Embed Size (px)
Citation preview

Methadone increases intracellular calcium in SH-SY5Y andSH-EP1-ha7 cells by activating neuronal nicotinic acetylcholinereceptors
Jukka S. Pakkanen,* Heli Nousiainen,* Jari Yli-Kauhaluoma,� Irene Kylanlahti,�Tommi Moykkynen,� Esa R. Korpi,� Jian-Hong Peng,§ Ronald J. Lukas,§ Liisa Ahtee*and Raimo K. Tuominen*,�
*Division of Pharmacology and Toxicology, �Viikki Drug Discovery Technology Center, Faculty of Pharmacy and �Institute of
Biomedicine, Pharmacology, Biomedicum Helsinki, University of Helsinki, Helsinki, Finland
§Division of Neurobiology, Barrow, Neurological Institute, Phoenix, Arizona, USA
Abstract
(–)-Methadone acts as an agonist at opioid receptors. Both
(+)- and (–)-enantiomers of methadone have been suggested
to be potent non-competitive antagonists of a3b4 neuronal
nicotinic acetylcholine receptors (nAChRs). In the present
study, we have examined interactions of methadone with
nAChRs by using receptor binding assays, patch-clamp
recording and calcium fluorometry imaging with SH-SY5Y
cells naturally expressing a7 and a3* nAChR subtypes and
SH-EP1-ha7 cells heterologously expressing human a7
nAChRs. Methadone potently inhibited binding of [3H]methyl-
lycaconitine to a7 nAChRs and that of [3H]epibatidine to a3*
nAChRs. Methadone pretreatment induced up-regulation of
epibatidine binding sites in SH-SY5Y cells. Using whole-cell
patch-clamp recording, both isomers of methadone activated
cation currents via mecamylamine-sensitive nAChRs in SH-
SY5Y cells. Nicotine and both (+)- and (–)-methadone evoked
increases in [Ca2+]i in both fluo-3AM loaded cell lines, and
these effects were blocked by mecamylamine and by the a7
selective antagonist methyllycaconitine, suggesting effects of
methadone as a7-nAChR agonist. Sensitivity of sustained
nicotine and methadone effects to blockade by CdCl2, ry-
anodine and xestospongin-c implicates voltage-operated Ca2+
channels and intracellular Ca2+ stores as downstream mod-
ulators of elevated [Ca2+]i. Collectively, our results suggest
that methadone engages in complex and potentially pharma-
cologically significant interactions with nAChRs.
Keywords: addiction, calcium fluorometry, nicotine, opioid.
J. Neurochem. (2005) 94, 1329–1341.
Neuronal nicotinic acetylcholine receptors (nAChRs) arepentameric ligand-gated ion channels, composed of a or aand b subunit combinations from a family of nine a (a2–a10) and three b (b2–b4) subunits. nAChRs are widelydistributed in the central nervous system as well asautonomic and sensory ganglia. In the brain, many of thesereceptors are presynaptic, and they modulate the release of anumber of neurotransmitters (Wonnacott 1997). Activationof nAChRs causes behavioural changes, which can be relatedto these modulatory effects. Nicotine affects locomotoractivity via altered dopaminergic activity, cognitive functionsvia cholinergic stimulation, and pain perception via alteredopioidergic and serotonergic activity (Gotti et al. 1997).
Nicotine shares the ability to activate the dopaminergicsystem with other addictive drugs, including opioids
(Stolerman and Jarvis 1995). Although it is evident that thesedrugs have their independent mechanisms of action, which are
Received March 11, 2005; revised manuscript received April 27, 2005;accepted April 29, 2005.Address correspondence and reprint requests to Jukka Pakkanen,
Division of Pharmacology and Toxicology, Faculty of Pharmacy, Uni-versity of Helsinki, Helsinki, POB 56, FI-00014, Finland.E-mail: [email protected] used: a-bgt, a-bungarotoxin; AChR(s), nicotinic acetyl-
choline receptor(s); [Ca2+]i, amount of intracellular calcium; DHbE,dihydro-b-erythroidine; DMSO, dimethylsulfoxide; FCS, foetal calfserum; 5-HT, 5-hydroxytryptamine; lit, literature; MLA, methylly-caconitine; mp, melting point; Nal, naloxone; Nic, nicotinic; NMDA,N-methyl-D-aspartate; PBS, phosphate-buffered saline; TSS, Tyrode’ssalt solution.
Journal of Neurochemistry, 2005, 94, 1329–1341 doi:10.1111/j.1471-4159.2005.03279.x
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341 1329

mediated through specific receptors, there are interactionsbetween nicotine and opioids. In humans, opioid receptorantagonists such as naltrexone have been reported to modulatecigarette consumption (Epstein and King 2004). Animalexperiments in vivo show that nicotine attenuates morphinewithdrawal induced by naloxone (Zarrindast and Farzin1996). This may be as a result of release of endogenousopioid peptides, induced by nicotinic receptor stimulation(Suh et al. 1995). Moreover, in l-opioid receptor knock-outmice, the rewarding effects of nicotine are reduced (Berren-dero et al. 2002). Evidence of an interaction at the molecularlevel comes from in vitro experiments where methadone, asynthetic l-opioid agonist, has been suggested to be a non-competitive antagonist of a3b4 nAChRs (Xiao et al. 2001).
Methadone exists as (+)- and (–)-isomers, of which(–)-methadone may be more important as an analgesic,because (–)-methadone but not (+)-methadone acts as anagonist at opioid receptors (Horng et al. 1976). In addition toopioid receptors, methadone seems to have other targets, as itinhibits the uptake of 5-HT and noradrenaline (Ahtee andSaarnivaara 1973), and both isomers of methadone blockNMDA receptors (Stringer et al. 2000).
The analgesic effects of nicotine have been known fordecades (Mattila et al. 1968). Epibatidine, an agonist at a4b2nAChRs, has been shown to be 200 times more potent thanmorphine in experimental pain models (Badio and Daly1994). In mice lacking a4 and b2 nAChR subunits, theanalgesic effects of nicotine are reduced (Marubio et al.1999). Choline, which is an agonist at the a7 nAChRsubtype, also has analgesic properties (Damaj et al. 2000).Release of neurotransmitters, such as acetylcholine, dopam-ine, glutamate, noradrenaline, c-aminobutyric acid and 5-HT,appears to play a critical role in producing the anti-nociceptive effects of nAChR activation (Wonnacott 1997).
As both methadone and nicotine have anti-nociceptiveproperties (Hildebrand et al. 1999; Marubio et al. 1999), itwas quite surprising to us that methadone acts as anantagonist at a3b4 nAChRs (Xiao et al. 2001). The aim ofthis study was to determine the effects of the isomers ofmethadone on different nAChR subtypes, and especiallywhether methadone acts as an antagonist at nAChRs otherthan the a3b4 subtype. To address these questions, twodifferent cell lines were used: SH-SY5Y and SH-EP1-ha7cells. SH-SY5Y cells naturally express several nAChRsubunits (a3, a5, a7, b2, b4) (Peng et al. 1997) as well asl- and d-opioid receptors (Kazmi and Mishra 1986). SH-EP1-ha7 cells are transfected with the a7 nAChR gene (Penget al. 1999; Zhao et al. 2003) and thus express homopenta-meric a7 nAChRs, but not other nAChRs. They are epithelialderivates of the SK-N-SH human line, with native expressionof l- and d-opioid receptors (Baumhaker et al. 1993). Ligandbinding studies were carried out to study the binding sites formethadone on nAChRs. The effects of chronic methadonetreatment on nAChRs were studied by using ligand binding.
In functional studies, changes in intracellular Ca2+ concen-tration and ion currents induced by nicotine and/or meth-adone were measured by using calcium fluorometry andwhole-cell patch clamp, respectively.
Materials and methods
Drugs and reagents
Tissue culture plasticware was from Nunc (Nalge Nunc, Roskilde,
Denmark). Media and media supplements were from Gibco BRL
(Paisley, UK), nifedipine, xestospongin-c, ryanodine (–)-nicotine
hydrogen tartrate, dihydro-b-erythroidine (DHbE) and mecamylam-
ine were purchased from Sigma (Steinheim, Germany).
(±)-Methadone hydrochloride was from Star, Tampere, Finland.
Methyllycaconitine (MLA) was purchased from Tocris (Bristol,
UK). Fluo-3AM was purchased from Molecular Probes (Eugene,
OR, USA) and stored at )20�C as stock solutions in dimethylsulf-
oxide (DMSO). [3H]Epibatidine was purchased from NENTM
(Boston, MA, USA). Specific activity of the batch was 48.0
Ci/mmol. [3H]MLA was purchased from Tocris Cookson Ltd
(Avonmouth, UK). Specific activity of the batch was 25.0 Ci/mmol.
