Embed Size (px)
Citation preview

Molecular and Cellular Determinants of Triglyceride Availability"
E. SEBOKOVA AND I. KLIMES Diabetes and Nutrition Research Group Institute of Experimental Endocrinology
Slovak Academy of Sciences SK-833 06 Bratislava, Slovak Republic
Dyslipidemia, including hypertriglyceridemia, low HDL concentrations, and alter- ations in LDL composition, is part of the insulin resistance (IR) syndrome and is regarded as an independent risk factor for coronary heart disease in subjects with impaired glucose tolerance or diabetes. I-5 It has been shown in humans6 and in that both increased triglyceride (TG) levels in circulation and increased muscle TG stores are directly associated with IR.
While the genetic background is important, environmental factors play an essential role in the development of hypertriglyceridemia and IR.Z Thus, recently, much atten- tion has been paid to the composition of macronutrients in the diet, with special focus on fat composition. Increasing evidence exists demonstrating that dietary intake of polyunsaturated fatty acids (PUFA), particularly of the n-3 class, is associated with suppression of hypertriglyceridemia and improvement of insulin
The plasma levels and tissue content of TG are determined by the balance between de novo synthesis of fatty acids (and consequently of TG), TG clearance in circulation (by lipoprotein lipase), and TG hydrolysis in tissues (by lipoprotein and/or hormone- sensitive lipase in adipose tissue, skeletal muscle, and the heart).I8-z0 Thus, a defect at any step of this enzyme network may initiate a cascade of pathological events leading to hypertriglyceridemia.
REGULATION OF LIPOGENESIS
Understanding of the biochemistry and regulation of lipogenesis has resulted mostly from in vitro studies in rodents.'* However, the biological importance, activity, and tissue distribution of the lipogenic pathways vary greatly among species. Whereas active lipogenesis occurs in liver and adipose tissue in the rat21 de novo lipogenesis is an inefficient process in humans.Z2 It occurs predominantly in the liver and is of
(I This work was supported by research grants of the Institute of Experimental Endocrinology, Slovak Academy of Sciences, Bratislava, provided by the Slovak Grant Agency for Sciences (GAV) (No. 2/544/92-95); the Natural Sciences and Engineering Research Council of Canada; the Canadian Diabetes Association; and a COST B5 grant.
200

SEBOKOVA & KLIMES: TRIGLYCERIDES 201
minor significance in human adipose tissue, even under conditions of massive car- bohydrate 0verfeeding.~3 The nutritional status of the organism, closely allied to changes in circulating insulin concentrations, appears to be the major determinant of the rate of lipogenesis. Lipogenesis is high in the fed state and following carbo- hydrate administration,l4-41 whereas it is suppressed by fasting,*' high-fat diets,29-31 and insulin deficiency.38
Insulin stimulates de novo lipogenesis in liver and adipose tissue by several mech- anisms. 18.21 In adipose tissue, insulin stimulates glucose uptake, thereby increasing the supply of lipogenic substrate. Furthermore, several key enzymes in the lipogenic pathway, including fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and malic enzyme (ME), are induced by insulin. In addition, the antilipolytic action of insulin reduces free fatty acid availability to the liver and thereby reduces the inhib- itory effects of FA on hepatic lipogenesis. Similarly, the inhibition of lipolysis and the stimulatory effect of insulin on reesterification of FA in adipose tissue reduce the intra-adipocyte concentration of fatty acyl-CoA, an inhibitor of lipogenesis. Thus, the many actions of insulin serve to increase the biosynthesis of FA for subsequent storage in the form of TG. It should be emphasized, however, that the predominant source of FA substrate for TG synthesis in both the adipose tissue and liver comes from circulating free fatty acids derived either from adipose tissue lipolysis or from intravascular hydrolysis of lipoprotein TG by lipoprotein lipase.21
The stimulation of lipogenesis by insulin in response to nutritional changes involves a rapid activation of the key enzymes involved in FA synthesis. This phenomenon, however, is preceded and/or modulated by long-term transcriptional and translational events. It has been shown that food intake, in particular composition of the diet, may represent an important factor for the regulation of lipogenesis in liver and adipose tissue at the gene level.19q2"28 Although techniques for evaluation of the molecular basis of lipogenesis are available, it is still rather complicated to carry out appropriate studies in humans, particularly due to ethical reasons. Therefore, various animal models of IR have been widely used to elucidate the molecular and cellular mechanisms that may be responsible for hypertriglyceridemia in human IR. Theregulation of lipogenesis in these animal models will be discussed first.
REGULATION OF LIPOGENESIS IN ANIMAL MODELS OF INSULIN RESISTANCE
It is now generally accepted that feeding the animals a diet high in fat, particularly when the fat is saturated, is associated with development of IR and accumulation of TG in muscle tiss~es.~-8 There was a variable effect on circulating TG levels, which may depend on the fat content and FA composition of the diet.8 Despite an evident effect of the high-fat diet on muscle TG levels, the role of dietary fat content on de novo fat synthesis and storage is not so transparent. It has been demonstrated that the inclusion of large amounts of saturated fat (from 5% to 25%) has a minimal effect on the activity of lipogenic enzymes in liver or on the rate of in vivo fatty acid syn-

202 ANNALS NEW YORK ACADEMY OF SCIENCES
thesis.29 Other data, however, have demonstrated that the de novo fatty acid synthesis in liver and lipogenesis are suppressed when the animals are fed a high-fat diet con- taining at least 30% fat.303 The divergence in these observations may be explained either by various fat content or by the fact that as much as 70% of the de novo fatty acid synthesis in the animals fed the high-fat diet takes place in the adipose tissue, as demonstrated earlier by Romsos and L e ~ e i l l e ~ ~ and later by Nelson et af.33 Sup- portive evidence for the inhibition of lipogenesis by a high-fat diet comes from recent data showing a decreased rate of fa@ acid synthesis in the liver and in white adipose tissue of adult rats, which is paralleled with decreased expression of FAS and ACC.MJ5
During the entire suckling period, the expression and activity of the major liver and white adipose tissue lipogenic enzymes directly involved in the lipogenic pathway- FAS, ACC, and ATP citrate lyase (ATP-CL) - were found to be negligible.28 The tran- sition from the high-fat diet of suckling to a high-complex-carbohydrate diet of lab- oratory chow led to increased TG in both tissues. The dramatic increase in liver FAS content after weaning to a highcarbohydrate/low-fat diet was associated with an ac- celeration of enzyme synthesis as measured by increased levels of tissue mRNA, but without a change in the rate of enzyme degradation. However, when the animals were weaned on a high-fat diet, the increase in lipogenesis was prevented by decreased expression and protein content of FAS, ACC, and ATP citrate lyase in adipose tissue and liver.28
Prolonged fasting (1-4 days) was found to be associated with a sharp (95%) de- crease of FAS mRNA in white epididymal adipose tissue27-36,37 and with a moderate decline in the liver.
