Embed Size (px)
Citation preview

Loss of PKC�/� impairs Th2 establishment and allergicairway inflammation in vivoJun-Qi Yanga,1, Michael Leitgesb,1, Angeles Durana, Maria T. Diaz-Mecoa, and Jorge Moscata,2
aDepartment of Cancer and Cell Biology, University of Cincinnati College of Medicine, Cincinnati, OH 45267; and bThe Biotechnology Centre of Oslo,University of Oslo, N-0317, Oslo, Norway
Edited by Michael Karin, University of California at San Diego School of Medicine, La Jolla, CA, and approved December 8, 2008 (received for reviewJune 18, 2008)
The differentiation of T cells along different lineages is central to
the control of immunity. Here we have used a conditional gene
knockout system to delete PKC�/� selectively in activated T cells.
With this system we have demonstrated that PKC�/� is necessary
for T-helper cell (Th2) cytokine production and optimal T-cell
proliferation and allergic airway inflammation in vivo. Our data
demonstrate that the activation of the transcription factors nuclear
factor of activated T cells and NF-�B is impaired in PKC�/�-deficient
activated T cells. In addition, we present genetic knockout evidence
in ex vivo experiments with primary T cells that PKC�/� is critical for
the control of cell polarity during T-cell activation. Therefore
PKC�/� emerges as a critical regulator of Th 2 activation.
Asthma � NF-kappaB � T-cell activation � polarity � NF-AT
CD4� T cells are central in the control of immunity thanks totheir ability to differentiate into different subsets of T-helper
(Th) cells (1, 2). It now is well established that naïve CD4� T cellscan differentiate in response to antigen stimulation into 3 distinctsubsets of effector cells, Th1, Th2, or Th17 cells, which displaydistinct cytokine profiles and immune regulatory functions (3).Th17 cells are the latest addition to the group of effector T cells andare induced in vitro by the combined actions of TGF� and IL-6, arecharacterized by the secretion of the proinflammatory cytokineIL-17, and have been shown to play an essential role in autoim-munity (4, 5). In contrast, Th1 cells mainly produce IFN-� and areessential for cell-mediated immune responses against intracellularpathogens. Th2 cells produce a different set of cytokines, includingIL-4, IL-5, IL-10, and IL-13, and are important in the control ofhumoral immunity and allergy (6). In this regard, the pathology ofasthma, a chronic lung inflammatory disease with increased prev-alence in developed countries (7), is associated with aberrantactivation and/or differentiation of CD4� Th2 lymphocytes (8). Themolecular mechanisms controlling Th2 differentiation and functionhave been the object of much research because of their relevanceto inflammation disorders, such as asthma, for which better ther-apies are sorely needed, and because these mechanisms are a veryinteresting model system of crosstalk between different signalingcascades during a complex biological process.
IL-4 is important for induction and maintenance of differentiatedTh2 cells (9). IL-4 and IL-13 share interactions with the IL-4receptor chain and activate the transcription factor Stat6 through aJak1/Jak3 signaling pathway (6, 10). We recently have presented invitro, ex vivo, and in vivo genetic evidence that the atypical PKC(aPKC) isoform, PKC�, is necessary for optimal activation of theIL-4 signaling cascade upstream of Jak1 (11). Furthermore, in vivoadoptive transfer experiments using PKC��/� mice and cells dem-onstrated that PKC� in Th2 cells is required for allergy-inducedairway inflammation (11). These findings established PKC� as animportant mediator of Th2 differentiation in vivo in the IL-4signaling pathway. However, the fact that IL-4 is required for Th2differentiation but at the same time must be produced by Th2 cellsseems paradoxical. It suggests that other signals must trigger theinitial activation of the Th2 polarization event, and that the Th2-produced IL-4 serves to amplify and maintain that response.
In fact, recent studies propose that activated antigen-presenting cells (APCs) trigger a new set of signals that serveto instruct T cells toward the different differentiated lineages(2). In dendritic cells, for example, Notch ligands, as well as thereceptor tyrosine kinase, c-Kit, and its ligand, SCF, signal Tlymphocytes to polarize to the Th1 or Th2 lineages, dependingon the type of APC-stimulating trigger (12–15).
The role of the other aPKC member, termed PKC�/�, in thecontrol of the immune response has not yet been investigated usingin vivo genetic systems. PKC�/� is highly homologous to PKC� (16),and because the latter has been shown to be important in Th2differentiation, our goal in this study was to test whether PKC�/�plays a role in this process. Our data shown here clearly establishthat PKC�/� is required for optimal Th2 cytokine production andallergic airway inflammation and that its loss correlates withimpaired activation of key transcriptional factors and cell polarity.
Results
Generation of Conditional PKC�/� Knockout Mice on Activated T Cells.
Because PKC�/��/� mice die at very early embryonic stages,probably because of defects in cell polarity (Diaz-Meco, Leitges,and Moscat, unpublished observations), we generated a conditionalPKC�/� knockout (PKC�/�fl/fl) mouse line in which PKC�/� isspecifically deleted in activated T cells. To do so, we crossedPKC�/�fl/fl mice (17) with CreOX40 mice in which the expression ofCre is under the control of the Tnfrsf4 locus (18). OX-40 isexpressed almost exclusively in activated T cells, especially CD4�
cells, upon stimulation (18). In this mutant mouse line, PKC�/� isexpressed at normal levels in immature thymocytes and naïve Tcells and, as predicted, is deleted only upon T-cell activation. Thisstrategy is advantageous in that it avoids embryonic lethality andprevents potential confounding effects resulting from the deletionof PKC�/� during development or in resting cells. This approach hasbeen used previously to delete the GATA3 gene specifically inactivated T cells during Th2 differentiation experiments (18). PCRgenotyping was used to screen for homozygous conditional PKC�/�-deficient (PKC�/�fl/f lCreOX40), heterozygous (PKC�/�fl/wt
-
CreOX40), and wild-type (PKC�/�fl/fl) mice. No Cre-mediated ef-fects were detected when PKC�/�wt/wtCreOX40 and PKC�/�fl/fl micewere compared (data not shown). The deletion of PKC�/� inactivated CD4� T cells was confirmed by Western blot. That is,upon anti-CD3 plus anti-CD28 stimulation under non-skewedconditions or after differentiation of the cells under Th0, Th1, orTh2 conditions, we observed a significant up-regulation of PKC�/�
Author contributions: J.-Q.Y., M.T.D.-M., and J.M. designed research; J.-Q.Y. and A.D.