Naloxone was from Endo laboratories (New York, NY, USA).
Nicotine used for chronic treatment of SH-SY5Y cells was made up
freshly and directly in Dulbecco’s modified Eagle’s medium
(DMEM) : Ham’s F12 supplemented medium on day 1 of drug
treatment. Phosphate-buffered saline (PBS) was composed of
150 mM NaCl, 8 mM K2HPO4, 2 mM KH2PO4, pH 7.4 and stored
at 4�C. Tyrode’s salt solution (TSS) was composed of 137 mM
NaCl, 2.7 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 0.2 mM NaH2CO3
and 5.5 mM glucose, pH was adjusted to 7.4 and stored at )20�C.Probenecid was prepared fresh every day at a stock concentration of
250 mM, and was used at a working concentration of 2.5 mM.
Probenecid acid was solubilized 1 : 1 in 1.0 N NaOH and TSS. The
pH of the loading medium was adjusted to 7.4 with 37% HCl.
Markwell A was composed of 189 mM Na2CO3, 100 mM NaOH,
5.7 mM KNa tartrate and 1% sodium dodecyl sulfate (SDS) and
stored at room temperature (20-23�C). SH-SY5Y cells were from
the European Collection of Cell Cultures (ECACC, Salisbury, UK).
SH-EP1-ha7 cells were created as described in Zhao et al. (2003).
Resolution of (±)-methadone
Preparation of (–)-methadone (–)-3-bromocamphor-8-sulfonateA mixture of (±)-methadone hydrochloride (8.3 g, 24 mmol) and
(–)-ammonium 3-bromocamphor-8-sulfonate (4.1 g, 12.5 mmol) in
80% ethanol (21 mL)was stirred for 30 min, and added portions-wise
to crushed ice (185 g) with vigorous stirring. The resulting precipitate
was filtered, washed with ice water, and dried to give the crude salt
(5.9 g, mp. 127–129�C). The collected crude salt was dissolved
in ethanol and precipitated with ice. The resulting (–)-methadone
(–)-3-bromocamphor-8-sulfonate was filtered, washed with cold
water, and dried in vacuo [4.8 g, mp. 132–134�C, lit. 135–138�C(Howe and Sletzinger 1949)].
Preparation of (+)-methadone (–)-tartrateThe concentrated filtrate from the preparation of (–)-methadone
(–)-3-bromocamphor-8-sulfonate was extracted with ethyl acetate
(3 · 50 mL). Four molar NaOH (12 mL) was added to the aqueous
1330 J. S. Pakkanen et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

phase at 5�C. Vigorous stirring was continued until the precipitate
was loose and distinctly granular. Precipitates were filtered, washed
with ice water, and dried to give the free methadone base consisting
of approximately 75% of (+)-methadone and 25% of (–)-methadone
[2.73 g, mp. 76�C, lit. 79–81�C (Elks and Ganellin 1990)]. The
crude (+)-methadone base was dissolved in 1-propanol (5.5 mL) at
70�C, and treated with D-(–)-tartaric acid monopotassium salt
(1.66 g, 8.82 mmol). The resulting mixture was cooled to 4�C.Precipitated salt was filtered, washed with cold 50% 1-propanol/n-hexane (3 mL) and n-hexane (2 · 5 mL), and dried to give the
crude salt (mp. 80–81�C). The mp. of the reference (+)-methadone
(–)-tartrate was 98�C. The crude salt was dissolved in methanol
(150 mL) at reflux, and water was added to dissolve salt completely.
The resulting solution was cooled, 2-propanol (20 mL) added, and
methanol evaporated slowly under reduced pressure. The crystals of
(+)-methadone (–)-tartrate formed upon cooling were filtered and
dried (3.23 g, mp. 99�C).
Preparation of (–)- and (+)-methadone hydrochloride
Both (–)-methadone (–)-3-bromocamphor-8-sulfonate and (+)-meth-
adone (–)-tartrate were separately treated with 4 M NaOH (1.5
equiv.) to give free (–)- and (+)-methadone base, respectively.
Aqueous phases were extracted with ethyl acetate. The organic
layers were washed with brine and dried with anhydrous Na2SO4.
The solutions of (–)- and (+)-methadone in ethyl acetate were
concentrated and treated with a 4-M solution of hydrogen chloride in
1,4-dioxane (2 equiv.). Both (–)- and (+)-methadone hydrochloride
were filtered and dried in vacuo. (–)-Methadone hydrochloride
1.6 g, mp. 241�C, lit. 237–239�C (Howe and Sletzinger 1949),
[a]25 ¼ )127� (c ¼ 1.0, H2O). (+)-Methadone hydrochloride 0.8 g,
mp. 241�C, lit. 243–244�C (Howe and Sletzinger 1949), [a]25 ¼+127� (c ¼ 1.0, H2O). The course of the resolution of
(±)-methadone was also monitored by means of capillary electro-
phoresis.
Cell culture
SH-SY5Y cellsCell culture was carried out as described in detail by Ridley et al.(2002). In brief, SH-SY5Y human neuroblastoma stock cultures
were routinely maintained in a Dulbecco’s modified Eagle
medium : Ham’s F12 (1 : 1) modified medium containing 1%
non-essential amino-acids and supplemented with foetal calf
serum (FCS, 15%), glutamine (2 mM), penicillin (50 IU/mL)
and streptomycin (50 lg/mL). Stock cultures were passaged
1 : 5 weekly. Cell cultures were discarded when passage number
was above 20. For calcium fluorometry, cultures were subcultured
and grown to confluency in 175-cm2 flasks containing 25 mL of
supplemented medium and maintained at 37�C in 5% CO2/
humidified air. For ligand binding assays, cells were grown near
to confluency in 24-well plates, coated with poly D-lysine (10
lg/mL), as described previously (Ridley et al. 2001). The cells
contained supplemented medium 1 mL/well. Drugs: nicotine
(10 lM), (+)- or (–)-methadone (1–100 lM) were made up in
cell culture medium and were placed 1 mL/well. After 3 days,
prior to radioligand binding assay, the cultures were washed six
times with warm medium (1 mL/well) over 4 h to remove all the
traces of drugs from the culture, as described previously with
chronic nicotine treatments (Ridley et al. 2001). Between the
washes the cells were kept at 37�C. For electrophysiology the
cells were split on 60-mm Petri dishes.
SH-EP1-ha7 cells
SH-EP1 cells were transfected with the human nAChR a7 subunit
gene (Zhao et al. 2003). SH-EP1-ha7 cells were grown in
Dulbecco’s modified Eagle’s medium supplemented with horse
serum (10%), penicillin (100 IU/mL), streptomycin (100 lg/mL)
and amphotericin B (0.25 lg/mL) (Calbiochem, Darmstadt,
Germany, otherwise all the supplements were from the same sources
as in SH-SY5Y cells), hygromycin (0.25 mg/mL), and 5% FCS in a
humidified atmosphere containing 5% CO2 in air at 37�C (Peng
et al. 1999). Untransfected SH-EP1 cells were grown in the same
medium without hygromycin. SH-EP1-ha7 and SH-EP1 cells were
split weekly �1 : 50. Passage numbers 11–20 were used in assays.
Ligand binding
Competition binding assaysSH-SY5Y and SH-EP1-ha7 cell membranes were incubated with
serial dilutions of methadone and [3H]MLA (2 nM) or [3H]epibat-
idine (150 pM). [3H]Epibatidine binding was carried out by using
96-well plates (Millipore, MultiscreenTM-FC Opaque Plates,
Moisheim, France). To reduce non-specific binding, incubations
with [3H]MLA were performed in separate tubes, after which the
solutions were placed on 96-well plates. Incubations were otherwise
carried out as described previously (Sharples et al. 2002). Assaymixtures were incubated for 2.5 h at room temperature. Non-specific
binding was determined in the presence of 1 mM nicotine. Optiphase
‘SuperMix’ (Wallac, Turku, Finland) scintillant was added to the
wells (30 lL/well). Total and non-specific binding were determined
in triplicates.
In situ binding assayCulture medium was removed from the wells and was replaced by
250 lL of medium containing 1 nM [3H]epibatidine as described
previously (Ridley et al. 2001). Non-specific binding was deter-
mined in the presence of 1 mM nicotine. Cultures were incubated for
2 h at room temperature. The cultures were subsequently washed
quickly three times with 1 mL warm PBS/well, pH 7.4. They were
solubilised in Markwell A (0.5 mL/well), counted for radioactivity
(100 lL) and assayed for protein by a BCA–protein assay. The
binding assays were done by using Wallac 24-well Sample Plates
(1450–420) (Wallac), and the radioactivity was counted with Wallac
1450 Microbeta counter (Wallac). Optiphase ‘SuperMix’ (Wallac)
scintillant was added to the wells (400 lL/well). Total and non-
specific binding were determined in triplicates.
