Embed Size (px)
Citation preview

FULLPA
T
p
w
a
th
a
lu
5
is
o
re
co
D
so
T
DOI: 10.1002/adfm.200700697
PERFine-Tuning of Yellow or Red Photo- and Electroluminescence ofFunctional Difluoro-boradiazaindacene Films**
By Laure Bonardi, Hani Kanaan, Franck Camerel, Pascale Jolinat, Pascal Retailleau, and Raymond Ziessel*
he present work describes the synthesis of difluoro-boradiazaindacenes (Bodipy) functionalized at the central 8-position by
henylamino moieties easily transformable into phenyl amide scaffoldings. Molecules carrying three linear or branched chains
ere prepared and characterized. AnX-ray crystal structure for the pivotal trimethoxyphenyl-Bodipy derivative was determined,
nd the packing is discussed in terms of molecular interactions; a key feature for the formation of thin films. All of the dyes are
ermally stable up to 170 8C but no liquid-crystalline phases are observed. Reversible reduction and oxidation processes occur
round þ0.97 and �1.34 V, respectively, versus saturated calomel electrode in solution and the electroactivity and photo-
minescence are maintained in thin films produced by vacuum evaporation. Interestingly, two distinct emissions are observed at
50 and 635 nm by electroluminescence of the trimethoxyphenyl-Bodipy derivative, corresponding to the luminescence of
olated molecules and dimers, respectively. Doping Alq3 films with this Bodipy molecule by vacuum evaporation produces
rganic light-emitting diodes (OLEDs) in which very efficient energy transfer from the Alq3 matrix to the Bodipy occurs by a
sonance mechanism involving the first Bodipy excited state. Yellow light (550 nm, 344 cd m�2 at 15 V) is emitted at low doping
ncentration (7 mol %), whereas red light (635 nm, 125 cd m�2 at 15 V) is emitted at higher concentration (19 mol %).
ispersion of the Bodipy into a fluorescent poly(N-vinylcarbazole) polymer (PVK) (�3 mol % per repeating unit of PVK) by
lution processing exclusively produces yellow emission owing to the isolated Bodipyfluorophore (550 nm, 213 cd m�2 at 15 V).
he second excited state of the Bodipy dye is likely involved during energy transfer from the PVK matrix.
1. Introduction
The engineering of robust molecular materials with
improved luminescence, electron exchange, and charge-
transport properties is a fruitful subject, attracting worldwide
interest and undergoing rapid development. This widespread
interest stems from potential applications as light-emitting
films and electroluminescent materials[1,2] and molecular
[*] Dr. R. Ziessel, L. Bonardi, Dr. F. CamerelLaboratoire de Chimie MoleculaireEcole Chimie, Polymeres, Materiaux (ECPM), ULP-CNRS25 rue Becquerel, 67008 Strasbourg Cedex (France)E-mail: [email protected]
H. Kanaan, Dr. P. JolinatLaboratoire Plasma et Conversion d’Energie (LAPLACE)Universite Paul Sabatier, Bat 3R331062 Toulouse Cedex 09 (France)
Dr. P. RetailleauLaboratoire de Cristallochimie, ICSN – CNRS, Bat 271 avenue de la Terrasse, 91198 Gif-sur-Yvette (France)
[**] This work was jointly supported by the Louis Pasteur Universitythrough the European School of Polymer and Materials Chemistry(ECPM), by the Centre National de la Recherche Scientifique (CNRS)of France, and ANR Contract ANR-05-BLAN-0004-01, FCP-OLEDs. Wewarmly thank Dr Bertrand Donnio from the IPCMS in Strasbourg forperforming low- and wide-angle X-ray diffraction experiments on thesenovel compounds. We are also indebted to Professor Jack Harrowfield(ISIS Strasbourg) for careful reading and commenting thismanuscriptprior to journal submission. Supporting information is availableonline from Wiley InterScience or from the author.
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag
probes.[3] Many different electroluminescent, organic, low-
molecular-weight or polymeric materials have been prepared
and tested, among which are tetracene,[4] pentacene,[5]
a-sexithiophene,[6] polyphenylvinylene, polyacetylene, poly-
fluorene, polycarbazole, pyridine- or oxadiazole-containing
conjugated polymers, polyquinolines, polyquinoxalines, and
cyanopolymers.[7] Recently, the harvesting of triplet excitons
using metal-containing complexes (Pt or Ir) has proved to be a
very successful way of making efficient organic light-emitting
diodes (OLEDs), since these devices incorporate a heavy-
metal atom with strong spin–orbit coupling that enhances
intersystem crossing, favoring the triplet state. In this way, the
lowest triplet state is efficiently populated and can produce
phosphorescence. By using a phosphorescent dye that captures
both singlet and triplet excitons, the internal efficiency can be
increased.[8] The doping of Alq3 (q¼ 8-hydroxyquinolate) with
organic dyes has also been found to be a means of increasing
the overall electroluminescence efficiency by allowing efficient
energy transfer from the metal-containing matrix to the
organic light-emitting dye.[9,10]
Of major importance in the field of effective organic
fluorophores are the difluoro-boradiazaindacenes, commonly
named boron-dipyrromethene dyes (Bodipy).[11] These dyes
are widely employed as useful fluorescence probes for labeling
biomolecules[12] and for the sensing of calcium ions,[13] nitric
oxides,[14] protons,[15] or various cations[16,17] by optoelectronic
switching. The attractive features of these dyes stem from
the facile modification of their structures, providing opportu-
nities to fine-tune their properties and to incorporate recog-
GmbH & Co. KGaA, Weinheim 401

FULLPAPER
L. Bonardi et al. / Electroluminescence of Functional Difluoro-boradiazaindacene Films
402
nition sites for a variety of analytes. These dyes combine sharp
absorption bands (full width at half-maximum (FWHM)�25–35 nm), high molar absorption coefficients (e¼ 40 000 to
110 000 M�1 cm�1), large fluorescence quantum yields
(f¼ 60–90%), and relatively long excited-state lifetimes
(1 to 10 ns) with excellent chemical and photochemical
stability in solution and in the solid state, and valuable
charge-transfer properties. Furthermore, their optical proper-
ties are sensitive to modifications around the pyrrole core,[18]
the central meso position,[19,20] and the boron atom.[21] In
addition, the dyes have good solubility in organic solvents and
are thus amenable towards chromatography on silica or
alumina. The high purity and neutrality of these molecules
allow vacuum deposition in conventional equipment. Exten-
sive efforts have therefore been devoted to the design and
preparation of sophisticated dyes for use as chromogenic
probes,[22] fluorescent switches,[23] electro-chemiluminescent
molecules (in solution)[24] and electroluminescent materials (in
the solid state),[25] laser dyes,[26] sensitizers for solar cells,[27]
drug-delivery agents,[28] and as electron-transfer probes for
radical ion pairs generated by local electric fields.[29]
In some exceptional cases, Bodipy has been used as an
antenna transferring its excitonic energy to acceptor frame-
works such as zinc porphyrins[30–32] or perylene dyes.[33] Long-
range charge separation induced by photoinduced electron
transfer processes has also been highlighted in different
accounts.[34,35] Such photoinduced electron transfer has been
extensively exploited in chemosensor systems.[36,37] Electro-
luminescence from borodipyrromethene dyes doped into a
polyvinyl–carbazole host polymer[25a] or thin films of alumi-
nium complexes has previously been observed in OLEDs.[25b]
Electrogenerated luminescence has also been investigated in
acetonitrile solution by pulsing the working electrode between
the first oxidation and reduction peaks of the Bodipy.[24] The
well-defined molecular structure of Bodipy makes it relatively
easy to establish firm structure–property relationships. Recen-
tly, novel Bodipy architectures enabled their incorporation
into supramolecular assemblies, such as liquid-crystalline
materials or organo-gelators,[38–40] and luminescent phenol-
pyridyl-boron complexes displaying bright luminescence have been
exploited in white and blue electroluminescent devices.[41]
Scheme 1. i) H2 (1 atm), 5 % Pd/C (0.1 equiv. of Pd), EtOH/CH2Cl2, r.t., 1
www.afm-journal.de � 2008 WILEY-VCH Verlag GmbH
The design of luminescent and photoresponsive liquid
crystals is particularly appealing owing to their potential
applications in photoelectronic devices.[42] Advantages of
liquid crystals are their ease of processing (via solution
techniques), the possibility of alignment by shear forces or by
application of electrical or magnetic fields, and their capacity
for self-healing. For these reasons, we have synthesized new
Bodipy derivatives bearing gallate-substituted residues with
aliphatic chains (8, 10, 12, 16, and 20 carbons) or with methoxy
groups as model compounds, and these were well characterized
by X-ray diffraction on single crystals, NMR, absorption and
fluorescence spectroscopies, and electrochemistry. Their remar-
kable optoelectronic properties led us to test these materials in
electroluminescent devices. The methoxy-substituted deriva-
tive gives stable electroluminescent films as pure material,
while incorporation of the long-chain compounds produces
unstable devices. To improve the efficiency of the methox-
y-derivative device, this compound was sublimated in an Alq3matrix or incorporated by solution-processing in PVK polymer
to produce multilayered electroluminescent devices. Owing to
the propensity of Bodipy to form luminescent aggregates, it
was possible to tune the electrogenerated light from yellow to
red.
2. Results and Discussion
2.1. Synthesis
The starting p-nitrophenyl-4,4-difluoro-4-bora-3a,4a-diaza-
s-indacene derivative 1 was obtained by condensation of
2,4-dimethyl-3-ethylpyrrole (also called Kryptopyrrole) with
the p-nitrophenylacylchloride in dichloromethane at room
temperature, followed by a complexation to a BF2 fragment
after deprotonation with triethylamine.[43] By means of a
catalytic hydrogenation over Pd on charcoal, this nitro
derivative can be easily reduced into the corresponding amine
(Scheme 1).[44] Reaction of the p-aminophenyl-4,4-difluoro-4-
bora-3a,4a-diaza-s-indacene derivative with a series of
3,4,5-tris-alkyloxy-benzoic acids leads to the synthesis of a
wide range of amido-Bodipy derivatives functionalized with
day. ii) 3,4,5-tris-alkyloxy-benzoic acid chloride (1 equiv.), TEA, CH2Cl2, r.t.
