Embed Size (px)
Citation preview

EMBRYONIC STEM CELLS/INDUCED PLURIPOTENT STEM CELLS
Human Embryonic Stem Cells Are Capable of Executing G1/S
Checkpoint Activation
TOMAS BARTA,a,b
VLADIMIR VINARSKY,a,b
ZUZANA HOLUBCOVA,aDASA DOLEZALOVA,
a,bJAN VERNER,
c
SARKA POSPISILOVA,c PETR DVORAK,a,b ALES HAMPLa,b
aDepartment of Biology, Faculty of Medicine, Masaryk University, Brno, Czech Republic; bDepartment of
Molecular Embryology, Institute of Experimental Medicine, Academy of Sciences of the Czech Republic, Brno,
Czech Republic; cCenter of Molecular Biology and Gene Therapy, Department of Internal Medicine–
Hematooncology, Faculty of Medicine and University Hospital Brno, Czech Republic
Key Words. Human embryonic stem cells • DNA damage • Checkpoint activation • UVC • Cdc25A • p53
ABSTRACT
Embryonic stem cells progress very rapidly through thecell cycle, allowing limited time for cell cycle regulatory
circuits that typically function in somatic cells. Mecha-nisms that inhibit cell cycle progression upon DNA dam-
age are of particular importance, as their malfunctionmay contribute to the genetic instability observed inhuman embryonic stem cells (hESCs). In this study, we
exposed undifferentiated hESCs to DNA-damaging ultra-violet radiation-C range (UVC) light and examined their
progression through the G1/S transition. We show thathESCs irradiated in G1 phase undergo cell cycle arrest
before DNA synthesis and exhibit decreased cyclin-de-pendent kinase two (CDK2) activity. We also show that
the phosphatase Cdc25A, which directly activates CDK2,is downregulated in irradiated hESCs through the action
of the checkpoint kinases Chk1 and/or Chk2. Importantly,the classical effector of the p53-mediated pathway, proteinp21, is not a regulator of G1/S progression in hESCs.
Taken together, our data demonstrate that cultured undif-ferentiated hESCs are capable of preventing entry into S-
phase by activating the G1/S checkpoint upon damage totheir genetic complement. STEM CELLS 2010;28:1143–1152
Disclosure of potential conflicts of interest is found at the end of this article.
INTRODUCTION
In the past decade, pluripotent stem cells of both embryonicand adult origin have emerged as a promising tool for variousbiomedical applications, particularly cell replacement therapy.For most therapeutic purposes, in vitro amplification of stemcells will be necessary. Specifically for embryonic stem cells,immortal cell lines derived from the inner cell mass of blasto-cysts, propagation in culture is an inevitable part of their routinemanipulation. However, a growing body of evidence indicatesthat in vitro culture of human embryonic stem cells (hESCs)results in the accumulation of multiple changes to their genome[1, 2]. Although the biological consequences of these changesare largely unknown [3, 4], understanding how and why theyoccur is necessary to eliminate risk factors, which may dependon the cell type or cell line, or on culture conditions.
To prevent DNA mutations, such as point mutations,translocations, and aneuploidy, cells have developed a com-plex network of checkpoints at strategic points in the cellcycle to monitor various aspects of chromosomal metabolism.
Upon DNA damage, checkpoint pathways are activated andhalt the cell cycle to allow the cell to repair its genetic mate-rial. The G1/S checkpoint ensures DNA integrity and is gov-erned by two key pathways, which both converge uponcyclin-dependent kinase two (CDK2). However, these path-ways differ in their speed of execution, due to differences intheir molecular components. One pathway involves p53 stabi-lization followed by increased transcription and translation ofthe CDK inhibitor p21, and mediates a slow response. In con-trast, degradation of the CDK2-activating phosphataseCdc25A mediates a rapid and transient response to DNAdamage [5]. The kinases ataxia telangiectasia mutated (ATM),ataxia telangiectasia and Rad3 related (ATR), Chk1, andChk2, quickly and directly transduce signals from damagedDNA by phosphorylating Cdc25 family members, such asCdc25A. Under normal growth conditions, Cdc25A removesinhibitory phosphates from Thr14 and Tyr15 of CDK2, whichforms a complex with cyclin E [6]. When Cdc25A is phos-phorylated and subsequently undergoes proteasomal degrada-tion [7], the activity of CDK2 decreases, and the cell cyclearrests before DNA synthesis is initiated.
Author contributions: T.B.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing;V.V.: collection and assembly of data; Z.H.: collection and assembly of data; D.D.: collection and assembly of data; J.V.: collection andassembly of data; S.P.: data analysis and interpretation; P.D.: data analysis and interpretation; A.H.: conception and design, data analysisand interpretation, manuscript writing, final approval of manuscript.
Correspondence: Ales Hampl, D.V.M., Ph.D., Department of Biology, Faculty of Medicine, Masaryk University, Kamenice 5, 62500Brno, Czech Republic. Telephone: þ420-54949-3514; Fax: þ420-54949-8001; e-mail: [email protected] Received March 8, 2010;accepted for publication May 13, 2010; first published online in STEM CELLS EXPRESS June 1, 2010; available online without subscriptionthrough the open access option. VC AlphaMed Press 1066-5099/2009/$30.00/0 doi: 10.1002/stem.451
STEM CELLS 2010;28:1143–1152 www.StemCells.com

Maintaining the integrity of the genome is important forall cells, but it is especially crucial for stem cells, as muta-tions in their DNA can propagate throughout the tissue viatransmission to differentiated progeny. Surprisingly, severalstudies have recently reported that hESCs fail to fully executethe G1/S checkpoint, which may account for the fast mitogen-independent (‘‘automated’’) progression of hESCs through G1[8–10].
Given the critical importance of hESCs genetic stability,we further investigated the functional and molecular mecha-nisms of the DNA damage response in these cells. Usingsynchronized populations of hESCs, we show that these cellsprevent progression from G1 to S-phase in response to DNAdamage and demonstrate the involvement of Cdc25A, Chk1,and Chk2 in this process.
MATERIALS AND METHODS
Cell Culture and Chemical Treatments
Two independent hESC lines, CCTL12 and CCTL14, were usedthroughout this study [11]. Cells were maintained on mitoticallyinactivated mouse embryonic fibroblasts (MEFs) in culture mediaconsisting of Dulbecco’s modified Eagle medium (DMEM)/F12supplemented with 15% knockout serum replacement (KSR), L-Glutamine, MEM nonessential amino acids, 0.5% penicillin-strep-tomycin, (Invitrogen, Carlsbad, CA), b-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, www.sigmaaldrich.com) and 10 ng/mlFGF-2 (PeproTech, Rocky Hill, NJ, www.peprotech.com). Chem-ical inhibitors of Chk1 (UCN01) (Sigma-Aldrich), Chk2 (Chk2inhibitor II) (Calbiochem, San Diego, CA, http://www.merck-chemicals.com), and p38 (SB202190) (Sigma-Aldrich) were pre-pared as �1,000 stocks in DMSO and were added directly intothe culture media. DMSO (0.1%) was used as a vehicle control.hESC colonies were harvested by manual scraping to avoid con-tamination by MEFs.
