Embed Size (px)
Citation preview

doi:10.1006/jmbi.2001.4906 available online at http://www.idealibrary.com on J. Mol. Biol. (2001) 311, 625±638
COMMUNICATION
Four-body Potentials Reveal Protein-specificCorrelations to Stability Changes Caused byHydrophobic Core Mutations
Charles W. Carter Jr1*, Brendan C. LeFebvre1, Stephen A. Cammer2
Alexander Tropsha2 and Marshall Hall Edgell1,3
1Department of Biochemistryand Biophysics, University ofNorth Carolina at Chapel HillChapel Hill, NC 27599-7260, USA2The Laboratory for MolecularModeling, Division ofMedicinal Chemistry andNatural Products, School ofPharmacy, University of NorthCarolina at Chapel Hill, ChapelHill, NC 27599-7360, USA3Department of Microbiologyand Immunology, University ofNorth Carolina at Chapel HillChapel Hill, NC 27599-7290, USA
E-mail address of the [email protected]
Present addresses: B. C. LeFebvreCollege, Providence RI, USA; S. A.GeneFormatics, Inc., 5830 Oberlin RDiego, CA 92121, USA.
Abbreviations used: CI2, chymotrSNAPP, simplicial neighborhood anpacking; MuSE, mutation with SNA
0022-2836/01/040625±14 $35.00/0
Mutational experiments show how changes in the hydrophobic cores ofproteins affect their stabilities. Here, we estimate these effects computa-tionally, using four-body likelihood potentials obtained by simplicialneighborhood analysis of protein packing (SNAPP). In this procedure,the volume of a known protein structure is tiled with tetrahedra havingthe center of mass of one amino acid side-chain at each vertex. Log-likeli-hoods are computed for the 8855 possible tetrahedra with equivalentcompositions from structural databases and amino acid frequencies. Thesum of these four-body potentials for tetrahedra present in a given pro-tein yields the SNAPP score. Mutations change this sum by changing thecompositions of tetrahedra containing the mutated residue and theirrelated potentials. Linear correlation coef®cients between experimentalmutational stability changes, �(�Gunfold), and those based on SNAPPscoring range from 0.70 to 0.94 for hydrophobic core mutations in ®vedifferent proteins. Accurate predictions for the effects of hydrophobiccore mutations can therefore be obtained by virtual mutagenesis, basedon changes to the total SNAPP likelihood potential. Signi®cantly, slopesof the relation between �(�Gunfold) and �SNAPP for different proteinsare statistically distinct, and we show that these protein-speci®c effectscan be estimated using the average SNAPP score per residue, which isreadily derived from the analysis itself. This result enhances the predic-tive value of statistical potentials and supports previous suggestions that``comparable'' mutations in different proteins may lead to different�(�Gunfold) values because of differences in their ¯exibility and/orconformational entropy.
# 2001 Academic Press
Keywords: Delaunay tessellation; database-derived potentials; elementarytertiary motifs; multivariate statistics; conformational entropy
*Corresponding authorIt is generally agreed that proteins derive a con-siderable portion of their thermodynamic stabilityfrom interactions between non-polar amino acid
ing author:
, ProvidenceCammer,oad, Suite 200, San
ypsin inhhibitor 2;alysis of proteinPP evaluation.
side-chains sequestered from the aqueous environ-ment by the fold of the polypeptide chain. Thenature of these interactions has been the subjectof considerable experimentation,1 ± 5 theoreticalexploration6 ± 14 and debate.15 Experimental investi-gations have been based largely on mutagenesis,principally employing ®ve model proteins: T4lysozyme;16 ± 21 barnase;22 ± 24 staphylococcal nucle-ase;25,26 the chymotrypsin inhibitor, CI2;27,28 and,more recently, calbindin29 combined with exper-imental measurements of the associated change instability, as judged by unfolding free-energy differ-ences, �(�Gunfold), between mutant and wild-typeproteins.
# 2001 Academic Press

626 Four-body Potentials, �(�Gunfold), and Entropy Changes
Attempts to relate �(�Gunfold) values to speci®cstructural changes have been most successful incases where the mutant protein structures havebeen determined, so that their altered propertiescan be used explicitly in the correlations.30,31
Mutational stability changes estimated using dis-tance-derived statistical potentials, given only thestructure of the wild-type protein and the nature ofthe mutation, correlate less well withexperiment.32 ± 34 The best correlations34 combinedistance-derived and torsional preference poten-tials and explain only �62 % of the total variationin �(�Gunfold).
Use of knowledge-based potentials has beenwidely debated.35,36 For our purpose, those discus-sions can be summarized brie¯y. Distance-derivedpotentials substitute for a true, chemical equili-brium a virtual equilibrium between the observedfrequencies in the database of static structures andreference frequencies based on a null hypothesis,i.e. that the amino acids present in the sequence actindependently and interact randomly. The relation-ship between such statistical frequency ratios andfree energies is unclear, because the properties ofthe database have only heuristic connections to theBoltzmann distribution law. Arguing by analogywith the Boltzmann statistical formulation thusbegs the important question of what temperatureshould be chosen for the evaluation of statisticalpotentials derived from log-likelihood ratios.36
Moreover, adding such potentials to estimatemutational free energy changes is of questionablevalidity, given the potentially protein-speci®c,entropic consequences of mutation.37
Contributions to protein stability estimatedusing such potentials to represent tertiary packinginteractions also are conceptually problematicbecause they estimate, indirectly, interactionswhose stabilizing impact arises from the hydro-phobic effect, which is temperature-dependent. Thehydrophobic effect owes much to entropy changesin the aqueous phase, and is generally consideredto be beyond direct estimation from structural dataalone. For this reason, the contribution of database-derived statistical potentials to stability may actu-ally be different in proteins having different aminoacid compositions and chain lengths, N, which
{ Voronoi tessellation was introduced to the ®eld ofprotein structure by Finney42 and Richards.10
A collection of points is used as centers for polyhedrawhose faces form perpendicular bisectors of the linesconnecting them to their neighbors. The Voronoipolyhedra thus partition all points into those that liecloser to a given center than to any of the others in theset. The equivalent tessellation of Delaunay comprisespolyhedra whose edges connect the centers of Voronoipolyhedra and meet at a common vertex. AlthoughVoronoi and Delaunay tessellations represent the sameinformation, for bioinformatics purposes the latter hasthe decisive advantage that three-dimensional Delaunaysimplices are always tetrahedra, whereas Voronoipolyhedra have variable numbers of faces.
determine conformational entropies via lnN. Thisnotion was demonstrated explicitly using a simplelattice model to show that extracted pairwisepotentials were indeed proportional to both chainlength and composition.36 The absence of tempera-ture from statistical potentials therefore suggeststhat their application should be scaled according tothe total net entropy changes underlying proteinstability.36
Finally, Betancourt & Thirumalai38 concludedthat although knowledge-based potentials may beappropriate for the study of protein stability, pair-wise additive potentials are not suf®cient and thatreliable prediction of protein structure will requiremore complex, higher-order potentials. As thetetrahedron is the three-dimensional simplex, itseemed likely that four-body potentials wouldbetter represent tertiary packing interactions inproteins.
Description of the method
Simplicial neighborhood analysis of proteins byDelaunay tessallation and compositional likelihoodscoring (SNAPP)39±41
Several years ago, we embarked on the develop-ment of alternative, four-body potentials based onDelaunay tessellation{ of protein structures andcompositional likelihood scoring39 ± 41,43,44 and theirapplication for fold recognition45 and ab initiostructure prediction.46 In three dimensions, theDelaunay simplex is a tetrahedron that is bothnecessary and suf®cient to describe nearest-neigh-bor interactions present in any set of points. Forthe present work, we use a reduced, residue-basedrepresentation of a protein, in which each residuein a PDB ®le is replaced by its side-chain centroidas the basis for tetrahedron formation. With thisset of united residues, Delaunay tessellation natu-rally partitions a protein's tertiary structure into anaggregate of space-®lling, irregular tetrahedra, orsimplices, whose edges represent all nearest neigh-bors and thereby reduce the complex three-dimen-sional interactions in a protein to explicit,elementary tertiary motifs.39 We therefore call thisapproach simplicial neighborhood analysis of pro-tein packing (SNAPP).43,41
The invariant number of contributors to a Delau-nay simplex and the identi®cation of vertices withspeci®c amino acids facilitate comparisons of theircompositions to the database of known structures,as described.39,40 The amino acid types at the fourvertices de®ne the composition of each Delaunaysimplex. If the order of the contributing residues isunimportant, there are 8855 possible compositions.Delaunay tessellation has been applied to a non-redundant set of 1200 high-resolution proteinstructures47 selected for structural diversity.41 Log-likelihood scores have been assigned to nearly allof the 8855 possible compositions using theobserved frequencies of the tetrahedra, correctedfor overall amino acid compositions. These poten-

Figure 1. Delaunay tessallation and virtual mutagen-esis. (a) Delaunay tessellation of an X-ray crystal struc-ture of chymotrypsin inhibitor 2.48 The polypeptidebackbone is shown in brown; tessellation is based onthe set of points (not shown) representing the centroidsof each side-chain. Delaunay tetrahedra are indicated byspheres at their centers, and by their six edges. As indi-cated by the key, the color of each simplex re¯ects thelog-likelihood score, qijkl, of the compositional family towhich it belongs: red, qijkl > 0.9; magenta, qijkl > 0.6;yellow, qijkl > 0.3, green qijkl > 0.0; blue qijkl > 0.0;. Imagegenerated by ProCAM41 and MAGE75,76 from PDB struc-ture 2CI2.pdb.47 The asterisk marks the location of leu-cine 68, which is the site of virtual mutation illustratedin (b). (b) The local environment of Leu68, and its evol-ution under the virtual mutation to Ala. Only tetrahedrafrom the two, highest-scoring classes, are illustrated.Log-likelihoods for each Delaunay tetrahedron, andwhich contribute to the total �SSNAPP score, areindicated in bold for both native and virtual mutantdiagrams.
Four-body Potentials, �(�Gunfold), and Entropy Changes 627
tials differ from other, distance-derived potentials,because they re¯ect only compositional likelihoods.For this reason, we will refer to them as likelihoodpotentials.