& Co. KGaA, Weinheim Adv. Funct. Mater. 2008, 18,401–413

FULLPAPER
L. Bonardi et al./Electroluminescence of Functional Difluoro-boradiazaindacene Films
Figure 1. Oak Ridge Thermal Ellipsoid Plot (ORTEP) view of 3 withprincipal atomic numbering (thermal ellipsoids at 50% probability level).
straight or branched paraffin chains. Its formation results from
reacting the amino-Bodipy compound with 3,4,5-tris-alkyloxy-
benzoic acid chlorides in presence of triethylamine (TEA)
(Scheme 1). For the phytol strands a racemic mixture of
3,4,5-tris-(3,7,11,15-tetramethyl-hexadecyl) was used, whereas
for the citronellol strands the pure (S)-(þ)-citronellyl deriva-
tive was used.[45] All compounds were fully characterized by
means of NMR spectroscopy, Fourier transform infrared
(FTIR) spectroscopy, electrospray ionization mass spectro-
metry (ESI-MS), and elemental analysis. The introduction of
long carbon chains improves the solubility in common organic
solvents, facilitates evaporation under high vacuum, and
results in well-organized solid-state phases by microsegrega-
tion between the polar Bodipy core and the apolar chains.
The amide bond connecting the fluorophore to the gallate
platform was especially introduced to control and stabilize the
molecular organization through hydrogen bonding (vide infra).
2.2. X-Ray Characterization
Single-crystals of compound 3 suitable for X-ray structure
determinations were obtained by slow evaporation of a
CH2Cl2/hexane solvent mixture. This compound crystallized
in the monoclinic space group P21/c (a¼ 15.036(1) A; b¼21.641(2) A; c¼ 10.101(1) A; V¼ 3145.8(5) A3, Z¼ 4). Figure 1
shows the atomic numbering and labeling scheme. The
geometry around the boron atom is tetrahedral and the
observed distances and angles compare well with the ones
usually observed on similar compounds (distances being: B-F1,
1.382(4) A; B-F2, 1.388(4) A; B-N1, 1.540(4) A; B-N2, 1.549(4)
A and angles being: N1-B-F1, 110.5(3)8; N1-B-F2, 110.4(2)8;N2-B-N1, 106.8(2)8; N2-B-F2, 109.6(3)8; F1-B-F2, 109.0(3)8and N2-B-F1, 110.4(3)8).[46] The dipyrromethene subunit
(including the two pyrrole rings, the four methyl carbons,
and the two methylene carbons) is planar. The central phenyl
ring Ar1 is almost orthogonal to the Bodipy fragment (angle
Ar1-Bodipy¼ 80.08) and twisted by 39.18 from the plane of the
terminal phenyl ring Ar2. The twist between the Bodipy
fragment and the terminal phenyl ring Ar2 is 42.28.Different views of the crystal packing are presented in
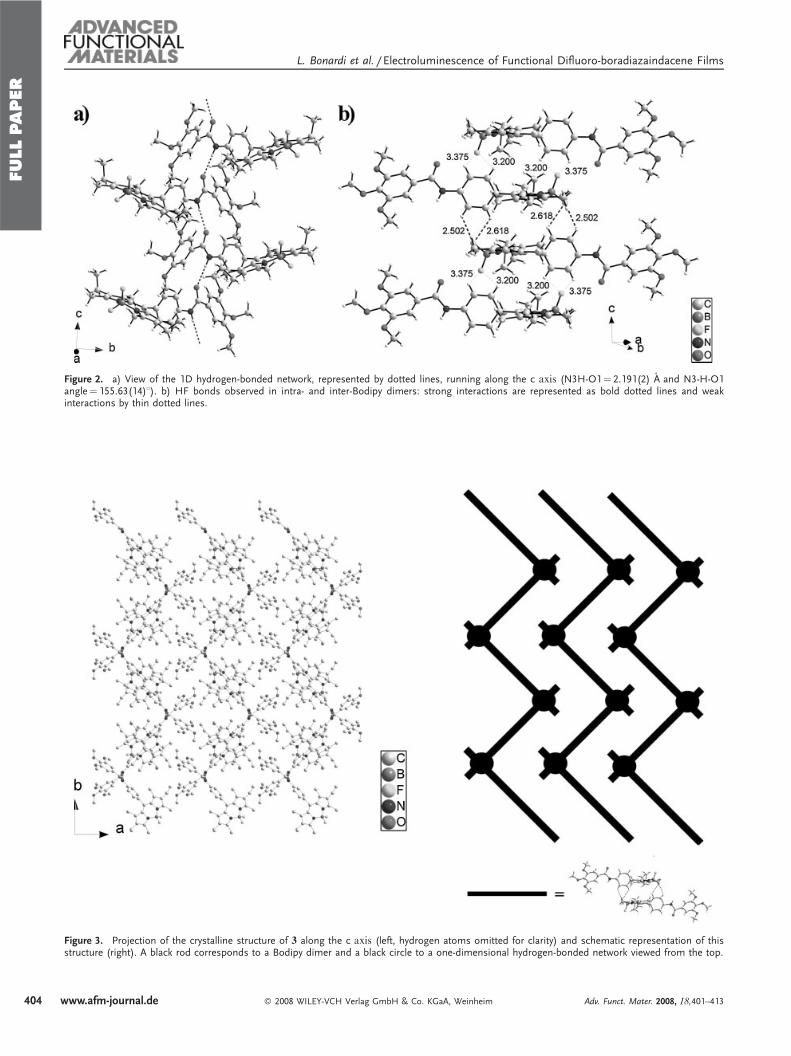
Figures 2 and 3. The molecules are connected by a uniform
unidimensional hydrogen-bonded network running along the c
axis (represented by dashed lines in Fig. 2). These inter-
molecular hydrogen bonds involve the hydrogen atom on N3
on one molecule and the oxygen atom O1 of the adjacent
molecule. In this way, all the molecules are engaged in two
hydrogen bonds pointing in opposite directions (N3H-O1¼2.19 A and N3-H-O1 angle¼ 155.78; Fig. 2a). FTIR spectro-
scopy is a powerful tool to probe hydrogen bonding and,[47–48]
at room temperature, this compound has its amide group
involved in strong hydrogen bonding, as clearly evidenced by
the nNH, nCO stretching vibrations at 3308 and 1650 cm�1
respectively. Note that corresponding values for free amides
usually lie at 3500-3400 cm�1 for nNH and around 1680 cm�1 for
nCO.[38,39] The presence of single nNH, and nCO stretching bands
in the IR spectra confirms a well-organized hydrogen-bonded
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag GmbH &
network in which only one type of hydrogen bond is effective.
The structure determination shows that this supramolecular
hydrogen-bonded edifice adopts a V-shape. These hydrogen-
bonded strands are further connected via the Bodipy fragments
by H . . .F interactions (Fig. 2b), which result in strongly
stabilized zig-zag layers (Fig. 3). Closer examination of the
structure reveals that the Bodipy fragments are dimerized in a
centrosymmetric fashion by the formation of two hydrogen
bonds between one fluorine atom of the BF2 fragments (F2) on
the first molecule and two hydrogen atoms on the phenyl ring
Ar1 of the second molecule (C6AH----F1¼ 2.618 A; C6AHF1
angle¼ 117.38; C5AH----F1¼ 2.502 A; C6AHF1 angle¼122.08; Fig. 2, bold dashed lines). These Bodipy dimers are
further connected by weaker H . . .F interactions along the c
direction (C2AH----F2¼ 3.200 A; C2AHF2 angle¼ 98.38;C3AH----F2¼ 3.375 A; C3AHF2 angle¼ 102.28; Fig. 2, thindashed lines). The formation of Bodipy dimers orients the ethyl
groups of one fluorophore in the same direction. No strong
interactions are apparent between the different zig-zag layers.
2.3. Thermal Properties
All of the compounds display a clear melting point, corres-
ponding to a phase transition from a birefringent powder to an
Co. KGaA, Weinheim www.afm-journal.de 403
FULLPAPER
L. Bonardi et al. / Electroluminescence of Functional Difluoro-boradiazaindacene Films
Figure 2. a) View of the 1D hydrogen-bonded network, represented by dotted lines, running along the c axis (N3H-O1¼ 2.191(2) A and N3-H-O1angle¼ 155.63(14)8). b) HF bonds observed in intra- and inter-Bodipy dimers: strong interactions are represented as bold dotted lines and weakinteractions by thin dotted lines.
Figure 3. Projection of the crystalline structure of 3 along the c axis (left, hydrogen atoms omitted for clarity) and schematic representation of thisstructure (right). A black rod corresponds to a Bodipy dimer and a black circle to a one-dimensional hydrogen-bonded network viewed from the top.
404 www.afm-journal.de � 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Funct. Mater. 2008, 18,401–413

FULLPAPER
L. Bonardi et al./Electroluminescence of Functional Difluoro-boradiazaindacene Films
Figure 4. Compound 4 viewed by optical microscopy below 150 8C on cooling from the isotropicliquid (left, without polarizer; right, same area between crossed polarizers).
isotropic liquid. Isotropization temperatures decrease as the
chain lengths increase (Table S1). The high enthalpy values
observed for the last transition upon heating confirmed these
observations. Compounds 7 and 8 directly melt from the crystal
to the isotropic liquid, but the other compounds (4, 5, and 6)
exhibit several successive crystal-to-crystal phase transforma-
tions before melting to the isotropic liquid. Unfortunately,
despite the presence of long carbon chains, none of these
compounds form liquid-crystalline phases. However, on cool-
ing from the isotropic state, it was observed by optical micros-
copy between crossed polarizers that these compounds have a
marked tendency to recrystallize uniformly as thin films. In
this way, large monodomains of well-aligned molecular
assemblies can be formed, as observed in Figure 4 where
three monodomains with different orientations are presented.
The tendency of these fluorophores to form well-organized,
crystalline thin films may be useful for optoelectronic appli-
cations such as OLEDs or solar cells.
2.4. Optical and Electrochemical Properties in Solution
Spectroscopic data for compounds 3 to 8 are gathered in
Table 1 and for compounds 1, 2 in Table S2. All of the
compounds show similar absorption patterns which are
characteristic of Bodipy fluorophores, and a representative
example is depicted in Figure 5. The absorption spectrum is
composed of a strong S0! S1 (p!p�) transition located
around 525 nm, with molar extinction coefficients ranging from
70 000 to 100 000 M�1 cm�1, in keeping with classical Bodipy
Scheme 2. Molecular structures of Bodipy A and reference compound B.
derivatives.[49] A second absorption band
centered around 370 nm, is assigned to
the S0! S2 transition of the Bodipy sub-
unit.[50] The third absorption at around
275 nm is likely due to p-p� and n-p�
transitions localized on the phenyl and
dipyrromethene fragments.[51] The amido
compounds have high fluorescence quantum
yields, ranging from 58 to 75%. The weak
Stokes’ shifts (about 500 cm�1) observed
over the whole series of fluorophores is in
good agreement with a singlet emitting
state. Excitation spectra perfectly match the
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
absorption spectra (Fig. 5 as a typical
example), which is in keeping with a
unique excited state, excluding the exis-
tence of a CT transition, despite the
presence of both electron-donating and
electron-attracting fragments on these
fluorophores. Moreover, no quenching
of the luminescence by molecular oxygen
is observed, excluding the presence of a
possible triplet emitting state. The fluor-
escence decay profiles of these molecules
can be fitted by a single exponential decay,
with fluorescence lifetimes ranging from
4.9 to 6.2 ns (Table 1 and in Fig. 5b), in line with a singlet
emissive state. All of the fluorescence spectra exhibit nice
mirror symmetry with the lowest-energy absorption band,
meaning that the corresponding transitions involve the same
excited state.