Western Blot Analysis
Cells were washed three times with phosphate buffer solution(PBS) (pH 7.4) and lysed in 100 mM Tris-HCl (pH 6.8) contain-ing 20% glycerol and 1% SDS. Protein concentrations were deter-mined using the DC Protein Assay Kit (Bio-Rad, Hercules, CA,www.bio-rad.com). Lysates were supplemented with bromophenolblue (0.01%) and b-mercaptoethanol (1%). Equal amounts oftotal protein were separated by SDS-polyacrylamide gel electro-phoresis (PAGE), electrotransferred onto polyvinylidene flouride(PVDF) membrane (Millipore, Billerica, MA, www.millipore.com), immunodetected using appropriate primary and secondaryantibodies, and visualized by ECL Plus reagent (GE HealthcareLife Sciences, Little Chalfont, U.K., www.gelifesciences.com)according to the manufacturer’s instructions. The intensity ofbands was quantified by ImageJ software (http://rsbweb.nih.gov/ij/). Primary antibodies used were as follows: mouse monoclonalantibodies against Cdc25A (sc-7389) and Chk2 (sc-5278), andrabbit polyclonal antibodies against p21 (sc-397) and poly(ADP-ribose) polymerase (PARP) (sc-7150) were purchased from SantaCruz Biotechnology (Santa Cruz, CA, www.scbt.com); rabbit pol-yclonal antibodies against p53 (Ser15) (9284), p53 (Ser33)(2526), p53 (Ser392) (9281), p38 (9212), p38 (Thr180/Tyr182)(9215), and MAPKAP-2 (Thr334) (3007) were purchased fromCell Signaling Technology (Danvers, MA, www.cellsignal.com);mouse monoclonal antibody against c-H2AX (Ser139) (05-636)was purchased from Millipore; mouse monoclonal antibodyagainst Chk1 (C9358) was purchased from Sigma-Aldrich; mousemonoclonal antibody against a-tubulin (11-250-C100) was pur-chased from Exbio (Vestec, Czech Republic, www.exbio.cz);mouse monoclonal antibody against cyclin E1 (Ab-2 HE12) waspurchased from Neomarkers (Fremont, CA, www.labvision.com);
mouse monoclonal antibody against p53 (DO-1) was generouslyprovided by Dr. Borivoj Vojtesek (Masaryk Memorial CancerInstitute, Brno, Czech Republic).
Flow Cytometry, Cell Cycle Synchronization, andUVC Irradiation
Cells were harvested by manual scraping and immediately fixedfor at least 30 minutes in ice-cold 70% ethanol. After fixation,cells were washed three times with PBS (pH 7.4) and incubatedwith RNase (final concentration 0.02 mg/ml) (Boehringer, Ingel-heim, Germany, http://www.boehringer-ingelheim.com/) for 30minutes at 37�C. DNA was stained with propidium iodide (40lg/ml in PBS) (Sigma-Aldrich). Flow cytometric measurement offluorescence was done using the Beckman Coulter flow cytometerCytomics FC500 (Beckman Coulter, CA, http://www.beckman-coulter.com). The distribution of cells in different phases of thecell cycle was determined using FlowJo software (http://www.flowjo.com).
To synchronize their cell cycles, hESCs were first arrested inmitotic prophase by exposure to 200 ng/ml nocodazole (Sigma-Aldrich) in culture media for 16 hours. Then the cells werewashed three times with fresh culture media and incubated at37�C for 30 minutes. The cells were washed again three times inmedia to allow them to enter G1 phase as a synchronous popula-tion. Two hours after the final wash, the cells were in early G1phase (supporting information Fig. S1).
For hESCs irradiation, the culture media was removed andwashed off with PBS (pH 7.4). The cells were then irradiatedwith UVC light, and were immediately allowed to recover in pre-warmed culture media.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 20 minutes onice, permeabilized for 15 minutes with 0.1% Triton-X, blockedfor 1 hour with 1% bovine serum albumin in PBS (pH 7.4), andincubated overnight at 4�C with primary antibody. Primary anti-bodies used were as follows: rabbit polyclonal antibody againstpericentrin (ab4448) was purchased from Abcam (Cambridge,MA, www.abcam.com); mouse monoclonal antibodies againstCdc25A (sc-7389), Chk1 (C9358), Chk2 (sc-5278), and c-H2AX(Ser139) (05-636) were the same as used for western blot analy-sis. Cells were washed with PBS (pH 7.4) and incubated for 1hour at room temperature with appropriate secondary antibody:Alexa594-conjugated A11005 (Invitrogen) or FITC-conjugatedF9887 (Sigma-Aldrich). Cell nuclei were counterstained with40,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich), and cellswere mounted in Mowiol containing 1,4-diazobicyclo[2.2.2.]-oc-tane to prevent fading. Microscopy was performed using anOlympus FluoView 500 laser scanning microscope (OlympusC&S Ltd., Prague, Czech Republic, http://www.olympus-global.com).
Inhibition of Gene Expression by siRNAs
For transfection with siRNAs, hESCs were cultured in media con-ditioned by MEFs (MEF-CM) on dishes coated with Matrigel(BD Biosciences, San Jose, CA, www.bdbiosciences.com) accord-ing to the manufacturer’s instructions. Two hours before transfec-tion, the media was replaced with MEF-CM without antibiotics.Immediately prior to transfection, MEF-CM was replaced withOpti-MEM I reduced serum medium (Invitrogen). Complexes ofsiRNA (50 pmol) (Santa Cruz Biotechnology) and X-tremeGENEreagent (Roche, Inc., Madison, WI, www.roche.com) were pre-pared in Opti-MEM I reduced serum medium and used to trans-fect cells. Silencer FAM-Labeled Negative Control (Applied Bio-systems, Inc., Foster City, CA, www.appliedbiosystems.com) wasemployed to control nonspecific inhibition. Cells were incubatedwith siRNA/X-tremeGENE complexes for 4 hours and then refu-eled with fresh MEF-CM without antibiotics. hESCs were col-lected for analysis at 48–72 hours after transfection.
