Embed Size (px)
Citation preview

Oliver Wirths 1; Gerd Multhaup 4; Christian Czech 2;Nicole Feldmann 1; Véronique Blanchard 2; GünterTremp 3; Konrad Beyreuther 4; Laurent Pradier 2;Thomas A. Bayer 1
1 Department of Psychiatry, University of Bonn Medical Center,Germany
2 Aventis Pharma, Neurodegenerative Disease Group France and3Functional Genomics, Vitry sur Seine, France
4 Center for Molecular Biology University of Heidelberg, Germany
Neuropil deposition of �-amyloid peptides A �40and A �42 is believed to be the key event in the neu-rodegenerative processes of Alzheimer’s disease(AD). Since A � seems to carry a transport signal thatis required for axonal sorting of its precursor �-amyloid precursor protein (APP), we studied theintraneuronal staining profile of A � peptides in atransgenic mouse model expressing human mutantAPP751 (KM670/671NL and V717I) and humanmutant presenilin-1 (PS-1 M146L) in neurons. Usingsurface plasmon resonance we analyzed the A �antibodies and defined their binding profile to APP,A�40 and A �42. Immunohistochemical stainingrevealed that intraneuronal A �40 and A �42 stainingpreceded plaque deposition, which started at 3months of age. A � was observed in the somatoden-dritic and axonal compartments of many neurons.Interestingly, the striatum, which lacks transgenicAPP expression harbored many plaques at 10months of age. This is most likely due to an APP/A �transport problem and may be a model region tostudy APP/A � trafficking as an early pathologicalevent.
Brain Pathol 2002;12:275-286.
Introduction�-amyloid precursor protein (APP) and its prote-
olytic product A� are major players in the etiology ofAlzheimer’s disease (AD). Loss of synapses andpyramidal neurons, induction of neuritic plaques, neu-rofibrillary tangles, and gliosis characterize AD. Whiletangles are predominantly composed of an abnormallyphosphorylated Tau protein, �-amyloid has been identi-fied as a major component of both neuritic plaques andblood vessels with amyloid angiopathy in AD. The
amyloid deposits in AD brains are aggregates of the 40-to 42-residue A� peptides (16, 37), which originatefrom the large precursor protein APP (29, 55). Pointmutations of the APP gene have been identified in somefamilies with a rare autosomal dominant form of earlyonset AD (18, 41). A further subset of familial AD casesis caused by mutations in the presenilin 1 (PS-1) gene onchromosome 14 (1, 50) and the presenilin 2 (PS-2) geneon chromosome 1 (35, 44). The gain-of-function of PSmutations modifies APP processing and enhance gener-ation of A�42 (14, 46).
A plethora of studies report that aberrant traffickingand/or processing of APP may be the key event in AD (forreview, see 11, 47, 49, 52). Enhanced secretion of A� maybe beneficial for proper functioning of neurons, sinceabnormal accumulation of intraneuronal A� may inter-fere with APP sorting (4, 49). Increasing evidence indi-cates that A�42 is generated intracellularly (9, 19, 20, 28,33, 42, 53, 59). Immunohistochemical studies in humanpostmortem brain tissue demonstrated that intraneu-ronal accumulation of A�42 precedes plaque formation(19) and may be the important step in extracellular �-amyloid deposition in human brain (28, 32, 33). Theenigmatic role of A�42 (46) is underscored by cell bio-logical studies, showing intracellular generation from theendoplasmatic reticulum (ER) (9, 23) to the trans-Golginetwork (TGN) (61), and the endosomal-lysosomalsystem (42). Moreover, intracellular staining of A�42 wasshown in cultured primary neurons by confocalmicroscopy (20).
Transgenic mice proved to be an ideal model systemto study the development of pathological events in AD.Several mutant APP (8, 15, 25, 26, 39, 54), mutant PS-1 (14, 34) and double APP/PS-1 (6, 8, 12, 24, 38, 60)transgenic lines have been published so far, reflectingsome aspects of AD pathology. In the present report, westudied the intracellular and extracellular A� and APPstaining pattern in double-transgenic mice expressingmutant human APP751 (carrying the Swedish and Lon-don mutations) and mutant PS-1 (M146L) proteins.Employing surface plasmon resonance analysis, theused A� antibodies were further characterized in detail.
Intraneuronal APP/A� Trafficking and PlaqueFormation in �-Amyloid Precursor Protein andPresenilin-1 Transgenic Mice
Corresponding author:Thomas A. Bayer, PhD, Department of Psychiatry, University of Bonn Medical Center, Sigmund-Freud-Strasse 25, 53105 Bonn, Ger-many (e-mail: [email protected])
RESEARCH ARTICLE

Materials and Methods
Animals. Generation and characterization of mutanthuman PS-1 (mutation M146L) (50) transgenic miceunder the control of a modified HMG-CoA reductase pro-moter was described elsewhere (34). Using this pro-moter, expression of the PS-1 transgene was shown to beessentially neuronal in the brain. The generation ofThy1-APP751 transgenic mice was previouslydescribed by Sturchler-Pierrat et al (54). Mutagenesis ofAPP was described previously (10). In brief, mutated APPsequences were introduced into the APP751 cDNA byinserting the APP SmaI/BglII fragment with APP exon8 into the Bluescript vector containing the mutations. Tooptimize the translation initiation site of APP, an opti-mized Kozak consensus sequence was introduced usingPCR directed mutagenesis. The primer combinationwas: sense primer: CCC GGG TCC ACC ATG COGCCC GGT TTG G, antisense primer: TTC AGG GTAGAC TTC TTG GC. The PCR product was subclonedinto pCR2 (Invitrogen, France), sequenced and subse-quently cloned into the Bluescript vector containing theAPP 751 SL (Swedish mutation KM670/671NL [41];London mutation V717I [18]) cDNA using SmaI andAccI thereby deleting the 5� UTR of APP and introduc-ing the Kozak consensus sequence. To generate theAPP751 Kozak SL Thy1 expression construct, the mod-ified APP cDNA extending 3� to ClaI (2699) was sub-cloned into a modified Bluescript vector containing twoSalI sites 5� and 3� of the insert. The vector was cutwith SalI and the insert cloned into the murine Thy1expression construct using the XhoI site (36). The cor-rect orientation was verified by restriction analysis andsequencing of the joining regions. For microinjection, thecassette was linearised with NotI/PvuI digest.