performed research; J.-Q.Y., A.D., M.T.D.-M., and J.M. analyzed data; M.L. contributed new
reagents/analytic tools; and J.-Q.Y., M.T.D.-M., and J.M. wrote the paper.,
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
1J.-Q.Y. and M.L. contributed equally to this work.
2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/
0805907106/DCSupplemental.
© 2009 by The National Academy of Sciences of the USA
www.pnas.org�cgi�doi�10.1073�pnas.0805907106 PNAS � January 27, 2009 � vol. 106 � no. 4 � 1099–1104
CELL
BIO
LOG
Y

in T cells from wild-type mice but not in those from conditionalPKC�/�-knockout mice (Fig. 1A and B). The heterozygous activatedT cells display PKC�/� levels that are intermediate to those ofwild-type and PKC�/�-deficient activated T cells (supporting infor-mation (SI) Fig. S1). Therefore, PKC�/� normally is induced duringsustained T-cell stimulation and differentiation and is deletedeffectively in the mutant cells. This observation in itself is interest-ing because it suggests that in unstimulated conditions PKC�/�levels are very low and that this kinase is induced only when T cellsare activated for a relatively long period (Fig. 1 A and B) and thatPKC�/� might be required for the sustained signaling leading toT-cell differentiation but not for early cell activation.
Of note, conditional PKC�/�-deficient mice appeared to developnormally (data not shown), and the percentages of T and B cells(CD4�, CD8�, and B220� cells) in the spleens and lymph nodes ofthese mice were similar to those in wild-type mice (Table S1). Thenumber of double-positive cells (CD4�CD8�) and single-positivecells (CD4� or CD8�) in the thymus also did not differ inconditional PKC�/�-deficient and wild-type mice (Table S2).
Reduced Th2 Cytokine Secretion by CD4� T Cells from Conditional
PKC�/�-Deficient Mice. Because PKC� has been shown to play acritical role during Th2 differentiation (11), and because of thehigh homology between the 2 aPKCs, we sought to determinewhether the conditional deletion of PKC�/� in activated T cellswould produce an effect similar to that of PKC� deficiency. Todo so, we stimulated purified naïve CD4� T cells from mice ofdifferent genotypes with anti-CD3 in the absence or in thepresence of anti-CD28; the levels of different secreted cytokineswere detected by ELISA. Interestingly, reduced secretion of IL-4was observed in PKC�/�-deficient cells as compared with wild-type littermate controls when both types of cells were activatedwith anti-CD3 plus anti-CD28 (Fig. 1C). In the case of heterozy-gous PKC�/� mice (PKC�/�fl/wtCreOX40), this reduction was ap-parent only under anti-CD3 activating conditions (Fig. 1C).These results indicate that PKC�/� is required for Th2 cytokineproduction. Of note, when IFN� secretion was determined underthe same conditions, it was clear that its production was unaf-
fected in the PKC�/�-deficient T cells (Fig. 1C). Consistently,secretion of IL-13, another Th2 cytokine, also was reduced in thePKC�/�-deficient T cells (Fig. 1C) stimulated under the sameconditions. Because these data suggest that PKC�/� is requiredfor the control of the balance between Th2 and Th1 cytokines,we next wanted to test whether PKC�/� likewise is necessaryduring Th2/Th1 differentiation in vitro. To address this possi-bility, isolated CD4� T cells were cultured for 4 days undernon-skewing (Th0) or under Th1 or Th2 conditions. Afterwards,cells were washed and restimulated with anti-CD3 for 24 hours,and cytokine secretion was determined by ELISA. Surprisingly,the loss of PKC�/� in activated T cells did not impair IL-4 orIL-13 secretion in Th2-polarized T cells or the secretion of INF-�in Th1-polarized T cells (Fig. 1D). Collectively these resultscould be interpreted to mean that PKC�/� is required for tiltingthe balance of Th2 vs. Th1 cytokine secretion but that, under theconditions of in vitro T-cell differentiation, the PKC�/� pathway
PKC fl/fl
PKCCre
fl/wt
OX40
PKCCre
fl/fl
OX40
1.2 37.4
0.5 27.1 0.8 26.3
4.1 47.2
3.9 43.4 0.6 53.9
Anti-CD3/CD28Medium
Splenic CD4+ LN CD4+
Anti-CD3/CD28Medium
Udr
B
CD4
Fig. 2. Deletion of PKC�/� impairs CD4� T-cell proliferation. Purified naïve
CD4� T cells from spleens or whole lymph node (LN) cells were stimulated with
plate-bound anti-CD3 plus anti-CD28 for 3 days. Then cell proliferation was
assessed by BrdU intracellular staining by FACS. Percentages of BrdU-positive
cells are shown.
