Embed Size (px)
Citation preview
This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copychemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
Contents lists available at ScienceDirect
Chemical Engineering Research and Design
j ourna l h omepage: www.elsev ier .com/ locate /cherd
Iron incorporated rice husk silica as a sorbent forhexavalent chromium attenuation in aqueous system
N.A. Oladoja ∗, I.A. Ololade, O.A. Alimi, T.A. Akinnifesi, G.A. OlaremuDepartment of Chemistry, Adekunle Ajasin University, Akungba Akoko, Nigeria
a b s t r a c t
An environmentally benign metal oxide, Fe, was incorporated into silica derived from rice husk via the sol gel route
and the physiognomies of both the modified (IRS) and raw rice husk derived silica (RHS) were studied via FTIR and
XRD analysis and pHPZC and surface area determinations. The stability of the Fe incorporated into the silica matrix,
determined via the toxicity characteristic leaching procedure, showed that the integrity of the sorbent was intact
only in basic medium but got vitiated in acidic medium. The sorption process conformed to the pseudo second order
model than reversible first order and pseudo first order kinetic models and the rate of sorption of Cr(VI) onto either
sorbents was determined by film diffusion. Process variables optimization showed that the amount of Cr(VI) removed
per gram of sorbent reduced with increase in initial solution pH and the negative impact of the anionic interference
was more in the presence of SO32− than NO3
2− and Cl−. The predicted Langmuir monolayer sorption capacity (mg/g)
of the IRS (63.69) was higher than that of the RHS (61.35). The value of the mean free energy (kJ/mol) of sorption,
obtained for IRS (267.26) and RHS (100.00), and the significant changes in the peak positions of specific functional
groups on the Cr(VI) laden sorbents showed that chemisorption was the dominant mechanism of Cr(VI) uptake.
© 2013 The Institution of Chemical Engineers. Published by Elsevier B.V. All rights reserved.
Keywords: Rice husk; Silica; Metal incorporated silica; Cr(VI) removal; Adsorption
1. Introduction
Owing to the wide use of chromium (Cr) containing materialsin diverse industrial processes (e.g. leather tanning, electro-plating, metal finishing and chromate preparation) and theeventual entry into the discharged industrial effluents, atten-tion is now focused on ameliorating the presence in suchwaste streams. Cr occurs in aqueous environment in triva-lent (Cr(III)) and hexavalent (Cr(VI)) forms (Horitsu et al., 1987).Cr(VI) is more toxic than Cr(III) because of its carcinogenicand mutagenic effects. Adsorption is a simple and low costtertiary water treatment procedure that is being exploredfor Cr(VI) attenuation in aqua system. Thus, waste materi-als such as apple residues (Chong et al., 1998), olive pomace(Martin-Lara et al., 2008; Pagnanelli et al., 2003), rice millingby-products (Tarley and Arruda, 2004), Pinus sylvestris sawdust(Taty-Costodes et al., 2005), beer yeast (Han et al., 2006), olivestone (Fiol et al., 2006), neem sawdust (Vinodhini and Das,2009) and CaO derived from snail shell (Oladoja et al., 2011)
∗ Corresponding author. Tel.: +234 8055438642.E-mail address: [email protected] (N.A. Oladoja).Received 1 December 2012; Received in revised form 1 March 2013; Accepted 10 March 2013
have been screened as effective and low cost sorbent in Cr(VI)removal from aqueous solutions in batch system.
In recent years, there has been pronounced tendency toutilize mechanically stable synthetic or natural solid matri-ces in many applications, such as chemically bonded phasein chromatography, extraction of cations from aqueous andnon-aqueous solvents, catalytic or ion-exchange reactions,electronics, ceramics and bioengineering. One of the impor-tant properties explored is related to the adsorption of traceelements onto solid surface, taking pre concentration or sep-aration into account, where from a complex mixture, a singleelement or a series of similar elements can be separated andquantitatively determined (Jal et al., 2004). Silica gels providean excellent surface for the pre-concentration of variousmaterials including metals from aqueous systems. Chemicalmodification of silica gels with organic and inorganic entitiesstems matrices that can host a wide variety of materials andcan be applied in modern techno arenas. These novel materi-als are environmentally friendly, provide a long term service
0263-8762/$ – see front matter © 2013 The Institution of Chemical Engineers. Published by Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.cherd.2013.03.001

Author's personal copy2692 chemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
and can efficiently substitute traditional catalysts, particularlyin liquid phase reactions (Jal et al., 2004).
Adam et al. (2004) and Adam and Hann (2004) have reportedthe use of series of metals, supported on rice husk ash, inmany catalytic reactions. The incorporation of iron into therice husk derived silica matrix was first carried out using thesol–gel technique by Adam et al. (2006) as a heterogeneouscatalyst in the benzylation reaction of toluene with benzylchloride. Chang et al. (2003a, b) also published the use of ricehusk ash as a matrix for metal supported catalyst. Prasetyoko(2001) has also described the preparation and catalytic activityof silica–alumina from rice husk ash.
It is noteworthy that these silica supported metal ions solidmatrices are used only in catalytic applications but rarelyas adsorbent in water pollution control. In the present stud-ies, the use of iron impregnated silica as a novel adsorbentfor Cr(VI) removal is being explored. The choice of iron asthe metal oxide to be incorporated onto the silica matrixwas predicated on the substantial affinity of oxides of ironfor anionic pollutants, namely, arsenite/arsenate (Jang andDempsey, 2008), selenite (Parida et al., 1997), or phosphate(Ryden et al., 1977; Khare et al., 2005), from contaminatedwaters. Furthermore, they are environmentally benign, chem-ically stable over a wide pH range, and cost effective. However,iron oxides are usually present as fine particles and difficultto use in a continuous flow system. In order to subjugate thistechnical bottleneck, researchers now focused on doping par-ticulate iron oxides within the traditional adsorbents of largerparticle size (Jang et al., 2008; Xiong and Peng, 2008; Zhanget al., 2008b). All these hybrid adsorbents combine the excel-lent handling, flow characteristics of adsorbent supports withthe specific affinity of iron oxides toward the targeted anionicpollutants.
The present studies aimed at enhancing the ability ofrice-husk–silica derived from rice husk in the removal ofCr(VI) from aqueous solution by incorporating iron oxideinto the silica matrix. Sorption data, for process scale-up,shall be obtained via equilibrium isotherm studies and thetime–concentration profile shall be studied to obtain thekinetic parameter and also to elucidate the possible mech-anism of sorption. Two reaction process variables (pH andanionic interference) shall be optimized by method of con-tinuous variation and the mechanism of sorbent–sorbateinteractions shall be explicated via FTIR analysis of the virginand Cr(VI) laden sorbent.
2. Materials and methods
2.1. Sorbent preparation
The rice husk derived silica (RHS) and the iron modified ricehusk silica (IRS) were produced by adopting the method ofAdam et al. (2006) accordingly: rice husk, collected from a ricemill, was washed with copious amount of water, rinsed withdistilled water and then dried in the open air. A white silicapowder was produced from the dried rice husk by pyrolysisin a muffle furnace at 700 ◦C for 5 h. The white silica pow-der was treated with 1.0 M HNO3 for 24 h, filtered and washedthoroughly with deionized water until a constant pH valuewas obtained and dried in the oven. The preparation of theIRS proceeded by the addition of 35 g of the dried silica pow-der into 1750 cm3 of 6.0 M NaOH, stirred for 12 h to producesodium silicate solution. The solution was filtered, to remove
undissolved materials, and the filtrate was divided into two.The first portion was titrated with 3.0 M HNO3, via a buret,at fast flow rate until pH 8 was reached then the flow ratewas reduced to drop by drop until a final pH 5 was obtained.The soft gel formed was aged for 4 days, filtered through suc-tion filtration, washed with distilled water and dried in theoven at 110 ◦C for 24 h and then calcined at 700 ◦C for 5 h toproduce RHS. The second portion of the filtrate was titratedwith 3.0 M HNO3 which contained 10 wt% Fe2+ (18.10 g FeSO4
was dissolved in 1000 mL of 3.0 M HNO3). The acid was addedvia a buret at a fast flow rate till pH 8 was reached and thetitration rate was reduced to drop by drop until a final pH5was obtained. The soft gel formed was aged for 4 days, filteredthrough suction filtration and washed with distilled water. Itwas dried in an oven at 110 ◦C for 24 h and then calcined at700 ◦C for 5 h to produce IRS.
The RHS and the IRS produced were characterized thus:The surficial functional group was determined by FTIR(Perkin–Elmer System 2000) spectroscopy, the crystallinitywas determined by powder X-ray diffractometry (Siemensdiffractometer D5000, Kristalloflex), and the surface area wasdetermined by a protocol designed by Sears (1956) for thedetermination of surface area of siliceous materials. The PZCwas determined by the solid addition method (Balistrieri andMurray, 1981). The stability of the iron incorporated into thesilica matrix in the IRS was assessed via batch leaching testby adopting the toxicity characteristics leaching procedure(TCLP), as described in USEPA protocol (1992).
2.2. Preparation and analysis of Cr(VI) solution
A stock solution of hexavalent chromium (1000 mg/L) was pre-pared in deionized double distilled water using potassiumdichromate (K2Cr2O7). All working solutions of varying con-centrations were obtained by serial dilution. The residualCr(VI) concentration in the solution was determined spec-trophotometrically at 540 nm using a UV–vis spectrophotome-ter (UV-1601, Shimadzu), following the 1,5-diphenylcarbazideprotocol (APHA, 1998).
2.3. Sorption studies
The time-concentration profiles of the sorption of Cr(VI) byeither sorbent (i.e. RHS or IRS) were studied to obtain thekinetic parameters by the addition of 2.0 g of either adsorbentinto a liter of Cr(VI) solution of different initial concentrations.Solution samples were withdrawn at intervals between 0 and4 h, of sorption, centrifuged and the supernatant was analyzedfor residual Cr(VI). The amount of chromium sorbed per unitmass of the adsorbent (in mg/g) was calculated using the massbalance procedure.
The equilibrium sorption isotherm was studied via theaddition of 0.l g of the adsorbent into 50 mL of working solutionof varying concentrations (50, 100, 150, 200, 250 and 300 mg/L).The mixture was agitated in a thermostatic shaker for 120 min,at a constant agitation speed. At the end of each experiment,samples were removed, centrifuged and the supernatant wasanalyzed for residual Cr(VI).