Sequence analysis of all PCR products verified thepresence of the different mutations and showed noadditional mutations due to errors of the Taq-poly-merase. The hybrid gene was injected in one of the 2pronuclei of fertilized mouse eggs (B6BCA-F2 hybrids,IFFA CREDO). Rodents harboring the transgene weredetected by Southern blot analysis of DNA preparedfrom tail samples. Animals were handled according toFrench and German guidelines for animal care. In total;Thy1-APP/PS-1 double-transgenic mice (n = 15), Thy1-APP single transgenic mice (n = 5), PS-1 transgenicmice (n = 10), as well as age and sex-matched wildtypecontrols were employed (n = 15).
Antibodies. The monoclonal antibody 8E5 was gen-erously supplied by ELAN (formerly Athena Neuro-
science). It was raised against the fragment betweenamino acids 444 to 592 of human APP 770. 8E5 reactsstrongly with secreted and full-length APP and does notcross react with the murine APP protein (3). The poly-clonal antibody pab27576 was raised against the 43 C-terminal amino acids of human APP. For the detection ofhuman A� peptides, the following monoclonal antibod-ies were employed: W0-2, G2-4 (A�40 and 42) (60), G2-10 (A�40) (27), and 22F9 (A�42).
Tissue preparation and immunohistochemistry.Transgenic and control mice were sacrificed by cervicaltranslocation. After removal of the brain, one hemi-sphere was formalin-fixed and paraffin-embedded forimmunohistochemical analysis, and the other hemi-sphere was snap-frozen for biochemical analysis.Immersion fixation was carried out using 4% buffered for-malin at 4°C. Immunohistochemistry was performed on4 �m paraffin sections according to standard protocols(3). In brief, sections were deparaffinized in xylene andrehydrated. After treatment with 1% H2O2 in methanol toblock endogenous peroxidase-activity, sections wereheated in a microwave oven in 0.01 M Citrate buffer pH6.0. Pretreatment with 88% formic acid enhancedimmunoreactivity. Sections were treated with fetal calfserum prior to the addition of the primary antibodies toblock non-specific binding sites. This was followed byan overnight incubation with the primary antibody at RT.Monoclonal primary antibodies W0-2 (1:2000), G2-10(1:500), G2-4 (1:500), 22F9 (1:100), 8E5 (1:4000) andpolyclonal antibody pab27576 (1:500) were incubatedovernight in a humid chamber at RT. Staining was visu-alized using the ABC method, with a Vectastain kit(Vector Laboratories, Burlingame, USA) anddiaminobenzidine as chromogen. Counterstaining wascarried out with hematoxylin. Double-labelingimmunohistochemical analysis was performed usingthe polyclonal antiserum pab27576 against human APPand the monoclonal A� antibody G2-10. Staining wasvisualized using a conventional double-labeling proto-col with goat anti-mouse Ig AP (NBT/BCIP, blue stain-ing) and goat anti-rabbit Ig HRP (DAB, brown staining)from DAKO as secondary antibodies. No immuno-staining was observed without the primary antibodies orwith unrelated rat and mouse IgG fractions.
Thioflavin S staining of brain sections. The remain-ing brain hemisphere was formalin-fixed and paraffin-embedded, cut into 4 �m sections and stained withThioflavin-S to detect aggregated forms of amyloid A�.
276 Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
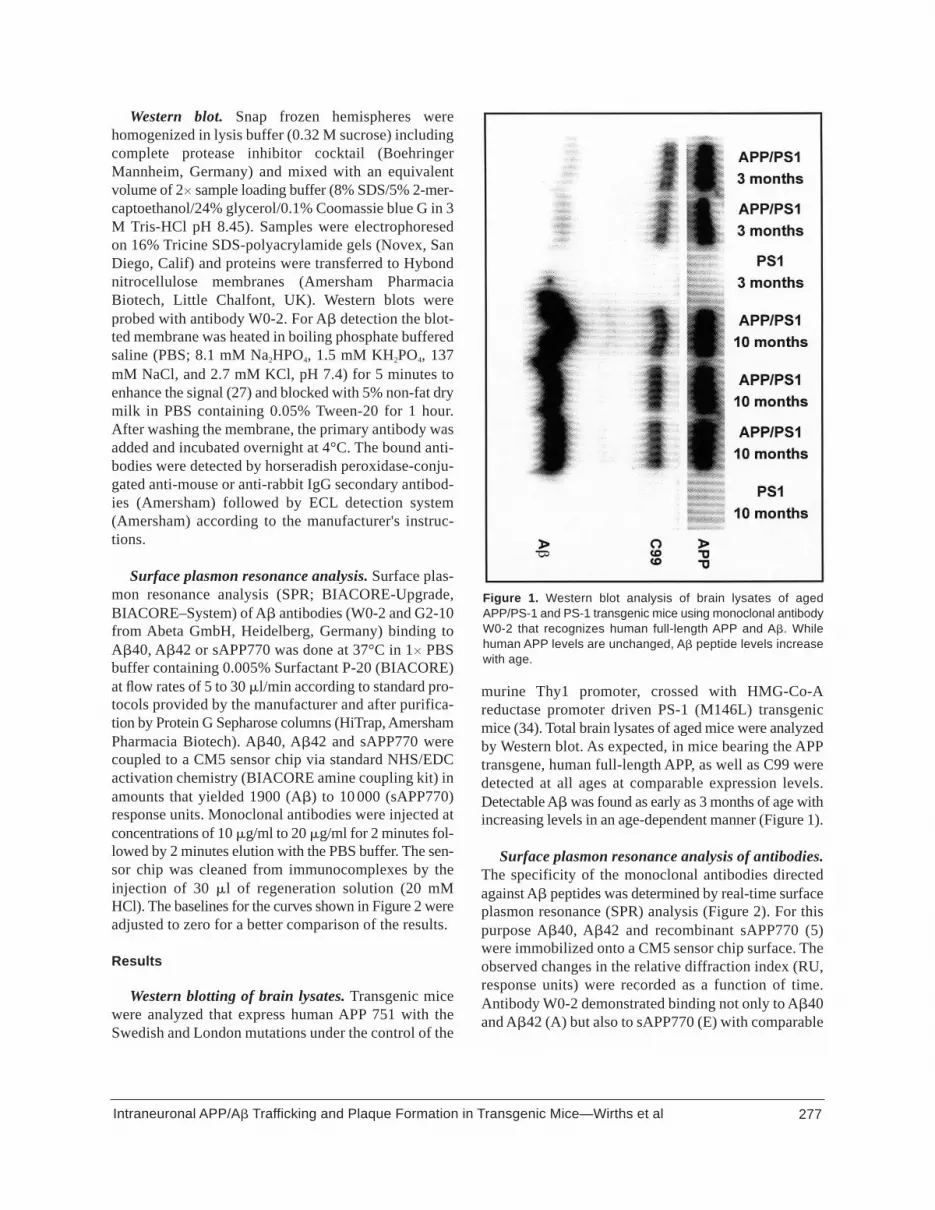