1144 DNA Damage Response in Human Embryonic Stem Cells

Immunoprecipitation and Kinase Assay
Cells were extracted in ice-cold extraction buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM ethylenediamine-tetraacetic acid, 8 mM b-glycerophosphate, 50 mM NaF, 1 mMphenylmethylsulphonyl fluoride, 1 mM tosylphenylalanine chloro-methane, 1 mM dithiothreitol, 1 lg/ml leupeptin, 1 lg/ml aproti-nin, 10 lg/ml soybean trypsin inhibitor), and the extracts werestored at �80�C until use. Before use, extracts were thawed andcleared by centrifugation at 15,000g for 5 minutes at 4�C andprotein concentrations were determined using the DC ProteinAssay Kit (Bio-Rad). Extracts were incubated with appropriateantibody for 1 hour on ice. Rabbit polyclonal antibody againstCDK2 (sc-163) (Santa Cruz Biotechnology) was used to immuno-precipitate CDK2. Immunoprecipitates were collected on ProteinG agarose beads (Sigma-Aldrich) by overnight rotation at 4�Cand washing three times with extraction buffer and twice with ki-nase assay buffer (50 mM N-(2-hydroxyethyl) piperazine-N0-(2-ethanesulfonic) acid (HEPES) (pH 7.5) 10 mM MgCl2, 10 mMMnCl2, 8 mM b-glycerophosphate, 1 mM dithiothreitol). Kinasereactions were carried out for 30 minutes at 37�C in a total vol-ume of 25 ll of kinase assay buffer supplemented with 100 lg/ml histone H1 (Sigma-Aldrich) and 40 lCi/ml [32P]ATP. Reac-tions were terminated by addition of �2 Laemmli sample buffer,and each reaction mix was subjected to SDS-PAGE andautoradiography.
Quantitative Reverse Transcription PolymeraseChain Reaction
Total RNA was extracted using RNeasy Mini Kit (Qiagen, CA,www.qiagen.com). Synthesis of cDNA from 500 ng total RNAwas performed using SuperScriptTM II Reverse Transcriptasewith oligo(dT)12–18 primer (Invitrogen). Each sample was ana-lyzed in triplicate using the TaqMan
VRGene Expression Assay for
CDKN1gene (Applied Biosystems) according to the manufac-turer’s instructions. DNA amplification was detected using the7,300 Real Time PCR
System (Applied Biosystems). Data were analyzed usingSequence Detection System software version 1.3.1 (Applied Bio-systems). Relative gene expression was calculated by normaliza-tion to glyceraldehyde 3-phosphate dehydrogenase (GAPDH)expression.
RESULTS
UVC Light Causes DNA Damage in hESCs, AsShown by Histone H2AX Phosphorylation
Various means have been used to induce damage to DNA ofin vitro cultured cells with UVC being one of the most com-mon. Here, we used UVC irradiation to induce DNA damagein hESCs in order to investigate their response at the level ofcell cycle regulation. In somatic cells with damaged DNA,histone phosphorylation allows repair proteins to access DNAand initiates DNA damage signaling [12]. To confirm thatUVC induces DNA damage in cultured hESCs, we examinedthe phosphorylation of serine 139 of histone H2AX (c-H2AX)by immunofluorescence (Fig. 1A) and by western blot (Fig.1B). We found that c-H2AX is phosphorylated in the vast ma-jority of hESCs exposed to as little as 1.5 J/m2 UVC, demon-strating that UVC induces double-stranded breaks in thesecells (Fig. 1A). The response is even more pronounced athigher doses of UVC light. There is a noticeable nonhomoge-neity in phosphorylation of H2AX in UVC-irradiated hESCs.Therefore, total amount of c-H2AX are shown in Figure 1Bto provide better quantification of this phenomenon. Impor-tantly, the gross morphology of the cells and cleavage of
PARP both indicated that very little cell death occurred incultures exposed to 1.5 J/m2 dose of UVC (Fig. 1C, 1D).
hESCs Arrest at G1 upon UVC Irradiation andShow Reduced CDK2 Activity
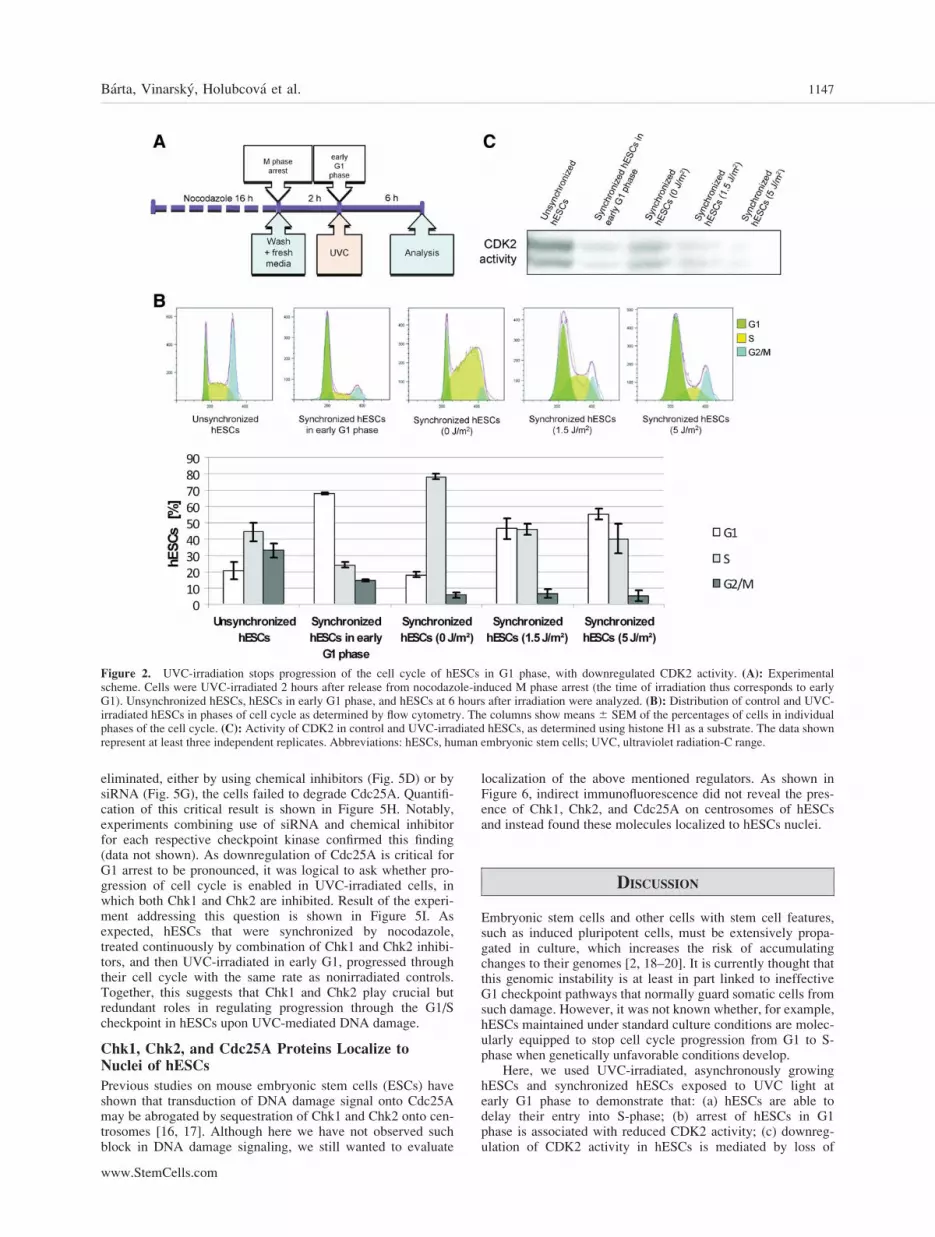
The progression of hESCs through G1 is very fast, so undernormal culture conditions, the proportion of G1 phase cells isbelow 25%. Therefore, we synchronized hESCs using nocoda-zole to enrich for cells in G1 (Fig. 2A). As shown in Figure2B, 2 hours after the release from M phase block, about 70%of hESCs reach G1. If not irradiated, these cells transit syn-chronously into S-phase after another 6 hours of culture (Fig.2B, supporting information Fig. S1). However, when hESCsare exposed during G1-1.5 J/m2 of UVC light, an increasedproportion of cells fail to progress to S-phase, compared withcontrols (47% of UVC-irradiated cells arrest in G1 phase,compared to 19% in untreated controls), with the proportionincreasing even further with a higher UVC dose (55% G1arrested cells with 5 J/m2 UVC) (Fig. 2B). Cells normallyprogress from G1 to S-phase primarily due to the activity ofCDK2 (bound to cyclin E) [13]. Accordingly, UVC-irradiatedhESCs arrested in G1 phase show reduced CDK2 activitycompared with nonirradiated controls (Fig. 2C). Thus, hESCsreduce the activity of a major initiator/driver of S-phase whentheir DNA is damaged by external means, allowing for G1arrest/delay to be executed.
