor and surface immobilised platelets at physiological shear rates. Br
J Haematol, 2007. 138(4): p. 527-33.
433. Leroyer, A.S., A. Tedgui, and C.M. Boulanger, Role of microparticles in
atherothrombosis. J Intern Med, 2008. 263(5): p. 528-37.
434. Berckmans, R.J., et al., Cell-derived microparticles circulate in healthy humans and
support low grade thrombin generation. Thromb Haemost, 2001. 85(4): p. 639-46.
435. Lechner, D. and A. Weltermann, Circulating tissue factor-exposing microparticles.
Thromb Res, 2008. 122 Suppl 1: p. S47-54.
436. Yu, J.L. and J.W. Rak, Shedding of tissue factor (TF)-containing microparticles
rather than alternatively spliced TF is the main source of TF activity released from
human cancer cells. J Thromb Haemost, 2004. 2(11): p. 2065-7.
437. Biró, E., et al., Human cell-derived microparticles promote thrombus formation in
vivo in a tissue factor-dependent manner. J Thromb Haemost, 2003. 1(12): p. 2561-
8.
438. Sadallah, S., et al., Ectosomes released by platelets induce differentiation of CD4+T
cells into T regulatory cells. Thromb Haemost, 2014. 112(6): p. 1219-29.
439. Dinkla, S., et al., Platelet microparticles inhibit IL-17 production by regulatory T
cells through P-selectin. Blood, 2016. 127(16): p. 1976-86.
440. Sprague, D.L., et al., Platelet-mediated modulation of adaptive immunity: unique
delivery of CD154 signal by platelet-derived membrane vesicles. Blood, 2008.
111(10): p. 5028-36.
441. Yari, F., et al., Interaction of Platelet-Derived Microparticles with a Human B-
Lymphoblast Cell Line: A Clue for the Immunologic Function of the Microparticles.
Transfus Med Hemother, 2018. 45(1): p. 55-61.
442. Tessandier, N., et al., Platelets Disseminate Extracellular Vesicles in Lymph in
Rheumatoid Arthritis. Arterioscler Thromb Vasc Biol, 2020: p.
ATVBAHA119313698.
443. Milasan, A., et al., Extracellular vesicles are present in mouse lymph and their level
differs in atherosclerosis. J Extracell Vesicles, 2016. 5: p. 31427.

209
444. Laffont, B., et al., Activated platelets can deliver mRNA regulatory
Ago2*microRNA complexes to endothelial cells via microparticles. Blood, 2013.
122(2): p. 253-61.
445. van der Grein, S.G. and E.N. Nolte-'t Hoen, "Small Talk" in the Innate Immune
System via RNA-Containing Extracellular Vesicles. Front Immunol, 2014. 5: p. 542.
446. Boon, R.A. and K.C. Vickers, Intercellular transport of microRNAs. Arterioscler
Thromb Vasc Biol, 2013. 33(2): p. 186-92.
447. Mittelbrunn, M. and F. Sanchez-Madrid, Intercellular communication: diverse
structures for exchange of genetic information. Nat Rev Mol Cell Biol, 2012. 13(5):
p. 328-35.
448. Marcoux, G., et al., Revealing the diversity of extracellular vesicles using high-
dimensional flow cytometry analyses. Sci Rep, 2016. 6: p. 35928.
449. Marcoux, G., et al., Microparticle and mitochondrial release during extended
storage of different types of platelet concentrates. Platelets, 2017. 28(3): p. 272-280.
450. Robert, S., et al., High-sensitivity flow cytometry provides access to standardized
measurement of small-size microparticles--brief report. Arterioscler Thromb Vasc
Biol, 2012. 32(4): p. 1054-8.
451. Poncelet, P., et al., Tips and tricks for flow cytometry-based analysis and counting
of microparticles. Transfus Apher Sci, 2015. 53(2): p. 110-26.
452. Qiu, P., et al., Extracting a cellular hierarchy from high-dimensional cytometry
data with SPADE. Nat Biotechnol, 2011. 29(10): p. 886-91.
453. Anchang, B., et al., Visualization and cellular hierarchy inference of single-cell
data using SPADE. Nat Protoc, 2016. 11(7): p. 1264-79.
454. Mair, F., et al., The end of gating? An introduction to automated analysis of high
dimensional cytometry data. Eur J Immunol, 2016. 46(1): p. 34-43.
455. Alkhatatbeh, M.J., et al., Strategies for enumeration of circulating microvesicles on
a conventional flow cytometer: Counting beads and scatter parameters. J Circ
Biomark, 2018. 7: p. 1849454418766966.
456. Ayre, D.C., et al., CD24 induces changes to the surface receptors of B cell
microvesicles with variable effects on their RNA and protein cargo. Sci Rep, 2017.
7(1): p. 8642.
457. Benmoussa, A., et al., A subset of extracellular vesicles carries the bulk of
microRNAs in commercial dairy cow's milk. J Extracell Vesicles, 2017. 6(1): p.
1401897.
458. Bodega, G., et al., Microvesicles: ROS scavengers and ROS producers. J Extracell
Vesicles, 2019. 8(1): p. 1626654.
459. Denis, H.L., et al., Platelet-derived extracellular vesicles in Huntington's disease. J
Neurol, 2018. 265(11): p. 2704-2712.
460. Boilard, E., Extracellular vesicles and their content in bioactive lipid mediators:
more than a sack of microRNA. J Lipid Res, 2018. 59(11): p. 2037-2046.
461. Cossarizza, A., et al., Guidelines for the use of flow cytometry and cell sorting in
immunological studies (second edition). Eur J Immunol, 2019. 49(10): p. 1457-
1973.
462. Hasilo, C.P., et al., Presence of diabetes autoantigens in extracellular vesicles
derived from human islets. Sci Rep, 2017. 7(1): p. 5000.

210
463. Kondratov, K., et al., Heterogeneity of the nucleic acid repertoire of plasma
extracellular vesicles demonstrated using high-sensitivity fluorescence-activated
sorting. J Extracell Vesicles, 2020. 9(1): p. 1743139.
464. Kwizera, E.A., et al., Molecular Detection and Analysis of Exosomes Using
Surface-Enhanced Raman Scattering Gold Nanorods and a Miniaturized Device.
Theranostics, 2018. 8(10): p. 2722-2738.
465. Li, K., et al., Advances, challenges, and opportunities in extracellular RNA biology:
insights from the NIH exRNA Strategic Workshop. JCI Insight, 2018. 3(7).
466. Taverna, S., et al., Breast Cancer Derived Extracellular Vesicles in Bone Metastasis
Induction and Their Clinical Implications as Biomarkers. Int J Mol Sci, 2020.
21(10).
467. Tucher, C., et al., Extracellular Vesicle Subtypes Released From Activated or
Apoptotic T-Lymphocytes Carry a Specific and Stimulus-Dependent Protein Cargo.
Front Immunol, 2018. 9: p. 534.
468. Vagner, T., et al., Protein Composition Reflects Extracellular Vesicle
Heterogeneity. Proteomics, 2019. 19(8): p. e1800167.
469. Vazquez, J., I.M. Ong, and A.K. Stanic, Single-cell technologies in reproductive
immunology. Am J Reprod Immunol, 2019. 82(3): p. e13157.
470. Veziroglu, E.M. and G.I. Mias, Characterizing Extracellular Vesicles and Their
Diverse RNA Contents. Front Genet, 2020. 11: p. 700.
471. Zhang, X., M.J. Hubal, and V.B. Kraus, Immune cell extracellular vesicles and their
mitochondrial content decline with ageing. Immun Ageing, 2020. 17: p. 1.
472. Marcoux, G., et al., Platelet-derived extracellular vesicles convey mitochondrial
DAMPs in platelet concentrates and their levels are associated with adverse
reactions. Transfusion, 2019. 59(7): p. 2403-2414.
473. Picca, A., et al., Mitochondrial Signatures in Circulating Extracellular Vesicles of
Older Adults with Parkinson's Disease: Results from the EXosomes in PArkiNson's
Disease (EXPAND) Study. J Clin Med, 2020. 9(2).
474. Picca, A., et al., Generation and Release of Mitochondrial-Derived Vesicles in
Health, Aging and Disease. J Clin Med, 2020. 9(5).
475. Jang, S.C., et al., Mitochondrial protein enriched extracellular vesicles discovered
in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles,
2019. 8(1): p. 1635420.
476. Poveda, E., et al., Massive release of CD9+ microvesicles in HIV infection,
regardless of virologic control. J Infect Dis, 2020.
477. Ha, B.G., et al., Depletion of Mitochondrial Components from Extracellular
Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome. Int J
Mol Sci, 2021. 22(1).
478. Puhm, F., et al., Mitochondria Are a Subset of Extracellular Vesicles Released by
Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial
Cells. Circ Res, 2019. 125(1): p. 43-52.
479. McVey, M.J., et al., Acid sphingomyelinase mediates murine acute lung injury
following transfusion of aged platelets. Am J Physiol Lung Cell Mol Physiol, 2017.
312(5): p. L625-L637.
480. McVey, M.J., et al., Platelet Extracellular Vesicles mediate Transfusion-related
Acute Lung
Injury by imbalancing the Sphingolipid Rheostat. 2020: Blood.

211
481. Todkar, K., et al., Selective packaging of mitochondrial proteins into extracellular
vesicles prevents the release of mitochondrial DAMPs. Nat Commun, 2021. 12(1):
p. 1971.
482. Phinney, D.G., et al., Mesenchymal stem cells use extracellular vesicles to
outsource mitophagy and shuttle microRNAs. Nat Commun, 2015. 6: p. 8472.
483. Fergie, N., et al., Hypercapnic acidosis induces mitochondrial dysfunction and
impairs the ability of mesenchymal stem cells to promote distal lung epithelial
repair. FASEB J, 2019. 33(4): p. 5585-5598.
484. Abraham, A. and A. Krasnodembskaya, Mesenchymal stem cell-derived
extracellular vesicles for the treatment of acute respiratory distress syndrome. Stem
Cells Transl Med, 2020. 9(1): p. 28-38.
485. Ikeda, G., et al., Mitochondria-Rich Extracellular Vesicles From Autologous Stem
Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J Am
Coll Cardiol, 2021. 77(8): p. 1073-1088.
486. Peruzzotti-Jametti, L., et al., Neural stem cells traffic functional mitochondria via
extracellular vesicles. PLoS Biol, 2021. 19(4): p. e3001166.
487. Zhang, Z., et al., Mesenchymal Stem Cell-Conditioned Medium Improves
Mitochondrial Dysfunction and Suppresses Apoptosis in Okadaic Acid-Treated SH-
SY5Y Cells by Extracellular Vesicle Mitochondrial Transfer. J Alzheimers Dis,
2020. 78(3): p. 1161-1176.
488. Maouia, A., et al., The Immune Nature of Platelets Revisited. Transfus Med Rev,
2020.
489. Korenevskii, A.V., et al., Mass-Spectrometric Analysis of Proteome of
Microvesicles Produced by NK-92 Natural Killer Cells. Bull Exp Biol Med, 2018.
165(4): p. 564-571.
490. Zhu, Y., et al., A Comprehensive Proteomics Analysis Reveals a Secretory Path-
and Status-Dependent Signature of Exosomes Released from Tumor-Associated
Macrophages. J Proteome Res, 2015. 14(10): p. 4319-31.
491. Rody, W.J., et al., The proteome of extracellular vesicles released by clastic cells
differs based on their substrate. PLoS One, 2019. 14(7): p. e0219602.
492. Dekel, E., et al., 20S proteasomes secreted by the malaria parasite promote its
growth. Nat Commun, 2021. 12(1): p. 1172.
493. Robbins, P.D. and A.E. Morelli, Regulation of immune responses by extracellular
vesicles. Nat Rev Immunol, 2014. 14(3): p. 195-208.
494. Kalra, H., G.P. Drummen, and S. Mathivanan, Focus on Extracellular Vesicles:
Introducing the Next Small Big Thing. Int J Mol Sci, 2016. 17(2): p. 170.

212
Annexe I: Mitochondrial damage-associated
molecular patterns in blood transfusion products
Genevieve Marcoux1,2,3 et Eric Boilard1,2,3 Affiliations
1 Axe Maladies Infectieuses et Inflammatoires, Centre de Recherche du CHU de Québec, Université Laval, Québec, QC, Canada.
2 Département de Microbiologie-infectiologie et D'immunologie and Centre ARThrite, Université Laval, Québec, QC, Canada.
3 Department of Infectious Diseases and Immunity, Centre de Recherche du CHU de Québec, Québec, QC, Canada.

213
Abstract
Mitochondria are organelles in charge of energy supply and the control of apoptosis.
Owing to their similarities with bacteria, however, extracellular mitochondria are
considered damage‐associated molecular patterns (DAMPs) capable of activating
the immune system and non‐immune cells. Studies revealed that diverse blood
products contain extracellular mitochondria, which could account for the adverse
reactions that can occur in transfusion. In this review, we discuss how mitochondrial
DAMPs can trigger inflammatory responses, we highlight conditions relevant to
transfusion during which mitochondria have been identified outside cells, and we
discuss their potential as biomarkers for the assessment of blood product quality.

214
Mitochondria and inflammation
The mitochondrion is a small organelle localized within cells that mediates the
oxidative phosphorylation necessary for ATP production1. In addition to its role in
energy supply, the mitochondrion also plays an instrumental role in the control of cell
division and apoptosis1, 2. The number of mitochondria present in different cellular
lineages is variable: there are no mitochondria in erythrocytes, approximately 4–7
mitochondria in platelets and leucocytes, and up to 2000 mitochondria in
hepatocytes1, 3. Depending on the metabolic activities necessary for cell functions,
the size of mitochondria can vary (0.5 μm to 10 μm)4, 5, with platelets presenting
smaller mitochondria, and cardiomyocytes demonstrating changes in mitochondrial
dimensions depending on their activities and hypoxia3, 6. Moreover, mitochondria are
dynamic organelles; they can divide, a process known as fission, and can merge
together, a process known as fusion7.
Studies suggest that mitochondria share with bacteria several similarities8, 9. Both
bacteria and mitochondria can consume O2 and produce CO2, express formylated
peptides10, 11 and bear a circular genome composed of hypomethylated CpG DNA
motifs12, 13. Hence, the genome of mitochondria shares significant levels of homology
with the one of Rickettsia prowatzekii, a gram‐negative alphaproteobacteria,
suggesting that they might have a common ancestor12-14. While the mitochondrion
in the cytosol contributes to activation of the inflammasome, its components can also
activate pathogen recognition receptor (PRR), thus providing distinct mechanisms
potentially leading to inflammatory responses15, 16. Reactive oxygen species (ROS)
and ATP derived from mitochondria are known triggers of inflammasomes and were
shown to activate Type I interferon regulated genes17. Its mitochondrial DNA
(mtDNA) can activate toll‐like receptors (TLR) present in endosomes whereas its
formylated peptides can activate formyl peptide receptors (Fig. 1)10, 18-20.
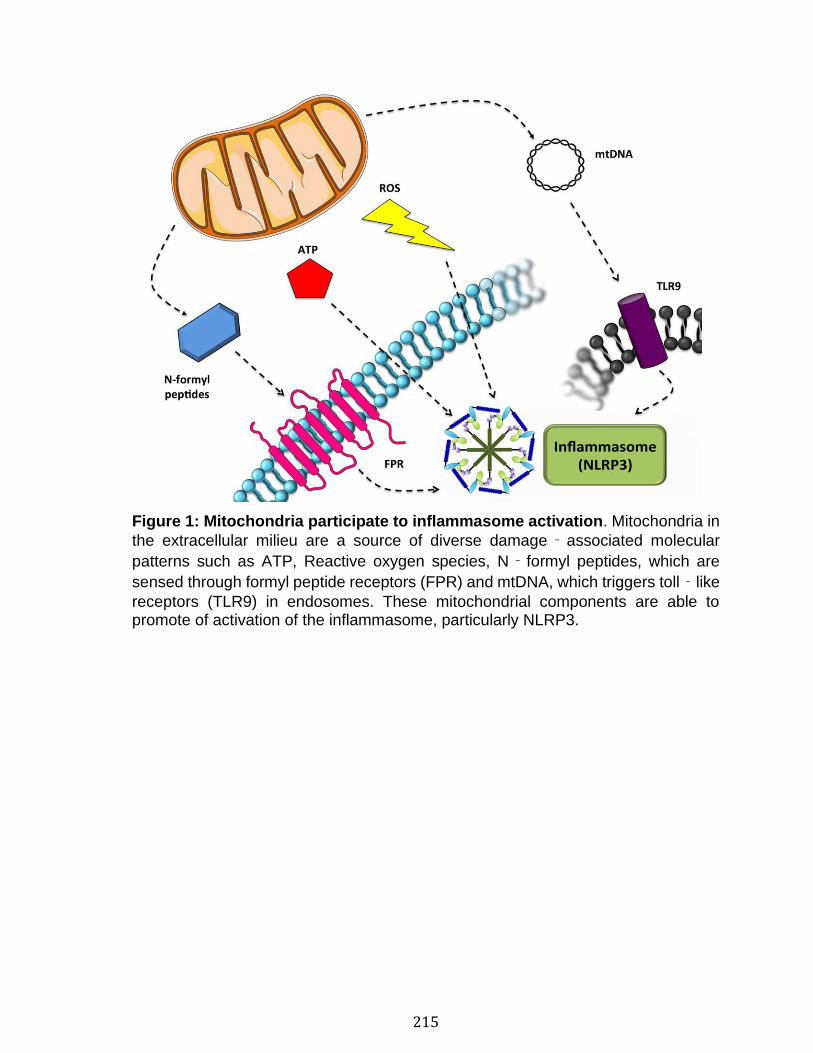
215
Figure 1: Mitochondria participate to inflammasome activation. Mitochondria in
the extracellular milieu are a source of diverse damage‐associated molecular
patterns such as ATP, Reactive oxygen species, N‐formyl peptides, which are
sensed through formyl peptide receptors (FPR) and mtDNA, which triggers toll‐like
receptors (TLR9) in endosomes. These mitochondrial components are able to promote of activation of the inflammasome, particularly NLRP3.

216
Mitochondrial release
Mitochondria are released from different cellular lineages in multiple conditions:
Damaged organs or tissues (e.g. liver and brain)11, 21-23, neutrophils24-26,
hepatocytes21, mast cells27, eosinophils28, T cells29 and platelets3, 30 can release
mitochondria. Extracellular mitochondria have been described in brain trauma31, in
synovial fluid of patients with rheumatoid arthritis3, 32, in systemic lupus
erythematosus25, 26, in steatohepatitis21, burn injury33 and in blood of patients at the
intensive care unit34. As parasites also expulse their mitochondria33, it points to a
well‐conserved mechanism and suggests that it might have relevance to different
biological processes yet to determine.
With their great abundance in blood, platelets are the main pool of mitochondria in
blood circulation. Thus, the presence of mitochondria in the extracellular milieu in
blood transfusion products, such as stored red blood cells, plasma and platelet
concentrates3, 35-37, is highly likely to reflect platelet activation or lesions, which might
have occurred during blood collection, blood processing, storage condition and
duration or treatment with agents (e.g. pathogen inactivation systems and platelet
additive solutions)36-40. Evidence of mitochondrial release by platelets originate from
studies on platelet extracellular vesicles, submicron vesicles released by cells on
activation or apoptosis. Extracellular vesicles produced by platelets, known as
microparticles, were revealed to contain mitochondrial peptides41, pointing to a
mitochondrial content. Whether mitochondria were actually present in microparticles
was however unknown. Studies were, therefore, undertaken to determine the impact
of platelet activation on mitochondrial release. Boudreau et al.3 showed that various
stimuli (collagen, thrombin and ionophore) could induce release of mitochondria.
Mitochondria were identified in the extracellular milieu either as naked organelle (free
mitochondria) or encapsulated within microparticles3. These structures were
quantified using flow cytometry and visualized using electron microscopy. Given that
platelets represent an important source of mitochondria in blood circulation, platelet
concentrates were searched for the presence of extracellular mitochondria.
Mitochondria, both as free organelle and inside microparticles, were found in platelet

217
concentrates prepared in blood bank conditions3. Interestingly, a simultaneous study
confirmed that mtDNA is present in various blood products, such as plasma, platelet
concentrates and stored red blood cells33. It is noteworthy to mention that the
quantification of mtDNA in extracellular milieu is an actual measure of potential
mtDNA in solution, free mitochondria and microparticle‐containing mitochondria. A
complete characterization of the extracellular mitochondria is thus needed in order
to determine the mechanism leading to mitochondrial release and to understand their
impact on other cells. Although the origin of mtDNA in these products is unknown, it
is tempting to speculate that most originate from platelets.

218
Extracellular mitochondria and adverse reactions
There are studies in which the potential contribution of extracellular mitochondria to
adverse reactions was examined. Boudreau et al. quantified mtDNA in the
extracellular milieu of platelet concentrates that has induced adverse reactions and
compared the levels within samples that were transfused without incidents3.
Interestingly, the authors confirmed that significantly higher levels of extracellular
mitochondria correlated with adverse reactions. Using a decisional tree based on an
algorithm to compare cytokine levels and mtDNA, Cognasse et al. found that mtDNA
did not correlate with levels of cytokines, suggesting that it might be an independent
risk factor40, 42. Importantly, these observations were corroborated by Yasui et al.
who further confirmed that elevated levels of mtDNA were present in platelet
concentrates that induced nonhemolytic transfusion reactions (NHTRs) in platelet
transfusion43. Moreover, levels of mtDNA present in plasma and platelet
concentrates correlate well with levels of mtDNA in serum of post‐transfusion
patients and are associated with higher risk of acute respiratory distress syndrome35,
44.
How exactly mitochondria may mediate their inflammatory role in transfusion is
unknown, but different studies on transfusion research provided mechanistic
insights. Boudreau et al. suggested that an enzyme, a secretory phospholipase A2
(sPLA2) called IIA (sPLA2‐IIA), could hydrolyse naked mitochondria, due to its
bactericidal properties, and thereby release mtDNA and lysophospholipids, which
are known triggers of transfusion related acute lung injury3, 45. Hence sPLA2‐IIA is
present in blood, in platelet concentrates, and is induced in blood in inflammatory
conditions3. Yasui et al. found that mtDNA could activate neutrophils, monocytes and
basophils43. The authors showed that when neutrophils were primed with formylated
peptides or anti‐HLA antibodies, mtDNA at concentrations found in nonhaemolytic
transfusion reactions could induce oxidative burst and Mac‐ 1 expression43.
Although studies are still needed to confirm whether or not platelet concentrates also
contain extracellular formylated peptides, mitochondrial formylated peptides were

219
found to induce neutrophil infiltration and airway contraction, further demonstrating
that extracellular mitochondria could contribute to adverse reactions through diverse
components, other than mtDNA46.

220
Extracellular mitochondria as a biomarker
What induces mitochondrial release in blood transfusion products is not clear at the
moment. Given that mtDNA is present in frozen plasma, it suggests that
mitochondria were released prior to plasma preparation. The combination of flow
cytometry and spanning‐tree progression analysis of density‐normalized events
(SPADE) as computational approach permits a better appreciation of the
heterogeneity of microparticles (i.e. cellular source, compartment origin, organelle
content and surface markers expression)47. Using flow cytometry and SPADE to
assess presence of naked organelles and mitochondria encapsulated in
microparticles, it was found that the preparation method had a significant impact on
extrusion of mitochondria and that storage duration had no or very modest effect on
release37, 38, 47. Platelets prepared using the platelet‐rich plasma method contained
more extracellular mitochondria than those prepared using apheresis and buffy coat
methods, even before storage37. Furthermore, treatments of platelets with pathogen
inactivation systems, which can induce release of microparticles39, 48, also
engendered mitochondrial release36. There is currently no study that verified whether
mitochondrial damage‐associated molecular patterns (DAMPs)16, 20 were actually
present in blood of certain blood donors, thus prior blood processing and storage.

221
Summary
Although more work is necessary to confirm whether mitochondrial DAMPs are the
actual cause of adverse reactions, the measurement of mtDNA in blood transfusion
products is a simple inexpensive approach to determine its quality. Using a
quantitative PCR, it was found that pathogen inactivation systems could cross‐link
mtDNA, thus impeding polymerase reactions when longer amplification fragments
are generated49. While these observations confirm that Intercept and Mirasol are
both capable of damaging mtDNA50, 51, the approach reported in this study49 permits
to confirm efficiency of the inactivation process. Quantitative PCR can also be
performed to assess the presence of mtDNA in the extracellular milieu. In sum,
quantification of mtDNA using PCR or assessment of extracellular mitochondria
using flow cytometry are powerful strategies to determine the quality of blood
transfusion products.
There is a growing number of studies that report mitochondrial release in
pathological conditions. Future studies are thus needed to determine whether
extracellular mitochondria are more abundant in blood of certain blood donors,
thereby explaining their presence in only a fraction of blood products. While
mitochondrial release might be part of well‐regulated mechanisms of intercellular
communication in differential biological processes, the presence of extracellular
mitochondria in blood transfusion products, however, may contribute to adverse
reactions. The assessment of extracellular mitochondria in blood transfusion
products will indicate the usefulness of this biomarker in transfusion and will
contribute to determine conditions underlying mitochondrial release.

222
Acknowledgements
The authors declare no competing financial interests. EB and GM wrote the review.
This work was supported by the Canadian Institutes of Health Research and the
Canadian Blood Services (EB). EB is the recipient of a Canadian Institutes of Health
Research new investigator award and is a Canadian National Transplant Research
Program researcher. GM is a recipient of awards from the Canadian Blood Services.
The views expressed herein do not necessarily represent the view of the federal
government.

223
References
1 Andersen JL, Kornbluth S: The tangled circuitry of metabolism and apoptosis. Mol Cell 2013; 49:399–410 2 Wang C, Youle RJ: The role of mitochondria in apoptosis*. Annu Rev Genet 2009; 43:95–118 3 Boudreau LH, Duchez AC, Cloutier N, et al.: Platelets release mitochondria serving as substrate for bactericidal group IIAsecreted phospholipase A2 to promote inflammation. Blood 2014; 124:2173–2183 4 Palade GE: An electron microscope study of the mitochondrial structure. J Histochem Cytochem 1953; 1:188–211 5 Palade GE: The fine structure of mitochondria. Anat Rec 1952; 114:427–451 6 Neary MT, Ng KE, Ludtmann MH, et al.: Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J Mol Cell Cardiol 2014; 74:340–352 7 van der Bliek BA, Shen Q, Kawajiri S: Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol2013; 5:a011072 8 Gray MW, Burger G, Lang BF: Mitochondrial evolution.Science 1999; 283:1476–1481 9 Arechaga I: Membrane invaginations in bacteria and mitochondria: common features and evolutionary scenarios. J Mol Microbiol Biotechnol 2013; 23:13–23 10 Zhang Q, Raoof M, Chen Y, et al.: Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104–107 11 McDonald B, Pittman K, Menezes GB, et al.: Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010; 330:362–366 12 Lang BF, Burger G, O’Kelly CJ, et al.: An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature 1997; 387:493–497 13 Friedman JR, Nunnari J: Mitochondrial form and function. Nature 2014; 505:335–343 14 Andersson SG, Zomorodipour A, Andersson JO, et al.: The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998; 396:133–140 15 Sandhir R, Halder A, Sunkaria A: Mitochondria as a centrally positioned hub in the innate immune response. Biochem Biophys Acta 2017; 1863:1090–1097 16 Mills EL, Kelly B, O’Neill LAJ: Mitochondria are the powerhouses of immunity. Nat Immunol 2017; 18:488–498 17 Zhou R, Yazdi AS, Menu P, et al.: A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221–225 18 Weinberg SE, Sena LA, Chandel NS: Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015; 42:406–417 19 West AP, Shadel GS, Ghosh S: Mitochondria in innate immune responses. Nat Rev Immunol 2011; 11:389–402 20 Krysko DV, Agostinis P, Krysko O, et al.: Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011; 32:157– 164 21 Garcia-Martinez I, Santoro N, Chen Y, et al.: Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 2016; 126:859–864 22 Marques PE, Amaral SS, Pires DA, et al.: Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012; 56:1971–1982 23 Oka T, Hikoso S, Yamaguchi O, et al.: Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012; 485:251–255 24 Yousefi S, Mihalache C, Kozlowski E, et al.: Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 2009; 16:1438–1444 25 Lood C, Blanco LP, Purmalek MM, et al.: Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016; 22:146–153 26 Caielli S, Athale S, Domic B, et al.: Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 2016; 213:697–713 27 Zhang B, Asadi S, Weng Z, et al.: Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS One 2012; 7: e49767

224
28 Yousefi S, Gold JA, Andina N, et al.: Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 2008; 14:949–953 29 Maeda A, Fadeel B: Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis 2014; 5:e1312 30 Boilard E, Duchez AC, Brisson A: The diversity of platelet microparticles. Curr Opin Hematol 2015; 22:437–444 31 Zhao Z, Wang M, Tian Y, et al.: Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood 2016; 127:2763–2772 32 Hajizadeh S, DeGroot J, TeKoppele JM, et al.: Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther 2003; 5:R234–R240 33 Simmons JD, Lee YL, Mulekar S, et al.: Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg 2013; 258:591–596; discussion 6–8 34 Nakahira K, Kyung SY, Rogers AJ, et al.: Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 2013; 10: e1001577; discussion e 35 Simmons JD, Lee YL, Pastukh VM, et al.: Potential contribution of mitochondrial DNA damage associated molecular patterns in transfusion products to the development of acute respiratory distress syndrome after multiple transfusions. J Trauma Acute Care Surg 2017; 82:1023–1029 36 Chen Z, Schubert P, Bakkour S, et al.: p38 mitogen-activated protein kinase regulates mitochondrial function and microvesicle release in riboflavin- and ultraviolet light-treated apheresis platelet concentrates. Transfusion 2017; 57:1199–1207 37 Marcoux G, Duchez AC, Rousseau M, et al.: Microparticle and mitochondrial release during extended storage of different types of platelet concentrates. Platelets 2017; 28:272–280 38 Bakkour S, Acker JP, Chafets DM, et al.: Manufacturing method affects mitochondrial DNA release and extracellular vesicle composition in stored red blood cells. Vox Sang 2016; 111:22–32 39 Magron A, Laugier J, Provost P, et al.: Pathogen reduction technologies: the pros and cons for platelet transfusion. Platelets 2017. doi: 10.1080/09537104.2017.1306046. [Epub ahead of print] 40 Cognasse F, Aloui C, Anh NK, et al.: Platelet components associated with adverse reactions: predictive value of mitochondrial DNA relative to biological response modifiers. Transfusion 2016; 56:497–504 41 Dean WL, Lee MJ, Cummins TD, et al.: Proteomic and functional characterisation of platelet microparticle size classes. Thromb Haemost 2009; 102:711–718 42 Burnouf T, Chou ML, Goubran H, et al.: An overview of the role of microparticles/microvesicles in blood components: are they clinically beneficial or harmful? Transfus Apher Sci 2015; 53:137–145 43 Yasui K, Matsuyama N, Kuroishi A, et al.: Mitochondrial damage-associated molecular patterns as potential proinflammatory mediators in post-platelet transfusion adverse effects. Transfusion 2016; 56:1201–1212 44 Lee YL, King MB, Gonzalez RP, et al.: Blood transfusion products contain mitochondrial DNA damage-associated molecular patterns: a potential effector of transfusion-related acute lung injury. J Surg Res 2014; 191:286–289 45 Silliman CC, Bjornsen AJ, Wyman TH, et al.: Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion 2003; 43:633–640 46 Wenceslau CF, Szasz T, McCarthy CG, et al.: Mitochondrial N-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm Pharmacol Ther 2016; 37:49–56 47 Marcoux G, Duchez AC, Cloutier N, et al.: Revealing the diversity of extracellular vesicles using high-dimensional flow cytometry analyses. Sci Rep2016; 6:35928 48 Osman A, Hitzler WE, Meyer CU, et al.: Effects of pathogen reduction systems on platelet microRNAs, mRNAs, activation, and function. Platelets 2015; 26:154–163 49 Bakkour S, Chafets DM, Wen L, et al.: Development of a mitochondrial DNA real-time polymerase chain reaction assay for quality control of pathogen reduction with riboflavin and ultraviolet light. Vox Sang 2014; 107:351–359 50 Bruchmuller I, Losel R, Bugert P, et al.: Effect of the psoralen-based photochemical pathogen inactivation on mitochondrial DNA in platelets. Platelets 2005; 16:441–445

225
51 Bakkour S, Chafets DM, Wen L, et al.: Assessment of nucleic acid modification induced by amotosalen and ultraviolet A light treatment of platelets and plasma using real-time polymerase chain reaction amplification of variable length fragments of mitochondrial DNA. Transfusion 2016; 56:410–420

226
Annexe II: Role of platelets and megakaryocytes in
adaptive immunity
Genevieve Marcoux 1 ,2 , 3 , Audrée Laroche 1, 2 , 3 , Jenifer Espinoza Romero1 , 2 , 3
and Eric Boilard 1 , 2 ,3
Affiliations
1 Axe Maladies Infectieuses et Inflammatoires, Centre de Recherche du CHU de Québec, Université Laval , Québec, QC, Canada.
2 Département de Microbiologie-infectiologie et D'immunologie and Centre ARThrite, Université Laval , Québec, QC, Canada.
3 Department of Infectious Diseases and Immunity, Centre de Recherche du CHU de Québec , Québec, QC, Canada.

227
Abstract
The immune system is comprised of two principal interconnected components called
innate and adaptive immunity. While the innate immune system mounts a
nonspecific response that provides protection against the spread of foreign
pathogens, the adaptive immune system has developed to specifically recognize a
given pathogen and lead to immunological memory. Platelets are small fragments
produced from megakaryocytes in bone marrow and lungs. They circulate
throughout the blood to monitor the integrity of the vasculature and to prevent
bleeding. Given their large repertoire of immune receptors and inflammatory
molecules, platelets and megakaryocytes can contribute to both innate and adaptive
immunity. In adaptive immunity, platelets and megakaryocytes can process and
present antigens to lymphocytes. Moreover, platelets, via FcγRIIA, rapidly respond
to pathogens in an immune host when antibodies are present. This manuscript
reviews the reported contributions of platelets and megakaryocytes with emphasis
on antigen presentation and antibody response in adaptive immunity.

228
Introduction
The ability of an organism to defend itself against foreign substances and infectious
agents principally requires contributions from two components of the immune
system, namely innate or natural immunity and acquired, also known as adaptive,
immunity. The innate immune response implicates the recognition of well-conserved
molecular motifs, known as pathogen-associated or damage-associated molecular
patterns (respectively PAMPs or DAMPs). Pathogen-associated molecular patterns
are exogenous components conserved among a large spectrum of microorganisms
and allergens, while DAMPs, derived from host cells, are endogenous alarm signals
produced during cellular stress or death and lead to sterile inflammatory responses
[1,2]. PAMPs and DAMPs bind to cell surface receptors and intracellular sensors
known as pattern recognition receptors (PRRs), which include the Toll-like receptors
(TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and C-type lectin-
like receptors (CLRs). Pattern recognition receptors are expressed by innate
immune cells and nonimmune cells. Innate immune responses are nonspecific and
do not confer long-lasting immunity.
In contrast, the adaptive immune system has evolved to confer immunological
memory. It involves a process called antigen presentation that occurs during the
initial response and provides a stronger and more rapid immune response to a
subsequent encounter. More advanced acquired immunity is present in vertebrates,
but microorganisms such as prokaryotes have developed acquired immune memory
that provides protection against bacteriophages through a system called CRISPR
[3,4]. The adaptive immune system requires B lymphocytes, involved in the humoral
immune response, and T lymphocytes involved in the cell-mediated immune
response. T cells are further divided into subcategories according to their receptor
repertoire and functions.
Platelets are small (2–4 µm in diameter) anucleated fragments produced by
megakaryocytes. Nearly one trillion platelets patrol the blood vessels to monitor and
maintain the integrity of the latter. Thrombus formation occurs in response to blood

229
vessel damage in order to prevent bleeding [5]. However, the role of platelets is not
restricted to the hemostatic response [6,8]. Platelets are capable of phagocytosis,
which is suggested to be a conserved innate mechanism from thrombocytes in lower
vertebrates such as birds and fish [9,11]. Platelets also express immune and
inflammatory molecules, and a set of immune receptors such as TLRs, CD40L [6]
and the Fc receptor for immunoglobulin G (IgG) IIA (FcγRIIA) [12]. Upon activation,
platelets also release extracellular vesicles (EVs) that harbor some of these platelet
components. Given the vast number of platelets in blood (40:1 ratio to immune cells)
and their extensive array of immune receptors, the capacity of platelets to support,
promote and to actively participate in immunity is the focus of numerous
investigations.

230
Platelet-leukocyte Interactions and Implications for
Adaptive Immunity
Megakaryocytes (MK) and platelets, as well as their EVs, are suggested to play a
role in the innate immune response (reviewed in [6,8,13,16]). During adaptive
immunity, platelets can support the activities of classical antigen presenting cells.
While it has been demonstrated that platelet-derived molecules (PF4, CD40L, P-
selectin, serotonin,) are capable of enhancing dendritic cell (DC) differentiation and
antigen presentation, platelets can also promote recruitment of lymphocytes to sites
of inflammation [17,25]. Different molecules participate in platelet and leukocyte
interactions. The activated form of platelet αIIbβ3, the fibrinogen receptor, can bind
the solute carrier family 44 member 2 (SLC44A2) on neutrophils, which can enhance
their activation and netosis [26]. This receptor is also likely expressed by other
cellular lineages from the bone marrow (myeloid and non-myeloid), and whether it
contributes to platelet–lymphocyte interactions remains to be established. Through
its binding to fibrinogen, αIIbβ3 also indirectly interacts with Mac-1 [27]. Platelets and
neutrophils interact through CD62P or glycoprotein Ib, found on platelets, as well as
P-selectin glycoprotein ligand-1 (PSGL-1) and integrin CD11b/CD18 (Mac-1), found
on neutrophils [28,29].
Through direct interactions of platelet P-selectin and PSGL1 expressed by
monocytes, platelets mediate DC maturation. The contribution of platelet P-selectin
also enhances antigen cross presentation by DC, as they were more potent than
cytokine-derived DC in generating tumor-specific T cell immunity [20]. The effect of
another platelet molecule, serotonin, appears to have opposite effect on DC
differentiation: The DC capacity to stimulate T cells is decreased by serotonin, as
the latter reduces expression DC co-stimulatory molecules and enhances IL-10
production [30]. Similarly, platelet factor 4 (PF4) increases the DC responsiveness
to Toll-like receptor ligands [31], reduces the processing of antigens [17]. Moreover,
platelet-derived CD40L was shown to induce monocyte differentiation into DC, DC
maturation and upregulation of costimulatory molecules [23,25]. This function of
CD40L derived by platelets may be highly relevant to autoimmune diseases, such

231
as systemic lupus erythematosus in which platelets induce DC differentiation and
type-I interferon release, thereby promoting B-cell secretion of antibodies [32].
Platelets also impact lymphocyte functions. For instance, platelet interactions with
CD40 bearing cells, such as lymphocytes, via CD40L have already been covered
thoroughly in several reviews (for instance see [33,36]). In vivo and in vitro
investigations revealed a role of platelet CD40L in B cell isotype switching and the
increased CD8+ T cell function during infection [37]. Platelets expressing CD40L
were also identified in different pathologies where they directly activate the
endothelium [38], or in which they contribute in the recruitment of neutrophils and T
cells to the damaged endothelium, more particularly in the intima and in plaque
ruptures in atherosclerosis [39]. Upon activation, platelets release molecules such
as PF4, P-selectin or serotonin, which can modulate lymphocyte responses. For
example, PF4 inhibits proliferation of activated human T cells and their release of
cytokines [40]. While P-selectin expressed by platelets plays a role in the recruitment
of lymphocytes to sites of inflammation [18], the interaction of CD4+ T cells (but not
CD8+ T cells) with platelet P-selectin mediates the platelet recruitment in the liver,
where they activate the endothelium and enhance hepatocellular damage [19].
Serotonin, not synthesize by platelets but captured and stored via their serotonin
transporter in platelet dense granules [41], is also able to promote the recruitment of
CD4+ and CD8+ T cells [21,22]. With their PF4 and TGF-β content, platelets are
also involved in induction of immune tolerance as they can control the differentiation
of Th17 and the expression of Foxp3+ Tregs [42,43].
In sum, platelets and the platelet-derived molecules impact DC and both B- and T-
cells in adaptive immunity. This review, however, focuses on the role played by
platelets and megakaryocytes in antigen presentation and antibody response.

232
Antigen Processing and Presentation by Platelets
The proteasome is a high molecular weight multicatalytic protease primarily known
for its function in the maintenance of cellular protein hemostasis. It was first purified
from the cytosolic fraction of human platelets in the early nineties [44,45]. The
presence of all catalytically active subunits of the standard 20 S proteasome in
human platelets has since been confirmed, including the immunoproteasome
subunit β5 [46]. It was demonstrated that all of these components could assemble
into the constitutively active standard proteasome or immunoproteasome complex
in platelets [46]. The relevance of this organelle in platelets is not yet fully
understood, but a number of key functions such as platelet production by
megakaryocytes [47,48], delimiting platelet lifespan [49], activation [50,51]
(challenged by Koessleret al [52].), upregulation of proteasome activity during
bacterial sepsis [53] and release of extracellular vesicles (EVs) [54] have been
identified (reviewed recently [55]). These findings are of clinical relevance, as
bortezomib, a proteasome inhibitor used to treat patients with multiple lymphoma,
reportedly induces thrombocytopenia [47,48].
Another function of the proteasome in adaptive immunity is to hydrolyze proteins into
smaller peptides. This process is critically important for peptide loading into the major
histocompatibility complex (MHC) class I and their subsequent antigenic
presentation to CD8+ T cells. MHC I molecules are found on both murine and human
platelets [56,57], but a large proportion (70% to 80%) was shown to be adsorbed
from circulating MHC I molecules in plasma [58]. MHC II molecule, in contrast, is
absent on normal platelets although it was identified on platelets in immune
thrombocytopenia (ITP) [59,61]. In addition, platelets are also able to transfer their
MHC class I onto the tumor cell surface, conferring the tumor cell with an apparently
normal phenotype thereby providing protection from deletion by natural killer (NK)
cells [62]. This insidious role of platelet MHC I allows tumor growth and cancer
establishment.

