Embed Size (px)
Citation preview

Review Detection of potential microbial antigens byimmuno-PCR (PCR-amplified immunoassay)
Promod K. Mehta,1 Ankush Raj,13 Netra Pal Singh13
and Gopal K. Khuller2
Correspondence
Promod K. Mehta
Received 6 November 2013
Accepted 21 February 2014
1Centre for Biotechnology, Maharshi Dayanand University, Rohtak-124001 (Haryana), India
2Department of Biochemistry, Postgraduate Institute of Medical Education and Research,Chandigarh-160014, India
Immuno-PCR (PCR-amplified immunoassay; I-PCR) is a novel ultrasensitive method combining
the versatility of ELISA with the sensitivity of nucleic acid amplification of PCR. The enormous
exponential amplification power of PCR in an I-PCR assay leads to at least a 102–104-fold
increase in sensitivity compared with an analogous ELISA. I-PCR has been used to detect many
biological molecules such as proto-oncogenes, toxins, cytokines, hormones, and biomarkers for
autoimmune and Alzheimer’s diseases, as well as microbial antigens and antibodies, and it can be
adapted as a novel diagnostic tool for various infectious and non-infectious diseases. Quantitative
real-time I-PCR has the potential to become the most analytically sensitive method for the
detection of proteins. The sensitivity and specificity of a real-time I-PCR assay can be enhanced
further with the use of magnetic beads and nanoparticles. This review is primarily focused on the
detection of potential viral, bacterial and parasitic antigens by I-PCR assay, thus enabling their
application for immunological research and for early diagnosis of infectious diseases.
Introduction
Immuno-PCR (I-PCR) combines the versatility of ELISAwith the exponential amplification power and sensitivity ofPCR, thus leading to an increase in sensitivity comparedwith an analogous ELISA. I-PCR is basically similar to ELISA(Fig. 1a), which detects an antigen–antibody reaction, butinstead of using an enzyme-conjugated antibody, the anti-body is labelled with a DNA fragment, which can be amp-lified by PCR. Although ELISA is the most commonly usedmethod for the detection of antigens, it fails when there isa low concentration of target antigen. PCR is widely used asa routine laboratory technique for the detection of nucleicacid molecules, but it cannot detect non-nucleic acidmolecules (Niemeyer et al., 2007; Mehta et al., 2012a).
This novel ultrasensitive I-PCR method has been widelyexplored for the detection of a variety of biological moleculesincluding cytokines, tumour markers, T-cell receptors,angiotensinogen, toxins, hormones, and biomarkers forautoimmune and Alzheimer’s diseases, and it can be adaptedas a novel diagnostic tool for the detection of microbialantigens and antibodies (Niemeyer et al., 2005, 2007; Malou& Raoult, 2011; Potuckova et al., 2011; Mehta et al., 2012b).I-PCR was originally developed by Sano et al. (1992) by usinga detection antibody coupled to a biotinylated DNA moleculethrough a streptavidin–protein A chimaeric molecule andcould detect as few as 580 BSA antigen molecules. However,the limited availability of streptavidin–protein A fusion pro-tein, and the widely varied affinities of protein A with anti-bodies of various classes and subclasses from different speciesrestricted its immediate applications. Therefore, Zhou et al.(1993) modified I-PCR to ‘universal I-PCR’ in which anantigen (to be detected) is coupled subsequently with anantigen-specific primary antibody, followed by a species-specific biotinylated detection antibody, streptavidin and abiotinylated DNA (Fig. 1b). The resulting procedure retainsthe sensitivity and specificity of the original I-PCR but can beuniversally applied without any immunoglobulin source or(sub)class restrictions.
Several commonly used formats of universal I-PCR includedirect I-PCR, indirect I-PCR, sandwich I-PCR and indirectsandwich I-PCR (Fig. 1b). In direct I-PCR, the antigen tobe detected is coated on wells of a microtitre plate, and
3These authors contributed equally to this work.
Abbreviations: BoNT/A, Clostridium botulinum neurotoxin A; EIA,enzyme immunoassay; EID50, 50% egg infectious dose; FMDV, foot-and-mouth disease virus; GNP-I-PCR, gold nanoparticle I-PCR; HBcAg,hepatitis B virus core antigen; HBsAg, hepatitis B virus surface antigen;HBV, hepatitis B virus; HIV, human immunodeficiency virus; IL-PCR,immunoliposome-PCR; I-PCR, immuno-PCR; LOD, limit of detection;NPA-I-PCR, nanoparticle-amplified I-PCR; NV, norovirus; PD-I-PCR,phage display-mediated I-PCR; PrP, prion protein; RD, region ofdifference; rNV-VLP, recombinant norovirus virus-like particles;RT-PCR, reverse transcription PCR; SEA, Staphylococcus aureusenterotoxin A; SEB, S. aureus enterotoxin B; SMCC, succinimidyl4-[N-maleimidomethyl]-cyclohexane-1-carboxylate; STEC, Shiga toxin-producing Escherichia coli; Stx, Shiga toxin.
Journal of Medical Microbiology (2014), 63, 627–641 DOI 10.1099/jmm.0.070318-0
070318 G 2014 The Authors Printed in Great Britain 627

biotinylated detection antibody is then added, which isfurther attached to a biotinylated reporter DNA throughstreptavidin (Liang et al., 2003; Zhang et al., 2008). Inindirect I-PCR, the antigen to be detected is coated on thewells of a microtitre plate and the detection antibody is thenadded, followed by the addition of biotinylated antibodyagainst the detection antibody (anti-detection antibody),
which is attached to biotinylated reporter DNA throughstreptavidin (Chao et al., 2004). In sandwich I-PCR, theantigen is sandwiched between a capture antibody onthe wells of a microtitre plate and a biotinylated detectionantibody, which is attached to biotinylated reporter DNAthrough streptavidin (Chye et al., 2004). In indirectsandwich I-PCR, the antigen is sandwiched between a
(a)
(b)
ES
E
S
(i)
Microtitre plate Colour reaction Biotin
Streptavidin
Reporter DNA
Detection antibody
Capture antibody
Anti-detection antibody
Antigen
Enzyme
Substrate
Indirect I-PCR Sandwich I-PCRDirect I-PCR
PCR/real-time PCR
PCR
Indirect sandwich I-PCR(ii) (iii) (iv)
ELISA I-PCR
Fig. 1. (a) Evolution of I-PCR. The general set-up of I-PCR is almost similar to that of antigen detection with ELISA, but in I-PCR, a conjugate comprising an antibody and a reporter DNA is amplified by PCR for signal generation. (b) Different formats ofuniversal I-PCR. Streptavidin acts as a linker molecule between the biotinylated detection antibody and the biotinylated reporterDNA; the biotinylated reporter DNA is amplified by PCR/real-time PCR. (i) Direct I-PCR: the antigen is captured on the wells ofa microtitre plate and detected directly by biotinylated antibody, which is attached to biotinylated DNA through streptavidin. (ii)Indirect I-PCR: a biotinylated antibody against the detection antibody (anti-detection antibody) is attached to the detectionantibody, and is further attached to biotinylated DNA through streptavidin. (iii) Sandwich I-PCR: the antigen is sandwichedbetween a capture antibody and the detection antibody, which is further attached to biotinylated DNA through streptavidin. (iv)Indirect sandwich I-PCR: the antigen is sandwiched between a capture antibody and the detection antibody, and an anti-detection biotinylated antibody is attached to the detection antibody, which is further attached to biotinylated DNA throughstreptavidin.
P. K. Mehta and others
628 Journal of Medical Microbiology 63

capture antibody on the wells of a microtitre plate and adetection antibody, and the biotinylated anti-detectionantibody is attached to the detection antibody and linkedto biotinylated reporter DNA through streptavidin (Mehtaet al., 2012b). Sandwich I-PCR is advantageous over thedirect I-PCR method for antigen detection as it eliminatesthe need for the direct coating of biological samples as asource of antigen, which may decrease non-specific bindingwithout compromising its stability (Niemeyer et al., 1997;Mehta et al., 2012b). Real-time I-PCR has emerged as themost sensitive assay for the detection of protein targets.Sims et al. (2000) were the first researchers to use real-timeI-PCR for the detection of vascular endothelial growthfactor within human and mouse sera. In contrast to thehigh sensitivities reported from the other laboratories, nofurther increase in ELISA sensitivity was observed by theseresearchers, probably due to matrix effects attributed tonon-specific binding of serum constituents that increasedthe background noise and hence influenced the perform-ance of the I-PCR. The non-specific binding occurring withthe use of classical microtitre plates (solid format) canbe reduced by using appropriate blocking solutions. Theintroduction of a liquid format, for example the useof magnetic beads/gold nanoparticles (Adler et al., 2008;Barletta et al., 2009; Perez et al., 2011; Adams et al., 2012),has been a major breakthrough in devising I-PCR assays, asit allows more thorough washing of the captured antigenand nanoparticles to decrease non-specific binding andreduce the background signals (Malou & Raoult, 2011;Perez et al., 2011). The large surface area of nanoparticles incomparison with that of microtitre plates also permits ahigher and a faster interaction between the capture antibodyand respective antigen.