The sorption process variables were optimized on the eachof the sorbents, viz. the effect of pH on the sorption processwas investigated by varying the pH of the initial Cr(VI) solutionbetween pH 2 and 10; the presence of anionic interference wassimulated by the addition of different concentrations (0.01, 0.1,
Author's personal copychemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702 2693
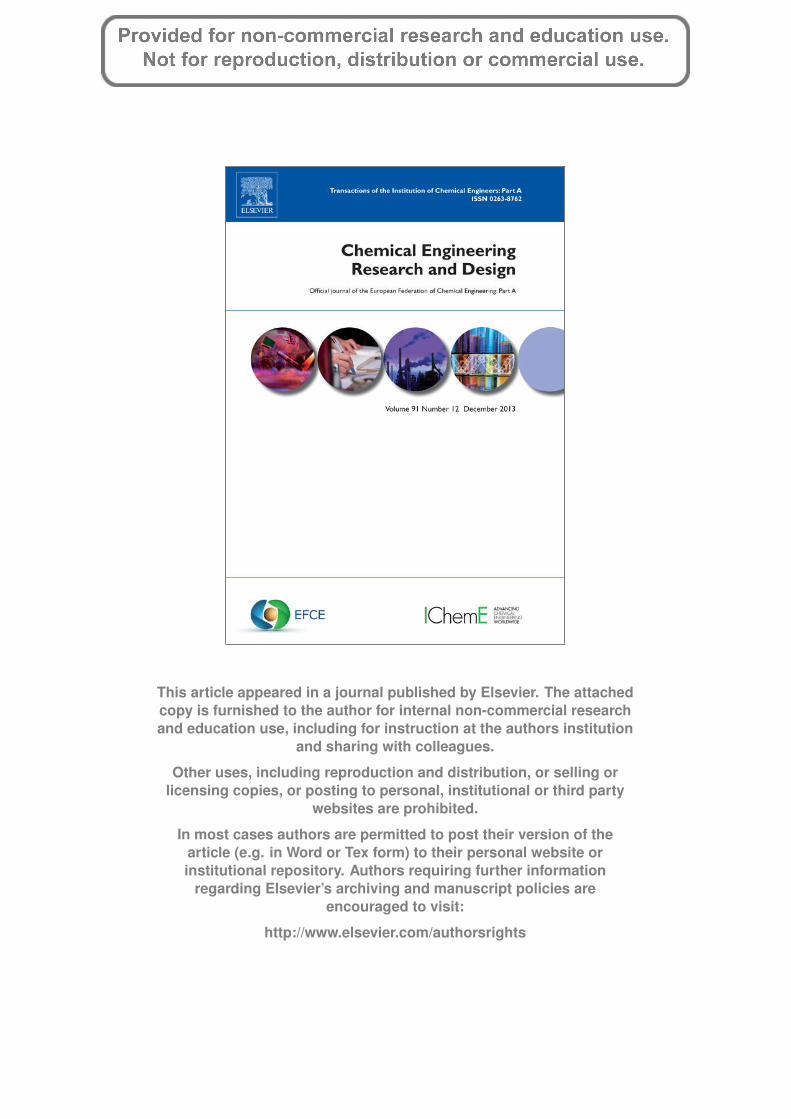
Fig. 1 – (a) Results of the FTIR analysis of the RHS. (b)Results of the FTIR analysis of the IRS.
0.25 M) of the potassium salts of different anions, viz. NO3−,
Cl− and SO32−.
3. Results and discussion
3.1. Sorbent characterization
The silica surface consists of two types of functional groups,siloxane ( Si O Si ) and silanol ( Si OH). Thus, silica gelmodification has been performed via the reaction of aparticular molecule with either the siloxane (nucleophilic sub-stitution at the Si) or silanol (direct reaction with the hydroxylgroup) functional group, although it is generally accepted thatit is the reaction with the silanol group that constitutes themain modification pathway (Dash et al., 2008).
The results of the FTIR analysis of the RHS and IRS are pre-sented in Fig. 1a and b. The RHS showed broad bands at 3385and 3234 which were ascribed to the stretching vibration of theOH group. The presence of the OH group has been ascribed tothe OH on the silanol group and adsorbed water molecules,bonded to the silica surface (Adam et al., 2006). The peak at1647 was ascribed to the bonding vibration of water moleculetrapped in the silica matrix. The band at 1014, 989 and 976were ascribed to the structural siloxane band, Si O Si, whilethe peaks at 866 and 478 were ascribed to the deformation ofthe Si O group.
The IRS spectra showed a broader band between 3001 and3545 which is an indication of interaction of the silanol OHor the adsorbed water molecule with the incorporated Fe.
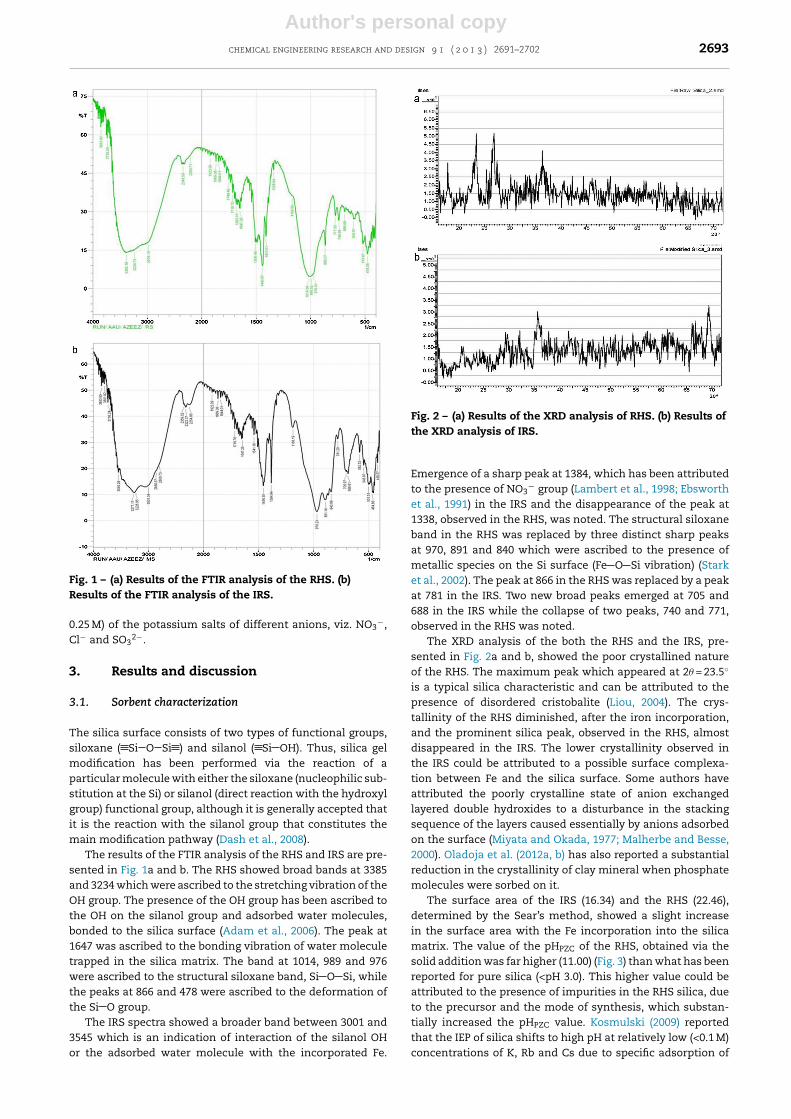
Fig. 2 – (a) Results of the XRD analysis of RHS. (b) Results ofthe XRD analysis of IRS.
Emergence of a sharp peak at 1384, which has been attributedto the presence of NO3
− group (Lambert et al., 1998; Ebsworthet al., 1991) in the IRS and the disappearance of the peak at1338, observed in the RHS, was noted. The structural siloxaneband in the RHS was replaced by three distinct sharp peaksat 970, 891 and 840 which were ascribed to the presence ofmetallic species on the Si surface (Fe O Si vibration) (Starket al., 2002). The peak at 866 in the RHS was replaced by a peakat 781 in the IRS. Two new broad peaks emerged at 705 and688 in the IRS while the collapse of two peaks, 740 and 771,observed in the RHS was noted.
The XRD analysis of the both the RHS and the IRS, pre-sented in Fig. 2a and b, showed the poor crystallined natureof the RHS. The maximum peak which appeared at 2� = 23.5◦
is a typical silica characteristic and can be attributed to thepresence of disordered cristobalite (Liou, 2004). The crys-tallinity of the RHS diminished, after the iron incorporation,and the prominent silica peak, observed in the RHS, almostdisappeared in the IRS. The lower crystallinity observed inthe IRS could be attributed to a possible surface complexa-tion between Fe and the silica surface. Some authors haveattributed the poorly crystalline state of anion exchangedlayered double hydroxides to a disturbance in the stackingsequence of the layers caused essentially by anions adsorbedon the surface (Miyata and Okada, 1977; Malherbe and Besse,2000). Oladoja et al. (2012a, b) has also reported a substantialreduction in the crystallinity of clay mineral when phosphatemolecules were sorbed on it.
