Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
008) 4139–4144www.elsevier.com/locate/tsf
Thin Solid Films 516 (2
Fine tuning of color via copolymerization and its electrochromicdevice application
Pinar Camurlu a, Ertuğrul Şahmetlioğlu b, Elif Şahin c, İdris Mecidoglu Akhmedov d,Cihangir Tanyeli d, Levent Toppare d,⁎
a Akdeniz University, Department of Chemistry, 07058, Antalya, Turkeyb Nigde University, Department of Chemistry, 51100, Nigde, Turkey
c Harran University, Department of Chemistry, 63300, Sanliurfa, Turkeyd Middle East Technical University, Department of Chemistry, 06531, Ankara, Turkey
Received 29 March 2007; received in revised form 15 October 2007; accepted 19 October 2007Available online 26 November 2007
Abstract
In this study electrochemical copolymerization of 1-(perfluorophenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole and 3,4-ethylenedioxythiophene wasused to fulfill a strategy in achieving desired multichromic properties. The resultant polymer displayed distinct color changes between red–violet,amber, green and blue color with good switching times and optical contrast. A direct correlation between the color of the polymer in neutral stateand the applied potential during synthesis was observed. Hence, it was possible to achieve colors from maroon, red–violet, and eggplant to indigodepending on the applied potential. A dual type electrochromic device of the copolymer and poly(3,4-ethylenedioxythiophene) was constructed toevaluate the possible use of such copolymer in device applications. The device displayed multichromism, with 1.1 s switching time and goodelectrochromic memory.© 2007 Elsevier B.V. All rights reserved.
Keywords: Electrochemical copolymerization; 1-(perfluorophenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole; 3,4-ethylenedioxythiophene; Multichromism; Electrochromicdevices
1. Introduction
Electrochromic materials have aroused a great deal ofinterest owing to their possible use in optical devices such asin information display and storage, in automotive industry andarchitecture [1]. These materials reveal reversible and visiblechange in reflected or transmitted light upon electrochemicaloxidation and/or reduction. Till now, a wide variety of elec-trochromic materials have been developed, including inorganicmetal oxides such as tungsten trioxide (WO3) or iridium dioxide(IrO2), mixed-valence metal complexes [2], small organicmolecules as viologens and phthalocyanine derivatives, andconducting polymers [3].
Among organic molecules, conducting polymers haveattached significant interest in the field of electrochromism,
⁎ Corresponding author. Tel: +903122103251; fax: +903122103200.E-mail address: [email protected] (L. Toppare).
0040-6090/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.tsf.2007.10.098
since they offer additional advantages, such as, low processingcost, no dependence with angle of vision, good UV stability,high coloration efficiency, fast switching ability and fine-tuningof the band gap through the modification of polymer's chemicalstructure [4]. Conducting polymers generally reveal an opticalabsorption band in the visible region due to their extendeddelocalization of the π electrons along the polymer backbone.Upon doping, charge carriers are formed on the conjugatedbackbone, namely polaron and bipolarons, which result in achange in the optical spectra [5]. By adjusting the electroniccharacter of the π system along the neutral polymer backbone,the π−π⁎ transition can be adjusted across the electromagneticspectrum [6]. However, most of the electroactive polymers arelimited with two colors, where only few show multiple colorstates [7]. Although there are some reports on multichromicmaterials [8,9], it is still important to achieve materials thatdisplay distinctive color changes covering the entire visibleregion upon applied potential.

Scheme 1. Schematical representation for the electrochemical polymerization of FPTPy with EDOT.
4140 P. Camurlu et al. / Thin Solid Films 516 (2008) 4139–4144
The electrochromic properties of conducting polymers canbe varied over a wide range by controlling the band gap ofthe polymer via proper choice of heteroaromatic ring and sub-stituents. Blending and copolymerization are among thefrequently used methods to tune the electrochromic propertiesof the materials. Copolymerization is an easy, facile method tocombine the electrochromic properties of the comonomers [10].The copolymerization of distinct monomers or homopolymer-ization of hybrid monomers containing several distinct units,can lead to an appealing combination of the properties of theparent polymers [2].