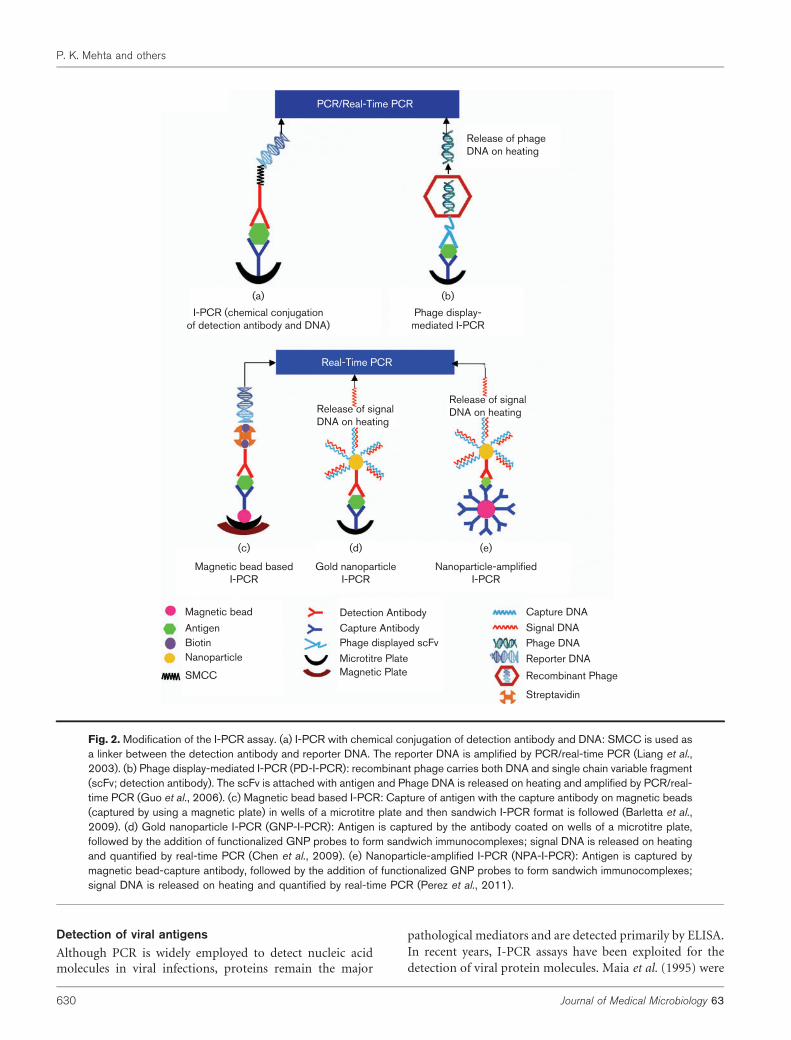
There can be direct conjugation of detection antibodywith the reporter DNA through covalent binding with achemical compound such as succinimidyl 4-[N-maleimi-domethyl]-cyclohexane-1-carboxylate (SMCC) (Wu et al.,2001; Liang et al., 2003) as shown in Fig. 2(a). This assay isquicker to perform than a stretptavidin–biotin-based I-PCR,as washing is required only after the addition of sampleantigen and after the addition of the DNA–antibody con-jugate. Another important strategy is phage display-mediated I-PCR (PD-I-PCR; Fig. 2b). Instead of usingmAb and streptavidin–biotin/chemically linked DNA in theconventional I-PCR, a recombinant phage particle is used asa ‘ready reagent’ for I-PCR (Guo et al., 2006). The surface-displayed single chain variable fragment (scFv) and phageDNA themselves can serve directly as detection antibody andPCR template, respectively. The target antigen is capturedby the immobilized capture antibody coated on a microtitreplate, whilst the recombinant phage particles are anchoredthrough the interaction between the displayed scFv andbound target antigen. The phage DNA is released by heatlysis and serves as PCR template for amplification. PD-I-PCR has merits over conventional I-PCR, as the recombin-ant phage particles carry both scFv and DNA to be amplifiedand they can easily be obtained by simply centrifuging
the overnight Escherichia coli culture broth. However, thesensitivity of PD-I-PCR is not as high as that of mAb-basedI-PCR due to the lower affinity of scFv towards the antigen.
In recent years, nanobiotechnology has emerged as the mostpromising tool for the development of a powerful strategyfor drug delivery diagnostics (Griffiths et al., 2010). Besidescircumventing the background noise, the use of magneticbeads and nanoparticle-based I-PCR can further improvedetection limits and reduce washing steps, as well asreducing incubation steps, thus improving the assay (Adleret al., 2008; Chen et al., 2009; Perez et al., 2013). In themagnetic bead-based I-PCR assay (Fig. 2c), the captureantibody is adsorbed to the magnetic beads to capture theantigen (Barletta et al., 2009) and the sandwich I-PCR formatis then followed using streptavidin as a bridge betweenthe detection antibody and the reporter DNA. Briefly, themagnetic beads are captured (using a magnetic plate placedunder the microtitre plate) periodically after each washingphase; the magnetic beads are then released from themagnetic force and allowed to circulate freely in solutionduring incubation with antigen or antibody. Another I-PCRformat is gold nanoparticle I-PCR (GNP-I-PCR; Fig. 2d).The antigen is captured by the specific polyclonal antibodycoated on the wells of a microtitre plate, followed by theaddition of gold nanoparticle (GNP) probes [functionalizedwith thiolated oligonucleotides (capture DNA) and mAb] toform sandwich immunocomplexes (Chen et al., 2009). Theoligonucleotides on the GNP contain two strands: one as acapture DNA immobilized on the surface of the GNP and theother as a signal DNA, which is partially complementary tothe capture DNA. The immunocomplexes are heated torelease the signal DNA, which is quantified by real-time PCR.In another I-PCR format, known as nanoparticle-amplifiedI-PCR (NPA-I-PCR) (Fig. 2e), antigen is captured usingantibody-functionalized magnetic beads (Perez et al., 2011,2013). The gold nanoparticles are functionalized with thedetection antibodies and thiolated DNA complementaryto the hybridized tag DNA. The magnetic bead–antigencomplex is reacted with antibody–DNA-functionalizedGNPs. The hybridized tag DNA (signal DNA) is releasedfrom the GNPs by heating and quantified by real-time PCR.
I-PCR assay is not restricted to proteins: many small moleculartargets are also detected up to femtogram levels; for example,biogenic amine serotonin has been detected by sandwichI-PCR with a 1000-fold increase in sensitivity compared withan analogous ELISA (Adler et al., 2005). In addition, the assayhas been translated into a commercial kit (Chimera Biotech;http://www.chimera-biotec.com) (Wacker, 2006), thus dem-onstrating that I-PCR is progressing from the research anddevelopment platform to a routine diagnostic method.Recently, Malou & Raoult (2011) and previously Niemeyeret al. (2005, 2007) meticulously reviewed the detection ofvarious antigens and antibodies using an ultrasensitive I-PCRassay. In this review, we have further updated our knowledgeof the detection of potential viral, bacterial and parasiticantigens by I-PCR assay, and its application in immunologicalresearch and clinical diagnosis of infectious diseases.
Detection of potential microbial antigens by immuno-PCR
http://jmm.sgmjournals.org 629
Detection of viral antigens
Although PCR is widely employed to detect nucleic acidmolecules in viral infections, proteins remain the major
pathological mediators and are detected primarily by ELISA.
In recent years, I-PCR assays have been exploited for the
detection of viral protein molecules. Maia et al. (1995) were
PCR/Real-Time PCR
Real-Time PCR
Release of phageDNA on heating
I-PCR (chemical conjugationof detection antibody and DNA)
Phage display-mediated I-PCR
Release of signalDNA on heating
Magnetic bead basedI-PCR
Gold nanoparticleI-PCR
Nanoparticle-amplifiedI-PCR
Release of signalDNA on heating
Magnetic bead Detection Antibody Capture DNA
Signal DNAPhage DNA
Reporter DNA
Recombinant Phage
Streptavidin
Capture AntibodyPhage displayed scFv
Microtitre PlateMagnetic Plate
AntigenBiotinNanoparticle
SMCC
(a)
(c) (d) (e)
(b)
Fig. 2. Modification of the I-PCR assay. (a) I-PCR with chemical conjugation of detection antibody and DNA: SMCC is used asa linker between the detection antibody and reporter DNA. The reporter DNA is amplified by PCR/real-time PCR (Liang et al.,2003). (b) Phage display-mediated I-PCR (PD-I-PCR): recombinant phage carries both DNA and single chain variable fragment(scFv; detection antibody). The scFv is attached with antigen and Phage DNA is released on heating and amplified by PCR/real-time PCR (Guo et al., 2006). (c) Magnetic bead based I-PCR: Capture of antigen with the capture antibody on magnetic beads(captured by using a magnetic plate) in wells of a microtitre plate and then sandwich I-PCR format is followed (Barletta et al.,2009). (d) Gold nanoparticle I-PCR (GNP-I-PCR): Antigen is captured by the antibody coated on wells of a microtitre plate,followed by the addition of functionalized GNP probes to form sandwich immunocomplexes; signal DNA is released on heatingand quantified by real-time PCR (Chen et al., 2009). (e) Nanoparticle-amplified I-PCR (NPA-I-PCR): Antigen is captured bymagnetic bead-capture antibody, followed by the addition of functionalized GNP probes to form sandwich immunocomplexes;signal DNA is released on heating and quantified by real-time PCR (Perez et al., 2011).
P. K. Mehta and others
630 Journal of Medical Microbiology 63

the first researchers to develop a sandwich I-PCR assay forthe detection of viral antigens, including the hepatitis Bsurface antigen (HBsAg) of hepatitis B virus (HBV), whichcombined the specificity of two high-affinity anti-HBsAgmAbs employed both as capture and as detection antibody(Table 1). The use of two-site mAbs was found to greatlyenhance the specificity and sensitivity of the assay such thatit could detect as little as 0.5 pg HBsAg in serum samples(Maia et al., 1995).
Several laboratories have demonstrated high-sensitivitydetection (100–700-fold increase) of HBsAg by I-PCRcompared with ELISA (Niemeyer et al., 1997; Cao et al.,2000). Magneto I-PCR was devised by Wacker et al. (2007)for the better detection of recombinant HBsAg in humanserum. The HBsAg-specific antibody–magnetosome con-jugate and the DNA–antibody conjugate were incubatedsimultaneously with a serum sample containing HBsAgresulting in a signal-generating detection complex. Thedetection complex was concentrated using an externalmagnetic field to quantify the immobilized HBsAg by real-time PCR with a detection limit of 320 pg ml21 comparedwith 40 ng ml21 detected by Magneto ELISA. As well asHBsAg, the core antigen of HBV (HBcAg) is anotherpotential biomarker for the identification of HBV infec-tion. Recently, a TaqMan real-time PCR assay based onPD-I-PCR has been developed for the detection of HBcAgin human serum samples (Monjezi et al., 2013). In thisassay, a constrained peptide displayed on the surface ofan M13 recombinant bacteriophage that interacted tightlywith HBcAg was employed as a diagnostic reagent andcould detect as little as 10 ng HBcAg with 108 p.f.u. M13recombinant phage ml21, which was about 10 000 timesmore sensitive than the phage ELISA.