Cdc25A, Not p21, Is Involved in CDK2 Inactivationupon DNA Damage in hESCs
In somatic and cancer cells, two fundamental molecularmechanisms have evolved to downregulate CDK2 activitywhen conditions are unfavorable for production of cell prog-eny, including loss of DNA integrity. The first mechanism isrelatively slow and occurs via p53-mediated transactivation ofthe CDK inhibitor p21. We found that in UVC-irradiatedhESCs, p53 protein accumulates to high levels in cell nuclei(Fig. 3A, 3B) and is phosphorylated on residues critical for itstransactivating function (Ser15, Ser33, and Ser392) (Fig. 3A).However, although p21 mRNA levels are increased in irradi-ated hESCs (Fig. 3C), the p21 protein level is similar to thatof nonirradiated hESCs (Fig. 3D). The inability of hESCs toaccumulate p21 protein was observed: (a) for both doses ofUVC light tested (1.5 and 5 J/m2) (Fig. 3D), (b) after bothshort (3 hours) and long (48 hours) periods of time after irra-diation of asynchronously growing hESCs (Fig. 3E), (c) andat 6 hours after irradiation of hESCs in G1 phase (Fig. 3F).Thus, it is unlikely that this slow pathway is used to regulatethe cell cycle response to DNA damage in hESCs.
The second mechanism is much faster and involves the re-moval of inhibitory phosphates from Thr14 and Tyr15 ofCDK2. In somatic cells, the phosphatase responsible for this,Cdc25A, becomes degraded upon DNA damage, thus leavingthe inhibitory phosphates of CDK2 intact. As shown in Figure4A, dose-dependent downregulation of Cdc25A was observedin asynchronously growing hESCs exposed to UVC light. Tomore precisely examine Cdc25A regulation and CDK2 activ-ity in hESCs as they progressed from G1 to S-phase, the lev-els of Cdc25A and cyclin E1, a key activator of CDK2, weredetermined in hESCs that were synchronized as describedabove. Importantly, although cyclin E1 levels undergo theexpected cell cycle-dependent fluctuations and are almostundetectable in nonirradiated hESCs passing through early G1phase, UVC-irradiated hESCs in G1 phase show high levelsof cyclin E1 (Fig. 4B). In contrast, Cdc25A is decreased inirradiated hESCs compared to nonirradiated controls, whichare progressing normally in their cell cycle (Fig. 4B).
Barta, Vinarsky, Holubcova et al. 1145
www.StemCells.com

Downregulation of Cdc25A in hESCs can be effectivelyachieved by siRNA [14]. Here, we used this approach to eval-uate a nature and extent of the change that develops in oursetting to the cell cycle progression of hESCs upon reductionof the level of Cdc25A. As shown in Figure 4C, decreasingthe amount of Cdc25A in asynchronously growing hESCs bysiRNA leads to delayed transit of cells into S-phase and theiraccumulation in G1. Notably, such change to cell cycle pro-gression takes place at the level of Cdc25A that is about thesame as that detected in asynchronously growing hESCsexposed to UVC light (Fig. 4A). Thus, as Cdc25A decidesabout the progression through G1/S border similarly to so-matic cells and downregulation of Cdc25A but not of cyclinE1 parallels inactivation of CDK2 in irradiated hESCs weassume that Cdc25A is a key molecule in the DNA damageresponse machinery of hESCs.
Chk1 and Chk2, but Not p38, Are Key Mediators ofG1/S Checkpoint Signaling in hESCs
To gain further insight into the signaling events involved inDNA damage-induced G1 arrest in hESCs, we assessed thefunction of kinases that phosphorylate Cdc25A and mark itfor degradation. Two types of kinases are currently known toperform this task in somatic cells: p38 MAP kinase [15] andthe checkpoint kinases Chk1 and Chk2 [5]. We found that all
three Cdc25A-phosphorylating kinases are activated by phos-phorylation in hESCs, and at least in the case of p38, this isdetectable by an anti-phosphoresidue antibody. As shown inFigure 5A, phosphorylation of p38 and the p38 substrateMAPKAPK-2 (threonine 334) in UVC-irradiated hESCs isincreased. To further test if p38 mediates the DNA damageresponse in hESCs, we used the chemical inhibitor SB202190to inhibit its kinase activity. Human ES cells were cultured inthe presence or absence of inhibitor (10 lM) and wereexposed to two different doses of UVC light, as before.Although p38 activity was completely abolished inSB202190-treated hESCs, as evidenced by the lack of MAP-KAPK-2 kinase phosphorylation, these cells were still able todegrade Cdc25A upon UVC-induced damage (Fig. 5A). Thus,although p38 activity increases during the hESCs response toDNA damage, this change is not critical for the cells to de-grade Cdc25A.