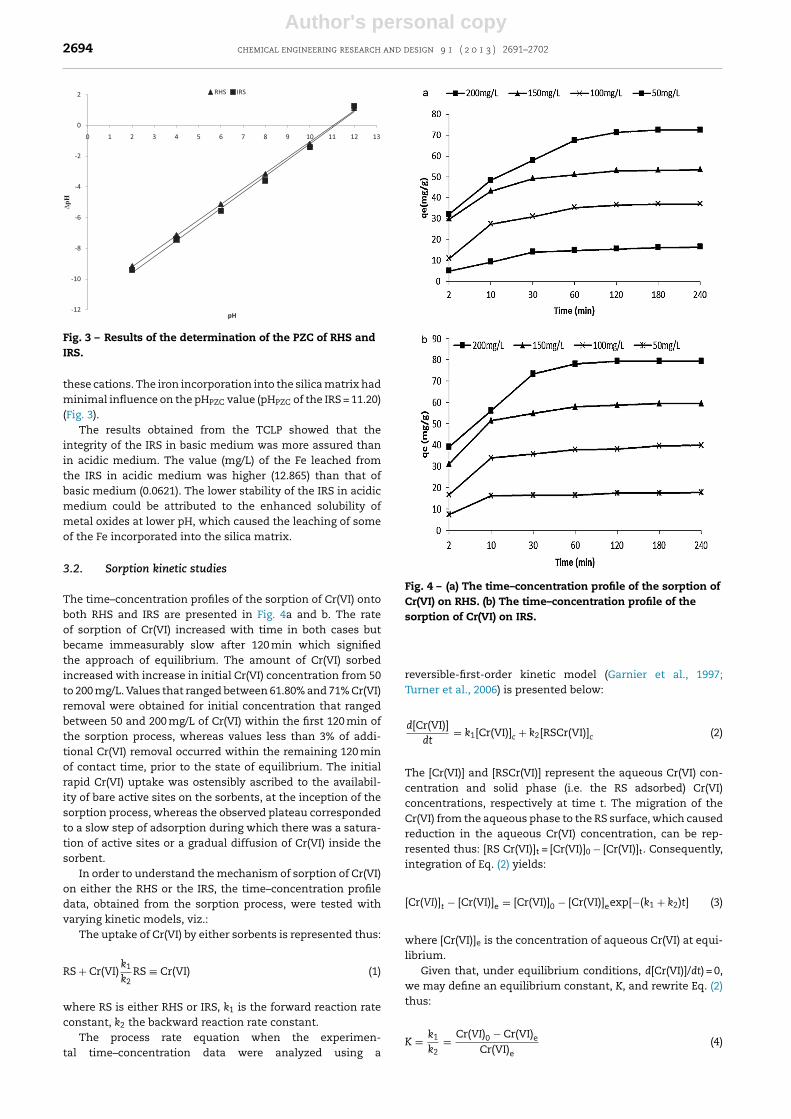
The surface area of the IRS (16.34) and the RHS (22.46),determined by the Sear’s method, showed a slight increasein the surface area with the Fe incorporation into the silicamatrix. The value of the pHPZC of the RHS, obtained via thesolid addition was far higher (11.00) (Fig. 3) than what has beenreported for pure silica (<pH 3.0). This higher value could beattributed to the presence of impurities in the RHS silica, dueto the precursor and the mode of synthesis, which substan-tially increased the pHPZC value. Kosmulski (2009) reportedthat the IEP of silica shifts to high pH at relatively low (<0.1 M)concentrations of K, Rb and Cs due to specific adsorption of
Author's personal copy2694 chemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
-12
-10
-8
-6
-4
-2
0
2
0 1 2 3 4 5 6 7 8 9 10 11 12 13
∆pH
pH
RHS IRS
Fig. 3 – Results of the determination of the PZC of RHS andIRS.
these cations. The iron incorporation into the silica matrix hadminimal influence on the pHPZC value (pHPZC of the IRS = 11.20)(Fig. 3).
The results obtained from the TCLP showed that theintegrity of the IRS in basic medium was more assured thanin acidic medium. The value (mg/L) of the Fe leached fromthe IRS in acidic medium was higher (12.865) than that ofbasic medium (0.0621). The lower stability of the IRS in acidicmedium could be attributed to the enhanced solubility ofmetal oxides at lower pH, which caused the leaching of someof the Fe incorporated into the silica matrix.
3.2. Sorption kinetic studies
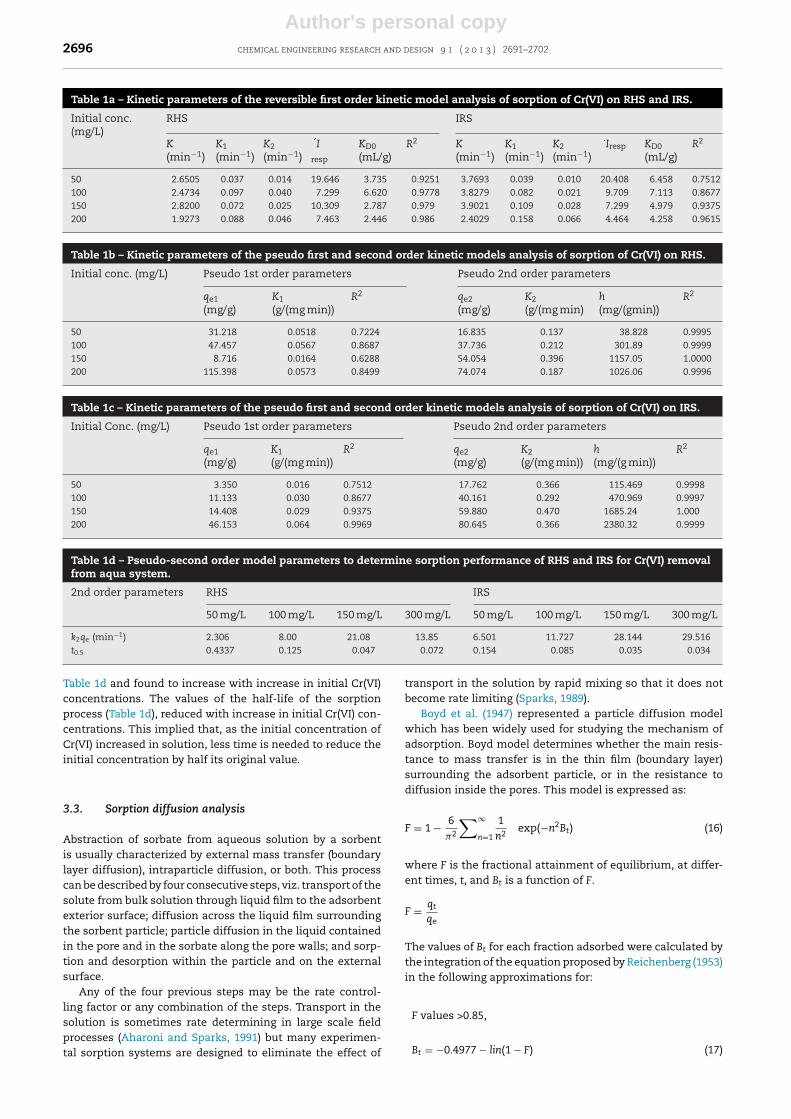
The time–concentration profiles of the sorption of Cr(VI) ontoboth RHS and IRS are presented in Fig. 4a and b. The rateof sorption of Cr(VI) increased with time in both cases butbecame immeasurably slow after 120 min which signifiedthe approach of equilibrium. The amount of Cr(VI) sorbedincreased with increase in initial Cr(VI) concentration from 50to 200 mg/L. Values that ranged between 61.80% and 71% Cr(VI)removal were obtained for initial concentration that rangedbetween 50 and 200 mg/L of Cr(VI) within the first 120 min ofthe sorption process, whereas values less than 3% of addi-tional Cr(VI) removal occurred within the remaining 120 minof contact time, prior to the state of equilibrium. The initialrapid Cr(VI) uptake was ostensibly ascribed to the availabil-ity of bare active sites on the sorbents, at the inception of thesorption process, whereas the observed plateau correspondedto a slow step of adsorption during which there was a satura-tion of active sites or a gradual diffusion of Cr(VI) inside thesorbent.
In order to understand the mechanism of sorption of Cr(VI)on either the RHS or the IRS, the time–concentration profiledata, obtained from the sorption process, were tested withvarying kinetic models, viz.:
The uptake of Cr(VI) by either sorbents is represented thus:
RS + Cr(VI)k1
k2RS ≡ Cr(VI) (1)
where RS is either RHS or IRS, k1 is the forward reaction rateconstant, k2 the backward reaction rate constant.
The process rate equation when the experimen-tal time–concentration data were analyzed using a
Fig. 4 – (a) The time–concentration profile of the sorption ofCr(VI) on RHS. (b) The time–concentration profile of thesorption of Cr(VI) on IRS.
reversible-first-order kinetic model (Garnier et al., 1997;Turner et al., 2006) is presented below:
d[Cr(VI)]dt
= k1[Cr(VI)]c + k2[RSCr(VI)]c (2)
The [Cr(VI)] and [RSCr(VI)] represent the aqueous Cr(VI) con-centration and solid phase (i.e. the RS adsorbed) Cr(VI)concentrations, respectively at time t. The migration of theCr(VI) from the aqueous phase to the RS surface, which causedreduction in the aqueous Cr(VI) concentration, can be rep-resented thus: [RS Cr(VI)]t = [Cr(VI)]0 − [Cr(VI)]t. Consequently,integration of Eq. (2) yields:
[Cr(VI)]t − [Cr(VI)]e = [Cr(VI)]0 − [Cr(VI)]eexp[−(k1 + k2)t] (3)
where [Cr(VI)]e is the concentration of aqueous Cr(VI) at equi-librium.
Given that, under equilibrium conditions, d[Cr(VI)]/dt) = 0,we may define an equilibrium constant, K, and rewrite Eq. (2)thus:
K = k1
k2= Cr(VI)0 − Cr(VI)e
Cr(VI)e(4)

Author's personal copychemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702 2695
Rearranging Eq. (4) in terms of k2 and substituting this into Eq.(3) results in the following:
k1t =(
Cr(VI)0 − Cr(VI)eCr(VI)0
)ln
(Cr(VI)0 − Cr(VI)eCr(VI)t − Cr(VI)e
)(5)
The gradient of a plot of the term on the right hand sideof Eq. (5) versus time affords an estimate of the first order for-ward rate constant for the reaction, and thence from Eq. (4)an estimate of the reverse reaction constant may be obtained.The linearity of the reversible first order kinetic model, pre-sented in Tables 1a–1d, showed that a poorer description of thesorption process was obtained in the Cr(VI) sorption on IRS (r2
range: 0.7512–0.9615) than with RHS (r2 range: 0.9251–0.986).The overall reaction rate, K, (min−1) was higher in the IRS(range: 2.4029–3.9212) than in the RHA (range: 1.9273–2.8200).The higher values of the overall reaction rate in the IRS couldbe attributed to an increased surface activity on the IRS thanon the RHS, during the sorption process. The rate of forwardreaction, k1, was faster, in both the IRS and the RHS, than thatof the backward reaction, k2 (min−1), at all the concentrationsstudied and both rates were found to increase with increasein initial sorbate concentration (Table 1a).
The concentration dependence of the sorption of Cr(VI), oneither the IRS or the RHS, to the state of equilibrium attain-ment was assessed using the system response time ( Iresp),defined as the reciprocal of the summed rate constants.
′Iresp = (k1 + k2)−1 (6)
I
resp represents the time required for the system to reach thenew equilibrium with the optimization of initial Cr(VI) con-centration (Turner et al., 2006; Anirudhan and Rijith, 2009).An increase in the initial Cr(VI) concentration from 50 to200 mg/L, witnessed a reduction in the Iresp value from 19.646to 7.463 min and 20.408 to 4.464 min with the use of RHS andIRS, respectively for Cr(VI) sorption (Table 1a). This revealedthat the attainment of new equilibrium occurred faster athigher initial Cr(VI) concentrations than lower Cr(VI) concen-trations. The tendency of the system for rapid attainment ofequilibrium at higher initial Cr(VI) concentrations could beascribed to the increase in the driving force of overcomingall mass transfer resistance between the Cr(VI) species andthe sorbent (RHS or IRS) with the increase in the initial Cr(VI)concentration. Consequently, an increase in the initial Cr(VI)concentration caused an increase in the amount of Cr(VI)uptake per gram of the sorbent.
