Embed Size (px)
Citation preview

A
ac
PbPiP
tgrptp
aIb©
K
R
0d
Colloids and Surfaces B: Biointerfaces 59 (2007) 194–207
Impact of bulk and surface properties of some biocompatible hydrophobicpolymers on the stability of methylene chloride-in-water mini-emulsions
used to prepare nanoparticles by emulsification–solvent evaporation
Valery G. Babak a,b,∗, Francis Baros b, Omar Boulanouar c, Frank Boury d, Michel Fromm c,Nathalie R. Kildeeva e, Nathalie Ubrich f, Philippe Maincent f
a DCPR, CNRS UMR 7630, 1 rue Grandville, BP 451, 54001 Nancy Cedex, Franceb INEOS RAS, Vavilova str. 28, Moscow 117813, Russian Federation
c LMN-AC, UMR CEA E4, Universite de Franche-Comte, 16 route de Gray, 25030 Besancon, Franced INSERM Unite 646 “Ingenierie de la Vectorisation Particulaire” Immeuble IBT, 10 rue Andre Boquel, 49100 Angers, France
e MSTU, 1 M.Kaluzhskaya str., 117918 Moscow, Russian Federationf Laboratoire de Pharmacie Galenique, Faculte de Pharmacie, Universite Henri Poincare, 54001 Nancy Cedex, France
Received 28 December 2006; received in revised form 4 May 2007; accepted 9 May 2007Available online 18 May 2007
bstract
The emulsifying and stabilizing ability of several hydrophobic (insoluble in water and soluble in volatile organic solvents) polymers, suchs Eudragit RL, Eudragit RS, PLGA, PCL, and their mixtures, with regard to the methylene chloride (MC)-in-water mini-emulsions, has beenompared to the viscosity of MC solutions and to the properties of adsorption and spread monolayers of these polymers.
Eudragits RS and RL contain ∼2.5 and ∼5 mol% of pendent cationic trimethylammonium (TMA) groups per ∼164 g/mol segments, whereasLGA and PCL contain 1 and 2 polar carbonyl groups per 130 and 114 g/mol, respectively. The electrostatic attraction between the dipoles, formedy TMA groups and the condensed counter ions in the MC solutions, leads to the contraction of macromolecular coils of Eudragits, whereas theLGA and PCL macromolecules, interacting by low polar carbonyl groups (with dipole moment μ= 2.7 D) retain more extended conformation
n MC. This explains why the characteristic viscosities [η] of MC solutions are much lower for the former polymers (∼0.1 dL/g) with regard toLGA and PCL solutions whose [η] is equal to 0.3 and 0.6 dL/g, respectively.The ionization of TMA groups in contact with the water phase leads to the irreversible adsorption of Eudragits at the MC/water interface and
o high decrease of the interfacial tension γ (down to 4 mN/m for the 5% MC solutions). Whereas PLGA and PCL possessing low polar carbonylroups adsorb poorly at the MC/water interface exhibiting γ ∼= 28 mN/m. Higher stability of spread monolayers of Eudragits (π* ∼ 40 mN/m) withegard to PLGA and PCL (π* < 20 mN/m) correlates well with higher interfacial activity of the former with regard to the later. The higher surfaceotential �V of Eudragits (0.9 V) with regard to PLGA (0.3 V) and PCL (0.4 V) is explained by the formation of electric double layer (DL) byhe former, whereas the later contribute to the �V only by cumulative dipole moments of carbonyl groups. The experimental values of surfaceotentials correlate well with the Gouy–Chapman model of the DL and the Helmholtz model of the monolayer.
The ensemble of experimental results leads to the conclusion that higher emulsifying and stabilizing ability of Eudragits with regard to PLGA
nd PCL is due to higher adsorption activity of the former which form the corona of polymeric chains with ionized TMA groups around the droplets.t can be postulated that Eudragit polymers have good surface active properties which may allow manufacturing of biocompatible nanoparticlesy emulsification–solvent evaporation method without surfactants. 2007 Elsevier B.V. All rights reserved.eywords: Stability of mini-emulsions; Hydrophobic biocompatible polymers; Inter
1
∗ Corresponding author at: INEOS RAS, 28 Vavilova str., Moscow 117813,ussian Federation. Tel.: +7 495 1356502; fax: +7 495 1355085.
E-mail address: [email protected] (V.G. Babak).
sg
927-7765/$ – see front matter © 2007 Elsevier B.V. All rights reserved.oi:10.1016/j.colsurfb.2007.05.010
facial tension; Spread monolayers; Surface and zeta potentials
. Introduction
Biocompatible and biodegradable hydrophobic polymersuch as poly(�-caprolactone) (PCL), poly(d,l-lactide-co-lycolides) (PLGA) and acrylic polymers (Eudragits RL, RS and

ces B
oamosoemdwsw
trptfaw[eatp
tteioaRiwsbptafnwutttp
tatppcte
smacv
2
2
aG4(wQ
2
2
pmwaddbenamtt(cp
mmtcito
2
Msa
V.G. Babak et al. / Colloids and Surfa
thers) are widely used in medicine for the preparation of micro-nd nanoparticles: vectors of drugs, vaccines, enzymes, geneticaterials, etc. The most wide-spreading method of the synthesis
f vectors is simple oil/water or multiple water/oil/water emul-ification followed by solvent evaporation (extraction) of therganic solvent [1,2]. The crucial step of this method is themulsification of the organic solutions of hydrophobic poly-ers containing additional bioactive substances dissolved or
ispersed in the organic phase or in the internal water phaseith the subsequent solidification of emulsion droplets by diffu-
ion or evaporation of the organic solvent in or through externalater phase [3–6].The physico-chemical parameters which have an impact on
he functional properties of nano- and microparticles (e.g. theelease rate of drugs from these vectors) are interfacial and bulkroperties of the wall forming polymers, the composition ofhe polymeric blends, rate of the organic solvent evacuationrom the emulsion droplets during evaporation (extraction) step,dsorption activity of drug and their ability to form complexesith polymers, presence of surfactants in the formulation, etc.
7–9]. The size of vectors considered as a very important param-ter responsible for drug distribution, absorption and biologicalctivity after administration [10,11], is particularly sensitive tohe interfacial activity and emulsifying ability of hydrophobicolymers [9,12].
In our previous study [12] effectuated under a strict con-rol of physico-chemical parameters (concentration of polymers,ype of organic solvent, volume fraction of dispersed phase inmulsions, applied dispersing power, time of evaporation, etc.)t has been found that size order of the solidified nanoparticlesbtained by evaporation of methylene chloride (MC) used asn organic solvent for polymers, was PCL > PLGA > EudragitS > Eudragit RL. For example, in one of these series of exper-
ments the size of Eudragits nanoparticles was 280–290 nmhereas PLGA and PCL lead to bigger particles having the mean
ize of ∼350 and ∼450 nm, respectively [12]. Moreover, it haseen found that the nanoparticles produced from Eudragits hadositive zeta-potential of the order of +60 to +70 mV, whereashe zeta-potentials of PCL and PLGA nanoparticles were neg-tive of the order of −20 mV [12]. The positive zeta-potentialor Eudragits nanoparticles is due to pendent trimethylammo-ium (TMA) groups which are ionized in contact with theater phase. But the slightly negative zeta-potential for thencharged hydrophobic polymers was not expected. The mix-ures of cationic polymers, Eudragits, and PLGA, or PCL, leadso the nanoparticles with variable zeta-potential going from posi-ive to negative sign with increasing the content of the unchargedolymers in the mixture [13].
Considering the possibility of control the size and the elec-ric charge of nanoparticles as promising for the medicalpplications (mucoadhesivity, stealth properties) we have under-aken an attempt to understand why and how the functionalroperties of nanoparticles depend on the choice of different
olymers. Special attention was drawn to the effect of vis-osity of organic solutions of polymers and of the interfacialension and electric charge of droplets on the stability of mini-mulsions—precursors of nanoparticles.(dwq
: Biointerfaces 59 (2007) 194–207 195
The aim of this work is to bring more insight to the pos-ibility of control the stability of methylene chloride-in-waterini-emulsions, particularly, the droplet size of these emulsions
nd consequently the size of polymeric nano- and microparti-les, obtained by emulsification/solvent evaporation method, byarying physico-chemical parameters.
. Materials and methods
.1. Materials
The following materials were used as received: Eudragit RSnd RL (acrylic polymers, Rohm Pharma GmbH, Darmstadt,ermany), poly(d,l-lactide-co-glycolide) 50/50 (PLGA) (MW0,000 Da) (Medisorb Technologies), poly(�-caprolactone)PCL) (MW 42,000 Da) (Aldrich). Methylene chloride (Sigma)ere of analytical grade purity. Water was purified using a Milli-plus 185 system (Millipore).
.2. Methods
.2.1. Preparation of mini-emulsionsMini-emulsions with the droplet size of ∼10–100 nm were
repared according to a procedure described in [14,15]. Fortyicroliters of polymer solution in methylene chloride (2.5%,/v) was dispersed in 4 mL of water using an ultrasound probe
t 15 W for 30 s (1 s pulses at 1 s intervals). To prevent theissolution of methylene chloride (MC) from the emulsionroplets into water phase, the water was previously saturatedy MC, and during light scattering measurements the contain-rs were hermetically corked up. So, the emulsion droplets didot undergo solidification during size measurement. It has beenssumed that the osmotic pressure due to the dissolved poly-ers inside the dispersed MC was sufficiently high to prevent
he Ostwald ripening of droplets during relatively short laps ofime ∼1 h. All mini-emulsions were treated in the same waytime and sonication power) and were prepared with similaroncentration of polymers and volume of organic and aqueoushases.
Point out that in order to optimize the dynamic light scatteringeasurements of the droplets size of emulsion (to prevent theultiple diffusion of the light beam from emulsion droplets),
he volume fraction ϕMC = VMC/(VMC + Vw) of the methylenehloride in the mini-emulsions was reduced to ∼0.1%. Whereasn the standard formulation of nanoparticles by emulsifica-ion/solvent evaporation method [3,4] the value of ϕMC is oner two order of magnitude much higher.
