Embed Size (px)
Citation preview

FULL PAPER
DOI:10.1002/ejic.201300635
Helical Oxidovanadium(IV) Salen-Type Complexes:Synthesis, Characterisation and Catalytic Behaviour
Sanmitra Barman,[a] Smita Patil,[a] John Desper,[a]
Christine M. Aikens,[a] and Christopher J. Levy*[a]
Keywords: Helical structures / Metallofoldamers / Vanadium / Schiff base ligands / Circular dichroism
Chiral [VO(salen)] complexes (R,R)-7 and (R,R)-8 were syn-thesised by ligand replacement from [VO(acac)2] with the re-solved (1R,2R)-cyclohexyl ligands 3,3�-[(1R,2R)-1,2-cyclohex-ane-diylbis(nitrilomethylidyne)]bis-4-phenanthrenol [(R,R)-3] and 3,3�-[(1R,2R)-1,2-cyclohexanediylbis(nitrilomethylidy-ne)]bisbenz[a]anthracen-1-ol [(R,R)-4], respectively. Simi-larly, complexes (R)-9 and (R)-10 were synthesised using the(R)-binaphthyl ligands 3,3�-[(1R)-(1,1�-binaphthalene)-2,2�-diylbis(nitrilomethylidyne)]bis-4-phenanthrenol [(R)-5] and3,3�-[(1R)-(1,1�-binaphthalene)-2,2�-diylbis(nitrilomethyl-idyne)]bis-benz[a]anthracen-1-ol [(R)-6], respectively. These
Introduction
Catalytic asymmetric oxidations are important reactionsin the fine chemical and pharmaceutical industries.[1] Manymedicinally useful compounds have chiral sulfoxide moie-ties in their active structures,[2] yet high enantioselectivityin the catalytic oxidation of sulfides to sulfoxides hasproven to be difficult. Improved selectivity and the develop-ment of methods that have reduced environmental impact,such as through the use of benign oxidants, are importantgoals in this field.[3]
Salen and related Schiff base ligands with a central unit(backbone) derived from a chiral diamine have been studiedas catalysts for asymmetric sulfoxidations.[4] Nakajima et al.used organic peroxides and VIVO(salen) complexes derivedfrom (1R,2R)-diaminocyclohexane to oxidise prochiral sulf-ides to sulfoxides and achieved up to 42% ee.[5] Corre-sponding VVO(salen)+ complexes gave much lower levels ofasymmetric induction. Adão et al. used several VO(salen)and VO(salan) complexes to oxidise thioanisole with hydro-gen peroxide and achieved up to 50 % ee.[4f,4g] Bolm et al.improved the selectivity (up to 85% ee) by incorporatingbulky tert-butyl groups in the ortho positions and electron-withdrawing nitro groups at the para positions of the aro-matic rings.[6] Recently, Katsuki et al. introduced a water-
[a] Dept. of Chemistry, Kansas State University,213 CBC Bldg., Manhattan, KS 66506, USAE-mail: [email protected]://www.k-state.edu/chem/people/faculty/levy.htmlSupporting information for this article is available on theWWW under http://dx.doi.org/10.1002/ejic.201300635.
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5708
complexes were characterised by IR spectroscopy, ESI-TOF-MS, electronic absorption spectroscopy and circular dichro-ism (CD). Single crystal X-ray analysis of (R,R)-7 and (R,R)-8revealed distorted square pyramidal geometries and a 1:1 ra-tio of diastereomeric M and P helical conformers. SolutionCD studies in THF combined with DFT calculations indicatethat the M conformer dominates in solution for (R,R)-7. Pre-liminary catalytic oxidation of thioanisole with H2O2 and 1%[VO(salen)] showed good selectivity for sulfoxides over sulf-ones but low enantioselectivities (8 to 33%) which are sol-vent dependent but insensitive to the catalyst used.
soluble highly twisted salen ligand system, the metal com-plexes of which gave sulfoxide ee values up to 98 %.[7] In thecurrent study helical vanadyl salen complexes are reportedand their characterisation and use as catalysts for the asym-metric oxidation of thioanisole with aqueous hydrogen per-oxide are discussed.
Results and Discussion
Synthesis
The resolved diamines (1R,2R)-diaminocyclohexane(DAC) and (R)-BINAM were condensed with the phen-anthryl and benz[a]anthryl aldehydes 1 and 2 (Scheme 1)to give four tetradentate salen ligands, 3–6, as previouslyreported by our group.[8] Metallation of the ligands withvanadyl acetylacetonate in the presence of excess base(NaOMe) produced the vanadyl complexes 7–10 [Equa-
Scheme 1. Ligand precursors used in this work.

www.eurjic.org FULL PAPER
tions (1), (2), (3) and (4)]. A 2:1 mixture of CH2Cl2 andEtOH provided sufficient solubility for the deprotonated li-gand and VO(acac)2 to react in a reasonable amount oftime. The excess base resulted in better yields and higherinitial purities. Vanadyl complexes were normally purifiedby Soxhlet extraction with CH2Cl2 which was effective atdissolving the products away from the NaOMe, Na(acac)and any unreacted VO(acac)2. Complexes (R,R)-7 and(R,R)-8 were isolated as green powders whereas (R)-9 and(R)-10 were brown powders.
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5709
Characterisation of the Complexes
The compositions of the complexes were determined bycombustion analysis with mass spectrometry confirming themolecular species (vide infra). Single crystal X-ray struc-tural analysis was also obtained for (R,R)-7 and (R,R)-8.IR spectroscopy identified the V=O stretches for the com-plexes in the 1008–1036 cm–1 range, consistent with pre-vious studies.[9] 1H NMR spectra of all the complexes hadbroadened signals due to the paramagnetic VIV centre anddid not provide significant structural information. EPRdata confirmed the paramagnetic nature of the complexes.Details of the IR, UV/Vis and electronic CD spectroscopicstudies are presented below.
The high-resolution ESI-TOF mass data (Table 1) ofthese complexes showed the M+ and/or M + H+ molecularion peaks and expected isotopic patterns. The dimer M2
+
was observed for (R,R)-7 and (R,R)-8, and this is likely theresult of μ-oxido bridging of vanadium centres in the gasphase. The mass data for (R)-9 and (R)-10 provide strongevidence for the proposed molecular compositions of thesecomplexes in the absence of crystal structures.
Table 1. ESI-TOF-MS data for vanadyl complexes.
