Embed Size (px)
Citation preview

of February 17, 2016.This information is current as
Gram-Negative PneumoniaActivation Impairs Host Defense against Ablation of Leptin Receptor-Mediated ERK
Aronoff and Marc Peters-GoldenSerezani, Edmund O'Brien, Jared Goldberg, David M. Peter Mancuso, Martin G. Myers, Jr, Deepti Goel, Carlos H.
http://www.jimmunol.org/content/189/2/867doi: 10.4049/jimmunol.12004652012;
2012; 189:867-875; Prepublished online 8 JuneJ Immunol
Referenceshttp://www.jimmunol.org/content/189/2/867.full#ref-list-1
, 22 of which you can access for free at: cites 44 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2012 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
at H
enry Ford Health System
/Sladen Library on February 17, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Ablation of Leptin Receptor-Mediated ERK ActivationImpairs Host Defense against Gram-Negative Pneumonia
Peter Mancuso,*,† Martin G. Myers, Jr.,‡,x,{ Deepti Goel,* Carlos H. Serezani,‖
Edmund O’Brien,*,# Jared Goldberg,* David M. Aronoff,†,{,** and Marc Peters-Golden†,{,††
The adipocyte-derived hormone leptin plays an important role in regulation of energy homeostasis and the innate immune response
against bacterial infections. Leptin’s actions are mediated by signaling events initiated by phosphorylation of tyrosine residues on
the long form of the leptin receptor. We recently reported that disruption of leptin receptor-mediated STAT3 activation augmented
host defense against pneumococcal pneumonia. In this report, we assessed leptin receptor-mediated ERK activation, a pathway
that was ablated in the l/l mouse through a mutation of the tyrosine 985 residue in the leptin receptor, to determine its role in host
defense against bacterial pneumonia in vivo and in alveolar macrophage (AM) antibacterial functions in vitro. l/l mice exhibited
increased mortality and impaired pulmonary bacterial clearance after intratracheal challenge with Klebsiella pneumoniae. The
synthesis of cysteinyl-leukotrienes was reduced and that of PGE2 enhanced in AMs in vitro and the lungs of l/l mice after infection
with K. pneumoniae in vivo. We also observed reduced phagocytosis and killing of K. pneumoniae in AMs from l/l mice that was
associated with reduced reactive oxygen intermediate production in vitro. cAMP, known to suppress phagocytosis, bactericidal
capacity, and reactive oxygen intermediate production, was also increased 2-fold in AMs from l/lmice. Pharmacologic blockade of
PGE2 synthesis reduced cAMP levels and overcame the defective phagocytosis and killing of bacteria in AMs from l/lmice in vitro.
These results demonstrate that leptin receptor-mediated ERK activation plays an essential role in host defense against bacterial
pneumonia and in leukocyte antibacterial effector functions. The Journal of Immunology, 2012, 189: 867–875.
Pneumonia is a common consequence of malnutrition,a leading threat to human health throughout the worldregardless of socioeconomic status (1). Rapid depletion of
energy storage in the form of adipose tissue often occurs duringperiods of famine in the developing world and in hospitalizedpatients suffering from chronic and critical illness (2–6). Associ-ated with the decline in fat mass is a decrease in leptin, an adi-pokine produced by white adipose tissue and known to regulateenergy homeostasis. Under normal circumstances, leptin levels arecorrelated with adipose tissue mass (7). However, during acute
bacterial infections and after endotoxin administration in labora-tory animals, leptin levels increase disproportionately to fat mass(8–12). An important role for leptin in the regulation of immunefunction during periods of fasting, obesity, and in disease statesmediated by inflammation is emerging.We and others have observed that leptin plays a protective role
in the host response against infectious disease (13–18). Using mu-rine models of Klebsiella and pneumococcal pneumonia, we havefound that leptin deficiency induced by genetic means or byfasting compromised pulmonary bacterial clearance and survival.This defect in pulmonary host defense was associated with abro-gated alveolar macrophage (AM) and polymorphonuclear neu-trophil (PMN) phagocytosis and killing of bacteria in vitro (12–14,19). The mechanisms underlying defective leukocyte effectorfunction in cells from leptin-deficient mice were associated witha reduction in leukotriene (LT) synthesis in AMs, reduced com-plement receptor (CR3) expression, and decreased H2O2 synthesisin PMNs (12, 14, 19). Other studies have revealed that the pro-duction of cytokines IL-6, MIP-2, and MCP-1 in leptin-deficientor leptin receptor-deficient mice was lower than that observed forwild-type animals (13, 15). The intracellular signaling eventsdownstream of the leptin receptor (LepR) that regulate leukocyteeffector functions, in the context of bacterial pneumonia, have notbeen determined.LepR signaling is mediated by the long isoform of the leptin
receptor (LepRb) via the JAK–STAT and MAPK signaling path-ways. Upon binding to its ligand (Fig. 1), the LepRb activates theconstitutively associated JAK2 tyrosine kinase to induce tyrosinephosphorylation-dependent signaling via several divergent path-ways. JAK2 mediates phosphorylation of Tyr1138, which binds andmediates the phosphorylation-dependent activation of the latenttranscription factor, STAT3. After nuclear translocation, STAT3activates transcription of SOCS-3, a protein that inhibits JAK2and STAT3 signaling during prolonged stimulation of the LepRb(20). LepRb-mediated phosphorylation of Tyr1077 activates STAT5
*Department of Environmental Health Sciences, School of Public Health, Universityof Michigan, Ann Arbor, MI 48109; †Graduate Program in Immunology, Universityof Michigan, Ann Arbor, MI 48109; ‡Division of Molecular Endocrinology andDiabetes, University of Michigan School of Medicine, Ann Arbor, MI 48109; xDe-partment of Integrative Physiology, School of Medicine, University of Michigan, AnnArbor, MI 48109; {Department of Internal Medicine, University of Michigan, AnnArbor, MI 48109; ‖Department of Microbiology and Immunology, Indiana UniversitySchool of Medicine, Indianapolis, IN 46202; #Toxicology Program, School of PublicHealth, University of Michigan, Ann Arbor, MI 48109; **Division of InfectiousDiseases, University of Michigan, Ann Arbor, MI 48109; and ††Division of Pulmo-nary and Critical Care Medicine, University of Michigan, Ann Arbor, MI 48109
Received for publication February 6, 2012. Accepted for publication May 8, 2012.
This work was supported by grants from the National Institutes of Health (HL077417to P.M., HL058897 to M.P.-G., DK56731 to M.G.M., T32ES007062 supporting E.O.,and HL10377701 to C.H.S.) and a grant from the Flight Attendants Medical ResearchInstitute (CIA-103071 to P.M.).
Address correspondence and reprint requests to Dr. Peter Mancuso, Department ofEnvironmental Health Sciences, School of Public Health, University of Michigan,6627 SPH1A, Ann Arbor, MI 48109. E-mail address: [email protected]
Abbreviations used in this article: AM, alveolar macrophage; BAL, bronchoalveolarlavage; COX-1, cyclooxygenase-1; COX-2, cyclooxygenase-2; cysLT, cysteinyl-leu-kotriene; GRB2, growth factor binding 2; LepR, leptin receptor; LepRb, long isoformof the leptin receptor; 5-LO, 5-lipoxygenase; LT, leukotriene; LTB4, leukotriene B4;MOI, multiplicity of infection; mPGEs-1, microsomal prostaglandin E synthase-1;Pen/Strep, penicillin–streptomycin; PMN, polymorphonuclear neutrophil; RFU, rel-ative fluorescence unit; ROI, reactive oxygen intermediate; SHP-2, SH2-containingtyrosine phosphatase; WT, wild-type.
