Embed Size (px)
Citation preview

MEASUREMENT AND PARAMETERIZATION OF SPIN-SPIN COUPLING
CONSTANTS AND POTENTIAL APPLICATIONS IN CONFORMATIONAL
ANALYSIS OF OLIGOSACCHARIDES
A Dissertation
Submitted to the Graduate School
of the University of Notre Dame
in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
by
Hongqiu Zhao
Anthony S. Serianni, Director
Graduate Program in Chemistry and Biochemistry
Notre Dame, Indiana
December 2010

MEASUREMENT AND PARAMETERIZATION OF SPIN-SPIN COUPLING
CONSTANTS AND POTENTIAL APPLICATIONS IN CONFORMATIONAL
ANALYSIS OF OLIGOSACCHARIDES
Abstract
by
Hongqiu Zhao
Recent studies showed that car bohydrates play critical roles in a lot o f biological
activities, and conformations of carbohydrates and hydrogen bonds were believed to hold
the key to understanding these biological functions of oligosaccharides. Since the pioneer
work of Karplus, spin-spin coupling constants (J-coupling constants) had been the mos t
important and reliable tool in conformational analysis.
In this thes is, dens ity functional th eory ( DFT) was used to calculate J-coupling
constants involving hydroxyl protons, while systematically changes the dihedral angles of
interest. Kar plus-like equations par amerized wer e in good agr eement with pr eviously
reported equations , in which s mall non- carbohydrate molecules wer e us ed a s model
compounds. Newly obtained Kar plus equations we re us ed to inter pret exper imentally
collected 3JHCOH and 3JCCOH coupling constants in methyl- D- - and -lactoside. Low

Hongqiu Zhao
temperature (- 20 °C) and mixed solvent (aceton-d6 and water) were applied to reduce the
exchange between hydroxyl protons and bulk solvent. Most H-C-O-H coupling constants
measured were in the 4-6 Hz range, which should be expected from a f ree C-O rotation.
However, an exceptionall y s mall coupling ( 2.8 Hz) f or H3’ -C3’-O3’-H s uggested a
rotational bias of C 3’-O3’ bond. C hanging s olvent to DM SO r educed the coupling
constants further to ~ 1.3 Hz, indicating stronger bias in non-protic solvent. This bias was
consistent with the gau che c onformation of C 3’-O3’ in cr ystal s tructure and was
interpreted as potential hydr ogen bonding betwe en O3’- H in the glucos e r ing and r ing
oxygen (O5) in the galactos e ring. The hydrogen bond was studied theoretically, and its
strength (~ 4 kcal/mol) was in line with normal hydrogen bond. A few experiments were
designed to f ind direct evidence of this H-bonding. Some interesting observations were
made, however, none of them could firmly prove the persistency of this H-bond.
Trans-glycoside linkage coupling pa thways C -O-C-H and C -O-C-C ar e another
focus of this thesis. Early parameterizations of C -O-C-H and C -O-C-C assumed similar
Karplus behavior as vicinal H -H couplings . Ho wever, ther e is no r igorous theor etical
support of this as sumption. I n this thes is, it is clear ly s hown that electr onegative
substituents c an s ignificantly af fect the Kar plus-like behavior of thes e coupling
pathways; hence s ubstituent ef fects s hould be cons idered when inter preting the
experimental data. Subs tituent ef fects ar e systematically s tudied f or C-O-C-C coupling
constants. The results support the usage of Karplus-like equations for C-O-C-C coupling
pathway, with some necessary modifications when electronegative substituents exist.

ii
This is dedicated to my wife and my beautiful daughter, Aleen.

iii
CONTENTS
Figures..........................................................................................................................vii
Schemes .......................................................................................................................xiv
Tables.... ......................................................................................................................xvi
Acknowledgments .......................................................................................................xvii
Chapter 1: Introduction....................................................................................................1 1.1 Overview .......................................................................................................1 1.2 Carbohydrates ................................................................................................1 1.3 Structural varieties of carbohydrates...............................................................2 1.4 Conformational analysis of oligosaccharides..................................................3
1.4.1 Conformations within oligosaccharides .................................................4 1.4.2 NMR and conformational analysis of carbohydrate................................6 1.4.3 Discrete model and conformational analysis in oligosaccharides............8 1.4.4 Obtaining 3JHH and 3JCH vicinal coupling constants...........................9 1.4.5 Carbohydrate modeling and conformational analysis ...........................11
1.5 Density Functional Theory and J-coupling Calculations...............................12 1.5.1 Introduction to Density Functional Theory ..........................................12 1.5.2 LSDA, GGA and hybrid DFT methods................................................14 1.5.3 Advantages and disadvantages of DFT methods ..................................15 1.5.4 Perturbation and J-coupling constants..................................................16 1.5.5 DFT computations of J-coupling constants ..........................................19
1.6 Some challenges of carbohydrate conformational study and aims of this thesis...................................................................................................................19
1.6.1 Collection and interpretation of J-coupling constants of hydroxyl protons ..................................................................................................22
1.6.2 Substituent effect on trans-glycoside vicinal couplings and Karplus equations...............................................................................................23
1.6.3 Aims of this thesis ...............................................................................24 1.7 References ...................................................................................................25
Chapter 2: Density Functional Theory Calculations and Parameterization of 2JCOH, 3JHCOH and 3JCCOH Spin-Spin Coupling Constants in Saccharides...............32 2.1 Introduction .................................................................................................32

iv
2.2 Computational Method.................................................................................34 2.3 Results and Discussion.................................................................................37
2.3.1 H-C-O-H coupling pathway.................................................................37 2.3.1.1 P arameterization............................................................................37 2.3.1.2 Non-Fermi Contact Contributions to 3JHCOH. .............................39 2.3.1.3 Effect of extra OH group on 3JH2,O2H.........................................40
2.3.1.4 Other 3JHCOH coupling pathway in methyl monoglycosides........41 2.3.1.5 3JHCOH involving the C1-O1 bond..............................................41 2.3.2 C-C-O-H coupling pathway.................................................................44 2.3.2.1 Coupling Between C1 and O2H in 1-4...........................................44 2.3.2.2 C-C-O-H Coupling Pathways in 5-12.............................................47 2.3.2.3 C ouplings Involving C1-O1 bond ..................................................50 2.3.2.4 Studies of 3JCCOH in Simpler Model Systems. ............................51 2.3.3 Coupling involving O6H .....................................................................52 2.3.4 Solvation Effects on 3JHCOH and 3JCCOH. ......................................54 2.3.5 C-O-H Coupling Pathways. .................................................................56
2.4 Conclusions .................................................................................................56 2.5 References ...................................................................................................59
Chapter 3: Application of 3JHCOH and 3JCCOH Karplus equations in oligosaccharides in DMSO and in aqueous solution......................................................................62 3.1 Introduction .................................................................................................62 3.2 Experimental Methods .................................................................................64 3.3 Results and Discussion.................................................................................66
3.3.1 Quality of spectra and further treatment of samples .............................66 3.3.2 Experimental studies of monosaccharides............................................67 3.3.2.1 3JHCOH and 3JCCOH values in monosaccharides: DMSO-d6
solvent ..................................................................................................67 3.3.2.2 Populations of hydroxyl groups calculated from Karplus
relationships..........................................................................................69 3.3.2.3 Comparisons with molecular dynamics simulation data .................73 3.3.2.4 3JHCOH and 3JCCOH spin-couplings in monosaccharides in
aqueous solution....................................................................................73 3.3.3 Experimental studies of disaccharides..................................................76 3.3.3.1 3JHCOH and 3JCCOH in 3 and 6. ..................................................76 3.3.3.1 Is the bias in the C3’-O3’ bond torsion in 3 caused by intramolecular
H-bonding? ...........................................................................................80 3.3.3.2 Hydroxyl proton exchange rates in 3 and bridging water................83 3.3.4 Further discussion................................................................................85
3.4 Conclusions .................................................................................................87 3.5 References ...................................................................................................90

v
Chapter 4: probing the presence and strength of hydrogen bonding in Saccharides via J-Couplings: A DFT Study ...................................................................................93 4.1 Introduction .................................................................................................93 4.2 Computational Method:................................................................................95 4.3 Results and Discussion:................................................................................97 4.4 Conclusion.................................................................................................109 4.5 References .................................................................................................110
Chapter 5: An nmr investigation of putative inter-residue H-bonding in methyl -cellobioside in solution ....................................................................................112 5.1 Introduction ...............................................................................................112 5.2 Results and Discussion:..............................................................................115 5.3 Conclusion.................................................................................................129 5.4 References .................................................................................................130
Chapter 6: Oligosaccharide Trans-glycoside 3JCOCC Karplus Curves Are Not Equivalent: Effect of Internal Electronegative Substituents .............................133 6.1 Introduction ...............................................................................................133 6.2 Calculational Section .................................................................................135 6.3 Results and Discussion...............................................................................136
6.3.1 3JCOCC in 2 - 5................................................................................136
6.3.2 3JCOCC in 6 and 7 ...........................................................................138 6.3.3 Further Discussion.............................................................................139
6.4 Conclusion.................................................................................................141 6.5 References .................................................................................................141
Chapter 7: Vicinal 13C-13C nmr spin-spin coupling constants in oligosaccharides: towards generalized equations for o-glycoside linkage conformational analysis142 7.1 Introduction ...............................................................................................142 7.2 Calculational Methods................................................................................144
7.2.1 Model Systems. ..................................................................................144 7.2.2 Spin-Spin Coupling Constant Calculations. .......................................145
7.3 Results and Discussion...............................................................................146 7.3.1 3JCCOC values in reference structure 1 (CH3-CH2-O-CH3) ............146
7.3.2 Electronegative Substituent Effects on 3JCOCC................................147 7.3.2.1 Inte rnal Substituent Effects..........................................................147 7.3.2.2 Te rminal Substituent Effects........................................................149 7.3.2.3 Par ametrization of 3JCOCC ( , , ,M).....................................149
7.3.3 3JCCOC Spin-couplings in Model Structure 1...................................150 7.3.4 3JCCOC in Model Structure 2: Internal Electronegative Substituent
Effect ..................................................................................................151

vi
7.3.5 3JCCOC in Model Structure 3: Terminal Electronegative Substituent Effect ..................................................................................................153
7.3.6 Parametrization of 3JCCOC in O-Glycosidic Linkages of Oligosaccharides .................................................................................158
7.3.7 Investigations of 3JC2’,Cn in O-Glycosidic Linkages........................159 7.4 Conclusion.................................................................................................163 7.5 Reference...................................................................................................166

vii
FIGURES
Figure 1.1 An example of monosaccharides in 4C1 chair form ........................................3
Figure 1.2 Conformational domains of Oligosaccharides. Key: (a) ring conformation; (b) O-glycoside conformation; (c) hydroxymethyl group conformation; (d) exocyclic C-O bond conformation. ......................................................................................4
Figure 1.3 Idealized Rotamers about the C5-C6 bond of Aldohexopyranosyl Rings.........6
Figure 1.4 t, g+ and g- staggered structure for A-X-X-B. A and B and other atoms involved vary according to different conformational domain studied....................9
Figure 1.5 Schematic representation of the backbone (left), showing the binding site of hevein domains for chito-oligosaccharides. The site is rather solvent exposed. The existence of three aromatic residues that provide interactions to the sugar is highlighted (right). (Figure from ref. 94) ............................................................20
Figure 1.6 Calculated pKa of 2-OH in 3,5-dideoxy-1-O-Methyl ribose as a function of C2-C1-O1-CH3 torsion angle. The ring conformation is restricted to 2E, E2, 3E and E3 respectively. ...........................................................................................21
Figure 2.1 Dependence of 3JH2,O2H on and determined by DFT for 1 (A), 2 (B), 3 (C) and 4 (D). Note the strong dependence on and minimal dependence on . 38
Figure 2.2 (A) Dependence of 3JH2, O2H on the H2-C2-O2-H torsion angle in 1-4; all data points are superimposed to illustrate similar J-couplings in each configuration. The curve fit to these data (equation 2.2) is indicated by the black line. Curves in red, green and blue are those reported by Fraser et al.,[3] Alkorta and Elguero[5], and Fukui et al.,[4] respectively. (B) Curve showing the dependence of 3JH2,O2H on the H2-C2-O2-H torsion angle in 1-4 (equation 2.2, blue curve) on which are superimposed computed 3JH1,O1H in 5-8 and 3JH2,O2H in 9-12 for H-C-O-H torsion angles of 60°, -60° and 180°. ...............39
Figure 2.3 Fermi (FC) and non-Fermi (NFC) contact contributions to 3JH2,O2H in 2 computed by Gaussian94 (G94) and Gaussian03 (G03) as a function of the H2-

viii
C2-O2-H torsion angle. Black filled circles, FC (G94). Open black circles, FC (G03). Filled black squares, SD term (G03). Black open squares, PSO term (G03). Blue filled triangles, DSO term (G03). Filled blue circles, FC + NFC. Green open circles, DSO + PSO. Blue open squares, total NFC. .......................40
Figure 2.4 (A) Dependence of 3JH1,O1H on the C1-O1 bond torsion in 5/7 (blue triangles) and 6/8 (black squares). Data are superimposed on the curve derived from equation 2.4 (red circles). (B) Dependence of 3JC2,O1H on the C1-O1 torsion angle in 5 (green circles), 6 (blue triangles), 7 (black squares), and 8
(purple diamonds). .............................................................................................43
Figure 2.5 Dependence of 3JC1,O2H on and determined by DFT for 1 (A), 2 (B), 3 (C) and 4 (D). Note the strong dependence on and minimal dependence on . The maximal coupling observed in 4 (~8 Hz) is considerably smaller than observed in 1-3 (~11 Hz). ..................................................................................44
Figure 2.6 (A) Dependence of 3JC1,O2H on the C1-C2-O2-H torsion angle in 1 (open black circles) and 2 (closed blue circles) superimposed on the curve defined by eq 11. (B) Dependence of 3JC1,O2H on in 3 (open black circles) and 4 (closed blue circles), superimposed on the curves defined by eqs 9 and 8, respectively. The scatter at discrete torsion angles is due to the effect of . ............................45
Figure 2.7 Plot of the Fermi contact (FC) and non-Fermi contact (NFC) contributions to calculated 3JC1,O2H values in 2 as a function of the C1-C2-O2-H torsion angle. FC = black squares; NFC, filled blue squares; total, open blue diamonds. ..........47
Figure 2.8 Dependence of 3JC1,O2H on the C1-C2-O2-H torsion angle based on
equation 2.11 on which are superimposed 3JC2,O1H in 5-8, 3JC5,O4H in 14, 3JC1,O2H in 6 and 8, and 3JC1,O2H in 9-12 for perfectly staggered C-C-O-H rotamers (60°, -60° and 180°). Couplings are reduced at 180° for pathways lacking an “in-plane” terminal oxygen substituent on the coupled carbon (see text). ..................................................................................................................48
Figure 2.9 Dependence of 3JC1,O2H on the C1-C2-O2-H torsion angle based on
equation 2.11 on which are superimposed 3JC2,O3H and 3JC3,O2H values in 9-12 computed for perfectly staggered C-C-O-H rotamers (60°, -60° and 180°). Couplings are reduced at 180° for those pathways that lack an “in-plane” OH substituent on the coupled carbon (see text). ......................................................50

ix
Figure 2.10 Calculated 3JCCOH in ethylene glycol (open circles) and n-propanol (closed circles) as a function of the O-C-C-O (glycol) or C-C-C-O (propanol) torsion angle. In both cases the C-C-O-H torsion angle was fixed at 180°. ....................52
Figure 2.11 Calculated 3JH6R,O6H (green circles) and 3JH6S,O6H (blue diamonds) at staggered comformations are superimposed on equation 2.4 (black line)............53
Figure 2.12 Effects of the C5-C6-O6-H (small blue open circles) and O5-C5-C6-O6 (large black open circles) torsion angles on 3JC5,O6H in 13. The terminal “in-plane” O5 enhances the anti coupling in the tg conformation, whereas the effect on gauche couplings is negligible.......................................................................54
Figure 2.13 Hypersurface showing the dependence of 2JC2,O2H on and in 2. ........56
Figure 2.14 Plot of 3JH2,O2H (blue line), 3JC1,O2H (red line) and 3JC3,O2H (green line) for b-Glc 2 as a function of the H2-C2-O2-H torsion angle. .......................59
Figure 3.1 The partial 600 MHz 1H NMR spectrum of methyl -D-[4-13C]glucopyranoside in DMSO-d6 at 25 °C showing only the OH region of the spectrum. The assignments of the four signals (left to right) are as follows: O2H, O3H, O4H and O6H. .........................................................................................68
Figure 3.2 Partial 600 MHz 1H NMR spectrum of 2 in 1:1 H2O/acetone-d6 at –20 °C showing signals due to the four exchangeable hydroxyl protons. The signal marked with an “X” is due to the OH protons of acetone gem-diol. ....................75
Figure 3.3 Partial 600 MHz 1H NMR spectrum of 3 in 2:3 H2O/acetone-d6 at –20 °C showing signals due to the seven exchangeable hydroxyl protons. The signal marked with an “X” is assigned to the OH protons of acetone gem-diol. ............77
Figure 3.4 Partial 600 MHz 1H-1H TOCSY spectrum of 3 in 2:3 H2O/acetone-d6 at -20 °C showing hydroxyl proton signal assignments based on correlations to the carbon bound protons. The Gal signals are shown in blue, and the Glc signals are shown in red. Hydroxyl proton assignments are shown at the top of the spectrum. ........78
Figure 3.5 Partial 600 MHz 1H NMR spectrum of 5 in 1:1 H2O/acetone-d6 at –20 °C showing signals due to the four hydroxyl protons. Note the additional splitting of the O2H signal relative to that shown in Figure 3.2 caused by the presence of 3JC1,O2H (see text)...........................................................................................79

x
Figure 3.6 (A) Partial 600 MHz 1H NMR spectrum of 3 in 2:3 H2O/acetone-d6 at –20 °C showing signals from O2H, O2’H and O6H. (B) Expansion and resolution enhancement of the O2H signal showing the presence of the small J-coupling to the enriched C1 (3JC1,O2H) (see text)...............................................................80
Figure 3.7 Partial 600 MHz 1H NMR spectrum of 50 mM 3 in DMSO-d6 at ~ 22 °C. Signal assignments of the hydroxyl protons shown adjacent to each signal, are based on the TOCSY data shown in Figure 3.8. Measured 3JHCOH values are shown in blue. Note that the small value of 3JH3’,O3’H. Resonance line-width were ~ 1.3 Hz for the OH protons, compared to ~ 1 Hz for CH protons..............81
Figure 3.8 Partial 600 MHz 1H-1H TOCSY spectrum of 50 mM 3 in DMSO-d6 at ~ 22 °C. Gal signals are shown in red, and Glc signals are shown in blue. The hydroxyl proton assignments are shown at the top of the spectrum. These data were used to make the OH signal assignments shown in Figure 3.7. The lack of correlations beyond H3’ arising from O3’H, and the single hydroxyl proton correlation to O2’H arising from H1’, are caused by the small value of 3JH3’,O3’H. ..............84
Figure 3.9 Partial 600 MHz 1H NMR spectrum of 50 mM 9 in 2:3 H2O/acetone-d6 at – 20 °C showing signal assignments and 3JHCOH values (in blue). The small 3JH3’,O3’H (2.1 ± 0.3 Hz) is similar in magnitude to the corresponding coupling observed in 3 (see Table 3.4)..............................................................................87
Figure 4.1 Dependence of rO3,O5’ and total energy in 2 as a function of (A) and (B). The scatter of points at discrete or torsions is due to rotation about the C3-O3 bond. Blue points, rO3,O5’; green points, total energy......................................96
Figure 4.2 (A) Effect of the H3-C3-O3-H torsion angle on calculated 3JH3,O3H values in 2 and 3. (B) Effect of the C2-C3-O3-H torsion angle on calculated 3JC2,O3H values in 2 and 3. (C) Effect of the C4-C3-O3-H torsion angle on calculated 3JC4,O3H values in 2 and 3. Blue symbols = 2; purple symbols = 3. ................97
Figure 4.3 (A) Effects of the H3-C3-O3-H torsion angle on rO5’-O3H (blue circles) and the O5’-H-O3 pseudo bond angle (red circles) in 2. The minimum distance and maximum angle are observed at a torsion of ~285°. (B) Effect of the H3-C3-O3-H torsion angle on calculated rC3,H3 in 2 (blue circles) and 3 (green circles). (C) Effect of the H3-C3-O3-H torsion angle on calculated 1JC3,H3 in 2 (blues circles) and 3 (green circles). .............................................................................98

xi
Figure 4.4 Dependence of calculated total energy on the H3-C3-O3-H torsion angle in 2 (blue symbols) and 3 (green symbols). ...............................................................99
Figure 4.5 The effect of the H3-C3-O3-H torsion angle on (A) rC1’,O5’ (open circles) and rC5’,O5’ (filled circles) in 2, and (B) rC1’,C6’ (open circles) and rC5’,C6’ (filled circles) in 3............................................................................................100
Figure 4.6 (A) Effects of the H3-C3-O3-H torsion angle on rC3,H3 in 2 (blue circles) and 3 (green circles). (B) Effects of the H3-C3-O3-H torsion angle on calculated 1JC3,H3 in 2 (blue circles) and 3 (green circles). .............................................103
Figure 4.7 Difference plots of calculated 1JC3,H3 in 2 and 3 (1JC3,H3 (2) – 1JC3,H3 (3)), showing the significant reduction in coupling in H-bonded forms of 2 relative to that observed in 3. In vacuo = blue circles; solvated = green symbols.........................................................................................................................104
Figure 4.8 (A) Effect of rHB in 4 on calculated energy. The lowest energy structure was observed at rHB near 2.8 Å, as expected for a conventional H-bond.13b (B) Effect of rHB on calculated rCH,donor (blue circles) and rCH,acceptor (green circles) in 4. (C) Effect of rHB on calculated 1JCH,donor (blue circles) and 1JCH,acceptor (green circles) in 4. ..................................................................106
Figure 4.9 The effects of rHB on rO,H (A) and rC,O (B) in model structure 4. Blue symbols = donor alcohol; green symbols, acceptor alcohol...............................107
Figure 4.10 Effect of the H3-C3-O3-H torsion angle in 2 and 3 on calculated 1JC1’,H1’ values (A) and calculated 1JC5’,H5’ values (B). Black squares, 2; purple squares, 3. (C) Difference plots generated from the subtraction of plots in (A) and (B); black points, 1JC1’,H1’; purple points, 1JC5’,H5’...........................................109
Figure 5.1The 13C{1H} NMR spectrum (150 MHz) of 4’ in 2H2O at 22 °C, showing signal assignments and splittings due to 13C-13C spin-spin coupling (Table 5.1).........................................................................................................................116
Figure 5.2 1H NMR spectrum (600 MHz) of 4’ in 2H2O at 22 °C showing signal assignments (Table 5.2). ..................................................................................118
Figure 5.3 The partial 1H NMR spectrum (600 MHz) of 4’ in DMSO-d6 at 22 °C showing signal assignments of the OH and anomeric protons. The inset is an

xii
expansion of the O6H/O6’H and O3H signals, showing the additional small splitting of the latter. 3JC4’,O3’H = ~ 2.7 Hz; 2JC4’,O4’H = ~ 2 Hz...............120
Figure 5.4 1H-1H COSY spectrum of 4’ in DMSO-d6 at 22 °C showing signal assignments of the OH protons. Assignments of the on-diagonal signals based on interpretation of the 1D spectrum (Table 5.3) allowed identification of the specific OH signals through the indicated crosspeaks....................................................122
Figure 5.5 The partial 1H NMR spectrum (600 MHz) of 4 in DMSO-d6 at 22 °C showing signal assignments of the hydroxyl and anomeric protons. The inset is an expansion of the O6H/O6’H and O3H signals, showing a small splitting in the latter. ...............................................................................................................123
Figure 5.6 The partial 1H NMR spectrum of 4’ in DMSO-d6 at 22 °C showing only the H4, OCH3 and H2 signals. Note the broadened H4 signals, in contrast to the relatively sharp H2 signals. ..............................................................................124
Figure 5.7 Partial 1H-1H COSY spectrum (600 MHz) of 4 in DMSO-d6 at 22 °C showing weak cross peaks between H1’-H4 (4JHCOCH) and H4-O3H (4JHCCOH). The H1-H2, H1’-H2’, H3-O3H and H6’-O6’H cross peaks are also shown. .............................................................................................................126
Figure 5.8 (A) DFT-calculated 3JH3,O3H values in 5 as a function of the H3-C3-O3-H torsion angle (blue symbols); red curve is derived from a generalized 3JHCOH Karplus equation [6]. (B) DFT-calculated 4JH2,O3H (blue symbols) and 4JH4,O3H (red symbols) as a function of the H3-C3-O3-H torsion angle in 5. Area enclosed by the green box approximates the allowed torsion angles for inter-residue O5’…HO3 H-bonding. ........................................................................128
Figure 6.1 (A) Calculated 3JCOCC values in 2a (green symbols) and 2b (blue symbols) as a function of the central C-O bond torsion. Pertinent coupling pathways are highlighted in green in 2a and blue in 2b. (B) Calculated 3JCOCC values in 3 (green symbols) and 4 (blue symbols) as a function of the central C-O bond torsion. Pertinent coupling pathways are highlighted in green in 3 and blue in 4.........................................................................................................................137
Figure 6.2 (A) Calculated 3JCOCC values in 2b (blue symbols) and 5 (green symbols) as a function of the central C-O bond torsion. Pertinent coupling pathways are

xiii
highlighted in blue in 2b and green in 5. (B) Calculated 3JCOCC values in 6 (blue symbols) and 7 (green symbols for 3JCCCC and red symbols for 3JCOCC) as a function of the central C-O or C-C bond torsion. Pertinent coupling pathways are highlighted in color in structures 6 and 7..........................................................138
Figure 7.1 Dependence of calculated 3JCCOC values in model compounds 1 and 2 on the C-C-O-C torsion angle. Black circles and red diamonds are computed coupling constants and curves correspond to the fitted equations. Black circles/blue dashed curve (eq 7.13), 1; red diamonds/red solid curve (eq 7.14), 2............................151
Figure 7.2 A plot of the curve described by eq 7.15, showing the effect of a single internal electronegative substituent on 3JCOCC values....................................152
Figure 7.3 A 3D hypersurface of calculated 3JCCOC values in 3 as a function of torsion angles and , showing the effect of a terminal electronegative substituent on coupling magnitude..........................................................................................153
Figure 7.4 2D Slices of 3JCOCC data in Figure 3 as a function of at given and/or as a function of at given . A. 3JCOCC as a function of at fixed staggered . Black line and dots are for = 180° (or trans); blue line and triangles are for = 60° (or g+); red line and diamonds are for = 60° (or g ). Note that 3JCCOC shows a clear Karplus-like behavior. B. 3JCOCC as a function of at fixed . Red line and open diamonds are for = 180° (or trans); green line and triangles are for = 60° (or g+); black line and open circles are for = 60° (or g ); blue line and filled diamonds are for = 0° (eclipsed). Note the maximum enhancement when is in the trans region. ....................................................154
Figure 7.5 A. Data in Figure 7.3 shown in two dimensions as a function of . The bandwidth for each given corresponds to dependence. B. The bandwidth in A fitted to a Karplus-like equation (eq. 7.16)....................................................157
Figure 7.6 Plots of Karplus curves for 3JC2,C1’ in 4 - 7 based on eqs 7.21-24. Blue, green, black and red curves are for model compounds 4, 5, 6 and 7 respectively.........................................................................................................................163

xiv
SCHEMES
Scheme 2.1 Model compounds used in calculating J-coupling constants involving hydroxyl protons. ...............................................................................................36
Scheme 2.2 Effect of ring oxygen as an internal electronegative substituent on the coupling pathways between H1 and O1H or C2 and O1H. .................................42
Scheme 2.3 Terminal electronegative substituent effect. In 1-3, the terminal oxygen, either ring oxygen O5, or O-methyl oxygen O1, is “in-plane” with the coupling pathway. No “in plane” oxygen in 4...................................................................46
Scheme 3.1 Structures of compounds 1 - 6 and locations of the 13C labeling.................64
Scheme 3.2 Definition of rotamers about the C3’-O3’ bond in 3....................................82
Scheme 3.3 DFT-generated bridging water complexed with 3. The H2O molecule (blue box) straddles the glycosidic linkage and is in H-bonding contact with O5 and O3’H..................................................................................................................85
Scheme 3.4 Structures of -chitobiose (7), methyl -D-lactoside (9) and methyl -L-lactoside (10). ....................................................................................................86
Scheme 4.1 Model structures used in this chapter. .........................................................95
Scheme 4.2 Effects of H-bonding on proximal bond lengths in 2. ................................101
Scheme 4.3 2-proponal molecules. One will serve as H-bond donor and another as H-bond acceptor...................................................................................................105
Scheme 4.4 The effects of H-bonding on rCH and 1JCH in both donor side and acceptor side. .................................................................................................................107
Scheme 5.1 Structure of model compound 1 – 4 and 4’ and their H-bonding pattern according to x-ray crystal structures. ................................................................114
Scheme 5.2 Model structure 5 and one of its snapshots when rotating H3-C3-O3-H torsion angle. ...................................................................................................127

xv
Scheme 6.1 Methyl -lactoside and definitions of Type I and Type II trans-glycoside COCC (or CCOC) pathways. ...........................................................................134
Scheme 6.2 A generalized Type I and Type II CCOC pathways...................................135
Scheme 6.3 Strucures of 2, 3, 4 and 5 and the coupling pathways. ...............................136
Scheme 6.4 Couplings that are sensitive to either or in 1. ......................................139
Scheme 6.5 Three staggered structures for rotation. All angles are referenced to H1-C1-O1-C4’.............................................................................................................140
Scheme 7.1 Definition of dihedral angles and substituents on O-methyl ethane (1) and model compounds used. ...................................................................................144
Scheme 7.2 Model compounds used to mimic transglycosidic linkage. ........................146

xvi
TABLES
Table 2.1 J-Couplings in 13 Calculated In Vacuo and in Water.....................................55
Table 3.1 J-couplings (IN Hz) involving THE hydroxyl PROTONS in methyl -D-glucoPYRANOSIDE and methyl -D-glucoPYRANOSIDE in DMSO-d6 at 25 °C ......................................................................................................................69
Table 3.2 Calculated Rotamer Populations (+ 0.10) for methyl glucopyranosides in DMSO-d6 ..........................................................................................................72
Table 3.3 C-O rotational Populations based on solvated Amber and Charmm Molecular dynamics simulations of methyl - and -D-glucopyranosides ..........................74
Table 3.4 Experimental 3JHCOH and 3JCCOH spin-coupling constantsa in H2O/Acetone-d6 solutionb of 1-6 ......................................................................76
Table 4.1 Calculated 1JCH and C-H bond length in methanol and methoxide anion ...102
Table 5.1 13C Chemical shifts and 13C-13C spin-spin coupling constants in 4/4’ .......117
Table 5.2 1H Chemicalshifts and 1H-1H and 13C-1H spin-spin coupling constnats in 4/4’ ..................................................................................................................119
Table 5.3 1H Chemicalshifts and 1H-1H spin-spin coupling constnats in 4..................121
Table 7.1 Summary of Relevant Structural Variables and Identification of Pertinent Equations to Treat Trans-glycoside 3JCOCC Values in Different O-Glycosidic Linkages ..........................................................................................................162

xvii
ACKNOWLEDGMENTS
First of all, I would l ike to thank my wif e, wh o continuous ly s upport me and
sacrifice her academic career for our family.
I want acknowledge my advis or, D r. Anthony S . Ser ianni, for his s upport, not
only f inancially, but als o per sonally. I am es pecially gr ateful to his extr eme
consideration, and his help to lead me out of my difficult times.
Another two persons I want to acknowledge are Dr. Ian Carmichael and Dr . Seth
Brown. Dr. Carmichael provided a lot of help on my dissertation throughout my stay at
Notre Dame. He is mor e like my second advis or. Dr . B rown ins pired me thr ough his
enthusiastic in teaching and dedication to undergraduate education.
I would like to thank the colleagues in Serianni’s lab for their help, discussion and
collaboration. I also want to give my thanks to my committee members and all others at
University of Notre Dame who helped me.

1
CHAPTER 1:
INTRODUCTION
1.1 Overview
In this chapter, the following topics are briefly reviewed:
1. Carbohydrates and unique structural and conformational properties related to carbohydrates and oligosaccharides.
2. NMR and computational modeling as tools in conformational analysis.
3. Density functional theory (DFT) and J-coupling constants calculations.
Then the challenges of conf ormational anal ysis and the aims of this thes is are
introduced.
1.2 Carbohydrates
Carbohydrates ar e polyhydr oxyaldehydes or polyhydroxyketones or their
derivatives. They constitute a la rge and var ied class of organic compounds consisting of
carbon, hydrogen, and oxygen in the ratio of n-2n-n, respectively. They can be f ound in
all living systems, from plants to humans. Probably the most well known carbohydrate is
the structurally simple polysaccharide cellulose ( -(1-4)-linked glucopyranose), which is
also the most abundant biomolecule in nature.
Most of earlier studies of carbohydrates were centered on plant carbohydrate such
as cellulose, starch, pectins, etc., largely due to their wide range of industrial and natural
abundance. M etabolism of car bohydrates is anot her active f ield in ear ly days . C urrent

2
interests of chemis ts and biologis ts, however , ar e f ocused on s accharides having
biological pr operties or f unctions. Once thought as a s upporting c ast, the r ole of
carbohydrates in biological ev ents ha s be en r ecognized [1-4] and glycobiology has
emerged as a new and challenging research area at the interface of biology and chemistry.
Oligosaccharides and glyco conjugates, i. e., glycolipids , glycopr oteins, and
glycosaminoglycans pr esent at the c ell s urface and dis play diver sity in glycos ylation
pattern between s pecies. [5] They play cr itical roles in biological metaboli sm, including
protein folding, stability and processing, cell adhesion, growth and differentiation, signal
transduction, s ubcellular localization, anti -inflammatory and anticoagulant pr operties,
angiogenesis, and protein/enzyme functions. [6] This large variety of properties originates
from the structural arrangement of carbohydrates.
1.3 Structural varieties of carbohydrates
Carbohydrate can f orm linear polymer ic f orms, like the pr evious mentioned
cellulose, but als o ver y compl ex br anched s tructures or conjugates with other type of
molecules, mos t impor tantly, pr oteins. I n glycoconjugates , s uch as glycopr oteins, the
saccharide part can be very small (consisting of even a single monosaccharide unit), or up
to conjugates where the saccharide part is larger than the protein structure itself.
Multiple hydr oxyl gr oups, which have dif ferent conf igurations and potential
derivatives, provide much mo re building b locks for carbohydrates, contrasted to only 4
building blocks f or DNA and/or RNA, and 20 for pr oteins. Bes ides, the structure of
monosaccharides, pyr anoses or f uranoses, allows cr eation of linkages in var ious
positions. F or example, hexopyr anoses c an be l inked with other monos accharides in

3
positions 1, 2, 3, 4, and 6 ( Figure 1. 1), thus a lar ge number of pos sible gly cosidic
linkages (one or more linkage s for one mono saccharide r esidue) can b e formed among
monosaccharide unites . For example, the number of all pos sible linear and br anched
isomers of a hexas accharides exceed s 10 12. [7] The ability to c reate complex branched
forms and the d ifferent linkage types is unique structural property of carbohydrates with
respect to peptides and nucleic acids, and consequently, result in their functional diversity
in biological systems.
O
H
HO
H
HO
H
H
OHHOH
OH
Figure 1.1 An example of monosaccharides in 4C1 chair form
1.4 Conformational analysis of oligosaccharides
Conformation is another f eature th at ha s the substantial inf luence on biological
activities. Three-dimensional structures of carboh ydrates are complicated and originates
from their complex cons titution. In order to f ully des cribe their three -dimensional
structures and dynamics in s olution, s everal conformational domains mus t be def ined.
These domains , which include c onformation of the cons tituent pyr anosyl or f unosyl
rings, O-glycoside linkage conformation, hydroxymethyl conformation, and exocyclic C-
O bond conformation (Figure 1.2), [8, 9] are expected to be inter- dependent and to exhibit
varied degrees of flexibility.

4
O
O
HO
OH
O
OH
O
HO
OH
OH
OH
a
b c
c d
Figure 1. 2 Conf ormational do mains of Oligos accharides. Key: ( a) ring conformation; (b) O -glycoside conf ormation; ( c) hydr oxymethyl gr oup conformation; (d) exocyclic C-O bond conformation.
1.4.1 Conformations within oligosaccharides
As carbohydrates are nearly exclusively present in the ring forms, stereo-chemical
arrangement of carbohydrates is affected by ring puckering. [10] Pyranoses adopt the 4C1
(Figure 1.1) or 1C4 chair forms in most cases whereas other conformations are relatively
rare. Large s ubstituents, however , can have a pr onounced inf luence on the ring
conformation and the boat o r the skew conformers can present in substituted derivatives.
Furanose f orms ar e mor e flexible compar ing to pyranose f orms and the populations of
conformers largely depend on local s tructures. This is also valid f or deoxy der ivatives,
such a s 2- deoxy- -D-ribose, which is the s ugar component of DNA s tructure.
Furthermore, both D and L forms exist in oligo - and polysaccharides, though D form is
more abundant naturally. The L form is typical for some residues, e.g. L-fucose in blood
group oligosaccharides or L-iduronic acid in glycosaminoglycan compounds.
Probably the mos t complicated, and the mos t dis cussed, is conf ormation at t he
glycosidic linkages. The mutual orientation of neighboring saccharide unites is described
by the torsion angles ( = O5’-C1’-O1’-Cx ) and ( = C1’-O1’-Cx-Cx-1 ) (Figure 1.2).
The orientation defined by H ( = H1’-C1’-O1’-Cx ) and H ( = C1’-O1’-Cx-Hx ) is also

5
often presented in literature. [11] The , angles can substantially affect the overall shape
of oligo- and polys accharides, therefore, the dete rmination of their values is one of the
key elements analyzed when dealing with 3D carbohydrate structure. This also comprises
an ans wer for the qu estion wheth er s ingle or multiple conformers are pres ent at th e
glycosidic linkages. The above-mentioned phenomenon has been thoroughly studied and
discussed in terms of “flexibility” and “rigidity” and nume rous theoretical and
experimental studies [4, 12, 13] have been devoted to solve this dilemma.
Conformation of the hydr oxymethyl gr oup is de scribed by the tor sion angle
(O5-C5-C6-O6). Three stable conformers at the C5-C6 linkage are designated as gauche-
gauche, or gg ( = - 60), gauche-trans, or gt ( = 60), and trans-gauche, or tg ( = 180).
The first symbol refers to the tor sion angle O5-C5-C6-O6, and the s econd to C4-C5-C6-
O6 ( Figure 1. 3). T he pr eference of thes e c onformers depends on the type of
monosaccharide and substituents. For example, the gauche ef fect, which s tates the non -
bonded interaction between the bonding or bital of ring oxygen O5 with the anti- bonding
orbital of one of the C 6-Hs or vic e versa, influe nces the conformational equilibrium a t
this linkage. [9, 1 2] As a result, gt and gg ar e pr eferred conf ormations over tg in
hexopyranoses, s uch as gluco se or mannos e. Hydr oxymethyl gr oup conf ormation
modulates the hydr ogen bonding char acteristics of oligos accharides ( both intr a- and
intermolecular) and the dipole moment o f the mo lecule ( as does C-O rotation), both of
which will affect overall physical and chemical properties. [14, 15]

6
C4
H5
gggauche-gauche = 60°
C4
H5
gtgauche-trans = 60°
O6H6S H6R H6S
O5
O6
O5
H6R
C4
H5
tgtrans-gauche = 180°
O6
H6S
O5
H6R
Figure 1. 3 I dealized R otamers about th e C 5-C6 bond of Aldohexopyranosyl Rings
More subtle C-O rotations, including exo-cyclic C-O conformations, are normally
ignored, at leas t for molecules in solution, but these conformations are expected to pla y
important roles in dictating structure and, presumably, reactivity in the bond state, where
C-O bond r otations ar e either f ixed or highly co nstrained, ther eby enhancing lone -pair
effects on C-H and C-C bond lengths. [16-19]
1.4.2 NMR and conformational analysis of carbohydrate
For s mall oligos accharide molecule s, the di fference in ener gy between
conformers is typically s mall. T hey ther efore un dergo f ast inter conversion in s olution.
The f ast inter conversion of rotamers in solution means individual lif etimes in the
picosecond range. Due to the short period, rotamers are inseparable, virtually coexisting
species. [20]
Nuclear M agnetic R esonance ( NMR) exper iments have alway s played a major
role in the ar ea of oligosaccharide conformational analysis. [20-36] The greatest advantage
of NM R is its ability to yield s tructural da ta in aqueous s olution, and to pe rmit the

7
analysis of the dynamic pr operties of glycan chains. His torically, s tructural s tudies of
biomolecules have been r elying on NOE ef fects. However , the pr oton dens ity of
carbohydrates is r elatively s mall comparing to pr oteins. As a r esult, NOE s i n
oligosaccharides ar e ver y limited. B esides, the N OEs that can be obs erved ar e mainly
intra-residue NOEs that do not supply information about the conformations on glycosidic
linkages, which determine the molecular flexibility. Due to the non -linear dependence of
NOEs and the av eraging ef fect r esulted f rom hi gh f lexibility of oligos accharides, it is
very challenging to interpret NO Es. [30, 3 1] T hough r esidual dipolar couplings ( RDCs)
showed pr omising r esults r ecently, [32-36] J-coupling cons tants [14-19] are s till the majo r
NMR parameters used in oligosaccharides conformational study,
In 1958, Pople published his NMR study on substituted ethane and established the
fact that NM R parameters, such as chemical shifts and s pin-spin coupling con stants, are
weighted average values of staggered rotamers. [37] The application of NMR spectroscopy
in s tereochemical analys is began with the pion eering wor ks of Kar plus, [38-40] who
described the relationship between 3-bond coupling constants (3J) and the dihedral angle
( ) between the two C-H vectors in a HCCH fragment as follows:
3JHH = A cos2 + B cos + C 1.1
where A, B and C are adjustable empirical parameters.
Following that clas sical equation, Karp lus-type equations f or other coupling
pathways were der ived. [41 - 48 ] Although attemp ts have been made to elucidate r otamer
populations from other types of measurements, there is no doubt that today this is the best
established, most reliable, and most often used methodology for this purpose. [49, 50]

8
1.4.3 Discrete model and conformational analysis in oligosaccharides
Most deter minations of r otamer populations ar e based on the as sumption that a
limited number of dis crete, low -energy conformers of a flexible molecule cov er the
whole conformational space. This is called “d iscrete” model of rotamer analysis. In six-
member ring, the r otation around the c entral bond (C-C or C-O bond, depending on the
rotamer s tudied) res ults in th ree s taggered conformers. Des ignation of the conformers
may appear differently in the literature for different rotamers. For example, gg (gauche-
gauche), gt ( gauche-trans), and tg (trans-gauche) wer e us ed in hydr oxymethyl gr oup
conformation in s accharides (Figure 1.3). [15, 51, 52] t (trans-, = 180°), g+ (gauche, =
60°) and g- (gauche, = - 60°), where is the d ihedral angle of coupled spins (Figure
1.4), ar e mor e commonly us ed in other r otamers. Since tr ansglycosidic linkage an d
hydroxyl groups will be f ocused rather than hydroxyl methyl group, t, g+ and g- will be
used in the following discussion.
Because of rapid interconversion of the rotamers, the obs erved vicinal couplings
are the weighted aver age of the individual cou plings of conf ormers, [37] als o called
limiting coupling, where weighting f actors are the appropriate mole f ractions or rotamer
populations (P). By assuming three dis tinct conformers around a s ingle bond, only two
observed couplings are needed to obtain rotamer populations.
3J1 = P1, t J1, t + P1, g+ J1, g+ + P1, g- J1, g- 1. 2
3J2 = P2, t J2, t + P2, g+ J2, g+ + P2, g- J2, g- 1. 3
Pt + Pg+ + Pg- = 1 1.4

9
Here Jt, Jg+ and Jg- are limiting couplings at per fect s taggered conformations t,
g+ and g- r espectively. Pt, Pg+ and Pg- ar e the r otamer populations of these staggered
conformations. Subscript 1 and 2 indicate two co upling pathways centering on the same
bond of interest.
g gauche = 60°
B
t trans = 180°
B
A A
B
g+ gauche = 60°
A
Figure 1. 4 t, g+ and g- s taggered structure f or A- X-X-B. A and B an d other atoms involved var y accor ding to di fferent conf ormational domain studied.
1.4.4 Obtaining 3JHH and 3JCH vicinal coupling constants
To determine the r otamer populations of either C-C or C-O bond, two obs erved
coupling constants and six limiting coupling constants are needed, according to equations
1.2, 1.3 and 1.4. Determination of observed vicinal coupling constants can be achieved by
a variety of one-dimensional (1D) and two -dimensional (2D) NMR techniques. [53, 54] In
most cases, however, 1D techniques are sufficient, especially for small oligosaccharides.
The obs erved couplings s hould be meas ured as pr ecisely as possible and s ometimes
spectrum s imulation is neces sary to extr act the r eal coupling values f rom the
experimental spectrum.

10
Traditional as sessments of oligos accharide conf ormational domains by N MR
have r elied heavily on 3JHH ( for r ing conf ormations) and 1H-1H NOE ( for linkage
geometry), but thes e parameters are not witho ut the ir limitations . For example, O-
glycosidic linkage conf ormation cannot be as sessed via 3JHH, and inter- residue 1H-1H
NOEs are frequently few in number and their interpretation complicated by the pr esence
of significant resonance overlap that precludes reliable NOE measurements and/or by the
presence of conformational averaging. [55] In this case, carbon based vicinal couplings has
to be obtained to accurately analyze the rotamer populations.
Obtaining car bon-based 3J, however , is not a trivial tas k. The intr oduction of
stable is otopes into s accharides of fers oppor tunities to mea sure the se coupling s
accurately using simple 1D techniques and thus addresses the problems described above.
13C-enrichment facilitate s the meas urement of trans-glycoside 2JCOC, 3JCOCH, a nd
3JCOCC value s. I n addition, 13C-enrichment enables the meas urement of JCH and JCC
values within f uranosyl and pyr anosyl rings. [19, 56-59] Being considerably more abundant
than 3JHH, thes e car bon-based J-couplings can be u seful when inv estigating
conformationally flexible structures. [17, 56]
Currently, site-selected 13C enrichment method is generally used. There are a few
advantages to us e s ite-selected lab eling ov er enr ichment of all car bons in
oligosaccharides. First of all, since oligosaccharides cannot be expressed in a lar ge scale
as pr otein, their s ynthesis r elies on ch emical methods . L abeling all car bons will be
extremely expens ive. Second, s ince thos e car bons ar e coupling with r ing pr otons in
saccharides through 2-bonds and/or 3-bonds, too many 13C will significantly complicate
the 1H-spectra and make it even mor e dif ficult to interpre t. T hird, ring protons for

11
oligosaccharides gener ally r eside in a ver y nar row r ange in NM R s pectra, and s pectra
overlapping can be a big problem. Site-selected 13C enrichment can split the proton on
the enriched car bon and s eparate the s ignals f rom other p rotons, thus help p roton
assignment and/or obtaining accurate coupling constants.
1.4.5 Carbohydrate modeling and conformational analysis
Solution NMR data represent only the average molecular structure over the time
course of the NM R exper iment, and for f lexible molecules it is extremely di fficult to
decompose the data into contr ibutions f rom the individual conf ormations. I t is in this
situation that modeling methods are often used to complement the NMR data. [60, 61] Two
approaches ar e f requently applied in car bohydrate modeling: M onte C arlo ( MC)
sampling, and Molecular Dynamics (MD) simulation.
Monte Carlo (MC) methods [62] generate configurations by r andomly per turbing
the geometr ies of inter est. T he acceptan ce of the new conf iguration will f ollow a
Metropolis procedure [63] to ensure Boltzmann distribution. The ability to accept higher
energy configura tion allows M C method s to cl imb uphill and es cape from a local
minimum. So ideally M C methods can s ample all energy minimum. However, since the
new configuration is randomly generated, MC methods are non-deterministic.
Molecular Dynamics (MD) methods, [64] on the other hand, are deterministic. New
configuration is generated by propagating a s tarting set of coordinates and velocities (or
momentum) by a s eries of finite time s teps. These time-correlated steps/points are called
trajectory. However , when the ener gy of new conf iguration in M D methods increase, a
force will be generated to p ull the system back. As a res ult, it can eas ily trapped ne ar a
local energy minimum and not able to sample all possibilities.