We next tested whether the checkpoint kinases Chk1 andChk2 are involved in the DNA damage response in hESCs byusing specific chemical inhibitors and siRNA to abrogate theirfunction, using degradation of Cdc25A as a readout. When ei-ther Chk1 or Chk2 function was abrogated by chemical inhib-itors (Chk1 inhibitor UCN01 or Chk2 inhibitor II) or bysiRNA, degradation of Cdc25A in UVC-irradiated hESCsappeared normal (Fig. 5B, 5C, 5E, and 5F). However, whenthe activity of both checkpoint kinases was simultaneously
Figure 1. Irradiation of hESCs with UVC causes dose-dependent damage to their DNA, as shown by increased phosphorylation of histone H2AXat serine 139 (c-H2AX), with only a modest level of apoptosis. Undifferentiated hESCs were irradiated with 1.5 and 5 J/m2 UVC, and 3 hours laterthey were analyzed for histone H2AX phosphorylation, and for molecular and morphological signs of apoptosis. (A): Phosphorylated histone H2AX(red) as shown by indirect immunofluorescence. Blue indicates chromatin stained by DAPI. Scale bar ¼ 50 lm. The inserts with higher magnificationare to visualize punctuate pattern of c-H2AX. (B): Western blot quantification of c-H2AX. (C): Western blot visualizing the extent of PARP proteincleavage. (D):Morphology of control and irradiated hESCs, as observed by light microscopy. Alpha-tubulin was used as a loading control. The graphshows means 6 SEM of three independent replicates. The data represent at least three independent replicates. Abbreviations: DAPI, 40,6-diamidino-2-phenylindole; hESCs, human embryonic stem cells; PARP, poly(ADP-ribose) polymerase; UVC, ultraviolet radiation-C range.
1146 DNA Damage Response in Human Embryonic Stem Cells
eliminated, either by using chemical inhibitors (Fig. 5D) or bysiRNA (Fig. 5G), the cells failed to degrade Cdc25A. Quantifi-cation of this critical result is shown in Figure 5H. Notably,experiments combining use of siRNA and chemical inhibitorfor each respective checkpoint kinase confirmed this finding(data not shown). As downregulation of Cdc25A is critical forG1 arrest to be pronounced, it was logical to ask whether pro-gression of cell cycle is enabled in UVC-irradiated cells, inwhich both Chk1 and Chk2 are inhibited. Result of the experi-ment addressing this question is shown in Figure 5I. Asexpected, hESCs that were synchronized by nocodazole,treated continuously by combination of Chk1 and Chk2 inhibi-tors, and then UVC-irradiated in early G1, progressed throughtheir cell cycle with the same rate as nonirradiated controls.Together, this suggests that Chk1 and Chk2 play crucial butredundant roles in regulating progression through the G1/Scheckpoint in hESCs upon UVC-mediated DNA damage.
Chk1, Chk2, and Cdc25A Proteins Localize toNuclei of hESCs
Previous studies on mouse embryonic stem cells (ESCs) haveshown that transduction of DNA damage signal onto Cdc25Amay be abrogated by sequestration of Chk1 and Chk2 onto cen-trosomes [16, 17]. Although here we have not observed suchblock in DNA damage signaling, we still wanted to evaluate
localization of the above mentioned regulators. As shown inFigure 6, indirect immunofluorescence did not reveal the pres-ence of Chk1, Chk2, and Cdc25A on centrosomes of hESCsand instead found these molecules localized to hESCs nuclei.
DISCUSSION
Embryonic stem cells and other cells with stem cell features,such as induced pluripotent cells, must be extensively propa-gated in culture, which increases the risk of accumulatingchanges to their genomes [2, 18–20]. It is currently thought thatthis genomic instability is at least in part linked to ineffectiveG1 checkpoint pathways that normally guard somatic cells fromsuch damage. However, it was not known whether, for example,hESCs maintained under standard culture conditions are molec-ularly equipped to stop cell cycle progression from G1 to S-phase when genetically unfavorable conditions develop.
Here, we used UVC-irradiated, asynchronously growinghESCs and synchronized hESCs exposed to UVC light atearly G1 phase to demonstrate that: (a) hESCs are able todelay their entry into S-phase; (b) arrest of hESCs in G1phase is associated with reduced CDK2 activity; (c) downreg-ulation of CDK2 activity in hESCs is mediated by loss of
Figure 2. UVC-irradiation stops progression of the cell cycle of hESCs in G1 phase, with downregulated CDK2 activity. (A): Experimentalscheme. Cells were UVC-irradiated 2 hours after release from nocodazole-induced M phase arrest (the time of irradiation thus corresponds to earlyG1). Unsynchronized hESCs, hESCs in early G1 phase, and hESCs at 6 hours after irradiation were analyzed. (B): Distribution of control and UVC-irradiated hESCs in phases of cell cycle as determined by flow cytometry. The columns show means 6 SEM of the percentages of cells in individualphases of the cell cycle. (C): Activity of CDK2 in control and UVC-irradiated hESCs, as determined using histone H1 as a substrate. The data shownrepresent at least three independent replicates. Abbreviations: hESCs, human embryonic stem cells; UVC, ultraviolet radiation-C range.
Barta, Vinarsky, Holubcova et al. 1147
www.StemCells.com

Cdc25A rather than by accumulation of p21 cyclin dependentkinase inhibitor (CKI); (d) Chk1 and Chk2 initiate Cdc25Aphosphorylation, which triggers its degradation in hESCs; and(e) preventing degradation of Cdc25A by simultaneous inhibi-tion of Chk1 and Chk2 abrogates UVC-induced G1/S block.