Copyright� 2012 by The American Association of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1200465
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

signaling (21). Finally, phosphorylation of Tyr985 recruits bindingpartners SH2-containing tyrosine phosphatase (SHP-2) and growthfactor binding 2 (GRB2), which activate ERK1/2 (22). The gen-eration of the l/l mouse was first described by Bjornholm et al.(23), who reported that these animals lack the ability to activatethe ERK1/2 pathway via the LepRb due to a substitution mutationat Tyr985 with L985. Although a recent report by Guo et al. (17)demonstrated that l/l mice exhibit greater susceptibility to entericEntamoeba histolytica infection, the mechanism responsible forthis defect is unknown, and bacterial infections have not beenstudied in these mice.The role of LepRb-mediated (LepRb→) signaling events in the
innate immune response against bacterial infections is complex anddifficult to study in vivo. Not all LepR mutations result in impairedimmunity. For example, we recently reported that disruption ofLepR-mediated STAT3 signaling improved AM phagocytosis andkilling of bacteria in vitro and host defense against pneumococcalpneumonia in s/s mice in vivo (18). In the current report, weassessed the contribution of intracellular signals initiated by theLepRb→Tyr985 by comparing the responses of wild-type (WT) andl/lmice in a murine model of bacterial pneumonia. We demonstratefor the first time, to our knowledge, that l/l mice exhibit increasedsusceptibility to Gram-negative pneumonia and that this pathwayplays an essential role in the innate immune response against bac-terial pneumonia.
Materials and MethodsAnimals
Heterozygous C57BL/6 (back-crossed for eight generations or greater)LeprTm2mgmj/+ mice were intercrossed in the University of Michigan Unitfor Laboratory Animal Medicine (Ann Arbor, MI) to generate age- andgender-matched male and female (LeprTm2mgmj/Tm2mgmj) l/l and WT (+/+)littermates 8–14 wk of age (23). Animals were genotyped by TaqMan SNPallelic discrimination assays and were treated according to NationalInstitutes of Health guidelines for the use of experimental animals with theapproval of the University of Michigan Committee for the Use and Care ofAnimals.
Cell isolation and culture
Resident AMs were recovered from mice by bronchoalveolar lavage (BAL)as previously described, resuspended in RPMI 1640 (Life Technologies,Invitrogen, Carlsbad, CA) to a concentration of 23 106 cells per milliliter,and allowed to adhere to tissue-culture plates for 1 h (37˚C, 5% CO2) (24).After replacing the media with RPMI 1640 containing 10% FBS (LifeTechnologies, Invitrogen) and 1% penicillin–streptomycin (Pen/Strep)(Invitrogen), the cells were cultured overnight. PMNs were obtained frommice by peritoneal lavage 5 h after an i.p. injection of a 1% glycogensolution in PBS as previously described (25).
Immunoblot analysis
AMs obtained fromWTand l/lmicewere plated at 43 106 cells per well andcultured overnight in RPMI 1640 containing FBS and Pen/Strep. On thefollowing day, the cells were prepared for lysis or cultured with media aloneor leptin (50 ng/ml) for 5, 15, or 30min for the assessment of ERK activation.The macrophages were then washed with HBSS and scraped with ice-coldlysis buffer (RIPA buffer; Sigma), and cells were disrupted with sonication(10 bursts at 20% duty/cycle). Twenty micrograms of protein, as determinedby a modified Coomassie blue binding assay (Pierce Chemical, Rockford,IL), was separated by SDS-PAGE under reducing conditions and transferredto nitrocellulose membranes. Membranes were probed with the rabbitpolyclonal Abs against phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204)(1:2000), p44/42MAPK (ERK1/2) (1:2000), 5-lipoxygenase (5-LO) (1:250),GAPDH (1:1000) (Cell Signaling Technology, Danvers, MA), cyclooxy-genase-1 (COX-1) (Bio-Rad, Hercules, CA) (1:200) and cyclooxygenase-2(COX-2) (1:200), or microsomal PGE synthase-1 (mPGEs-1) (1:200)(Cayman Chemical, Ann Arbor, MI). Primary Abs were detected using al-kaline phosphatase-conjugated goat anti-rabbit secondary Ab (titer 1:5000)and visualized with the ECF detection system (Amersham PharmaciaBiotech, Piscataway, NJ). The densities of the luminescent bands werequantitated in appropriately exposed nitrocellulose by using Image Reader
(Fuji Film). The density value of the p-ERK1/2, 5-LO, COX-1, COX-2, andmPGEs-1 blots were divided by the density value of the ERK1/2 (p-ERK1/2)or GAPDH (5-LO, COX-1, COX-2, and mPGEs-1) blots, respectively, tonormalize the relative band densities.
Klebsiella pneumoniae preparation and inoculation
K. pneumoniae strain 43816, serotype 2, was obtained from the AmericanType Culture Collection (Manassas, VA), and aliquots were grown intryptic soy broth (Difco, Detroit, MI) for 18 h at 37˚C. The concentrationof bacteria in culture was determined spectrophotometrically (A600). K.pneumoniae was then pelleted by centrifugation (13,000 rpm for 3 min)two times, resuspended in PBS, and serially diluted in PBS to obtain theappropriate concentration. Mice were anesthetized with ketamine andxylazine as previously described (12). A midline incision was made toexpose the trachea, a 30-ml inoculum containing 5 3 103 CFU K. pneu-moniae was administered via the trachea using a 26-gauge needle, and thewound was closed using surgical glue (Nexaband, Phoenix, AZ) (12).
Determination of survival and lung and spleen K. pneumoniaeCFUs
After intratracheal inoculation with K. pneumoniae, mice were evaluatedfor survival daily for 7 d. Four and twenty-four hours after K. pneumoniaechallenge, mice were euthanized by CO2 asphyxiation, and lungs andspleen were harvested for CFU determinations as previously described(26). Briefly, lungs and spleen were homogenized in 0.5 ml of sterile sa-line, serially diluted, and plated on soy-based blood agar plates (Difco).After 18 h at room temperature, CFUs were enumerated.
Blood and lung leukocyte differential and total cell count
Four and twenty-four hours postinfection, lung leukocytes were obtainedfrom mice by BAL after CO2 asphyxiation, and differential counts wereperformed on cells after staining with a modified Wright–Giemsa stain(American Scientific Products, McGraw Park, IL). Blood was collected bycardiac puncture for peripheral WBC counts using a Hemavet cell analyzer(Drew Scientific) operated by the University of Michigan Unit for Labo-ratory Animal Medicine Animal Diagnostic Laboratory.
Determination of cytokines, cysteinyl-leukotrienes, leukotrieneB4, PGE2, and leptin
In a separate group of mice, lungs obtained from euthanized mice 4 and 24 hpostinfection were homogenized, and cytokine [CXCL2 (MIP-2), CCL2(MCP-1), IL-6, IL-10, IL-12, and TNF-a] (R&D Duoset, R&D Systems,Minneapolis, MN) and cysteinyl-leukotrienes (cysLTs), leukotriene B4
(LTB4), and PGE2 (Cayman Chemical, Ann Arbor, MI) levels were de-termined using commercially available EIA kits according to the manu-facturer’s instructions. Blood leptin levels were determined using an EIAkit from R&D Systems according to the manufacturer’s instructions.