Poly(2,5-dithienyl pyrrole) derivatives have been investi-gated for their electronic properties, reaction kinetics [11] alongwith electrochemical processing of the conducting polymer[12]. Among those 1-(perfluorophenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole (FPTPy) derivative is shown to exhibit color changebetween yellow and light purple [13]. In 1991, Jonas et al. [14]synthesized 3,4-ethylenedioxythiophene (EDOT) by lockingthe 3- and 4-positions of thiophene with an ethylenedioxy groupyielding a highly electron-rich fused heterocycle which has lowoxidation potential and free from the possible α, β and β, βlinkages [15]. Poly(3,4-ethylenedioxythiophene) (PEDOT)exhibits an optical band gap of 1.6 eV. Doped PEDOT isalmost transparent in the visible region and the neutral polymeris dark blue. Thus, this material is significant for its cathodicallycoloring electrochromic properties in device applications.
Here we report the synthesis the copolymer of 1-(perfluor-ophenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole and 3,4-ethylene-dioxythiophene via electropolymerization under differentexperimental conditions (Scheme 1). The copolymer revealedmultichromism throughout the entire visible region. To explorethe use of thismaterial in electrochromic devicewe constructed anabsorptive/transmissive type electrochromic device with ITO(indium tin oxide coated glass)/copolymer/gel electrolyte/PEDOT/ITO configuration, where the copolymer and PEDOTfunctioned as the anodically and the cathodically coloring layersrespectively. Characterizations of the electrochromic behavior ofthe copolymer and the device were achieved by spectroelec-trochemistry, kinetic, colorimetry studies.
2. Experimental details
2.1. Materials
Propylene carbonate (PC), NaClO4, LiClO4, poly(methylmethacrylate) (PMMA) and 3,4-ethylenedioxythiophene werepurchased from Aldrich and used without further purification.
Acetonitrile (AN) (Merck) was distilled prior to use. 1-(perfluorophenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole was synthe-sized as reported previously [13]. The gel electrolytewas preparedfrom an acetonitrile solution containing PMMA, NaClO4 andLiClO4. Acetonitrile was slowly evaporated under stirring andpropylene carbonate was added to decrease the vapor pressure ofthe gel electrolyte yielding a highly conducting transparent gel.The ratio of the composition of NaClO4:LiClO4:PMMA:PC:ACN was 1.5:1.5:7:20:70 by weight [16].
2.2. Electrochemical synthesis of P(FPTPy-co-EDOT)
A three-electrode cell containing ITO-coated glass slide asthe working electrode, a platinum foil as the counter electrode,and a silver wire as the pseudo-reference electrode were used forthe electrodeposition of polymer films by potentiodynamicmethods (cyclic voltammetry) using a Voltalab 50 potentiostat/galvanostat. 5 mM FPTPy, 5 mM EDOT, 0.1 M NaClO4, andLiClO4 containing AN solution were placed into the cell. Cyclicvoltammetry runs were performed between −0.5 Vand 1.1, 1.2,1.3,1.4 or 1.5 V with a scan rate of 100 mV/s.
2.3. Instrumentation
UV–Vis–NIR (Ultraviolet–Visible–Near Infrared) spectrawere recorded on a Varian Cary 5000 spectrophotometer at ascan rate of 2000 nm/min. The potentials were controlled usingSolartron 1285 potentiostat/galvanostat. In situ colorimetryanalyses were performed by Minolta CS-100A Chroma Meter.During measurement, samples were placed in a light boothsystem where it was illuminated from behind by a D65 lightsource (Philips).
2.4. Construction of electrochromic device
The cathodically coloring layer, poly(3,4-ethylenedioxythio-phene), was synthesized at 1.2 V in single compartment cell inthe presence of 0.1 M NaClO4, and LiClO4 in AN. Copolymerwas synthesized on ITO according to the above procedure andused as the anodically coloring layer. The redox sites ofanodically and cathodically coloring films were matched bystepping the potentials between the ultimate oxidized andreduced states by chronoamperometry. Electrochromic devices(ECDs) were built by arranging oxidized (PEDOT) and neutral(copolymer) electrochromic polymer films facing each otherseparated by a gel electrolyte. The devices were dried underatmospheric conditions.