Ion Formula Mass Error(obs)[a] (ppm)[b]
[(R,R)-7]+ C36H28N2O3V 587.1567 4.6[(R,R)-7 + H]+ C36H29N2O3V 588.1624 1.0[(R,R)-7]2+ C72H54N4O6V2 1174.3140 5.2[(R,R)-8]+ C44H32N2O3V 687.1856 0.4[(R,R)-8 + H]+ C44H33N2O3V 688.1919 1.7[(R,R)-8]2+ C88H64N4O6V2 1374.3749 3.2[(R)-9]+ C50H30N2O3V 757.1611 11.2[(R)-9 + H]+ C50H31N2O3V 758.2069 38.9[(R)-10]+ C58H34N2O3V 857.2039 3.5
[a] Mass in Daltons. [b] Error = 106·(measured mass – theoreticalmass)/theoretical mass.
X-ray Crystal Structures of Complexes (R,R)-7 and (R,R)-8
Single crystals of (R,R)-7 and (R,R)-8 were grown byslow solvent diffusion of hexane into dichloromethane solu-tions of the complexes. The crystal data are presented inTable 2. In each case, the salen ligand is bound in a tetra-dentate manner to the vanadium centre and two dia-stereomeric helical conformers (P and M) are present in a1:1 ratio. Selected metric parameters for the two structuresare presented in Table 3.
The thermal ellipsoid plot of the M conformer of (R,R)-7 is presented in Figure 1. The geometry is approximatelysquare pyramidal and vanadium sits above the ONNOplane by 0.58 Å. Reedijk’s structural index parameter, τ =0.29, indicates a small distortion towards trigonal bipyrami-dal geometry.[10] Here N52–V1–O12 (136.2°) is substantiallysmaller than N51–V1–O32 (153.8°), at least partially due tothe steric repulsion between the phenanthryl groups.
The thermal ellipsoid plot of the P conformer of (R,R)-7 is presented in Figure 2. The vanadium sits above theONNO plane by 0.57 Å, similar to that seen for the M con-

www.eurjic.org FULL PAPER
Table 2. Crystal data for (R,R)-7 and (R,R)-8.
Compound (R,R)-7 (R,R)-8·CH2Cl2
Empirical formula C36H28N2O3V1 C45H34Cl2N2O3V1
Mr 587.54 772.58Crystal system monoclinic triclinica [Å] 9.668(3) 12.0364(9)b [Å] 10.827(3) 16.8357(12)c [Å] 25.450(7) 17.9974(13)α [°] 90 89.1610(10)β [°] 99.717(14) 77.6150(10)γ [°] 90 85.9340(10)Unit cell vol. [Å3] 2625.8(13) 3553.2(4)Space group P2(1) P1Z 4 4T [K] 173(2) 100(2)μ [mm–1] 0.422 0.476N 24584 37413Nind 10225 23786Rint 0.0695 0.0384R1
[a] [I � 2σ(I)] 0.0579 0.0690wR2
[a] [I � 2σ(I)] 0.1553 0.1670R1 (all data) 0.0830 0.1107wR2 (all data) 0.1752 0.1905GoF 1.012 1.068Flack parameter 0.03(3) 0.02(3)
[a] R1 = Σ||Fo| – |Fc||/Σ|Fo| for Fo � 2σ(Fo) and wR2 = {Σ[w(Fo2 –
Fc2)2]/Σ[w(Fc
2)]}1/2.
Figure 1. Thermal ellipsoid plots (50%) for the M helical isomerof (R,R)-7. This is designated as molecule 1 in the CIF file and thelabels carry the _1 suffix. Hydrogens are omitted for clarity.
former. On the other hand τ = 0.03 indicates essentially notrigonal-bipyramidal character. Here the basal angles arenearly the same and the geometry is square pyramidal, withthe vanadium well above the plane. The shapes of the twoisomers are shown more clearly by the space-filling repre-sentations in Figure 3. The isomers are only pseudo-mirrorimages since both contain the (1R,2R)-cyclohexyl group.
Selected bond lengths, bond angles, torsion angles andcalculated parameters for (R,R)-7 are presented in Table 3.The vanadium–oxido bond lengths are consistent withthose seen for other VO(salen) complexes.[11] The vana-dium–phenoxido bonds are significantly shorter that the va-nadium–imine bonds, consistent with the anionic nature ofthe phenoxido ligands. The imine groups (C25–N51) have
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5710
Table 3. Selected bond lengths, bond angles, torsion angles and cal-culated parameters for (R,R)-7.[a]
Bond Length [Å] (R,R)-7 (R,R)-7 (R,R)-8 (R,R)-8M isomer P isomer M isomers[b] P isomers[b]
V–Oax[c] 1.581(4) 1.574(4) 1.598[27] 1.590[27]
V–O12 1.930(3) 1.942(3) 1.924[3] 1.932[24]V–O32 1.944(4) 1.935(4) 1.923[10] 1.930[10]V–N51 2.030(4) 2.052(4) 2.051[14] 2.028[20]V–N52 2.038(4) 2.032(4) 2.066[5] 2.053[2]C25/29�N51[d] 1.287(7) 1.261(7) 1.293[3] 1.317[16]C45/49�N52[d] 1.271(7) 1.279(7) 1.290[9] 1.298[18]C51�N51 1.468(5) 1.478(5) 1.495[6] 1.503[33]C52�N52 1.494(6) 1.469(6) 1.485[10] 1.470[1]
Angles [°]
Oa–V–O12 112.84(18) 107.05(18) 109.3[93] 108.0[41]Oax�V–O32 106.58(18) 109.48(17) 108.4[24] 108.3[11]Oax–V–N51 98.7(2) 104.5(2) 106.8[6] 106.4[18]Oax–V–N52 109.89(18) 104.62(18) 105.9[42] 106.2[37]O12–V–O32 90.16(16) 91.07(16) 86.85[23] 87.7[38]O12–V–N51 86.34 (15) 85.40(14) 87.15[7] 87.1[3]O32–V–N52 86.59(16) 86.85(16) 86.8[9] 86.4[4]N51–V–N52 78.25(17) 78.07(16) 78.6[8] 78.8[17]O32–V–N51 153.82(17) 145.37(18) 144.4[84] 145.0[27]O12–V–N52 136.21(16) 147.04(16) 145[13] 145.5[0]V–O12�C12 133.4(3) 130.3(3) 131.8[16] 131.6[19]V–O32�C32 131.1(3) 132.7(3) 130.0[59] 131.6[24]V–N51�C25/29[d] 126.5(3) 122.4(3) 126.4[4] 126.7[15]V–N52�C45/49[d] 126.1(3) 127.1(3) 125.5[11] 125.8[3]N52–C52– 42.4(5) 44.1(5)C51�N51 44.6[1] 41.2[44]arm-arm[e] 32.7 29.4 22.0[55] 21.6[47]curv. A[f] 3.96 7.52 17.4[45] 14.2[5]curv. B[f] 5.54 4.19 22.0[43] 19.9[25]
[a] (R,R)-7: vanadium is V1 for M and V2 for P, (R,R)-8: vanadiumis V1 and V2 for M and V3 and V4 for P. [b] For (R,R)-8 theaverage values (VC1 and VC2 for M, VC3 and VC4 for P) are givenwith the difference between the two values in brackets. [c] Oax isthe axial oxygen (the numbering changes depending on the mole-cule). [d] C25 and C45 for (R,R)-7, C29 and C49 for (R,R)-8. [e]The absolute value of the angle between the mean planes for thephenanthryl or benz[a]anthryl groups: C11-C24 and C31–C44 for(R,R)-7, C11–C28 and C31–C48 for (R,R)-8. [f] Curvature of thephenanthryl group defined as the angle between the first and lastC6 ring planes in the system. Curv. A is for the ring curved towardthe axial oxido group and curv. B is for the ring curved away fromthe axial oxido group.