Fluorometric assay of AM phagocytosis
AM phagocytosis of K. pneumoniae was assessed using a previouslypublished protocol for determining the ingestion of fluorescent, FITC-labeled Streptococcus pneumoniae (24). Briefly, AMs obtained by BALwere adhered and seeded in replicates of eight to 384-well tissue cultureplates with opaque sides and optically clear bottoms (Costar, Corning LifeSciences, Lowell, MA) and cultured overnight with RPMI 1640 with 1%Pen/Strep and 10% FCS (Invitrogen). On the following day, FITC-labeledK. pneumoniae were opsonized with 3% immune serum as previouslydescribed (27). AMs pretreated with RPMI 1640 media alone, with in-domethacin (10 mM) (Cayman Chemical) for 30 min, or with cysLTs (100nM) and LTB4 (1 nM) (alone or together) for 15 min were incubated withopsonized FITC–K. pneumoniae using a multiplicity of infection (MOI) of150:1 for 60 min to allow phagocytosis to occur. Trypan blue (250 mg/ml;Molecular Probes) was added for 1 min to quench the fluorescence ofextracellular bacteria, and fluorescence was determined using a Spec-tramax Gemini EM fluorometer with 485-nm excitation/535-nm emission(Molecular Devices, Sunnyvale, CA). The phagocytic index was calculatedas previously described in relative fluorescence units (RFUs) (18, 24).Three separate experiments were conducted with eight replicate wells forevery experimental condition, and the RFUs were normalized to the con-trol condition (untreated AMs from WT animals) in each experiment.
Bactericidal assays
The survival of internalized K. pneumoniae within the AM was quantifiedusing a tetrazolium dye reduction assay, as described previously (27).
868 LEPTIN RECEPTOR SIGNALING MUTATION IMPAIRS HOST DEFENSE
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

Briefly, 2 3 105/ml AMs, prepared as described previously, were adheredin quintuplicate in 96-well, half-area, tissue culture dishes (Corning,Lowell, MA). After overnight culture, K. pneumoniae were opsonized with3% anti-K. pneumoniae rat specific immune serum, as previously described(28). Cells were then treated with either cell culture media alone or in-domethacin (10 mM) for 30 min and were infected with an 0.1-ml sus-pension of opsonized K. pneumoniae (1 3 107 CFU/ml; MOI, 50:1) for 30min to allow phagocytosis to occur. The AMs were then washed threetimes with PBS to remove extracellular bacteria and incubated for anadditional 60 min to permit intracellular killing. The remainder of theassay was completed, as described elsewhere (27). On the basis of thisassay, it has been determined that the intensity of the absorbance at 595 nmis directly proportional to the number of intracellular bacteria associatedwith the macrophages. Results were expressed as percentage of survival ofingested bacteria, where the survival of ingested bacteria = 100% 3 A595
control plate/A595 experimental plate.
Reactive oxygen intermediate production
AMs were adhered to 384-well plates at a concentration of 1.253 105 cells/well and cultured overnight in RPMI 1640 containing 10% FCS andantibiotics. On the next day, the medium was replaced with PBS containing10 mM H2DCF, and the cells were cultured for 1 h. The medium was thenreplaced with warmed HBSS, and the cells were stimulated with heat-killed K. pneumoniae opsonized with 3% specific immune serum usingan MOI of 50:1. Reactive oxygen intermediate (ROI) production wasassessed every 30 min for 2 h by measuring fluorescence using a Spec-tramax Gemini XS fluorometer (Molecular Devices) with excitation/emission setting at 493/522 nm.
Assessment of NO production
AMs were adhered to 96-well plates at a concentration of 23 105 cells/welland cultured with DMEM supplemented with 1% sodium pyruvate (Invi-trogen) containing 10% FCS and penicillin–streptomycin with or without10 ng/ml LPS from E. coli (Sigma-Aldrich) and 10 ng/ml IFN-g (R&DSystems) for 24 h. NO production was determined by measuring stablenitrite (NO2
–) concentrations using a modified Griess reaction with acommercially available assay kit according to the manufacturer’s instruc-tions (Cayman Chemical).
Measurement of intracellular cAMP by AMs in vitro
AMswere cultured overnight in 96-well plates in RPMI 1640with 10% FCSand 1% penicillin–streptomycin at concentrations of 23 106 cells/well. Onthe following day, the cell culture media was replaced with warm RPMI1640, and AMs were incubated for 30 min in the presence or absence ofindomethacin (10 mM) with the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (EMD Biosciences) for 30 min prior to stimulation withheat-killed K. pneumoniae opsonized with 3% rat specific immune serumusing an MOI of 50:1. After 1 h, culture supernatants were aspirated andthe cells were lysed by incubation for 20 min with 0.1 M HCl (22˚C) forcAMP experiments. The cells were then disrupted using a cell scraper, andintracellular cAMP levels were determined by ELISA kit according to themanufacturer (Cayman Chemicals).
Statistical analyses
Where appropriate, mean values were compared using a paired Student ttest, a one-way or a two-way ANOVA followed by the Bonferroni cor-rection. Survival was evaluated for differences using a log-rank test. Dif-ferences were considered significant if p # 0.05, and the actual p valuesare mentioned in the results section. All experiments were performed on atleast three separate occasions unless otherwise specified. Data are pre-sented as mean values 6 SEM unless otherwise noted.
ResultsSubstitution of LepRb Tyr985 with L985 in l/l mice abrogatesLepRb-mediated ERK1/2 activation
To confirm that l/l mice lack the ability to signal via LepRb Tyr985,we assessed ERK1/2 activation using immunoblot analysis of p-ERK1/2 in AMs obtained from WT and l/l mice cultured withleptin. As shown in Fig. 1C, levels of total ERK1/2 were the samefor both groups of mice. However, when AMs from WT mice werecultured with exogenous leptin for 30 min, we observed an in-crease in p-ERK1/2 as determined by a 50% increase in p-ERK1/2(p = 0.0002). We conducted time-course experiments for ERK
activation (p-ERK) (i.e., 5, 15, and 30 min after stimulation withleptin), and only the blots from cells stimulated for 30 min areshown as this represents the peak of this response. In contrast, wedid not observe any increases in p-ERK1/2 levels in AMs from l/lmice after leptin treatment for 30 min or at any other time point(p = 0.12). Other signaling events initiated by this mutant receptorsuch as LepRb→STAT3 or STAT5 are normal as previously re-ported (21, 23). In addition, hypothalamic p-ERK activation wasnot observed in a previous report using l/l mice treated with muchhigher doses of leptin (5 mg/g of body weight) (23). Blood leptinlevels were slightly lower (p = 0.04) in the l/l mice (2.9 6 0.5 ng/ml) compared with that of WT animals (4.5 6 0.9 ng/ml) aspreviously reported (23). These data indicate that leptin inducesphosphorylation of ERK1/2 via the LepRb Tyr985 and that thispathway is abrogated in AMs from l/l mice.
l/l mice exhibit greater mortality and reduced pulmonarybacterial clearance after K. pneumoniae challenge
We have previously demonstrated that ob/ob mice, which lackfunctional leptin, or mice rendered leptin deficient by fasting aremore susceptible to both Gram-negative and Gram-positive pneu-monia (12, 13). To determine if intracellular signals arising from theLepRb Tyr985 play a role in pulmonary host defense against Gram-negative pneumonia, we compared the responses ofWTand l/lmiceafter an intratracheal challenge with K. pneumoniae. As shown inFig. 2A, l/l mice exhibited substantially lower survival (27%)compared with that of WT (65%, p = 0.04) after K. pneumoniaechallenge 7 d postinfection. Because the differences in survival mayindicate impaired pulmonary host defense in l/l mice, we assessedthe bacterial burdens in the lungs and spleen of mice 4 and 24 hpostinfection. We chose these time points because we observed thatthe first death recorded for an l/l mouse occurred 48 h after K.pneumoniae challenge. As shown in Fig. 2B, bacterial burdens were∼1-log fold greater after 4 h (p = 0.02) and 4-log fold higher at 24 h(p = 0.0001) in l/l compared with WT animals. We did not find anybacterial CFUs in spleens harvested from any of these animals 4 h(p = 0.38) and 24 h (p = 0.73) postinfection. These results indicatea defective pulmonary innate immune response against pulmonarybacterial infection in l/l mice.