The electrochemical properties were determined by cyclic
voltammetry in dichloromethane solution. Table 1 lists the
potentials (relative to the saturated chemical electrode (SCE)
reference) for the waves that were observed in the þ1.6 to
�2.0 Vwindow. Firstly, for all of the compounds, the reversible
anodic wave around þ0.97 V is assigned to the (Bodipy/
Bodipyþ�) couple. Note that this wave is less anodic with
respect to phenyl- or toluyl-substituted Bodipy’sA (Scheme 2)
[þ1.11 (60) V and �1.26 (70) V versus SCE].[53] This likely
reflects the fact that the trialkoxy-gallate substituents are
better electron-donating groups compared to the other
derivatives. There is no indication of oxidation to the
(Bodipy2þ) dication within the given electrochemical window
as previously observed for pyridine-linked Bodipy com-
pounds.[53] The second oxidation is irreversible at around
þ1.52 V and is likely localized on the trialkoxyphenyl subunit
in keeping with literature data[54,55] and the electrochemistry of
the reference compound B (Scheme 2) (þ1.55 V irrev.).
The single reduction is attributed to the Bodipy radical
anion (Bodipy��), which was in all cases reversible and more
cathodic compared to the toluyl compound A. This observa-
tion is also in keeping with the increase of electron density
imparted by the trialkoxyphenyl fragments.
Note that the LUMO–HOMO gap (at about 2.30 eV)
remains similar along the series, as reflected by the constancy of
www.afm-journal.de 405
FULLPAPER
L. Bonardi et al. / Electroluminescence of Functional Difluoro-boradiazaindacene Films
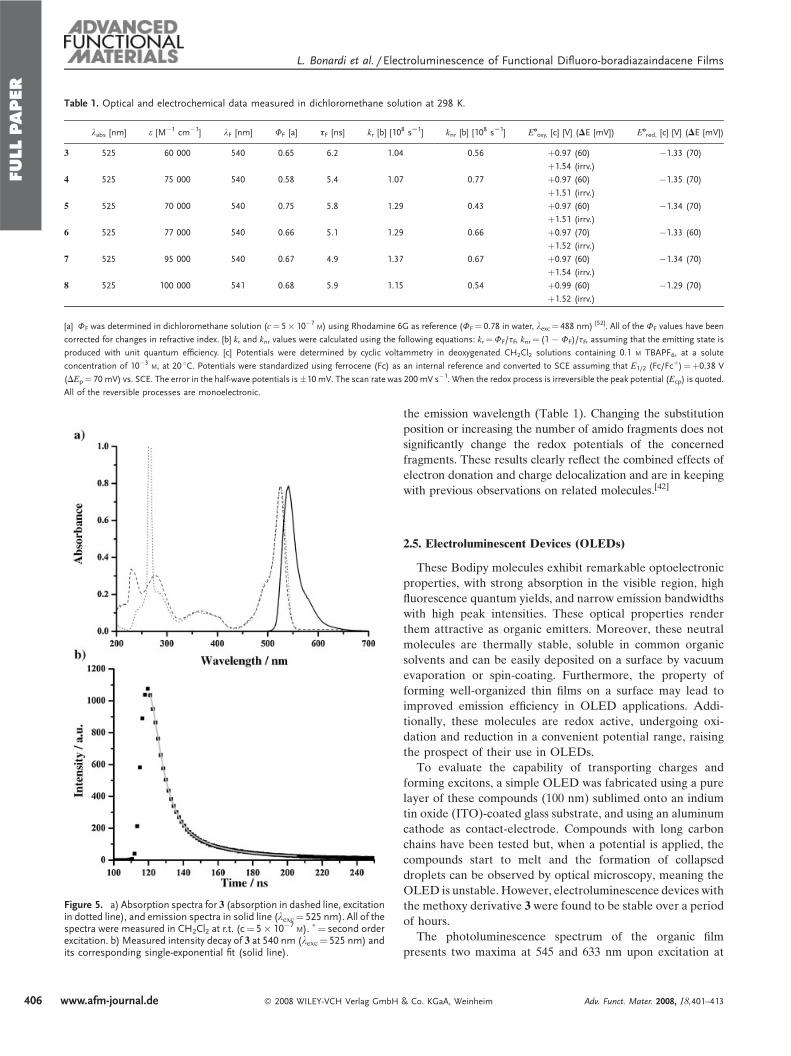
Table 1. Optical and electrochemical data measured in dichloromethane solution at 298 K.
labs [nm] e [M�1 cm�1] lF [nm] FF [a] tF [ns] kr [b] [108 sS1] knr [b] [10
8 sS1] E-oxy, [c] [V] (DE [mV]) E-red, [c] [V] (DE [mV])
3 525 60 000 540 0.65 6.2 1.04 0.56 þ0.97 (60) �1.33 (70)
þ1.54 (irrv.)
4 525 75 000 540 0.58 5.4 1.07 0.77 þ0.97 (60) �1.35 (70)
þ1.51 (irrv.)
5 525 70 000 540 0.75 5.8 1.29 0.43 þ0.97 (60) �1.34 (70)
þ1.51 (irrv.)
6 525 77 000 540 0.66 5.1 1.29 0.66 þ0.97 (70) �1.33 (60)
þ1.52 (irrv.)
7 525 95 000 540 0.67 4.9 1.37 0.67 þ0.97 (60) �1.34 (70)
þ1.54 (irrv.)
8 525 100 000 541 0.68 5.9 1.15 0.54 þ0.99 (60) �1.29 (70)
þ1.52 (irrv.)
[a] FF was determined in dichloromethane solution (c¼ 5� 10�7M) using Rhodamine 6G as reference (FF¼ 0.78 in water, lexc¼ 488 nm) [52]. All of the FF values have been
corrected for changes in refractive index. [b] kr and knr values were calculated using the following equations: kr¼FF/tF, knr¼ (1 � FF)/tF, assuming that the emitting state is
produced with unit quantum efficiency. [c] Potentials were determined by cyclic voltammetry in deoxygenated CH2Cl2 solutions containing 0.1 M TBAPF6, at a solute
concentration of 10�3M, at 20 8C. Potentials were standardized using ferrocene (Fc) as an internal reference and converted to SCE assuming that E1/2 (Fc/Fc
þ)¼þ0.38 V
(DEp¼ 70 mV) vs. SCE. The error in the half-wave potentials is�10 mV. The scan rate was 200 mV s�1. When the redox process is irreversible the peak potential (Ecp) is quoted.
All of the reversible processes are monoelectronic.
Figure 5. a) Absorption spectra for 3 (absorption in dashed line, excitationin dotted line), and emission spectra in solid line (lexc¼ 525 nm). All of thespectra were measured in CH2Cl2 at r.t. (c¼ 5� 10�7
M). �¼ second orderexcitation. b) Measured intensity decay of 3 at 540 nm (lexc¼ 525 nm) andits corresponding single-exponential fit (solid line).
406 www.afm-journal.de � 2008 WILEY-VCH Verlag GmbH
the emission wavelength (Table 1). Changing the substitution
position or increasing the number of amido fragments does not
significantly change the redox potentials of the concerned
fragments. These results clearly reflect the combined effects of
electron donation and charge delocalization and are in keeping
with previous observations on related molecules.[42]
2.5. Electroluminescent Devices (OLEDs)
These Bodipy molecules exhibit remarkable optoelectronic
properties, with strong absorption in the visible region, high
fluorescence quantum yields, and narrow emission bandwidths
with high peak intensities. These optical properties render
them attractive as organic emitters. Moreover, these neutral
molecules are thermally stable, soluble in common organic
solvents and can be easily deposited on a surface by vacuum
evaporation or spin-coating. Furthermore, the property of
forming well-organized thin films on a surface may lead to
improved emission efficiency in OLED applications. Addi-
tionally, these molecules are redox active, undergoing oxi-
dation and reduction in a convenient potential range, raising
the prospect of their use in OLEDs.
To evaluate the capability of transporting charges and
forming excitons, a simple OLED was fabricated using a pure
layer of these compounds (100 nm) sublimed onto an indium
tin oxide (ITO)-coated glass substrate, and using an aluminum
cathode as contact-electrode. Compounds with long carbon
chains have been tested but, when a potential is applied, the
compounds start to melt and the formation of collapsed
droplets can be observed by optical microscopy, meaning the
OLED is unstable. However, electroluminescence devices with
the methoxy derivative 3 were found to be stable over a period
of hours.
The photoluminescence spectrum of the organic film
presents two maxima at 545 and 633 nm upon excitation at
& Co. KGaA, Weinheim Adv. Funct. Mater. 2008, 18,401–413
FULLPAPER
L. Bonardi et al./Electroluminescence of Functional Difluoro-boradiazaindacene Films
390 nm. The first emission peak can be safely attributed, in
regard to the results obtained in solution, to the emission of
monomeric and isolated Bodipy molecules. The second
emission peak at 633 nm is attributed to the formation of
dimers. Based on the crystal structure of the tris-amide
derivative bearing methoxy groups in place of the aliphatic
chains, we suggest that the aggregation process is likely driven
by formation of dimers stabilized by hydrogen bonding. In fact,
in the solid state, strongly stabilized Bodipy dimers are formed
through H-F bonds (Fig. 2b). Such behavior has already been
observed by fluorescencemeasurements performed on gels and
liquid-crystalline materials based on Bodipy cores.[39] It has
been demonstrated that the emission at 635 nm originates from
the formation of excited dimers (excimers). Larger and less-
organized aggregates are virtually not observed because they
should emit at lower energies and the width of the emission
band should be larger. The excitation spectra measured at both
emission wavelengths are identical and perfectly match the
absorption spectra over the entire spectral range.