.2.2. Properties of Langmuir monolayers of polymersLangmuir monolayer of polymers were studied with a KSV
inithrough (Finland) equipped with a surface potential mea-urement device. The experimental surface pressure–area (π–A)nd surface potential–area (�V–A) isotherms were presented as
π–Γ ) and (�V–Γ ) isotherms, where Γ = m/A is the surfaceensity of hydrophobic (non-soluble in water) polymers at theater–air interface (for the sake of brevity we will denote thisuantity as adsorption amount in spite of the fact that these lay-
1 faces B: Biointerfaces 59 (2007) 194–207
et
2
MwFldttu
2
mce(oeo
3
pmtopptomw
3
1Mmcϕ
pot((se
3
s
Fig. 1. Kinetic curves of the cube of the inverse size, d−3(t), for the dropletsoc(
ec(ETomdeow
3
bcasPlaE
d[
wnslo
Krd
96 V.G. Babak et al. / Colloids and Sur
rs are formed by spreading and evaporation of organic solventechnique), m is the mass of the polymer.
.2.3. Dynamic tensiometry at the MC/water interfaceThe interfacial tension versus time was measured at the
C/water interface by using a dynamic pendant drop methodith a drop tensiometer (Traker®, ITConcept, Longessaigne,rance) [16]. The interfacial tension was determined by ana-
yzing the axial symmetric shape (Laplacian profile) of pendantrops of organic solutions of polymers immersed in water. Allhe measurements of the interfacial tension were recorded in realime (five measurements per second) at constant drop area bysing the motor to control the volume of the drop.
.2.4. ViscosimetryThe viscosity of each MC solution of polymer was deter-
ined with a Ubhellode viscosimeter at 20 ◦C with a glassapillary (internal diameter 0.56 mm) and a flowing time of MCqual to 35 s. In the polymer concentration range from 0 to 5%w/v), the isotherms of the reduced viscosities ηred(C) of eachrganic solution of polymers could be linearized and allowed thestimation of the characteristic viscosity [η] by the extrapolationf ηred(C) to C → 0.
. Results and discussion
As it has been pointed out in Section 1, the physico-chemicalarameters including the type of polymers may influence enor-ously on the stability of mini-emulsions and consequently on
he nanoparticles size. The mini-emulsions studied here were/w emulsion types which are widely used in the one stepreparation of nanoparticles but also correspond to the secondart of the manufacturing process of nanoparticles accordingo the w/o/w technique. The study of the stability of water-in-il mini-emulsions in connection with the internal structure oficroparticles is in progress [17] and will be published else-here.
.1. Stability of MC-in-water mini-emulsions
Fig. 1 displays the cube of the inverse mean diameter,/d3, of the droplets in function of the storage time, t, for theC/water mini-emulsions stabilized by different types of poly-ers. Note that all mini-emulsions were prepared in identical
onditions (concentration of polymers in MC, volume fractionMC = VMC/(VMC + Vw) of the methylene chloride, sonicationower and time, etc., see Section 2.2.1). It must be pointedut, that the value 1/d3 is proportional to the number concentra-ion n = N/V = 6ϕMC/πd3 of emulsion droplets in the emulsionn ∼= 2 × 1018/d3), where n and d are expressed in (m−3) andnm), respectively. Smaller is the mean droplet size of an emul-ion, higher is the concentration n of liquid droplets in themulsions.
.1.1. Emulsifying ability of polymersFor example, according to Fig. 1, the initial mean droplet
ize d0 of emulsions stabilized by PCL and Eudragit RS are
ftit
f MC-in-water emulsions, stabilized by different polymers of the same bulkoncentration in MC (C = 5%, w/v): Eudragit RL (curve 1, �), Eudragit RScurve 2, �), PLGA (curve 3, ©); PCL (curve 4, �).
qual to d0,PCL ∼= 270 nm and d0,RS ∼= 40 nm, respectively, thatorresponds to the initial droplets concentration n0,PCL ∼= 1011
m−3) and n0,RS ∼= 3 × 1012 (m−3), respectively. This means thatudragit RS is ∼30 times best emulsifier with regard to the PCL.he emulsifying ability of the PLGA is ∼8 times better than thatf PCL, while the initial concentration of the droplets for the for-er polymer is n0,PLGA ∼= 8 × 1011 (m−3). Methylene chloride
roplets in water (without any polymer) were the largest and themulsion appeared instable since a total phase separation wasbserved after ∼300 s. So, the emulsifying property of polymersas in the order Eudragit RL ∼= Eudragit RS > PLGA > PCL.
.1.2. Stabilizing ability of polymersThe slopes of the kinetic curves d−3(t) characterize the sta-
ility of mini-emulsions: the lower the slope, the smaller theoalescence rate of the droplets, the more stable the emulsionnd the highest stabilizing ability of the polymer. The highestlopes are observed for the mini-emulsions containing PCL andLGA, whereas the slopes for the Eudragits systems were much
ower. So, we can note that the rank order of the stabilizingbility of polymers was the same as for the emulsifying ability:udragit RL ∼= Eudragit RS > PLGA > PCL.
If one formally applies to the coalescence rate of emulsionroplets the kinetic equation of the second order, i.e.:
1
n(t)− 1
n0
]= Kt (1)
here K is the constant of the reaction of second order, it isot difficult to verify that the experimental curves n−1(t) for allystems are linearized after t > 300 s. This signifies that in thisap of time the process of the coalescence obeys to the conditionsf bimolecular reactions.
For example, for Eudragit RS and PCL one findsEud RS ∼= 6 × 10−26 (m3 s−1) and KPCL ∼= 10−23 (m3 s−1),
espectively. Although, the rate of coalescence of emulsionroplet stabilized by Eudragit RS is ∼100 times lower than
or PCL, i.e. the Eudragit RS is ∼100 times better stabilizerhan PCL, nevertheless the order of value of these constantss much lower than that predicted by the von Smolukhowskyheory of fast coagulation of sols: KSm = (4/3)(kBT/η) ∼= 6 ×
V.G. Babak et al. / Colloids and Surfaces B
Fig. 2. Kinetic curves of the cube of the inverse size, d−3(t), for the dropletsof MC-in-water emulsions containing as a dispersed phase the 5% MC solu-tϕ
3
1tTlblamap
3
tcFan75stp
tcpaoitcctbe
3h
3h
oPIt
η
wsammwptt
RcrPtfehcroI(pFig. 3). In this representation, the PLGA macromolecule mustbe also imagined as a rather compact coil similar to the Eudragitpolymers.
ions of the mixture of Eudragit RL and PCL with different concentration ratio= CPCL/(CRL + CPCL): ϕ = 0 (curve 1, �), ϕ = 0.5 (curve 2, ), ϕ = 0.75 (curve, ), ϕ = 1.0 (curve 4, �).
0−18 (m3 s−1), where kB is the Boltzmann constant, T theemperature, η is the viscosity of MC solutions of polymers.his means that in both case we have a slow coagulation (coa-
escence) of droplets which is characterized by the energeticarrier, or by the activation energy Ea of the coagulation (coa-escence). If one formally expresses the experimental constants K ∼ KSm exp(−Ea/kBT), one obtains the following approxi-ate values: EEud RS ∼= 18 kT and EPCL ∼= 13 kT. By the way one
scertains that the difference of 5kBT in the activation energyroduces so remarkable difference in the stability of emulsions.
.1.3. Mini-emulsions stabilized by the polymers blendIt is interesting to note that emulsifying and stabilizing abili-
ies of the mixtures of polymers vary no monotonically with theoncentration ratio of these polymers in the mixed MC solution.or example, in the case of the mixture of cationic, Eudragit RL,nd uncharged, PCL, polymers the droplet size practically doesot vary with the increase of PCL content in the mixture up to5% and is similar to that observed with Eudragit RL taken at% in the MC solution (Fig. 2). These two polymers were cho-en because they display the highest dissimilitude with regardo emulsifying and stabilizing abilities for the MC in the waterhase.
In general, the droplet size of emulsions prepared under iden-ical physico-chemical conditions (time and sonication power,oncentration of polymers, volumes of organic and aqueoushases, etc.) must decrease when both, the interfacial tension γnd the viscosityηof the dispersed phase, decrease. The decreasef both factors leads to the decrease of the dispersive work whichs spending to emulsify the internal phase (in our case this ishe MC solution of polymers). To verify which of these factorsontributes mainly to the mini-emulsion stability we have pro-
eeded to the measurement of the viscosity of MC solutions ofhese polymers, of the interfacial tension of MC/water interfacesearing the adsorption layers of polymers, and of surface andlectric properties of spread monolayers of polymers.FE
: Biointerfaces 59 (2007) 194–207 197
.2. Impact of viscosity of methylene chloride solutions ofydrophobic polymers on droplet size of mini-emulsions
.2.1. Intrinsic viscosity of organic solutions ofydrophobic polymers
The isotherms of the reduced viscosities ηred of MC solutionsf hydrophobic polymers, Eudragits RL and RS, PLGA andCL, in function of their concentration C are presented in Fig. 3.n the studied concentration range, all the isotherms ηred(C) obeyo the Huggins equation:
red = [η] + kH[η]2C (2)
here [η] is the intrinsic viscosity and kH is the Huggins con-tant [18]. The critical overlap concentrations values estimatedccording to the relationship C* = 1/[η] show that for the poly-er PCL the semi-dilution concentration threshold (where theacromolecular coils begin to sterically interact) is at ∼2%hereas for the PLGA it is equal to ∼3%. For the Eudragitolymers the studied concentration range 0–5% corresponds tohe dilute regime because the corresponding values of C* forhese polymers are equal to ∼10%.