In order to clarify the mechanism of a sorption process, theinclusion of instantaneous uptake of sorbate, as a precursor tothe first-order reaction was suggested by Martino et al. (2003),viz.:
RS’ + Cr(VI)KD0RS’ ≡ Cr(VI); RS + Cr(VI)k1
k2RS ≡ Cr(VI)
In this case, sorption sites, RS′, on the sorbent are immedi-ately occupied as RS particles are introduced into the system,an effect that is quantified by an instantaneous distributioncoefficient, (KD)0 (mL/g). This constant is calculated from thedifference in sorbate aqueous concentration at the start ofthe experiment, [Cr(VI)]0, and at the “effective” start of the
experiment, [Cr(VI)′]0, defined as the first measurement in theadsorption time course.
KD0 = [Cr(VI)]0 − [Cr(VI)′]0[Cr(VI)′]0[m]
(7)
where [m] represents the amount of RS particles (g) in thereactor. The value of the instantaneous distribution coeffi-cient, (KD)0 (mL/g), reduced with increase in the initial Cr(VI)concentration (Table 1a).
The Lagergren (1898) pseudo first-order model representedby Eq. (8) and pseudo-second order model (McKay, 1984) rep-resented by Eq. (9) were also employed to analyze the kineticdata obtained from the sorption of Cr(VI) on RHS and IRS.
log[qe − qt] = log[qe] −[
k1
2.303
]t (8)
t
qt= 1
kqe
+ 1qe
t (9)
where the initial sorption rate, h, for the pseudo second orderkinetic model is represented by
h = k2q2e (10)
The results from the fitting of the data obtained from thesorption of Cr(VI) on RHS and IRS into the aforesaid kineticmodels are presented in Tables 1b and 1c respectively. Theexperimental data showed better fitting to the pseudo secondorder kinetic equations (r2 > 0.99) than the pseudo first order.This is a pointer to the fact that the sorption of the Cr(VI) oneither sorbent occurred via chemisorption.
Wu et al. (2009), surmised that the parameter, k2qe (min−1)can be used to define kinetic performance; consequently, thisparameter was obtained by rearranging Eq. (11) to give Eq. (12)viz.:
dqt
dt= k2(qe − qt) (11)
dqt/qe
dt= k2qe
[1 − qt
qe
]2(12)
Eq. (12) reveals that the time changes in dimensionless solid-phase concentration, d(qt/qe)/dt, which is another form ofsorption rate, is proportional to the square of the residualamount of sorbate, 1 − (qt/qe). Consequently, the proportion-ality constant k2qe can be defined as the second order rateindex. Eq. (9) can be rewritten as:(
1qt
− 1qe
)t = 1
k2q2e
(13)
and
t = qt/(qe − qt)k2qe
(14)
At the half-life of the sorption process (i.e. t = t0.5), we haveqt = 0.5qe and
t0.5 = 1k2qe
(15)
The values for pseudo-second order index k2qe, determinedfor RHA-Cr(VI) and IRS-Cr(VI) systems are presented in
Author's personal copy2696 chemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
Table 1a – Kinetic parameters of the reversible first order kinetic model analysis of sorption of Cr(VI) on RHS and IRS.
Initial conc.(mg/L)
RHS IRS
K(min−1)
K1(min−1)
K2(min−1)
Iresp
KD0(mL/g)
R2 K(min−1)
K1(min−1)
K2(min−1)
Iresp KD0(mL/g)
R2
50 2.6505 0.037 0.014 19.646 3.735 0.9251 3.7693 0.039 0.010 20.408 6.458 0.7512100 2.4734 0.097 0.040 7.299 6.620 0.9778 3.8279 0.082 0.021 9.709 7.113 0.8677150 2.8200 0.072 0.025 10.309 2.787 0.979 3.9021 0.109 0.028 7.299 4.979 0.9375200 1.9273 0.088 0.046 7.463 2.446 0.986 2.4029 0.158 0.066 4.464 4.258 0.9615
Table 1b – Kinetic parameters of the pseudo first and second order kinetic models analysis of sorption of Cr(VI) on RHS.
Initial conc. (mg/L) Pseudo 1st order parameters Pseudo 2nd order parameters
qe1(mg/g)
K1(g/(mg min))
R2 qe2(mg/g)
K2(g/(mg min)
h(mg/(gmin))
R2
50 31.218 0.0518 0.7224 16.835 0.137 38.828 0.9995100 47.457 0.0567 0.8687 37.736 0.212 301.89 0.9999150 8.716 0.0164 0.6288 54.054 0.396 1157.05 1.0000200 115.398 0.0573 0.8499 74.074 0.187 1026.06 0.9996
Table 1c – Kinetic parameters of the pseudo first and second order kinetic models analysis of sorption of Cr(VI) on IRS.
Initial Conc. (mg/L) Pseudo 1st order parameters Pseudo 2nd order parameters
qe1(mg/g)
K1(g/(mg min))
R2 qe2(mg/g)
K2(g/(mg min))
h(mg/(g min))
R2
50 3.350 0.016 0.7512 17.762 0.366 115.469 0.9998100 11.133 0.030 0.8677 40.161 0.292 470.969 0.9997150 14.408 0.029 0.9375 59.880 0.470 1685.24 1.000200 46.153 0.064 0.9969 80.645 0.366 2380.32 0.9999
Table 1d – Pseudo-second order model parameters to determine sorption performance of RHS and IRS for Cr(VI) removalfrom aqua system.
2nd order parameters RHS IRS
50 mg/L 100 mg/L 150 mg/L 300 mg/L 50 mg/L 100 mg/L 150 mg/L 300 mg/L
k2qe (min−1) 2.306 8.00 21.08 13.85 6.501 11.727 28.144 29.516t0.5 0.4337 0.125 0.047 0.072 0.154 0.085 0.035 0.034
Table 1d and found to increase with increase in initial Cr(VI)concentrations. The values of the half-life of the sorptionprocess (Table 1d), reduced with increase in initial Cr(VI) con-centrations. This implied that, as the initial concentration ofCr(VI) increased in solution, less time is needed to reduce theinitial concentration by half its original value.
3.3. Sorption diffusion analysis
Abstraction of sorbate from aqueous solution by a sorbentis usually characterized by external mass transfer (boundarylayer diffusion), intraparticle diffusion, or both. This processcan be described by four consecutive steps, viz. transport of thesolute from bulk solution through liquid film to the adsorbentexterior surface; diffusion across the liquid film surroundingthe sorbent particle; particle diffusion in the liquid containedin the pore and in the sorbate along the pore walls; and sorp-tion and desorption within the particle and on the externalsurface.
Any of the four previous steps may be the rate control-ling factor or any combination of the steps. Transport in thesolution is sometimes rate determining in large scale fieldprocesses (Aharoni and Sparks, 1991) but many experimen-tal sorption systems are designed to eliminate the effect of
transport in the solution by rapid mixing so that it does notbecome rate limiting (Sparks, 1989).
Boyd et al. (1947) represented a particle diffusion modelwhich has been widely used for studying the mechanism ofadsorption. Boyd model determines whether the main resis-tance to mass transfer is in the thin film (boundary layer)surrounding the adsorbent particle, or in the resistance todiffusion inside the pores. This model is expressed as:
F = 1 − 6�2
∑∞
n=1
1n2
exp(−n2Bt) (16)
where F is the fractional attainment of equilibrium, at differ-ent times, t, and Bt is a function of F.
F = qt
qe
The values of Bt for each fraction adsorbed were calculated bythe integration of the equation proposed by Reichenberg (1953)in the following approximations for:
F values >0.85,
Bt = −0.4977 − lin(1 − F) (17)
Author's personal copychemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702 2697
0
1
2
3
4
5
6
7a
b
0 20 40 60 80 10 0 12 0 14 0 16 0 18 0 20 0
Bt
Time (min)
200mg/L 150mg/L 100mg /L 50mg/L
0
2
4
6
8
10
12
14
0 20 40 60 80 10 0 12 0 14 0 16 0 18 0 20 0
Bt
Time (min)
200mg/L 150mg /L 100mg /L 50mg/L
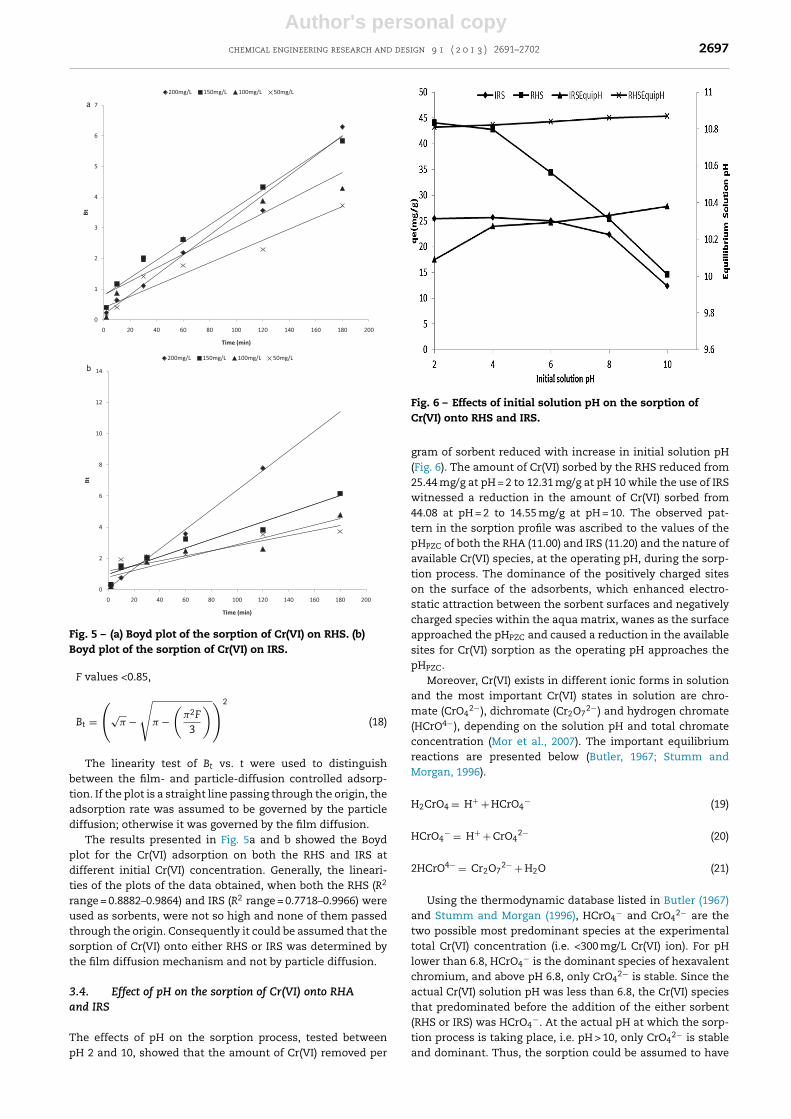
Fig. 5 – (a) Boyd plot of the sorption of Cr(VI) on RHS. (b)Boyd plot of the sorption of Cr(VI) on IRS.