lengths consistent with C=N bonds. The angles between thesalen donor atoms (D) and the oxido group (Oxido-V–D)are all significantly larger than 90°, consistent with a metalcentre that is sitting well above the basal plane. The N51–V–N52 angle of the five-membered chelate ring in each iso-mer is substantially smaller than 90°, while the N–V–Oangles of the six-membered chelates are (86�1)°. Theangles are similar to what has been seen for other five-coor-dinate complexes of this ligand.[16]
The angle between the averaged planes of the phenan-thryl rings is 32.7° for the M isomer and 29.4° for the Pisomer. These values are intermediate between what wasseen for [MII(L)(py)] (47.9–54.0° M = Zn, Fe) and [FeIII(L)-Cl] (11.2–12.0°).[16] The angles correlate with the strengthof ligand bonding as measured by the M–donor distances.For a given complex, the phenanthryl groups have a smaller

www.eurjic.org FULL PAPER
Figure 2. Thermal ellipsoid plots (50%) for the P helical isomer of(R,R)-7. This is designated as molecule 2 in the CIF file and thelabels carry the _2 suffix. Hydrogens are omitted for clarity.
Figure 3. Space-filling plots (50%) of the diastereomeric helical iso-mers of (R,R)-7.
interplanar angle as their termini are drawn together bystronger ligand binding and they are forced into parallelstacking. Weaker binding, such as seen with ZnII, resultsis reduced steric repulsion and greater freedom for largerinterplanar angles between the phenanthryl groups.
Curvature of fused aromatic systems is common if sig-nificant inter- or intramolecular interactions are present.The phenanthryl groups in (R,R)-7 show modest curvature(4.19–7.52°) as measured by the angle between the calcu-lated planes for the first and third rings in the carbonframework. In the crystal lattice the molecules are arrangedin stacks along the a axis. Figure 4 shows nine pairings,each consisting of columns of M and P isomers that areinterleaved. The relationship between the isomers in one ofthese stacks is shown in Figure 5 which looks along the[111] direction. For a given closest M/P pair the oxidogroups are anti and the cyclohexyl group on one isomer sitsover the open area between the phenanthryl groups of theother.
The molecular structure of (R,R)-8 (Figure 6) with thebenz[a]anthryl sidearm also showed a 1:1 mixture of helicalisomers. In this case there are four independent vanadylcomplexes in the unit cell (VC1–VC4, Figure 7) as well astwo dichloromethanes of solvation. VC1 and VC2 have theM conformation while VC3 and VC4 have the P. Complexesof the same conformation have similar structures. The ge-ometry at the metal centres is approximately square pyrami-
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5711
Figure 4. View down the a cell axis for (R,R)-7. The P isomers havea grey carbon framework and the M isomers have a black carbonframework.
Figure 5. View of one molecular stack viewed along the [111] direc-tion of (R,R)-7. The P isomers (bottom row) have a grey carbonframework and the M isomers (top row) have a black carbonframework.
dal with the vanadium sitting above the plane (M, 0.577and 0.623 Å; P, 0.574 and 0.599 Å). The M isomer showsgreater distortion (τ = 0.18 for molecules 1 and 2) than doesthe P isomer (τ = 0.01, 0.03) as was seen for (R,R)-7. Thebenz[a]anthryl groups are overlapping in each of the iso-mers (Figure 8) but there is no strong π-π interaction sincethe centroid�C distances (3.86–4.30 Å) are outside the nor-mal 3.4–3.6 Å range. The shortest distances are 3.89 Å(VC1, M) and 3.86 Å (VC3, P). The benz[a]anthryl grouprelationships are intermediate between what was seen forthe ZnII and FeII complexes of this ligand.[8]
The structures of (R,R)-7 and (R,R)-8 are similar butwith a few notable differences. On average, the bonds tovanadium in (R,R)-8 are longer for V–Oax (+0.017 Å) andV–N (+0.012 Å) while the V–O (phenoxido) lengths areshorter (–0.011 Å). This latter difference is somewhat sur-prising, given the more sterically-demanding nature of theextended arms in (R,R)-8 and this points to stronger phen-

www.eurjic.org FULL PAPER
Figure 6. Thermal ellipsoid plot (50 % probability) of the M isomerof (R,R)-8 (VC1).
Figure 7. Unit cell of (R,R)-8 (P1) showing the four independentvanadyl complexes. The dichloromethanes of solvation have beenremoved for clarity.
Figure 8. Space-filling plots (50%) of the diastereomeric helical iso-mers of (R,R)-8: M = VC1, P = VC3.
oxido donors for the benz[a]anthryl arms. The angle be-tween the aromatic arms is smaller for (R,R)-8 than for(R,R)-7 by an average of 9.3°. This reflects the fact that thelonger arms are forced in to a more parallel arrangementin order to avoid strong edge to face repulsions. The curva-
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5712
ture of the aromatic system is larger by 13° on average forthe benz[a]anthryl arms: the range is 14.0 to 24.2° as mea-sured by the angle between the calculated planes for thefirst and fourth rings in the carbon framework. This is con-sistent with a longer and more flexible aromatic system thathas significant steric repulsions on one face.