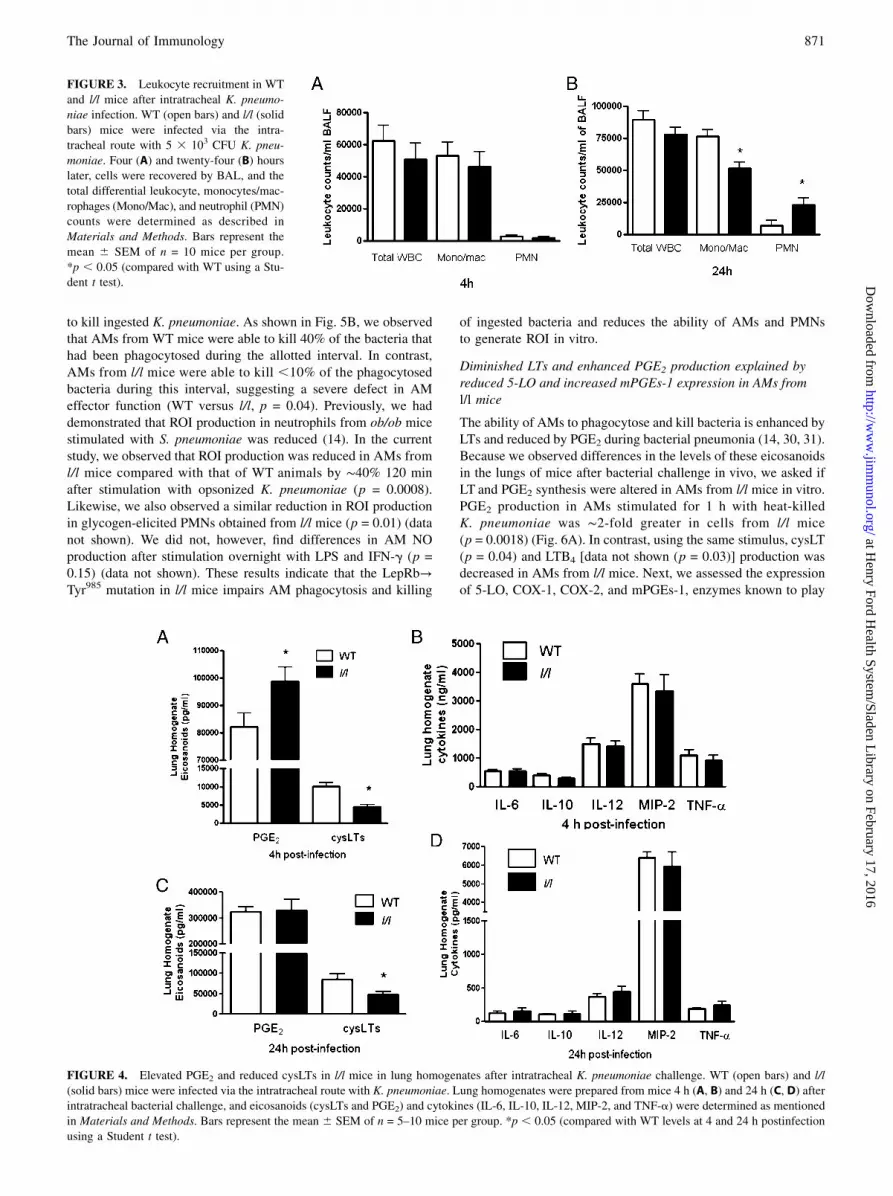
Modest differences between WT and l/l mice in leukocyterecruitment after K. pneumoniae challenge
The recruitment of leukocytes to the lungs is essential for effectivehost defense during bacterial pneumonia. We have previouslyobserved increased PMN recruitment to the lungs of leptin-deficient (ob/ob) mice and attenuated leukocyte recruitment inmice rendered leptin deficient by fasting in response to pneumo-coccal pneumonia (12, 13). To determine if the ablation of LepRbTyr985 signaling alters leukocyte recruitment to the lung duringbacterial pneumonia, we recovered leukocytes by lavage in mice 4and 24 h postinfection as these were the time points when weobserved differences in pulmonary bacterial burdens. As shown inFig. 3, there were no differences (p = 0.29) between WT and l/lmice in total or differential leukocyte counts in BAL fluid 4 h afterK. pneumoniae challenge. However, we did find lower BAL fluidmonocyte/macrophage (p = 0.04) and higher neutrophil (p = 0.04)counts in l/l mice at this time point. We also observed higher totalperipheral blood leukocyte counts (l/l 6.7 3 106 WBC/ml versusWT 4.1 3 106 WBC/ml) (p = 0.04) and elevated PMN counts (l/l3.8 3 106 versus WT 1.9 3 106) (p = 0.04) in the l/l mice 24 hpostinfection. These results indicate that differences in PMN re-cruitment, known to play a critical role in host defense againstGram-negative pneumonia (29), do not explain the limitations inKlebsiella clearance from the lungs of l/l mice.
The Journal of Immunology 869
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

Pulmonary cytokine and eicosanoid levels after K. pneumoniaechallenge
To determine whether the impairment in pulmonary bacterialclearance was due to differences in proinflammatory mediators
produced in the lung during infection, we assessed lung homog-
enate cysLTs, PGE2, and cytokine levels after bacterial challenge.
As shown in Fig. 4A, there were no differences in lung homog-
enate cytokines known to play an important role in antibacterial
host defense (IL-6, TNF-a, IL-10, IL-12, MIP-2, TNF-a) 4 h
postinfection. However, we did find major differences in lipid
mediators. Cyclooxygenase-derived PGE2 was elevated (p = 0.02),
whereas the 5-LO–derived cysLTs were reduced (p = 0.01) in l/l
mice at this time point. Reduced cysLT levels persisted 24 h
postinfection in the lungs of l/l mice (p = 0.02) (Fig. 4C), whereas
no differences in PGE2 or cytokines were observed (p = 0.93)(Fig. 4D).
Impaired phagocytosis, killing, and ROI production in AMsfrom l/l mice
The observed 1-log fold elevation of bacterial CFUs in l/l mice 4 hpostinfection suggests a defect in the pulmonary innate immuneresponse and implicates the resident AM, which plays a criticalrole in the early stages of K. pneumoniae clearance. To test thispossibility, we compared the ability of cells from WT and l/l miceto phagocytose and kill opsonized K. pneumoniae in vitro. Asshown in Fig. 5A, phagocytosis of K. pneumoniae opsonized withimmune serum was 25% less in AMs from l/l mice than thatobserved in cells from WT animals (p = 0.03). Next, we asked ifthere were differences in the ability of AMs from WT and l/l mice
FIGURE 1. LepRb signaling events.
(A) In WT mice, leptin binding activates
the LepRb-associated JAK2, a tyrosine
kinase that mediates tyrosine phosphor-
ylation of LepRb to promote several in-
tracellular signaling events: 1) LepRb
Tyr985 recruits SHP-2 and GRB2 to pro-
mote the activation of ERK1/2. ERK1/2
phosphorylates downstream targets and
induces the transcription of genes. 2)
Phosphorylated Tyr1107 binds and me-
diates the phosphorylation-dependent
activation of STAT5, and 3) Tyr1138 re-
cruits STAT3, which promotes the tran-
scription of many different genes. (B) In
l/l mice, substitution of LepRb Tyr985
with LepRb L985 prevents leptin-medi-
ated ERK1/2 activation in AMs from l/l
mice. (C) AMs obtained from WT and
l/l mice were cultured with media alone
or with exogenous leptin (50 ng/ml) for
30 min and evaluated for p-ERK1/2 and
total ERK1/2 by immunoblot analysis.
Protein levels were determined by den-
sitometric analysis of immunoblots from
five separate experiments. *p , 0.001
(versus untreated WT using the Student
t test).