Patch clamp recordings of SH-SY5Y cells
Whole-cell patch-clamp recordings (Hamill et al. 1981) were
performed under an inverted microscope and the ion currents were
evoked by agonist application using a stepper motor solution
exchanger (model SF-77B, Warner Instrument, Hamden, CT, USA).
During the recordings SH-SY5Y cells were continuously superfused
with a recording solution containing (in mM): NaCl2 120, KCl 3,
MgCl2 2, CaCl2 2, HEPES 10 and D-glucose 20, pH adjusted to 7.4
with Tris base. All stock solutions of the drugs were also diluted
with this solution. Patch-clamp electrodes had a resistance of
4–6 MW when filled with intracellular solution containing (in mM):
Effects of the isomers of methadone on nAChRs 1331
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

Tris phosphate dibasic 110, Tris base 28, EGTA 11, MgCl2 2,
CaCl2 0.1 and Na-ATP 4, pH adjusted to 7.3 with Tris base (Zhao
et al. 2003). All the recordings were carried out in voltage clamp
mode at the holding potential of )60 mV. To decrease the solution
exchange time, the cells were lifted from the bottom of the culture
dish with the recording electrode after entering to the whole-cell
configuration of patch clamp. After each 1-s drug application, 2 min
was waited until the next application in order to avoid the rundown
of the ion currents. Currents were recorded using Axopatch 200B
patch-clamp amplifier (Axon Instruments, Foster City, CA, USA),
filtered with 1 kHz and stored to the hard disk of a personal
computer (PC) at the sampling rate of 20 kHz. Data were analyzed
with pClamp 8.2 software (Axon Instruments) and Prism 3.02
software (GraphPad Software Inc. San Diego, CA, USA).
Calcium fluorometry
Wallac IsoplateTM 1450–516 plates, coated with poly D-lysine
(10 lg/mL), were used. Ca2+-mediated increases in fluo-3 fluores-
cence were monitored as described previously (Ridley et al. 2002).Culture medium was removed from the wells and the cells were
washed twice with Tyrode’s salt solution (TSS). The loading
medium was composed of fluo-3AM (10 lM) and probenecid acid
(2.5 mM) in TSS, and the pH was adjusted to 7.4. The cells were
incubated (40 lL/well) at room temperature for 1 h in the dark.
After incubation, the cells were washed once in TSS (200 lL/well)and were left in TSS (200 lL/well) for 15 min. After washings,
80 lL of TSS with or without antagonists were added, and the plate
was transferred to Wallac 1420 Multilabel counter. When antago-
nists were used, the cells were pre-incubated for 10 min in the dark.
The studied compound was injected (20 lL/well), and the fluores-
cence was monitored for 50 s at 485/535 nM (1 s). Average of the
fluorescence values observed 40–45 s after injection were used in
calculations. The magnitudes of the signals were calculated by
reducing from the signal (average from 40 to 45 s) the magnitude
(average from 0 to 5 s) of the signal measured before injecting. As a
control in each experiment, the cells were also stimulated by
injecting vehicle (TSS). Tomeasure intracellular Ca2+-concentrations,
the maximum and minimum fluorescence values were determined by
adding 0.2% Triton (Fmax), which was followed by 40 mM MnCl2(Fmin). Data were calculated as a percentage of Fmax–Fmin. The
changes in [Ca2+]i were expressed as a percentage of nicotine-evoked
increase in fluorescence. In SH-SY5Y cells, all the responses
were compared with the response evoked by 30 lM nicotine, which
was taken as 100%. In SH-EP1-ha7 cells, the responses were
compared with the response evoked by 10 lM nicotine. In each
experiment, there were at least four replicates for each condition.
Data analysis
Dissociation constant (Kd) and maximum binding (Bmax) values from
saturation binding experiments were determined by non-linear
regression, fitting data points to a single-site ligand binding model.
The affinity constant (Ki) values were derived from IC50 values using
the method of Cheng and Prussoff 1973); Ki ¼ IC50/(1 + [L]/KD),
where [L] is the concentration of radioactive compound, Kd is the
affinity of radioactive compound and IC50 is ligand concentration,
which reduces radioligand binding to half maximum. Changes in
ligand binding values were expressed as mean ± SEM (fmol/mg
bound protein) of independent experiments. For dose–response
curves, agonist responses were calculated as a percentage of the
increase in [Ca2+]i produced by a close to maximally effective
concentration of nicotine (30 lM with SH-SY5Y cells and 10 lMwith SH-EP1-ha7 cells). IC50 values were calculated by fitting the
data points to the single site Hill equation using a non-linear
regression curve fitting facility of GraphPad Prism (version
3.02; GraphPad Software) Y ¼ Bottom + (Top–Bottom)/
1 +10(logEC50–X)Hillslope). All data were analysed statistically using
an analysis of variance (ANOVA) for repeated experiments using
GraphPad Prism (version 3.02; GraphPad Software). Values of
p < 0.05 were taken to be statistically significant.
Results
Ligand binding
Competition binding assaysTo address the possibility of a competitive interaction atreceptor level, methadone was studied for its ability todisplace nicotinic radioligands from their binding sites(Fig. 1). Methadone did compete for [3H]MLA (2 nM) and[3H]epibatidine (150 pM) binding sites. The Ki values arepresented in Table 1. Binding of [3H]epibatidine to SH-SY5Y cells membranes was saturable with a maximumbinding capacity (Bmax) of 38.1 ± 8.8 fmol/mg protein and aKd value of 0.39 ± 0.05 nM (n ¼ 3). Bmax for binding of[3H]MLA to SH-SY5Y cell membranes was 19.3 ± 6.9fmol/mg protein with a Kd of 2.41 ± 0.39 nM (n ¼ 3). Therewas evidence for saturability for binding of [3H]MLA toSH-EP1-ha7 cell membranes, but, because of the high levelsof a7 nAChR expression, the apparent Kd value of32.32 ± 5.15 nM (n ¼ 3) is likely an overestimate of true Kd.
Up-regulation of nAChRs evoked by prolonged incubationwith methadoneIn SH-SY5Y cells, 500 pM [3H]epibatidine binds mostly toa3b2-subunits containing nAChRs (Wang et al. 1998).Up-regulation evoked by the isomers of methadone (5 lM),153 ± 31% (n ¼ 4) for (+)-methadone and 182 ± 8% (n ¼4) for (–)-methadone, were similar in magnitude toup-regulation induced in nicotine (10 lM)-treated cells(155 ± 9%; n ¼ 5) (Fig. 2). Up-regulation evoked by meth-adone increased with higher concentrations of methadone(100 lM) and was 364 ± 61% (n ¼ 4) for (+)-methadoneand 290 ± 68% (n ¼ 4) for (–)-methadone.
Patch-clamp electrophysiology
Acute challenge of SH-SY5Y cells with 100 lM (+)-meth-adone, (–)-methadone or nicotine evoked fast inward ioncurrents assessed using whole-cell patch-clamp recording.These responses desensitized slightly during drug exposureand returned to the baseline shortly after the end of drugapplication (Fig. 3a). Co-exposure to mecamylamine (10 lM)inhibited the peak response to 100 lM (+)-methadone
1332 J. S. Pakkanen et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

by 92.1 ± 8.3%, n ¼ 6, (–)-methadone by 91.9 ± 11.0%(n ¼ 8), or nicotine by 93.5 ± 5.9% (n ¼ 5) (Fig. 3a). Theamplitude of the peak whole-cell current response wasconcentration dependent, as detectable currents were firstevident at 10 lM agonist for each drug, and concentration–response curves for the agonists were almost overlapping,with EC50-values of 34 lM for (+)-methadone, 38 lM for (–)-methadone and 24 lM for nicotine (Fig. 3b). There was nosignificant difference in maximal currents for differentagonists [-70 ± 58 pA for (+)-methadone, )58 ± 38 pA for
(–)-methadone, and )84 ± 72 pA for nicotine; data from thesame set of cells; n ¼ 4].
