Embed Size (px)
Citation preview

www.fems-microbiology.org
FEMS Microbiology Ecology 52 (2005) 115–128
Microbial community dynamics based on 16S rRNA gene profilesin a Pacific Northwest estuary and its tributaries
Anne E. Bernhard a,*,1, Debbie Colbert b, James McManus b, Katharine G. Field a
a Department of Microbiology, Oregon State University, Corvallis, OR 97331, USAb College of Oceanic and Atmospheric Sciences, Oregon State University, Corvallis, OR 97331, USA
Received 8 June 2004; received in revised form 12 October 2004; accepted 28 October 2004
First published online 28 November 2004
Abstract
We analyzed bacterioplankton community structure in Tillamook Bay, Oregon and its tributaries to evaluate phylogenetic var-
iability and its relation to changes in environmental conditions along an estuarine gradient. Using eubacterial primers, we amplified
16S rRNA genes from environmental DNA and analyzed the PCR products by length heterogeneity polymerase chain reaction (LH-
PCR), which discriminates products based on naturally occurring length differences. Analysis of LH-PCR profiles by multivariate
ordination methods revealed differences in community composition along the estuarine gradient that were correlated with changes in
environmental variables. Microbial community differences were also detected among different rivers. Using partial 16S rRNA
sequences, we identified members of dominant or unique gene fragment size classes distributed along the estuarine gradient. Gam-
maproteobacteria and Betaproteobacteria and members of the Bacteroidetes dominated in freshwater samples, while Alphaproteobac-
teria, Cyanobacteria and chloroplast genes dominated in marine samples. Changes in the microbial communities correlated most
strongly with salinity and dissolved silicon, but were also strongly correlated with precipitation. We also identified specific gene frag-
ments that were correlated with inorganic nutrients. Our data suggest that there is a significant and predictable change in microbial
species composition along an estuarine gradient, shifting from a more complex community structure in freshwater habitats to a com-
munity more typical of open ocean samples in the marine-influenced sites. We also demonstrate the resolution and power of LH-
PCR and multivariate analyses to provide a rapid assessment of major community shifts, and show how these shifts correlate with
environmental variables.
� 2004 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Estuary; 16S ribosomal RNA; Microbial community; Salinity gradient; LH-PCR; Tillamook bay
1. Introduction
The mixing of marine and fresh waters in estuaries
creates steep physico-chemical gradients, which are
undoubtedly accompanied by shifts in the biological
0168-6496/$22.00 � 2004 Federation of European Microbiological Societies
doi:10.1016/j.femsec.2004.10.016
* Corresponding author. Tel.: +1 860 439 2580; fax: +1 860 439
2519.
E-mail address: [email protected] (A.E. Bernhard).1 Current address: Connecticut College, Department of Biology,
New London, CT, USA.
communities. The ephemeral nature of microbes pre-
dicts that these communities, in particular, will undergo
dynamic changes along the estuarine gradient. Rapid
fluctuations in environmental conditions demand meta-
bolic plasticity of the resident microbes, or, alterna-
tively, genetically diverse microbial assemblages that
exploit niche differentiation. Shifts in estuarine micro-bial community structure may be regulated by the abil-
ity of individual populations to overcome various
environmental stresses. These stresses include changes
in nutrient concentrations, tidal currents, and predation,
. Published by Elsevier B.V. All rights reserved.

116 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
but the primary factor is thought to be changes in salin-
ity [1]. Most freshwater bacteria cannot survive even in
oligohaline waters [2,3], and salinity effects have been
observed on bacterial abundance and activity [4].
Changes in salinity also affect chemical constituents
[5], which may, in turn, affect the microbialcommunities.
In addition to the effects of salinity, seasonally driven
changes in microbial communities in estuaries [6] and
rivers [7] also occur. Changes may result from variations
in freshwater flow, which would covary with changes in
salinity. In addition, high flow may supply large pulses
of nutrient-rich material, driving bacterial productivity
up within the estuary and ultimately leading to a net het-erotrophic system [8]. Since bacteria are primarily
responsible for degradation and remineralization of dis-
solved organic material, changes in nutrient concentra-
tion and composition may lead to differences in
distributions of major bacterial populations [9,10].
Extensive studies of microbial diversity and commu-
nity structure have been carried out in ocean and lake
ecosystems (e.g. [11,12]), but only recently haveresearchers begun to address this subject in estuaries
[13–17]. Other studies in estuaries have focused on the
structure and function of bacterioplankton as a group
(e.g. [8,18]), disregarding population dynamics within
the communities. Because of the substantial contribu-
tion of bacterial production to ecosystem function in
estuaries, it is important to investigate the structure of
these bacterial communities and what factors contributemost to microbial species composition.
Recent advances in molecular techniques provide
necessary tools for microbial ecologists to assess com-
munity structure using culture-independent methods
[19]. One such technique, length-heterogeneity polymer-
ase chain reaction (LH-PCR) [20], discriminates among
numerically dominant 16S rRNA genes based solely on
length polymorphisms of the PCR products. The resultis a community profile of 16S rRNA gene products rep-
resenting different populations of bacteria. Because the
method is based on length of DNA sequences, presump-
tive identifications can be assigned to each different peak
in the profile by comparisons to sequences in the public
databases or by comparison to sequences obtained from
clone libraries. A major advantage of LH-PCR over
other molecular methods, such as gene cloning andsequencing, is that it provides a quick way to analyze
and compare communities from a large number of sam-
ples. Recently, microbial communities in soil [21] and
coastal [20,22,23] ecosystems have been characterized
by LH-PCR.
We applied LH-PCR to characterize the microbial
community structure along a salinity gradient in Tilla-
mookBay,Oregonand itsmajor tributaries, and to followmicrobial community dynamics over several seasons. Til-
lamook Bay is a drowned-river estuary in the Pacific
Northwest and is prone to high levels of fecal pollution
and sedimentation from five major rivers that drain into
the bay. The watershed is primarily forested, but the
bay is significantly influenced by agricultural, urban,
and rural land uses. TillamookBay experiences large tidal
changes, with low tides exposing more than 50% of thebay. This twice daily flux may have a significant impact
on the microbial communities, shifting from halophilic
and halotolerant organisms dominating at high tide to
halophobic organisms at low tide. Seasonal changes, such
as fluctuations in freshwater inflow, nutrient inputs, sun-
light, and temperature, may also be important factors in
determining microbial community composition.
The objectives of this study were fourfold. First, wecharacterized the major microbial populations in the
estuary based on 16S rRNA gene profiles and identified
major environmental factors correlated with the com-
munity changes. Second, we used 16S rRNA sequences
from clone libraries to identify potential members of
these communities. Third, because LH-PCR is a rela-
tively new technique, we evaluated its efficacy in produc-
ing ecologically meaningful community data that couldbe analyzed using classical community ecology methods.
And finally, we assessed the impact of including rare
species (or gene products) in the community analysis.
2. Methods and materials
2.1. Sampling site
Tillamook Bay is a shallow estuary on the northwest
coast of Oregon (Fig. 1). The Tillamook watershed cov-
ers nearly 1500 km2, and is drained by five major rivers.
A large floodplain created by drainage from the Tilla-
mook, Trask, Wilson, and Kilchis Rivers is mostly
developed, with the majority in dairy farms. The remain-
der of the drainage basin consists mainly of steep for-ested slopes. The bay is also the site of commercial
and recreational fisheries, including shellfish.
The climate in the Tillamook watershed is under a
strong marine influence from the Pacific Ocean, leading
to very wet winters and dry summers. Average annual
rainfall is approximately 250 cm, with most of this fall-
ing during the fall and winter months, especially during
large winter storms.
2.2. Sample collection
Water samples were collected in Tillamook Bay and
its tributaries in June, July, October, and November
1998, and February and April 1999, at sites marked in
Fig. 1 and Table 1. Not all sites were sampled on all
dates due to inclement weather or time constraints. Sam-ples were collected at approximately one meter below
the surface using a modified 5-L Niskin sampling bottle.