FIGURE 2. Reduced survival and impaired pulmonary bacterial clearance in l/l mice after an intratracheal challenge with K. pneumoniae. (A) WT and l/l
mice were challenged with 53 103 CFU K. pneumoniae via the intratracheal route and monitored for survival for 7 d. Survival curves represent n = 11 to 15
mice per group from 3 separate experiments. (B) In another group of animals, WT (open bars) and l/l (solid bars) mice were euthanized 4 and 24 h
postinfection, lungs were harvested, and bacterial burdens in tissue homogenates were determined by counting CFUs as described in Materials and
Methods. Bars represent the mean 6 SEM of n = 10 mice per group from three separate experiments. p = 0.04 (compared with WT using a log-rank test),
*p , 0.05 (compared with WT levels at 4 and 24 h postinfection using a Student t test).
870 LEPTIN RECEPTOR SIGNALING MUTATION IMPAIRS HOST DEFENSE
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
to kill ingested K. pneumoniae. As shown in Fig. 5B, we observedthat AMs from WT mice were able to kill 40% of the bacteria thathad been phagocytosed during the allotted interval. In contrast,AMs from l/l mice were able to kill ,10% of the phagocytosedbacteria during this interval, suggesting a severe defect in AMeffector function (WT versus l/l, p = 0.04). Previously, we haddemonstrated that ROI production in neutrophils from ob/ob micestimulated with S. pneumoniae was reduced (14). In the currentstudy, we observed that ROI production was reduced in AMs froml/l mice compared with that of WT animals by ∼40% 120 minafter stimulation with opsonized K. pneumoniae (p = 0.0008).Likewise, we also observed a similar reduction in ROI productionin glycogen-elicited PMNs obtained from l/l mice (p = 0.01) (datanot shown). We did not, however, find differences in AM NOproduction after stimulation overnight with LPS and IFN-g (p =0.15) (data not shown). These results indicate that the LepRb→Tyr985 mutation in l/l mice impairs AM phagocytosis and killing
of ingested bacteria and reduces the ability of AMs and PMNsto generate ROI in vitro.
Diminished LTs and enhanced PGE2 production explained byreduced 5-LO and increased mPGEs-1 expression in AMs froml/l mice
The ability of AMs to phagocytose and kill bacteria is enhanced byLTs and reduced by PGE2 during bacterial pneumonia (14, 30, 31).Because we observed differences in the levels of these eicosanoidsin the lungs of mice after bacterial challenge in vivo, we asked ifLT and PGE2 synthesis were altered in AMs from l/l mice in vitro.PGE2 production in AMs stimulated for 1 h with heat-killedK. pneumoniae was ∼2-fold greater in cells from l/l mice(p = 0.0018) (Fig. 6A). In contrast, using the same stimulus, cysLT(p = 0.04) and LTB4 [data not shown (p = 0.03)] production wasdecreased in AMs from l/l mice. Next, we assessed the expressionof 5-LO, COX-1, COX-2, and mPGEs-1, enzymes known to play
FIGURE 3. Leukocyte recruitment in WT
and l/l mice after intratracheal K. pneumo-
niae infection. WT (open bars) and l/l (solid
bars) mice were infected via the intra-
tracheal route with 5 3 103 CFU K. pneu-
moniae. Four (A) and twenty-four (B) hours
later, cells were recovered by BAL, and the
total differential leukocyte, monocytes/mac-
rophages (Mono/Mac), and neutrophil (PMN)
counts were determined as described in
Materials and Methods. Bars represent the
mean 6 SEM of n = 10 mice per group.
*p , 0.05 (compared with WT using a Stu-
dent t test).
FIGURE 4. Elevated PGE2 and reduced cysLTs in l/l mice in lung homogenates after intratracheal K. pneumoniae challenge. WT (open bars) and l/l
(solid bars) mice were infected via the intratracheal route with K. pneumoniae. Lung homogenates were prepared from mice 4 h (A, B) and 24 h (C, D) after
intratracheal bacterial challenge, and eicosanoids (cysLTs and PGE2) and cytokines (IL-6, IL-10, IL-12, MIP-2, and TNF-a) were determined as mentioned
in Materials and Methods. Bars represent the mean 6 SEM of n = 5–10 mice per group. *p , 0.05 (compared with WT levels at 4 and 24 h postinfection
using a Student t test).
The Journal of Immunology 871
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

essential roles in LT and PGE2 synthesis in AMs, respectively. Incomparison with cells from WT animals, 5-LO protein expressionin AMs from l/l mice was reduced by ∼40% (p = 0.008). Althoughthere were no differences in COX-1 (p = 0.86) or COX-2(p = 0.90) (data not shown) expression, we did find a 90% in-crease in the levels of mPGEs-1 in AMs from l/l mice (p = 0.04).These changes in enzyme expression therefore explain the reducedLT and enhanced PGE2 production in l/l mice.
Elevated PGE2 production mediates enhanced cAMP levels inAMs from l/l mice
An important mechanism by which LTs and PGE2 differentiallyregulate AM phagocytosis and killing of K. pneumoniae in vitrois via decreases and increases, respectively, in the levels of thesecond messenger cAMP, which is known to inhibit phagocytosisand bacterial killing (30, 31). First, we confirmed that AMs from l/lmice produced more PGE2 than cells from WT mice (p = 0.001)after stimulation with heat-killed K. pneumoniae and this response
could be blocked with indomethacin (Fig. 6C). Next, we assessedintracellular cAMP levels and observed that AMs from l/l micestimulated with K. pneumoniae for 1 h produced twice as muchcAMP compared with AMs from WT animals (p = 0.001). Theincrease in intracellular cAMP was completely blocked when cellsfrom l/l mice were pretreated with indomethacin (Fig. 6D). AMsfrom l/l mice pretreated with LTB4 did not significantly affectcAMP production after stimulation with K. pneumoniae (p = 0.37)(data not shown). These results suggest that the increased pro-duction of PGE2 mediates the increased cAMP levels in AMs froml/l mice.
Indomethacin restores phagocytosis and bacterial killing inAMs from l/l mice
We next assessed the ability of addition or blockade of lipidmediators to restore AM effector functions in vitro. As shown inFig. 7A and 7B, we again observed deficient bacterial phagocy-tosis (p = 0.002) and killing (p = 0.002) in AMs from l/l mice.
FIGURE 5. Impaired phagocytosis, bacterial kill-
ing, and ROI production in AMs from l/l mice. AMs
obtained from WT (open bars) and l/l (solid bars)
mice were assessed for their ability to phagocytose
(A) and kill ingested K. pneumoniae opsonized with
immune serum (B) according to the procedures de-
scribed in Materials and Methods. Bars represent the
mean 6 SEM of at least three separate experiments.
*p , 0.05 (compared with WT using a Student t test).
(C) AMs from WT (open circles) and l/l (closed cir-
cles) mice were cultured with H2DCF for 1 h and
then stimulated with heat-killed K. pneumoniae
opsonized with immune serum using an MOI of 100:1
to assess ROI production. ROI production was as-
sessed using a fluorometer and expressed as RFUs.
The data represent the mean of three experiments
completed in quadruplicate for each time point using
a two-way ANOVA. *p , 0.05, ***p , 0.001
(compared with WT).