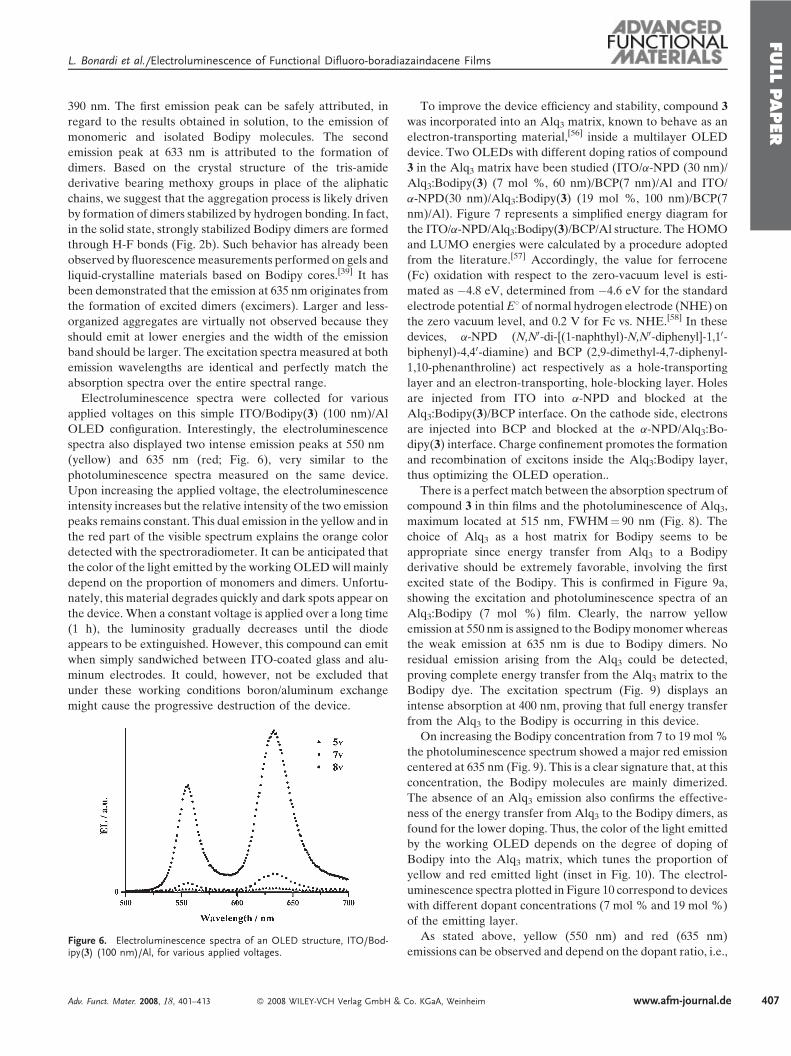
Electroluminescence spectra were collected for various
applied voltages on this simple ITO/Bodipy(3) (100 nm)/Al
OLED configuration. Interestingly, the electroluminescence
spectra also displayed two intense emission peaks at 550 nm
(yellow) and 635 nm (red; Fig. 6), very similar to the
photoluminescence spectra measured on the same device.
Upon increasing the applied voltage, the electroluminescence
intensity increases but the relative intensity of the two emission
peaks remains constant. This dual emission in the yellow and in
the red part of the visible spectrum explains the orange color
detected with the spectroradiometer. It can be anticipated that
the color of the light emitted by the working OLEDwill mainly
depend on the proportion of monomers and dimers. Unfortu-
nately, this material degrades quickly and dark spots appear on
the device. When a constant voltage is applied over a long time
(1 h), the luminosity gradually decreases until the diode
appears to be extinguished. However, this compound can emit
when simply sandwiched between ITO-coated glass and alu-
minum electrodes. It could, however, not be excluded that
under these working conditions boron/aluminum exchange
might cause the progressive destruction of the device.
Figure 6. Electroluminescence spectra of an OLED structure, ITO/Bod-ipy(3) (100 nm)/Al, for various applied voltages.
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag GmbH &
To improve the device efficiency and stability, compound 3
was incorporated into an Alq3 matrix, known to behave as an
electron-transporting material,[56] inside a multilayer OLED
device. Two OLEDs with different doping ratios of compound
3 in the Alq3 matrix have been studied (ITO/a-NPD (30 nm)/
Alq3:Bodipy(3) (7 mol %, 60 nm)/BCP(7 nm)/Al and ITO/
a-NPD(30 nm)/Alq3:Bodipy(3) (19 mol %, 100 nm)/BCP(7
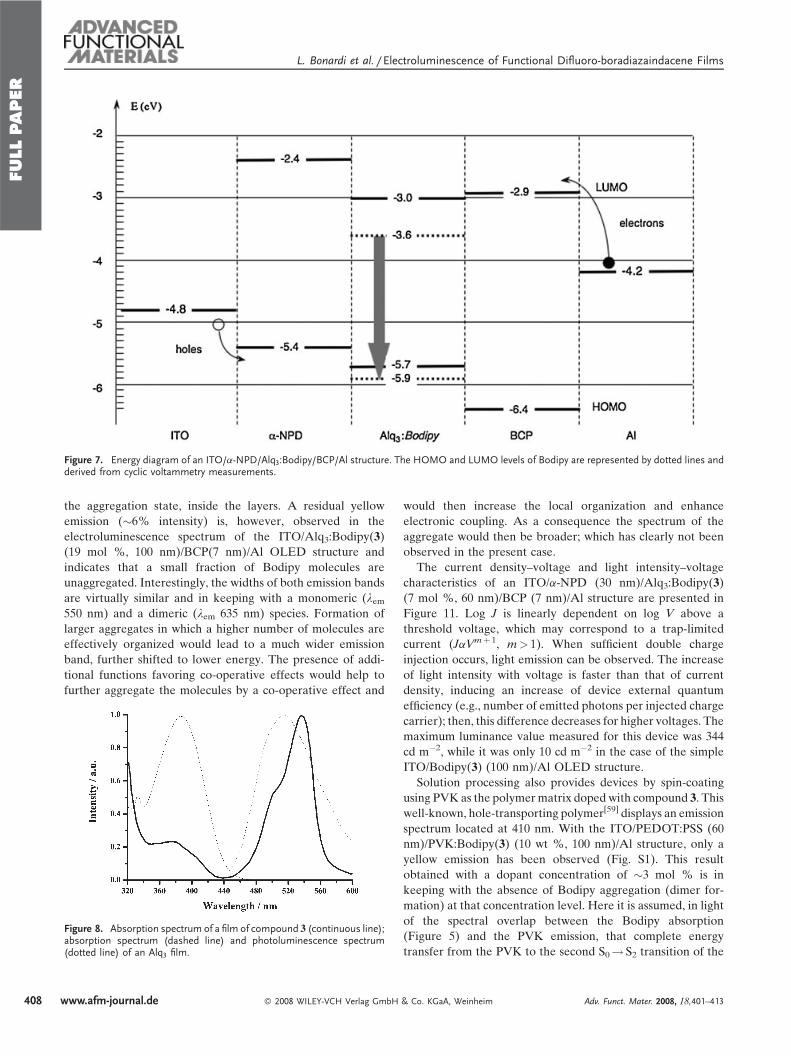
nm)/Al). Figure 7 represents a simplified energy diagram for
the ITO/a-NPD/Alq3:Bodipy(3)/BCP/Al structure. The HOMO
and LUMO energies were calculated by a procedure adopted
from the literature.[57] Accordingly, the value for ferrocene
(Fc) oxidation with respect to the zero-vacuum level is esti-
mated as �4.8 eV, determined from �4.6 eV for the standard
electrode potentialE8 of normal hydrogen electrode (NHE) on
the zero vacuum level, and 0.2 V for Fc vs. NHE.[58] In these
devices, a-NPD (N,N0-di-[(1-naphthyl)-N,N0-diphenyl]-1,10-
biphenyl)-4,40-diamine) and BCP (2,9-dimethyl-4,7-diphenyl-
1,10-phenanthroline) act respectively as a hole-transporting
layer and an electron-transporting, hole-blocking layer. Holes
are injected from ITO into a-NPD and blocked at the
Alq3:Bodipy(3)/BCP interface. On the cathode side, electrons
are injected into BCP and blocked at the a-NPD/Alq3:Bo-
dipy(3) interface. Charge confinement promotes the formation
and recombination of excitons inside the Alq3:Bodipy layer,
thus optimizing the OLED operation..
There is a perfect match between the absorption spectrum of
compound 3 in thin films and the photoluminescence of Alq3,
maximum located at 515 nm, FWHM¼ 90 nm (Fig. 8). The
choice of Alq3 as a host matrix for Bodipy seems to be
appropriate since energy transfer from Alq3 to a Bodipy
derivative should be extremely favorable, involving the first
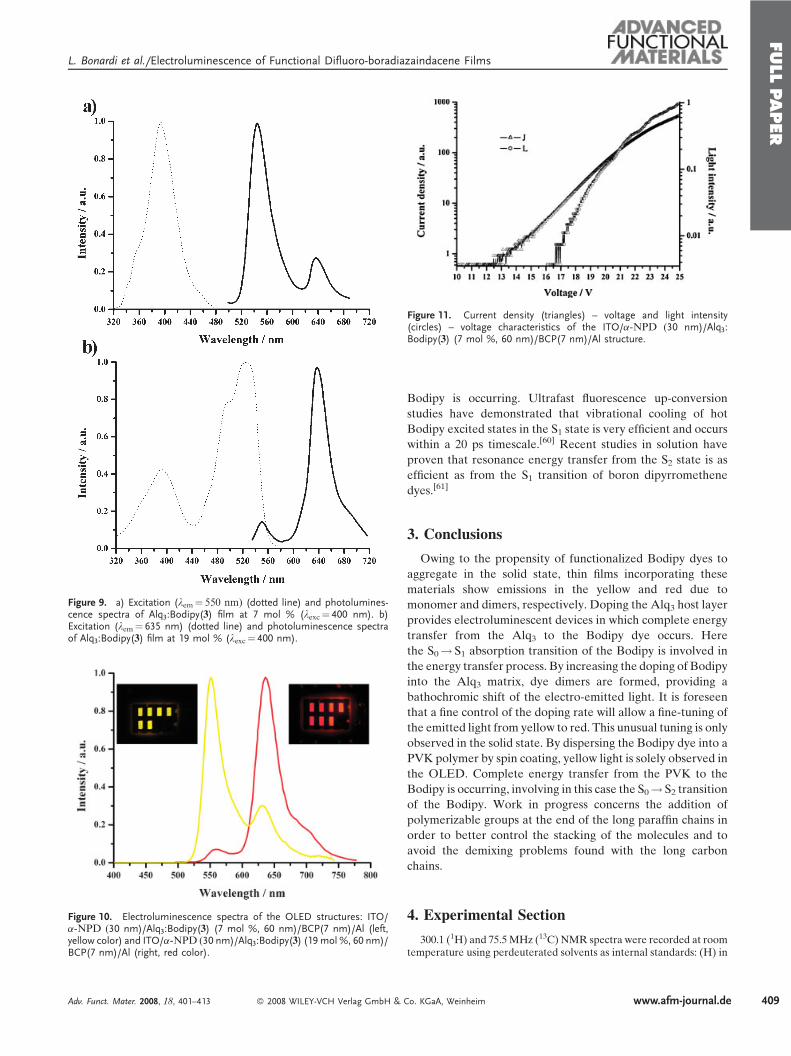
excited state of the Bodipy. This is confirmed in Figure 9a,
showing the excitation and photoluminescence spectra of an
Alq3:Bodipy (7 mol %) film. Clearly, the narrow yellow
emission at 550 nm is assigned to the Bodipymonomer whereas
the weak emission at 635 nm is due to Bodipy dimers. No
residual emission arising from the Alq3 could be detected,
proving complete energy transfer from the Alq3 matrix to the
Bodipy dye. The excitation spectrum (Fig. 9) displays an
intense absorption at 400 nm, proving that full energy transfer
from the Alq3 to the Bodipy is occurring in this device.