12
1.5 Density Functional Theory and J-coupling Calculations
As s tated in 1. 4.4, enr ichment of 13C widened the availability of vicinal C -H
coupling cons tants. Unf ortunately, is otopically la beled oligos accharides ar e not r eadily
available and ther efore, many heter onuclear NMR experiments are not applicable to the
study of oligosaccharides. Although a lot of efforts have been made towards synthesis of
isotopically labele d oligos accharides, s tructural and/or conf ormational s tudies of
oligosaccharides have to rely heavily on computational methods.
Another big challenge is to obtai n limi ting coup ling cons tants, both trans and
gauche, which are needed to elicit rotamer populations from equations 1.2, 1.3 and 1.4. In
order to der ive limiting coupli ng constants, the r otamer should exist overwhelmingly in
one conformation, either gauche or trans. Model compounds with special constraint have
to be s ynthesized f or this pu rpose. C urrently th e mos t power ful method to obtaining
limiting coupling cons tants is the us e of appropriate Karplus-like equations. Due to lack
of exper imental 3Js in oligos accharides to de rive accur ate Kar plus-like equations ,
computational method s, es pecially Dens ity Functional T heory (DF T), are impo rtant
sources of obtaining Karplus-like equations, and hence limiting coupling cons tants. Here
DFT theory and their applications in J-coupling cons tants c alculations ar e br iefly
reviewed.
1.5.1 Introduction to Density Functional Theory
Density functional theory is based on th e proof by Hohenberg and Kohn that the
electronic ener gy is deter mined completely by t he electr on dens ity. [65] Or , in another
word, there is a one -on-one connection between electron density and ener gy. Since the
electron dens ity only depends on thr ee coor dinates, but not the number of electr ons, it

13
will significantly simplify energy calculations. Finding suitable functionals to connect the
electron density and the energy became the primary task for the next a few decades.
The foundation for the us e of DFT methods in c omputational chemistry was the
Kohn and Sham For malism, [66] which is splitting the kinetic energy functional into two
parts, one that can be calculated exactly f rom S later deter minant, TS and a s mall
correction term, or kinetic cor relation energy Tcorr (equation 1.6). The potential ener gy,
on the other hand, is divided into 3 par ts: attr action between nuclei and electr ons Vne,
repulsion between electr ons Vee, and nuclear- nuclear r epulsion Vnn. However , the
nuclear-nuclear r epulsion is a cons tant in the B orn-Oppenheimer appr oximation, and
generally dropped out of consideration (equation 1.7). Similar to Hartree-Fock approach,
the electron-electron repulsion is further divided into a Coulomb part VC and an exchange
part Vxc. The exchange part also includes all terms of correlation potential energies. [67-69]
E = T + V 1.5
T = TS + Tcorr 1.6
V = Vne + Vee = Vne + VC + Vxc 1. 7
EDFT ( ) = TS( ) +Vne( ) + VC( ) + Exc( ) 1. 8
In above equations, T represents kinetic energy and V represents potential energy
whereas is the electron density. Equation 1.8 is a gener al DFT energy expression. TS,
Vne and VC terms can be calculated r elatively easily and accurately, and all exchange and
correlation energies are summarized into an exchange-correlation term Exc.
Exc ( ) = Tcorr ( ) + Vxc ( ) 1. 9

14
Now the new ta sk f or DFT methods is to der ive s uitable f unctionals f or the
exchange-correction term. Some of the early efforts focused on exchange part of equation
1.10, whereas others focused on the cor relation part, though the exchange ener gy is the
largest contributor to Exc term. The difference between DFT methods is the choice of the
functional form of the exchange-correlation energy.
1.5.2 LSDA, GGA and hybrid DFT methods
A lot of DFT functionals have been proposed so far. To judge which functional is
better, however, will have to rely on comparing the perfo rmance with experiments or
high-level wave mechanics calculations. In general there are three classes of functionals:
local dens ity method s, gr adient cor rected metho ds and hybr id methods . I n the L ocal
Density Appr oximation ( LDA) and/or Local Spin Dens ity Appr oximation ( LSDA), the
electrons ar e tr eated as a unif orm electr on gas, in which electr on dens ity is as sumed
localized. However , s implicity of the fundamental as sumption in L SDA appr oximation
underestimates the exchange ener gy, but s imultaneously over estimates the cor relation
energy. For example, bond strengths are generally overestimated. [70]
Generalized Gradient Approximation (GGA) can be considered as corrections to
LSDA methods . Non- uniform electr on gas is used ins tead, and the ex change and
correlation energies depend not only on the el ectron density, but als o on der ivatives of
the dens ity. Per dew and Wang ( PW86) [71] pr oposed the f irst G GA f unctional by
introducing a dimens ionless gr adient var iable in to L SDA exchange ter m. T here have
been var ious gr adient cor rected f unctional f orms pr oposed f or the cor relation ener gy
since then, and the most popular is due to Lee, Yang and Parr (LYP). [72]

15
Without wave f unctions, however , DFT methods inher ently have pr oblems
treating exchange energies, which account f or ~ 90% of exchange-correlation term. [67-69]
Currently, hybr id methods ar e mor e widely us ed in DFT calculatio ns by intr oducing
Hartree-Fock calculations to tr eat exchange energy. Hybrid methods used both Har tree-
Fock calculations and DFT calculations for exchange-correlation term. The most known
hybrid method is Becke 3 parameter functional (B3): [73]
ExcB3 = (1 – a ) ExLSDA + a ExHF + b ExB88 + EcorrLSDA + c EcorrGGA 1. 11
The parameters a, b , and c are determined by fitting to experimental data and depend
on the form chosen for EcorrGGA.
The specification of a DFT method r equires selection of a s uitable form for the
exchange and cor relation ener gies. T ypically ei ther B 88 or B 3 hybr id ar e us ed f or
exchange and either the L YP or PW91 for correlation. Associated acronyms are BLYP,
BPW91, B3LYP and/or B3PW91.
1.5.3 Advantages and disadvantages of DFT methods
The s trength of DF T is that its formalism is exact yet efficient, wi th one
determinant des cribing the electr on dens ity, though it s till needs multi -dimensional
orbitals to get accurate results. All of the complexity is hidden in one term, the exchange-
correlation functional. With a computational cost similar to HF theory, DFT methods can
sometimes obtain s imilar or mor e accur ate r esults as of high-level quantum mechanic s
calculations. For example, hybr id methods like B 3PW91 per form almost as well as the
elaborate G2 model in some enthalpy calculations. [74]

16
DFT methods in s ome c ases have it s clear adv antages over quantum mech anics
methods. In general, GGA methods give geometries and vibrational frequencies for stable
molecules of the s ame or better quality than MP2 (Møller-Plesset perturbation theory ).
However, f or s ystems containing multi- reference char acter, wher e M P2 us ually f ails,
DFT methods can still be used and generate results with reasonable quality. [75]
Despite the wides pread popular ity and s uccess, DF T met hods have its own
limitations, and their applications sometimes suffer from large pervasive errors that cause
qualitative failures in predicted properties. For example, DFT methods are not well suited
for excited s tates of the s ame s ymmetry a s the gr ound s tate bec ause, without wav e
functions, it is di fficult to ens ure excited s tate or thogonal to gr ound s tate. C urrent
functionals also describe weak interactions poorly, especially when di spersion forces are
significant. [76] This is inherited from K ohn-Sham formalism that is widely used in DFT.
The f ailures, however , ar e not br eakdowns of the theor y its elf but ar e only due t o
deficiencies of the appr oximate exchange-correlation f unctionals cur rently us ed. T he
failures ar e r eviewed and s ummarized into tw o sources. T he firs t one is delocalization
error, due to the dominating C oulomb term that pushes electr ons ap art. T his er ror was
understood as a per spective of f ractional char ges. T he s econd one i s static cor relation
error, which was understood in perspective of fractional spins. [77]
1.5.4 Perturbation and J-coupling constants
Most molecular properties, such as indirect nuclear-nuclear coupling (J-coupling)
as dis cussed her e, may be def ined as the r esponse of a w ave f unction, an ener gy or
expectation value of an operator to a perturbation. For perturbation strength , the energy
can be expanded in a Taylor series. [67]

17
E( ) = E(0) +E
+1
2
2E2
2+
1
6
3E3
3+K 1. 11
where E(0) is the energy when perturbation is absent. Any nuclear spin I will result in an
internal magnetic moment M:
M = hI 1. 12
As a result of M as the perturbation, equation 1.12 becomes:
E(M1, M2,L) = E(0) +E
M1
M1 +1
2
2E
M1 M2
M1M2 +L 1. 13
The second der ivative with r espect to two dif ferent nuclear spins is the r educed
NMR coupling cons tant K12, and as shown in the clas sical papers by Pople et. al. [78-80],
the disappears since only distinct pairs of nuclei will be considered:
K12 =2E
M1 M2
1. 14
The normal nuclear indirect spin-spin coupling constant between two nuclei 1 and
2, J12 will be:
J12 = h 1
22
2K12 = h 1
22
2
2E
M1M2
1. 15
When magnetic f ields ar e involved, the Hamiltonia oper ator can be ver y
complicated, and not intended to r eview her e. F our mechanis ms, Fer mi-contact ( FC),
spin-dipole ( SD), diamagnetic s pin-orbit ( DSO) and par amagnetic s pin-orbit (PSO)
respectively, ar e int roduced in Rams ey’s expr ession f or the r educed spin-spin coupling
constants: [81]

18
K12 = 0 h12DSO
0 + 20 h1
PSOs s h2
PSO0
T
E0 Ess>0
+ 20 h1
FC+ h1
SDt t h2
FC+ h2
SD0
T
E0 Ett
1.16
where the f irst summation is over all s inglet states different from the r eference state and
the s econd s ummation over all tr iplet s tates. hDSO, hPSO, hFC, a nd hSD ar e the
Hamiltonia operators for DSO, PSO, FC, and SD terms as defined below: [67]
hDSO=
gAgBμN2
2c 4 IA
(riAt riB riB riA
t )
riA3 riB
3 IB 1. 17
hPSO=μN
mc 2 gA
IA (riA pi)
riA3
A=1
Nn
i=1
Ne
1. 18
hFC=
8 gegAμBμN
3c 2 (riA )(si IA ) 1. 19
hSD=
gegAμBμN
c 2 si
riAt riA 3riAriA
t
riA5
IA 1. 20
In many cas es, the F ermi-contact ( FC) cont ributions dominate the s pin-spin
couplings, with pr obably the only exc eption for f luorine-based couplings. Paramagnetic
spin-orbit (PSO) contributions generally rank the second, followed by diamagnetic s pin-
orbit (DSO). These two terms are magnetic interaction through space between the nuclear
magnetic moments (spin) and th e orbital motion (orbit) of electrons. However, PSO and
DSO generally have dif ferent signs and they nearly cancel e ach other. Spin-dipole (SD)
represents the clas sical magnetic dipole-dipole interaction between electr ons and nuclei.
Generally, non-Fermi-contact contributions are very small. [82] As a result, early spin-spin
coupling calculations only included Fer mi-contact term, while ignor ing PSO, DSO and
SD terms. [83]

19
1.5.5 DFT computations of J-coupling constants
Although R amsey’s expr ession ( equation 1. 16) clearly expr esses the dif ferent
mechanisms that contribute to the s pin-spin coupling constants, it is not us eful for DFT
calculations, s imply becaus e it r equires a s ummation of the f ull s et of excited s tates,
which are not accessible in DFT.
Using f inite per turbation theor y ( FPT), Pople did the pioneer wor k [78-80] in
theoretical calculations of s pin-spin coupling constants. I t is f urther impr oved by
Kowalewski et. al. [84] and the following equation is obtained:
K12 =[E( 1, 2) E( 1, 2)]
2 1 2
1. 18
Here 1, 2 are finite (arti ficially large) values of nuclear magnetic moments , typica lly
104 – 105 times larger than the real ones.
Traditional Hartree-Folk (or post-Hartree-Fock) methods were first used in finite
perturbation calculations of s pin-spin coup ling cons tants. L ater, it was f ound that,
coupling DFT and FP T ( DFT-FPT) could p rovide even mor e pr omising r esults. [85- 8 8]
Some variations recently developed include SOS-DFPT (Sum-over-states Density Finite
Perturbation Theory) [89] and CPDTF (Coupled perturbation DFT) [90-93]. All these works
led to including of s pin-spin coupling cons tants calculations in s ome commerc ially
available programs, notably, Gaussian.
1.6 Some challenges of carbohydrate conformational study and aims of this thesis
Interaction of car bohydrates with pr oteins s uch as enzym es, antibodies and
lectins, is a topic of major interes t. [94] Besides possibilities to form covalent bond, there

20
are thr ee dif ferent mechanis ms that car bohydrates can bind to pr otein r eceptors and
enzymes. T he f irst one is thr ough hydr ogen b onding between hydr oxyl gr oups in
carbohydrates and polar groups in protein.[95, 96] The second very common one is through
stacking of hydr ophobic s ugar f aces againt ar omatic ( CH/ interaction) and aliphatic
amino acid side chains. [97 - 100] In some cases, cations such as Ca2+ and Mg2+ can work as
salt bridges to connect carbohydrates and proteins. [101] Occasionally water molecules can
be used to mediate carbohydrate-protein recognition. [102]
Figure 1. 5 Schematic r epresentation of the back bone ( left), s howing the binding s ite of hevein domains f or chito -oligosaccharides. T he s ite is rather s olvent expos ed. T he existence of thr ee ar omatic r esidues that provide inter actions to the s ugar is highlighted ( right). ( Figure f rom r ef. 94)
Studies on the recognition of carbohydrate and proteins or protein analogues have
shown that dis tortion of normal conformation of saccharide is very common. [103 – 1 05] It
can be either r ing dis tortion o r abnor mal tr ansglycosidic linkage conf ormations. These
distortions can be a r esult of car bohydrate-protein binding. T here is an alter natively
understanding, however , that conf ormational ch ange may f acilitate the binding and
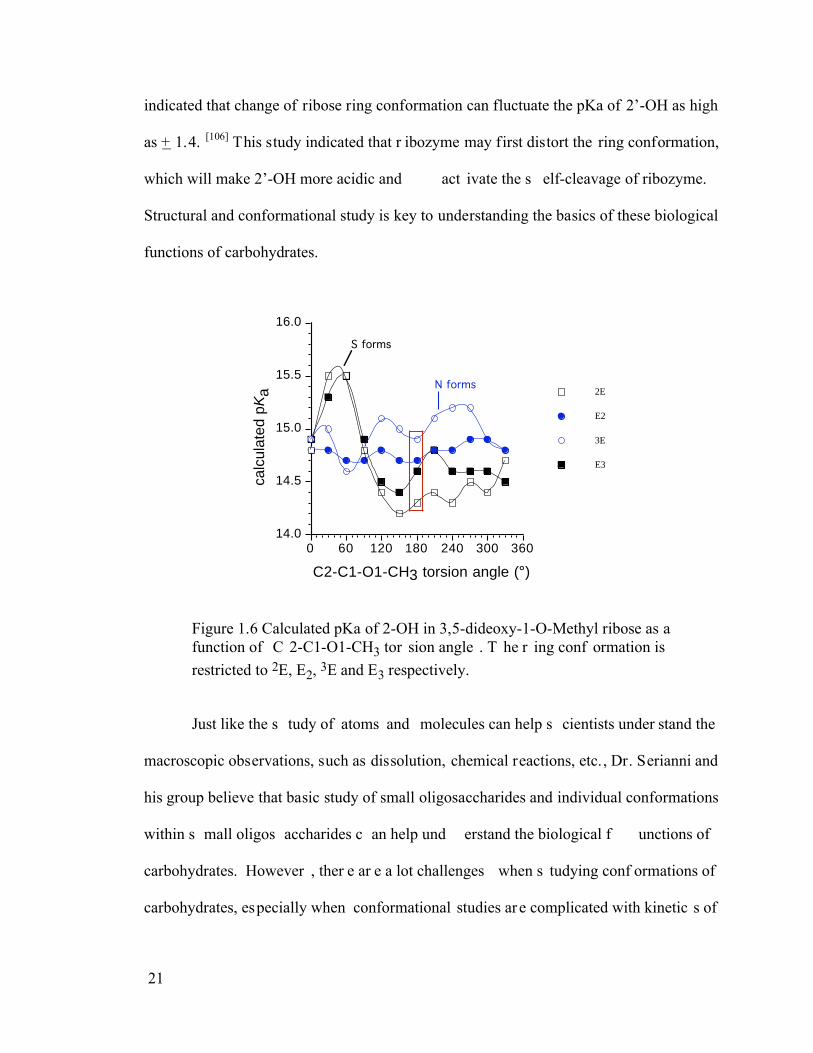
biological f unctions. F or ex ample, a r ecent s tudy in S erianni’s gr oup ( figure 1. 6)
21
indicated that change of ribose ring conformation can fluctuate the pKa of 2’-OH as high
as + 1.4. [106] This study indicated that r ibozyme may first distort the ring conformation,
which will make 2’-OH more acidic and act ivate the s elf-cleavage of ribozyme.
Structural and conformational study is key to understanding the basics of these biological
functions of carbohydrates.
14.0
14.5
15.0
15.5
16.0
calc
ulat
ed p
Ka
0 60 120 180 240 300 360
C2-C1-O1-CH3 torsion angle (°)
E3
3E
E2
2E
S forms
N forms
Figure 1.6 Calculated pKa of 2-OH in 3,5-dideoxy-1-O-Methyl ribose as a function of C 2-C1-O1-CH3 tor sion angle . T he r ing conf ormation is restricted to 2E, E2, 3E and E3 respectively.
Just like the s tudy of atoms and molecules can help s cientists under stand the
macroscopic observations, such as dissolution, chemical reactions, etc., Dr. Serianni and
his group believe that basic study of small oligosaccharides and individual conformations
within s mall oligos accharides c an help und erstand the biological f unctions of
carbohydrates. However , ther e ar e a lot challenges when s tudying conf ormations of
carbohydrates, especially when conformational studies are complicated with kinetic s of

22
carbohydrates. A f ew of these challenges are lis ted as follows and will be add ressed in
this thesis.
1.6.1 Collection and interpretation of J-coupling constants of hydroxyl protons
Despite their s tructural diver sity, all car bohydrates car ry a common f unctional
group, the hydr oxyl gr oup (-OH). E ven thou gh many hydr oxyl gr oups exis t in
carbohydrates, it can be stated that th ey have not been at the core of exper imental an d
theoretical exercises. However, the high density of hydroxyl groups in these carbohydrate
structures confers uni que chemical and phy sical properties due to their ability to form
hydrogen bond intr a- and inter molecularly, notably in the latter case with s olvent water.
Recent studies suggest that hydr ogen bonding may dictate the conf ormations of
oligosaccharids. [107 – 110]
The detection of hydroxyl protons is an immediate challenge, primarily due to the
fast exchange between hydr oxyl pr otons and bul k s olvent. T he cla ssic appr oach i s to
eliminate the pr oblem of exchange between hydr oxyl pr otons and the bulk s olvent b y
using aprotic s olvent. With dimethyl s ulfoxide (DM SO) as a suitable s olvent for
carbohydrates, hydroxyl proton resonances were recorded as sharp peaks. [111] Along with
the benefits us ing aprotic s olvent, there are ar guments that the conf ormations of
carbohydrates may be alter ed f rom the r eal co nformations in aqueous s olution. An
alternative appr oach is to us e s uper cooled aqu eous s olution, which can significantly
reduces the exchange r ate between hydr oxyl gr oups and s olvent. [112] T o accommodate
the drastic decrease in temperature, mixed solvent of acetone-d6 and water is commonly
used. [113] Other techniques, such as water gate, are also used to s uppress the exchang e

23
rates. Absence of traces of ionic contamination and adjustment to a neutral pH value are
further adjustable parameters to minimize the exchange rate.1
Generally three vicinal coupling constants are available for each OH groups, one
3JHCOH and two 3JCCOH (Figure 1.1). [114] Following the above-mentioned techniques,
these coupling constants are relatively easy to obtain, given the availability of selectively
13C labeled oligosaccharides, which is an ongoing task in Serianni group.
Karplus equations for 3JHCOH based on experimental and computational
approaches have been reported using small, non-carbohydrate molecules such as
methanol as model compounds. [41-43] 3JCCOH values have been used qualitatively to
determine C-O torsion angles, [115, 116] although little is known about their dependence on
saccharide structure. Parameterization of 3JHCOH and 3JCCOH is important in hydroxyl
group conformational analysis, which can provide potential useful information, such as
hydrogen bonding in carbohydrates.
1.6.2 Substituent effect on trans-glycoside vicinal couplings and Karplus equations
Since the discovery of the dihedral angle dependence of vicinal proton-proton
coupling constants,[38-40] many factors were proven to affect coupling constants, including
electronegativity of substituents, orientation of substituents relative to the coupled nuclei,
bond lengths, and bond angles. Recent Karplus-type relationships took these factors into
account and some generalized Karplus equations were proposed.[117-126]
1 The exceptions include hydroxyl methyl OH, in which two 3JHCOH and one 3JCCOH are
available, and 1-OH at the reducing end of oligosaccharides, in which only one 3JHCOH and one 3JCCOH are available. Generally there is only one reducing end per oligosaccharides. Hydroxyl methyl groups are studied exclusively elsewhere and are not a focus of this thesis.

24
Though including di fferent f actors can s ignificantly impr ove the accur acy of
Karplus equation, it als o limits the applicati ons. The oppos ite tr end is to s implify the
analysis. For example, in the analy sis of tr ansglycoside linkage conf ormation, one
assumption is widely used that the limiting coupli ng values belonging to = + 60° (two
gauche structures) are of the s ame value. This assumption is applied to parameter ization
of 3JCOCC and 3JCOCH involving the linkage, r esulting in s ymmetrical Kar plus-like
curves around 180°. [59, 127] This assumption bears several shortcomings, one of which i s
that it ignores the spatial orientation of various gauche substituents. Since the accuracy of
the Karplus-like equations directly affect the accu racy of the con formational analysis, it
is impor tant to r evisit the s ubstituent ef fect on car bohydrate-based Kar plus-like
equations.
1.6.3 Aims of this thesis
Aim #1: Density functional theory (DFT) wil l be used to calculate 3JHCOH and
3JCCOH and Kar plus-like equ ations will be par ameterized in monos accharides model
compounds.
Aim #2: With the aid of 13C isotopic labeling, J-coupling constants involving OH
groups ( 3JHCOH, 3JCCOH and 2JCOH) of mono- and dis accharides will be meas ured in
DMSO and in mixed solvent of ac eton-d6 and water and b e inter preted us ing
parameterized Karplus-like equations.
Aim #3: Substituent effect on tr ans-glycoside vicinal coupling cons tants will be
studied and Karplus-like equa tions related to trans-glycoside linkages will be re -
parameterized.

25
1.7 References
1. Kobata, A. Eur. J. Biochem. 1992, 209, 483-501.
2. Varki, A. Glycobiology 1993, 3, 97-130.
3. Dwek, R. A. Chem. Rev. 1996, 96, 683-720.
4. Imberty, A.; Perez, S. Chem. Rev. 2000, 100, 4567-4588.
5. Gagneux, P.; Varki, A. Glycobiology 1999, 9, 747-755.
6. Brooks, S. A.; Dwek, M. V.; Schumacher, V. Functional and Molecular
Glycobiology, BIOS Scientific Publishing: Oxford, 2002.
7. Laine, R. A. Glycobiology. 1994, 4, 759-767.
8. Serianni, A. S. In Glycoconjugates: Composition, Structure and Function. Marcel-Dekker, 1992, p71.
9. Serianni, A. S. In Bioorganic Chemistry: Carbohydrates. Oxford University Press: New York, 1999, p244.
10. Stoddart, J. F. Stereochemistry of Carbohydrates. Wiley-Interscience: New York, 1971.
11. Hricovini, M. Current Medicinal Chemistry. 2004, 11, 2565-2583.
12. French, A. D.; Brady, J. W. Computer Modeling of Carbohydrate Molecules. ACS series 430: Washington DC, 1990.
13. Rao, V. S. R.; Qasba, P. K.; Balaji, P. V.; Chandrasekaran, R. Conformation of
Carbohydrates. Harwood Academic Publishers: Amsterdam, 1998.
14. Rochwell, G. D.; Grindley, T. B. J. Am. Chem. Soc. 1998, 120, 10953-10963.
15. Bock, K.; Duus, J. O. J. Carbohydr. Chem. 1994, 13, 513-543.
16. Serianni, A. S.; Wu, J.; Carmichael, I. J. Am. Chem. Soc. 1995, 117, 8645-8650.
17. Podlasek, C. A. Stripe, W. A.; Carmichael, I.; Shang, M.; Basu, B.; Serianni, A. S. J. Am. Chem. Soc. 1996, 118, 1413-1425.
18. Kennedy, J.; Wu, J.; Drew, K.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1997, 119, 8933-8945.

26
19. Cloran, F.; Zhu, Y.; Osborn, J.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 2000, 122, 6435-6448.
20. Kraszni, M.; Szakacs, Z.; Noszal, B. Anal. Bioanal. Chem. 2004, 378, 1449-1463.
21. Weimar, T.; Woods, R. J. In NMR Spectroscopy of Glycoconjugates. Wiley-VCH: Weinheim, 2003.
22. Van De Ven, F. J. M.; Hilbers, C. W. Eur. J. Biochem. 1988, 178, 1-38.
23. Chary, K. V. R.; Modi, S. FEBS Lett. 1988, 233, 319-325.
24. Radhakrishnan, I.; Patel, D. J.; Gao X. Biochemistry. 1992, 31, 2514-2523.
25. Kim, S. G.; Lin, L. J.; Reid, B. R. Biochemistry. 1992, 31, 3564-3574.
26. Salazar, M.; Fedoroff, O. Y.; Miller, J. M.; Ribeiro, N. S.; Reid, B. R. Biochemistry, 1993, 32, 4207-4215.
27. Majumdar, A.; Hosur, R. V. Prog. Nucl. Magn. Reson. Spectrosc. 1992, 24, 109-158.
28. Clore, G. M.; Gronenborn, A. M. Crit. Rev. Biochem. Mol. Biol. 1989, 24, 479-564.
29. Altona, C.; Sundaralingam, M. J. J. Am. Chem. Soc. 1973, 95, 2333-2344.
30. Noggel, J. H.; Schirmer, R. E. The Nuclear Overhauser Effect. Chemical
applications. Academic Press: New York, 1971.
31. Neuhaus D.; Williamson, M. The Nuclear Overhauser Effect in Structural and
Conformational Analysis. Wiley-VCH: New York, 2000.
32. Brochier-Salon, M. C.; Morin C. Magn. Reson. Chem. 2000, 38, 1041-1042.
33. Rundlof, T.; Eriksson, L.; Widmalm, G. Chem. Eur. J. 2001, 7, 1750-1758.
34. Kiddle, G. R.; Homans, S. W. FEBS Lett. 1998, 436, 128-130.
35. Thompson, G. S.; Shimizu, H.; Homans, S. W.; Donohue-Rolfe, A. Biochemistry. 2000, 39, 13153-13156.
36. Prestegard, J. H.; Glushka, J. In NMR Spectroscopy of Glycoconjugates. Wiley-VCH: Weinheim, 2003.
37. Pople, J. A. Mol. Phys. 1958, 1, 3-8.
38. Karplus, M. J. Chem. Phys. 1959, 30, 11-15.

27
39. Karplus, M. J. Phys. Chem. 1960, 64, 1793-1798.
40. Karplus, M. J. Am. Chem. Soc. 1963, 85, 2870-2871.
41. Fraser, R.R.; Kaufman, M.; Morand, P.; Govil, G. Can. J. Chem. 1969, 47, 403-409.
42. Fukui, H.; Baba. T.; Inomata, H.; Miura, K.; Matsuda, H. Mol. Phys. 1997, 92, 161-165.
43. Alkorta, I.; Elguero, J. Theor. Chem. Acc. 2004, 111, 31-35.
44. Vuister, G.W.; Bax, A. J. Am. Chem. Soc. 1993, 115, 7772-7777.
45. Tvaroska, I.; Gadjos, J. Carbohydr. Res. 1995, 271, 151-162.
46. Mulloy, B.; Frenkiel, T.A.; Davies, D.B.; Carbohydr. Res. 1988, 184, 39-46.
47. Tvaroska, I.; Hricovini, H.; Petrakova, E. Carbohydr. Res. 1989, 189, 359-362.
48. Hennig, M.; Bermel, W.; Schwalbe, H.; Griesinger, C. J. Am. Chem. Soc. 2000, 122, 6268-6277.
49. Chachaty, C.; Perly, B.; Langlet, G. J. Magn. Reson. 1982, 50, 125-141.
50. Spassov, S.; Stefanova, R. J. Mol. Struct. 1977, 42, 109-115.
51. Stenutz, R.; Carmichael, I.; Widmalm, G.; Serianni, A. S. J. Org. Chem. 2002, 67, 949-958.
52. Cloran, F.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 2001, 123, 4781-4791.
53. Eberstadt, M.; Gemmecker, G.; Mierke, D. F.; Kessler, H. Angew. Chem. Int.
Engl. 1995, 34, 1671-1695.
54. Braun, S.; Kalinowski, H. O.; Berger S. 150 and more basic NMR experiments. Wiley-VCH: Weinheim, 1998.
55. Homans, S. W. Prog. NMR Spectrosc. 1990. 22, 55-81.
56. Church, T.; Carmichael, I.; Serianni A. S. Carbohydr. Res. 1996, 280, 177-186.
57. Serianni, A. S.; Bondo, P. B.; Zajicek, J. J. Magn Reson. Ser. B. 1996, 112, 69-74.
58. Basu, B.; Zhao, S.; Bondo, P.; Bondo, G.; Gloran, F.; Carmichael, I.; Stenutz, R.; Hertz, B.; Serianni, A. S. J. Am. Chem. Soc. 1998, 120, 11158-11173.

28
59. Cloran, F.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1999, 121, 9843-9851.
60. Bukowski, R.; Morris, L. M.; Woods, R. J. Eur. J. Org. Chem. 2001, 2697-2705.
61. Woods, R. J.; Pathiaseril, A.; Wormald, M. R.; Edge, C. J.; Dwek, R. A. Eur. J.
Biochem. 1998, 258, 372-386.
62. Hammond, B. L.; Lester, W. A. and Reynolds, P. J. Monte Carlo Methods in Ab
Initio Quantum Chemistry. 1994, World Scientific.
63. Metroplis, N. A.; Rosenbluth, A. W.; Teller, A. H. and Teller, E. J. Chem. Phys., 1953, 21, 1087-1092.
64. Marx, D. and Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and
Adanced Methods. 2009, Cambridge University Press.
65. Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864-871.
66. Kohn, W.; Sham, L. J. Phys. Rev., 1965, 140, A1133-1138.
67. Jensen, F. Introduction to Computational Chemistry. 2nd ed, 2007, John Wiley & Sons, Ltd.
68. Cramer, C. J. Essentials of Computational Chemistry. 2002, John Wiley & Sons, Ltd.
69. Koch, W. and Holthausen, M. C. A Chemist’s Guide to Density Functional
Theory. 2000, Wiley-VCH
70. Donnerberg, H. J. Phys.: Condens. Matter. 1995, 7, L689-L694.
71. Perdew, J. P.; Wang Y. Phys. Rev. B, 1986, 33, 8800-8802.
72. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B, 1988, 37, 785-789.
73. Becke, A. D. J. Chem. Phys., 1993, 98, 5648-5652.
74. Curtiss, L. A.; Raghavachari, K.; Redfern, P. C.; Pople, J. A. J. Chem. Phys., 1997, 106, 1063-1079.
75. Oliphant, N.; Bartlett, R. J. J. Chem. Phys., 1994, 100, 6550-6561.
76. Meijer, E. J.; Sprik, M. J. Chem. Phys., 1996, 105, 8684-8689.
77. Cohen, A. J.; Mori-Sánchez, P.; Yang, W. Science, 2008, 321, 792-794.
78. Pople, J. A.; McIver Jr., J. W.; Ostlund, N. S. Chem. Phys. Lett. 1967, 1, 465-466.

29
79. Pople, J. A.; McIver Jr., J. W.; Ostlund, N. S. J. Chem. Phys. 1968, 49, 2960-2964.
80. Pople, J. A.; McIver Jr., J. W.; Ostlund, N. S. J. Chem. Phys. 1968, 49, 2965-2970.
81. Ramsey, N. F. Phys. Rev. 1953, 91, 303-307.
82. Helgaker, T.; Watson, M.; Handy, N. C. J. Chem. Phys., 2000, 113, 9402-9409.
83. Perera, S. A.; Nooijen, M.; Bartlett, R. J. J. Chem. Phys., 1996, 104, 3290-3305.
84. Kowalewski, J.; Laaksonen, A.; Roos, B.; Siegbahn, P. J. Chem. Phys., 1979, 71, 2896-2902.
85. Fukui, H. J. Chem. Phys., 1976, 65, 844-846.
86. Onak, T.; Jaballas, J.; Barfield, M. J. Am. Chem. Soc., 1999, 121, 2850-2856.
87. Francis, C.; Carmichael, I.; Serianni, A. S. J. Phys. Chem. A, 1999, 103, 3783-3795.
88. Peralta, J. E.; Barone, V.; De Azua, M. C. R.; Contreras, R. H. Molecular Phys., 2001, 99, 655-661.
89. Malkin, V. G.; Malkina, O. L.; Salahub, D. R. Chem. Phys. Lett., 1994, 221, 91-99.
90. Sychrovsky, V.; Grafenstein, J.; Cremer, D. J. Chem. Phys., 2000, 113, 3530-3547.
91. Peralta, J. E.; Barone, V.; Contreras, R. H.; Zaccari, D. G.; Snyder, J. P. J. Am.
Chem. Soc., 2001, 123, 9162-9163.
92. Barone, V.; Peralta, J. E.; Contreras, R. H.; Snyder, J. P. J. Phys. Chem. A, 2002, 106, 5607-5612.
93. Peralta, J. E.; Scuseria, G. E.; Cheeseman, J. R.; Frisch, M. J. Chem. Phys. Lett., 2003, 375, 452-458.
94. Gabius, H. J.; Siebert H. C.; André, S.; Jiménez-Barbero, J.; Rüdiger, H. Chem.
Biochem. 2004, 5, 740 – 764.
95. Wang, M. and Boddy, C. N. Biochemistry, 2008, 47, 11793 – 11803.
96. Xie, H.; Bolam, D. N.; Nagy, T.; Szabo, L.; Cooper, A.; Simpson, P. J.; Lakey, J. H.; Williamson, M. P.; Gilbert, H. J. Biochemistry, 2001, 40, 5700 – 5707.
97. Lia, H. and Sharon, N. Chem. Rev., 1998, 98, 637 – 674.

30
98. Muraki, M. Protein and Peptide Letters. 2002, 9, 195 – 209.
99. Fernandez-Alonso, M. C.; Canada, F. J.; Jiménez-Barbero, J.; Cuevas, G. J. Am.
Chem. Soc., 2005, 127, 7379 – 7386.
100. Chavez, M. I.; Andreu, C.; Vidal, P.; Aboitiz, N.; Freire, F.; Groves, P.; Asensio, J. L.; Asensio, G.; Mraki, M.; Canada, F. J.; Jiménez-Barbero, J. Chemistry – A
European Journal, 2005, 11, 7060 – 7074.
101. Sharon, N.; Lia, H. Advances in Experimental Medicine and Biology. 2001, 491, 1 – 16.
102. Toone, E. Curr. Opin. Struct. Biol., 1994, 4, 719 – 728.
103. Shidu, G.; Withers, S. G.; Nguyen, N. T.; McIntosh, L. P.; Ziser, L.;Brayer, G. D. Biochemistry, 1999, 38, 5346 – 5354.
104. Heightam, T. D. and Vasella, A. T. Angew. Chem. Int. Ed., 1999, 38, 750 – 770.
105. Notenboom, V.; Cilliams, S. J.; Hoos, R.; Withers, S. G.; Rose, D. R. Biochemistry, 2000, 39, 11553 – 11563.
106. A commercial available program Jaguar was used in pKa calculations.
107. Carcabal, P.; Jockusch, R. A.; Hunig, I.; Snoek, L. C.; Kroemer, R. T.; Davis, B. G.; Gamblin, D. P.; Compagnon, I.; Oomens, J.; Simons, J. P. J. Am. Chem. Soc., 2005, 127, 11414-11425.
108. Shim, G.; Shin, J.; Kim, Y. Bull. Korean Chem. Soc. 2004, 25, 198-202.
109. Kirschner, K. N.; Woods, R. J. Prog. Natl. Aced. Sci. 2001, 98, 10541-10545.
110. Casu, B., Reggiani, M., Gallo, G. G.; Vigevani, A. Tetrahedron, 1966, 22, 3061-3083.
111. Poppe, L.; van Halbeek, H. Nat. Struct. Biol. 1994, 1, 215-216.
112. Adams, B.; Lerner, L. J. Am. Chem. Soc. 1992, 114, 4827-4829.
113. Bekiroglu, S.; Sandstrom, C.; Norberg, T.; Kenne, L. Carbohydr. Res. 2000, 328, 409-418.
114. Betta, G.; Kover, K. E. Carbohydr. Res. 1999, 320, 267-272.
115. Carmichael, I.; Chipman, D. M.; Podlasek, C. A.; Serianni, A. S. J. Am. Chem.
Soc. 1993, 115, 10863-10870.
116. Thomas, W. A. Prog. NMR Spectr. 1997, 30, 183-207.

31
117. Contrereas, R. H.; Peralta, J. E. Prog. NMR Spectr. 2000, 37, 321-425.
118. Haasnoot, C. A. G. De Leeuw, F. A. A. M.; Altona, C. Tetrahedron, 1980, 36, 2783-2792.
119. Huggins, M. L. J. Am. Chem. Soc. 1953, 75, 4123-4126.
120. Altona, C.; Ippel, J. H.; Hoekzema, A. J. A. W.; Erkelens, C.; Groesbeek, M.; Donders, L. A. Magn. Reson. Chem. 1989, 27, 564-576.
121. Altona, C.; Francke, R.; de Haan, R.; Ippel, J. H.; Daalmans, G. J.; Hoekzema, A. J. A. Q.; can Wijk, J. Magn. Reson. Chem. 1994, 32, 670-678.
122. Colucci, W. J.; Jungk, S. J.; Gandour, R. D. Magn. Reson. Chem. 1985, 23, 335-343.
123. Imai, K.; Osawa, E. Magn Reson. Chem. 1990, 28, 668-674.
124. Mullay, J. J. Am. Chem. Soc. 1985, 107, 7271-7275.
125. Zhao, H.; Carmichael, I.; Serianni, A. S. J. Org. Chem. 2008
126. Zhao, H.; Pan, Q.; Zhang, W.; Carmichael, I.; Serianni, A. S. J. Org. Chem. 2007, 72, 7071-7082.
127. Bose, B.; Zhao, S.; Stenuta, R.; Cloran, F.; Bondo, P. B.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1998, 120, 11158-11173.

32
CHAPTER 2:
DENSITY FUNCTIONAL THEORY CALCULATIONS AND PARAMETERIZATION
OF 2JCOH, 3JHCOH AND 3JCCOH SPIN-SPIN COUPLING CONSTANTS IN
SACCHARIDES
2.1 Introduction
Karplus [1] firs t reported that v icinal 3JHCCH NMR spin-spin coupling cons tants
(J-couplings) could be app roximately f it to a co s2 f unction ( equation 2. 1), whe re
represents the H -C-C-H tors ion angle, thu s pr oviding a theor etical ba sis f or ear lier
empirical observations made by Lemieux and coworkers. [2]
3JHH = A cos2 + B cos + C 2.1
Since then, additional Karplus -like relati onships have been r eported f or other
homonuclear J-couplings such as 3JHCOH [3-5] and 3JHCNH [6], and f or heteronuclear J-
couplings such as 3JCCCH [7], 3JCOCH [8,9] and 3JCCNH.[10] 3JHCOH and 3JCCOH values are
potentially us eful par ameters to inve stigate in tra- and/or inter molecular hyd rogen
bonding in OH- containing structures in s olution, but f ew studies, especially of 3JCCOH,
have been reported on their parameterization and application in carbohydrates. [11-13]
Mono- and oligos accharides contain s everal conf ormational domains , including
ring, linkage, exocyclic hydr oxymethyl, a nd/or exocyclic C -O bond conf ormations.

33
Redundant tr ans-glycoside J-couplings ( 2JCOC, 3JCOCH, and 3JCOCC) hav e been
investigated as probes of C-O bond conformation in glycosidic linkages. [14-16] Studies of
redundant JHH, JCH and JCC within the exo cyclic hydr oxymethyl ( CH2OH) gr oup of
saccharides have r evealed a dual dependen ce o f s ome J-couplings on C -C and C -O
torsion angles, leading to their application to ev aluate correlated conformations. [15] For
example, 2JC5,H6R/S, 2JC6,H5 and 2JH6R,H6S serve as probes of C6-O6 bond conformation
in aldohexopyr anosyl r ings even though they d o not involve the exch angeable O6 H
proton as a coupled nucleus.
2JCOH, 3JHCOH and 3JCCOH involving exch angeable hydr oxyl pr otons ar e
available to conf irm and/or refine conclusions based on the above -noted indirect probes
of C -O bond conf ormation. Kar plus-like equatio ns f or 3JHCOH ba sed on exper imental
and computational approaches [3-5]
have been r eported us ing s mall, non -carbohydrate
molecules s uch as methanol as model s tructures. 3JCCOH value s have been us ed
qualitatively to determin e C-O torsion angles, [13, 18]
although little is known about their
dependence on saccharide structure.
In this inves tigation, dens ity f unctional theor y (DFT) has been applied to s tudy
the effects of molecular structure on 2JCOH, 3JHCOH and 3JCCOH in model aldopyranosyl
rings, leading to new Kar plus equations f or 3JHCOH and 3JCCOH. Anomer ic
configuration, r ing conf iguration, and pa thway l ocation wer e f ound to ex ert minimal
effects on 3JHCOH. On the othe r hand, while 3JCCOH values show a pr imary dependence
on the C -C-O-H tor sion angl e a s expect ed, the y als o show a s ignificant systematic

34
secondary dependence on the orientation of terminal electronegative substituents attached
to the coupled carbon, reminiscent of that observed for 3JCCOC. [18, 19]
2.2 Computational Method
Model compounds wer e chos en to mimic methyl - a nd -D-
aldohexopyranosides (1-4, 9-14) and - and -D-aldohexopyranoses (5-8) (Scheme 2.1).
Torsion angles , and , are def ined as O5-C1-O1-CH3 (or O5-C1-O1-H) and C1-C2-
O2-H, respectively (Scheme 2.1). Density functional theory (DFT) calculations using the
B3LYP functional [20]
and 6-31G* basis set [21]
were conducted within Gaussian98 [22] for
geometric optimization of molecular s tructures. [15, 23- 25] J-Couplings were calculated by
DFT us ing a modif ied ver sion of Gaussian94 [26] and an extended bas is set
([5s2p1d|3s1p]) [27]
designed to recover only the Fermi contact (FC) contr ibution to the
coupling.
Three series of calculations were conducted using the above protocol. In the first,
3JH2,O2H and 3JC1,O2H were calculated in the methyl glycos ides 1-4. Torsion angles,
and , w ere v aried fro m 0 ° - 360 ° in 30 ° incr ements by holding both angles at f ixed
values, giving 144 s tructures f or each compound . All r emaining molecular par ameters
were geometrically optimized. 3JH2,O2H and 3JC1,O2H were calculated in each s tructure,
yielding a hypersurface from which the relative sensitivities of the J-couplings to and
were determined.
The s econd s eries of calculations wa s conducted on 5-8, which contains a f ree
anomeric hydr oxyl gr oup, to deter mine the ef fect of hydr oxyl cha racter (1º, 2º or

35
anomeric) on 3JH1,O1H and 3JC2,O1H. Initial values of ~180º were chosen to eliminate
potential intramolecular H-bonding between O1 a nd O2. Dur ing geometric optimization,
r emained at ~180º in all s tructures. The tor sion angle was f ixed at 60º , –60º and
180º, and all remaining molecular parameters were optimized.
The third series of calculations was conducted on 9-12 to s tudy the effect of O3
on 3JH2,O2H, 3JC1,O2H and/or 3JC3,O2H. Initial C2-C1-O1-CH3 and C2-C3-O3-H torsion
angles were set at 180º and optimized (these torsions remained at ~180º in the optimized
geometries). Values of were f ixed at 60º , –60º and 180º , and all r emaining molecular
parameters were optimized.
J-Couplings were also calculated using Gaussian03 [28] in whic h both the Fermi
and non-Fermi contact contributions were evaluated. DFT -optimized s tructures of 2
obtained from Gaussian98 were used, and 3JH2,O2H, 3JC1,O2H and 3JC3,O2H values were
calculated. Gaussian03 wa s al so u sed to calculat e 3JH6R,O6H, 3JH6S,O6H and 3JC5,O6H
values in 13. In this case, the C5-C6-O6-H and O5-C5-C6-O6 torsion angles were fixed
at 60º, –60º and 180º, yielding 9 structures in which the three couplings were computed.
The ef fect of s olvent on 3JHCOH, 3JCCOH and other J-couplings in 13 wa s
evaluated us ing the Self- Consistent R eaction Fie ld ( SCRF) and the Integral Equation
Formalism (polarizable continuum) model (IEFPCM) and the above-noted [5s2p1d|3s1p]
basis set as implemented in Gaussian03. A s ingle optimized s tructure was studied
containing the following exocyclic torsion angles: C2-C1-O1-CH3, 170.4º; C1-C2-O2-H,
-167.7º; C2-C3-O3-H, -165.2º; O5-C5-C6-O6, 180º; and C 5-C6-O6-H, 180º (the former
three torsions were relaxed and the latter two were fixed). A similar set of calculations on
13 was conducted in vacuum.

36
3JC5,OH2 calculations were conducted us ing model compound 14. Initial C2-C1-
O1-CH3 and C1-C2-O2-H torsion angles were set at 180º and allowed to optimize (in the
optimized geometries, these torsion angles remained in anti orientations), and the C5-C4-
O4-H torsion angle was fixed at 60º, -60º or 180º.
OCH3
OCH3
H
H
OH
OCH3
OCH3
H
H
OH
OCH3
OCH3
H
H
OHO
CH3
OCH3
HH
OH
1 2 3 4
OCH3
OH
H
H
OH
OCH3
OH
H
H
OH
OCH3
OH
H
H
OHO
CH3
OH
HH
OH
5 6 7 8
OCH3
OCH3
H
H
OH
OCH3
OCH3
HH
OH
9 10
HO HO
OCH3
OCH3
H
H
OH
OCH3
OCH3
HH
OH
11
12
OH
OH
OHOH2C
OCH3
H
H
OH
13
HO
OH3C
OCH3
H
H
OH
14
HO
Scheme 2. 1 M odel compounds us ed in calculati ng J-coupling cons tants involving hydroxyl protons.

37
2.3 Results and Discussion
2.3.1 H-C-O-H coupling pathway.
2.3.1.1 Parameterization
Compounds 1-4 were s elected as glyco side mimics to reduce complications
caused by intramolecular H-bonding between adjacent hydroxyl groups. 3JH2,O2H values
were calculated in thes e s tructures as a f unction of C2-O2 ( ) and C 1-O1 ( ) (Scheme
2.1) bond rotation (Figure 2.1). As expected, 3JH2,O2H exhibits a primary dependence on
, and a small secondary dependence on . Plots for 1 and 2, and for 3 and 4, were nearly
identical, demonstrating that 3JH2,O2H values are unaffected by anomeric configuration.
All hypersurfaces show a global maximum of ~15 Hz at an H2 -C2-O2-H torsion angle
of 180º and a local maximum of ~10 Hz at = 0º. Portions of the hypersurfaces from
= 0 - 180º and = 0 - -180º were superimposable.
3JH2,O2H values in 1-4 are combined and plotted in two-dimensions as a function
of in Figur e 2.2A (black line) . The four data s ets are superimposable, indicating that
3JH2,O2H is un affected by C 2-O2 bond or ientation. Us ing data in Figur e 2. 1, a lea st-
square fitted Karplu s equation (equation 2 .2) w as obt ained in which only the Fermi
contact contribution to the coupling was c onsidered (s ee below). T he fitted curve is
shown in Figure 2.2A. This equation is similar to equation 2.3 reported by Fraser et al. [3]
(Figure 2. 2A) bas ed on theor etical calculations on C H3OH and exper imental dat a
obtained on an H -bonded system in C 2HCl3 solvent to cons train C-O torsions, although
noticeable differences exist at the larger dihedral angles.
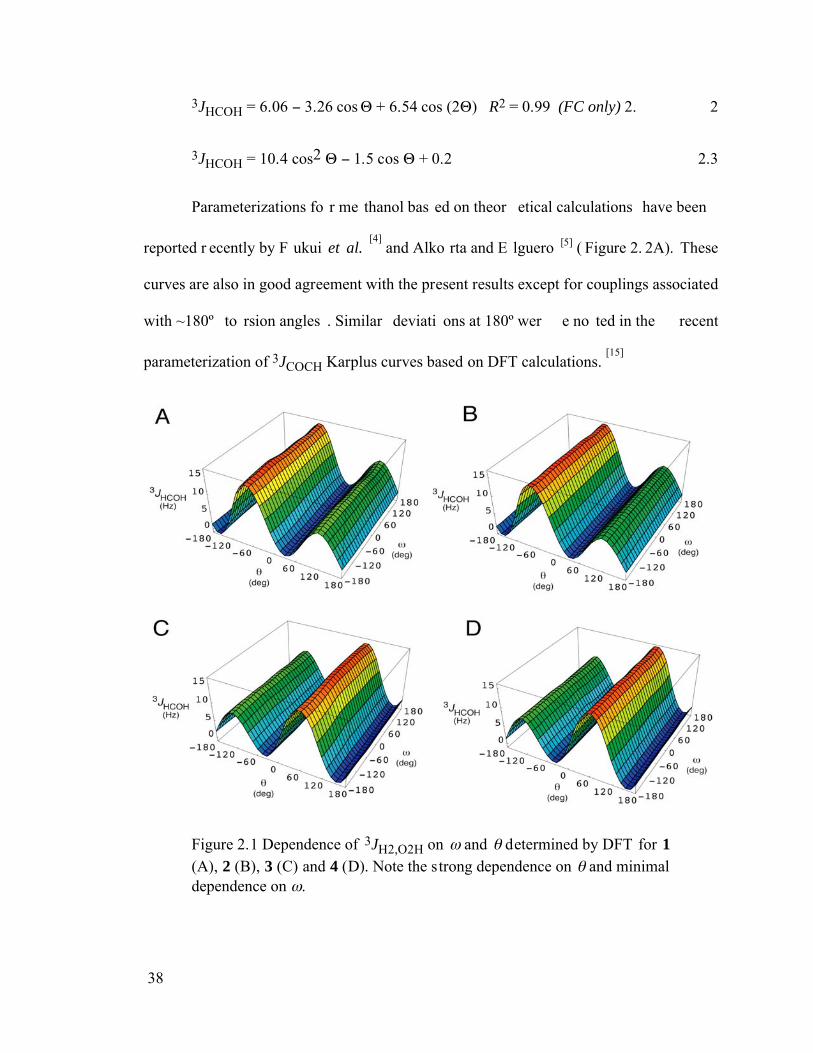
38
3JHCOH = 6.06 3.26 cos + 6.54 cos (2 ) R2 = 0.99 (FC only) 2. 2
3JHCOH = 10.4 cos2 1.5 cos + 0.2 2.3
Parameterizations fo r me thanol bas ed on theor etical calculations have been
reported r ecently by F ukui et al. [4]
and Alko rta and E lguero [5] ( Figure 2. 2A). These
curves are also in good agreement with the present results except for couplings associated
with ~180º to rsion angles . Similar deviati ons at 180º wer e no ted in the recent
parameterization of 3JCOCH Karplus curves based on DFT calculations. [15]
Figure 2.1 Dependence of 3JH2,O2H on and determined by DFT for 1 (A), 2 (B), 3 (C) and 4 (D). Note the strong dependence on and minimal dependence on .
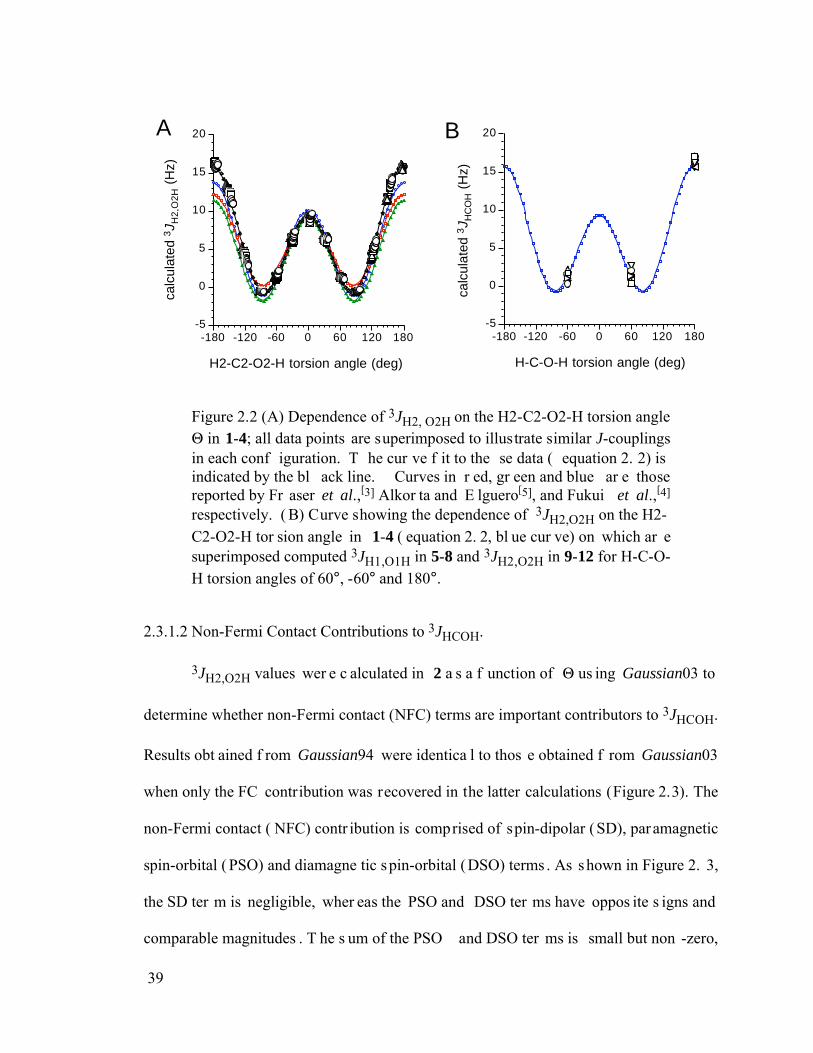
39
-5
0
5
10
15
20
calc
ulat
ed 3
J H2,
O2H
(H
z)
-180 -120 -60 0 60 120 180
H2-C2-O2-H torsion angle (deg)
-5
0
5
10
15
20
calc
ulat
ed 3
J HC
OH (
Hz)
-180 -120 -60 0 60 120 180
H-C-O-H torsion angle (deg)
A B
Figure 2.2 (A) Dependence of 3JH2, O2H on the H2-C2-O2-H torsion angle in 1-4; all data points are superimposed to illustrate similar J-couplings
in each conf iguration. T he cur ve f it to the se data ( equation 2. 2) is indicated by the bl ack line. Curves in r ed, gr een and blue ar e those reported by Fr aser et al.,[3] Alkor ta and E lguero[5], and Fukui et al.,[4] respectively. ( B) Curve showing the dependence of 3JH2,O2H on the H2-C2-O2-H tor sion angle in 1-4 ( equation 2. 2, bl ue cur ve) on which ar e superimposed computed 3JH1,O1H in 5-8 and 3JH2,O2H in 9-12 for H-C-O-H torsion angles of 60°, -60° and 180°.
2.3.1.2 Non-Fermi Contact Contributions to 3JHCOH.
3JH2,O2H values wer e c alculated in 2 a s a f unction of us ing Gaussian03 to
determine whether non-Fermi contact (NFC) terms are important contributors to 3JHCOH.
Results obt ained f rom Gaussian94 were identica l to thos e obtained f rom Gaussian03
when only the FC contribution was recovered in the latter calculations (Figure 2.3). The
non-Fermi contact ( NFC) contr ibution is comprised of spin-dipolar (SD), paramagnetic
spin-orbital ( PSO) and diamagne tic spin-orbital (DSO) terms . As shown in Figure 2. 3,
the SD ter m is negligible, wher eas the PSO and DSO ter ms have oppos ite s igns and
comparable magnitudes . T he s um of the PSO and DSO ter ms is small but non -zero,
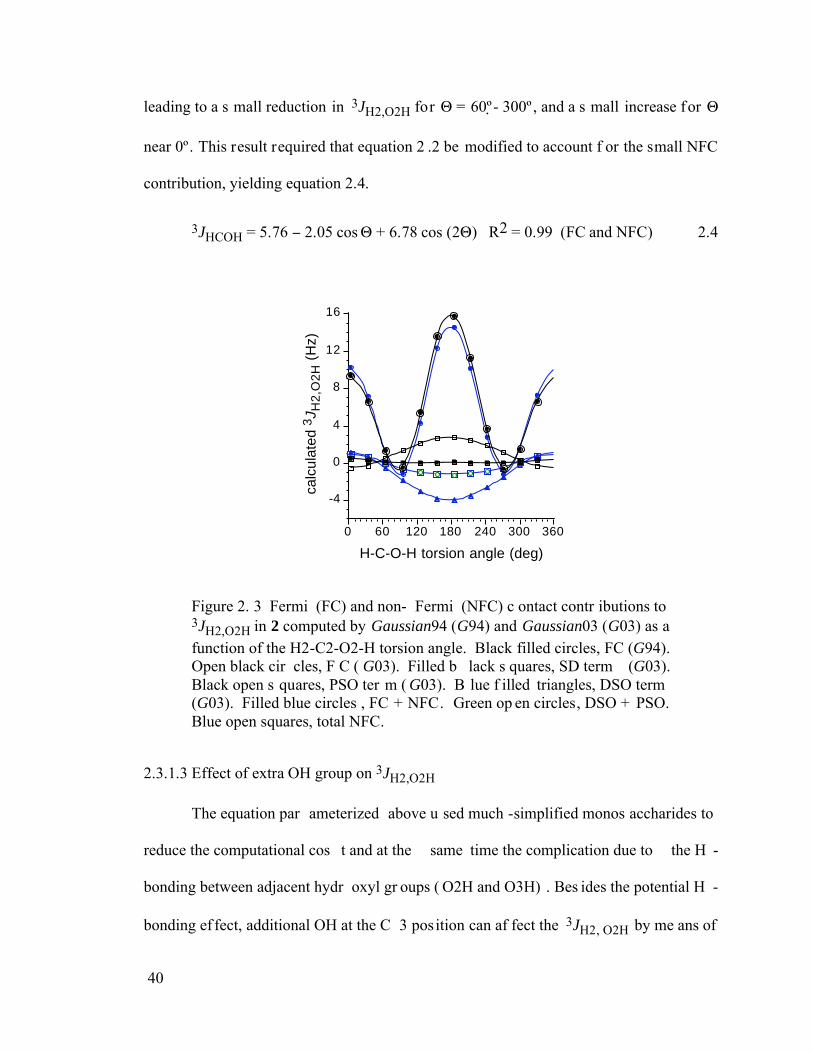
40
leading to a s mall reduction in 3JH2,O2H for = 60º- 300º , and a s mall increase for
near 0º. This result required that equation 2 .2 be modified to account f or the small NFC
contribution, yielding equation 2.4.
3JHCOH = 5.76 2.05 cos + 6.78 cos (2 ) R2 = 0.99 (FC and NFC) 2.4
-4
0
4
8
12
16ca
lcul
ated
3J H
2,O
2H
(H
z)
0 60 120 180 240 300 360
H-C-O-H torsion angle (deg)
Figure 2. 3 Fermi (FC) and non- Fermi (NFC) c ontact contr ibutions to 3JH2,O2H in 2 computed by Gaussian94 (G94) and Gaussian03 (G03) as a function of the H2-C2-O2-H torsion angle. Black filled circles, FC (G94). Open black cir cles, F C ( G03). Filled b lack s quares, SD term (G03). Black open s quares, PSO ter m ( G03). B lue f illed triangles, DSO term (G03). Filled blue circles , FC + NFC. Green op en circles, DSO + PSO. Blue open squares, total NFC.
2.3.1.3 Effect of extra OH group on 3JH2,O2H
The equation par ameterized above u sed much -simplified monos accharides to
reduce the computational cos t and at the same time the complication due to the H -
bonding between adjacent hydr oxyl gr oups ( O2H and O3H) . Bes ides the potential H -
bonding ef fect, additional OH at the C 3 position can af fect the 3JH2, O2H by me ans of