FIGURE 6. Eicosanoid synthesis, eicosanoid bio-
synthetic enzyme expression, and cAMP levels in AMs
fromWT (open bars) and l/l (solid bars) mice. (A) AMs
from WT (open bars) and l/l (solid bars) mice were
stimulated with heat-killed K. pneumoniae opsonized
with specific immune serum for 1 h, and cysLTs and
PGE2 were determined by ELISA as described in
Materials and Methods. (B) Immunoblots for 5-LO,
COX-1, mPGEs-1, and GAPDH (protein loading con-
trol) in AM lysates from a single experiment that is
representative of a total of three independent experi-
ments. Bars represent the mean arbitrary densitometric
units 6 SEM of immunoblots from three separate
experiments. AMs cultured with and without indo-
methacin (10 mM) for 30 min were stimulated with or
without heat-killed K. pneumoniae opsonized with
immune serum for 1 h, and the cell culture media was
assayed for PGE2 (C) and AM lysates were assayed for
cAMP as described in Materials and Methods (D).
*p , 0.05 (versus WT using the Student t test). Indo,
Indomethacin; K.p., heat-killed K. pneumoniae.
872 LEPTIN RECEPTOR SIGNALING MUTATION IMPAIRS HOST DEFENSE
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

Although we have previously reported that exogenous adminis-tration of LTB4 or cysLTs augments phagocytosis and killing inAMs from WT mice and rats (27, 32), these lipids failed to im-prove these endpoints in cells from l/l mice (data not shown).However, blocking of PGE2 production with indomethacin re-stored defective antimicrobial responses in AMs from l/l mice.These results suggest that the defects in pulmonary host defenseagainst K. pneumoniae in vivo were mainly due to the enhancedproduction of PGE2 in cells from l/l mice. They also imply that theLepR mutation in l/l mice impairs responsiveness to LTs.
DiscussionIn this study, we report the novel observation that l/l mice, whichlack LepRb→ERK1/2 activation via Tyr985, exhibit greater sus-ceptibility to Gram-negative pneumonia. The defect in pulmonaryhost defense in l/l mice was associated with impaired AMphagocytosis and killing of bacteria in vitro. In addition, we alsoobserved increased PGE2 and reduced LTs after bacterial chal-lenge in the lung in vivo and in AMs after culture with heat-killedbacteria in vitro. Phagocytosis and killing could be restored ifAMs from l/l mice were pretreated with the cyclooxygenase in-hibitor indomethacin, which normalized eicosanoid synthesis andintracellular cAMP levels. These results provide novel insightsinto the role of LepR-mediated signaling in the innate immuneresponse against bacterial infection.There are now a number of reports demonstrating that leptin or
LepR deficiency disables host defense against bacterial infections(12–15, 19, 33–35). Unlike other models of leptin or LepR defi-ciency, the l/l mouse is neither obese nor hyperglycemic and thusprovides an excellent model for assessing the importance of dis-tinct LepR-mediated signaling events in host defense againstbacterial infection in the absence of endocrine abnormalities. Werecently reported that LepRb→STAT3 activation is not essentialfor host defense, as ablating this pathway in s/s mice, whichpossess a mutant LepRb, were obese and resistant to pneumo-coccal pneumonia in vivo, and this was associated with improvedAMs antibacterial functions in vitro (18). Notably, these micewere protected by an enhanced ability to produce LTs, and thisimproved AM antibacterial functions. In the current study, weobserved that ablation of the LepRb→Tyr985 pathway in l/l mice,which were lean, resulted in defective host defense against K.pneumoniae in vivo and diminished AM effector functionsin vitro. These differences were also most likely due to alterationsin eicosanoid production.One novel observation in this report was the enhanced synthesis
of the immunosuppressive eicosanoid PGE2 in the lungs and inAMs of l/l mice postinfection.
This enhancement was the result of the increased expression ofmPGEs-1 as demonstrated by immunoblot analysis. The mecha-nism underlying this enhancement is unknown and beyond thescope of this report. PGE2 suppresses bacterial phagocytosis, ROIgeneration, and bacterial killing in AMs, and these effects aremediated through the E-prostanoid 2 receptor, a Gas protein-coupled receptor that activates adenylate cyclase and increasesintracellular cAMP levels (30, 31). We also demonstrated elevatedcAMP levels in AMs from l/l mice stimulated with bacteriain vitro, and this response could be blocked with the cyclo-oxygenase inhibitor indomethacin. This approach also normalizedimpaired phagocytosis and killing of K. pneumonia in AMs froml/l mice in vitro. Other reports have also demonstrated that ele-vated pulmonary PGE2 synthesis suppresses host defense againstbacterial pneumonia in vivo, and this defect can be rescued withindomethacin or through genetic ablation of the E-prostanoid 2receptor (36, 37).These results suggest that the defects in host defense in l/l mice
were largely due to the overproduction of PGE2 during bacterialpneumonia.Another unexpected and novel finding in this report was the
lower levels of LTs produced by l/l mice after pulmonary bacterialchallenge in vivo and in AMs in vitro. This result was likely dueto reduced 5-LO protein whose expression is regulated by tran-scription factors Sp1 and Egr1 (38). Leptin is known to enhancethe expression of both these transcription factors, and the lack ofLepRb→Tyr985 signaling may have reduced their expression in l/lmice (22, 39, 40). The LTs play a protective role in Klebsiellapneumonia, as 5-LO knockout mice exhibited increased lethalityand reduced pulmonary bacterial clearance (41). Furthermore, LTproduction was diminished in AMs from leptin-deficient (ob/ob)mice, and exogenous leptin restored LT synthesis and AMphagocytosis and killing of K. pneumoniae in vitro (12, 27, 32).However, the provision of exogenous LTs did not reduce cAMPlevels or restore antibacterial responses in AMs from l/l mice,suggesting a defect in LT receptor responsiveness or signaling.Further evaluation of this possibility is a focus of future investi-gation but beyond the scope of the current report.Our data implicate dysregulated eicosanoid generation in AMs in
the phenotype observed in l/l mice after K. pneumoniae challengein vivo and in vitro. On the basis of p-STAT3 staining as a surro-gate marker for the expression of the LepRb in the murine lunginflated with PBS containing leptin, we have shown that theLepRb is expressed primarily in AMs and to a much lesser extentin alveolar epithelial cells (18). Therefore, only those cells thatexpress high levels of this receptor are likely to be influenced bythe lack of LepRb→Tyr985 signaling. The primary sources of LTs
FIGURE 7. Inhibition of PGE2 synthesis with indomethacin restores bacterial phagocytosis and killing in AMs from l/l mice. AMs obtained from WT
and l/l mice were cultured with and without indomethacin (Indo) (10 mM) for 30 min prior to the addition of K. pneumoniae opsonized with immune serum.
Phagocytosis (A) and bacterial killing of K. pneumoniae ingested by AMs from WT and l/l mice (B) were assessed according to procedures mentioned in
Materials and Methods. Bars represent the mean 6 SEM of at least three separate experiments. *p , 0.01 (versus WT and l/l + Indo by ANOVA using the
Bonferroni test for mean separation).