4141P. Camurlu et al. / Thin Solid Films 516 (2008) 4139–4144
3. Results and discussion
3.1. Cyclic voltammetry
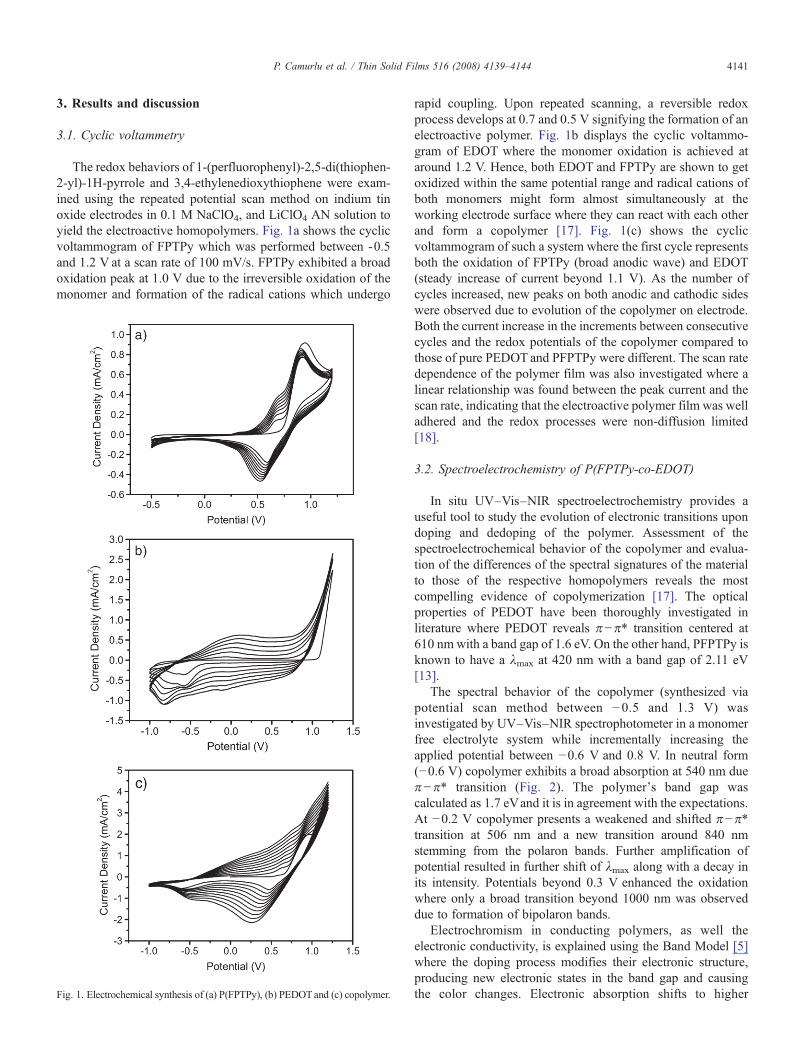
The redox behaviors of 1-(perfluorophenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole and 3,4-ethylenedioxythiophene were exam-ined using the repeated potential scan method on indium tinoxide electrodes in 0.1 M NaClO4, and LiClO4 AN solution toyield the electroactive homopolymers. Fig. 1a shows the cyclicvoltammogram of FPTPy which was performed between -0.5and 1.2 V at a scan rate of 100 mV/s. FPTPy exhibited a broadoxidation peak at 1.0 V due to the irreversible oxidation of themonomer and formation of the radical cations which undergo
Fig. 1. Electrochemical synthesis of (a) P(FPTPy), (b) PEDOTand (c) copolymer.
rapid coupling. Upon repeated scanning, a reversible redoxprocess develops at 0.7 and 0.5 V signifying the formation of anelectroactive polymer. Fig. 1b displays the cyclic voltammo-gram of EDOT where the monomer oxidation is achieved ataround 1.2 V. Hence, both EDOT and FPTPy are shown to getoxidized within the same potential range and radical cations ofboth monomers might form almost simultaneously at theworking electrode surface where they can react with each otherand form a copolymer [17]. Fig. 1(c) shows the cyclicvoltammogram of such a system where the first cycle representsboth the oxidation of FPTPy (broad anodic wave) and EDOT(steady increase of current beyond 1.1 V). As the number ofcycles increased, new peaks on both anodic and cathodic sideswere observed due to evolution of the copolymer on electrode.Both the current increase in the increments between consecutivecycles and the redox potentials of the copolymer compared tothose of pure PEDOT and PFPTPy were different. The scan ratedependence of the polymer film was also investigated where alinear relationship was found between the peak current and thescan rate, indicating that the electroactive polymer film was welladhered and the redox processes were non-diffusion limited[18].
