Embed Size (px)
Citation preview

ORIGINAL PAPER
Molecular phylogeography of two sibling species of Euremabutterflies
Satoko Narita Æ Masashi Nomura Æ Yoshiomi Kato ÆOsamu Yata Æ Daisuke Kageyama
Received: 3 November 2006 / Accepted: 12 December 2006 / Published online: 11 January 2007� Springer Science+Business Media B.V. 2007
Abstract The common yellow butterfly Eurema hec-
abe is widely distributed in East Asia, and is one of the
most burdensome species for taxonomists due to the
numerous geographic and seasonal wing colour pat-
terns. Moreover, within this species, individuals with a
yellow wing fringe that occur in temperate regions of
Japan (Y type) proved to be biologically different from
others that occur widely in subtropical regions of Japan
and all over East Asia (B type). To unveil the genetic
variation within and between the two types, a total of
50 butterflies collected at 18 geographic localities in
East Asia were examined for nucleotide sequence
variation of three mitochondrial regions: cytochrome c
oxidase subunit I (COI), cytochrome c oxidase subunit
III (COIII) and NADH dehydrogenase subunit 5
(ND5). In addition, they were also examined for
infection status with the endosymbiotic bacteria
Wolbachia. The three mitochondrial sequences con-
sistently showed that (i) Y type and B type were highly
divergent, (ii) nucleotide variation within B type was
very small although sampled from a geographically
wide range, and (iii) a weak association existed be-
tween mitochondrial DNA haplotypes and Wolbachia
infection status.
Keywords Cytochrome c oxidase subunit I (COI) �Cytochrome c oxidase subunit III (COIII) � NADH
dehydrogenase subunit 5 (ND5) � Butterfly � Eurema
hecabe � Mitochondrial DNA � Phylogeography �Sibling species � Wolbachia
Introduction
The common yellow butterfly, Eurema hecabe
(Linnaeus, 1758) (Lepidoptera: Pieridae), is distributed
almost all over the Oriental, Australian and Afro-
tropical Regions, and extends into the temperate zone
of the Eastern Palaearctic Region such as the northern
part of Honshu, Japan (up to 40�N) (Yata 1989, 1995).
Numerous seasonal and geographic variations of wing
colour pattern in E. hecabe have confounded taxono-
mists, and have resulted in many subspecies and forms
(Yata 1995). But Yata (1989), found that the black
distal border on the forewing upperside and the
markings on the underside show a continuous clinal
variation with latitude. Based on this finding, he inte-
grated these subspecies into the nominotypical E.
hecabe hecabe (Yata 1989).
However, Kato and Handa (1992) found that tem-
perate and subtropical populations of Japan differed
in expression of the polyphenism in response to
S. Narita � M. NomuraLaboratory of Applied Entomology and Zoology,Faculty of Horticulture, Chiba University, Matsudo, Chiba271-8510, Japan
Y. KatoDepartment of Biology, International Christian University,Mitaka, Tokyo 181-8585, Japan
O. YataBiosystematics Laboratory, Faculty of Social and CulturalStudies, Kyushu University, Ropponmatsu, Fukuoka 810-8560, Japan
D. Kageyama (&)Insect-Microbe Research Unit, National Institute ofAgrobiological Sciences (NIAS), Owashi 1-2, Tsukuba,Ibaraki 305-8634, Japane-mail: [email protected]
123
Genetica (2007) 131:241–253
DOI 10.1007/s10709-006-9134-1

photoperiod and temperature. Following this discov-
ery, it was demonstrated that the temperate popula-
tions and the subtropical ones of E. hecabe are distinct
from each other in a number of traits: use of host
plants (Kato et al. 1992), colouration of wing fringe
(Kato 1999), reflection pattern against ultra-violet
rays (Matsuno 1999), allelic frequencies of allo-
zymes (Nomura and Kato 1993), and nuclear locus
Tpi (Narita et al. 2006); moreover, they are repro-
ductively isolated through selective mate choice by
females, although hybridization can occur when
reared in the laboratory (Kato 2000b; Kobayashi et al.