0
3
6
9
12
)lm /
gn(
-N
F I)l
m/g
n(31-
LI) l
m/g
p (4-
LI
0
1000
2000
3000
0
500
1000
1500
0
50
100
150
0
40
80
120
160
0
10
20
30
Anti-CD3Anti-CD28
C D
PKC
Actin
Spleen
Spleen
LN
PKC
PKC
Actin
Actin
A
B
PKC fl/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PKC Crefl/fl OX40
PK
C Cre
fl/f
l
OX
40
PK
C Cre
fl/f
l
OX
40
PK
C Cre
fl/f
l
OX
40
0 24 48 0 24 48 h
Th0 Th1 Th2
– + +– – +
Th0 Th1 Th2
PKC fl/fl
PKCCre
fl/wt
OX40
PKCCre
fl/fl
OX40
*
*
**
*
Fig. 1. PKC�/� is up-regulated upon T-cell activation and differentiation and is required for Th2-cytokine secretion. (A) Purified naïve splenic CD4� T cells or
cells from cervical, axillary, inguinal, and popliteal lymph nodes (LN) were stimulated with anti-CD3/CD28 for 24 or 48 hours. (B) Splenic CD4� T were
differentiated under Th0, Th1, and Th2 conditions for 4 days. Cells in A and B were restimulated with anti-CD3/CD28 for 2 days. Cells were collected for Western
blot analysis. Results shown are from a single experiment representative of 2 independent experiments. (C and D) Purified naïve splenic CD4� T cells were
stimulated with (C) anti-CD3/CD28 for 3 days or (D) differentiated under Th0/Th1/Th2 conditions for 4 days and restimulated with plate-bound anti-CD3 for 1
day. Supernatants from cell cultures were collected for ELISA assays to detect cytokines. Data (mean � SE) are from 4 experiments with triplicated determinations
in each sample. * P � 0.05.
1100 � www.pnas.org�cgi�doi�10.1073�pnas.0805907106 Yang et al.

is overridden by the presence of IL-4 (the trigger for Th2polarization). Of note, as determined by BrdU incorporation,T-cell proliferation in response to stimulation with anti-CD3/CD28 was reduced significantly in both splenic and lymph nodePKC�/�-deficient CD4� T cells (Fig. 2). The same results wereobtained when PKC�/�wt/wtCreOX40 mice were used as a controlto rule out any unspecific effect of the Cre line (data not shown).
PKC�/� Is an Important Mediator in Ovalbumin-Induced Allergic Air-
way Inflammation. As demonstrated in the previous discussion,although the lack of PKC�/� in activated T cells does not impair Th2differentiation under the standard conditions established in vitro,PKC�/� is important for IL-4 and IL-13 secretion in vitro undernon-skewing conditions. Therefore, it is conceivable that pathwaysmay be set in motion in vivo that could require PKC�/� for an
adequate Th2 response. For these reasons, we sought to determinewhether PKC�/� is necessary for an optimal Th2 inflammatoryresponse in vivo, although it does not seem to be necessary for Th2differentiation driven by the exogenous addition of IL-4 understandard in vitro conditions. In this regard, it is well established thatTh2 cells play a critical role in the development of allergic airwayinflammation (11). Because our data show that PKC�/� is requiredfor a proper balance of Th2/Th1 cytokines in vitro, we reasoned thatPKC�/� might affect the airway allergy response in vivo. Therefore,in the next series of experiments we tested the requirement forPKC�/� in the ovalbumin (OVA)-induced model of allergic airwayinflammation. Thus, wild-type and conditional PKC�/�-deficientmice were immunized by i.p. injection of OVA and then challenged3 times with aerosolized OVA or PBS as a negative control.Twenty-four hours after the last aerosol challenge, mice were killedand subjected to bronchoalveolar lavage (BAL) to determine therecruitment of inflammatory cells. Also, lungs were fixed andexamined histologically by H&E staining for cellular infiltration.There was a robust increase in total BAL cell numbers in wild-typemice that had been OVA immunized and challenged with aero-solized OVA, as compared with unimmunized naïve mice (Fig. 3A).Eosinophils accounted for most of this increase in the recruitmentof inflammatory cells (Fig. 3B). However, these increases werereduced dramatically in identically treated conditional PKC�/�-mutant mice (Fig. 3 A and B). H&E histological analysis of lungsections from these experiments consistently showed that chal-lenged wild-type mice displayed a prominent inflammatory re-sponse with massive perivascular and peribronchial infiltration,whereas the conditional PKC�/�-mutant mice displayed a highlyattenuated response (Fig. 3C). These data indicate that in vitroobservations of impaired Th2 cytokine secretion by T cells derivedfrom these mutant mice (see Fig. 1) are relevant to airway inflam-mation in vivo. Consistent with this idea, ELISA data on thecytokine levels in BAL from these in vivo experiments showed thatIL-4, IL-5, and IL-13, which were undetectable in naïve mice, wereincreased significantly in OVA-challenged wild-type mice but wereinhibited in identically treated conditional PKC�/�-mutant mice(Fig. 3D). IFN-� was slightly increased in BAL from PKC�/�-mutant mice as compared with identically treated wild-type mice,consistent with the in vitro data of Fig. 1. Interestingly, lung mRNAlevels of IL-4, IL-5, and eotaxin also were up-regulated in chal-lenged wild-type mice as compared with unimmunized mice, andthis up-regulation was reduced significantly in conditional PKC�/�-mutant mice (Fig. 3E).