The intrinsic viscosity [η] order PCL > PLGA > EudragitS ≥ Eudragit RL signifies that the size of the macromolecularoils of these polymers in the MC solutions (their hydrodynamicadius Rh) ranges in the same order, i.e. it is the highest for theCL and the smallest for the Eudragit polymers. This signifies
hat the thermodynamic quality of the MC as a solvent is highestor the PCL and diminishes for the PLGA and Eudragits. In gen-ral, the better is the solvent thermodynamic quality for a givenydrophobic polymer, the higher the size of the macromolecularoil and the higher the characteristic viscosity [η] will be. Thisule holds even in spite of the much smaller molecular massf the PCL (42 kDa) with regard to the Eudragits (∼150 kDa).n the MC solution, the PCL behaves as a rather extendedswelled) statistical coil whereas the Eudragits admit very com-act (collapsed) conformation (see the right side schema in
ig. 3. Reduced viscosity vs. concentration of MC solutions of polymers: (1)udragit RL (�); (2) Eudragit RS (�); (3) PLGA (©); (4) PCL (�).

1 faces B: Biointerfaces 59 (2007) 194–207
3o
MttdttMwgto
obtesaasotmtcticgctio
bsueb(tmimblr
3h
tis
Fig. 4. Specific viscosity ηexpsp (ϕ), of MC solutions of Eudragit RL and PCL
blends vs. the PCL content ϕ in comparison with the analogical dependenceoTp
wdiη
mtaMltdη
dt
3p
bRcttd
ds
η
w[
98 V.G. Babak et al. / Colloids and Sur
.2.2. Effect of electrostatic interaction on the conformationf hydrophobic polymers in methylene chloride solutions
The compactness of the Eudragit macromolecules in theC may be explained by the electrostatic interaction between
he pendent polar TMA groups. These bulky cationic groupsogether with the condensed counter ions (anions Cl−) behave asipoles and interact by electric forces in MC solution. The elec-rostatic interaction between these dipoles is ∼10 times higherhan in the water phase because the relative dielectric constant of
C being equal to εMC = 9.1 is ∼10 times lower than that for theater, εw = 81. The dipole–dipole interaction between the TMAroups in the MC is responsible for their attraction and associa-ion in aggregates in the MC solution that leads to the reductionf the size of the macromolecular coils of the Eudragits.
This aggregation is analogous to the micelle formationf alkylated polymers in water via hydrophobic interactionetween the alkyl chains which is a driving force of this aggrega-ion. Point out that the electrostatic interaction between chargedlectric groups of water soluble polymers in water is negligiblymall with regard to the hydrophobic interaction between thelkyl chains while the Bjerrum length for the electrostatic inter-ction in water is of the order of lB = e2/4πε0εkBT ∼ 0.2 nm. Thisignifies that at the mean distance between the polymeric chainsf the polyelectrolytes in the water which is higher than ∼1 nmhe energy of the electrostatic attraction between the charges is
uch lower than kBT and thereby may be neglected with regardo the hydrophobic interaction between long alkyl chains and theontribution of the translational entropy of counter ions. Unlikehe water, the Bjerrum length for the electrostatic interactionn MC is of the order of lB = e2/4πε0εkBT ∼ 6 nm, and therebyontributes considerably to the interaction between the chargedroups situated at the distance much lower than lB. One of theonsequences of this increased electrostatic interaction betweenhe dipoles formed by TMA groups and their condensed counterons is the aggregation of the Eudragits in MC and the reductionf the viscosity of the MC solutions.
It must be pointed out that the considerable differenceetween the intrinsic viscosities of the methylene chlorideolutions of PLGA and PCL which have comparable molec-lar masses, 40 and 42 kDa, respectively, may be rationallyxplained by the electrostatic interactions between the car-onyl groups whose dipole moment value is μ0 = 2.7 D1 D = 1.336 × 10−30 C m). Although the dipole–dipole interac-ion between carbonyl groups is relatively low, nevertheless the
ultiplicity of these groups in the macromolecular backbones susceptible to produce the effect of the globulisation of the
acromolecule. For example, twofold higher density of the car-onyl groups of PLGA with regard to PCL produces ∼ twofoldower intrinsic viscosity of the MC solution of the PLGA withegard to the PCL (Fig. 3).
.2.3. Initial droplet size of MC individual solutions ofydrophobic polymers
Comparison between Figs. 1 and 3 shows that the ini-ial droplet size d0 of mini-emulsion gradually increases withncreasing viscosity ηp of organic (methylene chloride, MC)olutions of polymer. This effect is not surprising while it is
va
s
f the initial droplet size, d0, of mini-emulsions of these solutions in water.he linear function ηidsp(ϕ) corresponds to the specific viscosity of ideal mixedolymeric solution constructed according to Eq. (3).
ell known that the viscosity is the factor that increases theispersive power which is necessary to break droplets dur-ng the emulsifying stage. For example, the specific viscositiessp =(ηp − η0)/η0 of 5% MC solutions of PCL and PLGA (whoseolecular mass is ∼40 kDa) are equal to 8.5 and 2.8, respec-
ively, for the corresponding initial droplets size equal to 270nd 140 nm, respectively, where η0 is the cinematic viscosity ofC. The Eudragit polymers which have much higher molecu-
ar mass (M ∼= 150 kDa) exhibit much lower specific viscosity ofheir 5% MC solutions (ηsp ∼= 1.6), and lowest initial droplet size0 ∼= 40 nm. Although the strict proportionality between d0 andsp is not fulfilled, nevertheless a quite pronounced monotonicependence between droplet size and viscosity is observed forhe solutions of individual polymers.
.2.4. Initial droplet size of MC solutions of hydrophobicolymers blends
The monotonic dependence d0(ηsp) is disturbed when thelends of polymers with different structure, such as EudragitL and PCL, are used. Fig. 4 illustrates the effect of the con-entration ratio ϕ = CPCL/(CPCL + CEud) = CPCL/C of the PCL onhe specific viscosity ηsp of C = CPCL + CEud = 5% solutions ofhese polymers in MC, and on the initial droplet size d0 of theirect mini-emulsions.
The experimental curve ηexpsp (ϕ) undergoes a slight positive
eviation from the theoretical curve ηidsp(ϕ) corresponding to thepecific viscosity of ideal mixed polymeric solution [19]:
idsp(ϕ) = ([ηEud](1 − ϕ) + [ηPCL]ϕ)C
+ (KEud(1 − ϕ)2 + K2PCLϕ
2)C2 (3)
here KEud = kEud[ηEud]2 and KPCL = kPCL[ηPCL]2,ηEud] = 0.08 dL/g and [ηPCL] = 0.57 dL/g are characteristic
iscosities of polymers, and kEud = 11 g/dL and kPCL = 0.76 g/dLre the corresponding Huggins constants.Usually, the positive deviation of physical properties of mixedolutions or blends from the ideal conditions is interpreted as

ces B
amtisi
icrttildlvo
3m
rm
3ii
ictiftoto(f
Ff((
io
siatdawwopmir
pMthctbttcseowsn
ffirm that the adsorption of Eudragit polymers is much higherthan that of PLGA and PCL. Regarding the strongly adsorb-
V.G. Babak et al. / Colloids and Surfa
n indication on the less intensive attraction between macro-olecules of different type with regard to the attraction between
he macromolecules of the same type in the solution [20]. Thisnterpretation seems realistic while we have found the phaseeparation in the mixed Eudragit/PCL solutions in MC withncreasing the total polymer concentration up to ∼10% [17].
As concerned the initial droplet size d0 of emulsions, its noteworthy that with increasing the PCL content (bottomurve in Fig. 4) the d0 value increases very slowly in theange ϕ = 0–0.75. In this ϕ range the relative viscosity ofhe mixed solution remarkably increases from η
expsp (0) = 2.2
o ηexpsp (0.75) = 6.6 whereas the initial droplet size d0 was
ncreased from ∼40 to 65 nm only. With approaching to theimit ϕ→ 100% the droplet size increases drastically up to0 ∼ 270 nm when ηexp
sp varies only from 6.5 to 8.5. This noinear dependence d0(ηsp) obviously signifies that the specificiscosity is not a main factor which determines the droplets sizef MC/water mini-emulsions of the mixtures of these polymers.
.3. Interfacial tension of polymer solutions at theethylene chloride/water boundary
Consider now how the interfacial tension of methylene chlo-ide solutions of polymers acts on the droplet size of directini-emulsions.
.3.1. Dynamic interfacial tension and isotherms ofnterfacial tension of individual polymers at MC/waternterface
The dynamic interfacial tension curves γ(t) for differentndividual polymers solutions in MC taken at the same bulk con-entration C = 8 × 10−3% and recorded during relatively longime tf > 103 s of formation of adsorption layers are presentedn Fig. 5. It is to note a remarkable difference between the sur-ace activities of cationic polymers, Eudragits RL and RS, onhe one hand, and uncharged polymers, PLGA and PCL, on thether hand. The former group of polymers remarkably decreases
he interfacial tension γ (curves 1 and 2) whereas the later pairf polymers is characterized by very poor interfacial activitycurves 3 and 4). Fig. 5 proves that the adsorption of polymersrom the organic phase to the MC/water interface is obviouslyig. 5. Kinetic curves of the interfacial tension γ(t) at the MC/water inter-ace for the solutions in MC of different polymers with the bulk concentrationC = 8 × 10−3%, w/v): Eudragit RL (curve 1), Eudragit RS (curve 2), PLGAcurve 3); PCL (curve 4).
ip
Fi
: Biointerfaces 59 (2007) 194–207 199
nfluenced by the presence of charged TMA functional groupsf the polymers.
Another remarkable feature of the dynamic interfacial ten-ion curves γ(t) of Eudragits is that at the time tf = 103 s thenterfacial tension ceases to decrease sharply and is stabilizedt some quasi-equilibrium value. Nevertheless γ(t) continueso decrease with low but measurable rate of the order ofγ/dt ∼ −10−3 (mN/m) s−1. The decrease of γ for irreversiblydsorbing Eudragits with measurable rate during tens of hoursas reported in [21]. Similar interfacial behavior exhibit proteinshich adsorb irreversibly from water to the boundary with airr oil [22]. This tendency is less pronounced for the PLGA andractically imperceptible for PCL. So, each hydrophobic poly-er behaves very differently at the methylene chloride/water
nterface, and the interfacial tension values γ confirm the orderank PCL > PLGA > Eudragit RS ≥ Eudragit RL.