2001). These data consistently and strongly suggested
that the temperate populations with a yellow wing
fringe (Y type) and the subtropical populations with
a brown wing fringe (B type) represent two closely
related but biologically different species (Kato and
Yata 2005).
Molecular phylogenetic analyses of Y-type and
B-type populations in Japan showed that, unlike
nuclear DNA sequences, mitochondrial DNA
(mtDNA) sequences did not represent the diver-
gence of the two types. Indeed, mtDNA clearly di-
verged into two clades, and one of them contained
only the Y type. Curiously, however, the other clade
contained both types. All the butterflies in the latter
clade were infected with a single strain of endo-
symbiont Wolbachia (wHec1), while all the butter-
flies in the former clade were uninfected. These
findings suggest that the B-type mitochondria have
introgressed into Y type through hybridization in
the past (Narita et al. 2006). Similarly, Wolbachia
and its associated mitochondria were suggested to
have experienced introgression in other insect species
such as Acraea butterflies, Drosophila fruit flies and
Solenopsis fire ants (Jiggins 2003; Dean et al. 2003;
Shoemaker et al. 2003).
Wolbachia, belonging to the alpha subdivision of
Proteobacteria, is ubiquitously found in arthropods
and filarial nematodes. Wolbachia can affect repro-
duction or sex determination of its insect hosts in
various ways; e.g. via cytoplasmic incompatibility,
parthenogenesis induction, male killing or feminiza-
tion (O’Neill et al. 1997; Werren 1997; Stouthamer
et al. 1999: Bourtzis and Miller 2003, 2006). In
general, Wolbachia are transmitted exclusively from
mothers to offspring, i.e. not from fathers. It can be
considered that these reproductive alterations are
elegant strategies adopted by Wolbachia in order to
increase their transmission efficiency through host
generations.
In a Y-type population in Okinawa, a subtropical
southwestern island of Japan, feminized genetic males
producing almost exclusively female progeny were
found (Hiroki et al. 2002). These females were infected
with two different strains of Wolbachia, wHec1 and
wHec2, while normal females were infected with only
wHec1. It has been known that wHec1 is indistin-
guishable by means of a wsp sequence from the cyto-
plasmic incompatibility-inducing Wolbachia strain
found on mainland Japan (Hiroki et al. 2004; Narita
et al. 2006).
The purpose of this study was to answer the fol-
lowing questions. (1) What is the extent of divergence
between the Y and B types? (2) What is the extent of
variability of mtDNA sequences within the B type? (3)
Is mtDNA variation within the B type associated with
biogeography? (4) Is Wolbachia infection associated
with mtDNA variation?
Keeping these questions in mind, we conducted
molecular phylogenetic analyses of Y and B type but-
terflies collected from 18 geographic localities of seven
countries in East Asia using three mitochondrial
regions. Furthermore, all samples used in this study
were diagnosed to ascertain the status of Wolbachia
infection and examined for its possible effects on the
phylogenetic relationship within and between the two
types of E. hecabe.
Materials and methods
Butterfly sampling and DNA extraction
In total, 49 adults of E. hecabe were collected from 18
geographic localities in East Asia (Table 1, Fig. 1).
Two or more butterflies were obtained from each
locality except for Japan where a single butterfly was
available per location. Field-collected adults were
stored at –20�C until DNA extraction; a small number
of dry specimens were also used. The butterflies were
divided into Y or B type based on the fringe colour
of the forewings (Kato 1999, 2000a). E. blanda from
Iriomote Island and Kanchanaburi (Table 1) was used
as outgroup. Dissected thoracic muscles were subjected
to DNA extraction by using the DNeasy Tissue Kit
(Qiagen).