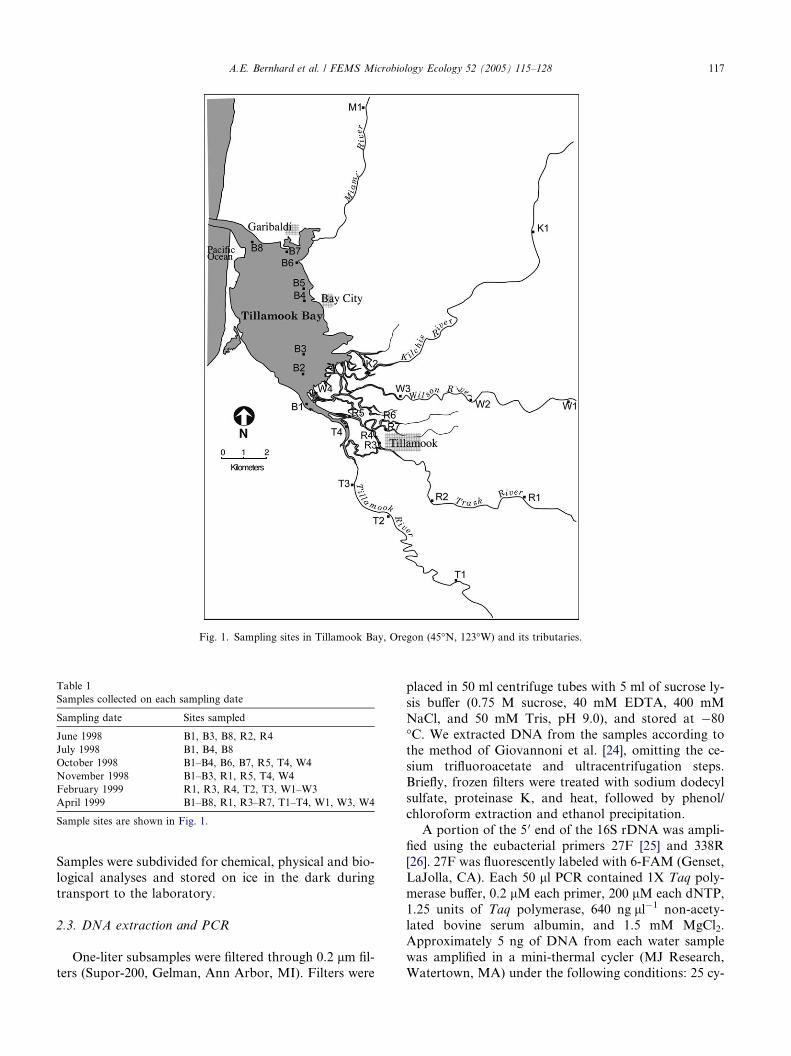
Fig. 1. Sampling sites in Tillamook Bay, Oregon (45�N, 123�W) and its tributaries.
Table 1
Samples collected on each sampling date
Sampling date Sites sampled
June 1998 B1, B3, B8, R2, R4
July 1998 B1, B4, B8
October 1998 B1–B4, B6, B7, R5, T4, W4
November 1998 B1–B3, R1, R5, T4, W4
February 1999 R1, R3, R4, T2, T3, W1–W3
April 1999 B1–B8, R1, R3–R7, T1–T4, W1, W3, W4
Sample sites are shown in Fig. 1.
A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128 117
Samples were subdivided for chemical, physical and bio-
logical analyses and stored on ice in the dark during
transport to the laboratory.
2.3. DNA extraction and PCR
One-liter subsamples were filtered through 0.2 lm fil-
ters (Supor-200, Gelman, Ann Arbor, MI). Filters were
placed in 50 ml centrifuge tubes with 5 ml of sucrose ly-
sis buffer (0.75 M sucrose, 40 mM EDTA, 400 mM
NaCl, and 50 mM Tris, pH 9.0), and stored at �80
�C. We extracted DNA from the samples according tothe method of Giovannoni et al. [24], omitting the ce-
sium trifluoroacetate and ultracentrifugation steps.
Briefly, frozen filters were treated with sodium dodecyl
sulfate, proteinase K, and heat, followed by phenol/
chloroform extraction and ethanol precipitation.
A portion of the 5 0 end of the 16S rDNA was ampli-
fied using the eubacterial primers 27F [25] and 338R
[26]. 27F was fluorescently labeled with 6-FAM (Genset,LaJolla, CA). Each 50 ll PCR contained 1X Taq poly-
merase buffer, 0.2 lM each primer, 200 lM each dNTP,
1.25 units of Taq polymerase, 640 ng ll�1 non-acety-
lated bovine serum albumin, and 1.5 mM MgCl2.
Approximately 5 ng of DNA from each water sample
was amplified in a mini-thermal cycler (MJ Research,
Watertown, MA) under the following conditions: 25 cy-

118 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
cles of 94 �C for 30 s, 55 �C for 1 min, 72 �C for 1 min,
followed by a final extension at 72 �C for 6 min. PCR
products were analyzed by electrophoresis in a 1% aga-
rose gel stained with 1 lg ml�1 ethidium bromide. Con-
centrations of the PCR products were estimated by
comparison of band intensities to DNA mass standards(Gibco-BRL, Gaithersburg, MD).
2.4. Gene scan analysis
Approximately 10–15 fmol of PCR product were re-
solved on a Long Ranger polyacrylamide gel (FMC,
Rockland, ME) on an ABI 377 automated DNA
sequencer using GeneScan software (Applied Biosys-tems Inc., Fremont, CA). The internal size standard,
GS400HD-ROX (ABI), was loaded in each lane. Sizes
of the PCR products were estimated using the third or-
der cubic spline method in GeneScan software v. 3.1
(ABI). We used the software to convert fluorescence
data from the gel into electropherograms, in which the
bands were represented by peaks and the integrated
fluorescence of each band was the area under the peaks.The relative abundance of each different size PCR prod-
uct was estimated by calculating the ratio of the area un-
der each peak to the total area for each sample. Each
peak in the profile represented one PCR product size
in base pairs (LH-PCR gene fragment) and the area un-
der the peak represented the relative abundance of the
gene fragment. Artifactual peaks were minimized by
using a threshold of 100 fluorescence units. High repro-ducibility of this method using the same primers, PCR
conditions, and the same ABI system at Oregon State
University has been previously determined [21].
2.5. Clone library construction, sequencing and
phylogenetic analyses
Samples collected during February, June, July, andOctober from sites B1, B7, B8, B9, T4, and R3 (Fig.
1) were chosen to represent a range of salinities (0–30
ppt) and environmental conditions. Environmental
DNAs were amplified with eubacterial primers 27F
and 1522R [27]. Pooled PCR products were gel purified
using a gel purification kit (Qiagen, Valencia, CA) and
cloned into the pGEM-T Easy vector (Promega, Madi-
son, WI) according to the manufacturer�s directions.Transformants were randomly selected and inoculated
into 100 ll LB broth with 100 lg ampicillin ml�1 in
96-well microtiter plates. The plates were incubated for
six hours and then one replica plate was made from each
original microtiter plate. All plates were incubated over-
night at 37 �C. LH-PCR peak sizes were determined for
138 clones by the methods described above.
Thirty-three plasmid DNAs from clones correspond-ing to dominant or unique LH-PCR fragments in marine
and freshwater sites were prepared using the Qiaprep
Spin Column Purification Kit (Qiagen) according to
the manufacturer�s directions. DNAs were quantified
spectrophotometrically on a UV/Vis Spectrophotometer
(Shimadzu, Columbia, MD). Bidirectional sequences
were obtained using the primers 27F and 700R [12]. Se-
quences were determined on an ABI 377 DNA Sequencerusing dye-terminator chemistry. We aligned the se-
quences using the sequence editor and Fast Align in
ARB [28] and checked all alignments manually. Regions
of ambiguous alignments were excluded from the analy-
sis. All phylogenetic analyses were done using PAUP* v.
4.0 [29]. Phylogenetic trees were constructed using neigh-
bor-joining and maximum parsimony. The relative con-
fidence in branching order was evaluated byperforming 100 bootstrap replicates. Sequences were
checked for possible chimeric structure by using the
CHECK_CHIMERA program available from the
RDP [30] and by comparison of neighbor-joining trees
based on different regions of the sequence.
2.6. Environmental variables
Nutrient analyses are discussed in detail elsewhere
[31]. Briefly, dissolved inorganic phases of nitrogen
ðNHþ4 and NO�
3 þNO�2 Þ, dissolved silicon, and phos-
phorous were determined within 12 h of filtration using
standard analytical techniques [32] adapted for a seg-
mented-flow autoanalyzer [33]. Dissolved organic car-
bon was determined by high temperature combustion
on a Shimadzu TOC-5000 analyzer. Salinity was calcu-lated from measured chloride concentration using the
expression S = 1.80655 · chlorinity [34]. Chloride con-
centration was determined using conductivity detection
with a DIONEX ED40 Electrochemical Detector. Salin-
ity was measured for all samples except during Novem-
ber, and is reported in Practical Salinity Scale units.
Because salinity and dissolved silicon were linearly re-
lated (r2 = 0.97), salinity values for November sampleswere predicted from dissolved silicon concentrations.
Samples with salinity values less than 1 were classified
as freshwater; samples with salinity values greater than
or equal to 1 were classified as marine. Precipitation
data were obtained from the Oregon Climate Service
Website (www.ocs.orst.edu/) for the Tillamook Station
1W (45�2700W and 123�5200N). We used the sum of pre-
cipitation over the five days prior to and including theday of sampling.