F values <0.85,
Bt =(
√� −√
� −(
�2F
3
))2
(18)
The linearity test of Bt vs. t were used to distinguishbetween the film- and particle-diffusion controlled adsorp-tion. If the plot is a straight line passing through the origin, theadsorption rate was assumed to be governed by the particlediffusion; otherwise it was governed by the film diffusion.
The results presented in Fig. 5a and b showed the Boydplot for the Cr(VI) adsorption on both the RHS and IRS atdifferent initial Cr(VI) concentration. Generally, the lineari-ties of the plots of the data obtained, when both the RHS (R2
range = 0.8882–0.9864) and IRS (R2 range = 0.7718–0.9966) wereused as sorbents, were not so high and none of them passedthrough the origin. Consequently it could be assumed that thesorption of Cr(VI) onto either RHS or IRS was determined bythe film diffusion mechanism and not by particle diffusion.
3.4. Effect of pH on the sorption of Cr(VI) onto RHAand IRS
The effects of pH on the sorption process, tested betweenpH 2 and 10, showed that the amount of Cr(VI) removed per
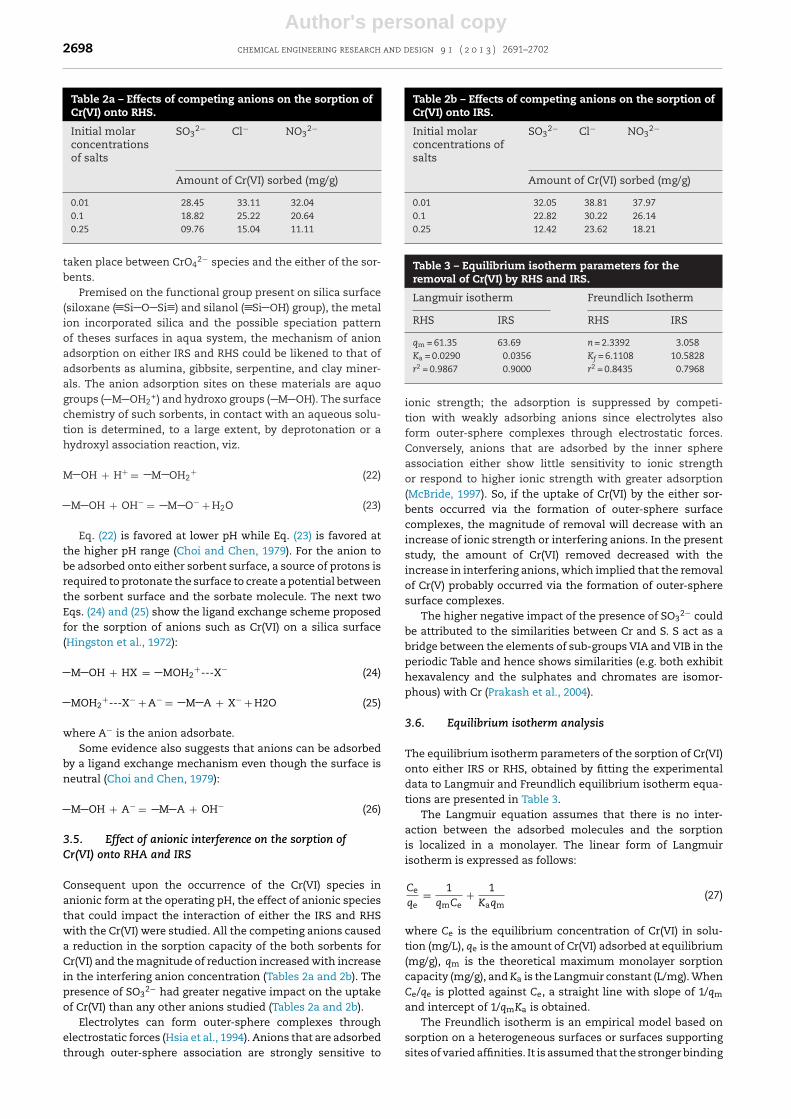
Fig. 6 – Effects of initial solution pH on the sorption ofCr(VI) onto RHS and IRS.
gram of sorbent reduced with increase in initial solution pH(Fig. 6). The amount of Cr(VI) sorbed by the RHS reduced from25.44 mg/g at pH = 2 to 12.31 mg/g at pH 10 while the use of IRSwitnessed a reduction in the amount of Cr(VI) sorbed from44.08 at pH = 2 to 14.55 mg/g at pH = 10. The observed pat-tern in the sorption profile was ascribed to the values of thepHPZC of both the RHA (11.00) and IRS (11.20) and the nature ofavailable Cr(VI) species, at the operating pH, during the sorp-tion process. The dominance of the positively charged siteson the surface of the adsorbents, which enhanced electro-static attraction between the sorbent surfaces and negativelycharged species within the aqua matrix, wanes as the surfaceapproached the pHPZC and caused a reduction in the availablesites for Cr(VI) sorption as the operating pH approaches thepHPZC.
Moreover, Cr(VI) exists in different ionic forms in solutionand the most important Cr(VI) states in solution are chro-mate (CrO4
2−), dichromate (Cr2O72−) and hydrogen chromate
(HCrO4−), depending on the solution pH and total chromateconcentration (Mor et al., 2007). The important equilibriumreactions are presented below (Butler, 1967; Stumm andMorgan, 1996).
H2CrO4 = H+ + HCrO4− (19)
HCrO4− = H+ + CrO4
2− (20)
2HCrO4− = Cr2O72− + H2O (21)
Using the thermodynamic database listed in Butler (1967)and Stumm and Morgan (1996), HCrO4
− and CrO42− are the
two possible most predominant species at the experimentaltotal Cr(VI) concentration (i.e. <300 mg/L Cr(VI) ion). For pHlower than 6.8, HCrO4
− is the dominant species of hexavalentchromium, and above pH 6.8, only CrO4
2− is stable. Since theactual Cr(VI) solution pH was less than 6.8, the Cr(VI) speciesthat predominated before the addition of the either sorbent(RHS or IRS) was HCrO4
−. At the actual pH at which the sorp-tion process is taking place, i.e. pH > 10, only CrO4
2− is stableand dominant. Thus, the sorption could be assumed to have
Author's personal copy2698 chemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
Table 2a – Effects of competing anions on the sorption ofCr(VI) onto RHS.
Initial molarconcentrationsof salts
SO32− Cl− NO3
2−
Amount of Cr(VI) sorbed (mg/g)
0.01 28.45 33.11 32.040.1 18.82 25.22 20.640.25 09.76 15.04 11.11
taken place between CrO42− species and the either of the sor-
bents.Premised on the functional group present on silica surface
(siloxane ( Si O Si ) and silanol ( Si OH) group), the metalion incorporated silica and the possible speciation patternof theses surfaces in aqua system, the mechanism of anionadsorption on either IRS and RHS could be likened to that ofadsorbents as alumina, gibbsite, serpentine, and clay miner-als. The anion adsorption sites on these materials are aquogroups ( M OH2
+) and hydroxo groups ( M OH). The surfacechemistry of such sorbents, in contact with an aqueous solu-tion is determined, to a large extent, by deprotonation or ahydroxyl association reaction, viz.
M OH + H+ = M OH2+ (22)
M OH + OH− = M O− + H2O (23)
Eq. (22) is favored at lower pH while Eq. (23) is favored atthe higher pH range (Choi and Chen, 1979). For the anion tobe adsorbed onto either sorbent surface, a source of protons isrequired to protonate the surface to create a potential betweenthe sorbent surface and the sorbate molecule. The next twoEqs. (24) and (25) show the ligand exchange scheme proposedfor the sorption of anions such as Cr(VI) on a silica surface(Hingston et al., 1972):
M OH + HX = MOH2+---X− (24)
MOH2+---X− + A− = M A + X− + H2O (25)
where A− is the anion adsorbate.Some evidence also suggests that anions can be adsorbed
by a ligand exchange mechanism even though the surface isneutral (Choi and Chen, 1979):
M OH + A− = M A + OH− (26)
3.5. Effect of anionic interference on the sorption ofCr(VI) onto RHA and IRS
Consequent upon the occurrence of the Cr(VI) species inanionic form at the operating pH, the effect of anionic speciesthat could impact the interaction of either the IRS and RHSwith the Cr(VI) were studied. All the competing anions causeda reduction in the sorption capacity of the both sorbents forCr(VI) and the magnitude of reduction increased with increasein the interfering anion concentration (Tables 2a and 2b). Thepresence of SO3
2− had greater negative impact on the uptakeof Cr(VI) than any other anions studied (Tables 2a and 2b).
Electrolytes can form outer-sphere complexes throughelectrostatic forces (Hsia et al., 1994). Anions that are adsorbedthrough outer-sphere association are strongly sensitive to
Table 2b – Effects of competing anions on the sorption ofCr(VI) onto IRS.
Initial molarconcentrations ofsalts
SO32− Cl− NO3
2−
Amount of Cr(VI) sorbed (mg/g)
0.01 32.05 38.81 37.970.1 22.82 30.22 26.140.25 12.42 23.62 18.21
Table 3 – Equilibrium isotherm parameters for theremoval of Cr(VI) by RHS and IRS.
Langmuir isotherm Freundlich Isotherm
RHS IRS RHS IRS
qm = 61.35 63.69 n = 2.3392 3.058Ka = 0.0290 0.0356 Kf = 6.1108 10.5828r2 = 0.9867 0.9000 r2 = 0.8435 0.7968
ionic strength; the adsorption is suppressed by competi-tion with weakly adsorbing anions since electrolytes alsoform outer-sphere complexes through electrostatic forces.Conversely, anions that are adsorbed by the inner sphereassociation either show little sensitivity to ionic strengthor respond to higher ionic strength with greater adsorption(McBride, 1997). So, if the uptake of Cr(VI) by the either sor-bents occurred via the formation of outer-sphere surfacecomplexes, the magnitude of removal will decrease with anincrease of ionic strength or interfering anions. In the presentstudy, the amount of Cr(VI) removed decreased with theincrease in interfering anions, which implied that the removalof Cr(V) probably occurred via the formation of outer-spheresurface complexes.