PCR and sequencing
The mitochondrial COI region, encoding cytochrome
c oxidase subunit I, was amplified by the primers
C1-J-1718 (5¢-GGGGGGTTTGGAAATTGATTAGT
GCC-3¢) and TL2-N-3014 (5¢-TCCATTGCACTAA
TCTGCCATATTA-3¢) (Simon et al. 1994). The mito-
242 Genetica (2007) 131:241–253
123
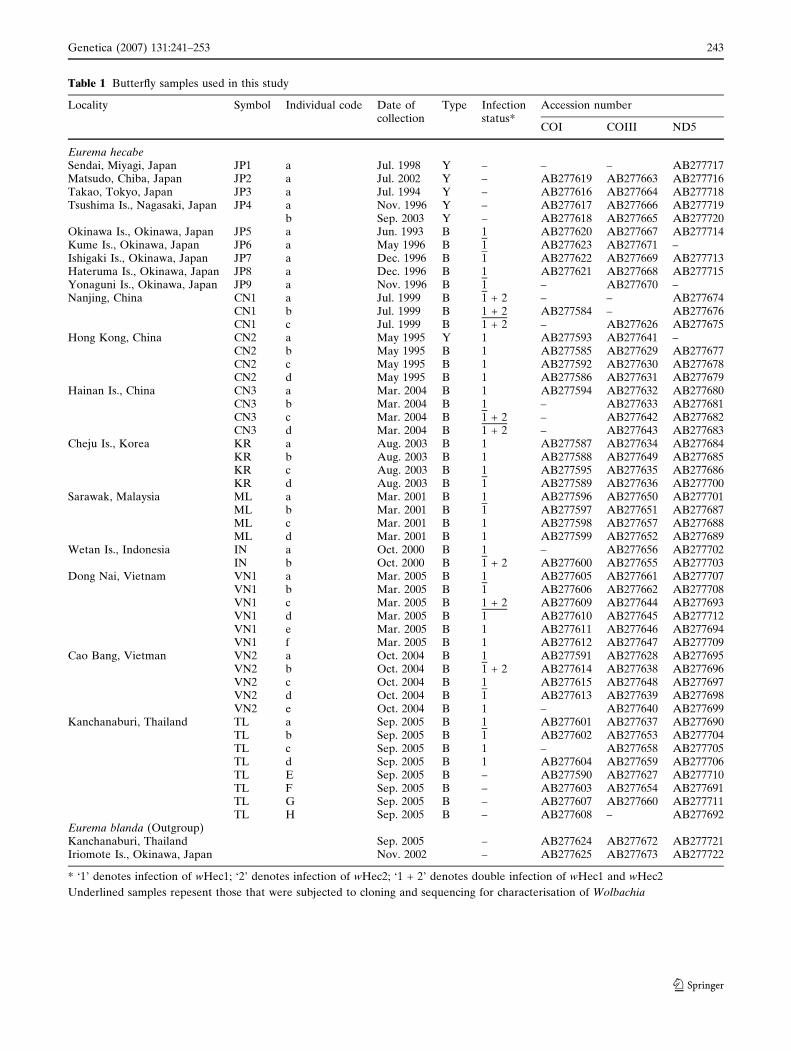
Table 1 Butterfly samples used in this study
Locality Symbol Individual code Date ofcollection
Type Infectionstatus*
Accession number
COI COIII ND5
Eurema hecabeSendai, Miyagi, Japan JP1 a Jul. 1998 Y – – – AB277717Matsudo, Chiba, Japan JP2 a Jul. 2002 Y – AB277619 AB277663 AB277716Takao, Tokyo, Japan JP3 a Jul. 1994 Y – AB277616 AB277664 AB277718Tsushima Is., Nagasaki, Japan JP4 a Nov. 1996 Y – AB277617 AB277666 AB277719
b Sep. 2003 Y – AB277618 AB277665 AB277720Okinawa Is., Okinawa, Japan JP5 a Jun. 1993 B 1 AB277620 AB277667 AB277714Kume Is., Okinawa, Japan JP6 a May 1996 B 1 AB277623 AB277671 –Ishigaki Is., Okinawa, Japan JP7 a Dec. 1996 B 1 AB277622 AB277669 AB277713Hateruma Is., Okinawa, Japan JP8 a Dec. 1996 B 1 AB277621 AB277668 AB277715Yonaguni Is., Okinawa, Japan JP9 a Nov. 1996 B 1 – AB277670 –Nanjing, China CN1 a Jul. 1999 B 1 + 2 – – AB277674
CN1 b Jul. 1999 B 1 + 2 AB277584 – AB277676CN1 c Jul. 1999 B 1 + 2 – AB277626 AB277675
Hong Kong, China CN2 a May 1995 Y 1 AB277593 AB277641 –CN2 b May 1995 B 1 AB277585 AB277629 AB277677CN2 c May 1995 B 1 AB277592 AB277630 AB277678CN2 d May 1995 B 1 AB277586 AB277631 AB277679
Hainan Is., China CN3 a Mar. 2004 B 1 AB277594 AB277632 AB277680CN3 b Mar. 2004 B 1 – AB277633 AB277681CN3 c Mar. 2004 B 1 + 2 – AB277642 AB277682CN3 d Mar. 2004 B 1 + 2 – AB277643 AB277683
Cheju Is., Korea KR a Aug. 2003 B 1 AB277587 AB277634 AB277684KR b Aug. 2003 B 1 AB277588 AB277649 AB277685KR c Aug. 2003 B 1 AB277595 AB277635 AB277686KR d Aug. 2003 B 1 AB277589 AB277636 AB277700