233
In addition to MHC I and beta-2 microglobulin (B2-MG), a molecular chaperone for
the MHC I complex, mass spectrometry approaches identified 43 proteins related to
the “antigen processing and presentation” pathway within the platelet α-granule
proteome [63]. Among those proteins, the authors identified antigen peptide
transporters 1 and 2 (TAP1 and TAP2), which are involved in the translocation of
processed peptides into the platelet ER before they are loaded into MHC I molecules
[63]. Components of the peptide loading complex (Erp57, calreticulin, calnexin and
tapasin), as well as the T cell co-stimulatory molecules CD40, ICOSL and CD86 (the
latter in humans only) [63,64], are found in platelets, suggesting that platelets bear
the entire machinery concerned with antigen processing and presentation to T-cells.
While most molecules were identified in the intracellular compartment, surface
expression by platelets is necessary for the formation of immunological synapse with
T cells. Confocal microscopy analysis revealed that MHC I, B2-MG, TAP1 and TAP2
could localize on the surface of thrombin-activated platelets, pointing to their
secretion upon activation [63]. Moreover, young murine platelets displayed higher
levels of MHC I molecules on their plasma membrane than older platelets,
suggesting that MHC I surface expression is modulated throughout the platelet
lifespan [65]. Platelet activation during infection can also lead to the expression of
MHC I on the plasma membrane: both murine and human platelets increase
expression of MHC I molecules in response to infection by Plasmodium berghei [64]
and dengue virus [66].
Pioneer work by Chapman et al. unequivocally demonstrated the capacity of
platelets to process and present antigens to T-cells [64]. The authors loaded
platelets with ovalbumin (OVA) protein and confirmed that the machinery in platelets
could efficiently process OVA into peptides, including the antigenic peptide
sequence SIINFEKL, which can be recognized by the T cell receptor (TCR)
expressed by all T cells in transgenic OT-1 mice when presented by MHC I
molecules. Using both in vitro and in vivo approaches, they further demonstrated
that platelets could indeed present processed ovalbumin antigen through MHC I to

234
T-cells and, given the expression of co-stimulatory molecules by platelets, could
stimulate IL-2 production by T cells and their activation [64].
While these findings point to a potential use for platelets in a cell-based vaccine, this
hypothesis was tested with an actual pathogen, Plasmodium berghei, the parasite
that causes malaria. Using a Plasmodium berghei parasite transgenic for the C-
terminal amino acids 150–368 of OVA (Pba-OVA), it was shown that platelets, via
their MHC I, are able to acquire and present PbA-OVA-derived antigens to generate
protective T cells [64]. Together, these observations demonstrate that platelets are
capable of antigen processing and presentation (Figure 1). While platelets can
internalize pathogens other than Plasmodium berghei, such as dengue virus,
influenza virus and HIV [67,70] through phagocytosis or endocytosis, they also
increase bacterial clearance in septic mice [71]. However, it remains to be
established whether processed microbial antigens can be presented by MHC I
molecules on activated platelets to fulfill a role in adaptive immunity against these
pathogens.
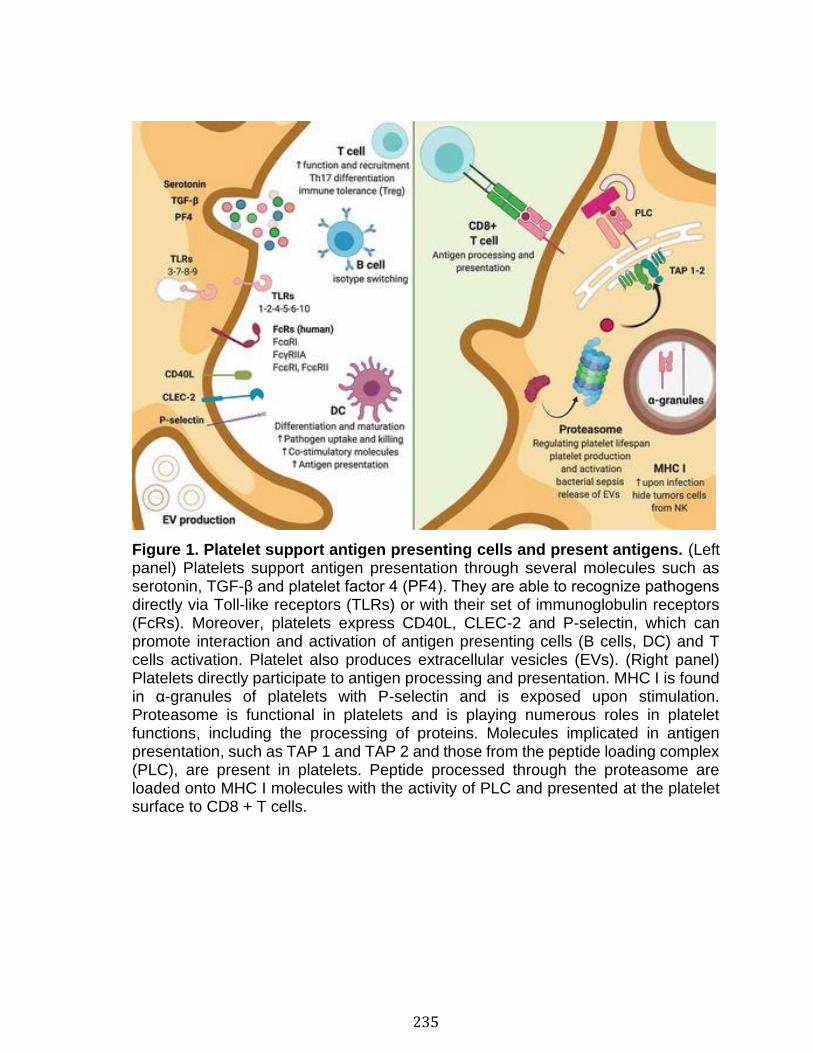
235
Figure 1. Platelet support antigen presenting cells and present antigens. (Left panel) Platelets support antigen presentation through several molecules such as serotonin, TGF-β and platelet factor 4 (PF4). They are able to recognize pathogens directly via Toll-like receptors (TLRs) or with their set of immunoglobulin receptors (FcRs). Moreover, platelets express CD40L, CLEC-2 and P-selectin, which can promote interaction and activation of antigen presenting cells (B cells, DC) and T cells activation. Platelet also produces extracellular vesicles (EVs). (Right panel) Platelets directly participate to antigen processing and presentation. MHC I is found in α-granules of platelets with P-selectin and is exposed upon stimulation. Proteasome is functional in platelets and is playing numerous roles in platelet functions, including the processing of proteins. Molecules implicated in antigen presentation, such as TAP 1 and TAP 2 and those from the peptide loading complex (PLC), are present in platelets. Peptide processed through the proteasome are loaded onto MHC I molecules with the activity of PLC and presented at the platelet surface to CD8 + T cells.

236
Antigen Processing and Presentation by Megakaryocytes
Megakaryocytes are large (50 to 100 µM) heterogeneous cells with a multilobular
nucleus that can contain several copies of each chromosome (up to 128 N).
Megakaryocytopoiesis in the bone marrow is under the control of a complex process
primarily regulated by thrombopoietin. Megakaryocytes in the bone marrow niche
are in contact with hemopoietic stem cells. By secretion of several molecules like
thrombopoietin, PF4 or TGF-β, they are thus able to regulate hemopoietic stem cells
quiescence or proliferation [72,73].
Megakaryocytes express numerous immune receptors (reviewed in [16]), including
TLR 1 through 6 (TLR 5 mRNA was identified in lung MK), Ig receptors (FcγRIIA and
FcεRI for human MKs, and FcγRI on a subpopulation of murine MKs), and co-
stimulatory molecules, such as CD40L. Moreover, neutrophils can migrate through
MKs by a mechanism known as emperipolesis, which can permit membrane
exchange between the two cellular lineages [74].
In contrast to platelets, which do not normally express MHC II molecules, early MK
progenitors derived from human hematopoietic stem cells express MHC II. These
MKs were suggested to play roles reminiscent of professional antigen presenting
cells. They have been shown to support T cell activation and to increase the
expansion of Th17, Th1, and Th17/Th1 double-positive cells when incubated in the
presence of lymphocytes from normal subjects or systemic lupus erythematosus
(SLE) patients [75]. Megakaryocyte progenitors were efficient at mounting an
immune response against the opportunistic pathogen Candida albicans, thereby
suggesting that the mobilization of hematopoietic stem cells can lead to the
generation of a certain population of MK with immune functions [76]. MHC II
molecules classically present peptides derived from extracellular pathogens
trafficking through endosomes, which is a site of virus entry. Dengue virus can grow
in MK progenitors [77], and mature MKs can generate antiviral immunity implicating
interferon molecules in the response to dengue virus as well as influenza virus.
However, it remains to be established whether MKs can process and present viral
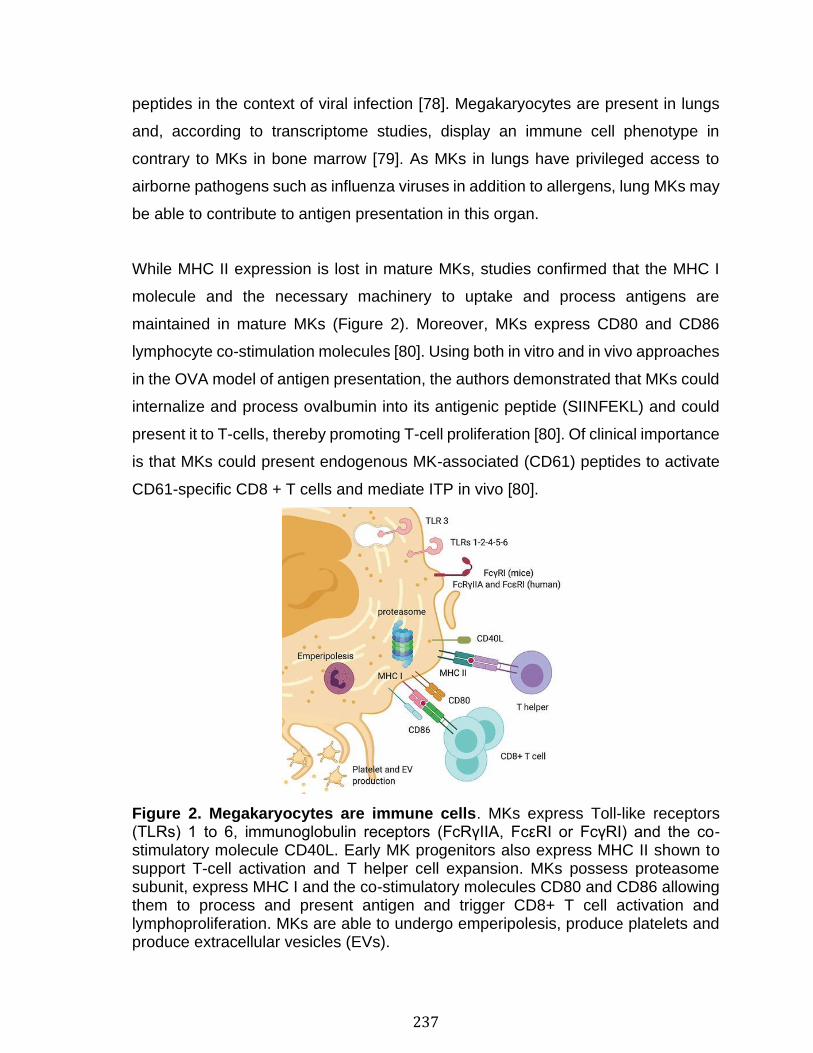
237
peptides in the context of viral infection [78]. Megakaryocytes are present in lungs
and, according to transcriptome studies, display an immune cell phenotype in
contrary to MKs in bone marrow [79]. As MKs in lungs have privileged access to
airborne pathogens such as influenza viruses in addition to allergens, lung MKs may
be able to contribute to antigen presentation in this organ.
While MHC II expression is lost in mature MKs, studies confirmed that the MHC I
molecule and the necessary machinery to uptake and process antigens are
maintained in mature MKs (Figure 2). Moreover, MKs express CD80 and CD86
lymphocyte co-stimulation molecules [80]. Using both in vitro and in vivo approaches
in the OVA model of antigen presentation, the authors demonstrated that MKs could
internalize and process ovalbumin into its antigenic peptide (SIINFEKL) and could
present it to T-cells, thereby promoting T-cell proliferation [80]. Of clinical importance
is that MKs could present endogenous MK-associated (CD61) peptides to activate
CD61-specific CD8 + T cells and mediate ITP in vivo [80].
Figure 2. Megakaryocytes are immune cells. MKs express Toll-like receptors (TLRs) 1 to 6, immunoglobulin receptors (FcRγIIA, FcεRI or FcγRI) and the co-stimulatory molecule CD40L. Early MK progenitors also express MHC II shown to support T-cell activation and T helper cell expansion. MKs possess proteasome subunit, express MHC I and the co-stimulatory molecules CD80 and CD86 allowing them to process and present antigen and trigger CD8+ T cell activation and lymphoproliferation. MKs are able to undergo emperipolesis, produce platelets and produce extracellular vesicles (EVs).

238
Platelets Response to Antibodies
Fc Receptors
Fc receptors (FcRs) regulate antibody-mediated responses by binding to the
fragment crystallizable (Fc) region of immunoglobulins. Fc receptors are at the
interface between the innate and adaptative immune systems as they activate
immune cells and can promote internalization of pathogens captured by antibodies.
Fc receptors are divided according to their affinity for different immunoglobulin (Ig)
classes or subclasses, their interaction with different forms of Ig (monomeric,
aggregated, immune complexes IC) and their function (activatory/inhibitory).
According to their heterogeneity, many biological functions can be regulated by
FcRs, such as degranulation, proliferation, phagocytosis, transcytosis and
immunoglobulin recycling [81].
Both mice and humans express FcRs although many differences can be observed
with regard to expression pattern or the affinities of murine and human FcRs. For
example, humans express some FcRs that mice do not, such as FcαRI and FcγRIIA,
the specific receptors for IgA and IgG, respectively [82]. Moreover, humans possess
more IgG-binding receptors FcγR than mice [82]. Conversely, mice express FcγRIV
that has no human counterpart [83], although human FcγRIIIA and FcγRI are
suggested as functional orthologs of murine FcγRIV.
Like many other immune cells, human platelets express FcRs. It has been shown
that human platelets express four activatory FcRs: FcαRI [84], FcεRI, FcεRII [85,87],
and FcγRIIA [88]. Expressed FcRs allow platelets to interact with different
immunoglobulin classes (IgA, IgE and IgG), which further highlights the role of
platelets in the immune response. For example, platelets can be activated by IgE
(via FcεRI and FcεRII), which points to a potential role for platelets in allergic
disorders, or against parasites [85,87]. In addition, platelets contribute to thrombosis
in human inflammatory diseases and in response to infection via their receptors
FcαRI (via IgA) and FcγRIIA (via IgG) [84,88].

239
The most studied platelet FcR is FcγRIIA. FcγRIIA is the sole IgG-specific FcR
expressed by platelets in humans and is thereby the most abundantly expressed
FcR in blood [89,90]. This receptor binds with low affinity to monomeric IgGs but
induces signaling when antibodies are aggregated or bound to an antigen. Immune
complexes are known to make a pathogenic contribution to autoimmune diseases
such as rheumatoid arthritis, SLE, antiphospholipid syndrome and heparin-induced
thrombocytopenia (HIT) [91].
FcγRIIA
The FcγRIIA receptor is a 40 kDa type I transmembrane protein with two extracellular
Ig-like domains: one transmembrane domain and a cytoplasmic tail [92]. Unlike the
majority of FcγRs, FcγRIIA does not require association with an FcRγ subunit for
activation. FcγRIIA possesses its own two immunoreceptor tyrosine-based
activation motif (ITAMs) domains on its cytoplasmic tail, similar to the FcRγ subunit.
Immunoreceptor tyrosine-based activation motifs are signaling motifs recognized by
their classical YxxL sequences, which possess two tyrosines that can be
phosphorylated by Src kinases after initiation of FcγRIIA activation. Phosphorylation
creates a docking site for tyrosine kinase Syk via its SH2 domains [93,94]. In
platelets, phosphatidylinositide3-kinases are phosphorylated by Syk and propagate
the intracellular signaling cascade through the phosphorylation of PLCγ2. PLCγ2
activation leads to the production of second messengers, an increase in intracellular
calcium and activation of protein kinase C [95]. As a result, platelet FcγRIIA
activation leads to granule content secretion, ROS generation, integrin activation,
upregulation of P-selectin, platelet aggregation and thrombus formation [96,101].
The P2X1 channel, which is the main calcium entry site in platelets, has been shown
to contribute to the increase in calcium after initiation of FcγRIIA activation and leads
to platelet aggregation [102].
FcγRIIA is encoded by the FCGR2A gene on chromosome 1q23.3 [103]. Two
different alleles of FcγRIIA are well studied and can be co-dominantly expressed.

240
The two alleles differ by the presence of an arginine (R) or a histidine (H) at position
131 in the amino acid chain [104]. The mutation lies in the second Ig-like domain,
which provides a different binding affinity to the allelic variants. A histidine at position
131 (FcγRIIA-H131) confers a higher affinity for IgG2 than the FcγRIIA-R131 variant
[104,106]. These FcγRIIA polymorphisms have been widely studied for their roles in
susceptibility to several diseases or the severity of these diseases. For example,
FcγRIIA polymorphisms are suggested to play a role in sepsis [107], SLE [108], HIT
[109,110], sickle cell disease [111], malaria, HIV [112], Epstein-Barr virus [113],
dengue virus [114,115], systemic meningococcal infections [116] and cardiac
diseases [117]. However, an actual contribution of the different FcγRIIA
polymorphisms remains controversial and further mechanistic insights are
necessary to fully assess their contribution to disease [118,123].
Platelets and Antibody Response
Immune Complexes
Immune complexes can mediate inflammation and have been intensively studied for
their role in pathogeneses such as SLE, rheumatoid arthritis and HIT. These
pathologies are all associated with thrombotic events and, consequently, possible
involvement of platelet activation mediated by IC. Activation of FcγRIIA is initiated
following an encounter with IC and has been shown to subsequently activate αIIbβ3,
thereby leading to full platelet activation and degranulation [124,125]. The integrin
αIIbβ3 changes to its activated conformation following signaling from activated
FcγRIIA leading to positive feedback (outside-in signaling), thus initiating α- and
dense granule secretion, which is important for thrombus formation [98,125,126].
Transgenic mice expressing FcγRIIA (FcγRIIATGN) have been generated given that
this receptor is absent in mice [127]. Immune complexes induce thrombosis in the
lungs and thrombocytopenia in FcγRIIATGN mice [124,128,131]. Many studies have
also shown that following activation of FcγRIIA, transgenic mice experienced
systemic shock [124,131,133]. Shock requires the release of serotonin from dense
granules, which relies on the activation of platelet αIIbβ3 and fibrinogen binding.

241
Moreover, serotonin was shown to be a mediator of IgG-dependent anaphylaxis and
to contribute to the severity of the reaction in a humanized mouse that expressed all
human FcγRs in place of all murine FcγRs [134]. However, although the systemic
shock induced by IC had a critical requirement for serotonin, the latter is dispensable
in thrombosis and thrombocytopenia observed in FcγRIIATGN mice, thus revealing
the heterogeneity of functions mediated by the activation of platelet FcγRIIA.
Heparin-induced thrombocytopenia, an immune reaction against heparin, is also
characterized by thrombocytopenia and implicates circulating IgG antibodies that
target PF4 and heparin complexes. PF4 changes its structural conformation after
binding to charged polyanions, such as those present in heparin, lipopolysaccharide
(LPS) or Gram-negative bacteria. PF4 exposes neoepitopes after conformational
change, which promotes the binding of anti-PF4/heparin antibodies (anti-PF4/P)
present in certain individuals [135]. As a result, anti-PF4/P and PF4 complexes
promote interaction with FcγRIIA. These IC in HIT stimulate platelet FcγRIIA, thereby
activating platelets to release procoagulant factors and EVs and promote platelet
clearance [136].
Bacterial Infection
PF4 binding to endogenous polyanions and the recognition of the complex by
antibodies can play a role in the protection of the host against pathogens [135].
Therefore, the clearance of platelets by anti-PF4/PF4 in HIT could be explained by
a disturbed host defense mechanism, where bacteria may be the target of anti-PF4
[137]. Antibody-opsonized bacteria also bind to FcγRIIA, possibly implicating
platelets in the combat of infections. Bacteria are widely studied for their interactions
with platelets and many differences have been observed between bacterial strains
with regard to the manner in which they bind and activate platelets. A wide range of
bacterial strains (Staphylococcus aureus, Streptococcus sanguinis, Streptococcus
gordonii, Streptococcus oralis, Streptococcus pneumoniae, Helicobacter pylori,
Escherichia coli, Streptococcus pyogenes) appears to share a common property:
they all induce platelet aggregation through an involvement with FcγRIIA
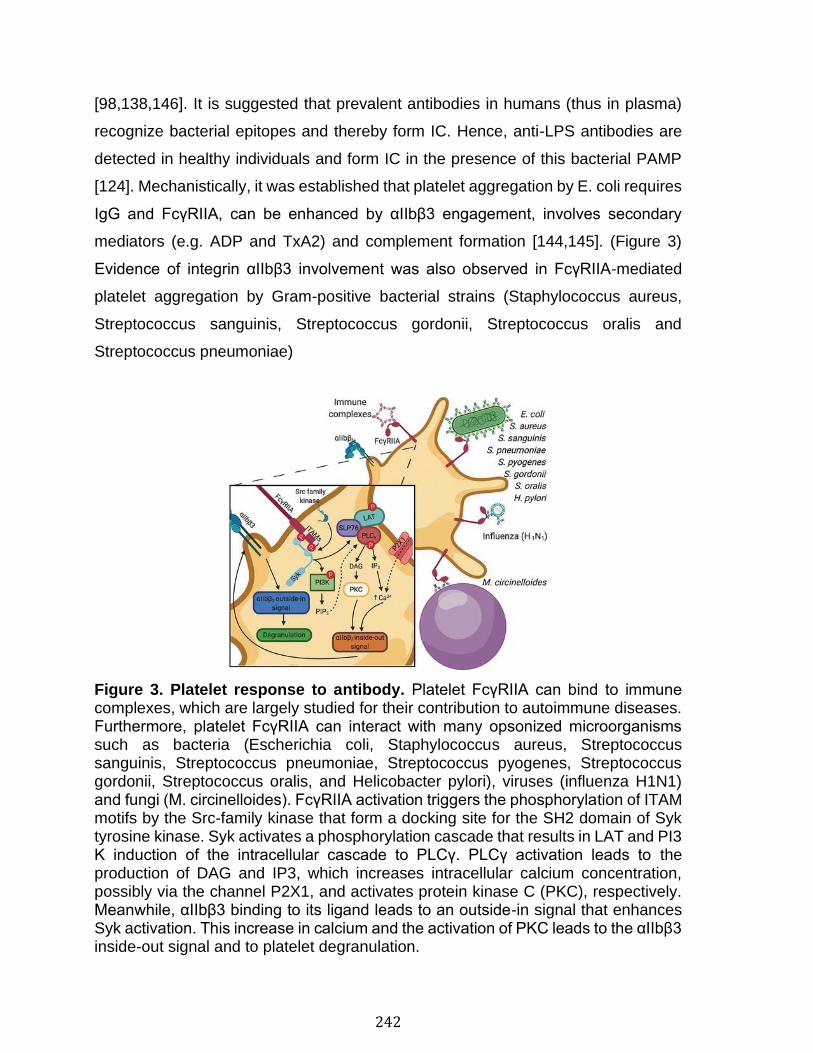
242
[98,138,146]. It is suggested that prevalent antibodies in humans (thus in plasma)
recognize bacterial epitopes and thereby form IC. Hence, anti-LPS antibodies are
detected in healthy individuals and form IC in the presence of this bacterial PAMP
[124]. Mechanistically, it was established that platelet aggregation by E. coli requires
IgG and FcγRIIA, can be enhanced by αIIbβ3 engagement, involves secondary
mediators (e.g. ADP and TxA2) and complement formation [144,145]. (Figure 3)
Evidence of integrin αIIbβ3 involvement was also observed in FcγRIIA-mediated
platelet aggregation by Gram-positive bacterial strains (Staphylococcus aureus,
Streptococcus sanguinis, Streptococcus gordonii, Streptococcus oralis and
Streptococcus pneumoniae)
Figure 3. Platelet response to antibody. Platelet FcγRIIA can bind to immune complexes, which are largely studied for their contribution to autoimmune diseases. Furthermore, platelet FcγRIIA can interact with many opsonized microorganisms such as bacteria (Escherichia coli, Staphylococcus aureus, Streptococcus sanguinis, Streptococcus pneumoniae, Streptococcus pyogenes, Streptococcus gordonii, Streptococcus oralis, and Helicobacter pylori), viruses (influenza H1N1) and fungi (M. circinelloides). FcγRIIA activation triggers the phosphorylation of ITAM motifs by the Src-family kinase that form a docking site for the SH2 domain of Syk tyrosine kinase. Syk activates a phosphorylation cascade that results in LAT and PI3 K induction of the intracellular cascade to PLCγ. PLCγ activation leads to the production of DAG and IP3, which increases intracellular calcium concentration, possibly via the channel P2X1, and activates protein kinase C (PKC), respectively. Meanwhile, αIIbβ3 binding to its ligand leads to an outside-in signal that enhances Syk activation. This increase in calcium and the activation of PKC leads to the αIIbβ3 inside-out signal and to platelet degranulation.

243
It was shown that bacteria-mediated FcγRIIA activation could have a possible
negative impact on host recovery, for example, by colonization of the heart valves
by platelets aggregated with Streptococcus oralis [147]. In addition, studies
confirmed that thrombocytopenia in sepsis is associated with increased risk of
mortality [148,151]. E. coli is involved in sepsis and in other pathologies where
thrombi are detrimental, such as in the development of hemolytic uremic syndrome
(HUS). On the other hand, platelet activation may play a role in the protection against
bacteria. Platelets possess the capacity to kill bacteria in an FcγRIIA-dependent
manner through the internalization of bacteria or by release of granules containing
antimicrobial proteins [152,154]. FcγRIIA-mediated PF4 release is also implicated in
the killing capacity of opsonized bacteria by platelets [98]. Moreover, aggregation
was suggested to restrict pathogens to a site with a high concentration of
antibacterial substances implying that platelet aggregation plays a possible
protective role against infection [152].
Viral, Fungal and Parasitic Infection
Although thrombocytopenia occurs in viral, fungal and parasitic infections, only a
limited number of studies have examined the role of platelet FcγRIIA in these
scenarios [69,155,160]. H1N1 promotes platelet activation through FcγRIIA.
Activation requires the presence of antibodies against influenza virus and leads to
release of granule components and EVs [68], which could contribute to
overwhelming inflammation [68]. Thrombocytopenia was induced by influenza virus
injection into FcγRIIATGN mice, only if the mice had developed antibodies to a
previous influenza virus infection. While this observation suggests a platelet
contribution to the defense of the host against infection, platelet activation may
contribute to an exacerbated inflammation, known as a cytokine storm, which can
be lethal in influenza infection [161]. In the case of Ebola virus, FcγRIIA has been
shown to enhance the infection of a cell line after its activation by virus–antibody
complexes, it is however unknown whether the mechanism takes place in platelets
[162].

244
Dengue virus has been shown to bind to FcγRIIA after the production of platelet
associated IgG, suggesting a possible role for FcγRIIA platelets in the clearance of
the viruses. Dengue virus is also able to replicate in platelets [67]. While interactions
between platelets and other viruses have been observed, the FcγRIIA contribution
was generally not verified [163].
Very little is known regarding platelet FcγRIIA in parasitic disease. It has been shown
that platelets can induce lysis of parasite digestive vacuole membrane via PF4,
which results in elimination of the parasite [164]. Thus, degranulation following
platelet activation is an important platelet function in parasitic infection, and it
remains to be determined whether this pathway is mediated via FcγRIIA.
In fungal infections, platelet FcγRIIA-mediated activation shares several similarities
with bacterial FcγRIIA activation. Mucormycete spores induce platelet aggregation
through FcγRIIA and αIIbβ3 [165]. The interaction with fungi is dependent on the
developmental stage of the fungus, and initiates the release of secondary mediators
by platelets, such as ADP and TxA2 [165]. As platelets are able to inhibit the
mucormycete germination process [166], this points to a potential role for FcγRIIA in
the protection against fungal infection.

245
Conclusion and Perspectives
Platelets and their mother cell, the megakaryocyte, can play roles in adaptive
immunity. In addition to their support of professional APC and in lymphocyte
functions, they can process and present antigens (Figures 1 and 2). Platelets bind
IC and microorganisms through FcγRIIA (Figure 3), most likely because of prevalent
antibodies against most common microorganisms even in healthy individuals.
Consistent with this, FcγRIIA is the dominant receptor activated by Zika virus if in the
presence of sera from convalescent patients infected by dengue or West Nile virus,
pointing to the formation of IC in blood of patients with preexisting antiflavivirus
immunity [167]. It is not completely understood how platelet FcγRIIA plays a role
against infections or in autoimmune diseases, but in response to antibodies, platelets
have the potential to control the proliferation of, or to kill, certain microorganisms
through the release of their vast arsenal of inflammatory molecules. It is important to
note that this activation pathway by antibodies is absent in laboratory mice given the
absence of FcγRIIA in mice and the fact that mice (1) are used at a relatively young
age, (2) are housed in overly clean environments, (3) never develop any infectious
diseases, (4) are never vaccinated, and (5) therefore may not have antibodies
against these microbes.
As a result of their abundance in blood and lungs, platelets and megakaryocytes are
ideally positioned to act as the primary responder and to convey antigens to
lymphocytes. This includes their potential response to airway-infecting viruses such
as the SARS-Cov2, which generates overwhelming inflammatory responses and
cardiovascular manifestations in certain individuals [168]. However, in contrast to
leukocytes, which can circulate through lymph to reach lymphoid organs, platelets
are not observed in lymph. They may directly interact with lymphocytes in the spleen
or secondary and tertiary lymphoid organs through high endothelial venules.
Activated platelets also release EVs. Platelet EVs are the most abundant EV in the
bloodstream, representing approximately 80% of circulating EVs [169]. Platelet EVs
are suggested to play numerous roles in regulating both physiological and

246
pathological functions [170]. Platelet EVs can interact with lymphocytes and regulate
regulatory T cell differentiation and activity [171,172]. Moreover, they can promote
the formation of germinal centers and the production of IgG by B-cells as a function
of their CD40L load [173,174]. As platelet EVs can circulate in lymph [175,176], they
may be able to transport adaptive immunity molecules to lymphoid organs through
this circulatory system. In autoimmune disease implicating IC, platelet EVs are
shown to harbor autoantigens such as non-histone nuclear protein high mobility
group box 1 (HMGB1) and citrullinated proteins (e.g. vimentin and fibrinogen) that
are the targets of prevalent antibodies in SLE and rheumatoid arthritis [177,178].
Whether FcγRIIA can remain exposed on certain platelet EVs to contribute to
dissemination of IC in these pathologies is not known. Future studies are necessary
to highlight whether platelet EVs play roles in adaptive immunity, similarly to platelets
and MK.

247
References
1. Minnicozzi M, Sawyer RT, Fenton MJ. Innate immunity in allergic disease. Immunol Rev 2011;242:106–127. 2. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol 2020;15:493–518. 3. Koonin EV, Makarova KS, Wolf YI. Evolutionary genomics of defense systems in archaea and bacteria. Annu Rev Microbiol 2017;71:233–261. 4. Sorek R, Lawrence CM, Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 2013;82:237–266. 5. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007;357:2482–2494. Epub 2007/ 12/14. 6. Semple JW, Italiano JE Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011;11:264–274. 7. Rick Kapur AZ, Boilard E, Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol 2015;194:12. 8. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood 2014;123:2759–2767. 9. Lewis JC, Maldonado JE, Mann KG. Phagocytosis in human platelets: localization of acid phosphatase-positive phagosomes following latex uptake. Blood 1976;47:833–840. 10. Male R, Vannier WE, Baldeschwieler JD. Phagocytosis of liposomes by human platelets. Proc Natl Acad Sci U S A 1992;89:9191–9195. 11. Nagasawa T, Nakayasu C, Rieger AM, Barreda DR, Somamoto T, Nakao M. Phagocytosis by thrombocytes is a conserved innate immune mechanism in lower vertebrates. Front Immunol 2014;5:445. 12. Karas SP, Rosse WF, Kurlander RJ. Characterization of the IgG-Fc receptor on human platelets. Blood 1982;60:1277–1282. Epub 1982/ 12/01. 13. Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of innate immune responses by platelets. Front Immunol 2019;10:1320. 14. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost 2020;4:23–35. 15. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol 2015;194:5579–5587. 16. Cunin P, Nigrovic PA. Megakaryocytes as immune cells. J Leukoc Biol 2019;105:1111–1121. 17. Silva-Cardoso SC, Affandi AJ, Spel L, Cossu M, JAG VR, Boes M, Radstake T. CXCL4 exposure potentiates TLR-driven polarization of human monocyte-derived dendritic cells and increases stimulation of T cells. J Immunol 2017;199:253–262. 18. Moore KL, Thompson LF. P-selectin (CD62) binds to subpopulations of human memory T lymphocytes and natural killer cells. Biochem Biophys Res Commun 1992;186:173–181. 19. Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology 2006;43:306–315. 20. Han P, Hanlon D, Arshad N, Lee JS, Tatsuno K, Robinson E, Filler R, Sobolev O, Cote C, Rivera-Molina F, et al. Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci Adv 2020;6:eaaz1580. 21. Laberge S, Cruikshank WW, Beer DJ, Center DM. Secretion of IL-16 (lymphocyte chemoattractant factor) from serotonin-stimulated CD8 + T cells in vitro. J Immunol 1996;156:310–315. 22. Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M, Cervantes-Barragan L, Ludewig B, Calzascia T, Bolinger B, et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med 2008;14:756–761. 23. Martinson J, Bae J, Klingemann HG, Tam Y. Activated platelets rapidly up-regulate CD40L expression and can effectively mature and activate autologous ex vivo differentiated DC. Cytotherapy 2004;6:487–497.

248
24. Kaneider NC, Kaser A, Tilg H, Ricevuti G, Wiedermann CJ. CD40 ligand-dependent maturation of human monocyte-derived dendritic cells by activated platelets. Int J Immunopathol Pharmacol 2003;16:225–231. 25. Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to antigen-presenting cells: a potential bridge between injury and immune activation. Exp Hematol 2004;32:135–139. 26. Constantinescu-Bercu A, Grassi L, Frontini M, Salles C II, Woollard K, Crawley JT. Activated alphaIIbbeta3 on platelets mediates flow-dependent NETosis via SLC44A2. Elife 2020;9:e53353. 27. Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, Thornton S, Degen JL. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest 2007;117:3224–3235. 28. Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, Lopez J, Plow E, Simon DI. Targeting platelet-leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med 2003;198:1077–1088. 29. Furie B, Furie BC. The molecular basis of platelet and endothelial cell interaction with neutrophils and monocytes: role of P-selectin and the P-selectin ligand, PSGL-1. Thromb Haemost 1995;74:224–227. 30. Katoh N, Soga F, Nara T, Tamagawa-Mineoka R, Nin M, Kotani H, Masuda K, Kishimoto S. Effect of serotonin on the differentiation of human monocytes into dendritic cells. Clin Exp Immunol 2006;146:354–361. 31. Xia CQ, Kao KJ. Effect of CXC chemokine platelet factor 4 on differentiation and function of monocyte-derived dendritic cells. Int Immunol 2003;15:1007–1015. 32. Duffau P, Seneschal J, Nicco C, Richez C, Lazaro E, Douchet I, Bordes C, Viallard JF, Goulvestre C, Pellegrin JL, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med 2010;2:47ra63. 33. Sowa JM, Crist SA, Ratliff TL, Elzey BD. Platelet influence on T- and B-cell responses. Arch Immunol Ther Exp (Warsz) 2009;57:235–241. 34. Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive immunity. Cell Immunol 2005;238:1–9. 35. Blumberg N, Spinelli SL, Francis CW, Taubman MB, Phipps RP. The platelet as an immune cell-CD40 ligand and transfusion immunomodulation. Immunol Res 2009;45:251–260. 36. Sprague DL, Sowa JM, Elzey BD, Ratliff TL. The role of platelet CD154 in the modulation in adaptive immunity. Immunol Res 2007;39:185–193. 37. Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, Stein CS, Nieswandt B, Wang Y, Davidson BL, et al. Platelet mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 2003;19:9–19. 38. Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med 2002;8:247–252. 39. Buchner K, Henn V, Grafe M, de Boer OJ, Becker AE, Kroczek RA. CD40 ligand is selectively expressed on CD4+ T cells and platelets: implications for CD40-CD40L signalling in atherosclerosis. J Pathol 2003;201:288–295. 40. Fleischer J, Grage-Griebenow E, Kasper B, Heine H, Ernst M, Brandt E, Flad HD, Petersen F. Platelet factor 4 inhibits proliferation and cytokine release of activated human T cells. J Immunol 2002;169:770–777. 41. Adnot S, Houssaini A, Abid S, Marcos E, Amsellem V. Serotonin transporter and serotonin receptors. Handb Exp Pharmacol 2013;218:365–380. 42. Mudd JC, Panigrahi S, Kyi B, Moon SH, Manion MM, Younes SA, Sieg SF, Funderburg NT, Zidar DA, Lederman MM, et al. Inflammatory function of CX3CR1+ CD8+ T cells in treated HIV infection is modulated by platelet interactions. J Infect Dis 2016;214:1808–1816. 43. Shi G, Field DJ, Ko KA, Ture S, Srivastava K, Levy S, Kowalska MA, Poncz M, Fowell DJ, Morrell CN. Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J Clin Invest 2014;124:543–552. 44. Yukawa M, Sakon M, Kambayashi J, Shiba E, Kawasaki T, Uemura Y, Murata K, Tanaka T, Nakayama T, Shibata H, et al. Purification and characterization of endogenous protein activator of human platelet proteasome. J Biochem 1993;114:317–323.

249
45. Yukawa M, Sakon M, Kambayashi J, Shiba E, Kawasaki T, Ariyoshi H, Mori T. Proteasome and its novel endogeneous activator in human platelets. Biochem Biophys Res Commun 1991;178:256–262. 46. Klockenbusch C, Walsh GM, Brown LM, Hoffman MD, Ignatchenko V, Kislinger T, Kast J. Global proteome analysis identifies active immunoproteasome subunits in human platelets. Mol Cell Proteomics 2014;13:3308–3319. 47. Shi DS, Smith MC, Campbell RA, Zimmerman PW, Franks ZB, Kraemer BF, Machlus KR, Ling J, Kamba P, Schwertz H, et al. Proteasome function is required for platelet production. J Clin Invest 2014;124:3757–3766. 48. Murai K, Kowata S, Shimoyama T, Yashima-Abo A, Fujishima Y, Ito S, Ishida Y. Bortezomib induces thrombocytopenia by the inhibition of proplatelet formation of megakaryocytes. Eur J Haematol 2014;93:290–296. 49. Nayak MK, Kulkarni PP, Dash D. Regulatory role of proteasome in determination of platelet life span. J Biol Chem 2013;288:6826–6834. 50. Grundler K, Rotter R, Tilley S, Pircher J, Czermak T, Yakac M, Gaitzsch E, Massberg S, Krotz F, Sohn HY, et al. The proteasome regulates collagen-induced platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb Res 2016;148:15–22. 51. Nayak MK, Kumar K, Dash D. Regulation of proteasome activity in activated human platelets. Cell Calcium 2011;49:226–232. 52. Koessler J, Schuepferling A, Klingler P, Koessler A, Weber K, Boeck M, Kobsar A. The role of proteasome activity for activating and inhibitory signalling in human platelets. Cell Signal 2019;62:109351. 53. Grundler Groterhorst K, Mannell H, Pircher J, Kraemer BF. Platelet proteasome activity and metabolism is upregulated during bacterial sepsis. Int J Mol Sci 2019;20(23). 54. Gupta N, Li W, Willard B, Silverstein RL, McIntyre TM. Proteasome proteolysis supports stimulated platelet function and thrombosis. Arterioscler Thromb Vasc Biol 2014;34:160–168. 55. Colberg L, Cammann C, Greinacher A, Seifert U. Structure and function of the ubiquitin-proteasome system in platelets. J Thromb Haemost 2020;18(4):771–780. 56. Boegel S, Lower M, Bukur T, Sorn P, Castle JC, Sahin U. HLA and proteasome expression body map. BMC Med Genomics 2018;11:36. 57. Semple JW, Speck ER, Milev YP, Blanchette V, Freedman J. Indirect allorecognition of platelets by T helper cells during platelet transfusions correlates with anti-major histocompatibility complex antibody and cytotoxic T lymphocyte formation. Blood 1995;86:805–812. 58. Kao KJ, Cook DJ, Scornik JC. Quantitative analysis of platelet surface HLA by W6/32 anti-HLA monoclonal antibody. Blood 1986;68:627–632. 59. Boshkov LK, Kelton JG, Halloran PF. HLA-DR expression by platelets in acute idiopathic thrombocytopenic purpura. Br J Haematol 1992;81:552–557. 60. Semple JW, Milev Y, Cosgrave D, Mody M, Hornstein A,Blanchette V, Freedman J. Differences in serum cytokine levels in acute and chronic autoimmune thrombocytopenic purpura: relationship to platelet phenotype and antiplatelet T-cell reactivity. Blood 1996;87:4245–4254. 61. Lazarus AH, Ellis J, Semple JW, Mody M, Crow AR, Freedman J. Comparison of platelet immunity in patients with SLE and with ITP. Transfus Sci 2000;22:19–27. 62. Placke T, Orgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, Salih HR. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res 2012;72:440–448. 63. Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J Proteomics 2014;101:130–140. 64. Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, Topham DJ, Baldwin WM 3rd, Morrell CN. Platelets present antigen in the context of MHC class I. J Immunol 2012;189:916–923. 65. Angenieux C, Dupuis A, Gachet C, de la Salle H, Maitre B. Cell surface expression of HLA I molecules as a marker of young platelets. J Thromb Haemost 2019;17:1511–1521. 66. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME, Corona-de la Pena NA, Salazar MI. Dengue virus induces the release of sCD40L and changes in levels of membranal CD42b and CD40L molecules in human platelets. Viruses 2018;10:357.

250
67. Simon AY, Sutherland MR, Pryzdial EL. Dengue virus binding and replication by platelets. Blood 2015;126:378–385. 68. Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, Borgeat P, Flamand L. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood 2014;123:2854–2863. 69. Terada H, Baldini M, Ebbe S, Madoff MA. Interaction of influenza virus with blood platelets. Blood 1966;28:213–228. 70. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun 2019;10:1780. 71. Carestia A, Mena HA, Olexen CM, Ortiz Wilczynski JM, Negrotto S, Errasti AE, Gomez RM, Jenne CN, Carrera Silva EA, Schattner M. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep 2019;28:896–908 e895. 72. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol 2019;20:303–320. 73. Heazlewood SY, Neaves RJ, Williams B, Haylock DN, Adams TE, Nilsson SK. Megakaryocytes co-localise with hemopoietic stem cells and release cytokines that up-regulate stem cell proliferation. Stem Cell Res 2013;11:782–792. 74. Cunin P, Bouslama R, Machlus KR, Martinez-Bonet M, Lee PY, Wactor A, Nelson-Maney N, Morris A, Guo L, Weyrich A, et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. Elife 2019;8:e44031. 75. Kang HK, Chiang MY, Ecklund D, Zhang L, Ramsey-Goldman R, Datta SK. Megakaryocyte progenitors are the main APCs inducing Th17 response to lupus autoantigens and foreign antigens. J Immunol 2012;188:5970–5980. 76. Finkielsztein A, Schlinker AC, Zhang L, Miller WM, Datta SK. Human megakaryocyte progenitors derived from hematopoietic stem cells of normal individuals are MHC class II-expressing professional APC that enhance Th17 and Th1/Th17 responses. Immunol Lett 2015;163:84–95. 77. Clark KB, Hsiao HM, Bassit L, Crowe JE Jr., Schinazi RF, Perng GC, Villinger F. Characterization of dengue virus 2 growth in megakaryocyte-erythrocyte progenitor cells. Virology 2016;493:162–172. 78. Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, Hunter-Mellado R, Tolley ND, Christensen M, Eustes AS, et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood 2019;133:2013–2026. 79. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017;544:105–109. 80. Zufferey A, Speck ER, Machlus KR, Aslam R, Guo L, McVey MJ, Kim M, Kapur R, Boilard E, Italiano JE Jr., et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv 2017;1:1773–1785. 81. Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol 2007;96:179–204. 82. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev 2015;268:25–51. 83. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity 2005;23:41–51. 84. Qian K, Xie F, Gibson AW, Edberg JC, Kimberly RP, Wu J. Functional expression of IgA receptor FcalphaRI on human platelets. J Leukoc Biol 2008;84:1492–1500. 85. Hasegawa S, Pawankar R, Suzuki K, Nakahata T, Furukawa S, Okumura K, Ra C. Functional expression of the high affinity receptor for IgE (FcepsilonRI) in human platelets and its’ intracellular expression in human megakaryocytes. Blood 1999;93:2543–2551. 86. Joseph M, Gounni AS, Kusnierz JP, Vorng H, Sarfati M, Kinet JP, Tonnel AB, Capron A, Capron M. Expression and functions of the high-affinity IgE receptor on human platelets and megakaryocyte precursors. Eur J Immunol 1997;27:2212–2218. 87. Capron M, Jouault T, Prin L, Joseph M, Ameisen JC, Butterworth AE, Papin JP, Kusnierz JP, Capron A. Functional study of a monoclonal antibody to IgE Fc receptor (Fc epsilon R2) of eosinophils, platelets, and macrophages. J Exp Med 1986;164:72–89. 88. King M, McDermott P, Schreiber AD. Characterization of the Fc gamma receptor on human platelets. Cell Immunol 1990;128:462–479.