The cell cycle progression that underlies cell proliferationis controlled by a series of highly orchestrated actions ofCDK family members in association with their partneringcyclins. In somatic cells, the transition from G1 to S-phase,which is followed by DNA synthesis, is initiated by CDK4and CDK6 in conjunction with D-type cyclins, and is furtherpropelled mainly by CDK2 complexed with cyclin E andcyclin A, with the key target of these CDKs being retinoblas-toma protein (RB)/E2F-mediated transcriptional regulation
[21]. In mouse ESCs, the RB/E2F pathway is constitutivelyactivated, thus eliminating effective regulation of the G1/Stransition [22–24]. In contrast, several lines of evidence pub-lished by four independent laboratories and unpublished datafrom our laboratory indicate that hESCs can modify CDK ac-tivity to promote and/or inhibit cell cycle progression. Theseinclude the following: (a) cyclin quantities and CDK activitiesfluctuate during the cell cycle of hESCs [25, 26, our unpub-lished data]; (b) CDK2 activity is controlled by Cdc25A [14];(c) both hypo- and hyperphosphorylated forms of RB proteinare detectable in hESCs [27, our unpublished data]; (d) miR-92b regulates passage through the G1/S checkpoint by target-ing the CDK inhibitor p57 [28]; and (e) regulation of the S-phase-associated CDK target, transcriptional coactivator
Figure 3. Cell cycle arrest of UVC-irradiated hESCs at G1 is not mediated by p21 accumulation. (A): Quantity and phosphorylation status of p53protein in asynchronously growing control and UVC-irradiated hESCs, as determined by western blot analysis 3 hours after irradiation. (B): Accu-mulation of p53 protein (red) in nuclei of asynchronously growing control and UVC-irradiated hESCs, as determined by indirect immunofluores-cence 3 hours after irradiation. Blue, chromatin stained by DAPI. (C): The level of p21 mRNA in asynchronously growing control and UVC-irradiated hESCs, as determined by quantitative reverse transcription polymerase chain reaction 3 hours after irradiation. The graph shows means 6SEM of the fold change relative to nonirradiated controls. (D): The level of p21 protein in asynchronously growing control and UVC-irradiatedhESCs, as determined by western blot analysis 3 hours after irradiation. (E): The level of p21 protein in UVC-irradiated hESCs, as determined bywestern blot analysis 3 and 48 hours after irradiation. (F): The level of p21 protein in hESCs UVC-irradiated in G1 phase. The level of p21 wasdetermined by western blot analysis 6 hours after irradiation (see Fig. 2 for the experimental design). (D–F): Human foreskin fibroblasts (HFF areincluded as positive controls. Alpha tubulin was used as a loading control. The data represent at least three independent replicates. Abbreviations:DAPI, 40,6-diamidino-2-phenylindole; hESC, human embryonic stem cell; HFF, human foreskin fibroblasts; UVC, ultraviolet radiation-C range.
1148 DNA Damage Response in Human Embryonic Stem Cells

p220NPAT, is operative in hESCs [26]. In this study, we addyet another line of evidence by showing that when DNA dam-age occurs, hESCs can regulate CDKs to interrupt cell cycleprogression from G1 phase.
Curiously, no such cell cycle block was noted in a previ-ous study of UVC-irradiated hESCs; rather, massive celldeath under DNA-damaging conditions was observed [29].However, as this study did not use synchronized hESCs anddid not focus on cell cycle regulation, rigorous comparisonsto our data are difficult. To determine whether cell death wasaffected under our experimental conditions, we examined thegross morphology of the cells and cleavage of PARP follow-ing UVC exposure. As shown in Figure 1, under our irradia-tion conditions (1.5 J/m2), hESCs exhibited very little celldeath. Thus, it is unlikely that cell death contributed signifi-cantly to the overall outcome of our experiments.
For cells to stop cell cycle progression before entering S-phase, there are two possible strategies that may at least theo-retically be employed individually or in combination. Bothconverge upon CDK, but one involves increased binding ofthe CDK inhibitor p21 to CDKs, while the other employsdownregulation of Cdc25A, which normally keeps CDK2 inan active state by removing its inhibitory phosphates [5, 6].p21 is a transcriptional target of p53, the well-known gate-keeper that operates in somatic cells. The function of p53-p21axis has been studied by several groups in both mouse andhuman ESCs and in pluripotent human embryonal carcinomacells using various strategies [10, 29–32]. Surprisingly, therehave been many discrepancies between their findings, includ-
ing differences in the ability of cells to transactivate the p21gene or to produce increased levels of p21 protein when p21transcripts are upregulated [10, 29–31, 33]. When we exam-ined p53 expression in hESCs with UVC-induced G1 delay,we found that at 3 hours after UVC irradiation, p53 protein isalready dramatically increased, is phosphorylated on serines15, 33, and 392, and localizes completely to cell nuclei.Moreover, p21 mRNA is increased 10-fold above its basallevel in nonirradiated cells. Despite these obvious signs ofp53 activation, however, the level of p21 protein is low orundetectable in all experimental regimens and at all UVCdoses we tested. Although genetic lesions to p53 are unlikelyto underlie the lack of p21 translation, it should be noted thatin all hESC lines used here, we have excluded p53 mutationsusing a functional assay in yeast (data not shown). Thus,although p21 metabolism is associated with regulation of theG1/S transition in hESCs, our findings still support the notionthat the relationship between p53 activation and cell cycleregulators in hESCs does not follow the typical scenarioobserved in somatic cells. In fact, the p53-p21 pathway maybe used by hESCs to regulate entry into differentiation ratherthan to maintain intact DNA [30].
Regulation of CDK activity by CKIs depends on their denovo synthesis. Given the fast progression of hESCs throughG1 phase, it may not be surprising that this process is regu-lated via mechanisms that do not require protein synthesis.Indeed, we demonstrated that hESCs can activate the check-point kinases Chk1 and Chk2, which phosphorylate Cdc25A,thus directing this CDK2 activator to a degradation pathway.
Figure 4. UVC-induced G1arrest of hESCs is associatedwith downregulation of Cdc25Aprotein. (A): The level ofCdc25A protein in asynchro-nously growing control andUVC-irradiated hESCs, as deter-mined by western blot analysis 3hours after irradiation. The graphshows means 6 SEM of the foldchange relative to nonirradiatedcontrols. (B): The levels ofcyclin E1 and Cdc25A protein inhESCs UVC-irradiated in G1phase. Protein levels were deter-mined by western blot analysis 6hours after irradiation (see Fig. 2for the experimental design).(C): The effect of Cdc25Adownregulation by siRNA oncell cycle progression of asyn-chronously growing hESCs asdetermined by flow cytometry.Alpha tubulin was used as aloading control. The data repre-sent at least three independentreplicates. Abbreviation: hESC,human embryonic stem cell;UVC, ultraviolet radiation-Crange.