The prominence of tetrahedra composed ofhydrophobic residues and their possiblerelationship to stabilizing tertiary interactions
Tetrahedra with different compositions do notoccur with random frequencies in this database.Rather, those composed entirely of four non-polarside-chains occur far more frequently thanexpected under the null hypothesis of equal fre-quencies, and most of the high-scoring simplicesconsist entirely of hydrophobic residues. Thisphenomenon is a natural consequence of hydro-phobes' tendency to pack together. Their high log-likelihoods suggest that tetrahedra composed offour hydrophobic side-chains may encode the mostinformation about thermodynamically importanttertiary interactions. Indeed, in related work wehave observed a quite similar ordering of four-body potentials derived for the same, high-scoringtetrahedra from estimates of relative free energiesof transfer.41
If the unexpectedly frequent tetrahedra invol-ving four non-polar residues play dominant rolesin stabilizing protein folds, they might be effectivein predicting experimental consequences of hydro-phobic core mutations. We have investigated thishypothesis by speci®cally evaluating correlationsbetween SNAPP score and protein-stabilitychanges for speci®c mutations made in the hydro-phobic cores of several proteins.
Virtual mutagenesis (MuSE; mutation withSNAPP evaluation41)
Any protein with a known or model structurecan be tessellated to produce the set of elementarytetrahedral tertiary motifs de®ned by its structure.The SNAPP score is determined by summing thelog-likelihoods for all such motifs present in theprotein. Mutations will change this sum, becauseeach mutation affects compositions of all tetrahe-dra that share a mutated residue. To calculate adifference in SNAPP scoring of native and mutantproteins, we assume that discrete mutations do notchange the structure.
An example is useful to illustrate how we usethe SNAPP procedure in a virtual mutagenesisexperiment. A modi®ed Delaunay tessellationobtained for the chymotrypsin inhibitor, CI2,48 isshown in Figure 1(a). Technically, this is only asample of the complete tessellation. A large num-ber of simplices involving adjacent residues in theprimary sequence have been ®ltered out becausethey do not represent tertiary interactions, and sim-plices with a vertex-to-vertex distance of >10 AÊ areomitted on the grounds that signi®cant direct inter-actions will not occur at greater distances.
Amino acids contributing to the highest-scoringtetrahedra are shown explicitly in Figure 1(b), inorder to illustrate the consequences of mutatingLeu68, a residue central to the CI2 hydrophobiccore. The L68A mutation changes the compositionof three of the four highest-scoring core tetrahedraand removes multiple van der Waals contactsholding them together. The (red) core tetrahedrainvolving Leu68 have all been replaced by tetrahe-dra with lower log-likelihoods than their wild-typecounterparts. The total SNAPP score is reduced byÿ2.36 log-likelihood units in the L68A variant, ofwhich 1.00 log-likelihood units arise from changesin the three highest-scoring Delaunay simplices

628 Four-body Potentials, �(�Gunfold), and Entropy Changes
illustrated in Figure 1(b). Experimentally, thismutation reduces the CI2 stability by ÿ3.84 kcal/mol.49
To facilitate ef®cient processing of a large num-ber of virtual protein variants for different pro-teins, we developed an intuitive, interactive Webinterface to a suite of programs, MuSE{ thatimplement the SNAPP method.41 The necessarygeometric calculations and PDB code47 databasequeries are performed quickly and automatically.This virtual-mutagenesis algorithm proceeds as fol-lows. (1) Create the tessellation pattern for thenative protein. This is done directly from the PDB®le. (2) Calculate the wild-type SNAPP score. Thefour-body SNAPP potential, q, is precalculatedfrom non-redundant subsets of the PDB as a loglikelihood:
qijkl � logfijkl
pijkl�1�
where i, j, k, l are any four amino acid residues, fijkl
is the frequency of occurrence of a given quadru-plet as a Delaunay tetrahedron in the structuraldatabase, and pijkl is the expected frequency ofoccurrence of a given quadruplet based on aminoacid frequencies in the same database. The qijkl
shows the likelihood of ®nding four particular resi-dues in one simplex. The SNAPP score is obtainedas the sum of q factors for all quadruplets of aminoacids observed in a protein after tessellation, i.e.:
SSNAPP �Xnq
i�1
qi �2�
where qi is the statistical potential for ith quadru-plet and nq is the total number of Delaunay tetra-hedra in the protein. The current version of theSNAPP potential is derived from 1200 single-chainprotein structures in the CullPDB data set{
(3) Change residues in tessellated protein model.No structural changes are made to the modelduring this step. The identity of a wild-typeresidue(s) is changed to that of a mutant, so thatthe underlying mutant residue(s) participate inprecisely the same tetrahedra as the wild-typeresidues.
(4) Re-calculate SNAPP score. The ``mutant''model's set of Delaunay simplices will be the samein terms of the tessellation pattern, as the positionsof points in 3D space are assumed to beunchanged. However, the compositions of sim-plices in which the mutated residue(s) participatewill be different, resulting in different SNAPPscores for the given position in the proteinsequence and hence for the protein as a whole(Figure 1).
{ http://mmlsun4.pha.unc.edu/3dworkbench.html{ http://www.fccc.edu/research/labs/dunbrack/
culledpdb.html
(5) Calculate �SSNAP � [SSNAPP(mut) ÿ SSNAPP
(wt)]. In practice, as indicated above, this differ-ence is de®ned only by the new compositions fortetrahedra in which the mutated residue partici-pates.
Experimental �(�G) values
To assess the utility of this procedure in predict-ing consequences of mutation within hydrophobiccores of proteins, we compared experimental�(�Gunfold) values with �SSNAPP scores estimatedfor the same mutants. Seventy-six mostly hydro-phobic core mutants (some multiple mutations alsoinvolved partially exposed residues) were collectedfrom reports of studies of ®ve proteins: T4 lyso-zyme (1L63), 30 mutants,16,17,50 ± 54 barnase (1B2X)nine mutants,2,55 ± 57 chymotrypsin inhibitor 2(2CI2), nine mutants,27,28,49 staphylococcal nuclease(1EY0), 19 mutants,25,58,59, and calbindin (6ICB),29
nine mutants. These mutants are summarized inTable 1.
The literature data were restricted to mutationsof ``core'' residues. Where possible (CI-2, barnase)mutations were chosen that had been identi®ed inthe original literature citations to involve core resi-dues. Where the authors made no such formal dis-tinction (as with T4 lysozyme, staphylococcalnuclease, and calbindin) we de®ned a residue as``core'' if its individual residue score (i.e. the sumof scores for all quadruplets that share this residue)exceeded a threshold value of 1.5, chosen to beconsistent with the selections for CI2 and barnase.We also reasoned that virtual tessellation changesbased on the native state could only report onnative-state behavior, and hence eliminated, whenpossible, mutants that might have a �(�G)unfold
component due to changes in the denatured state.Hence, we omitted from consideration single pointmutations that involved either glycine or proline inwild-type or variant, as these residues probably dochange the entropy of the denatured state. Finally,we narrowed the extensive list of staphylococcalmutants to those whose response to denaturantm-value was within 5 % of the native value, againbecause outliers in this respect are likely to showeffects in the denatured state.26
Reproducibility of Delaunay tessellation frommultiple coordinate sets
We assessed the variance of the Delaunay tessel-lation and SNAPP scoring due to crystallographiccoordinate errors by two tests. The unligandedforms of Bacillus stearothermophilus tryptophanyl-tRNA synthetase provide 18 copies of the TrpRSmonomer, owing to non-crystallographic sym-metry equivalence in the crystals. Each monomerwas tessellated. The mean total SNAPP score was91.1(�2.1), suggesting that the error in tessellationarising from coordinate variance is roughly 2 % ofthe total value. An independent veri®cation of thereliability of the tessellation procedures arises in
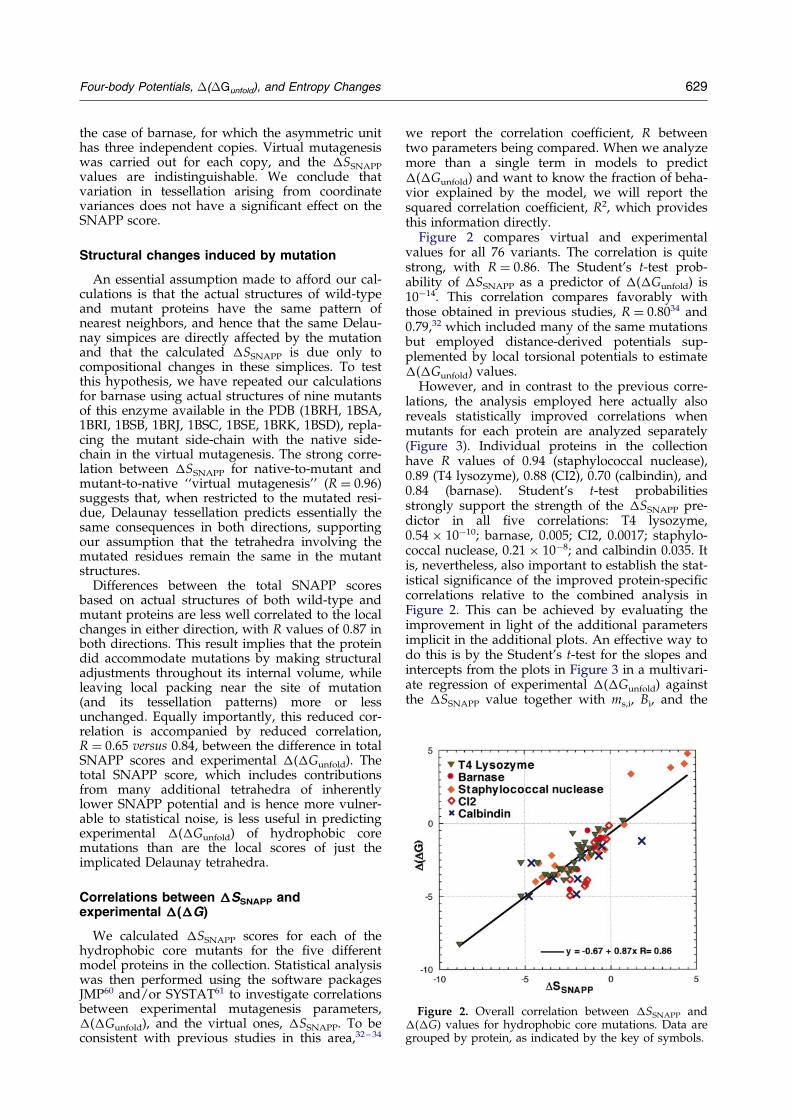
Figure 2. Overall correlation between �SSNAPP and�(�G) values for hydrophobic core mutations. Data aregrouped by protein, as indicated by the key of symbols.