2.7. Statistical analysis
All multivariate analyses were performed using PC-
ORD version 4 [35]. The relative abundance data were
transformed by an arcsine square root function to reduce
skew. Beta-diversity (a measure of compositional hetero-geneity), average half changes (amount of change in spe-
cies composition along a gradient), skewness, and

A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128 119
coefficient of variation (CV) for rows (samples) and col-
umns (gene fragments) were determined before and after
the transformation. The transformation of the raw data
did not affect the beta-diversity and produced a 42% and
35% reduction in row and column skewness, respectively,
and a 25% reduction in the CV for gene fragment totals.Non-metric multidimensional scaling (NMS) [36] was
used to ordinate samples in gene fragment space, using
the SØrenson�s distance measure. Nonmetric scaling is
an ordination method based on ranked distances and
uses an iterative optimization procedure. The effect of
rare fragments on the ordination results was assessed
by progressively deleting fragments that occurred in
fewer than X% of samples (values for X were 0, 5, 10,and 20) and re-running the ordination. In a separate
analysis, we also evaluated the effect of excluding peaks
that were less than 1% of the total area. The autopilot
option was set to the slow and thorough level for all
ordinations. Dimensionality (the optimal number of
dimensions or axes required to explain a sufficient pro-
portion of the variance) was assessed by choosing the
number of axes that minimized final stress and maxi-mized interpretability of the results. The axes have no
real significance and can be rotated or mirrored without
influencing the relative distances between the points.
Stress is a measure of departure from monotonicity in
the relationship between the dissimilarity in the original
p-dimensional space and the dissimilarity in the reduced
k-dimensional ordination space and is a measure of
goodness of fit. Stability of the solutions was assessedby the final instability output and by plotting final stress
versus number of iterations. Monte Carlo tests were run
to confirm that results obtained were significantly better
than would be obtained from randomized data. Addi-
tionally, the proportion of variance explained by each
axis and the cumulative variance explained was deter-
mined for each analysis by calculating the coefficient
of determination between distances in ordination spaceand distances in the original 57-dimensional space. Cor-
relation coefficients in the ordination space were deter-
mined for each environmental variable and gene
fragment by rotating the ordination to maximize the
coefficient on one axis (using the varimax rotation op-
tion in PC-ORD) and were used to improve the inter-
pretability of the solution, not the quality of the
mathematical fit.
2.8. GenBank accession numbers
Nucleotide sequences for 16S rRNA gene clones can
be found under Accession Nos.: AY628650, AY628651,
AY628652, AY628653, AY628654, AY628655,
AY628656, AY628657, AY628658, AY628659,
AY628660, AY628661, AY628662, AY628663,AY628664, AY628665, AY628666, AY628667,
AY628668, AY628669, AY628670, AY628671,
AY628672, AY628673, AY628674, AY628675,
AY628676, AY628677, AY628678, AY628679,
AY628680.
3. Results
We compared 16S rRNA gene profiles of planktonic
microbial communities in Tillamook Bay, Oregon, to
evaluate spatial and temporal differences and correlated
these differences to changes in environmental conditions
by using multivariate statistics. The community profiles
were generated by PCR, which is known to have poten-
tial kinetic biases, partly due to the number of cyclesperformed [20]. To test the effects of different cycle num-
bers of the PCR on the relative abundance of each frag-
ment, we performed 15, 20, 25, and 30 cycles of
amplification and compared the results. The CV of rela-
tive abundance of each peak between PCRs of different
cycle numbers was much lower (12.4%) when we used
the data set that exluded peaks representing less than
1% of the total area than when all peaks were included(42%). This CV is very similar to the PCR variation
found by Ritchie et al. [21]. These data suggest no signif-
icant kinetic bias from 15 to 30 cycles, so we chose 25
cycles because it was the lowest cycle number that con-
sistently produced easily observable PCR product in a
gel.
We also evaluated the effect of deleting rare fragment
lengths to simulate the common, and often recom-mended, practice of deleting rare species in community
composition analyses [37]. We progressively deleted
fragment lengths that occurred in fewer than 5%, 10%,
and 20% of the samples. Our analyses, however, re-
vealed little effect on the ordination, as determined by fi-
nal stress, and instability (Table 2). Based on these
results, the data matrix containing all fragment lengths
representing greater than 1% of the total area was usedfor additional analyses, so that maximal data were re-
tained without increasing noise. It should be noted that
each LH-PCR peak may represent many different bacte-
rial populations. Additionally, the relative abundance of
each gene fragment does not necessarily represent the
abundance of that gene in a natural sample due to po-
tential PCR bias.
Community profiles of 16S rRNA genes revealed ashift from communities dominated by LH-PCR frag-
ments longer than 330 bp in the freshwater samples to
communities dominated by shorter fragments (313–329
bp) in the marine samples (Fig. 2). Differences in micro-
bial community structure along the salinity gradient
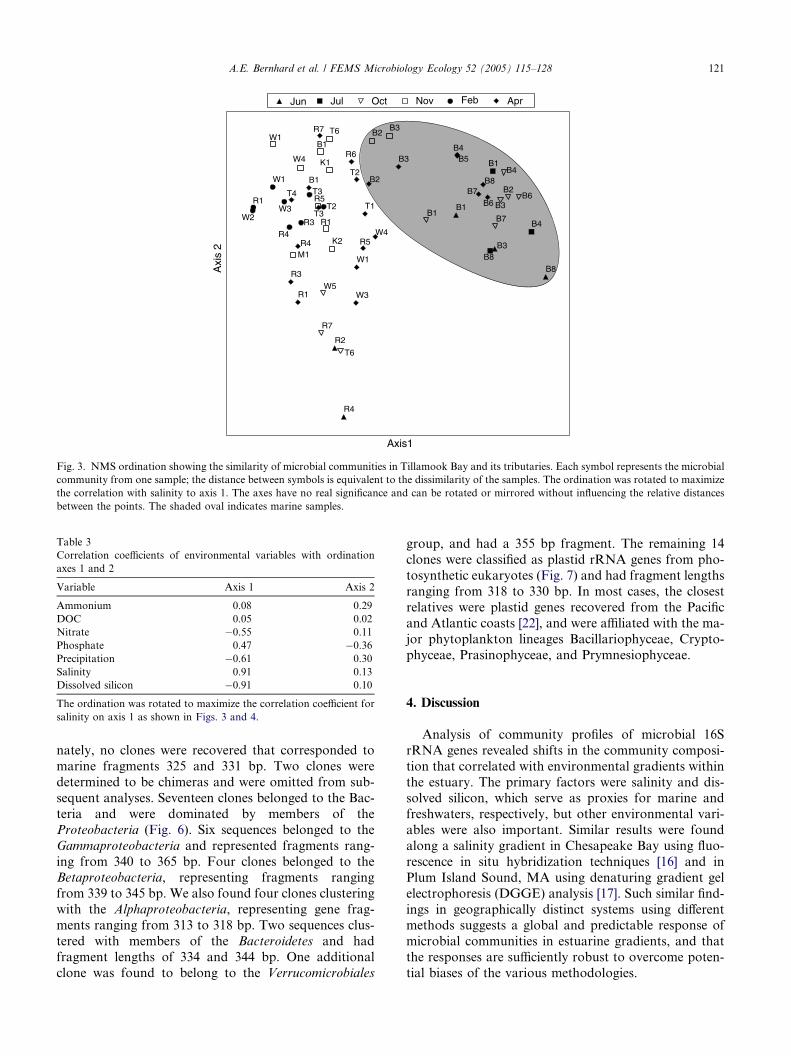
were obvious in the NMS ordination (Fig. 3), which
produced stronger axes than expected by chance as eval-
uated by the Monte Carlo test, with 85.1% of the vari-ability explained by the first three axes (Table 2). Axis
1 accounted for 46.5% of the variability in community
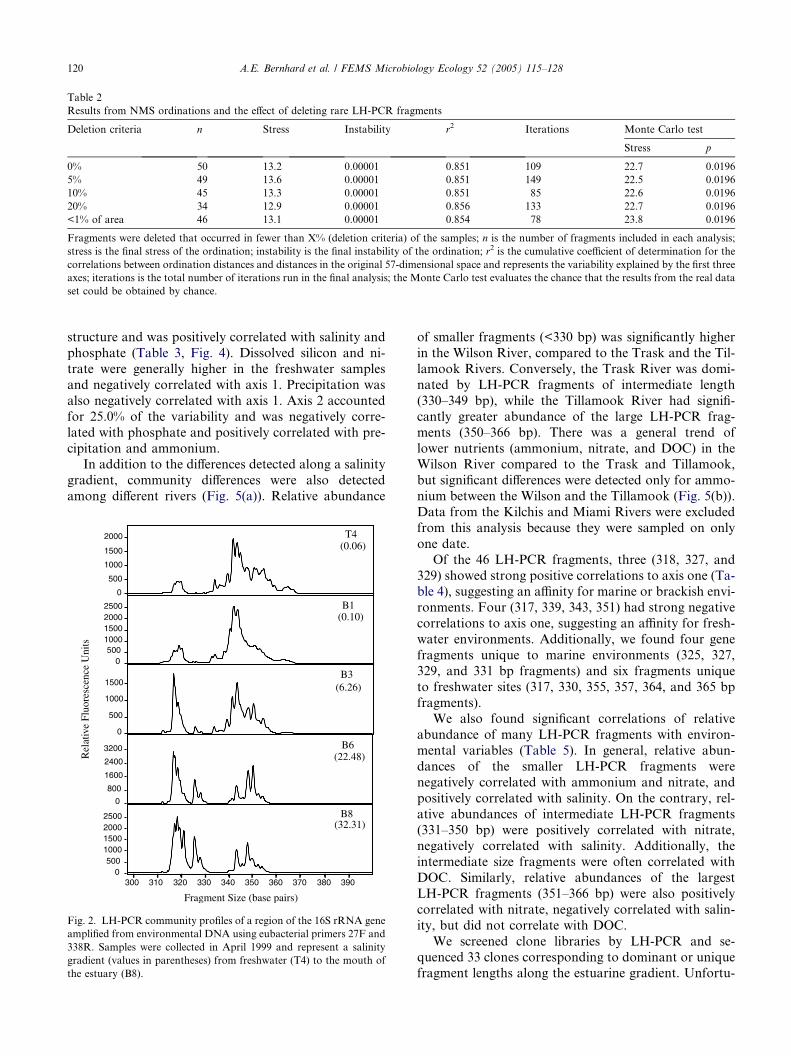
Table 2
Results from NMS ordinations and the effect of deleting rare LH-PCR fragments
Deletion criteria n Stress Instability r2 Iterations Monte Carlo test
Stress p
0% 50 13.2 0.00001 0.851 109 22.7 0.0196
5% 49 13.6 0.00001 0.851 149 22.5 0.0196
10% 45 13.3 0.00001 0.851 85 22.6 0.0196
20% 34 12.9 0.00001 0.856 133 22.7 0.0196
<1% of area 46 13.1 0.00001 0.854 78 23.8 0.0196
Fragments were deleted that occurred in fewer than X% (deletion criteria) of the samples; n is the number of fragments included in each analysis;
stress is the final stress of the ordination; instability is the final instability of the ordination; r2 is the cumulative coefficient of determination for the
correlations between ordination distances and distances in the original 57-dimensional space and represents the variability explained by the first three
axes; iterations is the total number of iterations run in the final analysis; the Monte Carlo test evaluates the chance that the results from the real data
set could be obtained by chance.