In streptozotocin-induced diabetes associated with altered TG levels, gene expres- sion for lipogenic enzymes in both liver and white adipose tissue (WAT) was markedly decreased.38 On the contrary, the improvement of diabetes with vanadate treatment normalized mRNA levels only in liver, but not in WAT,3* supporting the role of tissue specificity for the regulation of lipogenesis. Refeeding and insulin treatment of streptozotocin-treated (STZ) diabetic rats normalized the activity and gene expression of lipogenic enzymes in both the adipose tissue and liver.38
Neonatal streptozotocin rat model of type I[ diabetes: In spite of increased plasma and TG content in skeletal muscle and in the heart, we did not observe any change in liver lipogenesis when indexed by ACC activity14 (TABLE 1).
In the hereditary hypertriglyceridemic (hHTG) rat, increased lipogenesis from tri- tiated water in the liver together with a mild rise in the secretion rate of TG from the liver (increase in post-Triton triglyceridemia) and the unchanged activity of the postheparin lipase indicate the hyperproductive nature of this form of hypertriglycer- idemia.39.40 In harmony with the data obtained in vivo, we also found increased ex- pression of FAS and ME in the liver of these animals. On the contrary, the activity of the other lipogenic enzyme ACC and the activity of ME were not changed in the liver of hHTG rats when compared to controls (TABLES 2 and 3).
Feeding the animals high-fructose or high-sucrose diets leads to hypertriglycer- idemia4Iq3 and to the accumulation of TG in mu~c les .9J~J5~~ The cascade of changes in lipid metabolism at high-sucrose or high-fructose intake is triggered in the liver
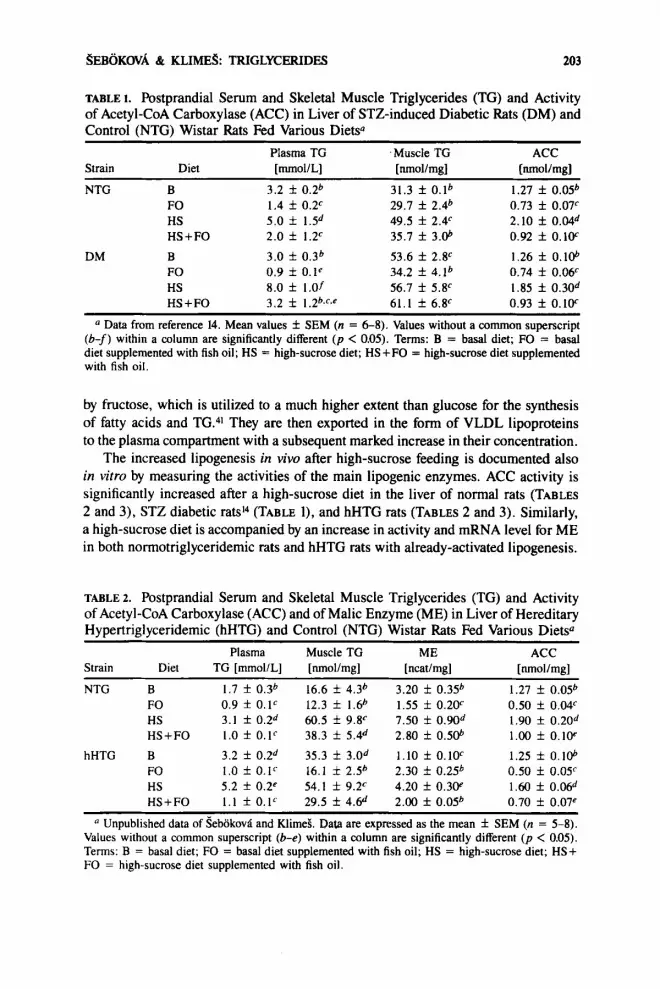
SEBOKOVA & IUIMES: TRIGLYCERIDES 203
TABLE 1. Postprandial Serum and Skeletal Muscle Triglycerides (TG) and Activity of Acetyl-CoA Carboxylase (ACC) in Liver of STZ-induced Diabetic Rats (DM) and Control (NTG) Wistar Rats Fed Various Diets0
Strain Diet [mmol/L] [nmol/mg] [nmol/mg] Plasma TG Muscle TG ACC
NTG B FO HS HS + FO
DM B FO HS HS+FO
3.2 f 0.2b 1.4 f 0.2c 5.0 f 1.5d 2.0 f 1.2c 3.0 f 0.3b 0.9 f O . l e 8.0 f 1.0f 3.2 f 1.2b~f~e
31.3 f O.lb 29.7 f 2.4b 49.5 f 2.4c 35.7 f 3.06
53.6 f 2 . F 34.2 f 4.1b 56.7 f 5.V 61.1 f 6.F
1.27 f 0.05b 0.73 f 0.07c 2.10 f 0.04d 0.92 f 0.1W 1.26 f 0. 106 0.74 f 0.mc 1.85 f 0.30d 0.93 f 0.1W
~~~~
Data from reference 14. Mean values f SEM (n = 6-8). Values without a common superscript (b-f) within a column are significantly different ( p < 0.05). Terms: B = basal diet; FO = basal diet supplemented with fish oil; HS = high-sucrose diet; HS+FO = high-sucrose diet supplemented with fish oil.
by fructose, which is utilized to a much higher extent than glucose for the synthesis of fatty acids and TG.41 They are then exported in the form of VLDL lipoproteins to the plasma compartment with a subsequent marked increase in their concentration.
The increased lipogenesis in vivo after high-sucrose feeding is documented also in vitro by measuring the activities of the main lipogenic enzymes. ACC activity is significantly increased after a high-sucrose diet in the liver of normal rats (TABLES 2 and 3), STZ diabetic ratsi4 (TABLE l), and hHTG rats (TABLES 2 and 3). Similarly, a high-sucrose diet is accompanied by an increase in activity and mRNA level for ME in both normotriglyceridemic rats and hHTG rats with already-activated lipogenesis.