3.2. Spectroelectrochemistry of P(FPTPy-co-EDOT)
In situ UV–Vis–NIR spectroelectrochemistry provides auseful tool to study the evolution of electronic transitions upondoping and dedoping of the polymer. Assessment of thespectroelectrochemical behavior of the copolymer and evalua-tion of the differences of the spectral signatures of the materialto those of the respective homopolymers reveals the mostcompelling evidence of copolymerization [17]. The opticalproperties of PEDOT have been thoroughly investigated inliterature where PEDOT reveals π−π⁎ transition centered at610 nm with a band gap of 1.6 eV. On the other hand, PFPTPy isknown to have a λmax at 420 nm with a band gap of 2.11 eV[13].
The spectral behavior of the copolymer (synthesized viapotential scan method between −0.5 and 1.3 V) wasinvestigated by UV–Vis–NIR spectrophotometer in a monomerfree electrolyte system while incrementally increasing theapplied potential between −0.6 V and 0.8 V. In neutral form(−0.6 V) copolymer exhibits a broad absorption at 540 nm dueπ−π⁎ transition (Fig. 2). The polymer's band gap wascalculated as 1.7 eVand it is in agreement with the expectations.At −0.2 V copolymer presents a weakened and shifted π−π⁎transition at 506 nm and a new transition around 840 nmstemming from the polaron bands. Further amplification ofpotential resulted in further shift of λmax along with a decay inits intensity. Potentials beyond 0.3 V enhanced the oxidationwhere only a broad transition beyond 1000 nm was observeddue to formation of bipolaron bands.
Electrochromism in conducting polymers, as well theelectronic conductivity, is explained using the Band Model [5]where the doping process modifies their electronic structure,producing new electronic states in the band gap and causingthe color changes. Electronic absorption shifts to higher
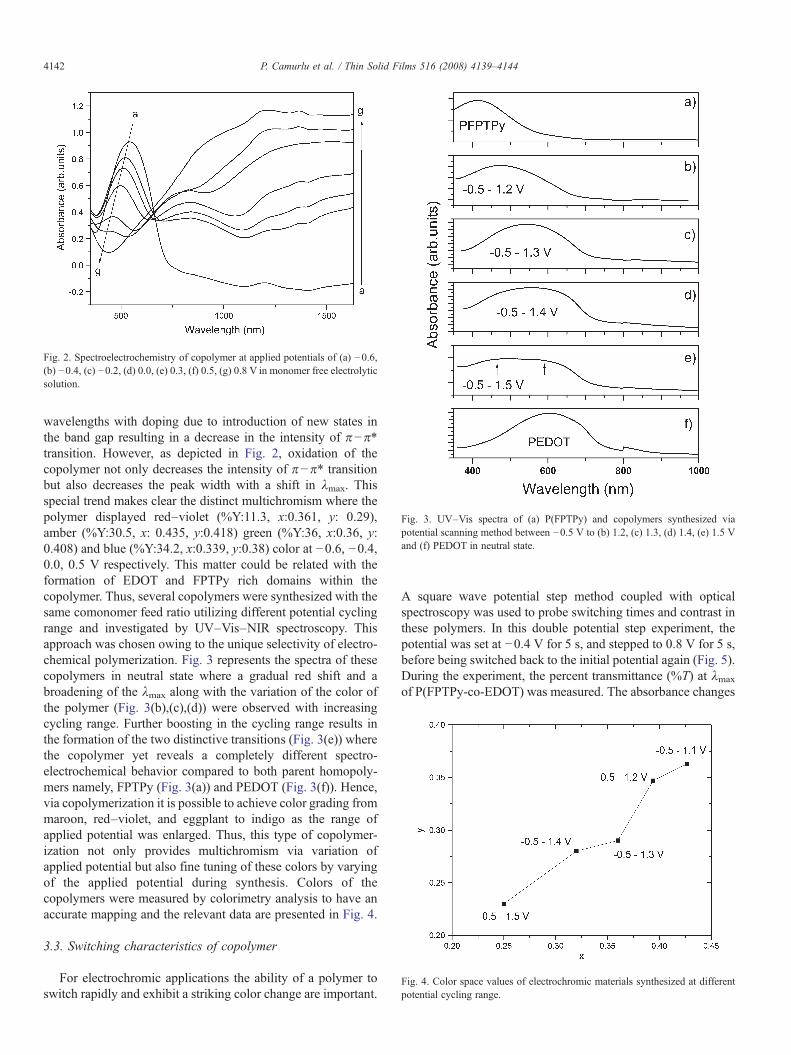
Fig. 3. UV–Vis spectra of (a) P(FPTPy) and copolymers synthesized viapotential scanning method between −0.5 V to (b) 1.2, (c) 1.3, (d) 1.4, (e) 1.5 Vand (f) PEDOT in neutral state.