Barta, Vinarsky, Holubcova et al. 1149
www.StemCells.com

Figure 5. Chk1 and Chk2, but not p38, are responsible for Cdc25A downregulation and block of cell cycle progression in UVC-irradiatedhESCs. (A): The effect of chemical inhibition of p38 kinase by SB202190 on the quantity of Cdc25A in asynchronously growing control andUVC-irradiated hESCs, as determined by western blot analysis 3 hours after irradiation. The level of phoshorylation of p38 itself (p-p38) andphosphorylation status of MAPKAPK-2 (substrate of p38) are shown. (B–D): The effect of chemical inhibition of Chk1 (UCN01), Chk2 (Chk2inhibitor II), and both Chk1 and Chk2 (UCN01 þ Chk2 inhbitor II) on the quantity of Cdc25A in asynchronously growing control and UVC-irra-diated hESCs, as determined by western blot analysis 3 hours after irradiation. (E–G): The effect of siRNA-mediated downregulation of Chk1,Chk2, and both Chk1 and Chk2 on the quantity of Cdc25A in asynchronously growing control and UVC-irradiated hESCs, as determined bywestern blot analysis 3 hours after irradiation. Quantities of Chk1 and/or Chk2, as determined by western blot analysis, are shown to documentthe effectiveness of siRNA-mediated downregulation. (H): Quantification of the effects of simultaneous inhibition of Chk1 and Chk2 from panels(D) and (G). The graph shows means 6 SEM of the fold change in quantity of Cdc25A protein, compared to nonirradiated controls. (I): Theeffect of simultaneous chemical inhibition of Chk1 and Chk2 on the progression of cell cycle of hESCs UVC-irradiated in G1 phase. The experi-mental design was the same as described in Figure 2, with the cells being exposed to the inhibitors since the time of nocodazole removal. Thecolumns show means 6 SEM of the percentages of cells in individual phases of the cell cycle. Phosphorylation of H2AX at serine 139 (c-H2AX) is shown for all experiments to demonstrate damage to DNA. Alpha tubulin was used as a loading control. The data represent at leastthree independent replicates. Abbreviations: DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell.

Although Chk1 and Chk2 can functionally substitute for oneanother, since the degradation of Cdc25A is prevented andUVC-induced cell cycle block is abrogated only when the ac-tivity of both is eliminated, two other kinases that are capableof phosphorylating Cdc25A, p38 and GSK-3b [15, 34], do notphosphorylate Cdc25A in UVC-treated hESCs (inhibition ofGSK-3b by lithium chloride, data not shown). In this context,the recent finding that GSK-3b is responsible for Cdc25Aphosphorylation in c-irradiated mouse ESCs is of note [17].These authors of this study also indicate that mouse andhuman ESCs differ from one another in the sequestration ofboth checkpoint kinases at centrosomes, but we have not beenable to document this phenomenon in our experiments (Fig.6). Why these and many other fundamental, cell cycle-relateddifferences exist between mouse and human ESCs remainsunresolved [35]. One might speculate that they reflect differ-ences between the developmental stages of the embryos fromwhich mouse and human ESCs are derived [36, 37]. Althoughsuch interspecies differences are interesting, it is crucial to
understand the basis for the discrepancies between observa-tions made by different research groups. There are many plau-sible explanations that can be experimentally addressed,including the use of different cell lines (enormous variabilityin differentiation potential has already been shown [38]), theuse of different DNA damaging agents, the use of differentexperimental regimens (synchronized vs. asynchronous cellpopulations), and others. In our study, we have unexpectedlybeen able to activate the G1/S checkpoint in hESCs, with thesame results observed in more than one hESC line. As thedose of UVC light that caused this response was quite low,we propose that hESCs differentially regulate pathways thatensure DNA integrity delicately according to the magnitudeof the damage. It is possible that G1 delay associated withsuch a limited amount of damage may allow hESCs to initiatedifferentiation program. However, as we did not find anychanges to the expression of cell surface markers typical forundifferentiated hESCs at 48 hours after irradiation (ourunpublished data), this scenario is either not valid or moretime and better assays are required to reveal differentiation-associated changes.
CONCLUSION
This study shows that under a low-dose of DNA-damagingUVC light, hESCs respond by activating classical G1/Scheckpoint molecules to prevent entry into S-phase with unre-paired DNA. This G1 block in hESCs is mediated by Chk1and Chk2, which phosphorylate Cdc25A, thereby marking itfor degradation. Lack of CDK-activating Cdc25A results inlow CDK2 activity, without contribution from the p53-p21pathway.
ACKNOWLEDGMENTS
We thank to Dr. Borivoj Vojtesek (Masaryk Memorial CancerInstitute, Brno, Czech Republic) for providing us with anti-p53antibody, to Dr. Jana Smardova for assaying functionality of p53gene, and to Dr. Martina Vodinska, Dr. Klara Koudelkova, andIveta Peterkova, M.S. for excellent technical assistance. Thiswork was supported by the Ministry of Education, Youth, andSports of the Czech Republic (MSM0021622430, MUNI/E/0118/2009, 1M0538), the Academy of Sciences of the CzechRepublic (AV0Z50390512, AV0Z50390703), the EU (LSHG-CT-2006-018739), and the Ministry of Health of the CzechRepublic (IGAMZCRNS10439-3/2009).
DISCLOSURE OF POTENTIAL CONFLICTS
OF INTEREST
The authors indicate no potential conflicts of interest.
REFERENCES
1 Maitra A, Arking DE, Shivapurkar N et al. Genomic alterations incultured human embryonic stem cells. Nat Genet 2005;37:1099–1103.
2 Baker DEC, Harrison NJ, Maltby E et al. Adaptation to culture ofhuman embryonic stem cells and oncogenesis in vivo. Nat Biotechnol2007;25:207–215.
3 Gertow K, Cedervall J, Unger C et al. Trisomy 12 in HESC leads tono selective in vivo growth advantage in teratomas, but induces anincreased abundance of renal development. J Cell Biochem 2007;100:1518–1525.
4 Blum B, Bar-Nur O, Golan-Lev T et al. The anti-apoptotic gene survi-vin contributes to teratoma formation by human embryonic stem cells.Nat Biotechnol 2009;27:281–287.
5 Bartek J, Lukas J. Pathways governing G1/S transition and theirresponse to DNA damage. FEBS Lett 2001;490:117–122.
Figure 6. Chk1, Chk2, and Cdc25A do not localize to centrosomesin proliferating human embryonic stem cells. Green, centrosomes, asvisualized by indirect immunofluorescence for pericentrin. Red, Chk1,Chk2, and Cdc25A, as visualized by indirect immunofluorescence.Blue, chromatin stained by DAPI. Scale bars ¼ 20 lm. Abbreviation:DAPI, 40,6-diamidino-2-phenylindole.