PKC fl/fl PKC Crefl/fl OX40C
PB
SA
VO
A
)01x(
sllecL
ABlat
oT-4
)01x(
sllecL
AB
-4
B
)lm/
gp(
enik
otyC
0
10
20
30
40
50
D
0
10
20
30
40
s tin
uyra rt i
brA
IL-5
Eotaxin
0
5
10
15
20Gob-5MUC5-AC
E
OVA
OVA OVA
OVA
OVA
PBS
PBS PBS
PBS
PBS
Eo M Neu Lym
IL-4
IL-4
IFN-
IL-5
IL-13
PKC fl/fl
PKCCre
fl/fl
OX40
PKC fl/fl
PKC
PKC Crefl/fl OX40
****
**
*
** **
***
*
F
Actin
0 48 0 48 h
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
C Cre
fl/f
lO
X40
PK
C Cre
fl/f
lO
X40
PK
C Cre
fl/f
lO
X40
PK
C Cre
fl/f
lO
X40
PK
C Cre
fl/f
lO
X40
0
20
40
60
0
25
50
75
100
0
10
20
30
40
0
6
12
18
24
0
2
4
6
8
10
0
40
80
120
160
0
30
60
90
0
25
50
75
Fig. 3. Role of PKC �/� in OVA-induced allergic airway inflammation. Con-
ditional PKC�/�-deficient mice and their wild-type littermates were immu-
nized with OVA twice and then challenged with aerosolized OVA. Mice were
killed 25 hours after the last challenge. (A) The total cells in BAL fluid were
counted using a hemocytometer. (B) Differential cell counts of � 300 cells
were performed on cytospins stained with Kwik-Diff. The numbers of eosin-
ophils (Eo), macrophages (M�), neutrophils (Neu), and lymphocytes (Lym) in
BAL are shown. (C) Representative H&E staining of lung histology. (D) Cyto-
kine levels in BAL fluid were determined by ELISA. (E) mRNA levels of various
molecules in OVA-induced airway hyperresponsiveness (AHR). Total RNA was
extracted from the right lower lobe of the lungs for real-time PCR analysis. The
mRNA levels of various molecules are expressed as arbitrary units. All samples
were determined by triplicate; the data are normalized to an 18S reference. (F)
Representative Western blot results from trachea lymph node cells stimulated
with anti-CD3/CD28 for 48 hours from mice with OVA-induced AHR. Results in
A, B, D, and E are expressed as mean � SE from 3 independent experiments
with 3–5 mice per group in each experiment. *P � 0.05; **P � 0.01.
)m
nD
O(gI
AV
O-itn
A45
0
IgE IgM
IgG
IgG2a
IgG1
IgG3
**
OVA OVAPBS PBS
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
Cfl
/fl
PK
C Cre
fl/f
lO
X40
PK
C Cre
fl/f
lO
X40
0
0.4
0.8
1.2
0
1.0
2.0
0
1
2
3
0
0.1
0.2
0.3
0
0.1
0.2
0.3
0
0.1
0.2
Fig. 4. Serum OVA-specific IgE is reduced in conditional PKC�/�-deficient
mice. In the same experiments as Fig. 3, serum levels of OVA-specific IgE, IgM,
IgG, and IgG subclasses were assayed by ELISA (mean � SE). **P � 0.01.
Yang et al. PNAS � January 27, 2009 � vol. 106 � no. 4 � 1101
CELL
BIO
LOG
Y
Hypersecretion of mucus plays an important role in thepathogenesis and severity of asthma. The primary proteins inmucus are mucin glycoproteins, with MUC-5AC being the pri-mary airway mucin gene. The calcium chloride-activated channelgene hCLCA1 (gob-5 in the mouse) has been suggested toincrease MUC-5AC gene expression (19). Both MUC-5AC andgob-5 were increased dramatically in challenged wild-type mice,but, importantly, more than 2/3rds of this increase was lost in theconditional PKC�/�-mutant mice (Fig. 3E). Results in Fig. 3Fconfirm the deletion of PKC�/� in this in vivo experiment.
IgE is an important mediator of allergic airway inflammation (7).In mice, IL-4 preferentially induces immunoglobulin isotype switch-ing to IgE and IgG1, whereas IFN-� preferentially induces switch-ing to IgG2a and IgG3 (7). We reasoned that the reduced IL-4response observed in OVA-induced allergic airway inflammation inconditional PKC�/�-mutant mice might correlate with reducedOVA-specific IgE in vivo. Consistent with this prediction, serumlevels of OVA-specific IgE were reduced significantly in conditionalPKC�/�-mutant mice as compared with their wild-type controllittermates (Fig. 4), whereas OVA-specific IgM, total IgG, andIgG1 were reduced only slightly in the PKC�/�-deficient mice.OVA-specific total IgG2a and IgG3 did not differ significantlybetween the 2 mouse genotypes (Fig. 4). Taken together, theseresults indicate that the loss of PKC�/� in activated T cells leads toa significant inhibition of the Th2 response in vivo, consistent witha relevant role for this aPKC in Th2 cell differentiation.