Fig. 4. Color space values of electrochromic materials synthesized at differentpotential cycling range.
Fig. 2. Spectroelectrochemistry of copolymer at applied potentials of (a) −0.6,(b) −0.4, (c) −0.2, (d) 0.0, (e) 0.3, (f) 0.5, (g) 0.8 V in monomer free electrolyticsolution.
4142 P. Camurlu et al. / Thin Solid Films 516 (2008) 4139–4144
wavelengths with doping due to introduction of new states inthe band gap resulting in a decrease in the intensity of π−π⁎transition. However, as depicted in Fig. 2, oxidation of thecopolymer not only decreases the intensity of π−π⁎ transitionbut also decreases the peak width with a shift in λmax. Thisspecial trend makes clear the distinct multichromism where thepolymer displayed red–violet (%Y:11.3, x:0.361, y: 0.29),amber (%Y:30.5, x: 0.435, y:0.418) green (%Y:36, x:0.36, y:0.408) and blue (%Y:34.2, x:0.339, y:0.38) color at −0.6, −0.4,0.0, 0.5 V respectively. This matter could be related with theformation of EDOT and FPTPy rich domains within thecopolymer. Thus, several copolymers were synthesized with thesame comonomer feed ratio utilizing different potential cyclingrange and investigated by UV–Vis–NIR spectroscopy. Thisapproach was chosen owing to the unique selectivity of electro-chemical polymerization. Fig. 3 represents the spectra of thesecopolymers in neutral state where a gradual red shift and abroadening of the λmax along with the variation of the color ofthe polymer (Fig. 3(b),(c),(d)) were observed with increasingcycling range. Further boosting in the cycling range results inthe formation of the two distinctive transitions (Fig. 3(e)) wherethe copolymer yet reveals a completely different spectro-electrochemical behavior compared to both parent homopoly-mers namely, FPTPy (Fig. 3(a)) and PEDOT (Fig. 3(f)). Hence,via copolymerization it is possible to achieve color grading frommaroon, red–violet, and eggplant to indigo as the range ofapplied potential was enlarged. Thus, this type of copolymer-ization not only provides multichromism via variation ofapplied potential but also fine tuning of these colors by varyingof the applied potential during synthesis. Colors of thecopolymers were measured by colorimetry analysis to have anaccurate mapping and the relevant data are presented in Fig. 4.
3.3. Switching characteristics of copolymer
For electrochromic applications the ability of a polymer toswitch rapidly and exhibit a striking color change are important.