Western blot. Snap frozen hemispheres werehomogenized in lysis buffer (0.32 M sucrose) includingcomplete protease inhibitor cocktail (BoehringerMannheim, Germany) and mixed with an equivalentvolume of 2� sample loading buffer (8% SDS/5% 2-mer-captoethanol/24% glycerol/0.1% Coomassie blue G in 3M Tris-HCl pH 8.45). Samples were electrophoresedon 16% Tricine SDS-polyacrylamide gels (Novex, SanDiego, Calif) and proteins were transferred to Hybondnitrocellulose membranes (Amersham PharmaciaBiotech, Little Chalfont, UK). Western blots wereprobed with antibody W0-2. For A� detection the blot-ted membrane was heated in boiling phosphate bufferedsaline (PBS; 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 137mM NaCl, and 2.7 mM KCl, pH 7.4) for 5 minutes toenhance the signal (27) and blocked with 5% non-fat drymilk in PBS containing 0.05% Tween-20 for 1 hour.After washing the membrane, the primary antibody wasadded and incubated overnight at 4°C. The bound anti-bodies were detected by horseradish peroxidase-conju-gated anti-mouse or anti-rabbit IgG secondary antibod-ies (Amersham) followed by ECL detection system(Amersham) according to the manufacturer's instruc-tions.
Surface plasmon resonance analysis. Surface plas-mon resonance analysis (SPR; BIACORE-Upgrade,BIACORE–System) of A� antibodies (W0-2 and G2-10from Abeta GmbH, Heidelberg, Germany) binding toA�40, A�42 or sAPP770 was done at 37°C in 1� PBSbuffer containing 0.005% Surfactant P-20 (BIACORE)at flow rates of 5 to 30 �l/min according to standard pro-tocols provided by the manufacturer and after purifica-tion by Protein G Sepharose columns (HiTrap, AmershamPharmacia Biotech). A�40, A�42 and sAPP770 werecoupled to a CM5 sensor chip via standard NHS/EDCactivation chemistry (BIACORE amine coupling kit) inamounts that yielded 1900 (A�) to 10 000 (sAPP770)response units. Monoclonal antibodies were injected atconcentrations of 10 �g/ml to 20 �g/ml for 2 minutes fol-lowed by 2 minutes elution with the PBS buffer. The sen-sor chip was cleaned from immunocomplexes by theinjection of 30 �l of regeneration solution (20 mMHCl). The baselines for the curves shown in Figure 2 wereadjusted to zero for a better comparison of the results.
Results
Western blotting of brain lysates. Transgenic micewere analyzed that express human APP 751 with theSwedish and London mutations under the control of the
murine Thy1 promoter, crossed with HMG-Co-Areductase promoter driven PS-1 (M146L) transgenicmice (34). Total brain lysates of aged mice were analyzedby Western blot. As expected, in mice bearing the APPtransgene, human full-length APP, as well as C99 weredetected at all ages at comparable expression levels.Detectable A� was found as early as 3 months of age withincreasing levels in an age-dependent manner (Figure 1).
Surface plasmon resonance analysis of antibodies.The specificity of the monoclonal antibodies directedagainst A� peptides was determined by real-time surfaceplasmon resonance (SPR) analysis (Figure 2). For thispurpose A�40, A�42 and recombinant sAPP770 (5)were immobilized onto a CM5 sensor chip surface. Theobserved changes in the relative diffraction index (RU,response units) were recorded as a function of time.Antibody W0-2 demonstrated binding not only to A�40and A�42 (A) but also to sAPP770 (E) with comparable
277Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
Figure 1. Western blot analysis of brain lysates of agedAPP/PS-1 and PS-1 transgenic mice using monoclonal antibodyW0-2 that recognizes human full-length APP and A�. Whilehuman APP levels are unchanged, A� peptide levels increasewith age.

affinities, whereas G2-4 showed the same specificitybut with considerably less avidity (C, E). The epitopeslocated along the C-terminal ends of the peptide mole-cules A�40 and A�42 were responsible for the specificantibody recognition of G2-10 (B) and 22F9 (D),respectively. Reactivity against individual peptides of G2-10 has already been described as a useful tool in West-ern blots to differentiate between A�40 and A�42 (27).It is evident from B that G2-10 associates quickly to A�40and dissociates faster from its target than W0-2 (A).22F9 is characterized by slow association and fast dis-sociation from the A�40 surface showing a weak cross-reactivity with A�40 (D). G2-10 and 22F9 did not showany cross-reactivity with sAPP770 (E).