TABLE 2. Postprandial Serum and Skeletal Muscle Triglycerides (TG) and Activity of Acetyl-CoA Carboxylase (ACC) and of Malic Enzyme (ME) in Liver of Hereditary Hypertriglyceridemic (hHTG) and Control (NTG) Wistar Rats Fed Various Diets0
Strain Diet TG [mmollL] [nmol/mg] [ncatlmg] [nmol/mg]
NTG B 1.7 f 0.3b 16.6 f 4.3b 3.20 f 0.35~~ 1.27 f 0.05b FO 0.9 f 0. lc 12.3 f 1.6b 1.55 f 0.2W 0.50 f 0.04c HS 3.1 f 0.2d 60.5 f 9 . F 7.50 f 0.90d 1.90 f 0.20d HS+FO 1.0 f O.lc 38.3 f 5.4d 2.80 f 0.506 1.00 f 0.1w
hHTG B 3.2 f 0.2d 35.3 f 3.0d 1.10 f O.lOc 1.25 f 0.1@ FO 1.0 f 0.lc 16.1 i 2.56 2.30 f 0.25b 0.50 f 0.0Y HS 5.2 f 0.2p 54.1 f 9.2E 4.20 f 0.3W 1.60 f 0.Md HS+FO 1.1 f O.Ic 29.5 f 4.6d 2.00 f 0.05b 0.70 f 0.07e
Unpublished data of sebokovii and KlimeS. Data are expressed as the mean f SEM (n = 5-8). Values without a common superscript (b-e) within a column are significantly different ( p < 0.05). Terms: B = basal diet; FO = basal diet supplemented with fish oil; HS = high-sucrose diet; HS + FO = high-sucrose diet supplemented with fish oil.
Plasma Muscle TG ME ACC

204 ANNALS NEW YORK ACADEMY OF SCIENCES
TABLE I Relative Abundance of mRNA for Malic Enzyme (ME) and Fatty Acid Syn- thase (FAS) in Liver and of Lipoprotein Lipase (LPL) in Skeletal Muscle of Hereditary Hypertriglyceridemic (hHTG) and Control (NTG) Wistar Rats Fed Various Diets"
ME mRNA FAS mRNA LPL mRNA Strain Diet [AUI [AUI WJI NTG B 0.47 f O.OSb 1.23 * 0.47b 6.6 f I . l b
FO 0.81 f 0.07b 1.85 * 0.45b 24.2 f 1.F HS 7.70 f 0.3W 14.1 f 0.68' 2.6 f 0.7d HS+FO 2.90 f 0.20d 5.64 f 0.34d 46.2 f 2Se
hHTG B 1.66 f 0.2W 2.76 f 0.22p 7.9 f 0.7b FO 2.48 f 0.4W 1.06 f O.3lb 26.2 f 2 .Y HS 10.4 f 0.28f 5.80 f 0.4Id 7.3 f 0.5b HS+FO 7.50 f 0.808 5.92 f 0.43d 24.4 f 1.Y
" Data from reference 37, except for the LPL data, which has not yet been published. Mean values f SEM (n = 4). Values without a common superscript (b-g) within a column are significantly different ( p < 0.05). Terms: B = basal diet; FO = basal diet supplemented with fish oil; HS = high-sucrose diet; HS+FO = high-sucrose diet supplemented with fish oil; AU = arbitrary units.
In animal models of IR associated with obesity, similar data on increased lipo- genesis in liver have been reported. Thus, an overexpression of genes together with increased activities for FAS, ACC, and ME have been found in the liver of ob/ob mice." In the genetically obese Zucker fa/fa rat, the increased lipogenic capacity of adipose tissue is due to the preferential channeling of nutrients into fatty acids and due to the activation of lipid storage-related enzymes. In particular, the expression of ME, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and FAS is stimulated in the adipose tissue of these rats. The overexpression of these genes is an early feature of the onset of obesity and is adipose tissue-specific since no differences are found in liver in comparison to normal rats.45
Taken together, the excessive dietary lipid supply associated with decreased ac- tivity and expression of lipogenic enzymes and/or increased endogenous lipid synthesis (after high-sucrose feeding or in other animal models of IR) may contribute to the development of hypertriglyceridemia. It should be noted here that the dietary manipu- lations lead to more pronounced changes in the activation of the iipogenic machinery than those that are hereditary-fixed.
REGULATION OF LIPOLYSIS
Alternatively, or in addition to an effect on the lipid biosynthetic pathway, the con- trol of TG and fatty acid availability may be accomplished through changes in lipolytic enzymes involved in TG metabolism. The main role for lipases is to release fatty acids and make them available as an alternative fuel in order to meet the energy demands of tissues. In IR, preferential oxidation of fatty acids instead of glucose is demonstrated in m ~ s c l e . ~ . ~ ~ The potential sources for the lipid oxidation in muscle are (1) free fatty acids (FFA) from adipose tissue lipolysis through the action of the adipocyte protein

SEBOKOVA & KLIMES: TRIGLYCERIDES 205
hormone-sensitive lipase, (2) plasma lipoprotein TG fatty acids released via lipoprotein lipase (LPL), and (3) fatty acids derived from the intramuscular TG droplet likely provided through the action of lipoprotein and hormone-sensitive lipase, but within the muscle tissue.48 Although it is assumed that most of the fatty acids oxidized by muscle come from adipose tissue in the form of FFA,49 all three sources may con- tribute and the relative role of each needs to be delineated. Recently, a growing body of evidence is in favor of the assumption that modulation of the tissue LPL activity is a major mechanism controlling the destination of TG transport.50
Following an overnight fast (when blood insulin concentrations are low), fat is mobilized from adipose tissue stores by adipose tissue hormone-sensitive lipase to release FFA into plasma, where they circulate complexed with albumin and are avail- able for uptake by tissues such as liver, skeletal muscle, and heart. Here, they can undergo oxidation or reesterification into TG.2' After a mixed meal, the rise in blood insulin levels inhibits adipose tissue lipolysis and restrains FFA mobilization from endogenous fat stores. Exogenous fat from the meal is packaged by the intestine and released as TG-rich chylomicrons. Lipoprotein lipase (LPL), a secretory glycoprotein made in a number of tissues including adipose tissue and skeletal and cardiac muscle,20 is transported to the glycocalyx of the capillary endothelium in the tissue of origin. Here, it carries out its hydrolytic function against the TG core of the TG-rich lipo- proteins (chylomicrons and VLDL). Following the release of lipolysis products, the generated FFA are taken up by muscle and adipose tissue where they can be oxidized or stored after reesterification, respec t i~e ly .~~.~~ The highest activities of LPL are found in tissues that oxidize (heart, various types of skeletal muscle) and esterify (adipose tissue, lactating mammary gland) large quantities of fatty acids.20
Feeding, particularly of carbohydrate-rich meals,53 is accompanied by elevated postprandial insulin levels, which results in activation of adipose tissue lipoprotein lipase (AT-LPL) and clearance of TG-rich lipoproteins. In contrast, the activity of LPL in skeletal muscle is decreased by i n ~ u l i n . ~ * ~ ~ ~ It is of interest that the magnitude of insulin-induced suppression of skeletal muscle LPL (delta SM-LPL) is inversely related to the insulin-mediated glucose uptake by the muscle.54 As both carbohydrate and lipid oxidation are balanced in skeletal muscle, these data suggest that the reg- ulation of LPL by insulin in skeletal muscle is appropriate for the metabolic needs of the tissue.