Barta, Vinarsky, Holubcova et al. 1151
www.StemCells.com

6 Mailand N, Falck J, Lukas C et al. Rapid destruction of humanCdc25A in response to DNA damage. Science 2000;288:1425–1429.
7 Ray D, Kiyokawa H. Cdc25A phosphatase: A rate-limiting oncogenethat determines genomic stability. Cancer Res 2008;68:1251–1253.
8 Becker KA, Ghule PN, Therrien JA et al. Self-renewal of human em-bryonic stem cells is supported by a shortened G1 cell cycle phase.J Cell Physiol 2006;209:883–893.
9 Becker KA, Stein JL, Lian JB et al. Establishment of histone generegulation and cell cycle checkpoint control in human embryonic stemcells. J Cell Physiol 2007;210:517–526.
10 Filion TM, Qiao M, Ghule PN et al. Survival responses of human em-bryonic stem cells to DNA damage. J Cell Physiol 2009;220:586–592.
11 Adewumi O, Aflatoonian B, Ahrlund-Richter L et al. Characterizationof human embryonic stem cell lines by the International Stem CellInitiative. Nat Biotechnol 2007;25:803–816.
12 Paull TT, Rogakou EP, Yamazaki V et al. A critical role for histoneH2AX in recruitment of repair factors to nuclear foci after DNA dam-age. Curr Biol 2000;10:886–895.
13 Morgan DO. Cyclin-dependent kinases: Engines, clocks, and micro-processors. Annu Rev Cell Dev Biol 1997;13:261–291.
14 Zhang X, Neganova I, Przyborski S et al. A role for NANOG in G1to S transition in human embryonic stem cells through direct bindingof CDK6 and Cdc25A. J Cell Biol 2009;184:67–82.
15 Goloudina A, Yamaguchi H, Chervyakova DB et al. Regulation ofhuman Cdc25A stability by Serine 75 phosphorylation is not sufficientto activate a S phase checkpoint. Cell Cycle 2003;2:473–478.
16 Hong Y, Stambrook PJ. Restoration of an absent G1 arrest and protec-tion from apoptosis in embryonic stem cells after ionizing radiation.Proc Natl Acad Sci USA 2004;101:14443–14448.
17 Koledova Z, Kafkova LR, Kramer A et al. DNA Damage-induceddegradation of Cdc25A does not lead to inhibition of Cdk2 activity inmouse embryonic stem cells. Stem Cells 2010;28:450–461.
18 Imreh MP, Gertow K, Cedervall J et al. In vitro culture conditionsfavoring selection of chromosomal abnormalities in human ES cells.J Cell Biochem 2006;99:508–516.
19 Catalina P, Montes R, Ligero G et al. Human ESCs predisposition to karyo-typic instability: Is a matter of culture adaptation or differential vulnerabilityamong hESC lines due to inherent properties? Mol Cancer 2008;7:76–84.
20 Wu H, Kim KJ, Mehta K et al. Copy number variant analysis ofhuman embryonic stem cells. Stem Cells 2008;26:1484–1489.
21 Nevins JR, Leone G, DeGregori J et al. Role of the Rb/E2F pathwayin cell growth control. J Cell Physiol 1997;173:233–236.
22 Savatier P, Huang S, Szekely L et al. Contrasting patterns of retino-blastoma protein expression in mouse embryonic stem cells and em-bryonic fibroblasts. Oncogene 1994;9:809–818.
23 Stead E, White J, Faast R et al. Pluripotent cell division cycles aredriven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene2002;21:8320–8333.
24 White J, Stead E, Faast R et al. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-depend-ent kinase activity during embryonic stem cell differentiation. MolBiol Cell 2005;16:2018–2027.
25 Neganova I, Zhang X, Atkinson S et al. Expression and functionalanalysis of G1 to S regulatory components reveals an important rolefor CDK2 in cell cycle regulation in human embryonic stem cells.Oncogene 2009;28:20–30.
26 Becker KA, Ghule PN, Lian JB et al. Cyclin D2 and the CDK sub-strate p220(NPAT) are required for self-renewal of human embryonicstem cells. J Cell Physiol 2010;222:456–464.
27 Filipczyk AA, Laslett AL, Mummery C et al. Differentiation iscoupled to changes in the cell cycle regulatory apparatus of humanembryonic stem cells. Stem Cell Res 2007;1:45–60.
28 Sengupta S, Nie J, Wagner RJ et al. MicroRNA 92b controls the G1/Scheckpoint gene p57 in human embryonic stem cells. Stem Cells 2009;27:1524–1528.
29 Qin H, Yu T, Qing T et al. Regulation of apoptosis and differentiationby p53 in human embryonic stem cells. J Biol Chem 2007;282:5842–5852.
30 Maimets T, Neganova I, Armstrong L et al. Activation of p53 by nut-lin leads to rapid differentiation of human embryonic stem cells.Oncogene 2008;27:5277–5287.
31 Momcilovic O, Choi S, Varum S et al. Ionizing radiation inducesataxia telangiectasia mutated-dependent checkpoint signaling and G(2)but not G(1) cell cycle arrest in pluripotent human embryonic stemcells. Stem Cells 2009;27:1822–1835.
32 Aladjem MI, Spike BT, Rodewald LW et al. ES cells do not activatep53-dependent stress responses and undergo p53-independent apopto-sis in response to DNA damage. Curr Biol 1998;8:145–155.
33 Wang X, Lui VCH, Poon RTP et al. DNA damage mediated s andg(2) checkpoints in human embryonal carcinoma cells. Stem Cells2009;27:568–576.
34 Kang T, Wei Y, Honaker Y et al. GSK-3 beta targets Cdc25A forubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlateswith Cdc25A overproduction in human cancers. Cancer Cell 2008;13:36–47.
35 Conklin JF, Sage J. Keeping an eye on retinoblastoma control ofhuman embryonic stem cells. J Cell Biochem 2009;108:1023–1030.
36 Brons IGM, Smithers LE, Trotter MWB et al. Derivation of pluripo-tent epiblast stem cells from mammalian embryos. Nature 2007;448:191–195.
37 Tesar PJ, Chenoweth JG, Brook FA et al. New cell lines from mouseepiblast share defining features with human embryonic stem cells. Na-ture 2007;448:196–199.
38 Osafune K, Caron L, Borowiak M et al. Marked differences in differ-entiation propensity among human embryonic stem cell lines. Nat Bio-technol 2008;26:313–315.
See www.StemCells.com for supporting information available online.
1152 DNA Damage Response in Human Embryonic Stem Cells





