Because of the low concentration of p24 antigen in the earlystage of human immunodeficiency virus (HIV) infection,these viral proteins in particular have become a primarytarget in I-PCR developments (Barletta et al., 2004, 2009).Detection of HIV-1 p24 antigen increases the probabilityof HIV-1 detection, as free HIV-1 p24 antigen circulates inthe blood and/or is sequestered in immune complexesand tissues in the absence of virions (Barletta et al., 2004).Barletta et al. (2004) documented a quantitative real-timeI-PCR assay to determine HIV-1 infection by detecting thepresence of HIV-1 p24 antigen in serum samples with anelevated sensitivity in comparison with RNA detection byreverse transcription-PCR (RT-PCR), which had a sensitiv-ity of 50 viral copies ml21. The HIV-1 p24 antigen detectionwas more sensitive for the detection of virus, as this targetprotein is present in the virion at much higher numbersthan the viral RNA copies (approx. 3000 HIV-1 p24 antigenmolecules versus two RNA copies per virion). Of 52 samplesof HIV-1-infected patients (with ,50 RNA copies ml21),22 samples were found to be positive by real-time I-PCR.Later, a modified quantitative real-time I-PCR assay basedon magnetic beads was devised by Barletta et al. (2009).The HIV-1 p24 antigen was captured by an anti-HIV-1p24 antibody adsorbed to the magnetic beads, which were
captured by a magnetic plate. The p24 antigen was thendetected by a biotinylated anti-p24 antibody linked to abiotinylated reporter DNA via streptavidin–horseradishperoxidase. The use of streptavidin–horseradish peroxidaseallowed ELISA to be performed simultaneously, as well asthe amplification and quantification of the reporter DNA.This assay had a limit of quantification of 10–100 p24antigen molecules, enabling a detection limit of less thanone HIV-1 virion in a complex sample matrix (plasma)and providing better potential to monitor an early HIV-1infection than detecting the nucleic acid molecules by PCR(Barletta et al., 2009). The ultralow level of detection ofp24 antigen by I-PCR is not just of academic interestbut might also have potential implications in examiningHIV pathogenesis and anti-retroviral treatment strategies.Standardization and optimization of this method areneeded to move the method to a framework that wouldmake it a candidate for routine use in clinical diagnosis.
Recently, an immunoliposome-PCR (IL-PCR) assay wasintroduced to quantify recombinant HIV-1 p24 antigen inbuffer and carcinoembryonic antigen in human serum witha detection limit of 2.4 pg ml21 for p24 antigen (He et al.,2012). This method used a liposome preparation incorp-orating reporter DNA encapsulated inside and a biotin-labelled polyethylene glycol phospholipid conjugate as adetection reagent in the outer surface of a liposome, whichis coupled to real-time PCR for antigen detection. This assayhas a number of merits over other I-PCR formats, as thereporter DNA and detection reagent are incorporated intothe liposomes, thus simplifying preparation of the detectionreagent. The ability to encapsulate multiple reporters perliposome could also overcome the effect of polymeraseinhibitors present in many biological specimens and thusmight have wide applications for the diagnosis of infectiousdiseases.
Rotavirus infection causes serious diarrhoeal disease inchildren at an early stage of their life. To minimize the riskof rotavirus infection, a highly sensitive quantitative real-time I-PCR was developed by Adler et al. (2005) for earlydetection of the rotavirus antigen VP6. The VP6 antigenwas immobilized between mouse anti-rotavirus mAb andCHI-Rota (a chimaeric conjugate of mouse anti-rotavirusmAb and marker DNA) to attain a detection limit of 100virus particles ml21 by quantitative real-time I-PCR assayusing standardized I-PCR protocols (Imperacer; ChimeraBiotec). Similar results were also obtained with a PCR-ELISA, in which the amplified products were labelled withbiotin as well as with DIG, and later the labelled amplifiedproducts were attached to a streptavidin-coated microtitreplate for quantification using anti-DIG–alkaline phospha-tase antibody–enzyme conjugate and fluorescence-generat-ing substrate (Adler et al., 2005).
Recently, Perez et al. (2011, 2013) described an NPA-I-PCRassay that combined traditional ELISA with a 50-foldnanoparticle valence amplified step, followed by real-timePCR amplification. The assay could detect respiratory
Detection of potential microbial antigens by immuno-PCR
http://jmm.sgmjournals.org 631
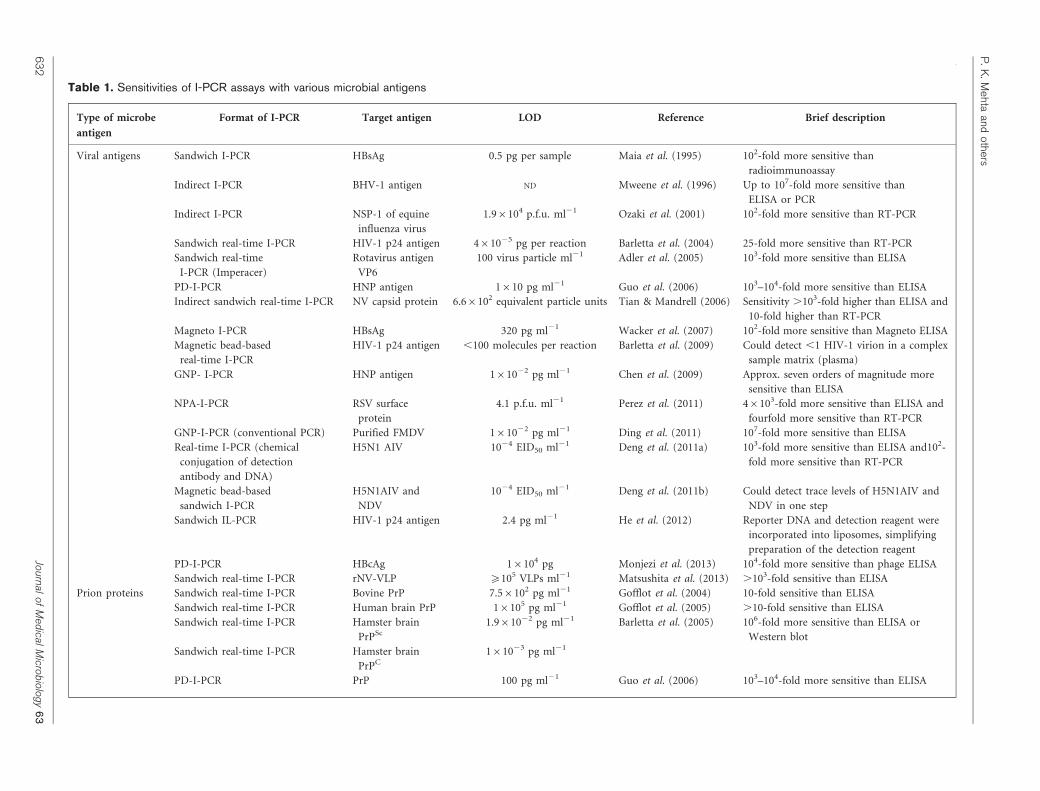
Table 1. Sensitivities of I-PCR assays with various microbial antigens
Type of microbe
antigen
Format of I-PCR Target antigen LOD Reference Brief description
Viral antigens Sandwich I-PCR HBsAg 0.5 pg per sample Maia et al. (1995) 102-fold more sensitive than
radioimmunoassay
Indirect I-PCR BHV-1 antigen ND Mweene et al. (1996) Up to 107-fold more sensitive than
ELISA or PCR
Indirect I-PCR NSP-1 of equine
influenza virus
1.96104 p.f.u. ml21 Ozaki et al. (2001) 102-fold more sensitive than RT-PCR
Sandwich real-time I-PCR HIV-1 p24 antigen 461025 pg per reaction Barletta et al. (2004) 25-fold more sensitive than RT-PCR
Sandwich real-time
I-PCR (Imperacer)
Rotavirus antigen
VP6
100 virus particle ml21 Adler et al. (2005) 103-fold more sensitive than ELISA
PD-I-PCR HNP antigen 1610 pg ml21 Guo et al. (2006) 103–104-fold more sensitive than ELISA
Indirect sandwich real-time I-PCR NV capsid protein 6.66102 equivalent particle units Tian & Mandrell (2006) Sensitivity .103-fold higher than ELISA and
10-fold higher than RT-PCR
Magneto I-PCR HBsAg 320 pg ml21 Wacker et al. (2007) 102-fold more sensitive than Magneto ELISA
Magnetic bead-based
real-time I-PCR
HIV-1 p24 antigen ,100 molecules per reaction Barletta et al. (2009) Could detect ,1 HIV-1 virion in a complex
sample matrix (plasma)
GNP- I-PCR HNP antigen 161022 pg ml21 Chen et al. (2009) Approx. seven orders of magnitude more
sensitive than ELISA
NPA-I-PCR RSV surface
protein
4.1 p.f.u. ml21 Perez et al. (2011) 46103-fold more sensitive than ELISA and
fourfold more sensitive than RT-PCR
GNP-I-PCR (conventional PCR) Purified FMDV 161022 pg ml21 Ding et al. (2011) 107-fold more sensitive than ELISA
Real-time I-PCR (chemical
conjugation of detection
antibody and DNA)
H5N1 AIV 1024 EID50 ml21 Deng et al. (2011a) 103-fold more sensitive than ELISA and102-
fold more sensitive than RT-PCR
Magnetic bead-based
sandwich I-PCR
H5N1AIV and
NDV
1024 EID50 ml21 Deng et al. (2011b) Could detect trace levels of H5N1AIV and
NDV in one step
Sandwich IL-PCR HIV-1 p24 antigen 2.4 pg ml21 He et al. (2012) Reporter DNA and detection reagent were
incorporated into liposomes, simplifying
preparation of the detection reagent
PD-I-PCR HBcAg 16104 pg Monjezi et al. (2013) 104-fold more sensitive than phage ELISA
Sandwich real-time I-PCR rNV-VLP ¢105 VLPs ml21 Matsushita et al. (2013) .103-fold sensitive than ELISA
Prion proteins Sandwich real-time I-PCR Bovine PrP 7.56102 pg ml21 Gofflot et al. (2004) 10-fold sensitive than ELISA
Sandwich real-time I-PCR Human brain PrP 16105 pg ml21 Gofflot et al. (2005) .10-fold sensitive than ELISA
Sandwich real-time I-PCR Hamster brain
PrPSc
1.961022 pg ml21 Barletta et al. (2005) 106-fold more sensitive than ELISA or
Western blot
Sandwich real-time I-PCR Hamster brain
PrPC
161023 pg ml21
PD-I-PCR PrP 100 pg ml21 Guo et al. (2006) 103–104-fold more sensitive than ELISA
P.K
.Mehta
andothers
63
2Jo
urnalo
fM
edical
Micro
bio
logy
63
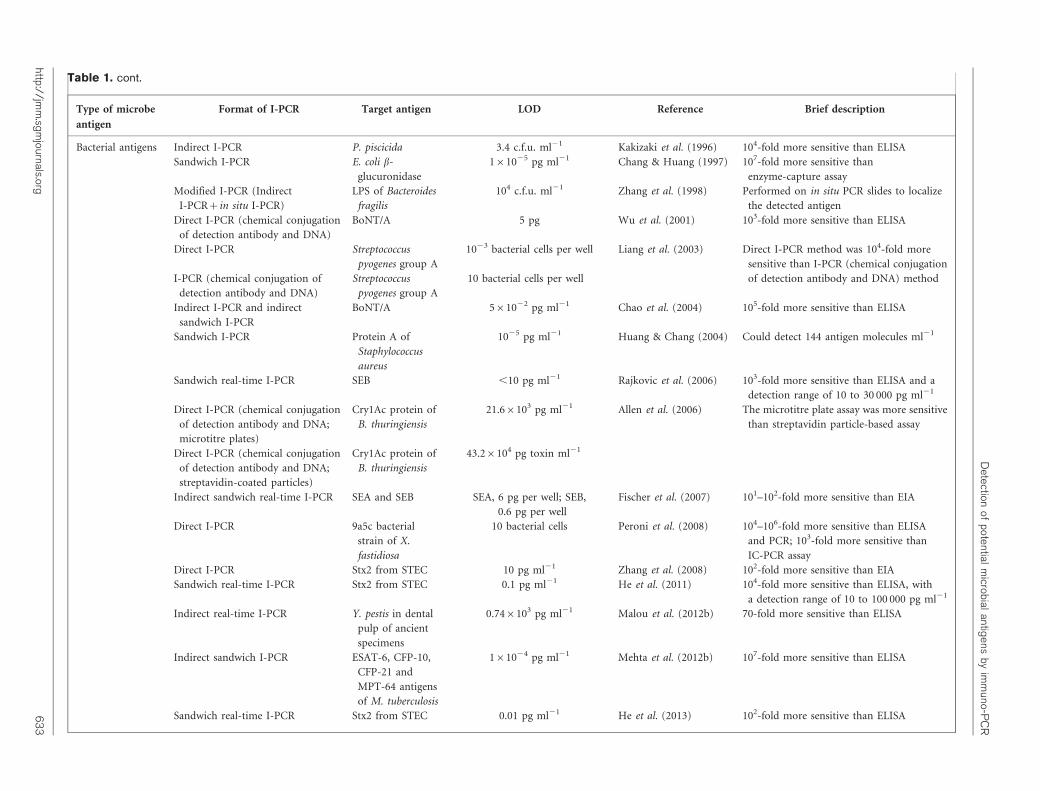
Table 1. cont.