Role of PKC�/� in T-Cell Activation. Because PKC�/� is important forTh2 cytokine secretion ex vivo and in vivo, we next sought to
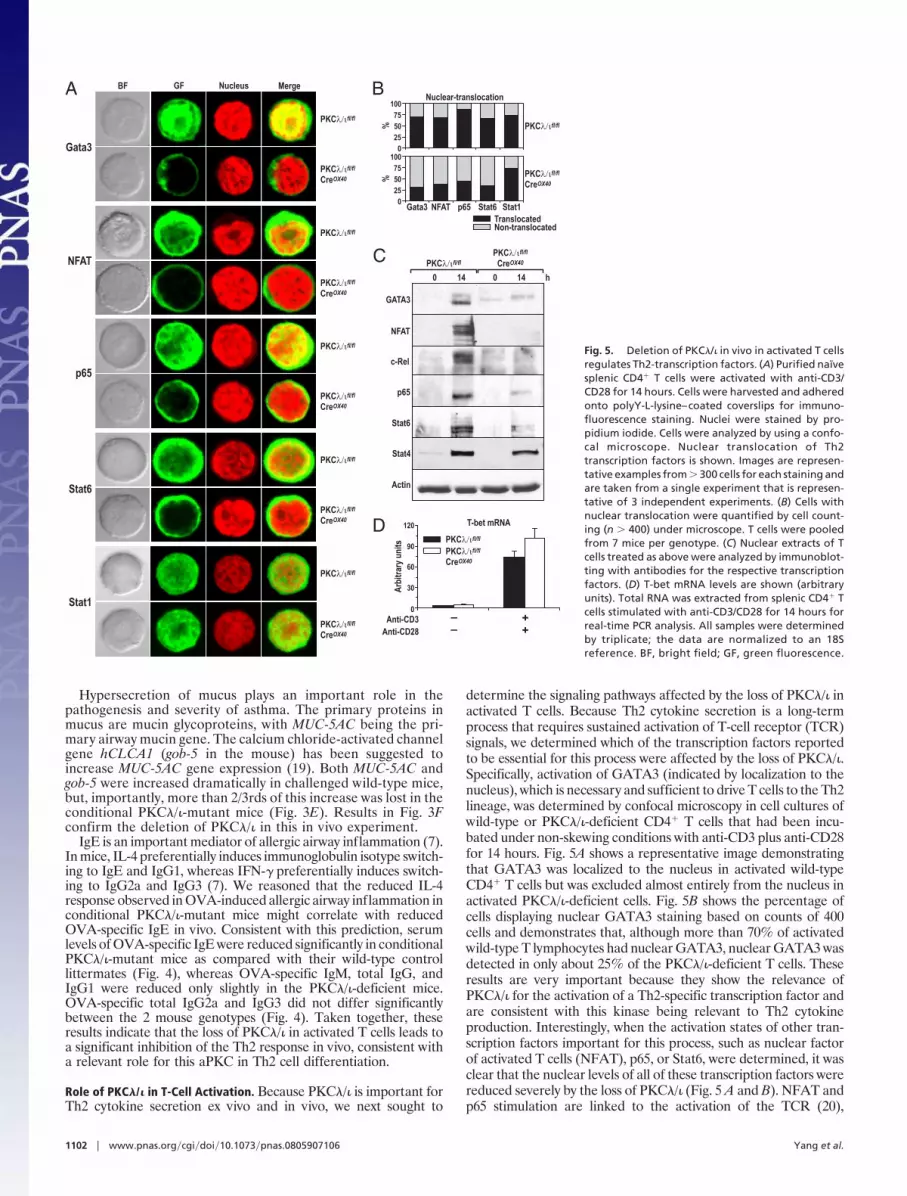
determine the signaling pathways affected by the loss of PKC�/� inactivated T cells. Because Th2 cytokine secretion is a long-termprocess that requires sustained activation of T-cell receptor (TCR)signals, we determined which of the transcription factors reportedto be essential for this process were affected by the loss of PKC�/�.Specifically, activation of GATA3 (indicated by localization to thenucleus), which is necessary and sufficient to drive T cells to the Th2lineage, was determined by confocal microscopy in cell cultures ofwild-type or PKC�/�-deficient CD4� T cells that had been incu-bated under non-skewing conditions with anti-CD3 plus anti-CD28for 14 hours. Fig. 5A shows a representative image demonstratingthat GATA3 was localized to the nucleus in activated wild-typeCD4� T cells but was excluded almost entirely from the nucleus inactivated PKC�/�-deficient cells. Fig. 5B shows the percentage ofcells displaying nuclear GATA3 staining based on counts of 400cells and demonstrates that, although more than 70% of activatedwild-type T lymphocytes had nuclear GATA3, nuclear GATA3 wasdetected in only about 25% of the PKC�/�-deficient T cells. Theseresults are very important because they show the relevance ofPKC�/� for the activation of a Th2-specific transcription factor andare consistent with this kinase being relevant to Th2 cytokineproduction. Interestingly, when the activation states of other tran-scription factors important for this process, such as nuclear factorof activated T cells (NFAT), p65, or Stat6, were determined, it wasclear that the nuclear levels of all of these transcription factors werereduced severely by the loss of PKC�/� (Fig. 5 A and B). NFAT andp65 stimulation are linked to the activation of the TCR (20),
Gata3
NFAT
p65
Stat6
Stat1
A
0
25
50
75
100
0
25
50
75
100
B
TranslocatedNon-translocated
Gata3 NFAT p65 Stat6
Nuclear-translocation
%%
GATA3
c-Rel
Stat4
Actin
0 14 0 14 h
NFAT
p65
Stat6
C
D
Stat1
0
30
60
90
120
Anti-CD3 –
Anti-CD28 –
+
+
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKC fl/fl
PKC fl/fl
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
T-bet mRNA
stin
uyr arti
b rA
BF GF Nucleus Merge
Fig. 5. Deletion of PKC�/� in vivo in activated T cells
regulates Th2-transcription factors. (A) Purified naïve
splenic CD4� T cells were activated with anti-CD3/
CD28 for 14 hours. Cells were harvested and adhered
onto polyY-L-lysine–coated coverslips for immuno-
fluorescence staining. Nuclei were stained by pro-
pidium iodide. Cells were analyzed by using a confo-
cal microscope. Nuclear translocation of Th2
transcription factors is shown. Images are represen-
tative examples from � 300 cells for each staining and
are taken from a single experiment that is represen-
tative of 3 independent experiments. (B) Cells with
nuclear translocation were quantified by cell count-
ing (n � 400) under microscope. T cells were pooled
from 7 mice per genotype. (C) Nuclear extracts of T
cells treated as above were analyzed by immunoblot-
ting with antibodies for the respective transcription
factors. (D) T-bet mRNA levels are shown (arbitrary
units). Total RNA was extracted from splenic CD4� T
cells stimulated with anti-CD3/CD28 for 14 hours for
real-time PCR analysis. All samples were determined
by triplicate; the data are normalized to an 18S
reference. BF, bright field; GF, green fluorescence.