The Journal of Immunology 873
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

in the lung during bacterial pneumonia are the resident AMs andPMNs known to express high levels of 5-LO. As a consequence,we observed reduced production of LTs at both time points afterK. pneumoniae challenge in vivo and in AMs vitro. In contrast, theexpression mPGEs-1 is not limited to AMs and would be presentin alveolar epithelial cells that do not express high levels of theLepRb (18, 42). Consistent with this, we observed increased PGE2
production in AMs stimulated in vitro and 4 h postinfection in thelungs of l/l mice in vivo. Under these circumstances, the AM is themajor source of PGE2. Twenty-four hours after Klebsiella chal-lenge, the alveolar epithelial cells are the major producers of PGE2
in vivo, and there were no differences in lung PGE2 levels betweenWT and l/l mice. The impairment in pulmonary bacterial clear-ance in l/l mice in vivo was therefore most likely due to the ele-vated levels of PGE2 produced by AMs. PGE2, by increasingintracellular cAMP, is known to impair AM phagocytosis andkilling of bacteria and to reduce ROI production, all of which arerequired for the elimination of K. pneumoniae (27, 30, 31).The reduced number of monocyte/macrophages recovered from
the lungs of l/l mice 24 h after Klebsiella challenge was not due toimpairments in either chemokine (MCP-1) production or periph-eral blood monocyte counts (data not shown), which did not differfrom those of WT mice. It is also unlikely that the reduction ofLTs in the lungs of l/l mice was responsible for lower monocyte/macrophage counts 24 h after K. pneumoniae challenge, as nodifferences in lung leukocytes counts were reported in 5-LOknockout mice after K. pneumoniae challenge (41). Althoughwe did not assess cell viability, we speculate that the reducedmonocyte/macrophage population in the lung of l/l mice 24 h afterK. pneumoniae challenge may reflect increased apoptosis of thesecells, as leptin is known to enhance the survival of humanmonocytes via an ERK1/2-dependent pathway (43). In support ofthis speculation, Guo et al. (17) reported increased cell death anddisruption of the intestinal epithelium in l/l mice after infectionwith E. histolytica. In contrast to monocytes/macrophages, theincreased numbers of PMNs in BAL fluid and the peripheral bloodof l/l mice 24 h after K. pneumoniae challenge were likely due tothe higher pulmonary bacterial burdens in these animals. On thebasis of this result, it appears that the observed defect in ROIgeneration in PMNs (data not shown), rather than recruitment,may have contributed to the impairment in pulmonary bacterialclearance in l/l mice at this later time point. Finally, it is ac-knowledged that other bactericidal mechanisms may be dysfunc-tional in leukocytes from the l/l animals.In summary, we report for the first time, to our knowledge, that
LepRb→Tyr985 intracellular signaling plays a critical role in thehost response against Gram-negative pneumonia in vivo and inleukocyte antibacterial functions in vitro. At present, defects inhuman LepRb→ERK activation have not been identified. How-ever, a leptin receptor mutation was associated with greater sus-ceptibility to intestinal parasitic infections in humans (44). Agreater understanding of the role of leptin receptor signaling inhost defense against infection will facilitate the development oftargeted therapeutic interventions for the prevention and treatmentof bacterial pneumonia.
AcknowledgmentsWe thank Justin Jones for assistance in genotyping the l/l mice for these
studies.
DisclosuresThe authors have no financial conflicts of interest.
References1. Mizgerd, J. P. 2006. Lung infection—a public health priority. PLoS Med. 3: e76.2. Palacio, A., M. Lopez, F. Perez-Bravo, F. Monkeberg, and L. Schlesinger. 2002.
Leptin levels are associated with immune response in malnourished infants. J.Clin. Endocrinol. Metab. 87: 3040–3046.
3. Rodrıguez, L., J. Graniel, and R. Ortiz. 2007. Effect of leptin on activation andcytokine synthesis in peripheral blood lymphocytes of malnourished infectedchildren. Clin. Exp. Immunol. 148: 478–485.
4. Ben-Ari, Z., Z. Schafer, J. Sulkes, V. Manhaim, R. Tur-Kaspa, and M. Fainaru.2002. Alterations in serum leptin in chronic liver disease. Dig. Dis. Sci. 47: 183–189.
5. Scholze, A., D. Rattensperger, W. Zidek, and M. Tepel. 2007. Low serum leptinpredicts mortality in patients with chronic kidney disease stage 5. Obesity (SilverSpring) 15: 1617–1622.
6. Langouche, L., S. Vander Perre, J. Frystyk, A. Flyvbjerg, T. K. Hansen, andG. Van den Berghe. 2009. Adiponectin, retinol-binding protein 4, and leptin inprotracted critical illness of pulmonary origin. Crit. Care 13: R112.
7. Maffei, M., J. Halaas, E. Ravussin, R. E. Pratley, G. H. Lee, Y. Zhang, H. Fei,S. Kim, R. Lallone, S. Ranganathan, et al. 1995. Leptin levels in human androdent: measurement of plasma leptin and ob RNA in obese and weight-reducedsubjects. Nat. Med. 1: 1155–1161.
8. Grunfeld, C., C. Zhao, J. Fuller, A. Pollack, A. Moser, J. Friedman, andK. R. Feingold. 1996. Endotoxin and cytokines induce expression of leptin, theob gene product, in hamsters. J. Clin. Invest. 97: 2152–2157.
9. Sarraf, P., R. C. Frederich, E. M. Turner, G. Ma, N. T. Jaskowiak, D. J. Rivet, III,J. S. Flier, B. B. Lowell, D. L. Fraker, and H. R. Alexander. 1997. Multiplecytokines and acute inflammation raise mouse leptin levels: potential role ininflammatory anorexia. J. Exp. Med. 185: 171–175.
10. Bornstein, S. R., J. Licinio, R. Tauchnitz, L. Engelmann, A. B. Negrao, P. Gold,and G. P. Chrousos. 1998. Plasma leptin levels are increased in survivors of acutesepsis: associated loss of diurnal rhythm, in cortisol and leptin secretion. J. Clin.Endocrinol. Metab. 83: 280–283.
11. Moshyedi, A. K., M. D. Josephs, E. K. Abdalla, S. L. D. Mackay, C. K. Edwards,III, E. M. Copeland, III, and L. L. Moldawer. 1998. Increased leptin expressionin mice with bacterial peritonitis is partially regulated by tumor necrosis factoralpha. Infect. Immun. 66: 1800–1802.
12. Mancuso, P., A. Gottschalk, S. M. Phare, M. Peters-Golden, N. W. Lukacs, andG. B. Huffnagle. 2002. Leptin-deficient mice exhibit impaired host defense inGram-negative pneumonia. J. Immunol. 168: 4018–4024.
13. Mancuso, P., G. B. Huffnagle, M. A. Olszewski, J. Phipps, and M. Peters-Golden. 2006. Leptin corrects host defense defects after acute starvation inmurine pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 173: 212–218.
14. Hsu, A., D. M. Aronoff, J. Phipps, D. Goel, and P. Mancuso. 2007. Leptinimproves pulmonary bacterial clearance and survival in ob/ob mice duringpneumococcal pneumonia. Clin. Exp. Immunol. 150: 332–339.
15. Ikejima, S., S. Sasaki, H. Sashinami, F. Mori, Y. Ogawa, T. Nakamura, Y. Abe,K. Wakabayashi, T. Suda, and A. Nakane. 2005. Impairment of host resistance toListeria monocytogenes infection in liver of db/db and ob/ob mice. Diabetes 54:182–189.
16. Wieland, C. W., S. Florquin, E. D. Chan, J. C. Leemans, S. Weijer, A. Verbon,G. Fantuzzi, and T. van der Poll. 2005. Pulmonary Mycobacterium tuberculosisinfection in leptin-deficient ob/ob mice. Int. Immunol. 17: 1399–1408.
17. Guo, X., M. R. Roberts, S. M. Becker, B. Podd, Y. Zhang, S. C. Chua, Jr.,M. G. Myers, Jr., P. Duggal, E. R. Houpt, and W. A. Petri, Jr. 2011. Leptinsignaling in intestinal epithelium mediates resistance to enteric infection byEntamoeba histolytica. Mucosal Immunol. 4: 294–303.
18. Mancuso, P., M. Peters-Golden, D. Goel, J. Goldberg, T. G. Brock,M. Greenwald-Yarnell, and M. G. J. Myers, Jr. 2011. Disruption of leptinreceptor-STAT3 signaling enhances leukotriene production and pulmonary hostdefense against pneumococcal pneumonia. J. Immunol. 186: 1081–1090.
19. Moore, S. I., G. B. Huffnagle, G. H. Chen, E. S. White, and P. Mancuso. 2003.Leptin modulates neutrophil phagocytosis of Klebsiella pneumoniae. Infect.Immun. 71: 4182–4185.