251
89. Gardiner EE, Andrews RK. Structure and function of platelet receptors initiating blood clotting. Adv Exp Med Biol 2014;844:263–275. 90. Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, Fiebiger BM, Ravetch JV. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol 2014;15:707–716. 91. Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding Fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front Immunol 2019;10:811. 92. Hibbs ML, Bonadonna L, Scott BM, McKenzie IF, Hogarth PM. Molecular cloning of a human immunoglobulin G Fc receptor. Proc Natl Acad Sci U S A 1988;85:2240–2244. 93. Yanaga F, Poole A, Asselin J, Blake R, Schieven GL, Clark EA, Law CL, Watson SP. Syk interacts with tyrosine-phosphorylated proteins in human platelets activated by collagen and cross-linking of the Fc gamma-IIA receptor. Biochem J 1995;311 (Pt 2):471–478. 94. Rowley RB, Burkhardt AL, Chao HG, Matsueda GR, Bolen JB. Syk protein-tyrosine kinase is regulated by tyrosine-phosphorylated Ig alpha/Ig beta immunoreceptor tyrosine activation motif binding and autophosphorylation. J Biol Chem 1995;270:11590–11594. 95. Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiology (Bethesda) 2017;32:162–177. 96. Kerrigan SW, Clarke N, Loughman A, Meade G, Foster TJ, Cox D. Molecular basis for Staphylococcus aureus-mediated platelet aggregate formation under arterial shear in vitro. Arterioscler Thromb Vasc Biol 2008;28:335–340. 97. Mitrugno A, Williams D, Kerrigan SW, Moran N. A novel and essential role for FcgammaRIIa in cancer cell-induced platelet activation. Blood 2014;123:249–260. 98. Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, Kerrigan SW, Watson SP. Amplification of bacteria-induced platelet activation is triggered by FcgammaRIIA, integrin alphaIIbbeta3, and platelet factor 4. Blood 2014;123:3166–3174. 99. Zhi H, Rauova L, Hayes V, Gao C, Boylan B, Newman DK, McKenzie SE, Cooley BC, Poncz M, Newman PJ. Cooperative integrin/ITAM signaling in platelets enhances thrombus formation in vitro and in vivo. Blood 2013;121:1858–1867. 100. Boulaftali Y, Hess PR, Kahn ML, Bergmeier W. Platelet immunoreceptor tyrosine-based activation motif (ITAM) signaling and vascular integrity. Circ Res 2014;114:1174–1184. 101. Arthur JF, Qiao J, Shen Y, Davis AK, Dunne E, Berndt MC, Gardiner EE, Andrews RK. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J Thromb Haemost 2012;10:1133–1141. 102. Ilkan Z, Watson S, Watson SP, Mahaut-Smith MP. P2X1 receptors amplify FcgammaRIIa-induced Ca2+ increases and functional responses in human platelets. Thromb Haemost 2018;118:369–380. 103. Li X, Ptacek TS, Brown EE, Edberg JC. Fcgamma receptors: structure, function and role as genetic risk factors in SLE. Genes Immun 2009;10:380–389. 104. Qiao J, Al-Tamimi M, Baker RI, Andrews RK, Gardiner EE. The platelet Fc receptor, FcgammaRIIa. Immunol Rev 2015;268:241–252. 105. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009;113:3716–3725. 106. Warmerdam PA, van de Winkel JG, Vlug A, Westerdaal NA, Capel PJ. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol 1991;147:1338–1343. 107. Beppler J, Koehler-Santos P, Pasqualim G, Matte U, Alho CS, Dias FS, Kowalski TW, Velasco IT, Monteiro RC, Pinheiro da Silva F. Fc gamma receptor IIA (CD32A) R131 polymorphism as a marker of genetic susceptibility to sepsis. Inflammation 2016;39:518–525. 108. Vigato-Ferreira IC, Toller-Kawahisa JE, Pancoto JA, Mendes- Junior CT, Martinez EZ, Donadi EA, Louzada-Junior P, Del Lama JE, Marzocchi-Machado CM. FcgammaRIIa and FcgammaRIIIb polymorphisms and associations with clinical manifestations in systemic lupus erythematosus patients. Autoimmunity 2014;47:451–458. 109. Rollin J, Pouplard C, Sung HC, Leroux D, Saada A, Gouilleux- Gruart V, Thibault G, Gruel Y. Increased risk of thrombosis in FcgammaRIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood 2015;125:2397–2404.

252
110. Carlsson LE, Santoso S, Baurichter G, Kroll H, Papenberg S, Eichler P, Westerdaal NA, Kiefel V, van de Winkel JG, Greinacher A. Heparin-induced thrombocytopenia: new insights into the impact of the FcgammaRIIa-R-H131 polymorphism. Blood 1998;92:1526–1531. 111. Kangne HK, Jijina FF, Italia YM, Jain DL, Nadkarni AH, Gupta M, Pradhan V, Mukesh RD, Ghosh KK, Colah RB. The Fc receptor polymorphisms and expression of neutrophil activation markers in patients with sickle cell disease from Western India. Biomed Res Int 2013;2013:457656. 112. Forthal DN, Landucci G, Bream J, Jacobson LP, Phan TB, Montoya B. FcgammaRIIa genotype predicts progression of HIV infection. J Immunol 2007;179:7916–7923. 113. Diamantopoulos PT, Kalotychou V, Polonyfi K, Sofotasiou M, Anastasopoulou A, Galanopoulos A, Spanakis N, Vassilakopoulos T, Angelopoulou M, Siakantaris M, et al. Correlation of Fc-gamma RIIA polymorphisms with latent Epstein-Barr virus infection and latent membrane protein 1 expression in patients with low grade B-cell lymphomas. Leuk Lymphoma 2013;54:2030–2034. 114. Mohsin SN, Mahmood S, Amar A, Ghafoor F, Raza SM, Saleem M. Association of FcgammaRIIa polymorphism with clinical outcome of dengue infection: first insight from Pakistan. Am J Trop Med Hyg 2015;93:691–696. 115. Pereira A, de Siqueira TR, de Oliveira Prado AA, CAV DS, de Fatima Silva Moraes T, Aleixo AA, de Magalhaes JC, de Souza GAP, Drumond BP, Ferreira GP, et al. High prevalence of dengue antibodies and the arginine variant of the FcgammaRIIa polymorphism in asymptomatic individuals in a population of Minas Gerais State, Southeast Brazil. Immunogenetics 2018;70:355–362. 116. Bredius RG, Derkx BH, Fijen CA, de Wit TP, de Haas M, Weening RS, van de Winkel JG, Out TA. Fc gamma receptor IIa (CD32) polymorphism in fulminant meningococcal septic shock in children. J Infect Dis 1994;170:848–853. 117. Kroupis C, Theodorou M, Chaidaroglou A, Dalamaga M, Oliveira SC, Cokkinos DV, Degiannis D, Manginas A. The association between a common FCGR2A polymorphism and C-reactive protein and coronary artery disease revisited. Genet Test Mol Biomarkers 2010;14:839–846. 118. Connolly S, Wall KM, Tang J, Yu T, Kilembe W, Kijak G, Allen S, Hunter E. Fc-gamma receptor IIA and IIIA variants in two African cohorts: lack of consistent impact on heterosexual HIV acquisition, viral control, and disease progression. Virology 2018;525:132–142. 119. Arepally G, McKenzie SE, Jiang XM, Poncz M, Cines DB. Fc gamma RIIA H/R 131 polymorphism, subclass-specific IgG anti-heparin/platelet factor 4 antibodies and clinical course in patients with heparin-induced thrombocytopenia and thrombosis. Blood 1997;89:370–375. 120. Rhodes B, Vyse TJ. The genetics of SLE: an update in the light of genome-wide association studies. Rheumatology (Oxford) 2008;47:1603–1611. 121. Schallmoser K, Rosin C, Knittelfelder R, Sailer T, Ulrich S, Zoghlami C, Lehr S, Pabinger I, Panzer S. The Fc gammaRIIa polymorphism R/H131, autoantibodies against the platelet receptors GPIb alpha and Fc gammaRIIa and a risk for thromboembolism in lupus anticoagulant patients. Thromb Haemost 2005;93:544–548. 122. Botto M, Theodoridis E, Thompson EM, Beynon HL, Briggs D, Isenberg DA, Walport MJ, Davies KA. Fc gamma RIIa polymorphism in systemic lupus erythematosus (SLE): no association with disease. Clin Exp Immunol 1996;104:264–268. 123. Smyth LJ, Snowden N, Carthy D, Papasteriades C, Hajeer A, Ollier WE. Fc gamma RIIa polymorphism in systemic lupus erythematosus. Ann Rheum Dis 1997;56:744–746. 124. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, Levesque T, Becker Y, Tessandier N, Melki I, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S A 2018;115:E1550–E1559. 125. Zhi H, Dai J, Liu J, Zhu J, Newman DK, Gao C, Newman PJ. Platelet activation and thrombus formation over IgG immune complexes requires integrin alphaIIbbeta3 and lyn kinase. PLoS One 2015;10:e0135738. 126. Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood 2008;112:2780–2786. 127. McKenzie SE, Taylor SM, Malladi P, Yuhan H, Cassel DL, Chien P, Schwartz E, Schreiber AD, Surrey S, Reilly MP. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol 1999;162:4311–4318. 128. Arman M, Krauel K. Human platelet IgG Fc receptor FcgammaRIIA in immunity and thrombosis. J Thromb Haemost 2015;13:893–908.

253
129. Amirkhosravi A, Boulaftali Y, Robles-Carrillo L, Meyer T, McKenzie SE, Francis JL, Bergmeier W. CalDAG-GEFI deficiency protects mice from FcgammaRIIa-mediated thrombotic thrombocytopenia induced by CD40L and beta2GPI immune complexes. J Thromb Haemost 2014;12:2113–2119. 130. Stolla M, Stefanini L, Andre P, Ouellette TD, Reilly MP, McKenzie SE, Bergmeier W. CalDAG-GEFI deficiency protects mice in a novel model of Fcgamma RIIA-mediated thrombosis and thrombocytopenia. Blood 2011;118:1113–1120. 131. Reilly MP, Taylor SM, Hartman NK, Arepally GM, Sachais BS, Cines DB, Poncz M, McKenzie SE. Heparin-induced thrombocytopenia/ thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcgammaRIIA. Blood 2001;98:2442–2447. 132. Meyer T, Robles-Carrillo L, Davila M, Brodie M, Desai H, Rivera-Amaya M, Francis JL, Amirkhosravi A. CD32a antibodies induce thrombocytopenia and type II hypersensitivity reactions in FCGR2A mice. Blood 2015;126:2230–2238. 133. Jonsson F, Mancardi DA, Zhao W, Kita Y, Iannascoli B, Khun H, van Rooijen N, Shimizu T, Schwartz LB, Daeron M, et al. Human FcgammaRIIA induces anaphylactic and allergic reactions. Blood 2012;119:2533–2544. 134. Beutier H, Hechler B, Godon O, Wang Y, Gillis CM, de Chaisemartin L, Gouel-Cheron A, Magnenat S, Macdonald LE, Murphy AJ, et al. Platelets expressing IgG receptor FcgammaRIIA/CD32A determine the severity of experimental anaphylaxis. Sci Immunol 2018;3:22. 135. Palankar R, Kohler TP, Krauel K, Wesche J, Hammerschmidt S, Greinacher A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcgammaRIIA. J Thromb Haemost 2018;16:1187–1197. 136. Chong BH, Chong JJ. Heparin-induced thrombocytopenia. Expert Rev Cardiovasc Ther 2004;2:547–559. 137. Helin I, Axenram M, Grubb A. Serum cystatin C as a determinant of glomerular filtration rate in children. Clin Nephrol 1998;49:221–225. 138. Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, Cox DM. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003;124:1846–1854. 139. Ford I, Douglas CW, Cox D, Rees DG, Heath J, Preston FE. The role of immunoglobulin G and fibrinogen in platelet aggregation by Streptococcus sanguis. Br J Haematol 1997;97:737–746. 140. Kerrigan SW, Douglas I, Wray A, Heath J, Byrne MF, Fitzgerald D, Cox D. A role for glycoprotein Ib in Streptococcus sanguis-induced platelet aggregation. Blood 2002;100:509–516. 141. Miajlovic H, Loughman A, Brennan M, Cox D, Foster TJ. Both complement- and fibrinogen-dependent mechanisms contribute to platelet aggregation mediated by Staphylococcus aureus clumping factor B. Infect Immun 2007;75:3335–3343. 142. Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, Visai L, Speziale P, Cox D, Foster TJ. Fibronectinbinding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol Microbiol 2006;59:212–230. 143. Shannon O, Hertzen E, Norrby-Teglund A, Morgelin M, Sjobring U, Bjorck L. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol 2007;65:1147–1157. 144. Watson CN, Kerrigan SW, Cox D, Henderson IR, Watson SP, Arman M. Human platelet activation by Escherichia coli: roles for FcgammaRIIA and integrin alphaIIbbeta3. Platelets 2016;27:535–540. 145. Moriarty RD, Cox A, McCall M, Smith SG, Cox D. Escherichia coli induces platelet aggregation in an FcgammaRIIa-dependent manner. J Thromb Haemost 2016;14:797–806. 146. Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, Foster TJ. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 2005;57:804–818. 147. Tilley DO, Arman M, Smolenski A, Cox D, O’Donnell JS, Douglas CW, Watson SP, Kerrigan SW, Ibalpha G. FcgammaRIIa play key roles in platelet activation by the colonizing bacterium, Streptococcus oralis. J Thromb Haemost 2013;11:941–950. 148. Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, Bobbaers H. Thrombocytopenia and prognosis in intensive care. Crit Care Med 2000;28:1871–1876.

254
149. Claushuis TA, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PM, Hoogendijk AJ, Ong DS, Cremer OL, Horn J, Franitza M, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood 2016;127:3062–3072. 150. Strauss R, Wehler M, Mehler K, Kreutzer D, Koebnick C, Hahn EG. Thrombocytopenia in patients in the medical intensive care unit: bleeding prevalence, transfusion requirements, and outcome. Crit Care Med 2002;30:1765–1771. 151. Dewitte A, Lepreux S, Villeneuve J, Rigothier C, Combe C, Ouattara A, Ripoche J. Blood platelets and sepsis pathophysiology: A new therapeutic prospect in critically [corrected] ill patients? Ann Intensive Care 2017;7:115. 152. Krijgsveld J, Zaat SA, Meeldijk J, van Veelen PA, Fang G, Poolman B, Brandt E, Ehlert JE, Kuijpers AJ, Engbers GH, et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem 2000;275:20374–20381. 153. Yeaman MR. The role of platelets in antimicrobial host defense. Clin Infect Dis 1997;25:951–968; quiz 969–970. 154. Riaz AH, Tasma BE, Woodman ME, Wooten RM, Worth RG. Human platelets efficiently kill IgG-opsonized E. coli. FEMS Immunol Med Microbiol 2012;65:78–83. 155. Hudson JB, Weinstein L, Chang TW. Thrombocytopenic purpura in measles. J Pediatr 1956;48:48–56. 156. Hirst GK. The quantitative determination of influenza virus and antibodies by means of red cell agglutination. J Exp Med 1942;75:49–64. 157. Chomette G, Auriol M, Guilbert F. [Pleomorphic adenoma. Diagnostic difficulties. Value of ultrastructural and immunohistochemical studies. Apropos of 2 cases]. Rev Stomatol Chir Maxillofac 1987;88:356–358. 158. Ojha A, Nandi D, Batra H, Singhal R, Annarapu GK, Bhattacharyya S, Seth T, Dar L, Medigeshi GR, Vrati S, et al. Platelet activation determines the severity of thrombocytopenia in dengue infection. Sci Rep 2017;7:41697. 159. Kelton JG, Keystone J, Moore J, Denomme G, Tozman E, Glynn M, Neame PB, Gauldie J, Jensen J. Immune-mediated thrombocytopenia of malaria. J Clin Invest 1983;71:832–836. 160. Charkes ND. Purpuric chickenpox: report of a case, review of the literature, and classification by clinical features. Ann Intern Med 1961;54:745–759. 161. Hsieh YC, Wu TZ, Liu DP, Shao PL, Chang LY, Lu CY, Lee CY, Huang FY, Huang LM. Influenza pandemics: past, present and future. J Formos Med Assoc 2006;105:1–6. 162. Furuyama W, Marzi A, Carmody AB, Maruyama J, Kuroda M, Miyamoto H, Nanbo A, Manzoor R, Yoshida R, Igarashi M, et al. Fcgamma-receptor IIa-mediated Src signaling pathway is essential for the antibody-dependent enhancement of ebola virus infection. PLoS Pathog 2016;12:e1006139. 163. Eisinger F, Langer HF. The mutual relation of platelet activation and innate immunity. Hamostaseologie 2018;38:186–202. 164. Love MS, Millholland MG, Mishra S, Kulkarni S, Freeman KB, Pan W, Kavash RW, Costanzo MJ, Jo H, Daly TM, et al. Platelet factor 4 activity against P. falciparum and its translation to nonpeptidic mimics as antimalarials. Cell Host Microbe 2012;12:815–823. 165. Ghuman H, Shepherd-Roberts A, Watson S, Zuidscherwoude M, Watson SP, Voelz K. Mucor circinelloides induces platelet aggregation through integrin alphaIIbbeta3 and FcgammaRIIA. Platelets 2019;30:256–263. 166. Perkhofer S, Kainzner B, Kehrel BE, Dierich MP, Nussbaumer W, Lass-Florl C. Potential antifungal effects of human platelets against zygomycetes in vitro. J Infect Dis 2009;200:1176–1179. 167. Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, Nachbagauer R, Foster GA, Krysztof D, Tortorella D, et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 2017;356:175–180. 168. Akhmerov A, Marban E. COVID-19 and the heart. Circ Res 2020;126(10):1443–1455. 169. Benameur T, Osman A, Parray A, Ait Hssain A, Munusamy S, Agouni A. Molecular mechanisms underpinning microparticle-mediated cellular injury in cardiovascular complications associated with diabetes. Oxid Med Cell Longev 2019;2019:6475187. 170. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets 2017;28:214–221. 171. Sadallah S, Amicarella F, Eken C, Iezzi G, Schifferli JA. Ectosomes released by platelets induce differentiation of CD4 +T cells into T regulatory cells. Thromb Haemost 2014;112:1219–1229.

255
172. Dinkla S, van Cranenbroek B, van der Heijden WA, He X, Wallbrecher R, Dumitriu IE, van der Ven AJ, Bosman GJ, Koenen HJ, Joosten I. Platelet microparticles inhibit IL-17 production by regulatory T cells through P-selectin. Blood 2016;127:1976–1986. 173. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 2008;111:5028–5036. 174. Yari F, Motefaker M, Nikougoftar M, Khayati Z. Interaction of platelet-derived microparticles with a human B-lymphoblast cell line: a clue for the immunologic function of the microparticles. Transfus Med Hemother 2018;45:55–61. 175. Tessandier N, Melki I, Cloutier N, Allaeys I, Miszta A, Tan S, Milasan A, Michel S, Benmoussa A, Levesque T, et al. Platelets disseminate extracellular vesicles in lymph in rheumatoid arthritis. Arterioscler Thromb Vasc Biol 2020;40(4):929–942. 176. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell Vesicles 2016;5:31427. 177. Burbano C, Villar-Vesga J, Orejuela J, Munoz C, Vanegas A, Vasquez G, Rojas M, Castano D. Potential involvement of platelet-derived microparticles and microparticles forming immune complexes during monocyte activation in patients with systemic lupus erythematosus. Front Immunol 2018;9:322. 178. Cloutier N, Tan S, Boudreau LH, Cramb C, Subbaiah R, Lahey L, Albert A, Shnayder R, Gobezie R, Nigrovic PA, et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes. EMBO Mol Med 2013;5:235–249.

256
Annexe III: Platelets release pathogenic serotonin
and return to circulation after immune complex-
mediated sequestration
Nathalie Cloutier 1 , Isabelle Allaeys 1 , Genevieve Marcoux 1, Kellie R Machlus 2
, Benoit Mailhot 3 , Anne Zufferey 1 , Tania Levesque 1 , Yann Becker 1 , Nicolas Tessandier 1 , Imene Melki 1 , Huiying Zhi 4 , Guy Poirier 5 , Matthew T Rondina 6
, Joseph E Italiano 2 , Louis Flamand 1 , Steven E McKenzie 7 , Francine Cote 8
, Bernhard Nieswandt 9 , Waliul I Khan 10 ,11 , Matthew J Flick12 , Peter J Newman4
, Steve Lacroix 3 , Paul R Fortin 13 , Eric Boilard14 ,15
Affiliations
1 Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval, Département de microbiologie et immunologie, Faculté de Médecine, Université Laval, QC, Canada G1V 4G2.
2 Division of Hematology, Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA 02115.
3 Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval, Département de Médecine Moléculaire, Faculté de Médecine, Université Laval, QC, Canada G1V 4G2.
4 Blood Research Institute, Milwaukee, WI 53213.
5 Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval, Département d'Oncologie, Faculté de Médecine, Université Laval, QC, Canada G1V 4G2.
6 Department of Internal Medicine, University of Utah, Salt Lake City, UT 84112.
7 Cardeza Foundation for Hematological Research, Thomas Jefferson University, Philadelphia, PA 19107.
8 Institut Imagine, INSERM U1163, CNRS ERL8254, Université Paris Descartes, Hôpital Necker, 75006 Paris, France.
9 Department of Experimental Biomedicine, University Hospital and Rudolf Virchow Center, 97080 Wuerzburg, Germany.

257
10 Farncombe Family Digestive Health Research Institute, McMaster University, Hamilton, ON, Canada L8S 4L8.
11 Department of Pathology & Molecular Medicine, McMaster University, Hamilton, ON, Canada L8S 4L8.
12 Cancer and Blood Diseases Institute, Division of Experimental Hematology and Cancer Biology, Cincinnati Children's Hospital, Cincinnati, OH 45229.
13 Centre de recherche du Centre Hospitalier Universitaire de Québec-Université Laval, Département de médecine, Faculté de médecine, Université Laval, QC, Canada G1V 4G2.
14 Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval, Département de microbiologie et immunologie, Faculté de Médecine, Université Laval, QC, Canada G1V 4G2; [email protected].
15 Canadian National Transplantation Research Program, Edmonton, AB, Canada T6G 2E1.

258
Abstract
There is a growing appreciation for the contribution of platelets to immunity; however,
our knowledge mostly relies on platelet functions associated with vascular injury and
the prevention of bleeding. Circulating immune complexes (ICs) contribute to both
chronic and acute inflammation in a multitude of clinical conditions. Herein, we
scrutinized platelet responses to systemic ICs in the absence of tissue and
endothelial wall injury. Platelet activation by circulating ICs through a mechanism
requiring expression of platelet Fcγ receptor IIA resulted in the induction of systemic
shock. IC-driven shock was dependent on release of serotonin from platelet-dense
granules secondary to platelet outside-in signaling by αIIbβ3 and its ligand
fibrinogen. While activated platelets sequestered in the lungs and leaky vasculature
of the blood-brain barrier, platelets also sequestered in the absence of shock in mice
lacking peripheral serotonin. Unexpectedly, platelets returned to the blood circulation
with emptied granules and were thereby ineffective at promoting subsequent
systemic shock, although they still underwent sequestration. We propose that in
response to circulating ICs, platelets are a crucial mediator of the inflammatory
response highly relevant to sepsis, viremia, and anaphylaxis. In addition, platelets
recirculate after degranulation and sequestration, demonstrating that in adaptive
immunity implicating antibody responses, activated platelets are longer lived than
anticipated and may explain platelet count fluctuations in IC-driven diseases.

259
Introduction
Platelets are best known for their involvement in hemostasis. Their abundance in
blood underlies their perfect positioning for constant surveillance of the endothelium
to ensure vascular integrity. The role of platelets, however, is not restricted to the
hemostatic response (1). Not only do platelets express a vast array of mediators
serving wound repair but they also possess immune receptors and inflammatory
molecules (1–3). Moreover, following tissue injury or infection, platelets are a central
effector in driving an inflammatory process for recruiting leukocytes, principally
neutrophils, to the affected site (4–6). The platelet contribution at an inflamed site
also includes the prevention of bleeding (7, 8), as platelets seal the breaches formed
during the extravasation of neutrophils (9).
Platelet activation occurs at the interface with the vasculature, is elicited by injury or
inflammation, and is restricted to the endothelium or tissue. However, given the great
number of platelets in blood and their extensive set of immune receptors, systemic
immune triggers can also activate platelets while in circulation (i.e., in the absence
of endothelial or tissue injury). In rheumatic diseases, such as rheumatoid arthritis
(RA), systemic lupus erythematosus (SLE), and antiphospholipid syndrome, there is
a prevalence of pathogenic antibody–antigen scaffolds, called immune complexes
(ICs), in circulation in patients (10). Anaphylactic reactions due to allergens (11, 12)
and i.v. administration of drugs, such as in heparin-induced thrombocytopenia (HIT)
(13), also implicate ICs, thereby provoking acute inflammatory responses and a rapid
drop in platelet count (thrombocytopenia). ICs can also form in blood during sepsis
and viremia, when antibodies in the immune host recognize microbial antigens or
their toxins.
The pathological link between ICs and cellular responses is mediated by members
of the Fcγ receptor (FcγR) family. The low-affinity receptor FcγRIIA is the sole FcγR
expressed by platelets in humans, and is thereby the most abundantly expressed
receptor in blood (12, 14, 15). Of note, it is the dominant receptor activated by Zika
virus if in the presence of sera from convalescent patients infected by dengue or

260
West Nile virus, pointing to the formation of ICs in blood in patients with preexisting
antiflavivirus immunity (16). Moreover, diverse strains of bacteria and the influenza
virus H1N1 trigger platelet FcγRIIA activation after forming ICs in an immune host
(17, 18). In addition, studies with human platelets show that ICs from patients with
SLE also activate platelets through FcγRIIA (19). However, mice do not express
FcγRIIA, and murine platelets are completely devoid of any FcγRs (12). Thus, IC-
mediated responses in mice strongly favor leukocytes, which express other FcγRs,
while platelets play small or accessory roles in mouse models implicating ICs.
To more accurately model FcγRIIA reactions in mice, transgenic FcγRIIA
(FcγRIIATGN) mice that display FcγRIIA expression on platelets and certain
leukocytes, including monocytes and neutrophils as in humans, were developed (12,
20). Notably, stimulation of FcγRIIA in transgenic mice induces a shock response
that is reminiscent of anaphylaxis in humans, with a strong reliance on neutrophil
activity and implicating release of platelet-activating factor (PAF) (14, 21–24).
How ICs trigger systemic responses is not completely understood, as no study has
definitively determined the contribution of platelets to this response in vivo. Here, we
employed FcγRIIATGN mice to elucidate the precise mechanisms by which platelets
contribute to IC-mediated shock.

261
Results
Platelets Are Critical During the Systemic Response.
ICs injected into FcγRIIATGN mice trigger systemic shock (22–24). Although
thrombosis in the lungs and thrombocytopenia accompany systemic shock in these
mice (22, 25–27), the platelet contribution to IC-induced shock has never been
formally assessed. We used heat-aggregated (HA) IgG as an IC surrogate (28, 29),
as it permits precise investigation of the role of FcγR activation with no bias owing
to the nature of antigen specificities. HA-IgG (160 ± 7 nm in diameter) was injected
i.v. into WT mice lacking FcγRIIA (FcγRIIAnull) and FcγRIIATGN mice. Significant
temperature loss (15 min) preceded by rapid significant systemic shock (3 min) in all
of the FcγRIIATGN mice was confirmed, but it was never observed in the FcγRIIAnull
mice (Fig. 1A and Movie S1). All of the FcγRIIATGN mice collapsed, underwent
profound immobility, and lost consciousness (Fig. S1A). Mice recovered from the
shock and hypothermia within 180 min, after which no residual signs were apparent
(Fig. S1B). The occurrence of shock was dependent on the concentration of ICs
present in blood, not induced by monomeric IgG, similarly induced when murine ICs
were used, and more profound in male FcγRIIATGN mice compared with females (Fig.
S1 C–E).
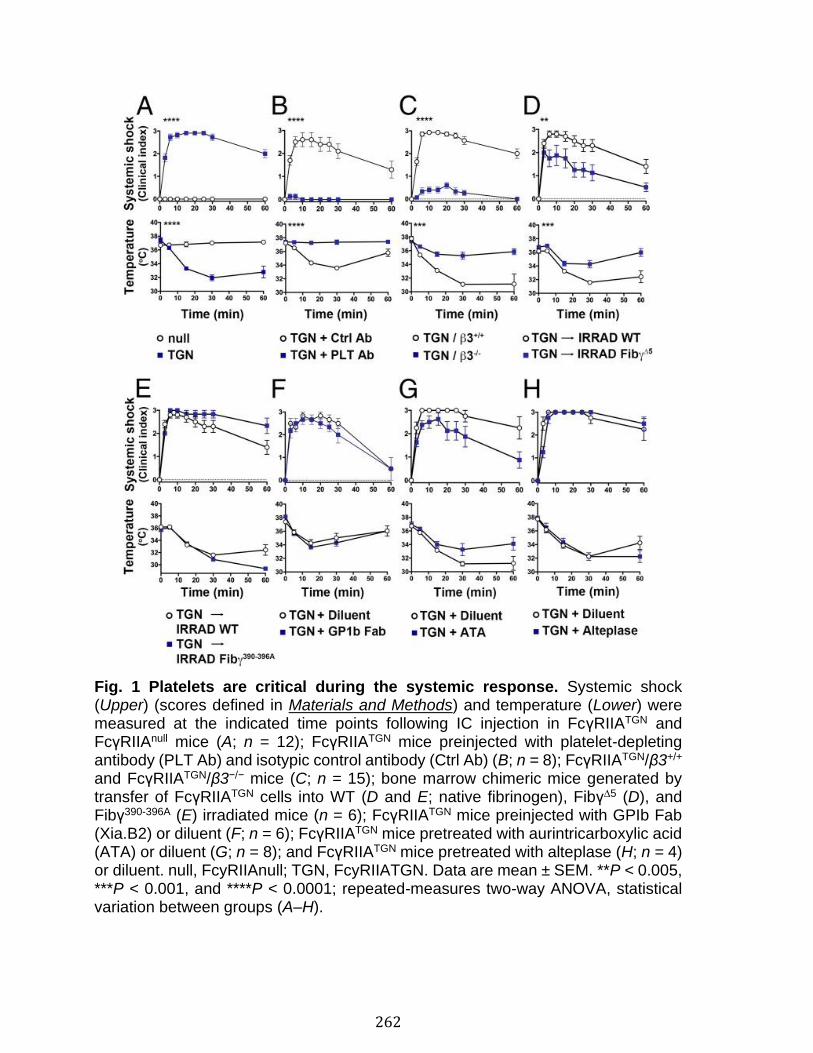
262
Fig. 1 Platelets are critical during the systemic response. Systemic shock (Upper) (scores defined in Materials and Methods) and temperature (Lower) were measured at the indicated time points following IC injection in FcγRIIATGN and FcγRIIAnull mice (A; n = 12); FcγRIIATGN mice preinjected with platelet-depleting antibody (PLT Ab) and isotypic control antibody (Ctrl Ab) (B; n = 8); FcγRIIATGN/β3+/+ and FcγRIIATGN/β3−/− mice (C; n = 15); bone marrow chimeric mice generated by transfer of FcγRIIATGN cells into WT (D and E; native fibrinogen), Fibγ∆5 (D), and Fibγ390-396A (E) irradiated mice (n = 6); FcγRIIATGN mice preinjected with GPIb Fab (Xia.B2) or diluent (F; n = 6); FcγRIIATGN mice pretreated with aurintricarboxylic acid (ATA) or diluent (G; n = 8); and FcγRIIATGN mice pretreated with alteplase (H; n = 4) or diluent. null, FcyRIIAnull; TGN, FcyRIIATGN. Data are mean ± SEM. **P < 0.005, ***P < 0.001, and ****P < 0.0001; repeated-measures two-way ANOVA, statistical variation between groups (A–H).

263
To formally assess the contribution of platelets to the systemic shock, we depleted
platelets (>98%) before injection of ICs. Thrombocytopenic mice were completely
protected from IC-mediated systemic responses (Fig. 1B and Fig. S1F), identifying
a critical role for platelets in shock induced by circulating ICs. In platelets, efficient
αIIbβ3 signaling relies on the FcγRIIA immunoreceptor tyrosine-based activation
motif, and, accordingly, the expression of FcγRIIA by platelets enhances fibrinogen-
mediated platelet activation (30). Thus, we verified the functional association of
FcγRIIA and the αIIbβ3 receptor in vivo by comparing the response in
FcγRIIATGN/β3−/− and FcγRIIATGN/β3+/+ mice. The FcγRIIATGN/β3−/− mice were
completely resistant to IC challenge (Fig. 1C), further emphasizing the role of
platelets and pointing to the requirement of αIIbβ3 in the mechanism of IC-mediated
shock.
While αIIbβ3 may amplify FcγRIIA signaling through common kinases located in the
platelet cytoplasm, it is predominantly known as the fibrinogen receptor, which may
contribute upon binding to platelet activation (31–33). Fibrinogen also mediates
platelet–neutrophil interactions by bridging αIIbβ3 and macrophage-1 antigen (Mac-
1) (34). Hence, platelet–neutrophil aggregates formed in blood in the presence of
ICs (Fig. S2A), as well as neutrophils, were also necessary in the systemic
responses (Fig. S2B), in agreement with prior studies (23, 28). To verify the specific
role of fibrinogen, we used mutant fibrinogen chimera mice. We lethally irradiated
fibrinogenγΔ5 (FibγΔ5) and fibrinogenγ390-396A (Fibγ390-396A) mutant mice, which
express mutant fibrinogen unable to bind αIIbβ3 or Mac-1 (34, 35), respectively, and
engrafted the mice with bone marrow from FcγRIIATGN mice. After injection with ICs,
we found that fibrinogen binding to αIIbβ3 was necessary for the systemic response,
whereas bridging platelets with leukocytes through fibrinogen binding to Mac-1 was
dispensable (Fig. 1 D and E). Consistent with these data, the blockade of Mac-1 as
well as PSGL-1, the counterreceptor of platelet P-selectin on leukocytes (36), had
no effect on shock (Fig. S2C).
Like αIIbβ3, GPIb is critical in the prevention of bleeding and thrombosis, and the
ablation of the gene coding for GPIb in mice leads to severe bleeding defects (37).

264
As GPIb is localized within lipid raft membrane microdomains in physical proximity
with FcγRIIA (38), we used a GPIb-blocking Fab to probe the contribution of GPIb,
and found that it is dispensable in shock mediated by ICs (Fig. 1F). Furthermore, the
pharmacological blockade of von Willebrand factor (vWF) binding to GPIb (ATA;
aurintricarboxylic acid) or administration of recombinant tissue plasminogen
activator (tPA; alteplase) to promote the lysis of any existing thrombi also had no
effect on shock (Fig. 1 G and H). These data further demonstrate that platelet
activation by circulatory ICs implicates pathways distinct from those promoting vaso-
occlusion and vascular injury.
Identification of the Shock Mediator.
Consistent with the rapid response, an increase in platelet factor 4 (PF4) and
serotonin, components stored in α- and δ-granules, respectively, was detected in
blood within 10 min (at the time of mouse collapse and before hypothermia) of the
IC trigger in FcγRIIATGN mice (Fig. 2A). We therefore hypothesized that a component
released from platelet granules may be responsible for induction of systemic shock.
Intravital microscopy of the microvasculature using two-photon microscopy revealed
significant vessel leakage and vasodilatation in response to IC injection in
FcγRIIATGN mice (Fig. 2 B and C). Vessel leakage was systematically observed in
response to IC injection in both FcγRIIATGN mice and FcγRIIAnull mice, but was
absent in FcRγ−/− mice, pointing to the role of other receptor(s) for ICs (Fig. S3A). It
was also maintained in the absence of neutrophils, further suggesting that vascular
leakage is not the key pathological feature downstream of FcγRIIA driving
IC/platelet-mediated shock (Fig. S3A).
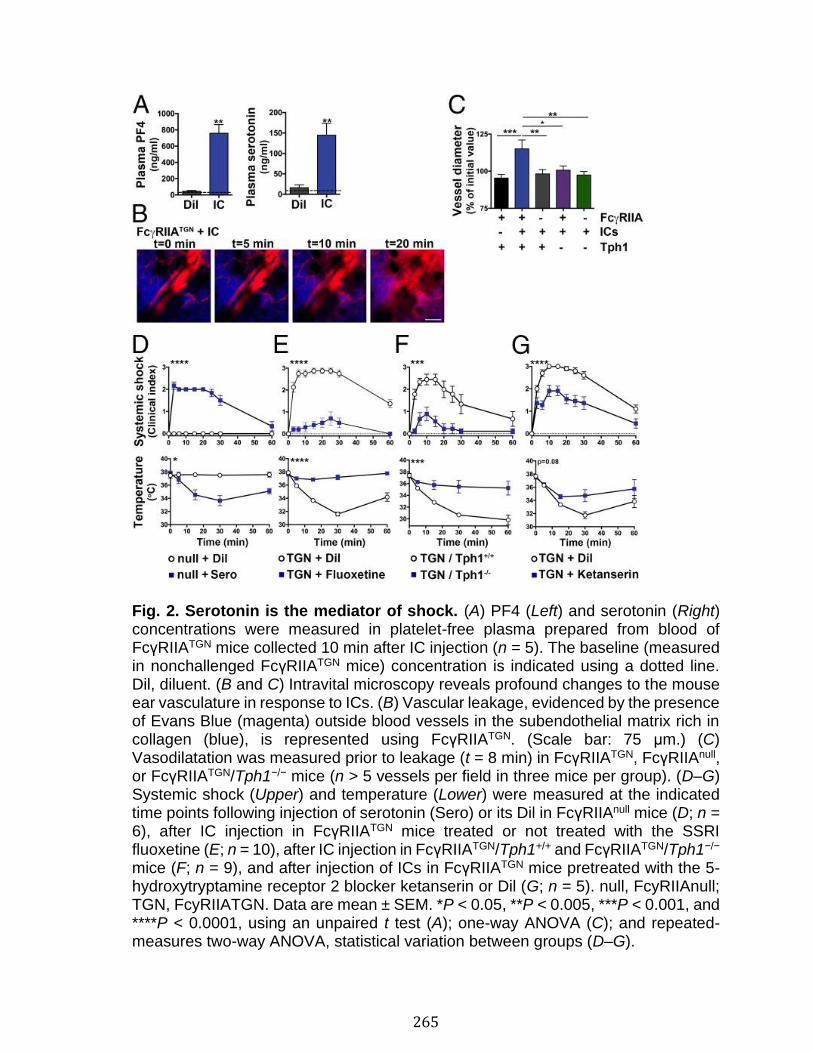
265
Fig. 2. Serotonin is the mediator of shock. (A) PF4 (Left) and serotonin (Right) concentrations were measured in platelet-free plasma prepared from blood of FcγRIIATGN mice collected 10 min after IC injection (n = 5). The baseline (measured in nonchallenged FcγRIIATGN mice) concentration is indicated using a dotted line. Dil, diluent. (B and C) Intravital microscopy reveals profound changes to the mouse ear vasculature in response to ICs. (B) Vascular leakage, evidenced by the presence of Evans Blue (magenta) outside blood vessels in the subendothelial matrix rich in collagen (blue), is represented using FcγRIIATGN. (Scale bar: 75 μm.) (C) Vasodilatation was measured prior to leakage (t = 8 min) in FcγRIIATGN, FcγRIIAnull, or FcγRIIATGN/Tph1−/− mice (n > 5 vessels per field in three mice per group). (D–G) Systemic shock (Upper) and temperature (Lower) were measured at the indicated time points following injection of serotonin (Sero) or its Dil in FcγRIIAnull mice (D; n = 6), after IC injection in FcγRIIATGN mice treated or not treated with the SSRI fluoxetine (E; n = 10), after IC injection in FcγRIIATGN/Tph1+/+ and FcγRIIATGN/Tph1−/− mice (F; n = 9), and after injection of ICs in FcγRIIATGN mice pretreated with the 5-hydroxytryptamine receptor 2 blocker ketanserin or Dil (G; n = 5). null, FcyRIIAnull; TGN, FcyRIIATGN. Data are mean ± SEM. *P < 0.05, **P < 0.005, ***P < 0.001, and ****P < 0.0001, using an unpaired t test (A); one-way ANOVA (C); and repeated-measures two-way ANOVA, statistical variation between groups (D–G).