41
induction effects. To check this , J-Coupling calculations were conducted on 9 - 12 for a
limited s et of C 1-O1, C 2-O2 and C 3-O3 tor sion angles to s tudy the ef fect of a C 3
hydroxyl gr oup on 3JH2,O2H. T hese data, s hown in Figur e 2. 2B s uperimposed on th e
parameterized cur ve des cribed by equation 2. 2, indicate that O3, whether axial or
equatorial, exerts a minimal effect on 3JH2,O2H.
2.3.1.4 Other 3JHCOH coupling pathway in methyl monoglycosides
The above results show that structure and relative configuration at C1 and C2 do
not inf luence 3JH2,O2H values significantly. Other HC OH vicinal couplings constants,
including 3JH3,O3H in model compounds 9 – 12, 3JH4,O4H in 14 and 3JH6R/S,O6H in 13,
were computed as well. T hree per fectly s taggered conf ormations with respect to the
interested torsion angles were f ixed. The C-O torsion angles of the adjacent OH groups
are f ixed at a s taggered pos ition so that no hydr ogen bond will f orm. T he r est of the
structures are optimi zed. All computed 3JHCOH are superimposed on the Kar plus curve
derived f rom 3JH2,O2H, as shown in Figur e 2.2. Clearly, 3JHCOH of dif ferent pathways
within a methyl monoglyco side obeys the same Karplus behavior as 3JH2,O2H. The only
exception is the pathway involving C1-O1 bond, which will be discussed next.
2.3.1.5 3JHCOH involving the C1-O1 bond
A unique feature of the H1-C1-O1-H coupling pathway (see Scheme 2.2 Ia) is the
presence of an ele ctronegative substituent (O5) that aligns anti to the coupled pr oton in
one of the two gauche conformations (see Scheme 2.2 Ib and Ic). This anti alignment (Ib)

42
reduces the 3JHCOH in this s tructure, thus r endering the magnitudes of the two gauche
couplings non- equivalent and caus ing the der ived Kar plus cur ve to be phas e-shifted
relative to that predicted by equation 2.4 (Figure 2.4A). The direction of the shift depends
on anomer ic conf iguration. O5 in a conf iguration causes the cur ve shifted to the lef t,
compared to a right s hift in a configuration. This shifting leads to modif ied equations
2.5 and 2. 6 to t reat 3JH1,O1H in and -pyranoses, where is the H1 -C1-O1-H torsion
angle. Two additional sine terms are introduced to treat the unique shifting in H1-C1-O1-
H coupling pathway, cont rast to the no rmal K arplus equation in other pathways in
saccharides.
3JH1,O1H ( ) = 5.52 2.93 cos + 6.71 cos (2 ) + 0.14 sin 1.00 sin (2 ) 2. 5
R2 = 0.99
3JH1,O1H ( ) = 5.71 2.88 cos + 6.65 cos (2 ) 0.076 sin + 1.17 sin (2 ) 2. 6
R2 = 0.99
Scheme 2. 2 E ffect of r ing oxygen as an inter nal electr onegative substituent on the coupling pathways between H1 and O1H or C 2 and O1H.
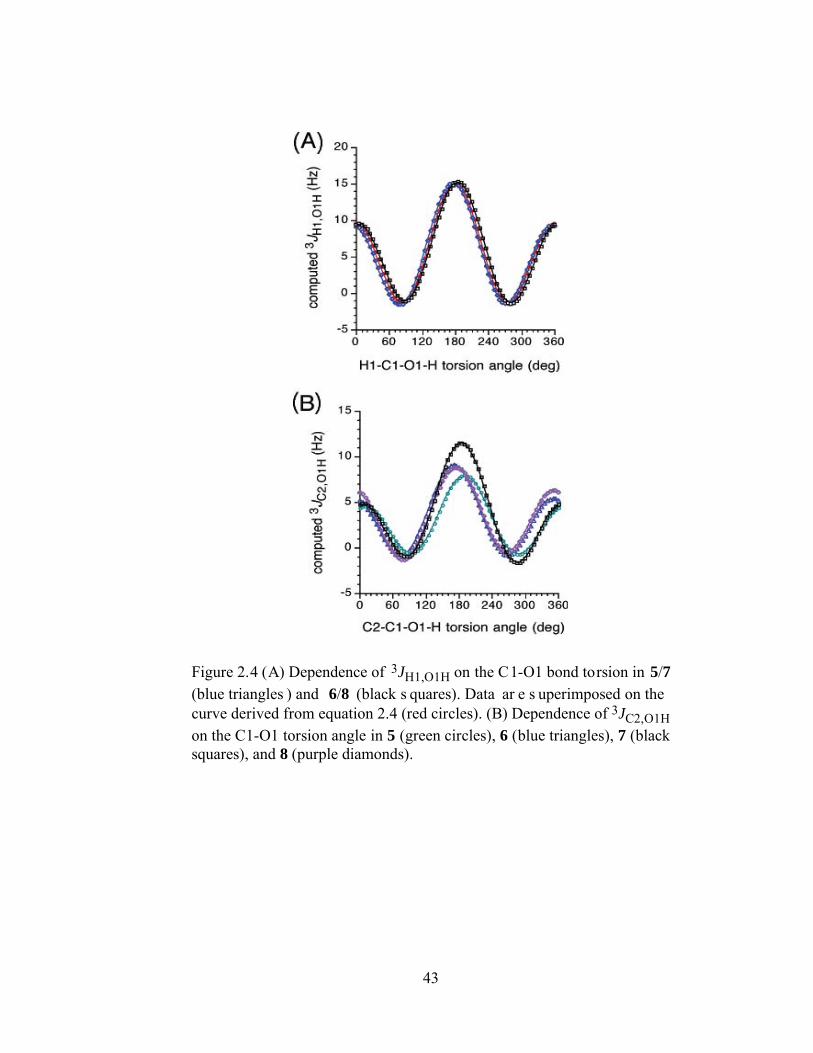
43
Figure 2.4 (A) Dependence of 3JH1,O1H on the C1-O1 bond torsion in 5/7
(blue triangles ) and 6/8 (black s quares). Data ar e s uperimposed on the curve derived from equation 2.4 (red circles). (B) Dependence of 3JC2,O1H on the C1-O1 torsion angle in 5 (green circles), 6 (blue triangles), 7 (black squares), and 8 (purple diamonds).
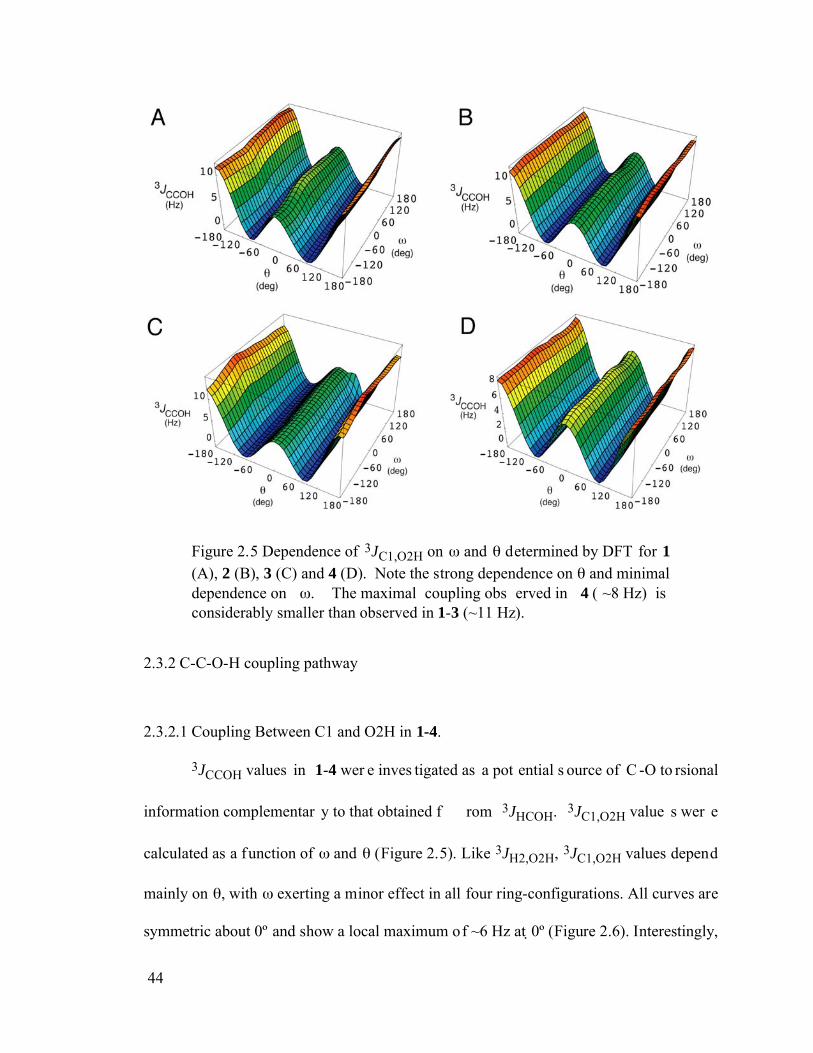
44
Figure 2.5 Dependence of 3JC1,O2H on and determined by DFT for 1 (A), 2 (B), 3 (C) and 4 (D). Note the strong dependence on and minimal dependence on . The maximal coupling obs erved in 4 ( ~8 Hz) is considerably smaller than observed in 1-3 (~11 Hz).
2.3.2 C-C-O-H coupling pathway
2.3.2.1 Coupling Between C1 and O2H in 1-4.
3JCCOH values in 1-4 wer e inves tigated as a pot ential s ource of C -O to rsional
information complementar y to that obtained f rom 3JHCOH. 3JC1,O2H value s wer e
calculated as a function of and (Figure 2.5). Like 3JH2,O2H, 3JC1,O2H values depend
mainly on , with exerting a minor effect in all four ring-configurations. All curves are
symmetric about 0º and show a local maximum of ~6 Hz at 0º (Figure 2.6). Interestingly,
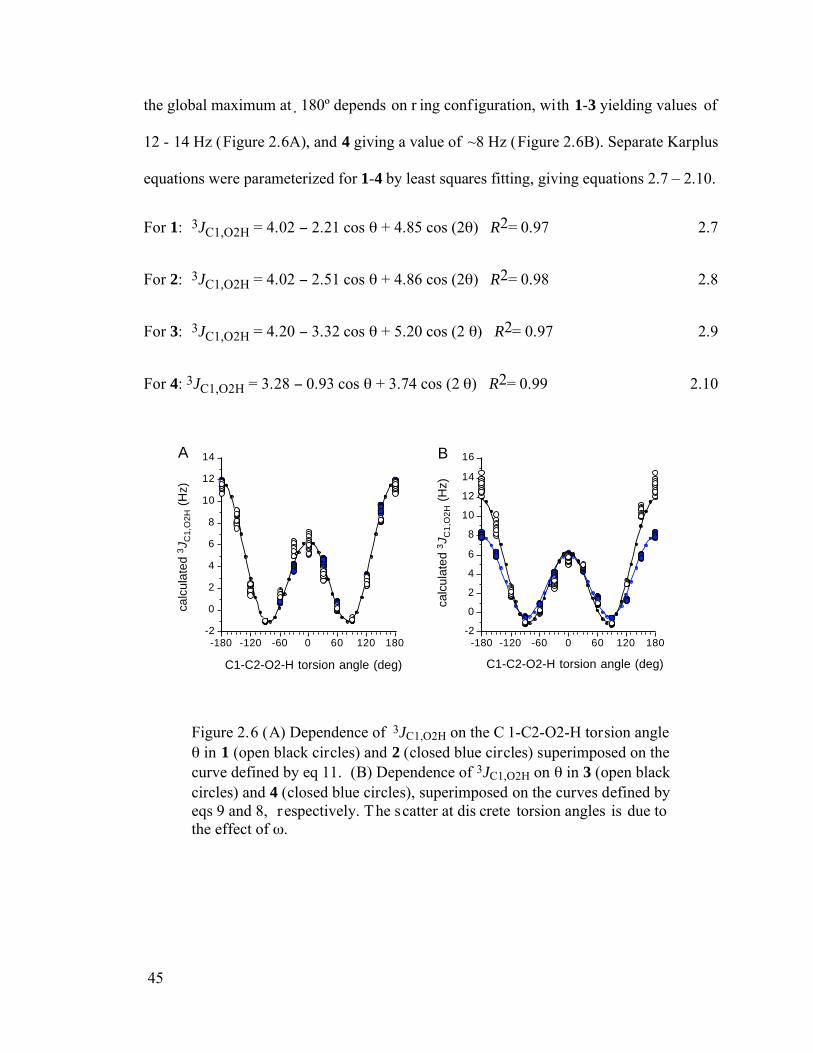
45
the global maximum at 180º depends on r ing configuration, with 1-3 yielding values of
12 - 14 Hz (Figure 2.6A), and 4 giving a value of ~8 Hz (Figure 2.6B). Separate Karplus
equations were parameterized for 1-4 by least squares fitting, giving equations 2.7 – 2.10.
For 1: 3JC1,O2H = 4.02 2.21 cos + 4.85 cos (2 ) R2= 0.97 2.7
For 2: 3JC1,O2H = 4.02 2.51 cos + 4.86 cos (2 ) R2= 0.98 2.8
For 3: 3JC1,O2H = 4.20 3.32 cos + 5.20 cos (2 ) R2= 0.97 2.9
For 4: 3JC1,O2H = 3.28 0.93 cos + 3.74 cos (2 ) R2= 0.99 2.10
-2
0
2
4
6
8
10
12
14
calc
ulat
ed 3
J C1,
O2H
(H
z)
-180 -120 -60 0 60 120 180
C1-C2-O2-H torsion angle (deg)
-2
0
2
4
6
8
10
12
14
16ca
lcul
ated
3J C
1,O
2H (
Hz)
-180 -120 -60 0 60 120 180
C1-C2-O2-H torsion angle (deg)
A B
Figure 2.6 (A) Dependence of 3JC1,O2H on the C 1-C2-O2-H torsion angle in 1 (open black circles) and 2 (closed blue circles) superimposed on the
curve defined by eq 11. (B) Dependence of 3JC1,O2H on in 3 (open black circles) and 4 (closed blue circles), superimposed on the curves defined by eqs 9 and 8, r espectively. The scatter at dis crete torsion angles is due to the effect of .

46
OCH3
OCH3
H
H
O
OCH3
OCH3
H
H
O
OCH3
OCH3
H
H
O
OCH3
OCH3
HH
O
1 2 3 4
"in-plane" oxygen enhances 3JC1,O2H "in-plane" oxygen absent
H H
H H
Scheme 2. 3 T erminal electronegative s ubstituent effect. In 1-3, t he terminal oxygen, either ring oxygen O5 , or O-methyl oxygen O1, is “in-plane” with the coupling pathway. No “in plane” oxygen in 4.
Since equations 2. 7 – 2. 9 are very s imilar, they were combined to give a
generalized equation (equation 2.11).
For 1-3: 3JC1,O2H = 4.10 2.72 cos + 5.01 cos (2 ) R2= 0.97 2.11
The unique behavior of 3JC1,O2H in 4 appears to derive from the effect of terminal
electronegative substituents on the coupled carbon, which is a factor known to influence
other car bon-based J-couplings ( e.g., 3JCOCC). [14]
For 1-3, an oxygen s ubstituent is
antito O2 (“in -plane” substituent): O5 in 1-2 and O1 in 3 (Scheme 2.3). An “in- plane”
orientation is abs ent in 4 ( Scheme 2. 3). B y an alogy to 3JCOCC, ter minal “in- plane”
electronegative s ubstituents are expected to enha nce 3JCCOH values when the coupled
atoms are approximately anti. 3JCCOH values exhibit this behavior, and the enhancement
is considerably larger (3-4 Hz) than observed for 3JCOCC (~ 0.7 Hz). [14]
3JC1,O2H values in 2 were computed using Gaussian03 to determine the effect of
NFC ter ms on the computed coupling s ( Figure 2. 7). Unlike 3JHCOH, the calculated
couplings were virtually unaffected by the inclusion of these terms. Thus, equations 2.10
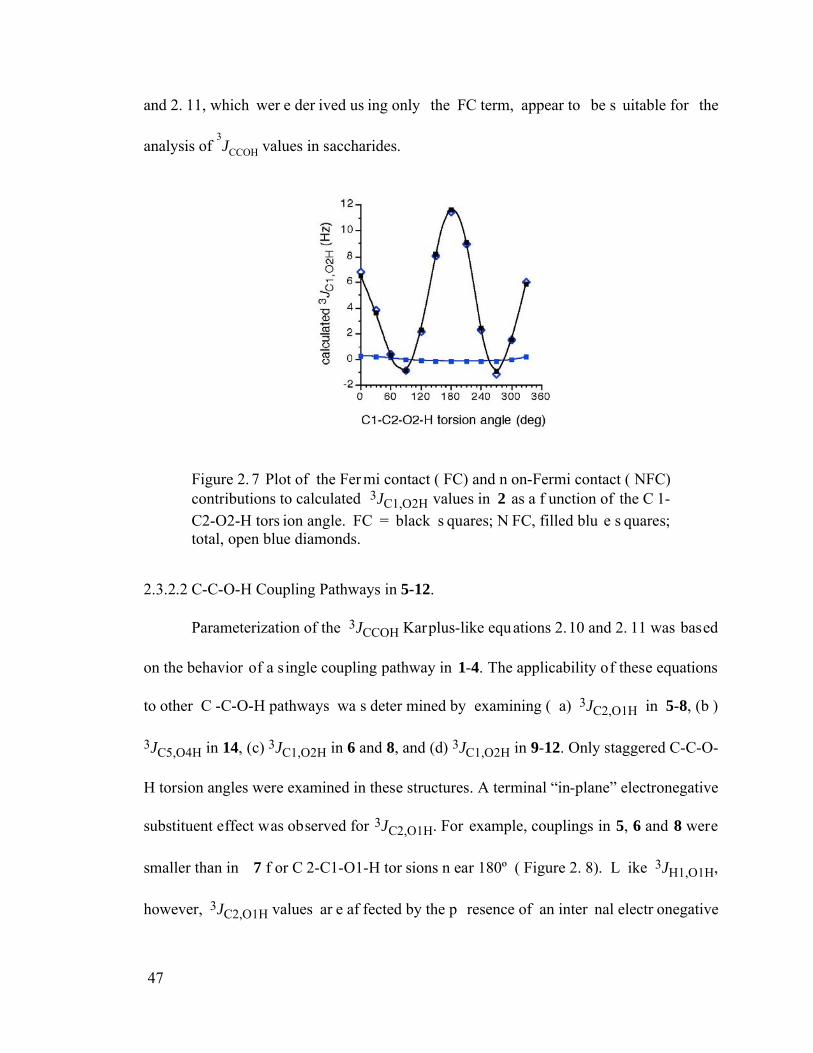
47
and 2. 11, which wer e der ived us ing only the FC term, appear to be s uitable for the
analysis of 3JCCOH values in saccharides.
Figure 2. 7 Plot of the Fer mi contact ( FC) and n on-Fermi contact ( NFC) contributions to calculated 3JC1,O2H values in 2 as a f unction of the C 1-C2-O2-H tors ion angle. FC = black s quares; N FC, filled blu e s quares; total, open blue diamonds.
2.3.2.2 C-C-O-H Coupling Pathways in 5-12.
Parameterization of the 3JCCOH Karplus-like equations 2.10 and 2. 11 was based
on the behavior of a single coupling pathway in 1-4. The applicability of these equations
to other C -C-O-H pathways wa s deter mined by examining ( a) 3JC2,O1H in 5-8, (b )
3JC5,O4H in 14, (c) 3JC1,O2H in 6 and 8, and (d) 3JC1,O2H in 9-12. Only staggered C-C-O-
H torsion angles were examined in these structures. A terminal “in-plane” electronegative
substituent effect was observed for 3JC2,O1H. For example, couplings in 5, 6 and 8 were
smaller than in 7 f or C 2-C1-O1-H tor sions n ear 180º ( Figure 2. 8). L ike 3JH1,O1H,
however, 3JC2,O1H values ar e af fected by the p resence of an inter nal electr onegative
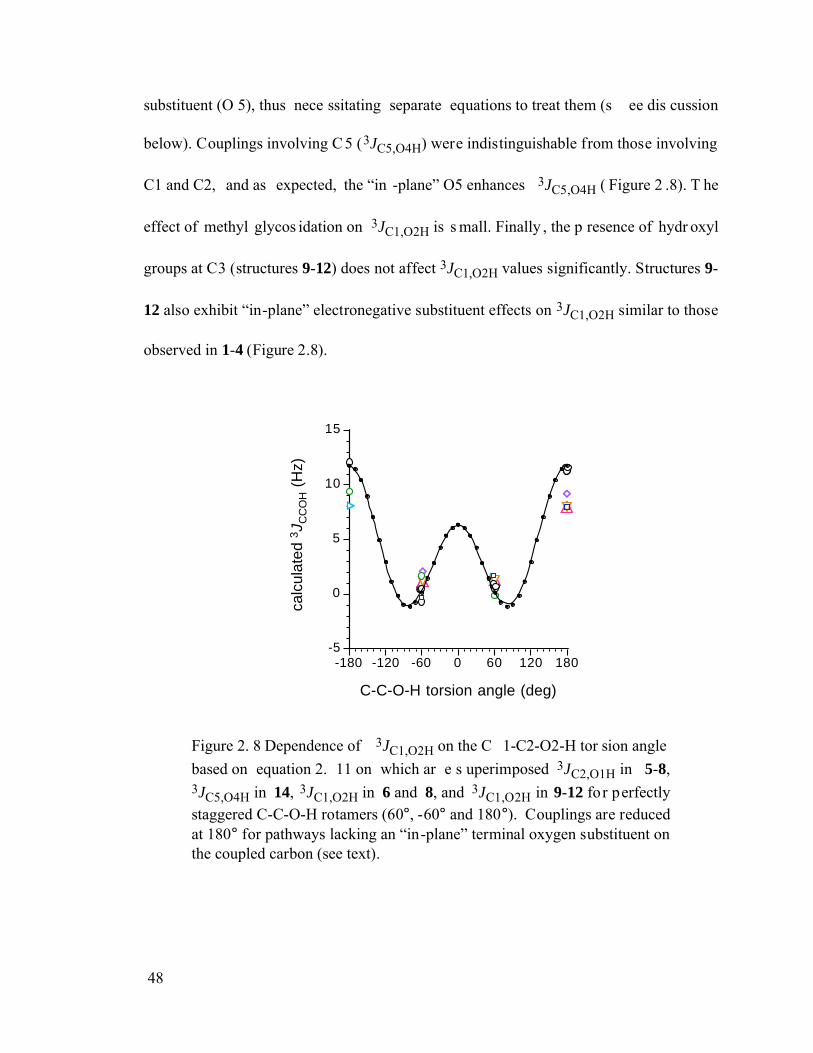
48
substituent (O 5), thus nece ssitating separate equations to treat them (s ee dis cussion
below). Couplings involving C 5 (3JC5,O4H) were indistinguishable from those involving
C1 and C2, and as expected, the “in -plane” O5 enhances 3JC5,O4H ( Figure 2 .8). T he
effect of methyl glycos idation on 3JC1,O2H is s mall. Finally , the p resence of hydr oxyl
groups at C3 (structures 9-12) does not affect 3JC1,O2H values significantly. Structures 9-
12 also exhibit “in-plane” electronegative substituent effects on 3JC1,O2H similar to those
observed in 1-4 (Figure 2.8).
-5
0
5
10
15
calc
ulat
ed 3
J CC
OH (
Hz)
-180 -120 -60 0 60 120 180
C-C-O-H torsion angle (deg)
Figure 2. 8 Dependence of 3JC1,O2H on the C 1-C2-O2-H tor sion angle based on equation 2. 11 on which ar e s uperimposed 3JC2,O1H in 5-8, 3JC5,O4H in 14, 3JC1,O2H in 6 and 8, and 3JC1,O2H in 9-12 for perfectly staggered C-C-O-H rotamers (60°, -60° and 180°). Couplings are reduced at 180° for pathways lacking an “in-plane” terminal oxygen substituent on the coupled carbon (see text).

49
The behavior of 3JC2,O3H and 3JC3,O2H in 9-12 is cons istent with that of
3JC1,O2H, 3JC2,O1H and 3JC5,O4H in the s ame s tructures ( Figure 2 .9). T he former
couplings exhibit “in-plane” electronegative substituent effects (e.g., 3JC3,O2H is smaller
in 9, 10 and 11 than in 12).
A closer examination of 3JC3,O2H in 9-11 suggests the exi stence of a small “in-
plane” effect due to car bon. For C-Cx-C-O-Hx coupling pathways (subscript denotes the
coupled nuclei) containing two inter nal tr ans dihedral angles ( a zig -zag coplanar
arrangement; e.g., C4-C3-C2-O2-H in 9), a modification of equation 2.10, which applies
to pathways devoid of ter minal car bon and ox ygen “in -plane” s ubstituents, appear s
necessary, yielding equation 2.12.
3JCCOH = 3.49 1.41 cos + 4.18 cos (2 ) R2= 0.99 2.12
3JC1,O2H = 3.38 1.24 cos + 3.98 cos (2 ) R2= 0.99 2.13
An additional equation ( equation 2. 13) was also der ived, which r epresents an
average of equations 2. 10 and 2 .12. Since the “in-plane” car bon ef fect is s mall, this
averaged equation could be applie d to simplify the treatment of 3JCCOH in s accharides
(i.e., only equations 2.11 and 2.13 are needed instead of three), with only a s mall loss in
accuracy.
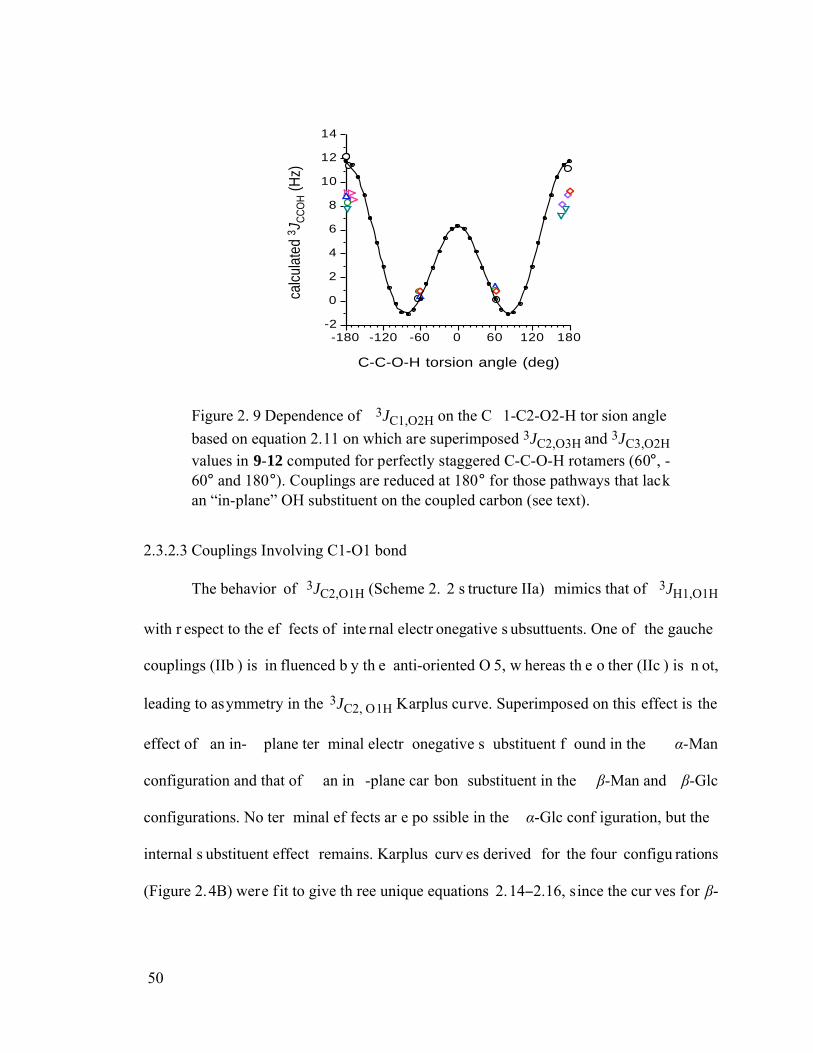
50
-2
0
2
4
6
8
10
12
14
calc
ulat
ed 3 J
CC
OH (H
z)
-180 -120 -60 0 60 120 180
C-C-O-H torsion angle (deg)
Figure 2. 9 Dependence of 3JC1,O2H on the C 1-C2-O2-H tor sion angle based on equation 2.11 on which are superimposed 3JC2,O3H and 3JC3,O2H
values in 9-12 computed for perfectly staggered C-C-O-H rotamers (60°, -60° and 180°). Couplings are reduced at 180° for those pathways that lack an “in-plane” OH substituent on the coupled carbon (see text).
2.3.2.3 Couplings Involving C1-O1 bond
The behavior of 3JC2,O1H (Scheme 2. 2 s tructure IIa) mimics that of 3JH1,O1H
with r espect to the ef fects of inte rnal electr onegative s ubsuttuents. One of the gauche
couplings (IIb ) is in fluenced b y th e anti-oriented O 5, w hereas th e o ther (IIc ) is n ot,
leading to asymmetry in the 3JC2, O1H Karplus curve. Superimposed on this effect is the
effect of an in- plane ter minal electr onegative s ubstituent f ound in the -Man
configuration and that of an in -plane car bon substituent in the -Man and -Glc
configurations. No ter minal ef fects ar e po ssible in the -Glc conf iguration, but the
internal s ubstituent effect remains. Karplus curv es derived for the four configu rations
(Figure 2.4B) were f it to give th ree unique equations 2.14 2.16, since the cur ves for -

51
Man and -Glc ar e vir tually identical ( separated equations f or -Man and -Glc a re
plotted but not shown here).
3JC2,O1H ( -Glc) = 2.85 1.67 cos + 3.20 cos (2 ) 0.25 sin + 1.22 sin (2 ) 2. 14
R2= 0.99
3JC2,O1H ( -Man) = 3.60 3.35 cos + 4.51 cos (2 ) 0.029 sin + 0.85 sin (2 ) 2. 15
R2= 0.99
3JC2,O1H ( -Glc/ -Man) = 3.33 1.56 cos + 3.94 cos (2 ) 1.11 sin (2 ) 2. 16
R2= 0.99
2.3.2.4 Studies of 3JCCOH in Simpler Model Systems.
The effect of ter minal substituents on 3JCCOH values was studied in two s impler
model systems, ethylene glycol and n-propanol. In the former, the C-C-O-H torsion angle
was f ixed at 180º and the O- C-C-O torsion angle was varied in 30º increments through
360º. T he calculated 3JCCOH was enhanced s ignificantly by 4 -5 Hz at an O- C-C-O
torsion angle of 180º (J ~ 14 Hz) compared to co uplings at 60º and –60º (J ~ 9- 10 Hz)
(Figure 2.10). This result is consistent with that observed in the more complex saccharide
mimics. Calculations were conducted on n-propanol by s ystematically varying the C-C-
C-O tor sion angle in 30º increments while keepi ng the C -C-O-H tor sion angle f ixed at
180º. An enhanced coupling at a C -C-C-O torsion angle of 180º was observed ( Figure
2.10), although the ef fect is s maller ( ~2 Hz) than f ound in the diol. T hese r esults
demonstrate that “in-plane” terminal substituents enhance 3JCCOH values, but the degr ee
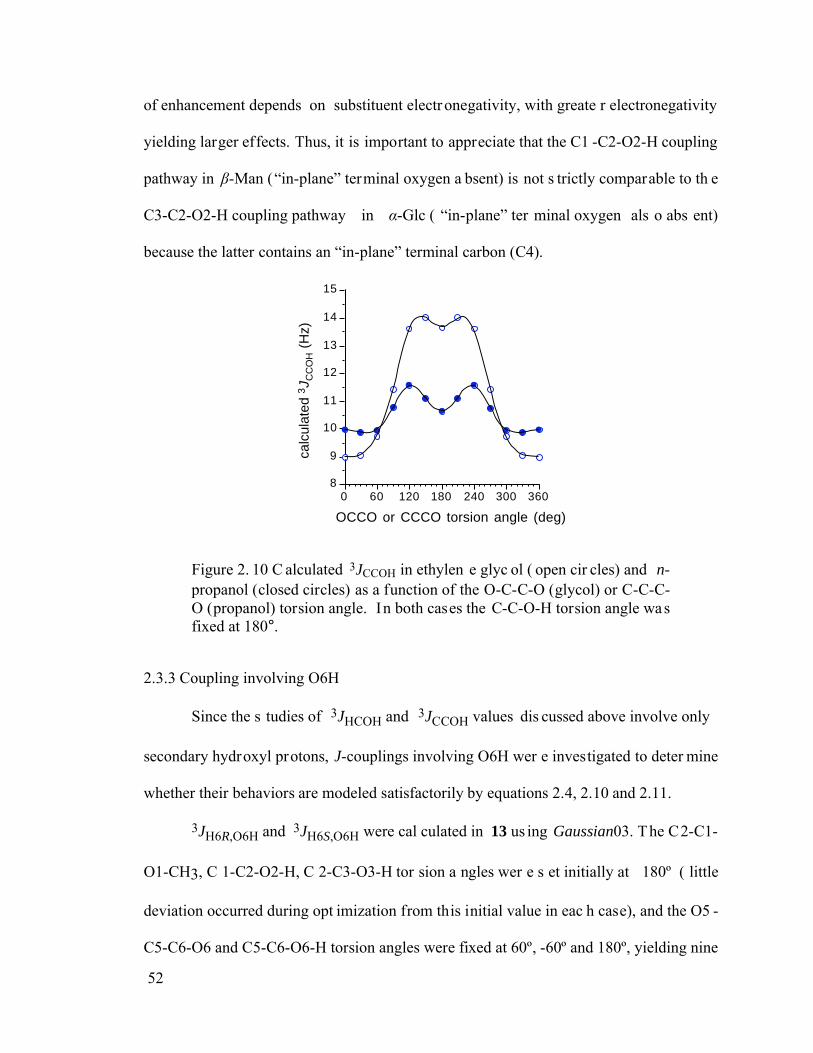
52
of enhancement depends on substituent electronegativity, with greate r electronegativity
yielding larger effects. Thus, it is important to appreciate that the C1 -C2-O2-H coupling
pathway in -Man (“in-plane” terminal oxygen a bsent) is not s trictly comparable to th e
C3-C2-O2-H coupling pathway in -Glc ( “in-plane” ter minal oxygen als o abs ent)
because the latter contains an “in-plane” terminal carbon (C4).
8
9
10
11
12
13
14
15ca
lcul
ated
3J C
CO
H (
Hz)
0 60 120 180 240 300 360
OCCO or CCCO torsion angle (deg)
Figure 2. 10 C alculated 3JCCOH in ethylen e glyc ol ( open cir cles) and n-propanol (closed circles) as a function of the O-C-C-O (glycol) or C-C-C-O (propanol) torsion angle. In both cases the C-C-O-H torsion angle was fixed at 180°.
2.3.3 Coupling involving O6H
Since the s tudies of 3JHCOH and 3JCCOH values dis cussed above involve only
secondary hydroxyl protons, J-couplings involving O6H wer e investigated to deter mine
whether their behaviors are modeled satisfactorily by equations 2.4, 2.10 and 2.11.
3JH6R,O6H and 3JH6S,O6H were cal culated in 13 us ing Gaussian03. The C2-C1-
O1-CH3, C 1-C2-O2-H, C 2-C3-O3-H tor sion a ngles wer e s et initially at 180º ( little
deviation occurred during opt imization from this initial value in eac h case), and the O5 -
C5-C6-O6 and C5-C6-O6-H torsion angles were fixed at 60º, -60º and 180º, yielding nine
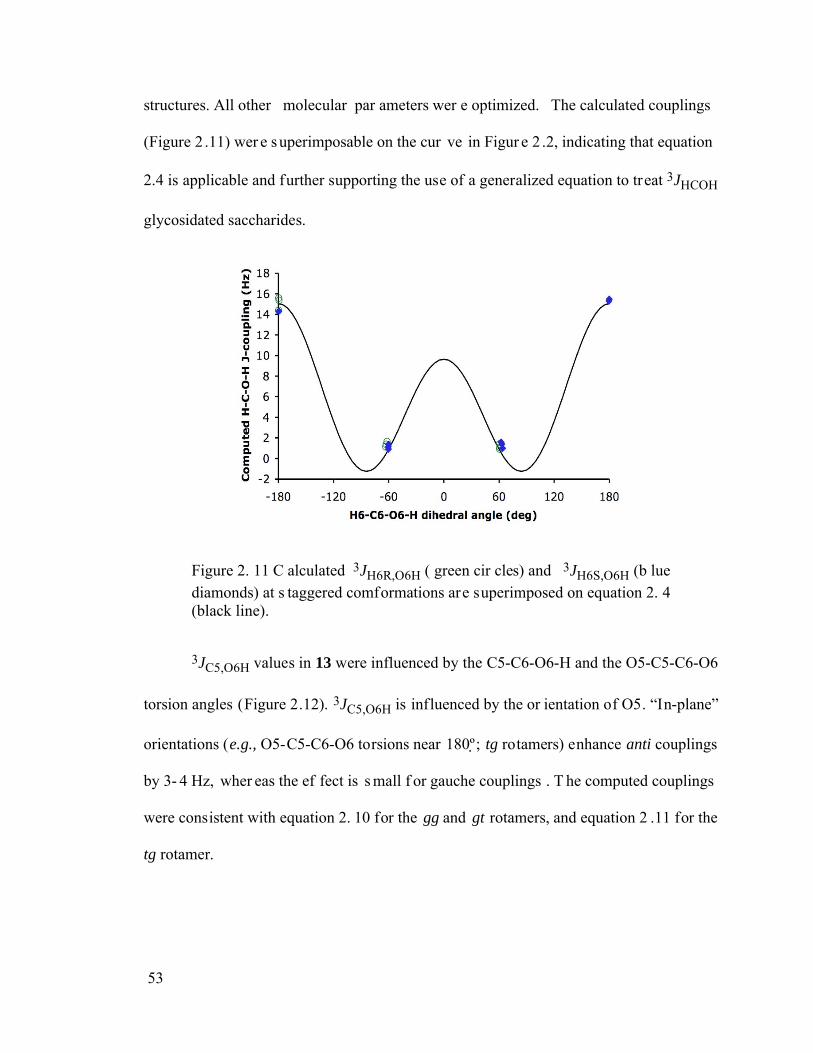
53
structures. All other molecular par ameters wer e optimized. The calculated couplings
(Figure 2 .11) wer e superimposable on the cur ve in Figur e 2 .2, indicating that equation
2.4 is applicable and further supporting the use of a generalized equation to treat 3JHCOH
glycosidated saccharides.
Figure 2. 11 C alculated 3JH6R,O6H ( green cir cles) and 3JH6S,O6H (b lue diamonds) at s taggered comformations are superimposed on equation 2. 4 (black line).
3JC5,O6H values in 13 were influenced by the C5-C6-O6-H and the O5-C5-C6-O6
torsion angles (Figure 2.12). 3JC5,O6H is influenced by the or ientation of O5. “In-plane”
orientations (e.g., O5-C5-C6-O6 torsions near 180º; tg rotamers) enhance anti couplings
by 3- 4 Hz, wher eas the ef fect is s mall f or gauche couplings . T he computed couplings
were consistent with equation 2. 10 for the gg and gt rotamers, and equation 2 .11 for the
tg rotamer.
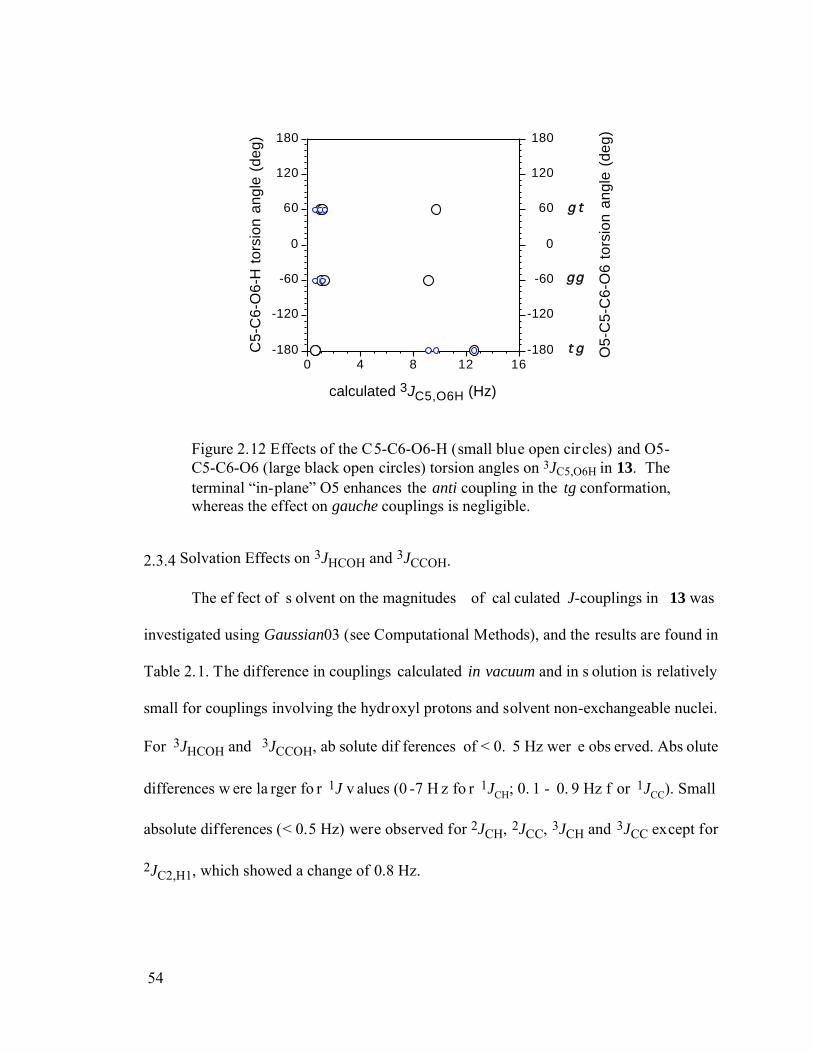
54
-180
-120
-60
0
60
120
180
O5-
C5-
C6-
O6
tors
ion
angl
e (d
eg)
-180
-120
-60
0
60
120
180
C5-
C6-
O6-
H t
orsi
on a
ngle
(de
g)
0 4 8 12 16
calculated 3JC5,O6H (Hz)
t g
gg
g t
Figure 2.12 Effects of the C5-C6-O6-H (small blue open circles) and O5-C5-C6-O6 (large black open circles) torsion angles on 3JC5,O6H in 13. The terminal “in-plane” O5 enhances the anti coupling in the tg conformation, whereas the effect on gauche couplings is negligible.
2.3.4 Solvation Effects on 3JHCOH and 3JCCOH.
The ef fect of s olvent on the magnitudes of cal culated J-couplings in 13 was
investigated using Gaussian03 (see Computational Methods), and the results are found in
Table 2.1. The difference in couplings calculated in vacuum and in s olution is relatively
small for couplings involving the hydroxyl protons and solvent non-exchangeable nuclei.
For 3JHCOH and 3JCCOH, ab solute dif ferences of < 0. 5 Hz wer e obs erved. Abs olute
differences w ere la rger fo r 1J v alues (0 -7 H z fo r 1JCH; 0. 1 - 0. 9 Hz f or 1JCC). Small
absolute differences (< 0.5 Hz) were observed for 2JCH, 2JCC, 3JCH and 3JCC except for
2JC2,H1, which showed a change of 0.8 Hz.
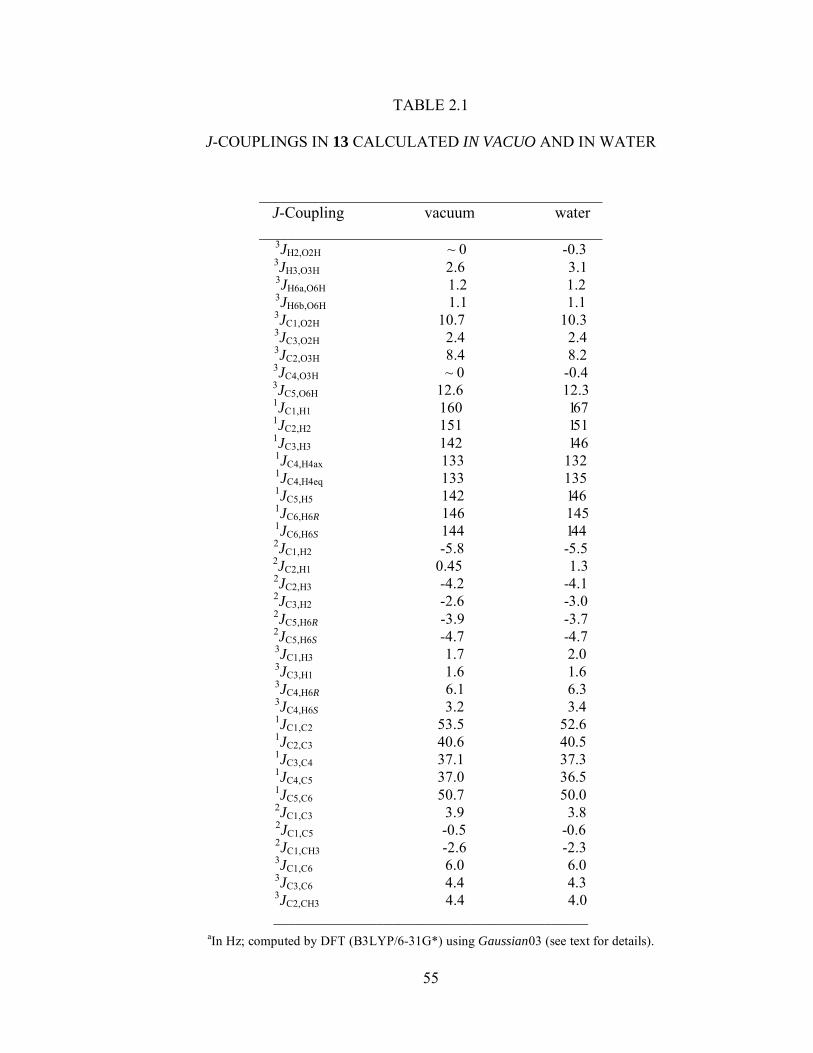
55
TABLE 2.1
J-COUPLINGS IN 13 CALCULATED IN VACUO AND IN WATER
___________________________________________ J-Coupling vacuum water
___________________________________________ 3JH2,O2H ~ 0 -0.3 3JH3,O3H 2.6 3.1 3JH6a,O6H 1.2 1.2 3JH6b,O6H 1.1 1.1 3JC1,O2H 10.7 10.3 3JC3,O2H 2.4 2.4 3JC2,O3H 8.4 8.2 3JC4,O3H ~ 0 -0.4 3JC5,O6H 12.6 12.3 1JC1,H1 160 167 1JC2,H2 151 151 1JC3,H3 142 146 1JC4,H4ax 133 132 1JC4,H4eq 133 135 1JC5,H5 142 146 1JC6,H6R 146 145 1JC6,H6S 144 144 2JC1,H2 -5.8 -5.5 2JC2,H1 0.45 1.3 2JC2,H3 -4.2 -4.1 2JC3,H2 -2.6 -3.0 2JC5,H6R -3.9 -3.7 2JC5,H6S -4.7 -4.7 3JC1,H3 1.7 2.0 3JC3,H1 1.6 1.6 3JC4,H6R 6.1 6.3 3JC4,H6S 3.2 3.4 1JC1,C2 53.5 52.6 1JC2,C3 40.6 40.5 1JC3,C4 37.1 37.3 1JC4,C5 37.0 36.5 1JC5,C6 50.7 50.0 2JC1,C3 3.9 3.8 2JC1,C5 -0.5 -0.6 2JC1,CH3 -2.6 -2.3 3JC1,C6 6.0 6.0 3JC3,C6 4.4 4.3 3JC2,CH3 4.4 4.0 ___________________________________________
aIn Hz; computed by DFT (B3LYP/6-31G*) using Gaussian03 (see text for details).