Sarawak, Malaysia ML a Mar. 2001 B 1 AB277596 AB277650 AB277701ML b Mar. 2001 B 1 AB277597 AB277651 AB277687ML c Mar. 2001 B 1 AB277598 AB277657 AB277688ML d Mar. 2001 B 1 AB277599 AB277652 AB277689
Wetan Is., Indonesia IN a Oct. 2000 B 1 – AB277656 AB277702IN b Oct. 2000 B 1 + 2 AB277600 AB277655 AB277703
Dong Nai, Vietnam VN1 a Mar. 2005 B 1 AB277605 AB277661 AB277707VN1 b Mar. 2005 B 1 AB277606 AB277662 AB277708VN1 c Mar. 2005 B 1 + 2 AB277609 AB277644 AB277693VN1 d Mar. 2005 B 1 AB277610 AB277645 AB277712VN1 e Mar. 2005 B 1 AB277611 AB277646 AB277694VN1 f Mar. 2005 B 1 AB277612 AB277647 AB277709
Cao Bang, Vietman VN2 a Oct. 2004 B 1 AB277591 AB277628 AB277695VN2 b Oct. 2004 B 1 + 2 AB277614 AB277638 AB277696VN2 c Oct. 2004 B 1 AB277615 AB277648 AB277697VN2 d Oct. 2004 B 1 AB277613 AB277639 AB277698VN2 e Oct. 2004 B 1 – AB277640 AB277699
Kanchanaburi, Thailand TL a Sep. 2005 B 1 AB277601 AB277637 AB277690TL b Sep. 2005 B 1 AB277602 AB277653 AB277704TL c Sep. 2005 B 1 – AB277658 AB277705TL d Sep. 2005 B 1 AB277604 AB277659 AB277706TL E Sep. 2005 B – AB277590 AB277627 AB277710TL F Sep. 2005 B – AB277603 AB277654 AB277691TL G Sep. 2005 B – AB277607 AB277660 AB277711TL H Sep. 2005 B – AB277608 – AB277692
Eurema blanda (Outgroup)Kanchanaburi, Thailand Sep. 2005 – AB277624 AB277672 AB277721Iriomote Is., Okinawa, Japan Nov. 2002 – AB277625 AB277673 AB277722
* ‘1’ denotes infection of wHec1; ‘2’ denotes infection of wHec2; ‘1 + 2’ denotes double infection of wHec1 and wHec2
Underlined samples repesent those that were subjected to cloning and sequencing for characterisation of Wolbachia
Genetica (2007) 131:241–253 243
123

chondrial COIII region, encoding cytochrome c oxidase
subunit III, was amplified by the primers Cox3-F2 (5¢-TCAGCTGTTGCTATAATTCAA-3¢) and Cox3-R2
(5¢-TATGATTGGAAGTCAAATATA-3¢) (designed
by N. Haruyama). The mitochondrial ND5 region,
encoding NADH dehydrogenase subunit 5, was ampli-
fied by using the primers V1 (5¢-CCTGTTT
CTCTGCTTTAGTTTAGTTCA-3¢) and A1 (5¢-AA-
TATDA GGTATAAATCATAC-3¢) (Yagi et al.