On increasing the Bodipy concentration from 7 to 19 mol %
the photoluminescence spectrum showed a major red emission
centered at 635 nm (Fig. 9). This is a clear signature that, at this
concentration, the Bodipy molecules are mainly dimerized.
The absence of an Alq3 emission also confirms the effective-
ness of the energy transfer from Alq3 to the Bodipy dimers, as
found for the lower doping. Thus, the color of the light emitted
by the working OLED depends on the degree of doping of
Bodipy into the Alq3 matrix, which tunes the proportion of
yellow and red emitted light (inset in Fig. 10). The electrol-
uminescence spectra plotted in Figure 10 correspond to devices
with different dopant concentrations (7 mol % and 19 mol %)
of the emitting layer.
As stated above, yellow (550 nm) and red (635 nm)
emissions can be observed and depend on the dopant ratio, i.e.,
Co. KGaA, Weinheim www.afm-journal.de 407
FULLPAPER
L. Bonardi et al. / Electroluminescence of Functional Difluoro-boradiazaindacene Films
Figure 7. Energy diagram of an ITO/a-NPD/Alq3:Bodipy/BCP/Al structure. The HOMO and LUMO levels of Bodipy are represented by dotted lines andderived from cyclic voltammetry measurements.
408
the aggregation state, inside the layers. A residual yellow
emission (�6% intensity) is, however, observed in the
electroluminescence spectrum of the ITO/Alq3:Bodipy(3)
(19 mol %, 100 nm)/BCP(7 nm)/Al OLED structure and
indicates that a small fraction of Bodipy molecules are
unaggregated. Interestingly, the widths of both emission bands
are virtually similar and in keeping with a monomeric (lem550 nm) and a dimeric (lem 635 nm) species. Formation of
larger aggregates in which a higher number of molecules are
effectively organized would lead to a much wider emission
band, further shifted to lower energy. The presence of addi-
tional functions favoring co-operative effects would help to
further aggregate the molecules by a co-operative effect and
Figure 8. Absorption spectrum of a film of compound 3 (continuous line);absorption spectrum (dashed line) and photoluminescence spectrum(dotted line) of an Alq3 film.
www.afm-journal.de � 2008 WILEY-VCH Verlag GmbH
would then increase the local organization and enhance
electronic coupling. As a consequence the spectrum of the
aggregate would then be broader; which has clearly not been
observed in the present case.
The current density–voltage and light intensity–voltage
characteristics of an ITO/a-NPD (30 nm)/Alq3:Bodipy(3)
(7 mol %, 60 nm)/BCP (7 nm)/Al structure are presented in
Figure 11. Log J is linearly dependent on log V above a
threshold voltage, which may correspond to a trap-limited
current (JaVmþ 1, m> 1). When sufficient double charge
injection occurs, light emission can be observed. The increase
of light intensity with voltage is faster than that of current
density, inducing an increase of device external quantum
efficiency (e.g., number of emitted photons per injected charge
carrier); then, this difference decreases for higher voltages. The
maximum luminance value measured for this device was 344
cd m�2, while it was only 10 cd m�2 in the case of the simple
ITO/Bodipy(3) (100 nm)/Al OLED structure.
Solution processing also provides devices by spin-coating
using PVK as the polymermatrix doped with compound 3. This
well-known, hole-transporting polymer[59] displays an emission
spectrum located at 410 nm. With the ITO/PEDOT:PSS (60
nm)/PVK:Bodipy(3) (10 wt %, 100 nm)/Al structure, only a
yellow emission has been observed (Fig. S1). This result
obtained with a dopant concentration of �3 mol % is in
keeping with the absence of Bodipy aggregation (dimer for-
mation) at that concentration level. Here it is assumed, in light
of the spectral overlap between the Bodipy absorption
(Figure 5) and the PVK emission, that complete energy
transfer from the PVK to the second S0! S2 transition of the
& Co. KGaA, Weinheim Adv. Funct. Mater. 2008, 18,401–413
FULLPAPER
L. Bonardi et al./Electroluminescence of Functional Difluoro-boradiazaindacene Films
Figure 9. a) Excitation (lem¼ 550 nm) (dotted line) and photolumines-cence spectra of Alq3:Bodipy(3) film at 7 mol % (lexc¼ 400 nm). b)Excitation (lem¼ 635 nm) (dotted line) and photoluminescence spectraof Alq3:Bodipy(3) film at 19 mol % (lexc¼ 400 nm).
Figure 10. Electroluminescence spectra of the OLED structures: ITO/a-NPD (30 nm)/Alq3:Bodipy(3) (7 mol %, 60 nm)/BCP(7 nm)/Al (left,yellow color) and ITO/a-NPD (30 nm)/Alq3:Bodipy(3) (19 mol %, 60 nm)/BCP(7 nm)/Al (right, red color).
Figure 11. Current density (triangles) – voltage and light intensity(circles) – voltage characteristics of the ITO/a-NPD (30 nm)/Alq3:Bodipy(3) (7 mol %, 60 nm)/BCP(7 nm)/Al structure.
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag GmbH &
Bodipy is occurring. Ultrafast fluorescence up-conversion
studies have demonstrated that vibrational cooling of hot
Bodipy excited states in the S1 state is very efficient and occurs
within a 20 ps timescale.[60] Recent studies in solution have
proven that resonance energy transfer from the S2 state is as
efficient as from the S1 transition of boron dipyrromethene
dyes.[61]
3. Conclusions
Owing to the propensity of functionalized Bodipy dyes to
aggregate in the solid state, thin films incorporating these
materials show emissions in the yellow and red due to
monomer and dimers, respectively. Doping the Alq3 host layer
provides electroluminescent devices in which complete energy
transfer from the Alq3 to the Bodipy dye occurs. Here
the S0! S1 absorption transition of the Bodipy is involved in
the energy transfer process. By increasing the doping of Bodipy
into the Alq3 matrix, dye dimers are formed, providing a
bathochromic shift of the electro-emitted light. It is foreseen
that a fine control of the doping rate will allow a fine-tuning of
the emitted light from yellow to red. This unusual tuning is only
observed in the solid state. By dispersing the Bodipy dye into a
PVK polymer by spin coating, yellow light is solely observed in
the OLED. Complete energy transfer from the PVK to the
Bodipy is occurring, involving in this case the S0!S2 transition
of the Bodipy. Work in progress concerns the addition of
polymerizable groups at the end of the long paraffin chains in
order to better control the stacking of the molecules and to
avoid the demixing problems found with the long carbon
chains.
4. Experimental Section
300.1 (1H) and 75.5MHz (13C) NMR spectra were recorded at roomtemperature using perdeuterated solvents as internal standards: (H) in
Co. KGaA, Weinheim www.afm-journal.de 409

FULLPAPER
L. Bonardi et al. / Electroluminescence of Functional Difluoro-boradiazaindacene Films
410
ppm relative to residual protiated solvent; (C) in ppm relative to thesolvent. A ZAB-HF-VB-analytical apparatus in FABþ mode was usedwith m-nitrobenzyl alcohol (m-NBA) as the matrix. Chromatographicpurification was conducted using 40–63 mm silica gel. Thin-layerchromatography (TLC) was performed on silica-gel plates coated withfluorescent indicator. FTIR spectra were recorded using a Perki-n-Elmer ‘‘spectrum one’’ spectrometer from thin films deposited ontodry KBr pellets. UV-vis spectra were recorded using a UVIKON 940/941 dual-beam grating spectrophotometer (Kontron Instruments)with a 1 cm quartz cell. Fluorescence spectra were recorded on aPerkin-Elmer LS50B spectrofluorimeter. All of the fluorescencespectra were corrected. The fluorescence quantum yield (Fexp) wascalculated fromEquation 1. In Equation 1, F denotes the integral of thecorrected fluorescence spectrum, A is the absorbance at the excitationwavelength, and n is the refractive index of the medium. The referencesystem used was rhodamine 6G (Fref¼ 0.78, lexc¼ 488 nm) [52] inair-equilibrated water.
Fexp ¼ Fref
Ff1� expð�Aref ln 10Þn2gFref f1� expð�A ln 10Þn2ref g
(1)
Luminescence lifetimes were measured on a PTI QuantaMasterspectrofluorimeter, using TimeMaster software with time-correlatedsingle photon mode coupled to a stroboscopic system. The excitationsource was a thyratron-gated flash lamp filled with nitrogen gas. Nofilter was used for the excitation. An interference filter centred at550 nm selected the emission wavelengths. The instrument responsefunction was determined by using a light-scattering solution(LUDOX). Differential scanning calorimetry (DSC) was performedon a Netzsch DSC 200 PC/1/M/H Phox instrument equipped with anintracooler, allowing measurements from �65 8C up to 450 8C. Thesamples were examined at a scanning rate of 10 K min�1 by applyingtwo heating cycles and one cooling cycle. The apparatus was calibratedwith indium (156.6 8C). Phase behavior was studied by polarized-lightoptical microscopy (POM) on a Leica DMLB microscope equippedwith a Linkam LTS350 hot-stage and a Linkam TMS94 centralprocessor.