Comparatively low interfacial tension obtained with Eudragitolymers may be explained by their strong adsorption at theC/water interface. The driving force of this adsorption is
he important gain in free energy due to the ionization andydration of the hydrophilic quaternary ammonium groups andounter ions in contact with the water phase [21]. Moreover,he adsorption of Eudragit polymers is practically irreversibleecause of the multiple number of ionized groups anchored athe interface which increases with the storage time because ofhe rearrangement of polymer molecules from coil toward flatonformation as represents Fig. 6a. The lowest interfacial ten-ion value of Eudragit RL with regards to Eudragit RS may bexplained by the higher quaternary ammonium group’s densityf Eudragit RL. On the other hand, PLGA and PCL polymershich have not ionizable hydrophilic groups, do not display any
ignificant adsorption at the MC/water interface, explaining theon-significant decrease in the interfacial tension (Fig. 5).
Fig. 7 illustrates the isotherms of the interfacial tension γ(C)or the MC solutions of these polymers. These isotherms con-
ng Eudragit polymers which “anchor” to the water phase byendent hydrophilic TMA groups (Fig. 6a) we could try to
ig. 6. Schematic representation of Eudragit, PLGA and PCL macromoleculesn the bulk of MC solution and at the MC/water interface.

200 V.G. Babak et al. / Colloids and Surfaces B: Biointerfaces 59 (2007) 194–207
Fig. 7. Isotherms of the interfacial tension of different MC solutions of polymeri(s
eaFdtolpec
G
Γ
tptgrtAwtotToi
ftug∼m
Fvd
alcvrfltt(o
Tcm
3b
pnTopt
iϕ
spWpddo
n contact with water: Eudragit RL (curve 1,�), Eudragit RS (curve 2, �), PLGAcurve 3, ©); PCL (curve 4, �). The dashed line AB is drawn to visualize thelope dγ/dlog C corresponding to Eudragit polymers.
stimate the surface density of these adsorbed TMA groupst the MC/water interface under some reasonable assumptions.or example, we can assume that the main contribution to theecrease of the interfacial tension is given by the adsorption ofhese TMA groups that allows us to neglect by the adsorptionf hydrophobic segments of Eudragit macromolecules behavingike the hydrophobic backbone of non-adsorbing PCL or PLGAolymers. Analogical assumption has been made concerning thestimation of the density of adsorbed dodecyl chains which wereovalently grafted to hydrophilic polyelectrolytes [23].
Under above mentioned assumption, we can try to apply theibbs adsorption equation:
= 0.43
RT
dγ
dlogC(4)
o estimate the adsorption amount Γ (or the area aTMA = 1/ΓNAer one pending TMA group of the Eudragit) in the adsorp-ion layer. From Fig. 7 on obtains: dγ/dlog C ∼= 17 mN/m thatives for the adsorption amount of TMA groups in the satu-ated adsorption layer the value Γ ∼= 7 × 10−6 mol/m2, and forhe mean area per one TMA group the value aTMA = 0.25 nm2.lthough the obtained value for the area aTMA is consistentith the geometric (steric) dimension of a TMA group, never-
heless one must bear in mind that the produced estimation isbtained under realistic but nevertheless rather artificial assump-ions such as an assumption of a quasi independent motion ofMA groups in the solution and the absence of the adsorption ofther segments of the Eudragit macromolecule at the MC/waternterface.
The first assumption may be justified if one takes into accountor low content (2.5–5 mol%) of TMA groups statistically dis-ributed along the Eudragit macromolecules whose repeating
nits have the mass ∼164 g/mol. The total number of TMAroups per macromolecular chain (M ∼ 150 kDa) is equal to20 and ∼40 for Eudragit RS and RL, respectively, and theean distance between these TMA groups corresponds to ∼25wi
ϕ
ig. 8. Interfacial tension γ of 5% MC solutions of Eudragit RL and PCL blendss. the PCL contentϕ in comparison with the analogical dependence of the initialroplet size, d0, of mini-emulsions of these solutions in water.
nd ∼50 repeating segments, respectively. On account for theength of one repeating segment ∼3 nm, one has for the meanontour length lTMA between the neighboring TNA groups thealues ∼75 and ∼150 nm, respectively, that is much higher withegard to the persistent (Kuhn) length lK ∼= 2 nm for the chainexible polymethacrylate polymers [24]. So, we can admit that
he TMA groups behave as quasi-independent in the MC solu-ion, and the application of the Gibbs adsorption isotherm (Eq.3)) is admissible for these rough estimation of the parametersf adsorption layers of Eudragits.
It is to note, that the rough estimation of the area per oneMA groups in a saturated adsorption layer aTMA = 0.25 nm2
orrelates well with the corresponding area inside the Langmuironolayers of the Eudragit polymers (see Section 3.4).
.3.2. Interfacial tension of MC solution of polymericlends at MC/water interface
As concerned the interfacial tension γ of mixed 5% MColymeric solutions Eudragit RL/PCL, we have found a flagranton-linear dependence of γ on the concentration ratio ϕ (Fig. 8).he γ(ϕ) curve practically coincides with the dependence d0(ϕ)f the initial droplet size on ϕ that allows us to conclude for theroportionality between the droplet size d0 and the interfacialension γ .
A somewhat unusual behavior of the function γ(ϕ) when thenterfacial tension value does not practically vary in the range= 0–0.75 being equal to that of the Eudragit, and afterwards
harply increases to the value γ ∼= 28 mN/m corresponding toure PCL solution, requires to be explained and interpreted.e anticipate that the more surface active Eudragit RL tends to
referentially adsorb at the MC/water interface that leads to theecrease of the interfacial tension. Unlike Eudragit RL, the PCLoes not posses strong adsorbing groups, and remains in the bulkf the MC solution. This conclusion seems quite reasonable if
e compare the γ(ϕ) values of Fig. 8 with the isotherm of thenterfacial tension for the Eudragit RL (curve 1, Fig. 7).Effectively, the mixed 5% MC solution of Eudragit/PCL with
= 0.75 contains the Eudragit RL with the partial concentration
ces B: Biointerfaces 59 (2007) 194–207 201
CtwictmlbbhaitmEgmoiCt
idc(st
3
pdtwtpmgwplc
aftifoscdtm
Fs2
btπ
aoiip
3i
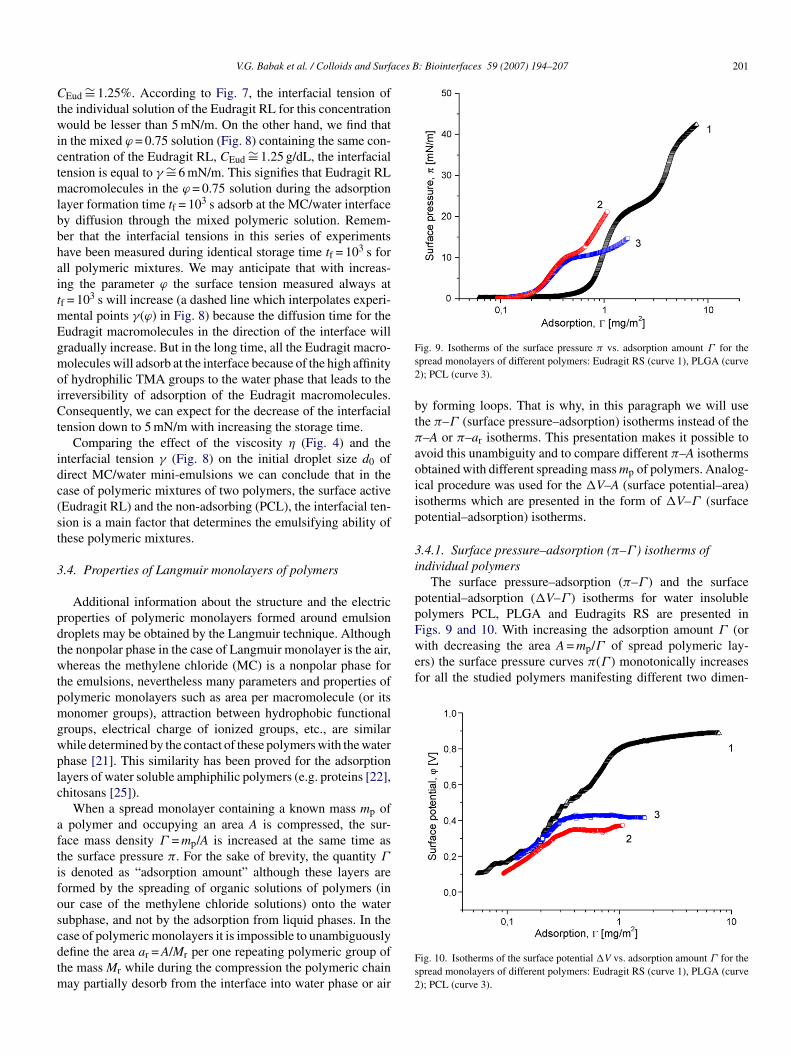
ppFigs. 9 and 10. With increasing the adsorption amount Γ (orwith decreasing the area A = mp/Γ of spread polymeric lay-ers) the surface pressure curves π(Γ ) monotonically increasesfor all the studied polymers manifesting different two dimen-
V.G. Babak et al. / Colloids and Surfa
Eud ∼= 1.25%. According to Fig. 7, the interfacial tension ofhe individual solution of the Eudragit RL for this concentrationould be lesser than 5 mN/m. On the other hand, we find that
n the mixed ϕ = 0.75 solution (Fig. 8) containing the same con-entration of the Eudragit RL, CEud ∼= 1.25 g/dL, the interfacialension is equal to γ ∼= 6 mN/m. This signifies that Eudragit RL
acromolecules in the ϕ = 0.75 solution during the adsorptionayer formation time tf = 103 s adsorb at the MC/water interfacey diffusion through the mixed polymeric solution. Remem-er that the interfacial tensions in this series of experimentsave been measured during identical storage time tf = 103 s forll polymeric mixtures. We may anticipate that with increas-ng the parameter ϕ the surface tension measured always atf = 103 s will increase (a dashed line which interpolates experi-
ental points γ(ϕ) in Fig. 8) because the diffusion time for theudragit macromolecules in the direction of the interface willradually increase. But in the long time, all the Eudragit macro-olecules will adsorb at the interface because of the high affinity
f hydrophilic TMA groups to the water phase that leads to therreversibility of adsorption of the Eudragit macromolecules.onsequently, we can expect for the decrease of the interfacial
ension down to 5 mN/m with increasing the storage time.Comparing the effect of the viscosity η (Fig. 4) and the
nterfacial tension γ (Fig. 8) on the initial droplet size d0 ofirect MC/water mini-emulsions we can conclude that in thease of polymeric mixtures of two polymers, the surface activeEudragit RL) and the non-adsorbing (PCL), the interfacial ten-ion is a main factor that determines the emulsifying ability ofhese polymeric mixtures.