The higher negative impact of the presence of SO32− could
be attributed to the similarities between Cr and S. S act as abridge between the elements of sub-groups VIA and VIB in theperiodic Table and hence shows similarities (e.g. both exhibithexavalency and the sulphates and chromates are isomor-phous) with Cr (Prakash et al., 2004).
3.6. Equilibrium isotherm analysis
The equilibrium isotherm parameters of the sorption of Cr(VI)onto either IRS or RHS, obtained by fitting the experimentaldata to Langmuir and Freundlich equilibrium isotherm equa-tions are presented in Table 3.
The Langmuir equation assumes that there is no inter-action between the adsorbed molecules and the sorptionis localized in a monolayer. The linear form of Langmuirisotherm is expressed as follows:
Ce
qe= 1
qmCe+ 1
Kaqm(27)
where Ce is the equilibrium concentration of Cr(VI) in solu-tion (mg/L), qe is the amount of Cr(VI) adsorbed at equilibrium(mg/g), qm is the theoretical maximum monolayer sorptioncapacity (mg/g), and Ka is the Langmuir constant (L/mg). WhenCe/qe is plotted against Ce, a straight line with slope of 1/qm
and intercept of 1/qmKa is obtained.The Freundlich isotherm is an empirical model based on
sorption on a heterogeneous surfaces or surfaces supportingsites of varied affinities. It is assumed that the stronger binding
Author's personal copychemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702 2699
Table 4 – Comparison of monolayer sorption capacity(qmax, mg/g) of Cr(VI) on various adsorbents with RHSand IRS.
Adsorbent qmax (mg/g) References
RHS 61.35 Present studiesIRS 63.69 Present studiesNeem leaves 62.97 Babu and Gupta (2008)Trichoderma Fungal 48.9 Liliana and Eliseo (2008)Weed Salvina cucullata 13 Soroj and Surendra (2008)Black tea leaves 45.5 Mohammed and Mikio (2005)Alligator weed 82.57 Wang et al. (2008)Reed lignin 52.6 Young et al. (2006)Activated lignin 75.75 Tazerouti and Amrani (2009)Sugarcane bagasse 13.4 Shamah and Forster (1994)Nano calcium oxide 125 Oladoja et al. (2012a,b)
sites are occupied first and that the binding strength decreaseswith the increasing degree of site occupation. The linear formof Freundlich isotherm is written as follows:
log qe = log Kf + 1n
log Ce (28)
where Ce is the equilibrium concentration of Cr(VI) in solu-tion (mg/L), qe is the amount of Cr(VI) adsorbed at equilibrium(mg/g), Kf is the Freundlich constant (mg/g)(L/mg)1/n and 1/nis the heterogeneity factor. Therefore, a plot of log qe versuslog Ce enables the constant Kf and exponent 1/n to be deter-mined.
The conformity of the experimental data with each of theisotherm equation was tested via the determination of thecorrelation coefficient (r2) value of the linear plot. The Lang-muir equilibrium isotherm model gave better description tothe sorption of Cr(VI) onto both RHS and IRS than Freundlichisotherm model. The enhanced sorption capacity of the mod-ified sorbent for Cr(VI) over the unmodified sorbent could beseen in the qm (mg/g)value obtained for the IRS (63.69) whichwas higher than that of RHS (61.35). The value of the Lang-muir constant, Ka (L/mg), was higher in the IRS (0.0356) thanin the RHS (0.0290). The comparison of the Langmuir param-eter, qm (mg/g), obtained from the present studies with thevalues obtained by other investigators is presented in Table 4.
The values of Kf and 1/n obtained from the fitting of theexperimental data to the Freundlich isotherm were higher inthe RHS-Cr(VI) system than in the RHS-Cr(VI) system but thelinearity was relatively poor (<0.9).
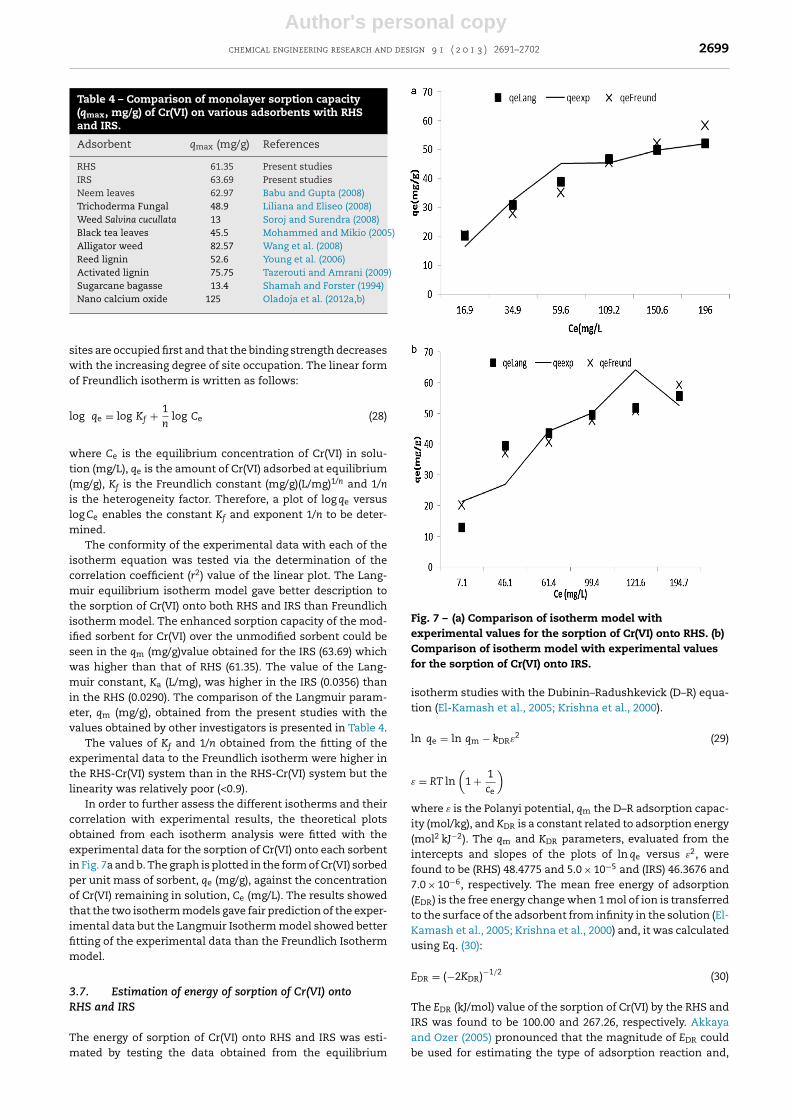
In order to further assess the different isotherms and theircorrelation with experimental results, the theoretical plotsobtained from each isotherm analysis were fitted with theexperimental data for the sorption of Cr(VI) onto each sorbentin Fig. 7a and b. The graph is plotted in the form of Cr(VI) sorbedper unit mass of sorbent, qe (mg/g), against the concentrationof Cr(VI) remaining in solution, Ce (mg/L). The results showedthat the two isotherm models gave fair prediction of the exper-imental data but the Langmuir Isotherm model showed betterfitting of the experimental data than the Freundlich Isothermmodel.
3.7. Estimation of energy of sorption of Cr(VI) ontoRHS and IRS
The energy of sorption of Cr(VI) onto RHS and IRS was esti-mated by testing the data obtained from the equilibrium
Fig. 7 – (a) Comparison of isotherm model withexperimental values for the sorption of Cr(VI) onto RHS. (b)Comparison of isotherm model with experimental valuesfor the sorption of Cr(VI) onto IRS.
isotherm studies with the Dubinin–Radushkevick (D–R) equa-tion (El-Kamash et al., 2005; Krishna et al., 2000).
ln qe = ln qm − kDRε2 (29)
ε = RT ln(
1 + 1ce
)where ε is the Polanyi potential, qm the D–R adsorption capac-ity (mol/kg), and KDR is a constant related to adsorption energy(mol2 kJ−2). The qm and KDR parameters, evaluated from theintercepts and slopes of the plots of ln qe versus ε2, werefound to be (RHS) 48.4775 and 5.0 × 10−5 and (IRS) 46.3676 and7.0 × 10−6, respectively. The mean free energy of adsorption(EDR) is the free energy change when 1 mol of ion is transferredto the surface of the adsorbent from infinity in the solution (El-Kamash et al., 2005; Krishna et al., 2000) and, it was calculatedusing Eq. (30):
EDR = (−2KDR)−1/2 (30)
The EDR (kJ/mol) value of the sorption of Cr(VI) by the RHS andIRS was found to be 100.00 and 267.26, respectively. Akkayaand Ozer (2005) pronounced that the magnitude of EDR couldbe used for estimating the type of adsorption reaction and,
Author's personal copy2700 chemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
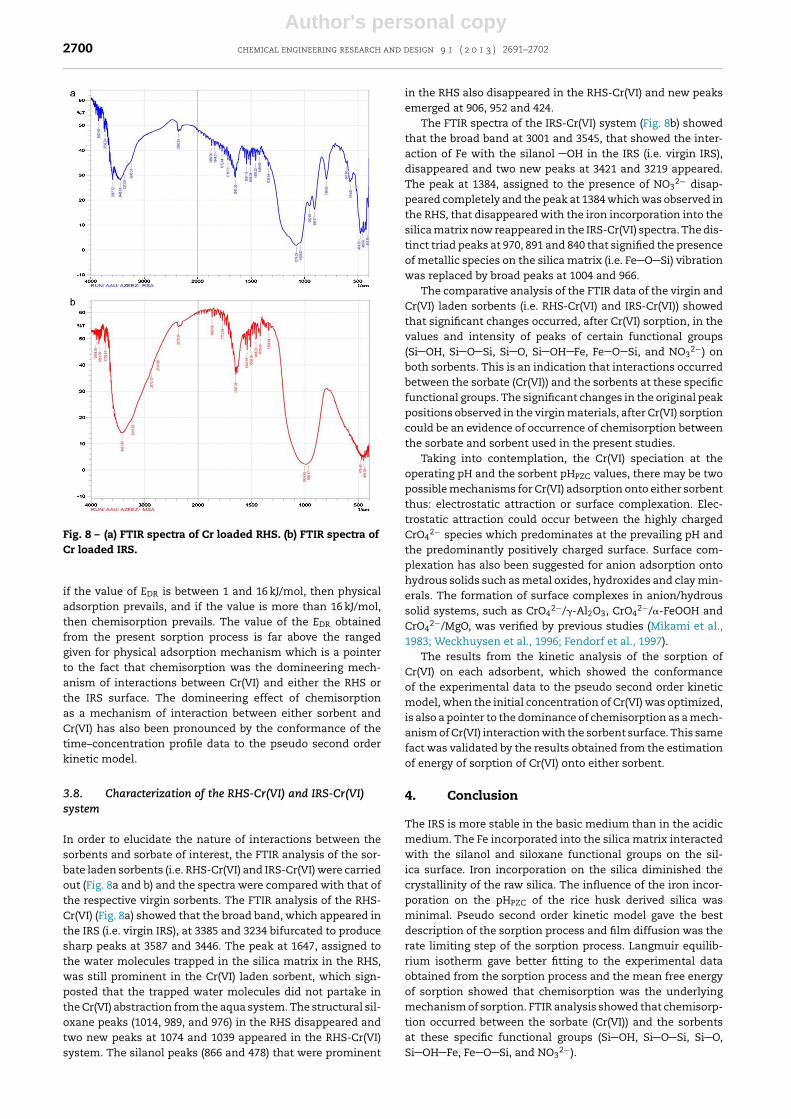
Fig. 8 – (a) FTIR spectra of Cr loaded RHS. (b) FTIR spectra ofCr loaded IRS.
if the value of EDR is between 1 and 16 kJ/mol, then physicaladsorption prevails, and if the value is more than 16 kJ/mol,then chemisorption prevails. The value of the EDR obtainedfrom the present sorption process is far above the rangedgiven for physical adsorption mechanism which is a pointerto the fact that chemisorption was the domineering mech-anism of interactions between Cr(VI) and either the RHS orthe IRS surface. The domineering effect of chemisorptionas a mechanism of interaction between either sorbent andCr(VI) has also been pronounced by the conformance of thetime–concentration profile data to the pseudo second orderkinetic model.