266
In stark contrast, vasodilatation was a response observed exclusively in FcγRIIATGN
mice (Fig. 2C). Identified first as a serum agent mediating vascular tone, serotonin
is a powerful vasoconstrictor when added to smooth muscle cells. However, it is a
potent vasodilator on endothelial cells (39). In the case of systemic activation in the
absence of vascular injury, and thus an intact endothelium, we hypothesized that
serotonin might be the key platelet component driving vasodilatation. This is
supported by the fact that the majority of peripheral serotonin, which represents 95%
of the total body serotonin pool, is stored in platelet δ-granules (40, 41). Platelets do
not synthesize serotonin; they utilize their serotonin transporter (SERT) to capture
circulatory serotonin, with the latter being generated by enterochromaffin cells from
the digestive tract by the enzyme tryptophan hydroxylase 1 (Tph1) (42, 43).
Consistent with our hypothesis, vasodilatation was abrogated in FcγRIIATGN/Tph1−/−
mice in response to ICs (Fig. 2C). It was also maintained in absence of neutrophils,
further confirming the direct role of peripheral serotonin in IC-induced vasodilatation
of microvessels (Fig. S3B).
Serotonin levels in plasma were back to normal 1 h postshock (Fig. S3C). To directly
verify its role in systemic shock, serotonin was injected into FcγRIIAnull mice, which
resulted in a shock response reminiscent of the responses observed in IC-injected
mice (Fig. 2D). We then used a selective serotonin reuptake inhibitor (SSRI), a SERT
blocker used as an antidepressant, to inhibit serotonin storage by platelets.
Administration of the SSRI for 3 wk to deplete the serotonin content of δ-granules
(41) revealed the important contribution of serotonin uptake in IC-mediated
inflammation: SSRI treatment nearly abolished the systemic shock response (Fig.
2E). Furthermore, FcγRIIATGN/Tph1−/− mice lacking platelet serotonin (43) showed
almost complete resistance to shock (Fig. 2F), confirming the critical role of
peripheral serotonin in IC-mediated shock. In addition, blockade of the 5-
hydroxytryptamine 2 receptor family, which is expressed in the periphery by
platelets, but also by monocytes and macrophages, dendritic cells, eosinophils, B
and T lymphocytes, endothelial cells, fibroblasts, cells from the cardiovascular
system, and neurons in the peripheral nervous system (40), was effective at reducing
shock (Fig. 2G). In support of the potential role of αIIbβ3 in degranulation, serotonin

267
was not released in FcγRIIATGN/β3−/− mice after IC injection, in agreement with the
absence of shock in those mice (Fig. S3D). Key actors in the prevention of bleeding
and thrombosis, the platelet-derived mediators ADP and thromboxane A2, were
dispensable for shock (Fig. S3 E and F). In addition, neutrophil extracellular traps,
which can be present in thrombosis (44), were detected during shock according to
quantifications of circulating nucleosomes (Fig. S3G); however, the injection of
DNase did not protect mice from shock (Fig. S3H). Together, these observations
highlight the importance of platelet-derived serotonin in the systemic response to ICs
in vivo, a platelet response that is distinct from that traditionally observed in
hemostasis and thrombosis.
And the Platelet Count?
Circulating platelets were also monitored throughout the systemic response and
beyond. We observed that FcγRIIATGN mice rapidly underwent profound
thrombocytopenia (∼10% of total normal platelet count) in response to ICs, whereas
FcγRIIAnull mice presented with only very minimal or no changes in platelet counts
(Fig. 3A). In fact, thrombocytopenia occurred rapidly (<10 min), and the platelet
count gradually increased, reaching ∼20% by 60 min, and it was 50% resolved within
24 h (Fig. 3A). The occurrence of thrombocytopenia was critically dependent on the
expression of FcγRIIA and was similarly induced in males and females (Fig. S4A).
Only platelets were affected, as the levels of RBCs, monocytes, and neutrophils
were unchanged in response to ICs (Fig. S4 B–D). Thrombocytopenia was as
profound in native fibrinogen mice as in FibγΔ5 and Fibγ390-396A mice (Fig. 3B),
suggesting that fibrinogen binding was dispensable, and that thrombocytopenia only
modestly involved β3, potentially through binding to its other ligands. Conversely,
thrombocytopenia was unaltered by the inhibition of GPIb and vWF interactions, or
the blockade of thromboxane A2 synthesis and ADP (Fig. 3B). Thrombin–
antithrombin complexes and D-dimers, which are evidence of coagulation activation
and thrombus degradation, respectively, were not significantly elevated 24 h after
shock (Fig. S4 E and F), and consistent with this, the destruction of potential thrombi
by the injection of alteplase did not impact platelet count (Fig. 3B). These data further
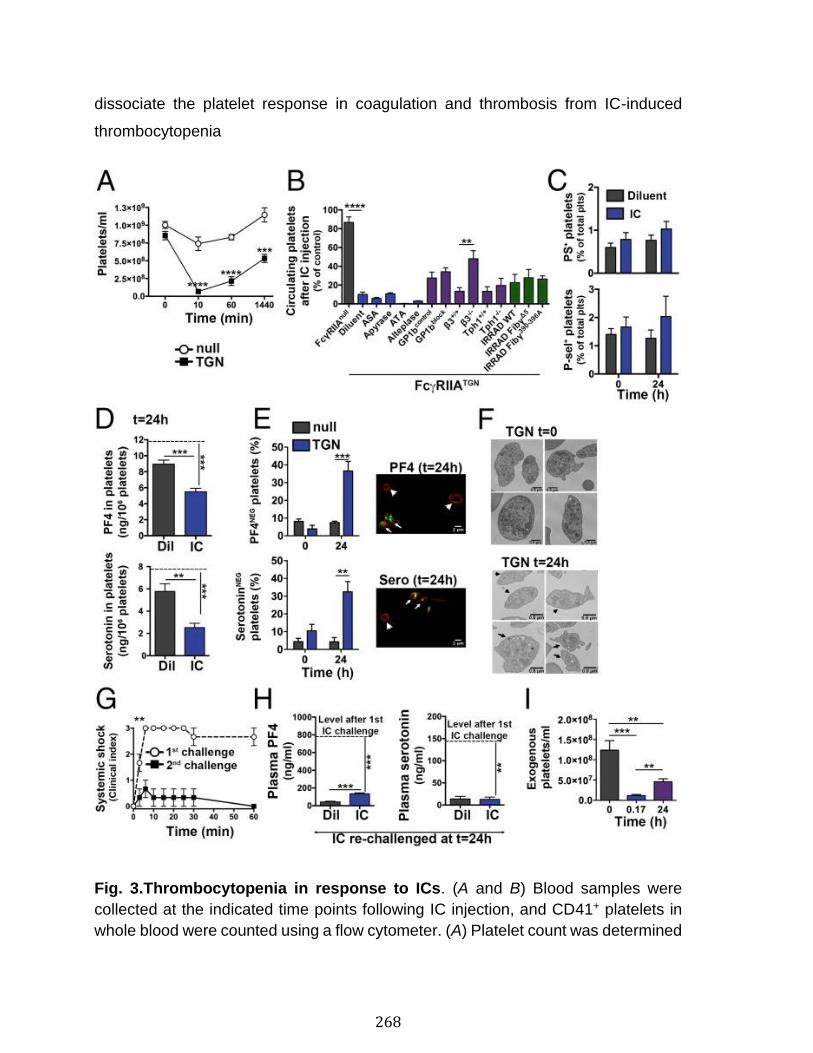
268
dissociate the platelet response in coagulation and thrombosis from IC-induced
thrombocytopenia
Fig. 3.Thrombocytopenia in response to ICs. (A and B) Blood samples were
collected at the indicated time points following IC injection, and CD41+ platelets in
whole blood were counted using a flow cytometer. (A) Platelet count was determined

269
in FcγRIIATGN and FcγRIIAnull mice (n = 11). (B) Percentages of circulating platelets
10 min after IC injection compared with initial platelet count were calculated in
FcγRIIAnull mice (black) and the indicated three different groups of FcγRIIATGN mice.
Mice were pretreated with acetylsalicylic acid (ASA) (n = 4), apyrase (n = 6),
aurintricarboxylic acid (ATA) (n = 3), alteplase (n = 5), or diluent (n = 14) (all shown
in blue). FcγRIIATGN mice pretreated with GPIb Fab or control (n = 4),
FcγRIIATGN/β3+/+ or FcγRIIATGN/β3−/− mice (n = 10); FcγRIIATGN/Tph1+/+ mice; or
FcγRIIATGN/Tph1−/− mice (n = 4) are represented (all shown in purple). Bone marrow
chimeric mice generated by transfer of FcγRIIATGN cells into WT (native fibrinogen),
Fibγ∆5, and Fibγ390-396A irradiated (IRRAD) mice are represented (n = 3 per group)
(shown in green). (C and D) Mice were injected with diluent or IC, and blood was
collected 24 h later. (C) Exposition of PS and P-selectin (P-sel) was assessed on
CD41+ platelets using flow cytometry in FcγRIIATGN mice (n = 7). (D) Platelet content
of PF4 (n = 6) and serotonin (n = 5) was measured by ELISA in 106 platelets retrieved
24 h after shock. Baseline (measured in nonchallenged FcγRIIATGN mice) contents
are indicated using dotted lines. (E) PF4 and serotonin-negative cells were counted
using immunofluorescence microscopy (n = 3 different mice). Representative image
of Z-stack projections using confocal microscopy. Platelets that returned to
circulation 24 h after IC injection were used for quantification. Tubulin (red) was used
as a platelet marker. PF4 (green) and serotonin (green) were observed in less than
60% of platelets. Empty platelets (arrowheads) and platelets (arrows) are
represented. (Scale bars: 2 μm.) As a negative control, serotonin labeling was
performed on Tph1−/− platelets (Fig. S9). (F) Electron microscopy of FcγRIIATGN
platelets before IC injection (Upper) and 24 h after IC injection (Lower). Empty
platelets (arrowheads) and platelets (arrows) are represented. (Scale bars: 0.8 μm.)
(G) Systemic shock was measured in FcγRIIATGN mice injected with ICs at t = 0 and
rechallenged at t = 24 h (n = 3). (H) Plasma levels of PF4 (n = 4) and serotonin (n =
5) were determined in mice rechallenged 24 h after the first challenge with ICs.
Results were compared with the level after the first challenge (dotted lines). (I)
Platelets were purified from FcγRIIATGN mouse blood, fluorescently labeled, and
adoptively transferred i.v. in FcγRIIATGN mice. One hour later, mice were injected
with ICs, and fluorescent platelets in circulation were determined at the indicated
time points following injection of ICs (n = 10). Dil, diluent; null, FcyRIIAnull; TGN;
FcyRIIATGN. Data are mean ± SEM. **P < 0.005, ***P < 0.001, and ****P < 0.0001,
using an unpaired t test (A, D, and H); one-way ANOVA (B, C, E, and I); and
repeated-measures two-way ANOVA, statistical variation between groups (G).

270
Platelet conversion to microparticles also did not explain the profound
thrombocytopenia, as microparticle levels only increased 60 min after the IC trigger
(Fig. S4G). The decrease in platelet number could also not be attributed to platelet
interaction with leukocytes, as neutrophil depletion, or blockade of Mac-1 and PSGL-
1, had no effect on thrombocytopenia (Fig. S4 H and I). Given that
FcγRIIATGN/Tph1−/− mice also underwent profound thrombocytopenia (Fig. 3B), and
that exogenous serotonin did not induce thrombocytopenia (Fig. S4J), this suggests
that serotonin serves as the effector of IC-mediated shock but is not the driver of the
resulting thrombocytopenia. Thus, although thrombocytopenia occurs concurrently
with shock, it is not causative in the mechanism of inducing the shock.
The unanticipated rapid recovery in platelet count prompted a detailed analysis of
the platelets present in blood 24 h after IC challenge. We found that platelets were
not activated or apoptotic, as evidenced by the lack of phosphatidylserine (PS) and
P-selectin on their surface (Fig. 3C). However, the platelet granule content was
markedly reduced (Fig. 3D), as ∼30% of the circulating platelets contained no
detectable serotonin or PF4 (Fig. 3E), suggesting significant degranulation. Electron
microscopy further confirmed that circulating platelets were frequently devoid of any
granules (Fig. 3F). Notably, platelets still expressed surface FcγRIIA 24 h postshock
(Fig. S5 A and B), and still underwent thrombocytopenia if challenged a second time
with ICs (Fig. S5C). This is in contrast to shock, where FcγRIIATGN mice were
resistant to shock induced by further challenges (Fig. 3G), and neither serotonin nor
PF4 was induced in the blood of these mice (Fig. 3H). Thus, we hypothesized that
platelets circulating 24 h postshock were, in fact, platelets that had already
degranulated and had undergone temporary sequestration. To verify this, we
performed fluorescent labeling of FcγRIIATGN platelets, which we adoptively
transferred into FcγRIIATGN mice. As expected, 87 ± 12% of fluorescent platelets,
which were negative for PS (Fig. S5D), rapidly became undetectable from the blood
circulation after an IC trigger. Of particular importance, 42 ± 16% of the fluorescently
labeled FcγRIIA platelets, devoid of any surface PS, were identified in blood 24 h
after shock (Fig. 3I), confirming a temporary sequestration of platelets following
FcγRIIA activation and their return to the blood circulation after degranulation.
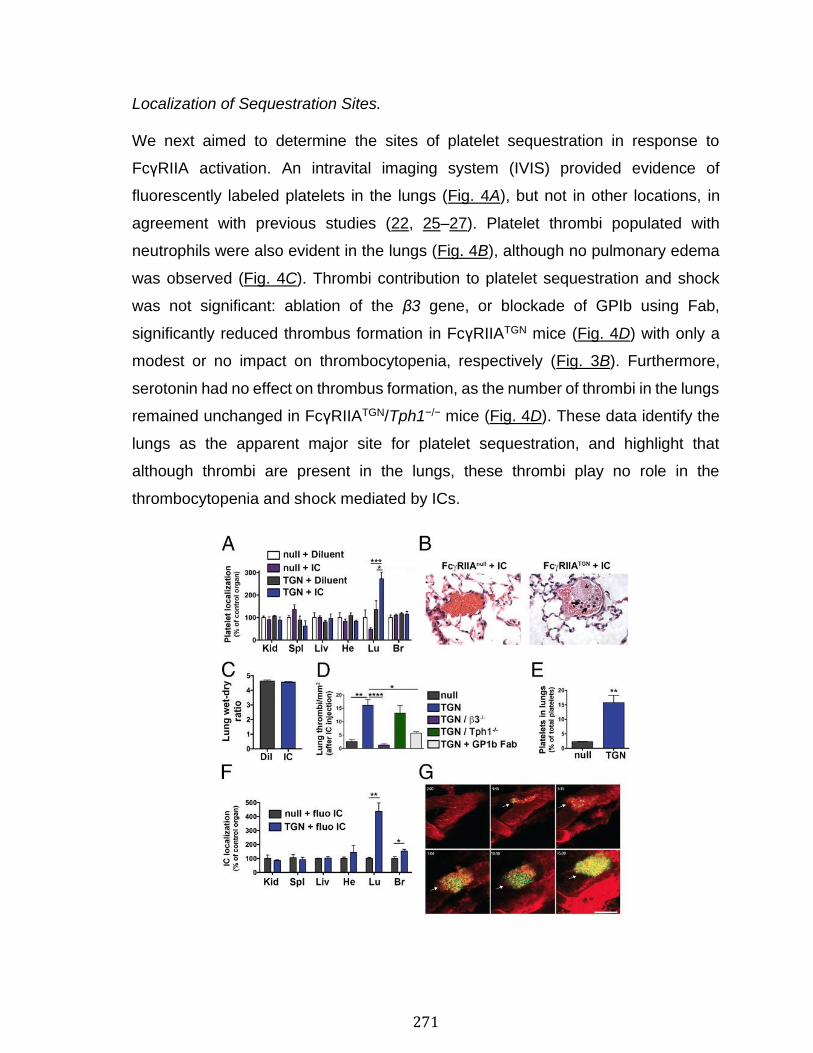
271
Localization of Sequestration Sites.
We next aimed to determine the sites of platelet sequestration in response to
FcγRIIA activation. An intravital imaging system (IVIS) provided evidence of
fluorescently labeled platelets in the lungs (Fig. 4A), but not in other locations, in
agreement with previous studies (22, 25–27). Platelet thrombi populated with
neutrophils were also evident in the lungs (Fig. 4B), although no pulmonary edema
was observed (Fig. 4C). Thrombi contribution to platelet sequestration and shock
was not significant: ablation of the β3 gene, or blockade of GPIb using Fab,
significantly reduced thrombus formation in FcγRIIATGN mice (Fig. 4D) with only a
modest or no impact on thrombocytopenia, respectively (Fig. 3B). Furthermore,
serotonin had no effect on thrombus formation, as the number of thrombi in the lungs
remained unchanged in FcγRIIATGN/Tph1−/− mice (Fig. 4D). These data identify the
lungs as the apparent major site for platelet sequestration, and highlight that
although thrombi are present in the lungs, these thrombi play no role in the
thrombocytopenia and shock mediated by ICs.

272
Fig. 4.Localization of sites of sequestration. (A) Mouse platelets from whole blood of FcγRIIATGN mice were labeled in vivo before IC injection. The kidneys (Kid), spleen (Spl), liver (Liv), heart (He), lungs (Lu), and brain (Br) were harvested 10 min after IC injection, and fluorescence was measured in each organ using an IVIS to determine platelet localization (n = 3). Results were compared with FcγRIIAnull mice injected with diluent, and are presented as the percentage of fluorescence in diluent-injected FcγRIIAnull mice. (B) Lungs were collected 10 min after IC injection. Thrombi (star) and neutrophils (arrows) were examined by microscopy after hematoxylin and eosin coloration. (Magnification: 400×.) (C) FcγRIIATGN mice were injected with diluent (Dil) or ICs, and lungs were collected after 10 min. The lung wet-to-dry ratio was evaluated. (D) Number of lung thrombi per square millimeter was quantified in FcγRIIAnull (n = 4), FcγRIIATGN (n = 12), FcγRIIATGN/β3−/− (n = 7), and FcγRIIATGN/Tph1−/− (n = 3) mice, as well as in FcγRIIATGN mice pretreated with GP1b Fab antibody (n = 4). (E) Mouse platelets from FcγRIIAnull and FcγRIIATGN mice were labeled in vivo. Lungs were collected 10 min after the IC trigger, and the number of fluorescent platelets was estimated (n = 4) using a standard curve designed with known numbers of fluorescent platelets spiked into nonfluorescent control lung homogenates. Percentages were obtained by comparison with platelet count obtained before the experiment and are presented as the percentage of total platelet number in the whole-mouse body. (F) ICs were labeled with DyLight-647 anti-Human IgG (fluo ICs) before injection in mice. The Kid, Spl, Liv, He, Lu, and Br were harvested 10 min later, and fluorescence was measured in each organ using an IVIS to determine IC localization (n = 3). Results are presented as the percentage of fluo determined in FcγRIIAnull mice. (G) Two-photon intravital microscopy in brain microvasculature of FcγRIIATGN/CD41-YFP mice injected with ICs. Thrombi were observed 5 min after IC injection (arrows), whereas brain vasculature leakage occurred after 10 min. (Scale bar: 50 μm.) null, FcyRIIAnull; TGN; FcyRIIATGN. Data are mean ± SEM. *P < 0.05, **P < 0.005, ***P < 0.001, and ****P < 0.0001 using one-way ANOVA (A and D) and an unpaired t test (C, E, and F).
However, we suspected that other anatomical sites of platelet sequestration might
exist, as the IVIS approach may not permit an optimal distinction between
immobilized platelet aggregates and circulating platelets in the microvasculature,
where small platelet aggregates are to be expected. In addition, if the entire platelet
population was sequestered in the lungs, it was puzzling that none of the mice died
from the IC trigger. Using a quantitative approach to measure fluorescently labeled
CD41+ platelets, we estimated that the lungs, in fact, contained only 16% of the total
platelet load (Fig. 4E), confirming that other sites of platelet sequestration likely
existed.
The presence of platelets outside the vasculature was excluded first, as no platelets
were detected in the thoracic lymph (Fig. S6A). Furthermore, assessment using

273
whole-mouse imaging of yellow fluorescent protein (YFP) in FcγRIIATGN/CD41-YFP
mice, which constitutively express YFP in CD41-expressing cells, efficiently
identified thrombi in the lungs and megakaryocytes in the bone marrow, but did not
reveal platelets outside blood vessels or in other tissues (Fig. S6B). Therefore, we
speculated that tracking ICs in mice might be a more efficient means of leading us
to platelet sequestration sites. We found that ICs not only localized to the lungs but
were also observed in the brain vasculature (Fig. 4F). Thus, the brain
microvasculature was examined using two-photon microscopy in live
FcγRIIATGN/CD41-YFP mice, with the inclusion of FcγRIIAnull/CD41-YFP mice for
comparison. We observed profound leakage of the brain vasculature in both
FcγRIIAnull and FcγRIIATGN mice when injected with ICs (Fig. 4G and Movies S2 and
S3). Of importance, in the presence of ICs, small platelet aggregates readily formed
in the leaky brain microvasculature, but only if FcγRIIA was expressed by platelets
(Fig. 4G and Movie S2). Thrombi were not detected in the microvasculature of the
kidney, liver, or spleen of FcγRIIATGN mice injected with diluent or ICs (Fig. S6C),
and two-photon microscopy of the femurs in live FcγRIIATGN/CD41-YFP mice did not
reveal any platelet aggregation in sinusoids in the bone marrow (Fig. S6D). These
data suggest that in the presence of ICs, platelets sequester in certain microvascular
beds, notably in the lungs and brain, and that this event occurs independent of
thrombosis and leakage.
Role of Platelet FcγRIIA and Serotonin in Acute Inflammatory Responses.
During microbial invasion, foreign antigens are recognized by host antibodies and
form ICs. Hence, incubation of human platelets in the presence of influenza virus or
various strains of bacteria leads to platelet activation, which strictly requires the
presence of plasma and FcγRIIA (17, 18). While these observations suggest that
motifs on pathogens contribute to the formation of ICs, whether they can trigger
platelet activation in vivo has not been established.
Antibodies against lipopolysaccharide (LPS), a common gram-negative pathogen-
associated molecular pattern (PAMP), were detected in the blood of healthy
volunteers similar to patients with septic shock due to confirmed gram-negative
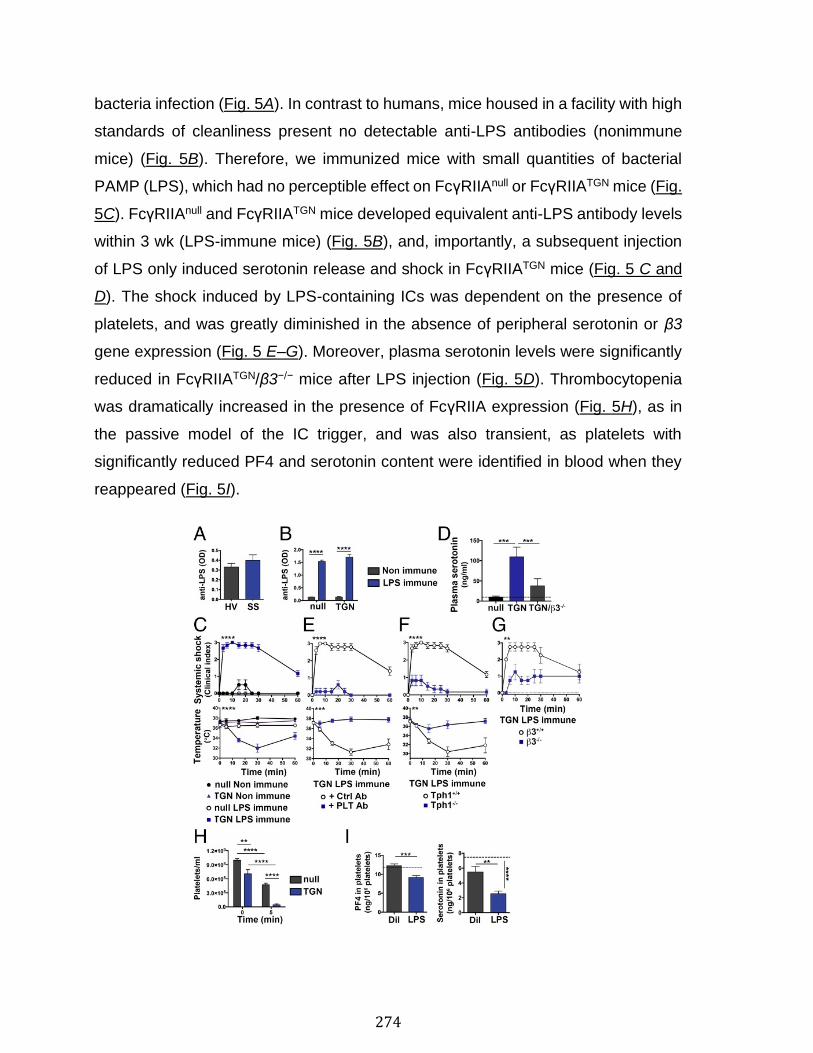
274
bacteria infection (Fig. 5A). In contrast to humans, mice housed in a facility with high
standards of cleanliness present no detectable anti-LPS antibodies (nonimmune
mice) (Fig. 5B). Therefore, we immunized mice with small quantities of bacterial
PAMP (LPS), which had no perceptible effect on FcγRIIAnull or FcγRIIATGN mice (Fig.
5C). FcγRIIAnull and FcγRIIATGN mice developed equivalent anti-LPS antibody levels
within 3 wk (LPS-immune mice) (Fig. 5B), and, importantly, a subsequent injection
of LPS only induced serotonin release and shock in FcγRIIATGN mice (Fig. 5 C and
D). The shock induced by LPS-containing ICs was dependent on the presence of
platelets, and was greatly diminished in the absence of peripheral serotonin or β3
gene expression (Fig. 5 E–G). Moreover, plasma serotonin levels were significantly
reduced in FcγRIIATGN/β3−/− mice after LPS injection (Fig. 5D). Thrombocytopenia
was dramatically increased in the presence of FcγRIIA expression (Fig. 5H), as in
the passive model of the IC trigger, and was also transient, as platelets with
significantly reduced PF4 and serotonin content were identified in blood when they
reappeared (Fig. 5I).

275
Fig. 5.Role of the FcγRIIA/serotonin axis in acute inflammation. Endogenous antibodies directed against LPS (anti-LPS) were measured by ELISA in plasma of healthy volunteers (HV) and patients with ongoing septic shock (SS) (A; n = 11) and in FcγRIIAnull and FcγRIIATGN mice that were immunized with LPS (LPS-immune) or diluent (nonimmune) (B; n = 4). OD, optical density. (C) Systemic shock and temperature were evaluated in nonimmune (n = 4) or LPS-immune mice (n = 6) expressing or not expressing FcγRIIA. (D) Serotonin levels in plasma were determined in LPS-immune FcγRIIAnull, FcγRIIATGN, and FcγRIIATGN/β3−/− mice immediately after LPS injection (n = 5) and compared with nonimmune FcγRIIATGN mice (baseline level, indicated as a dotted line). Systemic shock and temperature were measured after LPS injection in LPS-immune FcγRIIATGN mice preinjected with platelet-depleting (PLT Ab) or control (Ctrl Ab) antibodies (E; n = 5) and in LPS-immune FcγRIIATGN/Tph1+/+ mice and FcγRIIATGN/Tph1−/− mice (F; n = 6). (G) Systemic shock was measured after LPS injection in LPS-immune FcγRIIATGN/β3+/+ mice and FcγRIIATGN/β3−/− mice (n = 4). (H) CD41+ platelets in whole blood were counted using flow cytometry at the indicated time points after injection of LPS in LPS-immune FcγRIIAnull and FcγRIIATGN mice (n = 5). (I) Platelet content in PF4 (n = 5; Left) and serotonin (n = 6; Right) were determined 48 h after LPS injection in LPS-immune FcγRIIAnull and FcγRIIATGN mice and compared with nonimmune FcγRIIATGN mouse levels (dotted lines). Dil, diluent; null, FcyRIIAnull; TGN, FcyRIIATGN. Data are mean ± SEM. **P < 0.005, ***P < 0.001, and ****P < 0.0001, using an unpaired t test (A and I); repeated-measures two-way ANOVA, statistical variation between groups (C and E–G); and one-way ANOVA (B, D, and H).
We further verified whether the same conclusions hold in viremia and in a well-
described model of active systemic anaphylaxis (23, 28, 29). The i.v. injection of
herpes simplex virus-1 (HSV-1), capable of blood dissemination in immunized
humans (45, 46), in HSV-1–immunized mice induced shock reminiscent of the
responses seen in IC-injected mice and dependent on FcγRIIA (Fig. S7A). Moreover,
BSA injection in BSA-immunized mice initiated systemic anaphylactic responses,
and a proportion of the FcγRIIATGN mice, but never the FcγRIIAnull mice, died. Modest
shock response and moderate thrombocytopenia were also observed in FcγRIIAnull
mice (Fig. S7B), possibly due to the high antigenicity of BSA and the potent
immunization protocol implicating adjuvant, consistent with other studies implicating
neutrophils and FcγRIIA-independent responses in these experimental conditions
(23, 28, 29). Of importance is that in all models, platelet FcγRIIA and serotonin were
involved in shock, while FcγRIIA was implicated in thrombocytopenia (Fig. S7 C–E).
Thus, as in our passive model of IC-mediated immune reaction, active immunization
with gram-negative PAMPs, virus, or protein antigen dominantly implicates platelet
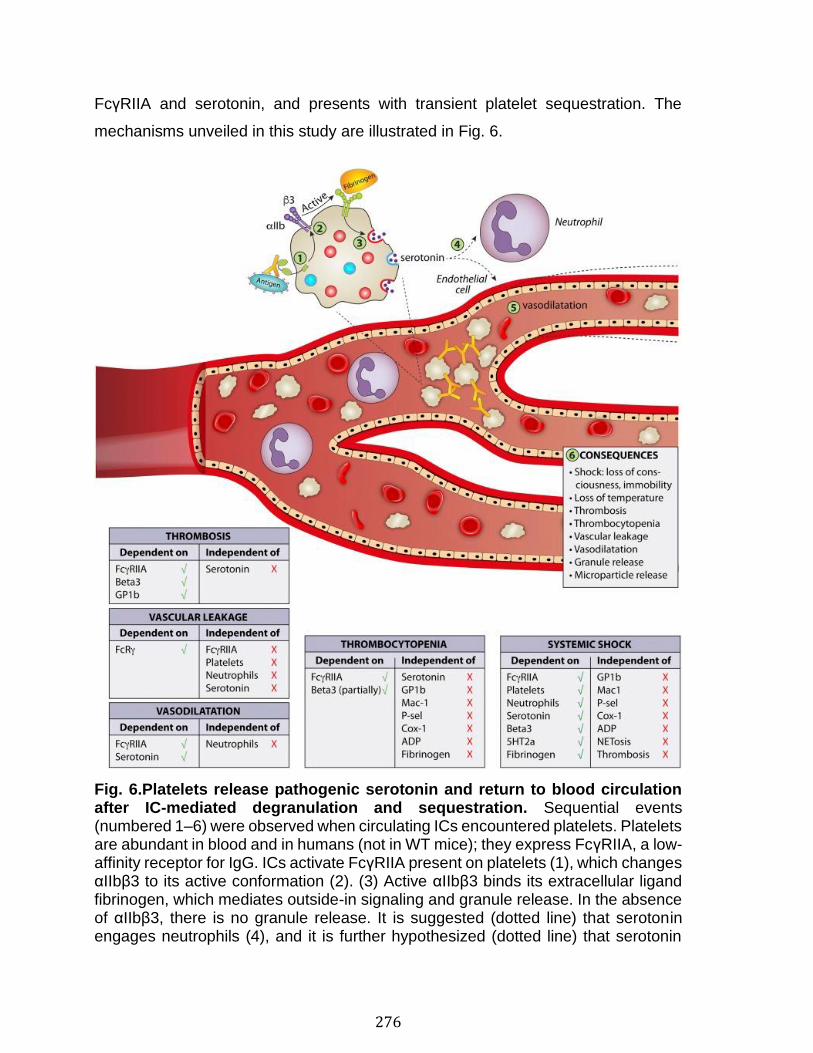
276
FcγRIIA and serotonin, and presents with transient platelet sequestration. The
mechanisms unveiled in this study are illustrated in Fig. 6.
Fig. 6.Platelets release pathogenic serotonin and return to blood circulation after IC-mediated degranulation and sequestration. Sequential events (numbered 1–6) were observed when circulating ICs encountered platelets. Platelets are abundant in blood and in humans (not in WT mice); they express FcγRIIA, a low-affinity receptor for IgG. ICs activate FcγRIIA present on platelets (1), which changes αIIbβ3 to its active conformation (2). (3) Active αIIbβ3 binds its extracellular ligand fibrinogen, which mediates outside-in signaling and granule release. In the absence of αIIbβ3, there is no granule release. It is suggested (dotted line) that serotonin engages neutrophils (4), and it is further hypothesized (dotted line) that serotonin

277
induces vasodilatation through its action on endothelial cells (5). (6) Multiple manifestations are observed when ICs form in blood. Shock, characterized by loss of consciousness, immobility, shallow respiration, and hypothermia, strictly implicates platelets, αIIbβ3 binding to fibrinogen, and serotonin release. In the absence of neutrophils, serotonin is released but shock is abolished. It is surmised that neutrophils produce PAF in response to serotonin, which may contribute to shock downstream of serotonin release. Mediators of shock are indicated in the figure. Thrombocytopenia is due, at least in part, to platelet sequestration in certain vascular beds, notably in the lung and brain microvasculature. Sequestration implicates FcγRIIA but, in contrast to shock, occurs independent of serotonin and neutrophils, and only partially implicates β3. Thrombocytopenia is only transient, and platelets return to blood circulation with emptied granules. Microparticle release is observed in blood before return of platelets. Roles of different molecules in thrombocytopenia are indicated in the figure. Vasodilatation is implicating platelet-derived serotonin and is occurring independent of the presence of neutrophils. Thrombosis was characterized in lungs following IC administration. It implicates FcγRIIA and β3, and, in contrast to shock, it involves GPIb and not serotonin. These data support the notion that shock and thrombosis are independent events. Vascular leakage occurs independent of platelets and FcγRIIA. Molecules implicated in leakage are presented in the figure. NETosis, neutrophil extracellular traps; P-sel, P-selectin.

278
Discussion
In this study, we examined platelet activity in IC-induced systemic inflammation in
the absence of vascular insult. We confirmed a central role for platelet activation and
identified serotonin as a critical platelet component mediating mechanisms of shock.
Albeit artificial, models utilizing ferric chloride, laser-induced injury, or injection of
cytokines or PAMP in tissue/organs (e.g., cremaster muscle, liver, lungs)–(6, 9, 47)
have provided crucial insights to key pathways implicated in inflammation. Herein,
we used HA-IgG as a surrogate model of ICs and revealed critical components in
the in vivo response to systemic ICs, which were further confirmed using active
immunization with endotoxin, virus, or a commonly used antigen in the study of
anaphylaxis. Our observations were only possible using FcγRIIATGN mice, where we
confirmed the functional association of FcγRIIA and αIIbβ3 in platelet activation, and
suggest that targeting these receptors may have clinical benefits in severe conditions
involving ICs. Conversely, other molecules (i.e., P-selectin, GPIb, ADP,
thromboxane) that classically play a dominant role in platelet activation in
thrombosis, or favor platelet and neutrophil interactions, were dispensable. The
contribution of neutrophils to the systemic response to ICs is, however, not excluded,
as neutrophils have also been previously implicated, possibly through the release of
PAF (12, 23, 28). Hence, although serotonin release was maintained in the absence
of neutrophils, shock was dramatically reduced (Figs. S2B and S8A). We thus
propose that the release of serotonin precedes the neutrophil contribution to shock,
consistent with the reported role of serotonin in neutrophil activation (48). As
vasodilation was also present in the absence of neutrophils (Fig. S3B), these data
suggest that platelets, through serotonin, orchestrate neutrophil activation and
endothelial cell functions, and that these events can occur independently (Fig. 6).
Vascular leakage is another feature systematically observed when ICs were present
in blood. Intriguingly, leakage and vasodilatation were not connected, as leakage
took place independent of FcγRIIA but vasodilation critically required FcγRIIA and
serotonin. As such, vessel permeability likely involved smaller postcapillaries

279
vessels and more subtle changes in endothelial cell interactions, and was also
insufficient to induce significant edema in the lungs, although platelets and
neutrophils accumulated in great number in the lungs. In addition, leakage critically
involved IC-mediated signaling, given that it was totally absent in FcRγ−/− mice (Fig.
S3A). While neutrophils express other IgG receptors (other than FcγRIIA), they were
dispensable in the process (Fig. S3A). Therefore, leakage could be attributed to mast
cells or basophils, for instance, which also express an array of receptors, such as
FcγRI and FcγRIII, capable of responding to ICs and can mediate permeability,
potentially through histamine release (49).
Whereas serotonin is mostly known for its role in mood, anxiety, psychosis, or
memory in the central nervous system, more than 95% of total body serotonin is
present in the periphery (39). The majority of peripheral serotonin is stored in
platelets, and our observations further revealed that platelets from female mice
contain less serotonin than those from males (Fig. S8B), which may explain, in part,
the aggravated phenotype observed in male mice compared with females following
IC challenge. Moreover, although platelets in females and males expressed similar
levels of FcγRIIA, female FcγRIIATGN mice presented significantly lower platelet
counts than males, which could also partially explain the reduced shock in females
in comparison to male mice (Fig. S8 C–E). Other immune components, such as
complement C5a, are also more abundant in males than in females (50). Whether
the serotonin reservoir in platelets explains a fundamental gender-related dichotomy
in susceptibility to inflammatory responses to ICs remains to be established.
The function of serotonin in platelets is not clear; studies suggest that it is important
for the serotonylation of proteins necessary in platelet aggregation (51). However,
SSRIs are typically used by patients during the perioperative period and mice lacking
peripheral serotonin present only mild bleeding defects (43). Thus, the present study
sheds light on a major role of platelet serotonin in response to a systemic stimulus,
which occurs independent of other molecules typically implicated in the prevention
of bleeding. The advantages for an organism to release bulk serotonin in response
to systemic ICs are unclear. We can speculate that in response to a microbial

280
invasion in an immune host, it might be preferable to reduce blood flow to prevent
dissemination of the pathogen to vital organs and to facilitate its capture by
phagocytes. As serotonin also mediates organ regeneration (52), its liberation may
be pivotal to regrowth following insults caused by pathogen invasion.
Thrombocytopenia, which coincides with thrombi formation in the lungs, has been
reported to occur in FcγRIIATGN mice, and requires the expression of guanine
nucleotide exchange factor CalDAG-GEF1 and 12-lipoxygenase (22, 25–27, 53).
We showed here that the formation of thrombi or occlusion of blood vessels was not
the primary cause of shock; thrombi formed normally in FcγRIIATGN/Tph1−/− mice,
whereas shock was nearly totally abrogated in these mice. Furthermore, reduction
of thrombosis by blockade of GPIb was without effect on shock and
thrombocytopenia, thereby revealing that thrombocytopenia resulted mainly from
platelet sequestration, not thrombosis. Whereas platelets were observed in the lungs
and brain, principally in the smaller and intertwined vessels, platelets may also be
hiding in other yet-to-be-discovered vascular beds despite our careful investigations.
In thrombotic thrombocytopenic purpura, profound thrombocytopenia is explained by
the failure of ADAMTS13 to perform proteolysis of vWF attached to the endothelium
(54), a mechanism distinct from what is observed in IC-induced thrombocytopenia,
as the blockade of vWF and GPIb had no effects on thrombocytopenia in our study.
As in-depth whole-mouse imaging uniquely identified megakaryocytes and thrombi,
which comprise more stable and adherent platelets, it suggests that the majority of
the sequestered platelets were dislodged by the perfusion procedure. Therefore, we
propose that platelets might be bridged together by ICs, and that platelet–IC
scaffolds may be loosely trapped in the microvasculature.
Degranulated platelets recirculate, a finding of potential significance for elucidating
mechanisms underlying thrombocytopenia. The absence of PS at the surface
suggests that they are not procoagulant platelets, known as balloon- or zombie-like
platelets (55). How platelets return to the circulation after sequestration is unclear,
but it is reasonable to speculate that disengagement of FcγRIIA after its
desensitization by yet unknown mechanisms (e.g., unidentified immunoreceptor

281
tyrosine-based inhibitory motif-containing receptors or phosphatases) or IC
internalization by platelets (56) might release platelets from platelet–IC scaffolds and
permit their liberation from the microvasculature. FcγRIIA expression was
maintained, however, on recirculating platelets, suggesting that it might be recycled
if the internalization of ICs is implicated. Of interest is that platelets at thrombi
surfaces visualized in vascular injury models appear loosely packed and lightly
activated (57, 58), and might also return to the circulation. As platelets activated in
vitro with thrombin can also circulate after degranulation if transfused (59, 60), our
study reveals that thrombotic and immunological triggers can induce degranulation
independent of platelet elimination. These models contrast with the general belief
that platelets “have only one life,” and may not recirculate after undergoing activation
in vivo.
The insertion of human activating (FcγRIIA/IIIA/IIIB) and inhibitory (FcγRIIB) FcγR
into the equivalent murine locus confirmed the predominance of FcγRIIA in systemic
shock in the humanized mouse model (28), suggesting that our findings may well be
translatable to humans as platelets from transgenic mice and humans express
equivalent levels of FcγRIIA (18). While these approaches cannot fully recapitulate
all of the subtleties of IC-driven inflammation in humans, it is very likely that the
mechanisms revealed in our study may, at least in part, take place in disease states
such as rheumatic disease, HIT, sepsis, viremia, anaphylaxis, and adverse reactions
following i.v. IgG therapy.
PAMPs trigger innate immune responses through activation of pattern-recognition
receptors, but the recognition of PAMPs by antibodies in adaptive immunity can
modulate different responses (61). We found that LPS, a Toll-like receptor 4 (TLR-
4) trigger, dominantly implicates platelets and serotonin when involving the adaptive
immune response in mice that had been preexposed to LPS. All adult volunteers we
examined also displayed antibodies directed against the endotoxin. Interestingly,
humans lacking TLR signaling molecules are extremely susceptible to infections in
infancy and childhood, and thereafter develop significant resistance, consistent with
the prevalent role of adaptive immunity in adults (62). Sepsis susceptibility is

282
associated with FcγRIIA polymorphism (63) and is accompanied by elevated
serotonin and endothelial hyperpermeability (64), which could be attributed to
platelets. Furthermore, thrombocytopenia measured in patients with sepsis is a
strong predictor of mortality (65, 66), and our data suggest that platelets may be
sequestered through FcγRIIA activation in some patients. It is thus important to note
that most mouse models of sepsis or viral infection may be suboptimal, as they
overlook the contribution of FcγRIIA in a nonimmune host. Observations from trials
during which LPS was injected into volunteers revealed that humans responded
more promptly to small doses of LPS than mice (67), pointing to the involvement of
ICs and FcγRIIA in humans, a pathway absent in mice.
Circulating platelets presenting reduced granule content are reported in different
contexts, such as cardiovascular diseases, type-2 diabetes mellitus, preeclampsia,
autoimmune diseases, and sepsis (68–72). In patients with severe sepsis, ADP and
serotonin contents in platelets were reduced by 40% and 50%, respectively (71).
However, very little is known about the impact of in vivo platelet degranulation on
platelet life span. In chronic conditions implicating ICs, such as rheumatic diseases,
ICs constantly trigger platelet FcγRIIA, consistent with the frequent shifts in platelet
count. Hence, microthrombi are recognized in these diseases, including in the lungs,
and the platelet content of serotonin is reportedly reduced in patients with SLE and
RA by 25% and 27%, respectively (72, 73), suggesting that platelets might indeed
circulate in their degranulated form in various contexts implicating ICs (70).
Neuropsychiatric SLE is the least understood, yet the most prevalent, manifestation
of SLE (74). Our identification of localized platelet activation in the brain
microvasculature and leakage of the blood–brain barrier may thus have implications
in the important and poorly understood neurological manifestations in rheumatic
diseases.
In summary, our study reveals platelet contributions to inflammation in reactions
involving ICs. It appears that the FcγRIIA signaling and serotonin release are unique
in regard to their major role in inflammation and minor roles in the prevention of

283
bleeding, suggesting that interference in this process might be a promising avenue
for further research.
Materials and Methods
Mice.
C57BL/6J (FcγRIIAnull mice), FcγRIIATGN hemizygous mice (12, 20), and FcRγ−/−
mice were obtained from The Jackson Laboratory. FcγRIIATGN hemizygous mice
described to express human FcγRIIA on platelets, megakaryocytes, monocytes,
macrophages, neutrophils, eosinophils, basophils, mast cells, and dendritic cells (12,
20) were backcrossed to C57BL/6J more than 10 times. The β3−/− mice (30), Tph1−/−
mice (43), and CD41-YFP mice (75) were crossed with FcγRIIATGN mice to obtain
FcγRIIATGN/β3−/− (30), FcγRIIATGN/Tph1−/−, and FcγRIIATGN/CD41-YFP mice.
Chimeric mice were generated by transfer of bone marrow cells of FcγRIIATGN mice
into FcγRIIAnull (WT), Fibγ390-396A, and Fibγ∆5 irradiated mice (34, 35, 76). Guidelines
of the Canadian Council on Animal Care were followed in a protocol approved by
the Animal Welfare Committee at Laval University (2013-106-3).
HA-IgG as an IC Model.
Human IgG (IC) (MP-biological, Sigma–Aldrich and Innovative Research) were i.v.
injected (600 μg and 750 μg) in males and females, respectively. In some
experiments, human IgG was dissolved but nonaggregated (monomeric) or mouse
IgG was aggregated and injected in mice (29).
Measure of Shock.
Temperature was measured using a rectal probe thermometer at indicated time
points. Signs of apparent shock were assessed as described previously (24, 77)
using a score from 0 to 3 (24, 77). A score of 3 represents completely immobilized
and unconscious mice (mice collapse and do not react to sound or touch), a score
of 2 represents mice with impaired mobility and irregular respiration, a score of 1
corresponds to mice with slow motions and shallow respiration, and a score of 0

284
describes normal mice. In anaphylaxis experiments implicating BSA injection in
BSA-immunized FcγRIIATGN mice, death was sometimes observed. In those
exceptional cases, shock was scored as 4. Scores measured in experimental groups
were averaged and are presented as a function of time (minutes). Scores for
individual mice for key experiments are provided in SI Materials and Methods (Fig.
S1 A and F).
LPS Immunization Model.
Mice were immunized with i.v. injection of 1 mg/kg LPS (Escherichia coli 0111:B4;
Sigma–Aldrich) at days 0, 14, 28, and 42. At the first and the last immunizations,
shock and temperature were monitored for 1 h. Mouse blood was drawn by cardiac
puncture 10 min after the fourth injection and used for platelet count and ELISA.
Lungs were also collected as described below.
Flow Cytometry.
Flow cytometry was performed using a BD FACSCanto II instrument with forward
scatter coupled to a photomultiplier tube “small particles option” flow cytometer (BD
Biosciences). Platelets, platelets interacting with neutrophils, and platelet
microparticles were analyzed.
Histology.
In some experiments, organs were collected at the end of the experiment.
Intratracheal instillation with 1 mL of 4% paraformaldehyde in lungs was performed
before collection. Brain, kidneys, spleen, heart, liver, and lungs were collected and
then fixed in 4% paraformaldehyde for 24 h (lungs) or 72 h (other organs); they were
then washed and stored in PBS at 4 °C before histology. After fixation, paraffin-
embedded organs were cut into 5-μm sections and stained with hematoxylin and
eosin. Thrombi were observed on five different spots at 400× resolution, they were
counted using light microscopy (BX51; Olympus) by a blinded investigator, and the
numbers of lung thrombi per square millimeter were calculated. Neutrophils were
identified in lungs of IC-injected FcγRIIATGN mice.