120 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
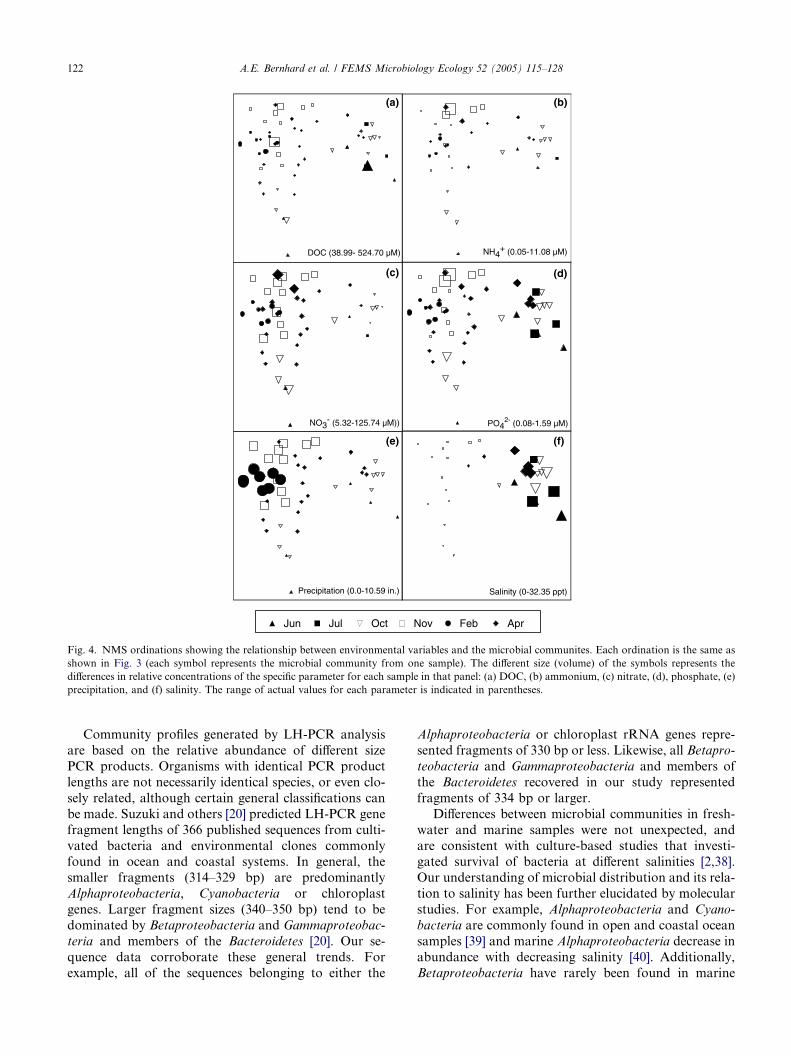
structure and was positively correlated with salinity and
phosphate (Table 3, Fig. 4). Dissolved silicon and ni-
trate were generally higher in the freshwater samples
and negatively correlated with axis 1. Precipitation was
also negatively correlated with axis 1. Axis 2 accounted
for 25.0% of the variability and was negatively corre-
lated with phosphate and positively correlated with pre-
cipitation and ammonium.In addition to the differences detected along a salinity
gradient, community differences were also detected
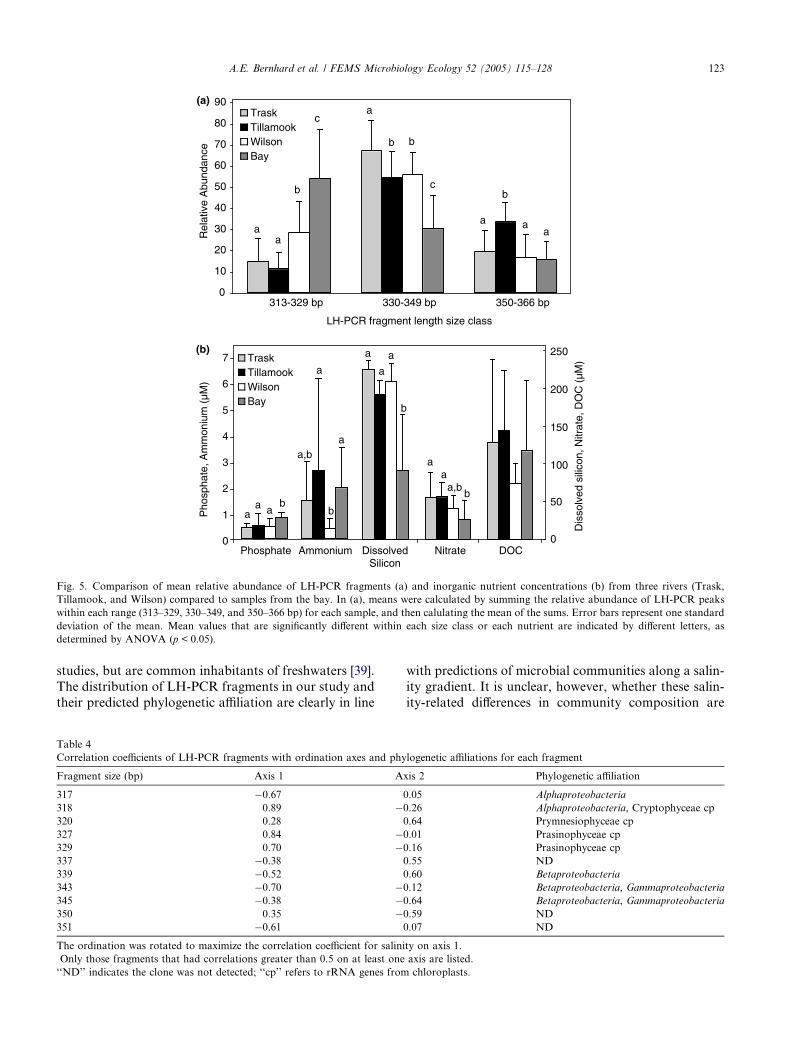
among different rivers (Fig. 5(a)). Relative abundance
0
500
1000
1500
2000
0500
1000150020002500
0
500
1000
1500
Rel
ativ
e Fl
uore
scen
ce U
nits
300 310 320 330 340 350 360 370 380 390
0
800
1600
2400
3200
0500
1000150020002500
T4
B1
B3
B6
B8
Fragment Size (base pairs)
(0.06)
(0.10)
(6.26)
(22.48)
(32.31)
Fig. 2. LH-PCR community profiles of a region of the 16S rRNA gene
amplified from environmental DNA using eubacterial primers 27F and
338R. Samples were collected in April 1999 and represent a salinity
gradient (values in parentheses) from freshwater (T4) to the mouth of
the estuary (B8).
of smaller fragments (<330 bp) was significantly higher
in the Wilson River, compared to the Trask and the Til-
lamook Rivers. Conversely, the Trask River was domi-
nated by LH-PCR fragments of intermediate length
(330–349 bp), while the Tillamook River had signifi-
cantly greater abundance of the large LH-PCR frag-
ments (350–366 bp). There was a general trend of
lower nutrients (ammonium, nitrate, and DOC) in theWilson River compared to the Trask and Tillamook,
but significant differences were detected only for ammo-
nium between the Wilson and the Tillamook (Fig. 5(b)).
Data from the Kilchis and Miami Rivers were excluded
from this analysis because they were sampled on only
one date.