.4. Properties of Langmuir monolayers of polymers
Additional information about the structure and the electricroperties of polymeric monolayers formed around emulsionroplets may be obtained by the Langmuir technique. Althoughhe nonpolar phase in the case of Langmuir monolayer is the air,hereas the methylene chloride (MC) is a nonpolar phase for
he emulsions, nevertheless many parameters and properties ofolymeric monolayers such as area per macromolecule (or itsonomer groups), attraction between hydrophobic functional
roups, electrical charge of ionized groups, etc., are similarhile determined by the contact of these polymers with the waterhase [21]. This similarity has been proved for the adsorptionayers of water soluble amphiphilic polymers (e.g. proteins [22],hitosans [25]).
When a spread monolayer containing a known mass mp ofpolymer and occupying an area A is compressed, the sur-
ace mass density Γ = mp/A is increased at the same time ashe surface pressure π. For the sake of brevity, the quantity Γs denoted as “adsorption amount” although these layers areormed by the spreading of organic solutions of polymers (inur case of the methylene chloride solutions) onto the waterubphase, and not by the adsorption from liquid phases. In the
ase of polymeric monolayers it is impossible to unambiguouslyefine the area ar = A/Mr per one repeating polymeric group ofhe mass Mr while during the compression the polymeric chainay partially desorb from the interface into water phase or air
Fs2
ig. 9. Isotherms of the surface pressure π vs. adsorption amount Γ for thepread monolayers of different polymers: Eudragit RS (curve 1), PLGA (curve); PCL (curve 3).
y forming loops. That is why, in this paragraph we will usehe π–Γ (surface pressure–adsorption) isotherms instead of the–A or π–ar isotherms. This presentation makes it possible tovoid this unambiguity and to compare different π–A isothermsbtained with different spreading mass mp of polymers. Analog-cal procedure was used for the �V–A (surface potential–area)sotherms which are presented in the form of �V–Γ (surfaceotential–adsorption) isotherms.
.4.1. Surface pressure–adsorption (π–Γ ) isotherms ofndividual polymers
The surface pressure–adsorption (π–Γ ) and the surfaceotential–adsorption (�V–Γ ) isotherms for water insolubleolymers PCL, PLGA and Eudragits RS are presented in
ig. 10. Isotherms of the surface potential �V vs. adsorption amount Γ for thepread monolayers of different polymers: Eudragit RS (curve 1), PLGA (curve); PCL (curve 3).

2 faces
s((iπ
ramsfap
mriiTGtoftimpfhi
optot(mha∼oispctspπ
pPwi
mw∼
i(π
tottcoThPep
3i
ltcilro�
(pit(
tfatAvlt
�
wdpto(att
02 V.G. Babak et al. / Colloids and Sur
ional phase states and obeying to the usual sequences: gaseousG) → liquid extended (LE) → liquid condensed (LC) → solidS) states. The transfer G → LE corresponds to the sharpncrease of the surface pressure π from near zero values to∼ 10 mN/m. The transfer LE → LC corresponds to the plateau
egion occurring at πG→LE ∼ 10 mN/m for PLGA and PCL andt πG→LE ∼ 20 mN/m for Eudragit polymers. Compression ofonolayers later on produces the transfer from LC to solid like
tate that is accompanied by a later sharp increase of the sur-ace pressure which is ended by the collapse of the monolayerst some critical value π*. Some difference between the surfaceressure–adsorption (π–Γ ) isotherms must be pointed out.
First of all, the G → LE transfer for the non-charged poly-ers, PLGA and PCL, occurs when the adsorption amount
eaches the value ΓG→LE ∼ 0.2 mg/m2 whereas the correspond-ng adsorption amount for the cationic polymer ∼Eudragit RSs ∼3 times higher being equal to ΓG→LE ∼ 0.6 mg/m2 (Fig. 9).he areas a1 per one repeating unit of these polymers at the→ LE transfer are equal to 1.1, 0.94 and 0.45 nm2, respec-
ively, that approximately correlates with the steric dimensionsf these repeating groups. Somewhat lower value a1 = 0.45 nm2
or the Eudragit could be related to the more hydrophilic charac-er of this polymer: the charged TMA groups produce a partialmmersion of hydrophobic chains into water phase with the for-
ation of loops. This diminishes a real mass of the Eudragitolymer remaining at the air–water surface which is responsibleor the surface pressure effect. Unlike the Eudragit, the moreydrophobic chains of PLGA and PCL remain wholly at thenterface and contribute to the surface pressure.
Secondly, the LE → LC transfer for the studied polymersccurs at different surface pressure values corresponding to thelateau of π(Γ ) isotherms. The plateau region corresponds tohe phase transition between extended and condensed statesf the macromolecules at the interface. It is not surprisinghat the πLE→LC values for PLGA and PCL are much lowerπLE→LC ∼ 10 mN/m) with regard to that for Eudragit poly-er (πLE→LC ∼ 22 mN/m) while the former polymers are less
ydrophilic than the later (Fig. 9). The corresponding areas1 per one repeating unit are equal to ∼0.5 nm2 (PLGA),0.45 nm2 (PCLA) and ∼0.2 nm2 (Eudragit). These values are
bviously lesser than the steric dimensions of these repeat-ng groups testifying for the aggregation processes inside thepread monolayers. Effectively, when polymeric chains are com-ressed, the attraction acting between hydrophobic segments inontact with the water (the hydrophobic interaction) produceshe aggregation between the chains at some degree of compres-ion. More hydrophobic is polymer, lower will be the surfaceressure πLE→LC corresponding to the plateau region in the(Γ ) isotherm. Shorter interval of Γ which corresponds to thelateau of the π(Γ ) isotherm for the PLGA with regard to theCL may be explained by higher stiffness of the former polymerhich has much higher linear density of polar carbonyl groups
n the macromolecular backbone.
And finally, the collapse under compression of Eudragitonolayers occurs at the critical surface pressureπ* ∼ 40 mN/mhereas PLGA and PCL monolayers are collapsed only at20 and ∼15 mN/m, respectively (Fig. 9). The correspond-
gour
B: Biointerfaces 59 (2007) 194–207
ng areas a1 per one repeating unit are equal to ∼0.2 nm2
PLGA) ∼0.13 nm2 (PCLA) and ∼0.03 nm2 (Eudragit). The* value characterizes the stability of solid-like monolayers
o the compression and correlates with the adsorption energyf polymer in contact with the water subphase. The adsorp-ion energy of ionized TMA groups of Eudragit polymers athe air/water interface is much higher with regard to that ofarbonyl groups. So, it is not surprising that the monolayersf Eudragit polymers which are anchored to the water by itsMA groups are much stable with regard to the non-chargedydrophobic PLGA and PCL polymers. Higher π* value ofLGA with regard to PCL is explained by much higher lin-ar density of carbonyl polar groups interacting with the waterhase.
.4.2. Electric surface potential–adsorption (�V–Γ )sotherms of individual polymers
As concerned the surface potential �V of polymeric mono-ayers (Fig. 10), it is much higher for Eudragit RS with regardo PLGA and PCL. The general trend of the surface potentialurves �V(Γ ) for all polymers is the linear dependence on Γn gaseous (G) and liquid extended (LE) regions, and the stabi-ization at some practically constant�V value after having beeneached the adsorption amount corresponding to the beginningf the LE → LC transfer. With the later increase of Γ , the curveV(Γ ) does not practically vary even in the LC → S region
Fig. 10). The maximum value of the surface potential�V at thelateau region is equal to ∼0.85 V for the Eudragit RS whereast is equal only to ∼0.3 and ∼0.4 V for PLGA and PCL, respec-ively. The corresponding value for the Eudragit RL is ∼0.90 Vdata not shown) [17].
The surface potential �V arises from the dipole moments ofhe polar functional groups of polymers, from the double layerormed by ionized groups and corresponding counter ions of thequeous bulk phase, and also from the change in orientation ofhe water molecules of the subphase near the interface [26,27].ccording to the Demchak–Fort model [28] and its modifiedariant [29] the surface potential of insoluble monolayers ofow molecular weight amphiphiles is commonly presented ashe sum:
V = n
ε0
[μw
εw+ μsw
εsw+ μsa
εsa
]+ ψ0 (5)
here n is the numerical surface density (expressed in m−2) ofipoles (of surfactant molecules) in the layer, ε0 the dielectricermittivity of vacuum, μw, μsw and μsa are the contribu-ions to the surface dipole moment from normal componentsf dipoles moments of water molecules, spread film terminalin the water) and spread film terminal (in the air), respectively,nd εw, εsw and εsa are local effective dielectric constants ofhe respective regions in which the dipoles are embedded, ψ0 ishe double layer potential provided from the ionized functional
roups of the monolayer. It has been found that the contributionf water molecules being equal to μw/εw = −0.065 D in Debyenits (1 D = 3.33564 × 10−30 C m) is relatively much low withegard to the contributions of other components. That is why
V.G. Babak et al. / Colloids and Surfaces B
Fig. 11. Schematic representation of spread monolayers of hydrophobic poly-mers in the extended and condensed states. The ionized TMA groups of theEudragits together with their counter ions Cl− form an electric double layerwith a relatively high effective dipole moment μ , whereas the carbonylgs
i[
cvhmwo[tta1ct
aibaicpipfdtVho
s
uc
�
woGumac2
3bmE
�
wctmapLb(t�
iatamotbada
P0hu(iPedwhen passing from gaseous to liquid condensed state (Fig. 11).