Type of microbe
antigen
Format of I-PCR Target antigen LOD Reference Brief description
Bacterial antigens Indirect I-PCR P. piscicida 3.4 c.f.u. ml21 Kakizaki et al. (1996) 104-fold more sensitive than ELISA
Sandwich I-PCR E. coli b-
glucuronidase
161025 pg ml21 Chang & Huang (1997) 107-fold more sensitive than
enzyme-capture assay
Modified I-PCR (Indirect
I-PCR+in situ I-PCR)
LPS of Bacteroides
fragilis
104 c.f.u. ml21 Zhang et al. (1998) Performed on in situ PCR slides to localize
the detected antigen
Direct I-PCR (chemical conjugation
of detection antibody and DNA)
BoNT/A 5 pg Wu et al. (2001) 103-fold more sensitive than ELISA
Direct I-PCR Streptococcus
pyogenes group A
1023 bacterial cells per well Liang et al. (2003) Direct I-PCR method was 104-fold more
sensitive than I-PCR (chemical conjugation
of detection antibody and DNA) methodI-PCR (chemical conjugation of
detection antibody and DNA)
Streptococcus
pyogenes group A
10 bacterial cells per well
Indirect I-PCR and indirect
sandwich I-PCR
BoNT/A 561022 pg ml21 Chao et al. (2004) 105-fold more sensitive than ELISA
Sandwich I-PCR Protein A of
Staphylococcus
aureus
1025 pg ml21 Huang & Chang (2004) Could detect 144 antigen molecules ml21
Sandwich real-time I-PCR SEB ,10 pg ml21 Rajkovic et al. (2006) 103-fold more sensitive than ELISA and a
detection range of 10 to 30 000 pg ml21
Direct I-PCR (chemical conjugation
of detection antibody and DNA;
microtitre plates)
Cry1Ac protein of
B. thuringiensis
21.66103 pg ml21 Allen et al. (2006) The microtitre plate assay was more sensitive
than streptavidin particle-based assay
Direct I-PCR (chemical conjugation
of detection antibody and DNA;
streptavidin-coated particles)
Cry1Ac protein of
B. thuringiensis
43.26104 pg toxin ml21
Indirect sandwich real-time I-PCR SEA and SEB SEA, 6 pg per well; SEB,
0.6 pg per well
Fischer et al. (2007) 101–102-fold more sensitive than EIA
Direct I-PCR 9a5c bacterial
strain of X.
fastidiosa
10 bacterial cells Peroni et al. (2008) 104–106-fold more sensitive than ELISA
and PCR; 103-fold more sensitive than
IC-PCR assay
Direct I-PCR Stx2 from STEC 10 pg ml21 Zhang et al. (2008) 102-fold more sensitive than EIA
Sandwich real-time I-PCR Stx2 from STEC 0.1 pg ml21 He et al. (2011) 104-fold more sensitive than ELISA, with
a detection range of 10 to 100 000 pg ml21
Indirect real-time I-PCR Y. pestis in dental
pulp of ancient
specimens
0.746103 pg ml21 Malou et al. (2012b) 70-fold more sensitive than ELISA
Indirect sandwich I-PCR ESAT-6, CFP-10,
CFP-21 and
MPT-64 antigens
of M. tuberculosis
161024 pg ml21 Mehta et al. (2012b) 107-fold more sensitive than ELISA
Sandwich real-time I-PCR Stx2 from STEC 0.01 pg ml21 He et al. (2013) 102-fold more sensitive than ELISA
Detection
ofpotential
microbial
antigens
byim
muno-P
CR
http://jmm
.sgm
journals.org6
33

syncytial virus surface protein in paediatric infection usingantibody-functionalized magnetic beads and a Synagis an-tibody bound to a GNP co-functionalized with thiolatedDNA. This assay showed a 4000-fold improvement in thelimit of detection (LOD) compared with ELISA and afourfold improvement in the LOD compared with RT-PCRfrom HEp-2 cell extracts, suggesting that the NPA-I-PCRassay could offer a viable platform for the development ofearly-stage diagnostics requiring exceptionally low levels ofdetection.
Norovirus (NV) is an important human pathogen thatcauses epidemic acute non-bacterial gastroenteritis on aglobal scale (Glass et al., 2001). A sensitive real-time I-PCRmethod was devised by Tian & Mandrell (2006) for thedetection of NV capsid protein in stool and food samples,which showed an improved LOD compared with ELISAand RT-PCR assays. The removal of micro-organisms bydrinking-water treatment processes has been examined inlaboratory-scale experiments using artificially propagatedmicro-organisms; however, this method cannot be appliedto NV removal because of the lack of a cell-culture systemor an animal model for this virus. This fact has restrictedthe ability to examine its behaviour during drinking-watertreatment. To overcome this problem, Shirasaki et al.(2010) previously used recombinant NV virus-like particles(rNV-VLPs), which comprised an artificially expressed NVcapsid protein, in laboratory-scale drinking-water treat-ment experiments. The ELISA method used to detecttherNV-VLPs was not sensitive enough to examine highremoval ratios such as those obtained by ultrafiltration. Ahighly sensitive real-time I-PCR assay has been describedfor the quantification of rNV-VLPs to investigate NVremoval by microfiltration, ultrafiltration and hybrid pre-coagulation–microfiltration processes (Matsushita et al.,2013).
Mweene et al. (1996) earlier introduced an I-PCR methodspecific for the detection of antigens and antibodies tobovine herpes virus type 1, which attained a sensitivity upto 107 times higher than that of ELISA for antigen detectionand 105 times higher than that of ELISA for antibodydetection. For the diagnosis of equine influenza virus, viralnon-structural protein 1 was detected in nasal swab samplesby I-PCR with 10–100 times better sensitivity than RT-PCR(Ozaki et al., 2001), and 107–108 times better sensitivity wasobserved by I-PCR than from virus isolation or RT-PCRfor the detection of the haemagglutinin subtype-specificantigen.
Avian influenza virus H5N1 and Newcastle disease virus aretwo major causes of high mortality in poultry. Therefore,rapid identification of these two viruses has importantclinical and epidemiological implications. A highly sensitive,quantitative real-time I-PCR assay has been documented byDeng et al. (2011a) to detect an ultralow concentration ofavian influenza virus in clinical samples of allantoic fluidfrom eggs and tracheal swab specimens in the poultry. Thisassay could detect up to 1024 50% egg infectious doseT
ab
le1.
cont
.
Typ
eo
fm
icro
be
anti
gen
Fo
rmat
of
I-P
CR
Tar
get
anti
gen
LO
DR
efer
ence
Bri
efd
escr
ipti
on
Par
asit
ican
tige
nS
and
wic
hI-
PC
R2
04
kD
aA
cL5
of
A.
can
ton
ensi
s(f
ifth
-
stag
ela
rvae
)
10
0p
gm
l21
Ch
yeet
al.
(20
04
)1
02–
10
3-f
old
mo
rese
nsi
tive
than
EL
ISA
Myc
oto
xin
San
dw
ich
real
-tim
eI-
PC
R(c
hem
ical
con
juga
tio
no
fd
etec
tio
nan
tib
od
y
and
DN
A)
Afl
ato
xin
B1
0.1
p.p
.b.
Bab
u&
Mu
rian
a(2
01
1)
Det
ecti
on
ran
geo
faf
lato
xin
B1
fro
m
10
to0
.1p
.p.b
.
HB
sAg,
hep
atit
isB
viru
ssu
rfac
ean
tige
n;
BH
V-1
,b
ovi
ne
her
pes
viru
s-1
;N
D,
no
td
eter
min
ed;
NS
P-1
,n
on
-str
uct
ura
lp
rote
in-1
;R
T-P
CR
,re
vers
etr
ansc
rip
tio
nP
CR
;H
IV,
hu
man
imm
un
od
efic
ien
cy
viru
s;H
NP
,H
anta
anvi
rus
nu
cleo
cap
sid
pro
tein
;N
V,
no
rovi
rus;
RS
V,
resp
irat
ory
syn
cyti
alvi
rus;
FM
DV
,fo
ot-
and
-mo
uth
dis
ease
viru
s;A
IV,
avia
nin
flu
enza
viru
s;E
ID5
0,
50
%eg
gin
fect
iou
sd
ose
;
ND
V,
New
cast
led
isea
sevi
rus;
IL-P
CR
;im
mu
no
lip
oso
me-
PC
R;
HB
cAg,
core
anti
gen
of
hep
atit
isB
viru
s;rN
V-V
LP
,re
com
bin
ant
no
rovi
rus-
viru
s-li
ke
par
ticl
es;
PrP
,p
rio
np
rote
in;
PrP
C,
cell
ula
r
pri
on
pro
tein
s;P
rPS
c,p
rio
np
rote
insc
rap
ie;
Bo
NT
/A,
C.
botu
lin
um
neu
roto
xin
typ
eA
;S
EA
,S
tap
hyl
ococ
cus
au
reu
sen
tero
toxi
nA
;S
EB
,S
tap
hyl
ococ
cus
au
reu
sen
tero
toxi
nB
;E
IA,
enzy
me
imm
un
oas
say;
IC-P
CR
,im
mu
no
cap
ture
PC
R;
ES
AT
-6,
earl
yse
cret
ory
anti
gen
icta
rget
-6;
CF
P-1
0,
cult
ure
filt
rate
pro
tein
-10
;C
FP
-21
,cu
ltu
refi
ltra
tep
rote
in-2
1,
MP
T-6
4,
myc
ob
acte
rial
pro
tein
fro
msp
ecie
stu
ber
culo
sis-
64
.