IR Spectra
The coordination environment of the VIVO complexes isnormally reflected ny the V=O stretching frequency,νV=O.[12] For octahedral complexes with similar tetradentateONNO ligands, the stretching frequency is lower due to theexpected weakening of the V=O bond by the trans axialligand. The V=O stretch for octahedral complexes generallycomes in the 970 to 1000 cm–1 range.[13] It has been seenthat more electron-withdrawing ligands lead to VIVO com-plexes with significantly lower νV=O values due to···V=O···V=O··· interactions that weaken the V=O bond. Aseries of VV=O(salen)X complexes showed νV=O in a nar-row 960–990 cm–1 range.[5]
For complexes (R,R)-7 and (R,R)-8, the V=O stretch isobserved at 1036.8 and 1035.7 cm–1, respectively (Table 4).These are characteristic of green-coloured monomericsquare-pyramidal VIVO(salen) complexes studied pre-viously.[14] The νV=O values of the binaphthyl complexes(R)-9 and (R)-10 are lower than seen for the cyclohexyl-containing complexes (Table 4). These lower frequencies areconsistent with the more electron-withdrawing nature of thebinaphthyl unit and suggest that some ···V=O···V=O··· as-sociation may be present in these complexes.[13]
Table 4. IR stretching frequencies and bond lengths of V=O andC=N groups in complexes.[a]
νV=O / cm–1 rV=O / Å νC=N / cm–1 rC=N / Å
(R,R)-7 1036.8 1.581(4) 1608.0 1.271 (7)(R,R)-8 1035.7 1.584(5) 1609.5 1.291(9)(R)-9 1016.0 1613.0(R)-10 1008.5 1606.0
[a] Crystallographic bond lengths are not available for (R)-9 and(R)-10.
EPR Spectra
The X-band EPR spectrum of (R,R)-7 was recorded atroom temperature in dichloromethane. A broad spectrumwith g = 2.0, typical of a paramagnetic d1 configuration, isobserved (Figure 9). The broadness is likely due to the rapidinterconversion between the M and the P isomers. For com-plex (R)-9, the X-band EPR spectrum of the V1VO complexwas collected in DMSO at 77 K (Figure 10) and the spec-trum simulated using software from Rockenbauer and Ko-recz.[15] The system was simulated by considering the mole-cule as having octahedral geometry where the fifth coordi-nation site is occupied by the axial oxygen and the sixthcoordination site by the DMSO. For the V1VO system, weused the additivity rule to estimate the hyperfine couplingconstant A�
est, based on the contributions of A�,i of each of
www.eurjic.org FULL PAPER
the four equatorial donor groups [A�est = ΣA�,i (i = 1 to 4)].
The estimated accuracy of the A�est is�3 �10–4 cm–1. The
data can be used to establish the most probable bindingmode of the V1V complex. The influence of the axial ligandwas not taken into account, since it is not relevant in thepresent system. Table 5 shows the comparison between thesimulated and the experimental data. The complex was en-visioned as being in an octahedral environment with
Figure 9. X-band EPR spectrum of (R,R)-7 in dichloromethane atroom temperature.
Figure 10. X-band first derivative EPR spectrum of (R)-9 inDMSO at 77 K.
Table 5. Spin-Hamiltonian parameters calculated for the EPR spec-trum of (R)-9 in DMSO.
g� A� � 104 cm–1 g� A� � 104 cm–1 Binding mode(A�
est � 104 cm–1)
1.97 160 1.97 174 α-cis (165)β-cis (174.3)trans (160)
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5713
DMSO occupying the sixth coordination site. Among thethree possible geometric isomers, complex (R)-9 shows thebest match with the β-cis geometry.
Solution Property Studies: Electronic and CD Spectra forComplexes (R,R)-7 and (R,R)-8
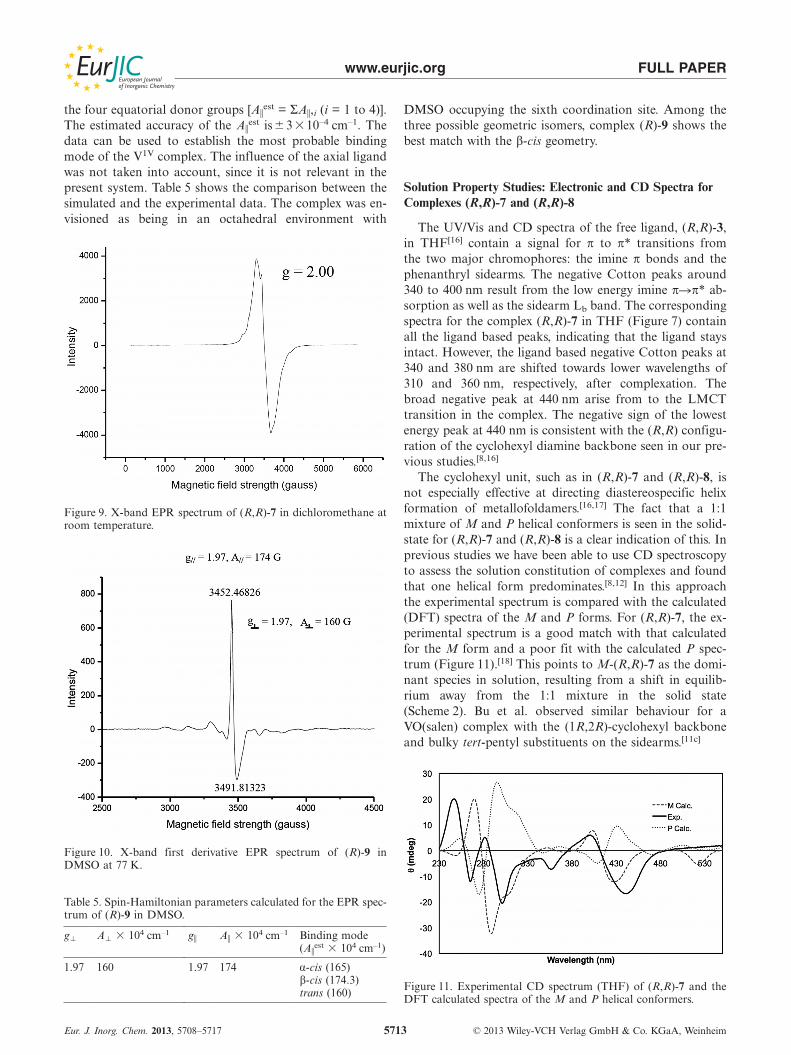