56
2.3.5 C-O-H Coupling Pathways.
3JHCOH and 3JCCOH values exhibit strong dep endencies on the c entral C -O
torsion angle, with dynamic r anges of 8-15 Hz. In contrast, 2JCOH values are relatively
small (-2 to -4 Hz), are negative, and display nonsystematic dependencies on C-C and C-
O tor sion angles ( Figure 2. 13). These r esults s uggest that 2JCOH will not be a us eful
parameter to assess C-O conformation in solution.
Figure 2.13 Hypersurface showing the dependence of 2JC2,O2H on and in 2.
2.4 Conclusions
Hydroxyl pr oton orientation in s olution is an impor tant component in
conformational studies of saccharides. The high density of OH groups in these structures
confers unique chemical and phys ical properties due to their ability to H -bond intra- and
intermolecularly, especially in the latter cas e with solvent water. Prior work has shown
that 1JCC [18] and 2JCCH [17]
in saccharides provide useful information about exocyclic C-

57
O rotamers in aqueous s olution. T his inf ormation, when us ed in conjunction with the
3JHCOH and 3JCCOH values s tudied her ein, s hould pr ovide firmer as signments of C -O
rotamer populations in solution.
The present investigation shows that 3JHCOH values (a) depend predominantly on
the H- C-O-H tor sion angle, ( b) a re lar gely unaf fected by adjacent C -O bond
conformation, (c) are largely unaffected by the type or location of the carbon bearing the
OH gr oup ( except f or thos e couplings s ensitive to the C 1-O1 tor sion), and ( d) ar e
modestly affected by non-Fermi contact contributions. These results provide firm support
for the us e of a gener alized Karplus equation to interpret 3JHCOH in s accharides.
However, s eparate equations ar e needed to treat 3JH1,O1H, because these couplings ar e
subject to the additional effect of in ternal electro negative s ubstituents. T he later ef fect
causes phases shifting of the Karplus curve partly due to the nonequivalent values of the
gauche couplings. While the n eed for a separate set of equations to treat the se couplings
prevents us e of a tr uly gener alized 3JHCOH eq uation, it s hould be appr eciated that
analysis of H1-C1-O1-H torsion are only per tinent in reducing sugars; most analyses are
likely to involve glycos ides wher ein only non -anomeric s ites will be the f ocus of
attention.
3JCCOH values are determined mainly by C -C-O-H tor sion angles , but the
orientation of ter minal s ubstituents on the cou pled car bon, s pecifically oxygen and
carbon, plays an impor tant s econdary r ole, with “in -plane” s ubstituents enhancing th e
coupling when the coupled atoms ar e anti. T he degr ee of enhancement depends on
substituent electronegativity, with greater electro negativity yi elding large r effects . Fo r
3JCCOH, two gener alized equations wer e der ived, one per tinent to coupling pathways

58
containing an “in -plane” OH or OR s ubstituent and one for cas es lacking this
arrangement. This effect is reminiscent of the beh avior of 3JCCOC, which i s affected by
the or ientation of ter minal electr onegative s ubstituents on both coupled c arbons. [14]
3JCOCH values may be influenced in a similar manner in saccharides, although the effect
is small and presently less well defined. [19]
Internal substutuent ef fects inf luence 3JC2,O1H in a manner s imilar to 3JH1,O1H,
and thus s eparate equations ar e r equired to tr eat this coupling. T he exact f orm o f the
equation depends on r elative conf iguration at C 1 and C 2, s ince the mix of s tructural
factors influencing this coupling varies with local structure.
The combined us e of 3JHCOH and 3JCCOH value s is required to determine C -O
rotamer populations in s olution, and the pa rameterized 3JCCOH equations des cribed
herein make thes e treatments possible. Fo r example, for the H2-C2-O2-H torsion in -
Glc 2, 3JH2,O2H, 3JC1,O2H and 3JC3,O2H exhibit the phase-shifted dependencies shown in
Figure 2. 14. E xperimental deter minations of th e thr ee OH -based couplings pr ovide
redundant information to determine rotameric populations with gr eater confidence. This
information can be combined with that obtained f rom 1JCC [17] and 2JCCH [14-16]
values to
improve these analyses. The latter two types of J-coupling provide indirect C-O torsional
information in that they do not depend on dir ect obs ervation of the OH pr oton. Site -
specific incorporation of 13C at one or more sites in the saccharide facilitates not only the
measurement of 3JCCOH value s, but als o the as signment of OH signals and the
measurement of complementary 1JCC and 2JCCH values.
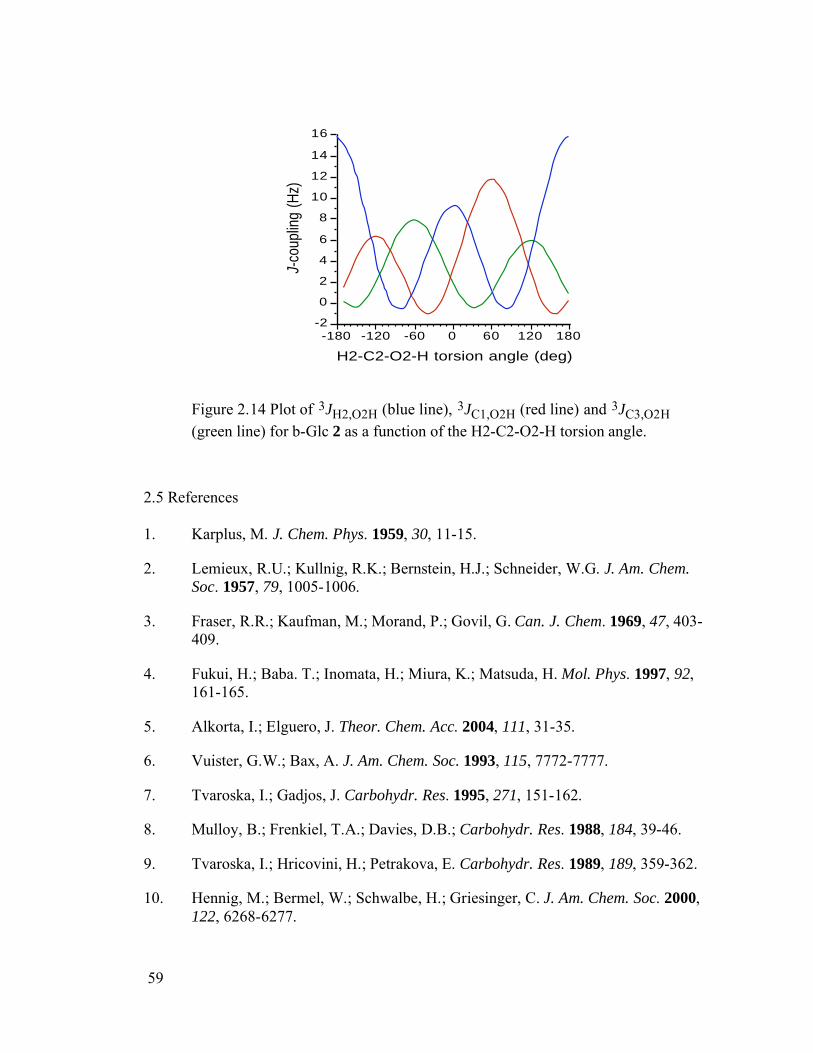
59
-2
0
2
4
6
8
10
12
14
16
J-co
uplin
g (H
z)
-180 -120 -60 0 60 120 180
H2-C2-O2-H torsion angle (deg)
Figure 2.14 Plot of 3JH2,O2H (blue line), 3JC1,O2H (red line) and 3JC3,O2H (green line) for b-Glc 2 as a function of the H2-C2-O2-H torsion angle.
2.5 References
1. Karplus, M. J. Chem. Phys. 1959, 30, 11-15.
2. Lemieux, R.U.; Kullnig, R.K.; Bernstein, H.J.; Schneider, W.G. J. Am. Chem.
Soc. 1957, 79, 1005-1006.
3. Fraser, R.R.; Kaufman, M.; Morand, P.; Govil, G. Can. J. Chem. 1969, 47, 403-409.
4. Fukui, H.; Baba. T.; Inomata, H.; Miura, K.; Matsuda, H. Mol. Phys. 1997, 92, 161-165.
5. Alkorta, I.; Elguero, J. Theor. Chem. Acc. 2004, 111, 31-35.
6. Vuister, G.W.; Bax, A. J. Am. Chem. Soc. 1993, 115, 7772-7777.
7. Tvaroska, I.; Gadjos, J. Carbohydr. Res. 1995, 271, 151-162.
8. Mulloy, B.; Frenkiel, T.A.; Davies, D.B.; Carbohydr. Res. 1988, 184, 39-46.
9. Tvaroska, I.; Hricovini, H.; Petrakova, E. Carbohydr. Res. 1989, 189, 359-362.
10. Hennig, M.; Bermel, W.; Schwalbe, H.; Griesinger, C. J. Am. Chem. Soc. 2000, 122, 6268-6277.

60
11. Adams, B.; Lerner, L. J. Am. Chem. Soc. 1992, 114, 4827-4829.
12. Sandström, C.; Basumann, L.; Kenne, J. J. Chem. Soc. Perkin Trans. 2 1998, 809-815.
13. Batta, G.; Kövér, K.E. Carbohydr. Res. 1999, 320, 267-272.
14. Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F.; Bondo, P.B.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 1998, 120, 11158-11173.
15. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 1999, 121, 9843-9851.
16. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2000, 122, 396-397.
17. Thibaudeau, C.; Stenutz, R.; Hertz, B.; Klepach, T.; Zhao, S.; Wu, Q.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2004, 126, 15668-15685.
18. Carmichael, I.; Chipman, D.M.; Podlasek, C.A.; Serianni, A.S. J. Am. Chem. Soc. 1993, 115, 10863-10870.
19. Podlasek, C.A.; Wu, J.; Stripe, W.A.; Bondo, P.B.; Serianni, A.S. J. Am. Chem.
Soc. 1995, 117, 8635-8644.
20. Becke, A.D. J. Chem. Phys. 1993, 98, 5648-5652.
21. Hehre, W.J.; Ditchfield, R.; Pople, J.A. J. Chem. Phys. 1972, 56, 2257-2261.
22. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, Jr., J.A.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Baboul, A.G.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian98; Revision A.9, Gaussian, Inc.: Pittsburgh, PA, 1998.
23. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Phys. Chem. 1999, 19, 3783-3795.
24. Cloran, F.; Zhu, Y.; Osborn, J.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2000, 122, 6435-6448.
25. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2001, 123, 4781-4791.

61
26. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; AlLaham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Peng, C. Y.; Ayala, P.Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian94; Gaussian, Inc.: Pittsburgh, PA, 1995.
27. Stenutz, R.; Carmichael, I;. Widmalm, G.; Serianni, A.S. J. Org. Chem. 2002, 67, 949-958.
28. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, Jr., J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian03, Revision A.1, Gaussian, Inc., Pittsburgh PA, 2003.

62
CHAPTER 3:
APPLICATION OF 3JHCOH AND 3JCCOH KARPLUS EQUATIONS IN
OLIGOSACCHARIDES IN DMSO AND IN AQUEOUS SOLUTION
3.1 Introduction
The interaction between carbohydrates and proteins plays a critical role in protein
folding and p rocessing, cell -cell adhes ion and recognition, and def ense of immune
system.[1]. Hydr ogen bonding is one of the non- covalent bonding f orces responsible for
protein-carbohydrate binding. [2,3] C arbohydrates contain multiple hydr oxyl gr oups that
can s imultaneously donate and accept pr otons to f orm hydr ogen bonds .[4] T hese
hydrogen bonds can either be direct or solvent mediated, with the best example being the
binding of N-acetyl-D-galactosamine (GalNAc) or N-acetyl-D-glucosamine (GlcNAc) to
the Sclerotium rolfsii lectin (SR L).[5] T he co nformations of OH gr oups, or mor e
specifically, of the C -O bond, af fect the formation and s trength of inter- or intr a-
molecular hydrogen bonding in saccharides.[6]
NMR vicinal spin-spin coupling constants (3J) are useful experimental parameters
to investigate molecular conformation in solution since their magnitudes and signs can be
related to the molecular tor sion angles between coupled nuclei thr ough the Kar plus
relationship.[7] Although 3JHCOH and 3JCCOH values are potentially useful parameters to
investigate intra- and/or intermolecular hydrogen bonding in OH-containing structures in

63
solution,[6,8-10] few s tudies, es pecially of 3JCCOH, have been r eported on thei r
parameterization and application in carbohydrate systems. Par ameterization of 3JHCOH
and 3JCCOH values has relied heavily on comput ational methods due to limited access to
experimental data. C hapter 2 of this di ssertation des cribed the par ameterization of
3JHCOH and 3JCCOH values using the DFT method.
The applications of 3JHCOH and 3JCCOH values have been very limited partly due
to two r easons. Fir st, hydr oxyl pr otons ar e hig hly exchangeable with s olvent, s o the
measurements of J-coupling constants require the use of either an apr otic solvent, or a
supercooled water solvent to eliminate or reduce the exchange rate between the s olute
OH pr otons gr oups and the bulk s olvent. Di methyl s ulfoxide ( DMSO) o r a mixed
water/aprotic solvent at sub-zero temperatures have been commonly used.
Secondly, to obtain accur ate heter onuclear J-coupling cons tants, 3JCCOH, 13C-
labeled oligos accharides are either h ighly des irable or r equired. Due to complicated
vicinal and/or ger minal coupling pa tterns between car bon and hydr oxyl pr otons,
uniformly 13C-labeled oligosaccharides, however, will yield significantly complicated 1H
NMR spectra. T he pr esence of contiguous 13C-labeling in the s accharide r esidues no t
only complicates the as signment of OH gr oups, but also makes the meas urement of J-
coupling constants considerably more difficult. Thus, site-specific incorporation of 13C
at one or mor e s ites is the pr eferred strategy to enable not only the meas urement of
accurate 3JCCOH values but als o to allow the correct assignment of OH signals and th e
measurement of complementary 1JCC,[11] 2JCCH[12] and 2JCC
[13] spin-coupling constants,
which are also sensitive to C-O bond conformation.

64
In this chapter, vicinal spin-coupling constants involving the hydr oxyl protons in
singly 13C-labeled methyl glu copyranosides and s everal di -13C-labeled di saccharides
have been measured in either DMSO-d6 solvent or supercooled H2O. Karplus equations
parameterized in C hapter 2 have been a pplied to inves tigate C -O bond tor sions and
potential intramolecular H-bonding in a di-13C-labeled -(1 4)-linked disaccharide. We
show that C -O bond r otations in s olution ar e bia sed in both mono - and dis accharides.
The data also suggest that persistent inter-residue H-bonding might exist in both solvents,
a res ult having important implications fo r th e identif ication of s tructural factors
influencing the conformation and dynamics of glycosidic linkages in solution.
OHO
HOOH
OCH3
OH
O
OH
HOOH
OCH3
OH
OH
OO
HOOH
OH
1 2
3
OHO
HOOH
OCH3
OH
O
OH
HOOH
OCH3
OH
4 5
6
1'2'3'
4'5'
6'
O
OH
HOOH
OH
12
3
456
OH
OO
HOOH
OH
O
OH
HOOH
OH
= 13C
Scheme 3. 1 St ructures of compounds 1 - 6 an d locations of the 13C labeling.
3.2 Experimental Methods
Unlabeled monos accharides, methyl -D-glucopyranoside 1 and methyl -D-
galactopyranoside 2 were purchased f rom Sigma C hemical Company and used without
further purification. Methyl -lactoside 3, methyl -D-[4-13C]glucopyranoside 4, methyl
-D-[1-13C]galactopyranoside 5 and methyl -[1,4’-13C2]lactoside 6 were synthesized as

65
described previously.[14,15]
Acetone-d6 and DMSO-d6 (99.9 atom-% 2H) were purchased
from Cambridge Isotope Laboratories. Methyl - and -D-[1-13C], [2-13C], [3-13C], [4-
13C], [ 5-13C] and [6-13C]glucopyranosides wer e pr epared by methods des cribed by
Podlasek et al.[14]
(these syntheses were conducted by Mr. Wenhui Zhang in the Serianni
laboratory).
NMR samples in H2O/acetone-d6 solvent were prepared to minimize the presence
of contaminants that could catalyze hydr oxyl pr oton exchange.[16]
Dis tilled H 2O was
deionized and ultrafilte red to a res istance > 18 M using a M illipore Milli-Q apparatus,
degassed at a spirator pr essure in an ult rasonic b ath, and s aturated with N 2 gas. NMR
tubes (3 mm) were soaked for a minimum of 1 h in 50 mM sodium phosphate buffer (pH
7), rinsed with high resistance dis tilled water , and dr ied under a s tream of N 2. NMR
samples wer e pr epared in a s ealed glove b ox under an ar gon atmos phere us ing the
degassed H2O and NM R tubes described above. Solute concentr ation was ~50 m M for
all samples. A 1:1 (v/v) H2O/acetone-d6 solvent was used for the monosaccharides, and
1:1 or 2:3 (v/v) H2O/acetone-d6 solvents were used for the disaccharides.
NMR e xperiments w ere p erformed o n a 6 00-MHz FT-N MR s pectrometer
operating at –20 °C to r educe the exchange r ate between bulk H 2O and the s olute
hydroxyl protons. 1D 1H NMR spectra were collected with an 8000 Hz s pectral window
and a recycle time of 3 s . FIDs were zero-filled twice to give final digital res olutions of
~0.06 Hz/pt, and wer e Four ier-transformed with or without weighting f unctions
depending on the spectral resolution.

66
2D 1H-1H TOCSY spectra were obtained with a spectral window of 2400 Hz in
both dimensions, H2O pre-saturation, and a 65 ms mixing time. The initial matrix (2K x
256) was zero-filled to yield a f inal 4K x 2K matrix apodized with a gaussian function in
both dimensions.
Molecular dynamics (MD) simulations were conducted us ing both CHARMM[17]
(version c28b1) and Amber [18] (version 8.0) and were done by Mr. Xiaosong Xu in the
Serianni labor atory. C arbohydrate Solut ion F orce Field ( CSFF)[19] was us ed in
CHARMM program. GLYCAM-04 carbohydrate force f ield[20-22] was used in AM BER
simulations.
3.3 Results and Discussion
3.3.1 Quality of spectra and further treatment of samples
Since ions can catalyze the exchange rate betw een hydr oxyl pr otons and bulk
water s olvent, the r emoval of ions f rom s olution is cr itical. T hough mo st of s amples
obtained as de scribed in the E xperimental S ection wer e sufficiently pur e to give high -
quality NMR spectra, some initially contained significant ions that broadened the signals
noticeably. T o remove these ions, these samples were treated with ion-exchange resins
(H+ and OAc -). F irst the r esins w ere was hed e xtensively by DI water . T hen a s mall
amount of s ynthesized oligos accharide s ample was dis solved in DI water with added
washed resins. The mixture was stirred to allow maximum exchange between resins and
traces of ions in sample. The resulting solutions were concentrated to dryness to remove
acetic acid gener ated by the ion -exchange pr ocess. For s ome s amples, two or more

67
repeated treatments were done to r emove all ions. This treatment significantly improved
the quality of the NMR spectra, especially for the aqueous samples.
3.3.2 Experimental studies of monosaccharides
3.3.2.1 3JHCOH and 3JCCOH values in monosaccharides: DMSO-d6 solvent
To obtain 3JHCOH and 3JCCOH values in methyl - and -D-glucopyranosides,
twelve s ingly 13C-labeled ( six isotopomers f or e ach anomer ) wer e synthesized. T hese
singly-labeled is otopomers wer e cho sen to r emove the potential complications in the
measurement of these J-couplings caused by the presence of germinal J-couplings (2J) to
adjacent 13C nuclei. 3JHCOH and 3JCCOH valu es wer e m easured in DM SO-d6, with
special f ocus on the s econdary hydr oxyl gr oups at C 2, C 3 and C 4. E ach o f thes e
hydroxyl gr oups is pr obed by thr ee r edundant J-couplings to determine C -O rota meric
populations. T he 1H s ignal as signments of eac h hydr oxyl pr oton s ignal r eported in
previous wor k [6] wer e conf irmed by the obs erved ef fect of the individual 13C-labeled
sites on signal multiplicities.
Shown in Figur e 3. 1 is a r epresentative par tial 1H NM R spectrum of a 13C-
labeled methyl glucopyr anoside ( in this cas e, methyl -D-[4-13C]glucopyranoside) in
DMSO-d6, s howing the well- resolved s ignals f rom the f our OH p rotons. J-Couplings
involving the OH gr oups in the complete series of 13C-labeled methyl - and -
glucopyranosides are summarized in Table 3.1. Values shown in the first column are the

68
Figure 3. 1 T he par tial 600 M Hz 1H NM R s pectrum of methyl -D-[4-13C]glucopyranoside in DM SO-d6 at 25 °C showing only the OH r egion of the s pectrum. The assignments of the f our signals (left to right) are as follows: O2H, O3H, O4H and O6H.
3JHCOH values and thos e in the remaining co lumns ar e either vicinal C -C-O-H or
germinal C-O-H spin-coupling constants. 3JHCOH values range from 4.8 Hz to 6. 5 Hz,
and 3JCCOH values range from 1.5 Hz to 4.0 Hz; both are assumed to be positive in sign.
3JHCOH and 3JCCOH values differ from site to site in a given monosaccharide, indicating
qualitatively different rotational biases between sites. Interestingly, when comparing two
monosaccharides, 3JHCOH and 3JCCOH value s dif fer s ignificantly at the O2H and O3H
sites, but are s imilar at O4H and O6H. C onsidering the direct relationship between
vicinal J-couplings and molecular dihedr al angles ( Karplus r elation), anomer ic
configuration at C 1 appear s to aff ect C2-O2 bon d conformation, and this ef fect can b e
extended to the C3- O3 bnd, which suggests interactions between adjacent C -O groups in

69
saccharides. A change at C 1 conf iguration has little ef fect on C 4-O4 and C 6-O6 bond
conformation, presumably because these sites are remote from the anomeric center.
TABLE 3.1
J-COUPLINGS (IN Hz) INVOLVING THE HYDROXYL PROTONS IN METHYL -
D-GLUCOPYRANOSIDE AND METHYL -D-GLUCOPYRANOSIDE IN DMSO-d6
AT 25 °C
___________________________________________ J-Coupling M Glc M Glc
___________________________________________ 3JH2,O2H 6.5 4.8 3JH3,O3H 4.9 4.8 3JH4,O4H 5.6 5.2 3JH6,O6H 6.0 6.0 3JC1,O2H 2.9 2.4 3JC3,O2H 1.8 3.5 3JC2,O3H 2.6 2.3 3JC4,O3H 1.7 2.9 3JC3,O4H 1.8 1.9 3JC5,O4H 3.6 4.0 3JC5,O6H 1.8 1.8 2JC2,OH2 -2.5 -2.5 2JC3,OH3 -2.4 -2.5 2JC4,O4H -2.3 -2.3
__________________________________________________ In Hz; + 0.1 Hz for H-H couplings, + 0.2 for C-H couplings.
3.3.2.2 Populations of hydroxyl groups calculated from Karplus relationships
Karplus equations for 3JHCOH and 3JCCOH spin-couplings described in Chapter 2
were used to tr eat exper imental J-couplings. A three-state model ha s been commonly
invoked to treat rotational isomerism about single bonds by NMR on systems with rapid
rotamer inter conversion,[23] and was adopted her ein f or the tr eatment o f C -O bond
rotamers. T hree idealized s taggered conformers (Figure 1.4), defined as g+ (+60), g- (-

70
60) and t (180), were assumed to exist in fast chemical exchange on the NMR time-scale
and are in dynamic equilibrium in solution. This treatment of C-O bond conformation is
similar to the appr oach used for exocyclic hydr oxymethyl group analysis previously.[24]
The populations ( i.e., mole f ractions) of each s taggered rotamer are def ined as Pg+, Pg-
and Pt, respectively. Equations 3. 1 – 3. 4 wer e used to calculate the population (mole
fraction) of each rotamer:
3JHn,OnH = (PHn,g+)(3JHn,g+) + (PHn,g-)(3JHn,g-) + (PHn,t)(3JHn,t) 3. 1
3JCn+1,OnH = (PCn+1,g+)(3JCn+1,g+) + (PCn+1,g-)(3JCn+1,g-) + (PCn+1,t)(3JCn+1,t) 3. 2
3JCn-1,OnH = (PCn-1,g+)(3JCn-1,g+) + (PCn-1,g-)(3JCn-1,g-) + (PCn-1,t)(3JCn-1,t) 3. 3
Pg+ + Pg- + Pt = 1 3.4
The subscripts in each J and P term in eqs 3.1 – 3.4 denote the specific C-O bond
under consideration and the reference atoms used to describe it, and 3Jt, 3Jg+ and 3Jg- are
the J-couplings as sociated with each s taggered r otamer calculated f rom Kar plus
equations des cribed in C hapter 2. Since C -C-O-H pathways ar e not s tructurally
equivalent, two dif ferent Karplus equations for 3JCCOH were used. Due to contr ibutions
from in-plane electronegative substituent effects, the r ing oxygen can enha nce 3JC1,O2H
and 3JC5,O4H values, differentiating them from the other 3JCCOH values available in the
molecules.[6] Becaus e they de scribe the s ame C n-On bond tor sion, the f ollowing
equations hold:
Pg+ = PHn,g+ = PCn+1,g- = PCn-1, t 3. 5
Pg- = PHn, g- = PCn+1, t = PCn-1, g+ 3. 6

71
Pt = PHn, t = PCn+1, g+ = PCn-1, g- 3. 7
Pg+, Pg- and Pt are populations referenced to the vicinal ring proton, Hn. Thus,
equations 3.1 – 3.3 can be rewritten as:
3JHn, OnH = Pg+ 3JHn, g+ + Pg- 3JHn, g- + Pt 3JHn, t 3.8
3JCn+1, OnH = Pt 3JCn+1, g+ + Pg+ 3JCn+1, g- + Pg- 3JCn+1, t 3.9
3JCn-1, OnH = Pg- 3JCn-1, g+ + Pt 3JCn-1, g- + Pg+ 3JCn-1, t 3.10
The exper imental vicinal J-couplings s hown in T able 3. 1 wer e tr eated us ing
equations 3.4 and 3.8 – 3.10. Monte Carlo sampling within the ProFit program was used
to calculate populations that ga ve the s mallest over all er ror.2 T he populations o f each
bond rotamer were summarized in Table 3.2.
3JH2, O2H is larger in the -configuration (6.5 Hz) than in the -configuration (4.8
Hz), resulting in a higher trans HCOH population (44% vs 30%). Larger 3JHCOH values
have been pr eviously r eported f or equator ial hydroxyl gr oups adjacent to axial oxygen
subsitutents,[25] and electrostatics was suggested to be the underlying reason. In the trans
conformation, the OH dipole is anti- parallel to the adjacent axial C -O dipole, thus
minimizing the electrostatic energy. However, exceptions to this argument exist, such as
for O3H in - or -mannopyranosides,[25] indicating that multiple competing molecular
forces are probably responsible for the rotational bias.
2 P roFit 5. 4, c ommercial s oftware from Qua ntumSoft, wa s us ed. A bui lt-in Monte Ca rlo algorithms was used in all parameter fitting. The fitting was run a t least over night, and set of p arameters that had the smallest error was selected.

72
TABLE 3.2
CALCULATED ROTAMER POPULATIONS (+ 0.10) FOR METHYL
GLUCOPYRANOSIDES IN DMSO-d6
_____________________________________________________ Bonds and populations M Glc M Glc
_____________________________________________________ H2-C2-O2-H, Pt 0.44 0.30 H2-C2-O2-H, Pg+ 0.33 0.24 H2-C2-O2-H, Pg- 0.27 0.46 H3-C3-O3-H, Pt 0.32 0.30 H3-C3-O3-H, Pg+ 0.28 0.39 H3-C3-O3-H, Pg- 0.40 0.31 H4-C4-O4-H, Pt 0.37 0.33 H4-C4-O4-H, Pg+ 0.28 0.28 H4-C4-O4-H, Pg- 0.35 0.39
_____________________________________________________________ All calculations are using ProFit, and based on J-coupling constants from Table 3.1.
It is noteworthy that C -O rotamers within the aldopyranosyl ring of methyl -D-
glucopyranoside ( Table 3. 2) adop t a co unterclockwise ar rangement, namely,
O4H O3H O2H O1 in DM SO-d6 s olvent at 25 °C. T his behavior indicates
interactions, possibly H-bonding, between the adjacent C -O groups. This intramolecular
cooperativity is cons istent with pr ior r esults s howing tha t hydr oxyl gr oups in phenyl-
substituted -glucopyranosides ar e bias ed t owards a similar counter clockwise
arrangement in the gas phase.[24,26] In contrast, methyl -D-glucopyranoside prefers trans
conformations for the C2-O2 and C 4-O4 bonds, and a gauche conformation for C3-O3
bond (Table 3.2). It thus seems that the presence of axial C-O bonds in the aldopyranosyl
ring and/or solvent affect rotational preference. For example, phenyl -mannopyranoside
shows a bias towards a clockwise arrangement, while singly-hydrated phenyl-substituted

73
-glucopyranosiodes adopt either a clockwise or counterclockwise pattern, depending on
the binding preference of the water molecule.[26]
3.3.2.3 Comparisons with molecular dynamics simulation data
Bias in C-O tors ions in methyl D-glucopyranosides wa s f urther inves tigated by
molecular dynamics simulations in water solvent, with the populations s ummarized in
Table 3. 3. B oth the AM BER and CHARMM programs wer e us ed, and they yiel d
different results. CHARMM appears to overemphasize intramolecular interactions. As a
result, it return s much lower trans C -O r otamer populations than pr edicted f rom the
analysis of exper imental J-coupling data. C -O Rotamer populations r eturned f rom the
AMBER simulations appe ar to be in better agree ment with the J-coupling analysis; for
example, the AM BER s imulations s upport the counterclockwise O4H -O3H-O2H-O1
pattern in methyl -D-glucopyranoside suggested by NMR, and give a higher trans C2-
O2 rotamer population in methyl -D-glucopyranoside.
3.3.2.4 3JHCOH and 3JCCOH spin-couplings in monosaccharides in aqueous solution
Approximately one equivalent of wa ter molecule s was pres ent in the 1H NM R
samples pr epared in DM SO-d6 s olvent, as determined fro m the in tegration of a weak,
relatively broad signal at ~3 ppm in the spectra. These water molecules arose from either
the sample itself, or from the solvent. Importantly, however, this small amount of water
is highly detr imental to 1H NM R quality when the s ample contains ion s. T he latter
significantly enhance the r ate of exchange between the OH hydrogens of the saccharide
and those of water, reducing spectra signal-to-noise and spectral resolution.

74
TABLE 3.3
C-O ROTATIONAL POPULATIONS BASED ON SOLVATED AMBER AND
CHARMM MOLECULAR DYNAMICS SIMULATIONS OF METHYL - AND -D-
GLUCOPYRANOSIDES
______________________________________________________ Bonds and populations CHARMM AMBER
_____________________________________________________ Methyl -D-glucopyranosides
H2-C2-O2-H, Pt 0.10 0.40 H2-C2-O2-H, Pg+ 0.62 0.30 H2-C2-O2-H, Pg- 0.28 0.30 H3-C3-O3-H, Pt 0.02 0.25 H3-C3-O3-H, Pg+ 0.50 0.46 H3-C3-O3-H, Pg- 0.48 0.29 H4-C4-O4-H, Pt 0.04 0.21 H4-C4-O4-H, Pg+ 0.30 0.09 H4-C4-O4-H, Pg- 0.66 0.70
Methyl -D-glucopyranosides
H2-C2-O2-H, Pt 0.02 0.21 H2-C2-O2-H, Pg+ 0.53 0.34 H2-C2-O2-H, Pg- 0.45 0.45 H3-C3-O3-H, Pt 0.03 0.18 H3-C3-O3-H, Pg+ 0.52 0.50 H3-C3-O3-H, Pg- 0.45 0.32 H4-C4-O4-H, Pt 0.02 0.20 H4-C4-O4-H, Pg+ 0.28 0.11 H4-C4-O4-H, Pg- 0.70 0.69
_____________________________________________________________
The obs ervation of hydr oxyl pr otons in aqueous s olution by 1H NM R is even
more challenging, largely due to the much gr eater rate of exchange between the OH and
bulk water protons. Supercooled H2O/acetone-d6 at -20 °C was used as a solvent instead
of pure H2O in the pres ent work to reduce this e xchange rate. 3JHCOH spin-couplings
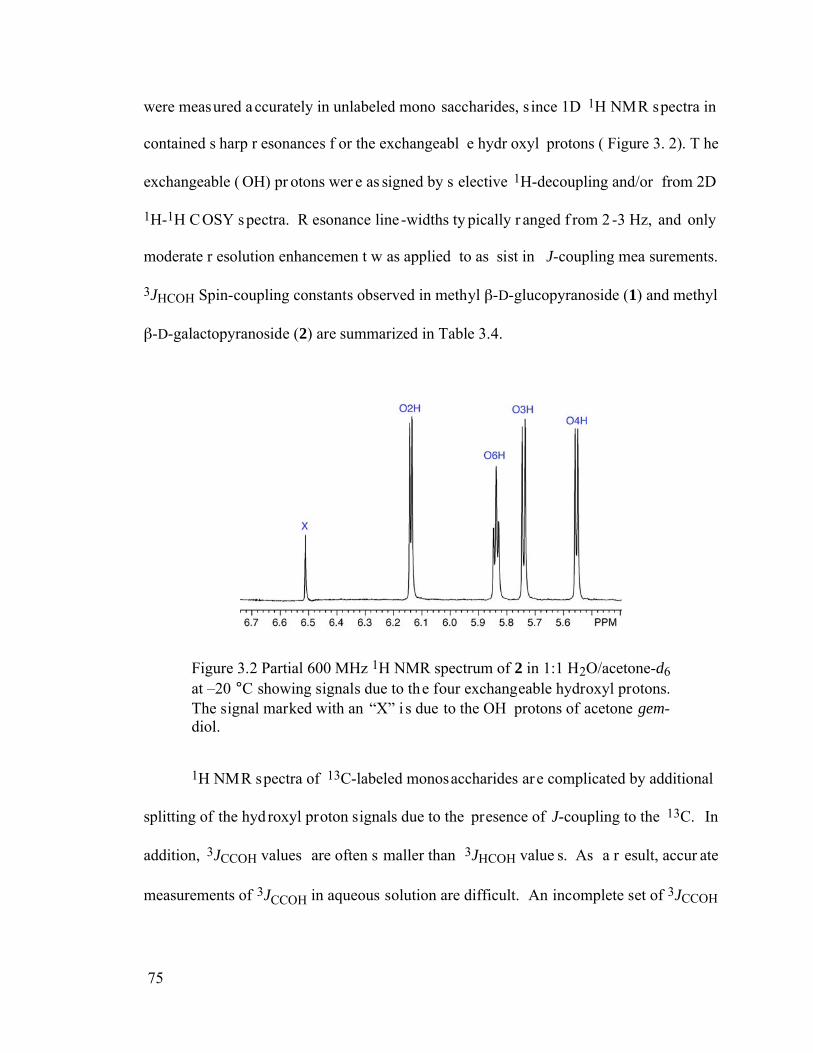
75
were measured a ccurately in unlabeled mono saccharides, s ince 1D 1H NMR spectra in
contained s harp r esonances f or the exchangeabl e hydr oxyl protons ( Figure 3. 2). T he
exchangeable ( OH) pr otons wer e as signed by s elective 1H-decoupling and/or from 2D
1H-1H COSY spectra. R esonance line -widths ty pically r anged f rom 2 -3 Hz, and only
moderate r esolution enhancemen t w as applied to as sist in J-coupling mea surements.
3JHCOH Spin-coupling constants observed in methyl -D-glucopyranoside (1) and methyl
-D-galactopyranoside (2) are summarized in Table 3.4.
Figure 3.2 Partial 600 MHz 1H NMR spectrum of 2 in 1:1 H2O/acetone-d6 at –20 °C showing signals due to the four exchangeable hydroxyl protons. The signal marked with an “X” i s due to the OH protons of acetone gem-diol.
1H NMR spectra of 13C-labeled monosaccharides are complicated by additional
splitting of the hydroxyl proton signals due to the presence of J-coupling to the 13C. In
addition, 3JCCOH values are often s maller than 3JHCOH value s. As a r esult, accur ate
measurements of 3JCCOH in aqueous solution are difficult. An incomplete set of 3JCCOH

76
values were measured in monos accharides in aqu eous solution, but wer e not us ed in the
rotational analysis.
TABLE 3.4
EXPERIMENTAL 3JHCOH AND 3JCCOH SPIN-COUPLING CONSTANTSa IN
H2O/ACETONE-d6 SOLUTIONb OF 1-6
________________________________________________________________________ J-couplings Methyl -lactoside (3/6) Methyl monosacharides Glc ring Gal ring M Glc (1/4) M Gal (2/5) 3JH2,O2H 4.5 5.1 4.6 4.5 3JH3,O3H 2.9 5.9 5.1 6.3 3JH4,O4H ---- 5.4 6.1 5.5 3JH6,O6H 5.6 4.7 6.6 5.5 3JC1,O2H ---- 1.5d ---- 2.2c
3JC4,O3H 1.1d ---- 1.9c ---- ______________________________________________________________________________
a In Hz + 0.1. b At – 20 °C; see Experimental Section for details. c + 0.2 Hz. d + 0.3 Hz
3.3.3 Experimental studies of disaccharides
3.3.3.1 3JHCOH and 3JCCOH in 3 and 6.
The measurement of 3JHCOH spin-couplings were the focus of attention in 3 and 6
due to the availability of 13C is otopomers, b ut s ome s everal 3JCCOH wer e als o
determined in thes e s tructures. M ethyl -lactoside ( 3) was chos en as the model
compound, and its 1D 1H NM R spectrum is shown in Figur e 3. 3. I n 3, the non-
exchangeable (CH) proton signals were assigned based on chemical s hifts data r eported
previously,[14,15]
and the hydroxyl proton signals were assigned from 2D 1H-1H TOCSY
spectra (Figure 3.4).

77
Figure 3.3 Partial 600 MHz 1H NMR spectrum of 3 in 2:3 H2O/acetone-d6 at –20 °C s howing s ignals due to the s even exchangeable hydr oxyl protons. The signal marked with an “X” is assigned to the OH p rotons of acetone gem-diol.
The 1D 1H NM R s pectrum of 3 in 1:1 H 2O/acetone-d6 at –20 °C contained
overlapping resonances for O2H and O2’H, and for O3H and O3’ H. A change in solvent
composition to 2:3 H2O/acetone-d6 eliminated these overlaps (Figure 3.3), yielding well-
resolved doublets f rom which 3JHCOH values co uld be meas ured r eliably ( Table 3. 4).
Similar analys es of s pectra of 1 and 2 gave th e J-couplings r eported in T able 3. 4.
3JH4,O4H and 3JH6,O6H in 2 are virtually identical to corresponding couplings observed in
the Gal moiety o f 3. Larger d ifferences w ere observed fo r 3JH2,O2H (~0.6 H z) a nd
3JC1,O2H (~0.7 Hz) , suggesting dif ferent preferred or ientations of the C 2-O2 bond in 2
and in the G al moiety of 3. 3JH2,O2H and 3JH6,O6H values in 1 are virtually identical to
corresponding values in the Glc moiety of 3, but 3JH3,O3H decreases significantly from
5.1 Hz in 1 to 2.9 Hz in 3.
With the exception of 3JH3’,O3’H in 3, exper imental 3JHCOH values involving
secondary alcohols range from 4.4 – 6.3 Hz (Table 3.4). Since theoretical studies show
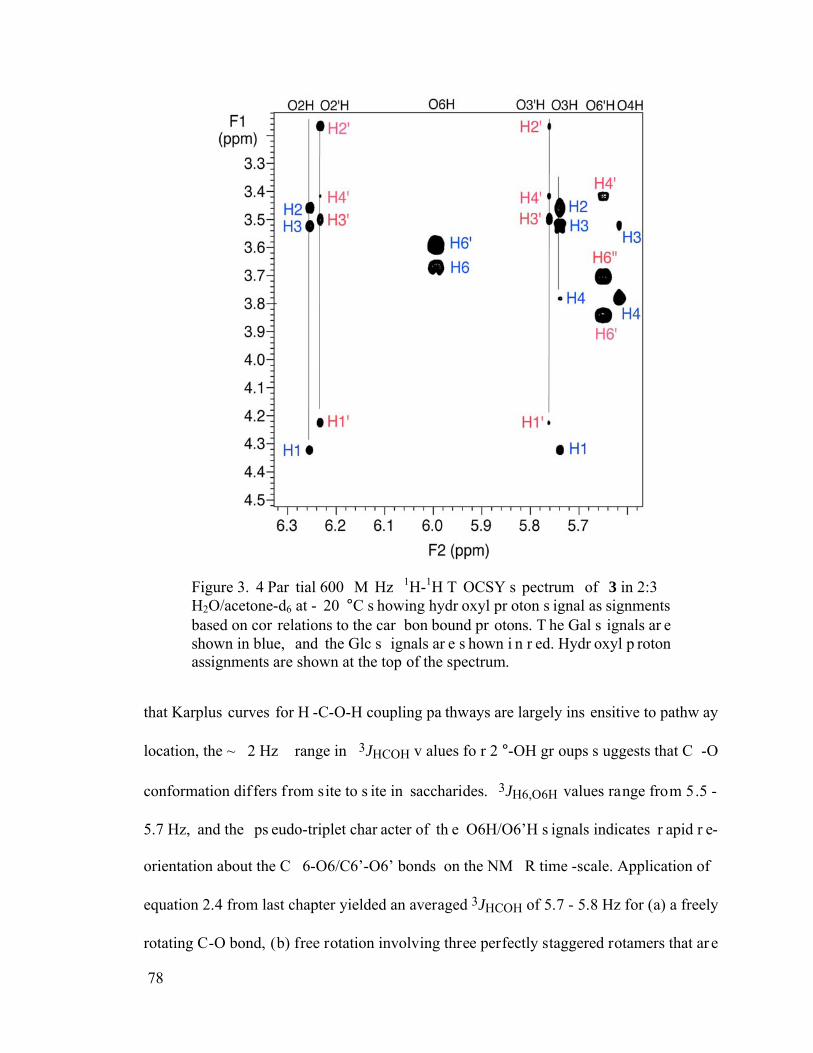
78
Figure 3. 4 Par tial 600 M Hz 1H-1H T OCSY s pectrum of 3 in 2:3 H2O/acetone-d6 at - 20 °C s howing hydr oxyl pr oton s ignal as signments based on cor relations to the car bon bound pr otons. T he Gal s ignals ar e shown in blue, and the Glc s ignals ar e s hown i n r ed. Hydr oxyl p roton assignments are shown at the top of the spectrum.
that Karplus curves for H -C-O-H coupling pa thways are largely ins ensitive to pathw ay
location, the ~ 2 Hz range in 3JHCOH v alues fo r 2 °-OH gr oups s uggests that C -O
conformation differs from site to s ite in saccharides. 3JH6,O6H values range from 5.5 -
5.7 Hz, and the ps eudo-triplet char acter of th e O6H/O6’H s ignals indicates r apid r e-
orientation about the C 6-O6/C6’-O6’ bonds on the NM R time -scale. Application of
equation 2.4 from last chapter yielded an averaged 3JHCOH of 5.7 - 5.8 Hz for (a) a freely
rotating C-O bond, (b) free rotation involving three perfectly staggered rotamers that ar e
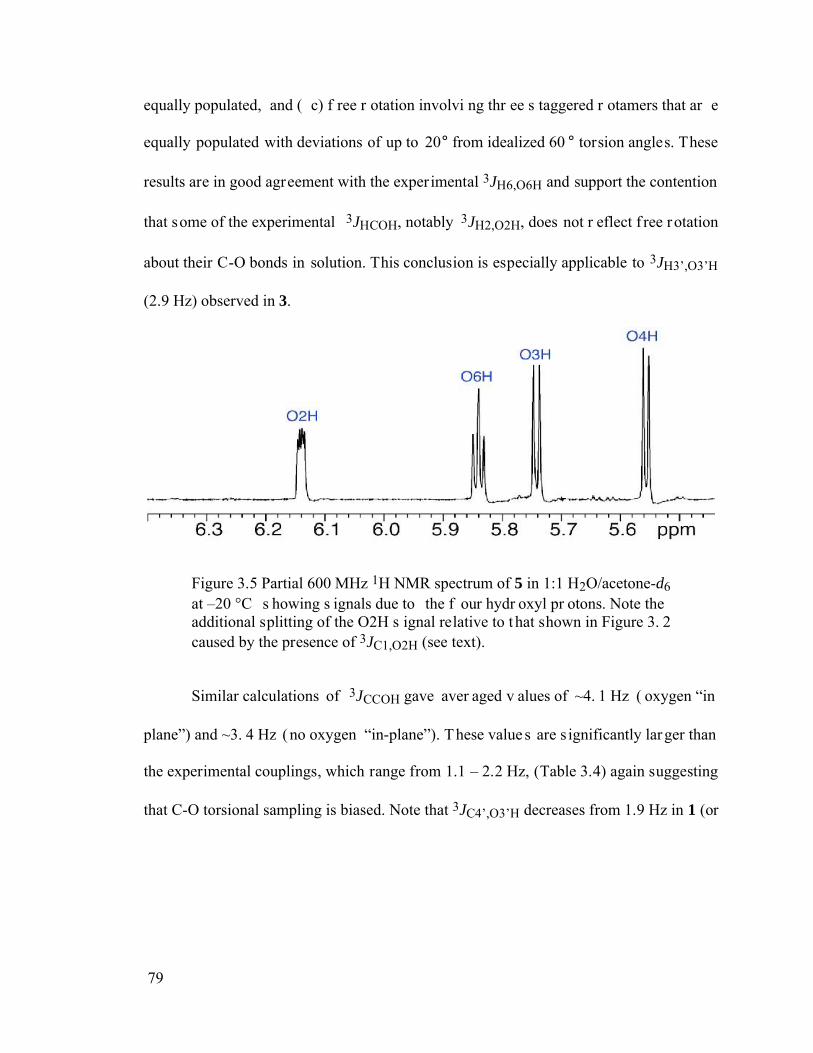
79
equally populated, and ( c) f ree r otation involvi ng thr ee s taggered r otamers that ar e
equally populated with deviations of up to 20° from idealized 60 ° torsion angles. These
results are in good agreement with the experimental 3JH6,O6H and support the contention
that some of the experimental 3JHCOH, notably 3JH2,O2H, does not r eflect f ree rotation
about their C-O bonds in solution. This conclusion is especially applicable to 3JH3’,O3’H
(2.9 Hz) observed in 3.
Figure 3.5 Partial 600 MHz 1H NMR spectrum of 5 in 1:1 H2O/acetone-d6 at –20 °C s howing s ignals due to the f our hydr oxyl pr otons. Note the additional splitting of the O2H s ignal relative to t hat shown in Figure 3. 2 caused by the presence of 3JC1,O2H (see text).
Similar calculations of 3JCCOH gave aver aged v alues of ~4. 1 Hz ( oxygen “in
plane”) and ~3. 4 Hz (no oxygen “in-plane”). These value s are s ignificantly larger than
the experimental couplings, which range from 1.1 – 2.2 Hz, (Table 3.4) again suggesting
that C-O torsional sampling is biased. Note that 3JC4’,O3’H decreases from 1.9 Hz in 1 (or
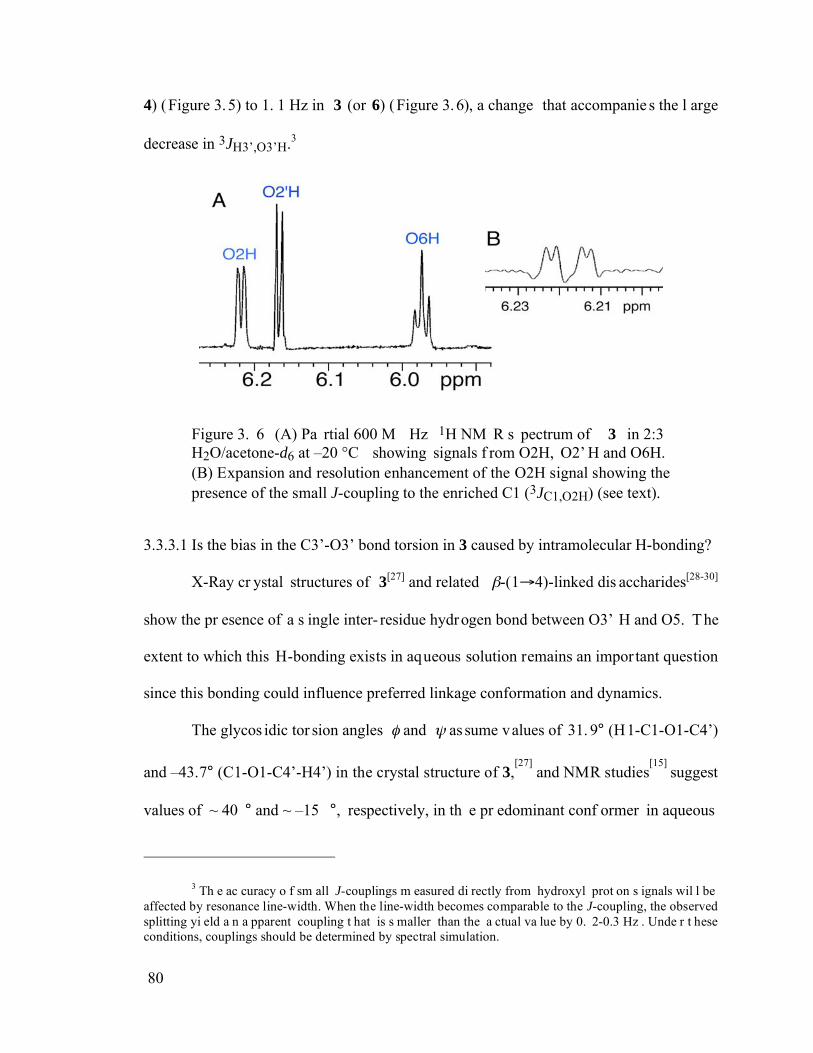
80
4) (Figure 3.5) to 1. 1 Hz in 3 (or 6) (Figure 3.6), a change that accompanie s the l arge
decrease in 3JH3’,O3’H.3
Figure 3. 6 (A) Pa rtial 600 M Hz 1H NM R s pectrum of 3 in 2:3 H2O/acetone-d6 at –20 °C showing signals f rom O2H, O2’ H and O6H. (B) Expansion and resolution enhancement of the O2H signal showing the presence of the small J-coupling to the enriched C1 (3JC1,O2H) (see text).
3.3.3.1 Is the bias in the C3’-O3’ bond torsion in 3 caused by intramolecular H-bonding?
X-Ray cr ystal structures of 3[27] and related -(1 4)-linked dis accharides[28-30]
show the pr esence of a s ingle inter- residue hydrogen bond between O3’ H and O5. The
extent to which this H-bonding exists in aqueous solution remains an impor tant question
since this bonding could influence preferred linkage conformation and dynamics.
The glycos idic tor sion angles and as sume values of 31. 9° (H1-C1-O1-C4’)
and –43.7° (C1-O1-C4’-H4’) in the crystal structure of 3,[27]
and NMR studies[15]
suggest
values of ~ 40 ° and ~ –15 °, respectively, in th e pr edominant conf ormer in aqueous
3 Th e ac curacy o f sm all J-couplings m easured di rectly from hydroxyl prot on s ignals wil l be affected by resonance line-width. When the line-width becomes comparable to the J-coupling, the observed splitting yi eld a n a pparent coupling t hat is s maller than the a ctual va lue by 0. 2-0.3 Hz . Unde r t hese conditions, couplings should be determined by spectral simulation.
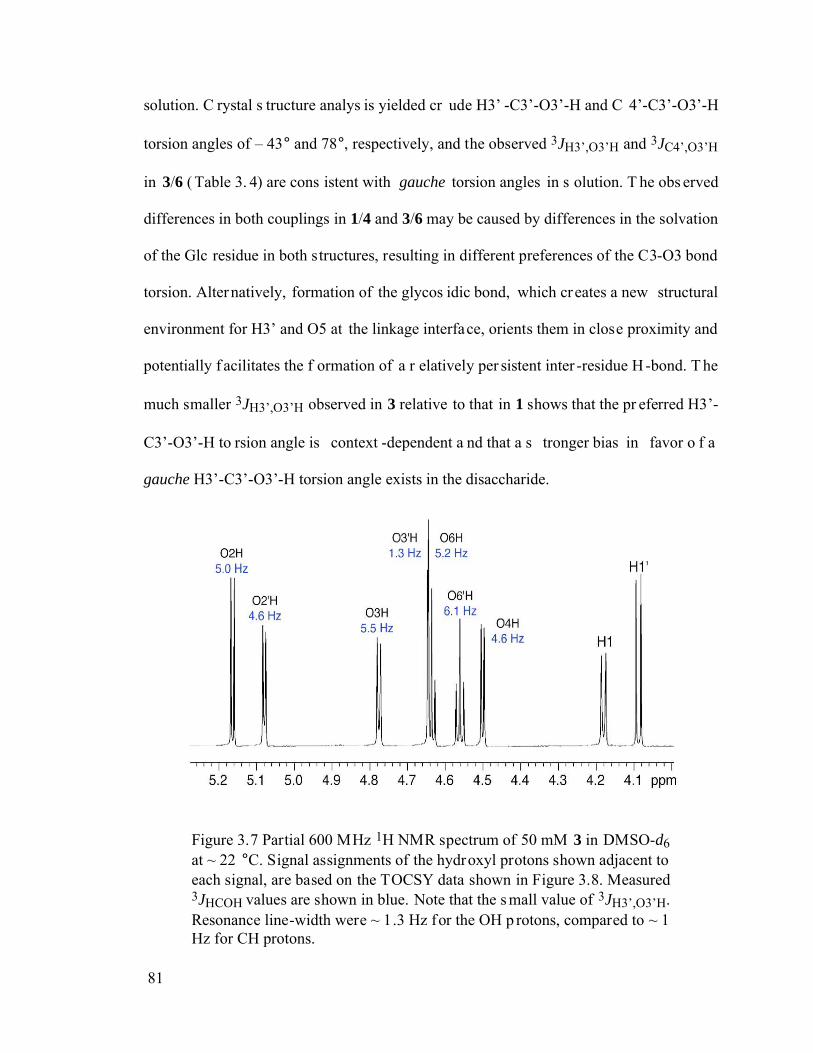
81
solution. C rystal s tructure analys is yielded cr ude H3’ -C3’-O3’-H and C 4’-C3’-O3’-H
torsion angles of – 43° and 78°, respectively, and the observed 3JH3’,O3’H and 3JC4’,O3’H
in 3/6 ( Table 3. 4) are cons istent with gauche torsion angles in s olution. T he obs erved
differences in both couplings in 1/4 and 3/6 may be caused by differences in the solvation
of the Glc residue in both structures, resulting in different preferences of the C3-O3 bond
torsion. Alternatively, formation of the glycos idic bond, which creates a new structural
environment for H3’ and O5 at the linkage interface, orients them in close proximity and
potentially f acilitates the f ormation of a r elatively per sistent inter -residue H-bond. T he
much smaller 3JH3’,O3’H observed in 3 relative to that in 1 shows that the pr eferred H3’-
C3’-O3’-H to rsion angle is context -dependent a nd that a s tronger bias in favor o f a
gauche H3’-C3’-O3’-H torsion angle exists in the disaccharide.
Figure 3.7 Partial 600 MHz 1H NMR spectrum of 50 mM 3 in DMSO-d6 at ~ 22 °C. Signal assignments of the hydroxyl protons shown adjacent to each signal, are based on the TOCSY data shown in Figure 3.8. Measured 3JHCOH values are shown in blue. Note that the s mall value of 3JH3’,O3’H. Resonance line-width were ~ 1.3 Hz for the OH p rotons, compared to ~ 1 Hz for CH protons.