1999).The PCR temperature profile for the mitochon-
drial genes was 94�C for 2 min, followed by 35 cycles of
94�C for 1 min, 47�C for 1.5 min and 72�C for 1.5 min,
and finally 72�C for 7 min. The PCR products were
purified using the PCR Purification Kit (Qiagen), and
directly subjected to DNA sequencing.
The nucleotide sequences of E. hecabe and E. blanda
were deposited in the DDBJ/EMBL/GenBank data-
bases. Accession numbers are listed in Table 1.
Phylogenetic analyses
Nucleotide sequences were aligned using the program
package ClustalW (Thompson et al. 1994). The se-
quence data of COI, COIII and ND5 regions was anal-
ysed separately. Phylogenetic trees were constructed by
the maximum likelihood (ML) and the maximum par-
simony (MP) methods using the program package
PAUP version 4.0b10 (Swofford 2001). For ML and MP
analyses, the F81 model was used to estimate the num-
ber of nucleotide substitutions. To visualize genealogical
relationships and possible population substructure, net-
works on the basis of sequence data were constructed by
the statistical parsimony algorithm (Templeton et al.
1992) implemented in the program package TCS version
1.13 (Clement et al. 2000). The TCS program calculates
the minimal number of mutational steps by which the
sequences can be joined with >95% confidence.
Fig. 1 Collection localities ofthe Y and B types of E.hecabe used in this study.Locality symbols representthose in Table 1. Scale barindicates 1000 km
244 Genetica (2007) 131:241–253
123

Detection and identification of Wolbachia
PCR detection of Wolbachia infection was performed
using the primers (5¢-TTGTAGCCTGCTATGGTA-
TAACT-3¢) and (5¢-GAATAGG TATGATTTT-
CATGT-3¢) for the 16S rRNA gene (O’Neill et al.
1992), and the primers wsp81F (5¢-TGGTCCAA-
TAAGTGATGAAGAAAC-3¢) and wsp691R (5¢-AA
AAATTAAACGCTACTCCA-3¢) for the wsp gene
(Zhou et al. 1998).
Infection with Wolbachia of either wHec1 or wHec2
strain was discriminated by specific PCR detection
targeting the wsp gene using the primers wsp81F and
WHecFem1 (5¢-ACTAACGTCGTTTTTGTTTAG-3¢:reverse) (designed by Y. Tagami) that amplify the 232
bp DNA fragment for wHec1, and primers WHecFem2
(5¢-TTACTCACAATTGGCTAAAGAT-3¢: forward)
(designed by Y. Tagami) and wsp691R that amplify the
398 bp DNA fragment for wHec2 under temperature
profile 35 cycles of 95�C for 1 min, 58�C for 1.5 min
and 72�C for 1.5 min, and then followed by 72�C for
7 min.
For characterization of Wolbachia strains, one or
two butterflies were used from each locality. A 0.6 kb
segment of wsp gene was amplified using the primers
wsp81F and wsp691R, and the PCR products were
cloned and subjected to DNA sequencing. In addition,
the diagnostic PCR product of the wsp gene (398 bp)
for wHec2 was ligated to the T-easy vector (Promega),
and transformed into E. coli JM109 competent cells.
Inserted plasmids were cloned and extracted from the
transformants, and subsequently subjected to DNA
sequencing using an ABI Prism Big DyeTM terminator
chemistry and ABI 3100 capillary sequencer (Applied
Biosystems).