The UV/Vis and CD spectra of the free ligand, (R,R)-3,in THF[16] contain a signal for π to π* transitions fromthe two major chromophores: the imine π bonds and thephenanthryl sidearms. The negative Cotton peaks around340 to 400 nm result from the low energy imine π�π* ab-sorption as well as the sidearm Lb band. The correspondingspectra for the complex (R,R)-7 in THF (Figure 7) containall the ligand based peaks, indicating that the ligand staysintact. However, the ligand based negative Cotton peaks at340 and 380 nm are shifted towards lower wavelengths of310 and 360 nm, respectively, after complexation. Thebroad negative peak at 440 nm arise from to the LMCTtransition in the complex. The negative sign of the lowestenergy peak at 440 nm is consistent with the (R,R) configu-ration of the cyclohexyl diamine backbone seen in our pre-vious studies.[8,16]
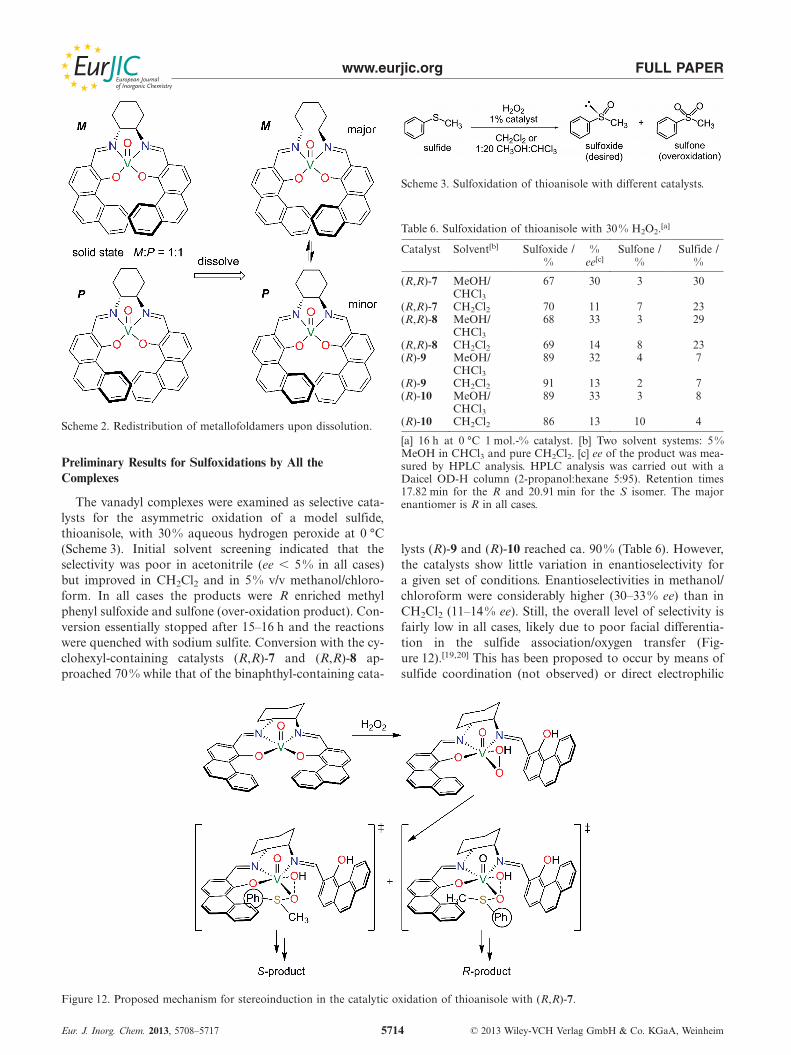
The cyclohexyl unit, such as in (R,R)-7 and (R,R)-8, isnot especially effective at directing diastereospecific helixformation of metallofoldamers.[16,17] The fact that a 1:1mixture of M and P helical conformers is seen in the solid-state for (R,R)-7 and (R,R)-8 is a clear indication of this. Inprevious studies we have been able to use CD spectroscopyto assess the solution constitution of complexes and foundthat one helical form predominates.[8,12] In this approachthe experimental spectrum is compared with the calculated(DFT) spectra of the M and P forms. For (R,R)-7, the ex-perimental spectrum is a good match with that calculatedfor the M form and a poor fit with the calculated P spec-trum (Figure 11).[18] This points to M-(R,R)-7 as the domi-nant species in solution, resulting from a shift in equilib-rium away from the 1:1 mixture in the solid state(Scheme 2). Bu et al. observed similar behaviour for aVO(salen) complex with the (1R,2R)-cyclohexyl backboneand bulky tert-pentyl substituents on the sidearms.[11c]
Figure 11. Experimental CD spectrum (THF) of (R,R)-7 and theDFT calculated spectra of the M and P helical conformers.
www.eurjic.org FULL PAPER
Scheme 2. Redistribution of metallofoldamers upon dissolution.
Preliminary Results for Sulfoxidations by All theComplexes
The vanadyl complexes were examined as selective cata-lysts for the asymmetric oxidation of a model sulfide,thioanisole, with 30% aqueous hydrogen peroxide at 0 °C(Scheme 3). Initial solvent screening indicated that theselectivity was poor in acetonitrile (ee � 5 % in all cases)but improved in CH2Cl2 and in 5% v/v methanol/chloro-form. In all cases the products were R enriched methylphenyl sulfoxide and sulfone (over-oxidation product). Con-version essentially stopped after 15–16 h and the reactionswere quenched with sodium sulfite. Conversion with the cy-clohexyl-containing catalysts (R,R)-7 and (R,R)-8 ap-proached 70 % while that of the binaphthyl-containing cata-
Figure 12. Proposed mechanism for stereoinduction in the catalytic oxidation of thioanisole with (R,R)-7.
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5714
Scheme 3. Sulfoxidation of thioanisole with different catalysts.
Table 6. Sulfoxidation of thioanisole with 30% H2O2.[a]
Catalyst Solvent[b] Sulfoxide / % Sulfone / Sulfide /% ee[c] % %
(R,R)-7 MeOH/ 67 30 3 30CHCl3
(R,R)-7 CH2Cl2 70 11 7 23(R,R)-8 MeOH/ 68 33 3 29
CHCl3(R,R)-8 CH2Cl2 69 14 8 23(R)-9 MeOH/ 89 32 4 7
CHCl3(R)-9 CH2Cl2 91 13 2 7(R)-10 MeOH/ 89 33 3 8
CHCl3(R)-10 CH2Cl2 86 13 10 4
[a] 16 h at 0 °C 1 mol.-% catalyst. [b] Two solvent systems: 5%MeOH in CHCl3 and pure CH2Cl2. [c] ee of the product was mea-sured by HPLC analysis. HPLC analysis was carried out with aDaicel OD-H column (2-propanol:hexane 5:95). Retention times17.82 min for the R and 20.91 min for the S isomer. The majorenantiomer is R in all cases.