Four-body Potentials, �(�Gunfold), and Entropy Changes 629
the case of barnase, for which the asymmetric unithas three independent copies. Virtual mutagenesiswas carried out for each copy, and the �SSNAPP
values are indistinguishable. We conclude thatvariation in tessellation arising from coordinatevariances does not have a signi®cant effect on theSNAPP score.
Structural changes induced by mutation
An essential assumption made to afford our cal-culations is that the actual structures of wild-typeand mutant proteins have the same pattern ofnearest neighbors, and hence that the same Delau-nay simpices are directly affected by the mutationand that the calculated �SSNAPP is due only tocompositional changes in these simplices. To testthis hypothesis, we have repeated our calculationsfor barnase using actual structures of nine mutantsof this enzyme available in the PDB (1BRH, 1BSA,1BRI, 1BSB, 1BRJ, 1BSC, 1BSE, 1BRK, 1BSD), repla-cing the mutant side-chain with the native side-chain in the virtual mutagenesis. The strong corre-lation between �SSNAPP for native-to-mutant andmutant-to-native ``virtual mutagenesis'' (R � 0.96)suggests that, when restricted to the mutated resi-due, Delaunay tessellation predicts essentially thesame consequences in both directions, supportingour assumption that the tetrahedra involving themutated residues remain the same in the mutantstructures.
Differences between the total SNAPP scoresbased on actual structures of both wild-type andmutant proteins are less well correlated to the localchanges in either direction, with R values of 0.87 inboth directions. This result implies that the proteindid accommodate mutations by making structuraladjustments throughout its internal volume, whileleaving local packing near the site of mutation(and its tessellation patterns) more or lessunchanged. Equally importantly, this reduced cor-relation is accompanied by reduced correlation,R � 0.65 versus 0.84, between the difference in totalSNAPP scores and experimental �(�Gunfold). Thetotal SNAPP score, which includes contributionsfrom many additional tetrahedra of inherentlylower SNAPP potential and is hence more vulner-able to statistical noise, is less useful in predictingexperimental �(�Gunfold) of hydrophobic coremutations than are the local scores of just theimplicated Delaunay tetrahedra.
Correlations between ���SSNAPP andexperimental ���(���G)
We calculated �SSNAPP scores for each of thehydrophobic core mutants for the ®ve differentmodel proteins in the collection. Statistical analysiswas then performed using the software packagesJMP60 and/or SYSTAT61 to investigate correlationsbetween experimental mutagenesis parameters,�(�Gunfold), and the virtual ones, �SSNAPP. To beconsistent with previous studies in this area,32 ± 34
we report the correlation coef®cient, R betweentwo parameters being compared. When we analyzemore than a single term in models to predict�(�Gunfold) and want to know the fraction of beha-vior explained by the model, we will report thesquared correlation coef®cient, R2, which providesthis information directly.
Figure 2 compares virtual and experimentalvalues for all 76 variants. The correlation is quitestrong, with R � 0.86. The Student's t-test prob-ability of �SSNAPP as a predictor of �(�Gunfold) is10ÿ14. This correlation compares favorably withthose obtained in previous studies, R � 0.8034 and0.79,32 which included many of the same mutationsbut employed distance-derived potentials sup-plemented by local torsional potentials to estimate�(�Gunfold) values.
However, and in contrast to the previous corre-lations, the analysis employed here actually alsoreveals statistically improved correlations whenmutants for each protein are analyzed separately(Figure 3). Individual proteins in the collectionhave R values of 0.94 (staphylococcal nuclease),0.89 (T4 lysozyme), 0.88 (CI2), 0.70 (calbindin), and0.84 (barnase). Student's t-test probabilitiesstrongly support the strength of the �SSNAPP pre-dictor in all ®ve correlations: T4 lysozyme,0.54 � 10ÿ10; barnase, 0.005; CI2, 0.0017; staphylo-coccal nuclease, 0.21 � 10ÿ8; and calbindin 0.035. Itis, nevertheless, also important to establish the stat-istical signi®cance of the improved protein-speci®ccorrelations relative to the combined analysis inFigure 2. This can be achieved by evaluating theimprovement in light of the additional parametersimplicit in the additional plots. An effective way todo this is by the Student's t-test for the slopes andintercepts from the plots in Figure 3 in a multivari-ate regression of experimental �(�Gunfold) againstthe �SSNAPP value together with ms,i, Bi, and the
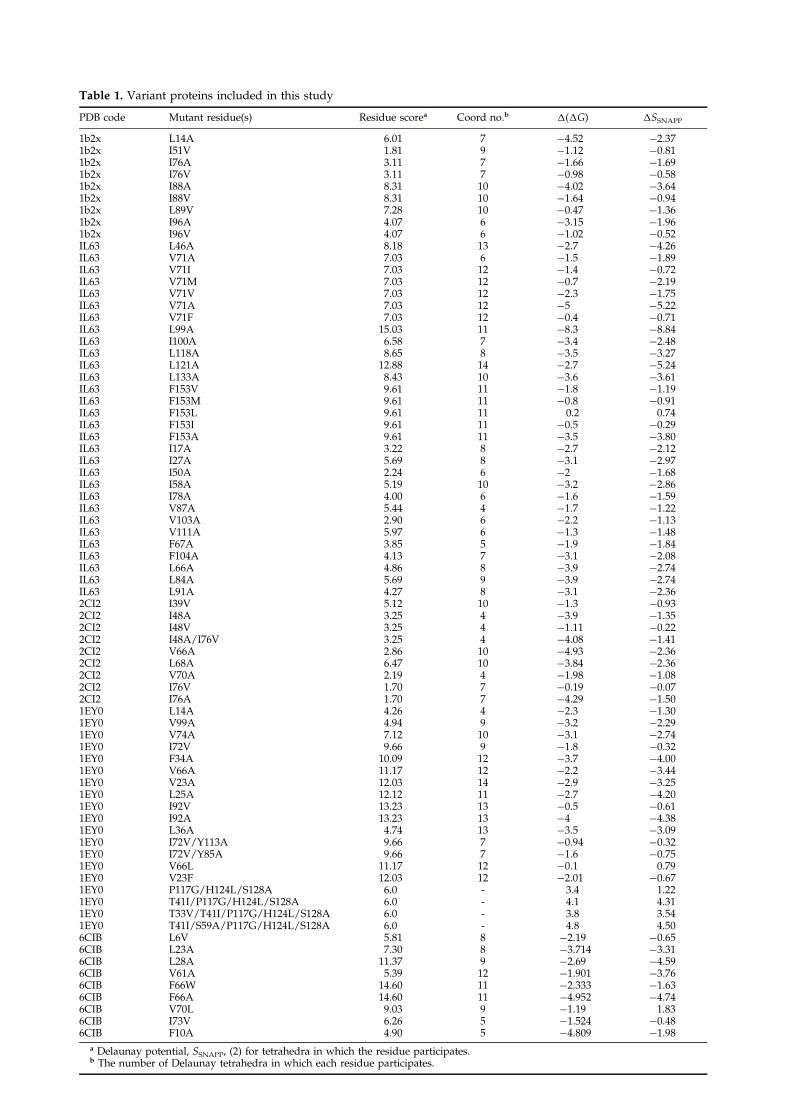
Table 1. Variant proteins included in this study
PDB code Mutant residue(s) Residue scorea Coord no.b �(�G) �SSNAPP