285
Two-Photon Intravital Microscopy.
For in vivo imaging of the mouse brain, FcγRIIATGN/CD41-YFP mice (8–12 wk old)
were anesthetized with 1–2% isoflurane (vol/vol) and a cranial window was made to
expose the vasculature of the sensorimotor cortex. Animals were imaged 2 wk after
the surgery. Briefly, for the imaging session, the head of the mouse was restrained
using a custom-built cranial stereotaxic apparatus (David Kopf Instruments) and
placed under the microscope. For the ear imaging, mice were anesthetized and the
hair recovering the ears was gently removed using Nair, a commercial depilatory
lotion. One ear was then gently flattened and fixed on a Plexiglas bloc using MSI-
EpiDermGlu (Medisav Services). Gelseal (GE Healthcare) was applied around the
tissues to form a watertight rim, and the imaging cavities were filled with sterile HBSS
without Ca2+/Mg2+ (Thermo Fisher Scientific). Blood vessels were stained by an i.v.
injection of 0.05% Evans Blue (Sigma–Aldrich) diluted in 0.9% sterile saline. Body
temperature was kept at 37 °C during all procedures with a heating pad. Four to six
different vessels per mouse were analyzed at 1 and 8 min postinjection.
Patients with Septic Shock.
Adult (age ≥ 21 y) patients with septic shock and confirmed, gram-negative
bacteremia were included. Each patient or an authorized family member provided
written informed consent. Septic shock was defined as the presence of sepsis
(requiring evidence of systemic infection and two or more of the following:
temperature >38 °C or <36 °C; heart rate >90 beats per minute; respiratory rate >20
breaths per minute or partial pressure of carbon dioxide in arterial blood <32 mmHg;
WBC count >12,000/mL, <4,000/mL, or >10% bands) and the need for vasopressors
to maintain a systolic blood pressure >90 mmHg or within 40 mmHg of baseline
despite adequate fluid resuscitation (5, 6). For identification of gram-negative
bacteremia, blood samples were obtained from patients upon intensive care unit
(ICU) admission as part of their routine clinical care. Blood samples underwent gram
staining and culturing in a clinical pathology laboratory. Gram-negative pathogens
were identified from the gram stain and/or cultures by the clinical laboratory. Healthy,

286
fasting adult (age ≥ 21 y) control subjects provided informed consent. Following
informed consent, demographic data, physiological parameters, and laboratory data
were recorded. Plasma was harvested by centrifugation on whole blood collected in
sterile ACD vacutainer tubes. Plasma was frozen at −80 °C until used for assays. In
patients with septic shock, plasma was obtained within 48 h of ICU admission.
Study Approval.
Informed consent was obtained from all human subjects in the study. The study was
approved by the institutional review board at University of Utah.
Statistical Analysis.
Results are presented as mean ± SEM. The statistical significance for comparisons
between groups was determined using one-way ANOVA, two-way repeated-
measures ANOVA, an unpaired Student’s t test, or a Mann–Whitney U test. The
D’Agostino–Pearson test was used as a normality test. All statistical analysis was
done using Prism software package 6 (GraphPad Software).

287
Acknowledgments
We thank Isabelle Dubuc for expert technical assistance with virus preparation. This
work was supported by a foundation grant from the Canadian Institutes of Health
Research (CIHR) (to E.B.); it was also supported, in part, by NIH/National Heart,
Lung, and Blood Institute Grant 5F32HL118865 (to K.R.M.), Grant HL130054 (to
P.J.N.), and Grant R01Hl68130 (to J.E.I.); by Grant 1K01DK111515 from the
National Institutes of Diabetes and Digestive and Kidney Diseases (to K.R.M.); by a
grant from the Canadian Funds for Innovation (to G.P.); by Grant HL112311 (to
M.T.R.); by Grant HL126547 (to M.T.R.); by National Institute on Aging Grant
AG048022 (to M.T.R.), by a CIHR foundation grant (to W.I.K.); by Grant
P01HL110860 (to S.E.M.), by a CIHR operating grant (to S.L.); and by CIHR Grant
MOP 93575 (to L.F.). E.B. is the recipient of an investigator award from the CIHR.
K.R.M. and J.E.I. are American Society of Hematology Fellow Scholars. G.M. has
an award from the Canadian Blood Services, N.T. and I.M. are recipients of
fellowships from The Arthritis Society (TAS), A.Z. is a recipient of fellowships from
the TAS and CIHR, and B.M. has a fellowship from the Fondation du Centre
Hospitalier Universitaire de Quebec. This material is also the result of work
supported with resources and the use of facilities at the George E. Wahlen Veterans
Affairs Medical Center (M.T.R.). The contents do not represent the views of the US
Department of Veterans Affairs or the US Government.

288
References
1. Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–274. 2. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123:2759–2767. 3. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: Platelets served with inflammation. J Immunol. 2015;194:5579–5587. 4. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. 5. Sreeramkumar V, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346:1234–1238 6. Imhof BA, et al. CCN1/CYR61-mediated meticulous patrolling by Ly6Clow monocytes fuels vascular inflammation. Proc Natl Acad Sci USA. 2016;113:E4847–E4856. 7. Goerge T, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111:4958–4964. 8. Boulaftali Y, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123:908–916. 9. Gros A, et al. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood. 2015;126:1017–1026. 10. Suurmond J, Zou YR, Kim SJ, Diamond B. Therapeutics to block autoantibody initiation and propagation in systemic lupus erythematosus and rheumatoid arthritis. Sci Transl Med. 2015;7:280ps5. 11. Jönsson F, et al. Mouse and human neutrophils induce anaphylaxis. J Clin Invest. 2011;121:1484–1496. 12. Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol Rev. 2015;268:25–51. 13. Cai Z, et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat Commun. 2015;6:8277. 14. Pincetic A, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014;15:707–716. 15. Gardiner EE, Andrews RK. Structure and function of platelet receptors initiating blood clotting. Adv Exp Med Biol. 2014;844:263–275. 16. Bardina SV, et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science. 2017;356:175–180. 17. Arman M, et al. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood. 2014;123:3166–3174. 18. Boilard E, et al. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood. 2014;123:2854–2863. 19. Duffau P, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med. 2010;2:47ra63. 20. McKenzie SE, et al. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: A transgenic mouse model. J Immunol. 1999;162:4311–4318. 21. Ishii S, et al. Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J Exp Med. 1998;187:1779–1788. 22. Reilly MP, et al. Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcγRIIA. Blood. 2001;98:2442–2447. 23. Jönsson F, et al. Human FcγRIIA induces anaphylactic and allergic reactions. Blood. 2012;119:2533–2544. 24. Meyer T, et al. CD32a antibodies induce thrombocytopenia and type II hypersensitivity reactions in FCGR2A mice. Blood. 2015;126:2230–2238. 25. Stolla M, et al. CalDAG-GEFI deficiency protects mice in a novel model of Fcγ RIIA-mediated thrombosis and thrombocytopenia. Blood. 2011;118:1113–1120. 26. Amirkhosravi A, et al. CalDAG-GEFI deficiency protects mice from FcγRIIa-mediated thrombotic thrombocytopenia induced by CD40L and beta2GPI immune complexes. J Thromb Haemost. 2014;12:2113–2119.

289
27. Arman M, Krauel K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J Thromb Haemost. 2015;13:893–908. 28. Gillis CM, et al. Mechanisms of anaphylaxis in human low-affinity IgG receptor locus knock-in mice. J Allergy Clin Immunol. 2016;139:1253–1265.e14. 29. Bruhns P, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. 30. Zhi H, et al. Cooperative integrin/ITAM signaling in platelets enhances thrombus formation in vitro and in vivo. Blood. 2013;121:1858–1867. 31. Nieswandt B, Varga-Szabo D, Elvers M. Integrins in platelet activation. J Thromb Haemost. 2009;7(Suppl 1):206–209. 32. Ren Q, Ye S, Whiteheart SW. The platelet release reaction: Just when you thought platelet secretion was simple. Curr Opin Hematol. 2008;15:537–541. 33. Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. 34. Flick MJ, et al. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest. 2007;117:3224–3235. 35. Flick MJ, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113:1596–1606. 36. Ehlers R, et al. Targeting platelet-leukocyte interactions: Identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med. 2003;198:1077–1088. 37. Kato K, et al. Genetic deletion of mouse platelet glycoprotein Ibbeta produces a Bernard-Soulier phenotype with increased alpha-granule size. Blood. 2004;104:2339–2344. 38. Shrimpton CN, et al. Localization of the adhesion receptor glycoprotein Ib-IX-V complex to lipid rafts is required for platelet adhesion and activation. J Exp Med. 2002;196:1057–1066. 39. Vanhoutte PM. Regenerated endothelium and its senescent response to aggregating platelets. Circ J. 2016;80:783–790. 40. Adnot S, Houssaini A, Abid S, Marcos E, Amsellem V. Serotonin transporter and serotonin receptors. Handb Exp Pharmacol. 2013;218:365–380.] 41. Cloutier N, et al. Platelets can enhance vascular permeability. Blood. 2012;120:1334–1343. 42. Zhang X, Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science. 2004;305:217. 43. Côté F, et al. Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci USA. 2003;100:13525–13530. 44. Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–15885. 45. Berrington WR, et al. Clinical correlates of herpes simplex virus viremia among hospitalized adults. Clin Infect Dis. 2009;49:1295–1301. 46. Zahariadis G, Jerome KR, Corey L. Herpes simplex virus-associated sepsis in a previously infected immunocompetent adult. Ann Intern Med. 2003;139:153–154. 47. McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. 48. Duerschmied D, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. 2013;121:1008–1015. 49. Binstadt BA, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol. 2006;7:284–292. 50. Strait RT, et al. MHC class I-specific antibody binding to nonhematopoietic cells drives complement activation to induce transfusion-related acute lung injury in mice. J Exp Med. 2011;208:2525–2544. 51. Walther DJ, et al. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell. 2003;115:851–862. 52. Lesurtel M, et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312:104–107. 53. Yeung J, et al. Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood. 2014;124:2271–2279. 54. Dong JF, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039.

290
55. Podoplelova NA, et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. 2016;128:1745–1755. 56. White JG, Krumwiede MD, Cocking-Johnson DJ, Escolar G. Uptake of vWF-anti-vWF complexes by platelets in suspension. Arterioscler Thromb Vasc Biol. 1996;16:868–877. 57. Stalker TJ, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121:1875–1885. 58. Welsh JD, et al. A systems approach to hemostasis: 4. How hemostatic thrombi limit the loss of plasma-borne molecules from the microvasculature. Blood. 2016;127:1598–1605. 59. Michelson AD, et al. In vivo tracking of platelets: Circulating degranulated platelets rapidly lose surface P-selectin but continue to circulate and function. Proc Natl Acad Sci USA. 1996;93:11877–11882. 60. Berger G, Hartwell DW, Wagner DD. P-Selectin and platelet clearance. Blood. 1998;92:4446–4452. 61. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat Immunol. 2010;11:373–384. 62. Picard C, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 2010;89:403–425. 63. Beppler J, et al. Fc gamma receptor IIA (CD32A) R131 polymorphism as a marker of genetic susceptibility to sepsis. Inflammation. 2016;39:518–525. 64. Li Y, et al. Sepsis-induced elevation in plasma serotonin facilitates endothelial hyperpermeability. Sci Rep. 2016;6:22747. 65. Claushuis TA, et al. Molecular Diagnosis and Risk Stratification of Sepsis Consortium Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. 2016;127:3062–3072. 66. Greinacher A, Selleng S. How I evaluate and treat thrombocytopenia in the intensive care unit patient. Blood. 2016;128:3032–3042. 67. Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D. Inflammation and the Host Response to Injury Investigators Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol. 2005;12:60–67. 68. Willoughby S, Holmes A, Loscalzo J. Platelets and cardiovascular disease. Eur J Cardiovasc Nurs. 2002;1:273–288. 69. Fateh-Moghadam S, et al. Platelet degranulation is associated with progression of intima-media thickness of the common carotid artery in patients with diabetes mellitus type 2. Arterioscler Thromb Vasc Biol. 2005;25:1299–1303. 70. Janes SL, Goodall AH. Flow cytometric detection of circulating activated platelets and platelet hyper-responsiveness in pre-eclampsia and pregnancy. Clin Sci (Lond) 1994;86:731–739. 71. Protti A, et al. Platelet mitochondrial dysfunction in critically ill patients: Comparison between sepsis and cardiogenic shock. Crit Care. 2015;19:39. 72. Zeller J, Weissbarth E, Baruth B, Mielke H, Deicher H. Serotonin content of platelets in inflammatory rheumatic diseases. Correlation with clinical activity. Arthritis Rheum. 1983;26:532–540. 73. Lood C, et al. Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLoS One. 2015;10:e0125109. 74. Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. 2014;10:338–347. 75. Zhang J, et al. CD41-YFP mice allow in vivo labeling of megakaryocytic cells and reveal a subset of platelets hyperreactive to thrombin stimulation. Exp Hematol. 2007;35:490–499. 76. Flick MJ, et al. The development of inflammatory joint disease is attenuated in mice expressing the anticoagulant prothrombin mutant W215A/E217A. Blood. 2011;117:6326–6337. 77. Robles-Carrillo L, et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol. 2010;185:1577–1583.

291
Annexe IV: Autoantibodies in Systemic Lupus
Erythematosus Target Mitochondrial RNA
Yann Becker 1, Geneviève Marcoux1, Isabelle Allaeys1 , Anne-Sophie
Julien2, Renée-Claude Loignon 3, Hadrien Benk-Fortin 1, Emmanuelle Rollet-
Labelle 1, Joyce Rauch 4, Paul R Fortin1,3,5 , Eric Boilard 1 , 5
Affiliations
1 Département de microbiologie et immunologie, Faculté de Médecine de l'Université Laval, Centre de Recherche du CHU de Québec-Université Laval, Québec City, QC, Canada.
2 Département de mathématiques et statistiques, Université Laval, Québec City, QC, Canada.
3 Division de Rhumatologie, Département de Médecine, CHU de Québec-Université Laval, Québec City, QC, Canada.
4 Division of Rheumatology, Department of Medicine, Research Institute of the McGill University Health Centre, Montreal, QC, Canada.
5 Axe maladies infectieuses et inflammatoires, Centre de Recherche du CHU de Québec-Université Laval, Québec City, QC, Canada.

292
Abstract
The mitochondrion supplies energy to the cell and regulates apoptosis. Unlike other
mammalian organelles, mitochondria are formed by binary fission and cannot be
directly produced by the cell. They contain numerous copies of a compact circular
genome that encodes RNA molecules and proteins involved in mitochondrial
oxidative phosphorylation. Whereas, mitochondrial DNA (mtDNA) activates the
innate immune system if present in the cytosol or the extracellular milieu, it is also
the target of circulating autoantibodies in systemic lupus erythematosus (SLE).
However, it is not known whether mitochondrial RNA is also recognized by
autoantibodies in SLE. In the present study, we evaluated the presence of
autoantibodies targeting mitochondrial RNA (AmtRNA) in SLE. We quantified
AmtRNA in an inducible model of murine SLE. The AmtRNA were also determined
in SLE patients and healthy volunteers. AmtRNA titers were measured in both our
induced model of murine SLE and in human SLE, and biostatistical analyses were
performed to determine whether the presence and/or levels of AmtRNA were
associated with clinical features expressed by SLE patients. Both IgG and IgM
classes of AmtRNA were increased in SLE patients (n = 86) compared to healthy
controls (n = 30) (p < 0.0001 and p = 0.0493, respectively). AmtRNA IgG levels
correlated with anti-mtDNA-IgG titers (r s = 0.54, p < 0.0001) as well as with both
IgG and IgM against β-2-glycoprotein I (anti-β2GPI; r s = 0.22, p = 0.05), and
AmtRNA-IgG antibodies were present at higher levels when patients were positive
for autoantibodies to double-stranded-genomic DNA (p < 0.0001). AmtRNA-IgG
were able to specifically discriminate SLE patients from healthy controls, and were
negatively associated with plaque formation (p = 0.04) and lupus nephritis (p = 0.03).
Conversely, AmtRNA-IgM titers correlated with those of anti-β2GPI-IgM (r s = 0.48,
p < 0.0001). AmtRNA-IgM were higher when patients were positive for anticardiolipin
antibodies (aCL-IgG: p = 0.01; aCL-IgM: p = 0.002), but AmtRNA-IgM were not
associated with any of the clinical manifestations assessed. These findings identify
mtRNA as a novel mitochondrial antigen target in SLE, and support the concept that
mitochondria may provide an important source of circulating autoantigens in SLE.

293
Introduction
The mitochondrion is an intracellular organelle involved in the regulation of
numerous cellular functions, among which the best known are ATP production and
programmed cell death (1, 2). Mitochondria are considered as deriving from the
endosymbiosis of an α-synfular; proteobacterium (3, 4), providing the organelles
many bacterial features (3, 5–9).
Different cellular lineages (10–18) may extrude their mitochondria upon activation.
Extracellular mitochondria have been identified in damaged tissues (8, 18–20);
diverse inflammatory conditions (11, 12, 14, 21–24); and in the blood of critical care
patients (22). As mitochondria retained several characteristics of their ancestral
prokaryotic origin, the release of mitochondrial components onto the extracellular
milieu can activate the innate immune system (25, 26). The efflux of mtDNA is
facilitated by megapores formed in the mitochondrial membrane during apoptosis,
and detected by the cytosolic DNA sensors cGAS and stimulator of interferon genes
(STING) pathway, thereby leading to type I interferon synthesis (27). Cardiolipin, N-
formylated peptides, mtDNA, ATP and reactive oxygen species are known
mitochondrial damage-associated molecular patterns (9, 28–30). They further
activate cells through nuclear oligomerization domain-like receptors (28, 29, 31), toll-
like receptors (TLR) (e.g., TLR9 for mtDNA), or formyl peptide receptors (9, 28–31).
Systemic lupus erythematosus is an autoimmune disease characterized by the
presence of circulating immune complexes and inflammation in multiple organs and
tissues. Recent evidence point to an involvement of mtDNA, liberated by neutrophils,
in the activation of STING and type-I IFN production in SLE (11, 12). Moreover,
extracellular mtDNA can enhance leukocyte migration and degranulation (32), and
promotes the secretion of the pro-inflammatory cytokine TNF-α by plasmacytoid
dendritic cells (33). Production of autoantibodies targeting several mitochondrial
components was reported in SLE as well as in other diseases [e.g., primary biliary
cirrhosis (PBC), antiphospholipid syndrome (APS), and cardiomyopathies] (Figure

294
1). Anti-mitochondrial autoantibodies recognize proteins, such as those involved in
oxidative phosphorylation, phospholipids or unidentified epitopes present in the
mitochondrial membrane. Despite the extensive literature regarding antibodies
targeting the cardiolipin (also known as the mitochondrial antigen M1) in SLE, the
anti-mitochondrial autoantibody repertoire and their antigenic targets remains mostly
uncharacterized (12, 34–37). Using intact mitochondria and mtDNA as antigens to
screen autoantibodies in SLE patients, we have shown that different sets of
autoantibodies also target the mitochondrial outer membrane and mtDNA (36).
Given the accumulating evidence for mitochondrial release during inflammatory
pathogenesis, these observations point to a role for mitochondria both in the
stimulation of the innate immune system and as a potential source of autoantigens.

295
Figure 1 Anti-mitochondrial antibodies and related diseases. Several types of anti-mitochondrial antibodies (AMA) have been reported in various diseases. The epitopes targeted by AMA cover all families of biomolecules: lipids (yellow background), proteins (red hues) or nucleic acids (blue hues). However, the precise nature of some mitochondrial epitopes targeted by AMA are still unclear. (gray hues). To date, the sole mitochondrion-specific phospholipid antigen reported in both APS and SLE is cardiolipin (M1). M1 is located within the mitochondrial inner membrane (MIM) in healthy organelles, but may be displayed on the outer membrane (MOM) upon damages to the organelle. Distinct AMA against an unknown antigen (M5) were also reported in both APS and SLE. Four antigens are associated with PBC; PDC-E2 (M2, MIM), sulfite oxidase (M4, MOM), M8 (MOM), and glycogen phosphorylase (M9, MOM). These mitochondrial antigens are peptidic, with the exception of M8, whose nature remains uncharacterized. Sarcosine dehydrogenase (M7) is another immunogenic protein that is targeted by autoantibodies in patients suffering from cardiac conditions (i.e., hypertrophic or idiopathic cardiomyopathies or acute myocarditis). Two types of AMA were reported as iatrogenically induced in human patients: AMA-M3 (unknown, MOM) and AMA-M6 (monoamine oxidase B, MOM). In addition to these autoantibodies, we have reported the presence of autoantibodies targeting whole mitochondria (AwMA) in patients with SLE, APS, and PBC (with higher titers found in SLE donors). Moreover, antibodies specific to the mtDNA were specific to SLE patients. In the present study, we describe autoantibodies against mtRNA in patients with SLE and APS.
Whereas, the mitochondrion has already been described as a source of mtDNA
during inflammation (17, 21, 32), it is not known whether its important RNA content
(mtRNA) can contribute to the autoantigenic load in SLE. Despite its presence at
high copy numbers, the mitochondrial genome is very compact (38–40) During its
translation into mitochondrial messenger RNA (38), a long polycistronic transcript is
generated from each strand of mtDNA prior to undergoing processing into mtRNA
molecules. This highly regulated process is thought to occur in a particular location
in the mitochondrion, called mitochondrial RNA granules (41), and requires key RNA
processing enzymes such as the members of the FASTK family of proteins (42). The
human mitochondrial transcriptome comprises 16S ribosomal RNA molecules
(78%), transfer (13%), messenger (8%) and small non-coding antisense (1%)
mtRNA molecules. The complete mitochondria transcriptome is controlled by the
cell's energy requirements, and therefore varies greatly depending on its tissue
distribution. In the heart, 30% of the total messenger RNA molecules are of
mitochondrial origin, whereas ~5% of the total messenger RNA load in less
metabolically active cells such as leukocytes is encoded by mitochondrial genes

296
(39). The important quantity of mtRNA may thus represent a major antigenic load for
the adaptive immune system upon release of mitochondria onto the extracellular
milieu.
With the accumulating evidence supporting the liberation of mitochondrial
components into the extracellular milieu in SLE (11, 12), it is crucial to identify the
various mitochondrial antigens. In the present study, we examined whether the RNA
molecules present in mitochondria are antigenic. The levels of anti-mtRNA
(AmtRNA) were measured in SLE sera, and we determined whether AmtRNA were
associated with antibodies against whole mitochondrial organelles (AwMA) and
mtDNA (AmtDNA). We also investigated the occurrence of AmtRNA in an induced
model of murine SLE. Finally, we determined whether AmtRNA were associated with
disease manifestations in patients with SLE.

297
Materials and Methods
Induced Model of Murine SLE
This study was carried out in accordance with the recommendations of the Canadian
Council on Animal Care. The protocol was approved by McGill University Animal
Care Committee. C57BL/6 mice were obtained from Harlan Sprague Dawley Inc.
(Indianapolis, IN, USA) and housed in a specific-pathogen-free animal facility at the
animal facility of the Research Institute of the McGill University Health Center.
Female (10–12-weeks-old) mice were injected intravenously (i.v.) with 100 μL
human β2-GPI (20 μg) (Crystal Chem Inc., Elk Grove Village, IL, USA), followed 24
h later by a 100 μL i.v. injection of lipopolysaccharide (LPS from E.coli, serotype
O111:B4; 10 μg) (List Biological Laboratories, Campbell, CA, USA). β2-GPI and LPS
injections were repeated every 2 weeks for a total of three rounds of immunizations,
and then at 2-month intervals for the fourth and the fifth immunizations. C57BL/6
mice injected i.v. with PBS and LPS following the same schedule were used as
controls. Mice were bled 1 week after the fifth immunization and serum was kept
frozen at −70°C until testing.
Mitochondria Isolation
Mitochondria were isolated from the livers of C57BL/6 mice as previously described
(43). In brief, cells and tissues were disrupted by grinding in a glass/Teflon tissue
potter containing 12 mL ice-cold mitochondrial isolation buffer (10 mM Tris, 1 mM
EGTA, 200 mM sucrose) for each gram of liver. Debris were pelleted twice at 700 g,
for 10 min at 4°C and the supernatants were transferred to fresh tubes. Mitochondria
were further separated from other cellular fractions by three centrifugation steps
(twice at 7,000 g and once at 10,000 g, for 10 min at 4°C). Between each step,
pelleted mitochondria were re-suspended in 12 mL isolation buffer. Samples were
kept at −80°C until required for RNA isolation.
Mitochondrial RNA Isolation
Mitochondrial RNA was isolated using the Aurum™ Total RNA Mini Kit (Bio-Rad,
Hercules, CA, USA), following the manufacturer's instructions. Ribonucleic acid

298
yields were quantified using a BioDrop μLITE and its proprietary software (BioDrop
Ltd., Cambridge, UK). The absence of contamination by mitochondrial DNA was
assessed by resolution of 1 μg untreated mtRNA and the same amount of RNAse
A-treated (QIAgen, 100 μg/mL) mtRNA on a 1.5% (w/v) agarose gel (Supplementary
Figure 1A). 15.09 ± 2.74 μg mtRNA were isolated for each mg of bicinchoninic acid
assay (BCA)-dosed mitochondria used (n = 3).
Enzyme-Linked Immunoassays for the Detection of Antibodies Targeting
Mitochondrial Antigens
Clear 96-well High Bind half-area flat bottom ELISA microplates (Corning, New York,
USA) were pre-coated with 100 μL per well of 1% protamine sulfate (Sigma-Aldrich)
in double-distilled water for 1 h at RT. Plates were then washed thrice with PBS and
loaded with mtRNA. Plates were coated overnight at 4°C, washed thrice and non-
specific binding was blocked for 4 h at 37°C with 100 μL per well of ELISA blocking
buffer (PBS−10% FBS−0.5% gelatin). Wells were rinsed three times with PBS and
incubated in duplicate with serum diluted (1:150 for human and 1:50 for mice) in
incubation buffer (PBS−10% FCS−0.3% gelatin). Plates were washed thrice with
PBS and incubated for 90 min at RT with either γ or μ chain-specific-alkaline
phosphatase-(AP) conjugated goat anti-human IgG or IgM (Sigma-Aldrich) for
human serum, or γ chain-specific-horseradish peroxidase (HRP)-conjugated goat
anti-mouse IgG (Sigma-Aldrich) for mice (1:1000) in secondary antibody buffer
(PBS−0.4% bovine serum albumin [BSA]). Unbound antibodies were washed thrice
with PBS. Signals from AP-conjugated antibodies were developed with para-
nitrophenol phosphate (p-NPP) for ~30 min at 37°C, and HRP-conjugated antibodies
were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) at RT. The reaction was
stopped with 2 N sulfuric acid (H2SO4). Optical densities (OD) were measured at
405 nm (p-NPP) or 450 nm (HRP) on a SpectraMax 190 microplate reader
(Molecular Devices, Sunnyvale, CA, USA), using SoftMax Pro 5.4.1 (Molecular
Devices). For each experiment, blank values (i.e., wells coated with mtRNA, but
without sera) were subtracted from each measurement.

299
The quantity of purified mitochondrial RNA (mtRNA) required for coating half-area
flat-bottom 96-well ELISA microplates (Corning, New York, USA) was optimized
following the aforementioned protocol, by using increasing concentrations from 0 to
1,600 ng of coating mtRNA. Pooled sera (1:150) from 6 SLE patients, who had
previously tested positive for AmtDNA and AwMA, were incubated after blocking
non-specific binding. The peak signal for optical densities at 405 nm was obtained
with 200 ng of coating mtRNA (Supplementary Figure 1B).
Ethics and Study Approval
This study was carried out in accordance with the recommendations of the Research
Ethics Board of the CHU de Québec—Université Laval with written informed consent
from all subjects. All subjects gave written informed consent in accordance with the
Declaration of Helsinki. The protocol was approved by Research Ethics Board of the
CHU de Québec—Université Laval.
Human Serum Samples
The human sera tested in this study were obtained from the Systemic Autoimmune
Rheumatic Disease (SARD) biobank and data repository (SARD-BDB) located at the
CHU de Québec-Université Laval (UL). This SARD-BDB and the specific use of the
sera for the present study were approved by the CHU de Québec-UL research ethics
board (#B13-06-1243 and #B14-08-2108, respectively). Patients with SLE met the
1982 ACR classification criteria for SLE (revised in 1997) (44, 45). A peripheral blood
sample was collected at the time of their first visit. Serum samples from 30 healthy
donors and 87 SLE patients included in the SARD-DBD cohort were used in the
present study. However, one patient had no clinical data available and was therefore
excluded for bio-statistical comparisons (i.e., n = 86 SLE donors for these tests).
Additional Serum Samples
Sera from a cohort of patients and controls from the University of Toronto Lupus
Clinic, as well as patients with primary biliary cirrhosis (PBC) from Quebec City, were
used in additional exploratory analyses to test the presence of AmtRNA in patients

300
with the antiphospholipid syndrome (APS, n = 12) and PBC (n = 12). APS patients
and healthy controls, distinct from those included in the SARD-BDB (n = 43), were
originally recruited between August 2010 and October 2011, and gave consent to
allow remaining biospecimens to be used for future studies on lupus biomarkers.
This study has been reviewed and approved by the Research Ethics Board of the
University Health Network (#10-0637-BE) and of the CHU de Québec—Université
Laval (#B14-08-2108). APS patients met 1999 Sapporo criteria for the disease
(revised in 2006) (46, 47), and healthy controls were recruited if they had no known
illnesses and had no infectious symptoms at the time of the blood draw. Donors gave
a single blood sample that was linked to their anonymized clinical data. PBC patients
were positive for anti-mitochondrial antibodies and presented clinical criteria for the
disease (47, 48).
Clinical Variables Collected in SLE Patients
Sociodemographic Variables
Information was collected concerning patient's age, gender, marital status, and
ethnicity at the first visit in the SARD-BDB.
Patient Characteristics Including Exposures to Cardiovascular Risk Factors
A body mass index (BMI) was calculated and reported as underweight, normal,
overweight and obese. Hypertension and diabetes mellitus were documented as
present or absent. Smoking history was reported as non-smokers, ex-smokers or
current smokers. Female patients were considered post-menopausal in the absence
of menstruations for more than 12 continuous months.
Disease Specific Characteristics
ACR classification criteria (44, 45) were documented for each of the 11 categories
and a total score calculated (5 ± 1.28). Disease duration, lupus disease activity using
the SLE Disease Activity Index–2000 (SLEDAI-2K) (49, 50) and lupus damage using
the Systemic Lupus International Collaborating Clinics (SLICC)/ACR damage index
(SDI) (51, 52) were collected during the clinical visit matched to the blood specimen

301
draw. Both the SLEDAI-2K and the SDI are reported as continuous variables and
they both have proven validity, reliability, and perform well in observational studies.
Medication Variables
Antimalarial use was defined as use of hydroxychloroquine or chloroquine at the
current visit. Steroid use was defined as prednisone use in the past year.
Clinical Outcomes
Clinically relevant lupus disease activity and damage were used as clinical outcome
in our analyses and were defined as a SLEDAI-2K of 4 or more to capture clinically
active lupus and a SDI of 1 or more to capture clinically significant damage. Other
outcome variables included arterial and venous thrombotic event ever in the past
and presence of lupus nephritis according to the presence or absence of the renal
item of the SLICC Classification criteria for SLE (53). Presence or absence of carotid
plaques, as well as average carotid-intima media thickness (CIMT) was also
documented by carotid ultrasound following a standard examination of both carotids
(standard carotid ultrasound research protocol using an Esaote MyLab Five
ultrasound machine with digital images sent for blind reading at the IMT Core
Laboratory of the Montreal Heart Institute).
Information From Clinical Laboratories
For SLE patients, an automated complete blood count was documented. The anti-
dsDNA, anticardiolipin antibodies (aCL) (IgG and IgM—laboratory cut-offs of 40 GPL
or MPL units) and anti-β2-GPI (IgG and IgM—laboratory cut-offs above the 99th
percentile of controls) were measured by ELISA. The lupus anticoagulant assay (LA)
followed international guidelines for the performance of this functional assay (54).
The above tests were performed in a clinical laboratory at CHU de Quebec-
Universite Laval as part of routine care.
Information From Research Laboratories

302
In addition to the measurements provided by the clinical laboratories, our research
laboratory performed antibody assays to detect AwMA and AmtDNA, following
previously described methods (36).
Statistical Analyses
Descriptive statistics are presented as mean with standard deviation or frequency
with percentage without missing values for continuous and categorical variables,
respectively. Comparisons between groups were performed using the Student's,
Wilcoxon or Kruskal-Wallis tests depending on the nature of the variables and their
distribution. Spearman correlations were calculated to assess association between
continuous variables. Associations between AmtRNA and clinical outcomes were
studied by bivariate and multivariate logistic regressions, for dichotomous and
continuous outcomes, respectively. The latter were adjusted for gender, disease
duration, age, BMI, antimalarial medication and prednisone use. ROC curves were
generated to assess the predictive ability of AmtRNA to discriminate between SLE
and controls, and their area under the curve (AUC) was calculated. Participants'
results were considered positive for AmtRNA when their value was above the cut-off
value identified after maximizing Youden's Index. A 95% confidence interval was
obtained for the cut-off using 10,000 bootstrap samples. Performance measures are
presented with their 95% exact confidence interval. Statistical analyses were
performed with Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA) and
SAS version 9.4 (SAS Institute Inc., Cary, NC, USA) and Figures were assembled
with Photoshop CS6 13.0 (Adobe Systems Inc., Mountain View, CA, USA).
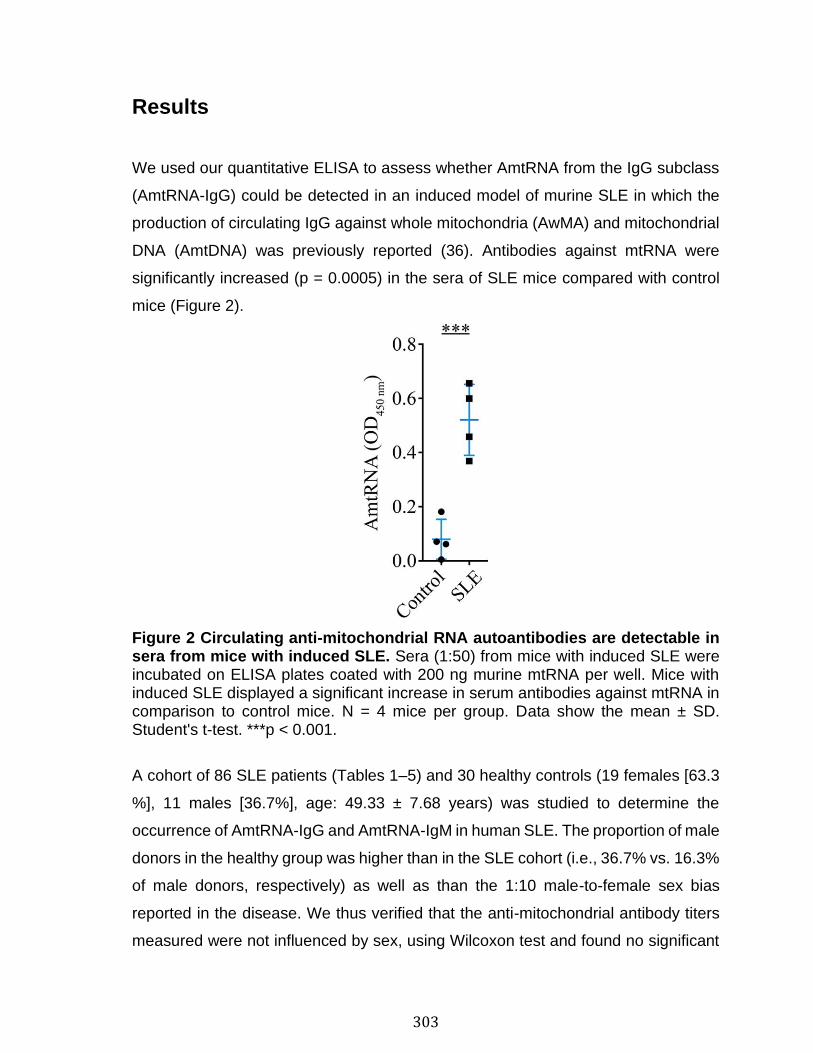
303
Results
We used our quantitative ELISA to assess whether AmtRNA from the IgG subclass
(AmtRNA-IgG) could be detected in an induced model of murine SLE in which the
production of circulating IgG against whole mitochondria (AwMA) and mitochondrial
DNA (AmtDNA) was previously reported (36). Antibodies against mtRNA were
significantly increased (p = 0.0005) in the sera of SLE mice compared with control
mice (Figure 2).
Figure 2 Circulating anti-mitochondrial RNA autoantibodies are detectable in sera from mice with induced SLE. Sera (1:50) from mice with induced SLE were incubated on ELISA plates coated with 200 ng murine mtRNA per well. Mice with induced SLE displayed a significant increase in serum antibodies against mtRNA in comparison to control mice. N = 4 mice per group. Data show the mean ± SD. Student's t-test. ***p < 0.001.
A cohort of 86 SLE patients (Tables 1–5) and 30 healthy controls (19 females [63.3
%], 11 males [36.7%], age: 49.33 ± 7.68 years) was studied to determine the
occurrence of AmtRNA-IgG and AmtRNA-IgM in human SLE. The proportion of male
donors in the healthy group was higher than in the SLE cohort (i.e., 36.7% vs. 16.3%
of male donors, respectively) as well as than the 1:10 male-to-female sex bias
reported in the disease. We thus verified that the anti-mitochondrial antibody titers
measured were not influenced by sex, using Wilcoxon test and found no significant

304
differences (p-values between 0.14 and 0.97). Both AmtRNA-IgG and -IgM were
significantly increased in SLE patients, compared with healthy individuals (p =
0.0002 and p = 0.0493, respectively) (Figure 3; Supplementary Figure 1). In healthy
donors, AmtRNA-IgM were higher than AmtRNA-IgG levels (0.32 ± 0.24 vs. 0.16 ±
0.12), suggesting that antibodies targeting mitochondrial epitopes may be present in
healthy individuals even in the absence of any detectable pathology.
Table 1: Sociodemographic characteristics in the SARD-BDB.
Variable n Mean ± SD [or n (%)] Female 86 72 (83.7) Age (years) 86 49.41 ± 14.60 Marital status 82 Single 14 (17.1) Married 55 (67.1) Tobacco intake 83 Non-smokers 48 (57.8) Smokers 14 (16.9) Ex-smokers 21 (25.3)