lysts (R)-9 and (R)-10 reached ca. 90% (Table 6). However,the catalysts show little variation in enantioselectivity fora given set of conditions. Enantioselectivities in methanol/chloroform were considerably higher (30–33% ee) than inCH2Cl2 (11–14 % ee). Still, the overall level of selectivity isfairly low in all cases, likely due to poor facial differentia-tion in the sulfide association/oxygen transfer (Fig-ure 12).[19,20] This has been proposed to occur by means ofsulfide coordination (not observed) or direct electrophilic

www.eurjic.org FULL PAPER
attack of the hydroperoxido group on the sulfur and thisproposal is supported by computational results.[21]
A key aspect of the proposed mechanism is the proton-ation and decoordination of a phenoxo donor of the VO-(salen) complex upon reaction with hydrogen peroxide. Thisbreaks the helical conformation of the complex and likelyreduces any impact the helical structure has on the selectiv-ity of the catalyst. The observed enantioselectivities are notsignificantly better than those with simpler vanadyl salencatalysts or catalysts with related tridentate Schiff-base li-gands.[4,5]
Conclusions
Helical vanadyl salen complexes with the (1R,2R)-cyclo-hexyl chiral unit do not have a strong thermodynamic pref-erence for the M helical conformer over the P conformer,as evidenced by the 1:1 ratio of diastereomers seen in thesolid-state structures. This is consistent with previous re-sults from our group and others. However, solution CDstudies suggest that the M conformer is favoured in solu-tion, at least in the case of (R,R)-7. This behaviour was alsoseen for ZnII, FeII and FeIII complexes of these ligands andhas been discussed recently by Akine et al.[22] Complexes(R)-9 and (R)-10, which are based on the binaphthyl chiralunit, are almost certainly M-helical, consistent with ourprevious findings. Despite this, the stereoselectivity of thesecatalysts for thioanisole oxidation with H2O2 are not signif-icantly better than for the catalysts with the cyclohexyl unit.This implies that it is the poor facial selectivity of the cata-lysts, which is similar in all cases, rather than the conforma-tional flexibility that leads to poor enantioselectivity. Sol-vent polarity and the presence of protic solvents have alarge impact on the catalytic outcome as well, although thisis not surprising given the hydroperoxido species present inthe reaction pathway, which can participate in hydrogen-bonding.
Experimental SectionGeneral: All reactions were carried out under inert atmospheresunless otherwise noted. Tetrahydrofuran and dichloromethane weredried with calcium hydride, ethanol was dried with magnesium,and chloroform was dried with 4 Å molecular sieves. Solvents weredegassed and vacuum transferred into storage flasks prior to use.The ligands were synthesised by previously reported methods.[8,16]
Elemental analyses were carried out by Desert Analytics of Tucson,Arizona. UV/Vis spectra were obtained on a Varian Cary 500 spec-trometer, and CD spectra (5 scans, 1.00 mm path length quartz cell)on JASCO 720 or J-815 spectropolarimeters. Solution samples forthese two techniques were prepared using dried spectroscopic gradeTHF at concentrations that ranged between 1.5 and 2.5 �10–5 m.1H and 13C NMR spectra were obtained on a Varian Unity400 MHz spectrometer employing residual solvent protons or TMSas an internal standard. Crystallographic data were collected usingeither a Bruker SMART 1000 CCD or a Bruker-AXS SMARTAPEX CCD diffractometer. Gas chromatographs were collected ona Varian 3900 instrument with a 39XL FID detector and a Supelco
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5715
Beta Dex 120 30 m �0.25 mm capillary column. HPLC was carriedout using a Gilson 322 Pump and a Hewlett–Packard 1100 UVdetector with a standard flowcell. A Daicel Chiracel OD-H columnwas used with 1.0 mLmin–1 of 5% 2-propanol in hexanes as eluent.EPR spectra of the vanadium complexes were collected by using aVarian E-4 EPR spectrometer. FTIR spectra were collected onpowders with an Agilent Cary 630 spectrometer with a diamondATR attachment. Some spectra were also collected using KBr pel-lets and a Nicolet 6700 spectrometer.
High-resolution electrospray ionisation spectra were acquired ona LCT Premier (Waters Corp., Milford MA) time of flight massspectrometer. The instrument was operated at 10000 resolution (Wmode) with dynamic range enhancement that attenuates large in-tensity signals. The cone voltage was 60 eV. Spectra were acquiredat a 16666 Hz pusher frequency covering the mass range 100 to1200 u and accumulating data for 2 seconds per cycle. Mass correc-tion for exact mass determinations was made automatically withthe lock mass feature in the MassLynx data system. A referencecompound in an auxiliary sprayer can be sampled every third cycleby toggling a “shutter” between the analysis and reference needles.The reference mass is used for a linear mass correction of the ana-lytical cycles. Samples were diluted in dichloromethane and deliv-ered to the instrument in MeOH 0.1 % formic acid through directinfusion. Lower resolution mass spectra were collected using aBruker Ultraflex MALDI-TOF/TOF instrument.
The Amsterdam Density Functional (ADF) program was used forall DFT calculations.[23] The Slater type full core basis sets em-ployed are of polarised triple-zeta (TZP) quality. The asymptoti-cally correct statistical average of orbital potentials (SAOP)[24,25]
was utilized in time-dependent density functional theory (TDDFT)computations to determine energetics and oscillator strengths ofexcited states. Rotatory strengths were also computed using theRESPONSE module in ADF.[26] The lowest 150 singlet excitedstates were evaluated. The circular dichroism spectra were fit witha scaled Gaussian function with a width at half-maximum of20 nm. When compared with experimental data, it is evident thatthe energies are overestimated by an average of ca. 0.1 eV, as hasbeen noted previously for these functionals.[27] In view of the over-estimation, a 0.1 eV correction was applied to the calculated ener-gies.
(R,R)-7: Ligand (R,R)-3 (0.100 g, 0.192 mmol) and sodium meth-oxide (0.103 g, 1.92 mmol) were dissolved into a mixture (2:1 ratio)of dichloromethane (10.0 mL) and ethanol (5.0 mL). Oxidovanadi-um(IV) acetylacetonate (0.0520 g, 0.192 mmol) was added to themixture and the solution was stirred for 16 h at room temperature.The green precipitate was isolated by filtration, washed with dichlo-romethane (5 mL) and dried in vacuo to yield (R,R)-7 (0.100 g,89% yield). Single-crystals suitable for X-ray analysis were grownby slow liquid-liquid diffusion: hexanes were layered over a solu-tion of (R,R)-7 in dichloromethane in a sealed tube. IR ν̃ = 1032.45(V=O) cm–1. ESI-TOF-MS: M+ (C36H28N2O3V)+1 587.1567 (δ =4.6 ppm), [M + H]+, (C36H29N2O3V)+1 588.1624 (δ = 1.0 ppm),[M2]+ (C72H54N4O6V2)+1 1174.3140 (δ = 5.2 ppm). C36H28N2O3V(587.57): calcd. C 63.54, H 3.79, N 4.01; found C 63.55, H 3.78, N4.00.