Nucleotide sequences were aligned with published
Wolbachia sequences from other insects using the
program package ClustalW. Phylogenetic trees were
constructed by the ML methods using the PAUP
4.0b10.
Estimation of nucleotide diversity
Nucleotide diversity (P), i.e. the degree of polymor-
phism within a population, is defined as the average
number of nucleotide differences per site between any
two DNA sequences chosen randomly from the sample
population (Nei and Li 1979). In each mitochondrial
region, nucleotide diversity within double-infected,
single-infected and uninfected butterflies was calcu-
lated using the program package DnaSP (Rozas et al.
2003).
Results
COI region
Haplotype sequences
Forty butterflies (5 of the Y type and 35 of the B type)
sequenced for the COI region, 1055 bp in size, were
polymorphic in 51 nucleotide sites including 34 parsi-
moniously informative sites and constituted 22 haplo-
types.
Maximum parsimony phylogeny
On the phylogeny, 19 haplotypes constituted a mono-
phyletic group exclusively containing B-type butter-
flies, whereas 3 haplotypes of the Y type constituted a
clade although the bootstrap value was low (52%). On
the other hand, Y-type haplotypes, except for that of
CN2-a (from Hong Kong), formed a strongly sup-
ported clade (98%) (Fig. 2a).
Maximum likelihood phylogeny
On the phylogeny, 19 haplotypes constituted a mono-
phyletic group exclusively containing B type butter-
flies, whereas 3 haplotypes of the Y type formed a
clade, although the bootstrap value was low (42%).
Conversely, Y-type haplotypes, except for that of CN2-
a, formed a strongly supported clade (98%) (Fig. 2b).
Statistical parsimony network
TCS calculated a 95% parsimony connection limit of
14 steps. Three clades containing the Y type, CN2-a,
and B-type haplotypes, could not be connected within
this limit. One position in the TCS network had a
closed loop that could not unambiguously be resolved.
This loop was caused by a single homoplastic align-
ment position (Fig. 2c).
COIII region
Haplotype sequences
Forty-eight butterflies (6 of the Y type and 42 of the B
type) sequenced for the COIII region, 736 bp in size,
were polymorphic in 44 nucleotide sites, including 29
parsimoniously informative sites and constituting 21
haplotypes.
Genetica (2007) 131:241–253 245
123

E. blanda (Kanchanaburi)
E. blanda (Iriomote Is.)
Hong KongTokyo
Matsudo
Tsushima Is. Tsushima Is.
Wetan Is. Kanchanaburi
NanjinHong KongHong KongCheju Is. Cheju Is. Cheju Is. KanchanaburiCao Bang
Hong KongHainan Is. Cheju Is.
SarawakCao Bang
Hateruma Is.
Kanchanaburi
Okinawa Is. Kume Is.
Kanchanaburi
KanchanaburiKanchanaburi
Dong Nai
Dong NaiDong NaiDong NaiCao Bang
Kanchanaburi e
pyt-
Ye
pyt-
B
SarawakSarawakSarawak
Dong NaiDong Nai
Cao Bang
Ishigaki Is.
CN1-b
CN2-b
CN2-d
KR-a
KR-b
KR-d
TL-e
VN2-a
CN2-c
CN3-a
KR-c
ML-a
ML-b
ML-c
ML-d
VN2-b
VN2-c
JP8
JP7
TL-g
TL-f
TL-b
TL-a
VN1-d
VN2-d
VN1-c
IN-b
TL-h
VN1-e
VN1-f
JP5
JP6
VN1-a
VN1-b
TL-d
JP2
JP4-b
JP4-a
JP3
CN2-a
99
52
98
62
87
86
10 changes
100
E.blanda (Kanchanaburi)E.blanda (Iriomote Is.)
TokyoTsushima Is.Tsushima Is.
Hong Kong
Matsudo
0.005 substitutions/site
SarawakSarawakSarawak
Dong NaiDong Nai
Sarawak
Okinawa Is. Kume Is.