1102 � www.pnas.org�cgi�doi�10.1073�pnas.0805907106 Yang et al.

whereas activation of Stat6 probably is caused by the secretion ofIL-4 in activated T cells. IL-4 production is reduced in PKC�/�-deficient activated T cells (see Fig. 1C), and this reduction is likelyto account for the reduced nuclear Stat6 levels detected in PKC�/�mutant T cells and is consistent with our data demonstrating thelack of a role for PKC�/� in IL-4 signaling in Stat6 activation(Diaz-Meco, et al., unpublished data). Immunoblot analysis of asimilar experiment demonstrates a dramatic inhibition of GATA3,NFAT, and p65 nuclear levels in the PKC�/�-deficient T cellsactivated as described earlier in this article (Fig. 5C), consistent withthe data in Fig. 5 A and B. Interestingly, the nuclear levels of the Th1transcription factors Stat1 and Stat4 were not affected by the lossof PKC�/� in these experiments (Fig. 5 A–C). Of note, the expres-sion of the Th1 master regulatory gene, T-bet, was induced inPKC�/�-deficient activated T cells to levels comparable to those ofthe wild-type cells (Fig. 5D). Results shown in Fig. S2 demonstratethat the OX40-Cre transgene is expressed at 14 hours of stimulationand, consistently, PKC�/� is up-regulated at this time point inwild-type T cells and is effectively depleted in the knockout cells.(Fig. S3). The activation of Akt and ERK was not affected by theloss of PKC�/� (Fig. S3). Deletion of PKC�/� in Jurkat T cells byRNA interference also inhibited NFAT and NF-�B activities (Fig.S4). Collectively, these results could be interpreted to mean thatPKC�/� plays a decisive role in the TCR activation of p65 andNFAT, which leads to GATA3 activation and the synthesis of IL-4,thus influencing the stimulation of Stat6. Therefore, we concludethat PKC�/� is induced during and is required for the sustainedactivation of key transcriptional events of Th2 differentiation.
Role of PKC�/� in T-Cell Polarity. The aPKCs have been implicatedin the control of cell polarity in several mammalian in vitrocell-culture experiments (21). More recently, the potential roleof polarity in T-cell activation has been shown also (22, 23).However, no genetic evidence from ex vivo experiments has beenproduced to test directly the involvement of the aPKCs in T-cellpolarity. A recent study identified Crtam as a scaffold proteinthat binds the cell polarity protein Scribble and through thisinteraction regulates cell polarity, proliferation, and the secre-tion of cytokines (24). Therefore, a potential explanation for thefindings reported here is that PKC�/� could be necessary forT-cell proliferation and Th2 cytokine secretion because of itspotential role in T-cell polarization. To address this question, weincubated CD4� T cells from either wild-type or PKC�/�-mutantmice with anti-CD3 plus anti-CD28 for 14 hours, followingexactly a previously described procedure to induce and assessT-cell polarization (24). We then determined T-cell polarity bymonitoring the ability of the polarity marker Scribble to localizeto one of the poles of the activated T cell (24). Fig. 6 A and Bstrongly suggests that the lack of PKC�/� during T-cell activationleads to a significant loss of T-cell polarity. To establish thisconclusion more firmly, we analyzed the localization of CD44and CD3 in double immunofluorescence analysis. From the dataof Fig. 6C it is clear that CD44 is asymmetrically polarizedrelative to CD3, consistent with the induction of cell polarity inthe activated T cells. More importantly, this polarization isseverely impaired in the PKC�/�-deficient T cells (Fig. 6 C andD). Interestingly, PKC�/� colocalizes with Crtam and is requiredfor its proper polarization in activated T cells (Fig. 6 E and F).Similar conclusions were obtained when the role of PKC�/� wasinvestigated in late T-cell polarity using polystyrene beadscoated with anti-CD3 plus anti-CD28 (Fig. S5). Of note, the lackof PKC�/� does not affect T-cell migration under these experi-mental conditions (Fig. S6). Collectively these results indicatethat PKC�/� deficiency in activated T cells leads to impairedpolarity during late T-cell activation.
Discussion
Unraveling the signaling pathways that promote T-cell differenti-ation along the different lineages is of paramount importance on 2levels: for the general insight it provides on the interplay betweendifferent cell-signaling pathways in the regulation of complexcellular processes and, more specifically, for an increased under-standing of T-cell biology under normal and pathological condi-tions. Here we show that PKC�/�, 1 of the 2 members of the aPKCfamily, plays a critical role in allergic airway inflammation in vivo,most likely because it is required for the production of Th2cytokines. Ex vivo experiments with PKC�/�-deficient T cells dem-onstrate that this kinase is required for the activation of transcrip-tion factors critical for adequate Th2 cell function and differenti-ation. We also provide genetic evidence using conditional knockoutcells that PKC�/� is essential for T-cell polarity, an event that hasbeen suggested to be relevant to T-cell function (22). Consideredcollectively, these results suggest that defects in cell polarity causedby the lack of PKC�/� in activated T cells, along with alterations ingene expression programs, are responsible for the defects in Th2cytokine production detected in the ex vivo experiments andaccount for the impaired lung inflammatory response observed inthe PKC�/� mutant mice when challenged with an allergic stimulus.