1b2x L14A 6.01 7 ÿ4.52 ÿ2.371b2x I51V 1.81 9 ÿ1.12 ÿ0.811b2x I76A 3.11 7 ÿ1.66 ÿ1.691b2x I76V 3.11 7 ÿ0.98 ÿ0.581b2x I88A 8.31 10 ÿ4.02 ÿ3.641b2x I88V 8.31 10 ÿ1.64 ÿ0.941b2x L89V 7.28 10 ÿ0.47 ÿ1.361b2x I96A 4.07 6 ÿ3.15 ÿ1.961b2x I96V 4.07 6 ÿ1.02 ÿ0.52IL63 L46A 8.18 13 ÿ2.7 ÿ4.26IL63 V71A 7.03 6 ÿ1.5 ÿ1.89IL63 V71I 7.03 12 ÿ1.4 ÿ0.72IL63 V71M 7.03 12 ÿ0.7 ÿ2.19IL63 V71V 7.03 12 ÿ2.3 ÿ1.75IL63 V71A 7.03 12 ÿ5 ÿ5.22IL63 V71F 7.03 12 ÿ0.4 ÿ0.71IL63 L99A 15.03 11 ÿ8.3 ÿ8.84IL63 I100A 6.58 7 ÿ3.4 ÿ2.48IL63 L118A 8.65 8 ÿ3.5 ÿ3.27IL63 L121A 12.88 14 ÿ2.7 ÿ5.24IL63 L133A 8.43 10 ÿ3.6 ÿ3.61IL63 F153V 9.61 11 ÿ1.8 ÿ1.19IL63 F153M 9.61 11 ÿ0.8 ÿ0.91IL63 F153L 9.61 11 0.2 0.74IL63 F153I 9.61 11 ÿ0.5 ÿ0.29IL63 F153A 9.61 11 ÿ3.5 ÿ3.80IL63 I17A 3.22 8 ÿ2.7 ÿ2.12IL63 I27A 5.69 8 ÿ3.1 ÿ2.97IL63 I50A 2.24 6 ÿ2 ÿ1.68IL63 I58A 5.19 10 ÿ3.2 ÿ2.86IL63 I78A 4.00 6 ÿ1.6 ÿ1.59IL63 V87A 5.44 4 ÿ1.7 ÿ1.22IL63 V103A 2.90 6 ÿ2.2 ÿ1.13IL63 V111A 5.97 6 ÿ1.3 ÿ1.48IL63 F67A 3.85 5 ÿ1.9 ÿ1.84IL63 F104A 4.13 7 ÿ3.1 ÿ2.08IL63 L66A 4.86 8 ÿ3.9 ÿ2.74IL63 L84A 5.69 9 ÿ3.9 ÿ2.74IL63 L91A 4.27 8 ÿ3.1 ÿ2.362CI2 I39V 5.12 10 ÿ1.3 ÿ0.932CI2 I48A 3.25 4 ÿ3.9 ÿ1.352CI2 I48V 3.25 4 ÿ1.11 ÿ0.222CI2 I48A/I76V 3.25 4 ÿ4.08 ÿ1.412CI2 V66A 2.86 10 ÿ4.93 ÿ2.362CI2 L68A 6.47 10 ÿ3.84 ÿ2.362CI2 V70A 2.19 4 ÿ1.98 ÿ1.082CI2 I76V 1.70 7 ÿ0.19 ÿ0.072CI2 I76A 1.70 7 ÿ4.29 ÿ1.501EY0 L14A 4.26 4 ÿ2.3 ÿ1.301EY0 V99A 4.94 9 ÿ3.2 ÿ2.291EY0 V74A 7.12 10 ÿ3.1 ÿ2.741EY0 I72V 9.66 9 ÿ1.8 ÿ0.321EY0 F34A 10.09 12 ÿ3.7 ÿ4.001EY0 V66A 11.17 12 ÿ2.2 ÿ3.441EY0 V23A 12.03 14 ÿ2.9 ÿ3.251EY0 L25A 12.12 11 ÿ2.7 ÿ4.201EY0 I92V 13.23 13 ÿ0.5 ÿ0.611EY0 I92A 13.23 13 ÿ4 ÿ4.381EY0 L36A 4.74 13 ÿ3.5 ÿ3.091EY0 I72V/Y113A 9.66 7 ÿ0.94 ÿ0.321EY0 I72V/Y85A 9.66 7 ÿ1.6 ÿ0.751EY0 V66L 11.17 12 ÿ0.1 0.791EY0 V23F 12.03 12 ÿ2.01 ÿ0.671EY0 P117G/H124L/S128A 6.0 - 3.4 1.221EY0 T41I/P117G/H124L/S128A 6.0 - 4.1 4.311EY0 T33V/T41I/P117G/H124L/S128A 6.0 - 3.8 3.541EY0 T41I/S59A/P117G/H124L/S128A 6.0 - 4.8 4.506CIB L6V 5.81 8 ÿ2.19 ÿ0.656CIB L23A 7.30 8 ÿ3.714 ÿ3.316CIB L28A 11.37 9 ÿ2.69 ÿ4.596CIB V61A 5.39 12 ÿ1.901 ÿ3.766CIB F66W 14.60 11 ÿ2.333 ÿ1.636CIB F66A 14.60 11 ÿ4.952 ÿ4.746CIB V70L 9.03 9 ÿ1.19 1.836CIB I73V 6.26 5 ÿ1.524 ÿ0.486CIB F10A 4.90 5 ÿ4.809 ÿ1.98
a Delaunay potential, SSNAPP, (2) for tetrahedra in which the residue participates.b The number of Delaunay tetrahedra in which each residue participates.
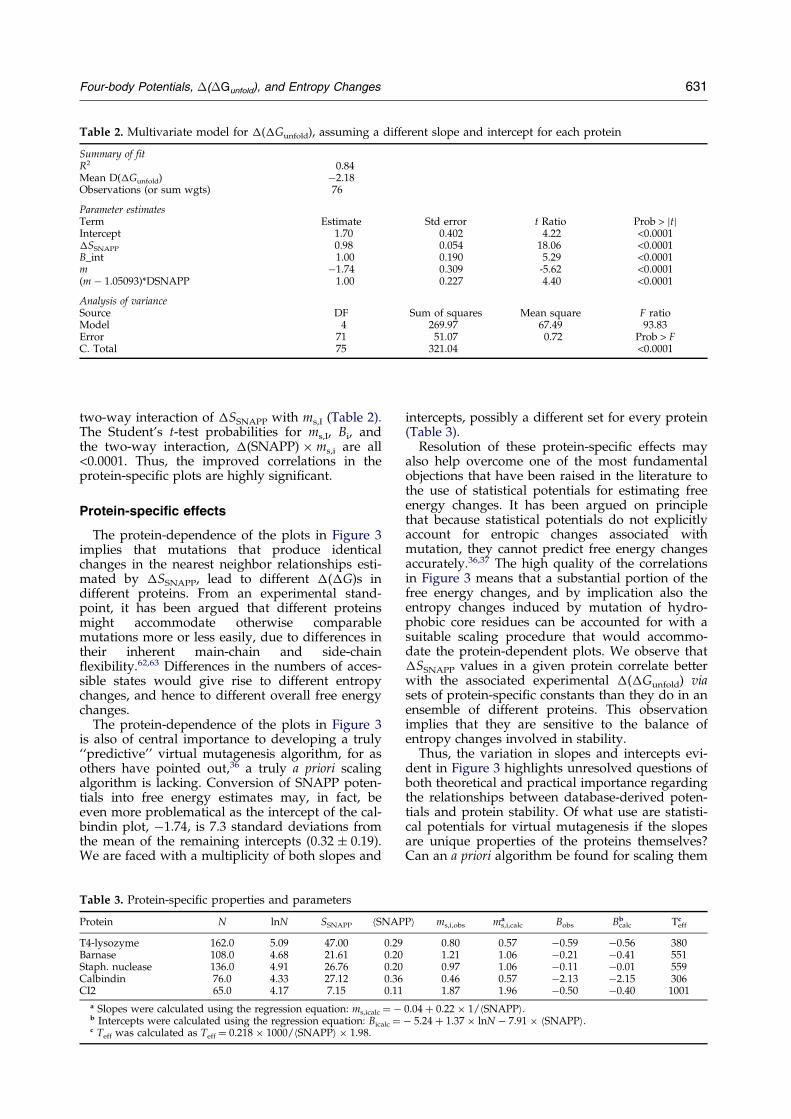
Table 2. Multivariate model for �(�Gunfold), assuming a different slope and intercept for each protein
Summary of fitR2 0.84Mean D(�Gunfold) ÿ2.18Observations (or sum wgts) 76
Parameter estimatesTerm Estimate Std error t Ratio Prob > jtjIntercept 1.70 0.402 4.22 <0.0001�SSNAPP 0.98 0.054 18.06 <0.0001B_int 1.00 0.190 5.29 <0.0001m ÿ1.74 0.309 -5.62 <0.0001(m ÿ 1.05093)*DSNAPP 1.00 0.227 4.40 <0.0001
Analysis of varianceSource DF Sum of squares Mean square F ratioModel 4 269.97 67.49 93.83Error 71 51.07 0.72 Prob > FC. Total 75 321.04 <0.0001
Four-body Potentials, �(�Gunfold), and Entropy Changes 631
two-way interaction of �SSNAPP with ms,I (Table 2).The Student's t-test probabilities for ms,I, Bi, andthe two-way interaction, �(SNAPP) � ms,i are all<0.0001. Thus, the improved correlations in theprotein-speci®c plots are highly signi®cant.
Protein-specific effects
The protein-dependence of the plots in Figure 3implies that mutations that produce identicalchanges in the nearest neighbor relationships esti-mated by �SSNAPP, lead to different �(�G)s indifferent proteins. From an experimental stand-point, it has been argued that different proteinsmight accommodate otherwise comparablemutations more or less easily, due to differences intheir inherent main-chain and side-chain¯exibility.62,63 Differences in the numbers of acces-sible states would give rise to different entropychanges, and hence to different overall free energychanges.
The protein-dependence of the plots in Figure 3is also of central importance to developing a truly``predictive'' virtual mutagenesis algorithm, for asothers have pointed out,36 a truly a priori scalingalgorithm is lacking. Conversion of SNAPP poten-tials into free energy estimates may, in fact, beeven more problematical as the intercept of the cal-bindin plot, ÿ1.74, is 7.3 standard deviations fromthe mean of the remaining intercepts (0.32 � 0.19).We are faced with a multiplicity of both slopes and
Table 3. Protein-speci®c properties and parameters
Protein N lnN SSNAPP hSNAP
T4-lysozyme 162.0 5.09 47.00 0.29Barnase 108.0 4.68 21.61 0.20Staph. nuclease 136.0 4.91 26.76 0.20Calbindin 76.0 4.33 27.12 0.36CI2 65.0 4.17 7.15 0.11
a Slopes were calculated using the regression equation: ms,icalc � ÿb Intercepts were calculated using the regression equation: Bicalc �c Teff was calculated as Teff � 0.218 � 1000/hSNAPPi � 1.98.
intercepts, possibly a different set for every protein(Table 3).
Resolution of these protein-speci®c effects mayalso help overcome one of the most fundamentalobjections that have been raised in the literature tothe use of statistical potentials for estimating freeenergy changes. It has been argued on principlethat because statistical potentials do not explicitlyaccount for entropic changes associated withmutation, they cannot predict free energy changesaccurately.36,37 The high quality of the correlationsin Figure 3 means that a substantial portion of thefree energy changes, and by implication also theentropy changes induced by mutation of hydro-phobic core residues can be accounted for with asuitable scaling procedure that would accommo-date the protein-dependent plots. We observe that�SSNAPP values in a given protein correlate betterwith the associated experimental �(�Gunfold) viasets of protein-speci®c constants than they do in anensemble of different proteins. This observationimplies that they are sensitive to the balance ofentropy changes involved in stability.