20. Bates, S. H., W. H. Stearns, T. A. Dundon, M. Schubert, A. W. Tso, Y. Wang,A. S. Banks, H. J. Lavery, A. K. Haq, E. Maratos-Flier, et al. 2003. STAT3signalling is required for leptin regulation of energy balance but not reproduc-tion. Nature 421: 856–859.
21. Gong, Y., R. Ishida-Takahashi, E. C. Villanueva, D. C. Fingar, H. Munzberg, andM. G. Myers, Jr. 2007. The long form of the leptin receptor regulates STAT5 andribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 282: 31019–31027.
22. Bjørbaek, C., R. M. Buchholz, S. M. Davis, S. H. Bates, D. D. Pierroz, H. Gu,B. G. Neel, M. G. Myers, Jr., and J. S. Flier. 2001. Divergent roles of SHP-2 inERK activation by leptin receptors. J. Biol. Chem. 276: 4747–4755.
23. Bjornholm, M., H. Munzberg, R. L. Leshan, E. C. Villanueva, S. H. Bates,G. W. Louis, J. C. Jones, R. Ishida-Takahashi, C. Bjørbaek, and M. G. Myers, Jr.2007. Mice lacking inhibitory leptin receptor signals are lean with normal en-docrine function. J. Clin. Invest. 117: 1354–1360.
24. Phipps, J. C., D. M. Aronoff, J. L. Curtis, D. Goel, E. O’Brien, and P. Mancuso.2010. Cigarette smoke exposure impairs pulmonary bacterial clearance and al-veolar macrophage complement-mediated phagocytosis of Streptococcus pneu-moniae. Infect. Immun. 78: 1214–1220.
25. Mancuso, P., P. Nana-Sinkam, and M. Peters-Golden. 2001. Leukotriene B4
augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect. Immun. 69:2011–2016.
874 LEPTIN RECEPTOR SIGNALING MUTATION IMPAIRS HOST DEFENSE
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

26. Greenberger, M. J., R. M. Strieter, S. L. Kunkel, J. M. Danforth, R. E. Goodman,and T. J. Standiford. 1995. Neutralization of IL-10 increases survival in a murinemodel of Klebsiella pneumonia. J. Immunol. 155: 722–729.
27. Serezani, C. H., D. M. Aronoff, S. Jancar, P. Mancuso, and M. Peters-Golden.2005. Leukotrienes enhance the bactericidal activity of alveolar macrophagesagainst Klebsiella pneumoniae through the activation of NADPH oxidase. Blood106: 1067–1075.
28. Mancuso, P., T. J. Standiford, T. Marshall, and M. Peters-Golden. 1998. 5-Lip-oxygenase reaction products modulate alveolar macrophage phagocytosis ofKlebsiella pneumoniae. Infect. Immun. 66: 5140–5146.
29. Greenberger, M. J., R. M. Strieter, S. L. Kunkel, J. M. Danforth, L. L. Laichalk,D. C. McGillicuddy, and T. J. Standiford. 1996. Neutralization of MIP-2attenuates neutrophil recruitment and bacterial clearance in murine Klebsiellapneumonia. J. Infect. Dis. 173: 159–165.
30. Aronoff, D. M., C. Canetti, and M. Peters-Golden. 2004. Prostaglandin E2
inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 173: 559–565.
31. Serezani, C. H., J. Chung, M. N. Ballinger, B. B. Moore, D. M. Aronoff, andM. Peters-Golden. 2007. Prostaglandin E2 suppresses bacterial killing in alve-olar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol.37: 562–570.
32. Mancuso, P., T. J. Standiford, T. Marshall, and M. Peters-Golden. 1998. 5-Lip-oxygenase reaction products modulate alveolar macrophage phagocytosis ofKlebsiella pneumoniae. Infect. Immun. 66: 5140–5146.
33. Conge, G. A., P. Gouache, Y. Joyeux, J. Goichot, and J. M. Fournier. 1988.[Influence of different types of experimental obesity on resistance of the mouseto infection by Salmonella typhimurium and Klebsiella pneumoniae]. Ann. Nutr.Metab. 32: 113–120.
34. Wehrens, A., T. Aebischer, T. F. Meyer, and A. K. Walduck. 2008. Leptin re-ceptor signaling is required for vaccine-induced protection against Helicobacterpylori. Helicobacter 13: 94–102.
35. Tschop, J., R. Nogueiras, S. Haas-Lockie, K. R. Kasten, T. R. Castaneda,N. Huber, K. Guanciale, D. Perez-Tilve, K. Habegger, N. Ottaway, et al. 2010.CNS leptin action modulates immune response and survival in sepsis. J. Neu-rosci. 30: 6036–6047.
36. Ballinger, M. N., D. M. Aronoff, T. R. McMillan, K. R. Cooke, K. Olkiewicz,G. B. Toews, M. Peters-Golden, and B. B. Moore. 2006. Critical role of pros-taglandin E2 overproduction in impaired pulmonary host response followingbone marrow transplantation. J. Immunol. 177: 5499–5508.
37. Medeiros, A. I., C. H. Serezani, S. P. Lee, and M. Peters-Golden. 2009. Effer-ocytosis impairs pulmonary macrophage and lung antibacterial function viaPGE2/EP2 signaling. J. Exp. Med. 206: 61–68.
38. Coffey, M. J., C. H. Serezani, S. M. Phare, N. Flamand, and M. Peters-Golden.2007. NADPH oxidase deficiency results in reduced alveolar macrophage 5-lipoxygenase expression and decreased leukotriene synthesis. J. Leukoc. Biol.82: 1585–1591.
39. de Lartigue, G., G. Lur, R. Dimaline, A. Varro, H. Raybould, and G. J. Dockray.2010. EGR1 Is a target for cooperative interactions between cholecystokinin andleptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology 151:3589–3599.
40. Mauvoisin, D., M. Prevost, S. Ducheix, M. P. Arnaud, and C. Mounier. 2010.Key role of the ERK1/2 MAPK pathway in the transcriptional regulation of theStearoyl-CoA Desaturase (SCD1) gene expression in response to leptin. Mol.Cell. Endocrinol. 319: 116–128.
41. Bailie, M. B., T. J. Standiford, L. L. Laichalk, M. J. Coffey, R. M. Strieter, andM. Peters-Golden. 1996. Leukotriene-deficient mice manifest enhanced le-thality from Klebsiella pneumonia in association with decreased alveolarmacrophage phagocytic and bactericidal activities. J. Immunol. 157: 5221–5224.
42. Lazarus, M., C. J. Munday, N. Eguchi, S. Matsumoto, G. J. Killian,B. K. Kubata, and Y. Urade. 2002. Immunohistochemical localization of mi-crosomal PGE synthase-1 and cyclooxygenases in male mouse reproductiveorgans. Endocrinology 143: 2410–2419.
43. Najib, S., and V. Sanchez-Margalet. 2002. Human leptin promotes survival ofhuman circulating blood monocytes prone to apoptosis by activation of p42/44MAPK pathway. Cell. Immunol. 220: 143–149.
44. Duggal, P., X. Guo, R. Haque, K. M. Peterson, S. Ricklefs, D. Mondal, F. Alam,Z. Noor, H. P. Verkerke, C. Marie, et al. 2011. A mutation in the leptin receptoris associated with Entamoeba histolytica infection in children. J. Clin. Invest.121: 1191–1198.
The Journal of Immunology 875
at Henry Ford H
ealth System/Sladen L
ibrary on February 17, 2016http://w
ww
.jimm
unol.org/D
ownloaded from











![[2013] 10 SCR 867 - Supreme Court of India](https://img.pdfslide.net/doc/110x75/6337a4f340a96001d40108c6/2013-10-scr-867-supreme-court-of-india.jpg)