Kanchanaburi
Wetan Is. Kanchanaburi
NanjinHong Kong
Hong KongCheju Is. Cheju Is. Cheju Is. KanchanaburiCao Bang
Hateruma Is.
Ishigaki Is.
Cao Bang
Cao Bang
Hong Kong
Kanchanaburi
KanchanaburiKanchanaburi
Dong NaiDong NaiDong NaiCao Bang
Dong Nai
Hainan Is. Cheju Is.
100
99
42
98
48
48
50
64
61
89
86
ep y
t-Y
ep y
t-B
CN1-bCN2-b
CN2-dKR-aKR-bKR-d
TL-e
VN2-aCN2-cCN3-aKR-c
ML-aML-bML-c
ML-d
VN2-b
VN2-cJP8
JP7
TL-g
TL-fTL-b
TL-aVN1-d
VN2-dVN1-c
IN-bTL-h
VN1-eVN1-f
JP5JP6
VN1-aVN1-b
TL-d
JP2JP4-bJP4-a
JP3
CN2-a
Kanchanaburi
B-type
JP2JP3
JP4-aJP4-b
CN2-a
Y-type
TL-d
CN1-b
CN2-b
CN2-d
TL-a
TL-g
ML-d
TL-h
VN1-c
IN-b
CN3-a
CN2-c
KR-c
JP7JP8
VN1-d
VN1-eVN1-f
VN2-d
VN2-bVN2-c
KR-a
KR-b
KR-d
TL-e
VN2-a
Eurasian Continent
Cheju-Is.
JP5
JP6
Polynesia
Japan
TL-bTL-fML-b
ML-c
ML-a
VN1-aVN1-b
a b
c
Fig. 2 Molecular phylogenetic analyses based on COI regionsequences (1055 aligned nucleotide sites) of the Y type and Btype of E. hecabe. (A) Maximum likelihood phylogenetic tree. A50% majority-rule consensus bootstrap tree is shown, withbootstrap values over 50% at the nodes. The tree has a loglikelihood of –1919.25. (B) Maximum parsimony phylogenetictree. The tree has consistency index of 0.865 and retention indexof 0.875. The bootstrap values over 50% are shown at the nodes.
(C) TCS parsimony network. Ellipses represent individualbutterflies (white: uninfected, grey: single-infected with wHec1,black: double-infected with wHec1 and wHec2). Rectanglesrepresent haplotypes. The size of each rectangle reflects anumber of butterflies. Small black circles represent missinghaplotypes. A network with 95% connection limit is shown. Thesequences are labelled according to the locality as shown inTable 1 and Fig. 1
246 Genetica (2007) 131:241–253
123

Maximum parsimony phylogeny
On the phylogeny, 18 haplotypes, constituted a
monophyletic group, exclusively containing B-type
butterflies, whereas 3 haplotypes of the Y type con-
stituted a clade which was weakly supported (59%).
On the other hand, Y-type haplotypes, except for that
of CN2-a, formed a strongly supported clade (100%)
(Fig. 3a).
Maximum likelihood phylogeny
On the phylogeny, 18 haplotypes, constituted a
monophyletic group, exclusively containing B-type
butterflies, whereas 3 haplotypes of the Y type con-
stituted a clade which was weakly supported (58%).
On the other hand, Y-type haplotypes, except for that
of CN2-a, formed a strongly supported clade (100%)
(Fig. 3b).
Statistical parsimony network
TCS calculated a 95% parsimony connection limit of
11 steps. Each of the three clades containing Y-type,
CN2-a, and B-type haplotypes could not be connected
within this limit. One position in the TCS network had
a closed loop that could not unambiguously be re-
solved. This loop was associated with combinations of
three homoplastic alignment positions (Fig. 3c).
ND5 region
Haplotype sequences
Forty-seven butterflies (five of the Y type and 42 of the
B type) sequenced for the ND5 region, 716 bp in size,
were polymorphic in 38 nucleotide sites, including 25
parsimoniously informative sites and constituted 21
haplotypes. A sample from Hong Kong (CN2-a) could
not be contained in this phylogenetic tree because we
failed to amplify the ND5 region.