In insulin-resistant states, an alternative regulation of LPL exists. Numerous studies have described a decrease in the LPL catalytic activity in adipose tissueSS and post- heparin plasma56 of NIDDM patients who are under poor glycemic control. Improved diabetes control reverses the defect in LPL activity-due to an increase in LPL synthesis - and reduces serum TG levels.57 Yost and EckeP have also demonstrated that the in- hibition of AT-LPL by fat feeding usually seen in normal-weight subjects is no longer seen after isocaloric maintenance of the reduced obese state. Moreover, the response of AT-LPL to insulin is shifted to the right in obesity and is further diminished in NIDDM patients, suggesting that the greater degree of IR in NIDDM generates the lesser response.48
In skeletal muscle, insulin action also relates to the lipase response to insulin.

206 ANNALS NEW YORK ACADEMY OF SCIENCES
SM-LPL is increased to a greater degree by insulin in obese versus lean subjects. Moreover, when SM-LPL is plotted against the amount of glucose needed to maintain euglycemia during the hyperinsulinemic euglycemic clamp, an inverse correlation is seen. This suggests the following: when SM-LPL is further increased by insulin, then a greater amount of lipid and less glucose are oxidized.48 Positive correlations between fasting LPL activity and the glucose infusion rate (amount of glucose needed to main- tain euglycemia) have been demonstrated in skeletal muscle, but not in adipose tissue, when the data of control, nondiabetic-obese, and obese NIDDM have been analyzed together. However, the relative contribution of the intramuscular pool of TG fatty acids to meet the needs of skeletal muscle for lipid oxidation is not apparent.
Other studies aimed at the identification of possible factors contributing to the development of hypertriglyceridemia in subjects with obesity and/or IR have revealed that the ability of noradrenaline to stimulate lipolysis in fat cells in normal-weight subjects with obese first-degree relatives is markedly decreased.” The decreased lipo- lytic function of catecholamines in adipose tissue of these subjects was probably due, at least in part, to the decreased function of hormone-sensitive lipase (HSL) as demon- strated by suppressed levels of HSL in fat cells.59 One may therefore speculate that impaired function of HSL is an early metabolic disturbance that may later lead to development of hyperlipidemia, obesity, and IR, especially under the conditions of increased energy intake.
A decreased ability of catecholamines to stimulate lipolysis was demonstrated also in fat cells from obese elderly men suffering from the metabolic syndrome.@ Marked reduction of the maximum lipolytic response found in these patients was due to mul- tiple defects in the lipolytic cascade. Both (i) reduced expression of 02 receptors leading to decreased binding of the hormone to the cell surface and (ii) decreased ability of CAMP to activate the hormone-sensitive lipase in adipocytes might contribute to the development of severe manifestations of the IR syndrome in these subjects.6o With the recent development of highly sophisticated methods of molecular biology,
using high-quality antibodies and the cDNA probes, more detailed studies on the reg- ulation of lipolysis have become possible. In harmony with this, it was demonstrated in both humans61 and dents62 that changes in tissue LPL activity can be accompanied by changes at the transcriptional and translational level.
There are several situations in humans and in rodents as well where LPL activity is controlled at the level of gene expression.63 The continuous modulation of adipose tissue LPL activity in response to feeding-fasting cycles is also associated with changes in mRNA levels, but these are often not of the same magnitude as the changes in LPL activity.@ Furthermore, the changes in mRNA levels occur more slowly than the changes in LPL activity.65 After a 16-hour fasting, LPL activity in guinea pig ad- ipose tissue was found to be decreased by 8056, while LPL mass dropped only by 30%,50 implying that posttranscriptional mechanisms are involved. In contrast, the increase of heart LPL activity during fasting occurred without any change in LPL mRNA nor in the rate of LPL synthesis, suggesting that in the heart other mechanisms are involved in the modulation of functional LPL activity.49

SEBOKOVA & KLIMES: TRIGLYCERIDES 207
REGULATION OF LIPOLYSIS IN ANIMAL MODELS OF INSULIN RESISTANCE
Low LPL activity and decreased LPL mRNA levels are found in the adipose tissue of streptozotocin-diabetic rats.66 Such defects in adipose tissue LPL are hypothesized to be the rate-limiting step in the development of hypertriglyceridemia in STZ-induced diabetes." The results on heart LPL activity and mRNA levels, which were not changed in STZ-diabetic rats, further confirm the previous observation on the tissue specificity for LPL regulation at the gene level. However, it is more likely that the LPL expressed in adipose tissue is more important than that in the heart for the de- velopment of hypert~iglyceridemia.~.~~ Other studies looking at the mechanisms of hypertriglyceridemia in STZ-diabetic rats have shown that the slow lipolysis of TG- rich particles with postheparin plasma lipase may be the basis of the increased TG levels in circulation of these animals in the fed state, but not during fasting.68
Acute administration of fructose in vivo, contrary to the administration of an equal amount of glucose, does not enhance the activity of LPL in adipose tissue of fasting rats,69 despite the fact that in v i m both glucose and fructose enhance the LPL activity in incubated adipose tissue of fasting ratsm Feeding the high-fructose or high-sucrose diets to rats in chronic experiments has revealed conflicting data, showing an increase, decrease, or no change in the adipose tissue LPL (for review, see reference 71). As these dietary treatments differed in the amount of sugar and in the duration of feeding, it may be speculated that different kinetics of LPL activation may be responsible for the aforementioned fact. This explanation is in agreement with the data of Vr i r~a ,~ ' who demonstrated a slight decrease in LPL activity in adipose tissue after fructose feeding to rats, which was dependent on the duration of feeding. However, when the data on LPL activity were compared to the data obtained in rats fed the basal lab- oratory chow, an increase in adipose tissue LPL activity was demonstrated.7' In con- trast, the activity of LPL in heart muscle and diaphragm of these animals was increased when compared to rats fed either glucose or standard laboratory chow, no matter the duration of the feeding. Since the activity of this lipolytic enzyme represents the acute activation under particular metabolic conditions of the organism, it seems more ap- propriate, especially in long-term dietary manipulations, to look at the expression of LPL in tissues.