P. K. Mehta and others
634 Journal of Medical Microbiology 63

(EID50) ml21 of avian influenza virus using SMCC as across-linker between the reporter DNA and the detectingantibody. In addition, the reporter DNA was cut with BamHI and quantified by real-time I-PCR, thus enabling animproved detection by at least 102-fold in comparison withRT-PCR and a 103-fold improvement in comparison withELISA. Later, Deng et al. (2011b) introduced an I-PCR assayutilizing magnetic particles as the adsorption surface andadopting the conventional method of streptavidin linkingthe detection antibody with the reporter DNA for thedetection of both avian influenza virus and Newcastledisease virus in one step. A detection limit of as low as1024 EID50 ml21 was observed for both the viruses, thusproviding a viable scaffold capable of mass screening as wellas detection of these infections in one step.
The RNA-binding nucleoprotein has been shown to be anexcellent target for the development of influenza A virusdiagnostics due to its high antigenicity and also its presencein large numbers in the virus particles. Interestingly, itbinds non-specifically to the sugar–phosphate backbone ofRNA as well as to ssDNA in an in vitro system. Recently,Morin & Schaeffer (2012) took advantage of this propertyto develop an ssDNA probe for the detection of recombinantinfluenza A virus nucleoprotein in the low picomolar rangeusing a quantitative PCR assay.
Foot-and-mouth disease virus (FMDV) causes contagiousinfection in artiodactyls, especially in cattle and swine,leading to huge economic losses (Ding et al., 2011). ELISAand conventional PCR are the standard laboratory tests todetect FMDV; however, these tests have their limitations. Ahighly sensitive GNP-I-PCR assay derived from the bio-barcode assay has been developed for the detection of FMDV(Nam et al., 2003; Ding et al., 2011). The viral particles weresandwiched between the polyclonal antibody and an FMDV-specific mAb (clone 1D11) attached to GNP along withthe surface-bound oligonucleotides to form an immunecomplex. The signal DNA was released by heating andconsequently evaluated by PCR/real-time PCR. The detec-tion limit of the GNP-I-PCR assay could reach up to 10 fgml21 for purified FMDV particles in comparison with adetection limit of 100 ng ml21 using ELISA. A modifiedform of bio-barcode assay has also been devised for theultrasensitive detection of Hantaan virus nucleocapsidprotein by GNP-I-PCR assay and could determine up to10 fg ml21 of the purified nucleocapsid protein in buffers aswell as in human serum samples (Chen et al., 2009).
Detection of prion proteins (PrPs)
PrPs are known to cause fatal neurological disorders(Gofflot et al., 2004, 2005). The cellular prion proteins(PrPC) are modified to pathogenic PrP by conformationalchanges of a-helices into b-sheets resulting in the formationof proteinase K-resistant PrP forms; this forms the basis forthe diagnosis of prion infections. The introduction of I-PCRhas been a major breakthrough for the detection of PrP. Asandwich real-time I-PCR assay has been demonstrated for
the detection of recombinant bovine PrP (Gofflot et al.,2004), and revealed at least a 10-fold higher sensitivity thanELISA. Later, Gofflot et al. (2005) diagnosed PrP in humansinfected with sporadic Creutzfeldt–Jakob disease by ELISA,Western blotting, immunohistochemistry and quantitativereal-time I-PCR methods. All these methods revealed 100 %sensitivity and 100 % specificity; however, real-time I-PCRdetected the presence of PrP at concentrations at least 10-fold lower than ELISA. Pathological prion protein scrapie(PrPSc), implicated in transmissible spongiform encepha-lopathies, is detected primarily by Western blot or ELISAto screen the brainstem in cattle. Using the I-PCR assay,recombinant hamster PrPC could be detected consistentlyat 1 fg ml21, and proteinase K-digested scrapie-infectedhamster brain homogenates diluted to 1028 (approx. 10–100 infectious units) were detected with a semi-quantitativedose–response (Barletta et al., 2005). The level of detectionwas 1 million-fold more sensitive than the levels detected byWestern blot or ELISA, and these researchers recommendedI-PCR as a method capable of detecting PrPSc in the pre-clinical phase of infection. Furthermore, their findingssuggested that, unless complete proteinase K digestion ofPrPC in biological materials is verified, an ultrasensitiveI-PCR assay might inaccurately classify a sample as positive.A sensitive PD-I-PCR assay has also been documented forthe detection of PrP as well as purified Hantaan virusnucleocapsid protein by indirect, sandwich and real-timePD-I-PCR formats with enhanced detection sensitivitycompared with PD-ELISA (Guo et al., 2006).
As explained above, background signals are a critical factorin I-PCR assays, particularly when using complex proteinsuspensions like PrP. Recently, Kuczius et al. (2012) wereable to reduce background noise by including two heatingsteps, the first for protein denaturation and the second fordetachment of immunocomplexed DNA, enabling optimalDNA amplification with an increased detection sensitivityfor the simultaneous detection of PrPC and central nervoussystem indicators.
Detection of bacterial antigens
Rapid and accurate diagnostic tests are required by theclinical laboratories for early diagnosis of bacterial infec-tions, especially for slow-growing and fastidious bacteriathat are difficult to isolate. In recent years, I-PCR assayshave been reasonably exploited to detect important bacterialantigens.
Kakizaki et al. (1996) earlier demonstrated an indirect I-PCR assay for the detection of the fish pathogen Pasteurellapiscicida in naturally infected yellowtail. The assay detected3.4 c.f.u. ml21 compared with 3.46104 c.f.u. ml21 detectedwith ELISA.
E. coli b-glucuronidase is a specific marker used to identifyE. coli in a variety of diverse samples such as urine andfood. Chang & Huang (1997) documented sandwichI-PCR for the detection of glucuronidase with an LOD of
Detection of potential microbial antigens by immuno-PCR
http://jmm.sgmjournals.org 635

1610217 g ml21 (equivalent to 40 glucuronidase mole-cules ml21), which was 108 times more sensitive thanELISA. The utility of I-PCR method as a generic strategyfor rapid screening of fruit juices and plant products forthe detection of enterohaemorrhagic E. coli O157 : H7 hasalso been demonstrated (Ogunjimi & Choudary, 1999).
Zhang et al. (1998) developed a modified I-PCR assay,which was a combination of indirect I-PCR and in situI-PCR for the detection of Bacteroides fragilis using a mAbthat recognizes a specific epitope in the LPS present inmany strains of Bacteroides fragilis. Interestingly, bacterialcells were coated on an in situ PCR slide rather than on amicrotitre plate and an indirect I-PCR format was thenfollowed, with an LOD of 104 c.f.u. ml21.
The use of biological toxins by terrorists and in warfare isan emerging threat. Rapid identification of these biothreatsby an ultrasensitive method like I-PCR is essential forappropriate treatment and containment. These toxin bio-threats primarily include botulinum toxin (neurotoxin)produced by Clostridium botulinum, Staphylococcus aureusenterotoxin B (SEB) and ricin produced by castor beanplant (Allen et al., 2006; He et al., 2010). For the detectionof C. botulinum neurotoxin A (BoNT/A), Wu et al. (2001)demonstrated a direct I-PCR assay with reasonably gooddetection sensitivity, in which SMCC was used as a linkermolecule to conjugate anti-BoNT/A mAb with the reporterDNA through a covalent binding. However, by usingindirect I-PCR and indirect sandwich I-PCR formats, Chaoet al. (2004) documented an improved LOD for BoNT/A ofup to 50 fg with both the assays, which was 104–105-foldlower than with ELISA. In contrast to the conventional useof polyclonal antibody as the capture antibody in sandwichI-PCR format, these researchers used anti-BoNT/A mAb asthe capture antibody because of its higher binding affinityfor BoNT/A toxin.
Staphylococcus aureus releases a potent virulence factor,protein A, which is well known to inhibit phagocytosisvia binding of the immunoglobulin Fc fragment. Huang &Chang (2004) described a sandwich I-PCR to identifyprotein A at 10217 g ml21 (144 antigen molecules ml21).In addition to their role in food poisoning, Staphylococcusaureus enterotoxins have significant effects on the immunesystem, as these are members of the family of pyrogenictoxin superantigens (Rajkovic et al., 2006; Fischer et al.,2007). A quantitative real-time I-PCR assay was developedby Rajkovic et al. (2006) for the detection of SEB in pureculture and various foods. The LOD of their I-PCR was lessthan 10 pg ml21, which was approximately 103-fold lowerthan ELISA and Vidas set-2 assays. Fischer et al. (2007)documented a quantitative real-time I-PCR assay for thedetection of SEB and Staphylococcus aureus enterotoxin A(SEA), in which there was covalent binding between thereporter DNA and secondary detection antibodies throughthe chemical linker N-succinimidyl-S-actyl-thioacetate.This assay could detect Staphylococcus aureus enterotoxinsin quantities as low as 0.6 pg for SEB and 6 pg for SEA.
Bacillus thuringiensis var. kurstaki serves as an importantreservoir of Cry1Ac toxin protein for the productionof biological insecticides and insect-resistant geneticallymodified crops (Romeis et al., 2006). The gene encodingthis toxin has also been genetically transformed into cropplants. A reliable method for detecting the Cry1Ac toxinwould facilitate monitoring for this protein in crop andnon-crop plants, as well as in foods from the transgenicplants. Allen et al. (2006) introduced an I-PCR assay for thedetection of purified Cry1Ac protein in which a polyclonalantibody was used as the detection antibody, which wascovalently coupled to a reporter DNA through SMCCon two different surfaces: (i) polyvinyl chloride microtitreplates and (ii) streptavidin-coated particles. In the poly-vinyl chloride microtitre plate, a direct I-PCR format wasfollowed, whilst in the second method, Cry1Ac protein wasconjugated directly onto the streptavidin-coated particlesand an I-PCR assay was then performed. Interestingly, thesensitivity of the polyvinyl chloride microtitre plate for thedetection of Cry1Ac protein was found to be higher thanthe streptavidin-coated particles.