82
If int ra-residue H -bonding is r esponsible f or t he above -noted J-couplings in
H2O/acetone solution, then this bonding should be more persistent in aprotic solvents like
DMSO. T he 1H NMR spectrum of 3 in DM SO-d6 (Figure 3.7) contained well -resolved
hydroxyl pr oton s ignals ( see Figur e 3. 8 f or a ssignments). 3JHCOH values meas ured in
H2O/acetone-d6 ( Table 3 .4) and in DM SO-d6 ( see couplings in Figur e 3 .7) wer e
essentially unchanged except for 3JH3’,O3’H, which is reduced to 1.3 Hz in DMSO. This
finding s uggests gr eater r estriction of the H3’- C3’-O3’-H tor sion angle in DM SO,
possibly due to enhanced persistence of the H-bond.
J-Couplings obs erved in 1 and 3 were t reated q uantitatively us ing eqs 2. 4 and
2.11 and a th ree-state model was as sumed f or th e H3 -C3-O3-H bond torsion ( Scheme
3.2). Us ing 3JH3,O3H and 3JC4,O3H values of 5.1 Hz and 1. 9 Hz, r espectively, the
following r otamer populations wer e deter mined in 1 in H 2O/acetone-d6: gg, ~58% ; gt,
~14%; tg, ~28 %. Thus, in the monos accharide, the calculations pr edict unequal
populations of the three rotamers. A similar treatment of the corresponding couplings in
3 gave the following populations: gg, ~85%; gt, ~4%; tg, ~11%. The significant increase
in the gg population in 3 is consistent with the c ontention that inter- residue H-bonding
between O3’H and O5 may exist in this structure.
C4' C2' C4' C2'
H3'
C4' C2'
H3'
g+g- anti
H3'
HH
H
gg gt tg
Scheme 3.2 Definition of rotamers about the C3’-O3’ bond in 3.

83
3.3.3.2 Hydroxyl proton exchange rates in 3 and bridging water
Hydroxyl hydr ogen exchange r ates f or O3H and O3’ H in 3 wi th s olvent water
showed no significant differences.4 This result contrasts with studies of -chitobiose 7 in
which a slower O3’H exchange rate was interpreted as support for an analogous H-bond
between O5 and O3’H in solution.[31]
Thus, if O3’H in 3 experiences an interaction with
O5 in H 2O/acetone-d6 solvent as suggested by th e J-coupling data, the inter action does
not reduce the solvent exchange rate as might be expected for a direct (i.e., no intervening
water molecule involved) , relatively persistent H-bond. T he lack of a solvent exchange
effect might be explained if only a small percentage of molecules in solution experience a
direct H- bond, or if the H -bonding is weak. Al ternatively, H -bonding mediated by a
solvent water molecule ( i.e., an ind irect H-bond) could occur, which may not af fect the
exchange r ate of th e H-bonded hydr oxyl pr oton. We ex amined whether a s ingle water
molecule could H -bond to 3 while s atisfying the s tructural requirements dictated by the
observed J-couplings. Us ing the cr ystal s tructure[27]
of 3 as a s tarting point, a water
molecule was inserted in the vicini ty of O3’H and O5 and the bimolecular complex was
geometrically optimized us ing DF T (B 3LYP/6-31G*). C omplex 8 (Scheme 3 .3)
emerged with the water molecule pos itioned above the plane of the Gal r ing and in H-
bonding contact with O5 and O3’ H. T he H3’- C3’-O3’-H and C 4’-C3’-O3’-H tor sion
4 Hydroxyl proton exchange ra tes we re measured by s aturation-transfer a s d escribed previously (Serianni, A. S.; Pierce, J .; Huang, S. G.; Barker, R. J. Am. Chem. Soc. 1982, 104, 4037-4044). Hydroxyl proton signal intensities decayed exponentially as a function of water signal saturation time, and the decay time constant, 1, was related to the desired exchange rate constant, kex, by 1/ 1 = 1/T1 + kex, where T1 is the s pin-lattice re laxation t ime of t he hydroxyl prot on. T1 w as d etermined f rom th e r elationship,
Mz( )/Mz(0) = 1/T1. Rate c onstants of 1. 1 - 1. 7 s -1 ( ± 0. 3 s -1) we re obs erved at –20 °C in 2:3 H2O:acetone-d6. Measurements were made on a 600 MHz NMR.
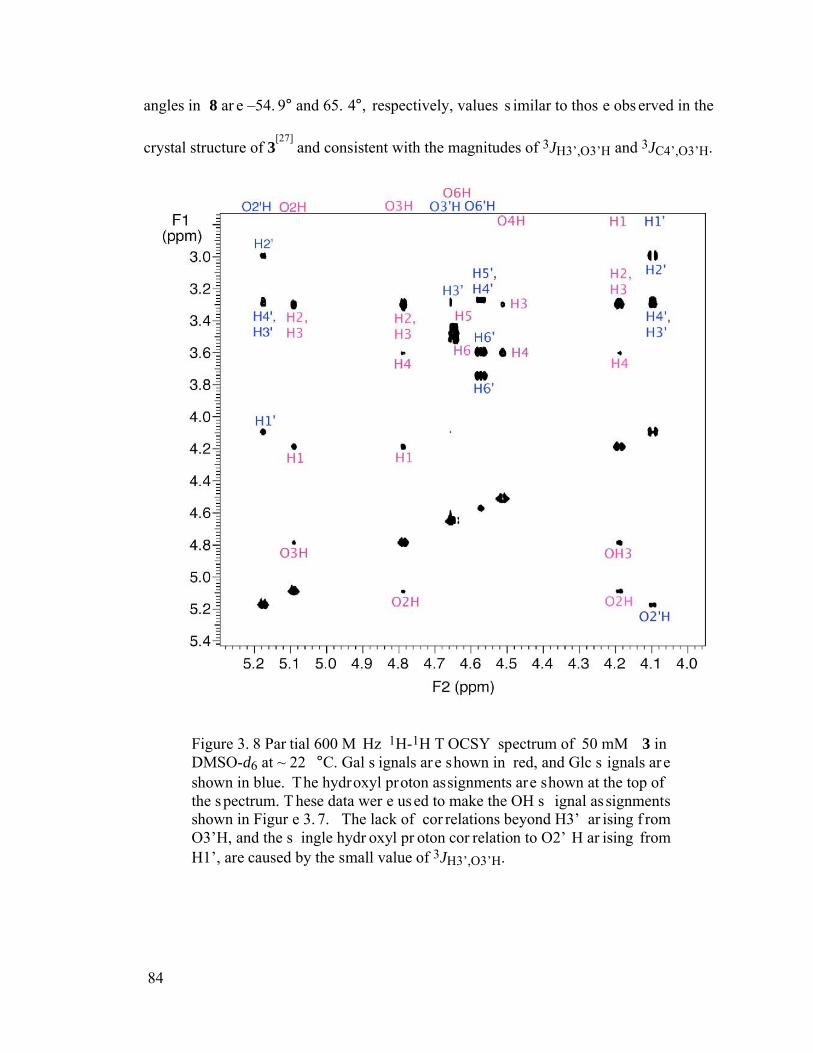
84
angles in 8 ar e –54. 9° and 65. 4°, respectively, values s imilar to thos e obs erved in the
crystal structure of 3[27]
and consistent with the magnitudes of 3JH3’,O3’H and 3JC4’,O3’H.
Figure 3. 8 Par tial 600 M Hz 1H-1H T OCSY spectrum of 50 mM 3 in DMSO-d6 at ~ 22 °C. Gal s ignals are shown in red, and Glc s ignals are shown in blue. The hydroxyl proton assignments are shown at the top of the spectrum. T hese data wer e used to make the OH s ignal as signments shown in Figur e 3. 7. The lack of cor relations beyond H3’ ar ising f rom O3’H, and the s ingle hydr oxyl pr oton cor relation to O2’ H ar ising from H1’, are caused by the small value of 3JH3’,O3’H.

85
Scheme 3. 3 DF T-generated br idging water comp lexed with 3. The H2O molecule (blue box) straddles the glycos idic linkage and is in H -bonding contact with O5 and O3’H.
3.3.4 Further discussion
The magnitudes of some of the 3JHCOH and 3JCCOH values observed in mono-
and disaccharides 1-6 are inconsistent with f ree rotational models about their constituent
C-O bonds , s uggesting that s ome bias f avoring specific C -O r otamers exi sts, at lea st
under the s olution conditions examined in this wor k. B y compa ring cor responding
couplings in di- and oligosaccharides to related couplings in their constituent monomers,
the ef fect of s tructural context can be ev aluated, and thus inf ormation about s olvent-
solute inter actions and/or the pr esence of inter -residue H -bonding may be obtained
indirectly.
J-Coupling tr ends wer e f ound to be cons istent w ith the pr esence of H -bonding
between O3’H and O5 in the -(1 4)linked dis accharide 3 in H 2O/acetone solvent at
low temperature. P rior s tudies of the -(1 4)linked chitobiose 7 by Kindahl et al.[31]
under similar solution conditions revealed an anomalously small 3JH3’,O3’H of 2.2 Hz,

86
Scheme 3. 4 Str uctures of -chitobiose ( 7), methyl -D-lactoside ( 9) and methyl -L-lactoside (10).
which is s imilar in magnitude to the cor responding 3JH3’,O3’H observed in 3/6. These
results were interpreted as suggesting the presence of weak H-bonding between O3’ and
O5, albeit without conf irmatory evidence pr ovided by 3JCCOH and in the abs ence of
appropriate Kar plus equations derived f or s accharides. 3JHCOH values as sociated with
highly pr eferred anti H- C-O-H tor sion angles ha ve als o been r eported as indicative of
intramolecular H-bonding in s acccharides.[32-34] Interestingly, NMR studies of methyl -
lactoside 9 in this laboratory have revealed a 3JH3’,O3’H similar to that ob served in 3, as
shown in Figur e 3. 9, s uggesting that the pr oposed inter- residue H -bonding may be a
general characteristic of -(1 4) linkages.

87
Figure 3. 9 Par tial 600 M Hz 1H NM R spectrum of 50 mM 9 in 2: 3 H2O/acetone-d6 at – 20 °C s howing signal a ssignments and 3JHCOH values ( in blue) . T he s mall 3JH3’,O3’H (2.1 ± 0. 3 Hz) is s imilar in magnitude to the corresponding coupling observed in 3 (see Table 3.4).
3.4 Conclusions
3JHCOH and 3JCCOH spin-couplings are valuable NMR probes for investigations
of intra- and intermolecular hydrogen bonding in solution, which plays a centr al role in
the chemis try and biochemis try of car bohydrates. Recent NM R s tudies have indicated
that H-bonds might exis t in dis accharides in D MSO solution.[25] A molecular dynamics
study suggests that O3’H is H-bonded to O5 in cellobiose up to 70% of time in water.[26]
In contr ast, OH gr oups dir ectly expos ed to solvent, es pecially w ater, s uch as tho se i n
monosaccharides and most of those in disaccharides, are expected to rotate freely without
any evidence of persistent intramolecular H-bonds.[16]
Access to complete s ets of singly 13C-labeled methyl - and -glucopyranosides
allows populations of C -O r otamers to b e evaluated in detail f or the f irst time.
Traditionally it was believed that per sistent hydr ogen bonding only exis ted in la rger
oligosaccharides at the interfaces between two residues in water. However, in this study,
we have s hown that C -O bonds in monosaccharides do not r otate freely. Instead, biases

88
are obs erved, which s uggests the pr esence of intr amolecular inter actions of s ome type
involving these OH groups.
The s trong bias in the H3’-C3’-O3’-H tor sion angle in 3 in f avor of a gauche
conformation is a pr erequisite f or intr a-residue H -bonding but does not pr ove its
existence. The preferred linkage conformation in 3 in aqueous solution, however, places
O3’H in clos e proximity to O5 in a very high percentage of molecules , thus increasing
the likelihood of H-bonding. The strength of this interaction in aqueous solution remains
uncertain, but r ecent s tudies[36] of 1JCH suggest t hat an ans wer to this question may be
possible. 1JC3’H3’ is expe cted to be inf luenced by C3’-O3’ bond conf ormation and by
the s trength of H- bonding involving O3’ H. An analys is of this J-coupling may shed
additional light on this problem.
Future s tudies of the potency of inter-residue H -bonding would benef it from
access to glycos idic linkage inter faces cont aining dif ferent potent ial modes of inter -
residue H-bonding. One way to achieve this variation is by incor porating L-sugars into
otherwise native di- or oligosaccharides. For example, the inter-residue interface present
in 3 is altered significantly when L-Gal is substituted for D-Gal, giving 10. In the crystal
structure of 10,[37] O2 and O3’, and O5 and O6’ , were found in close proximity, which is
clearly different from the O3’H-O5 interaction observed in 3. Measurements of 3JHCOH
and 3JCCOH in 10 labeled with 13C at C 1 in 2:3 H2O/acetone-d6 solvent at –20 °C gave
the following values: 3JH2,O2H, ~3.0 Hz; 3JC1,O2H, ~3.0 Hz; 3JH3,O3H, 6.0 Hz; 3JH4,O4H,
5.5 Hz; 3JH6,O6H, 5 .1 H z; 3JH2’,O2’H, 4 .6 H z; 3JH3’,O3’H, 4 .4 H z, 3JH6’,O6’H, 6 .3 H z.
These data r eveal an incr ease in 3JH6’,O6’H to 6.3 Hz relative to that obs erved in 1 (5.6

89
Hz). Likewise, 3JH2,O2H and 3JC1,O2H differ in 2/4 and 10, and 3JH3’,O3’H is reduced in
10 relative to the corresponding value in 1. Taken collectively, these results support the
presence of bias in the C3’-O3’, C 6’-O6’ and C 2-O2 bond tor sions in 10. T hese data
suggest H-bonding interactions between O6’H and O5, and between O2 and O3’.
Whether the detection of C -O tor sional bias a nd per sistent inter- residue H -
bonding in 3 are unique or will be obs erved more generally in di- and oligos accharides
remains an open and intr iguing question. Gl ycosidic linkage conf ormation, however, is
probably af fected not only by well known, albeit not f ully under stood, intr insic
stereoelectronic and steric factors, but also potentially by non-bonded and other extrinsic
interactions. I nter-residue H-bonding compr ises a subset of these extr insic f actors that
could influence preferred linkage conf ormation and dynamics . C ertainly multiple water
molecules will interact with saccharide solutes i n aqueous solution, and s ome of thes e
interactions may f unction as H-bond bridges between adjacent or remote residues. T his
bridging, if reasonably per sistent, could have significant implications f or pr eferred
linkage geometr ies and over all mole cular t opology. I ndeed, br idging water mo lecules
have been detected r ecently in molecular dynam ics s tudies of di saccharides, and DF T
calculations s uggest that water can f orm s pecific br idging H -bonded complexes with
disaccharides.[38]

90
3.5 References
1. Brooks, S. A.; Dwek, M. V.; Schumacher, U. Functional and Molecular Glycobiology, BIOS Scientific Publishers, 2002.
2. Hunter, C. A. Angew. Chem., Int. Ed. 2004, 73, 1019-1049.
3. Greenspan, N. S. Curr. Top. Microbiol. Immunol. 2001, 260, 65-85.
4. French, A. D.; Dowd, M. K.; Reilly, P. J. J. Mol. Struct.: THEOCHEM. 1997, 395-396. 271-287.
5. Leonidas, D. D.; Swamy, B. M.; Hatzopoulos, G. N.; Gonchigar, S. J.; Chachadi, V. B.; Inamdar, S. R.; Zographos, S. E.; Oikonomakos, N. G. J. Mol. Biol. 2007, 368, 1145-1161.
6. Zhao, H.; Pan, Q.; Zhang, W.; Carmichael, I.; Serianni, A. S. J. Org. Chem. 2007, 72, 7071-7082.
7. Karplus, M. J. J. Chem. Phys. 1959, 30, 11-15.
8. Fraser, R. R.; Kaufman, M.; Morand, P.; Govil, G. Can. J. Chem. 1969, 47, 403-409.
9. Fukui, H.; Baba, T.; Inomata, H.; Miura, K.; Matsuda, H. Mol. Phys. 1997, 92, 161-165.
10. Alkorta, I.; Elguero, J. Theor. Chem. Acc. 2004, 111, 31-35.
11. Carmichael, I.; Chipman, D. M.; Podlasek, C. A.; Serianni, A. S. J. Am. Chem. Soc. 1993, 115, 10863-10870
12. Klepach, T. E.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 2005, 127, 9781-9793.
13. Thibaudeau, C.; Stenutz, R.; Hertz, B.; Klepach, T. E.; Zhao, S.; Wu, Q.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 2004, 126, 15668-15685.
14. Podlasek, C.A.; Wu, J.; Stripe, W.A.; Bondo, P.B.; Serianni, A.S. J. Am. Chem. Soc. 1995, 117, 8635-8644.
15. Hayes, M.L.; Serianni, A.S.; Barker, R. Carbohydr. Res. 1982, 100, 87-101.
16. Adams, B.; Lerner, L. J. Am. Chem. Soc. 1992, 114, 4827-4829.

91
17. Brooks, B. R.; Bruccoleri, R. E.; Olafson, B. D.; States, D. J.; Swaminathan, S. and Karplus, M. J. Comp. Chem. 1983, 4, 187-217.
18. Case, D. A.; Cheatham, T. E.; Darden, T.; Gohlke, H.; Luo, R.; Merz Jr., K. M.; Onufriev, A.; Simmerling, C.; Wang, B. and Woods, R. J. Computat. Chem. 2005, 6, 1668-1688.
19. Kuttel, M.; Brady, J. W. and Naidoo, K. J. J. Comput. Chem. 2002, 23, 1236-1243.
20. Kirschner, K. N. and Woods, R. J. Proc. Natl. Acad. Sci. 2001, 98, 10541-10545.
21. Basma, B.; Sundara, S.; Calgan, D.; Vanali, T. and Woods, R. J. J. Comput. Chem. 2001, 22, 1125-1137.
22. Kirschner, K. N. and Woods, R. J. J. Phys. Chem. A. 2001, 105, 4150-4155.
23. M. Kraszni, Z. Szakacs, B. Noszal. Anal Bioanal. Chem. 2004, 378, 1449-1463.
24. Jockusch, R. A.; Kroemer, R. T.; Talbot, F. O.; Simons, J. P. J. Phys. Chem. A. 2003, 107, 10725-10732.
25. Adams, B.; Lerner, L. E. Magn. Res. Chem. 1994, 32, 225-330.
26. Carcabal, P.; Jockusch, R. A.; Hunig, I.; Snoek, L. C.; Kroemer, R. T.; Davis, B. G.; Gamblin, D. P.; Compagnon, I.; Oomens, J.; Simons, J. P. J. Am. Chem. Soc. 2005, 127, 11414-11425.
27. Stenutz, R.; Shang, M.; Serianni, A.S. Acta Cryst. 1999, C55, 1719-1721.
28. Ham, J.T.; Williams, D.G. Acta Cryst. 1970, B26, 1373-1383.
29. Fries, D.C.; Rao, S.T.; Sundaralingam, M. Acta Cryst. 1971, B27, 994-1005.
30. Hirotsu, K.; Shimada, A. Bull. Chem. Soc. Jpn. 1974, 47, 1872-1879.
31. Kindahl, L.; Sandström, C.; Norberg, T.; Kenne, L. J. Carbohydr. Chem. 2000, 19, 1291-1303.
32. Sandström, C.; Baumann, H.; Kenne, L. J. Chem. Soc. Perkin Trans. 2 1998, 809-815.
33. Sandström, C.; Baumann, H.; Kenne, L. J. Chem. Soc. Perkin Trans. 2 1998, 2385-2393.
34. Sandström, C.; Magnusson, G.; Nilsson, U.; Kenne, L. Carbohydr. Res. 1999, 322, 46-56.

92
35. Pereira, C. S.; Kony, D.; Baron, R.; Muller, M.; van Gunsteren, W. F. and Hunenberger, P. H. Biophys. J. 2006, 90, 4337-4344.
36. Maiti, N.C.; Zhu, Y.; Carmichael, I.; Serianni, A.S.; Anderson, V.E. J. Org. Chem. 2006, 71, 2878-2880.
37. Pan, Q.; Noll, B.; Serianni, A.S., Acta Cryst. 2006, C62, o82-o85.
38. Naidoo, K. J.; Chen, J. Y. Mol. Phys. 2003, 101, 2687-2694.

93
CHAPTER 4:
PROBING THE PRESENCE AND STRENGTH OF HYDROGEN BONDING IN
SACCHARIDES VIA J-COUPLINGS: A DFT STUDY
4.1 Introduction
Experimental methods to determine the strength of hydrogen bonding in s olution
have been dif ficult to develop. 1H NM R s pectroscopy pr ovides a means to detect H -
bonding in solution by exploiting the s ignificant deshielding that occurs when a proton is
present in an H -bonding envir onment.[1,2] S calar coupling ( J-coupling, s pin-spin
coupling) acr oss H -bonds pr ovides f irm evide nce f or the p resence of H -bonds,[3]
although thes e couplings ar e of ten s mall and may not be ob servable in all s uch
systems.[4] While thes e exper imental appr oaches allow detection of putative H -bonds,
they are not amenable to deter mining the strength of these bonds. Similar arguments can
be made for IR spectroscopy, where issues regarding the assignment of absorption bands
to specific O-H stretching and/or X-O-H bending modes must also be overcome.[5]
We s howed r ecently that NM R one -bond 13C-1H s pin-spin coupling cons tants
(1JCH) ar e sensitive to H -bond s trength.[6] U sing an experimental model system
comprised of hexaf luoroisopropanol as the H -bond donor and a gr oup of organic bases
having dif ferent pKb values , the alcoholic 1JCH was found to decr ease roughly linear ly

94
with increasing H-bond strength. A f it of the experimental data gave a s lope of ~-0.2 Hz
per kJ of H- bond s trength. T his behavior s uggests that N MR J-couplings c an be
classified into two groups, structural and functional, with the latter serving as quantitative
probes not only of molecular s tructure but also o f specific physical properties (e.g., H-
bonding) of molecules in solution.
Recent s tudies of the conf ormational p roperties of glycos idic linkages in
oligosaccharides have s uggested that, in s ome cas es, intr amolecular ( inter-residue) H-
bonding may play a f unctional r ole in dictatin g or bias ing the pr eferred s hapes and
dynamics of thes e molecules .[7-11] F or example, in cr ystal s tructures of methyl -
lactoside ( methyl -D-galactopyranosyl-(1 4)- -D-glucopyranoside, 1)[12] and
structurally r elated disaccharides,[13,14] an intr amolecular H-bond between O5 Gal (O5’)
and O3H Glc ( or equivalent nuclei) is obs erved. NM R s tudies of 1 in s olution at low
temperature, either in s upercooled water or in water/acetone mixtur es, s uggest that a
similar H-bond may persist, as revealed by anomalous 3JHCOH and 3JCCOH values.[15,16]
The latter couplings imply a bias in the H3-C3-O3-H tor sion angle f avoring
conformations cons istent with H -bonding. Ho wever, while thes e data ar e cons istent
structurally with the presence of H-bonding, they are not direct proof of its presence nor
do they allow a quantitative determination of its strength.
In this chapter, a theoretical study was undertaken using density functional theory
(DFT) to deter mine whether 1JCH values in a model dis accharide could be us ed to
measure inter -residue H-bond s trength and to f urther pr obe the ef fect o f H-bonding on
proximal molecular structure.
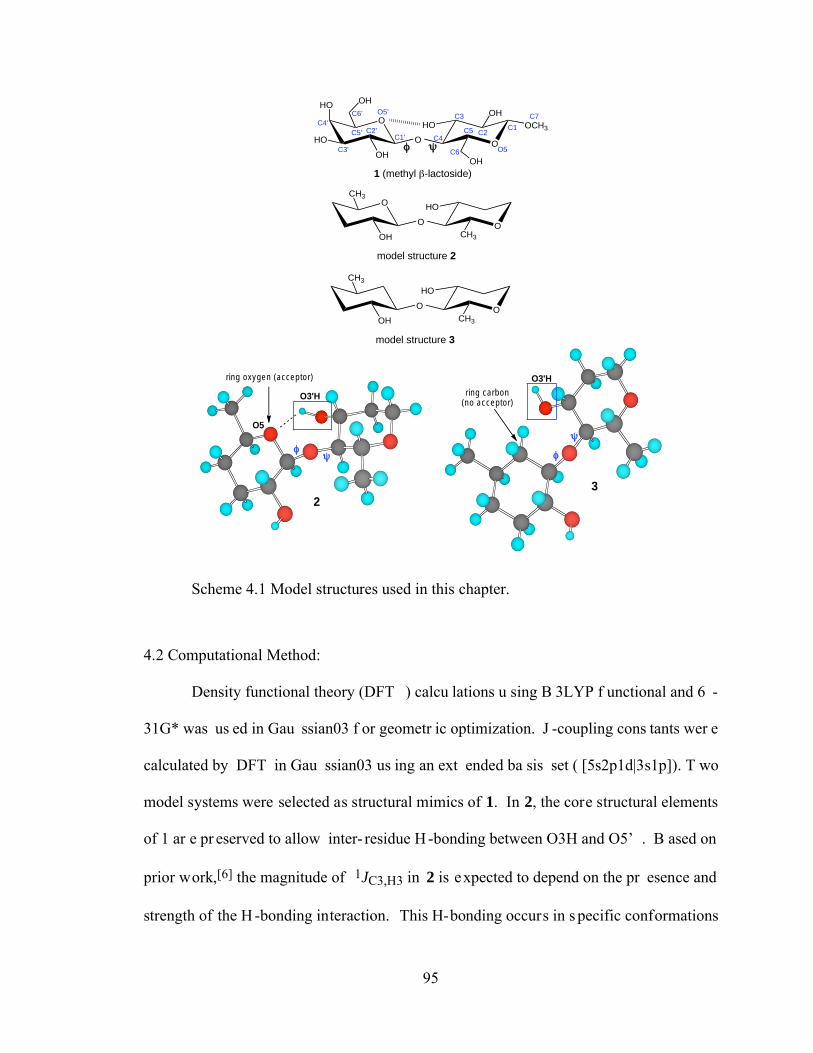
95
O
O OCH3
OH
HO
1 (methyl -lactoside)
O
OHHO
HO
OH
OH
C1'C2'
C3'
C4'C5'
C6' O5'
C4
C6 O5
C2C5
C3 C7
C1
O
O HO
model structure 2
O
OH
O
HO
model structure 3
O
OH
CH3
CH3
CH3
CH3
ring oxygen (acceptor)
ring carbon(no acceptor)
O5
23
O3'H
O3'H
Scheme 4.1 Model structures used in this chapter.
4.2 Computational Method:
Density functional theory (DFT ) calcu lations u sing B 3LYP f unctional and 6 -
31G* was us ed in Gau ssian03 f or geometr ic optimization. J -coupling cons tants wer e
calculated by DFT in Gau ssian03 us ing an ext ended ba sis set ( [5s2p1d|3s1p]). T wo
model systems were selected as structural mimics of 1. In 2, the core structural elements
of 1 ar e pr eserved to allow inter- residue H -bonding between O3H and O5’ . B ased on
prior work,[6] the magnitude of 1JC3,H3 in 2 is expected to depend on the pr esence and
strength of the H -bonding interaction. This H-bonding occurs in s pecific conformations
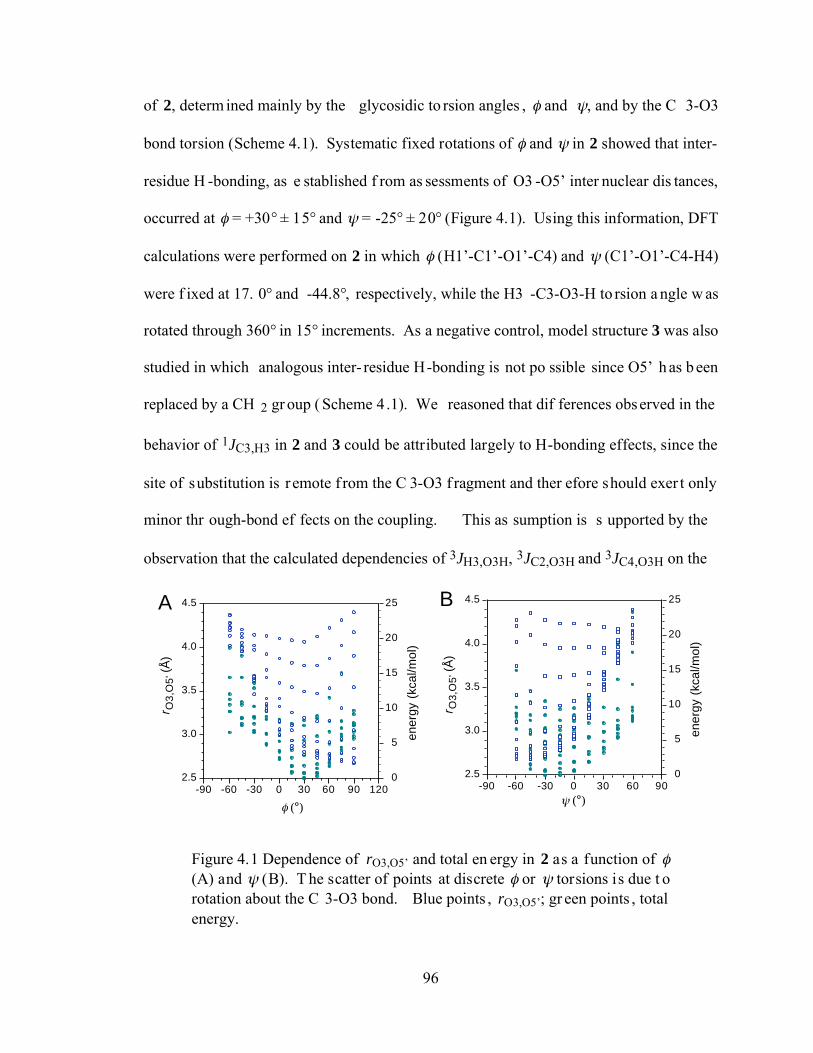
96
of 2, determ ined mainly by the glycosidic to rsion angles , and , and by the C 3-O3
bond torsion (Scheme 4.1). Systematic fixed rotations of and in 2 showed that inter-
residue H -bonding, as e stablished f rom as sessments of O3 -O5’ inter nuclear dis tances,
occurred at = +30° ± 15° and = -25° ± 20° (Figure 4.1). Using this information, DFT
calculations were performed on 2 in which (H1’-C1’-O1’-C4) and (C1’-O1’-C4-H4)
were f ixed at 17. 0° and -44.8°, respectively, while the H3 -C3-O3-H to rsion a ngle w as
rotated through 360° in 15° increments. As a negative control, model structure 3 was also
studied in which analogous inter- residue H-bonding is not po ssible since O5’ h as b een
replaced by a CH 2 gr oup ( Scheme 4 .1). We reasoned that dif ferences observed in the
behavior of 1JC3,H3 in 2 and 3 could be attributed largely to H-bonding effects, since the
site of substitution is r emote f rom the C 3-O3 f ragment and ther efore should exer t only
minor thr ough-bond ef fects on the coupling. This as sumption is s upported by the
observation that the calculated dependencies of 3JH3,O3H, 3JC2,O3H and 3JC4,O3H on the
0
5
10
15
20
25
ener
gy (
kcal
/mol
)
2.5
3.0
3.5
4.0
4.5
r O3,
O5'
(Å
)
-90 -60 -30 0 30 60 90 (°)
0
5
10
15
20
25
ener
gy (
kcal
/mol
)
2.5
3.0
3.5
4.0
4.5
r O3,
O5'
(Å
)
-90 -60 -30 0 30 60 90 120
(°)
A B
Figure 4.1 Dependence of rO3,O5’ and total en ergy in 2 as a function of (A) and (B). T he scatter of points at discrete or torsions is due t o rotation about the C 3-O3 bond. Blue points , rO3,O5’; gr een points , total energy.
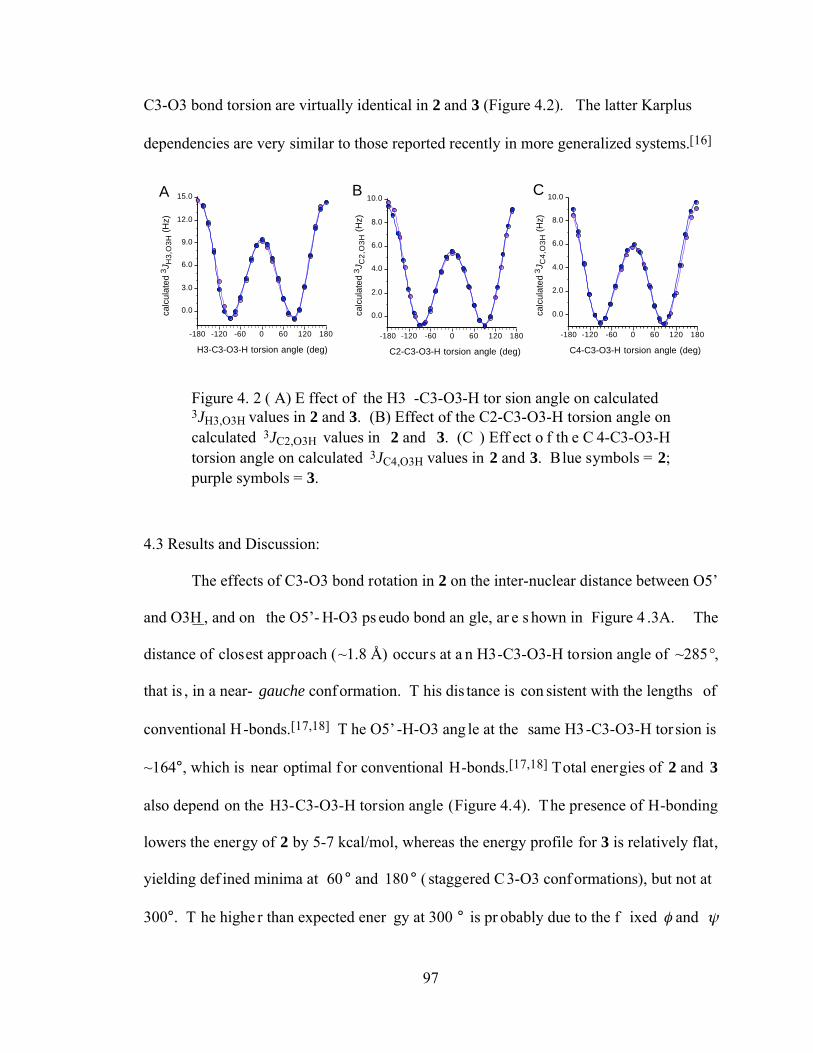
97
C3-O3 bond torsion are virtually identical in 2 and 3 (Figure 4.2). The latter Karplus
dependencies are very similar to those reported recently in more generalized systems.[16]
0.0
3.0
6.0
9.0
12.0
15.0
calc
ulat
ed 3
J H3
,O3
H (
Hz)
-180 -120 -60 0 60 120 180
H3-C3-O3-H torsion angle (deg)
0.0
2.0
4.0
6.0
8.0
10.0
calc
ulat
ed 3
J C2
,O3
H (
Hz)
-180 -120 -60 0 60 120 180
C2-C3-O3-H torsion angle (deg)
0.0
2.0
4.0
6.0
8.0
10.0
calc
ulat
ed 3
J C4
,O3
H (
Hz)
-180 -120 -60 0 60 120 180
C4-C3-O3-H torsion angle (deg)
A B C
Figure 4. 2 ( A) E ffect of the H3 -C3-O3-H tor sion angle on calculated 3JH3,O3H values in 2 and 3. (B) Effect of the C2-C3-O3-H torsion angle on calculated 3JC2,O3H values in 2 and 3. (C ) Eff ect o f th e C 4-C3-O3-H torsion angle on calculated 3JC4,O3H values in 2 and 3. Blue symbols = 2; purple symbols = 3.
4.3 Results and Discussion:
The effects of C3-O3 bond rotation in 2 on the inter-nuclear distance between O5’
and O3H , and on the O5’- H-O3 ps eudo bond an gle, ar e s hown in Figure 4 .3A. The
distance of closest approach (~1.8 Å) occurs at a n H3-C3-O3-H torsion angle of ~285°,
that is , in a near- gauche conf ormation. T his dis tance is con sistent with the lengths of
conventional H-bonds.[17,18] T he O5’ -H-O3 ang le at the same H3-C3-O3-H tor sion is
~164°, which is near optimal f or conventional H-bonds.[17,18] Total energies of 2 and 3
also depend on the H3-C3-O3-H torsion angle (Figure 4.4). The presence of H-bonding
lowers the energy of 2 by 5-7 kcal/mol, whereas the energy profile for 3 is relatively flat,
yielding def ined minima at 60 ° and 180 ° ( staggered C3-O3 conf ormations), but not at
300°. T he highe r than expected ener gy at 300 ° is pr obably due to the f ixed and
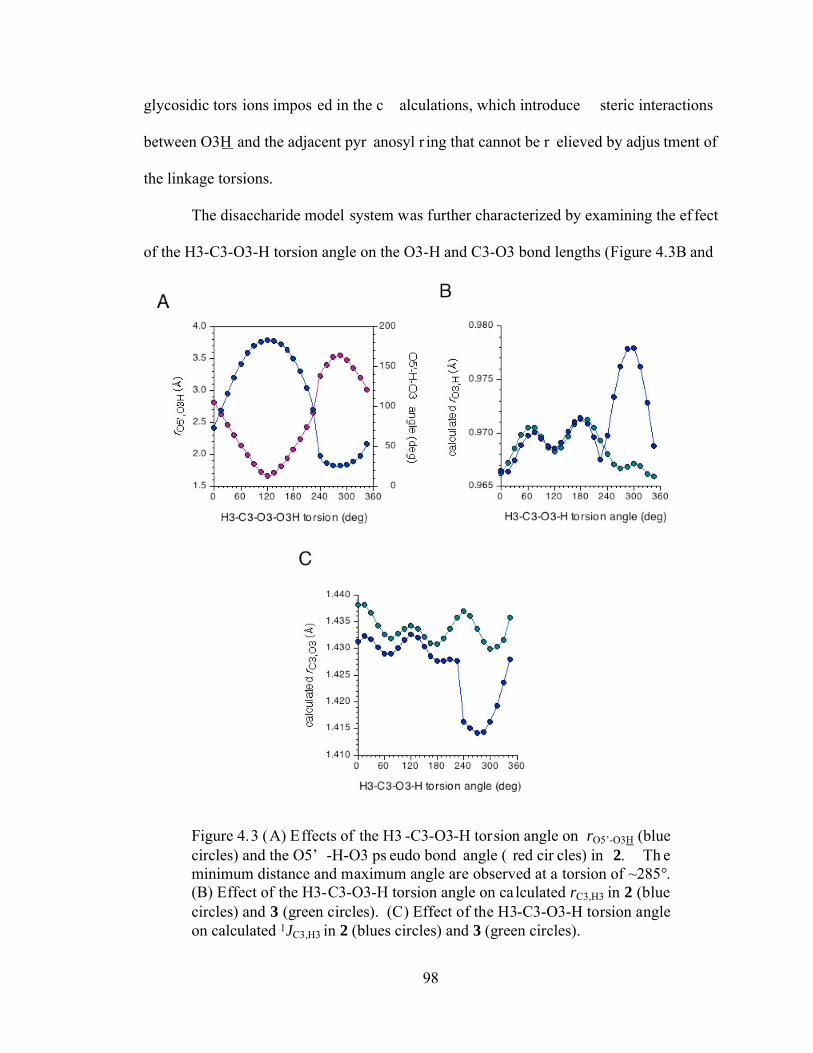
98
glycosidic tors ions impos ed in the c alculations, which introduce steric interactions
between O3H and the adjacent pyr anosyl r ing that cannot be r elieved by adjus tment of
the linkage torsions.
The disaccharide model system was further characterized by examining the ef fect
of the H3-C3-O3-H torsion angle on the O3-H and C3-O3 bond lengths (Figure 4.3B and
Figure 4.3 (A) Effects of the H3 -C3-O3-H torsion angle on rO5’-O3H (blue circles) and the O5’ -H-O3 ps eudo bond angle ( red cir cles) in 2. Th e minimum distance and maximum angle are observed at a torsion of ~285°. (B) Effect of the H3-C3-O3-H torsion angle on ca lculated rC3,H3 in 2 (blue circles) and 3 (green circles). (C) Effect of the H3-C3-O3-H torsion angle on calculated 1JC3,H3 in 2 (blues circles) and 3 (green circles).
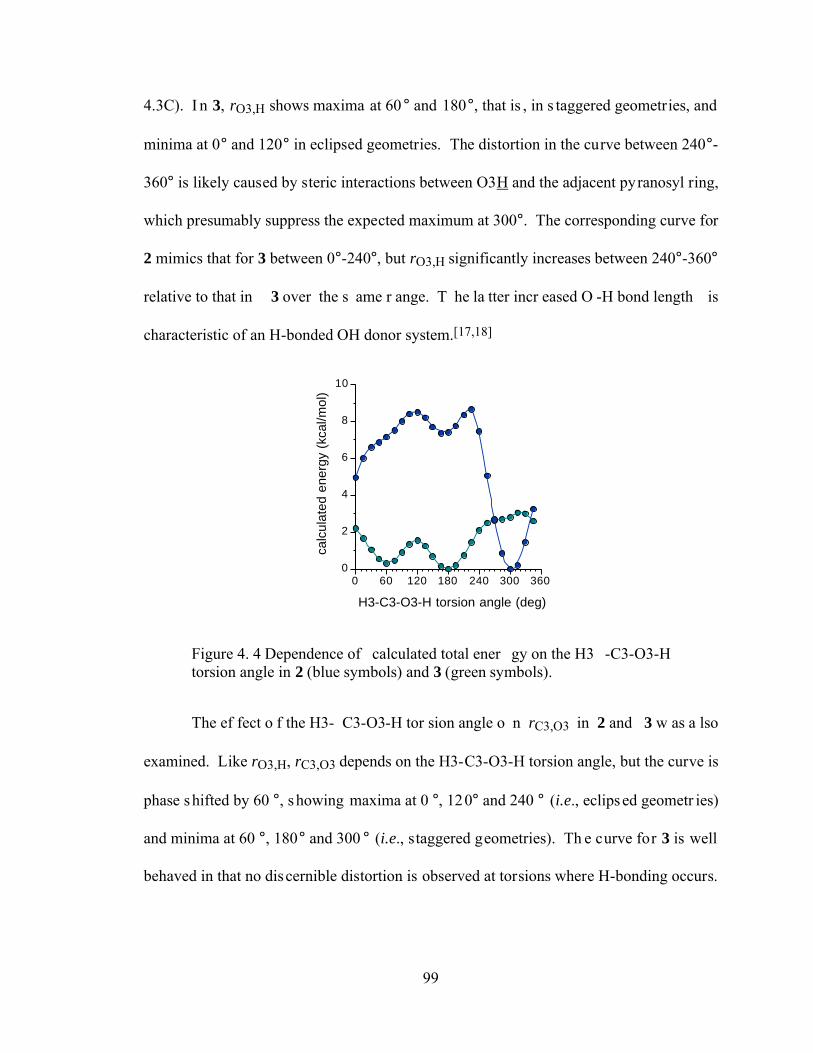
99
4.3C). I n 3, rO3,H shows maxima at 60 ° and 180°, that is , in s taggered geometries, and
minima at 0° and 120° in eclipsed geometries. The distortion in the curve between 240°-
360° is likely caused by steric interactions between O3H and the adjacent pyranosyl ring,
which presumably suppress the expected maximum at 300°. The corresponding curve for
2 mimics that for 3 between 0°-240°, but rO3,H significantly increases between 240°-360°
relative to that in 3 over the s ame r ange. T he la tter incr eased O -H bond length is
characteristic of an H-bonded OH donor system.[17,18]
0
2
4
6
8
10
calc
ulat
ed e
nerg
y (k
cal/m
ol)
0 60 120 180 240 300 360
H3-C3-O3-H torsion angle (deg)
Figure 4. 4 Dependence of calculated total ener gy on the H3 -C3-O3-H torsion angle in 2 (blue symbols) and 3 (green symbols).
The ef fect o f the H3- C3-O3-H tor sion angle o n rC3,O3 in 2 and 3 w as a lso
examined. Like rO3,H, rC3,O3 depends on the H3-C3-O3-H torsion angle, but the curve is
phase shifted by 60 °, showing maxima at 0 °, 120° and 240 ° (i.e., eclipsed geometr ies)
and minima at 60 °, 180° and 300 ° (i.e., staggered geometries). Th e curve for 3 is well
behaved in that no discernible distortion is observed at torsions where H-bonding occurs.
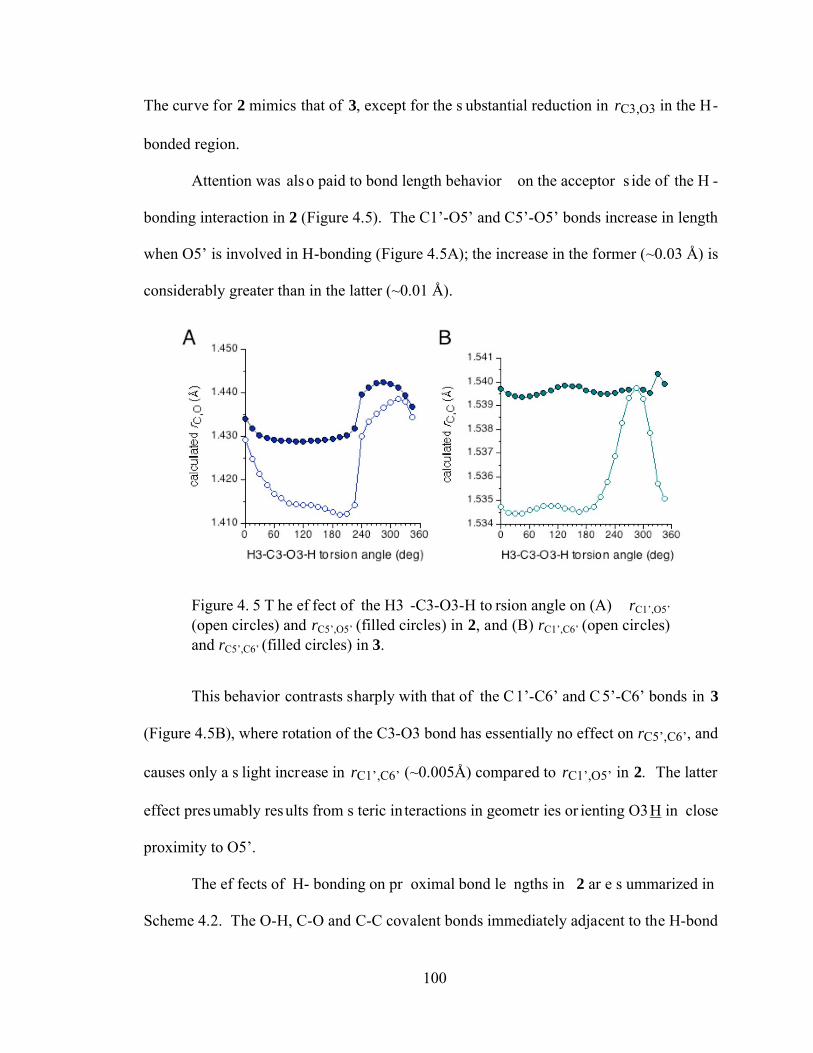
100
The curve for 2 mimics that of 3, except for the s ubstantial reduction in rC3,O3 in the H-
bonded region.
Attention was also paid to bond length behavior on the acceptor s ide of the H -
bonding interaction in 2 (Figure 4.5). The C1’-O5’ and C5’-O5’ bonds increase in length
when O5’ is involved in H-bonding (Figure 4.5A); the increase in the former (~0.03 Å) is
considerably greater than in the latter (~0.01 Å).
Figure 4. 5 T he ef fect of the H3 -C3-O3-H to rsion angle on (A) rC1’,O5’ (open circles) and rC5’,O5’ (filled circles) in 2, and (B) rC1’,C6’ (open circles) and rC5’,C6’ (filled circles) in 3.
This behavior contrasts sharply with that of the C1’-C6’ and C 5’-C6’ bonds in 3
(Figure 4.5B), where rotation of the C3-O3 bond has essentially no effect on rC5’,C6’, and
causes only a s light increase in rC1’,C6’ (~0.005Å) compared to rC1’,O5’ in 2. The latter
effect presumably results from s teric in teractions in geometr ies or ienting O3H in close
proximity to O5’.
The ef fects of H- bonding on pr oximal bond le ngths in 2 ar e s ummarized in
Scheme 4.2. The O-H, C-O and C-C covalent bonds immediately adjacent to the H-bond

101
are increasingly elongated as the H-bond forms, especially in the former two. In contrast,
the behavior of the penultimate C -O bond of the donor is truncated. These results are
consistent with pr ior s tudies,[17,18] ther eby vali dating this model s ystem f or us e in
probing the effects of H-bonding on NMR J-couplings.
R2CH O H .......... O CHR2
CHR2
Scheme 4.2 Effects of H-bonding on proximal bond lengths in 2.
The effect of C3-O3 bond rotation on the C3-H3 bond length, rC3,H3, in 2 and 3 is
shown in Figure 4 .6A. The two cu rves are s imilar with respect to overall s hape, as
expected, since rotation of the C 3-O3 bond changes the or ientation of the O3 lone -pairs
with respect to the C 3-H3 bond in a similar fashion in both s tructures. At to rsion angles
of 60° and 300°, an O3 lone-pair is anti to the C3-H3 bond, and n * donation elongates
the bond. [19] At 180°, th is ef fect is absent, s ince the O3 -H bond is anti to the C 3-H3
bond. The two cur ves track one another well f rom 0°-180°, with rC3,H3 in 3 consistently
larger than rC3,H3 in 2. This trend is reversed at torsion angles between 180° and 345°,
that is, in the region where inter-residue H-bonding is present in 2. In this region, rC3,H3
in 2 is lengthened r elative to rC3,H3 in 3. T his lengthening is cons istent with
expectations, s ince C-H bond lengths in C H3OH ar e s ignificantly shorter than those in
CH3O-; the H-bonded case, CH3OH----OR2, can be considered an intermediate along the
pathway of conver sion of C H3OH to CH 3O-, and thus inter mediate C -H bond
lengthening is expected in H-bonded structures (Table 4.1).