Of the 46 LH-PCR fragments, three (318, 327, and
329) showed strong positive correlations to axis one (Ta-ble 4), suggesting an affinity for marine or brackish envi-
ronments. Four (317, 339, 343, 351) had strong negative
correlations to axis one, suggesting an affinity for fresh-
water environments. Additionally, we found four gene
fragments unique to marine environments (325, 327,
329, and 331 bp fragments) and six fragments unique
to freshwater sites (317, 330, 355, 357, 364, and 365 bp
fragments).We also found significant correlations of relative
abundance of many LH-PCR fragments with environ-
mental variables (Table 5). In general, relative abun-
dances of the smaller LH-PCR fragments were
negatively correlated with ammonium and nitrate, and
positively correlated with salinity. On the contrary, rel-
ative abundances of intermediate LH-PCR fragments
(331–350 bp) were positively correlated with nitrate,negatively correlated with salinity. Additionally, the
intermediate size fragments were often correlated with
DOC. Similarly, relative abundances of the largest
LH-PCR fragments (351–366 bp) were also positively
correlated with nitrate, negatively correlated with salin-
ity, but did not correlate with DOC.
We screened clone libraries by LH-PCR and se-
quenced 33 clones corresponding to dominant or uniquefragment lengths along the estuarine gradient. Unfortu-
B1B2
B3T6
R1
R5
K1
K2
M1
W1
W4
B1 B2
B3B4
B5
B6B7
B8
T1
T2
T3
T4
R1
R3
R4 R5
R6
R7
W1
W3
W4
T2
T3R1
R3R4
W1
W2W3 B1
B3
B8
R2
R4
B1
B4
B8
B1
B2
B3
B4
B7
B6
T6
R7
W5
Jun Jul Oct Nov Feb Apr
Axis1
Axi
s 2
Fig. 3. NMS ordination showing the similarity of microbial communities in Tillamook Bay and its tributaries. Each symbol represents the microbial
community from one sample; the distance between symbols is equivalent to the dissimilarity of the samples. The ordination was rotated to maximize
the correlation with salinity to axis 1. The axes have no real significance and can be rotated or mirrored without influencing the relative distances
between the points. The shaded oval indicates marine samples.
Table 3
Correlation coefficients of environmental variables with ordination
axes 1 and 2
Variable Axis 1 Axis 2
Ammonium 0.08 0.29
DOC 0.05 0.02
Nitrate �0.55 0.11
Phosphate 0.47 �0.36
Precipitation �0.61 0.30
Salinity 0.91 0.13
Dissolved silicon �0.91 0.10
The ordination was rotated to maximize the correlation coefficient for
salinity on axis 1 as shown in Figs. 3 and 4.
A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128 121
nately, no clones were recovered that corresponded to
marine fragments 325 and 331 bp. Two clones were
determined to be chimeras and were omitted from sub-
sequent analyses. Seventeen clones belonged to the Bac-
teria and were dominated by members of the
Proteobacteria (Fig. 6). Six sequences belonged to the
Gammaproteobacteria and represented fragments rang-
ing from 340 to 365 bp. Four clones belonged to theBetaproteobacteria, representing fragments ranging
from 339 to 345 bp. We also found four clones clustering
with the Alphaproteobacteria, representing gene frag-
ments ranging from 313 to 318 bp. Two sequences clus-
tered with members of the Bacteroidetes and had
fragment lengths of 334 and 344 bp. One additional
clone was found to belong to the Verrucomicrobiales
group, and had a 355 bp fragment. The remaining 14
clones were classified as plastid rRNA genes from pho-
tosynthetic eukaryotes (Fig. 7) and had fragment lengths
ranging from 318 to 330 bp. In most cases, the closest
relatives were plastid genes recovered from the Pacific
and Atlantic coasts [22], and were affiliated with the ma-
jor phytoplankton lineages Bacillariophyceae, Crypto-
phyceae, Prasinophyceae, and Prymnesiophyceae.
4. Discussion
Analysis of community profiles of microbial 16S
rRNA genes revealed shifts in the community composi-
tion that correlated with environmental gradients within
the estuary. The primary factors were salinity and dis-solved silicon, which serve as proxies for marine and
freshwaters, respectively, but other environmental vari-
ables were also important. Similar results were found
along a salinity gradient in Chesapeake Bay using fluo-
rescence in situ hybridization techniques [16] and in
Plum Island Sound, MA using denaturing gradient gel
electrophoresis (DGGE) analysis [17]. Such similar find-
ings in geographically distinct systems using differentmethods suggests a global and predictable response of
microbial communities in estuarine gradients, and that
the responses are sufficiently robust to overcome poten-
tial biases of the various methodologies.
DOC (38.99- 524.70 µM) NH4+ (0.05-11.08 µM)
NO3- (5.32-125.74 µM)) PO4
2- (0.08-1.59 µM)
Precipitation (0.0-10.59 in.) Salinity (0-32.35 ppt)
Jun Jul Oct Nov Feb Apr
(a)
(c)
(e) (f)
(d)
(b)
Fig. 4. NMS ordinations showing the relationship between environmental variables and the microbial communites. Each ordination is the same as
shown in Fig. 3 (each symbol represents the microbial community from one sample). The different size (volume) of the symbols represents the
differences in relative concentrations of the specific parameter for each sample in that panel: (a) DOC, (b) ammonium, (c) nitrate, (d), phosphate, (e)
precipitation, and (f) salinity. The range of actual values for each parameter is indicated in parentheses.
122 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
Community profiles generated by LH-PCR analysis
are based on the relative abundance of different size
PCR products. Organisms with identical PCR product
lengths are not necessarily identical species, or even clo-
sely related, although certain general classifications can
be made. Suzuki and others [20] predicted LH-PCR gene
fragment lengths of 366 published sequences from culti-
vated bacteria and environmental clones commonlyfound in ocean and coastal systems. In general, the
smaller fragments (314–329 bp) are predominantly
Alphaproteobacteria, Cyanobacteria or chloroplast
genes. Larger fragment sizes (340–350 bp) tend to be
dominated by Betaproteobacteria and Gammaproteobac-
teria and members of the Bacteroidetes [20]. Our se-
quence data corroborate these general trends. For
example, all of the sequences belonging to either the
Alphaproteobacteria or chloroplast rRNA genes repre-
sented fragments of 330 bp or less. Likewise, all Betapro-
teobacteria and Gammaproteobacteria and members of
the Bacteroidetes recovered in our study represented
fragments of 334 bp or larger.
Differences between microbial communities in fresh-
water and marine samples were not unexpected, and
are consistent with culture-based studies that investi-gated survival of bacteria at different salinities [2,38].
Our understanding of microbial distribution and its rela-
tion to salinity has been further elucidated by molecular
studies. For example, Alphaproteobacteria and Cyano-
bacteria are commonly found in open and coastal ocean
samples [39] and marine Alphaproteobacteria decrease in
abundance with decreasing salinity [40]. Additionally,
Betaproteobacteria have rarely been found in marine
0
10
20
30
40
50
60
70
80
90
313-329 bp 330-349 bp 350-366 bp
Trask TillamookWilsonBay
Trask TillamookWilsonBay
a
a aaa
a
a
a
a
aa
a
a
a,b
a,b
a a
a
b
b
b
bb
b
b
b
c
c
0
1
2
3
4
5
6
7
0
50
100
150
200
250
LH-PCR fragment length size class
Pho
spha
te, A
mm
oniu
m (
µM)
Rel
ativ
e A
bund
ance
Dis
solv
ed s
ilico
n, N
itrat
e, D
OC
(µM
)
(a)
(b)
Phosphate Ammonium DissolvedSilicon
Nitrate DOC
Fig. 5. Comparison of mean relative abundance of LH-PCR fragments (a) and inorganic nutrient concentrations (b) from three rivers (Trask,
Tillamook, and Wilson) compared to samples from the bay. In (a), means were calculated by summing the relative abundance of LH-PCR peaks
within each range (313–329, 330–349, and 350–366 bp) for each sample, and then calulating the mean of the sums. Error bars represent one standard
deviation of the mean. Mean values that are significantly different within each size class or each nutrient are indicated by different letters, as
determined by ANOVA (p < 0.05).
A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128 123
studies, but are common inhabitants of freshwaters [39].
The distribution of LH-PCR fragments in our study and
their predicted phylogenetic affiliation are clearly in line
Table 4
Correlation coefficients of LH-PCR fragments with ordination axes and phy
Fragment size (bp) Axis 1 A
317 �0.67
318 0.89 �320 0.28
327 0.84 �329 0.70 �337 �0.38
339 �0.52
343 �0.70 �345 �0.38 �350 0.35 �351 �0.61
The ordination was rotated to maximize the correlation coefficient for salini
Only those fragments that had correlations greater than 0.5 on at least one
‘‘ND’’ indicates the clone was not detected; ‘‘cp’’ refers to rRNA genes from
with predictions of microbial communities along a salin-
ity gradient. It is unclear, however, whether these salin-
ity-related differences in community composition are
logenetic affiliations for each fragment
xis 2 Phylogenetic affiliation
0.05 Alphaproteobacteria
0.26 Alphaproteobacteria, Cryptophyceae cp
0.64 Prymnesiophyceae cp
0.01 Prasinophyceae cp
0.16 Prasinophyceae cp
0.55 ND
0.60 Betaproteobacteria
0.12 Betaproteobacteria, Gammaproteobacteria
0.64 Betaproteobacteria, Gammaproteobacteria
0.59 ND
0.07 ND
ty on axis 1.
axis are listed.
chloroplasts.