Immunohistochemical staining profile of APP,A�40 and A�42. Abundant transgenic expression ofhuman mutant APP751 was found in most if not alllarge pyramidal neurons of the subiculum, CA1, andcortex, as well as granule neurons of the dentate gyrus.Only some hippocampal pyramidal neurons of sectorCA2/3 were positive. The same held true for many neu-rons of subcortical structures. No expression wasdetected in the striatum. Subcellularly, many dendritic andaxonal projections harbored human APP. For example,APP was seen in apical dendrites of CA1 neurons andaxons of the corpus callosum, striatum, and mossyfibers of the dentate gyrus. APP exhibited a punctuatestaining pattern in the somatodendritic and axonal com-partments. Overall there was no significant age-dependent change in the subcellular, cellular, andregional distribution of APP expression. However,many APP positive neuritic plaques were seen in allbrain regions with APP expressing neurons. Brain
278 Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
Figure 2. Determination of binding specificity of monoclonal A�antibodies by surface plasmon resonance spectroscopy. A-D.overlays of sensorgrams from A�40 and A�42 flow cells for theinteraction of monoclonal antibodies W0-2 (A) and G2-4 (C) withA�40 and A�42, G2-10 (B) binding to A�40 and 22F9 bindingto A�42 (D). E. overlay of sensorgrams for the interaction ofmonoclonal antibodies with sAPP770 immobilized to a thirdflow cell. At 100 seconds antibodies diluted in running buffer wereinjected onto immobilized A�40, A�42 (A-D) or sAPP770 (E). At220 seconds the injection was stopped and dissociation followedin running buffer for 120 seconds. Note that rectangular suddenchanges in refractive indices for sensorgrams B (injection of G2-10 onto A�42), D (injection of 22F9 onto A�40) and E (injectionof 22F9 and G2-10 onto sAPP770) are due to differences in thecomposition between buffer and protein solution and by specificbinding to immobilized proteins. According to the kinetic constantseach of the monoclonal antibodies recognizes equally wellA�40 and A�42 (data not shown) due to the varying amountsof peptides A�40 and A�42 immobilized to the sensor surface.

regions lacking APP expressing cells, including striatum,also showed the appearance of APP positive neuriticplaques at later stages. The staining pattern was undis-tinguishable between human APP specific antibodies8E5 and 27576 (Figure 3).
The onset of A� plaque deposition in the APP/PS-1double-transgenic mice was at 3 months of age (Figures4, 5, 6) in subiculum, hippocampus, and neocortex,whereas the onset of A� neuropil deposits in APP singletransgenics was seen at 6 months (not shown). Mostinterestingly, there was an age-dependent regional andsubcellular differential distribution of intraneuronal andextracellular A�. At 3 months of age, the strongestintraneuronal staining was observed in the subiculum andcortical layers 4 and 5—the areas with earliest plaquedevelopment. At later stages, A� positive neuritic and dif-fuse plaques were seen in all brain regions with APPexpressing neurons. Brain regions lacking APP express-ing neurons, including striatum, also showed theappearance of A� positive plaques in older animals(Figures 4, 6). In this region, diffuse plaques dominat-ed neuritic plaques. A� staining could be detected invesicular structures in the somatodendritic and axonalcompartments at all stages, however, the overall intra-neuronal immunoreactivity of A� peptides was muchweaker or absent at later stages (Figure 5). All of the A�
antibodies (W0-2, G2-4, G2-10, 22F9) elicited stainingof neuropil deposits in APP/PS-1 transgenic mice,which were morphologically indistinguishable fromplaques in postmortem brain tissue of AD patients. Theintraneuronal staining pattern of the A� antibodies wascomparable, however, it varied in intensity (Figure 5). Theintraneuronal staining could be observed in the follow-ing order (ranked from strongest to weakest): W0-2(A�40 and A�42), G2-10 (A�40), G2-4 (A�40 andA�42), and 22F9 (A�42).
We performed several controls to insure that, usingimmunohistochemistry, the A� antibodies were specif-ic and did not cross-react with full-length APP. i) Inaged APP/PS-1 transgenic mice, the intraneuronal A�immunoreactivity was much weaker or undetectable,whereas the APP staining pattern was unchanged. Thisobservation argues against a strong cross-reactivity of theantibodies with human APP. ii) We have never detectedintraneuronal staining using W0-2, G2-4, 22F9, or G2-10 in formalin-fixed and paraffin-embedded humancontrol specimen (not shown). The fixation and embed-ding procedure of mouse brains was performed thesame way. iii) None of the A� antibodies showed cross-reactivity to murine APP in non-transgenic mouse con-trol brain sections (not shown). However, G2-4 andW0-2 did show weak intraneuronal staining in APP sin-
279Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
Figure 3. Immunohistochemical staining of human APP in APP/PS1 transgenic mice. A. Human APP expression (pab 27576) in pyram-idal neurons of the cortex at 3 months of age. B. Human APP expression (8E5) in axonal projections in the striatum at 3 months ofage. C. Double immunolabeling of human APP (brown) and human A� (blue) in the cortex of a Thy1-APP/PS1 transgenic mouse.D. Human APP (8E5) positive plaques in the striatum at ten months of age. E. higher magnification of C. Counterstaining with hema-toxylin (B, C). Scale bars 10 �m (A, B), 20 �m (E), 50 �m (C), 80 �m (D).

gle-transgenic mice, which might be due to cross-reac-tivity with human APP.
Histological staining against amyloid. As expectedThioflavin-S staining of brain sections showed thatamyloid plaques were found in hippocampus and cortexas early as 3 months of age, while striatum was constantlynegative. At 10 months of age, large plaques weredetected in hippocampus and cortex and smaller amyloiddeposits in the striatum, indicating a region-by-region pro-gression of extracellular amyloid formation (Figure 6).Striatal neurons do not express the human APP transgene;however, A�/APP is transported through the axons of pro-jecting neurons (see Figure 4).