3.8. Characterization of the RHS-Cr(VI) and IRS-Cr(VI)system
In order to elucidate the nature of interactions between thesorbents and sorbate of interest, the FTIR analysis of the sor-bate laden sorbents (i.e. RHS-Cr(VI) and IRS-Cr(VI) were carriedout (Fig. 8a and b) and the spectra were compared with that ofthe respective virgin sorbents. The FTIR analysis of the RHS-Cr(VI) (Fig. 8a) showed that the broad band, which appeared inthe IRS (i.e. virgin IRS), at 3385 and 3234 bifurcated to producesharp peaks at 3587 and 3446. The peak at 1647, assigned tothe water molecules trapped in the silica matrix in the RHS,was still prominent in the Cr(VI) laden sorbent, which sign-posted that the trapped water molecules did not partake inthe Cr(VI) abstraction from the aqua system. The structural sil-oxane peaks (1014, 989, and 976) in the RHS disappeared andtwo new peaks at 1074 and 1039 appeared in the RHS-Cr(VI)system. The silanol peaks (866 and 478) that were prominent
in the RHS also disappeared in the RHS-Cr(VI) and new peaksemerged at 906, 952 and 424.
The FTIR spectra of the IRS-Cr(VI) system (Fig. 8b) showedthat the broad band at 3001 and 3545, that showed the inter-action of Fe with the silanol OH in the IRS (i.e. virgin IRS),disappeared and two new peaks at 3421 and 3219 appeared.The peak at 1384, assigned to the presence of NO3
2− disap-peared completely and the peak at 1384 which was observed inthe RHS, that disappeared with the iron incorporation into thesilica matrix now reappeared in the IRS-Cr(VI) spectra. The dis-tinct triad peaks at 970, 891 and 840 that signified the presenceof metallic species on the silica matrix (i.e. Fe O Si) vibrationwas replaced by broad peaks at 1004 and 966.
The comparative analysis of the FTIR data of the virgin andCr(VI) laden sorbents (i.e. RHS-Cr(VI) and IRS-Cr(VI)) showedthat significant changes occurred, after Cr(VI) sorption, in thevalues and intensity of peaks of certain functional groups(Si OH, Si O Si, Si O, Si OH Fe, Fe O Si, and NO3
2−) onboth sorbents. This is an indication that interactions occurredbetween the sorbate (Cr(VI)) and the sorbents at these specificfunctional groups. The significant changes in the original peakpositions observed in the virgin materials, after Cr(VI) sorptioncould be an evidence of occurrence of chemisorption betweenthe sorbate and sorbent used in the present studies.
Taking into contemplation, the Cr(VI) speciation at theoperating pH and the sorbent pHPZC values, there may be twopossible mechanisms for Cr(VI) adsorption onto either sorbentthus: electrostatic attraction or surface complexation. Elec-trostatic attraction could occur between the highly chargedCrO4
2− species which predominates at the prevailing pH andthe predominantly positively charged surface. Surface com-plexation has also been suggested for anion adsorption ontohydrous solids such as metal oxides, hydroxides and clay min-erals. The formation of surface complexes in anion/hydroussolid systems, such as CrO4
2−/�-Al2O3, CrO42−/�-FeOOH and
CrO42−/MgO, was verified by previous studies (Mikami et al.,
1983; Weckhuysen et al., 1996; Fendorf et al., 1997).The results from the kinetic analysis of the sorption of
Cr(VI) on each adsorbent, which showed the conformanceof the experimental data to the pseudo second order kineticmodel, when the initial concentration of Cr(VI) was optimized,is also a pointer to the dominance of chemisorption as a mech-anism of Cr(VI) interaction with the sorbent surface. This samefact was validated by the results obtained from the estimationof energy of sorption of Cr(VI) onto either sorbent.
4. Conclusion
The IRS is more stable in the basic medium than in the acidicmedium. The Fe incorporated into the silica matrix interactedwith the silanol and siloxane functional groups on the sil-ica surface. Iron incorporation on the silica diminished thecrystallinity of the raw silica. The influence of the iron incor-poration on the pHPZC of the rice husk derived silica wasminimal. Pseudo second order kinetic model gave the bestdescription of the sorption process and film diffusion was therate limiting step of the sorption process. Langmuir equilib-rium isotherm gave better fitting to the experimental dataobtained from the sorption process and the mean free energyof sorption showed that chemisorption was the underlyingmechanism of sorption. FTIR analysis showed that chemisorp-tion occurred between the sorbate (Cr(VI)) and the sorbentsat these specific functional groups (Si OH, Si O Si, Si O,Si OH Fe, Fe O Si, and NO3
2−).

Author's personal copychemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702 2701
References
Adam, F., Hann, C.J., 2004. The adsorption of palmytic acid on ricehusk ash chemically modified with Al(III) ion using the sol–geltechnique. J. Colloid Interface Sci. 280, 55–61.
Adam, F., Kandasamy, K., Balakrishnan, S., 2006. Ironincorporated heterogeneous catalyst from rice husk ash. J.Colloid Interface Sci. 304, 137.
Adam, F., Saad, B., Saraswathy, B., Fazliawati, M., 2004. Silicasupported metal oxide catalysts from rice husk ash. In: PaperPresented at the Regional Conference of Young Chemist(RCYC 2004), 13–14th of April 2004, Universiti Sains Malaysia,Pulau Pinang, Malaysia.
Aharoni, C., Sparks, D.L., 1991. Kinetics of soil chemicalreactions—a theoretical treatment. In: Sparks, D.L., Suarez,D.L. (Eds.), Rates of Soil Chemical Processes. Soil ScienceSociety of America, Madison, WI, p. 1.
Akkaya, G., Ozer, A., 2005. Adsorption of acid red 274 (AR 274) onDicranella varia: determination of equilibrium and kineticmodel parameters. Proc. Biochem. 40, 3559–3568.
Anirudhan, T.S., Rijith, S., 2009. Glutaraldehyde cross-linkedepoxyaminated chitosan as an adsorbent for the removal andrecovery of copper (II) from aqueous media. Colloids Surf. A:Physicochem. Eng. Aspects 351, 52–59.
APHA, 1998. Standard Methods for the Examination of Water andWastewater, vol. 1., 20th ed. American Public HealthAssociation, Washington.
Babu, B.V., Gupta, S., 2008. Adsorption of Cr (VI) using activatedneem leaves: kinetic studies. Adsorption 14, 85–92.
Balistrieri, L.S., Murray, J.W., 1981. The surface chemistry ofgoethite (�-FeOOH) in major ion seawater. Am. J. Sci. 281 (6),788–806.
Boyd, G.E., Adamson, A.W., Myers Jr., L.S., 1947. The exchangeadsorption of ions from aqueous solution by organic zeolites.Part II. Kinetics. J. Am. Chem. Soc. 69, 2836–2848.
Butler, J.N., 1967. Ionic Equilibrium. Addison-Wesley, New York.Chang, F.W., Kuo, M.S., Tsay, M.T., Hsieh, M.C., 2003a. Appl. Catal.
A: Gen. 247, 309–320.Chang, F.W., Kuo, W.Y., Lee, K.C., 2003b. Appl. Catal. A: Gen. 246,
253–264.Choi, W.-W., Chen, K.Y., 1979. The removal of fluoride from
waters by adsorption. AWWA J. 71, 562–570.Chong, S.H., Jung, H., Chung, H., Lee, M.Y., Yang, J., 1998. Removal
of heavy metals from aqueous solution by apple residues.Process Biochem. 33 (2), 205–211.
Dash, S., Mishra, S., Patel, S., Mishra, B.K., 2008. Organicallymodified silica: synthesis and applications due to its surfaceinteraction with organic molecules. Adv. Colloid Interface Sci.140, 77–94.
Ebsworth, E.A.V., Rankin, D.W.H., Cradock, S., 1991. StructuralMethods in Inorganic Chemistry, 2nd ed. Blackwell ScientificPublication, Oxford, United Kingdom, p. 222.
El-Kamash, A.M., Zaki, A.A., El Geleel, M.A., 2005. Modeling batchkinetics and thermodynamics of zinc and cadmium ionsremoval from waste solutions using synthetic zeolite A. J.Hazard. Mater. 127, 211–220.
Fendorf, S., Eick, M.J., Grossl, P., Sparks, D.L., 1997. Arsenate andchromate retention mechanisms on goethite. 1. Surfacestructure. Environ. Sci. Technol. 31, 315–319.
Fiol, N., Villaescusa, I., Martínez, M., Miralles, N., Poch, J.,Serrarlos, J., 2006. Sorption of Pb(II), Ni(II), Cu(II) and Cd(II)from aqueous solution by olive stone waste. Sep. Purif.Technol. 50 (1), 132–140.
Garnier, J.M., Pham, M.K., Ciffroy, P., Martin, J.M., 1997. Kinetics oftrace metal complexation with suspended matter and withfilterable ligands in freshwater. Environ. Sci. Technol. 31,1597–1606.
Han, R., Li, H., Li, Y., Zhang, J., Xiao, H., Shi, J., 2006. Biosorption ofcopper and lead ions by waste beer yeast. J. Hazard. Mater. 137(3), 1569–1576.