Streptococcus pyogenes (also known as group A Streptoco-ccus) is associated with a number of infections in humans,including acute pharyngitis, pneumonia, sepsis, acuterheumatic fever and post-streptococcal glomerulonephritis(Musser et al., 1995). For the detection of Streptococcuspyogenes, two I-PCR formats were exploited by Liang et al.(2003). In the first format, streptavidin was used as a linkermolecule between the biotinylated reporter DNA and thebiotinylated anti-group A Streptococcus mAb with an LODof 1023 bacterial cells per well, which was much lower thancommercial Strep A test kits with LOD ranges of 16102–56105 bacterial cells per test. In the second format, therewas a covalent conjugation of the anti-group A mAb withthe reporter DNA through SMCC with an LOD of 10 cellsper well.
The Shiga toxin (Stx)-producing E. coli (STEC) serotypeO157 : H7 and several non-O157 : H7 serotypes have emergedworldwide, and cause diarrhoea, haemorrhagic colitisand the life-threatening haemolytic–uraemic syndrome inhumans (Zhang et al., 2008; He et al., 2011). STEC serotypesin the environment have been reported frequently, butdetection of STEC in these environmental samples remainsdifficult because the numbers of bacteria are often below thedetection threshold of the methods employed. Enzymeimmunoassay (EIA) fails to detect many of the Stx2 strainsbecause of the production of Stx2 below the level ofdetection (Zhang et al., 2005). A direct I-PCR was developedby Zhang et al. (2008) to detect purified Stx2 with enhanceddetection sensitivity. Moreover, this assay could detect Stx2and variants in STEC strains that produce the toxins at levelsthat are non-detectable by EIA and in EIA-negative enrichedstool cultures from patients. He et al. (2011) demonstrated areal-time I-PCR assay for the detection of Stx2 and variantsin environmental samples contaminated with soil, faeces andwater samples, whilst ELISA could not detect Stx2 in thesecontaminated samples. The quantitative detection limit of
P. K. Mehta and others
636 Journal of Medical Microbiology 63

the assay was 0.1 pg ml21 in buffer with a quantificationrange of 10–100 000 pg ml21, which was 10 000-foldmore sensitive than ELISA. Furthermore, there was directattachment of streptavidin-conjugated detection antibody tothe biotinylated DNA rather than using streptavidin as abridge between the biotinylated detection antibody and thebiotinylated DNA, which also facilitated reducing the non-specific signal.
Although outbreaks of haemolytic–uraemic syndrome dueto the consumption of dairy products occur frequently, fewreports are available on assays for the detection of Stx2in milk. A highly sensitive real-time I-PCR assay capableof detecting 0.01 pg Stx2a ml21 in milk has recently beenintroduced using mAb pair Stx2-1 and Stx2-2 (He et al.,2013). Such an assay can be a useful method for the routinediagnosis of Stx2 contamination in the milk-productionprocess, thus reducing the risk of STEC outbreaks.
Over the last two decades, the resurgence of tuberculosishas been documented throughout the world, and muchof this increase has coincided with HIV epidemics. An earlydiagnosis of tuberculosis is needed to initiate an early anti-tubercular therapy to avoid unnecessary morbidity andmortality (Mehta et al., 2012a). Recently, we developed anindirect sandwich I-PCR assay for the detection of Myco-bacterium tuberculosis specific region-of-difference (RD)antigens: early secretory antigenic target-6 (ESAT-6; Rvnumber Rv3875), culture filtrate protein-10 (CFP-10;Rv3874), mycobacterial protein from species tuberculosis(MPT-64; Rv1980c) and culture filtrate protein-21 (CFP-21; Rv1984c) in biological specimens of pulmonary aswell as extrapulmonary tuberculosis patients (Mehtaet al., 2012b). We could detect up to 0.1 fg purified M.tuberculosis RD antigens by I-PCR, which was 107 timesmore sensitive than an analogous ELISA.
Xylella fastidiosa, a bacterial plant pathogen, is known tocause significant losses in many economically importantcrops. Peroni et al. (2008) compared ELISA, PCR andimmuno-capture PCR assays, as well as I-PCR, to detect X.fastidiosa for an accurate diagnosis of Citrus variegatedchlorosis. As expected, I-PCR revealed the maximumsensitivity among the four assays examined with an LODof only 10 bacterial cells, whilst the LOD with the otherassays was 106 bacterial cells.
Palaeomicrobiology allows the identification of causativeagents of ancient diseases, their geographical distributionand study of the genetic evolution of micro-organisms(Tran et al., 2011). Molecular biology techniques are com-monly used to detect micro-organism DNA, but the riskof contamination, chemical modification/fragmentationof DNA and presence of PCR inhibitors in ancient sampleslimit the ability to detect ancient infections. However,good conservation of proteins has been described recentlywith the classification of mammalian species using thedental pulp of modern and ancient individuals, which ledthe researchers to explore alternative methods based onantigenic protein detection. Malou et al. (2012b) devised
an I-PCR method for the detection of Yersinia pestis proteinantigens in ancient dental pulp specimens collected fromBlack Death victims. This method could detect proteinconcentrations 70 times lower than ELISA. Moreover, I-PCRwas the most sensitive method, followed by PCR and ELISA,and a combination of these methods increased the overallcapacity to identify Y. pestis in ancient dental pulp specimens.
Detection of parasitic antigens
Angiostrongylus cantonensis causes angiostrongyliasis, themost common cause of eosinophilic meningitis andmeningoencephalitis in South-east Asia and the PacificBasin (Baheti et al., 2008). A reliable diagnostic measure-ment of infection with A. cantonensis is difficult, because theparasitic nematode is unidentified in the cerebrospinal fluidof most of the afflicted patients and the sensitivity of ELISAfor circulating worm antigens in patient sera is quite low(Chye et al., 1997). Chye et al. (2004) exploited a sandwich I-PCR assay in which both capture antibody and the detectionantibody were mAbs for the detection of circulating 204 kDaAcL5 (fifth-stage larvae) antigens of A. cantonensis in theserum of patients with eosinophilic meningitis/meningoen-cephalitis with 100 % specificity. In contrast to ELISA, thesensitivity of I-PCR was also 100 % for patients known tohave the parasitic worm in the cerebrospinal fluid and 97 %for patients who displayed only clinical syndromes. A single150 bp DNA band was routinely observed with 0.1 ngpurified antigen l21 with I-PCR, whilst ELISA could notdetect antigen concentrations .10 ng l21, thus demonstrat-ing that I-PCR was 102–103 times more sensitive than ELISA.
Detection of mycotoxins
Aflatoxin B1 is a natural mycotoxin that enters the foodchain by contamination of food grains and feedstuffs,potentially posing carcinogenic risks to animal and humanhealth. I-PCR has the potential to direct the need of meetingthe regulatory limits by detecting trace levels of aflatoxinpresent in food and animal feeds. Babu & Muriana (2011)described a real-time I-PCR assay for the quantification ofaflatoxin B1 suspended in a methanol : water solution thatcould also serve as an extraction solvent. I-PCR formatsincluded direct sandwich versus indirect sandwich assaysusing mAbs versus polyclonal antibodies. Their assay coulddemonstrate the sensitive detection of aflatoxin B1 from10 p.p.b. down to 0.1 p.p.b. and represents a useful modelsystem that can easily be adapted for aflatoxin detection ina variety of food or animal feed samples using a simplemethanol : water solution as an extraction solvent.
Some of the various microbial antigens detected by I-PCRassays are summarized in Table 1.
Detection of antibodies to microbial antigens
Unlike microbial antigen detection, there are few reportsavailable for the detection of circulating antibodies tomicrobial antigens by I-PCR assay. Mweene et al. (1996)
Detection of potential microbial antigens by immuno-PCR
http://jmm.sgmjournals.org 637

demonstrated an I-PCR assay for the detection of anti-bodies against bovine herpes virus type 1 in experimentallyinfected calf/rabbit serum samples with a 105-fold highersensitivity than ELISA.
The detection of mumps-specific IgG demonstrates animportant role in immunity surveillance, evaluating theefficacy of vaccination programmes, identifying susceptiblecohorts in the population and designing future vaccinationstrategies. However, this poses a problem when measuringmumps-specific IgG prevalence in populations in whichmumps virus infection is controlled by MMR (measles,mumps and rubella) vaccination. A quantitative real-timeI-PCR assay was developed by McKie et al. (2002a, b)for the detection of mumps-specific IgG antibodies. Theantibody responses to vaccination were lower than thosefollowing infection with WT virus, and the levels of antibodyin the population were no longer boosted by continuingcirculation of the virus. In contrast to results from otherlaboratories, the sensitivity and specificity of their real-timeI-PCR assay did not exceed that of ELISA.
The most commonly used methods for the diagnosis of Qfever are serological tests, and the interpretation of thesetests depends on the phenomenon of phase variationexhibited by Coxiella burnetii with a partial loss of LPS. Thelag phase in antibody response of 7–15 days after clinicalmanifestations is the major drawback regarding serologicaldiagnosis of acute Q fever. Recently, Malou et al. (2012a)compared the efficacy of I-PCR, PCR, ELISA and immuno-fluorescent antibody tests for the diagnosis of Q fever by thedetection of phase II IgM anti-Coxiella burnetii. It was foundthat the highest sensitivity was achieved with I-PCR (87 %),followed by PCR (58 %), ELISA (38 %) and immunofluor-escent antibody (32 %) tests.
Lyme disease caused by the slow-growing Borrelia burgdorferiis the fastest-growing zoonotic disease. Current assays for thedetection of Borrelia burgdorferi infection are challenged bythe standardization of antigen source and analysis subjec-tiveness. Therefore, Halpern et al. (2013) exploited an I-PCRassay using recombinant in vivo-expressed Borrelia burgdor-feri antigens for the detection of a host immune response toBorrelia burgdorferi infection. The I-PCR format was a liquid-phase protein detection method using magnetic beads coatedwith intact spirochaetes, which provided an effective antigenpresentation and demonstrated the detection of host-generated antibodies in experimentally infected mice at day11 post-inoculation, whereas host-generated antibodies weredetected at day 14 post-inoculation by ELISA and day 21post-inoculation by Western blotting. Furthermore, mag-netic beads coated with the recombinant Borrelia burgdorferiin vivo-expressed antigen outer surface protein C or Borreliamembrane protein A documented positive detection of host-generated antibodies in mice at day 7 post-inoculation withnoticeably enhanced I-PCR signals above the background,thus suggesting that their assay had the potential for thesensitive detection of multiple host-response antibodies andisotypes to Borrelia burgdorferi infection.