TMA
roups of PLGA and PCL contribute to the surface potential with relativelymall dipole moment μC O.
n the Vogel–Mobius model one neglects by this contribution30].
It must be pointed out some controversy in the literature con-erning the relative dielectric permittivity εsw and εsa whosealues depend on the type of the surfactant hydrophilic andydrophobic groups and on the degree of compression of theonolayers. Usually, one attributes to the permittivity of theater region the numerical value εsw = 6.4–7.6, whereas to thatf the air region the conventional value εsa = 2.8 is usually chosen27,29]. The dipole moments for different functional groups areabulated in [31]: for example μsa = 0.33 D for the methyl CH3erminal group, whereas the dipole moments μsw for the acid,lcohol, ester, and amine head groups are equal to 0.99, 1.00,.11, and 1.50 D, respectively. Note that the dipole moment of aarbonyl group in the case of PLGA and PCL polymers is equalo μsw = 2.7 D.
In our case of insoluble polymeric spread monolayers at their–water interface the hydrophobic macromolecular backbonesnclude pending TMA groups (Eudragit) and constitutive car-onyl groups (PLGA, PCL). It is assumed that the polar carbonylnd ionized TMA groups have the tendency to be in contact ormmersed in the water phase (Fig. 11), whereas hydrophobichains are preferentially located in the air phase but could beartially immersed in the water phase (as in the case of Eudrag-ts). It is more than likely that the contribution to the surfaceotential from these hydrophobic chains is much lower thanrom the electric dipoles of the carbonyl groups and the electricouble layer formed by ionized TMA groups. On account forhe obvious difficulty of applying the Demchak–Fort [28] andogel–Mobius [30] models for these polymeric monolayers, weave chosen the simplest Helmholtz model to analyze the effect
f compression of monolayers on their surface potential.In the frames of this Helmholtz model a monolayer is con-idered as a parallel plate condenser, comprising a sheet of
Idt
: Biointerfaces 59 (2007) 194–207 203
niformly distributed dipoles and ionized groups with theirounter ions, leading to a surface potential described by
V = nμ⊥ε0ε
+ ψ0 (6)
here μ⊥ is the normal component of the dipole momentsf a polar group, ε the relative dielectric constant, ψ0 is theouy–Chapman potential (see Eq. (5)). In many case, whensing this model, one assumes that ε= 1 [26] either becauseolecules are treated as isolated entities or owing to the lack ofknown value [29]. Schuhmann [32] suggested that for the short-hain hydrocarbons this dielectric permittivity must be taken as.
.4.2.1. Surface potential of uncharged polymers. Eq. (6) maye used to the numerical estimation of the surface potential�V ofonolayers formed by PCL or PLGA. For the PCL monolayersq. (6) may be rewritten in the form
VPCLΓNAμC O cos θ
MPCLε0ε∼= 2ΓμC O cos θ
ε(7)
here MPCL = 114 g/mol is the molecular mass of the monomerontaining one carbonyl group, NA the Avogadro number, Γhe adsorption amount (expressed in mg/m2), μC O the dipole
oment of one carbonyl group (expressed in Debyes), θ the tiltngle of the dipole from the normal to the surface. For exam-le, for the adsorption Γ = 0.3 mg/m2 which corresponds to theE–LC transfer in the PCL monolayers (Fig. 9) and to the sta-ilization of the potential in the plateau region (Fig. 10), Eq.7) gives �V ∗
PCL∼= 1.62 cos θ/ε. The numerical estimation of
he surface potential correlates well with the experimental valueV ∗
PCL∼= 0.4 V if one assume that ε= 2 (as has been found
n [32]) and cos θ = 0.5 (by averaging the dispersion of the tiltngle θ from the normal). The averaging has been made underhe assumption of the Gaussian dispersion of the angle θ onccount for the relatively high area par one carbonyl group in theonolayer: aC O = MC O/ΓNA ∼= 0.6 nm2. In general, because
f the complexity of the structure of polymeric monolayers,he obtained numerical estimation for the surface potential maye considered as quite satisfactory. Point out that with thesessumptions made, Eq. (7) describes well the experimental linearependence of the surface potential�VPCL(Γ ) of the adsorptionmount Γ (Fig. 10).
Interestingly, that the experimental surface potential of theLGA monolayers in the plateau region is equal to �V ∗
PLGA∼=
.3 V that is lesser than that of the PCL in spite of the ∼2 timesigher linear density of the carbonyl groups in the macromolec-lar backbone of the former (MPLGA = 65 g/mol) than of the laterMPCL = 114 g/mol). This may be explained by higher flexibil-ty of the PCL macromolecular chains with regard to those ofLGA. The former chains include relatively long –C5H10– spac-rs which could desorb by forming loops in the air that permit toipoles to remain in the water phase with the initial orientation
n the case of lesser flexible PLGA chains, the compressionestroys the normal orientation of dipoles. As the consequence,he compensation (neutralisation) of the electric champs of the

2 faces
nslsest
3smTe
ψ
webt
æ
ii
σ
imgtissft
rg(t�
tTTan
dw5i3
Eig
sIi6mraitamru∼δ
sct
3
tmpmrptϕ
tvc
ual variation of their shape with increasing the parameter ϕ.Although the curves with intermediate values of ϕ are locatedbetween those corresponding to ϕ = 0 (pure Eudragit) and ϕ = 1(pure PCL), nevertheless the shape of the isotherms (π–Γ ) cor-
04 V.G. Babak et al. / Colloids and Sur
eighboring dipoles occurs that produces the decrease of theurface potential. Only in the region of the collapse of the mono-ayers (at aPLGA ∼= 0.2 nm2) one observes some increase of theurface potential that may be explained by the effect of thelectrostatic induction between the dipoles inside the destroyedolid-like monolayer that favours the orientation of dipoles inhe normal direction.
.4.2.2. Surface potential of Eudragit RS. As concerned theurface potential of Eudragits monolayers, it is mainly deter-ined by the contribution of the double-layer of the ionizedMA groups. According to the Gouy–Chapman theory thexpression for this potential ψ0 may be written in the form
0 = 4kBT
etanh
(eσ
4kBTε0εæ
)∼= σδDH
ε0ε(8)
here kB the Boltzmann constant, T the absolute temperature,the elementary charge, ε the relative dielectric constant of theulk phase, σ the surface density of ionized groups, δDH = 1/æhe Debye–Huckel length:
= 1
δDH=
√e2NAC
ε0εkBT(9)
s the Debye–Huckel parameter, C the concentration of counterons in the bulk phase [18]. The product
δDH = nTMAμTMA (10)
n Eq. (8) may be formally considered as a cumulative dipoleoment of the elementary dipoles formed by the pairs of TMA
roups and their counter ions, whereμTMA =αeδDH is the effec-ive dipole moment of one TMA-ion pair, α the degree ofonization of the TMA group, nTMA the numerical surface den-ity of these TMA groups in the monolayer (Fig. 11). In thisemi-quantitative consideration, one neglects by the contributionrom constant dipoles of other functional groups of the Eudragito the surface potential.
It is interesting to note that Eudragit monolayers exhibitelatively high surface potential �V ∼ 0.5 V already in theaseous state where the adsorption is equal toΓ Eud ∼ 0.4 mg/m2
Fig. 10). At the moment of the LE → LC transfer the adsorp-ion is equal to Γ RS ∼ 1 mg/m2 and the surface potential to
V ∼ 0.8 V. On account for the 2.5 mol% of TMA groups ofhe Eudragit RS, the molecular mass of this polymers per oneMA group is equal to MTMA = 0.025. MRS/0.025 = 6650 g/mol.he surface densities of the TMA groups at G and LC statesre estimated asnG
TMA = ΓGRSNA/MTMA ∼= 3.6 × 1016 m−2 and
LCTMA
∼= 9.0 × 1016 m−2, respectively. This corresponds to theensities of the surface charges (under the assumption of thehole ionization of TMA groups) equal to σG = enG
TMA∼=
.8 C m−2 and σLC ∼= 14.5 C m−2, respectively. The correspond-ng areas per one TMA group are equal to aG
TMA = 1/nGTMA
∼=0 nm2 and aLC
TMA∼= 10 nm2, respectively.
Estimate now the Debye–Huckel lengths δGDH and δLC
DH inq. (4), or the effective dipole moments μG
TMA and μLCTMA
n Eq. (7), for the surface densities of the TMA groups inaseous G and LC states, using corresponding experimental
Fm2
B: Biointerfaces 59 (2007) 194–207
urface potentials ψG0 = 0.5 V and ψG
0 = 0.8 V, respectively.nterestingly, that one obtains for both states identical numer-cal values of the Debye–Huckel lengths δG
DH = ε0εψG0 /σ
G ∼=0 nm and δLC
DH∼= 60 nm, and also for of the effective dipole
oments μGTMA = ε0εψ
G0 /n
GTMA
∼= 30 D and μLCTMA
∼= 30 D,espectively. This coincidence testifies for the correctness of ourssumptions made in describing the double layer potential. Thencreased value for the effective dipole moment in the case ofhe double layer with regard to the uncharged polymers, PCLnd PLGA, is not surprising if one takes into account for theuch higher thickness of the double-layer δDC ∼ 60 nm with
egard to the characteristic lengths of the dipoles of polar molec-lar groups, e.g. of the carbonyl group, which is of the order of1 A. Note that according to Eq. (8), the Debye–Huckel lengths
DC ∼ 60 nm corresponds to the bulk concentration of ions in theubphase equal to C ∼ 5 × 10−5 mol/L. The later low ionic con-entration value is quite valuable for the local subphase layer ofhe pure water in the vicinity of the charged monolayer.