Hingston, F.J., Posner, A.M., Quirk, J.P., 1972. Anion adsorption bygeothite and gibbsite: I. The role of protonation indetermining adsorption envelopes. Soil Science 23 (2), 177.
Horitsu, H., Futo, S., Miyazawa, Y., Ogai, S., Kawai, K., 1987.Enzymatic reduction of hexavalent chromium by hexavalentchromium tolerant Pseudomonas ambigua G-1. Agric. Biol.Chem. 51, 2417–2420.
Hsia, T.H., Lo, S.L., Lin, C.F., Lee, D.Y., 1994. Characterization ofarsenate adsorption on hydrous iron-oxide using chemicaland physical methods. Colloid Surf. A 85, 1–7.
Jal, P.K., Patel, S., Mishra, B.K., 2004. Chemical modification ofsilica surface by immobilization of functional groups forextractive concentration of metal ions. Talanta 62, 1005–1028.
Jang, J.H., Dempsey, B.A., 2008. Coadsorption of arsenic(III) andarsenic(V) onto hydrous ferric oxide: effects on abioticoxidation of arsenic(III), extraction efficiency, and modelaccuracy. Environ. Sci. Technol. 42 (8), 2893–2898.
Jang, M., Chen, W.F., Cannon, F.S., 2008. Preloading hydrousferricoxide into granular activated carbon for arsenic removal.Environ. Sci. Technol. 42 (9), 3369–3374.
Khare, N., Hesterberg, D., Martin, J.D., 2005. XANES investigationof phosphate sorption in single and binary systems of ironand aluminum oxide minerals. Environ. Sci. Technol. 39 (7),2152–2160.
Kosmulski, M., 2009. Compilation of PZC and IEP of sparinglysoluble metal oxides and hydroxides from literature. Adv.Colloid Interface Sci. 152, 14–25.
Krishna, B.S., Murty, D.S.R., Jai Prakash, B.S., 2000.Thermodynamics of chromium(VI) anionic species sorptiononto surfactant-modified montmorillonite clay. J. ColloidInterface Sci. 229, 230–236.
Lagergren, S., 1898. About the theory of so-called adsorption ofsoluble substances. K. Sven. Vetenskapsakad. Handl. Band. 24,1–39.
Lambert, J.B., Shurvell, H.F., Lightner, D.A., Cooks, R.G., 1998.Organic Structural Spectroscopy. Prentice Hall, NJ, USA,p. 191.
Liliana, M.B., Eliseo, C.U., 2008. Hexavalent chromium removal bya Trichoderma inhhamatum fungal strain isolated from tanneryeffluent. Water Air Soil Pollut. 187, 327–336.
Liou, T.H., 2004. Preparation and characterization ofnano-structured silica from rice husk. Mater. Sci. Eng. A 364,313.
Malherbe, F., Besse, J.P., 2000. Investigating the effects ofguest–host interactions on the properties of anion-exchangedMg–Al–hydrotalcites. J. Solid State Chem. 155 (2),332–341.
Martin-Lara, M.A., Pagnanelli, F., Mainelli, S., Calero, M., Toro, L.,2008. Chemical treatment of olive pomace: effects onacid–basic properties and metal biosorption capacity. J.Hazard. Mater. 156 (1–3), 448–457.
Martino, M., Turner, A., Millward, G.E., 2003. Influence of organiccomplexation on the adsorption kinetics of nickel in riverwaters. Environ. Sci. Technol. 37, 2383–2388.
McBride, M.B., 1997. A critique of diffuse double layer modelsapplied to colloid and surface chemistry. Clays Clay Miner. 45,598–608.
McKay, G., 1984. The adsorption of basic dye onto silica fromaqueous solution–solid diffusion model. Chem. Eng. Sci. 39(1), 129–138.
Mikami, N., Sasaki, M., Klkuchi, T., Yasunaga, T., 1983. Kinetics ofadsorption–desorption of chromate on g-alumina surfaceusing the pressure-jump technique. J. Phys. Chem. 87 (25),5245.
Miyata, S., Okada, A., 1977. Synthesis of hydrotalcite-likecompounds and their physicochemical properties—thesystems Mg2+–Al3+–SO4
2− and Mg2+–Al3+–CrO42−. Clays Clay
Miner. 25, 14–18.Mohammed, A.H., Mikio, K., 2005. Optimisation of parameters for
Cr (VI) adsorption on used black tea leaves. Adsorption 11,561–568.
Mor, S., Ravindra, K., Bishnoi, N.R., 2007. Adsorption of chromiumfrom aqueous solution by activated alumina and activatedcharcoal. Bioresour. Technol. 98 (4), 954–957.
Oladoja, N.A., Ololade, I.A., Olaseni, S.E., Olatujoye, V.O., Jegede,O.S., Agunloye, A.O., 2011. Synthesis of nano calcium oxide

Author's personal copy2702 chemical engineering research and design 9 1 ( 2 0 1 3 ) 2691–2702
from a gastropod shell and the performance evaluation forCr(VI) removal from aqua system. Ind. Eng. Chem. Res.,http://dx.doi.org/10.1021/ie201189z.
Oladoja, N.A., Aboluwoye, C.O., Ololade, I.A., Adebayo, O.L.,Olaseni, S.E., Adelagun, R.O.A., 2012a. Intercalation ofgastropod shell derived calcium oxide in clay and applicationin phosphate abstraction from aqua medium. Ind. Eng. Chem.Res., http://dx.doi.org/10.1021/ie301520v.
Oladoja, N.A., Ololade, I.A., Olatujoye, V.O., Akinnifesi, T.A., 2012b.Performance evaluation of fixed bed of nano calcium oxidesynthesized from a gastropod shell (Achatina achatina) inhexavalent chromium abstraction from aqua system. WaterAir Soil Pollut. 223, 1861–1876.
Pagnanelli, F., Mainelli, S., Veglio, F., Toro, L., 2003. Heavy metalremoval by olive pomace: biosorbent characterisation andequilibrium modelling. Chem. Eng. Sci. 58 (20),4709–4717.
Parida, K.M., Gorai, B., Das, N.N., Rao, S.B., 1997. Studies on ferricoxide hydroxides: III. Adsorption of selenite (SeO3 2 ) ondifferent forms of iron oxyhydroxides. J. Colloid Interface Sci.185 (2), 355–362.
Prakash, S., Tuli, G.D., Basu, S.K., Madan, R.D., 2004. AdvancedInorganic Chemistry, vol. II. S. Chand and Company Ltd., NewDelhi.
Prasetyoko, D., 2001. Direct synthesis of �-zeolite from rice huskash and its use as a heterogeneous catalyst in theFriedel–Craft reaction, M.Sc. thesis, Universiti Teknologi,Malaysia.
Reichenberg, D., 1953. Properties of ion exchange resins inrelation to their structure. Part III. Kinetics of exchange. J. Am.Chem. Soc. 75, 589–598.
Ryden, J.C., Mclaughlin, J.R., Syers, J.K., 1977. Mechanisms ofphosphate sorption by soils and hydrous ferric-oxide gel.Journal of Soil Science 28 (1), 72–79.
Sears, G.W., 1956. Determination of specific surface area ofcolloidal silica by titration with sodium hydroxide. Anal.Chem. 28, 1981–1983.
Shamah, D.C., Forster, C.F., 1994. A preliminary examination intothe adsorption of Cr (VI) using low-cost adsorbents. Bioresour.Technol. 47, 257–264.
Soroj, S.B., Surendra, N.D., 2008. Adsorption of Cr (VI) by treatedweed Salvinia cucullata: kinetics and mechanism. Adsorption14, 111–121.
Sparks, D.L., 1989. Kinetics of Soil Chemical Processes. AcademicPress, New York.
Stark, W.J., Strobel, R., Gunther, D., Pratnis, S.E., Baiker, A., 2002.Titania-silica doped with transition metals via flamesynthesis: structural properties and catalytic behavior inepoxidation. J. Mater. Chem. 12, 3620–3625.
Stumm, W., Morgan, J.J., 1996. Aquatic Chemistry: AnIntroduction Emphasizing Chemical Equilibria in NaturalWaters. John Wiley & Sons, Inc., New York.
Tarley, C.R.T., Arruda, M.A.Z., 2004. Biosorption of heavy metalsusing rice milling byproducts characterisation and applicationfor removal of metals from aqueous effluents. Chemosphere54 (7), 987–995.
Taty-Costodes, V.C., Fauduet, H., Porte, C., Ho, Y.S., 2005. Removalof lead (II) ions from synthetic and real effluents usingimmobilized Pinus sylvestris sawdust: adsorption on afixed-bed column. J. Hazard. Mater. 123 (1–3), 135–144.
Tazerouti, Amrani, 2009. Chromium (VI) adsorption on activatedlignin. Chem. Prod. Process Model. 4 (1), Art. 37.
Turner, A., Crussel, M., Millward, G., Garcia, A.C., Fischer, A., 2006.Adsorption kinetics of platinum group elements in riverwater. Environ. Sci. Technol. 40, 1524–1531.
U.S. EPA, 1992. Test Methods for Evaluating Solid Waste,Physical/Chemical Methods, SW-846, 3rd ed. U.S. GovernmentPriniting Office, Washington, DC, Method 1311.
Vinodhini, V., Das, N., 2009. Biowaste materials as sorbents toremove chromium (VI) from aqueous environment—acomparative study. J. Agric. Biol. Sci. 4 (6), 19–23.
Wang, X.S., Tang, Y.P., Tao, S.R., 2008. Removal of Cr (VI) fromaqueous solutions by the non-living biomass of alligatorweed: kinetics and equilibrium? Adsorption 14 (6), 823–830.
Weckhuysen, B.M., Wachs, I.E., Schoonheydt, R.A., 1996. Surfacechemistry and spectroscopy of chromium in inorganic oxides.Chem. Rev. 96 (8), 3327–3349.
Wu, F.C., Tseng, R.L., Juang, R.S., 2009. Initial behavior ofintraparticle diffusion model used in the description ofadsorption kinetics. Chem. Eng. J. 153, 1–8.
Xiong, W.H., Peng, J., 2008. Development and characterization offerrihydrite-modified diatomite as a phosphorus adsorbent.Water Res. 42 (19), 4869–4877.
Young, S., Zhang, J.P., Yang, G., Li, Z.H., 2006. Removal ofpollutants with activated carbon produced from K2CO3
activated of lignin from reed black liquors. Chem. Biochem.Eng. 20 (4), 429–435.
Zhang, N., Lin, L.S., Gang, D.C., 2008b. Adsorptive seleniteremoval from water using iron-coated GAC adsorbents. WaterRes. 42 (14), 3809–3816.





