Thus, the variation in slopes and intercepts evi-dent in Figure 3 highlights unresolved questions ofboth theoretical and practical importance regardingthe relationships between database-derived poten-tials and protein stability. Of what use are statisti-cal potentials for virtual mutagenesis if the slopesare unique properties of the proteins themselves?Can an a priori algorithm be found for scaling them
Pi ms,i,obs ms,i,calca Bobs Bcalc
b Teffc
0.80 0.57 ÿ0.59 ÿ0.56 3801.21 1.06 ÿ0.21 ÿ0.41 5510.97 1.06 ÿ0.11 ÿ0.01 5590.46 0.57 ÿ2.13 ÿ2.15 3061.87 1.96 ÿ0.50 ÿ0.40 1001
0.04 � 0.22 � 1/hSNAPPi.ÿ 5.24 � 1.37 � lnN ÿ 7.91 � hSNAPPi.
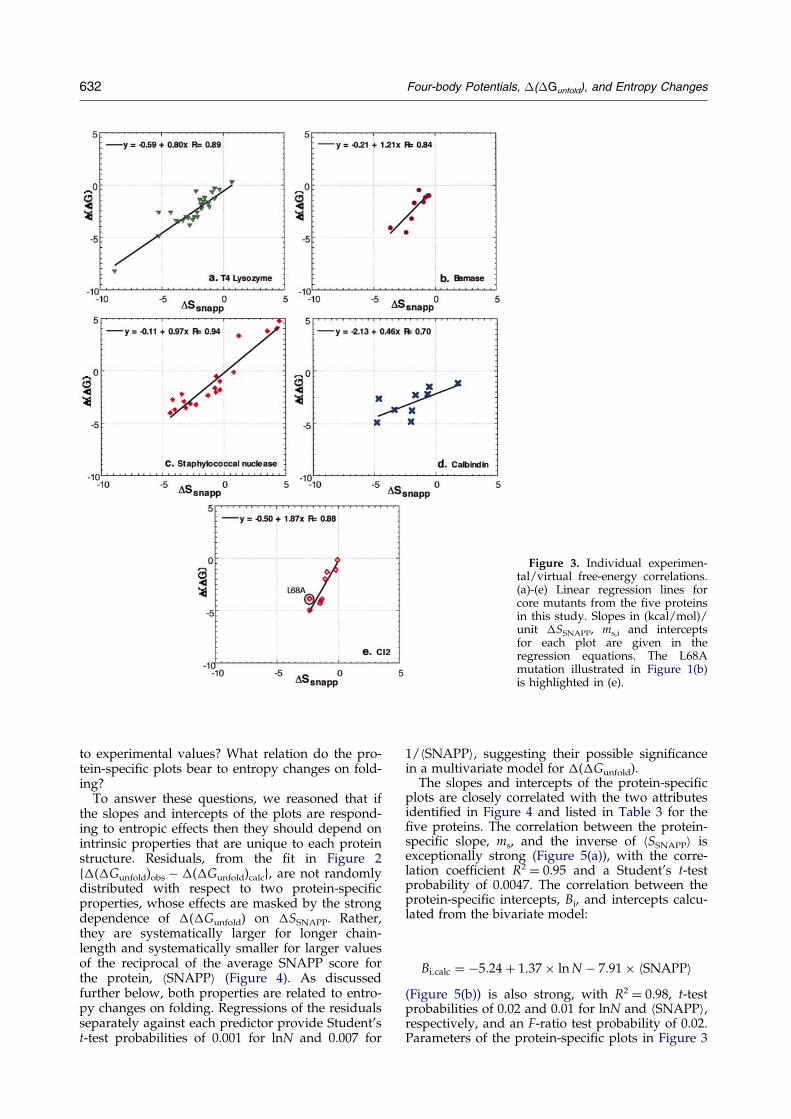
Figure 3. Individual experimen-tal/virtual free-energy correlations.(a)-(e) Linear regression lines forcore mutants from the ®ve proteinsin this study. Slopes in (kcal/mol)/unit �SSNAPP, ms,i and interceptsfor each plot are given in theregression equations. The L68Amutation illustrated in Figure 1(b)is highlighted in (e).
632 Four-body Potentials, �(�Gunfold), and Entropy Changes
to experimental values? What relation do the pro-tein-speci®c plots bear to entropy changes on fold-ing?
To answer these questions, we reasoned that ifthe slopes and intercepts of the plots are respond-ing to entropic effects then they should depend onintrinsic properties that are unique to each proteinstructure. Residuals, from the ®t in Figure 2{�(�Gunfold)obs ÿ �(�Gunfold)calc}, are not randomlydistributed with respect to two protein-speci®cproperties, whose effects are masked by the strongdependence of �(�Gunfold) on �SSNAPP. Rather,they are systematically larger for longer chain-length and systematically smaller for larger valuesof the reciprocal of the average SNAPP score forthe protein, hSNAPPi (Figure 4). As discussedfurther below, both properties are related to entro-py changes on folding. Regressions of the residualsseparately against each predictor provide Student'st-test probabilities of 0.001 for lnN and 0.007 for
1/hSNAPPi, suggesting their possible signi®cancein a multivariate model for �(�Gunfold).
The slopes and intercepts of the protein-speci®cplots are closely correlated with the two attributesidenti®ed in Figure 4 and listed in Table 3 for the®ve proteins. The correlation between the protein-speci®c slope, ms, and the inverse of hSSNAPPi isexceptionally strong (Figure 5(a)), with the corre-lation coef®cient R2 � 0.95 and a Student's t-testprobability of 0.0047. The correlation between theprotein-speci®c intercepts, Bi, and intercepts calcu-lated from the bivariate model:
Bi;calc � ÿ5:24� 1:37� ln N ÿ 7:91� hSNAPPi(Figure 5(b)) is also strong, with R2 � 0.98, t-testprobabilities of 0.02 and 0.01 for lnN and hSNAPPi,respectively, and an F-ratio test probability of 0.02.Parameters of the protein-speci®c plots in Figure 3
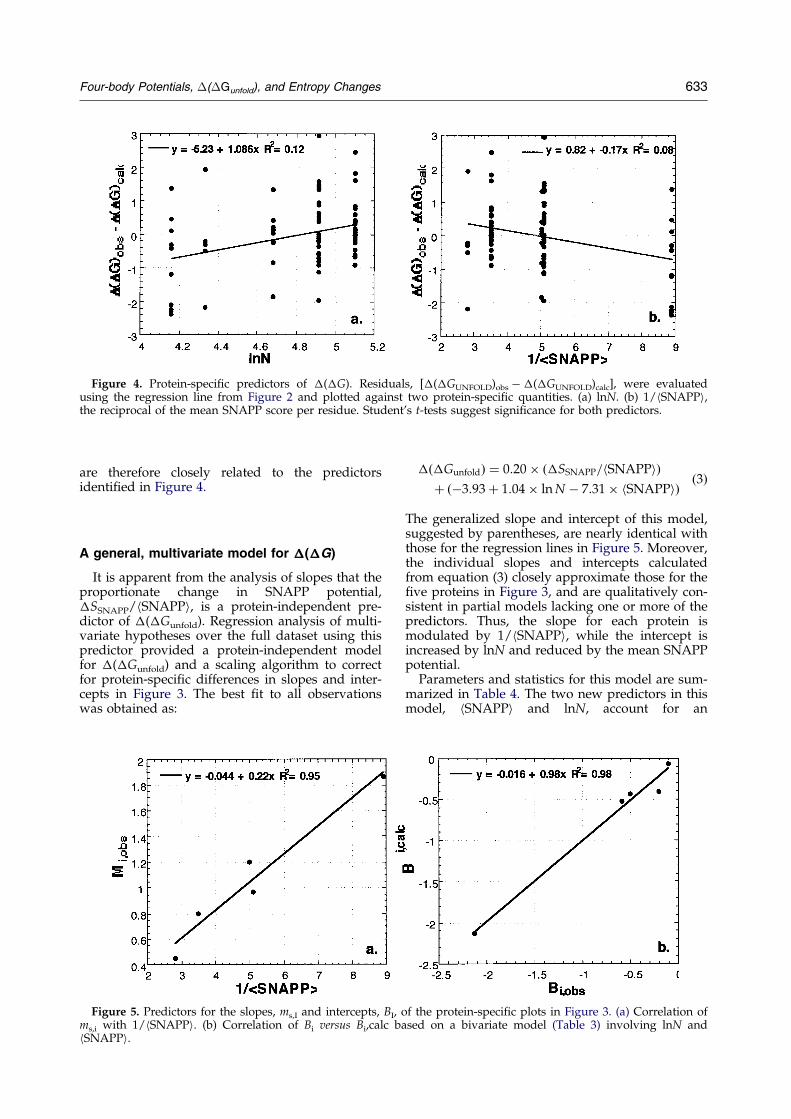
Figure 4. Protein-speci®c predictors of �(�G). Residuals, [�(�GUNFOLD)obs ÿ �(�GUNFOLD)calc], were evaluatedusing the regression line from Figure 2 and plotted against two protein-speci®c quantities. (a) lnN. (b) 1/hSNAPPi,the reciprocal of the mean SNAPP score per residue. Student's t-tests suggest signi®cance for both predictors.
Four-body Potentials, �(�Gunfold), and Entropy Changes 633
are therefore closely related to the predictorsidenti®ed in Figure 4.
A general, multivariate model for ���(���G)
It is apparent from the analysis of slopes that theproportionate change in SNAPP potential,�SSNAPP/hSNAPPi, is a protein-independent pre-dictor of �(�Gunfold). Regression analysis of multi-variate hypotheses over the full dataset using thispredictor provided a protein-independent modelfor �(�Gunfold) and a scaling algorithm to correctfor protein-speci®c differences in slopes and inter-cepts in Figure 3. The best ®t to all observationswas obtained as:
Figure 5. Predictors for the slopes, ms,I and intercepts, BI, oms,i with 1/hSNAPPi. (b) Correlation of Bi versus Bi,calc bahSNAPPi.
���Gunfold� � 0:20� ��SSNAPP=hSNAPPi�� �ÿ3:93� 1:04� ln N ÿ 7:31� hSNAPPi� �3�
The generalized slope and intercept of this model,suggested by parentheses, are nearly identical withthose for the regression lines in Figure 5. Moreover,the individual slopes and intercepts calculatedfrom equation (3) closely approximate those for the®ve proteins in Figure 3, and are qualitatively con-sistent in partial models lacking one or more of thepredictors. Thus, the slope for each protein ismodulated by 1/hSNAPPi, while the intercept isincreased by lnN and reduced by the mean SNAPPpotential.