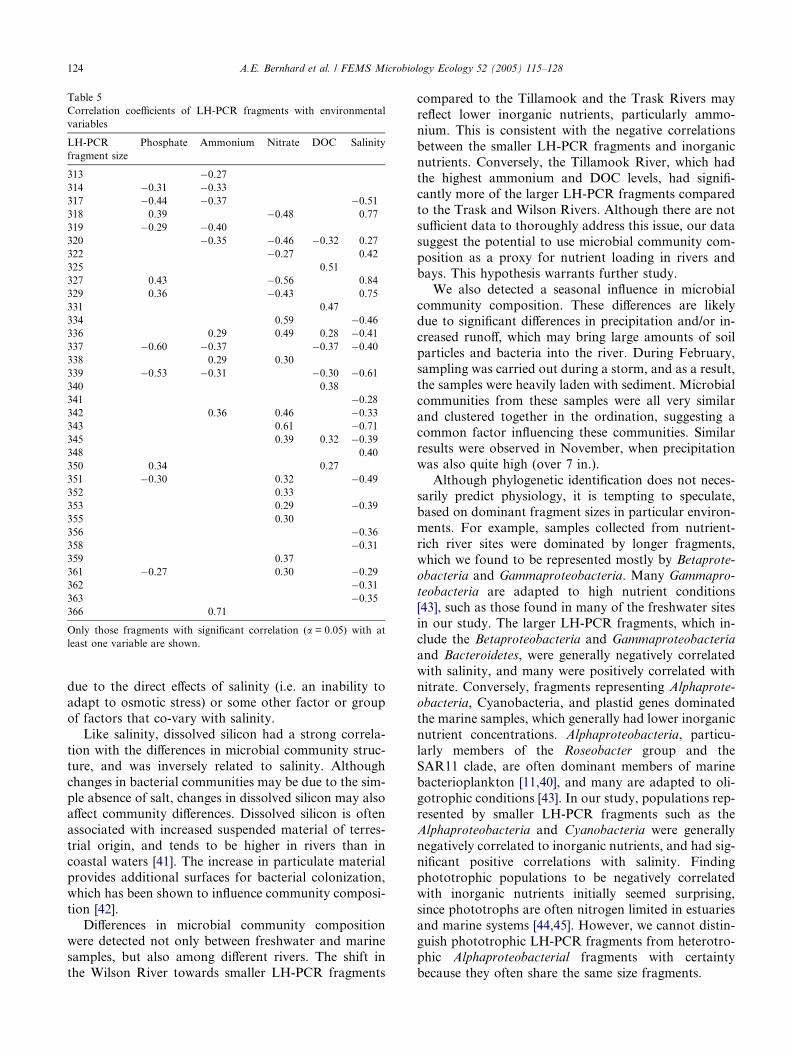
Table 5
Correlation coefficients of LH-PCR fragments with environmental
variables
LH-PCR
fragment size
Phosphate Ammonium Nitrate DOC Salinity
313 �0.27
314 �0.31 �0.33
317 �0.44 �0.37 �0.51
318 0.39 �0.48 0.77
319 �0.29 �0.40
320 �0.35 �0.46 �0.32 0.27
322 �0.27 0.42
325 0.51
327 0.43 �0.56 0.84
329 0.36 �0.43 0.75
331 0.47
334 0.59 �0.46
336 0.29 0.49 0.28 �0.41
337 �0.60 �0.37 �0.37 �0.40
338 0.29 0.30
339 �0.53 �0.31 �0.30 �0.61
340 0.38
341 �0.28
342 0.36 0.46 �0.33
343 0.61 �0.71
345 0.39 0.32 �0.39
348 0.40
350 0.34 0.27
351 �0.30 0.32 �0.49
352 0.33
353 0.29 �0.39
355 0.30
356 �0.36
358 �0.31
359 0.37
361 �0.27 0.30 �0.29
362 �0.31
363 �0.35
366 0.71
Only those fragments with significant correlation (a = 0.05) with at
least one variable are shown.
124 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
due to the direct effects of salinity (i.e. an inability to
adapt to osmotic stress) or some other factor or group
of factors that co-vary with salinity.
Like salinity, dissolved silicon had a strong correla-
tion with the differences in microbial community struc-
ture, and was inversely related to salinity. Although
changes in bacterial communities may be due to the sim-
ple absence of salt, changes in dissolved silicon may alsoaffect community differences. Dissolved silicon is often
associated with increased suspended material of terres-
trial origin, and tends to be higher in rivers than in
coastal waters [41]. The increase in particulate material
provides additional surfaces for bacterial colonization,
which has been shown to influence community composi-
tion [42].
Differences in microbial community compositionwere detected not only between freshwater and marine
samples, but also among different rivers. The shift in
the Wilson River towards smaller LH-PCR fragments
compared to the Tillamook and the Trask Rivers may
reflect lower inorganic nutrients, particularly ammo-
nium. This is consistent with the negative correlations
between the smaller LH-PCR fragments and inorganic
nutrients. Conversely, the Tillamook River, which had
the highest ammonium and DOC levels, had signifi-cantly more of the larger LH-PCR fragments compared
to the Trask and Wilson Rivers. Although there are not
sufficient data to thoroughly address this issue, our data
suggest the potential to use microbial community com-
position as a proxy for nutrient loading in rivers and
bays. This hypothesis warrants further study.
We also detected a seasonal influence in microbial
community composition. These differences are likelydue to significant differences in precipitation and/or in-
creased runoff, which may bring large amounts of soil
particles and bacteria into the river. During February,
sampling was carried out during a storm, and as a result,
the samples were heavily laden with sediment. Microbial
communities from these samples were all very similar
and clustered together in the ordination, suggesting a
common factor influencing these communities. Similarresults were observed in November, when precipitation
was also quite high (over 7 in.).
Although phylogenetic identification does not neces-
sarily predict physiology, it is tempting to speculate,
based on dominant fragment sizes in particular environ-
ments. For example, samples collected from nutrient-
rich river sites were dominated by longer fragments,
which we found to be represented mostly by Betaprote-
obacteria and Gammaproteobacteria. Many Gammapro-
teobacteria are adapted to high nutrient conditions
[43], such as those found in many of the freshwater sites
in our study. The larger LH-PCR fragments, which in-
clude the Betaproteobacteria and Gammaproteobacteria
and Bacteroidetes, were generally negatively correlated
with salinity, and many were positively correlated with
nitrate. Conversely, fragments representing Alphaprote-
obacteria, Cyanobacteria, and plastid genes dominated
the marine samples, which generally had lower inorganic
nutrient concentrations. Alphaproteobacteria, particu-
larly members of the Roseobacter group and the
SAR11 clade, are often dominant members of marine
bacterioplankton [11,40], and many are adapted to oli-
gotrophic conditions [43]. In our study, populations rep-
resented by smaller LH-PCR fragments such as theAlphaproteobacteria and Cyanobacteria were generally
negatively correlated to inorganic nutrients, and had sig-
nificant positive correlations with salinity. Finding
phototrophic populations to be negatively correlated
with inorganic nutrients initially seemed surprising,
since phototrophs are often nitrogen limited in estuaries
and marine systems [44,45]. However, we cannot distin-
guish phototrophic LH-PCR fragments from heterotro-phic Alphaproteobacterial fragments with certainty
because they often share the same size fragments.
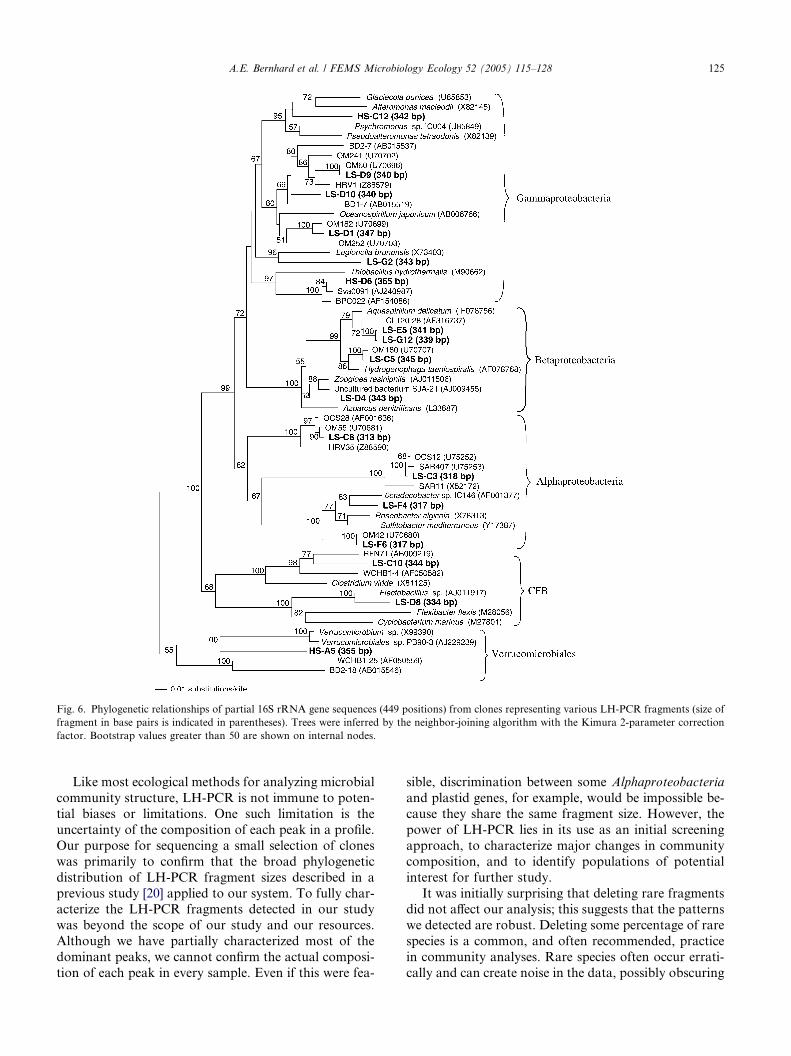
Fig. 6. Phylogenetic relationships of partial 16S rRNA gene sequences (449 positions) from clones representing various LH-PCR fragments (size of
fragment in base pairs is indicated in parentheses). Trees were inferred by the neighbor-joining algorithm with the Kimura 2-parameter correction
factor. Bootstrap values greater than 50 are shown on internal nodes.