OLED devices with pure Bodipy compounds were elaborated bysuccessively evaporating the organic compound and aluminum ontoITO-coated glass substrates (Visiontek Systems Ltd). MultilayerOLEDs of Bodipy compounds incorporated into an Alq3 matrix wereobtained by coevaporation ofAlq3 andBodipy from separate crucibles.The home-made evaporator used was located inside a nitrogen-filledglovebox comprising an oxygen and water-vapor elimination system(Jacomex). The materials were placed into silica crucibles andevaporated under a secondary vacuum (�10�5–10�4 Pa) by Jouleheating. Glass or ITO-coated glass substrates were placed �20 cmabove the evaporation sources. The deposition rates (typically 0.1 nms�1) and thicknesses were measured in situ with a quartz microbalance(InficonXTM/2). Due to the impossibility of simultaneously measuringthe Alq3 and Bodipy deposition rates, the Bodipy concentration in theAlq3 layer was measured after deposition by UV-visible spectroscopy.Prior to evaporation, the substrates were successively cleaned withacetone at 60 8C and ethanol, washed with deionized water and driedwith N2. N,N0-di-[(1-naphthyl)-N,N0-diphenyl]-1,10-biphenyl)-4,40-diamine (a-NPD, a hole-transporting material), tris(8-hydroxyquino-line)aluminium (Alq3, an electron-transporting and emitting material),2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (BCP, a hole-blockinglayer) and poly(N-vinylcarbazole) (PVK) were purchased fromSigma-Aldrich and used as received. Unlike the other organicmaterials, PVK was dissolved in chloroform and deposited by spincoating. The HOMO-LUMO levels of Bodipy were characterized bycyclic voltammetry. The ITO work function was measured with aKelvin probe (McAllister Technical Services KP-6500). MultilayerOLEDs were fabricated by successively depositing the organicmaterials on ITO-coated glass substrates (Visiontek Systems Ltd),
www.afm-journal.de � 2008 WILEY-VCH Verlag GmbH
constituting the transparent anode; an aluminium cathode was thendeposited, completing the structure. The electroluminescence intensityvariation versus voltage was measured with a silicon photodiode.Emission spectra and luminance were obtained respectively from aspectroradiometer (Minolta CS-1000) and a luminancemeter (MinoltaLS-110).
The X-ray-diffraction data were recorded from a single crystal ofdimensions 0.35� 0.35� 0.05 mm3, at ambient temperature on anEnraf-Nonius Kappa-CCD diffractometer, using graphite-mono-chromated Mo Ka radiation (l¼ 0.71073 A). Data reduction andcorrection for absorption were carried out using the HKL suite. 3609unique reflections were obtained from a total of 42 252 measuredreflections (Rint¼ 0.0732), in the range of h:�15 to 15; k:�21 to 22; andl: �10 to 10 with umax¼ 21.768. The structure was solved by Pattersonmethods and expanded to all non-hydrogen atoms by the Fouriermethod (PATTY) and refined by full-matrix least-squares on F2 valuesusing SHELX-L97 as implemented into CRYSTALBUILDER GUI.Convergence for 397 variable parameters by least-squares refinementon F2 with w¼ 1/[s2(F2
o)þ (0.0591P)2þ 1.5933P] where P¼(F2
o þ 2F2c)/3, for 2815 reflections with I> 2s (I) was reached at
R¼ 0.0524 and wR¼ 0.1302 with a goodness-of-fit of 1.062. The finaldifference Fourier map was featureless, with maximum positive andnegative peaks of 0.343 and �0.247 eA�3 respectively. CCDC-639641contains the supplementary crystallographic data for this paper. Thesedata can be obtained free of charge from the Cambridge Crystal-lographic Data Centre.
4.1. General Procedure for the Synthesis of
Compounds 3 to 10
Compound 2 and triethylamine (2 equiv.) were added to a
stirred solution of the benzoic acid chloride (1 equiv.) in
distilled CH2Cl2. The resulting mixture was stirred at room
temperature for 12 h, when total consumption of the starting
material was observed. The reaction mixture was then washed
with water, the organic layer dried over anhydrousMgSO4, and
the solvent evaporated. Purification was performed using a
chromatography column packed with silica gel, using CH2Cl2/
petroleum ether and CH2Cl2/MeOH as the eluents, and then
recrystallization from CH2Cl2/CH3CN, CH2Cl2/MeOH
or CH2Cl2/hexane.
Compound 3: The compound was prepared from 0.072 g of
3,4,5-tris-methoxy-benzoic acid (0.34 mmol), 3 mL of SOCl2,
0.0134 g of 2 (0.34 mmol), 0.09 mL of distilled triethylamine
(0.68 mmol) and 20 mL of distilled CH2Cl2 to give 0.181 g of 3
after crystallisation from CH2Cl2/hexane (90 %).1H NMR (300 MHz, CDCl3, d): 7.90 (s, 1H), 7.79 (dd, 2H,
3J¼ 8.5 Hz, 4J¼ 1.7 Hz), 7.29 (dd, 2H, 3J¼ 8.7 Hz, 4J¼ 1.9 Hz),
7.11 (s, 2H), 3.94 (s, 6H), 3.92 (s, 3H), 2.53 (s, 6H), 2.30 (q, 4H,3J¼ 7.5 Hz), 1.36 (s, 6H), 0.98 (t, 6H, 3J¼ 7.5Hz); 13CNMR (75
MHz, CDCl3, d): 165.7, 153.6, 141.6, 139.7, 138.7, 138.4, 133.0,
131.9, 131.1, 130.3, 129.3, 120.5, 104.7, 61.1, 56.6, 17.2, 14.8, 12.7,
12.1; 11B NMR (128 MHz, CDCl3, d): 3.87 (t, 1J¼ 32 Hz); IR
(KBr): n¼ 2960, 2932, 2870, 2839, 1675, 1648, 1583, 1539, 1497,
1474, 1413, 1399, 1373, 1363, 1334, 1318, 1295, 1273, 1235, 1184,
1124, 1072, 1045, 1007, 977 cm�1; UV-vis (CH2Cl2): l (e)¼525 (60 000), 492 (sh, 19 000), 367 (9400), 276 nm
(24 000M�1 cm�1); FABþ-MS (m/z (%)): 590.2 (100) [MþH]þ,
þ, 570.2 (15) [M-F]þ; Anal. calcd for C33H38BF2N3O4: C 67.24,
H 6.50; N 7.13; found: C 67.01, H 6.27, N 6.86.
& Co. KGaA, Weinheim Adv. Funct. Mater. 2008, 18,401–413

FULLPAPER
L. Bonardi et al./Electroluminescence of Functional Difluoro-boradiazaindacene Films
Compound 4: The compound was prepared from 0.115 g of
3,4,5-tris-octyloxy-benzoic acid (0.23 mmol), 3 mL of SOCl2,
0.090 g of 2 (0.23 mmol), 0.06 mL of distilled triethylamine
(0.46 mmol) and 20 mL of distilled CH2Cl2 to give 0.122 g of 4
after precipitation from CH2Cl2/CH3CN (61 %).1H NMR (400 MHz, CDCl3, d): 7.84 (s, 1H), 7.78 (dd, 2H,
3J¼ 8.3 Hz, 4J¼ 1.6 Hz), 7.28 (dd, 2H, 3J¼ 8.7 Hz, 4J¼ 2.1 Hz),
7.08 (s, 2H), 4.07–4.02 (m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H,3J¼ 7.5 Hz), 1.90–1.72 (m, 6H, CH2), 1.53–1.44 (m, 6H, CH2),
1.41-1.20 (m, 30H, CH3þCH2), 0.99 (t, 6H, 3J¼ 7.4 Hz),
0.92-0.85 (m, 9H, CH3);13C NMR (75 MHz, CDCl3, d): 165.9,
153.9, 153.5, 142.0, 139.8, 138.8, 138.5, 133.0, 131.8, 131.1, 129.8,
129.3, 120.4, 106.1, 73.7, 69.7, 32.04, 32.0, 31.0, 30.5, 29.7, 29.53,
29.51, 29.4, 26.2, 22.84, 22.81, 17.2, 14.7, 14.2, 12.6, 12.1;11B NMR (128 MHz, CDCl3, d): 3.87 (t, 1J¼ 32 Hz); IR
(KBr): n¼ 2971, 2926, 2856, 1649, 1582, 1547, 1512, 1495, 1398,
1337, 1264, 1238, 1114, 1084, 983 cm�1; UV-vis (CH2Cl2): l(e)¼ 525 (73 000), 493 (sh, 24 000), 367 (12 000), 278 nm
(32 000M�1 cm�1); FABþ-MS (m/z (%)): 884.1 (100) [MþH]þ,
864.2 (20) [M-F]þ; Anal. calcd for C54H80BF2N3O4: C 73.37, H
9.12, N 4.75; found: C 73.17, H 8.75, N 4.42.
Compound 5 : The compound was prepared from 0.128 g of
3,4,5-tris-dodecyloxy-benzoic acid (0.19 mmol), 3 mL of
SOCl2, 0.075 g of 2 (0.19 mmol), 0.05 mL of distilled triethyl-
amine (0.38 mmol) and 20 mL of distilled CH2Cl2 to give 0.109
g of 5 after precipitation from CH2Cl2/CH3CN (55 %).1H NMR (300 MHz, CDCl3, d): 7.82–7.76 (m, 3H), 7.28 (d,
2H, 3J¼ 8.7 Hz), 7.08 (s, 2H), 4.1–4.0 (m, 6H, OCH2), 2.53 (s,
6H), 2.31 (q, 4H, 3J¼ 7.5Hz), 1.88-1.72 (m, 6H, CH2), 1.51–1.27
(m, 60H, CH2þCH3), 0.98 (t, 6H, 3J¼ 7.5 Hz), 0.88 (m,
9H, CH3);13C NMR (75 MHz, CDCl3, d): 165.8, 157.9, 153.4,
141.9, 136.2, 132.9, 131.1, 129.3, 120.4, 113.7, 107.8, 106.0, 31.9,
29.74, 29.71, 29.6, 29.41, 29.37, 26.1, 22.7, 17.1, 14.8, 14.6, 14.1,
12.5, 11.9; 11B NMR (128 MHz, CDCl3, d): 3.86 (t,1J¼ 32 Hz);
IR (KBr): n¼ 2918, 2849, 1644, 1579, 1541, 1497, 1470, 1390,
1335, 1315, 1263, 1236, 1189, 1114, 1075, 976 cm�1; UV-vis
(CH2Cl2): l (e)¼ 525 (72 000), 492 (sh, 24 000), 370 (13 000),
278 nm (32 000 M�1 cm�1); FABþ-MS (m/z (%)): 1052.1
(100) [MþH]þ, 1032.1 (10) [M-F]þ; Anal. calcd for
C66H104BF2N3O4: C 75.33, H 9.96, N 3.99; found: C 75.18,
H 9.64, N 3.64.