Using a two-week dietary protocol, we have been able to show that the expression of LPL in skeletal muscle was decreased after high-sucrose feeding when compared to the data obtained in rats fed the basal diet (TABLE 3). When the hHTG rats (with increased TG availability in muscle) were fed the high-sucrose diet, LPL mRNA levels were not changed in skeletal muscle (TABLE 3). Moreover, compared with the data of Vrina,7' there is an uncoupling between the LPL activity and the LPL mRNA levels in muscle after high-sucrose feeding. This is in contrast to the studies on LPL reg- ulation during changes in nutritional status represented by fasting-feeding. Here, the activity of LPL in adipose tissue and in muscle was found to be related to the level of expression for LPL mRNA in those tissues.n

208 ANNALS NEW YORK ACADEMY OF SCIENCES
An alteration of LPL activity in adipose tissue and in the heart was demonstrated in rats with an intake of high-fat diets rich in saturated fatty acids.34~739~4 The data of Benhizia et al. 34 on high-lard diet-induced inhibition of adipose tissue LPL activity, together with the striking opposite increase in plasma chylomicron concentrations, further support the important role of adipose tissue LPL in the control of TG levels. In other experiments, in spite of no changes in the fasting LPL activity in white and brown adipose tissue, it was shown that the LPL activity in both types of fat was significantly stimulated postprandially in response to high-carbohydrate and high-fat diets, respectively." Tissue specificity of LPL regulation was further demonstrated in the heart and skeletal muscle, where increased fasting levels of LPL were found in high-k-fed animals, but where LPL was suppressed postprandially in skeletal muscle, although not in the heart.73
In the hereditary hypertriglyceridemic (hHTG) rat, it seems that overproduction of VLDL TG due to the increased lipogenesis in the liver,37~~~ together with a defect in adipose tissue lipolysis (without affecting the postheparin lipoprotein lipase activity), could be the cause of hype~triglyceridemia.~~ In harmony with this, increased adipose tissue lipolysis was found in the hHTG line, leading to an increase in glycerol and in FFA concentrations in the circulation. The markedly enhanced FFA mobilization, accompanied by only a mild rise in serum FFA concentrations, indicates that an in- creased turnover of FA, when combined with hypertriglyceridemia, may lead to pref- erential utilization of lipids and to IR. However, the expression of LPL measured as mRNA levels in skeletal muscle of hHTG rats (TABLE 3) was not changed even after feeding the animals a high-sucrose diet.
In animal models of IR associated with obesity, increased lipolysis in adipose tissue has been demonstrated. In addition, increased activity," together with increased gene expres~ion,~~ for LPL was found in adipose tissue of genetically obese Zucker falfa rats. Since the overexpression of the LPL gene is already present in adipose tissue in suckling rats, which do not have such a high degree of obesity, these data indicate that a disharmony in LPL regulation could be an early feature contributing later to the development of massive hyperlipidemia and obesity of these animals.
Since only about 50% of the FA that are oxidized in muscle originate from the circulation, FA released from the TG stored in muscle lipid droplets after the hydro- lysis are likely to be an important determinant of the preferential FA oxidation in the muscle in IR, especially in situations where increased TG storage is present. Evidence exists demonstrating that HSL, similar to that in adipose tissue, is present also in muscles and that this lipase could be responsible for intracellular lipolysis of TG.77 Furthermore, correlations between intracellular lipolysis and LPL activity suggest a coordinated regulation of HSL and LPL in muscle to provide FA as the fuel for meeting the energy demands of the muscle. However, at the time when HSL is ap- parently hydrolyzing intracellular TG, the LPL is sent to the capillary beds in search of substrate.77 Despite the availability of anti-HSL antibody and a specific cDNA, the regulation of this interesting enzyme with high potential for determining the muscle TG availability has not yet been extensively investigated at the molecular level.

SEBOKOVA & KLIMES: TRIGLYCERIDES 209
DIETARY FATTY ACIDS AS DETERMINANTS OF TG AVAILABILITY
Focus on n-3 PUFA
In addition to the amount of fat in the diet, it is generally accepted that FA com- position has a profound effect on the development of hypertriglyceridemia and IR.7-9J2 Thus, much attention has been paid to PUFA, with special focus on n-3 FA. It has been known for more than two decades that dietary fish oil (FO) rich in highly un- saturated eicosapentaenoic acid (205 n-3) and docosahexaenoic acid (22:6 n-3) de- creases plasma TG levels in healthyI6 as well as hypertriglyceridemic subjects. Con- cerning its hypolipidemic potential, it was also shown that it is much more effective than equal amounts of polyunsaturated vegetable oils, rich in n-6 fatty acids.79 In ad- dition to the hypotriglyceridemic effect of FO in serum, which is not associated with accumulation of TG in the liver,m these long-chain n-3 FA have the potential to sup- press the accumulation of TG in the heart and more importantly in skeletal muscle when included in high-sucrose or high-fat diets.8.9J4J5 The TG-lowering effect of n-3 fatty acids may result from decreased FA synthesis, decreased TG synthesis or se- cretion, increased clearance, or some combination of these factors.
Studies using diets with n-3 fatty acids fed to normal rats clearly indicate that the activities of several lipogenic enzymes are markedly reduced25*35s37 when compared with saturated FA diets and also when compared to diets rich in PUFA of the n-6 family. The reduced activities of lipogenic enzymes are a result of decreases in the quantity of enzyme present and, at least in the case of FAS and ACC, this is a result of a decline in the amount of mRNA for FAS and ACC due to decreased transcription of the respective genes.81.82
The composition of dietary FA seems to be of great importance also during the nutritional transition from the high-fat diet of suckling to the high-carbohydrate diet of weaning.28 In these experiments, weaning to the high-carbohydrate diet with a high content of long-chain PUFA prevented the increase in FAS and ACC mRNA and/or enzyme activities that occurred normally in the liver.28 Interestingly, there was no effect of dietary FA on lipogenic gene expression in white adipose tissue.
We have recently completed studies37 in which saturated beef tallow or fish oil (Activepa TG) was fed to rats in diets containing a high amount (70 cal W ) of sucrose (TABLES 2 and 3). We found that feeding diets high in n-3 PUFA led to a striking reduction of ME activity and of mRNA for ME and FAS in the liver of these high- sucrose diet-fed rats. In addition, although the inhibition of hepatic lipogenesis by PUFA was evident during the high-sucrose feeding, dietary FA composition had no effect on lipogenesis as indexed by ME gene expression in rat epididymal adipose tissue. 37
Further supportive evidence for the possibility that the hypotriglyceridemic effect of fish oil in sucrose-induced hypertriglyceridemia is due to the inhibition of lipo- genesis was obtained from correlation analyses of the circulating TG levels and the

210 ANNALS NEW YORK ACADEMY OF SCIENCES
activity of lipogenic enzymes. These analyses revealed positive correlations between the circulating TG levels and the activity of ACC (r = 0.65, n = 16, p < 0.01) and ME (r = 0.44, n = 16, p < 0.05) in the liver (FIGURES 1A and 1B).