A square wave potential step method coupled with opticalspectroscopy was used to probe switching times and contrast inthese polymers. In this double potential step experiment, thepotential was set at −0.4 V for 5 s, and stepped to 0.8 V for 5 s,before being switched back to the initial potential again (Fig. 5).During the experiment, the percent transmittance (%T) at λmax
of P(FPTPy-co-EDOT) was measured. The absorbance changes
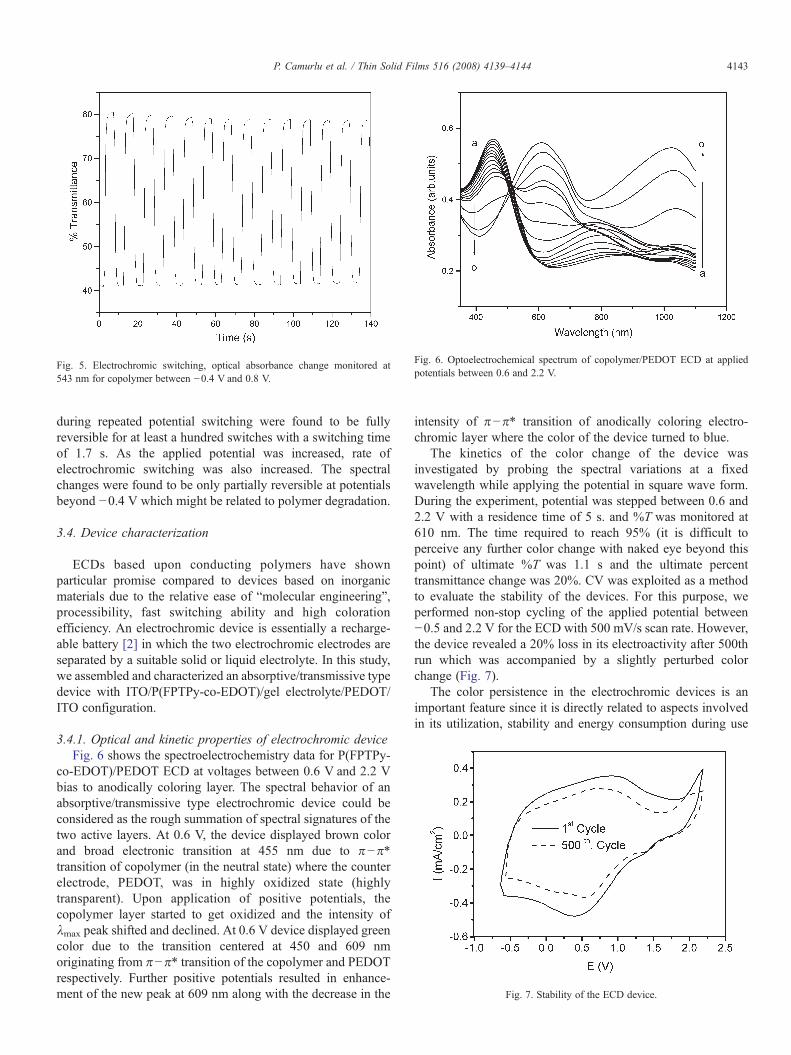
Fig. 6. Optoelectrochemical spectrum of copolymer/PEDOT ECD at appliedpotentials between 0.6 and 2.2 V.
Fig. 7. Stability of the ECD device.
Fig. 5. Electrochromic switching, optical absorbance change monitored at543 nm for copolymer between −0.4 V and 0.8 V.
4143P. Camurlu et al. / Thin Solid Films 516 (2008) 4139–4144
during repeated potential switching were found to be fullyreversible for at least a hundred switches with a switching timeof 1.7 s. As the applied potential was increased, rate ofelectrochromic switching was also increased. The spectralchanges were found to be only partially reversible at potentialsbeyond −0.4 V which might be related to polymer degradation.
3.4. Device characterization
ECDs based upon conducting polymers have shownparticular promise compared to devices based on inorganicmaterials due to the relative ease of “molecular engineering”,processibility, fast switching ability and high colorationefficiency. An electrochromic device is essentially a recharge-able battery [2] in which the two electrochromic electrodes areseparated by a suitable solid or liquid electrolyte. In this study,we assembled and characterized an absorptive/transmissive typedevice with ITO/P(FPTPy-co-EDOT)/gel electrolyte/PEDOT/ITO configuration.
3.4.1. Optical and kinetic properties of electrochromic deviceFig. 6 shows the spectroelectrochemistry data for P(FPTPy-
co-EDOT)/PEDOT ECD at voltages between 0.6 V and 2.2 Vbias to anodically coloring layer. The spectral behavior of anabsorptive/transmissive type electrochromic device could beconsidered as the rough summation of spectral signatures of thetwo active layers. At 0.6 V, the device displayed brown colorand broad electronic transition at 455 nm due to π−π⁎transition of copolymer (in the neutral state) where the counterelectrode, PEDOT, was in highly oxidized state (highlytransparent). Upon application of positive potentials, thecopolymer layer started to get oxidized and the intensity ofλmax peak shifted and declined. At 0.6 V device displayed greencolor due to the transition centered at 450 and 609 nmoriginating from π−π⁎ transition of the copolymer and PEDOTrespectively. Further positive potentials resulted in enhance-ment of the new peak at 609 nm along with the decrease in the
intensity of π−π⁎ transition of anodically coloring electro-chromic layer where the color of the device turned to blue.