DiscussionDespite extensive research on APP, the physiological
function is still enigmatic. The diverse functions of APPand its derivatives have been discussed earlier (7, 31, 48).A recent review from Dodart et al (13) focuses on thenormal function of APP and its secreted forms in the via-bility, growth and morphological, functional plasticity ofnerve cells, and in learning and memory processes.Modulation of memory function is tightly linked withAPP processing. A� was identified originally as anextracellular peptide (17, 22, 37), therefore any inter-ference with intracellular protein sorting seemed
unlikely. Wertkin et al (58) reported intracellular A� ina differentiated neuronal cell line. In a comparativestudy of neurons and peripheral cells, it was discoveredthat CNS neurons produce a larger amount of intracel-lular A� as compared to peripheral cells. The site ofintracellular A�42 production seems to be the endo-plasmatic reticulum, whereas the site for A�40 produc-tion has been reported to be the trans-Golgi network (9,23, 59). Unlike A�40, intracellular A�42 is not secret-ed immediately (9), but accumulates and converts intoinsoluble aggregates (53) preventing degradation (62).Recently, intraneuronal A�42 has been reported toaccumulate within pyramidal neurons in early and not inlate stage AD brains adding evidence that intraneuronalA�42 immunoreactivity appears to precede both NFT andplaque deposition (19). A�40 seems to be occurring inyoung patients with Downs syndrome predominately(21). In much earlier studies, the neuronal origin ofextracellular A� peptides has been proposed using post-mortem brains of AD and Downs syndrome patients (2,43). These findings are consistent with our observationsin the APP/PS-1 transgenic mouse model. Abundantintraneuronal A�40, rather than A�42, seemed to be thedominating intraneuronal peptide. This may be due to thetransgenic mouse model system used in the presentstudy and the low abundance of A�42 in neurons. Inagreement with other APP/PS-1 transgenic mice, A�40
280 Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
Figure 4. Immunohistochemical staining of human A� in APP/PS1 transgenic mice. A. Staining against A� (W0-2) in the hippocampusat 3 months of age. B. Staining against A� (W0-2) in the hippocampus at 10 months of age. Note that dendritic and axonal stain-ing is largely reduced at the later stage. C. Staining against A� (W0-2) in axonal projections in the striatum at 3 months of age. D.Staining against A� (G2-10) in pyramidal neurons of the subiculum at 3 months of age. E. Staining against A� (G2-10) in pyrami-dal neurons of the cortex at 3 months of age. F. Staining against A� (W0-2) in the striatum at 10 months of age. Counterstainingwith hematoxylin. Scale bars 200 �m (A, F), 100 �m (B, C), 50 �m (D, E).

281Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
Figure 5. Immunohistochemical staining of 3 (A, C, E, G) and 10 month old (B, D, F, H) APP/PS1 transgenic mice. This figure showsintraneuronal A� immunoreactivity as well as extracellular deposits using the antibodies W0-2 against A�40 and A�42 (A, B), G2-10 against A�40 (C, D), G2-4 against A�40 and A�42 (E, F) and 22F9 against A�42 (G, H). Counterstaining with hematoxylin. Scalebar 200 �m (A, B), 100 �m (C-H).

also seems to be the dominating deposited A� species.Borchelt et al, who used mice with the Swedish APPmutation crossed with PS-1 (A246E) mutant mice, andMc Gowan et al, who also used mice with the SwedishAPP mutation crossed with different PS-1 (M146V orM146L) mutant lines, showed that the majority ofextracellular A� deposits were reactive with A�40 butnot with A�42 specific antibodies (6, 38). The pattern ofplaque deposition was comparable to the mice of the pres-ent report.
Furthermore, the intraneuronal A� pools observedin the present APP/PS-1 model seem to be transient, anobservation reported earlier in another model expressingAPP (PDGF�-promoter) and PS-1 (HMG-Co reductasepromoter) (60). In the present APP/PS-1 transgenicmouse model, APP transgene expression was under thecontrol of the murine Thy1 promoter. These miceexhibited a strong A� immunohistochemical stainingpattern predominantly in pyramidal neurons of hip-pocampal formation and cortex. The robust intraneu-ronal staining was strongest in brain areas with earlyplaque formation as the subiculum and layers 4 and 5 ofthe cortex. Interestingly, A� peptides were detectedabundantly in the somatodendritic and axonal compart-ment at early stages, whereas the staining was muchweaker or absent at later stages. The striatum, which isa region without human APP expression, showedplaque formation at later stages pointing to an APP/A�
sorting problem in human APP positive axons. Thisfinding is consistent with previous reports. APP23mice, an APP transgenic model described by Sturchler-Pierrat et al, with human APP (Swedish mutant) expres-sion under the control of the Thy1-promoter, show acomparable pattern of plaque deposition; however,plaques in the striatum arise much later at the age of 24months (54). A recent report describes an APP transgenicmodel with a comparable sequence of plaque deposition,starting in subiculum and followed by frontal cortex, hip-pocampus, thalamus, and striatum even after 61/2
months. The authors used a different construct underthe control of the Prp-promoter with APP harboring theSwedish and Indiana mutations. A very rapid plaquedeposition after one month was achieved by crossingthese APP transgenic mice with double-mutant PS-1(M146L and L286V) mice (8). Early pathologicalchanges, possibly related to intraneuronal APP/A� mis-trafficking, have been described earlier in other transgenicmodels. Hsia et al previously reported deficits in synap-tic transmission in APP transgenic mice, harboring theSwedish and Indiana mutations under the control of thePDGF�-promoter, prior to plaque formation. Theauthors speculated that this disturbance could be due toneurotoxic effects induced by diffusible A� oligomers orintraneuronal accumulation of A� (25). In APP transgenicmice with the London mutation, described by Moecharset al, phenotypic changes as behavioral changes, differ-
282 Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al
Figure 6. Histological amyloid staining of 3 (A-C) and 10 month old (D-F) APP/PS1 transgenic mice using Thioflavin-S. Scale bar100 �m (A-F).

ential glutamate responses, deficits in long-term poten-tiation and premature death, occured prior to plaqueformation (39). Holcomb et al investigated single APP(Swedish mutant) and double transgenic mice (APPwith Swedish mutation and PS-1 M146L) and reportedreduced spontaneous alternation performance in a “Y”maze before substantial A� deposition was apparent.The authors suggested that some aspects of the behav-ioral phenotype in these mice could be due to a patho-logical event that preceded plaque formation (24). Theobservations in the present APP/PS-1 transgenic mousemodel are in good agreement with published reports ofother transgenic models, differing mainly in the onset ofpathology between 1 to 9 months, which is likely due tothe features of different promoters and/or APP muta-tions. In general, the pathology starts in the hippocam-pal area and neocortex, which is followed by subcorti-cal brain areas like thalamus and striatum depending onthe transgenic line.