.4.3. Surface properties of mixed Langmuir monolayersIn the context of the present work we have undertaken
he study of the properties of Langmuir monolayers for theixtures of polymers. As an example, we have tested the
air PCL and Eudragit RS as two hydrophobic polymers witharkedly different bulk and interfacial behavior. Figs. 12 and 13
epresent the surface pressure–adsorption (π–Γ ) and surfaceotential–adsorption (�V–Γ ) isotherms for the mixtures ofhese polymers corresponding to different concentration ratios=Γ PCL/(Γ PCL +Γ Eud) which vary from 0 to 1. It is to note that
he adsorption amount Γ = m/A of the mixtures has been con-entionally reported to the total mass m = mRS + mPCL of twoomponents.
The general trend of (π–Γ ) and (�V–Γ ) isotherms is a grad-
ig. 12. Effect of the composition ϕ = CPCL/(CRS + CPCL) of the mixed spreadonolayers on the form of the π–Γ isotherms: ϕ = 0 (curve 1), ϕ = 0.25 (curve
); ϕ = 0.5 (curve 3), ϕ = 0.75 (curve 4), ϕ = 1.0 (curve 5).

V.G. Babak et al. / Colloids and Surfaces B
Fm2
rltPi
Γ
caf(Ittthtfc
ccrtcpda4�
pia
�
sof
Maipotmoromtihi
psoClΓ
Ont
iorpthiCmssbi
3h
srofiawlp
ig. 13. Effect of the composition ϕ = CPCL/(CRS + CPCL) of the mixed spreadonolayers on the form of the�V–Γ isotherms: ϕ = 0 (curve 1), ϕ = 0.25 (curve
); ϕ = 0.5 (curve 3), ϕ = 0.75 (curve 4), ϕ = 1.0 (curve 5).
esponding to ϕ = 0.25 and 0.50 (curves 2 and 3, Fig. 12) areike that of pure Eudragit (curve 1, Fig. 12) whereas the shape ofhe curve 4 corresponding to ϕ = 0.75 is like that of the pureCL (curve 5, Fig. 12). The variations of the shapes of the
ntermediate isotherms (�V–Γ ) are more smooth (Fig. 13).For relatively low adsorption amounts of polymeric mixtures,< 0.8–0.9 mg/m2, the isotherm (π–Γ ) for ϕ = 0.75 practically
oincides with the curve corresponding to pure PCL (curves 4nd 5). These curves continue to be practically coincided withurther increase ofΓ up to 1–2 mg/m2, and afterward the curve 4ϕ = 0.75) sharply increases tending to reach the curve 1 (ϕ = 0).t is interesting to note that the curve 4 represented as the func-ion of the partial adsorption amount Γ Eud of the Eudragit inhe mixture, Γ Eud =(1 −ϕ) Γ =Γ /4, practically coincides withhe curve 1 for the pure Eudragit for the surface pressure valueigher than ∼10 mN/M (plateau region for PCL). At the sameime, at lower adsorption values, Γ < 0.8–0.9 mg/m2, the sur-ace pressure is determined by the PCL only (coincidence of theurves 4 and 5).
As concerned the electric properties of this mixed monolayer,orresponding to ϕ = 0.75, the curve 4 (Fig. 13) passes above theurve 5 (pure PCL). The surface potential�Vϕ=0.75 of the plateauegion exceeds that for the pure PCL (curve 5) by ∼0.1 V forhe adsorption Γ < 2 mg/m2. This value �V ∼ 0.1 V perfectlyorrelates with the surface potential isotherm �V –Γ for theure Eudragit (curve 1, Fig. 10) if one takes into account theifferences between the total and partial Γ Eud =Γ /4 adsorptionmounts of this polymer. In the region of Γ > 2 mg/m2 the curveexhibits a trend for increasing but the corresponding valueV ∼ 0.15 V of this increase is much lower than that of the
ure Eudragit (curve 1, Fig. 10). This may be considered as anndicative of the screening of ionized TMA groups as a result ofphase separation inside the mixed layer.
The fact that the surface pressure π and the electric potential
V of spread mixed polymeric layers are determined by lesserurface active and more hydrophobic PCL is a direct oppositef the case of mixed adsorption layers. For example, we haveound that the interfacial tension γ of the Eudragit/PCL mixed
pmc
: Biointerfaces 59 (2007) 194–207 205
C solution (Fig. 8) is determined mainly by the more surfacective and hydrophilic component, Eudragit. But we must bearn mind that in the case of the adsorption layers the lesser polarolymer, PCL, has the possibility to leave interface into the bulkf the methylene chloride solution that leads to the decrease ofhe local composition ratio ϕ =Γ PCL/(Γ RS +Γ PCL) inside this
ixed adsorption layers. This possibility is denied in the casef insoluble spread layers, where the local composition ratio ϕemains the same in the volume of this layer, but the contributionf polymers to the surface pressure is dependent on their confor-ation and phase state. Briefly, in the mixed spread monolayers
he Eudragit is displaced from the interface by the PCL, whereasn the mixed adsorption monolayer there is an opposite: the moreydrophilic Eudragit displaces the less hydrophilic PCL from thenterface.
If one estimates the local bulk concentration Csl of theolymer mixtures inside the spread layers (“sl”) by the relation-hip: Csl =Γ /δsl where δsl ∼ 1–5 nm is the effective thicknessf the monolayer, one obtains for Γ = 0.1 mg/m2 the valueml ∼ 2–10%. With further compression of a mixed layer the
ocal concentration Csl increases, and for the adsorption value∼ 1 mg/m2 the local concentration is increased moreover.bviously, that the proportionality between Cm and Γ doesot hold because of the possible simultaneous increase of thehickness δsl of the layer.
Previously, it has been shown [33] that the phase separationn the mixed MC solutions with the composition ϕ = 0.50–0.75ccurs in the total concentration rangeC∗
p ∼ 8–10% (the detailedeport on these results is preparing for the publication). The moreolar and less soluble in MC Eudragit separates from the solu-ion in form of nano-clusters. It has been shown as well that forigher ratiosϕ > 0.80 the critical concentration increases achiev-ng the value C∗
p ∼ 16% characteristic to the pure PCL solution.oncerning the mixed spread monolayers we may anticipate thatore polar Eudragit at such increased local concentrations could
eparate in form of clusters that decreases its contribution to theurface pressure and to the surface potential. This is confirmedy previous consideration and discussion of π–Γ and �V–Γsotherms (Figs. 12 and 13).
.5. Mechanism of the stabilization of mini-emulsions byydrophobic polymers
Considering together, the obtained results concerning thetabilizing ability of different polymers and their blends withegard to the stability of mini-emulsions and the zeta-potential ofbtained nanoparticles, on the one hand, and the bulk and inter-acial properties of these polymers (viscosity of MC solutions,nterfacial activity at the MC/water interface, surface pressurend electric potential of spread monolayers), on the other hand,e are able to propose a valuable mechanism of the stabi-
ization of direct (MC-in-water) mini-emulsions containing theolymers dissolved inside the internal organic phase.
The highest emulsifying and stabilizing ability of the cationicolymers, Eudragit RL and RS, compared to uncharged poly-ers, PCL and PLGA, is due to the formation of the corona of
harged pendent trimethylammonium (TMA) groups of cationic

206 V.G. Babak et al. / Colloids and Surfaces B: Biointerfaces 59 (2007) 194–207
of Eu
ptbpataigiohttota
htiTpeoboplg
E0EImMi∼p
tbwaϕ
tEtmbniadnfimtpf�
(
abebctc
4
b
Fig. 14. Scheme illustrating why the stabilizing ability
olymers around the droplets (Fig. 14a). In the MC solution,hese polar TMA groups with their condensed counter ionsehave as dipoles determining the associative properties of theseolymers, e.g. relatively low viscosity of MC solutions (Fig. 3),nd relatively high surface potentials (Fig. 10). On account forhe increased total gain in enthalpy due to the hydratation effectnd in translational entropy due to the implication of counter ionsnto the Brownian motion, the adsorption of these pendent TMAroups is practically irreversible [21]. The electrostatic repulsions a well-known stabilizing factor according to the DLVO the-ry. In addition, the steric repulsion between the loops bearingydrophilic charged groups of Eudragits may also contribute tohe overall droplet stability and act against the coalescence. So,he stability of emulsion droplets against coalescence in the casef Eudragits RL and RS is due the electro-sterical effect, i.e. tohe repulsion between ionized coronas of pendent TMA groupsnd polymeric loops.
Unlike the cationic polymers Eudragits, the unchargedydrophobic polymers, PCL and PLGA, adsorb very poor athe MC/water interface by putting their polar carbonyl croupsn contact with the water phase (as indicated in Fig. 14b) [21].his orientation of carbonyl groups in contact with the water isroved by the positive surface potential�V of spread monolay-rs of PLGA and PCL (Fig. 10) and low but finite zeta-potentialf nano-particles obtained by evaporation of mini-emulsions sta-ilized by PCL and PLGA [12]. Very low energy of adsorptionf these polymers explains why their stabilizing ability is veryoor. The stabilizing ability of PLGA is still low but neverthe-ess more important than PCL due to its higher content of polarroups and, consequently, to higher adsorption ability (Fig. 7).