We recently developed an indirect I-PCR assay for thedetection of anti-ESAT-6, anti-CFP-10, anti-MPT-64 andanti-CFP-21 antibodies in the sera of pulmonary tuber-culosis patients. However, paired sample analysis, that is,the detection of M. tuberculosis RD antigens and anti-RDantibodies together, in the biological samples of pulmonarytuberculosis patients by I-PCR assay exhibited bettersensitivity and specificity compared with the detection ofRD antigens or anti-RD antibodies (Mehta et al., 2012b).
As well as detecting microbial antigens and antibodiesin various infectious diseases, I-PCR assays have also beenexploited for the diagnosis of non-infectious diseases, forexample, for the detection of phosphorylated Tau epitopesand amyloid peptide biomarkers from the neurons forearly diagnosis of Alzheimer’s disease (Singer et al., 2009;Hashimoto et al., 2012).
Sensitivity and specificity are critical factors in I-PCRassays. Although I-PCR assays have been well documentedfor the ultralow detection of target antigens in biologicalspecimens, these assays are vulnerable to sense-non-specificsignals and compromise over the accuracy of the test. Thismethod remains unused as a routine clinical diagnostic test,as it does not meet the high criterion standards establishedby the Clinical and Laboratory Standards Institute requiring99 % sensitivity and specificity (levels easily achieved byELISA). The most serious problem preventing high sen-sitivity and specificity of I-PCR remains the high back-ground noise due to non-specific binding and the samplematrix effect, which can be resolved to some extent by theexploitation of a liquid format using a nanoparticle-basedassay.
Conclusion
This review has described the utility of the ultrasensitive I-PCR assay for the detection of potential microbial antigens,demonstrating its wide applications for early diagnosis ofinfectious diseases, especially for viral and prion infections,as well as for slow-growing and fastidious bacteria that aredifficult to isolate. Although I-PCR assays are mostlyrestricted to target proteins, we can detect any non-nucleicacid molecule, such as lipids or carbohydrates. To furtherimprove the I-PCR assay, a reduction in background noisein complex biological matrices, automation and a reduc-tion in various steps, as well as the use of diminutive formsof protocols, is needed. There are several formats of I-PCR assays; the preferred is the sandwich real-time I-PCR,followed by magnetic bead/GNP-based real-time I-PCR.The use of magnetic beads/GNPs might provide a concretesolution to the reduction in background noise, the esta-blishment of automated one-step I-PCR and hence areduction of the overall duration of the assay. Although thecost of I-PCR assays is high, the applicability of these assayshas been encouraged in recent years due to their ultra-sensitivity and robustness, which is required for the earlydetection of infectious as well as non-infectious diseases.However, further work is required to develop a reliable and
P. K. Mehta and others
638 Journal of Medical Microbiology 63

cost-effective I-PCR assay that can be used for routine usein resource-poor settings.
Acknowledgements
The financial assistance provided by the University Grant Commission,New Delhi, and the Department of Science and Technology, NewDelhi, to carry out this work is acknowledged.
References
Adams, N. M., Jackson, S. R., Haselton, F. R. & Wright, D. W.(2012). Design, synthesis, and characterization of nucleic-acid-functionalized gold surfaces for biomarker detection. Langmuir 28,1068–1082.
Adler, M., Schulz, S., Fischer, R. & Niemeyer, C. M. (2005). Detectionof Rotavirus from stool samples using a standardized immuno-PCR(‘‘Imperacer’’) method with end-point and real-time detection.Biochem Biophys Res Commun 333, 1289–1294.
Adler, M., Wacker, R. & Niemeyer, C. M. (2008). Sensitivity bycombination: immuno-PCR and related technologies. Analyst (Lond)133, 702–718.
Allen, R. C., Rogelj, S., Cordova, S. E. & Kieft, T. L. (2006). Animmuno-PCR method for detecting Bacillus thuringiensis Cry1Actoxin. J Immunol Methods 308, 109–115.
Babu, D. & Muriana, P. M. (2011). Immunomagnetic bead-basedrecovery and real time quantitative PCR (RT iq-PCR) for sensitivequantification of aflatoxin B1. J Microbiol Methods 86, 188–194.
Baheti, N. N., Sreedharan, M., Krishnamoorthy, T., Nair, M. D. &Radhakrishnan, K. (2008). Eosinophilic meningitis and an ocularworm in a patient from Kerala, south India. J Neurol NeurosurgPsychiatry 79, 271.
Barletta, J. M., Edelman, D. C. & Constantine, N. T. (2004). Loweringthe detection limits of HIV-1 viral load using real-time immuno-PCRfor HIV-1 p24 antigen. Am J Clin Pathol 122, 20–27.
Barletta, J. M., Edelman, D. C., Highsmith, W. E. & Constantine, N. T.(2005). Detection of ultra-low levels of pathologic prion protein inscrapie infected hamster brain homogenates using real-time immuno-PCR. J Virol Methods 127, 154–164.
Barletta, J., Bartolome, A. & Constantine, N. T. (2009). Immuno-magnetic quantitative immuno-PCR for detection of less than oneHIV-1 virion. J Virol Methods 157, 122–132.
Cao, Y., Kopplow, K. & Liu, G. Y. (2000). In-situ immuno-PCR todetect antigens. Lancet 356, 1002–1003.
Chang, T. C. & Huang, S. H. (1997). A modified immuno-polymerasechain reaction for the detection of b-glucuronidase from Escherichiacoli. J Immunol Methods 208, 35–42.
Chao, H. Y., Wang, Y. C., Tang, S. S. & Liu, H. W. (2004). A highlysensitive immuno-polymerase chain reaction assay for Clostridiumbotulinum neurotoxin type A. Toxicon 43, 27–34.
Chen, L., Wei, H., Guo, Y., Cui, Z., Zhang, Z. & Zhang, X. E. (2009).Gold nanoparticle enhanced immuno-PCR for ultrasensitive detec-tion of Hantaan virus nucleocapsid protein. J Immunol Methods 346,64–70.
Chye, S. M., Yen, C. M. & Chen, E. R. (1997). Detection of circulatingantigen by monoclonal antibodies for immunodiagnosis of angios-trongyliasis. Am J Trop Med Hyg 56, 408–412.
Chye, S. M., Lin, S. R., Chen, Y. L., Chung, L. Y. & Yen, C. M. (2004).Immuno-PCR for detection of antigen to Angiostrongylus cantonensiscirculating fifth-stage worms. Clin Chem 50, 51–57.
Deng, M. J., Xiao, X. Z., Zhang, Y. M., Wu, X. H., Zhu, L. H., Xin, X. Q. &Wu, D. L. (2011a). A highly sensitive immuno-PCR assay for detectionof H5N1 avian influenza virus. Mol Biol Rep 38, 1941–1948.
Deng, M., Long, L., Xiao, X., Wu, Z., Zhang, F., Zhang, Y., Zheng, X.,Xin, X., Wang, Q. & Wu, D. (2011b). Immuno-PCR for one step
detection of H5N1 avian influenza virus and Newcastle disease virus
using magnetic gold particles as carriers. Vet Immunol Immunopathol141, 183–189.
Ding, Y. Z., Liu, Y. S., Zhou, J. H., Chen, H. T., Zhang, J., Ma, L. N. & Wei,G. (2011). A highly sensitive detection for foot-and-mouth diseasevirus by gold nanopariticle improved immuno-PCR. Virol J 8, 148.
Fischer, A., von Eiff, C., Kuczius, T., Omoe, K., Peters, G. & Becker, K.(2007). A quantitative real-time immuno-PCR approach for detection
of staphylococcal enterotoxins. J Mol Med (Berl) 85, 461–469.
Glass, R. I., Bresee, J., Jiang, B., Gentsch, J., Ando, T., Fankhauser,R., Noel, J., Parashar, U., Rosen, B. & Monroe, S. S. (2001).Gastroenteritis viruses: an overview. Novartis Found Symp 238, 5–19,
discussion 19–25.
Gofflot, S., El Moualij, B., Zorzi, D., Melen, L., Roels, S., Quatpers, D.,Grassi, J., Vanopdenbosch, E., Heinen, E. & Zorzi, W. (2004).Immuno-quantitative polymerase chain reaction for detection andquantitation of prion protein. J Immunoassay Immunochem 25, 241–
258.
Gofflot, S., Deprez, M., El Moualij, B., Osman, A., Thonnart, J. F.,Hougrand, O., Heinen, E. & Zorzi, W. (2005). Immunoquantitative
PCR for prion protein detection in sporadic Creutzfeldt–Jakobdisease. Clin Chem 51, 1605–1611.
Griffiths, G., Nystrom, B., Sable, S. B. & Khuller, G. K. (2010).Nanobead-based interventions for the treatment and prevention oftuberculosis. Nat Rev Microbiol 8, 827–834.
Guo, Y. C., Zhou, Y. F., Zhang, X. E., Zhang, Z. P., Qiao, Y. M., Bi, L. J.,Wen, J. K., Liang, M. F. & Zhang, J. B. (2006). Phage display mediated
immuno-PCR. Nucleic Acids Res 34, e62.
Halpern, M. D., Jain, S. & Jewett, M. W. (2013). Enhanced detection ofhost response antibodies to Borrelia burgdorferi using immuno-PCR.
Clin Vaccine Immunol 20, 350–357.
Hashimoto, M., Aoki, M., Winblad, B. & Tjernberg, L. O. (2012).A novel approach for Ab1–40 quantification using immuno-PCR.
J Neurosci Methods 205, 364–367.
He, X., McMahon, S., Henderson, T. D., II, Griffey, S. M. & Cheng,L. W. (2010). Ricin toxicokinetics and its sensitive detection in mouse
sera or feces using immuno-PCR. PLoS ONE 5, e12858.
He, X., Qi, W., Quinones, B., McMahon, S., Cooley, M. & Mandrell,R. E. (2011). Sensitive detection of Shiga toxin 2 and some of itsvariants in environmental samples by a novel immuno-PCR assay.
Appl Environ Microbiol 77, 3558–3564.
He, J., Evers, D. L., O’Leary, T. J. & Mason, J. T. (2012). Immunoliposome-PCR: a generic ultrasensitive quantitative antigen detection system.
J Nanobiotechnology 10, 26.
He, X., McMahon, S., Skinner, C., Merrill, P., Scotcher, M. C. &Stanker, L. H. (2013). Development and characterization of mono-
clonal antibodies against Shiga toxin 2 and their application for toxindetection in milk. J Immunol Methods 389, 18–28.