Our findings bring up a number of questions that deservediscussion before a consensus model emerges on the links betweencell polarity, signaling, and function, at least in the immune system.How alterations in polarization correlate with defects in geneexpression signaling need more in-depth investigation, both in Tlymphocytes and in other cell systems in which polarity is essentialfor relevant cellular functions. In this regard, the identification of aScribble-interacting partner, Crtam, constitutes an important stepin linking polarity and T-cell function in a cause–effect type ofexperiment (24). Interestingly, the loss of Crtam leads to impairedT-cell polarity, which is accompanied by increased proliferation and
PolarizedNon-polarized
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKC fl/fl
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKCCre
fl/fl
OX40
PKC
PKCCre
fl/fl
OX40
B
0
25
50
75
100A
C D
EF
Scrib polarity
CD44 CD3 egreMsuelcuN
0
25
50
75
100CD3/CD44 polarity
Crtam egreMsuelcuN
0
25
50
75
100Crtam polarity
%%
%
ScribBF Nucleus Merge
Fig. 6. PKC�/� control cell polarity. (A, C, and E) Purified naïve splenic CD4�
T cells were activated with anti-CD3/CD28 for 14 hours. Cells were treated for
immunofluorescence staining as in Fig. 5. Polarity of Scrib (B), CD3/CD44 (D),
and Crtam/PKC� (F) are shown. Images are representative of � 300 cells for
each staining. Cells with polarity were quantified by cell counting (n � 400)
under a microscope. CD4� T cells were pooled from 7 conditional PKC�/�-
deficient mice and their wild-type littermates. The results are from a single
experiment that is representative of 2 independent experiments with identi-
cal results.
Yang et al. PNAS � January 27, 2009 � vol. 106 � no. 4 � 1103
CELL
BIO
LOG
Y

decreased production of the Th1 and Th17 cytokines, IFN� andIL-17, respectively, but not of the Th2 cytokine IL-4 (24). At firstglance, these published data could be interpreted to mean thatT-cell polarization is essential for T-cell differentiation toward Th1but not Th2 lineages. However, our results shown here establish thatthe loss of polarity as a consequence of PKC�/� ablation in activatedT cells does not correlate with increased proliferation, but ratherwith reduced proliferation, and leads to the impairment of theproduction of IL-4 but not of IFN�.
A potential explanation for these different observations is thatPKC�/�, in addition to binding the Phox and Bem1p-1 domain(PB1)-containing polarity adapter Par-6 (16), also binds a non-polarity PB1-containing signaling adaptor, termed p62, whosegenetic knockout gives a phenotype in vivo and ex vivo very similarto that of PKC�/� in terms of T-cell activation (3). This possibilitycould be interpreted as meaning that PKC�/�, because of its abilityto bind p62 through their respective PB1 domains (16), willinfluence late T-cell signaling, ultimately resulting in modulation ofTh2 cytokine production and of the response to allergic airwayinflammation. On the other hand, probably through its interactionwith polarity adapters such as the PB1 scaffold Par-6 and otherpolarity proteins (16), PKC�/� affects the localization of thesemolecules in polarized regions of the T cell. Therefore, PKC�/�generates 2 independent kinds of signals, depending on its differentbinding partner, and these signals direct PKC�/� participation todistinctive signaling cascades. These data are consistent with amodel in which the inactivation of different proteins with differentpolarity would have different cellular consequences because of theirassociation with different signaling complexes.
Materials and Methods
Mice. PKC�/�fl/fl mice were reported previously (17). CreOX40 mice were generated
in the laboratory of N. Killeen at the University of California, San Francisco (18).
Conditional PKC�/�-deficient mice on activated T cells were generated by crossing
PKC�/�fl/fl micewithCreOX40 mice.MousegenotypingwasperformedbyPCRusing
primers for PKC�/�fl/fl, OX-40-Cre, and PKC�/� deletion to screen homozygous
conditional PKC�/�-deficient (PKC�/�fl/fl CreOX40), heterozygous (PKC�/�fl/wt
CreOX40), and wild-type (PKC�/�fl/fl or PKC�/�wt/wt CreOX40) mice. Age- (9–12 weeks)
and sex-matched mice were used in each experiment.
CD4� T-Cell Isolation and Differentiation. Splenocytes were prepared by dis-
rupting spleens of 9- to12-week-old mice. Naive CD4� T cells were enriched
with a Mouse CD4� Isolation Kit using an AutoMACS Pro (Miltenyi Biotec). The
purity (� 95%) and naïve status of isolated CD4� T cells were confirmed by
FACS staining with conjugated mAbs to CD4, CD8, B220, CD44, and CD62L.
Naïve CD4� T cells (106/ml) were activated with plate-bound anti-CD3 (10
�g/ml) plus soluble anti-CD28 (2 �g/ml) or differentiated into Th0, Th1, or Th2
cells (11). The culture supernatants were collected at different times after
activation for cytokine assays by ELISA.
Cytokine Assay. Cytokines in the supernatants and BAL fluid were assayed by
ELISA. IL-4, IL-5, IL-6, IL-10, and IFN-� were assayed with OptEIA kits (BD PharM-
ingen); and IL-13 and eotaxin were assayed with DuoSet ELISA kits (R&D Systems).
ELISA plates were developed with TMB substrate (BD PharMingen), and read by
a microplate reader (model 550, Bio-Rad). Cytokine mRNA levels were measured
by real-time quantitative PCR.
FACS. Spleen and lymph node cells or purified CD4� cells were incubated with
anti-CD16/32 (2.4G2) to block FcR� II/III and then were stained with various
conjugated mAbs. Stained cells were analyzed by FACSCalibur with CellQuest
software (Becton Dickinson).
Proliferation. CD4� T-cell proliferation was assayed in vitro by measuring BrdU
incorporation with a BrdU Flow kit (BD PharMingen) following the manufac-
turers’ protocols. Briefly, purified naïve CD4� T cells or whole lymph node cells
were cultured with plate-bound anti-CD3 plus anti-CD28 for 2 to 3 days. BrdU
(10 mM) was added to the cultures in the last 18 hours. After fixation,
permeabilization, and DNase digestion, cells were stained with APC-
conjugated anti-BrdU mAb and analyzed by FACS.