305
Table 5: Laboratory measurements. Variable n Mean ± SD [or n (%)] Platelets (0.10∧9/L) 86 221.63 ± 72.68 White blood cells (0.10∧9/L) 86 5.80 ± 2.06 Creatinine clearance 26 91.88 ± 21.31 AmtDNA (OD 405 nm) IgG 86 0.49 ± 0.53 IgM 86 0.45 ± 0.38 AwMA (OD 405 nm) IgG 86 0.34 ± 0.37 IgM 86 0.56 ± 0.58 AmtRNA (OD 405 nm) IgG 86 0.42 ± 0.38 IgM 86 0.52 ± 0.47 Lupus anticoagulant (LA) 61 8.69 ± 22.76 Anticardiolipin antibodies (aCL) IgG 79 11.33 ± 12.39 IgM 79 6.92 ± 13.92 Anti-β2GPI antibodies IgG 79 2.78 ± 6.59 IgM 79 3.36 ± 3.89 Anti-dsDNA antibodies 22 31.01 ± 80.40
β2GPI, β-2-glycoprotein I; AmtDNA, anti-mitochondrial DNA antibodies; AmtRNA, anti-mitochondrial RNA antibodies; AwMA, anti-whole mitochondria antibodies; OD, optical density.
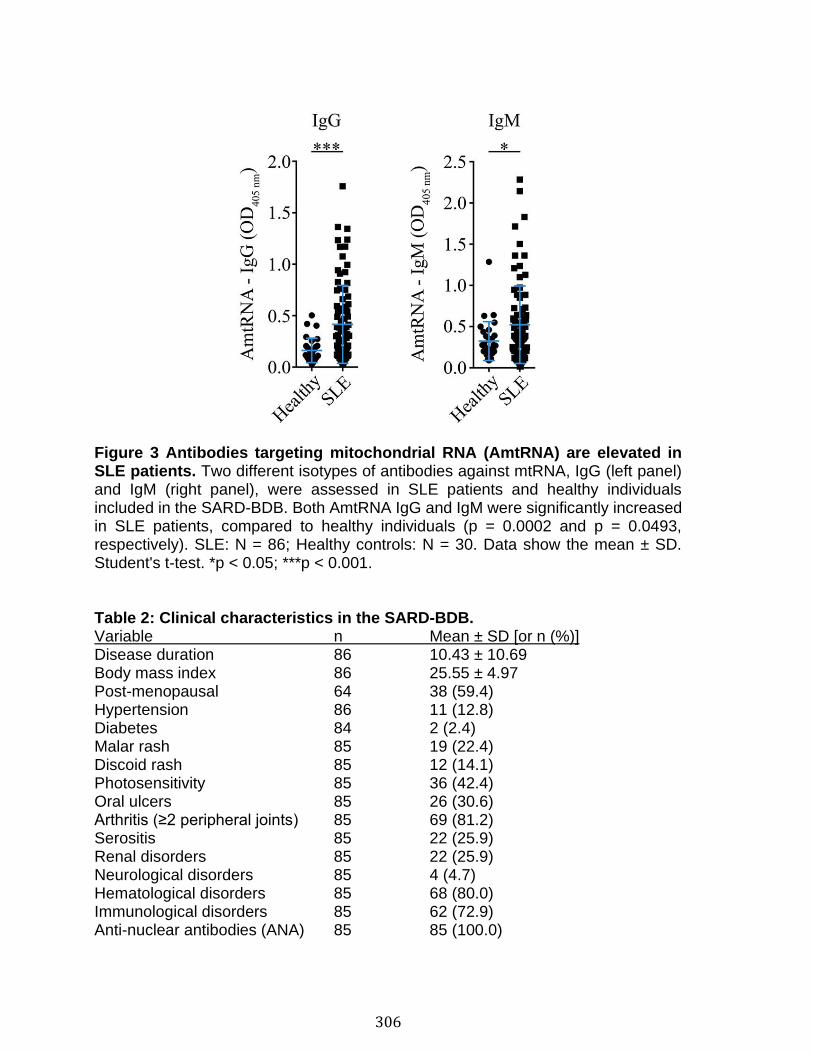
306
Figure 3 Antibodies targeting mitochondrial RNA (AmtRNA) are elevated in SLE patients. Two different isotypes of antibodies against mtRNA, IgG (left panel) and IgM (right panel), were assessed in SLE patients and healthy individuals included in the SARD-BDB. Both AmtRNA IgG and IgM were significantly increased in SLE patients, compared to healthy individuals (p = 0.0002 and p = 0.0493, respectively). SLE: N = 86; Healthy controls: N = 30. Data show the mean ± SD. Student's t-test. *p < 0.05; ***p < 0.001. Table 2: Clinical characteristics in the SARD-BDB. Variable n Mean ± SD [or n (%)] Disease duration 86 10.43 ± 10.69 Body mass index 86 25.55 ± 4.97 Post-menopausal 64 38 (59.4) Hypertension 86 11 (12.8) Diabetes 84 2 (2.4) Malar rash 85 19 (22.4) Discoid rash 85 12 (14.1) Photosensitivity 85 36 (42.4) Oral ulcers 85 26 (30.6) Arthritis (≥2 peripheral joints) 85 69 (81.2) Serositis 85 22 (25.9) Renal disorders 85 22 (25.9) Neurological disorders 85 4 (4.7) Hematological disorders 85 68 (80.0) Immunological disorders 85 62 (72.9) Anti-nuclear antibodies (ANA) 85 85 (100.0)
307
Table 3: Outcome variables of the study.
Variable n Mean ± SD [or n (%)] SLEDAI-2K (Score) 3.24 ± 3.96 SLEDAI-2K ≥ 4 86 36 (41.9) SDI (score) 3.24 ± 3.96 SDI ≥ 0 86 36 (41.9) Thrombosis 10 (11.6) Arterial events 86 3 (3.5) Venous events 4 (4.7) Presence of plaque in the carotid 63 24 (38.1) Carotid intima-media thickness (CIMT, μm) 34 0.63 ± 0.13 Nephritis 61 14 (23.0)
SDI, lupus severity disease index; SLEDAI-2K, systemic lupus erythematosus disease activity index−2000.
Table 4: Information about medications taken by SLE patients (n = 86) in the SARD-BDB. Variable n(%) Anticoagulation/anti-platelets 13 (15.1) Antimalarial 70 (81.4) Prednisone 18 (20.9) Lipid lowering 14 (16.3) Diabetes medication 2 (2.3) LUPUS TREATMENTS Hydroxychloroquine 65 (76) Chloroquine 6 (7) Azathioprine 15 (17) Methotrexate 15 (17) Leflunomide 1 (1) Mycophenolate mofetil 11 (13) Mycophenolic acid 1 (1) Cyclophosphamide (PO or IV) 3 (4)
IV: intravenous injection; PO: per os.
In a separate exploratory analysis using donors distinct from those included in the
SARD-BDB, AmtRNA-IgG were also significantly increased in patients with APS, an
autoimmune condition often associated with SLE (p < 0.001 vs. healthy controls).
However, no differences in AmtRNA-IgG were observed between patients with PBC,
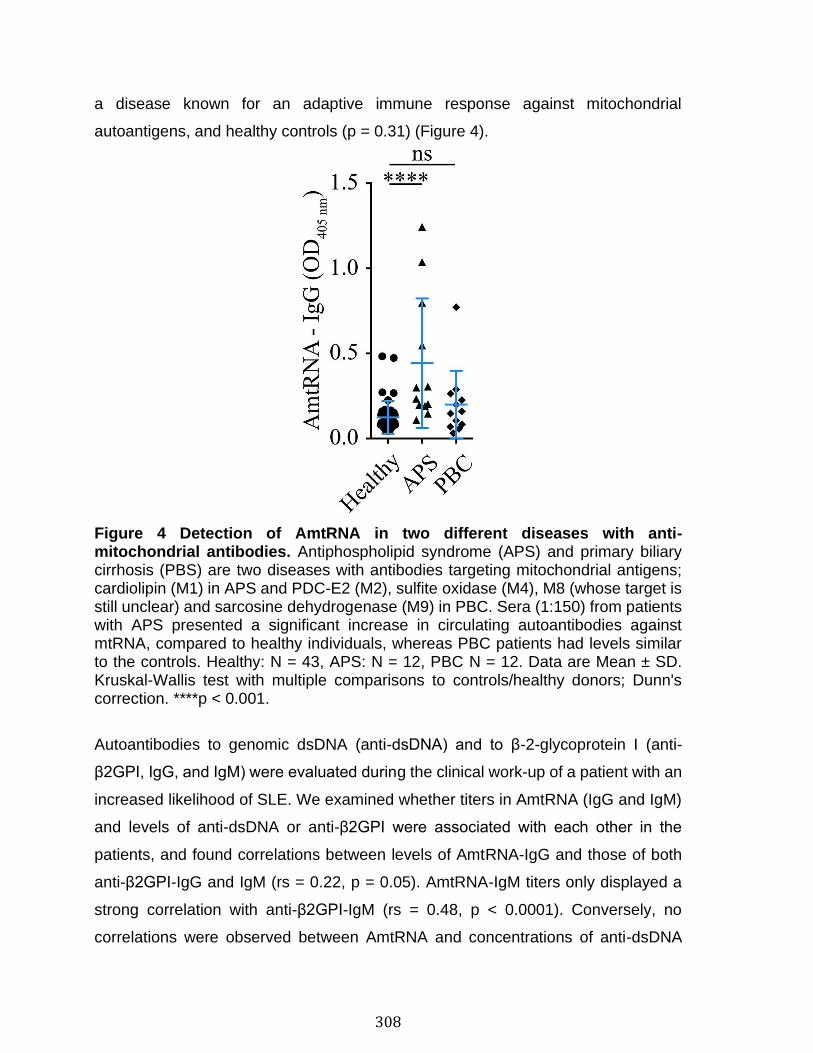
308
a disease known for an adaptive immune response against mitochondrial
autoantigens, and healthy controls (p = 0.31) (Figure 4).
Figure 4 Detection of AmtRNA in two different diseases with anti-mitochondrial antibodies. Antiphospholipid syndrome (APS) and primary biliary cirrhosis (PBS) are two diseases with antibodies targeting mitochondrial antigens; cardiolipin (M1) in APS and PDC-E2 (M2), sulfite oxidase (M4), M8 (whose target is still unclear) and sarcosine dehydrogenase (M9) in PBC. Sera (1:150) from patients with APS presented a significant increase in circulating autoantibodies against mtRNA, compared to healthy individuals, whereas PBC patients had levels similar to the controls. Healthy: N = 43, APS: N = 12, PBC N = 12. Data are Mean ± SD. Kruskal-Wallis test with multiple comparisons to controls/healthy donors; Dunn's correction. ****p < 0.001.
Autoantibodies to genomic dsDNA (anti-dsDNA) and to β-2-glycoprotein I (anti-
β2GPI, IgG, and IgM) were evaluated during the clinical work-up of a patient with an
increased likelihood of SLE. We examined whether titers in AmtRNA (IgG and IgM)
and levels of anti-dsDNA or anti-β2GPI were associated with each other in the
patients, and found correlations between levels of AmtRNA-IgG and those of both
anti-β2GPI-IgG and IgM (rs = 0.22, p = 0.05). AmtRNA-IgM titers only displayed a
strong correlation with anti-β2GPI-IgM (rs = 0.48, p < 0.0001). Conversely, no
correlations were observed between AmtRNA and concentrations of anti-dsDNA

309
(Table 6). We also determined whether the levels of AmtRNA correlated with IgG
and IgM antibodies targeting mitochondrial epitopes localized in diverse sub-
compartments of the organelle (36). Specifically, we measured antibodies
recognizing intact whole mitochondria (AwMA), which most likely bind epitopes
found on the outer mitochondrial membrane; aCL, which target cardiolipin, a
phospholipid located mainly within the mitochondrial inner membrane; and AmtDNA,
which recognize mitochondrial DNA. We found that AmtRNA-IgG levels correlated
with AmtDNA-IgG (rs = 0.54, p < 0.0001) and with AwMA-IgG (rs = 0.24, p = 0.03),
but not with aCL (IgG and IgM). AmtRNA-IgM concentrations correlated with
AmtDNA-IgM (rs = 0.83, p < 0.0001), AwMA-IgM (rs = 0.71, p < 0.0001), aCL-IgG
(rs = 0.27, p = 0.02), and aCL-IgM (rs = 0.57, p < 0.0001). Thus, in addition to the
newly described AmtRNA, different sets of anti-mitochondrial antibodies occur
conjointly in SLE.
Table 6: Correlations of anti-mtRNA levels, with titers of other auto-antibodies in SLE patients. AmtRNA IgG IgM AmtDNA IgG rs = 0.54 rs = 0.19 p < 0.0001 p = 0.08 IgM rs = −0.01 rs = 0.83 p = 0.92 p < 0.0001 AwMA IgG rs = 0.24 rs = 0.14 p = 0.03 p = 0.21 IgM rs = −0.03 rs = 0.71 p = 0.78 p < 0.0001 AmtRNA IgG rs = 0.16
p = 0.15 IgM rs = 0.16
p = 0.15 Anti-β2GPI antibodies IgG rs = 0.22 rs = 0.18 p = 0.05 p = 0.11 IgM rs = 0.22 rs = 0.48 p = 0.05 p < 0.0001 Anti-dsDNA antibodies rs = 0.13 rs = 0.11 p = 0.56 p = 0.62 Values are presented as Spearman correlation coefficient (rs) and p-value.

310
β2GPI, β-2-glycoprotein I; aCL, anti-cardiolipin antibodies; AwMA, anti-whole mitochondria antibodies. AmtDNA, anti-mitochondrial DNA antibodies; Anti-dsDNA, antibodies against double-stranded DNA. Data in bold are statistically significant (p < 0.05).
One of the main features of SLE is the expression of numerous autoantibodies in
patients (55), some of which are known to be associated with the clinical expression
of the disease (56). We assessed whether AmtRNA are qualitatively associated with
positivity to several autoantibodies commonly found in SLE, including anti-dsDNA,
aCL, and LA. AmtRNA-IgG levels were higher in presence of anti-dsDNA antibodies
(p < 0.0001), whereas AmtRNA-IgM titers were elevated in presence of aCL-IgG and
-IgM (p = 0.01 and p = 0.002, respectively) (Table 7). Of note, circulating AmtRNA-
IgM tended (p = 0.06) to be increased in the presence of LA in SLE patients.
Table 7: Association of AmtRNA with clinically relevant SLE autoantibodies. AmtRNA IgG IgM aCL IgG (–) 0.26 ± 0.43 (–) 0.34 ± 0.40 (+) 0.42 ± 0.91 (+) 0.56 ± 0.90 p = 0.14 p = 0.01 IgM (–) 0.26 ± 0.43 (–) 0.33 ± 0.38 (+) 0.53 ± 0.95 (+) 0.86 ± 1.43 p = 0.19 p = 0.002 Lupus anticoagulant (–) 0.25 ± 0.45 (–) 0.35 ± 0.40 (+) 0.40 ± 0.67 (+) 0.57 ± 0.98 p = 0.20 p = 0.06 Anti-dsDNA antibodies (–) 0.19 ± 0.28 (–) 0.34 ± 0.41 (+) 0.70 ± 0.71 (+) 0.55 ± 0.27 p < 0.0001 p = 0.10 Values presented as median ± IQR and Wilcoxon test p-value for patient positives (+) or negatives (–) for each variable. aCL, anti-cardiolipin antibodies; AmtRNA, anti-mitochondrial RNA antibodies; Anti-dsDNA, antibodies against double-stranded DNA. Data in bold are statistically significant (p < 0.05).
We examined whether AmtRNA were associated with disease manifestations in 86
SLE patients for whom detailed clinical information were available (Table 8). Higher
levels of AmtRNA-IgG were associated with a lower occurrence of plaque in the

311
carotid using a bivariate analysis [OR(95% CI) = 0.14 (0.02–0.91); p = 0.04], but this
significance was lost in the multivariate logistic regression [OR(95% CI) = 0.16
(0.01–1.81); p = 0.14]. We found no association between AmtRNA-IgG and two
clinical indices; one measuring SLE disease activity (SLEDAI-2K ≥ 4) and the other
indicating damages (SDI > 0), both by bi- and multivariate analyses. However, higher
concentrations of AmtRNA-IgG were positively associated with elevated anti-dsDNA
antibodies in both models. AmtRNA-IgG were not associated with lupus nephritis in
a bivariate analysis [OR(95% CI) = 0.17 (0.02–1.71); p = 0.13], but this association
became significant in the multivariate model [OR(95% CI) = 0.02 (0.00–0.68); p =
0.03]. In contrast, AmtRNA-IgM were not significantly associated with any of these
clinical outcomes by either the bi- or multi-variate analysis.
Table 8: Association of AmtRNA with clinical manifestations in SLE. AmtRNA IgG IgM OR (CI) p OR (CI) p Thrombotic events 1.28 (0.24;6.77) 0.77 0.93 (0.22;3.93) 0.92 [1.15 (0.17;7.87)] [0.88] [1.00 (0.18;5.61)] [1.00] Presence of plaque 0.14 (0.02–0.91) 0.04 0.83 (0.25–2.76) 0.76 [0.16 (0.01–1.81)] [0.14] [0.82 (0.23–2.91)] [0.76] SLEDAI-2K ≥ 4 2.30 (0.73–7.26) 0.16 0.86 (0.34–2.17) 0.75 [3.04 (0.78–11.77)] [0.11] [0.68 (0.25–1.88)] [0.46] SDI ≥ 0 0.95 (0.28–3.21) 0.94 0.50 (0.16–1.58) 0.24 [0.85 (0.15–4.92)] [0.85] [0.46 (0.11–1.86)] [0.28] Positivity to anti-dsDNA antibodies 34.97 (6.26–195.55) <0.0001 1.92 (0.67–5.50) 0.23 [70.60 (6.31–789.47)] [0.0005] [2.01 (0.50–8.11)] [0.33] Lupus nephritis 0.17 (0.02–1.71) 0.13 0.43 (0.08–2.30) 0.33 [0.02 (0.00–0.68)] [0.03] [0.25 (0.04–1.48)] [0.12]
Values presented as odds ratios (95% Wald Confidence Interval) and p-value from logistic regressions. In each instance, bivariate results are followed by multivariate analysis (between square brackets). Values in bold have a p-value ≤ 0.05. AmtRNA, anti-mitochondrial RNA antibodies; CI: 95% Wald Confidence Interval; OR, odds ratio; SDI, lupus severity disease index; SLEDAI-2K, SLE disease activity index−2000. Data in bold are statistically significant (p < 0.05).
Furthermore, we assessed if our conclusions were identical in patients with higher
disease activity by repeating our calculations with patients having a SLEDAI-2K
score > 6 (i.e., for 15 patients, compared to 36 with a cut-off value at a SLEDAI-2K
score ≥ 4). The associations between AmtRNA-IgG with SLEDAI-2K > 6 were
[OR(95% CI) = 2.71 (0.71–10.31)] for the bivariate logistic regression and [OR(95%
CI) = 1.99 (0.40–10.00)] for the multivariate regression model. Values for the

312
associations between AmtRNA-IgM and SLEDAI-2K > 6 for bi- and multivariate
analyses were [OR(95% CI) = 0.53 (0.12–2.25)] and [OR(95% CI) = 0.37 (0.07–
1.89)], respectively. Thus, the conclusions remain the same using either SLEDAI-2K
cut-off value.
To determine whether AmtRNAs might qualify as efficient predictors of SLE, we
optimized cut-off values by Youden's method (Table 9). Calculated cut-off values
were 0.30 for AmtRNA-IgG and 0.52 for AmtRNA-IgM. Both parameters were very
specific for SLE (0.90 for IgG and 0.87 for IgM). Even though both Ig isotypes
displayed a certain lack of sensitivity [43 SLE patients (49%) positive for AmtRNA-
IgG and 33 (38%) for IgM], their positive predictive values (0.93 and 0.89) suggest
that AmtRNAs may be considered as biomarkers of interest. Importantly, of all of the
anti-mitochondrial autoantibodies measured, AmtRNA-IgG was the most potent at
discriminating SLE patients from healthy donors. In this regard, AmtRNA-IgG was
closely followed by AmtDNA-IgM. In contrast, AwMA (IgG and IgM) and AmtDNA-
IgG failed to efficiently discriminate SLE patients from healthy controls.
Table 9:Performance of cut-off values for AmtRNA, AwMA, and AmtDNA (OD 405nm). Cutpoint Sensitivity Specificity PPV NPV AUC
(95% BCI) (95% ECI) (95% ECI) (95% ECI) (95% ECI) (95% ECI) AmtRNA IgG 0.30 (0.11–0.54) 0.49 (0.38–0.60) 0.90 (0.73–0.98) 0.93 (0.82–0.99) 0.38 (0.27–0.50) 0.72 (0.62–0.82) IgM 0.52 (0.24–0.64) 0.38 (0.28–0.49) 0.87 (0.69–0.96) 0.89 (0.75–0.97) 0.33 (0.23–0.44) 0.62 (0.51–0.72) AwMA IgG 0.30 (0.12–0.44) 0.36 (0.26–0.47) 0.80 (0.61–0.92) 0.84 (0.68–0.94) 0.30 (0.21–0.42) 0.57 (0.45–0.69) IgM 0.68 (0.19–1.37) 0.24 (0.16–0.35) 0.87 (0.69–0.96) 0.84 (0.64–0.96) 0.29 (0.20–0.39) 0.48 (0.37–0.60) AmtDNA IgG 0.44 (0.22–1.25) 0.35 (0.25–0.46) 0.77 (0.58–0.90) 0.81 (0.65–0.92) 0.29 (0.19–0.40) 0.51 (0.40–0.62) IgM 0.36 (0.24–0.57) 0.51 (0.40–0.62) 0.83 (0.65–0.94) 0.90 (0.78–0.97) 0.37 (0.26–0.50) 0.65 (0.55–0.75)
Values in bold have an AUC significantly different than 50%. AmtDNA, anti-mitochondrial DNA antibodies. AmtRNA, anti-mitochondrial RNA antibodies. AUC, area under the curve. AwMA, anti-whole mitochondria antibodies. BCI, Bootstrap Confidence Interval. ECI, Exact Confidence Interval. NPV, Negative Predictive Value. OD, optical density. PPV, Positive Predictive Value. Data in bold are statistically significant (p < 0.05)

313
Discussion
Although the interplay between extracellular mitochondria and innate immunity has
been well-described, the interactions between mitochondria and the adaptive
immune system are less appreciated. Mitochondrial components are generally seen
as potential damage-associated molecular pattern (DAMP) if released by cells, but
their inflammatory potential may be different if they are also recognized by
autoantibodies. Herein, we propose mtRNA as a novel source of mitochondrial
autoantigens with high relevance to SLE.
Mitochondrial RNA is not the only mitochondrial sub-component with antigenic
potential in SLE. The first descriptions of anti-mitochondrial antibodies (AMA) were
published in the 1980's. However, the actual epitope(s) of some AMA remain
unidentified (57). Thus, AmtRNA add to the more recently appreciated AmtDNA and
AwMA (36). Adaptive autoimmunity targeting mitochondrial motifs is not unique to
SLE: a humoral immune response against mitochondrial autoantigens was reported
in various diseases, and described as 9 different types of AMA targeting distinct
epitopes (namely, M1 to M9) (36, 57). While AMA have been observed in different
contexts such as in cardiovascular diseases, iatrogenic disorders, secondary
syphilis, APS and SLE, they are best characterized in PBC (57, 58). The latter is
characterized by progressive infiltration of autoreactive lymphocytes through the
hepatic portal system (48, 59, 60). These cells display targeted autoreactivities
directed against different mitochondrial antigens specifically expressed by bile ducts
(60) such as the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2, also
known as mitochondrial antigen M2) (61–64), sulfite oxidase (M4) (65), glycogen
oxidase (M9) (47, 66, 67) as well as other antigens that have not yet been described
(M8). Detection of these AMA in PBC by ELISA have a prognostic value: patients
positive for AMA-M4 and -M8 suffer from active and/or progressive forms of the
disease (68), whereas patients with only AMA-M2 and -M9 display diseases with
delayed evolutions (69) (recapitulated in Figure 1).

314
How exactly the mitochondrial antibodies are produced is not completely
understood, but mitochondrial antigens can be generated through the degradation
of old or damaged mitochondria by a specific form of autophagy known as
mitophagy. Autophagosomes containing mitochondria travel through the
endolysosomal system, leading to the degradation of its cargo and allowing the
production of mitochondrial peptides that can be processed and expressed by the
major histocompatibility complex (MHC). Both MHC-I and MHC-II have been
implicated (70), an involvement for the latter being suggested in the surveillance of
mitochondrial mutations occurring in cancer (71, 72). However, a recent study
revealed that mitochondrial antigen processing can also occur independently of
mitophagy. In this case, mitochondrial antigens are carried to endosomes by
mitochondrial-derived vesicles formed by a mechanism regulated by the proteins
PINK1 and Parkin (73, 74). Whether these mechanisms are involved in the
processing of mtRNA molecules remains to be established.
Mitochondrial RNA is a recognized trigger of TLR8, which similarly to bacterial RNA,
stimulates peripheral blood mononuclear cells (75). As the most metabolically active
cells express more mtRNA, they are more likely to contribute to mtRNA antigenic
load (39). Our study demonstrates that mtRNA is also recognized by antibodies,
suggesting that Fc receptors may be implicated in the internalization of mtRNA-IgG
complexes by endosomes, thereby favoring interactions with TLR8. Mitochondria
express various RNA species, the main one being ribosomal 16S RNA molecules
(39). However, the respective antigenicity of each mtRNA species was not assessed
in the present study. Moreover, the presence of certain nuclear messenger RNA has
been described within mitochondria (39), which could also account for the
antigenicity potential of the mitochondria. Considering the evidence for mitochondrial
release in different pathogeneses, our demonstration of the presence of antibodies
directed against mitochondrial RNA further confirms the role of mitochondria as a
source of autoantigens in autoimmunity.

315
We observed associations between the three sets of mitochondrial antibodies
(AwMA, AmtDNA, and AmtRNA), pointing to their common source. Moreover, both
AmtDNA-IgG and AmtRNA-IgG were associated with positivity for anti-dsDNA
antibodies, suggesting close relationships between auto-antibodies targeting distinct
nucleic acids.
While the IgG targeting mtRNA were significantly elevated in SLE patients, the IgG
recognizing mtDNA and whole mitochondria were not increased in these patients.
These observations contrast with our previous findings, which involved a different
cohort of patients and showed that AmtDNA and AwMA were significantly increased
in SLE (36). The patients included in our previous work were recruited by the
University of Toronto Lupus Clinic. The patients recruited in the present study
(SARD-BDB) are characterized by a shorter median duration of the disease (10 vs.
6 years) that may account for reduced organ damage as indicated by the SDI score
(median: Toronto = 1; SARD-BDB = 0) and the frequency of patients with lupus
nephritis (Toronto: 38.5%; SARD-BDB: 16.3%). These differences may reflect the
course of the disease with earlier titers of autoantibodies clearing detrimental
circulating autoantigens (i.e., in the SARD-BDB cohort) until other
pathophysiological processes such as epitope spread occur, eliciting immune
complex-mediated organ damage. Another interesting aspect is the discrepancy
between the representation of the various ethnicities included in both cohorts. The
SARD-BDB is almost exclusively composed of Caucasians (Caucasian: 97.7%,
Black: 1.2%, Other ethnicities: 1.2%), whereas the Toronto cohort includes a more
diverse ethnic panel (Caucasian: 57 %, Black: 18 %, Asian: 21%. Other: 5%). Such
differences between two groups of patients may also impact results such as the
incidence, prevalence and mortality rates (76–79). Together, these differences may
reflect upon the protective effects of AmtRNA-IgG reported in the present study.
Moreover, these elements suggest that the spectrum of anti-mitochondrial
antibodies may shift during the course of the disease.

316
The heterogeneity of the disease duration for the SLE patients included in the SARD-
BDB allows the optimization of cut-offs by Youden's method that discriminate
positive from negative samples. However, calculation of a universal cut-off requires
detection of AmtRNA in newly-diagnosed SLE patients. Additionally, associations
between AmtRNA and clinical features of the disease should be interpreted with
caution, as clinical outcomes identified might have occurred before or at the same
time than the blood draw. Verification of the temporal relationship between the
production of AmtRNA and clinical outcomes would require a study of the variation
in anti-mitochondrial antibodies titers over time and their levels at the onset of a
clinical outcome in a large prospective inception cohort.
Systemic lupus erythematosus is a highly complex disease, many aspects of which
still elude researchers (80). To date, only a limited number of biomarkers are
available (81, 82). There is an intense effort to discover new biomarkers that would
allow specific discrimination of SLE patients from both healthy individuals and those
with diseases that have clinical features close to those of SLE (83). From this
perspective, we present AmtRNA-IgG as antibodies present in SLE and APS, two
diseases that are often associated with each other. Interestingly, AmtRNA-IgG
appeared to be associated with less lupus nephritis and plaque formation in the
carotid. Together, these elements indicate that AmtRNA may have prognostic value
and help to identify patients with specific clinical profiles. Moreover, the different
associations of AmtDNA and AmtRNA with lupus nephritis (AmtDNA are positively
associated with nephritis, while AmtRNA display a negative association) may help
predict SLE patients at risk of kidney damage.
Our study highlights that expression of a broad repertoire of anti-mitochondrial
antibody subtypes (AMA; AMA-M1, AMA-M5, AwMA, AmtDNA, AmtRNA) is a major
feature of SLE, with specific targets being associated with different clinical features.
Future studies dedicated to the characterization of the mitochondrial autoantigens
recognized in SLE and their outcome on disease progression may provide useful

317
information that will ultimately help to improve diagnosis, prognosis, and stratification
of SLE patients.

318
References
1. Andersen JL, Kornbluth S. The tangled circuitry of metabolism and apoptosis. Mol Cell. (2013) 49:399–410. 10.1016/j.molcel.2012.12.026 2. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. (2009) 43:95–118. 10.1146/annurev-genet-102108-134850 3. Lang BF, Burger G, O'Kelly CJ, Cedergren R, Golding GB, Lemieux C, et al. . An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. (1997) 387:493–7. 10.1038/387493a0 4. Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UC, Podowski RM, et al. . The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. (1998) 396:133–40. 10.1038/24094 5. Friedman JR, Nunnari J. Mitochondrial form and function. Nature. (2014) 505:335–43. 10.1038/nature12985 6. Tincani A, Meroni PL, Brucato A, Zanussi C, Allegri F, Mantelli P, et al. . Anti-phospholipid and anti-mitochondrial type M5 antibodies in systemic lupus erythematosus. Clin Exp Rheumatol. (1985) 3:321–6. 7. Arechaga I. Membrane invaginations in bacteria and mitochondria: common features and evolutionary scenarios. J Mol Microbiol Biotechnol. (2013) 23:13–23. 10.1159/000346515 8. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. . Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. (2010) 330:362–6. 10.1126/science.1195491 9. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. . Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. 10.1038/nature08780 10. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. 10.1038/cdd.2009.96 11. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. . Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. 10.1038/nm.4027 12. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. . Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. 10.1084/jem.20151876 13. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. . Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. (2008) 14:949–53. 10.1038/nm.1855 14. Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, et al. . Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. (2014) 124:2173–83. 10.1182/blood-2014-05-573543 15. Marcoux G, Duchez AC, Rousseau M, Lévesque T, Boudreau LH, Thibault L, et al. . Microparticle and mitochondrial release during extended storage of different types of platelet concentrates. Platelets. (2017) 28:272–80. 10.1080/09537104.2016.1218455 16. Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS ONE. (2012) 7:e49767. 10.1371/journal.pone.0049767 17. Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. (2014) 5:e1312. 10.1038/cddis.2014.277 18. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. . Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. (2016) 126:859–64. 10.1172/JCI83885 19. Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, et al. . Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. (2012) 56:1971–82. 10.1002/hep.25801 20. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. . Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. 10.1038/nature10992

319
21. Zhao Z, Wang M, Tian Y, Hilton T, Salsbery B, Zhou EZ, et al. . Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood. (2016) 127:2763–72. 10.1182/blood-2015-12-688838 22. Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, et al. . Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. (2013) 10:e1001577. discussion: e1001577. 10.1371/journal.pmed.1001577 23. Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. . Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. (2013) 258:591–6. 10.1097/SLA.0b013e3182a4ea46 24. Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther. (2003) 5:R234–40. 10.1186/ar787 25. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. . Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. (2011) 32:157–64. 10.1016/j.it.2011.01.005 26. Calfee CS, Matthay MA. Clinical immunology: culprits with evolutionary ties. Nature. (2010) 464:41–2. 10.1038/464041a 27. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. . BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. (2018) 359:eaao6047. 10.1126/science.aao6047 28. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. 10.1016/j.immuni.2015.02.002 29. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. (2011) 11:389–402. 10.1038/nri2975 30. Sandhir R, Halder A, Sunkaria A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim Biophys Acta. (2017) 1863:1090–7. 10.1016/j.bbadis.2016.10.020 31. Mills EL, Kelly B, O'Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. (2017) 18:488–98. 10.1038/ni.3704 32. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. 10.1038/nature09663 33. Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFalpha release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS ONE. (2013) 8:e72354. 10.1371/journal.pone.0072354 34. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. (2015) 67:3190–200. 10.1002/art.39296 35. Reimer G, Rubin RL, Kotzin BL, Tan EM. Anti-native DNA antibodies from autoimmune sera also bind to DNA in mitochondria. J Immunol. (1984) 133:2532–6. 36. Becker Y, Loignon RC, Julien AS, Marcoux G, Allaeys I, Lévesque T, et al. . Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep. (2019) 9:4530. 10.1038/s41598-019-40900-3 37. Aatonen MT, Ohman T, Nyman TA, Laitinen S, Grönholm M, Siljander PR. Isolation and characterization of platelet-derived extracellular vesicles. J Extracell Vesicles. 3:10.3402/jev.v3.24692. 10.3402/jev.v3.24692 38. Rorbach J, Minczuk M. The post-transcriptional life of mammalian mitochondrial RNA. Biochem J. (2012) 444:357–73. 10.1042/BJ20112208 39. Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, et al. . The human mitochondrial transcriptome. Cell. (2011) 146:645–58. 10.1016/j.cell.2011.06.051 40. Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. (2001) 2:342–52. 10.1038/35072063 41. Jourdain AA, Boehm E, Maundrell K, Martinou JC. Mitochondrial RNA granules: compartmentalizing mitochondrial gene expression. J Cell Biol. (2016) 212:611–4. 10.1083/jcb.201507125 42. Jourdain AA, Popow J, de la Fuente MA, Martinou JC, Anderson P, Simarro M. The FASTK family of proteins: emerging regulators of mitochondrial RNA biology. Nucleic Acids Res. (2017) 45:10941–7. 10.1093/nar/gkx772

320
43. Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. (2007) 2:287–95. 10.1038/nprot.2006.478 44. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. . The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1982) 25:1271– 7 10.1002/art.1780251101 45. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1997) 40:1725. 10.1002/art.1780400928 46. Cicek G, Schiltz E, Hess D, Staiger J, Brandsch R. Analysis of mitochondrial antigens reveals inner membrane succinate dehydrogenase flavoprotein subunit as autoantigen to antibodies in anti-M7 sera. Clin Exp Immunol. (2002) 128:83–7 10.1046/j.1365-2249.2002.01816.x 47. Klein R, Berg PA. Characterization of a new mitochondrial antigen-antibody system (M9/anti-M9) in patients with anti-M2 positive and anti-M2 negative primary biliary cirrhosis. Clin Exp Immunol. (1988) 74:68–74. 48. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. . Primary biliary cirrhosis. Hepatology. (2009) 50:291–308. 10.1002/hep.22906 49. Gladman DD, Goldsmith CH, Urowitz MB, Bacon P, Fortin P, Ginzler E, et al. . The Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) damage index for systemic lupus erythematosus international comparison. J Rheumatol. (2000) 27:373–6. 50. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. (1992) 35:630–40. 10.1002/art.1780350606 51. Gladman DD, Urowitz MB, Goldsmith CH, Fortin P, Ginzler E, Gordon C, et al. . The reliability of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index in patients with systemic lupus erythematosus. Arthritis Rheum. (1997) 40:809–13. 10.1002/art.1780400506 52. Gladman D, Ginzler E, Goldsmith C, Fortin P, Liang M, Urowitz M, et al. . Systemic lupus international collaborative clinics: development of a damage index in systemic lupus erythematosus. J Rheumatol. (1992) 19:1820–1. 53. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. . Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2677–86. 10.1002/art.34473 54. Pengo V, Tripodi A, Reber G, Rand JH, Ortel TL, Galli M, et al. . Update of the guidelines for lupus anticoagulant detection. Subcommittee on lupus anticoagulant/antiphospholipid antibody of the scientific and standardisation committee of the international society on thrombosis and haemostasis. J Thromb Haemost. (2009) 7:1737–40. 10.1111/j.1538-7836.2009.03555.x 55. Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. . A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. (2015) 14:75–9. 10.1016/j.autrev.2014.10.003 56. Soliman S, Mohan C. Lupus nephritis biomarkers. Clin Immunol. (2017) 185:10–20. 10.1016/j.clim.2016.08.001 57. Berg PA, Klein R. Mitochondrial antigens and autoantibodies: from anti-M1 to anti-M9. Klin Wochenschr. (1986) 64:897–909 10.1007/BF01728613 [PubMed] [CrossRef] [Google Scholar] 58. Witt-Sullivan H, Heathcote J, Cauch K, Blendis L, Ghent C, Katz A, et al. . The demography of primary biliary cirrhosis in Ontario, Canada. Hepatology. (1990) 12:98–105. 10.1002/hep.1840120116 59. Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van de Water J, et al. . Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. (2002) 109:1231–40. 10.1172/JCI0214698 60. Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF, Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest. (2001) 108:223–32. 10.1172/JCI200110716 61. Yeaman SJ, Fussey SP, Danner DJ, James OF, Mutimer DJ, Bassendine MF. Primary biliary cirrhosis: identification of two major M2 mitochondrial autoantigens. Lancet. (1988) 1:1067–70. 10.1016/S0140-6736(88)91894-6

321
62. Lindenborn-Fotinos J, Baum H, Berg PA. Mitochondrial antibodies in primary biliary cirrhosis: species and nonspecies specific determinants of M2 antigen. Hepatology. (1985) 5:763–9. 10.1002/hep.1840050510 63. Gershwin ME, Ansari AA, Mackay IR, Nakanuma Y, Nishio A, Rowley MJ, et al. . Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. (2000) 174:210–25. 10.1034/j.1600-0528.2002.017402.x 64. Frazer IH, Mackay IR, Jordan TW, Whittingham S, Marzuki S. Reactivity of anti-mitochondrial autoantibodies in primary biliary cirrhosis: definition of two novel mitochondrial polypeptide autoantigens. J Immunol. (1985) 135:1739–45. 65. Klein R, Berg PA. Anti-M4 antibodies in primary biliary cirrhosis react with sulphite oxidase, an enzyme of the mitochondrial inter-membrane space. Clin Exp Immunol. (1991) 84:445–8. 66. Klein R, Berg PA. Anti-M9 antibodies in sera from patients with primary biliary cirrhosis recognize an epitope of glycogen phosphorylase. Clin Exp Immunol. (1990) 81:65–71. 10.1111/j.1365-2249.1990.tb05292.x 67. Klein R, Berg PA. Characterization of M9 by westernblot and relevance of anti-M9 as a marker antibody for early primary biliary cirrhosis (PBC). J Hepatol. (1987) 5:S36 10.1016/S0168-8278(87)80159-9 68. Berg PA, Wiedmann KH, Sayers T, Kloppel G, Lindner H. Serological classification of chronic cholestatic liver disease by the use of two different types of antimitochondrial antibodies. Lancet. (1980) 2:1329–32. 10.1016/S0140-6736(80)92397-1 69. Klein R, Klöppel G, Fischer R, Fintelmann V, Müting D, Berg PA. The antimitochondrial antibody anti-M9. A marker for the diagnosis of early primary biliary cirrhosis. J Hepatol. (1988) 6:299–306. 10.1016/S0168-8278(88)80046-1 70. Gu Y, Wang C, Roifman CM, Cohen A. Role of MHC class I in immune surveillance of mitochondrial DNA integrity. J Immunol. (2003) 170:3603–7. 10.4049/jimmunol.170.7.3603 71. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, et al. . Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. (2005) 102:7922–7. 10.1073/pnas.0501190102 72. Qi L, Rojas JM, Ostrand-Rosenberg S. Tumor cells present MHC class II-restricted nuclear and mitochondrial antigens and are the predominant antigen presenting cells in vivo. J Immunol. (2000) 165:5451–61. 10.4049/jimmunol.165.10.5451 73. Roberts RF, Fon EA. Presenting mitochondrial antigens: PINK1, Parkin and MDVs steal the show. Cell Res. (2016) 26:1180–1. 10.1038/cr.2016.104 74. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. . Parkinson's disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. (2016) 166:314–27. 10.1016/j.cell.2016.05.039 75. Krüger A, Oldenburg M, Chebrolu C, Beisser D, Kolter J, Sigmund AM, et al. . Human TLR8 senses UR/URR motifs in bacterial and mitochondrial RNA. EMBO Rep. (2015) 16:1656–63. 10.15252/embr.201540861 76. Liang MH, Partridge AJ, Daltroy LH, Straaton KV, Galper SR, Holman HR. Strategies for reducing excess morbidity and mortality in blacks with systemic lupus erythematosus. Arthritis Rheum. (1991) 34:1187–96. 10.1002/art.1780340918 77. Williams EM, Bruner L, Adkins A, Vrana C, Logan A, Kamen D, et al. . I too, am America: a review of research on systemic lupus erythematosus in African-Americans. Lupus Sci Med. (2016) 3:e000144. 10.1136/lupus-2015-000144 78. Molokhia M, McKeigue P. Risk for rheumatic disease in relation to ethnicity and admixture. Arthritis Res. (2000) 2:115–25. 10.1186/ar76 79. Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatol (Oxford). (2017) 56:1945–61. 10.1093/rheumatology/kex260 80. Peschken CA, Katz SJ, Silverman E, Pope JE, Fortin PR, Pineau C, et al. . The 1000 Canadian faces of lupus: determinants of disease outcome in a large multiethnic cohort. J Rheumatol. (2009) 36:1200–8. 10.3899/jrheum.080912 81. Liu CC, Kao AH, Manzi S, Ahearn JM. Biomarkers in systemic lupus erythematosus: challenges and prospects for the future. Ther Adv Musculoskelet Dis. (2013) 5:210–33. 10.1177/1759720X13485503

322
82. Arriens C, Wren JD, Munroe ME, Mohan C. Systemic lupus erythematosus biomarkers: the challenging quest. Rheumatol (Oxford). (2017) 56:i32–45. 10.1093/rheumatology/kew407 83. Nicolaou O, Kousios A, Hadjisavvas A, Lauwerys B, Sokratous K, Kyriacou K. Biomarkers of systemic lupus erythematosus identified using mass spectrometry-based proteomics: a systematic review. J Cell Mol Med. (2017) 21:993–1012. 10.1111/jcmm.13031

323
Annexe V: Anti-mitochondrial autoantibodies in
systemic lupus erythematosus and their
association with disease manifestations
Yann Becker,1 Renée-Claude Loignon,2 Anne-Sophie Julien,3 Geneviève Marcoux,1
Isabelle Allaeys,1 Tania Lévesque,1 Emmanuelle Rollet-Labelle,1 Hadrien Benk-Fortin,1 Nathalie Cloutier,1 Imène Melki,1 Lihi Eder,4 Éric Wagner,5 Martin Pelletier,1,6 Hassan El Hajj,7 Marie-Ève Tremblay,7 Clémence Belleannée,8 Marie-Josée Hébert,9 Mélanie Dieudé,9 Joyce Rauch,10 Paul R. Fortin,2,6 and Eric Boilard1,6
Author information 1Centre de Recherche du CHU de Québec – Université Laval, Département de microbiologie et immunologie, Faculté de Médecine de l′Université Laval, Québec, Qc Canada 2Division de Rhumatologie, Département de Médecine, CHU de Québec – Université Laval, Québec, Qc Canada 3Département de mathématiques et statistique, Université Laval, Québec, Qc Canada 4Women’s College Hospital and University of Toronto, Toronto, ON Canada 5Immunology and Histocompatibility Laboratory, Department of Medical Biology CHU de Québec - Université Laval; Department of Microbiology, Infectious Diseases and Immunology, Université Laval, Québec, Qc Canada 6Axe maladies infectieuses et inflammatoires, Centre de recherche du CHU de Québec – Université Laval, Québec, Qc Canada 7Axe Neurosciences, Centre de Recherche du CHU de Québec – Université Laval, Québec, Qc Canada 8Axe Reproduction, Santé de la mère et de l’enfant, Centre de recherche du CHU de Québec – Université Laval, Québec, Qc Canada 9Centre de recherche du CHU de Montréal, Montréal Québec, Québec, Qc Canada 10Division of Rheumatology, Department of Medicine, Research Institute of the McGill University Health Centre, Montreal, Qc H4A 3J1 Canada

324
Abstract
Mitochondria are organelles that govern energy supply and control cell death.
Mitochondria also express bacterial features, such as the presence of inner
membrane cardiolipin and a circular genome rich in hypomethylated CpG motifs.
While mitochondrial extrusion by damaged organs or activated cells is thought to
trigger innate immunity, it is unclear whether extracellular mitochondria also
stimulate an adaptive immune response. We describe the development of novel
assays to detect autoantibodies specific to two distinct components of the
mitochondrion: the mitochondrial outer membrane and mitochondrial DNA.
Antibodies to these two mitochondrial constituents were increased in both human
and murine systemic lupus erythematosus (SLE), compared to controls, and were
present at higher levels than in patients with antiphospholipid syndrome or primary
biliary cirrhosis. In both bi- and multi-variate regression models, antibodies to
mitochondrial DNA, but not whole mitochondria, were associated with increased
anti-dsDNA antibodies and lupus nephritis. This study describes new and optimized
methods for the assessment of anti-mitochondrial antibodies, and demonstrates
their presence in both human and murine SLE. These findings suggest that different
mitochondrial components are immunogenic in SLE, and support the concept that
extracellular mitochondria may provide an important source of circulating
autoantigens in SLE.