102
Calculated 1JC3,H3 values in 2 and 3 (Figure 4.6B) are maximal at H3 -C3-O3-H
torsion angles between 120 °-240°, and minimal at ~0 °. T he two curves have s imilar
shapes at tor sions from 0°- 225°, with 1JC3,H3 in 2 consistently larger than 1JC3,H3 in 3.
The pattern is reversed at torsions between 240°-300° where intra-residue H-bonding in 2
occurs. T he r educed J-coupling in the H- bonded s tructures of 2 is cons istent with
observations made in the CH3OH/CH3O- model system (Table 4.1).
TABLE 4.1
CALCULATED 1JCH AND C-H BOND LENGTH IN METHANOL AND
METHOXIDE ANION
aMP2/6-31G* geometries; J-coupling basis set, [5s2p1d,3s1p]. Data obtainedfrom calculations using Gaussian03.
compound rC,O(Å)
rCH(gauche)
(Å)
rC,H(anti)(Å)
rC,H (Å) 1JCH(gauche)
(Hz)
1JCH(anti)(Hz)
1JCH(Hz)
CH3OH 1.424 1.097 1.090 139.3 144.3CH3O- 1.325 1.149 115.4
Subtraction of the two cur ves shown in Figure 4.6B yielded a difference plot in
which the truncated 1JC3,H3 values in H-bonded structures of 2 relative to those in 3 can
be quantified (Figure 4.7). This subtraction eliminates the contr ibution of O3 lone -pair
effects on rC3,H3 and thus on 1JC3,H3.[19] T he dif ference plot contains thos e
contributions to 1JC3,H3 made solely by H-bonding effects. I f an av erage difference of
~2.5 Hz over H3- C3-O3-H tor sion angles of 0 °-180° is as sumed, then an overall
reduction in 1JC3,H3 of ~3.5 Hz is obtained. Us ing the exper imental calibration of ~0.2
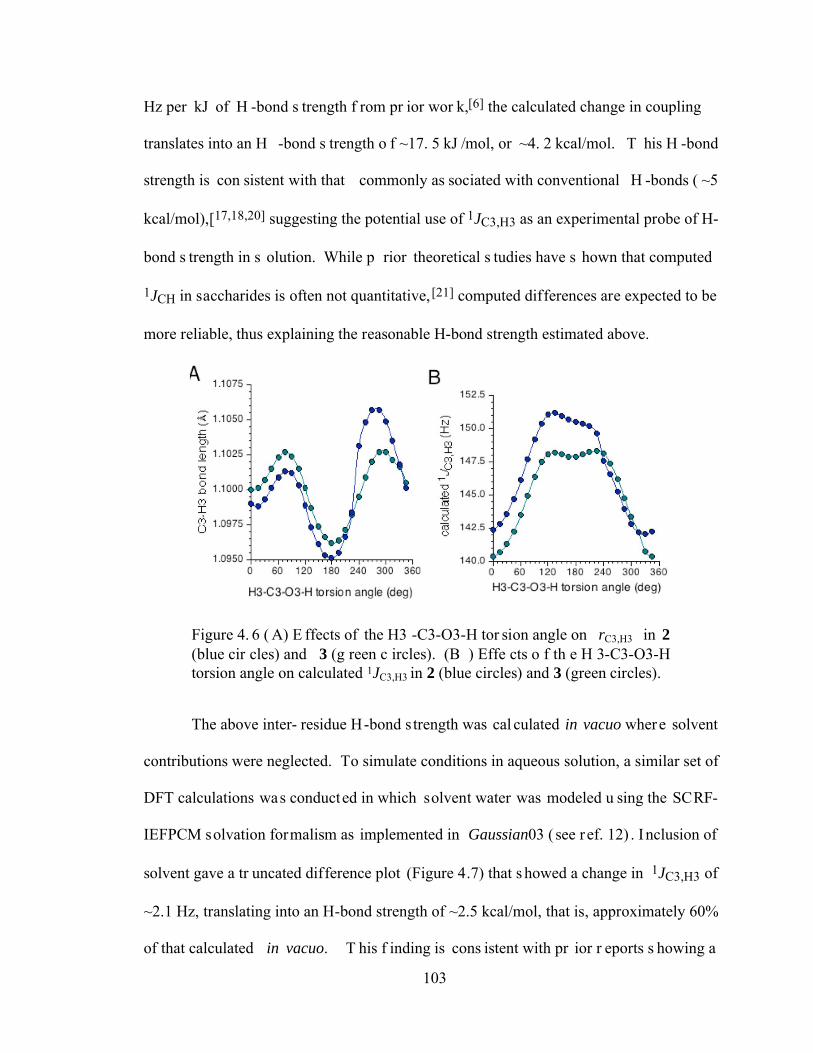
103
Hz per kJ of H -bond s trength f rom pr ior wor k,[6] the calculated change in coupling
translates into an H -bond s trength o f ~17. 5 kJ /mol, or ~4. 2 kcal/mol. T his H -bond
strength is con sistent with that commonly as sociated with conventional H -bonds ( ~5
kcal/mol),[17,18,20] suggesting the potential use of 1JC3,H3 as an experimental probe of H-
bond s trength in s olution. While p rior theoretical s tudies have s hown that computed
1JCH in saccharides is often not quantitative,[21] computed differences are expected to be
more reliable, thus explaining the reasonable H-bond strength estimated above.
Figure 4. 6 ( A) E ffects of the H3 -C3-O3-H tor sion angle on rC3,H3 in 2 (blue cir cles) and 3 (g reen c ircles). (B ) Effe cts o f th e H 3-C3-O3-H torsion angle on calculated 1JC3,H3 in 2 (blue circles) and 3 (green circles).
The above inter- residue H-bond s trength was cal culated in vacuo where solvent
contributions were neglected. To simulate conditions in aqueous solution, a similar set of
DFT calculations was conducted in which solvent water was modeled u sing the SCRF-
IEFPCM solvation formalism as implemented in Gaussian03 ( see r ef. 12) . Inclusion of
solvent gave a tr uncated difference plot (Figure 4.7) that s howed a change in 1JC3,H3 of
~2.1 Hz, translating into an H-bond strength of ~2.5 kcal/mol, that is, approximately 60%
of that calculated in vacuo. T his f inding is cons istent with pr ior r eports s howing a
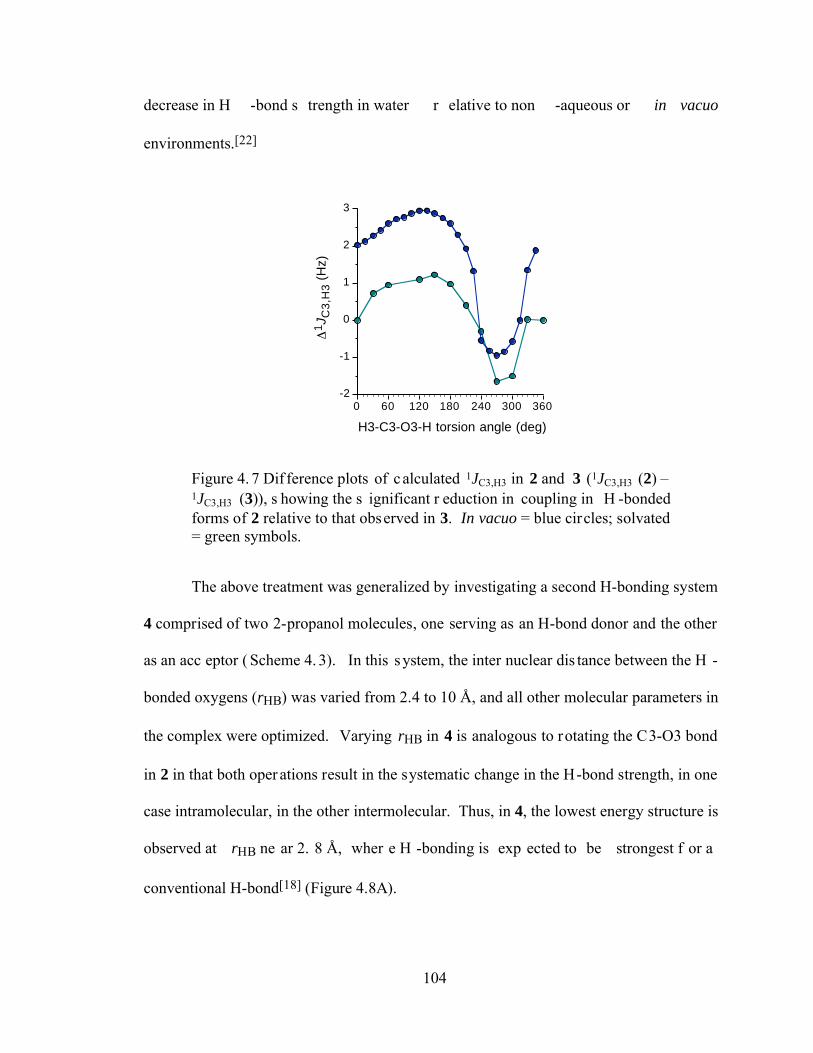
104
decrease in H -bond s trength in water r elative to non -aqueous or in vacuo
environments.[22]
-2
-1
0
1
2
3
1J C
3,H
3 (H
z)
0 60 120 180 240 300 360
H3-C3-O3-H torsion angle (deg)
Figure 4. 7 Dif ference plots of calculated 1JC3,H3 in 2 and 3 (1JC3,H3 (2) – 1JC3,H3 (3)), s howing the s ignificant r eduction in coupling in H -bonded forms of 2 relative to that observed in 3. In vacuo = blue circles; solvated = green symbols.
The above treatment was generalized by investigating a second H-bonding system
4 comprised of two 2-propanol molecules, one serving as an H-bond donor and the other
as an acc eptor ( Scheme 4. 3). In this system, the inter nuclear dis tance between the H -
bonded oxygens (rHB) was varied from 2.4 to 10 Å, and all other molecular parameters in
the complex were optimized. Varying rHB in 4 is analogous to rotating the C3-O3 bond
in 2 in that both oper ations result in the systematic change in the H-bond strength, in one
case intramolecular, in the other intermolecular. Thus, in 4, the lowest energy structure is
observed at rHB ne ar 2. 8 Å, wher e H -bonding is exp ected to be strongest f or a
conventional H-bond[18] (Figure 4.8A).

105
acceptor
donor
C-Hdonor
C-HacceptorrHB
4
Scheme 4. 3 2- proponal molecules . One will serve as H-bond donor an d another as H-bond acceptor.
The effects of H-bonding on the structure of the bimolecular model system 4 were
investigated by examining bond lengths in the vicinity of the H-bond. Decreasing rHB
from 10 Å to 2 .8 Å increas es the O -H bond o f the donor appreciably, whereas the O -H
bond length of the acceptor is virtually unaffected (Figure 4.9A). On the other hand, the
donor and acceptor C-O bond lengths are both in fluenced by a s imilar decrease in rHB,
with that of the dono r decr easing and that of t he acceptor incr easing ( Figure 4. 9B).
These results are internally consistent with behavior found in disaccharide model systems
2 and 3 (Figures 4.3 and 4.5).
The effect of rHB on the C -Hdonor and C -Hacceptor bond lengths in 4 is shown in
Figure 4.8B. As observed for rC3,H3 in 2, rCH,donor increases as rHB approaches 2.8 Å,
although the curve is not smooth and a minimum is observed at rHB of ~4.5 Å for reasons
that are unclear . I n contrast, rCH,acceptor smoothly decreases a s rHB approaches 2.8 Å.
Thus, the two effects are complementary, with the C-H bond length on the donor alcohol
increasing, and the C-H bondlength on the acceptor alcohol decr easing, with decr easing
rHB and thus increasing H-bond strength.
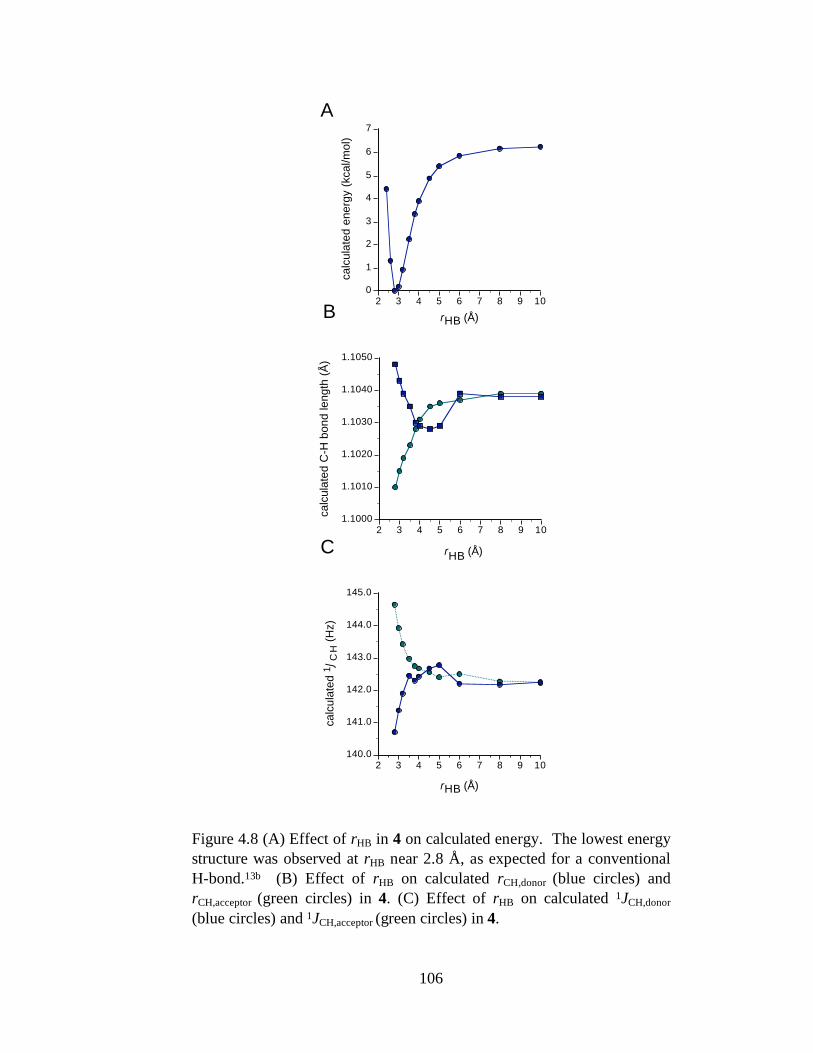
106
A
B
C
0
1
2
3
4
5
6
7
calc
ulat
ed e
nerg
y (k
cal/m
ol)
2 3 4 5 6 7 8 9 10
rHB (Å)
1.1000
1.1010
1.1020
1.1030
1.1040
1.1050
calc
ulat
ed C
-H b
ond
leng
th (
Å)
2 3 4 5 6 7 8 9 10
rHB (Å)
140.0
141.0
142.0
143.0
144.0
145.0
calc
ulat
ed 1
JC
H (
Hz)
2 3 4 5 6 7 8 9 10
rHB (Å)
Figure 4.8 (A) Effect of rHB in 4 on calculated energy. The lowest energy structure was observed at rHB near 2.8 Å, as expected for a conventional H-bond.13b (B) Effect of rHB on calculated rCH,donor (blue circles) and rCH,acceptor (green circles) in 4. (C) Effect of rHB on calculated 1JCH,donor
(blue circles) and 1JCH,acceptor (green circles) in 4.
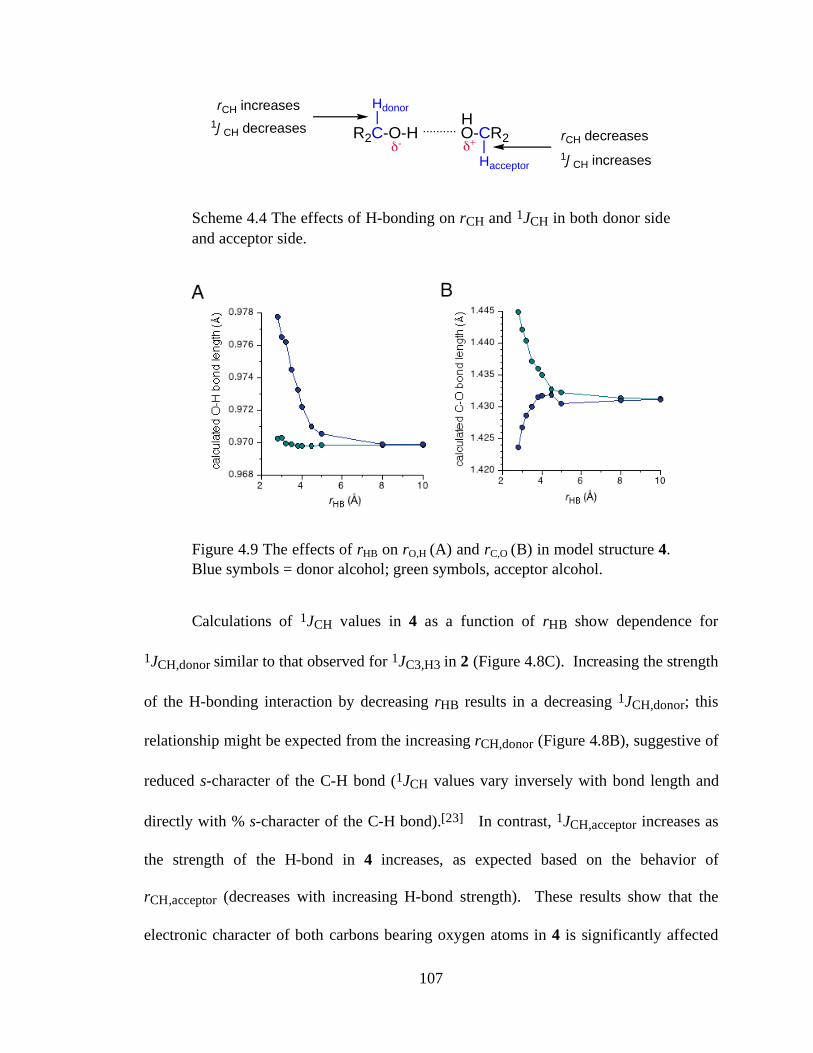
107
R2C-O-H .......... O-CR2
Hacceptor
HHdonor
!- !+
1JCH decreases
1JCH increases
rCH increases
rCH decreases
Scheme 4
Scheme 4.4 The effects of H-bonding on rCH and 1JCH in both donor side and acceptor side.
Figure 4.9 The effects of rHB on rO,H (A) and rC,O (B) in model structure 4. Blue symbols = donor alcohol; green symbols, acceptor alcohol.
Calculations of 1JCH values in 4 as a function of rHB show dependence for
1JCH,donor similar to that observed for 1JC3,H3 in 2 (Figure 4.8C). Increasing the strength
of the H-bonding interaction by decreasing rHB results in a decreasing 1JCH,donor; this
relationship might be expected from the increasing rCH,donor (Figure 4.8B), suggestive of
reduced s-character of the C-H bond (1JCH values vary inversely with bond length and
directly with % s-character of the C-H bond).[23] In contrast, 1JCH,acceptor increases as
the strength of the H-bond in 4 increases, as expected based on the behavior of
rCH,acceptor (decreases with increasing H-bond strength). These results show that the
electronic character of both carbons bearing oxygen atoms in 4 is significantly affected

108
by the strength of the H-bonding interaction, and that this changing character is reflected
in the 1JCH values involving these carbons. The overall effect is summarized in Scheme
4.4. As rHB decreases, the participating oxygens become increasingly polarized, with
that of the donor bearing a partial (-) charge, and that of the acceptor bearing a partial (+)
charge. These opposing charge characteristics translate into opposing trends in the 1JCH
values in the donor and acceptor. The enhanced (-) character (increased electron density)
of the donor lengthens (weakens) the C-Hdonor bond and reduces 1JCH,donor. On the
other hand, the concomitant enhanced (+) character (decreased electron density) of the
acceptor shortens (strengthens) the C-Hacceptor bond and increases 1JCH,acceptor.
These results suggest that, in disaccharide model system 2, not only is the C3-H3
bond a potential probe of inter-residue H-bonding (donor), but also potentially the C1’-
H1’ and C5’-H5’ bonds on the acceptor side of the interaction. Inspection of the
behaviors of 1JC1’,H1’ and 1JC5’,H5’ (Figure 4.10A and 4.10B) show a dependence on the
H3-C3-O3-H torsion angle, but unlike 1JC3,H3, the absolute values of these two couplings
in 2 and 3 differ substantially, as expected since the replacement of O5’ in 2 by a CH2
group in 3 occurs in the vicinity to the J-coupling pathways in question. This situation
complicates the analysis, although, as shown in Figure 4.10C, both couplings increase in
the H-bonded structures, consistent with predictions in Scheme 4.3.
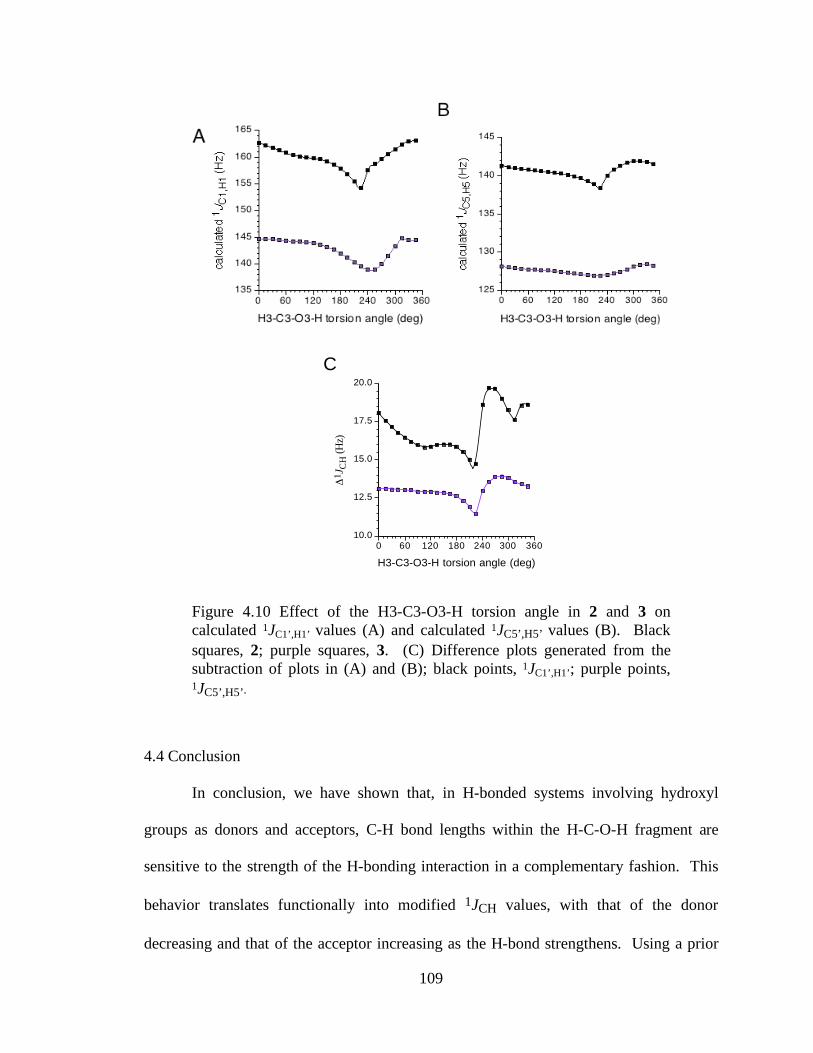
109
135
140
145
150
155
160
165
calc
ulat
ed 1
JC
1,H
1(H
z)
0 60 120 180 240 300 360
10.0
12.5
15.0
17.5
20.0
!1JC
H (
Hz)
0 60 120 180 240 300 360
H3-C3-O3-H torsion angle (deg)
125
130
135
140
145
calc
ulat
ed 1
JC
5,H
5(H
z)
0 60 120 180 240 300 360
H3-C3-O3-H torsion angle (deg)
C
Figure 4.10 Effect of the H3-C3-O3-H torsion angle in 2 and 3 on calculated 1JC1’,H1’ values (A) and calculated 1JC5’,H5’ values (B). Black squares, 2; purple squares, 3. (C) Difference plots generated from the subtraction of plots in (A) and (B); black points, 1JC1’,H1’; purple points, 1JC5’,H5’.
4.4 Conclusion
In conclusion, we have shown that, in H-bonded systems involving hydroxyl
groups as donors and acceptors, C-H bond lengths within the H-C-O-H fragment are
sensitive to the strength of the H-bonding interaction in a complementary fashion. This
behavior translates functionally into modified 1JCH values, with that of the donor
decreasing and that of the acceptor increasing as the H-bond strengthens. Using a prior

110
relationship that correlates the changes in 1JCH with H-bond strength, DFT calculations
yield reasonable H-bond strengths for inter-residue H-bonding in disaccharide models. It
should be appreciated, however, that while this observation suggests a potential role for
1JCH as an H-bond probe, in solution two factors will modulate their values, namely, the
C-O torsion itself (lone-pair effects on 1JCH) and H-bonding interactions. The former
may or may not be conformationally averaged in the absence of H-bonding, whereas in a
strong H-bond, this torsion will be more constrained. In order to properly interpret
changes in 1JCH to specific H-bonding interactions and strengths, it will be necessary to
take the C-O torsional contributions into account. This might be possible through the use
of other J-couplings that are sensitive to excyclic C-O torsions in saccharides,[16,24]
suggesting that a collective appraisal of multiple J-couplings will be needed to
deconvolute the probem and allow for a more quantitative interpretation of 1JCH in H-
bonded systems.
4.5 References
1. Pople, J. A.; Schneider, W. G.; Bernstein, H. J. High-Resolution Nuclear Magnetic Resonance, Chapter 15, McGraw-Hill, 1959, pp. 400-421.
2. Günther, H., NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry, 2nd Edition, John Wiley & Sons, Chichester, UK, pp. 97-99.
3. Dingley, A. J.; Cordier, F.; Grzesiek, S. Concepts in Magnetic Resonance 2001, 13, 103-127.
4. Zhang, W.; Zhao, H.; Carmichael, I.; Serianni, A. S. Carbohydr. Res. 2009, 344, 1582-1587.
5. Maiti, N. C.; Carey, P. R.; Anderson, V. E. J. Phys. Chem. A 2003, 107, 9910-9917.

111
6. Maiti, N. C.; Zhu, Y.; Carmichael, I.; Serianni, A. S.; Anderson, V. E. J. Org. Chem. 2006, 71, 2878-2880.
7. Kirschner, K. N.; Woods, R. J. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10541-10545.
8. Kroon, J.; Kroon-Batenburg, L. M.; Leeflang, B. R.; Vliegenthart, J. F. G. J. Mol. Struct. 1994, 322, 27-31.
9. Corzana, F.; Motawia, M. S.; Hervé du Penhoat, C.; van den Berg, F.; Blennow, A.; Pérez, S.; Engelsen, S. B. J. Am. Chem. Soc. 2004, 126, 13144-13155.
10. Almond, A. Carbohydr. Res. 2005, 340, 907-920.
11. Hansen, P. I.; Larsen, F. H.; Motawia, S. M.; Blennow, A.; Sprual, M.; Dvortsak, P.; Engelsen, S. B. Biopolymers 2008, 89, 1179-1193.
12. Stenutz, R.; Shang, M.; Serianni, A. S. Acta Cryst. 1999, C55, 1721-1725.
13. Pan, Q.; Noll, B.C.; Serianni, A. S. Acta Cryst. 2005, C61, o674-o677.
14. Hu, X.; Pan, Q.; Noll, B. C.; Oliver, A. G.; Serianni, A. S. Acta Cryst. 2010, C66, o67-o70.
15. Leeflang, B. R.; Vliegenthart, J. F. G.; Kroon-Batenburg, L. M. J.; van Eijck, B. P.; Kroon, J. Carbohydr. Res. 1992, 230, 41-61.
16. Zhao, H.; Pan, Q.; Zhang, W.; Carmichael, I.; Serianni, A. S. J. Org. Chem. 2007, 72, 7071-7082.
17. Steiner, T. Angew. Chem. Int. Ed. 2002, 41, 48.
18. Desiraju, G. R.; Steiner, T. The Weak Hydrogen Bond, IUCr Monographs on Crystallography, Vol. 9, Oxford Science Publications, 1999, p. 13.
19. Serianni, A. S.; Wu, J.; Carmichael, I. J. Am. Chem. Soc. 1995, 117, 8645-8650.
20. Cleland, W. W.; Frey, P. A.; Gerlt, J. A. J. Biol. Chem. 1998, 273, 25529-25532.
21. Stenutz, R.; Carmichael, I.; Widmalm, G.; Serianni, A. S. J. Org. Chem. 2002, 67, 949-958 (see Supporting Information)
22. Ben-Tal, N.; Sitkoff, D.; Topol, I. A.; Yang, A.-S.; Burt, S.K.; Honig, B. J. Phys. Chem. B 1997, 101, 450-457.
23. Ando, I.; Webb, G. A. Theory of NMR Parameters, Academic Press, New York, 1983, p. 106.
24. Klepach, T. E.; Carmichael, I.; Serianni, A. S., unpublished results.

112
CHAPTER 5:
AN NMR INVESTIGATION OF PUTATIVE INTER-RESIDUE H-BONDING IN
METHYL α-CELLOBIOSIDE IN SOLUTION
5.1 Introduction
Conformational analyses of oligosaccharides involve not only determinations of
the preferred geometries and time dependent motions of their constituent glycosidic
linkages and pyranosyl and furanosyl rings, but also assessments of the solution
behaviors of exocyclic substituents such as hydroxyl, hydroxymethyl (CH2OH) and N-
acetyl groups. [1-6] In recent years, considerable effort has been directed towards
improving these determinations partly through the development of more quantitative
analyses of NMR spin-spin coupling constants (J-couplings) involving 13C and 1H which
are abundant in saccharides. For example, homonuclear 13C-13C spin-couplings provide
unique and useful constraints not only to assess O-glycosidic linkage geometry [7,8] but
also to establish aldohexopyranosyl ring conformation (e.g., 3JC1,C6 and 3JC3,C6 values
in aldohexopyranosyl rings). [9-10] Karplus or Karplus-like relationships (the latter
defined as parameterization of J-couplings other than 3J in terms of one or more specific
molecular torsion angles) [11] have been described in which equations containing two
torsion angle variables are solved simultaneously to allow determinations of correlated

113
conformations in saccharides, information not easily obtained by other experimental
means. [12]
Establishing the conformational properties of oligosaccharides in solution is
important due to the presumed correlation between their 3D structures and biological
functions. Equally important, however, is determining why a specific oligosaccharide
behaves as it does: what intrinsic structural forces are at work and how do they interact,
and what extrinsic factors (e.g., solvation, context) play a role? Can these factors be
quantified?
The role of H-bonding in influencing oligosaccharide structural properties is an
example of an extrinsic factor whose importance in dictating oligosaccharide
conformation/dynamics in solution has received considerable attention [4,13-18] but still
remains unclear. Studies of single-crystal x-ray structures suggest that H-bonding
between contiguous residues of an oligosaccharide does occur. For example, in β-(1→4)-
linked disaccharides such as methyl α-lactoside (1), [19] methyl β-lactoside (2) [20] and
methyl β-cellobioside (3), [21] (Scheme 1) inter-residue H-bonding is inferred between
O3H (donor) and O5’ (acceptor) based on the O3-O5’ internuclear distance observed in
the crystal structures. The role of this H-bonding in dictating preferred linkage geometry
in aqueous solution is uncertain, given that competition is likely between this
intramolecular H-bonding and intermolecular H-bonding involving solvent water. It is
difficult to obtain definitive experimental evidence about specific saccharide H-bonding
behavior in solution, and more specifically, how strong/persistent this H-bonding might
be. Recent work suggests that 1JCH values involving alcoholic carbons might be useful in
this regard, since their magnitudes are not only influenced by vicinal lone-pair effects [22]

114
but also by the propensity of the OH group to H-bond in either a donor or acceptor
role.[23]
The present investigation approaches this general problem from a different point.
We reasoned that, if the type of inter-residue H-bonding observed in x-ray structures 1-3
were present (persistent) in solution, then J-coupling across this stable H-bond might be
O
O
methyl !-cellobioside (4)(methyl "-D-glucopyranosyl-(1#4)-!-D-glucopyranoside)
OCH3
O
OH
HOHO
HO
OOH
OH
H
O
O
1'
2'
3'
4'5'
6'1
2
3
4
5
6
OCH3
O
OH
HOHO
HO
OOH
OH
H
4'
$
O
O
methyl "-lactoside (2)(methyl "-D-galactopyranosyl-(1#4)-"-D-glucopyranoside)
OCH3
O
OHHO
HOHO
OOH
OH
H
O
O
methyl !-lactoside (1)(methyl "-D-galactopyranosyl-(1#4)-!-D-glucopyranoside)
1'
2'
3'
4'5'
6'1
2
3
4
5
6
OCH3
O
OHHO
HOHO
OOH
OH
H
Scheme 1
O
O
methyl "-cellobioside (3)(methyl "-D-glucopyranosyl-(1#4)-"-D-glucopyranoside)
OCH3O
OH
HOHO
HO
OOH
OH
H
$ = 13C
Scheme 5.1 Structure of model compound 1 – 4 and 4’ and their H-bonding pattern according to x-ray crystal structures.

115
observable, specifically between C4’ and the H-bonded O3H hydrogen (i.e., 3hJC4’,O3H).
To investigate this possibility, DFT calculations were initially performed on model
structures to estimate the magnitude of this coupling under conditions mimicking full
persistence. These calculations yielded predicted 3hJC4’,O3H values of ~ 0.1 Hz. The
uncertainty in these calculated couplings (~ ± 0.2 Hz) is relatively large, however, so we
decided to pursue an experimental investigation of the problem.
5.2 Results and Discussion:
Methyl α-cellobioside 4 (Scheme 5.1), whose solution conformation has been
studied previously, [24,25] was chosen as the target disaccharide, selectively labeled with
13C at C4’ (4’) (Scheme 5.1), because (a) synthesis of D-[4-13C]glucose [26] and
assembly of the labeled disaccharide are straightforward, and (b) the equatorial C4’-O4’
bond in 4/4’ is expected to enhance the 3hJC4’,O3H value, if it exists, due to the terminal
oxygen in-plane effect on 3JCH values. [6,27] In addition, in 4/4’, the C4’-C5’-O5’-O3H
pseudo-torsion angle approaches 180°, rendering this system ideally suited for detection
of a trans-H-bond 3JCCOH if conventional Karplus behavior applies.
The 600 MHz 13C{1H} NMR spectrum of 4’ in 2H2O is shown in Figure 5.1, and
13C spectral parameters are summarized in Table 5.1. Three intra-residue JCC values
were observed in the labeled β-Glcp residue: 1JC4’,C3’ = 39.5 Hz; 1JC4’,C5’ = 41.0 Hz;
2JC4’,C2’ = +2.6 Hz. These couplings are indistinguishable from those reported
previously in methyl β-D-glucopyranoside (Table 5.1). [12] The value of 2JC4’,C6’ was
very small or zero, consistent with the observed behavior of this coupling in
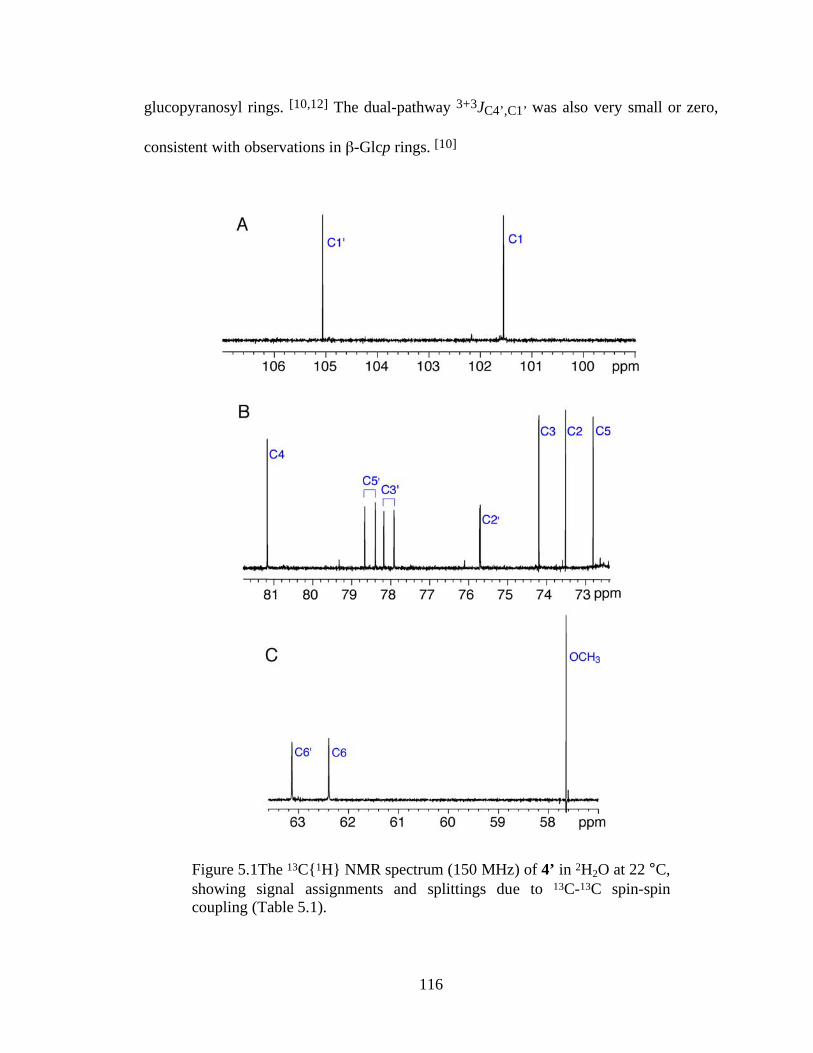
116
glucopyranosyl rings. [10,12] The dual-pathway 3+3JC4’,C1’ was also very small or zero,
consistent with observations in β-Glcp rings. [10]
Figure 5.1The 13C{1H} NMR spectrum (150 MHz) of 4’ in 2H2O at 22 °C, showing signal assignments and splittings due to 13C-13C spin-spin coupling (Table 5.1).
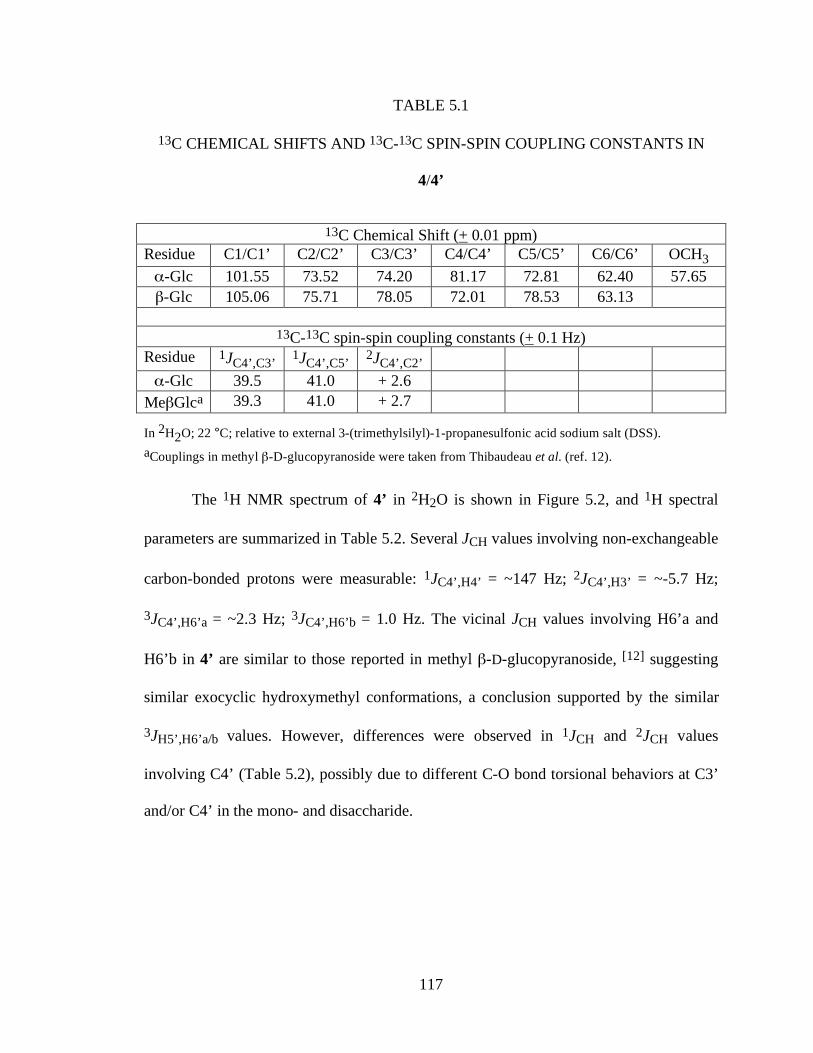
117
TABLE 5.1
13C CHEMICAL SHIFTS AND 13C-13C SPIN-SPIN COUPLING CONSTANTS IN
4/4’
13C Chemical Shift (+ 0.01 ppm) Residue C1/C1’ C2/C2’ C3/C3’ C4/C4’ C5/C5’ C6/C6’ OCH3 α-Glc 101.55 73.52 74.20 81.17 72.81 62.40 57.65 β-Glc 105.06 75.71 78.05 72.01 78.53 63.13
13C-13C spin-spin coupling constants (+ 0.1 Hz)
Residue 1JC4’,C3’ 1JC4’,C5’ 2JC4’,C2’ α-Glc 39.5 41.0 + 2.6
MeβGlca 39.3 41.0 + 2.7
In 2H2O; 22 °C; relative to external 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS). aCouplings in methyl β-D-glucopyranoside were taken from Thibaudeau et al. (ref. 12).
The 1H NMR spectrum of 4’ in 2H2O is shown in Figure 5.2, and 1H spectral
parameters are summarized in Table 5.2. Several JCH values involving non-exchangeable
carbon-bonded protons were measurable: 1JC4’,H4’ = ~147 Hz; 2JC4’,H3’ = ~-5.7 Hz;
3JC4’,H6’a = ~2.3 Hz; 3JC4’,H6’b = 1.0 Hz. The vicinal JCH values involving H6’a and
H6’b in 4’ are similar to those reported in methyl β-D-glucopyranoside, [12] suggesting
similar exocyclic hydroxymethyl conformations, a conclusion supported by the similar
3JH5’,H6’a/b values. However, differences were observed in 1JCH and 2JCH values
involving C4’ (Table 5.2), possibly due to different C-O bond torsional behaviors at C3’
and/or C4’ in the mono- and disaccharide.
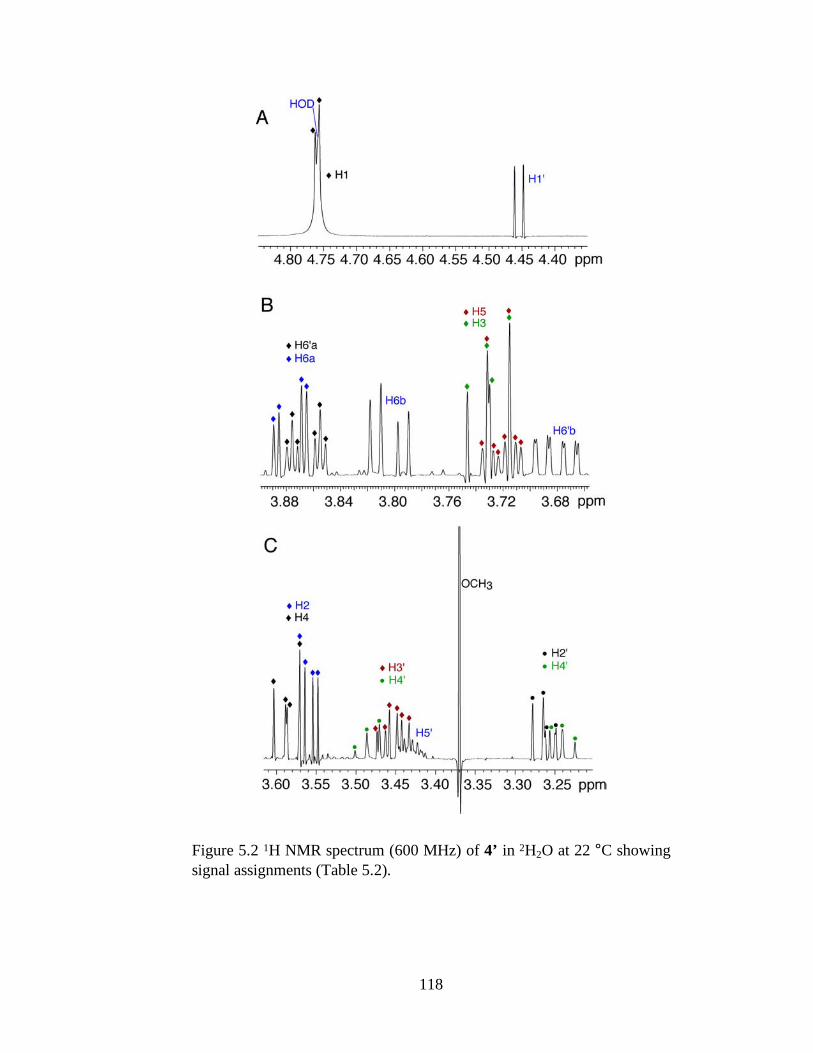
118
Figure 5.2 1H NMR spectrum (600 MHz) of 4’ in 2H2O at 22 °C showing signal assignments (Table 5.2).
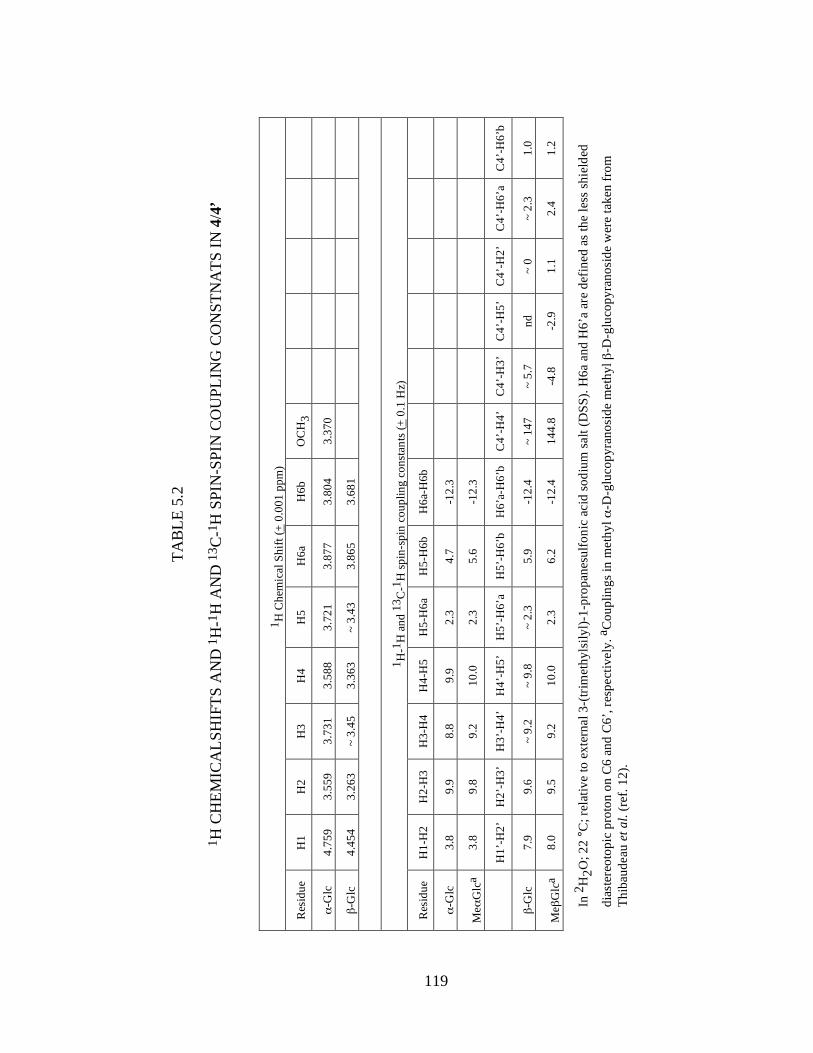
119
TAB
LE 5
.2
1 H C
HEM
ICA
LSH
IFTS
AN
D 1
H-1
H A
ND
13 C
-1H
SPI
N-S
PIN
CO
UPL
ING
CO
NST
NA
TS IN
4/4
’
1 H C
hem
ical
Shi
ft (+
0.0
01 p
pm)
Res
idue
H
1 H
2 H
3 H
4 H
5 H
6a
H6b
O
CH
3
α-G
lc
4.75
9 3.
559
3.73
1 3.
588
3.72
1 3.
877
3.80
4 3.
370
β-G
lc
4.45
4 3.
263
~ 3.
45
3.36
3 ~
3.43
3.
865
3.68
1
1 H-1
H a
nd 1
3 C-1
H sp
in-s
pin
coup
ling
cons
tant
s (+
0.1
Hz)
Res
idue
H
1-H
2 H
2-H
3 H
3-H
4 H
4-H
5 H
5-H
6a
H5-
H6b
H
6a-H
6b
α-G
lc
3.8
9.9
8.8
9.9
2.3
4.7
-12.
3
Meα
Glc
a 3.
8 9.
8 9.
2 10
.0
2.3
5.6
-12.
3
H
1’-H
2’
H2’
-H3’
H
3’-H
4’
H4’
-H5’
H
5’-H
6’a
H5’
-H6’
b H
6’a-
H6’
b C
4’-H
4’
C4’
-H3’
C
4’-H
5’
C4’
-H2’
C
4’-H
6’a
C4’
-H6’
b
β-G
lc
7.9
9.6
~ 9.
2 ~
9.8
~ 2.
3 5.
9 -1
2.4
~ 14
7 ~
5.7
nd
~ 0
~ 2.
3 1.
0
Meβ
Glc
a 8.
0 9.
5 9.
2 10
.0
2.3
6.2
-12.
4 14
4.8
-4.8
-2
.9
1.1
2.4
1.2
In 2
H2O
; 22
°C; r
elat
ive
to e
xter
nal 3
-(tri
met
hyls
ilyl)-
1-pr
opan
esul
foni
c ac
id so
dium
sal
t (D
SS).
H6a
and
H6’
a ar
e de
fined
as
the
less
shie
lded
dias
tere
otop
ic p
roto
n on
C6
and
C6’
, res
pect
ivel
y. a
Cou
plin
gs in
met
hyl α
-D-g
luco
pyra
nosi
de m
ethy
l β-D
-glu
copy
rano
side
wer
e ta
ken
from
Th
ibau
deau
et a
l. (r
ef. 1
2).
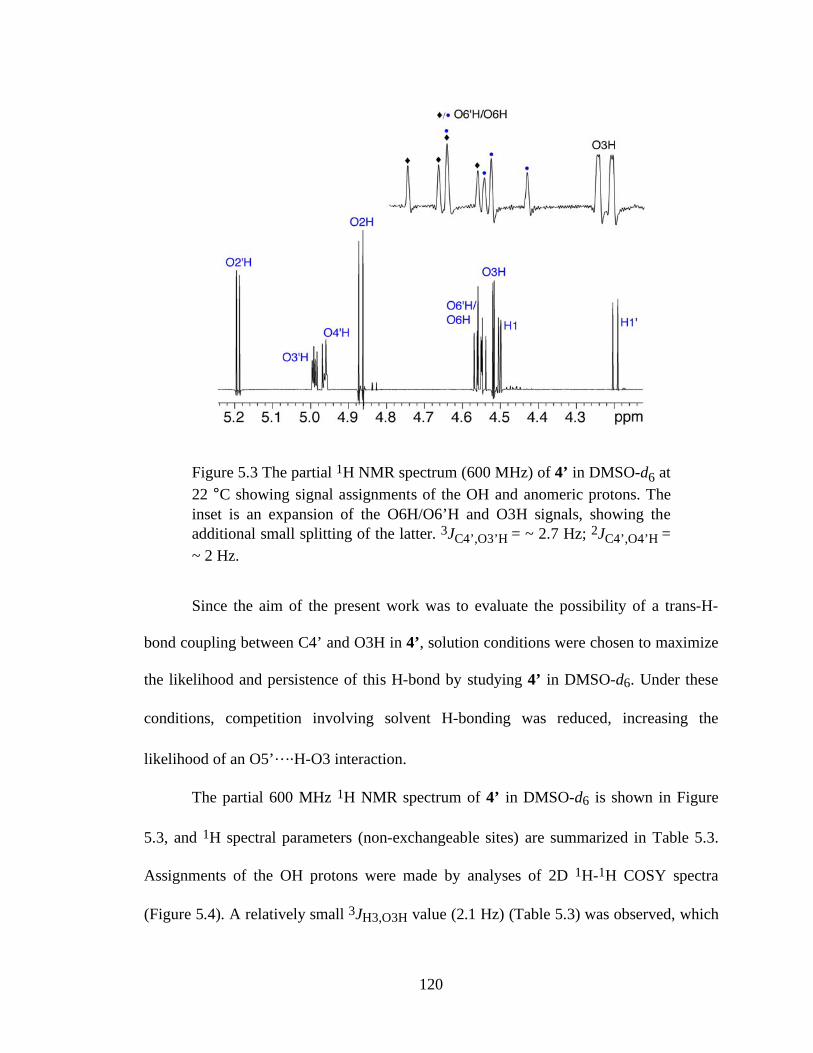
120
Figure 5.3 The partial 1H NMR spectrum (600 MHz) of 4’ in DMSO-d6 at 22 °C showing signal assignments of the OH and anomeric protons. The inset is an expansion of the O6H/O6’H and O3H signals, showing the additional small splitting of the latter. 3JC4’,O3’H = ~ 2.7 Hz; 2JC4’,O4’H = ~ 2 Hz.
Since the aim of the present work was to evaluate the possibility of a trans-H-
bond coupling between C4’ and O3H in 4’, solution conditions were chosen to maximize
the likelihood and persistence of this H-bond by studying 4’ in DMSO-d6. Under these
conditions, competition involving solvent H-bonding was reduced, increasing the
likelihood of an O5’….H-O3 interaction.
The partial 600 MHz 1H NMR spectrum of 4’ in DMSO-d6 is shown in Figure
5.3, and 1H spectral parameters (non-exchangeable sites) are summarized in Table 5.3.
Assignments of the OH protons were made by analyses of 2D 1H-1H COSY spectra
(Figure 5.4). A relatively small 3JH3,O3H value (2.1 Hz) (Table 5.3) was observed, which
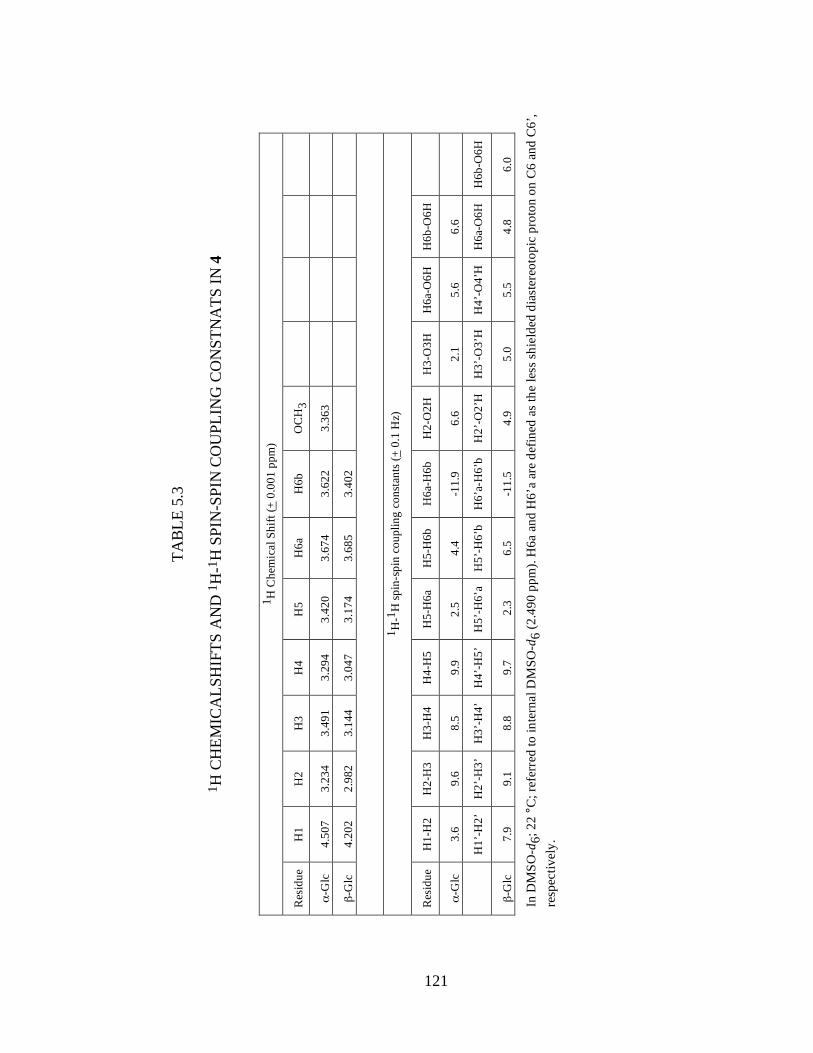
121
TAB
LE 5
.3
1 H C
HEM
ICA
LSH
IFTS
AN
D 1
H-1
H S
PIN
-SPI
N C
OU
PLIN
G C
ON
STN
ATS
IN 4
1 H C
hem
ical
Shi
ft (+
0.0
01 p
pm)
Res
idue
H
1 H
2 H
3 H
4 H
5 H
6a
H6b
O
CH
3
α-G
lc
4.50
7 3.
234
3.49
1 3.
294
3.42
0 3.
674
3.62
2 3.
363
β-G
lc
4.20
2 2.
982
3.14
4 3.
047
3.17
4 3.
685
3.40
2
1 H
-1H
spin
-spi
n co
uplin
g co
nsta
nts (
+ 0.
1 H
z)
Res
idue
H
1-H
2 H
2-H
3 H
3-H
4 H
4-H
5 H
5-H
6a
H5-
H6b
H
6a-H
6b
H2-
O2H
H
3-O
3H
H6a
-O6H
H
6b-O
6H
α-G
lc
3.6
9.6
8.5
9.9
2.5
4.4
-11.
9 6.
6 2.
1 5.
6 6.
6
H
1’-H
2’
H2’
-H3’
H
3’-H
4’
H4’
-H5’
H
5’-H
6’a
H5’
-H6’
b H
6’a-
H6’
b H
2’-O
2’H
H
3’-O
3’H
H
4’-O
4’H
H
6a-O
6H
H6b
-O6H
β-G
lc
7.9
9.1
8.8
9.7
2.3
6.
5 -1
1.5
4.9
5.0
5.5
4.8
6.0
In D
MSO
-d6;
22
°C; r
efer
red
to in
tern
al D
MSO
-d6
(2.4
90 p
pm).
H6a
and
H6’
a ar
e de
fined
as
the
less
shie
lded
dia
ster
eoto
pic
prot
on o
n C
6 an
d C
6’,
resp
ectiv
ely.
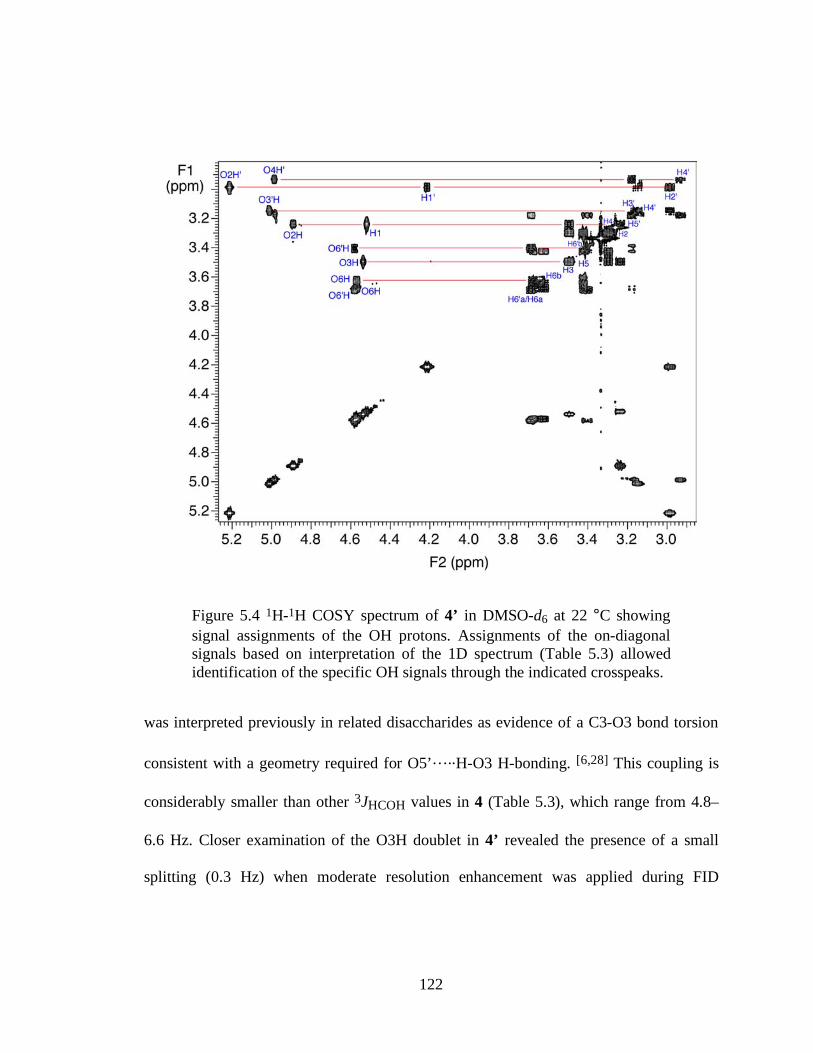
122
Figure 5.4 1H-1H COSY spectrum of 4’ in DMSO-d6 at 22 °C showing signal assignments of the OH protons. Assignments of the on-diagonal signals based on interpretation of the 1D spectrum (Table 5.3) allowed identification of the specific OH signals through the indicated crosspeaks.
was interpreted previously in related disaccharides as evidence of a C3-O3 bond torsion
consistent with a geometry required for O5’…..H-O3 H-bonding. [6,28] This coupling is
considerably smaller than other 3JHCOH values in 4 (Table 5.3), which range from 4.8–
6.6 Hz. Closer examination of the O3H doublet in 4’ revealed the presence of a small
splitting (0.3 Hz) when moderate resolution enhancement was applied during FID
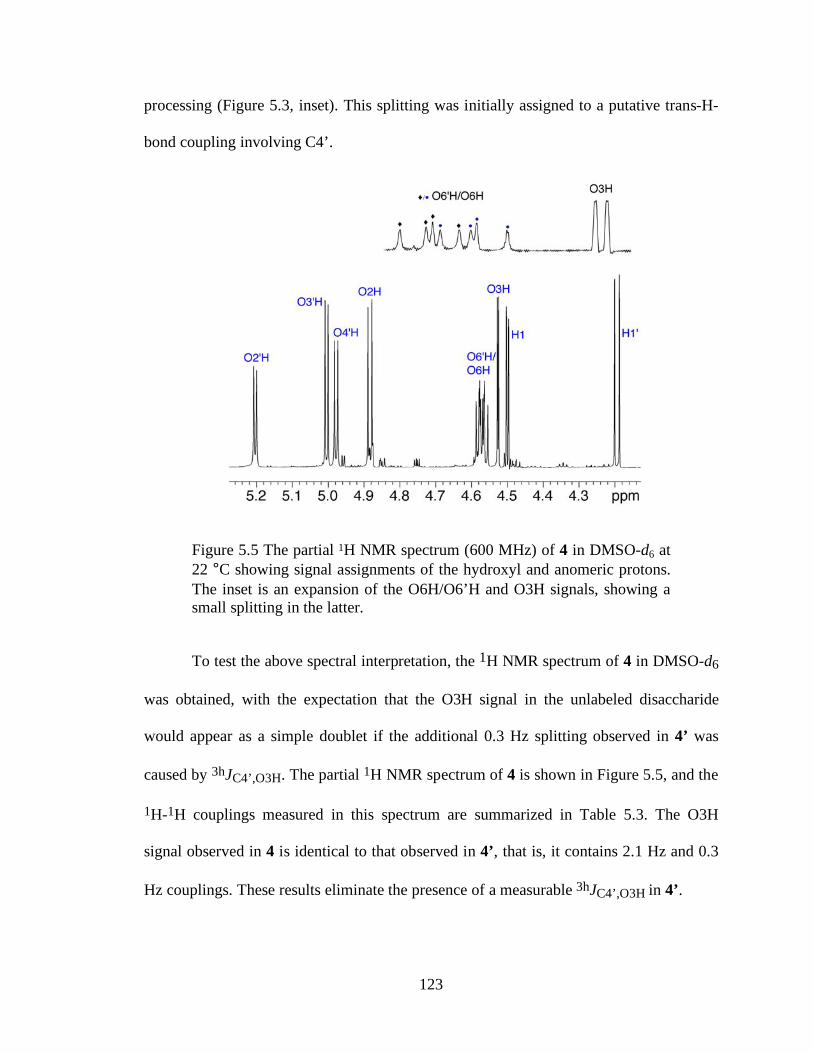
123
processing (Figure 5.3, inset). This splitting was initially assigned to a putative trans-H-
bond coupling involving C4’.
Figure 5.5 The partial 1H NMR spectrum (600 MHz) of 4 in DMSO-d6 at 22 °C showing signal assignments of the hydroxyl and anomeric protons. The inset is an expansion of the O6H/O6’H and O3H signals, showing a small splitting in the latter.
To test the above spectral interpretation, the 1H NMR spectrum of 4 in DMSO-d6
was obtained, with the expectation that the O3H signal in the unlabeled disaccharide
would appear as a simple doublet if the additional 0.3 Hz splitting observed in 4’ was
caused by 3hJC4’,O3H. The partial 1H NMR spectrum of 4 is shown in Figure 5.5, and the
1H-1H couplings measured in this spectrum are summarized in Table 5.3. The O3H
signal observed in 4 is identical to that observed in 4’, that is, it contains 2.1 Hz and 0.3
Hz couplings. These results eliminate the presence of a measurable 3hJC4’,O3H in 4’.
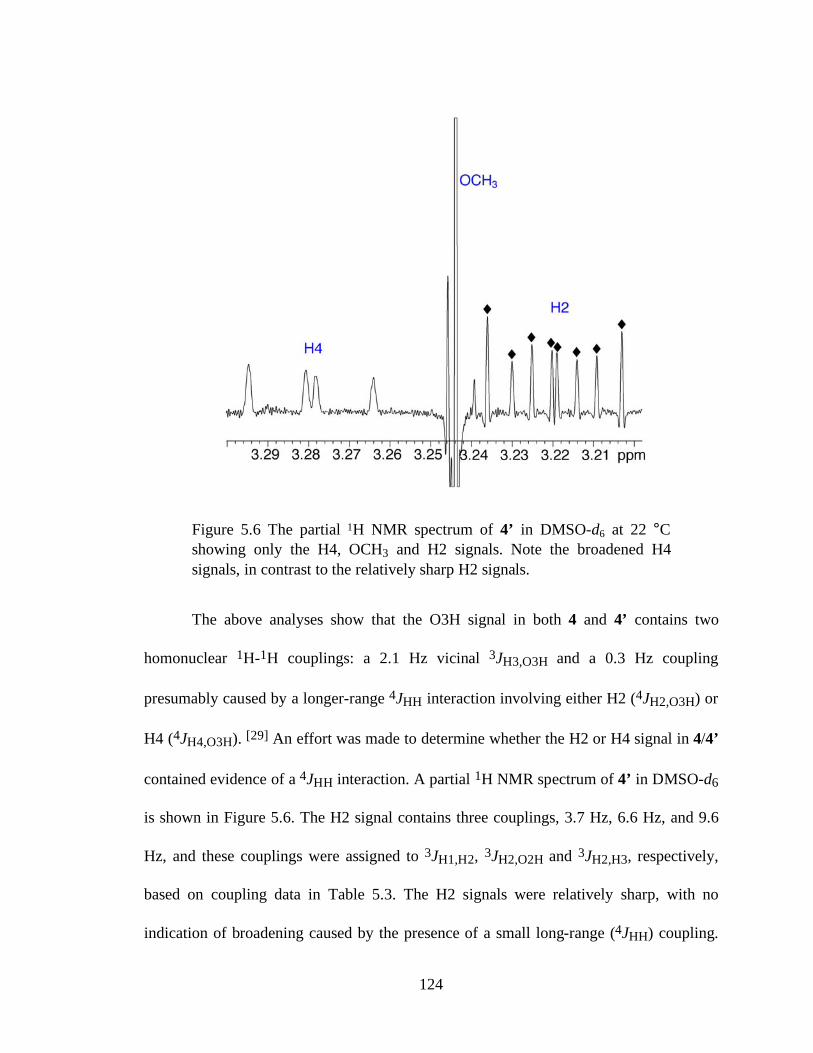
124
Figure 5.6 The partial 1H NMR spectrum of 4’ in DMSO-d6 at 22 °C showing only the H4, OCH3 and H2 signals. Note the broadened H4 signals, in contrast to the relatively sharp H2 signals.
The above analyses show that the O3H signal in both 4 and 4’ contains two
homonuclear 1H-1H couplings: a 2.1 Hz vicinal 3JH3,O3H and a 0.3 Hz coupling
presumably caused by a longer-range 4JHH interaction involving either H2 (4JH2,O3H) or
H4 (4JH4,O3H). [29] An effort was made to determine whether the H2 or H4 signal in 4/4’
contained evidence of a 4JHH interaction. A partial 1H NMR spectrum of 4’ in DMSO-d6
is shown in Figure 5.6. The H2 signal contains three couplings, 3.7 Hz, 6.6 Hz, and 9.6
Hz, and these couplings were assigned to 3JH1,H2, 3JH2,O2H and 3JH2,H3, respectively,
based on coupling data in Table 5.3. The H2 signals were relatively sharp, with no
indication of broadening caused by the presence of a small long-range (4JHH) coupling.