To explain why the mixtures of hydrophobic polymers,udragit and PCL, with the composition ratios ϕ in the range.25–0.75 stabilize mini-emulsions very well (as well as theudragit alone (Fig. 2)), we propose the following mechanism.
n the emulsions whose MC droplets contain 5 wt% of poly-ers, the more surface active Eudragit adsorbs irreversibly at the
C/water interface whereas the non-surface active PCL remainsn the core of emulsion droplets. According to Fig. 7, even the10 times lower concentration of the Eudragit, e.g. CEud = 0.5%,
roduces yet a remarkable decrease of the interfacial tension γ to
ptme
dragits droplets is higher than those stabilized by PCL.
he value lower than ∼5 mN/m. This CEud concentration shoulde equivalent to the concentration ratio ϕ∼ 0.9% in the mixture,hich is expected quite sufficient to make the droplets stable
gainst coalescence. So, it is not surprising that the mixture= 0.75 produces very stable mini-emulsions.It is remarkable that in spite of the same orientation at
he MC/water interface of the elementary dipoles μTMA forudragit and μC O for PCL (Fig. 11), and consequently of
he identical sign of the surface potential �V for both poly-ers (Fig. 10), nevertheless the sign of the zeta-potential for
oth polymers is different, being positive for the Eudragit andegative for the PCL. As it is known [18], the zeta-potentials the potential of the hydrodynamic shear surface appearingt some distance from the surface of the particle inside theiffuse Gouy–Chapman layer of counter ions during the Brow-ian motion of particles or their motion in the external electriceld. It is comprehensible why the zeta-potential of particlesade from Eudragit is positive and lower than the potential of
he surface of the particles, i.e. the Nernst potential. The laterotential is not accessible to the measurement, but its valueor the plane surfaces is comparable to the surface potentialV obtained for spread monolayers at the air/water interface
Fig. 13).As concerned the zeta-potential of PCL nanoparticles, its neg-
tive sign and relatively low value may be rationally explainedy the assumption that the positively charged ions (H+, Na+,tc.) form the Helmholtz layer around the surface of particlesearing oriented carbonyl dipoles (Fig. 14b), and correspondingounter ions (OH−, Cl−, etc.) form an electric double layer. Inhis case, the potential of the shear surface (the zeta-potential)ould be negative.
. Conclusion
The emulsifying and stabilizing ability of several hydropho-ic (insoluble in water and soluble in volatile organic solvents)
olymers, such as Eudragit RL, Eudragit RS, PLGA, PCL, andheir mixtures, with regard to the methylene chloride-in-waterini-emulsions have been compared to bulk and surface prop-rties of these polymers.

ces B
lEcmbawiPPd
aituteTwtfsbrra
iamapm
R
[
[
[
[
[[
[
[
[
[[
[
[[
[
[
[
[
[[[[
[32] D. Schuhmann, J. Colloid Interface Sci. 134 (1990) 152.
V.G. Babak et al. / Colloids and Surfa
The increase of the characteristic viscosity [η] of methy-ene chloride (MC) solutions of these polymers in the sequenceudragit RL ∼= Eudragit RS < PLGA < PCL, is explained by theontraction of macromolecular coils of these polymers in theethylene chloride due to the long range electrostatic attraction
etween the polar TMA and carbonyl groups. The irreversibledsorption of TMA groups at the MC/water interface explainshy the interfacial tension γ of 5% MC solutions of Eudrag-
ts RS and RL decreases down to 4 mN/m, whereas PLGA andCL possessing low polar carbonyl groups adsorb reversibly:LGA decrease γ only to 24 mN/m and PCL does not practicallyecrease the MC/water interfacial tension (γ = 28 mN/m).
Surface pressure π of spread monolayers of these polymerst the water subphase correlates well with their interfacial activ-ty: the most stable are Eudragit monolayers which exhibithe plateau pressure π∼= 20 mN/m corresponding to the liq-id extended (LE) to liquid condensed (LC) states, whereashis pressure is equal only to π∼= 10 mN/m for the monolay-rs of PLGA and PCL. The formation of the double layer byMA groups of Eudragits at the air–water interface explainshy the surface potential �V of monolayers corresponding to
he LE–LC transfer is equal to 0.8 V for the Eudragit, whereasor the monolayers of PLGA and PCL, which contribute to theurface potential by the cumulative dipole moment of polar car-onyl groups, the value of�V is equal only to 0.3 V and to 0.4 V,espectively. The experimental values of surface potentials cor-elate well with the Gouy–Chapman model of the double layernd the Helmholtz model of the monolayers.
Finally, we may conclude that the emulsifying and stabiliz-ng ability of hydrophobic polymers are explained by their highdsorption activity at the MC/water interface and by the for-ation of the corona of polymeric chains with ionized groups
round the droplets. It can be also concluded that Eudragitolymers have good surface active properties which may allowanufacturing of nanoparticles without surfactants.
eferences
[1] M. Kitajima, A. Kondo, Bull. Chem. Soc. Jpn. 44 (1971) 3201.[2] T. Heya, H. Okada, Y. Ogawa, H. Toguchi, Int. J. Pharm. 72 (1991) 199.[3] A. Lamprecht, N. Ubrich, M. Hombreiro Perez, C.-M. Lehr, M. Hoffman,
P. Maincent, Int. J. Pharm. 184 (1999) 97.[4] V. Hoffart, N. Ubrich, C. Simonin, V. Babak, C. Vigneron, M. Hoffman, T.
Lecompte, P. Maincent, Drug Dev. Ind. Pharm. 28 (2002) 1091.[5] R. Pignatello, C. Bucolo, P. Ferrara, A. Maltese, A. Puleo, G. Puglisi, Eur.
J. Pharm. Sci. 16 (2002) 53.
[
: Biointerfaces 59 (2007) 194–207 207
[6] Y. Sato, Y. Kawashima, H. Takeuchi, H. Yamamoto, Eur. J. Pharm. Bio-pharm. 55 (2003) 297.
[7] A. Lamprecht, N. Ubrich, M.H. Perez, C.-M. Lehr, M. Hoffman, P. Main-cent, Int. J. Pharm. 196 (2000) 177.
[8] B.B.C. Youan, T.L. Jackson, L. Dickens, C. Hernandez, G. Owusu-Ababio,J. Control. Rel. 76 (2001) 313.
[9] N. Nihant, C. Schugens, C. Grandfils, R. Jerome, P. Teyssie, J. ColloidInterface Sci. 173 (1995) 55.
10] P. Jani, G.W. Halbert, J. Langridge, A.T. Florence, J. Pharm. Pharmacol.42 (1990) 821.
11] M.J. Blanco-Prieto, E. Leo, F. Delie, A. Gulik, P. Couvreur, E. Fattal,Pharm. Res. 13 (1996) 1137.
12] Y.V. Chernysheva, V.G. Babak, N.R. Kildeeva, F. Boury, J.P. Benoit, N.Ubrich, P. Maincent, Mendeleev Commun. 2 (2003) 65.
13] Y.Y. Jiao, N. Ubrich, M. Marchand-Arvier, C. Vigneron, M. Hoffman, P.Maincent, Drug Deliv. 8 (2001) 135.
14] V.G. Babak, R. Gref, E. Dellacherie, Mendeleev Commun. (1998) 105–107.15] R. Gref, V. Babak, P. Bouillot, I. Lukina, M. Bodorev, E. Dellacherie,
Colloids Surf. A: Physicochem. Eng. Asp. 143 (1998) 413.16] P. Saulnier, F. Boury, A. Malzert, B. Heurtault, T. Ivanova, A. Cagna, I.
Panaiotov, J.E. Proust, Langmuir 17 (2001) 8104.17] V. Babak, F. Baros, F. Boury, J. Desbrieres, H. Huilier, N. Kildeeva, P.
Maincent, Proceedings of the 4th World Congress on Emulsions, Lyon,France, 2006, p. 270.
18] P.C. Hiemenz, Principles Colloids Surf. Chem., 2nd ed., Marcel Dekker,New York, 1986.
19] Z. Pingping, Y. Haiyang, W. Shiqiang, Eur. Polym. J. 34 (1998) 91.20] L.A. Utracki, Polymer alloys and blends, in: Thermodynamics and Rheol-
ogy, Hanser Publishers, Munich, Vienna, New York, 1989.21] V.G. Babak, F. Boury, Colloids Surf. A: Physicochem. Eng. Asp. 243 (2004)
33.22] C.J. Beverung, C.J. Radke, H.W. Blanch, Biophys. Chem. 81 (1999) 59.23] V.G. Babak, M. Rinaudo, J. Desbrieres, G.A. Vikhoreva, M.-C. Michalski,
Mendeleev Commun. 4 (1997) 149.24] A.A. Askadskii, Comput. Mater. Sci. Polym., Cambridge Int. Sci. Publish.,
Cambridge, 2003.25] V.G. Babak, J. Desbrieres, V.E. Tikhonov, Colloids Surf. A: Physicochem.
Eng. Asp. 255 (2005) 119.26] A. Ulman, An Introduction to Ultrathin Organic Films: From
Langmuir–Blodgett to Self-assembly, Academic Press, Inc., San Diego,USA, 1991.
27] P. Dynarowicz-Latka, A. Dhanabalan, O.N. Oliveira Jr., Adv. Colloid Inter-face Sci. 91 (2001) 221.
28] R.J. Demchak, T. Fort Jr., J. Colloid Interface Sci. 46 (1974) 191–202.29] O.N. Oliveira Jr., C. Bonardi, Langmuir 13 (1997) 5920.30] V. Vogel, D. Mobius, J. Colloid Interface 126 (1988) 408.31] O.N. Oliveira Jr., D.M. Taylor, L.T. J, S. Salvagno, C.J.M. Stirling, J. Chem.
Soc., Faraday Trans. 1 (85) (1989) 1009.
33] V.G. Babak, Y.V. Chernycheva, N.R. Kildeeva, V.E. Tikhonov, F. Baros,D. Dumas, N. Ubrich, P. Maincent, in: J.L. Pedras, G. Orive, D. Poncelet(Eds.), Proceedings of the XII International Workshop on Bioencapsula-tion, Universidad del Pais Vasco, Vittoria, Spain, 2004, pp. 41–44.





