A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128 125
Like most ecological methods for analyzing microbial
community structure, LH-PCR is not immune to poten-
tial biases or limitations. One such limitation is the
uncertainty of the composition of each peak in a profile.
Our purpose for sequencing a small selection of clones
was primarily to confirm that the broad phylogenetic
distribution of LH-PCR fragment sizes described in a
previous study [20] applied to our system. To fully char-acterize the LH-PCR fragments detected in our study
was beyond the scope of our study and our resources.
Although we have partially characterized most of the
dominant peaks, we cannot confirm the actual composi-
tion of each peak in every sample. Even if this were fea-
sible, discrimination between some Alphaproteobacteria
and plastid genes, for example, would be impossible be-
cause they share the same fragment size. However, the
power of LH-PCR lies in its use as an initial screening
approach, to characterize major changes in community
composition, and to identify populations of potential
interest for further study.
It was initially surprising that deleting rare fragmentsdid not affect our analysis; this suggests that the patterns
we detected are robust. Deleting some percentage of rare
species is a common, and often recommended, practice
in community analyses. Rare species often occur errati-
cally and can create noise in the data, possibly obscuring

Odontella sinensis (Z67753)
env. clone OM20 (U32670)
Skeletonema pseudocostatum (X82155)
LS-D11 (322 bp)
Chlorella vulgaris (X16579)
Chlorella kessleri (D11346)
LS-C12 (327 bp)
LS-H2 (327 bp)
LS-H10 (330 bp)
HS-C10 (329 bp)
env. clone OCS182 (AF001660)
LS-E12 (327 bp)
env. clone OM134 (AF001661)
env. clone OM5 (U70715)
Emiliana huxleyi (X82156)
Ochrosphaera neapolitana (X80390)
env. clone OM21 (U32671)
HS-A2 (320 bp)
Cryptomonas plastid (X56806)
Pyrenomonas salina (X55015)
LS-F10 (319 bp)
LS-F9 (319 bp)
LS-F7 (319 bp)
LS-C6 (318 bp)
LS-E11 (318 bp)
LS-F11 (318 bp)
LS-G10 (319 bp)
env. clone OCS20 (AF001654)
Unidentified Cryptomonad (U70724)
0.01 substitutions/site
100
100
100
100
100
100
100
100
100
9997
79
87
91
91
89
64
64
51
90
Fig. 7. Phylogenetic relationships of partial chloroplast rRNA sequences (501 positions) from clones representing various LH-PCR fragments (size
of fragment in base pairs is indicated in parentheses). Trees were inferred by the neighbor-joining algorithm with the Kimura 2-parameter correction
factor. Bootstrap values greater than 50 are shown on internal nodes.
126 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
important patterns [37]. Additionally, their presence can
elevate the variance among samples without contribut-
ing useful information about the sample site. However,
excluding rare species may violate general ecological
observations and theory [46]. Since the ordinations did
not differ with the exclusion of rare fragments, we left
them in to avoid losing any potentially usefulinformation.
Our ability to detect rare species, however, may be
compromised by the methods we used. Traditional mac-
roecology studies involve counting species directly,
which tends to increase the chances of detecting rare
species. In this study, estimates of DNA extraction effi-
ciencies were only about 30% (estimated by dividing the
total DNA recovered per sample by the expected yield
calculated from the average number of cells/ml
[2 · 105] and an average of 5 fg of DNA per cell).
Assuming that this 30% is representative of the sample,
then some rare species would likely be eliminated at this
step. Gene amplification by the PCR may be biased [47],
and rare sequences may not amplify as well as abundantones. Assuming rare sequences were amplified, they may
not be detected in the final GeneScan analysis. Gene
fragments that are below the detection limit will appear
only as background noise. The detection limit of Gene-
Scan is difficult to quantify because it is affected by
many variables, such as amount of template used in
the PCR, number of cycles, and amount of product

A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128 127
loaded in the gel. Since we were interested in major com-
munity shifts, we optimized the amount of DNA ana-
lyzed to maintain a maximum peak height below 6000
relative fluorescent units, to avoid cut-off peaks (with
flat tops). Additionally, each LH-PCR fragment likely
represents more than one species of bacteria, so that rarespecies may, in fact, be included in some of the frag-
ments, but we were unable to detect them individually.
In conclusion, our investigation of the diversity in
estuarine bacterioplankton and its relation to physical
and chemical parameters suggests that the differences
observed reflect the phylogenetic variability of the com-
munities under different conditions, rather than physio-
logical plasticity of the same microbial assemblage.Additionally, we have demonstrated that LH-PCR and
NMS are powerful tools for analyzing microbial com-
munity dynamics over spatial and temporal scales and
identifying factors that most influence these dynamics.
Although these methods alone do not provide informa-
tion on individual species within a sample, they equip
investigators with the ability to process many samples
quickly, thus providing information on heterogeneitywithin a system. Additionally, by identifying unique or
dominant gene fragments in a community, researchers
can streamline DNA sequencing, easily targeting only
those fragments of interest.
Acknowledgments
We thankMichael Rappe for comments on the manu-
script and phylogenetic expertise and Bruce McCune for
statistical expertise and guidance. We also thank the Ore-
gon State University Center for Gene Research and Bio-
technology Central Services Lab for GeneScan analyis
and DNA sequencing. Funding for A.B. and K.F. was
provided by Grant NA76RG0476 (project R/ECO-04)
from the National Oceanic and Atmospheric Adminis-tration to the Oregon State University Sea Grant College
Program, by Grant NCERQAR-827639-01 from the US
Environmental Protection Agency, and by appropria-
tions made by the Oregon state legislature. Funding for
J.M. and D.C. was provided by the EPA�s Water and
Watersheds program: GAD#R825751.
References
[1] Prieur, D., Troussellier, M., Romana, A., Chamroux, S., Mevel,
G. and Baleux, B. (1987) Evolution of bacterial communities in
the Gironde Estuary (France) according to a salinity gradient.
Estuarine Coastal Shelf Sci. 24, 95–108.
[2] Bordalo, A.A. (1993) Effects of salinity on bacterioplankton: field
and microcosm experiments. J. Appl. Bacteriol. 75, 393–398.
[3] Painchaud, J., Therriault, J.-C. and Legendre, L. (1995) Assess-
ment of salinity-related mortality of freshwater bacteria in the St.
Lawrence Estuary. Appl. Environ. Microbiol. 61, 205–208.
[4] Revilla, M., Iriarte, A., Madariaga, I. and Orive, E. (2000)
Bacterial and phytoplankton dynamics along a trophic gradient in
a shallow temperate estuary. Estuarine Coastal Shelf Sci. 50, 297–
313.
[5] Eyre, B. and Balls, P. (1999) A comparative study of nutrient
behavior along the salinity gradient of tropical and temperate
estuaries. Estuaries 22, 313–326.
[6] Soto, Y., Bianchi, M., Martinez, J. and Rego, J.V. (1993)
Seasonal evolution of microplanktonic communities in the estu-
arine front ecosystem of the Rhone River plume (North-western
Mediterranean Sea). Estuarine Coastal Shelf Sci. 37, 1–13.
[7] Lemke, M.J. and Leff, L.G. (1999) Bacterial populations in an
anthropogenically disturbed stream: comparison of different
seasons. Microb. Ecol. 38, 234–243.
[8] Findlay, S., Pace, M.L., Lints, D., Cole, J.J., Caraco, N.F. and
Peierls, B. (1991) Weak coupling of bacterial and algal production
in a heterotrophic ecosystem: the Hudson River estuary. Limnol.
Oceanogr. 36, 268–278.