325
Introduction
The roles of mitochondria in bioenergetics and the control of cell proliferation or
death are well-documented1,2. Furthermore, mitochondria share several similarities
with bacteria3,4. Like bacteria, mitochondria are formed of an outer and an inner
membrane (inner contains cardiolipin)4,5, express formylated peptides6,7, and contain
a circular genome with hypomethylated DNA CpG motifs, referred to as
mitochondrial DNA (mtDNA)8,9.
Various cellular lineages are capable of extruding their mitochondria. Activated mast
cells10, T-cells11, eosinophils12, hepatocytes13, neutrophils14–16 and platelets17,18, in
addition to damaged organs or tissues7,13,19,20, release extracellular mitochondria.
Mitochondria and their components (e.g. N-formylated peptides and mtDNA) are
recognized as damage-associated molecular patterns (DAMPs), which activate the
innate immune system and elicit an inflammatory response6,21–23. Moreover, ATP
and reactive oxygen species (ROS), produced by mitochondria are triggers of the
nuclear oligomerization domain (NOD)-like receptors and contribute to
inflammasome activation21,22,24. Extracellular mitochondria have been described in
various clinical conditions, including trauma25,26, burn injury27, cancer28, rheumatoid
arthritis17,29, systemic lupus erythematosus (SLE)15,16 and transfusion adverse
reactions17,18,30,31. Their pro-inflammatory potential is generally thought to occur
through activation of Toll-like receptors (TLR), formyl peptide receptors, and
cytosolic pathogen recognition receptors, all key components of the innate immune
system6,21–24.
The adaptive immune system can also recognize mitochondria. This concept is
important, as the immune response initiated by mitochondrial DAMPs may be
different if the adaptive immune system is also implicated32,33. Different sets of anti-
mitochondrial antibodies (AMA), namely AMA-M1 to -M9, have been characterized34
(recapitulated in Table 1). The AMA-M2, -M4, -M8, and -M9 classes are well-
described in primary biliary cirrhosis (PBC)35–37, an autoimmune disease
characterized by a progressive destruction of the bile ducts due to the infiltration of
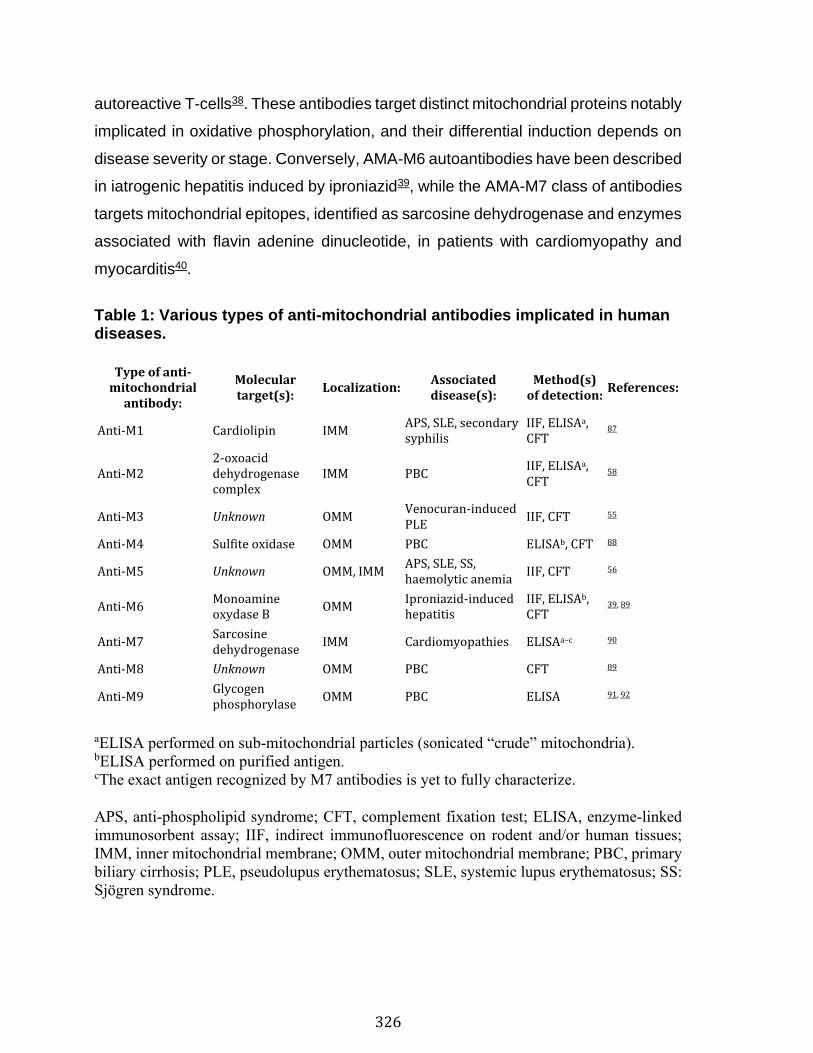
326
autoreactive T-cells38. These antibodies target distinct mitochondrial proteins notably
implicated in oxidative phosphorylation, and their differential induction depends on
disease severity or stage. Conversely, AMA-M6 autoantibodies have been described
in iatrogenic hepatitis induced by iproniazid39, while the AMA-M7 class of antibodies
targets mitochondrial epitopes, identified as sarcosine dehydrogenase and enzymes
associated with flavin adenine dinucleotide, in patients with cardiomyopathy and
myocarditis40.
Table 1: Various types of anti-mitochondrial antibodies implicated in human diseases.
Type of anti-mitochondrial
antibody:
Molecular target(s):
Localization: Associated disease(s):
Method(s) of detection:
References:
Anti-M1 Cardiolipin IMM APS, SLE, secondary syphilis
IIF, ELISAa, CFT
87
Anti-M2 2-oxoacid dehydrogenase complex
IMM PBC IIF, ELISAa, CFT
58
Anti-M3 Unknown OMM Venocuran-induced PLE
IIF, CFT 55
Anti-M4 Sulfite oxidase OMM PBC ELISAb, CFT 88
Anti-M5 Unknown OMM, IMM APS, SLE, SS, haemolytic anemia
IIF, CFT 56
Anti-M6 Monoamine oxydase B
OMM Iproniazid-induced hepatitis
IIF, ELISAb, CFT
39, 89
Anti-M7 Sarcosine dehydrogenase
IMM Cardiomyopathies ELISAa–c 90
Anti-M8 Unknown OMM PBC CFT 89
Anti-M9 Glycogen phosphorylase
OMM PBC ELISA 91, 92
aELISA performed on sub-mitochondrial particles (sonicated “crude” mitochondria). bELISA performed on purified antigen. cThe exact antigen recognized by M7 antibodies is yet to fully characterize.
APS, anti-phospholipid syndrome; CFT, complement fixation test; ELISA, enzyme-linked
immunosorbent assay; IIF, indirect immunofluorescence on rodent and/or human tissues;
IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; PBC, primary
biliary cirrhosis; PLE, pseudolupus erythematosus; SLE, systemic lupus erythematosus; SS:
Sjögren syndrome.

327
SLE is an autoimmune disease characterized by the presence of circulating antigen-
autoantibody immune complexes and inflammation in multiple organs and tissues.
In SLE, neutrophils were shown to release mtDNA through the generation of ROS,
a process leading to activation of the DNA sensor stimulator of interferon genes
(STING) and type-I IFN production15,16. Studies showed that anti-mtDNA antibodies
(AmtDNA) are induced in a subset of SLE patients, suggesting that extruded mtDNA
could be a relevant source of antigen for the anti-double stranded DNA (anti-dsDNA)
antibodies that prevail in SLE16,41,42. Clinically, anti-DNA antibodies are screened
initially by indirect immunofluorescence using human epithelial type 2 cells (HEp-2)
and enzyme immunoassay using double-stranded DNA43. Presence of anti-DNA
antibodies can also be assessed by indirect immunofluorescence using Crithidia
luciliae, an hemoflagellate parasite of blow-flies that possesses a uniquely large
mitochondrion that contains a high concentration of DNA, the kinetoplast44–46.
Another method commonly used is Farr radioimmunoassay, which involves the
precipitation of antibody-bound radiolabeled DNA and its detection with a scintillation
counter47.
Antibodies targeting mitochondrial components other than mtDNA are also found in
SLE. They include anti-cardiolipin [AMA-M1, also known as anti-cardiolipin
antibodies (ACA)], anti-60kDa heat shock protein (anti-HSP60), as well as AMA-M3
and AMA-M5 antibodies. AMA-M1 antibodies recognize cardiolipin and were
originally identified in syphilis-infected patients. Anti-cardiolipin antibodies are also
found in patients with SLE and antiphospholipid syndrome (APS), resulting in false
positive results in earlier syphilis detection assays34,48.
Interestingly, cardiolipin is a phospholipid that is found uniquely in the inner
mitochondrial membrane in eukaryotic cells. However, damaged mitochondria may
externalize their cardiolipin49, that may become accessible and induce the
development of antibodies to cardiolipin. Clinically, the presence of ACA is
associated with a greater risk of thrombotic events and thrombocytopenia50,51.

328
Another mitochondrial antigen, HSP60, is a mitochondrial chaperonin implicated in
mitochondrial protein import52. Patients with SLE have antibodies against HSP6053,
and their presence (when concomitant with anti-phospholid antibodies) is associated
with vascular events54.
AMA-M3 were described in patients using a drug called venocuran who developed
a drug-induced syndrome (“pseudolupus”) with clinical manifestations of arthralgia,
fever, serositis, and lymphopenia. The immunological profile of pseudolupus is
characterized by the absence of antinuclear antibody (ANA), but the presence of
AMA-M355. The antigenic target of AMA-M3 is different than that of the other AMA
classes described in PBC55. It is resistant to trypsin and it is extracted by solvent,
pointing to its lipid nature. AMA-M3 are no longer encountered clinically since the
withdrawal of the drug venocuran.
AMA-M5 comprise another class of anti-mitochondrial antibodies identified in
patients with SLE, as well in APS, Sjögren syndrome, recurrent fetal loss, and
hemolytic anemia56. The precise antigenic target(s) of AMA-M5 is undefined, but lack
of competition by cardiolipin-containing liposomes suggests that it is distinct from the
target of AMA-M157. Indirect immunofluorescence on human or rodent tissues is
used to identify anti-M5 antibodies58. While immunofluorescence and complement
fixation test revealed that only 2% of SLE patients were positive for AMA-M5,
approximately 40% were positive by enzyme-linked immunosorbent assay
(ELISA)59. However, in the latter approach, mitochondria were only partially purified
and were sonicated, thus revealing antigenic epitopes that might not be recognized
in tissues60.
Emerging evidence supports the liberation of mitochondria by activated cells and
their potential contribution to inflammation in SLE15,16. Identification of mitochondrial
antigens recognized by autoantibodies in SLE may provide information on the roles
of extracellular mitochondria in autoimmunity and systemic inflammation. Herein, we
developed new methods to detect the presence of two distinct types of circulating
AMA: anti-whole mitochondria antibodies (AwMA) and AmtDNA. We then

329
determined their usefulness in a murine model of SLE, as well as in a cohort of SLE
patients. We evaluated these AMA in parallel with antibodies to the mitochondrial
chaperonin HSP60 (a known mitochondrial target in SLE), and determined the
associations of these autoantibodies with disease characteristics. To our knowledge,
this is the first study that examines the association of these AMA with disease
manifestations in SLE.
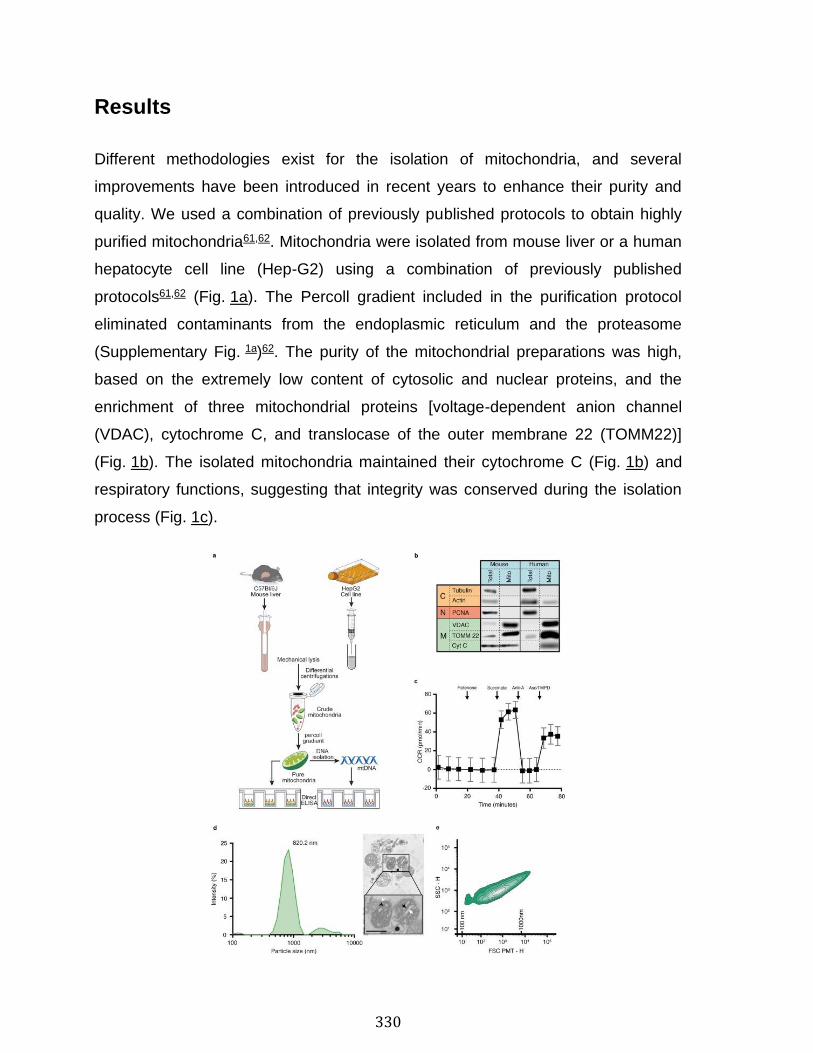
330
Results
Different methodologies exist for the isolation of mitochondria, and several
improvements have been introduced in recent years to enhance their purity and
quality. We used a combination of previously published protocols to obtain highly
purified mitochondria61,62. Mitochondria were isolated from mouse liver or a human
hepatocyte cell line (Hep-G2) using a combination of previously published
protocols61,62 (Fig. 1a). The Percoll gradient included in the purification protocol
eliminated contaminants from the endoplasmic reticulum and the proteasome
(Supplementary Fig. 1a)62. The purity of the mitochondrial preparations was high,
based on the extremely low content of cytosolic and nuclear proteins, and the
enrichment of three mitochondrial proteins [voltage-dependent anion channel
(VDAC), cytochrome C, and translocase of the outer membrane 22 (TOMM22)]
(Fig. 1b). The isolated mitochondria maintained their cytochrome C (Fig. 1b) and
respiratory functions, suggesting that integrity was conserved during the isolation
process (Fig. 1c).

331
Figure 1 : Assessment of the mitochondrial preparations. (a) Mitochondria were isolated from either mouse liver or human Hep-G2 cell line by differential centrifugations and further purified by ultracentrifugation against Percoll gradient. Alkaline lysis was performed to retrieve mtDNA. Pure mitochondria or mtDNA were used as coating antigens in direct ELISAs. (b) Cytoplasmic (C), nuclear (N) and mitochondrial (M) markers were assessed by western blotting in murine (left) and human (right) mitochondrial preparations (25 µg protein per lane). Results are representative of three distinct preparations. Blots separated by dashed lines are non-contiguous but from same membrane. Blots separated by full lines were performed on distinct membranes; (c) Functionality of murine mitochondria was determined by measurement of the oxygen consumption rate (OCR) of 10 µg mitochondria treated successively with 2 µM rotenone, 10 mM succinate, 40 µM antimycin A and 100 µM N,N,N’,N’-Tetramethyl-p-phenylenediamine (TMPD) along with 10 mM ascorbate (Asc). (d) The hydrodynamic size of the murine mitochondria was determined using zetasizer nano ZS (n = 3, left panel) and their morphology was visualized by electron microscopy (right panel). Inner membrane (black arrowhead), outer membrane (white arrowhead), cristae (black arrow), mitochondrial matrix (white arrow) are presented (scale bar account for 500 nm); (e) Size representation of purified murine mitochondria using high sensitivity-flow cytometry. Double-positive mitochondria (Mitotracker+ TOMM22+) were used for quantification. Silica beads were used to determine 100–1000 nm size scale. Data are mean ± SD. Anti-A: antimycin A; CytC: cytochtome C; FSC: forward scatter; Mito: mitochondria PCNA: proliferating cell nuclear antigen; SSC: side scatter; Total: total starting material; TOMM22: translocase of the outer mitochondrial membrane; VDAC: voltage-dependent anion channel.
Mitochondria were homogeneous in size, with a main peak detected at 820 nm by
dynamic light scattering (Fig. 1d, left panel), and retained their canonical morphology
when observed in electron microscopy (910 ± 210 nm) (Fig. 1d, right panel). High
sensitivity flow cytometry, which permits the identification of submicron particles, was
used as a quantitative approach. We estimated that 6.6 ± 1.9 × 106 mitochondria
(Mitotracker+ TOMM22+) (or 4.33 ± 1.17 µg mitochondrial proteins) could be isolated
per mg of mouse liver (n = 6), which was sufficient to prepare approximately 400
wells in half-area 96-well microplates using one mouse liver (Fig. 1e). The yield
obtained with Hep-G2 cells was lower, as 3.5 ± 0.18 µg of mitochondrial protein were
isolated per 106 Hep-G2 cells (7.0 × ± 2.1 × 108 Hep-G2 cells were harvested for
each 175 cm2 flask at confluence), which can be used to prepare approximately two
half-area 96-well microplates.
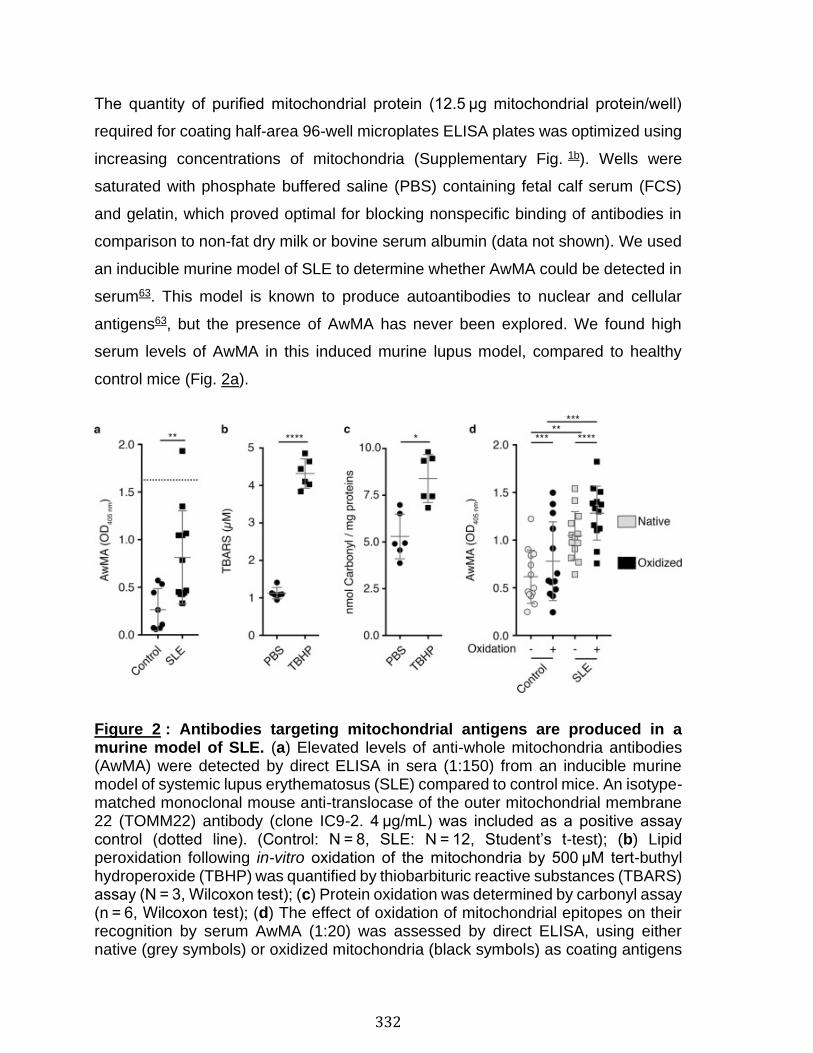
332
The quantity of purified mitochondrial protein (12.5 µg mitochondrial protein/well)
required for coating half-area 96-well microplates ELISA plates was optimized using
increasing concentrations of mitochondria (Supplementary Fig. 1b). Wells were
saturated with phosphate buffered saline (PBS) containing fetal calf serum (FCS)
and gelatin, which proved optimal for blocking nonspecific binding of antibodies in
comparison to non-fat dry milk or bovine serum albumin (data not shown). We used
an inducible murine model of SLE to determine whether AwMA could be detected in
serum63. This model is known to produce autoantibodies to nuclear and cellular
antigens63, but the presence of AwMA has never been explored. We found high
serum levels of AwMA in this induced murine lupus model, compared to healthy
control mice (Fig. 2a).
Figure 2 : Antibodies targeting mitochondrial antigens are produced in a murine model of SLE. (a) Elevated levels of anti-whole mitochondria antibodies (AwMA) were detected by direct ELISA in sera (1:150) from an inducible murine model of systemic lupus erythematosus (SLE) compared to control mice. An isotype-matched monoclonal mouse anti-translocase of the outer mitochondrial membrane 22 (TOMM22) antibody (clone IC9-2. 4 µg/mL) was included as a positive assay control (dotted line). (Control: N = 8, SLE: N = 12, Student’s t-test); (b) Lipid peroxidation following in-vitro oxidation of the mitochondria by 500 µM tert-buthyl hydroperoxide (TBHP) was quantified by thiobarbituric reactive substances (TBARS) assay (N = 3, Wilcoxon test); (c) Protein oxidation was determined by carbonyl assay (n = 6, Wilcoxon test); (d) The effect of oxidation of mitochondrial epitopes on their recognition by serum AwMA (1:20) was assessed by direct ELISA, using either native (grey symbols) or oxidized mitochondria (black symbols) as coating antigens

333
(N = 13, two-way ANOVA with multiple comparisons; Sidak’s correction). All experiment presented in the figure were performed using mouse mitochondria. Data are mean ± SD. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Reactive oxygen species are generated under inflammatory conditions, and were
reported during the release of mitochondria15,16. Thus, we assessed whether
oxidation of mitochondria could impact mitochondrial recognition by AwMA. Isolated
mitochondria were treated with increasing concentrations of the oxidant tert-butyl
hydroperoxide (TBHP), and the oxidized protein and lipid contents were confirmed
using commercial assays (Fig. 2b,c and Supplementary Fig. 2). We found that
oxidation had no or very little impact on recognition of mitochondria by SLE
antibodies (Fig. 2d) (Fold increase: 1.2 ± 0.2). The data suggest that mitochondria
are immunogenic in SLE regardless of the oxidation status of their antigens.
We next used our quantitative AwMA ELISA to screen human sera. We included 175
SLE patients and 43 healthy controls (76% female, mean age 42 ± 12) (Table 2). We
also evaluated sera from APS patients (n = 12), given the high levels of anti-
cardiolipin antibodies (AMA-M1) in APS, as well as sera from PBC patients (n = 12)
confirmed positive for AMA by indirect immunofluorescence on mouse
stomach/kidney slides (MSK).

334
Table 2: Demographics and clinical characteristics (ACR criteria) for SLE patients included in the study (n = 175). Characteristics SLE patients
Age
Range, years 20–78
Mean ± S.D, years 47 ± 15
Disease duration
Range, years 0–57
Mean ± S.D, years 18 ± 12
Gender, female, n (%) 175 (100)
Thrombotic events, n (%) 35 (20)
SLEDAI-2K ≥ 4, n (%) 57 (33)
SDI ≥ 1, n (%) 124 (71)
Increased anti-dsDNA, n (%) 59 (34)
Lupus nephritis, n (%) (n = 172) 67 (39)
Currently Prescribed Medication, n (%)
Anticoagulation or anti-platelet (n = 174) 40 (23)
Antimalarial 127 (73)
Prednisone 81 (46)
Lipid lowering 26 (15)
Diabetes medication 6 (3)
Malar rash 127 (72.6)
Discoid rash 24 (13.7)
Photosensitivity 113 (64.6)
Oral ulcers 108 (61.7)
Arthritis (≥2 peripheral joints) 151 (86.3)
Serositis 67 (38.3)
Neurologic disorder (seizure or psychosis) 24 (12.0)
Renal disorderA 100 (57.1)
eGFR (n = 160)
Range, mL/min/1.73 m2 17–121
Mean ± S.D, mL/min/1.73 m2 84.38 ± 24.70
<60 mL/min/1.73 m2, n (%) 26 (16.3)
Hematologic disorderB 155 (88.6)
Immunologic disorderC 159 (90.9)
Anti-nuclear antibodies (ANA) 170 (97.1)
American College of Rheumatology criteria (ACR) score
Range 03-Nov
Mean ± S.D 6.83 ± 1.62
A >0.5 g per day of protein in urine or cellular cast or end-stage renal disease. B Hemolytic anemia (low red blood cell count) or leukopenia (White blood cells < 4000/µl), lymphopenia (<100 000/µl) in the absence of offending drug. C Positive anti-Smith, anti-dsDNA, antiphospholipid antibody and/or false positive serological test for syphilis. eGFR: estimated glomerular filtration test.

335
Given the higher yield and purity of the mitochondria isolated from mouse liver, and
the fact that mice are readily accessible in most research laboratories, we used intact
murine mitochondria as coating antigens in our assay for the detection of
autoantibodies targeting the outer mitochondrial membrane in humans. We found
that AwMA were present in all healthy controls, but at much lower levels than those
encountered in a large proportion of the SLE patients. SLE patients were more
frequently positive and at higher levels for AwMA than healthy controls. APS and
PBC patients also presented a significant increase in AwMA compared to healthy
donors but signals detected for these patients were lower than those measured in
SLE patients (Fig. 3a). As the ELISA is performed on intact mitochondria, these
results suggest that AwMA are induced in SLE, and recognize autoantigens on the
outer mitochondrial membrane that are distinct from the epitopes in APS (cardiolipin)
and PBC (pyruvate dehydrogenase complex E2-component, PDC-E2), both located
in the mitochondrial inner membrane. Consistent with this, we identified PBC
patients positive for AMA when submitochondrial particles (i.e. sonicated
mitochondria) were used as coating antigens, suggesting that certain antigens
relevant to PBC may be exposed in these conditions (Supplementary Fig. 3). Human
and murine mitochondria were also compared in our assay using sera from a subset
of SLE patients. Both sources of mitochondria (human Hep-G2 cells and mouse
liver) were similarly recognized by human AwMA (Fig. 3b), suggesting that antigenic
epitopes may be conserved across these species. Hence, mitochondria from murine,
bovine and porcine tissues are routinely used to assess AwMA in humans,
suggesting that interspecies differences play a negligible role, if any, in mitochondrial
antigenicity.

336
Figure 3 : Detection of anti-mitochondrial antibodies in SLE patients and specificity of the assay. (a) Increased amounts of anti-whole mitochondria antibodies (AwMA) were detected by direct ELISA in sera (1:150) from systemic lupus erythematosus (SLE), anti-phospholipid syndrome (APS) and primary biliary cirrhosis (PBC) patients. Healthy: N = 43. SLE: N = 175. APS: N = 12, PBC: N = 12. The dotted line corresponds to the cutoff value as determined by Youden’s Index (see Table 5). Kruskal-Wallis test with multiple comparisons to healthy donors; Dunn’s correction. (b) No significant differences were detected by direct ELISA when either murine (Mm, gray symbols) or human mitochondria (Hs, black symbols) were used as coating antigens to detect AwMAs in control and SLE patient sera (1:100). Two-way ANOVA. (c) AwMA binding to coating mitochondria is inhibited in presence of mitochondria (filled circles) but not by red blood cells microparticles (filled squares). Two-way repeated measures ANOVA with multiple comparison (Dunnett’s correction) to signals detected without competitors (i.e. 100%). Experiments presented in panels 3 a and 3 c were performed using mouse mitochondria, the experiment presented in panel 3 b was performed in parallel on murine and human mitochondria. Data are mean ± SD. Not significant (ns): p > 0.05. *p < 0.05. ***p < 0.001. ****p < 0.0001. Hs: Homo sapiens; Mm: Mus musculus.
Membrane-bound vesicles in the extracellular milieu, known as extracellular vesicles
or microparticles, are proposed contributors to the antigenic load in SLE64–66. To
determine whether AwMA could also recognize microparticles derived from
membranes from cells or particles other than mitochondria, we utilized red blood cell
microparticles (RBCMP) as blood-borne microparticles devoid of mitochondria as a
competitor in our AwMA-ELISA, and compared it to extracellular mitochondria in
solution. Whereas increased concentrations of competing mitochondria decreased
AwMA binding by up to 49.84 ± 15.01%, RBCMP showed no inhibition of binding of
the SLE antibodies in our assay (Fig. 3c). While these results suggest that the
antibodies detected in the AwMA-ELISA might have a preferred substrate originating
in mitochondrial membrane, we cannot exclude the possibility that other membrane
bound microparticles or even cells may also be recognized by AwMA, given the
probable occurrence of numerous protein and non-protein antigens in mitochondria.
To determine whether mtDNA also represents an antigenic target of SLE
autoantibodies, we isolated mtDNA from crude mitochondria preparations, using
standard DNA extraction by alkaline lysis. Digestion of the mtDNA by Hae II, a
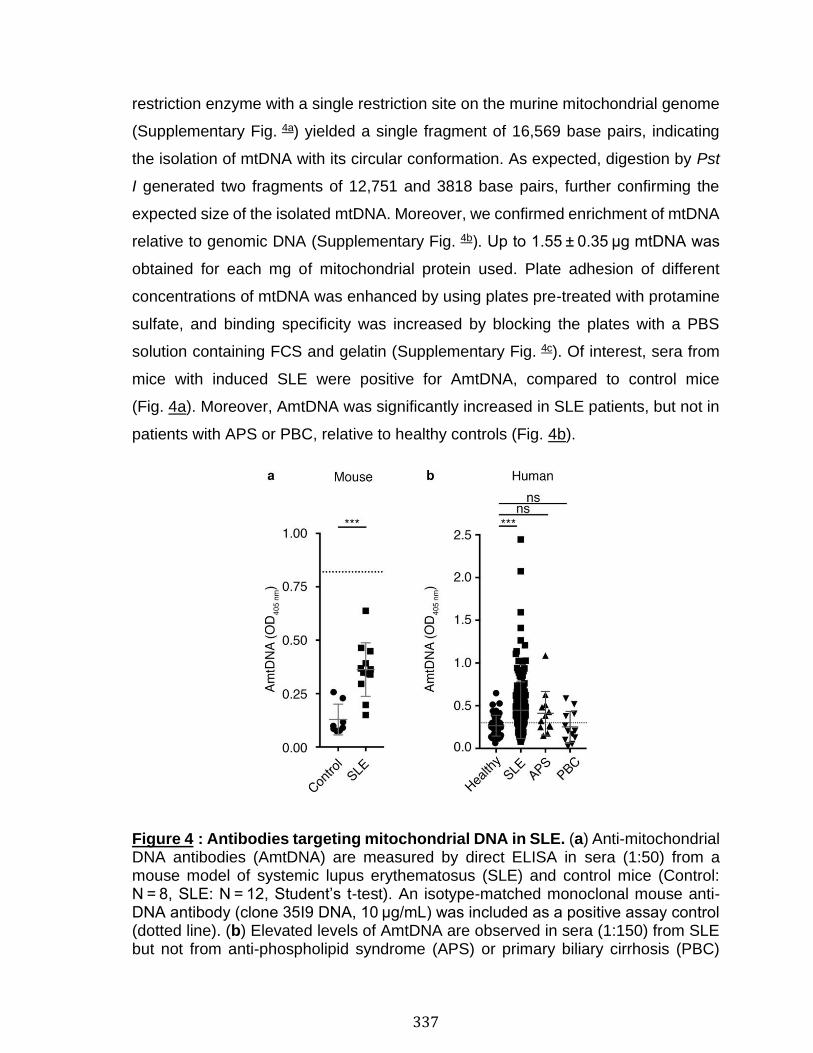
337
restriction enzyme with a single restriction site on the murine mitochondrial genome
(Supplementary Fig. 4a) yielded a single fragment of 16,569 base pairs, indicating
the isolation of mtDNA with its circular conformation. As expected, digestion by Pst
I generated two fragments of 12,751 and 3818 base pairs, further confirming the
expected size of the isolated mtDNA. Moreover, we confirmed enrichment of mtDNA
relative to genomic DNA (Supplementary Fig. 4b). Up to 1.55 ± 0.35 µg mtDNA was
obtained for each mg of mitochondrial protein used. Plate adhesion of different
concentrations of mtDNA was enhanced by using plates pre-treated with protamine
sulfate, and binding specificity was increased by blocking the plates with a PBS
solution containing FCS and gelatin (Supplementary Fig. 4c). Of interest, sera from
mice with induced SLE were positive for AmtDNA, compared to control mice
(Fig. 4a). Moreover, AmtDNA was significantly increased in SLE patients, but not in
patients with APS or PBC, relative to healthy controls (Fig. 4b).
Figure 4 : Antibodies targeting mitochondrial DNA in SLE. (a) Anti-mitochondrial DNA antibodies (AmtDNA) are measured by direct ELISA in sera (1:50) from a mouse model of systemic lupus erythematosus (SLE) and control mice (Control: N = 8, SLE: N = 12, Student’s t-test). An isotype-matched monoclonal mouse anti-DNA antibody (clone 35I9 DNA, 10 µg/mL) was included as a positive assay control (dotted line). (b) Elevated levels of AmtDNA are observed in sera (1:150) from SLE but not from anti-phospholipid syndrome (APS) or primary biliary cirrhosis (PBC)

338
patients. Healthy: N = 43. SLE: N = 175. APS: N = 12. PBC: N = 12. The dotted line corresponds to the cutoff value as determined by Youden’s index (see Table 5). Kruskal-Wallis test with multiple comparisons to controls/healthy donors; Dunn’s correction). All experiment presented in the figure were performed using mouse mtDNA. Data are mean ± SD. Not significant (ns): p > 0.05. ***p < 0.001. PBC: primary biliary cirrhosis.
Little is known about the association of AwMA and AmtDNA with the clinical
characteristics of SLE. We assessed whether AwMA and AmtDNA were associated
with disease manifestations in 175 SLE patients for whom detailed clinical
information was available. AwMA levels correlated with AmtDNA levels in SLE
patients (rs = 0.23, p = 0.003), but not in healthy controls (rs = 0.15, p = 0.33). In
contrast, AwMA did not correlate with antibodies against other mitochondrial
antigens (i.e. HSP60 and cardiolipin) and was not found to be associated with clinical
outcomes (Tables 3 and and4).4). Interestingly, AmtDNA was associated with both
increased anti-dsDNA antibodies (p = 0.02) and with a history of lupus nephritis
(p = 0.007), but not with any of the other clinical outcomes (Table 4). When the
duration of the disease, the age of the patients, their BMI, the use of prednisone
and/or antimalarial drugs, and circulating cholesterol LDL were taken into account,
the associations of AmtDNA with increased anti-dsDNA antibodies and lupus
nephritis remained significant in a multivariate logistic regression (p = 0.01 for both),
indicating an association between AmtDNA, and these two clinical parameters.
However, AmtDNA did not correlate with anti-dsDNA as measured by Farr assay in
the cohort, suggesting that the results measured by our AmtDNA-ELISA and those
obtained by Farr assay may not be redundant. Cut-off values were identified for
AwMA and AmtDNA (Figs 3, ,44 and Table 5). Our two ELISAs displayed high
specificities (AwMA: 0.88; AmtDNA: 0.74) and allowed us to efficiently discriminate
SLE patients from healthy donors (p < 0.001 for both AmtDNA and AwMA). Using
these values, we determined that 110 patients were positive for AmtDNA (62.86%)
and 101 patients (57.71%) were positive for AwMA.

339
Table 3: Intercorrelations of anti-mtDNA, anti-whole mitochondria, anti-dsDNA and anti-cardiolipin antibodies in SLE patients (n = 175). AmtDNAA AwMAA DNA (Farr)A ACA (+/−)B
Anti-HSP60 0.07 p = 0.37
0.10 p = 0.21
0.02 p = 0.81
(+) 0.28 ± 0.52 (−) 0.52 ± 0.53 p = 0.08
AmtDNA — 0.23, p = 0.003 0.05 p = 0.46
(+) 0.33 ± 0.17 (−) 0.37 ± 0.27 p = 0.40
AwMA — — 0.10 p = 0.19
(+) 0.32 ± 0.20 (−) 0.33 ± 0.23 p = 0.51
AValues are presented as Spearman correlation coefficient and p-value. BValues presented as median ± IQR and Wilcoxon test p-value for patient positives (+) or negatives (−) for ACA. ACA: anti-cardiolipin antibodies; AwMA: anti-whole mitochondria antibodies. AmtDNA: anti-mitochondrial DNA antibodies; DNA Farr: quantification of anti-dsDNA antibodies by Farr assay; HSP60: heat-shock protein 60 KDa.
Table 4: AmtDNA and AwMA associations with clinical manifestations in SLE patients (n = 175).
Clinical Outcomes AmtDNA AwMA
OR (CI) p OR (CI) p
Thrombotic events 0.43 (0.10–1.79) 0.25 0.27 (0.04–1.99) 0.2
[0.35 (0.07–1.67)]* [0.19] [0.21 (0.02–1.82)] [0.15]
SLEDAI-2K ≥ 4 0.96 (0.37–2.51) 0.93 1.34 (0.49–3.69) 0.57
[1.01 (0.33–3.05)] [0.99] [1.08 (0.37–3.14)] [0.89]
SDI ≥ 1 0.91 (0.35–2.42) 0.86 0.85 (0.30–2.40) 0.76
[0.93 (0.32–2.68)] [0.90] [0.69 (0.23–2.09)] [0.52]
Increased anti-dsDNAA 3.34 (1.22–9.16) 0.02 1.15 (0.42–3.16) 0.79
[3.94 (1.33–11.69)] [0.01] [1.16 (0.40–3.35)] [0.79]
Lupus nephritis 4.45 (1.50–13.20) 0.007 1.06 (0.39–2.90) 0.91
[4.60 (1.41–14.99)] [0.01] [0.97 (0.34–2.73)] [0.95]
AOccurrences of patients with anti-dsDNA antibodies above the clinical threshold. AwMA: anti-whole mitochondria antibodies. AmtDNA: anti-mitochondrial DNA antibodies. SDI: lupus severity disease index. SLEDAI-2K: systemic lupus erythematosus disease activity index - 2000. OR (CI): Odds ratios (95% Wald Confidence Interval). P from logistic regressions.
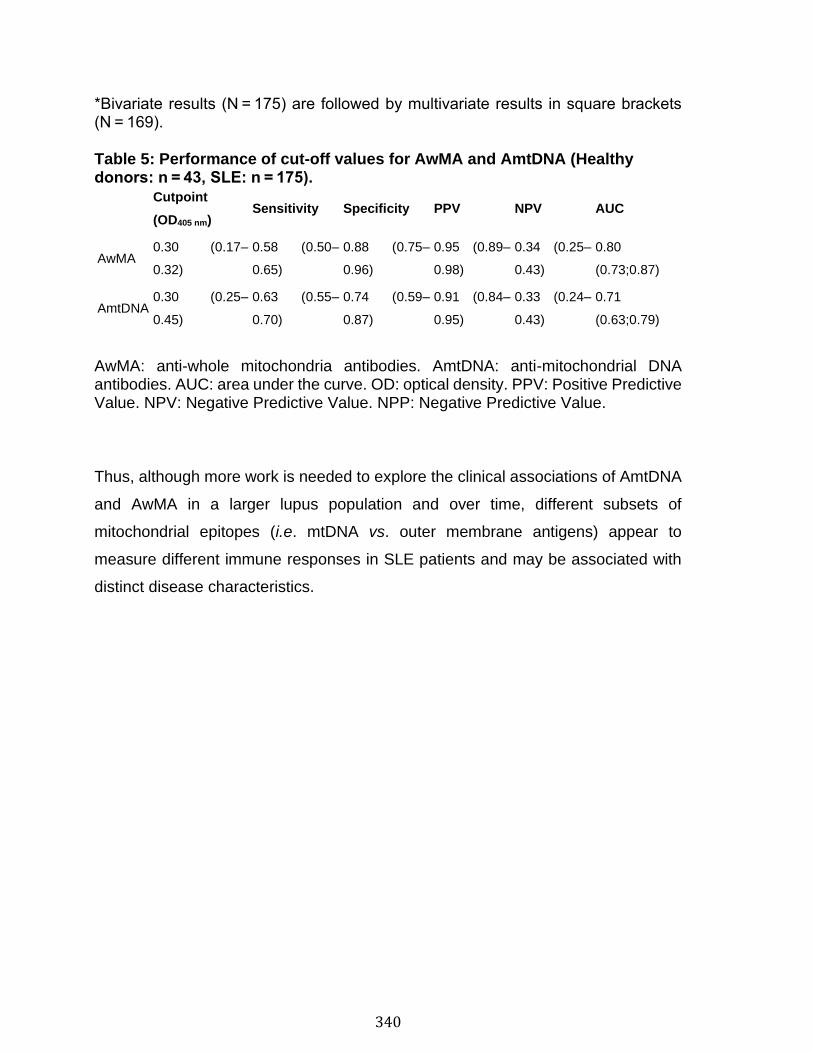
340
*Bivariate results (N = 175) are followed by multivariate results in square brackets (N = 169).
Table 5: Performance of cut-off values for AwMA and AmtDNA (Healthy donors: n = 43, SLE: n = 175).
Cutpoint
(OD405 nm) Sensitivity Specificity PPV NPV AUC
AwMA 0.30 (0.17–
0.32)
0.58 (0.50–
0.65)
0.88 (0.75–
0.96)
0.95 (0.89–
0.98)
0.34 (0.25–
0.43)
0.80
(0.73;0.87)
AmtDNA 0.30 (0.25–
0.45)
0.63 (0.55–
0.70)
0.74 (0.59–
0.87)
0.91 (0.84–
0.95)
0.33 (0.24–
0.43)
0.71
(0.63;0.79)
AwMA: anti-whole mitochondria antibodies. AmtDNA: anti-mitochondrial DNA antibodies. AUC: area under the curve. OD: optical density. PPV: Positive Predictive Value. NPV: Negative Predictive Value. NPP: Negative Predictive Value.
Thus, although more work is needed to explore the clinical associations of AmtDNA
and AwMA in a larger lupus population and over time, different subsets of
mitochondrial epitopes (i.e. mtDNA vs. outer membrane antigens) appear to
measure different immune responses in SLE patients and may be associated with
distinct disease characteristics.

341
Discussion
Mitochondrial DAMPs are known to stimulate the innate immune response, but it is
less clear whether mitochondrial antigens stimulate an antibody response and
whether these antibodies impact the inflammatory reaction. Interestingly, in PBC, a
paradigm of true organ-specific autoimmunity, evidence points to involvement of
both innate and adaptive immunity with a specific antibody response to mitochondrial
antigens67.
Although the prevalence of AMA in SLE was reported several decades ago59, this
observation was not pursued. Our study confirms the presence of AwMA (directed
against the mitochondrial surface and not redundant with that of PBC) and AmtDNA
(directed against mtDNA) in patients with SLE. However, it remains to be established
whether AwMA are pathogenic initiators of the auto-immune process or whether they
are consequences of an a priori cell activation or injury with subsequent release of
mitochondria in the extracellular milieu. These “free mitochondria” could
subsequently become antigenic in predisposed individuals and be a marker of cell
or tissue injury. They could also be a cause of further immune activation through the
formation of circulating or in-situ immune complexes, constituting a secondary
trigger of inflammation. The recognition of mitochondria by antibodies could also
implicate Fc receptors and thus modulate a distinct immune response. For example,
the intravenous injection of mtDNA into mice failed to induce proteinuria and kidney
damage68, but the response may differ in a recipient with circulating antibodies to
mitochondria.
Epitope modification, such as oxidation, can impact its antigenicity69. Notably, there
are reports suggesting that the oxidation of mtDNA occurs during its extrusion from
cells, and that the oxidized form is pathogenic15,16. Our findings using in vitro
oxidation of the mitochondria suggest that mitochondrial epitopes, regardless of
oxidation status, are targeted by autoantibodies in SLE. However, this does not
exclude the possibility that oxidized mitochondria may be more antigenic in other
pathogenesis, such as APS, or more efficient at promoting responses if recognized

342
by the innate immune system. Moreover, it is not excluded that following oxidation,
mitochondrial swelling and potential formation of pores within the mitochondrial
membrane might have revealed new antigens detected in our anti-whole
mitochondria ELISA using TBHP-treated mitochondria, but this treatment may also
have caused a release of existing antigens that could have been lost in this modified
assay, thus explaining the apparent absence of impact of oxidation of the antigenicity
of the mitochondria70. Future investigations will be needed to determine the relative
targeting of native versus oxidized forms of organelle components by immune cells.
We could not verify the role of oxidation on the recognition of mtDNA by
autoantibodies due to spontaneous oxidation of isolated mtDNA, an observation also
made by other groups despite extreme preventive measures taken to maintain the
molecules under their reduced form71. Thus, it is likely that AmtDNA and anti-dsDNA
antibodies routinely measured in clinical testing both evaluate antibodies to oxidized
DNA.
We observed that AwMA levels were more elevated in SLE than in PBC. PBC
patients, however, were positive for AMA when sonicated mitochondria were utilized
in our ELISA. These observations are consistent with the fact that pyruvate
dehydrogenase complex E2 (PDC-E2), the immunodominant epitope in PBC, is
located in the mitochondrial inner membrane. Our AwMA-ELISA measures
antibodies to epitopes on intact mitochondria, in which inner membrane epitopes
remain unavailable72. This contrasts with the sonicated mitochondria used in
previous studies on AMA in SLE59, which contained inverted membranous structures
and thus revealed antigens located in the mitochondrial inner membrane60.
Furthermore, we found that AwMA levels correlated with the levels of AmtDNA, but
not with other antibodies directed at antigens located within the inner membrane of
the organelle (e.g. cardiolipin or HSP60), pointing to the existence of mitochondrial
antigens that remain to be identified73. Our study provides simple and quantitative
assays for assessment of two types of AMA (i.e. AwMA and AmtDNA) likely specific
to SLE. Further research is required to identify the mitochondrial antigen(s) and
epitope(s) of our AMA assay in SLE.