125
The H4 signal contains two couplings, 8.5 Hz and 9.9 Hz, and these couplings were
assigned to 3JH3,H4 and 3JH4,H5, respectively, based on coupling data in Table 5.3.
Importantly, the H4 signals, relative to the H2 signals, were broadened, suggestive of the
presence of additional long-range (4JHH) coupling(s). The latter could arise from
4JH4,O3H, 4JH4,H1’ (trans-glycoside) [30] and/or 4JH4,H6a/4JH4,H6b. Closer examination of
the H4 signals revealed that they contain at least two long-range couplings based on their
line-shapes. Since the H2 signal shows no evidence of coupling to O3H, the only
remaining candidate is H4 whose signal exhibits the expected properties.
A 2D 1H-1H COSY spectrum was obtained on 4 (Figure 5.7) to further investigate
long-range coupling involving H4. Weak cross peaks correlating H4 with both H1’ and
O3H were observed, with the former considerably more intense than the latter. No
correlations were detected between H4 and the H6a/H6b sites. These findings indicate
that the line broadening observed in the H4 signal (Figure 5.6) is attributable to 4JH4,O3H
and 4JH4,H1’. Since there is no evidence of 4JH2,O3H in the H2 signal in the 1D 1H data
(Figure 5.6), the small 0.3 Hz splitting of the O3H signal (Figures 5.3 and 5.5) is
attributed to 4JH4,O3H.
The structural dependency of 4JHH involving hydroxyl protons in saccharides was
investigated in a limited set of DFT calculations performed on 5, a structural mimic of 4.
In 5, the H3-C3-O3-H torsion angle was rotated in 15° increments and 3JH3,O3H,
4JH2,O3H and 4JH4,O3H values were calculated. In these calculations, the following
torsion angles were held fixed: H1’-C1’-O1’-C4 = 31.9° (x-ray value) [21]; C1’-O1’-C4-
H4 = -43.7° (x-ray value) [21]. Initial values for the C2-C1-O1-CH3, C1-C2-O2-O2H, and
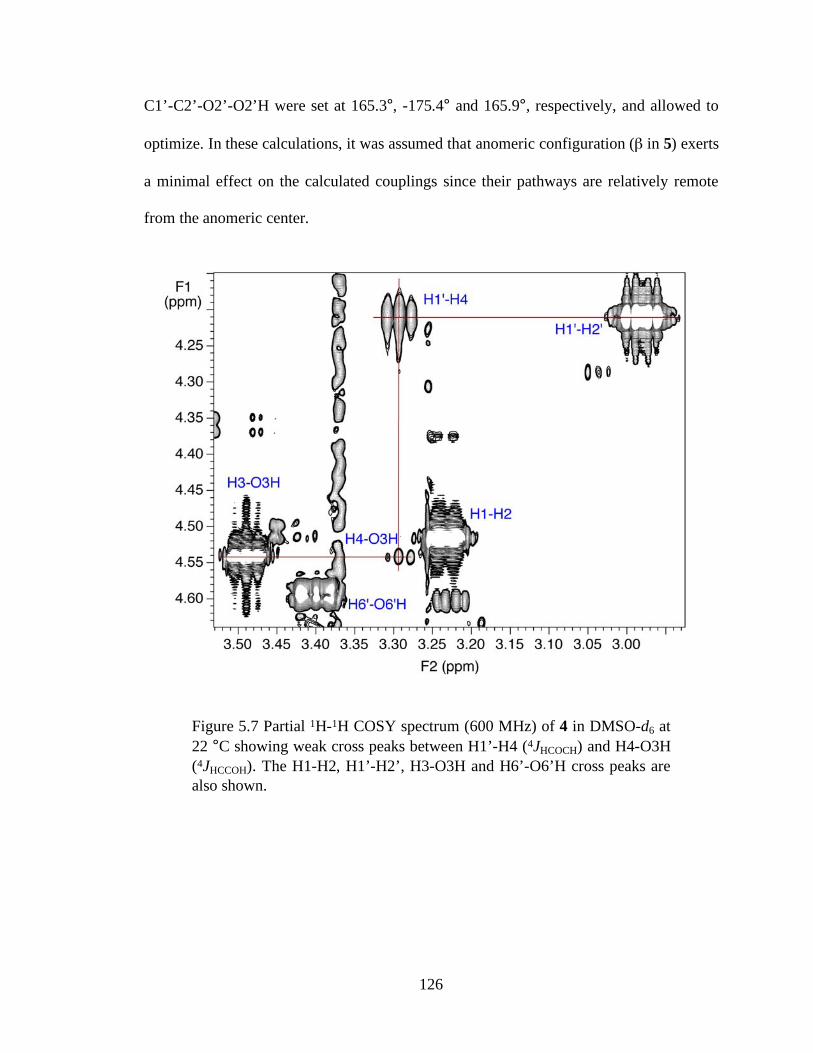
126
C1’-C2’-O2’-O2’H were set at 165.3°, -175.4° and 165.9°, respectively, and allowed to
optimize. In these calculations, it was assumed that anomeric configuration (β in 5) exerts
a minimal effect on the calculated couplings since their pathways are relatively remote
from the anomeric center.
Figure 5.7 Partial 1H-1H COSY spectrum (600 MHz) of 4 in DMSO-d6 at 22 °C showing weak cross peaks between H1’-H4 (4JHCOCH) and H4-O3H (4JHCCOH). The H1-H2, H1’-H2’, H3-O3H and H6’-O6’H cross peaks are also shown.

127
O
O OCH3
OOH
HOOHCH3
CH3
H3
H2
H45
Scheme 5.2 Model structure 5 and one of its snapshots when rotating H3-C3-O3-H torsion angle.
The dependence of 3JH3,O3H on the H3-C3-O3-H torsion angle in 5 is shown in
Figure 5.8A. Superimposed on these data is a generalized Karplus dependency for
3JHCOH reported recently by Zhao et al. [6] The close agreement between the two data
sets serves as an internal control for the present calculations, and validates the method for
use in the 4JHCCOH calculations.
The calculated dependencies of two 4JHCCOH, namely 4JH2,O3H and 4JH4,O3H,
are shown in Figure 5.8B. In both cases, the calculated couplings range from ~-0.7 Hz to
~+ 1.1 Hz, with maximum coupling observed at H3-C3-O3-O3H torsion angles near 165°
and 210° for 4JH2,O3H and 4JH4,O3H, respectively. The two curves have similar overall
shapes and are phase-shifted by ~45°.
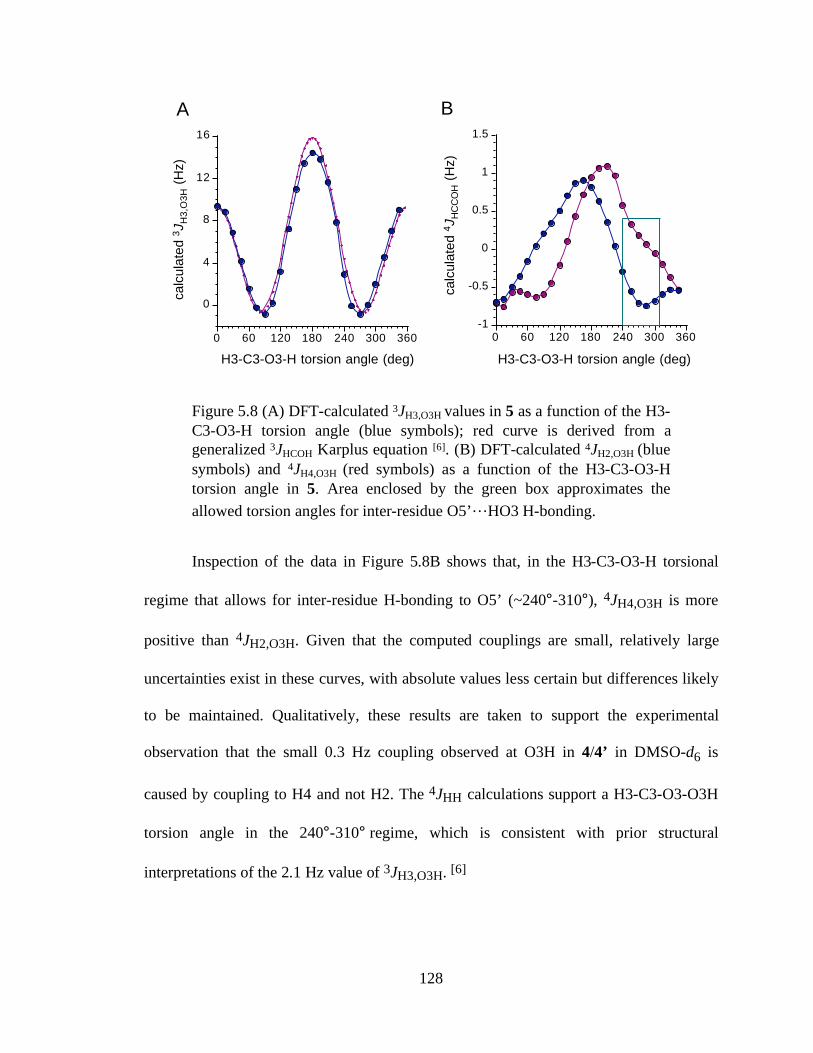
128
0
4
8
12
16
calc
ulat
ed 3
JH
3,O
3H (
Hz)
0 60 120 180 240 300 360
H3-C3-O3-H torsion angle (deg)
-1
-0.5
0
0.5
1
1.5
calc
ulat
ed 4
JH
CC
OH (
Hz)
0 60 120 180 240 300 360
H3-C3-O3-H torsion angle (deg)
A B
Figure 5.8 (A) DFT-calculated 3JH3,O3H values in 5 as a function of the H3-C3-O3-H torsion angle (blue symbols); red curve is derived from a generalized 3JHCOH Karplus equation [6]. (B) DFT-calculated 4JH2,O3H (blue symbols) and 4JH4,O3H (red symbols) as a function of the H3-C3-O3-H torsion angle in 5. Area enclosed by the green box approximates the allowed torsion angles for inter-residue O5’…HO3 H-bonding.
Inspection of the data in Figure 5.8B shows that, in the H3-C3-O3-H torsional
regime that allows for inter-residue H-bonding to O5’ (~240°-310°), 4JH4,O3H is more
positive than 4JH2,O3H. Given that the computed couplings are small, relatively large
uncertainties exist in these curves, with absolute values less certain but differences likely
to be maintained. Qualitatively, these results are taken to support the experimental
observation that the small 0.3 Hz coupling observed at O3H in 4/4’ in DMSO-d6 is
caused by coupling to H4 and not H2. The 4JHH calculations support a H3-C3-O3-O3H
torsion angle in the 240°-310° regime, which is consistent with prior structural
interpretations of the 2.1 Hz value of 3JH3,O3H. [6]

129
5.3 Conclusion
This investigation aimed to determine whether a trans-H-bond 3JCCOH value
could be detected in a β-(1→4)-linked disaccharide in DMSO-d6 solvent, thus providing
direct experimental evidence that this H-bonding is persistent under these solution
conditions. Prior evidence of this H-bonding has been indirect, either implied from x-ray
crystal structure studies based on observed C3-O3 bond torsions and O3-O5’ internuclear
distances, or from solution NMR studies in which aberrant 3JH3,O3H values were
interpreted as consistent with geometries that allow this H-bonding, and temperature
effects on hydroxyl proton chemical shifts were investigated. [6,28]
Despite optimizing the experiment to detect a potential 3hJC4’,O3H in 4’, no
coupling was detected. Initial observation of a small 0.3 Hz splitting in the O3H signal
was traced to long-range 4JHH coupling involving H4. The reasons for this failure may lie
in (a) the intrinsically very small values of trans-H-bond 3hJCCOH couplings, making
them inadequate for this type of characterization, or (b) the coupling is detectable but a
strong/persistent H-bond in 4 does not exist in DMSO-d6 solution. The former argument
is supported by DFT calculations that predict very small values of 3hJCCOH, but the
negative findings do not allow exclusion of the latter possibility. O’Leary and coworkers
[31] have reported analogous 2hJHH and 3hJHH couplings in conformationally constrained
1,3- and 1,4-diols in DMSO-d6, with values of < ~0.3 Hz.
DFT calculations of long-range 4JHH values involving hydroxyl protons are
predicted to be as large as +1 Hz, and in some cases may be negative in sign. The two H-
C-C-O-H coupling pathways examined in this report are structurally related in that both

130
involve an axial carbon-bound hydrogen (H2 or H4) coupled to a hydroxyl proton borne
by an equatorial hydroxyl group (O3H). This similarity is likely the root cause of the
similar C3-O3 bond torsion dependencies; when H3 is approximately anti to O3H,
4JH2,O3H and 4JH4,O3H are at or near their maximal values. Under these conditions, the
coupled hydrogens are in a 1,3-diaxial arrangement, which is known to give measurable
4JHH when carbon-bound hydrogens are involved in the coupling (four-bond H-C-C-C-H
pathways). [32] In the present work, the structural dependencies of the other two cases
were not examined, that is, when the coupled sites are 1,3-diequatorial or 1,3-
axial/equatorial. However, these cases, as found for the diaxial case, are expected to
mimic the behaviors of analogous H-C-C-C-H, H-C-O-C-H and H-C-C-O-C systems.
[30,32-33]
5.4 References
1. Homans, S.W. Prog. NMR Spectrosc. 1990, 22, 55-81.
2. Rao, V.S.R.; Qasba, P.K.; Balaji, P.V.; Chandrasekaran, R. Conformation of Carbohydrates, Harwood Academic Publishers, 1998.
3. Serianni, A.S. Carbohydrate Structure, Conformation and Reactivity: NMR Studies with Stable Isotopes, in Bioorganic Chemistry: Carbohydrates, S. Hecht, ed., Oxford University Press, New York, 1999.
4. Kirschner, K.N; Woods, R.J. Proc. Natl. Acad. Sci. 2001, 98, 10541-10545.
5. Stenutz, R.; Carmichael, I.; Widmalm, G.; Serianni, A.S. J. Org. Chem. 2002, 67, 949-958.
6. Zhao, H.; Pan, Q.; Zhang, W.; Carmichael, I.; Serianni, A.S. J. Org. Chem. 2007, 72, 7071-7082.
7. Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F.; Bondo, P.B.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 1998, 120, 11158-11173.

131
8. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 1999, 121, 9843-9851.
9. Wu, J.; Bondo, P.B.; Vuorinen, T.; Serianni, A.S. J. Am. Chem. Soc. 1992, 114, 3499-3505.
10. Bose-Basu, B.; Klepach, T.; Bondo, G.; Bondo, P.B.; Zhang, W.;Carmichael, I.; Serianni, A.S. J. Org. Chem. 2007, 72, 7511-7522.
11. Klepach, T.E.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2005, 127, 9781-9793.
12. Thibaudeau, C.; Stenutz, R.; Hertz, B.; Klepach, T.; Zhao, S.; Wu, Q.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2004, 126, 15668-15685.
13. Kroon, J.; Kroon-Batenburg, L.M.J.; Leeflang, B.R.; Vliegenthart, J.F.G. J. Mol. Struct. 1994, 322, 27-31.
14. Hemmingsen, L.; Madsen, D.E.; Esbensen, A.L.; Olsen, L.; Engelsen, S.B. Carbohydr. Res. 2004, 339, 937-948.
15. Engelsen, S.B.; Monteiro, C.; Hervé du Penhoat, C.; Pérez, S. Biophys. Chem. 2001, 93, 103-127.
16. Corzana, F.; Motawia, M.S.; Hervé du Penhoat, C.; van den Berg, F.; Blennow, A.; Pérez, S.; Engelsen, S.B. J. Am. Chem. Soc. 2004, 126, 13144-13155.
17. Almond, A. Carbohydr. Res. 2005, 340, 907-920.
18. Hansen, P.I.; Larsen, F.H.; Motawia, S.M.; Blennow, A.; Sprual, M.; Dvortsak, P.; Engelsen, S.B. Biopolymers 2008, 89, 1179-1193.
19. Pan, Q.; Noll, B.C.; Serianni, A.S. Acta Cryst. 2005, C61, o674-o677.
20. Stenutz, R.; Shang, M.; Serianni, A.S. Acta Cryst. 1999, C55, 1719-1721.
21. Ham, J.T.; Williams, D.G. Acta Cryst. 1970, B26, 1373-1383.
22. Serianni, A.S.; Wu, J.; Carmichael, I. J. Am. Chem. Soc. 1995, 117, 8645-8650.
23. Maiti, N.C.; Zhu, Y.; Carmichael, I.; Serianni, A.S.; Anderson, V.E. J. Org. Chem. 2006, 71, 2878-2880.
24. Larsson, E.A.; Staaf, M.; Söderman, P.; Höög, C.; Widmalm, G. J. Phys.Chem. A. 2004, 108, 3932-3937.
25. Olsson, U.; Serianni, A.S.; Stenutz, R. J. Phys. Chem. B 2008, 112, 4447-4453.
26. Serianni, A.S.; Vuorinen, T.; Bondo, P.B. J. Carbohydr. Chem. 1990, 9, 513- 541.

132
27. Podlasek, C.A.; Wu, J.; Stripe, W.A.; Bondo, P.B.; Serianni, A.S. J. Am. Chem. Soc. 1995, 117, 8635-8644.
28. Leeflang, B.R.; Vliegenthart, J.F.G.; Kroon-Batenburg, L.M.J.; van Eijck, B.P.; Kroon, J. Carbohydr. Res. 1992, 230, 41-61.
29. Larsson, E.A.; Ulicny, J.; Laaksonen, A.; Widmalm, G. Org. Lett. 2002, 4, 1831-1834.
30. Otter, A.; Bundle, D.R. J. Magn. Reson. 1995, B109, 194-201.
31. Fierman, M.; Nelson, A.; Khan, S.I.; Barfield, M.; O’Leary, D.J. Org. Lett. 2000, 2, 2077-2080.
32. Barfield, M.; Dean, A.M.; Fallick, C.J.; Spear, R.J.; Sternwell, S.; Westerman, P.W. J. Am. Chem. Soc. 1975, 97, 1482-1492.
33. Pan, Q.; Klepach, T.; Carmichael, I.; Reed, M.; Serianni, A.S. J. Org. Chem. 2005, 70, 7542-7549.

133
CHAPTER 6:
OLIGOSACCHARIDE TRANS-GLYCOSIDE 3JCOCC KARPLUS CURVES ARE NOT
EQUIVALENT: EFFECT OF INTERNAL ELECTRONEGATIVE SUBSTITUENTS
6.1 Introduction
Trans-glycoside (inter-residue) NMR 3JCOCC spin-spin coupling constants have
attracted increased attention in recent years as experimental parameters to improve the
assignment of linkage conformation of oligosaccharides in solution. [1, 2] These couplings
show the expected primary Karplus dependence on the central C-O bond torsion, but
secondary structural factors also influence them. An example of the latter is the effect of
terminal electronegative substituents, with in-plane oxygens on either of the terminal
(coupled) carbons increasing the observed coupling by ~0.7 Hz. [1] As part of efforts to
characterize these secondary dependencies, we posed the following question: Are the two
types of inter-residue C-O-C-C coupling pathways present in oligosaccharides equivalent,
that is, can both be treated adequately using a single, generalized Karplus equation that
accounts for the effects of terminal in-plane oxygen? We show herein that these pathways
are non-equivalent and should be treated using separate parametrized equations.

134
O
OOCH3
! "
OH
HO
O
OHHO
HO
HO
OH
C1C2
O5
C4'
C6'
C3'
O
OOCH3
OH
HO
1 (methyl #-lactoside)
O
OHHO
HO
HO
OH
Trans-glycoside COCC pathways
I: C1-O1-C4'-C3' C1-O1-C4'-C5'
II: C2-C1-O1-C4'
CaOCC
CCaOC
Type I
Type II
Scheme 6.1 Methyl β-lactoside and definitions of Type I and Type II trans-glycoside COCC (or CCOC) pathways.
Two different trans-glycoside C-O-C-C coupling pathways are present in
oligosaccharides, as illustrated using methyl β-lactoside 1. Type I involves C1 as a
terminal coupled carbon, is denoted a CaOCC pathway, where Ca denotes an anomeric
carbon, and is sensitive to the O1-C4’ bond torsion (ψ). Type II involves C2 as a terminal
coupled carbon, is denoted a CCaOC pathway, and is sensitive to the C1- O1 bond
torsion (φ). A generalized representation of both pathways (Scheme 6.2) illustrates that
the CaOCC pathway contains two terminal and no internal electronegative substituents,
whereas the CCaOC pathway contains one terminal and one internal electronegative
substituent. At issue is whether the internal substituent perturbs the coupling behavior.
Earlier work on 3JCOCC parametrization for O-glycosidic linkages hinted at
different behaviors for Type I and Type II pathways. Using β-(1→4)-linked disaccharide
mimics, 3JC1,C3’, 3JC1,C5’ and 3JC2,C4’ values were calculated by density functional
theory (DFT) as a function of the central torsion angle. [2] Noteworthy was an apparent
shift of the maximal 3JC2,C4’ value from the expected C2-C1-O1-C4’ torsion angle of

135
180°. However, since structures were confined to those having an optimal exoanomeric
effect, and data collection was confined to only one-half of the full 360° torsional range,
firm conclusions about the significance of this shift could not be made.
C1' O C4 C3/5
C2' C1' O C4
RO
RO
R'O
The Type I pathway contains only terminal electronegative substituents.
The Type II pathway contains terminal and internal electronegative substituents.
OR'
Scheme 6.2 A generalized Type I and Type II CCOC pathways.
In this study we showed that the two types of C-O-C-C coupling pathways across
O-glycosidic linkages of oligosaccharides are not equivalent, and that internal
electronegative substituents cause phase shifting of the corresponding Karplus curves.
This behavior will be examined in greater detail in the next chapter.
6.2 Calculational Section
Model structures 2-4 contain C-O-C-C coupling pathways that mimic those that
exist across O-glycosidic linkages of oligosaccharides. Model compounds 5-7 served as
structural analogs of 2. Density functional theory (DFT) [3] calculations using the B3LYP
functional [4] and 6-31G* basis set [5] were conducted within Gaussian03 [6] for geometric
optimization of molecular structures. J-Couplings were calculated by DFT within
Gaussian03 using an extended basis set ([5s2p1d|3s1p]). [7] Errors in the calculated
couplings are estimated at ± 0.2-0.4 Hz (absolute error), whereas relative changes in
couplings are expected to be more accurate. Different C-O-C-C torsion angles, denoted

136
by color in the structures, were varied from 0° to 360° in 15° increments by holding the
angles at fixed values while optimizing all other parameters. Geometry optimizations of
all model structures were initiated from reasonably low energy conformations except for
the fixed torsion angles indicated above. For example, the C2-C1-O1-CH3 torsion angles
in 3 and 4 started from an anti geometry and remained approximately anti throughout the
optimization.
OCH3
O
OH CH2
CH3
OCH3
O
OH CH2
CH32a
2b
OO
CH3
OO
OCH3
CH3
O
CH33
4
CH3
O
OH CH2
CH35
O
O
O
O
6
7
Scheme 6.3 Strucures of 2, 3, 4 and 5 and the coupling pathways.
6.3 Results and Discussion
6.3.1 3JCOCC in 2 - 5
3JCOCC values were calculated by DFT as a function of the central bond torsion
for the Type I and Type II coupling pathways in structures 2a and 2b, respectively
(Figure 6.1A). The overall shape of the two curves is conserved, but two differences are
noteworthy: curve amplitude is slightly reduced for the Type II pathway in 2b, and more
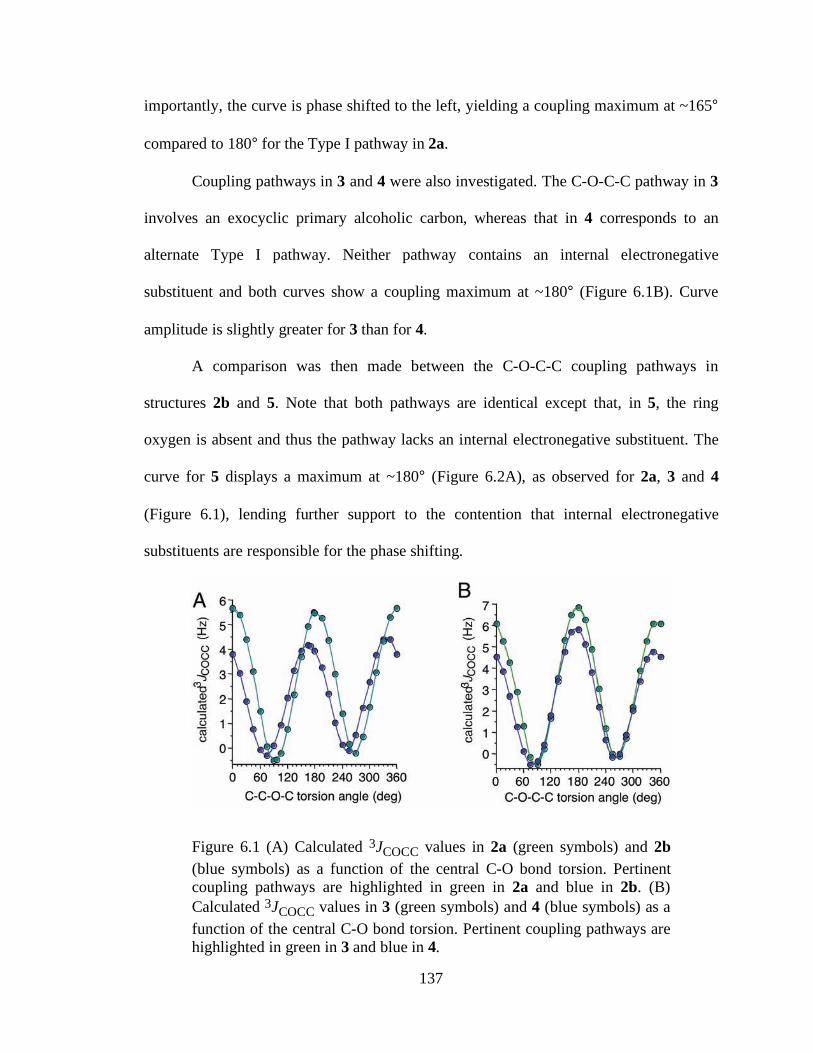
137
importantly, the curve is phase shifted to the left, yielding a coupling maximum at ~165°
compared to 180° for the Type I pathway in 2a.
Coupling pathways in 3 and 4 were also investigated. The C-O-C-C pathway in 3
involves an exocyclic primary alcoholic carbon, whereas that in 4 corresponds to an
alternate Type I pathway. Neither pathway contains an internal electronegative
substituent and both curves show a coupling maximum at ~180° (Figure 6.1B). Curve
amplitude is slightly greater for 3 than for 4.
A comparison was then made between the C-O-C-C coupling pathways in
structures 2b and 5. Note that both pathways are identical except that, in 5, the ring
oxygen is absent and thus the pathway lacks an internal electronegative substituent. The
curve for 5 displays a maximum at ~180° (Figure 6.2A), as observed for 2a, 3 and 4
(Figure 6.1), lending further support to the contention that internal electronegative
substituents are responsible for the phase shifting.
Figure 6.1 (A) Calculated 3JCOCC values in 2a (green symbols) and 2b (blue symbols) as a function of the central C-O bond torsion. Pertinent coupling pathways are highlighted in green in 2a and blue in 2b. (B) Calculated 3JCOCC values in 3 (green symbols) and 4 (blue symbols) as a function of the central C-O bond torsion. Pertinent coupling pathways are highlighted in green in 3 and blue in 4.
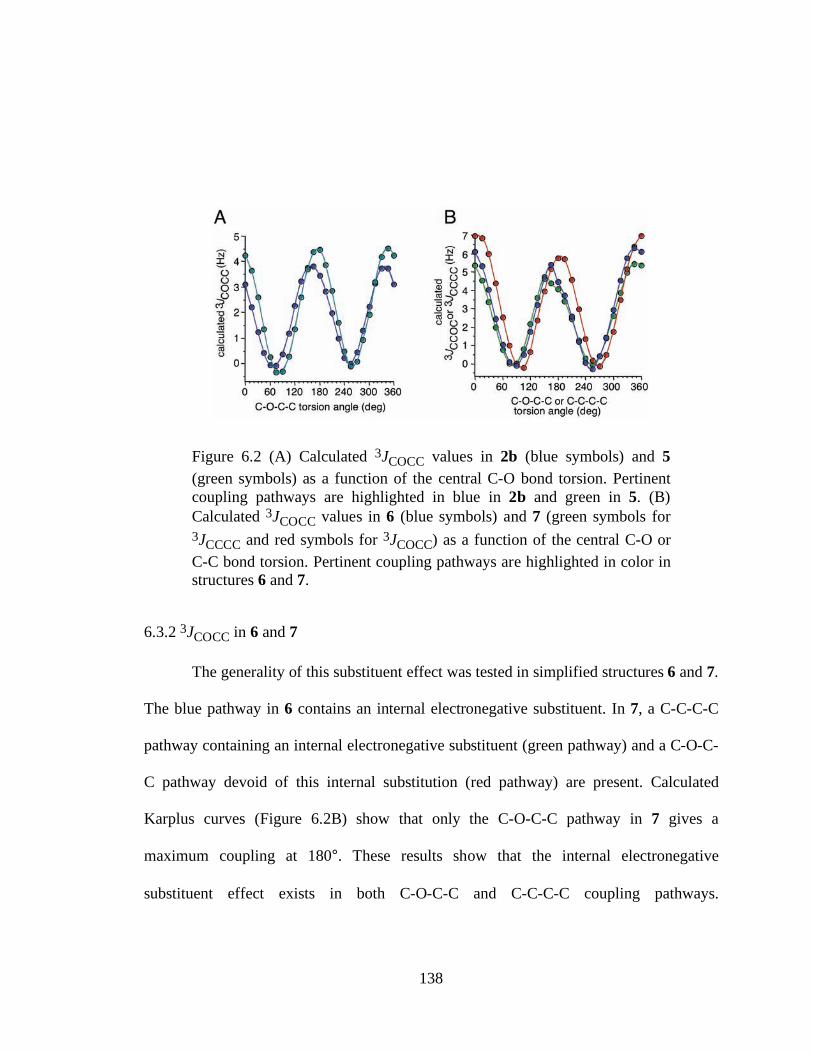
138
Figure 6.2 (A) Calculated 3JCOCC values in 2b (blue symbols) and 5 (green symbols) as a function of the central C-O bond torsion. Pertinent coupling pathways are highlighted in blue in 2b and green in 5. (B) Calculated 3JCOCC values in 6 (blue symbols) and 7 (green symbols for 3JCCCC and red symbols for 3JCOCC) as a function of the central C-O or C-C bond torsion. Pertinent coupling pathways are highlighted in color in structures 6 and 7.
6.3.2 3JCOCC in 6 and 7
The generality of this substituent effect was tested in simplified structures 6 and 7.
The blue pathway in 6 contains an internal electronegative substituent. In 7, a C-C-C-C
pathway containing an internal electronegative substituent (green pathway) and a C-O-C-
C pathway devoid of this internal substitution (red pathway) are present. Calculated
Karplus curves (Figure 6.2B) show that only the C-O-C-C pathway in 7 gives a
maximum coupling at 180°. These results show that the internal electronegative
substituent effect exists in both C-O-C-C and C-C-C-C coupling pathways.

139
6.3.3 Further Discussion
These findings have important implications for the interpretation of trans-
glycoside 3JCOCC values in oligosaccharides. Three trans-glycoside J-couplings in 1
(Scheme 6.4) depend on the φ torsion: 2JC1,C4’, 3JC4’,H1 and 3JC2,C4’. The geminal
2JC1,C4’, has limited dynamic range, and is mainly affected by φ but also by other
structural parameters, such as the C1-O1-C4’ bond angle. [3] The vicinal 3JC4’,H1 displays
a Karplus curve symmetric about the H1-C1-O1-C4’ torsion angle of 0°, and its
interpretation is problematic when discrimination between single and multiple state
conformational models is required. Thus, the remaining vicinal 3JC2,C4’ bears significant
weight in the assignment of linkage conformation about φ. The present results show that
coupling behavior for this CCaOC pathway is anomalous in that maximal couplings
cannot be expected when the coupled carbons are anti. This difference, in addition to an
apparent slight decrease in the amplitude of CCaOC curves relative to CaOCC curves,
must be taken into account in quantitative studies.
O
OOCH3
! "
OH
HO
O
OHHO
HO
HO
OH
C1C2
O5
C4'
C6'
C3'
1 (methyl #-lactoside)
!: H1-C1-O1-C4' ": C1-O1-C4'-H4'
Couplings sensitive to !2J
C1,C4',
3JC2,C4'
, 3J
C4',H1
Couplings sensitive to "3JC1,C3',
3JC1,C5',
3JC1,H4'
Scheme 6.4 Couplings that are sensitive to either φ or ψ in 1.

140
The underlying cause of the phase shift in Karplus curves for internally
substituted C-O-C-C pathways is beyond the scope of the present discussion. However,
inspection of Karplus curves calculated for 3JC2,C4’ in 1 shows that the two gauche
couplings are non-equivalent. Gauche rotamer II (with respect to 3JC2,C4’) yields a
smaller coupling (~0 Hz) than does gauche rotamer III (~1.5 Hz) (Scheme 6.5). This
behavior is attributed to the different orientations of O5 with respect to the coupled
carbons, being anti to C4’ in II and gauche to C4’ in III. By analogy to the behavior of
3JHH couplings, [8, 9] the anti arrangement is expected to reduce the value of 3JC2,C4’.
This reduction induces asymmetry in the Karplus curve and may be partly responsible for
the observed phase shift. However, the shifting is not solely the result of fitting an
asymmetric curve. DFT calculations show that maximal coupling is observed at torsions
displaced by ~15° from 180°, and thus the effect appears to be intrinsic and related to that
reported for 3JHCCH Karplus curves for fluoroethane. [8, 9]
H1
O5C2
C4'
g+
! = 60°
H1
O5C2
C4'
g"
! = "60°
H1
O5C2
C4'
anti
! = 180°
I II III
Scheme 6.5 Three staggered structures for φ rotation. All angles are referenced to H1-C1-O1-C4’.

141
6.4 Conclusion
We have shown that internal electronegative substituents perturb 3JCOCC Karplus
curves by phase shifting them with respect to analogous pathways devoid of this
substitution. Thus, the J-coupling maximum, which normally is observed near 180°, is
shifted by ~ 15°. These findings suggest that the two types of inter-residue C-O-C-C
coupling pathways observed in many oligosaccharides cannot be treated using a
generalized 3JCOCC Karplus equation. Quantitative interpretations of trans-glycoside J-
couplings to evaluate linkage conformations will need to take this effect into account.
6.5 References
1. Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F.; Bondo, P. B.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1998, 120, 11158-11173.
2. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 1999, 121, 9843-9851.
3. Cloran, F.; Carmichael, I.; Serianni, A.S. J. Am. Chem. Soc. 2000, 122, 396-397.
4. Becke, A. D. J. Chem. Phys. 1993, 98, 5648-5652.
5. Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257-2261.
6. Gaussian03, Revision C.02. Gaussian, Inc., Wallingford CT, 2004. Complete reference see chapter 2, reference 28.
7. Stenutz, R.; Carmichael, I.; Widmalm, G.; Serianni, A. S. J. Org. Chem. 2002, 67, 949-958.
8. Günther, H. NMR Spectroscopy, Wiley, New York, 1995, pp. 119-120.
9. Pachler, K.G.R. Tetrahedron 1971, 27, 187-199.

142
CHAPTER 7:
VICINAL 13C-13C NMR SPIN-SPIN COUPLING CONSTANTS IN
OLIGOSACCHARIDES: TOWARDS GENERALIZED EQUATIONS FOR O-
GLYCOSIDE LINKAGE CONFORMATIONAL ANALYSIS
7.1 Introduction
The pioneering work of Karplus[1] established three-bond (vicinal) 1H-1H NMR
spin-spin coupling constants (3JHH) as powerful tools for structural and conformational
analyses of molecules in solution.[2-4] Over the past fifty years, Karplus relationships
have been derived for other vicinal homo- and heteronuclear spin-couplings such as
3JCH,[5-8] 3JCC,[9.10] and 3JNH,[11] and equations have been parametrized experimentally
and theoretically. The reliable use of vicinal NMR spin-couplings depends on the
accuracy of the equations derived to interpret them. In addition to the dihedral angle
between the spin-coupled nuclei, the nature, position and orientation of substituents, bond
lengths, and bond angles are structural variables that influence 3J values. Equations with
multiple adjustable variables have been developed to improve accuracy, often at the
expense of limiting their use to very specific coupling pathway types. A prime example
of this approach was reported by Altona and coworkers[12,13] in their development of a
generalized Karplus equation for the treatment of 3JHCCH values in the furanosyl rings of
nucleic acids.