Compound 6: The compound was prepared from 0.139 g of
3,4,5-tris-hexadecyloxy-benzoic acid (0.16 mmol), 3 mL of
SOCl2, 0.065 g of 2 (0.16 mmol), 0.05 mL of distilled
triethylamine (0.32 mmol) and 20 mL of distilled CH2Cl2to give 0.135 g of 6 after precipitation from CH2Cl2/
CH3CN (89%).1H NMR (300 MHz, CDCl3, d): 7.84–7.76 (m, 3H), 7.28 (d,
2H, 3J¼ 8.5 Hz), 7.08 (s, 2H), 4.1–4.0 (m, 6H, OCH2), 2.53 (s,
6H), 2.31 (q, 4H, 3J¼ 7.5 Hz), 1.88–1.71 (m, 6H, CH2),
1.49–1.26 (m, 84H, CH2þCH3), 0.99 (t, 6H, 3J¼ 7.5 Hz), 0.88
(m, 9H, CH3);13C NMR (75 MHz, CDCl3, d):¼ 165.9, 153.9,
153.5, 145.2, 143.2, 142.0, 139.7, 138.8, 138.4, 132.9, 131.8, 131.1,
129.8, 129.3, 120.3, 106.0, 73.7, 69.7, 32.1, 30.5, 29.9, 29.8, 29.7,
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag GmbH &
29.6, 29.5, 26.2, 22.8, 17.2, 14.8, 14.2, 12.7, 12.1; 11B NMR
(128 MHz, CDCl3, d): 3.87 (t, 1J¼ 32 Hz); IR (KBr): n¼ 3285,
2951, 2916, 2849, 1673, 1648, 1584, 1540, 1524, 1497, 1467, 1423,
1403, 1383, 1337, 1313, 1276, 1264, 1179, 1158, 1115, 1082, 1057,
969 cm�1; UV-vis (CH2Cl2): l (e)¼ 525 (77 000), 493
(sh, 30 000), 362 (20 000), 276 nm (40 000 M�1 cm�1);
FABþ-MS (m/z (%)): 1220.2 (100) [MþH]þ, 1200.1 (25)
[M-F]þ; Anal. calcd for C78H128BF2N3O4: C 76.75, H 10.57, N
3.44; found: C 76.39, H 10.27, N 3.21.
Compound 7: This compound was prepared from 0.146 g of
3,4,5-tris-(3,7,11,15-tetramethyl-hexadecyloxy)-benzoic acid
(0.14 mmol), 3 mL of SOCl2, 0.057 g of 2 (0.14 mmol),
0.04 mL of distilled triethylamine (0.28 mmol) and 20 mL of
distilled CH2Cl2 to give 0.102 g of 7 after precipitation
from CH2Cl2/CH3CN (51%).1H NMR (300 MHz, CDCl3, d): 7.84 (s, br, 1H), 7.78 (d, 2H,
3J¼ 8.7 Hz), 7.28 (d, 2H, 3J¼ 8.4 Hz), 7.1 (s, 2H), 4.12–4.03 (m,
6H,OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J¼ 7.5 Hz), 1.93–0.83 (m,
129H); 13C NMR (75 MHz, CDCl3, d): 165.9, 153.9, 153.5,
142.0, 139.8, 138.8, 138.4, 133.0, 131.8, 131.1, 129.8, 129.3, 120.4,
106.0, 72.0, 68.1, 39.5, 37.7, 37.63, 37.59, 37.5, 37.4, 36.6, 36.5,
32.96, 30.07, 29.9, 28.1, 24.9, 24.6, 22.9, 22.8, 19.9, 19.83, 19.78,
19.7, 17.2, 14.7, 12.6, 12.1; IR (KBr): n¼ 2954, 2925, 2868, 1651,
1582, 1543, 1498, 1476, 1426, 1399, 1374, 1337, 1318,
1272, 1236, 1196, 1113, 1080, 1050, 982 cm�1; UV-vis
(CH2Cl2) l (e)¼ 525 (95 000), 492 (sh, 35 000), 363 (23 000),
276 nm (50 000 M�1 cm�1); FABþ-MS (m/z (%)): 1388.2
(100) [MþH]þ, 1368.2 (50) [M-F]þ; Anal. calcd for
C90H152BF2N3O4: C 77.82, H 11.03, N 3.03; found: C 77.44,
H 10.64, N 2.67.
Compound 8: The compound was prepared from 0.099 g of
3,4,5-tris-(3,7-dimethyl-octyloxy)-benzoic acid (0.17 mmol),
3 mL of SOCl2, 0.067 g of 2 (0.17 mmol), 0.05 mL of distilled
triethylamine (0.34 mmol) and 20 mL of distilled CH2Cl2 to
give 0.106 g of 8 after precipitation from CH2Cl2/CH3CN
(63 %).1H NMR (300 MHz, CDCl3, d): 7.85 (s, br, 1H), 7.78 (d, 2H,
3J¼ 8.5 Hz), 7.28 (d, 2H, 3J¼ 8.5 Hz), 7.09 (s, 2H), 4.15–4.0
(m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J¼ 7.5 Hz), 2.0–1.47
(m, 12H, CHþCH2), 1.36–1.15 (m, 24H, CH2þCH3), 1.02–0.86
(m, 33H, CH3);13C NMR (75 MHz, CDCl3, d): 165.9, 153.9,
153.5, 142.0, 139.8, 138.8, 138.4, 133.0, 131.8, 131.1, 129.8, 129.3,
120.4, 106.0, 72.0, 68.0, 39.5, 39.4, 37.7, 37.5, 36.5, 30.0, 29.8,
28.1, 24.9, 22.85, 22.76, 22.7, 19.7, 17.2, 14.7, 14.2, 12.6, 12.1; IR
(KBr): n¼ 3288, 2990, 2922, 3695, 1650, 1579, 1539, 1519, 1493,
1473, 1462, 1423, 1392, 1364, 1335, 1315, 1274, 1261, 1232, 1194,
1109, 1075, 1044, 980 cm�1; UV-vis (CH2Cl2): l (e)¼525 (100 000), 490 (sh, 33 000), 364 (20 000), 278 nm
(45 000M�1 cm�1); FABþ-MS (m/z (%)): 968.0 (100) [MþH]þ,
þ, 948.0 (15) [M-F]þ; Anal. calcd for C60H92BF2N3O4: C 74.43,
H 9.58, N 4.34; found: C 74.20, H 9.35, N 4.53.
Received: June 28, 2007
Revised: September 12, 2007
Co. KGaA, Weinheim www.afm-journal.de 411

FULLPAPER
lectroluminescence of Functional Difluoro-boradiazaindacene Films
412
L. Bonardi et al. / E
[1] a) U. Mitschke, P. Bauerle, J. Mater. Chem. 2000, 10, 1471. b) A. P.
Kulkarni, C. J. Tonzola, A. Babel, S. A. Jenekhe, Chem. Mater. 2004,
16, 4556.
[2] Special Issue on Organic Electronics and Optoelectronics (Eds: S. R.
Forrest, M. E. Thompson, Chem. Rev. 2007, 107, pp. 923–1386.
[3] a)M. Hissler, A. El-ghayoury, A. Harriman, R. Ziessel,Angew. Chem.
Int. Ed. 1998, 37, 1717. b) M. Hissler, A. Harriman, P. Jost, G. Wipff,
R. Ziessel, Angew. Chem. Int. Ed. 1998, 37, 3249. c) A. Harriman,
M. Hissler, P. Jost, G. Wipff, R. Ziessel, J. Am. Chem. Soc. 1999, 121,
14. d) H.Miyaji, W. Sato, J. L. Sessler,Angew. Chem. Int. Ed. 2000, 39,
1777. e) P. Anzenbacher, Jr., K. Jursikova, J. L. Sessler, J. Am. Chem.
Soc. 2000, 122, 9350. f) P. Zhang, T. Beck, W. Tan, Angew. Chem. Int.
Ed. 2001, 40, 402. g) T. D. James, S. Shinkai, Top. Curr. Chem. 2002,
218, 159. h) L. Charbonniere, R. Ziessel, M. Montalti, L. Prodi, N.
Zaccheroni, C. Boehme, G. Wipff, J. Am. Chem. Soc. 2002, 124, 7779.
i) F. Sancenon, R. Martinez-Manez, J. Soto, Angew. Chem. Int. Ed.
2002, 41, 1416.
[4] A. Hepp, H. Heil, W.Weise, M. Ahles, R. Schmechel, H. von Seggern,
Phys. Rev. Lett, 2003, 91, 157406.
[5] a) B. Prasad, P. K. Mishra, Indian J. Pure Appl. Phys. 1994, 32, 328.
b) M. Shengwei, A. Dongge, Semicond. Sci. Technol. 2005, 20, 1213.
[6] M. Muccini, M. Murgia, F. Biscarini, C. Taliani, Adv. Mater. 2001, 13,
355.
[7] The literature survey being too large we refer to this excellent review:
L. Akcelrud, Prog. Polym. Sci. 2003, 28, 875.
[8] a) S.-C. Chan, M. C. W. Chan, Y. Wang, C.-M. Che, K.-K. Cheung, N.
Zhu, Chem. Eur. J. 2001, 7, 4180. b) W. Lu, B.-X. Mi, M. C. W. Chan,
Z. Hui, C.-M. Che, N. Zhu, S.-T. Lee, J. Am. Chem. Soc. 2004, 126,
4958. c) W. Lu, M. C. W. Chan, N. Zhu, C.-M. Che, C. Li, Z. Hui,
J. Am. Chem. Soc. 2004, 126, 7639.
[9] J.-J. Shen, X.-L. Yang, T.-H. Huang, J. T. Lin, T.-H. Ke, L.-Y. Chen,
C.-C. Wu, M.-C. P. Yeh, Adv. Funct. Mater. 2007, 17, 983.
[10] Q.-D. Liu, J. Lu, J. Ding, M. Day, Y. Tao, P. Barrios, J. Stupak, K. C. J.
Li, Y. Chi, Adv. Funct. Mater. 2007, 17, 1028.
[11] R. P. Haugland, The Handbook: A Guide to Fluorescent Probes and
Labelling Technologies, 10th Ed., Invitrogen Molecular Probes Inc.,
2005.
[12] a) K. Yamada, T. Toyota, K. Takakura, M. Ishimaru, T. Sugawara,
New J. Chem. 2001, 25, 667. b) R. Reents, M.Wagner, J. Kuhlmann, H.
Waldmann, Angew. Chem. Int. Ed. 2004, 43, 2711.
[13] a) N. R. Cha, S. Y. Moon, S.-K. Chang, Tet. Lett. 2003, 44, 8265. (b) N.
Basaric, M. Baruah, W. Qin, B. Metten, M. Smet, W. Dehaen, N.
Boens, Org. Biomol. Chem. 2005, 3, 2755. (c) H. J. Kim, J. S. Kim,
Tetrahedron Lett. 2006, 47, 7051.