In another set of experiments, we have investigated whether altered activity and expression of lipogenic enzymes may account for the hypotriglyceridemic effect of FO under the condition of hereditary-fixed hypertriglyceridemia as seen in hHTG rats. Feeding the hHTG rat a diet high in n-3 PUFA suppressed the expression of FAS in liver. However, no effect on FAS gene expression was observed in hHTG rats when the hypertriglyceridemia was further exaggerated by high-sucrose feeding (TABLE 3). nor did the high content of n-3 FA in the diet suppress the ME mRNA level in liver of hHTG rats. In contrast, when the hHTG rats were fed a high-sucrose diet supple- mented with fish oil, a striking reduction of ME mRNA and activity in liver of hHTG rats was demonstrated. Also, here, the results of the correlation analysis ( r = 0.76, n = 16, p < 0.001) between ACC activity and plasma TG were consistent with the notion that the TG-lowering effect of FO may be mediated through the inhibition of lipogenic enzymes (data not shown).
A similar suppression of ACC in liver was also found when the n-3 FA were fed as a supplement to the basal or high-sucrose diet to rats with neonatal streptozotocin- induced type II diabetesI4 (TABLE 1). Moreover, the ACC activities correlated with the circulating TG levels (r = 0.75, n = 16, p < 0.001) in STZ-diabetic animals fed various FO-supplemented diets (data not shown).
The findings that diets enriched in n-3 FA reduce the activities and expression of enzymes required for FA synthesis are of particular interest because de novo syn- thesis of FA is a major determinant of hepatic TG s e ~ r e t i o n . ~ ~ The effect of dietary n-3 FA on synthesis and secretion of TG has been studied in vivo in both humansu and a n i m a l ~ , ~ ~ J ~ as well as in isolated hepatocytes87 and perfused livers.88 Most of these studies have agreed that n-3 FA suppress TG synthesis and secretion. Based on the results of Rustans9 and others, who found the inhibition of diacylglycerol acyl- transferase by n-3 FA, it was suggested that dietary FO reduces plasma TG by com- bining inhibition of FA synthesis (by suppression of the enzymes for de novo synthesis), inhibition of TG synthesis (probably through the inhibition of diacylglycerol acyltrans- ferase), and inhibition of TG secretion.
Another possible mechanism for lowering the TG level by dietary FO could be that the long-chain n-3 FA are preferentially oxidized and are thus unavailable for incorporation into the circulating or tissue TG pool.19 If increased oxidation is to play the key role in decreasing the levels of TG, then the activity of LPL in muscle has to be changed to allow for increased delivery of FA to the tissues for oxidation. In support of this hypothesis are the results of Herzberg and Rogerson,m who have doc- umented that LPL is increased in skeletal muscle and heart, but not in adipose tissue of rats fed a fructose-based diet containing FO. This information has been confirmed by Baltzell et al. ,91 who found that the activity of LPL was higher in the soleus muscle of rats fed menhaden oil compared with corn oil, although there was no effect on the hepatic lipase.
The role of dietary long-chain n-3 FA for modulation of the LPL activity has been further documented in our laboratory. We measured the expression of LPL in skeletal

A
n-1
6
r = 0
.a
p 0.
01
0 I
4
M+
FO
B 12
,
e 3 6
-
H f w
4-
2-
0
0 /
0,
I
I I
I
0 1
2 3
4 5
6 7
0 1
2 3
4 5
SrU
mTg
[mm
ol.C
‘] Se
rum
To
[mm
ol.r’
]
FIGURE 1.
Cor
rela
tion
anal
ysis
betw
een
hepa
tic a
ctiv
ity o
f ac
etyl
-CoA
car
boxy
lase
(A) o
r m
alic
enz
yme (B) a
nd s
erum
trig
lyce
ride
(Tg)
con
cent
ratio
n in
ra
ts fe
d fis
h oi
l-sup
plem
ente
d di
ets.
Term
s: FO
= b
asal
die
t sup
plem
ente
d with
fish
oil;
HS
= h
igh-
sucr
ose d
iet;
HS+
FO =
hig
h-su
cros
e die
t sup
plem
ente
d with
fish
oil.

212 ANNALS NEW YORK ACADEMY OF SCIENCES
muscle of rats fed FO with or without a high-sucrose diet. In this experiment, the expression of LPL was increased in the soleus muscle when the n-3 FA were included in the basal or high-sucrose diet (TABLE 3). In addition, similar stimulation of LPL mRNA levels by n-3 FA was found in hereditary hypertriglyceridemic rats (TABLE 3).
The mechanisms by which dietary PUFA may alter the activity and expression of lipogenic and lipolytic enzymes have been difficult to discover. These processes may be regulated directly at the gene affecting the nuclear mechanisms that change the expression of various genes encoding enzymes involved in lipid metab- olism by controlling transcription, RNA processing, or mRNA stability. 24-26 Another interpretation may involve a long-term, adaptive modulation of membrane composi- tion that leads to changes in hormone signaling.92 Feeding dietary fats high in PUFA enriches cellular membranes with long-chain PUFA, which subsequently may alter the signaling mechanisms leading to the modification of key enzymes involved in lipid synthesis and metabolism.92
In summary, a good deal of evidence at the molecular and cellular level has now been accumulated to show that the FA composition of the diet is important and may be one of the critical regulators of TG availability. The key enzymes involved in TG synthesis and metabolism are part of the membrane compartments in the cell. Thus, it might be expected that specific changes in lipids (particularly to FA composition) could be involved in the regulation of their activity and expression in both physiolog- ical and pathological conditions. However, whether the accumulation of specific long- chain FA into tissue phospholipids will have a causal relationship to the regulation of TG availability remains to be established.
As dietary PUFA may regulate, at the gene level, the activity of lipogenic and lipo- lytic enzymes under particular dietary and genetic circumstances, it may be speculated that feeding PUFA in the early stages of development might reduce subsequent hyper- triglyceridemia and/or IR.
REFERENCES
1. REAVEN, G. M. 1988. Diabetes 37: 1595-1607. 2. DEFRONZO, R. A., R. C. BONAWNNA & E. FERRANNINI. 1992. Diabetes Care 15: 318-368. 3. FRAYN, K. N. 1993. Curr. Opin. Lipidol. 4: 197-204. 4. HOWARD, B. V. 1995. Diabetes Rev. 3: 423-432. 5. TASKINEN, M . 1995. Curr. Opin. Lipidol. 6: 153-154. 6. STANDL, E., N. LOTZ, T. DEXEL, H. JANKA & H. J. KOLB. 1980. Diabetologia 18: 463-469. 7. STORLIEN, L. H., A. B. JENKINS, D. J. CHISHOLM, W. S. PASCOE, S. KHOURI & E. W. KRAEGEN.
8. STORLIEN, L. H., D. A. PAN, A. D. KRIKETOS & L. A. BAUR. 1993. Ann. N.Y. Acad. Sci. 683:
9. KLIMES, I., E. S E B ~ K O V ~ , A. V R ~ N A & L. KAZDOV~. 1993. Ann. N.Y. Acad. Sci. 683: 69-81. 10. KLIMES, I., E. S E B ~ K O V ~ , A. MINCHENKO, A. VRANA, M. FICKOV~, M. HROMADOV~, E.