143
Vicinal 3JHH and 3JCH coupling constants, and 1H-1H NOE, are useful parameters
for structural and conformational studies of saccharides. When these parameters are
unavailable, for example, in studies of trans-glycoside linkage conformation, use of
carbon-carbon vicinal couplings (3JCC) is highly desirable.[14,15] In contrast to 3JHH and
3JCH, however, present understanding of 3JCC in saccharides is incomplete mainly for two
reasons. First, fewer 3JCC data have been reported because their measurement generally
requires isotopic labeling. Secondly, Karplus dependencies of 3JCC on dihedral angle are
more complex than of 3JHH due to the more complex substituent patterns along the
coupling pathway.
Three-bond 13C-13C spin-couplings (3JCOCC) are important parameters in
conformational studies of the glycosidic linkages of oligosaccharides. Previous work has
shown that 3JCOCC values display a Karplus dependency, but few Karplus equations
applicable to oligosaccharides have been reported.[14,15] A significant terminal
substituent effect has been reported; a terminal oxygen substituent enhances these
couplings by ~0.7 Hz when the coupled carbons are anti and when the O-substituent is
“in-plane” (W shaped arrangement).[14] A systematic study of these terminal substituent
effects, however, is lacking. Equally important, internal electronegative substituent
effects on these couplings have been recognized but not fully quantified.[16]
In this chapter, 3JCOCC values have been calculated in an array of model stuctures
and the data used parameterize Karplus equations as a function of the three dihedral
angles along the coupling pathways. Substituent effects, both internal and terminal, have
been investigated and quantified. The various parameterizations of vicinal 13C-13C spin-
couplings across glycosidic linkages are rationalized, and an effort to parameterize

144
generalized equations pertinent to [1→2], [1→3], [1→4] and [1→6] linkages has been
made.
C!
C"
O
C#
A3
A2
A1
S1 S2
A1'
A3'
A2'
H3C!
C"
H2
O
C#H3
H3C!
C"H
O
C#H3
1
OH3C
C!
C"H
O
C#H3
OH3C
O
H3C
2 3
$
$!
$#
Scheme 7.1 Definition of dihedral angles and substituents on O-methyl ethane (1) and model compounds used.
7.2 Calculational Methods
7.2.1 Model Systems.
Several model systems were used to investigate the effects of specific substitution
patterns of 3JCOCC values; these systems are shown in Scheme 7.1 in which the notation
of carbons and dihedral angles is illustrated. A1, A2, and A3, and A1’, A2’, A3’, denote
the substituents attached to the terminal (coupled) carbons, Cα and Cγ, respectively. S1
and S2 are defined as the substituents attached to the internal carbon, Cβ. The dihedral
angles defining rotation about the Cα-Cβ, Cβ-O, and O-Cγ bonds along the coupling
pathway are denoted φ, φα and φγ, respectively. Due to the substituent effects, 3JCOCC
will show secondary dependences on φα and φγ in addition to a primary dependence on φ;

145
the intent of this investigation is to quantify both the primary and secondary
dependencies. Thus, 3JCOCC will be expressed as a function of φ, φα, φγ and the
substituents (generalized as X), or 3JCOCC(φ, φα,φγ,X).
7.2.2 Spin-Spin Coupling Constant Calculations.
Model structures 1, 2 and 3 (Scheme 7.1) were chosen to study the effects of
electronegative substituents on 3JCOCC spin-coupling constants in saccharides. In 1 and
2, the Cα-Cβ-O-Cγ (or φ) torsion angle was scanned in 15° increments through 360°,
yielding 24 structures for each model compound. In 3, O-Cα-Cβ-O and Cα-Cβ-O-Cγ (or
φα and φ, respectively) were each scanned in 30˚ increments through 360°, yielding 144
structures.
Model compounds 4-7 (Scheme 7.2) were used to specifically investigate the
structural dependency of 3JC2,C1’ in α- and β-linked O-glycosidic linkages. The C2-C1-
O1-C1’ torsion angle was scanned over 360° in 15° increments in 4 and 30° increments
in 5-7. H2-C2-O2-H torsion angles are fixed at 180 to remove any potential influence of
C2-O2 rotation on C2-C1-O1-C coupling constants. The resulting structures were
otherwise fully geometrically optimized, and the optimized structures used to calculate
3JC2,C1’ values as described below.
NMR spin-coupling constants were calculated in Gaussian03[17] using density
functional theory (DFT) and the B3LYP functional.[18] The 6-31G* basis set[19] was used
for geometric optimization of all molecular structures, and an extended basis set
([5s2p1d|3s1p])[20] was used for in vacuo calculations of NMR spin-spin coupling

146
constants. A Monte-Carlo fit of the calculated couplings was applied using Profit
software.
Scheme 7.2 Model compounds used to mimic transglycosidic linkage.
7.3 Results and Discussion
7.3.1 3JCCOC values in reference structure 1 (CH3-CH2-O-CH3)
Model structure 1 is defined as the standard “reference” structure in which all
carbon substituents along the C-C-O-C coupling pathway are protons (i.e., the minimally-
substituted structure). Under these conditions, the corresponding 3JCOCC is expected to
show little dependence on either φα or φγ, and thus its structural dependence can be
satisfactorily represented by a truncated Fourier series in φ,
!
3J
CCOC(","# ,"$ ,P)%3
JCCOC
(") = C0
+ (Cncosn" + S
nsinn")
n=1
m
& 7.1
where P denotes that all substituents are protons. Due to molecular symmetry, the
following relationship holds:
!
3J
CCOC(")=
3J
CCOC(#") 7.2

147
The cosine terms in eq 7.1 are not affected by eq 7.2, whereas the sine terms drop
out of this equation. The 3JCOCC relationship can therefore be further simplified to:
!=
+="m
nnCCOCCCOC nCCJPJ
10
33 cos)(),,,( ##### $% 7.3
If only the first three terms of eq 7.3 (up to the second order) are retained, the
equation for 3JCOCC in 1 conforms to the typical Karplus relationship:
!! 2coscos 2103 CCCJCCOC ++= 7.4
7.3.2 Electronegative Substituent Effects on 3JCOCC
The effect of a substituent X on a given carbon (Cα, Cβ or Cγ) of 1 on the
corresponding 3JCOCC is given by the difference, Δ3JCOCC (φ,φα,φγ,X).
),,,(),,,(),,,( 333 PJXJXJ CCOCCCOCCCOC !"!"!" ######### $=% 7.5
where 3JCOCC (φ,φα,φγ,X) and 3JCOCC (φ,φα,φγ,P) are the 3JCOCC values for X-mono-
substituted and non-substituted CH3-CH2-O-CH3, respectively. To differentiate between
terminal and internal substituent effects, X is replaced by A for terminal cases and by S
for internal cases.
7.3.2.1 Internal Substituent Effects
Similar internal substituent effects have been reported for 3JHCCH and for
3JCCCH.[21,22] The effect of internal substituents on 3JCCCH is described by the following

148
equation: [22]
!
" 3J
CH(#,#$ ,S) = "C
00
C(S) + ("C
n0
C(S)cosn# + "S
n0
C(S)sinn#)
n=1
m
%
+ [("Cnp
C(S)cos p#$ + "C
np
S(S)sin p#$ )cosn#
p= 3
k
%n= 0
m
%
+("Snp
C(S)cos p#$ + "S
np
S(S)sin p#$ )sinn#]
7.6
Where S in the parenthesis represents internal electronegative substituent.
Coefficient
!
"Cnp
C is for cosnφ cospφα term, where regular upper letter C and subscript n
indicates cosnφ, and superscript C and subscript p are for cospφα. Other coefficients are
defined in a similar way. Note that φα affects Δ3JCCCH in the third or higher-order terms,
thus showing that its effect is negligible. The same concept and treatment can be applied
to C-C-O-C vicinal coupling constants. The terms containing φα and φγ are of little
significance and are dropped. When same notation is adopted, Δ3JCOCC (φ,φα,φγ,S) can
be expressed as:
!=
"+"+"="m
n
Cn
Cn
CCCOC nSSnSCSCSJ
10000
3 )sin)(cos)(()(),,,( ##### $% 7.7
If the higher terms of φ are dropped, the internal substituent effects are further
simplified:
!!
!!!!! "#
2sin)(sin)(
2cos)(cos)()(),,,(
2010
2010003
SSSS
SCSCSCSJCC
CCCCCOC
$+$+
$+$+$=$ 7.8
When eq 7.8 is combined with eqs 7.4 and 7.5, the following equation results:
!!!!!!! "# 2sinsin2coscos),,,(3 EDCBASJCCOC ++++= 7.9

149
This is the same equation used to parameterize 3JHH when internal substituents
are present.[12,21] Owing to isodynamic operations, the following relationship holds,
which correlates the pro-R substituent S1 with the pro-S substituent S2:
),,,(),,,( 23
13 HSJHSJ CCOCCCOC !"!" ###### $$$%=% 7.10
In eq 7.8, the cosine terms are not affected by eq 7.10, but the sine terms change
sign when the internal substituent changes from the pro-R to pro-S position and vice
versa.
7.3.2.2 Terminal Substituent Effects
In a C-C-O-C coupling pathway, a terminal substituent can be located on either
terminal (coupled) carbons, Cα or Cγ (Scheme 7.1). Hereafter, a mono-substituted Cα is
assumed in the discussion, but a terminal substituent on Cγ can be treated similarly.
Because there is no substituent on Cγ in the present treatment, it is reasonable to assume
that 3JCOCC has little dependence on φγ. This assumption yields an equation similar to that
describing terminal substituent effects on 3JCH[22]:
!!
!
= =
=
"+"+
"+"="
m
n
k
p
Snp
Cnp
m
n
Cn
CCCOC
npASnpAC
nACACAJ
0 1
1000
3
]sinsin)(coscos)([
cos)()(),,,(
####
####
$$
%$
7.11
7.3.2.3 Parametrization of 3JCOCC (φ,φα,φγ,M)
An additive model has been used previously to treat the effects of substituents on
3JHCCH and 3JCCCH values, [22] and a similar approach was adopted to treat 3JCCOC.

150
3JCCOC spin-couplings for an M-poly-substituted CH3-CH2-O-CH3 coupling pathway are
described by
!!
!
==
=
"+"+
"+=
2
1
33
1
'3
3
1
333
),,,(),,,(
),,,(),,,(),,,(
kkCCOC
jjCCOC
iiCCOCCCOCCCOC
SJAJ
AJPJMJ
#$#$
#$#$#$
%%%%%%
%%%%%%%%%
7.12
where 3JCOCC (φ,φα,φγ,P) is the J-coupling in the non-substituted CH3-CH2-O-CH3,
Δ3JCOCC (φ,φα,φγ,Ai) is the terminal substituent effect of Ai bonded to Cα, Δ3JCOCC
(φ,φα,φγ,Aj’) is the terminal substituent effect of Aj’ bonded to Cγ, and Δ3JCOCC
(φ,φα,φγ,Sk) is the internal substituent effect of Sk bonded to Cβ.
The electronegativities of the pathway substituents are not discussed in detail in
this study. Instead, they are included in the coefficients of above terms. In addition to the
angular dependences, 3JCCOC also depends on a number of secondary effects, such as
bond lengths, bond angles, and so forth, and the empirical parametrization of 3JCOCC is
complicated due to these effects. A generalized equation is thus not plausible, and we
suggest specific fitted 3JCCOC for specific pathways.
7.3.3 3JCCOC Spin-couplings in Model Structure 1
Calculated 3JCCOC spin-couplings in 1 (Figure 7.1) display the expected
dependency on the C-C-O-C torsion angle, and the data were fitted to Karplus-like
equation 7.13:
!! 2cos51.3cos78.044.33++=CCOCJ (RMSE = 0.14 Hz) 7.13
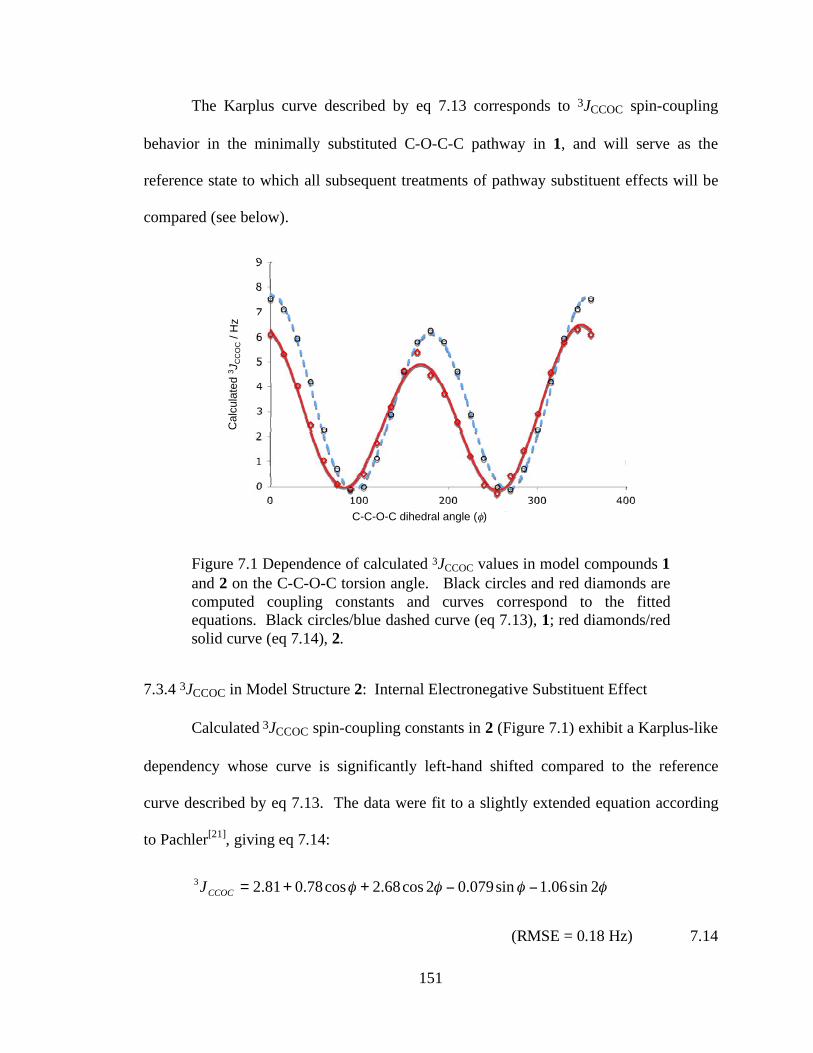
151
The Karplus curve described by eq 7.13 corresponds to 3JCCOC spin-coupling
behavior in the minimally substituted C-O-C-C pathway in 1, and will serve as the
reference state to which all subsequent treatments of pathway substituent effects will be
compared (see below).
C-C-O-C dihedral angle (φ)
Calc
ula
ted 3
J CC
OC / H
z
Figure 7.1 Dependence of calculated 3JCCOC values in model compounds 1 and 2 on the C-C-O-C torsion angle. Black circles and red diamonds are computed coupling constants and curves correspond to the fitted equations. Black circles/blue dashed curve (eq 7.13), 1; red diamonds/red solid curve (eq 7.14), 2.
7.3.4 3JCCOC in Model Structure 2: Internal Electronegative Substituent Effect
Calculated 3JCCOC spin-coupling constants in 2 (Figure 7.1) exhibit a Karplus-like
dependency whose curve is significantly left-hand shifted compared to the reference
curve described by eq 7.13. The data were fit to a slightly extended equation according
to Pachler[21], giving eq 7.14:
!!!! 2sin06.1sin079.02cos68.2cos78.081.23 ""++=CCOCJ
(RMSE = 0.18 Hz) 7.14
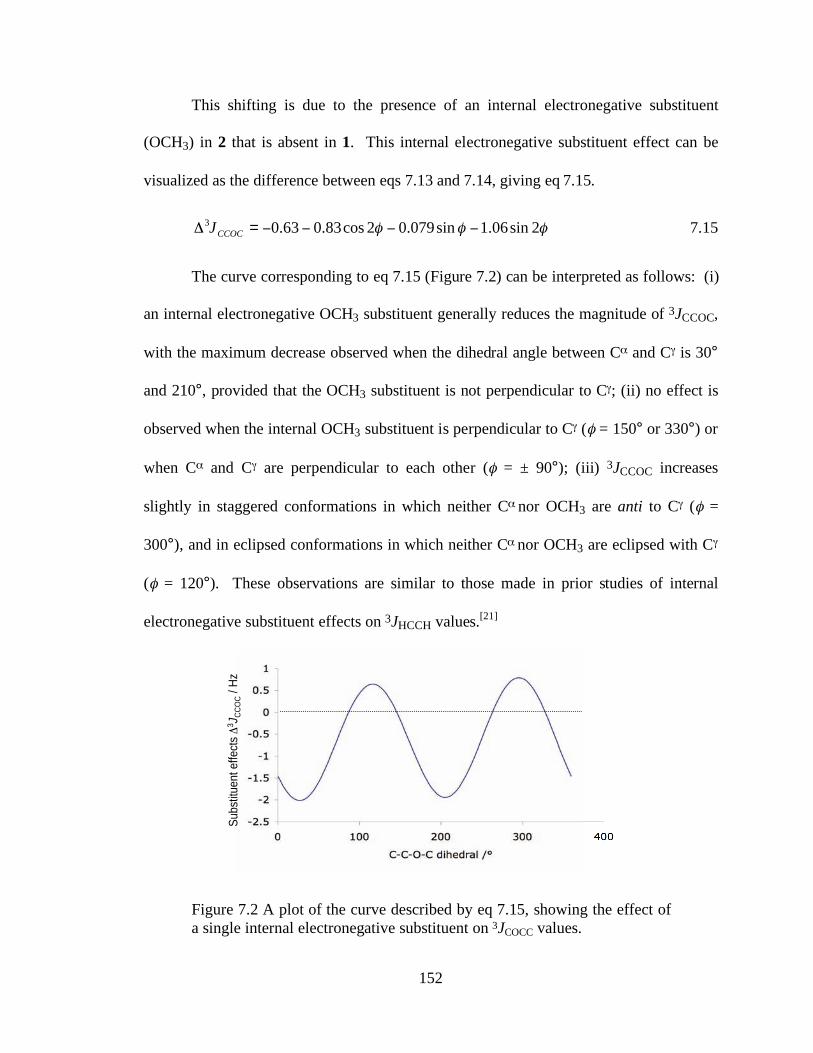
152
This shifting is due to the presence of an internal electronegative substituent
(OCH3) in 2 that is absent in 1. This internal electronegative substituent effect can be
visualized as the difference between eqs 7.13 and 7.14, giving eq 7.15.
!!! 2sin06.1sin079.02cos83.063.03 """"=# CCOCJ 7.15
The curve corresponding to eq 7.15 (Figure 7.2) can be interpreted as follows: (i)
an internal electronegative OCH3 substituent generally reduces the magnitude of 3JCCOC,
with the maximum decrease observed when the dihedral angle between C! and C" is 30°
and 210°, provided that the OCH3 substituent is not perpendicular to C"; (ii) no effect is
observed when the internal OCH3 substituent is perpendicular to C" (! = 150° or 330°) or
when C! and C" are perpendicular to each other (! = ± 90°); (iii) 3JCCOC increases
slightly in staggered conformations in which neither C! nor OCH3 are anti to C" (! =
300°), and in eclipsed conformations in which neither C! nor OCH3 are eclipsed with C"
(! = 120°). These observations are similar to those made in prior studies of internal
electronegative substituent effects on 3JHCCH values.[21]
Figure 7.2 A plot of the curve described by eq 7.15, showing the effect of a single internal electronegative substituent on 3JCOCC values.
Su
bstitu
en
t e
ffe
cts
!3J C
CO
C /
Hz
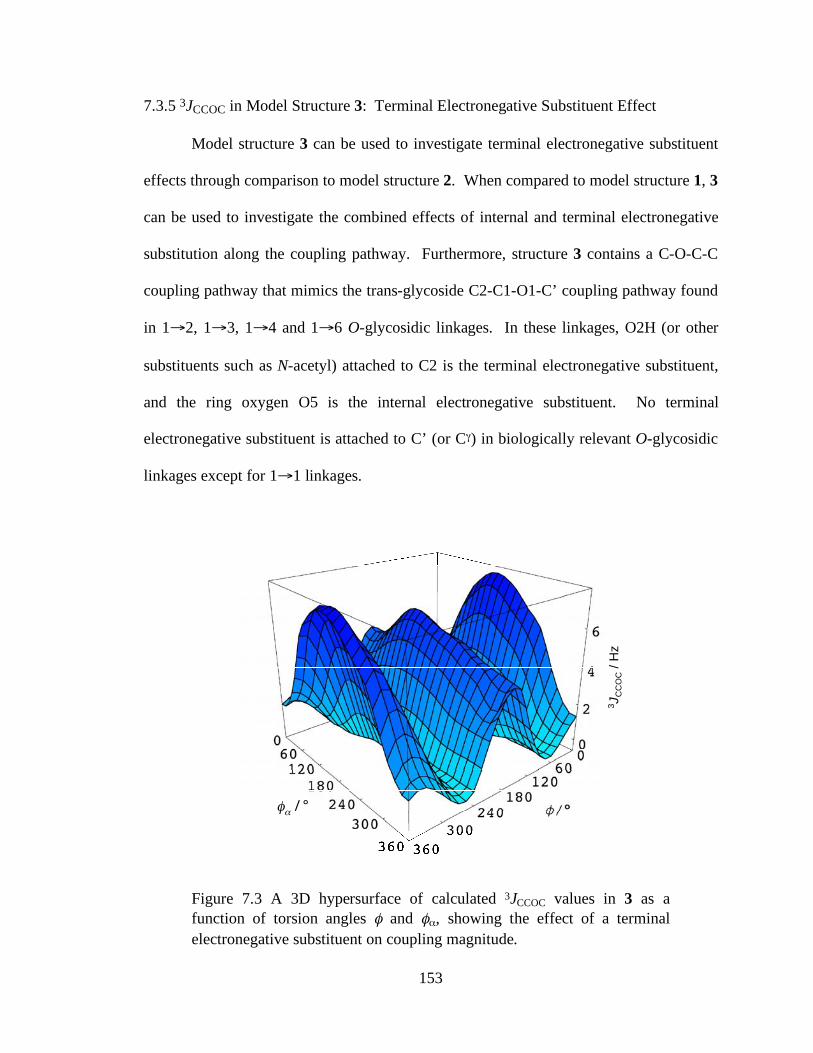
153
7.3.5 3JCCOC in Model Structure 3: Terminal Electronegative Substituent Effect
Model structure 3 can be used to investigate terminal electronegative substituent
effects through comparison to model structure 2. When compared to model structure 1, 3
can be used to investigate the combined effects of internal and terminal electronegative
substitution along the coupling pathway. Furthermore, structure 3 contains a C-O-C-C
coupling pathway that mimics the trans-glycoside C2-C1-O1-C’ coupling pathway found
in 1→2, 1→3, 1→4 and 1→6 O-glycosidic linkages. In these linkages, O2H (or other
substituents such as N-acetyl) attached to C2 is the terminal electronegative substituent,
and the ring oxygen O5 is the internal electronegative substituent. No terminal
electronegative substituent is attached to C’ (or Cγ) in biologically relevant O-glycosidic
linkages except for 1→1 linkages.
φα / °
3J C
CO
C /
Hz
Figure 7.3 A 3D hypersurface of calculated 3JCCOC values in 3 as a function of torsion angles φ and φα, showing the effect of a terminal electronegative substituent on coupling magnitude.
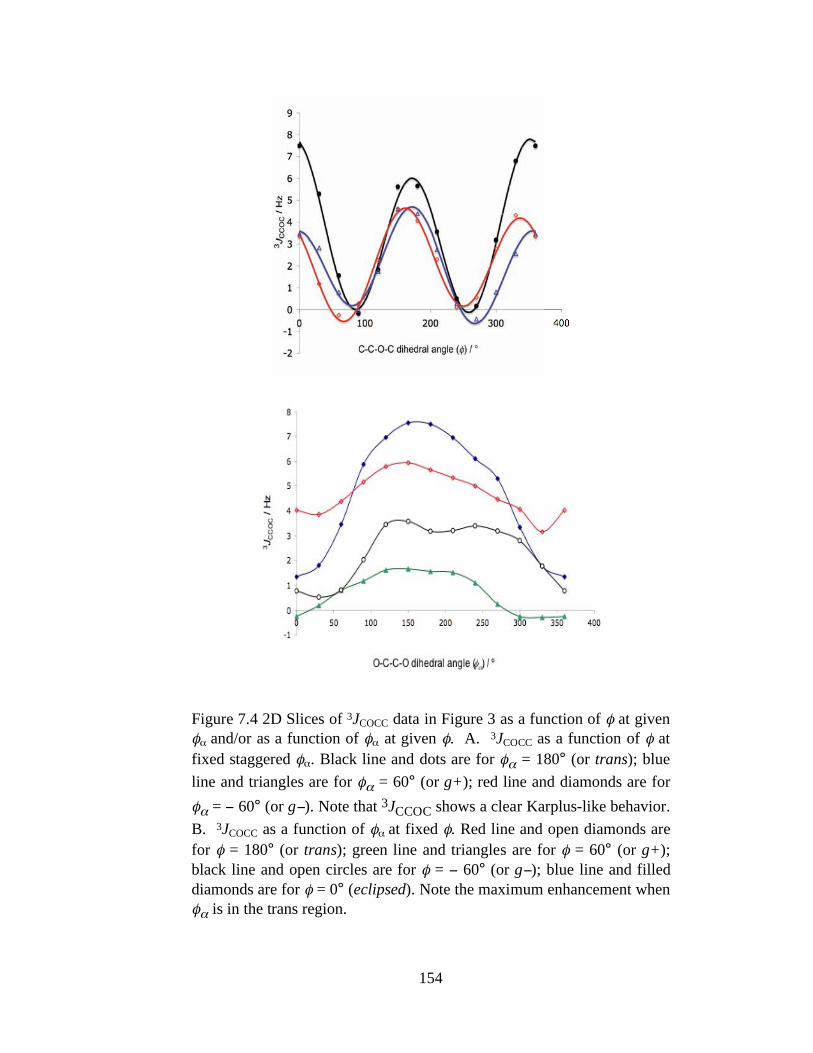
154
Figure 7.4 2D Slices of 3JCOCC data in Figure 3 as a function of φ at given φα and/or as a function of φα at given φ. A. 3JCOCC as a function of φ at fixed staggered φα. Black line and dots are for φα = 180° (or trans); blue line and triangles are for φα = 60° (or g+); red line and diamonds are for φα = − 60° (or g−). Note that 3JCCOC shows a clear Karplus-like behavior. B. 3JCOCC as a function of φα at fixed φ. Red line and open diamonds are for φ = 180° (or trans); green line and triangles are for φ = 60° (or g+); black line and open circles are for φ = − 60° (or g−); blue line and filled diamonds are for φ = 0° (eclipsed). Note the maximum enhancement when φα is in the trans region.

155
Vicinal 3JCCOC spin-coupling constants across the Cα-Cβ-O-Cγ pathway as a
function of φ and φα are shown in Figure 7.3. 3JCCOC depends primarily on φ, and the 2D
slices of Figure 7.3 at fixed values of φα (Figure 7.4a; only φα values for staggered
conformations are shown) show similar Karplus-like shapes as those found for 2. In
addition, 3JCCOC depend significantly on φα, as evidenced from the 2D slices of Figure
7.3 at fixed values of φ (Figure 7.4b). Previous work has shown[14] that 3JCCOC values
are enhanced by ~0.7 Hz when a terminal oxygen substituent is “in-plane” in a trans C-C-
O-C coupling pathway. The present results confirm this enhancement, which is
calculated here to be ~1.2 Hz. Interestingly, 3JCCOC shows a more significant terminal
electronegative substituent enhancement (~3.0 Hz) and a greater φα dependence at φ = 0°
than at φ = 180°. When φ is fixed at 0° and φα is rotated, a maximum coupling change of
6.2 Hz is observed, which is considerably greater than when φ is fixed at 180° (~1.8 Hz).
Equally noteworthy is that the maximum change due to rotation of φα at fixed φ (Figure
7.4.b, also as bandwidth in Figure 7.5A) shows Karplus-like behavior as a function of φ.
These changes have been fit to a typical Karplus equation and give the following
relationship:
!! 2cos69.1cos08.244.2 ++=BandWidth (RMSE = 0.81 Hz) 7.16
According to eq 7.11, to parameterize the 3JCCOC coupling data shown in Figure
7.3, extra terms have to be added to address the φα dependence in addition to the five
terms in eq 7.8. Nine extra terms, including several higher order terms, are added and the

156
following obtained:
!!!
!!!!
!
"""""""
"""""""
"""""
2sinsin13.02coscos21.0sin2sin32.03cos14.0cos2cos09.1sinsin24.0coscos05.12cos17.0
cos07.12sin03.1sin037.02cos47.2cos041.054.23
+#+#
#+##
##+++=CCOCJ
(RMSE = 0.38 Hz) 7.17
These terms, however, do not have equal importance; only the cosφα, cosφcosφα
and cos2φcosφα terms are significant, in agreement with the proposed equation
describing α-oxygen substituent effects on 3JCCCH in 1-propanol.[23,24] If the other six
terms that are insignificant are ignored, eq 7.17 can be rewritten as follows with little loss
of accuracy:
!!
!
""""
"""""
cos2cos08.1coscos03.1cos07.12sin03.1sin037.02cos47.2cos041.054.23
##
##+++=CCOCJ
(RMS = 0.44 Hz) 7.18
The last three terms in eq 7.18 can be rewritten as -cosφα (1.07 + 1.03 cosφ +
1.08 cos2φ). This in part explains the behavior observed in Figure 7.5. In addition,
because the above terms in parentheses are positive, an oxygen substituent in an anti or
near anti geometry is expected to enhance 3JCCOC. On the other hand, an oxygen
substituent in a cis or near cis geometry is expected to decrease 3JCCOC. This result is
consistent with previous observations that an anti electronegative substituent enhances
3JCCOC compared to a gauche electronegative substituent.[14]
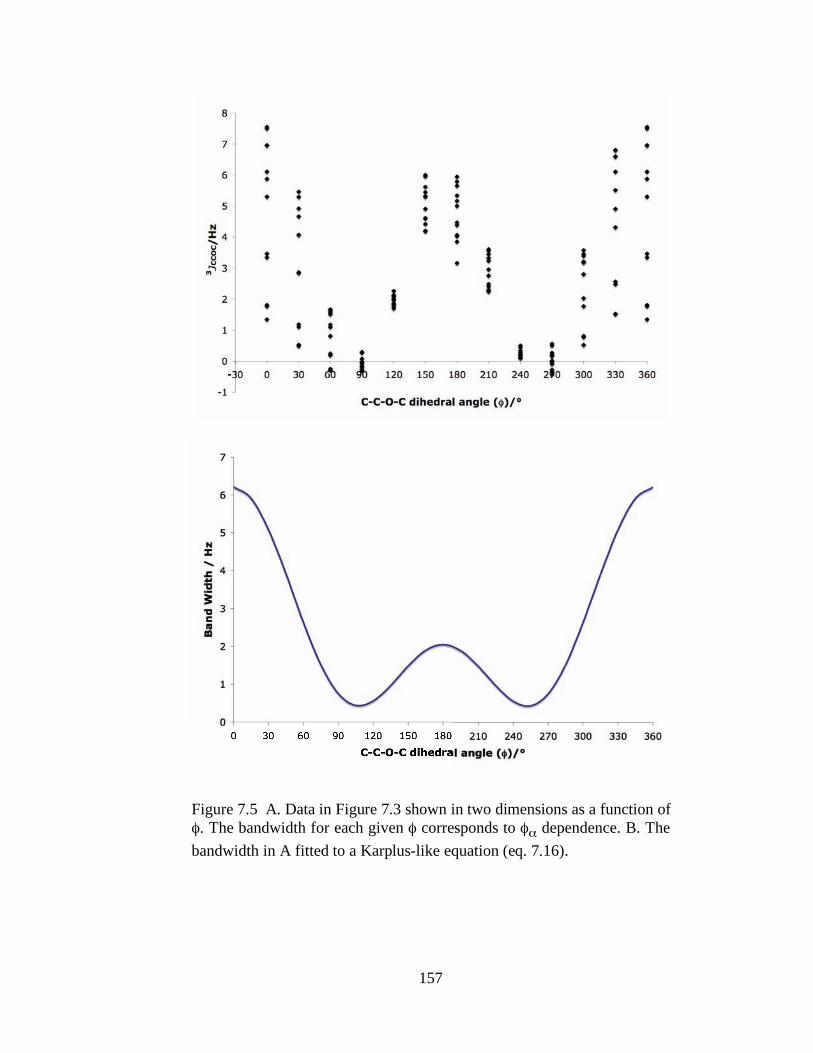
157
Figure 7.5 A. Data in Figure 7.3 shown in two dimensions as a function of φ. The bandwidth for each given φ corresponds to φα dependence. B. The bandwidth in A fitted to a Karplus-like equation (eq. 7.16).

158
Eq 7.18 and the 3JCCOC coupling data obtained on model structure 3 suggest the
following equation for 3JCCOC when one terminal and one internal oxygen subsitituent
are present in the coupling pathway:
)2coscos(cos2sinsin2coscos3
!!"
!!!!
HGFEDCBAJCCOC
+++
++++= 7.19
The first three terms of eq 7.19 are typical Karplus terms. The sinφ and sin2φ
terms are included to treat the internal substituent, and the remaining terms describe the
secondary dependence of 3JCCOC on either φα or φγ, depending on where the terminal
oxygen substituent is located.
If both terminal carbons are substituted by electronegative substituents, the
following equation should pertain if the additive rule is valid.
)2coscos(cos)2coscos(cos2sinsin2coscos3
!!!!!!
!!!!
"# KJIHGFEDCBAJCCOC
++++++
++++= 7.20
7.3.6 Parametrization of 3JCCOC in O-Glycosidic Linkages of Oligosaccharides
While the potential of multiple substituent orientations and the complicated
angular dependence render 3JCCOC parameterization difficult, the problem is simplified
for trans-glycoside coupling pathways in oligosaccharides because not all three torsions
φ, φα and φγ (Scheme 7.1) are rotatable. In O-glycosidic linkages (except for 1→6), the
Cα-Cβ bond in the C-C-O-C coupling pathway is commonly part of a pyranosyl ring and
is therefore not rotatable (assuming conformational rigidity of the ring). Thus, φα is
constrained to be either anti or gauche and is assumed to be constant for given linkage.
Inserting a constant φα simplifies eq 7.20 since φα-related terms can be dropped and/or

159
combined with other φ related terms. Additionally, substituent effects are largely
depending on the electronegativity of the substituents, [12,13] effects due to saturated
carbon derivatives are assumed to be small, except that this substituent is constantly anti
or “in-plane” to the coupling pathway. [25] This assumption further simplifies the
parametrization of 3JCCOC in trans-glycoside linkages. For example, the C2’-C1’-O1’-C4
coupling pathway in a 1→4 linkage contains five substituents: O2’H and C3’ on C2’;
O5’ (ring oxygen) on C1’; C3 and C5 on C4. Invoking the assumption above, 3JC2’,C4
should show little dependence on conformation about the O1’-C4 bond, so that φγ terms
can be dropped from eq 7.19. In addition, because the C2’-C1’ bond (φα) is not rotatable,
O2’ is either anti or gauche to the C1’-O1’ bond depending on anomeric configuration
and configuration at C2. If a constant φα is inserted into eq 7.19, then a simplified
equation similar to eq 7.9 can be used to parameterize 3JC2’,C4. Similar reasoning has
been applied to the substituent patterns in C-C-O-C coupling pathways across various O-
glycosidic linkages in oligosaccharides; these results are summarized in Table 7.1. In
this treatment, only oxygen substituents are considered and contributions from carbon
substituents are ignored.
7.3.7 Investigations of 3JC2’,Cn in O-Glycosidic Linkages
The C2’-C1’-O1’-Cn coupling pathway has the same substituent pattern in all
linkages and its corresponding spin-coupling constant, 3JC2’,Cn, depends only on torsion
angle φ (Table 7.1). 3JC2’,Cn also depends on configuration at C1’ (anomeric
configuration) and C2’. The contributions of these structural variables were investigated
in more detail with the use of model structures 4-7 (Scheme 7.2) which were chosen to

160
mimic the above-noted torsional and configurational variables. Calculated 3JC2’,C1’ (see
Scheme 7.2 for atom numbering) were fitted according to eq 7.9, giving eqs 7.21 - 24 for
model structures 4-7, respectively. These equations have low RMS errors (~0.1 Hz)
except that for 6 (0.5 Hz) in which O2’ is anti to O1’; in 6, the O2’-C2’-C1’-O1’ torsion
angle (φα) may not be constant due to small changes in ring conformation as φ is rotated.
This behavior is presumably due to the increased sensitivity of 3JC2’,C1’ when φα is anti.
!!!! 2sin83.0sin12.02cos30.1cos73.036.13+"+"=CCOCJ
(RMSE = 0.13 Hz) 7.21
!!!! 2sin28.1sin26.02cos82.1cos041.013.23 ""++=CCOCJ
(RMSE = 0.12 Hz) 7.22
!!! 2sin43.0sin19.02cos26.240.23+"+=CCOCJ
(RMSE = 0.50 Hz) 7.23
!!!! 2sin41.0sin38.02cos07.2cos21.093.13 "++"=CCOCJ
(RMSE = 0.15 Hz) 7.24
The 3JC2’,C1’ coupling data for 4-7 and the superimposed fitted curves from eqs
7.21 – 24 are shown in Figure 7.6. As expected, the curve for 6 has a largest amplitude
due primarily to the presence of an anti O2 in the coupling pathway. However, the
curves for 4, 5 and 7 also have different amplitudes, with 5 and 7 showing similar
behavior with curve amplitudes intermediate between those of 4 and 6. The intermediate
enhancement found for 5 and 7 may due to the terminal C3 carbon in the C2-C1-O1-C1’

161
coupling pathway. In 5 and 7, the β-configuration at C1 orients C3 always anti to the
coupling pathway (an “in-plane” C3), whereas in 4, C3 and O2 are both gauche to the
coupling pathway (“out-of-plane” substituents). This behavior is reminiscent of that for
3JCCOH spin-couplings in saccharides where an “in-plane” carbon has been shown
recently to enhance these spin-couplings by over 1 Hz.[25] This finding suggests that when
C, as terminal substituent, is constantly “in-plane” with the coupling pathway, its effect
may not be ignored, and a more complicated equation similar to 7.19 may be necessary.
All of the curves in Figure 7.6 display a global (or local) maximum near 165° or
195°, depending on anomeric configuration, rather than at the expected 180°. This
phase-shift has been shown recently to be caused the presence of an internal
electronegative substituent in the coupling pathway (O5; ring oxygen).[16] A comparision
of the coefficients in eqs 7.21- 24 reveals that the sine terms roughly change signs
between model structures 4 and 5, and between 6 and 7. Changing the anomeric
configuration is equivalent to changing the pro-R and pro-S positions of the internal
electronegative substituent as discussed earlier. Anomeric configuration does not appear
to be a significant factor influencing 3JC2’,C1’, behavior similar to that shown by 3JHCOH
and 3JCCOH spin-couplings in saccharides involving the anomeric carbon.
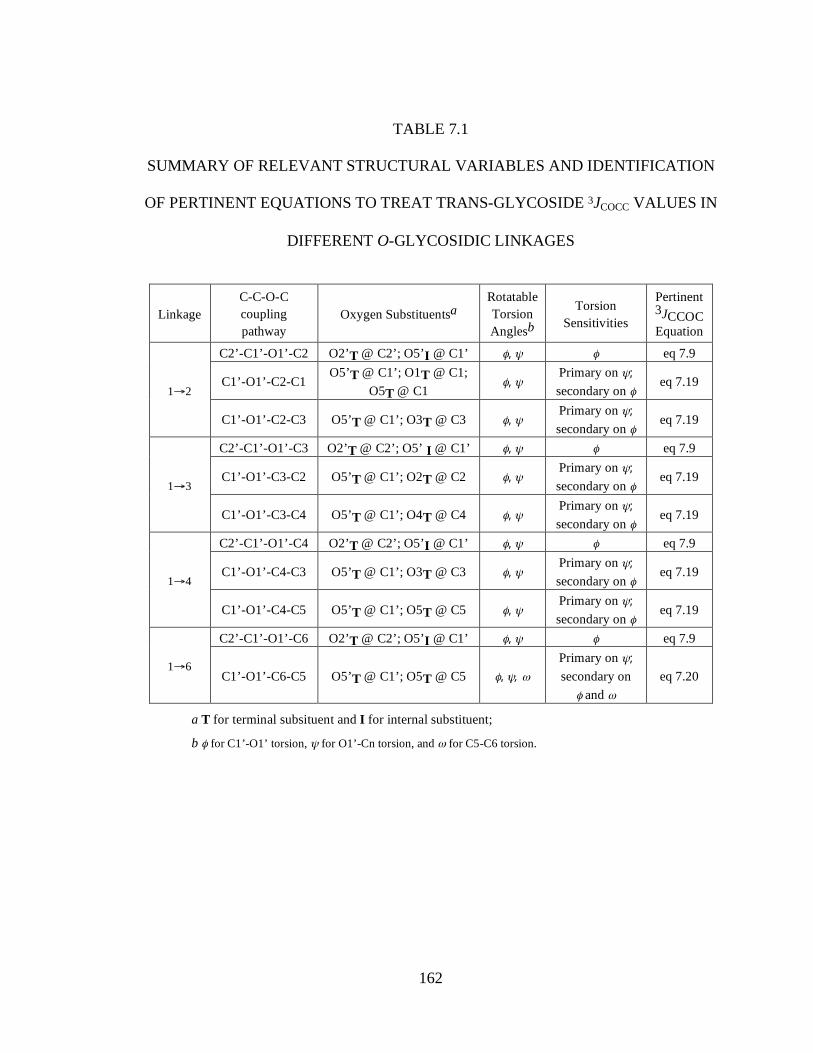
162
TABLE 7.1
SUMMARY OF RELEVANT STRUCTURAL VARIABLES AND IDENTIFICATION
OF PERTINENT EQUATIONS TO TREAT TRANS-GLYCOSIDE 3JCOCC VALUES IN
DIFFERENT O-GLYCOSIDIC LINKAGES
Linkage C-C-O-C coupling pathway
Oxygen Substituentsa Rotatable Torsion Anglesb
Torsion Sensitivities
Pertinent 3JCCOC Equation
C2’-C1’-O1’-C2 O2’T @ C2’; O5’I @ C1’ φ, ψ φ eq 7.9
C1’-O1’-C2-C1 O5’T @ C1’; O1T @ C1;
O5T @ C1 φ, ψ
Primary on ψ;
secondary on φ eq 7.19
1→2
C1’-O1’-C2-C3 O5’T @ C1’; O3T @ C3 φ, ψ Primary on ψ;
secondary on φ eq 7.19
C2’-C1’-O1’-C3 O2’T @ C2’; O5’ I @ C1’ φ, ψ φ eq 7.9
C1’-O1’-C3-C2 O5’T @ C1’; O2T @ C2 φ, ψ Primary on ψ;
secondary on φ eq 7.19
1→3
C1’-O1’-C3-C4 O5’T @ C1’; O4T @ C4 φ, ψ Primary on ψ;
secondary on φ eq 7.19
C2’-C1’-O1’-C4 O2’T @ C2’; O5’I @ C1’ φ, ψ φ eq 7.9
C1’-O1’-C4-C3 O5’T @ C1’; O3T @ C3 φ, ψ Primary on ψ;
secondary on φ eq 7.19
1→4
C1’-O1’-C4-C5 O5’T @ C1’; O5T @ C5 φ, ψ Primary on ψ;
secondary on φ eq 7.19
C2’-C1’-O1’-C6 O2’T @ C2’; O5’I @ C1’ φ, ψ φ eq 7.9
1→6 C1’-O1’-C6-C5 O5’T @ C1’; O5T @ C5 φ, ψ, ω
Primary on ψ;
secondary on
φ and ω
eq 7.20
a T for terminal subsituent and I for internal substituent;
b φ for C1’-O1’ torsion, ψ for O1’-Cn torsion, and ω for C5-C6 torsion.
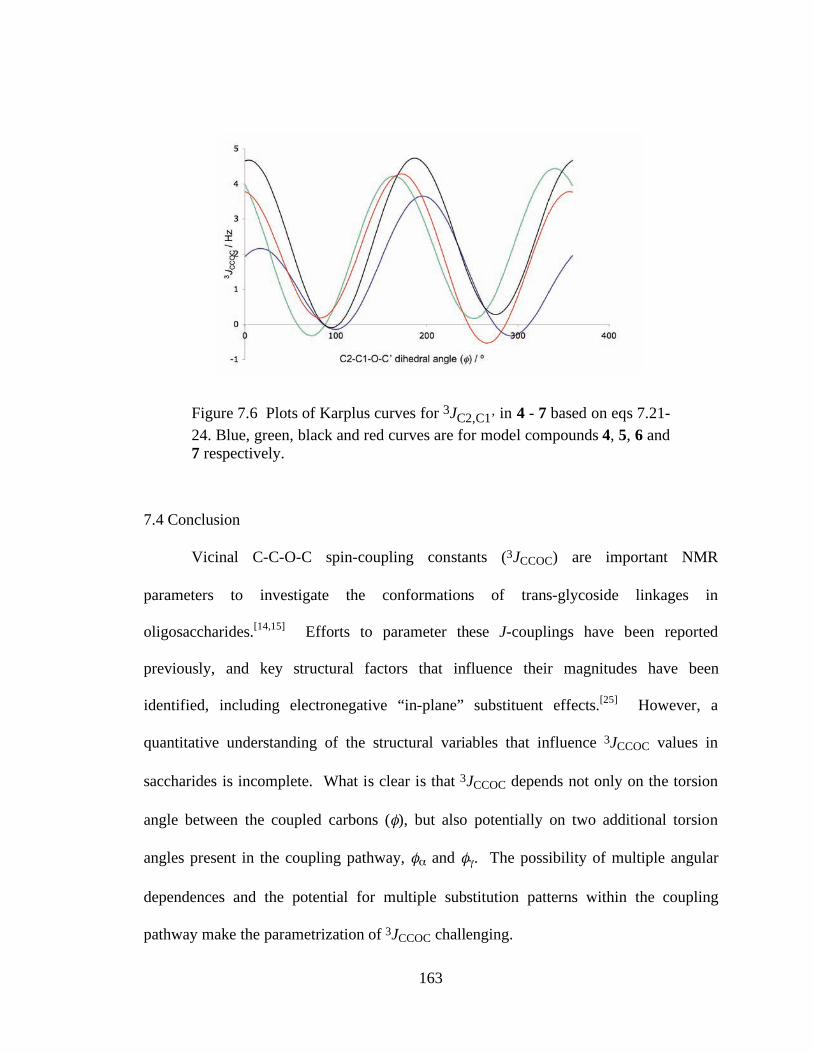
163
Figure 7.6 Plots of Karplus curves for 3JC2,C1’ in 4 - 7 based on eqs 7.21-24. Blue, green, black and red curves are for model compounds 4, 5, 6 and 7 respectively.
7.4 Conclusion
Vicinal C-C-O-C spin-coupling constants (3JCCOC) are important NMR
parameters to investigate the conformations of trans-glycoside linkages in
oligosaccharides.[14,15] Efforts to parameter these J-couplings have been reported
previously, and key structural factors that influence their magnitudes have been
identified, including electronegative “in-plane” substituent effects.[25] However, a
quantitative understanding of the structural variables that influence 3JCCOC values in
saccharides is incomplete. What is clear is that 3JCCOC depends not only on the torsion
angle between the coupled carbons (φ), but also potentially on two additional torsion
angles present in the coupling pathway, φα and φγ. The possibility of multiple angular
dependences and the potential for multiple substitution patterns within the coupling
pathway make the parametrization of 3JCCOC challenging.

164
Electronegative substituent effects and the angular dependencies of 3JCCOC have
been investigated in this report. When only internal electronegative substituents are
present, 3JCCOC depends primarily on φ, with little dependence on φα and φγ. In this
respect, 3JCCOC responds to internal electronegative substituent effects similar to 3JHCCH
and 3JCCCH; in general, an internal electronegative substituent decreases 3JCCOC values.
Sine terms must be included in the Karplus equation when internal substituents are
present.
When terminal electronegative substituents are present, 3JCCOC shows a
significant secondary angular dependence on φα and/or φγ. This study recommends the
use of – cosψ (A + B cosφ + C cos2φ) to describe the secondary angular dependence on
ψ when terminal oxygen substituents exist within the coupling pathway, where φ is the
dihedral angle between the coupled carbons, and ψ is φα and/or φγ depending on the
substituted sites. The term, (A + B cosφ + C cos2φ), is generally positive for any possible
value of φ. Thus, when ψ is in the range 90° – –90° (through 180°), a terminal oxygen
substituent enhances 3JCCOC, with the maximum enhancement occurring when the
oxygen substituent is trans to the pathway (180°). On the other hand, when ψ is in the
range –90° – 90° (through 0°), the terminal oxygen substituent reduces 3JCCOC, with a
greatest reduction occurring when the terminal oxygen is cis to the pathway (0˚). These
structural dependencies are consistent with the “in-plane” effects observed previously in
3JHCOH and 3JCCOH spin-couplings in saccharides.[25]
Studies of substituent effects and the angular dependencies of 3JCCOC provide
useful guidelines to assist in the structural interpretation of these J-couplings in
biomolecules, most notably oligosaccharides. Although we have shown herein that

165
3JCCOC shows a triple angular dependence, its treatment in oligosaccharides can be
simplified significantly because not all three torsion angles are rotatable. For example, in
the C-C-O-C pathway across O-glycosidic linkages, the C-C bond is commonly part of a
pyranosyl ring, and the latter’s rigidity constrains the former’s rotation. This effect
reduces the angular dependence of 3JCCOC by one degree.
The assumption that carbon substituent effects on 3JCCOC are small and can be
ignored further simplifies the parametrization. A survey of oxygen substituent patterns in
common trans-glycoside linkages, summarized in Table 7.1, shows that the substituents
in C2’-C1’-O1’-Cn pathways are similar for all pathways. Angularly, 3JC2’,Cn depends
primarily on φ, with little dependence on the remaining two torsion angles. 3JC2’,Cn is
also influenced by configuration at C1’ and C2’. Organizing all possible 3JC2’,Cn in O-
glycosidic linkages into four classes greatly simplifies the parametrization and, more
importantly, improves their application to studies of linkage conformation. The four
model structures used in this investigation (4-7) were chosen to mimic these four classes.
As expected, an anti O2’ enhances 3JC2’,Cn. Interestingly, an anti C3’ in β-anomeric
configurations also significantly enhances 3JC2’,Cn. These results suggest that anti
carbons cannot be ignored when interpreting experimental 3JCCOC, as discussed in a
recent study.[25]

166
7.5 Reference
1. Karplus, M. J. Chem. Phys. 1959, 30, 11-15.
2. Fraser, R.R.; Kaufman, M.; Morand, P.; Govil, G. Can. J. Chem. 1969, 47, 403-409.
3. Vuister, G.W.; Bax, A. J. Am. Chem. Soc. 1993, 115, 7772-7777.
4. Zhao, H. ; Pan, Q. ; Zhang, W.; Carmichael, I. and Serianni, A. S. J. Org. Chem. 2007, 72, 7071-7082.
5. Tvaroska, I.; Gadjos, J. Carbohydr. Res. 1995, 271, 151-162.
6. Mulloy, B.; Frenkiel, T.A.; Davies, D.B.; Carbohydr. Res. 1988, 184, 39-46.
7. Tvaroska, I.; Hricovini, H.; Petrakova, E. Carbohydr. Res. 1989, 189, 359-362.
8. Hennig, M.; Bermel, W.; Schwalbe, H. ; Griesinger, C. J. Am. Chem. Soc. 2000, 122, 6268-6277.
9. Severson, M. L. and Maciel, G. E. J. Magn. Reson. 1984, 57, 248-268.
10. Cho, J . H. ; Kles singer, M .; T ecklenborg, U. a nd Wilhelm, K. Magn. Reson.
Chem. 1985, 23, 95-100.
11. Lichter, R. L. and Roberts, J. D. J. Org. Chem. 1970, 35, 2806-2807;
12. Haasnoot, C . A. G.; De L eeuw, F. and Altona, C . Tetrahedron, 1980, 36 , 2783-2792.
13. Haasnoot, C . A. G.; De L eeuw, F. ; De L eeuw, H. and Altona, C . Org. Magn.
Reson., 1981, 15, 43-52.
14. Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F. ; Bondo, P. B.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1998, 120, 11158-11173.
15. Cloran, F. ; C armichael, I .; Serianni, A. S. J. Am. Chem. Soc. 1999, 121, 9843-9851.
16. Zhao, H.; Carmichael, I. and Serianni, A. S. J. Org. Chem., 2008, 73, 3255-3257.
17. Frisch, M .J.; T rucks, G .W.; Schlegel, H .B.; Scuseria, G. E.; R obb, M .A.; Cheeseman, J .R.; Montgomery, Jr., J .A.; Vreven, T .; Kudin, K.N.; Burant, J .C.; Millam, J .M.; I yengar, S. S.; T omasi, J .; B arone, V. ; M ennucci, B .; C ossi, M .; Scalmani, G. ; R ega, N .; Peters son, G. A.; Nakat suji, H. ; Hada, M .; Ehara, M .;

167
Toyota, K .; Fukuda, R .; Has egawa, J .; Ishida, M.; Nakajima, T.; Honda, Y. ; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C .; J aramillo, J .; Gomperts , R .; Str atmann, R .E.; Yazyev, O. ; Aus tin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P. ; Dannenber g, J .J.; Zakrzewski, V. G.; Dappr ich, S.; Daniels , A.D.; St rain, M.C.; Farkas, O. ; Malick, D. K.; Rabuck, A. D.; Raghavachari, K. ; Foresman, J .B.; Or tiz, J .V.; C ui, Q. ; B aboul, A. G.; C lifford S. ; C ioslowski, J .; Stefanov, B.B.; Liu, G.; Liashenko, A. ; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D. J.; Keith , T .; Al- Laham, M .A.; Pe ng, C. Y.; Nanayakkar a, A. ; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian03, Revision A.1, Gaussian, Inc., Pittsburgh PA, 2003.
18. Becke, A.D. J. Chem. Phys. 1993, 98, 5648-5652.
19. Hehre, W.J.; Ditchfield, R.; Pople, J.A. J. Chem. Phys. 1972, 56, 2257-2261.
20. Stenutz, R.; Carmichael, I;. Widmalm, G.; Serianni, A.S. J. Org. Chem. 2002, 67, 949-958.
21. Pachler, K. G. R. Tetrahedron Letters. 1970, 22, 1955-1958.
22. San Fabian, J.; Guilleme, J. and Diez, E. J. Mol. Struct. THEOCHEM. 1998, 426, 117-133.
23. Diez, E.; San Fabian, J.; Altona, C. and Donders, L. A. Mol. Phys. 1989, 68, 49-63.
24. Van Beuzekom, A. A.; De Leeuw, F. A. and Altona, C. Magnet. Reson. in Chem, 1990, 28, 68-74.
25. Zhao, H.; Pan, Q.; Zhang, W.; Carmichael, I. and Serianni, A. S. J. Org. Chem. 2007, 72, 7071-7082.










![A new measurement of the spin dependent structure function g[sub 1][sup p]](https://img.pdfslide.net/doc/110x75/634ce798b5aff40b380ec38a/a-new-measurement-of-the-spin-dependent-structure-function-gsub-1sup-p.jpg)


