Immunofluorescence Staining. Purified naïve CD4� T cells were cultured with
plate-bound anti-CD3 plus anti-CD28 for 14 hours. Cells were harvested and
adhered onto polyY-L-lysine (Sigma)–coated coverslips for 30 minutes at room
temperature, and nonadherent cells were washed off with PBS. Adherent cells
were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100,
and incubated with different mAbs. The signals were amplified by the Tyra-
mide Signal Amplification kit (Molecular Probes). For nuclear staining, cells
were incubated with propidium iodide (Sigma) or TOPRO-3 (Invitrogen).
Coverslips were mounted on Mowiol and examined with a Zeiss LSM-510 Meta
Confocal Laser Scanning Microscope (Carl Zeiss MicroImaging). Images from
single plane were taken.
Real-Time PCR. Total RNA was extracted from lung tissues or cultured cells with
the RNeasy Mini kit (Qiagen), and cDNA was prepared by the Omniscript
Reverse Transcription kit (Qiagen). Quantitative PCR was performed with the
SYBR Green PCR Master Mix (Applied Biosystems) on a Mastercycler ep real-
plex4 apparatus (Eppendorf). The data were normalized to the 18S reference.
Primers for IL-4, IL-5, IL-13, eotaxin, MUC-5AC, Gob-5, and T-bet were designed
with OLIG 4.0 software.
For more information, please see SI Materials and Methods.
ACKNOWLEDGMENTS. We thank Maryellen Daston for editing this manu-script, Glenn Doerman for the artwork, and Hongzhu Liu and Lyndsey Cheu-vront for technical assistance. We also thank Dr. Nigel Killeen (University ofCalifornia, San Francisco) for the generous gift of the CreOX40 mice. This workwas funded in part by the University of Cincinnati-CSIC Collaborative Agree-ment, and by National Institutes of Health Grant R01-AI072581.
1. Reiner SL (2007) Development in motion: Helper T cells at work. Cell 129:33–36.
2. Kubo M (2007) Notch: Filling a hole in T helper 2 cell differentiation. Immunity 27:3–5.
3. Martin P, Diaz-Meco MT, Moscat J (2006) The signaling adapter p62 is an important
mediator of T helper 2 cell function and allergic airway inflammation. EMBO J
25:3524–3533.
4. Ouyang W, Kolls JK, Zheng Y (2008) The biological functions of T helper 17 cell effector
cytokines in inflammation. Immunity 28:454–467.
5. McGeachy MJ, Cua DJ (2008) Th17 cell differentiation: The long and winding road.
Immunity 28:445–453.
6. Shuai K, Liu B (2003) Regulation of JAK-STAT signalling in the immune system. Nature
Reviews. Immunology 3:900–911.
7. Elias JA, et al. (2003) New insights into the pathogenesis of asthma. J Clin Invest
111:291–297.
8. Luster AD, Tager AM (2004) T-cell trafficking in asthma: Lipid mediators grease the
way. Nature Reviews. Immunology 4:711–724.
9. Paul WE, Seder RA (1994) Lymphocyte responses and cytokines. Cell 76:241–251.
10. O’Shea JJ, Gadina M, Schreiber RD (2002) Cytokine signaling in 2002: New surprises in
the Jak/Stat pathway. Cell 109 Suppl:S121–131.
11. Martin P, et al. (2005) Control of T helper 2 cell function and allergic airway inflam-
mation by PKC{zeta}. Proc Natl Acad Sci USA 102:9866–9871.
12. Lee GR, Kim ST, Spilianakis CG, Fields PE, Flavell RA (2006) T helper cell differentiation:
Regulation by cis elements and epigenetics. Immunity 24:369–379.
13. Sun J, Krawczyk CJ, Pearce EJ (2008) Suppression of Th2 cell development by notch
ligands delta1 and delta4. J Immunol 180:1655–1661.
14. Wiethe C, et al. (2008) Dendritic cell differentiation state and their interaction with
NKT cells determine Th1/Th2 differentiation in the murine model of Leishmania major
infection. J Immunol 180:4371–4381.
15. Amsen D, et al. (2004) Instruction of distinct CD4 T helper cell fates by different notch
ligands on antigen-presenting cells. Cell 117:515–526.
16. Moscat J, Diaz-Meco MT, Albert A, Campuzano S (2006) Cell signaling and function
organized by PB1 domain interactions. Mol Cell 23:631–640.
17. Farese RV, et al. (2007) Muscle-specific knockout of PKC-lambda impairs glucose
transport and induces metabolic and diabetic syndromes. J Clin Invest 117:2289–2301.
18. Zhu J, et al. (2004) Conditional deletion of Gata3 shows its essential function in
T(H)1-T(H)2 responses. Nat Immunol 5:1157–1165.
19. Busse PJ, et al. (2005) Chronic exposure to TNF-alpha increases airway mucus gene
expression in vivo. J Allergy Clin Immunol 116:1256–1263.
20. Schulze-Luehrmann J, Ghosh S (2006) Antigen-receptor signaling to nuclear factor �B.
Immunity 25:701–715.
21. Goldstein B, Macara IG (2007) The PAR proteins: Fundamental players in animal cell
polarization. Developmental Cell 13:609–622.
22. Krummel MF, Macara I (2006) Maintenance and modulation of T cell polarity. Nat
Immunol 7:1143–1149.
23. Chang JT, et al. (2007) Asymmetric T lymphocyte division in the initiation of adaptive
immune responses. Science 315:1687–1691.
24. Yeh JH, Sidhu SS, Chan AC (2008) Regulation of a late phase of T cell polarity and
effector functions by Crtam. Cell 132:846–859.
1104 � www.pnas.org�cgi�doi�10.1073�pnas.0805907106 Yang et al.





