(R,R)-8: Ligand (R,R)-4 (0.600 g, 0.965 mmol) and sodium meth-oxide (0.521 g, 9.65 mmol) were dissolved into a mixture (2:1 ratio)of dichloromethane (20.0 mL) and ethanol (10.0 mL). The solidswere dissolved by stirring. Oxidovanadium(IV) acetylacetonate(0.261 g, 0.965 mmol) was added to the mixture and the solutionwas stirred for 16 h at room temperature. The green precipitate wasisolated by filtration and purified by Soxhlet extraction with dichlo-

www.eurjic.org FULL PAPER
romethane. The solvent was removed by vacuum transfer and theresultant solid was dried in vacuo to give (R,R)-8 (0.49 g, 81%yield). ESI-TOF-MS: M+ (C44H32O3N2V)+1 687.1856 (δ =0.4 ppm), [M + H]+ (C44H33O3N2V)+1 688.1919 (δ = 1.7 ppm),[M2]+ (C88H64O4N6V2)+1 1374.3749 (δ = 3.2 ppm). IR ν̃ = 1008.50(V=O), 1606.50 (C=C) cm–1. UV (in THF) shows a signal forLMCT at 350 nm. The CD spectrum (in THF at 10–5 m) shows thepresence of the M helix in the solution. X-ray quality crystals weregrown from the diffusion method from dichloromethane and hex-ane. C44H32N2O3V (687.69): calcd. C 76.85, H 4.69, N 4.07; foundC 76.82, H 4.70, N 4.70.
(R)-9: Ligand (R)-5 (0.100 g, 0.145 mmol) and sodium methoxide(0.086 g, 1.60 mmol) were dissolved into a mixture (2:1 ratio) ofdichloromethane (10.0 mL) and ethanol (5.0 mL). Oxidovanadi-um(IV) acetylacetonate (0.039 g, 0.145 mmol) was added to themixture and the solution was stirred for 16 h at room temperature.The brown precipitate was isolated by filtration and purified bySoxhlet extraction with dichloromethane. The solvent wasremoved by vacuum transfer and the resultant solid was dried invacuo to give (R)-9 (0.089 g, 91% yield). ESI-TOF-MS: M+
(C50H30N2O3V)+1 757.1611 (δ = 11.2 ppm), [M + H]+
(C50H31N2O3V)+1 758.2069 (δ = 38.9 ppm). IR ν̃ = 1016.03 (V=O),1573.28 (C=C) cm–1). C50H30N2O3V (757.74): calcd. C 79.09, H3.99, N 3.80; found C 79.01, H 3.91, N 3.70.
(R)-10: Ligand (R)-6 (0.650 g, 0.821 mmol) and sodium methoxide(0.488 g, 9.03 mmol) were dissolved in a mixture (2:1 ratio) ofdichloromethane (20.0 mL) and ethanol (10.0 mL). Oxidovanadi-um(IV) acetylacetonate (0.222 g, 0.821 mmol) was added to themixture and the solution was stirred for 16 h at room temperature.The brown precipitate was isolated by filtration and purified bySoxhlet extraction with dichloromethane. The solvent was removedby vacuum transfer and the resultant solid was dried in vacuo togive (R, R)-10 (0.480 g, 80% yield). ESI-TOF-MS: M+
(C58H34N2O3V)+1 857.2039 (δ = 3.5 ppm). IR ν̃ = 1035.71(V=O) cm–1. C50H30N2O3V (757.74): calcd. C 81.21, H 3.99, N3.27; found C 80.91, H 4.02, N 3.31.
General Procedure for Sulfoxidations: 1 mol.-% of catalyst was dis-solved in dichloromethane (3.0 mL) in a two neck flask under ar-gon and stirred for 10 min. Thioanisole (0.591 mL, 5.00 mmol) wasadded and the solution was stirred for 15 min. The reaction wascooled to 0.0 °C in a circulating immersion bath fitted with a mag-netic stirrer. Hydrogen peroxide (30% in H2O, 0.187 mL,5.50 mmol) was added by means of a syringe pump over a 2 hperiod. The mixture was stirred for an additional 16 h. Saturatedaqueous sodium sulfite (5 mL) was added to quench the reaction.The organic components were extracted with dichloromethane,dried with anhydrous sodium sulfate and filtered. The solvent wasremoved in vacuo and the crude product was analysed for composi-tion by using 1H NMR spectroscopy and GC. The crude productmixture was purified by flash column chromatography in 1:3 ethylacetate:hexane. The isolated sulfoxide was analysed by 1H NMR(CDCl3, 400 MHz): 3.72 (s, 3 H, CH3), 7.5 (m, 3 H, CH), 7.6 (m,2 H, CH). The enantiomeric excess of the phenyl methyl sulfoxidewas determined by HPLC analysis. The R and S enantiomers eluteat 17.82 and 20.91 min, respectively, under the conditions used.
CCDC-954054 and -954055 contain the supplementary crystallo-graphic data for this paper. These data can be obtained free ofcharge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
Supporting Information (see footnote on the first page of this arti-cle): MS, IR, UV and CD spectra, and DFT simulated CD spectra.
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5716
Acknowledgments
This work is supported by the US National Science Foundation(NSF) under grant number CHE-0349258 and by Kansas StateUniversity. J. Tomich is thanked for use of the CD spectroscopyfacilities, L. Maurmann for assistance with NMR and HPLCanalyses and A. Borovik for the EPR spectra. The authors alsowish to thank Dr. Todd D. Williams, Mr. Robert Drake and Mr.Larry Seib of the University of Kansas mass spectrometry labora-tory for their efforts in acquiring the ESI spectra. The Waters LCTPremier mass spectrometer was purchased with support from theNational Institutes of Health (NIH) Shared Instrumentation GrantProgram (SIG S10 RR019398) (T. D. W.).
[1] R. A. Sheldon, F. C. W. I. Arends, D. V. F. Velde, Top. Catal.2000, 13, 259–265.
[2] G.-Q. Lin, X.-W. Sun, in: Chiral Drugs: Chemistry and Bio-logical Action (Eds: G.-Q. Lin, Q.-D. You, J.-F. Cheng), JohnWiley & Sons, Hoboken, 2011, p. 29–76.
[3] a) T. Katsuki, in: Science of Synthesis Water in Organic Synthe-sis (Ed.: S. Kobayashi), Thieme, New York, 2012, p. 52–93; b)G. E. O’Mahony, A. Ford, A. R. Maguire, J. Sulfur Chem.2013, 34, 301–341; c) K. P. Bryliakov, E. P. Talsi, Curr. Org.Chem. 2012, 16, 1215–1242.