Calcium fluorometry
Acute nicotine increases [Ca2+]i by a mechanism sensitive tonAChR antagonistsSH-SY5Y and SH-EP1-ha7 cell lines gave reproducible,concentration-dependent, increases in [Ca2+]i in response tonicotine (Fig. 4). The functionality of nAChRs in SH-SY5Ycells has been previously shown by using calcium fluoro-metry (Dajas-Bailador et al. 2002) and in SH-EP1-ha7 cellsby using patch-clamp recording (Dunckley et al. 2003; Zhaoet al. 2003). The increase in [Ca2+]i in SH-SY5Y cellsevoked by 30 lM nicotine was 93 ± 12 nM (n ¼ 7) andin SH-EP1-ha7 cells evoked by 10 lM nicotine was134 ± 26 nM (n ¼ 8). The half-maximum response (EC50)for nicotine-evoked increases in [Ca2+]i in SH-SY5Y cellswas 2.9 ± 0.4 lM (n ¼ 4; concentrations of 0.001–30 lM;single fit Hill equation; r2 ¼ 0.78) and in SH-EP1-ha7 cellswas 69 ± 7 nM (n ¼ 4; concentrations of 0.01 nM)30 lM;single fit Hill equation; r2 ¼ 0.83).
In SH-SY5Y cells, pre-incubation (10 min) with DHbE(3 lM), inhibited the response evoked by 30 lM nicotine by25% (n ¼ 3), pre-incubation with MLA (10 nM) inhibitedthe nicotine-evoked response by 26% (n ¼ 3), and
Table 1 Competition binding assay with tritiated nAChR ligands and
the isomers of methadone
Ki values
(lM)
[3H]MLA [3H]MLA [3H]epi
SH-SY5Y SH-EP1-ha7 SH-SY5Y
Nicotine 15.5 ± 4.7 12.3 ± 26 92 ± 26
(+)-Meth. 0.129 0.135 0.000127
(–)-Meth. 0.126 0.135 0.000091
Nicotine and the isomers of methadone replaced a7 nAChR subtype
selective ligand [3H]MLA from its binding sites in SH-SY5Y and
SH-EP1-ha7 cell membranes. In SH-SY5Y cells [3H]epibatidine,
binding to a3 subunit containing nAChRs, was also replaced by
nicotine and the isomers (n ¼ 3 in all experiments).
Fig. 1 Competition binding assays. Assays were performed in the
absence (total binding) or presence of nicotine (d) or (+)-methadone
(s) or (–)-methadone (e). (a, b) [3H]MLA (2 nM) binding to SH-SY5Y
(a) and SH-EP1-ha7 (b) cell membranes defined a7 nAChR. (c)
[3H]Epibatidine (150 pM) binding to SH-SY5Y cell membranes defined
a3* nAChR. (d, e, f) Saturation analysis for [3H]MLA binding to SH-
SY5Y (d) and SH-EP1-ha7 (e) cell membranes and for [3H]epibatidine
binding to SH-SY5Y (f) cell membranes. Note that high levels of
[3H]MLA binding to SH-EP1-ha7 cell membranes prevent saturation
from occurring at the concentrations used and likely force an over-
estimate of Ki value. Non-specific binding was determined in the
presence of 100 lM and 1 mM nicotine, respectively, and subtracted
from total binding to give specific binding. In SH-SY5Y cell mem-
branes, 2 nM [3H]MLA gave specific binding of 37% of total binding and
in SH-EP1-ha7 cell membranes 32% of total binding. In SH-SY5Y cell
membranes 150 pM [3H]epibatidine gave specific binding of 43% of
total binding. Values are the mean ± SEM from three assays, with
each point assayed in triplicate.
Effects of the isomers of methadone on nAChRs 1333
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

pre-incubation with mecamylamine (10 lM) produced 82%inhibition (n ¼ 3; Fig. 5). In addition, when cells were pre-incubated with CdCl2, a general blocker of voltage-operatedCa2+-channels, at a concentration that does not affectnAChRs (100 lM) (Rathouz and Berg 1994), the responsewas inhibited by 84% (n ¼ 3). Note that the response in thepresence of mecamylamine or CdCl2 was indistinguishablefrom that in the presence of the lowest concentrations ofnicotine (see Fig. 4a), suggesting that full block of thenicotine-evoked effect was achieved by either ligand.
In SH-EP1-ha7 cells, pre-incubation with MLA (1 lM) ormecamylamine (100 lM) reduced the nicotine (10 lM)-evoked increase in [Ca2+]i by 82 and 79%, respectively.Pre-incubation with CdCl2 inhibited the response by 81%(n ¼ 3). Again, each of these antagonists reduced theresponse to nicotine to a level observed at the lowestnicotine dose (see Fig. 4), suggesting full sensitivity of theeffect to any of these inhibitors. The concentration ofmecamylamine required to achieve maximum blockade ofthe nicotine response in SH-EP1-ha7 cells was higher(100 lM) than that required to block the nicotine-evokedresponse in SH-SY5Y cells (Fig. 6).
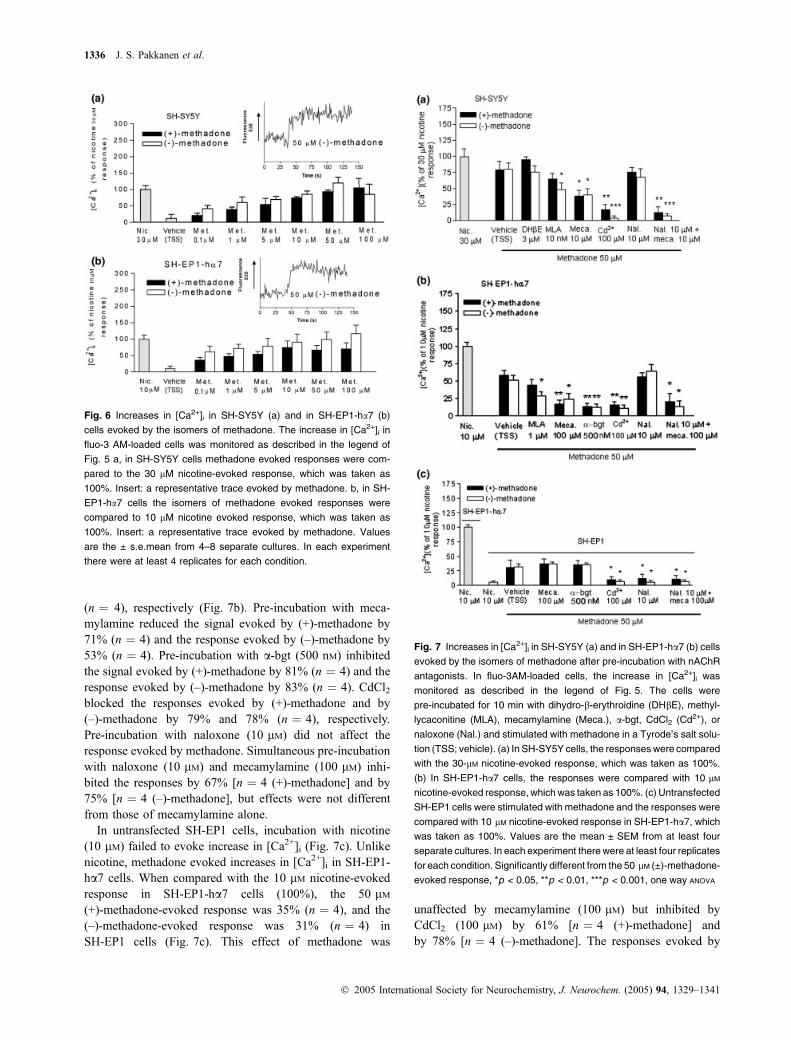
Methadone increases [Ca2+]i by activating nAChRs inSH-SY5Y and in SH-EP1-ha7 cellsBoth isomers of methadone evoked increases in [Ca2+]i inSH-SY5Y cells (Fig. 6a) and in SH-EP1-ha7 cells (Fig. 6b).Compared with 30 lM nicotine-evoked responses inSH-SY5Y cells, 50 lM methadone evoked similar increasesin [Ca2+]i.
Having characterised the methadone-evoked increases in[Ca2+]i in SH-SY5Y and in SH-EP1-ha7 cells, we
investigated the influx routes of Ca2+ activated by theisomers of methadone. The cells were stimulated withisomers of methadone (50 lM) after 10 min pre-incubationwith nAChR antagonists; DHbE, methyllycaconitine(MLA), mecamylamine, a-bgt, CdCl2 or the opioidreceptor antagonist naloxone.
In SH-SY5Y cells, pre-incubation with DHbE (3 lM) didnot change the responses evoked by the isomers of methadone(Fig. 7a). Previously, it has been shown that the predominantfunctional a3b4 nAChR in these cells is insensitive to DHbE(Lukas et al. 1993). Pre-incubation with the a7 nAChRselective antagonist MLA (10 nM) reduced the signal evokedby (+)-methadone by 19% (n ¼ 4) and that by (–)-methadoneby 40% (n ¼ 4). This suggests that there are functionalcontributions of the naturally expressed a7 nAChRs in SH-SY5Y cells to changes in evoked increases in [Ca2+]i. Pre-incubation with the non-selective nAChR antagonist meca-mylamine (10 lM) reduced the signal evoked by (+)-meth-
Fig. 2 Effects of chronic treatment of SH-SY5Y cells with isomers of
methadone on binding levels for 500 pM [3H]epibatidine. SH-SY5Y
cells were treated chronically (3 days) with either nicotine or meth-
adone, washed thoroughly as described in the Materials and methods,
and then binding of 500 pM [3H]epibatidine was measured in situ and
compared with the binding in untreated control cells, cultured and
assayed in parallel. Levels of 500 pM [3H]epibatidine binding are
expressed as percentages of the binding in control cells. Values are
the mean ± SEM of at least four separate cultures. In each experiment
there were at least four replicates for each condition. Significantly
different from control, *p < 0.05, **p < 0.01, one way ANOVA.