[14] a) Y. Gabe, Y. Urano, K. Kikuchi, H. Kojima, T. Nagano, J. Am.
Chem. Soc. 2004, 126, 3357. (b) T. Ueno, Y. Urano, H. Kojima, T.
Nagano, J. Am. Chem. Soc. 2006, 128, 10640.
[15] T. Gareis, C. Huber, O. S. Wolfbeis, J. Daub, Chem. Commun. 1997,
1717.
[16] M. Kollmannsberger, K. Rurack, U. Resch-Genger, J. Daub, J. Phys.
Chem. A 1998, 102, 10211.
[17] a) K. Rurack, M. Kollmannsberger, U. Resch-Genger, J. Daub, J. Am.
Chem. Soc. 2000, 122, 968. b) S. Y. Moon, N. R. Cha, Y. H. Kim, S.-K.
Chang, J. Org. Chem. 2004, 69, 181.
[18] a) R. P. Haugland, H. C. Kang, US 4, 774, 339, 1998. b) L. H. Thoresen,
H. Kim, M. B. Welch, A. Burghart, K. Burgess, Synlett 1998,
1276.
[19] C. Goze, G. Ulrich, L. Charbonniere, R. Ziessel,Chem. Eur. J. 2003, 9,
3748.
[20] W. Qin, M. Baruah, M. van der Auweraer, F. C. DeSchryver, N.
Boens, J. Phys. Chem. A 2005, 109, 7371.
[21] G. Ulrich, C. Goze, M. Guardigli, A. Roda, R. Ziessel, Angew. Chem.
Int. Ed. 2005, 44, 3694.
www.afm-journal.de � 2008 WILEY-VCH Verlag GmbH
[22] a) R.W.Wagner, J. S. Lindsey, Pure Appl. Chem. 1996, 68, 1373. b) G.
Beer, K. Rurack, J. Daub, Chem. Commun. 2001, 1138. c) G. Beer, C.
Niederalt, S. Grimme, J. Daub, Angew. Chem. Int. Ed. 2000, 39,
3252. d) M. Kollmannsberger, K. Rurack, U. Resch-Genger, W.
Rettig, J. Daub, Chem. Phys. Lett. 2000, 329, 363. e) Y. Gabe, Y.
Urano, K. Kikuchi, H. Kojima, T. Nagano, J. Am. Chem. Soc. 2004,
126, 3357.
[23] T. A. Golovkova, D. V. Kozlov, D. C. Neckers, J. Org. Chem. 2005, 70,
5545.
[24] a) M. Kollmannsberger, T. Gareis, S. Heinl, J. Breu, J. Daub, Angew.
Chem. Int. Ed. 1997, 36, 1333. b) R. Y. Lai, A. J. Bard, J. Phys. Chem. B
2003, 107, 5036. c) C. Trieflinger, H. Rohr, K. Rurack, J. Daub,Angew.
Chem. Int. Ed. 2005, 44, 6943.
[25] a) J. M. Brom, Jr., J. L. Langer, J. Alloys Compd 2002, 338, 112. b) A.
Hepp, G. Ulrich, R. Schmechel, H. von Seggern, R. Ziessel, Synth.
Met. 2004, 146, 11.
[26] a) T. Chen, J. H. Boyer, M. L. Trudell,Heteroat. Chem. 1997, 8, 51. b)
G. Sathyamoorthi, L. T. Wolford, A. M. Haag, J. H. Boyer, Heteroat.
Chem. 1994, 5, 245.
[27] S. Hattori, K. Ohkubo, Y. Urano, H. Sunahara, T. Nagano, Y. Wada,
N. V. Tkchanko, H. Lemmetyinen, S. Fukuzumi, J. Phys. Chem. B
2005, 109, 15368.
[28] C. McCusker, J. B. Carroll, V. M. Rotello, Chem. Commun. 2005, 996.
[29] M. P. Debreczeny, W. A. Svec, M. R. Wasielewski, Science 1996, 274,
584.
[30] P. G. Van Patten, A. P. Sheve, J. S. Lindsey, R. J. Donohoe, J. Phys.
Chem. B 1998, 102, 4209.
[31] a) F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin III, D. L. Singh,
D. Kim, R. R. Birge, D. F. Bocian, D. Holten, J. S. Lindsey, J. Am.
Chem. Soc. 1998, 120, 10001. b) R. K. Lammi, R. W. Wagner, A.
Ambroise, J. R. Diers, D. F. Bocian, D. Holten, J. S. Lindsey, J. Phys.
Chem. B 2001, 105, 5341.
[32] a) R. W. Wagner, J. S. Lindey, J. Am. Chem. Soc. 1994, 116, 9759.
b) R. W. Wagner, J. S. Lindsey, J. Seth, V. Palaniappan, D. F. Bocian,
J. Am. Chem. Soc. 1996, 118, 3996. c) R.W.Wagner, J. S. Lindsey,Pure
Appl. Chem. 1996, 68, 1373.
[33] M. D. Yilmaz, O. A. Bozdemir, E. U. Akkaya, Org. Lett. 2006, 8,
2871.
[34] H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki,
Y. Sakata, S. Fukuzumi, J. Am. Chem. Soc. 2001, 123, 100.
[35] F. D’Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou,
Y. Araki, O. Ito, J. Am. Chem. Soc. 2004, 126, 7898.
[36] a) B. Turfan, E. U. Akkaya, Org. Lett. 2002, 4, 2857. b) J. Wang,
X. Qian, Org. Lett. 2006, 8, 3721.
[37] a) L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff, C. J. Chang, J. Am.
Chem. Soc. 2006, 128, 10. b) X. Qi, E. Jin Jun, L. Xu, S.-J. Kim, J. S. J.
Hong, Y. J. Yoon, J. Yoon, J. Org. Chem. 2006, 71, 2881.
[38] F. Camerel, L. Bonardi, M. Schmutz, R. Ziessel, J. Am. Chem. Soc.
2006, 128, 4548.
[39] F. Camerel, L. Bonardi, G. Ulrich, L. Charbonniere, B. Donnio, C.
Bourgogne, D. Guillon, P. Retailleau, R. Ziessel, Chem. Mater. 2006,
18, 5009.
[40] F. Camerel, G. Ulrich, J. Barbera, R. Ziessel, Chem. Eur. J. 2007, 13,
2189.
[41] H. Zhang, C. Huo, K. Ye, P. Zhang, W. Tian, Y. Wang, Inorg. Chem.
2006, 45, 2788.
[42] S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hagele, G. Scalia,
R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel, M. Tosoni, Angew.
Chem. Int. Ed. 2007, 46, 4832.
[43] R. Ziessel, L. Bonardi, P. Retailleau, G. Ulrich, J. Org. Chem. 2006, 71,
3093.
[44] V. A. Azov, F. Diederich, Y. Lill, B. Hecht,Helv. Chim. Acta 2003, 86,
2149.
[45] F. Camerel, G. Ulrich, R. Ziessel, Org. Lett. 2004, 6, 4171.
& Co. KGaA, Weinheim Adv. Funct. Mater. 2008, 18,401–413

FULLPAPER
L. Bonardi et al./Electroluminescence of Functional Difluoro-boradiazaindacene Films
[46] a) C.-W.Wan, A. Burghart, J. Chen, F. Bergstrom, L. B.-A. Johanson,
M. F. Wolford, T. G. Kim, M. R. Topp, R. M. Hochstrasser, K.
Burgess, Chem. Eur. J. 2003, 9, 4430. b) Z. Shen, H. Rohr, K. Rurack,
H. Uno, M. Spieles, B. Schulz, G. Reck, N. Ono, Chem. Eur. J. 2004,
10, 4853.
[47] a) T. Kato, T. Kutsuna, K. Hanabusa, M. Ukon, Adv. Mater. 1998, 10,
606. b) K. Hanabusa, C. Koto,M. Kimura, H. Shirai, A. Kakehi,Chem.
Lett. 1997, 429.
[48] R. M. Silverstein, G. C. Bassler, T. C. Morrill, Spectrometric Identi-
fication of Organic Compounds, 3rd ed., Wiley, New York 1976.
[49] a) A. Burghart, H. Kim, M. B. Wech, L. H. Thorensen, J. Reibenspies,
K. Burgess, J. Org. Chem. 1999, 64, 7813. b) H. Kim, A. Burghart,
M. B. Welch, J. Reibenspies, K. Burgess, Chem. Commun. 1999,
1889. c) J. Chen, J. Reibenspies, A. Derecskei-Kovacs, K. Burgess,
Chem. Commun. 1999, 2501. d) K. Rurack, M. Kollmannsberger, J.
Daub,New J. Chem. 2001, 25, 289. e) K. Rurack, M. Kollmannsberger,
J. Daub, Angew. Chem. Int. Ed. 2001, 40, 385.
[50] J. Karolin, L. B.-A. Johansson, L. Strandberg, T. Ny, J. Am. Chem.
Soc. 1994, 116, 7801.
Adv. Funct. Mater. 2008, 18, 401–413 � 2008 WILEY-VCH Verlag GmbH &
[51] a)M.K.DeArmond, C.M. Carlin,Coord. Chem. Rev. 1981, 36, 325. b)
M. Klessinger, J. Michl, Excited States and Photochemistry of Organic
Molecules, VCH, Weinheim, Germany 1994.
[52] J. Olmsted, J. Phys. Chem. 1979, 83, 2581.
[53] G. Ulrich, R. Ziessel, J. Org. Chem. 2004, 69, 2070.
[54] F.Wurthner, C. Thalacker, S. Diele, C. Tschierske,Chem. Eur. J. 2001,
7, 2245.
[55] E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. Chen,
F. Wurthner, R. A. J. Janssen, J. Phys. Chem. A 2004, 108, 6933.
[56] Y. Hamada, IEEE Trans. Electron. Devices 1997, 44, 1208.
[57] J. Pommerehne, H. Vestweber, W. Guss, R. Mahrt, H. Bassler, M.
Porsch, J. Daub, Adv. Mater. 1995, 7, 551.
[58] A. Bard, R. Faulkner, Electrochemical Methods-Fundamentals and
Applications, Wiley, New York 1980, Ch. 14, pp 634.
[59] J. Kido, K. Hongawa, K. Okuyama, K. Nagai, Appl. Phys. Lett. 1993,
63, 2627.
[60] P. Toele, H. Zhang, C. Trieflinger, J. Daub, M. Glasbeek, Chem. Phys.
Lett. 2003, 368, 66.
[61] C. Goze, G. Ulrich, R. Ziessel, J. Org. Chem. 2007, 72, 313.
Co. KGaA, Weinheim www.afm-journal.de 413





