[4] a) T. Kurahashi, H. Fuji, Inorg. Chem. 2008, 47, 7556–7567; b)L. Canali, D. C. Sherrington, Chem. Soc. Rev. 1999, 28, 85–93; c) T. Katsuki, Chem. Soc. Rev. 2004, 33, 437–444; d) G.Romanowski, M. Wera, Polyhedron 2010, 29, 2747–2754; e) E.Kwiatkowski, G. Romanowski, W. Nowicki, M. Kwiatkowski,K. Suwinska, Polyhedron 2007, 26, 2559–2568; f) P. Adão, J.Costa Pessoa, R. T. Henriques, M. L. Kuznetsov, F. Avecilla,M. R. Maurya, U. Kumar, I. Correia, Inorg. Chem. 2009, 48,3542–3561; g) P. Adão, M. L. Kuznetsov, S. Barroso, A. M.Martins, F. Avecilla, J. Costa Pessoa, Inorg. Chem. 2012, 51,11430–11449.
[5] K. Nakajima, K. Kojima, M. Kojima, J. Fujita, Bull. Chem.Soc. Jpn. 1990, 63, 2620–2630.
[6] K. Bolm, Coord. Chem. Rev. 2003, 237, 245–256.[7] K. Matsumoto, B. Saito, T. Katsuki, Chem. Commun. 2007,
3619–3627.[8] a) A. V. Wiznycia, J. Desper, C. J. Levy, Chem. Commun. 2005,
4693–4695; b) A. V. Wiznycia, J. Desper, C. J. Levy, Inorg.Chem. 2006, 45, 10034–10036; c) A. V. Wiznycia, J. Desper,C. J. Levy, Dalton Trans. 2007, 1529–1527.
[9] a) D. M. Boghaei, A. Bezaatpour, M. Behzad, J. Coord. Chem.2007, 60, 973–983; b) D. E. Hamilton, Inorg. Chem. 1991, 30,1670–1671; c) M. Pasquali, F. Marchetti, C. Floriani, S. Mer-lino, J. Chem. Soc., Dalton Trans. 1977, 139–144; d) J. Selbin,L. H. Holmes Jr., S. P. McGlynn, J. Inorg. Nucl. Chem. 1963,25, 1359–1369.
[10] A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn, G. C. Ver-schoor, J. Chem. Soc., Dalton Trans. 1984, 1349–1356.
[11] a) E. Carter, I. A. Fallis, B. M. Kariuki, I. R. Morgan, D. M.Murphy, T. Tatchell, S. van Doorslaer, E. Vinck, Dalton Trans.2011, 40, 7454–7462; b) A. Horn Jr., C. A. L. Filgueiras, J. L.Wardell, M. H. Herbst, N. V. Vugman, P. S. Santos, J. G. S.Lopes, R. A. Howie, Inorg. Chim. Acta 2004, 357, 4240–4246;c) S. Liang, D. Van Derveer, S. Y. Qian, B. Sturgeon, X. R. Bu,Polyhedron 2002, 21, 2021–2025; d) P. Plitt, H. Pritzkow, T.Oeser, R. Kraemer, J. Inorg. Biochem. 2005, 99, 1230–1237; e)C. Wang, J.-H. Yuan, G. Xie, M.-J. Yu, J. Li, Acta Crystallogr.,Sect. E 2008, 64, m775–m776; f) P. Adao, M. R. Maurya, U.Kumar, F. Avecilla, R. T. Henriques, M. L. Kusnetsov, J. C.Pessoa, I. Correia, Pure Appl. Chem. 2009, 81, 1279–1296.
[12] L. V. Boas, J. C. Pessoa, in: Comprehensive Coordination Chem-istry (Ed.: G. Wilkinson), Pergamon, Oxford, UK, 1987, vol.3, p. 531–544.
[13] D. M. Boghaei, S. Mohebi, J. Mol. Catal. A 2002, 179, 41–51.

www.eurjic.org FULL PAPER
[14] D. L. Daughdrill, D. F. Martin, J. S. Binford Jr., J. Inorg. Nucl.Chem. 1970, 32, 2885–2890.
[15] A. Rockenbauer, L. Korecz, Appl. Magn. Reson. 1996, 10, 29–43.
[16] A. V. Wiznycia, J. Desper, C. J. Levy, Can. J. Chem. 2009, 87,224–231.
[17] a) Z. Dong, S. Bai, G. P. A. Yap, J. M. Fox, Chem. Commun.2011, 47, 3781–3783; b) L. A. Fisher, F. Zhang, G. P. A. Yap,J. M. Fox, Inorg. Chim. Acta 2010, 364, 259–260; c) Z. Dong,J. N. Plampin III, G. P. A. Yap, J. M. Fox, Org. Lett. 2010, 12,4002–4005; d) Z. Dong, G. P. A. Yap, J. M. Fox, J. Am. Chem.Soc. 2007, 129, 11850–11853; e) F. Zhang, S. Bai, G. P. A. Yap,V. Tarwade, J. M. Fox, J. Am. Chem. Soc. 2005, 127, 10590–10599.
[18] Comparisons of the CD spectrum of (R,R)-8 with DFT-calcu-lated spectra were inconclusive.
[19] H. Egami, T. Katsuki, J. Am. Chem. Soc. 2007, 129, 8940–8941.[20] A. Pasini, M. Gullotti, R. Ugo, J. Chem. Soc., Dalton Trans.
1977, 346–356.[21] a) C. J. Schneider, J. E. Penner-Hahn, V. L. Pecoraro, J. Am.
Chem. Soc. 2008, 130, 2712–2713; b) T. S. Smith II, V. L. Pecor-
Eur. J. Inorg. Chem. 2013, 5708–5717 © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5717
aro, Inorg. Chem. 2002, 41, 6754–6760; c) J. Y. Kravitz, V. L.Pecoraro, H. A. Carlson, J. Chem. Theory Comput. 2005, 1,1265–1274.
[22] S. Akine, S. Hotate, T. Matsumoto, T. Nabeshima, Chem. Com-mun. 2011, 47, 2925–2927.
[23] G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fon-seca Guerra, S. J. A. Van Gisbergen, G. J. Snijders, T. Ziegler,J. Comput. Chem. 2001, 22, 931–967.
[24] V. O. Gritsenko, T. R. P. Schipper, E. J. Baerends, Chem. Phys.Lett. 1999, 302, 199–207.
[25] T. R. P. Schipper, V. O. Gritsenko, A. J. S. Van Gisbergen, J. E.Baerends, J. Chem. Phys. 2000, 112, 1344.
[26] J. Autschbach, T. Ziegler, S. J. A. van Gisbergen, E. J. Baerends,J. Chem. Phys. 2002, 116, 6930–6940.
[27] A. Rosa, G. Ricciardi, O. Gritsenko, E. J. Baerends, Principlesand Applications of Density Functional Theory in InorganicChemistry I (Eds.: N. Kaltsoyannis, J. E. McGrady), Springer,Berlin, 2004, 112, p. 49.
Received: May 16, 2013Published Online: September 25, 2013





