Svk~ovk, P. BOHOV & L. K A Z M V ~ . 1991. In Lipoproteins and Atherosclerosis, p. 55-62. Fischer. Jena.
11. STORWEN, L. H., E. W. KRAEGEN, D. J. CHISHOLM, G. L. FORD, D. G. BRUCE & W. S. PASCOE. 1987. Science 32: 885-888.
12. STORLIEN, L. H., L. A. BAUR, A. D. KRIKETOS, D. A. PAN, G. J. COONEY, A. B. JENKINS, G. D. GALVERT & L. V. CAMPBELL. 1996. Diabetologia 39: 621-631.
1991. Diabetes 40: 280-289.
82-90.

SEBOKoVA & KLIMES: TRIGLYCERIDES 213
13.
14.
15.
16. 17. 18. 19. 20. 21.
22 23 24. 25, 26,
27. 28. 29. 30. 31. 32. 33. 34.
35.
36.
37.
38. 39. 40.
41. 42. 43 I
44. 45.
46. 47. 48. 49. 50.
51. 52. 53. 54.
SEBOKOV~, E., I. KLIMES, M. HERMANN, M. HROMADOV~, P. BOHOV & M. HUETTINGER. 1992.
SEBOKOVL, E., I. KLIMES, R. Moss, P. STOLBA, M. WIERSMA & A. MITKOV~. 1993. Ann. N.Y.
SEBOKOV~, E., I. KLIMES, M. HERMANN, A. MINCHENKO, A. MITKOV~ & M. HROMADOV~.
CONNOR, W. E., C. A. DEFRANCESCO & S. L. CONNOR. 1993. Ann. N.Y. Acad. Sci. 683: 16-34. SIMOPOULOS, A. P. 1994. Nutr. Today Januarylkbruary: 12-16. WAKIL, S. J., J . K. STOOPS & V. C. JOSHI. 1983. Annu. Rev. Biochem. 52: 53. HERZBERG, G. R. 1991. Physiol. Pharmacol. 69: 1637-1647. ECKEL, R. H. 1989. N. Engl. J. Med. 320: 1060-1068. FLAKOLL, P., M. G. CARLSON & A. CHERRINGTON. 1996. In Diabetes Mellitus, p. 121-131.
HELLERSTEIN, M. K., R. A. NEESE & J. M. SCHWARZ. 1993. Am. J. Physiol. 265: E814-E819. ACHESON, K. J., Y. SCHUTZ & T. BESSARD. 1988. Am. J. Clin. Nutr. 48: 240-256. CLARKE, S. D. & S. ABRAHAM. 1992. FASEB J. 6 3146-3152. CLARKE, S. D. & D. B. JUMP. 1994. Annu. Rev. Nutr. 14: 83-98. KATSUDARA, A., N. IRITANI, H. FUKUDA, Y. MATSUMURA & N. NISHIMOTO. 1990. Eur. J. Bio-
IRITANI, N. 1992. Eur. J. Biochem. 205: 433-442. GIRARD, J., D. PERDEREAU, F. FOUFELLE. C. PRIP-BUUS & P. FERRE. 1994. FASEB J. 8: 36-42. HERZBERG, G. R. & N. JANMOHAMED. 1980. Br. J. Nutr. 43: 571-579. GODBOLE, V. & D. A. YORK. 1978. Diabetologia 14: 191-197. CLANDININ, M. T. & S. K. CHEEMA. 1996. Biochim. Biophys. Acta 1299: 284-288. ROMSOS, D. R. & G. A. LEVEILLE. 1974. Biochim. Biophys. Acta 360: 1-11. NELSON, G. J.. D. S. KELLEY, P. C. SCHMIDT & C. M. SERRATO. 1987. Lipids 22: 338-344. BENHIZIA, F., I. HAINAULT, C. SEROUCNE, D. LAGRANCE, E. HAJDUCH, C. GUICHARD, M. I.
MALEWIAK, A. QUIGNARD-BOULANGE, M. LAVAU & S. GRIGLIO. 1994. Am. J. Physiol. 267
HAINAULT, I., M. CARLOTTI, E. HAJDUCH, C. GUICHARD & M. LAVAU. 1993. Ann. N.Y. Acad.
BECKER, D. J., L. N. ONGEMBA, V. BRICHARD, J. C. HENQUIN & S. M. BRICHARD. 1995. FEBS
Diabetes Nutr. Metab. 5: 249-257.
Acad. Sci. 683: 218-227.
1993. Ann. N.Y. Acad. Sci. 683: 183-191.
Lippincott-Raven. Philadelphia/New York.
chem. 190: 435-441.
ESn5-E982.
Sci. 683: 98-101.
Lett. 371. 324-328. SEEOKOVi, E., I. KLIMES, D. GASPERfKOVk, P. BOHOV, P. LANGER, M. LAVAU & M. T. CLAN-
DININ. 1996. Biochim. Biophys. Acta l303: 56-62. BRICHARD, S. M., L. N. ONGEMBA, J. GIRARD & J. C. HENQUIN. 1994. Diabetologia 3 7 1065- 1072. V ~ N A , A. & L. KAZDOV~. 1990. Transplant. P m . 22: 2579. V R ~ N A , A., L. KAZDOV~, Z. DOBESOV~, J. KUNES, V. K ~ E N , V. BfLi, P. STOLBA & I. KLIMES.
V R ~ N A , A. & L. KAZDOV~. 1986. Prop. Biochem. Pharmacol. 2 1 59-73. V R ~ N A , A. & P. F ~ B R Y . 1983. World Rev. Nutr. Diet. 42: 5_6-101. KLIME~, I., M. FICKOV~, A. VR~NA, P. BOHOV, E. S V ~ B O V ~ , S. Z6RAD. L. SIKUROV~, L. KAZ-
D O V ~ , V. BALL? & L. MACHO. 1989. In Insulin and the Cell Membrane, p. 413-427. Hawood. Chur.
1993. Ann. N.Y. Acad. Sci. 683: 57-68.
V R ~ N A , A. & L. KAZDOV~. 1982. Nutr. Rep. Int. 26: 743-749. DUGAIL, I., A. QUIGNARD-BOULANGE, X. LE LIEPVRE, B. ARDOUIN & M. LAVAU. 1992. Bio-
RANDLE, P. J., P. B. GARLAND, C. N. HALES & E. A. NEWSHOLME. 1963. Lancet 1: 785-789. GROOP, L. C., C. SALORANTA, M. SHANK er al. 1991. J. Clin. Endocrinol. Metab. 72: 96-107. ECKEL, R. H., T. J. YOST & D. R. JENSEN. 1995. Int. J. Obes. 19: S16-S21. COPPACK, S. W., R. L. JUDD & J. M. MILES. 1993. Diabetes 42: 38A.
chem. J. 281: 607-611.