Huang, S. H. & Chang, T. C. (2004). Detection of Staphylococcus
aureus by a sensitive immuno-PCR assay. Clin Chem 50, 1673–1674.
Kakizaki, E., Yoshida, T., Kawakami, H., Oseto, M., Sakai, T. & Sakai,M. (1996). Detection of bacterial antigens using immuno-PCR. LettAppl Microbiol 23, 101–103.
Kuczius, T., Becker, K., Fischer, A. & Zhang, W. (2012). Simultaneous
detection of three CNS indicator proteins in complex suspensionsusing a single immuno-PCR protocol. Anal Biochem 431, 4–10.
Detection of potential microbial antigens by immuno-PCR
http://jmm.sgmjournals.org 639

Liang, H., Cordova, S. E., Kieft, T. L. & Rogelj, S. (2003). A highly
sensitive immuno-PCR assay for detecting Group A Streptococcus.J Immunol Methods 279, 101–110.
Maia, M., Takahashi, H., Adler, K., Garlick, R. K. & Wands, J. R. (1995).Development of a two-site immuno-PCR assay for hepatitis B surface
antigen. J Virol Methods 52, 273–286.
Malou, N. & Raoult, D. (2011). Immuno-PCR: a promising ultra-sensitive diagnostic method to detect antigens and antibodies. Trends
Microbiol 19, 295–302.
Malou, N., Renvoise, A., Nappez, C. & Raoult, D. (2012a). Immuno-
PCR for the early serological diagnosis of acute infectious diseases: the
Q fever paradigm. Eur J Clin Microbiol Infect Dis 31, 1951–1960.
Malou, N., Tran, T. N., Nappez, C., Signoli, M., Le Forestier, C., Castex,D., Drancourt, M. & Raoult, D. (2012b). Immuno-PCR – a new tool for
paleomicrobiology: the plague paradigm. PLoS ONE 7, e31744.
Matsushita, T., Shirasaki, N., Tatsuki, Y. & Matsui, Y. (2013).Investigating norovirus removal by microfiltration, ultrafiltration,and precoagulation-microfiltration processes using recombinant
norovirus virus-like particles and real-time immuno-PCR. Water
Res 47, 5819–5827.
McKie, A., Samuel, D., Cohen, B. & Saunders, N. A. (2002a). A
quantitative immuno-PCR assay for the detection of mumps-specific
IgG. J Immunol Methods 270, 135–141.
McKie, A., Samuel, D., Cohen, B. & Saunders, N. A. (2002b).Development of a quantitative immuno-PCR assay and its use to
detect mumps-specific IgG in serum. J Immunol Methods 261, 167–175.
Mehta, P. K., Raj, A., Singh, N. & Khuller, G. K. (2012a). Diagnosis ofextrapulmonary tuberculosis by PCR. FEMS Immunol Med Microbiol
66, 20–36.
Mehta, P. K., Kalra, M., Khuller, G. K., Behera, D. & Verma, I. (2012b).Development of an ultrasensitive polymerase chain reaction-amp-
lified immunoassay based on mycobacterial RD antigens: implications
for the serodiagnosis of tuberculosis. Diagn Microbiol Infect Dis 72,166–174.
Monjezi, R., Tan, S. W., Tey, B. T., Sieo, C. C. & Tan, W. S. (2013).Detection of hepatitis B virus core antigen by phage display mediated
TaqMan real-time immuno-PCR. J Virol Methods 187, 121–126.
Morin, I. & Schaeffer, P. M. (2012). Combining RNA–DNA swappingand quantitative polymerase chain reaction for the detection of
influenza A nucleoprotein. Anal Biochem 420, 121–126.
Musser, J. M., Kapur, V., Szeto, J., Pan, X., Swanson, D. S. & Martin,D. R. (1995). Genetic diversity and relationships among Streptococcus
pyogenes strains expressing serotype M1 protein: recent intercontin-ental spread of a subclone causing episodes of invasive disease. Infect
Immun 63, 994–1003.
Mweene, A. S., Ito, T., Okazaki, K., Ono, E., Shimizu, Y. & Kida, H.(1996). Development of immuno-PCR for diagnosis of bovine
herpesvirus 1 infection. J Clin Microbiol 34, 748–750.
Nam, J. M., Thaxton, C. S. & Mirkin, C. A. (2003). Nanoparticle-based
bio-bar codes for the ultrasensitive detection of proteins. Science 301,
1884–1886.
Niemeyer, C. M., Adler, M. & Blohm, D. (1997). Fluorometric
polymerase chain reaction (PCR) enzyme-linked immunosorbent
assay for quantification of immuno-PCR products in microplates.Anal Biochem 246, 140–145.
Niemeyer, C. M., Adler, M. & Wacker, R. (2005). Immuno-PCR: highsensitivity detection of proteins by nucleic acid amplification. Trends
Biotechnol 23, 208–216.
Niemeyer, C. M., Adler, M. & Wacker, R. (2007). Detecting antigens byquantitative immuno-PCR. Nat Protoc 2, 1918–1930.
Ogunjimi, A. A. & Choudary, P. V. (1999). Adsorption of endogenous
polyphenols relieves the inhibition by fruit juices and fresh produce of
immuno-PCR detection of Escherichia coli O157 : H7. FEMS Immunol
Med Microbiol 23, 213–220.
Ozaki, H., Sugita, S. & Kida, H. (2001). A rapid and highly sensitive
method for diagnosis of equine influenza by antigen detection using
immuno-PCR. Jpn J Vet Res 48, 187–195.
Perez, J. W., Vargis, E. A., Russ, P. K., Haselton, F. R. & Wright, D. W.(2011). Detection of respiratory syncytial virus using nanoparticle
amplified immuno-polymerase chain reaction. Anal Biochem 410,
141–148.
Perez, J. W., Adams, N. M., Zimmerman, G. R., Haselton, F. R. &Wright, D. W. (2013). Detecting respiratory syncytial virus using
nanoparticle-amplified immuno-PCR. Methods Mol Biol 1026, 93–
110.
Peroni, L. A., Reis, J. R., Coletta-Filho, H. D., de Souza, A. A.,Machado, M. A. & Stach-Machado, D. R. (2008). Assessment of the
diagnostic potential of immmunocapture-PCR and immuno-PCR for
Citrus variegated chlorosis. J Microbiol Methods 75, 302–307.
Potuckova, L., Franko, F., Bambouskova, M. & Draber, P. (2011).Rapid and sensitive detection of cytokines using functionalized gold
nanoparticle-based immuno-PCR, comparison with immuno-PCR
and ELISA. J Immunol Methods 371, 38–47.
Rajkovic, A., El Moualij, B., Uyttendaele, M., Brolet, P., Zorzi, W.,Heinen, E., Foubert, E. & Debevere, J. (2006). Immunoquantitative
real-time PCR for detection and quantification of Staphylococcus
aureus enterotoxin B in foods. Appl Environ Microbiol 72, 6593–
6599.
Romeis, J., Meissle, M. & Bigler, F. (2006). Transgenic crops expressing
Bacillus thuringiensis toxins and biological control. Nat Biotechnol 24,
63–71.
Sano, T., Smith, C. L. & Cantor, C. R. (1992). Immuno-PCR: very
sensitive antigen detection by means of specific antibody–DNA
conjugates. Science 258, 120–122.
Shirasaki, N., Matsushita, T., Matsui, Y., Oshiba, A. & Ohno, K.(2010). Estimation of norovirus removal performance in a coagu-
lation–rapid sand filtration process by using recombinant norovirus
VLPs. Water Res 44, 1307–1316.
Sims, P. W., Vasser, M., Wong, W. L., Williams, P. M. & Meng, Y. G.(2000). Immunopolymerase chain reaction using real-time polymer-
ase chain reaction for detection. Anal Biochem 281, 230–232.
Singer, D., Soininen, H., Alafuzoff, I. & Hoffmann, R. (2009). Immuno-
PCR-based quantification of multiple phosphorylated tau-epitopes
linked to Alzheimer’s disease. Anal Bioanal Chem 395, 2263–2267.
Tian, P. & Mandrell, R. (2006). Detection of norovirus capsid proteins
in faecal and food samples by a real time immuno-PCR method.
J Appl Microbiol 100, 564–574.
Tran, T. N., Aboudharam, G., Raoult, D. & Drancourt, M. (2011).Beyond ancient microbial DNA: nonnucleotidic biomolecules for
paleomicrobiology. Biotechniques 50, 370–380.
Wacker, R. (2006). Infect Dis Workshop 1. Immuno-PCR- maximizing
immunoassay sensitivity. In Chimera Biotec Association for Molecular
Pathology. Annual Meeting & Exhibition, Orlando, FL, USA, 17
November 2006.
Wacker, R., Ceyhan, B., Alhorn, P., Schueler, D., Lang, C. & Niemeyer,C. M. (2007). Magneto immuno-PCR: a novel immunoassay based on
biogenic magnetosome nanoparticles. Biochem Biophys Res Commun
357, 391–396.
Wu, H. C., Huang, Y. L., Lai, S. C., Huang, Y. Y. & Shaio, M. F. (2001).Detection of Clostridium botulinum neurotoxin type A using immuno-
PCR. Lett Appl Microbiol 32, 321–325.
P. K. Mehta and others
640 Journal of Medical Microbiology 63

Zhang, G., Pan, Q. & Weintraub, A. (1998). Identification of Bacteroidesfragilis by a modified immuno polymerase chain reaction (mIPCR),using a specific monoclonal antibody. Anaerobe 4, 189–196.
Zhang, W., Bielaszewska, M., Friedrich, A. W., Kuczius, T. & Karch,H. (2005). Transcriptional analysis of genes encoding Shiga toxin 2and its variants in Escherichia coli. Appl Environ Microbiol 71, 558–561.
Zhang, W., Bielaszewska, M., Pulz, M., Becker, K., Friedrich, A. W.,Karch, H. & Kuczius, T. (2008). New immuno-PCR assay for detectionof low concentrations of Shiga toxin 2 and its variants. J Clin Microbiol46, 1292–1297.
Zhou, H., Fisher, R. J. & Papas, T. S. (1993). Universal immuno-PCRfor ultra-sensitive target protein detection. Nucleic Acids Res 21, 6038–6039.
Detection of potential microbial antigens by immuno-PCR
http://jmm.sgmjournals.org 641




![[IJCST-V3I2P37]: Mantoj Kaur, Malti Rani, Tulika Mehta](https://img.pdfslide.net/doc/110x75/633221c74e01430403008629/ijcst-v3i2p37-mantoj-kaur-malti-rani-tulika-mehta.jpg)
