The A� domain of APP has been shown to be of par-ticular importance for APP trafficking. Intraneuronalmisregulation of APP, which may lead to an accumula-tion of A� peptides in intracellular compartments hasbeen recognized earlier (49). Under normal conditionsAPP is first directed to the axon, later to the somato-den-dritic compartment. Tienari et al (57) reported that dele-tions inside the A� domain redirect APP polarized sort-ing by transcytosis (51, 56). The presence of abundantsoluble A� inside neurons could very well interferewith the targeting function of the A� domain inside ofAPP. APP-APP dimers are a preferred substrate forBACE (45), the rate limiting enzyme for A� generation.Any failure to transport APP-APP dimers antero- and ret-rogradly in axons could enhance the possibility ofBACE to cleave APP. APP-APP interactions may benegatively regulated by N-terminal APP ligands, such asCu(II), Zn(II) and heparin (5). Therefore, binding ofligands to APP-APP dimers could favor monomerizationof APP, which in turn is prone to �-secretase cleavagepreferentially occuring close to the plasma membrane.At 10 months of age, APP/PS-1 mice developedplaques in the striatum. The only source of extracellularA� could be the axons of projecting neurons, becausestriatal neurons lack human APP expression. Due to thehigh expression level of the human APP transgene, it islikely that APP transport and trafficking is down-regu-lated. Higher levels of APP-APP dimers may accumulatein intraneuronal compartments leading to increasedintraneuronal A� generation and subsequent releaseinto the neuropil. Mutant PS-1 is currently being dis-cussed to be involved not only in APP processing, but also
to have a role in APP trafficking (30, 63). Both of thesefunctions might contribute to the observed striatalaxonal mistrafficking.
Surface plasmon resonance (SPR) has been used tocharacterize the A� antibodies in detail. Monoclonalantibodies W0-2 and G2-4 recognize both peptidesA�40 and A�42, and secreted APP with lower strength.However, in human postmortem formalin-fixed andparaffin-embedded brain sections, we never observedintraneuronal immunohistochemical staining with theseantibodies (not shown). G2-10 is specific for A�40 and22F9 is specific for A�42, and neither antibody cross-reacts with APP by SPR (Figure 2) or Western blotanalysis (not shown). Moreover, while we found abun-dant intraneuronal staining at early stages with all anti-bodies against A�, in old transgenic mice this pattern wasmuch weaker or even absent. The APP expression pro-file, however, has not changed during aging. Overall, weconclude that all 4 A� antibodies are useful tools forimmunohistochemical depiction of intraneuronal A�.
Several models of plaque deposition have beendeveloped in the past few years (6, 12, 15, 24, 26, 38, 40,54, 60). Although major milestones, they do not repro-duce all features of AD. We demonstrate that intraneu-ronal A� accumulates transiently in APP/PS-1 trans-genic mice, as previously seen in early AD and Downssyndrome cases and another APP/PS-1 mouse model.Consequently, we propose that plaque formation isrelated to a sorting problem of APP/A� within neurons.
Acknowledgments With kind support from the Fritz Thyssen Foundation
and the Alzheimer Forschung Initiative e.V. (to TABand GM) and the Deutsche Forschungsgemeinschaft(Mu901 to GM and TAB and SFB 488 to KB). Theexpert technical assistance of Sophie Ligout, GwenaellRet, Brigitte Schombert, Nathalie Touchet, Ulrich Klattand Esther Barth is gratefully acknowledged.
References
1. The structure of the presenilin 1 (S182) gene and identi-fication of six novel mutations in early onset AD families.Alzheimer's Disease Collaborative Group. (1995) NatGenet 11:219-222.
2. Allsop D, Haga SI, Haga C, Ikeda SI, Mann DM, Ishii T(1989) Early senile plaques in Down’s syndrome brainsshow a close relationship with cell bodies of neurons.Neuropathol Appl Neurobiol 15:531-542.
3. Bayer TA, Fossgreen A, Czech C, Beyreuther K, WiestlerOD (1996) Plaque formation in brain transplants exposedto human beta-amyloid precursor protein 695. Acta Neu-ropathol (Berl) 92:130-137.
283Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al

4. Bayer TA, Wirths O, Majtenyi K, Hartmann T, Multhaup G,Beyreuther K, Czech C (2001) Key factors in Alzheimer'sdisease: beta-amyloid precursor protein processing,metabolism and intraneuronal transport. Brain Pathol11:1-11.
5. Beher D, Hesse L, Masters CL, Multhaup G (1996) Reg-ulation of amyloid protein precursor (APP) binding to col-lagen and mapping of the binding sites on APP and col-lagen type I. J Biol Chem 271:1613-1620.
6. Borchelt DR, Ratovitski T, van Lare J, Lee MK, GonzalesV, Jenkins NA, Copeland NG, Price DL, Sisodia SS(1997) Accelerated amyloid deposition in the brains oftransgenic mice coexpressing mutant presenilin 1 andamyloid precursor proteins. Neuron 19:939-945.
7. Buxbaum JD, Greengard P (1996) Regulation of APPprocessing by intra- and intercellular signals. Ann N YAcad Sci 777:327-331.
8. Chishti MA, Yang DS, Janus C, Phinney AL, Horne P,Pearson J, Strome R, Zuker N, Loukides J, French J,Turner S et al (2001) Early-onset amyloid deposition andcognitive deficits in transgenic mice expressing a doublemutant form of amyloid precursor protein 695. J BiolChem 276:21562-21570.
9. Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwat-subo T, Lee VM, Doms RW (1997) Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermedi-ate compartment of NT2N cells. Nat Med 3:1021-1023.
10. Czech C, Delaere P, Macq AF, Reibaud M, Dreisler S,Touchet N, Schombert B, Mazadier M, Mercken L,Theisen M, Pradier L et al(1997) Proteolytical processingof mutated human amyloid precursor protein in trans-genic mice. Brain Res Mol Brain Res 47:108-116.
11. De Strooper B, Annaert W (2000) Proteolytic processingand cell biological functions of the amyloid precursor pro-tein. J Cell Sci 113:1857-1870.
12. Dewachter I, Van Dorpe J, Smeijers L, Gilis M, Kuiperi C,Laenen I, Caluwaerts N, Moechars D, Checler F, Van-derstichele H, Van Leuven F (2000) Aging increasedamyloid peptide and caused amyloid plaques in brain ofold APP/V717I transgenic mice by a different mechanismthan mutant presenilin1. J Neurosci 20:6452-6458.
13. Dodart JC, Mathis C, Ungerer A (2000) The beta-amyloidprecursor protein and its derivatives: from biology tolearning and memory processes. Rev Neurosci 11:75-93.
14. Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J,Hutton M, Buee L, Harigaya Y, Yager D, Morgan D et al(1996) Increased amyloid-beta42(43) in brains of miceexpressing mutant presenilin 1. Nature 383:710-713.