Parameters and statistics for this model are sum-marized in Table 4. The two new predictors in thismodel, hSNAPPi and lnN, account for an
f the protein-speci®c plots in Figure 3. (a) Correlation ofsed on a bivariate model (Table 3) involving lnN and
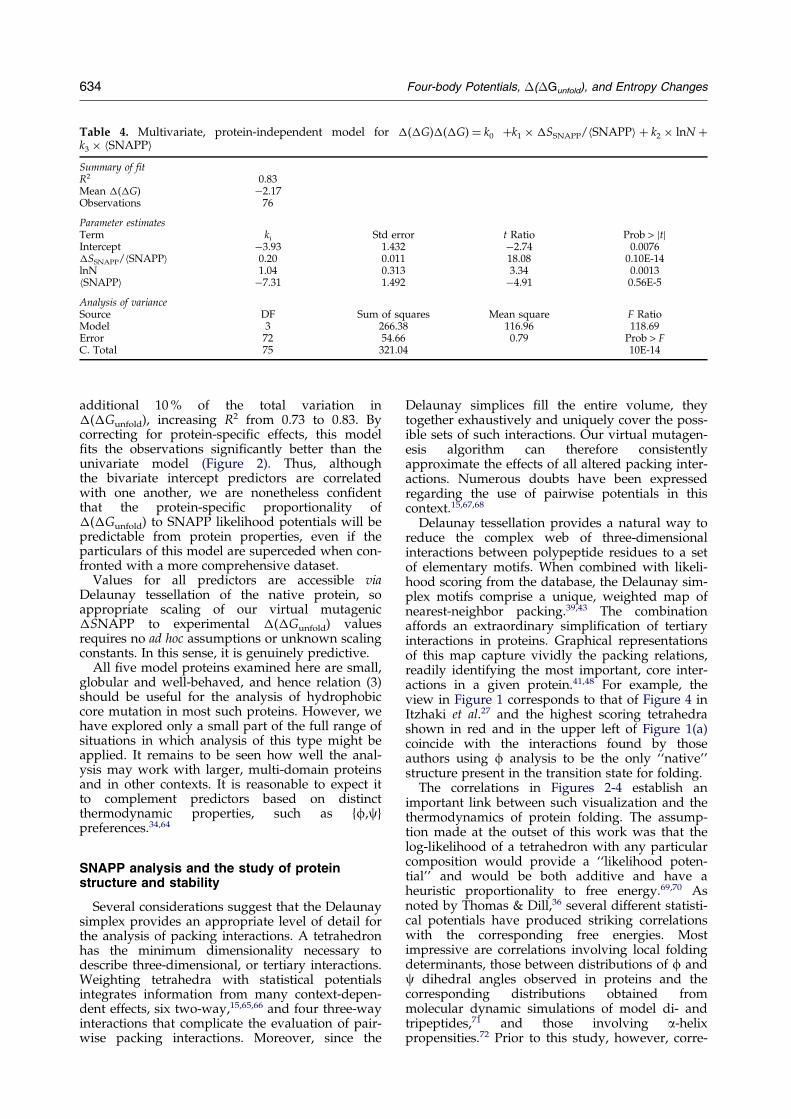
Table 4. Multivariate, protein-independent model for �(�G)�(�G) � k0 �k1 � �SSNAPP/hSNAPPi � k2 � lnN �k3 � hSNAPPiSummary of fitR2 0.83Mean �(�G) ÿ2.17Observations 76
Parameter estimatesTerm ki Std error t Ratio Prob > jtjIntercept ÿ3.93 1.432 ÿ2.74 0.0076�SSNAPP/hSNAPPi 0.20 0.011 18.08 0.10E-14lnN 1.04 0.313 3.34 0.0013hSNAPPi ÿ7.31 1.492 ÿ4.91 0.56E-5
Analysis of varianceSource DF Sum of squares Mean square F RatioModel 3 266.38 116.96 118.69Error 72 54.66 0.79 Prob > FC. Total 75 321.04 10E-14
634 Four-body Potentials, �(�Gunfold), and Entropy Changes
additional 10 % of the total variation in�(�Gunfold), increasing R2 from 0.73 to 0.83. Bycorrecting for protein-speci®c effects, this model®ts the observations signi®cantly better than theunivariate model (Figure 2). Thus, althoughthe bivariate intercept predictors are correlatedwith one another, we are nonetheless con®dentthat the protein-speci®c proportionality of�(�Gunfold) to SNAPP likelihood potentials will bepredictable from protein properties, even if theparticulars of this model are superceded when con-fronted with a more comprehensive dataset.
Values for all predictors are accessible viaDelaunay tessellation of the native protein, soappropriate scaling of our virtual mutagenic�SNAPP to experimental �(�Gunfold) valuesrequires no ad hoc assumptions or unknown scalingconstants. In this sense, it is genuinely predictive.
All ®ve model proteins examined here are small,globular and well-behaved, and hence relation (3)should be useful for the analysis of hydrophobiccore mutation in most such proteins. However, wehave explored only a small part of the full range ofsituations in which analysis of this type might beapplied. It remains to be seen how well the anal-ysis may work with larger, multi-domain proteinsand in other contexts. It is reasonable to expect itto complement predictors based on distinctthermodynamic properties, such as {f,c}preferences.34,64
SNAPP analysis and the study of proteinstructure and stability
Several considerations suggest that the Delaunaysimplex provides an appropriate level of detail forthe analysis of packing interactions. A tetrahedronhas the minimum dimensionality necessary todescribe three-dimensional, or tertiary interactions.Weighting tetrahedra with statistical potentialsintegrates information from many context-depen-dent effects, six two-way,15,65,66 and four three-wayinteractions that complicate the evaluation of pair-wise packing interactions. Moreover, since the
Delaunay simplices ®ll the entire volume, theytogether exhaustively and uniquely cover the poss-ible sets of such interactions. Our virtual mutagen-esis algorithm can therefore consistentlyapproximate the effects of all altered packing inter-actions. Numerous doubts have been expressedregarding the use of pairwise potentials in thiscontext.15,67,68
Delaunay tessellation provides a natural way toreduce the complex web of three-dimensionalinteractions between polypeptide residues to a setof elementary motifs. When combined with likeli-hood scoring from the database, the Delaunay sim-plex motifs comprise a unique, weighted map ofnearest-neighbor packing.39,43 The combinationaffords an extraordinary simpli®cation of tertiaryinteractions in proteins. Graphical representationsof this map capture vividly the packing relations,readily identifying the most important, core inter-actions in a given protein.41,48 For example, theview in Figure 1 corresponds to that of Figure 4 inItzhaki et al.27 and the highest scoring tetrahedrashown in red and in the upper left of Figure 1(a)coincide with the interactions found by thoseauthors using f analysis to be the only ``native''structure present in the transition state for folding.
The correlations in Figures 2-4 establish animportant link between such visualization and thethermodynamics of protein folding. The assump-tion made at the outset of this work was that thelog-likelihood of a tetrahedron with any particularcomposition would provide a ``likelihood poten-tial'' and would be both additive and have aheuristic proportionality to free energy.69,70 Asnoted by Thomas & Dill,36 several different statisti-cal potentials have produced striking correlationswith the corresponding free energies. Mostimpressive are correlations involving local foldingdeterminants, those between distributions of f andc dihedral angles observed in proteins and thecorresponding distributions obtained frommolecular dynamic simulations of model di- andtripeptides,71 and those involving a-helixpropensities.72 Prior to this study, however, corre-

Four-body Potentials, �(�Gunfold), and Entropy Changes 635
lations arising from tertiary folding determinantswere weaker. The high correlations in Figure 3indicate that, in a wide range of applications, four-body compositional likelihood potentials shouldhave a utility at least comparable to those invol-ving local folding determinants and may be comp-lementary to them.
More importantly, the evident proportionalitybetween sums of SNAPP potentials and exper-imental free energies, together with nature of thescaling algorithm in equation (3) provide a novelperspective on an important property of proteinstructures: folding equilibria for different proteinsmay involve different balances between the entro-pic restriction imposed by the native fold and theincrease in entropy associated with partitioninghydrophobic side-chains into the protein core. TheSNAPP score is the logarithm of a product of fre-quency ratios, and is therefore somewhat analo-gous to a product of equilibrium constants, whichare ratios of concentrations. From this heuristicconnection to the Boltzmann law, and becausethe slopes in Figure 3 are positive by our signconvention for �SSNAPP, we expect the proportion-ality between the estimated and experimentalfree energies to be equal to kTeff:�(�Gunfold) � kTeff � �SSNAPP, for some, effective,temperature, Teff. Thus, the observed variation inms,i implies that the ®ve proteins in this study areat different ``effective temperatures''36 with respectto the SNAPP four-body likelihood potential.
Remarkably, the key to scaling SNAPP poten-tials to the experimental free energies is the meanSNAPP potential of the protein itself. SNAPP pro-vides both a unique count and a statistical weightfor all tertiary interactions in a protein. Delaunaytetrahedra with the highest potentials coincidewith hydrophobic core regions; those with negativepotentials involve hydrophilic side-chains and aregenerally at the surface. Thus, a higher hSNAPPiimplies that a higher fraction of residues arehydrophobic side-chains that are withdrawn fromsolution into the core. The mean SNAPP potentialis thus a quantitative measure of the price paid inunits of buried non-polar contacts in order torestrict each residue to its native conformation.
Thomas & Dill36 showed that proteins in thestructural database manage to bury their hydro-phobic side-chains more or less well, and relatethis effect to a protein-speci®c, ``effective tempera-ture''. In general, larger proteins with few enoughhydrophobic residues that they can all be accomo-dated in the core have low apparent temperatures,whereas smaller proteins with more hydrophobicresidues have high apparent temperatures. Theyquantify these differences by de®ning a ``partitionpropensity'': p � 2nc/qhnh where, nc is the totalnumber of contacts in a given protein, qh is theaverage coordination number for hydrophobicside-chains, and nh is the number of hydrophobicresidues in the protein. Entirely consistent with itsscaling role, hSNAPPi is closely related to the reci-procal of the partition propensity. It is therefore
proportional to the fraction of the total stabilizationthat is due to the hydrophobic effect, and might becalled the ``hydrophobic moment'' in the stabiliz-ation free energy.