The kinetics of the color change of the device wasinvestigated by probing the spectral variations at a fixedwavelength while applying the potential in square wave form.During the experiment, potential was stepped between 0.6 and2.2 V with a residence time of 5 s. and %T was monitored at610 nm. The time required to reach 95% (it is difficult toperceive any further color change with naked eye beyond thispoint) of ultimate %T was 1.1 s and the ultimate percenttransmittance change was 20%. CV was exploited as a methodto evaluate the stability of the devices. For this purpose, weperformed non-stop cycling of the applied potential between−0.5 and 2.2 V for the ECD with 500 mV/s scan rate. However,the device revealed a 20% loss in its electroactivity after 500thrun which was accompanied by a slightly perturbed colorchange (Fig. 7).
The color persistence in the electrochromic devices is animportant feature since it is directly related to aspects involvedin its utilization, stability and energy consumption during use
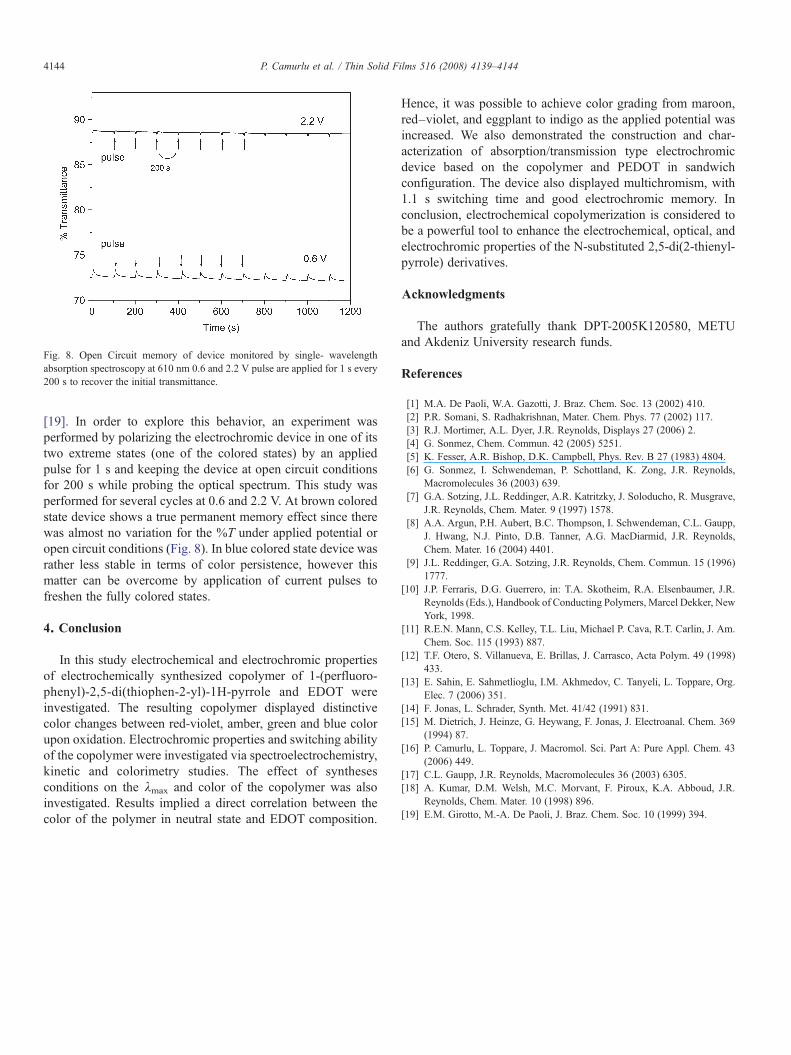
Fig. 8. Open Circuit memory of device monitored by single- wavelengthabsorption spectroscopy at 610 nm 0.6 and 2.2 V pulse are applied for 1 s every200 s to recover the initial transmittance.
4144 P. Camurlu et al. / Thin Solid Films 516 (2008) 4139–4144
[19]. In order to explore this behavior, an experiment wasperformed by polarizing the electrochromic device in one of itstwo extreme states (one of the colored states) by an appliedpulse for 1 s and keeping the device at open circuit conditionsfor 200 s while probing the optical spectrum. This study wasperformed for several cycles at 0.6 and 2.2 V. At brown coloredstate device shows a true permanent memory effect since therewas almost no variation for the %T under applied potential oropen circuit conditions (Fig. 8). In blue colored state device wasrather less stable in terms of color persistence, however thismatter can be overcome by application of current pulses tofreshen the fully colored states.