Fig. 3 Patch-clamp recording of nAChR-mediated responses in
SH-SY5Y cells. (a) Representative traces showing whole-cell current
responses evoked by 100 lM (+)-methadone, (–)-methadone or nico-
tine alone (left) or in the presence of 10 lM mecamylamine (right).
(b) Concentration–response curves for whole-cell peak current
responses normalized to the maximal response at 300 lM agonist for
(+)-methadone (j), (–)-methadone (h) or nicotine (d). Values are
mean ± SEM (n ¼ 6).
1334 J. S. Pakkanen et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

adone by 62% (n ¼ 4) and that by (–)-methadone by 60%(n ¼ 4). This is consistent with involvement of functionala3b4 nAChRs in addition to a7 nAChRs in these effects.
CdCl2 blocked the responses evoked by (+)-methadone by78% (n ¼ 4) and that by (–)-methadone by 95% (n ¼ 4). Pre-incubation with naloxone (10 lM) did not affect the responseevoked by methadone. Simultaneous pre-incubation withnaloxone (10 lM) and mecamylamine (10 lM) inhibited theresponses by 84% [n ¼ 4 for (+)-methadone] and by 91%[n ¼ 4 for (–)-methadone], which exceeded the effects ofmecamylamine alone.
In SH-EP1-ha7 cells, the responses evoked by(+)-methadone and by (–)-methadone were reduced afterpre-incubation with MLA (1 lM) by 24% and by 45%
Fig. 4 Increases in [Ca2+]i in SH-SY5Y (a) and in SH-EP1-ha7 (b)
cells evoked by nicotine. Fluo-3AM-loaded cells were stimulated with
nicotine, and the fluorescence signal was measured for 50 s. Fluor-
escence values observed 40–45 s after injection were used in calcu-
lations. The magnitudes of the signals were calculated by reducing
from the signal (average from 40 to 45 s) the magnitude (average from
0 to 5 s) of the signal measured before injecting. As a control in each
experiment, the cells were also stimulated with vehicle (TSS).
(a) Increases in [Ca2+]i above basal levels in response to nicotine in
SH-SY5Y cells as compared with response to 30 lM nicotine, which
was taken as 100%. About 23% of the resultant signal can be con-
sidered to be non-specific, in that it is evident even for the lowest
nicotine concentration. Data points (0.001–30 lM) were fitted to
the Hill equation giving an EC50 value of 2.9 ± 0.4 lM (n ¼ 4).
(b) Increases in [Ca2+]i above basal levels in response to nicotine in
SH-EP1-ha7 cells as compared with response to 10 lM nicotine, which
was taken as 100%. Note that about 18% of the resultant signal can be
considered to be non-specific, in that it is evident even for the lowest
nicotine concentration. Data points (0.01 nM)30 lM) were fitted to the
Hill equation giving an EC50 value of 69 ± 7 nM (n ¼ 4). Values are the
mean ± SEM from at least three separate cultures. In each experi-
ment there were at least four replicates for each condition.
Fig. 5 Effects of nicotinic antagonists on nicotine-evoked increases
on [Ca2+]i in SH-SY5Y cells (a) and in SH-EP1-ha7 (b) cells a, fluo-3
AM-loaded SH-SY5Y cells were incubated for 10 min with the nicotinic
AChR antagonists dihydro-b-erythroidine (DHbE), methyllycaconitine
(MLA) or mecamylamine (Meca.), and were stimulated with nicotine
(Nic.). b, fluo-3 AM-loaded SH-EP1-ha7 cells were incubated for
10 min with the nicotinic AChR antagonists methyllcaconitine (MLA)
and mecamylamine (Meca.), and were stimulated with nicotine (Nic.).
Responses are expressed as a percentage of the nicotine response
measured in parallel in the absence of antagonists. Values are the
mean ± s.e.mean of at least 3 separate cultures. In each experiment
there were at least 4 replicates for each condition. Significantly dif-
ferent from nicotine response, *p < 0.05, **p < 0.01, one way ANOVA.
Effects of the isomers of methadone on nAChRs 1335
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341
(n ¼ 4), respectively (Fig. 7b). Pre-incubation with meca-mylamine reduced the signal evoked by (+)-methadone by71% (n ¼ 4) and the response evoked by (–)-methadone by53% (n ¼ 4). Pre-incubation with a-bgt (500 nM) inhibitedthe signal evoked by (+)-methadone by 81% (n ¼ 4) and theresponse evoked by (–)-methadone by 83% (n ¼ 4). CdCl2blocked the responses evoked by (+)-methadone and by(–)-methadone by 79% and 78% (n ¼ 4), respectively.Pre-incubation with naloxone (10 lM) did not affect theresponse evoked by methadone. Simultaneous pre-incubationwith naloxone (10 lM) and mecamylamine (100 lM) inhi-bited the responses by 67% [n ¼ 4 (+)-methadone] and by75% [n ¼ 4 (–)-methadone], but effects were not differentfrom those of mecamylamine alone.
In untransfected SH-EP1 cells, incubation with nicotine(10 lM) failed to evoke increase in [Ca2+]i (Fig. 7c). Unlikenicotine, methadone evoked increases in [Ca2+]i in SH-EP1-ha7 cells. When compared with the 10 lM nicotine-evokedresponse in SH-EP1-ha7 cells (100%), the 50 lM(+)-methadone-evoked response was 35% (n ¼ 4), and the(–)-methadone-evoked response was 31% (n ¼ 4) inSH-EP1 cells (Fig. 7c). This effect of methadone was
unaffected by mecamylamine (100 lM) but inhibited byCdCl2 (100 lM) by 61% [n ¼ 4 (+)-methadone] andby 78% [n ¼ 4 (–)-methadone]. The responses evoked by
Fig. 7 Increases in [Ca2+]i in SH-SY5Y (a) and in SH-EP1-ha7 (b) cells
evoked by the isomers of methadone after pre-incubation with nAChR
antagonists. In fluo-3AM-loaded cells, the increase in [Ca2+]i was
monitored as described in the legend of Fig. 5. The cells were
pre-incubated for 10 min with dihydro-b-erythroidine (DHbE), methyl-
lycaconitine (MLA), mecamylamine (Meca.), a-bgt, CdCl2 (Cd2+), or
naloxone (Nal.) and stimulated with methadone in a Tyrode’s salt solu-
tion (TSS; vehicle). (a) In SH-SY5Y cells, the responses were compared
with the 30-lM nicotine-evoked response, which was taken as 100%.
(b) In SH-EP1-ha7 cells, the responses were compared with 10 lM
nicotine-evoked response, which was taken as 100%. (c) Untransfected
SH-EP1 cells were stimulated with methadone and the responses were
compared with 10 lM nicotine-evoked response in SH-EP1-ha7, which
was taken as 100%. Values are the mean ± SEM from at least four
separate cultures. In each experiment there were at least four replicates
for each condition. Significantly different from the 50 lM (±)-methadone-
evoked response, *p < 0.05, **p < 0.01, ***p < 0.001, one way ANOVA
Fig. 6 Increases in [Ca2+]i in SH-SY5Y (a) and in SH-EP1-ha7 (b)
cells evoked by the isomers of methadone. The increase in [Ca2+]i in
fluo-3 AM-loaded cells was monitored as described in the legend of
Fig. 5 a, in SH-SY5Y cells methadone evoked responses were com-
pared to the 30 lM nicotine-evoked response, which was taken as
100%. Insert: a representative trace evoked by methadone. b, in SH-
EP1-ha7 cells the isomers of methadone evoked responses were
compared to 10 lM nicotine evoked response, which was taken as
100%. Insert: a representative trace evoked by methadone. Values
are the ± s.e.mean from 4–8 separate cultures. In each experiment
there were at least 4 replicates for each condition.
1336 J. S. Pakkanen et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

(+)- or (–)-methadone were reduced after pre-incubation withnaloxone (10 lM) by 61% (n ¼ 4) or 83% (n ¼ 4), respect-ively. Simultaneous pre-incubation with naloxone (10 lM)and mecamylamine (100 lM) inhibited the responses simi-larly to naloxone alone by 65% [n ¼ 4 (+)-methadone] andby 81% [n ¼ 4 (–)-methadone].