OLIVECRONA, T., M. BERGO, M. HULTIN & G. OLIVECRONA. 1995. Can. J. Cardiol. ll(supp1.): 73G-78G.
COPPACK, S. W., M. D. JENSEN & J. M. MILES. 1994. J. Lipid Res. 35: 177-193. FARESE, R. V., JR., T. J. Yosr & R. H. ECKEL. 1991. Metabolism 40: 214-216. PYKALISTO, 0. J., P. H. SMITH & J. D. BRUNZELL. 19’75. J. Clin. Invest. 5 6 1108-1117. KIENS, B., H. LITHELL, K. J. MIKINES & E. A. RICHTER. 1989. J. Clin. Invest. 84: 1124-1129.

214 ANNALS NEW YORK ACADEMY OF SCIENCES
55. TASKINEN, M. & E. A. NIKKIW. 1979. Diabetologia 17: 351-356. 56. NIKKILA, E. A., J. K. HUTTUNEN & C. EHNHOLM. 1977. Diabetes 26: 11-21. 57. SIMSOLQ R. B., J. M. ONG, B. SAFFARI & P. A. KERN. 1992. J. Lipid Res. 33: 89-95. 58. YOST, T. J. & R. H. Ecm~. 1988. J. Clin. Endocrinol. Metab. 67: 259-264. 59. HELLSTR~M. L., D. LANGIN, S. REYNISDOTTIR, M. DAUZATS & P. ARNER. 1996. Diabetologia
60. REYNISDOTTIR, S., K. ELLERFELDT, H. WAHRENBERG, H. LITHELL & P. ARNER. 1994. J. Clin.
61. KERN, P. A., J. M. ONG, B. SAFFARI & J. CARTY. 1990. N. Engl. J. Med. 322: 1053-1059. 62. BESSESEN, D. H., A. D. ROBERTSON & R. H. ECKEL. 1991. Am. J. Physiol. 261: E246-E251. 63. BRAUN, J. E. A. & D. L. SEVERSON. 1992. Biochem. J. 287: 337-347. 64. DOOLITTLE, M. H., 0. BEN-ZEEV, J. ELOVSON, D. MARTIN & T. G. KIRCHGESSNER. 1990. J.
65. SEMB, H. & T. OIJVECRONA. 1989. Biochem. J. 262: 505-511. 66. TAVANGAR, K., Y. MURATA, M. E. PEDERSEN, J. F. GOERS, A. R. HOFFMAN & F. B. KRAEMER.
67. INADERA, H., J. TOSHIRO, Y. OKUBO, Y. ISHIKAWA, K. SHIRAI, Y. SAITO & S. YOSHIDA. 1992.
68. MAMO, J. C. L., T. HIRANO, A. SAINSBURY, A. K. FITZGERALD & T. G. REDGRAVE. 1992. Bio-
69. BAR-ON, H. & Y. STEIN. 1968. J. Nutr. 94: 95-105. 70. WEBB, W., P. J. NESTEL, C. FOXMAN & A. LYNCH. 1970. Nutr. Rep. Int. 1: 189-195. 71. V ~ N A , A., P. F ~ B R Y & L. KAZDOV~. 1974. Nutr. Metab. 17: 282-288. 72. LADU, M. J., H. KAPSAS & W. K. PALMER. 1991. Am. J. Physiol. 260: R953-R959. 73. BOIVIN, A., I. MONTPLAISIR & I. DESHAIES. 1994. Am. J. Physiol. 267: E620-E627. 74. DE GASQUET, P., S. GRIGLIO, E. PEQIJIGNOT-PLANCHE & M. I. MALEWIAK. 1979. J. Nutr. 1Cn:
75. VR~NA, A. & L. KAZDOVA. 1997. This volume. 76. BRAY, G. A., J. S. STER & T. W. CASTONGUAY. 1992. Am. J. Physiol. 261 E32-E39. 77. OSCAI, L. B., A. D. ESSIG & W. K. PALMER. 1990. J. Appl. Physiol. 6 9 1571-1577. 78. GOODNIGHT, S. H., JR., W. S. HARRIS, W. E. CONNOR & D. ILLINGWORTH. 1982. Arterioscle-
79. HARRIS, W. S., W. E. CONNOR & M. P. MCMURRY. 1983. Metab. CIin. Exp. 32: 179-184. 80. GARG, M. L., A. B. R. THOMSON & M. T. CLANDININ. 1989. Biochim. Biophys. Acta 1006:
81. BLAKE, W. L. & S. D. CLARKE. 1990. J. Nutr. 120 1727-1729. 82. GOODRIDGE, A. G. 1987. Annu. Rev. Nutr. 7: 157-185. 83. TOPPING, D. L., R. P. TRIMBLE & G. B. STORER. 1987. Biochim. Biophys. Acta 927: 423-428. 84. ILLINGWORTH, D., W. S. HARRIS & W. E. CONNOR. 1984. Arteriosclerosis 4 270-275. 85. LOMBARDO, Y. B., A. CHICCO, M. E. DALESSANDRO, M. MARTINELU, A. SORIA & R. GUTMAN.
86. HERZBERG, G. R. & M. ROGERSON. 1988. J. Nutr. 118: 1061-1067. 87. WONG, S., M. REARDON & P. NESTEL. 1985. Metabolism 34: 900-905. 88. WONG, S. H., P. H. NESTEL, R. P. TRIMBLE, G. B. STORER, R. I. ILLMAN & D. L. TOPPING.
89. RUSTAN, A. C., J. 0. NOSSEN, E. N. CHRISTIANSEN & C. A. DREVON. 1988. J. Lipid Res. 29:
90. HERZBERG, G. R. & M. ROGERSON. 1989. Lipids 24: 351-353. 91. BALTZELL, J. K., J. T. -EN & D. A. OTTO. 1991. Lipids 26: 289-294. 92. CLANDININ, M. T., S. CHEEMA, C. J. FIELD, M. L. GARG, J. VENKATRAMAN & T. R. CLAN-
39: 921-928.
Invest. 93: 2590-2599.
Biol. Chern. 265: 4570-4577.
1992. J. Clin. Invest. 90: 1672-1678
Scand. J. Clin. Lab. Invest. 52: 797-802.
chim. Biophys. Acta 1128: 132-138.
199-212.
rosis 2: 87-113.
127-130.
1996. Biochim. Biophys. Acta 1299: 175-182.
1984. Biochim. Biophys. Acta 792: 103-109.
1417-1426.
DININ. 1991. FASEB J. S: 2761-2769.





