343
Our analyses of AmtDNA antibodies in human sera showed an association with anti-
dsDNA as well as with lupus nephritis, consistent with the documented associations
between anti-dsDNA antibodies detected by Farr assay and lupus nephritis74. While
dsDNA used in Farr assays is usually isolated from plasmids (i.e. E. coli) and thus
may share similarities with mtDNA (e.g. hypomethylated CpG motives, circular
tertiary structure)75, the Farr assay has been described for its specificity in the
detection of high avidity antibodies of all classes (IgG and IgM, for instance). Our
data suggest that the Farr assay probably detects AmtDNA. Consistent with this,
nuclear DNA (nDNA) efficiently competed in our AmtDNA ELISA assay
(Supplementary Fig. 5a), further pointing to a certain degree of redundancy between
anti-dsDNA and AmtDNA. When nuclear and mtDNA were treated with S1 nuclease,
which digests potential single-stranded regions, both could compete in our AmtDNA
ELISA assay (Supplementary Fig. 5b), which suggests that most of the antigenicity
of mtDNA is provided by its double-stranded conformation. There might exist subsets
of antibodies that preferentially recognize mtDNA versus genomic DNA, but there is
no evidence of their occurrence at this stage. As extracellular mtDNA is reportedly
associated with various pathologies, including trauma25,26, burn injury27, cancer28
and rheumatoid arthritis17,29, it is tempting to postulate that extruded mtDNA, beyond
its role as a pro-inflammatory signal for the innate immune system, represents a
significant antigenic load available for the formation of immune complexes.
In this study, we focused our attention on immunoglobulin (Ig) G (IgG), but the
assays can be modified easily to quantify specific IgG subclasses (e.g. IgG1, IgG2a,
IgG2b) or other isotypes (e.g. IgA and IgM), which may reveal distinct functions of
these antibodies in disease. Both AwMA and AmtDNA were detected at low levels
in healthy individuals, suggesting that these antibodies are further induced during
SLE pathogenesis. These antibodies might be part of a pool of naturally occurring
autoantibodies that are thought to contribute to the continual clearance of membrane
vesicles in the circulation76,77. Protective natural autoantibodies have been described
predominantly as IgM, but other isotypes such as IgG or IgA have also been
reported78–80. It is thus tempting to postulate that antibodies targeting mitochondrial
epitopes are present in healthy individuals and might be involved in clearance of

344
extracellular mitochondria. A profound change in the balance of natural IgM and
pathogenic IgG against mitochondrial epitopes may in part explain the pathogenesis
in SLE.
While these observations require confirmation in an independent cohort of patients,
they suggest that, in SLE, the adaptive immune system recognizes mitochondrial
organelles. Furthermore, our findings suggest that clinical associations may differ
according to antibody recognition of inner (e.g., ACA) versus outer mitochondrial
(e.g., AwMA) membrane components. This leads to the postulation that
pathogenicity of these antibodies may depend on whether extracellular mitochondria
are intact or not. Evaluation of antibodies to mitochondrial components in SLE may
provide novel information on patients, such as their risk for developing nephritis. If
these findings are confirmed in a large prospective cohort of SLE patients, AwMA
and AmtDNA may prove useful in predicting disease activity and disease severity,
and in stratifying SLE patients. The quantification of mitochondrial antibodies may
thus open the way to novel directions in autoimmune disease research and may be
useful for achieving a better understanding of disease mechanisms.

345
Material and Methods
Study approval Patient population
The human sera tested in this study were obtained from a University Health Network
research ethics board (REB) approved study of SLE and APS patients in Toronto as
well as from healthy controls and PBC patients recruited from CHU de Québec –
Université Laval REB approved studies in Quebec City. SLE patients had to meet
the 1982 ACR classification criteria for SLE revised in 199781,82 and APS patients
met the 1999 Sapporo criteria for APS revised in 200683,84. For SLE, consecutive
female patients from the University of Toronto Lupus Clinic (UTLC) were approached
and provided consent between August 2010 and October 2011 to be part of a study
on the role of fatty acids and cardiovascular disease in lupus. They provided one
blood specimen and had their anonymized clinical data linked to their biospecimen.
Similarly, APS patients seen at the rheumatology clinic in Toronto were approached
following a similar procedure. All remaining biospecimen could be used for future
studies on biomarkers of lupus as per the original subjects’ consent. Healthy control
volunteers were recruited as part of study on markers of inflammation if they had no
known illnesses and did not have infectious symptoms at the time of the blood draw.
Age and sex were collected at that time. PBC patients met the 2009 PBC
classification criteria, revised in 201838,85 including the positivity for anti-
mitochondrial antibodies.
Data from clinical laboratories
For SLE patients, anti-dsDNA were measured using the Farr assay (laboratory cut-
off of 30%) and the anti-cardiolipin (IgG and IgM – laboratory cut-offs of 40 GPL or
MPL units) were measured by ELISA in a clinical laboratory.
Cell culture
Hep-G2 human hepatocarcinoma cells (ATCC, Manassas, VA, USA) were cultured
at 37 °C – 5% CO2 in Eagle’s Minimum Essential Medium (EMEM) (Wisent)

346
supplemented with 10% fetal bovine serum (FBS) (Wisent), non-essential amino-
acids (Wisent) and penicillin/streptomycin (Wisent).
Inducible mouse model of SLE
Recommendations of the Canadian Council on Animal Care were followed in a
protocol approved by the Animal Welfare Committee at Université Laval. C57BL/6 J
were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and housed in an
Elite-Plus specific-pathogen-free animal facility at CHU de Quebec. SLE
autoantibodies were induced in these mice as previously described63. In brief, 6–8-
week-old male mice received 100 µL i.v. injections of β2GPI (20 µg) (Crystal Chem,
Downers Grove, IL, USA) followed 24 h later by a 100 µL i.v. injection of
lipopolysaccharide (LPS from E. coli, O111:B4; 10 µg) (Sigma-Aldrich, Saint-Louis,
MO, USA). These injections were repeated after 2 and 4 weeks for a total of three
rounds of immunizations and mice were bled 48 h after the third immunization.
C57BL/6 J mice injected i.v. with PBS under the same schedule were used as
controls.
Ethics
Throughout the entire study, blood samples were obtained from patients under
informed consent. All the methods presented in this study were performed in
accordance with the relevant guidelines and regulations for both human and murine
donors.
Mitochondria isolation
Mitochondria were isolated from the livers of C57BL/6 J mice following a combination
of published protocols61,62. Mice were sacrificed by cervical dislocation, the liver was
swiftly removed, the gallbladder excised, and the liver was rinsed in ice-cold PBS
(137 mM NaCl, 3 mM KCl, 19 mM Na2HPO4, 2 mM KH2PO4). The liver was then
minced and transferred to a pre-cooled glass/Teflon tissue grinder containing 12 mL
of ice-cold mitochondrial isolation buffer (10 mM Tris, 1 mM EGTA, 200 mM sucrose)
per gram of liver then ground until an homogeneous suspension was obtained. Intact

347
cells and nuclei were pelleted twice at 700 g, 4 °C, 10 min. Contaminating
membranes, proteins and organelles were eliminated by two centrifugations at
7,000 g, 4 °C, 10 min followed by a centrifugation at 10,000 g, 4 °C, 10 min. The
crude mitochondrial suspension was further purified by ultracentrifugation against a
Percoll gradient (10 mM Tris, 1 mM EGTA, 30% v:v Percoll) at 95,000 g, 4 °C, 30 min.
The band containing mitochondria was collected in PBS. A similar approach was
used to isolate human mitochondria with the exception that up to 107 Hep-G2 cells
were lysed by repeated passages through a narrow-gauge needle. The commercial
kit QIAgen Qproteome (QIAgen, Hilden, Germany) was also used, following the
manufacturer’s instructions, to isolate mitochondria from Hep-G2 cells for quality
comparison by western blotting with the aforementioned protocol. Isolated
mitochondria were dosed by the bicinchoninic acid (BCA) method using BCA Protein
Assay Kit (Thermo Fisher scientific, Waltham, MA, USA). Freshly isolated
mitochondria were used in all experiments with the exception of the submitochondrial
particles preparations for which pelleted mitochondria were kept at −80 °C until
needed.
Submitochondrial particles (SMP) preparation
SMP were prepared as described elsewhere60. Briefly, frozen mitochondria were
thawed at room temperature and diluted to 10 mg of mitochondrial proteins in 10 mM
4-Morpholinepropanesulfonic acid (MOPS). Samples were then sonicated using a
Fisher Sonic Dismembrator Model 500 (Thermo Scientific) at 20% maximal output
for 20 seconds then kept on a salt-ice water bath for 10 minutes. This cycle was
repeated nine times. Samples were then centrifuged at 16,000 g, 4 °C, 10 minutes in
order to discard unbroken mitochondria and other unwanted debris. Supernatants
were collected in a fresh tube and their volumes adjusted to 2 mL in 10 mM MOPS.
Samples were then ultracentrifuged at 150,000 g, 4 °C for 45 minutes. Pelleted SMP
were resuspended in SMP buffer (225 mM mannitol, 75 mM sucrose, 10 mM
HEPES, 0.1 mM EDTA, pH 7.4) and dosed. SMP preparations were kept at −80 °C
until needed.

348
Red blood cells microparticles (RBCMP) preparation
Red blood cells (RBC) were isolated from the blood of healthy human volunteers by
centrifugation at 282 g for 10 minutes at RT. Platelet-rich plasma and buffy coat were
discarded, and the RBC fraction kept. RBC were counted (Cellometer Auto M10,
Nexcelom Bioscience, Lawrence, MA, USA) and adjusted to a concentration of
5 × 108 cells/mL in Tyrode’s buffer (12 mM NaHCO3, 127 mM NaCl, 5 mM KCl,
0.5 mM NaH2PO4, 1 mM MgCl2, 5 mM Glucose, 10 mM HEPES, pH 7.4). 109 cells
were then diluted in 45 mL double-distilled water and incubated for 5 minutes. The
tonicity of the buffer was then balanced by addition of 5 ml of PBS 10X (filtered
through a 0.22-μm membrane). Remnant RBC were removed by centrifugation at
1,300 g for 5 minutes at RT and supernatant was centrifugated at 18,000 g for
90 minutes at 18 °C. RBC microparticle pellet was then suspended in 500 μl PBS.
Protein concentration was measured with BCA assay.
Western blotting
After quantification, 25 µg of sample per lane were loaded onto a 12%
polyacrylamide gel and underwent migration for 1 h 30 min at 100 V (constant
voltage). Gels were then transferred overnight onto polyvinylidene difluoride
membranes (PVDF. BioRad, Hercules, CA, USA) at 4 °C and 60 mA (constant
current). Non-specific binding sites were blocked in Tris-buffered saline (TBS)−0.1%
Tween20 containing 5% non-fat dry milk for 4 h at room temperature. Proteins of
interest were labeled overnight at 4 °C with either mouse anti-actin (Clone AC-15,
1:5,000. Sigma-Aldrich), mouse anti-tubulin (Clone DM1A, 0.2 µg/mL. Abcam,
Cambridge, UK), mouse anti-proliferating cell nuclear antigen (PCNA. Clone PC10,
1 µg/mL. Santa Cruz biotechnology, Santa Cruz, CA, USA), rabbit anti-VDAC (Clone
D73D12, 1:1,000. Cell Signaling Technology, Danvers, MA, USA), mouse anti-
TOMM22 (Clone IC9-2, 2 µg/mL. Abcam) or mouse anti-cytochrome C antibody
(Clone 7H8.2C12, 0.5 µg/mL. BD Biosciences, Franklin Lakes, NJ, USA), polyclonal
rabbit anti-proteasome 20 S (0.8 µg/mL. Abcam), polyclonal rabbit anti-GRP94
(1:2,000, Abcam) diluted in superblock (BioRad). Following a 1-h incubation with
horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibody (1:10,000 in

349
superblock) (Jackson Immunoresearch, West Grove, USA), signals were visualized
with Western Lightning Chemiluminescence Reagent (Perkin Elmer, Waltham, MA,
USA) on a C-DiGit membrane scanner (LI-COR biotechnology, Lincoln, NE, USA).
Total proteins isolated from starting materials (i.e. mouse liver or Hep-G2 cells) were
used as controls. Full-length blots are presented in Supplementary Fig. 6a–c.
Mitochondrial size measurement by dynamic light scattering (DLS)
The size of the isolated mitochondria was measured by DLS, using a Zetasizer Nano
ZS device (Malvern instruments, Malvern, UK) equipped with the standard 4 mW,
633 nm He-Ne laser as a light source, set at a detection angle of 173°. Experiments
were replicated three times in 1 cm length disposable UV-Cuvettes (Brand GMBH,
Wertheim, Germany) containing 100 µL of isolated murine mitochondria diluted in
PBS at a concentration of 10 µg proteins/µL. The following parameters were taken
into account upon measuring the size of the mitochondrial: refraction index of the
solvent: 1.330, viscosity of the sample: 0.8872 mPas, refraction index of the proteins:
1.45, temperature: 25 °C.
Electron microscopy
Mitochondria (108) were fixed in 3.5% acrolein for 15 min at room temperature. Fixed
samples were rinsed twice in PBS then embedded in 4% agarose. 50 µm sections
were cut using a vibratome, post-fixed in 1% osmium tetroxide for 30 minutes and
embedded in Durcupan resin. Seventy-nanometer ultrathin sections were visualized
at 80 kV using a Tecnai G2 Spirit BioTWIN (FEI, Hillsboro, OR, USA) transmission
electron microscope.
Assessment of the mitochondrial oxygen consumption
Mitochondria were resuspended in mitochondrial assay solution (MAS: 70 mM
sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM
EGTA and 0.2%(w/v) fatty acid-free BSA, pH 7.4 at 37 °C) and supplemented with
10 mM pyruvate, 2 mM malate and 4 μM FCCP [carbonyl cyanide 4-
(trifluoromethoxy)phenylhydrazone], pH 7.4. An equivalent of 10 μg proteins was

350
seeded on XF-96 plates (Agilent, Santa Clara, CA, USA). Plates were then
centrifuged 2,000 g for 20 min at 4 °C. We visualized the distribution of mitochondria
under a bright-field microscope to ensure adherence and homogeneous repartition.
Plates were maintained at 37 °C without CO2 for approximately 40 minutes prior to
loading. Oxygen consumption rates (OCR) were measured in accordance with
manufacturer instructions (Agilent/Seahorse Bioscience). Experiments were
replicated in three wells and averaged for each experimental condition. A total of 3
measurements of oxygen consumption for each condition were made approximately
every 6 minutes under basal conditions and after sequential addition of rotenone
(2 μM), succinate (10 mM), antimycin A (40 μM) and TMPD (N,N,N′,N′-tetramethyl-
p-phenylenediamine)/Ascorbate (100 μM/10 mM). Succinate was used as the
electron donor for complex II, rotenone as a complex I inhibitor, antimycin A as a
complex III inhibitor and respiration through complex IV was measured using
TMPD/ascorbate.
High sensitivity flow cytometry
Due to their small size, mitochondria were detected by high sensitivity flow
cytometry, using a “small particle option” consisting of a forward scatter (FSC)
coupled to a photomultiplier tube (PMT) mounted on a BD FACS Canto II Special
Order Research Product (SORP, BD Biosciences). Mitochondria (0.5 µg) were
stained with 1 µg of anti-TOMM22-Atto 488 (clone IC9-2, Sigma-Aldrich) and 1 µM
of mitotracker deep red (Invitrogen, Carlsbad, CA, USA) for 30 minutes at 37 °C and
diluted with PBS to a final volume of 500 µl before flow cytometry analysis. To
quantitatively measure the number of mitochondria, we used 3 µm diameter
polystyrene microsphere (Polysciences, PA, USA). 80,000 microspheres were
added to each sample and 500 microspheres were acquired. Silica particles (Kisker
Biotech, Steinfurt, Germany) were used to determine 100–1,000 nm size scale.
Mitochondrial DNA extraction by alkaline lysis
Isolated mitochondria were pelleted at 10,000 g, 4 °C, 10 min and resuspended in
0.8 mL mtDNA isolation buffer A [50 mM Tris-HCl, 10 mM EDTA, 100 µg/mL RNAse

351
A (QIAgen), pH 7.5] for every 3 mg of mitochondrial proteins. Mitochondria were then
lyzed in one volume (v:v) mtDNA isolation buffer B (extemporaneously-prepared.
0.2 M NaOH, 1% SDS) for 3 min. on ice, under gentle agitation. mtDNA suspensions
were neutralized with one volume (v:v) mtDNA isolation buffer C (1.32 M potassium
acetate, pH 4.8 adjusted with ice-cold acetic acid) for 5 min. on ice, under gentle
agitation. Mitochondrial debris were pelleted by centrifugation for 10 minutes at
14,000 g, RT. Supernatants were transferred to fresh tubes. mtDNA was precipitated
overnight at −20 °C with 0.1 volume (v:v) potassium acetate (stock solution: 3 M
sodium acetate, pH adjusted to 5.2 with ice-cold acetic acid) and 0.7 volume (v:v)
absolute isopropanol. mtDNA was then pelleted at 14,000 g, washed thrice with
1.5 mL 70% ethanol and resuspended in DNA resuspension buffer (10 mM Tris-HCl,
10 mM EDTA, pH 8.0). Mitochondrial DNA concentration was determined by
spectrophotometry (Nanodrop 1000, Thermo Fisher Scientific).
Nuclei isolation from mouse hepatocytes
Nuclei were isolated from mouse livers using published methods86. Briefly, during
mitochondria isolation protocols, while supernatants were used for performing
mitochondria isolations, pellets from the first 700 g centrifugation were resuspended
in 10 mL of ice-cold nuclei isolation buffer A (5 mM MgCl2, 10 mM Tris–HCl, 250 mM
Sucrose. pH 7.4) and ground in a pre-cooled glass/Teflon tissue grinder. The
suspension was further disrupted by three passages through a 25 G5/8 gauge needle
followed by two passages through a 27 G ½ gauge needle. The suspension was
then filtered against a 40 µM nylon cell strainer (Thermo Fisher Scientific) and
centrifuged at 600 g, 4 °C, 10 min. Pellets were resuspended in 14 mL buffer A and
centrifuged at 600 g, 4 °C, 10 min. Pellets were then resuspended in 9 volumes of
ice-cold nuclei isolation buffer B (1 mM MgCl2, 10 mM Tris– HCl, 2.0 M sucrose. pH
7.4) and centrifuged at 16,000 g, 4 °C, 30 min. Pellets were resuspended in 200 µL
PBS and stored at −20 °C.

352
Whole-cell and nuclear DNA isolation
DNA from either 25 mg of whole mouse liver or the 200 µL or isolated nuclei were
extracted using QIAmp® DNA Mini Kit (QIAgen), following instructions from the
manufacturer. Samples were eluted in DNA resuspension buffer.
Mitochondrial DNA and nuclear DNA enrichments by qPCR
Thirty nanograms of mitochondrial DNA, nuclear DNA or the same amount of total
DNA extracted from whole mouse liver using the aforementioned protocols, were
amplified in a Rotor-Gene Q real time qPCR cycler (QIAgen) according to standard
protocols with SsoAdvanced Universal SYBR® Green Supermix (BioRad) in a 10 µL
reaction volume. Two distinct primer couples were used: one specific to
mitochondrial DNA (5′-GGAACAACCCTAGTCGAATGAA-3′/5′-
GCTAGGGCCGCGATAATAAA-3′) and the other to nuclear DNA (5′-
CCTGCTGCTTATCGTGGCTG-3′/5′-GCCAGGAGAATGAGGTGGTC-3′). The
experimental conditions were: 50 °C - 2 minutes in, 95 °C - 10 min and 40 cycles
(95 °C - 15 seconds, 60 °C - 1 minutes). Fold changes were calculated with the 2−ΔCt
method setting mean value for total liver DNA extracts as 1.
Mitochondrial DNA digestion by restriction enzymes
Purified mouse mtDNA was digested by Hae II or Pst I restriction endonucleases
(NEB, Whitby, ON, Canada) according to the manufacturer’s protocol. Digested
products (1.5 μg) were then resolved on a 0.5% (w/v) agarose gel. Images were
acquired on a Chemidoc MP gel documentation system (Bio-Rad). Full-length gel is
presented in Supplementary Fig. 6d.
S1 nuclease treatment of mitochondrial DNA and nuclear DNA
30 µg of mtDNA and nDNA were diluted in 200 µL of 1X S1-buffer and treated with
50 U S1 nuclease (Thermo Fischer Scientific) for 5 minutes at 37 °C. Reaction was
stopped by addition of 600 mM EDTA (final concentration) and incubation at 70 °C
for 10 minutes. Samples were precipitated by addition of 300 mM Sodium Acetate
(final concentration) and 2 volumes (v:v) absolute ethanol for 16 hours at −20 °C.

353
Samples were pelleted at 14,000 g, RT, 20 minutes, and washed 1 mL 70% ethanol
and resuspended in DNA resuspension buffer. For competition assays, untreated
nDNA and mtDNA followed the same treatment without addition of the enzyme.
Samples were dosed by qPCR.
Mitochondria oxidation in-vitro
Dosed mitochondria were pelleted and resuspended at a concentration of 1.5 mg of
mitochondrial proteins/mL in PBS containing 500 µM of the oxidant tertbutyl
hydroperoxide (TBHP) (Sigma-Aldrich). Mitochondria were oxidized for 1 h 30 min at
37 °C under gentle agitation then rinsed twice with ice-cold PBS. Oxidized
mitochondria were subsequently quantified by BCA.
Mitochondrial lipid oxidation
Thiobarbituric acid reactive substances (TBARS) formation was assessed, using the
TBARS Parameter Kit (R&D systems, Minneapolis, MN, USA), following the
manufacturer’s instructions. Two hundred micrograms of mitochondria were lysed,
the proteins were precipitated using trichloroacetic acid (TCA) and pelleted at
12,000 g, room temperature, 4 min. The supernatants were transferred to fresh
tubes. Samples (75 µL) were incubated with 37.5 µL of thiobarbituric acid in a 96-
well plate for 3 h at 50 °C. Absorbances were read at 532 nm on a SpectraMax 190
microplate reader (Molecular Devices, Sunnyvale, CA, USA) and TBARS
quantification was performed using a standard curve provided in the kit. Oxidized
samples were compared to control mitochondria that were incubated under the same
conditions in PBS devoid of TBHP.
Mitochondrial protein oxidation
The formation of carbonyl moieties following in-vitro mitochondrial oxidation was
measured using the Protein Carbonyl Content Assay Kit (Sigma Aldrich) following
the manufacturer’s instructions. Oxidized samples were compared to control
mitochondria that were incubated under the same conditions in PBS devoid of TBHP.

354
Detection of antibodies targeting mitochondrial epitopes by ELISA
For the detection of anti-whole mitochondrial antibodies (AwMA), murine
mitochondria were diluted (500 µg/mL) in 50 mM carbonate/bicarbonate buffer, pH
9.6 and 25 µL per well were loaded onto 96-well half-area clear flat bottom
polystyrene high-binding microplates (Corning, New York, USA). Plates were coated
for 18 h at 4 °C then blocked for 4 h at 37 °C with PBS containing 10% FBS and 0.5%
gelatin. After three washes with PBS, sera diluted 1:150 (unless otherwise specified)
in PBS-10% FBS-0.3% gelatin were incubated overnight at 4 °C in duplicate. After
three washes with PBS, plates were incubated for 1 h at room temperature with
alkaline phosphatase-(AP) conjugated goat anti-mouse or anti-human IgG (Sigma-
Aldrich) diluted 1:1,000 in PBS-0.4% bovine serum albumin (BSA). Plates were
washed thrice with PBS and developed with p-nitrophenol phosphate (p-NPP) for
~30 min at 37 °C and optical densities (OD) were read at 405 nm on a microplate
reader. The same protocol was used for the detection of autoantibodies targeting
submitochondrial particles by using 25 µL per well of SMP diluted (50 µg/mL) in
50 mM carbonate/bicarbonate buffer, pH 9.6. A similar approach was used for
human mitochondria with the following modification: plates were incubated with a
horseradish peroxidase-conjugated anti-human IgG secondary antibody (1:3,000),
peroxidase activity was revealed at room temperature for ~5 min with 3,3′,5,5′-
tetramethylbenzidine (TMB). The reaction was stopped with 2 N H2SO4 and the ODs
read at 450 nm. For the anti-mtDNA ELISA, plates were pre-coated with 1%
protamine sulfate in double-distilled water for 1 h at room temperature. Plates were
washed three times with PBS and coated overnight at 4 °C with 400 ng mtDNA in
PBS. All subsequent steps were identical to those used for AwMA-ELISAs. Blank
values (mitochondrial antigens and no sera) were substracted from measured values
for each patient. During the development of these assays, the proper coating of the
wells was tested by using isotype-matched mouse monoclonal antibodies (mAb). A
monoclonal antibody (Clone IV.3, 4 µg/mL) was used as a negative assay control in
each instance while different monoclonal antibodies were used, depending on the
coating antigens, as positive assay controls: an anti-TOMM22 mAb (Clone IC9-2,
4 µg/mL. Abcam) for intact mitochondria, an anti-DNA mAb (Clone 35I9 DNA,

355
10 µg/mL. Abcam) or an anti-Cytochrome C mAb (Clone 7H8.2C12, 5 µg/mL. BD
Biosciences).
Competition assay
AwMA-ELISAs were performed as described in the previous sections with the
following modifications: serum samples were pre-incubated in dilution buffer (1:150)
spiked with various concentrations (0.25, 1 and 3 mg/mL) of competitors (i.e.
mitochondria or red blood cells microparticles) for 3 hrs. at RT and incubated in
duplicate overnight at 4 °C. Data are presented as the percentage of signal
remaining after each competition, compared to the OD (405 nm) measured in
absence of competitors.
A similar procedure was used for AmtDNA-ELISA, using increased concentrations
(0, 3, 9 and 27 ng/µL) of competing DNA (extracted from nuclei or mitochondria, with
or without S1 nuclease treatment).
Statistics
Comparisons between groups were made using either Student’s t-test, Wilcoxon
test, Friedman or Kruskal-Wallis tests, one-way ANOVA, two-way or repeated
measures ANOVA depending on the outcome, as well as the number and type of
groups. When multiple comparisons were assessed, appropriate post-hoc correction
tests were used such as Dunn’s, Dunnett’s, Sidak’s or Bonferroni’s. Associations
between AwMA, AmtDNA and anti-HSP60 were computed with Spearman
correlations. Distribution of these antibodies according to ACA results were
compared using Wilcoxon test. Associations between AwMA or AmtDNA and clinical
outcomes were studied by bivariate and multivariate linear and logistic regressions.
Clinical outcomes studied are average intima media thickness, percent change of
the flow mediated dilatation (FMD) of the brachial artery, presence of FMD, presence
of plaque in the carotid, thrombotic event ever, white blood cells count, platelet count,
increased DNA binding by Farr assay above normal range for testing laboratory,
presence of damage according to SLICC Damage Index (SDI > 0), high activity
according to SLEDAI-2K activity score (SLEDAI ≥ 4), presence of lupus nephritis,

356
biopsy class, as well as chronicity and activity index from the biopsy. The latter were
adjusted for disease duration, age, body mass index (BMI), low-density lipoproteins
(LDL) cholesterol, antimalarial medication and prednisone. Logistic regressions are
presented with odd ratios and their 95% Wald confidence interval. Participants’
results were considered positive for AwMA and AmtDNA when their value was above
the cut-off value identified after maximizing Youden’s Index. A 95% confidence
interval was obtained for the cut-off using 10,000 bootstrap samples. Performance
measures are presented with their 95% exact confidence interval.
Software
Western blot images were acquired using Image Studio Digits 5.2 (LI-COR
biotechnology). mtDNA migration through agarose gel was imaged using Image Lab
6.0.1 (Bio-Rad). DLS studies were carried using the built-in Zetasizer 7.10 software
(Malvern Instruments). EM images were acquired with the Image Capture Software
601.384 (Advanced Microscopy Techniques Corp., Woburn, MA, USA). Flow
cytometry was performed using the BD FACSDiva™ 6.1.3 (BD Biosciences). Yields
from DNA isolations were quantified using the ND-1000 3.8.1 software (Thermo
Fisher Scientific) and qPCR were performed with RotorGene 6.1 (Corbett
Research/QIAgen). Optical densities were measured using SoftMax Pro 5.4.1
(Molecular Devices). Figures were assembled with ImageJ 1.47 (National Institutes
of Health, Rockville, MA, USA) and Photoshop CS6 13.0 (Adobe Systems Inc.,
Mountain View, CA, USA). Statistical analyses were carried with Prism 7 software
(GraphPad Software Inc., La Jolla, CA, USA) and SAS version 9.4 (SAS Institute
Inc., Cary, NC, USA).

357
Acknowledgements
This work was supported in part by an operating grant from The Arthritis Society (#
225638) (to PRF) and by a Canadian Institutes of Health Research (CIHR)
Foundation grant (EB). The authors have no competing interests. EB is recipient of
a new investigator award from the CIHR and is a Canadian National Transplant
Research Program (CNTRP) researcher. PRF is recipient of a tier 1 Canada
Research Chair on Systemic Autoimmune Rheumatic Diseases. GM is a recipient of
awards from the Canadian Blood Services. IM is recipient of a fellowship from the
Arthritis Society. CB is recipient of an award from the Fonds de Recherche en Santé
du Quebec. JR is the recipient of funding from the Division of Rheumatology and the
Department of Medicine (McGill University), and a CIHR Project Grant (PJT-
159652). The authors acknowledge Rebecca Subang for technical assistance in
preparing human healthy control samples. MET is grateful to the help of Julie-
Christine Lévesque and Nathalie Vernoux with TEM experiments. The views
expressed herein do not necessarily represent the view of the federal government.

358
References
1. Andersen JL, Kornbluth S. The tangled circuitry of metabolism and apoptosis. Mol. Cell. 2013;49:399–410. doi: 10.1016/j.molcel.2012.12.026. 2. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. 3. Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. 4. Arechaga I. Membrane invaginations in bacteria and mitochondria: common features and evolutionary scenarios. J Mol Microbiol Biotechnol. 2013;23:13–23. doi: 10.1159/000346515. 5. Tincani A, et al. Anti-phospholipid and anti-mitochondrial type M5 antibodies in systemic lupus erythematosus. Clin. Exp. Rheumatol. 1985;3:321–326. 6. Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. 7. McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. 8. Lang BF, et al. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387:493–497. doi: 10.1038/387493a0. 9. Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–343. doi: 10.1038/nature12985. 10. Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS One. 2012;7:e49767. doi: 10.1371/journal.pone.0049767. 11. Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis. 2014;5:e1312. doi: 10.1038/cddis.2014.277. 12. Yousefi S, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008;14:949–953. doi: 10.1038/nm.1855. 13. Garcia-Martinez I, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Invest. 2016;126:859–864. doi: 10.1172/JCI83885. 14. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–1444. doi: 10.1038/cdd.2009.96. 15. Lood C, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016;22:146–153. doi: 10.1038/nm.4027. 16. Caielli S, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016;213:697–713. doi: 10.1084/jem.20151876. 17. Boudreau LH, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–2183. doi: 10.1182/blood-2014-05-573543.

359
18. Marcoux G, et al. Microparticle and mitochondrial release during extended storage of different types of platelet concentrates. Platelets. 2017;28:272–280. doi: 10.1080/09537104.2016.1218455. 19. Marques PE, et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. 2012;56:1971–1982. doi: 10.1002/hep.25801. 20. Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. 21. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–417. doi: 10.1016/j.immuni.2015.02.002. 22. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011;11:389–402. doi: 10.1038/nri2975. 23. Sandhir R, Halder A, Sunkaria A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta. 2017;1863:1090–1097. doi: 10.1016/j.bbadis.2016.10.020. 24. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017;18:488–498. doi: 10.1038/ni.3704. 25. Zhao Z, et al. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood. 2016;127:2763–2772. doi: 10.1182/blood-2015-12-688838. 26. Nakahira, K. et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 10, e1001577; discussione1001577, 10.1371/journal.pmed.1001577 (2013). 27. Simmons, J. D. et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann. Surg. 258, 591–596; discussion 596–598, 10.1097/SLA.0b013e3182a4ea46 (2013). 28. Kohler C, et al. Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol. Cancer. 2009;8:105. doi: 10.1186/1476-4598-8-105. 29. Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003;5:R234–240. doi: 10.1186/ar787. 30. Yasui, K. et al. Mitochondrial damage-associated molecular patterns as potential proinflammatory mediators in post-platelet transfusion adverse effects. Transfusion (Paris), 10.1111/trf.13535 (2016). 31. Simmons JD, et al. Potential contribution of mitochondrial DNA damage associated molecular patterns in transfusion products to the development of acute respiratory distress syndrome after multiple transfusions. The journal of trauma and acute care surgery. 2017;82:1023–1029. doi: 10.1097/TA.0000000000001421. 32. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. 33. Cloutier N, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc. Natl. Acad. Sci. USA. 2018;115:E1550–E1559. doi: 10.1073/pnas.1720553115. 34. Berg PA, Klein R. Mitochondrial antigens and autoantibodies: from anti-M1 to anti-M9. Klin. Wochenschr. 1986;64:897–909. doi: 10.1007/BF01728613.

360
35. Meek F, Khoury EL, Doniach D, Baum H. Mitochondrial antibodies in chronic liver diseases and connective tissue disorders: further characterization of the autoantigens. Clin. Exp. Immunol. 1980;41:43–54. 36. Doniach D, Walker JG, Roitt IM, Berg PA. “Autoallergic” hepatitis. N. Engl. J. Med. 1970;282:86–88. doi: 10.1056/NEJM197001082820207. 37. Klein R, Berg PA. Demonstration of “naturally occurring mitochondrial antibodies” in family members of patients with primary biliary cirrhosis. Hepatology. 1990;12:335–341. doi: 10.1002/hep.1840120222. 38. Lindor KD, et al. Primary biliary cirrhosis. Hepatology. 2009;50:291–308. doi: 10.1002/hep.22906. 39. Homberg JC, et al. A new antimitochondria antibody (anti-M6) in iproniazid-induced hepatitis. Clin. Exp. Immunol. 1982;47:93–102. 40. Klein R, Maisch B, Kochsiek K, Berg PA. Demonstration of organ specific antibodies against heart mitochondria (anti-M7) in sera from patients with some forms of heart diseases. Clin. Exp. Immunol. 1984;58:283–292. 41. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis & rheumatology. 2015;67:3190–3200. doi: 10.1002/art.39296. 42. Reimer G, Rubin RL, Kotzin BL, Tan EM, Anti-native DNA. antibodies from autoimmune sera also bind to DNA in mitochondria. J. Immunol. 1984;133:2532–2536. 43. Bradwell A. R., Hughes R. G., Karim A. R. Immunofluorescent antinuclear antibody tests. Manual of Molecular and Clinical Laboratory Immunology. 7th edition, 995–1006 (2014). 44. Deegan MJ, Walker SE, Lovell SE. Antibodies to double-stranded DNA. A comparison of the indirect immunofluorescent test using crithidia luciliae and the DNA-binding assay. Am. J. Clin. Pathol. 1978;69:599–604. doi: 10.1093/ajcp/69.6.599. 45. Lakos G, et al. Detection of anti-dsDNA antibodies by computer-aided automated immunofluorescence analysis. J. Immunol. Methods. 2016;433:17–22. doi: 10.1016/j.jim.2016.02.019. 46. Crowe W, Kushner I. An immunofluorescent method using Crithidia luciliae to detect antibodies to double-stranded DNA. Arthritis Rheum. 1977;20:811–814. doi: 10.1002/art.1780200308. 47. Manthorpe R, Palit J, Bendixen G. Anti-DNA antibody in serum measured by radioimmunoassay (Farr technique). Description of method and recommended procedure. Allergy. 1978;33:42–49. doi: 10.1111/j.1398-9995.1978.tb01505.x. 48. Jessop S, Botha P. False-positive test results for syphilis in relatives of a patient with systemic lupus erythematosus. Br. J. Vener. Dis. 1979;55:292–294. 49. Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. 50. Hudson M, et al. The presence of multiple prothrombotic risk factors is associated with a higher risk of thrombosis in individuals with anticardiolipin antibodies. J. Rheumatol. 2003;30:2385–2391. 51. Neville C, et al. Thromboembolic risk in patients with high titre anticardiolipin and multiple antiphospholipid antibodies. Thromb. Haemost. 2003;90:108–115. doi: 10.1267/THRO03010108.

361
52. Cheng MY, Hartl FU, Horwich AL. The mitochondrial chaperonin hsp60 is required for its own assembly. Nature. 1990;348:455–458. doi: 10.1038/348455a0. 53. Dieude M, Senecal JL, Raymond Y. Induction of endothelial cell apoptosis by heat-shock protein 60-reactive antibodies from anti-endothelial cell autoantibody-positive systemic lupus erythematosus patients. Arthritis Rheum. 2004;50:3221–3231. doi: 10.1002/art.20564. 54. Dieude M, et al. Association of autoantibodies to heat-shock protein 60 with arterial vascular events in patients with antiphospholipid antibodies. Arthritis Rheum. 2011;63:2416–2424. doi: 10.1002/art.30411. 55. Grob PJ, Muller-Schoop JW, Hacki MA, Joller-Jemelka HI. Drug-induced pseudolupus. Lancet. 1975;2:144–148. doi: 10.1016/S0140-6736(75)90055-0. 56. Labro MT, Andrieu MC, Weber M, Homberg JC. A new pattern of non-organ- and non-species-specific anti-organelle antibody detected by immunofluorescence: the mitochondrial antibody number 5. Clin. Exp. Immunol. 1978;31:357–366. 57. Meroni PL, et al. Anti-mitochondrial type M5 and anti-cardiolipin antibodies in autoimmune disorders: studies on their association and cross-reactivity. Clin. Exp. Immunol. 1987;67:484–491. 58. Kizu R, Yamamoto T, Yokoyama T, Tanaka M, Miyazaki M. A sensitive postcolumn derivatization/UV detection system for HPLC determination of antitumor divalent and quadrivalent platinum complexes. Chem. Pharm. Bull. (Tokyo) 1995;43:108–114. doi: 10.1248/cpb.43.108. 59. Mouritsen S, Demant E, Permin H, Wiik A. High prevalence of anti-mitochondrial antibodies among patients with some well-defined connective tissue diseases. Clin. Exp. Immunol. 1986;66:68–76. 60. Gusdon AM, Chen J, Votyakova TV, Mathews CE. Chapter 24 Quantification, localization, and tissue specificities of mouse mitochondrial reactive oxygen species production. Methods Enzymol. 2009;456:439–457. doi: 10.1016/S0076-6879(08)04424-8. 61. Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. 62. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. 63. Levine JS, et al. Immunization with an apoptotic cell-binding protein recapitulates the nephritis and sequential autoantibody emergence of systemic lupus erythematosus. J. Immunol. 2006;177:6504–6516. doi: 10.4049/jimmunol.177.9.6504. 64. Ullal AJ, et al. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J. Autoimmun. 2011;36:173–180. doi: 10.1016/j.jaut.2011.02.001. 65. Mobarrez F, et al. Microparticles in the blood of patients with systemic lupus erythematosus (SLE): phenotypic characterization and clinical associations. Sci. Rep. 2016;6:36025. doi: 10.1038/srep36025. 66. Sisirak V, et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell. 2016;166:88–101. doi: 10.1016/j.cell.2016.05.034.

362
67. Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu. Rev. Pathol. 2013;8:303–330. doi: 10.1146/annurev-pathol-020712-164014. 68. He J, et al. Circulating Mitochondrial DAMPs Are Not Effective Inducers of Proteinuria and Kidney Injury in Rodents. PLoS One. 2015;10:e0124469. doi: 10.1371/journal.pone.0124469. 69. Eggleton P, Nissim A, Ryan BJ, Whiteman M, Winyard PG. Detection and isolation of human serum autoantibodies that recognize oxidatively modified autoantigens. Free Radic. Biol. Med. 2013;57:79–91. doi: 10.1016/j.freeradbiomed.2012.11.006. 70. Serbinova E, Kagan V, Han D, Packer L. Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radic. Biol. Med. 1991;10:263–275. doi: 10.1016/0891-5849(91)90033-Y. 71. Claycamp HG. Phenol sensitization of DNA to subsequent oxidative damage in 8-hydroxyguanine assays. Carcinogenesis. 1992;13:1289–1292. doi: 10.1093/carcin/13.7.1289. 72. Berg PA, Klein R. Mitochondrial antigen/antibody systems in primary biliary cirrhosis: revisited. Liver. 1995;15:281–292. doi: 10.1111/j.1600-0676.1995.tb00687.x. 73. Soltys BJ, Gupta RS. Immunoelectron microscopic localization of the 60-kDa heat shock chaperonin protein (Hsp60) in mammalian cells. Exp. Cell Res. 1996;222:16–27. doi: 10.1006/excr.1996.0003. 74. de Leeuw K, Bungener L, Roozendaal C, Bootsma H, Stegeman CA. Auto-antibodies to double-stranded DNA as biomarker in systemic lupus erythematosus: comparison of different assays during quiescent and active disease. Rheumatology (Oxford) 2017;56:698–703. doi: 10.1093/rheumatology/kew462. 75. Swaak AJ, Aarden LA, Statius van Eps LW, Feltkamp TE. Anti-dsDNA and complement profiles as prognostic guides in systemic lupus erythematosus. Arthritis Rheum. 1979;22:226–235. doi: 10.1002/art.1780220304. 76. Czompoly T, et al. Detailed analyses of antibodies recognizing mitochondrial antigens suggest similar or identical mechanism for production of natural antibodies and natural autoantibodies. Autoimmun Rev. 2008;7:463–467. doi: 10.1016/j.autrev.2008.03.006. 77. Binder CJ. Natural IgM antibodies against oxidation-specific epitopes. J. Clin. Immunol. 2010;30(Suppl 1):S56–60. doi: 10.1007/s10875-010-9396-3. 78. Avrameas S. Natural autoantibodies: from ‘horror autotoxicus’ to ‘gnothi seauton’ Immunol. Today. 1991;12:154–159. doi: 10.1016/S0167-5699(05)80045-3. 79. Mouthon L, et al. Analysis of the normal human IgG antibody repertoire. Evidence that IgG autoantibodies of healthy adults recognize a limited and conserved set of protein antigens in homologous tissues. J. Immunol. 1995;154:5769–5778. 80. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. 81. Tan EM, et al. The1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101.

363
82. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/1529-0131(199709)40:9<1725::AID-ART29>3.0.CO;2-Y. 83. Wilson WA, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309–1311. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. 84. Miyakis S, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J. Thromb. Haemost. 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. 85. Lindor, K. D., Bowlus, C. L., Boyer, J., Levy, C. & Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology, 10.1002/hep.30145 (2018). 86. Nagata T, Redman RS, Lakshman R. Isolation of intact nuclei of high purity from mouse liver. Anal. Biochem. 2010;398:178–184. doi: 10.1016/j.ab.2009.11.017. 87. Wright DJ, et al. New antibody in early syphilis. Lancet. 1970;1:740–743. doi: 10.1016/S0140-6736(70)90972-4. 88. Klein R, Kloppel G, Garbe W, Fintelmann V, Berg PA. Antimitochondrial antibody profiles determined at early stages of primary biliary cirrhosis differentiate between a benign and a progressive course of the disease. A retrospective analysis of 76 patients over 6–18 years. J. Hepatol. 1991;12:21–27. doi: 10.1016/0168-8278(91)90903-O. 89. Pons C, et al. Human anti-mitochondria autoantibodies appearing in iproniazid-induced immunoallergic hepatitis recognize human liver monoamine oxidase B. Biochem. Biophys. Res. Commun. 1996;218:118–124. doi: 10.1006/bbrc.1996.0021. 90. Cicek G, Schiltz E, Hess D, Staiger J, Brandsch R. Analysis of mitochondrial antigens reveals inner membrane succinate dehydrogenase flavoprotein subunit as autoantigen to antibodies in anti-M7 sera. Clin. Exp. Immunol. 2002;128:83–87. doi: 10.1046/j.1365-2249.2002.01816.x. 91. Klein R, Berg PA. Characterization of a new mitochondrial antigen-antibody system (M9/anti-M9) in patients with anti-M2 positive and anti-M2 negative primary biliary cirrhosis. Clin. Exp. Immunol. 1988;74:68–74 92. Klein R, Berg PA. Anti-M9 antibodies in sera from patients with primary biliary cirrhosis recognize an epitope of glycogen phosphorylase. Clin. Exp. Immunol. 1990;81:65–71. doi: 10.1111/j.1365-2249.1990.tb05292.x.





