Effects of simultaneous stimulation with nicotine andmethadone on [Ca2+]i in SH-SY5Y and in SH-EP1-ha7 cellsIn SH-SY5Y cells, simultaneous stimulation with nicotine andwith the isomers of methadone increased [Ca2+]i by 125 ± 9%[n ¼ 4; (+)-methadone] and by 128 ± 8% [n ¼ 4; (–)-meth-adone] as compared with nicotine alone (100%) (Fig. 8a).
In SH-EP1-ha7 cells, simultaneous stimulation with nico-tine (10 lM) and the isomers of methadone (50 lM)increased [Ca2+]i by 134 ± 14% [n ¼ 4 (+)-methadone]and by 150 ± 9% [n ¼ 4 (–)-methadone] as compared withnicotine alone (Fig. 8b).
Effects of VOCC and intracellular Ca2+ store antagonists onnicotine- and methadone-evoked increases in [Ca2+]i inSH-SY5Y and in SH-EP1-ha7 cellsIn the absence of extracellular Ca2+, no response wasobserved (data not shown), and thus the nicotine- andmethadone-evoked increase in fluorescence was dependenton extracellular Ca2+ in both cell lines. In SH-SY5Y cells,pre-incubation (10 min) with nifedipine (5 lM), inhibited theresponse evoked by 30 lM nicotine by 47% (n ¼ 3), pre-incubation with ryanodine (30 lM), inhibited the nicotine-evoked response by 61% (n ¼ 3), and pre-incubation withxestospongin-c (10 lM), produced 39% inhibition (n ¼ 3;Fig. 9a). Pre-incubation with nifedipine (5 lM) reduced thesignal evoked by (+)-methadone by 42% (n ¼ 3) and theresponse evoked by (–)-methadone by 49% (n ¼ 3)(Fig. 9b). Pre-incubation with ryanodine (30 lM) reducedthe signal evoked by (+)-methadone by 25% (n ¼ 3) and theresponse evoked by (–)-methadone by 35% (n ¼ 4; Fig. 9b).Pre-incubation with xestospongin-c (10 lM) reduced thesignal evoked by (+)-methadone by 17% (n ¼ 3) and theresponse evoked by (–)-methadone by 47% (n ¼ 4; Fig. 9b).
In SH-EP1-ha7 cells, pre-incubation (10 min) with nif-edipine (5 lM), inhibited the response evoked by 10 lMnicotine by 75% (n ¼ 3), pre-incubation with ryanodine(30 lM) inhibited the nicotine-evoked response by 42%(n ¼ 3), and pre-incubation with xestospongin-c (10 lM),produced 68% inhibition (n ¼ 3; Fig. 9c). Pre-incubationwith nifedipine (5 lM) reduced the signal evoked by(+)-methadone by 50% (n ¼ 3) and the response evokedby (–)-methadone by 71% (n ¼ 3; Fig. 9d). Pre-incubationwith ryanodine (30 lM) reduced the signal evoked by(+)-methadone by 43% (n ¼ 3) and the response evokedby (–)-methadone by 48% (n ¼ 3; Fig. 9d). Pre-incubationwith xestospongin-c (30 lM) reduced the signal evoked by(+)-methadone by 32% (n ¼ 3) and the response evoked by(–)-methadone by 46% (n ¼ 3; Fig. 9d).
Discussion
The principal findings from this study indicate that meth-adone interacts with nAChRs. Competition ligand bindingassays showed that methadone inhibits radioligand bindingto nAChRs. Chronic exposure of the cultured cells tomethadone for 3 days increased levels of 500 pM [3H]epi-batidine binding in the SH-SY5Y cell line. Nicotine and bothoptical isomers of methadone increased the level of freeintracellular Ca2+ ([Ca2+]i) in SH-SY5Y and SH-EP1-ha7cell lines. Patch-clamp studies showed that both isomers ofmethadone, similarly to nicotine, directly evoked nAChR-mediated inward currents in SH-SY5Y cells. The responsesto nicotine as well as to methadone were sensitive toblockade by nAChR antagonists, suggesting that methadonehas novel actions as a nAChR agonist. In untransfectedSH-EP1 cells, both isomers of methadone increased [Ca2+]i,
Fig. 8 Effects of co-exposure of the isomers of methadone and
nicotine on increases in [Ca2+]i in SH-SY5Y (a) and in SH-EP1-ha7 (b)
cells. Fluo-3AM-loaded cells were stimulated with methadone, with
nicotine or simultaneously with nicotine and methadone. (a) In
SH-SY5Y cells, the responses were compared with the 30-lM nicotine-
evoked response, which was taken as 100%. (b) In SH-EP1-ha7 cells,
the responses were compared with 10 lM nicotine-evoked response,
which was taken as 100%. Values are the mean ± SEM of at least four
independent experiments. In each experiment there were at least four
replicates for each condition. Significantly different from nicotine-
evoked response, *p < 0.05, one way ANOVA.
Effects of the isomers of methadone on nAChRs 1337
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

and this effect was blocked by opioid receptor antagonists,but not by nicotinic antagonists.
Initial studies concerning the interaction of methadonewith nAChRs used the human neuroblastoma cell lineSH-SY5Y, which endogenously expresses a3, a5, a7, b2 andb4 subunits, which in turn form several different nAChRsubtypes (Peng et al. 1997), including a7-nAChR and afunctionally important a3b4*-nAChR subtype based onpharmacological profiling (Lukas et al. 1993). The compe-tition ligand binding assays, carried out with SH-SY5Y cellmembranes by using tritiated methyllycaconitine ([3H]MLA,2 nM), a selective, competitive antagonist for the a7 nAChRsubtype at low nanomolar concentrations, and [3H]epibati-dine (150 pM), the agonist preferentially binding inSH-SY5Y cells to a3* subunit containing nAChRs, showedthat methadone was able to interact with both of thesenAChRs at the putative binding site for ACh. Underconditions where radioligand binding to a3*-nAChR onSH-SY5Y cells was preferentially queried (i.e. in thepresence of 150 pM [3H]epibatidine), the Ki value fornicotine of 92 nM was lower as expected than that forinteractions at a7 nAChRs. However, also much lower thanKi values for interaction with a7 nAChR were Ki values ofabout 100 pM for methadone interaction with a3* nAChR onSH-SY5Y cells. These findings indicate very high affinityinteractions of methadone with nAChRs. Moreover, althoughthere could be alternate mechanisms involved, up-regulationof epibatidine binding sites presumably corresponding to a3*nAChRs observed in SH-SY5Y cells (Wang et al. 1998) uponpretreatment with methadone also is consistent with inter-actions of the drug with nAChRs. We initially thought thatmethadone might bind to the site(s) for non-competitiveagonists on nAChRs, which is different from the ACh-bindingsite and is insensitive to agonists and competitive antagonists(Pereira et al. 1994). However, our direct demonstration ofmethadone blockade of radiolabelled agonist or competitiveantagonist binding supports some level of interaction ofmethadone at the agonist site(s) of nAChRs. Previous studieshave shown that agonists functionally activate nAChRs atconcentrations higher than those needed to block radioligandbinding at apparent steady state (Xiao et al. 1998; Sabey et al.1999). The current findings that methadones haveKi values forthe inhibition of radioligand binding to nAChRs lower than0.1 lM and lower than concentrations needed to activatenAChR function is consistent with these features.
In order to test the hypothesis that a7 nAChRscontribute to the effects observed in SH-SY5Y cells, weused the SH-EP1-ha7 cell line, stably expressing humana7 nAChRs (Peng et al. 1999; Zhao et al. 2003). Calciumfluorometry studies showed that mecamylamine and MLAblocked methadone-evoked [MLA only (–)-methadone]increases in [Ca2+]i in SH-EP1-ha7 cells, suggesting thatmethadone actions are mediated at least partly via a7nAChRs. Studies using untransfected SH-EP1 cells
Fig. 9 Effects of VOCC and intracellular Ca2+ store antagonists on
nicotine- and methadone-evoked increases in [Ca2+]i in SH-SY5Y
(a, b) and in SH-EP1-ha7 cells (c, d). Fluo-3AM-loaded SH-SY5Y and
SH-EP1-ha7 cells were incubated for 10 min with L-type VOCC ant-
agonist nifedipine (Nif.), ryanodine receptor antagonist ryanodine
(Rya.) and IP3 receptor antagonist xestospongin-c (Xe.-c), and were
stimulated with nicotine or with methadone. The increase in [Ca2+]i
was monitored as described in the legend of Fig. 4. Responses are
expressed as a percentage of the nicotine response measured in
parallel in the absence of antagonists. Values are the mean ± SEM of
at least three separate cultures. In each experiment there were at least
four replicates for each condition. Significantly different from nicotine
response, *p < 0.05, **p < 0.01, or methadone-evoked response,
#p < 0.05, ##p < 0.01, one way ANOVA.