15. Games D, Adams D, Alessandrini R, Barbour R,Berthelette P, Blackwell C, Carr T, Clemens J, DonaldsonT, Gillespie F, Guido T et al (1995) Alzheimer-type neu-ropathology in transgenic mice overexpressing V717Fbeta-amyloid precursor protein. Nature 373:523-527.
16. Glenner GG, Wong CW (1984) Alzheimer´s disease: Ini-tial report of the purification and characterization of anovel cerebrovasular amyloid protein. Biochem BiophysRes Commun 120:885-890.
17. Glenner GG, Wong CW, Quaranta V, Eanes ED (1984) Theamyloid deposits in Alzheimer's disease: their nature andpathogenesis. Appl Pathol 2:357-369.
18. Goate A, Chartier-Harlin MC, Mullan M, Brown J, CrawfordF, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al(1991) Segregation of a missense mutation in the amyloidprecursor protein gene with familial Alzheimer's disease.Nature 349:704-706.
19. Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M,Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, XuH, Greengard P, Relkin NR (2000) Intraneuronal Abeta42accumulation in human brain. Am J Pathol 156:15-20.
20. Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G,Checler F, Sisodia SS, Greengard P, Xu H (1999) Endo-plasmic reticulum and trans-Golgi network generate dis-tinct populations of Alzheimer beta-amyloid peptides.Proc Natl Acad Sci U S A 96:742-747.
21. Gyure KA, Durham R, Stewart WF, Smialek JE, TroncosoJC (2001) Intraneuronal abeta-amyloid precedes devel-opment of amyloid plaques in Down syndrome. ArchPathol Lab Med 125:489-492.
22. Haass C, Schlossmacher MG, Hung AY, Vigo Pelfrey C,Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, SchenkD, Teplow DB, Selkoe DJ (1992) Amyloid beta-peptide isproduced by cultured cells during normal metabolism.Nature 359:322-325.
23. Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, AllsopD, Roberts GW, Masters CL, Dotti CG, Unsicker K,Beyreuther K (1997) Distinct sites of intracellular produc-tion for Alzheimer's disease A beta40/42 amyloid pep-tides. Nat Med 3:1016-1020.
24. Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S,Jantzen P, Wright K, Saad I, Mueller R, Morgan D,Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM,Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerat-ed Alzheimer-type phenotype in transgenic mice carryingboth mutant amyloid precursor protein and presenilin 1transgenes. Nat Med 4:97-100.
25. Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, HuK, Kholodenko D, Malenka RC, Nicoll RA, Mucke L(1999) Plaque-independent disruption of neural circuits inAlzheimer's disease mouse models. Proc Natl Acad Sci US A 96:3228-233.
26. Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C,Yunis W, Xu S, Eckman C, Younkin S, Price D et al(1995) Age related CNS disorder and early death intransgenic FVB/N mice overexpressing Alzheimer amyloidprecursor proteins. Neuron 15:1203-1218.
27. Ida N, Hartmann T, Pantel J, Schröder J, Zerfass R,Förstl H, Sandbrink R, Masters CL, Beyreuther K (1996)Analysis of heterogeneous �A4 peptides in humancererbrospinal fluid and blood by a newly-developed sen-sitive Western blot assay. J Biol Chem 271:22908-22914.
28. Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N,Ihara Y (1994) Visualization of A beta 42(43) and A beta40 in senile plaques with end- specific A beta monoclon-als: evidence that an initially deposited species is A beta42(43). Neuron 13:45-53.
29. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, MastersCL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-HillB (1987) The precursor of Alzheimer´s disease amyloid A4protein resembles a cell surface receptor. Nature325:733-736.
284 Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al

30. Kim SH, Leem JY, Lah JJ, Slunt HH, Levey AI, Thi-nakaran G, Sisodia SS (2001) Multiple effects of aspartatemutant presenilin 1 on the processing and trafficking ofamyloid precursor protein. J Biol Chem 276:43343-43350.
31. Kosik KS, Qiu WQ, Greenberg S (1996) Cellular signalingpathways and cytoskeletal organization. Ann N Y Acad Sci777:114-120.
32. LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, JayG (1997) Neuronal cell death in Alzheimer's disease cor-relates with apoE uptake and intracellular Abeta stabi-lization. J Clin Invest 100:310-320.
33. Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T,Saido TC, Selkoe DJ (1996) Sequence of deposition of het-erogeneous amyloid beta-peptides and APO E in Downsyndrome: implications for initial events in amyloidplaque formation. Neurobiol Dis 3:16-32.
34. Leutner S, Czech C, Schindowski K, Touchet N, Eckert A,Muller WE (2000) Reduced antioxidant enzyme activity inbrains of mice transgenic for human presenilin-1 with sin-gle or multiple mutations. Neurosci Lett 292:87-90.
35. Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshi-ma J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD,Wang K, et al (1995) Candidate gene for the chromo-some 1 familial Alzheimer's disease locus. Science269:973-977.
36. Luthi A, Putten H, Botteri FM, Mansuy IM, Meins M, FreyU, Sansig G, Portet C, Schmutz M, Schroder M, Nitsch C,Laurent JP, Monard D (1997) Endogenous serine pro-tease inhibitor modulates epileptic activity and hip-pocampal long-term potentiation. J Neurosci 17:4688-4699.
37. Masters CL, Simms G, Weinman NA, Multhaup G,McDonald BL, Beyreuther K (1985) Amyloid plaque coreprotein in Alzheimer disease and Down syndrome. ProcNatl Acad Sci U S A 82:4245-4249.
38. McGowan E, Sanders S, Iwatsubo T, Takeuchi A, Saido T,Zehr C, Yu X, Uljon S, Wang R, Mann D, Dickson D, DuffK (1999) Amyloid phenotype characterization of trans-genic mice overexpressing both mutant amyloid precursorprotein and mutant presenilin 1 transgenes. NeurobiolDis 6:231-244.
39. Moechars D, Dewachter I, Lorent K, Reverse D, Baeke-landt V, Naidu A, Tesseur I, Spittaels K, Haute CV,Checler F, Godaux E, Cordell B, Van Leuven F (1999) Earlyphenotypic changes in transgenic mice that overexpressdifferent mutants of amyloid precursor protein in brain. JBiol Chem 274:6483-6492.