4. Conclusion
In this study electrochemical and electrochromic propertiesof electrochemically synthesized copolymer of 1-(perfluoro-phenyl)-2,5-di(thiophen-2-yl)-1H-pyrrole and EDOT wereinvestigated. The resulting copolymer displayed distinctivecolor changes between red-violet, amber, green and blue colorupon oxidation. Electrochromic properties and switching abilityof the copolymer were investigated via spectroelectrochemistry,kinetic and colorimetry studies. The effect of synthesesconditions on the λmax and color of the copolymer was alsoinvestigated. Results implied a direct correlation between thecolor of the polymer in neutral state and EDOT composition.
Hence, it was possible to achieve color grading from maroon,red–violet, and eggplant to indigo as the applied potential wasincreased. We also demonstrated the construction and char-acterization of absorption/transmission type electrochromicdevice based on the copolymer and PEDOT in sandwichconfiguration. The device also displayed multichromism, with1.1 s switching time and good electrochromic memory. Inconclusion, electrochemical copolymerization is considered tobe a powerful tool to enhance the electrochemical, optical, andelectrochromic properties of the N-substituted 2,5-di(2-thienyl-pyrrole) derivatives.
Acknowledgments
The authors gratefully thank DPT-2005K120580, METUand Akdeniz University research funds.
References
[1] M.A. De Paoli, W.A. Gazotti, J. Braz. Chem. Soc. 13 (2002) 410.[2] P.R. Somani, S. Radhakrishnan, Mater. Chem. Phys. 77 (2002) 117.[3] R.J. Mortimer, A.L. Dyer, J.R. Reynolds, Displays 27 (2006) 2.[4] G. Sonmez, Chem. Commun. 42 (2005) 5251.[5] K. Fesser, A.R. Bishop, D.K. Campbell, Phys. Rev. B 27 (1983) 4804.[6] G. Sonmez, I. Schwendeman, P. Schottland, K. Zong, J.R. Reynolds,
Macromolecules 36 (2003) 639.[7] G.A. Sotzing, J.L. Reddinger, A.R. Katritzky, J. Soloducho, R. Musgrave,
J.R. Reynolds, Chem. Mater. 9 (1997) 1578.[8] A.A. Argun, P.H. Aubert, B.C. Thompson, I. Schwendeman, C.L. Gaupp,
J. Hwang, N.J. Pinto, D.B. Tanner, A.G. MacDiarmid, J.R. Reynolds,Chem. Mater. 16 (2004) 4401.
[9] J.L. Reddinger, G.A. Sotzing, J.R. Reynolds, Chem. Commun. 15 (1996)1777.
[10] J.P. Ferraris, D.G. Guerrero, in: T.A. Skotheim, R.A. Elsenbaumer, J.R.Reynolds (Eds.), Handbook of Conducting Polymers, Marcel Dekker, NewYork, 1998.
[11] R.E.N. Mann, C.S. Kelley, T.L. Liu, Michael P. Cava, R.T. Carlin, J. Am.Chem. Soc. 115 (1993) 887.
[12] T.F. Otero, S. Villanueva, E. Brillas, J. Carrasco, Acta Polym. 49 (1998)433.
[13] E. Sahin, E. Sahmetlioglu, I.M. Akhmedov, C. Tanyeli, L. Toppare, Org.Elec. 7 (2006) 351.
[14] F. Jonas, L. Schrader, Synth. Met. 41/42 (1991) 831.[15] M. Dietrich, J. Heinze, G. Heywang, F. Jonas, J. Electroanal. Chem. 369
(1994) 87.[16] P. Camurlu, L. Toppare, J. Macromol. Sci. Part A: Pure Appl. Chem. 43
(2006) 449.[17] C.L. Gaupp, J.R. Reynolds, Macromolecules 36 (2003) 6305.[18] A. Kumar, D.M. Welsh, M.C. Morvant, F. Piroux, K.A. Abboud, J.R.
Reynolds, Chem. Mater. 10 (1998) 896.[19] E.M. Girotto, M.-A. De Paoli, J. Braz. Chem. Soc. 10 (1999) 394.





