1338 J. S. Pakkanen et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

indicate no responses to nicotine and a naloxone-sensitiveresponse to methadone that is less than observed inSH-EP1-ha7 cells, suggesting that methadone can evokeincreases in [Ca2+]i in the host cell line through non-nAChR-mediated mechanisms. The methadone effect inuntransfected SH-EP1 cells is insensitive to mecamylam-ine, demonstrating nAChR-specificity of major portion ofthe methadone response in SH-EP1-ha7 cells. These dataare consistent with our results showing that whenSH-SY5Y or SH-EP1-ha7 cells are simultaneously exposedto methadone (50 lM) and nicotine at their maximallyeffective doses (30 or 10 lM, respectively), there isadditivity of the effects compared with responses evokedby methadone or by nicotine alone. Thus, in SH-EP1 cells,methadone acts only through non-nAChR, but, whenmethadone is given alone to transfected SH-EP1-ha7 cellsor to SH-SY5Y cells, it could activate both nAChRs andnon-nAChR to cause an increase in intracellular calcium.
The electrophysiological results indicate direct agonismby methadone at nAChRs, but methadone increases [Ca2+]ialso through other mechanisms than nAChRs. InSH-SY5Y cells, opioids can mobilize Ca2+ from intracel-lular stores, but they require ongoing muscarinic, G-proteincoupled, receptor activation (Connor and Henderson 1996).Thus, an initial increase in free intracellular calcium is aprerequisite for the action of opioids. Indeed, the l-opioidagonist DAMGO (Tyr-D-Ala-Gyn-N-Me-Phe-Gly-ol enke-phalin) does not elevate [Ca2+]i when applied alone(Connor and Henderson 1996). They may even have anopposite effect, as l- and d-opioid receptor activationinhibits voltage-dependent Ca2+-currents, although thisnegative regulation can be overcome by strong membranedepolarization (Seward et al. 1991). In the present study,the strong depolarisation could be caused by the activationof nAChRs, especially the a7 nAChR, which has a highpermeability for Ca2+ (Broide et al. 1995) and thereforealso might provide the initial increase in [Ca2+]i that couldpermit an opioid-like action of methadone. However, beingnaloxone-sensitive, arguing against a role of opioidreceptors in the non-nAChR-mediated methadone-evoked[Ca2+]i increase, (+)-methadone also increased [Ca2+]i, andit is well established that (+)-methadone has much weakeropioid properties (Horng et al. 1976).
In our studies, a substantial blockade (80%) by CdCl2(100 lM), a general blocker of voltage-operated ion chan-nels, of nicotine- and methadone-evoked responses indicatesthat voltage-operated Ca2+ channels mediate most ofthe nicotine- and methadone-evoked responses. This isconsistent with previous studies, where it has been shownthat nicotine and opiates increase basal Ca2+-influx throughvoltage-operated calcium channels (Jin et al. 1992; Dajas-Bailador et al. 2002). The current observations that inhibitorsof intracellular calcium release channels also block eventsinitiated by nAChR activation clearly implicates release of
calcium from intracellular stores in the signal as well. Thefinding that doses of MLA higher than those needed to inhibitwhole-cell current responses of a7 nAChRs (see Zhao et al.2003) are required to prevent increases in [Ca2+]i identified inthis study suggests that activity in only a small fraction of a7nAChRs may be sufficient to sustain the calcium response,which is not surprising given the interplay between othermechanisms for [Ca2+]i accumulation. Even although a7nAChRs are quickly inactivated upon interaction with agonist,downstream consequences of their initial activation may notshow the same level of desensitisation, which might haveimportant implications for neuronal function.
The anti-nociceptive effects of nicotine involve spinalrelease of several neurotransmitters. Methadone, like otheropioids, powerfully enhances cerebral serotonergic transmis-sion by stimulating cerebral 5-HT synthesis and release and,through an opioid receptor-independent process, by inhibit-ing the uptake of 5-HT (Ahtee and Carlsson 1979).Methadone seems to increase [Ca2+]i, by which effect itcould also enhance the release of neurotransmitters, and thuspotentiate its anti-nociceptive effects. Methadone has a longhalf life (approximately 24 h), which coupled to its slowonset of action, blunts its euphoric effect, making it lessattractive to abuse than most other opioids. In maintenancetherapy, methadone is used in about 10-fold higher dosesthan as an analgesic. Our results indicate that methadone haseffects on nAChRs. We found that the EC50 values formethadone’s acute functional effects were in the 1–40 lMrange. In clinical studies, the plasma concentration ofmethadone following a single dose is approximately0.25 lM (Inturrisi and Verebely 1972), and the steady-stateconcentration in patients taking methadone chronically canexceed 1 lM (Dyer et al. 1999). Therefore, in vivo effects ofmethadone can be partially mediated by the interactions,including some level of agonism at nAChRs. The interac-tions of methadone with nAChRs may help explain why theprevalence of smoking has been 92% in a methadonemaintenance study population (Clemmey et al. 1997) com-pared with a smoking prevalence of 25% in the generalpopulation (US Department of Health and Human Services1996).
Taken together, our present results suggest that there is aneurochemical interaction between methadone and nicotineat a common molecular target, nAChRs. Further study isneeded to explain methadone–nicotine interactions related tothe treatment of pain, opioid abuse and more generally tonicotine and methadone addiction.
Acknowledgements
We thank Lic. Pharm. Paivi Lehtonen for professionally carrying out
the capillary electrophoretic analysis of methadone. This study was
supported financially by grants from the Academy of Finland, Sigrid
Juselius Foundation and Research Funds of the University of
Effects of the isomers of methadone on nAChRs 1339
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

Helsinki. J. Pakkanen was supported by grants from Research and
Science Foundation of Farmos and Finnish Cultural Foundation.
Work in Phoenix was supported by the National Institutes of Health
(NS040417). We want to acknowledge the late Dr Jiang-Hong Peng,
whose development of the SH-EP1-ha7 cell line made part of this
work possible.
References
Ahtee L. and Carlsson A. (1979) Dual action of methadone on 5-HTsynthesis and metabolism. Naunyn-Schmiedebergs Arch. Pharma-col. 307, 51–56.
Ahtee L. and Saarnivaara L. (1973) The effect of narcotic analgesics onthe uptake of 5-hydroxytryptamine and (-)-metaraminol by bloodplatelets. Br. J. Pharmacol. 47, 808–818.
Badio B. and Dady J. W. (1994) Epibatidine, a potent analgetic andnicotinic agonist. Mol. Pharmacol. 45, 563–569.
Baumhaker Y., Gafni M., Keren O. and Sarne Y. (1993) Selective andinteractive down-regulation of l- and d-opioid receptors in humanneuroblastoma SK-N-SH cells. Mol. Pharmacol. 44, 461–467.
Berrendero F., Kieffer B. L. and Maldonado R. (2002) Attenuation ofnicotine-induced anti-nociception, rewarding effects, and depend-ence in l-opioid receptor knock-out mice. J. Neurosci. 22, 10 935–10 940.
Broide R. S., O’Connor L. T., Smits M. A., Smits J. A. and Leslie F. M.(1995) Developmental expression of a7 neuronal nicotinic receptormessenger RNA in rat sensory cortex and talamus. Neuroscience67, 83–94.
Cheng Y. and Prussoff W. H. (1973) Relationship between the inhibitionconstant (K1) and the concentration of inhibitor which causes 50%inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22,3099–3108.
Clemmey P., Brooner R., Chutuape M. A., Kidor M. and Stitzer M.(1997) Smoking habits and attitudes in a methadone maintenancetreatment population. Drug Alcohol Depend. 44, 123–132.
Connor M. and Henderson G. (1996) d- and l-opioid receptor mobil-ization of intracellular calcium in SH-SY5Y human neuroblastomacells. Br. J. Pharmacol. 117, 333–340.
Dajas-Bailador F. A., Mogg A. J. and Wonnacott S. (2002) IntracellularCa2+ signals evoked by stimulation of nicotinic acetylcholinereceptors in SH-SY5Y cells: contribution of voltage-operated Ca2+
channels and Ca2+ stores. J. Neurochem. 81, 606–614.Damaj M. I., Meyer E. M. and Martin B. R. (2000) The anti-nociceptive
effects of a7 nicotinic agonists in an acute pain model. Neuro-pharmacology 39, 2785–2791.