40. Moechars D, Lorent K, De Strooper B, Dewachter I, VanLeuven F (1996) Expression in brain of amyloid precursorprotein mutated in the alpha- secretase site causes dis-turbed behavior, neuronal degeneration and prematuredeath in transgenic mice. Embo J 15:1265-1274.
41. Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Win-blad B, Lannfelt L (1992) A pathogenic mutation for prob-able Alzheimer's disease in the APP gene at the N-termi-nus of beta-amyloid. Nat Genet 1:345-347.
42. Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W,Selkoe DJ, Chen X, Stokin GB, Koo EH (1999) Mutage-nesis identifies new signals for beta-amyloid precursor pro-tein endocytosis, turnover, and the generation of secret-ed fragments, including Abeta42. J Biol Chem274:18851-18856.
43. Probst A, Langui D, Ipsen S, Robakis N, Ulrich J (1991)Deposition of beta/A4 protein along neuronal plasmamembranes in diffuse senile plaques. Acta Neuropathol(Berl) 83:21-29.
44. Rogaev EI, Sherrington R, Rogaeva EA, Levesque G,Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al(1995) Familial Alzheimer's disease in kindreds with mis-sense mutations in a gene on chromosome 1 related to theAlzheimer's disease type 3 gene. Nature 376:775-778.
45. Scheuermann S, Hambsch B, Hesse L, Stumm J,Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G(2001) Homodimerization of APP and its implication inthe amyloidogenic pathway of Alzheimers disease. J BiolChem 276:33923-33929.
46. Scheuner D, Eckman C, Jensen M, Song X, Citron M,Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, LarsonE, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P,Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D,Younkin S (1996) Secreted amyloid b-protein similar to thatin the senile plaques of Alzheimer's disease is increasedin vivo by the presenilin 1 and 2 and APP mutationslinked to familial Alzheimer's disease. Nature Med.2:864-870.
47. Selkoe DJ (1999) Translating cell biology into therapeu-tic advances in Alzheimer's disease. Nature 399:A23-31.
48. Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH,Teplow DB, Haass C (1996) The role of APP processingand trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci 777:57-64.
49. Sheetz MP, Pfister KK, Bulinski JC, Cotman CW (1998)Mechanisms of trafficking in axons and dendrites: impli-cations for development and neurodegeneration. ProgNeurobiol 55:577-594.
50. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA,Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al(1995) Cloning of a gene bearing missense mutations inearly-onset familial Alzheimer's disease. Nature 375:754-760.
51. Simons M, Ikonen E, Tienari PJ, Cid-Arregui A, MonningU, Beyreuther K, Dotti CG (1995) Intracellular routing ofhuman amyloid protein precursor: axonal delivery fol-lowed by transport to the dendrites. J Neurosci Res41:121-128.
52. Sinha S, Lieberburg I (1999) Cellular mechanisms ofbeta-amyloid production and secretion. Proc Natl Acad SciU S A 96:11049-11053.
53. Skovronsky DM, Doms RW, Lee VM (1998) Detection ofa novel intraneuronal pool of insoluble amyloid beta pro-tein that accumulates with time in culture. J Cell Biol141:1031-1039.
285Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al

54. Sturchler-Pierrat C, Abramowski D, Duke M, WiederholdKH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P,Paganetti PA, Waridel C, Calhoun ME, Jucker M, ProbstA, Staufenbiel M, Sommer B (1997) Two amyloid precur-sor protein transgenic mouse models with Alzheimer dis-ease-like pathology. Proc Natl Acad Sci U S A 94:13287-13292.
55. Tanzi RE, Gusella JF, Watkins PC, Bruns GAP,St.George-Hyslop PH, Van Keuren ML, Patterson D,Pagan S, Kurnit DM, Neve RL (1987) The amyloid � pro-tein gene: cDNA cloning, mRNA distribution, and geneticlinkage near the Alzheimer locus. Science 235:880-884.
56. Tienari PJ, De Strooper B, Ikonen E, Ida N, Simons M,Masters CL, Dotti CG, Beyreuther K (1996) Neuronalsorting and processing of amyloid precursor protein:implications for Alzheimer's disease. Cold Spring HarbSymp Quant Biol 61:575-585.
57. Tienari PJ, De Strooper B, Ikonen E, Simons M, Weide-mann A, Czech C, Hartmann T, Ida N, Multhaup G, Mas-ters CL, Van Leuven F, Beyreuther K, Dotti CG (1996) The�-amyloid domain is essential for axonal sorting of amy-loid precursor protein. EMBO J 15:5218-5229.
58. Wertkin AM, Turner RS, Pleasure SJ, Golde TE, YounkinSG, Trojanowski JQ, Lee VM (1993) Human neuronsderived from a teratocarcinoma cell line express solely the695-amino acid amyloid precursor protein and produceintracellular beta-amyloid or A4 peptides. Proc Natl AcadSci U S A 90:9513-9517.
59. Wild-Bode C, Yamazaki T, Capell A, Leimer U, Steiner H,Ihara Y, Haass C (1997) Intracellular generation andaccumulation of amyloid beta-peptide terminating atamino acid 42. J Biol Chem 272:16085-16088.
60. Wirths O, Multhaup G, Czech C, Blanchard V, MoussaouiS, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intra-neuronal Abeta accumulation precedes plaque formationin beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306:116-120.
61. Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Siso-dia SS, Greengard P, Gandy S (1997) Generation ofAlzheimer beta-amyloid protein in the trans-Golgi net-work in the apparent absence of vesicle formation. ProcNatl Acad Sci U S A 94:3748-3752.
62. Yang AJ, Chandswangbhuvana D, Shu T, Henschen A,Glabe CG (1999) Intracellular accumulation of insoluble,newly synthesized abeta-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42.J Biol Chem 274:20650-20656.
63. Yu G, Chen F, Levesque G, Nishimura M, Zhang DM,Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, StGeorge-Hyslop PH, Fraser PE (1998) The presenilin 1 pro-tein is a component of a high molecular weight intracellularcomplex that contains beta-catenin. J Biol Chem273:16470-16475.
286 Intraneuronal APP/A� Trafficking and Plaque Formation in Transgenic Mice—Wirths et al





