That the hydrophobic moment of a proteinshould provide the appropriate scaling to exper-imental free energies establishes a link to thenotion that ms,i is an effective temperature.hSNAPPi, like temperature, is an intensive quantityindependent of the size of the system, whereas�SSNAPP, is an extensive quantity dependent on thesum over contributing Delaunay tetrahedra. Ourfree energy estimate is thus a product of intensiveand extensive structural properties, in formalagreement with other components of free energy,like T�S.
It should be noted that ms,i decreases withincreasing hSNAPPi. hSNAPPi is related to hydro-phobic interactions that owe their stabilizing effectto the increased entropy of solvent water uponfolding. Proteins with a small hydrophobicmoment, like CI2 (hSNAPPi � 0.11) are at a highereffective temperature with respect to the likelihoodpotential than those with a large hydrophobicmoment, like T4 lyzozyme and calbindin(hSNAPPi � 0.36). In other words, they utilizehydrophobic bonding more ef®ciently, and aretherefore more vulnerable to structural changes intheir hydrophobic cores.
A physical rationalization for the observeddifferences in ms,i for different proteins, consistentwith the qualitative observation that proteins exhi-bit a rather narrow range of thermodynamic stabi-lities,73 is that the overall conformational entropychange on folding is smaller, i.e. less destabilizing,for proteins at higher effective temperatures. Thiswould be the case, for example, if the main-chainwere, on average, more ¯exible. Equivalently, anative fold which permitted more conformations inits native state, i.e. more microstates, would sup-port changes in its hydrophobic core with smallerstability changes.62 Effective temperatures indi-cated for the proteins in this study range from 318to 1001, and are included in Table 3.
Conclusions
The most novel and signi®cant result of thisstudy is the ®nding that four-body SNAPP poten-tials for different proteins scale to experimental�(�Gunfold) values according to their mean SNAPPpotential. The consistency of correlations inFigure 2 and the statistical signi®cance of the dis-tinct protein-speci®c plots have enabled us todetect a novel and important relationship betweenhydrophobic core packing structures and proteinstability. Resolution of protein-speci®c effects wasnot evident in previous studies using other typesof statistical potentials, and for which the corre-lations were of lower quality.32 ± 34 Conversely,reference free energies estimated using the SNAPPfour-body potentials are coherent enough to permitthe identi®cation of anticipated protein-speci®c

636 Four-body Potentials, �(�Gunfold), and Entropy Changes
effects. We consider the demonstration of, and cor-rection for, statistically different slopes also asimportant evidence for the value of four-bodycompositional log-likelihood potentials, whichappear remarkably versatile and well-suited forquantitative analysis of how tertiary packing inter-actions in proteins determine stability.
One reason for the enhanced performance offour-body potentials based on Delaunay simplicesmay be that they provide a natural, mathematicallyrigorous decomposition of the network of fullythree-dimensional interactions. In this context, wenote that Behe et al.15 failed to discern any signi®-cant pairwise interactions in their previous attemptto demonstrate them. Pairs and triples are lower-order packing decompositions, inherently linearand two-dimensional, and so cannot properly rep-resent three-dimensional interactions.
SNAPP ``building blocks'' should also provide astandard against which to measure the effects ofmutation quantitatively in combinatorial mutagen-esis experiments and hence a sound basis for theiterative identi®cation and eventual rationalizationof outliers in pursuit of a more complete basis setfor predicting protein stability. The precedent ofGilis & Rooman34 suggests that local conformation-al propensities can improve correlations betweenexperimental and virtual free energies. Multivariatemodels involving local and tertiary predictors, aswell as others such as changes in simplex volumeand shape may allow mathematical predictions toencompass those mutant types that tend to gener-ate less meaningful linear correlations. A morere®ned notion of this method's strengths willdoubtless grow as we test its ability to predict mul-tiple, combinatorial, and patterned-library74
mutations to the hydrophobic core. Whatever theiroutcome, such studies should contribute to ourunderstanding of protein-folding phenomena.
Acknowledgments
This work was supported by NIGMS-48519 (C.W.C.),01-SC-NSF-1010 (C.W.C.), NIGMS 58665 (M.H.E.) andNIH 1P01 DK58335. B.C.L. was supported by the UNCSURE program Summer Research Experiences forUndergraduates, funded by NSF DBI-9605149. Theauthors are grateful to O. Smithies, J. Hermans,M. Levitt, and P. Koehl for useful discussions and toShuxing Zhang for calculations.
References
1. Alonso, D. O. & Dill, K. A. (1991). Solvent denatura-tion and stabilization of globular proteins. Biochemis-try, 30, 5974-5985.
2. Buckle, A. M., Henrick, K. & Fersht, A. R. (1993).Crystal structural analysis of mutations in thehydrophobic cores of barnase. J. Mol. Biol. 234, 847-860.
3. Mateu, M. G. & Fersht, A. R. (1998). Nine hydro-phobic side-chains are key determinants of the
thermodynamic stability and oligomerization statusof tumour suppressor p53 tetramerization domain.EMBO J. 17, 2748-2758.
4. Matthews, B. W. (1993). Structural and geneticanalysis of protein stability. Annu. Rev. Biochem. 62,139-160.
5. Shortle, D. (1992). Mutational studies of proteinstructures and their stabilities. Quart. Rev. Biophys.25, 205-250.
6. Chothia, C. (1974). Surface area and hydrophobicfree energy. Nature, 248, 338-339.
7. Chothia, C. (1975). The nature of accessible andburied surfaces in proteins. J. Mol. Biol. 105, 1-14.
8. Dill, K. A. (1990). Dominant forces in protein fold-ing. Biochemistry, 29, 7133-7155.
9. Dill, K. A. & Stigter, D. (1995). Modeling proteinstability as heteropolymer collapse. Advan. ProteinChem. 46, 59-104.
10. Richards, F. M. (1974). The interpretation of proteinstructures: total volume, group volume distributions,and packing density. J. Mol. Biol. 82, 1-14.
11. Richards, F. M. (1977). Areas, volumes, packing andprotein structure. Annu. Rev. Biophys. Bioeng. 6, 151-176.
12. Richards, F. M. (1985). Calculation of molecularvolumes and areas for structures of known geome-try. Methods Enzymol. 115, 440-464.
13. Rose, G. D. & Wolfenden, R. (1993). Hydrogenbonding, hydrophobicity, packing, and protein fold-ing. Annu. Rev. Biophys. Biomol. Struct. 22, 381-415.
14. Baldwin, E. P. & Matthews, B. W. (1994). Core-pack-ing constraints, hydrophobicity and protein design.Curr. Opin. Biotechnol. 5, 396-402.
15. Behe, M. J., Lattman, E. E. & Rose, G. D. (1991). Theprotein-folding problem: The native fold determinespacking, but does packing determine the nativefold? Proc. Natl Acad. Sci. USA, 88, 4195-4199.
16. Matsumura, M., Becktel, W. J. & Matthews, B. W.(1988). Hydrophobic stabilization in T4 lysozymedetermined directly by multiple substitutions of Ile3. Nature, 334, 406-410.
17. Matsumura, M., Becktel, W. J. & Matthews, B. W.(1988). Structural studies of mutants of T4 lysozymethat alter hydrophobic stabilization. J. Biol. Chem.264, 16059-16066.
18. Matsumura, M., Wozniak, J. A., Sun, D. P. &Matthews, B. W. (1989). Structural studies ofmutants of T4 lysozyme that alter hydrophobicstabilization. J. Biol. Chem. 264, 16059-16066.
19. Alber, T. & Matthews, B. W. (1987). Structure andthermal stability of phage T4 lysozyme. MethodsEnzymol. 154, 511-533.
20. Alber, T., Sun, D. P., Nye, J. A., Muchmore, D. C. &Matthews, B. W. (1987). Temperature-sensitivemutations of bacteriophage T4 lysozyme occur atsites with low mobility and low solvent accessibilityin the folded protein. Biochemistry, 26, 3754-3758.
21. Karpusas, M., Baase, W. A., Matsumura, M. &Matthews, B. W. (1989). Hydrophobic packing in T4lysozyme probed by cavity-®lling mutants. Proc.Natl Acad. Sci. USA, 86, 8237-8241.
22. Serrano, L., Bycroft, M. & Fersht, A. R. (1991).Aromatic-aromatic interactions and protein stability.Investigation by double-mutant cycles. J. Mol. Biol.218, 465-475.
23. Kellis, J. T. J., Nyberg, K., Sali, D. & Fersht, A. R.(1988). Contribution of hydrophobic interactions toprotein stability. Nature, 333, 784-786.

Four-body Potentials, �(�Gunfold), and Entropy Changes 637
24. Kellis, J. T. J., Nyberg, K. & Fersht, A. R. (1989).Energetics of complementary side-chain packing in aprotein hydrophobic core. Biochemistry, 28, 4914-4922.
25. Shortle, D., Stites, W. & Meeker, A. K. (1990). Con-tributions of the large hydrophobic amino acids tothe stability of staphylococcal nuclease. Biochemistry,29, 8033-8041.
26. Shortle, D., Chan, H. S. & Dill, K. A. (1992). Model-ing the effects of mutations on the denatured statesof proteins. Protein Sci. 1, 201-215.
27. Itzhaki, L. S., Otzen, D. E. & Fersht, A. R. (1995).The structure of the transition state for folding ofchymotrypsin inhibitor 2 analysed by protein engin-eering methods: evidence for a nucleation-conden-sation mechanism for protein folding. J. Mol. Biol.254, 260-288.
28. Jackson, S. E., elMasry, N. & Fersht, A. R. (1993).Structure of the hydrophobic core in the transitionstate for folding of chymotrypsin inhibitor 2: acritical test of the protein engineering method ofanalysis. Biochemistry, 32, 11270-11278.
29. Julenius, K., Thulin, E., Linse, S. & Finn, B. E. (1998).Hydrophobic core substitutions in calbindin D9k:effects on stability and structure. Biochemistry, 37,8915-8925.