Dunckley T., Wu J., Zhao L. and Lukas R. J. (2003) Mutational analysisof roles for extracellular cysteine residues in the assembly andfunction of human a7-nicotinic acetylcholine receptors. Biochem.42, 870–876.
Dyer K. R., Foster D. J., White J. M., Somogyi A. A., Menelaou A. andBochner F. (1999) Steady-state pharmacokinetics and pharmaco-dynamics in methadone maintenance patients: comparison of thosewho do and do not experience withdrawal and concentration-effectrelationships. Clin. Pharmacol. Ther. 65, 685–694.
Elks J. and Ganellin C. R., (1990) Methadone, in Dictionary of Drugs(Elks, J. and Ganellin, C. R., eds), p. 781. London: Chapman &Hall.
Epstein A.M. and King A. C. (2004) Naltrexone attenuates acute cigarettesmoking behaviour. Pharmacol. Biochem. Behav. 77, 29–37.
Gotti C., Fornesari D. and Clementi F. (1997) Human neuronal nicotinicreceptors. Prog. Neurobiol. 53, 199–237.
Hamill O. P., Marty A., Neher E., Sakmann B. and Sigworth F. (1981)Improved patch-clamp techniques for high-resolution current
recording from cells and cell free membrane patches. PfluegersArch. 391, 85–100.
Hildebrand B. E., Panagis G., Svensson T. H. and Nomikos G. G. (1999)Behavioural and biochemical manifestations of mecamylamine-precipitated nicotine withdrawal in the rat: role of nicotinicreceptors in the ventral tegmental area. Neuropsychopharmacology21, 560–574.
Horng J. S., Smith S. E. andWong D. T. (1976) The binding of the opticalisomers of methadone, a-methadol, a-acetylmethadol and their N-demethylated derivatives to the opiate receptors of rat brain. Res.Commun. Chem. Pathol. Pharmacol. 14, 621–629.
Howe E. E. and Sletzinger M. (1949) Resolution of dl-methadone anddl-isomethadone. J. Am. Chem. Soc. 71, 2935–2936.
Inturrisi C. E. and Verebely K. (1972) The levels of methadone in theplasma in methadone maintenance. Clin. Pharmacol. Ther. 13,633–637.
Jin W., Lee N. M., Loh H. H. and Thayer S. A. (1992) Dual excitatoryand inhibitatory effects of opioids on intracellular calcium inneuroblastoma · glioma NG108-15 cells. Mol. Pharmacol. 42,1083–1089.
Kazmi S. M. and Mishra R. K. (1986) Opioid receptors in humanneuroblastoma SH-SY5Y cells: evidence for distinct morphine (l)and enkephalin (d) binding sites. Biochem. Biophys. Res. Commun.173, 813–820.
Lukas R. J., Norman S. A. and Lucero L. (1993) Characterization ofnicotinic acetylcholine receptors expressed by cells of the SH-SY5Yhuman neuroblastoma clonal line. Mol. Cell Neurosci. 4, 1–12.
Marubio L. M., Arroyo-Jimenez M., Cordero-Erausquin M., Lena C.,Le Novere N., De Kerchove D’Exaerde A., Huchet M., Damaj M. I.and Changeux J. (1999) Reduced anti-nociception in mice lackingneuronal nicotinic receptor subunits. Nature 398, 805–810.
Mattila M. J., Ahtee L. and Saarnivaara L. (1968) The analgesic andsedative effects of nicotine in white mice, rabbits and goldenhamsters. Ann. Med. Exp. Biol. Fenn. 46, 78–84.
Peng X., Gerzanich V., Anand R., Wang F. and Lindstrom J. (1997)Chronic nicotine treatment up-regulates a3 and a7 acetylcholinereceptor subtypes expressed by the human neuroblastoma cell lineSH-SY5Y. Mol. Pharmacol. 51, 776–784.
Peng J. H., Lucero L., Fryer J., Herl J., Leonard S. S. and Lukas R.(1999) Inducible, heterologous expression of human a7-nicotinicacetylcholine receptors in a native nicotinic receptor-null humanclonal line. Brain Res. 825, 172–179.
Pereira E. F., Alkodon M., Reinhard S., Maelicke A., Peng X., LindstromJ., Whiting P. and Albuquerque E. X. (1994) Physostigmine andgalantamine probes for a novel binding site on the a4b2 subtype ofneuronal nicotinic acetylcholine receptors stably expressed infibroblast cells. J. Pharmacol. Exp. Ther. 270, 768–778.
Rathouz M. M. and Berg D. K. (1994) Synaptic-type acetylcholinereceptors raise intracellular calcium levels in neurons by twomechanisms. J. Neurosci. 14, 6935–6945.
Ridley D. L., Rogers A. and Wonnacott S. (2001) Differential effects ofchronic drug treatment on a3* and a7 nicotinic receptor bindingsites, in hippocampal neurones and SH-SY5Y cells. Br. J. Phar-macol. 133, 1286–1295.
Ridley D. L., Pakkanen J. and Wonnacott S. (2002) Effects of chronicdrug treatments on increase in intracellular calcium mediated bynicotinic acetylcholine receptors in SH-SY5Y cells. Br. J. Phar-macol. 135, 1051–1059.
Sabey K., Paradiso K., Zhang J. and Steinbach J. H. (1999) Ligandbinding and activation of rat nicotinic a4b2 receptors stablyexpressed in HEK293 cells. Mol. Pharmacol. 55, 58–66.
Seward E. P., Hammond C. and Henderson G. (1991) l-Opioid receptor-mediated inhibition of the N-type calcium channel current. Proc. R.Soc. Lond. B 244, 129–135.
1340 J. S. Pakkanen et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341

Sharples C. G., Karig G., Simson G. L., Spencer J. A., Wright E., MillarN. S., Wonnacott S. and Gallagher T. (2002) Synthesis and phar-macological characterization of novel analogues of the nicotinicacetylcholine receptor agonist (+/–)-UB-165. J. Med. Chem. 45,3235–3245.
Stolerman I. P. and Jarvis M. J. (1995) The scientific case that nicotine isaddictive. Psychopharmacology 117, 2–10.
Stringer M., Makin M. K., Miles J. and Morley J. S. (2000) D-morphine,but not 1-morphine, has low micromolar affinity for the non-competitive N-methyl-D-aspartate site in rat forebrain. Possibleclinical implication for the management of neuropathic pain.Neurosci. Lett. 295, 21–24.
SuhH.W.,HudsonP.M.,McMillianM.K.,DasK. P.,WilsonB.C.,WuG.C. and Hong J. S. (1995) Long-term stimulation of nicotinic recep-tors is required to increase pre-enkephalin A mRNA levels and thedeleyed secretion of [met5]-enkephalin in bovine adrenal medullarycromaffin cells. J. Pharmacol. Exp. Ther. 275, 1663–1670.
US Department of Health and Human Services (USDHHS) (1996)Health Unites States 1995. National Center for Health Statistics,Hyatsville, MD. DHHS Pub. no. (PHS) 96–1232. US GovernmentPrinting Office, Washington, DC.
Wang F., Nelson M. E., Kururyatov A., Olale F., Cooper J., Keyser K.and Lindstrom J. (1998) Chronic nicotine treatment up-regulates
human a3b2 but not a3b4 acetylcholine receptors stably trans-fected in human embryonic kidney cells. J. Biol. Chem. 273,28 721–28 732.
Wonnacott S. (1997) Presynaptic nicotinic ACh receptors. Trends Neu-rosci. 20, 92–98.
Xiao Y., Meyer E. L., Thompson J. M., Surin A., Wroblewski J. andKellar K. J. (1998) Rat a3/b4 subtype of neuronal nicotinic acet-ylcholine receptor stably expressed in a transfected cell line:pharmacology of ligand binding and function.Mol. Pharmacol. 54,322–333.
Xiao Y., Smith R. D., Caruso F. S. and Kellar K. J. (2001) Blockadeof rat a3b4 nicotinic receptor function by methadone, itsmetabolites, and structural analogs. J. Pharmacol. Exp. Ther.299, 366–371.
Zarrindast M. R. and Farzin D. (1996) Nicotine attenuates naloxone-induced jumping behaviour in morphine-dependent mice. Eur. J.Pharmacol. 298, 1–6.
Zhao L., Kuo Y.-P., George A. A., Peng J. H., Purandare M. S.,Schroeder K. M., Lukas R. J. and Wu J. (2003) Functionalproperties of homomeric, human a7-nicotinic acetylcholinereceptors heterologously expressed in the SH-EP1 human epithelialcell line. J. Pharmacol. Exp. Ther. 305, 1132–1141.
Effects of the isomers of methadone on nAChRs 1341
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 94, 1329–1341





