[9] Murrell, M.C., Hollibaugh, J.T., Silver, M.W. and Wong, P.S.
(1999) Bacterioplankton dynamics in northern San Francisco
Bay: role of particle association and seasonal freshwater flow.
Limnol. Oceanogr. 44, 295–308.
[10] Cottrell, M.T. and Kirchman, D.L. (2000) Natural assemblages of
marine proteobacteria and members of the Cytophaga–Flavob-
acter cluster consuming low- and high-molecular-weight dissolved
organic matter. Appl. Environ. Microbiol. 66, 1692–1697.
[11] Giovannoni, S.J., Britschgi, T.B., Moyer, C.L. and Field, K.G.
(1990) Genetic diversity in Sargasso Sea bacterioplankton. Nature
345, 60–63.
[12] Urbach, E., Vergin, K.L., Young, L., Morse, A., Larson, G.L.
and Giovannoni, S.J. (2001) Unusual bacterioplankton commu-
nity structure in ultra-oligotrophic Crater Lake. Limnol. Oceangr.
46, 557–572.
[13] Benlloch, S., Rodriguez-Valera, F. and Martinez-Murcia, A.J.
(1995) Bacterial diversity in two coastal lagoons deduced from
16S rDNA PCR amplification and partial sequencing. FEMS
Microbiol. Ecol. 18, 267–280.
[14] Crump, B.C., Armbrust, V. and Baross, J.A. (1999) Phylogenetic
analysis of particle-attached and free-living bacterial communities
in the Columbia River, its estuary, and the adjacent coastal ocean.
Appl. Environ. Microbiol. 65, 3192–3204.
[15] Murray, A.E., Hollibaugh, J.T. and Orrego, C. (1996) Phyloge-
netic compositions of bacterioplankton from two California
estuaries compared by denaturing gradient gel electrophoresis of
16S rDNA fragments. Appl. Environ. Microbiol. 62, 2676–2680.
[16] Bouvier, T.C. and del Giorgio, P.A. (2002) Compositional
changes in free-living bacterial communities along a salinity
gradient in two temperate estuaries. Limnol. Oceanogr. 47, 453–
470.
[17] Crump, B.C., Hopkinson, C.S., Sogin, M.L. and Hobbie, J.E.
(2004) Microbial biogeography along an estuarine salinity gradi-
ent: combined influences of bacterial growth and residence time.
Appl. Environ. Microbiol. 70, 1494–1505.
[18] Hollibaugh, J.T. and Wong, P.S. (1999) Microbial processes in the
San Francisco Bay estuarine turbidity maximum. Estuaries 22,
848–862.
[19] Giovannoni, S.J., Mullins, T.D. and Field, K.G. (1995) Microbial
diversity in oceanic systems: rRNA approaches to the study of
unculturable microbes In: Molecular Ecology of Aquatic
Microbes (Joint, I., Ed.). Springer, Berlin.
[20] Suzuki, M.T., Rappe, M.S. and Giovannoni, S.J. (1998) Kinetic
bias in estimates of coastal picoplankton community structure
obtained by measurements of small subunit rRNA gene PCR
amplicon length heterogeneity. Appl. Environ. Microbiol. 64,
4522–4529.
[21] Ritchie, N.J., Schutter, M.E., Dick, R.P. and Myrold, D.D.
(2000) Use of length heterogeneity PCR and FAME to charac-

128 A.E. Bernhard et al. / FEMS Microbiology Ecology 52 (2005) 115–128
terize microbial communities in soil. Appl. Environ. Microbiol.
66, 1668–1675.
[22] Rappe, M.S., Suzuki, M.T., Vergin, K.L. and Giovannoni, S.J.
(1998) Phylogenetic diversity of ultraplankton plastid small-
subunit rRNA genes recovered in environmental nucleic acid
samples from the Pacific and Atlantic coasts of the United States.
Appl. Environ. Microbiol. 64, 294–303.
[23] Bernhard, A.E. and Field, K.G. (2000) Identification of nonpoint
sources of fecal pollution in coastal waters by using host-specific
16S ribosomal DNA genetic markers from fecal anaerobes. Appl.
Environ. Microbiol. 66, 1587–1594.
[24] Giovannoni, S.J., DeLong, E.F., Schmidt, T.M. and Pace, N.R.
(1990) Tangential flow filtration and preliminary phylogenetic
analysis of marine picoplankton. Appl. Environ. Microbiol. 56,
2572–2575.
[25] Lane, D.J. (1991) 16S/23S rRNA sequencing In: Nucleic acid
Techniques in Bacterial Systematics (Stackebrandt, E. and
Goodfellow, M., Eds.), pp. 115–175. Wiley, New York.
[26] Amann, R.I., Binder, B.J., Olson, R.J., Chisholm, S.W., Deve-
reux, R. and Stahl, D.A. (1990) Combination of 16S rRNA-
targeted oligonucleotide probes with flow cytometry for analyzing
mixed microbial populations. Appl. Environ. Microbiol. 56,
1919–1925.
[27] Giovannoni, S.J. (1991) The polymerase chain reaction In:
Sequencing and Hybridization Techniques in Bacterial Systemat-
ics (Stackebrandt, E. and Goodfellow, M., Eds.), pp. 177–201.
Wiley, New York.
[28] Ludwig, W. et al. (2004) ARB: a software environment for
sequence data. Nucleic Acids Res. 32, 1363–1371.
[29] Swofford, D.L. (1991) PAUP: Phylogenetic Analysis using Par-
simony. Sinauer Associates Inc., Fitchberg, MA.
[30] Maidak, B.L. et al. (2001) The RDP-II (Ribosomal Database
Project). Nucleic Acids Res. 29, 173–174.
[31] Colbert, D. and McManus, J. (2003) Nutrient biogeochemistry in
an upwelling-influenced estuary of the Pacific Northwest (Tilla-
mook Bay, Oregon, USA). Estuaries 26, 1205–1219.
[32] Stickland, J.D.H. and Parsons, T.R. (1972) A Practical Hand-
book of Seawater Analysis. Fisheries Research Board, Canada.
[33] Gordon, L.I., Jennings, J.C., Ross, A.A. and Krest, J.M. (1995) A
suggested protocol for continuous flow automated analysis of
seawater nutrients (phosphate, nitrate, nitrite, and silicic acid) In:
WOCE Hydrographic Program and the Joint Global Ocean
Fluxes Study, Methods Manual 91-1 WHOI, Woods Hole, MA.
[34] Knauss, J.A. (1978) Introduction to Physical Oceanography.
Prentice-Hall, Englewood Cliffs, NJ.
[35] McCune, B. and Mefford, M.J. (1999) PC-ORD, Multivariate
Analysis of Ecological Data.MjMSoftware, Gleneden Beach, OR.
[36] Kruskal, J.B. (1964) Nonmetric multidimensional scaling: a
numerical method. Psychometrika 29, 115–129.
[37] Gauch Jr., H.G. (1982) Multivariate Analysis in Community
Ecology. Cambridge Press, New York.
[38] Valdes, M. and Albright, L.J. (1981) Survival and heterotrophic
activities of Fraser River and Strait of Georgia bacterioplankton
within the Fraser River plume. Mar. Biol. 64, 231–241.
[39] Glockner, F.O., Fuchs, B.M. and Amann, R. (1999) Bacterio-
plankton compositions of lakes and oceans: a first comparison
based on fluorescence in situ hybridization. Appl. Environ.
Microbiol. 65, 3721–3726.
[40] Gonzalez, J.M., Whitman, W.B., Hodson, R.E. and Moran, M.A.
(1996) Identifying numerically abundant culturable bacteria from
complex communities: an example from a lignin enrichment
culture. Appl. Environ. Microbiol. 62, 4433–4440.
[41] Conley, D.J. and Malone, T.C. (1992) Annual cycle of dissolved
silicate in chesapeake bay – implications for the production and
fate of phytoplankton. Biomass 81, 121–128.
[42] DeLong, E.F., Franks, D.G. and Alldredge, A.L. (1993) Phylo-
genetic diversity of aggregate-attached vs. free-living marine
bacterial assemblages. Limnol. Oceanogr. 38, 924–934.
[43] Zavarzin, G.A., Stackebrandt, E. and Murray, R.G. (1991) A
correlation of phylogenetic diversity in the Proteobacteria with
the influences of ecological forces. Can. J. Microbiol. 37, 1–6.
[44] Bernhard, A.E. and Peele, E.R. (1997) Nitrogen limitation of
phytoplankton in a shallow embayment in northern Puget Sound.
Estuaries 20, 759–769.
[45] Howarth, R.W. (1988) Nutrient limitation of net primary
production in marine ecosystems. Annu. Rev. Ecol. 19, 89–110.
[46] Cao, Y., Williams, D.D. and Williams, N.E. (1998) How
important are rare species in aquatic community ecology and
bioassessment?. Limnol. Oceanogr. 43, 1403–1409.
[47] Suzuki, M.T. and Giovannoni, S.J. (1996) Bias caused by
template annealing in the amplification of mixtures of 16S rRNA
genes by PCR. Appl. Environ. Microbiol. 62, 625–630.





