30. Matthews, B. W. (1987). Genetic and structural anal-ysis of the protein stability problem. Biochemistry,26, 6885-6888.
31. Takano, K., Yamagata, Y. & Yutani, K. (1998).A general rule for the relationship between hydro-phobic effect and conformational stability of aprotein: stability and structure of a series of hydro-phobic mutants of human lysozyme. J. Mol. Biol.280, 749-761.
32. Topham, C. M., Srinivasan, N. & Blundell, T. L.(1997). Prediction of the stability of protein mutantsbased on structural environment-dependent aminoacid substitution and propensity tables. Protein Eng.10, 7-21.
33. Gilis, D. & Rooman, M. (1996). Stability changesupon mutation of solvent accessible resitudes in pro-teins evaluated by database-derived potentials.J. Mol. Biol. 257, 1112-1126.
34. Gilis, D. & Rooman, M. (1997). Predicting proteinstability changes upon mutation using database-derived potentials: solvent accessibility determinesthe importance of local versus non-local intgeractionsalong the sequence. J. Mol. Biol. 272, 276-290.
35. Rooman, M. & Wodak, S. (1995). Are database-derived potentials valid for scoring both forwardand inverted protein folding? Protein Eng. 8, 849-858.
36. Thomas, P. D. & Dill, K. A. (1996). Statistical poten-tials extracted from protein structures: how accurateare they? J. Mol. Biol. 257, 457-469.
37. Mark, A. E. & van Gunsteren, W. F. (1994).Decomposition of the free energy of a system interms of speci®c interactions. J. Mol. Biol. 240, 167-176.
38. Betancourt, M. R. & Thirumalai, D. (1999). Pairpotentials for protein folding: choice of referencestates and sensitivity of predicted native states tovariations in the interaction schemes. Protein Sci. 8,361-369.
39. Singh, R. K., Tropsha, A. & Vaisman, I. I. (1996).Delaunay tessellation of proteins. J. Comput. Biol. 2,213-221.
40. Tropsha, A., Singh, R. K., Vaisman, I. I. & Zheng,W. (1996). Statistical geometry analysis of proteins:implications for inverted structure prediction. InProceedings, 1st Paci®c Symposium on Biocomputing,Hawaii.
41. Cammer, S. (2000). Using Delaunay tessellationin the analysis of side-chain-side-chain packing inproteins. PhD, University of North Carolina atChapel Hill.
42. Finney, J. L. (1970). Random packing and the struc-tures of simple liquids I. The geometry of randomclose packing. Proc. Roy. Soc, 319, 479-493.
43. Cammer, S. A., Carter, C. W., Jr & Tropsha, A.(2001). Identi®cation of sequence-speci®c tertiarypacking motifs in protein structures using Delauneytessellation. In Lecture Notes in Computational Scienceand Engineering (Schlick, T., ed.), Springer-Verlag, inthe press.
44. Vaisman, I. I., Tropsha, A. & Zheng, W. (1998).Compositional preferences in quadruplets of nearestneighbor residues in protein structures: statisticalgeometry analysis. In Proceedings of the IEEE Sympo-sia on Intelligence and Systems, pp. 163-168.
45. Zheng, W., Cho, S. J., Vaisman, I. I. & Tropsha, A.(1997). A new approach to protein fold recognitionbased on Delaunay tessellation of protein structure.In: Paci®c Symposium on Biocomputing '97 (Altman, R.B. et al., eds), pp. 487-496, World Scienti®c,Singapore.
46. Gan, H. H., Tropsha, A. & Schlick, T. (2001). Latticefolding with two- and four-body statistical poten-tials. Proteins: Struct. Funct. Genet. In the press.
47. Berman, H. M., Westbrook, J., Feng, Z., Gilliland,G., Bhat, T. N., Weissig, H. N. S. I. & Bourne, P. E.(2000). The Protein Data Bank. Nucl. Acids Res. 28,235-242.
48. McPhalen, C. A. & James, M. N. G. (1987). Cystaland molecular structure of the serine proteinaseinhibitor CI-2 from barley seeds. Biochemistry, 26,261-269.
49. Jackson, S. E., Moracci, M., elMasry, N., Johnson,C. M. & Fersht, A. R. (1993). Effect of cavity-creatingmutations in the hydrophobic core of chymotrypsoininhibitor 2. Biochemistry, 32, 11259-11269.
50. Blaber, M. A. B. W., Nadine, G. & Matthews, B. W.(1995). Alanine scanning mutagenesis of the a-helix115-123 of phage T4 lysozyme: effects on structure,stability and the binding of solvent. J. Mol. Biol. 246,317-330.
51. Eriksson, A. E., Baase, W. A., Wozniak, J. A. &Matthews, B. W. (1992). A cavity-containing mutantof T4 lysozyme is stabilized by buried benzene.Nature, 355, 371-373.
52. Eriksson, A. E., Baase, W. A. & Matthews, B. W.(1993). Similar hydrophobic replacements of Leu99and Phe153 within the core of T4 lysozyme havedifferent structural and thermodynamic conse-quences. J. Mol. Biol. 229, 747-769.
53. Gassner, N. C., Baase, W. & Matthews, B. W. (1996).A test of the ``jigsaw puzzle'' model for protein fold-ing by multiple methionine substitutions within thecore of T4 lysozyme. Proc. Natl Acad. Sci. USA, 93,12155-12158.
54. Hurley, J. H., Baase, W. A. & Matthews, B. W.(1992). Design and structural analysis of alternativehydrophobic core packing arrangements in bacterio-phage T4 lysozyme. J. Mol. Biol. 224, 1143-1159.
55. Dalby, P. A., Clarke, J., Johnson, C. M. & Fersht,A. R. (1998). Folding intermediates of wild-type and

638 Four-body Potentials, �(�Gunfold), and Entropy Changes
mutants of barnase. II. Correlation of changes inequilibrium amide exchange kinetics with the popu-lation of the folding intermediate. J. Mol. Biol. 276,647-656.
56. Dalby, P. A., Oliveberg, M. & Fersht, A. R. (1998).Folding intermediates of wild-type and mutants ofbarnase. I. Use of phi-value analysis and m-valuesto probe the cooperative nature of the folding pre-equilibrium. J. Mol. Biol. 276, 625-646.
57. Johnson, C. M. & Fersht, A. R. (1995). Protein stab-ility as a function of denaturant concentration: thethermal stability of barnase in the presence of urea.Biochemistry, 34, 6795-6804.
58. Green, S. M., Meeker, A. & Shortle, D. (1992). Con-tributions of the polar, uncharged amino acids tothe stability of staphylococcal nuclease: evidence formutational effects on the free energy of thedenatured state. Biochemistry, 31, 5717-5728.
59. Green, S. M. & Shortle, D. (1993). Patterns of non-additivity between pairs of stability mutations instaphylococcal nuclease. Biochemistry, 32, 10131-10139.
60. JMP (2000). JMP 4, SAS, Cary, NC.61. Wilkinson, L. (1987). SYSTAT, The System for
Statistics 5.2.1, SYSTAT, Inc., Evanston, IL 60601.62. Cota, E., Hamill, S. J., Fowler, S. B. & Clarke, J.
(2000). Two proteins with the same structurerespond very differently to mutation: the role ofplasticity in protein stability. J. Mol. Biol. 302, 713-725.
63. Cordes, M. H. J., Davidson, A. R. & Sauer, R. T.(1996). Sequence space, folding and protein design.Curr. Opin. Struct. Biol. 6, 3-10.
64. MunÄ oz, V. & Serrano, L. (1994). Intrinsic secondarystructure propensities of the amino acids, using stat-istical f-c matrices: comparison with experimentalscales. Proteins: Struct. Funct. Genet. 20, 301-311.
65. Miyazawa, S. & Jernigan, R. L. (1985). Estimation ofeffective contact energies from protein crystalstructures: quasi-chemical approximation. Macro-molecules, 18, 534-552.
66. Miyazawa, S. & Jernigan, R. L. (1996). Residue-resi-due potentials with a favorable contact pair termand an unfavorable high packing density term forsimulation and threading. J. Mol. Biol. 256, 623-644.
67. Vendruscolo, M. & Domany, E. (1998). Pairwise con-tact potentials are unsuitable for protein folding.J. Chem. Phys. 109, 11101-11108.
68. Vendruscolo, M. & Domany, E. (2000). Can a pair-wise contact potentials stabilize native proteinagainst decoys obtained by threading? Proteins:Struct. Funct. Genet. 38, 134-148.
69. Sippl, M. (1990). Calculation of conformationalensembles from potentials of mean force. Anapproach to the knowledge-based prediction of localstructures in globular proteins. J. Mol. Biol. 213, 859-883.
70. Tanaka, S. & Scheraga, H. (1976). Medium- andlong-range interaction parameters between aminoacids for predicting three-dimensional structures ofproteins. Macromolecules, 9, 945-950.
71. O'Connell, T. M., Wang, L., Hermans, J. & Tropsha,A. (1999). The ``random-coil'' state of proteins: com-parison of database statistics and molecular simu-lations. Proteins: Struct. Funct. Genet. 36, 407-418.
72. Serrano, L., Sancho, J., Hirschberg, M. & Fersht,A. R. (1992). a-Helix stability in proteins I. Empiricalcorrelations concerning substitution of side-chains atthe N and C-caps and the replacement of alanine byglycine or serine at solvent-exposed surfaces. J. Mol.Biol. 227, 544-559.
73. Creighton, T. E. (1993). Proteins: Structures andMolecular Properties, W.H. Freeman and Company,New York.
74. Lahr, S. J., Broadwater, A., Carter, C. W., Jr, Collier,M., Hensley, L., Waldner, J., Pielak, G. J. & Edgell,M. H. (1999). Patterned library analysis: a methodfor the quantitative assessment of hypotheses con-cerning the determinants of protein structure. Proc.Natl Acad. Sci. USA, 96, 14860-14865.
Edited by A. R. Fersht
(Received 7 March 2001; received in revised form 5 July 2001; accepted 5 July 2001)





