Maximum parsimony phylogeny
On the phylogeny, the haplotypes were split into two
discrete clades. One clade with five haplotypes con-
sisted exclusively of Y type butterflies from Japan
(strongly supported by the bootstrap probability of
99%), whereas the other clade with 14 haplotypes
exclusively consisted of the B type (strongly supported
with bootstrap probability of 86%) (Fig. 4a).
Maximum likelihood phylogeny
On the phylogeny, the haplotypes were split into two
discrete clades. One clade with five haplotypes con-
sisted exclusively of the Y type from Japan (strongly
supported by the bootstrap probability of 97%),
whereas the other clade with 14 haplotypes exclusively
consisted of the B type (strongly supported with
bootstrap probability of 100%) (Fig. 4b).
Statistical parsimony network
TCS calculated a 95% parsimony connection limit of
11 steps. Two clades containing the Y-type and B-type
haplotypes could not be connected within this limit.
One position in the TCS network revealed a closed
loop that could not unambiguously be resolved. This
loop was associated with combinations of four homo-
plastic alignment positions (Fig. 4c).
Wolbachia endosymbiont
Diagnostic PCR
Of 43 B-type adults examined by diagnostic PCR for
Wolbachia, 39 (90.9%) were positive. Of 6 Y-type
adults, one from Hong Kong (CN2-a) was positive
(Table 1). In addition, of all the 40 infected samples
subjected to diagnostic PCR for discriminating be-
tween wHec1 and wHec2, seven were double-infected
with wHec1 and wHec2, while 33 were single-infected
with wHec1 (Table 1).
Characterisation of Wolbachia strains
One or two Wolbachia-infected adults from each
locality, i.e. 15 adults (JP5, JP6, JP8, JP9, CN1-b, CN3-
b, CN3-c, KR-c, ML-a, IN-a, VN1-a, VN1-c, VN2-a,
VN2-c, TL-a), were subjected to PCR that amplifies
the ca. 600 bp segment of wsp gene by using primer
pair wsp81F and wsp691R followed by cloning and
DNA sequencing.
About 71 clones in total, i.e. three to eight clones
from each of the 15 Wolbachia-infected butterflies
(including 12 single-infected and three double-infected
butterflies), were identical to that of wHec1 (Hiroki
et al. 2004). Since no wHec2-specific sequence was
found from the three double-infected butterflies (24
clones in total), the wHec2-specific PCR product
of the wsp gene (398 bp) in each of the double-
infected butterflies (Table 1) was directly subjected to
DNA sequencing. The sequences were identical to
Genetica (2007) 131:241–253 247
123

10 changes
Miyagi MatsudoTokyoTsushima Is.
Hong Kong
Tsushima Is.
Hong Kong Hong Kong Hong Kong Hainan Is. Hainan Is. Cheju Is. Cheju Is. Cheju Is. Kanchanaburi Cao Bang Cao Bang Cao Bang
Hainan Is. Cheju Is. Wetan Is.
SarawakSarawak Kanchanaburi Kanchanaburi Yonaguni Is.
Ishigaki Is. Hateruma Is.
Hainan Is. Dong NaiDong NaiDong NaiDong NaiCao Bang
SarawakWetan Is.
Sarawak
KanchanaburiDong NaiOkinawa Is.
Dong Nai
Kanchanaburi Kanchanaburi
Kume Is.
Nanjing KanchanaburiCao Bang
59
100
96
62
E.blanda (Kanchanaburi)E.blanda (Iriomote Is.)
epy
t -Y
epy
t -B
CN3-d
CN2-b
CN2-d
KR-a
KR-b
KR-d
TL-eVN2-a
CN2-c
CN3-a
KR-c
ML-a
ML-b
ML-c
ML-d
VN2-b
VN2-e
JP8JP7
JP4-a
TL-fTL-b
TL-a
VN1-d
VN2-d
VN1-c
IN-bTL-g
TL-c
VN1-a
JP5
JP6
VN1-e
VN1-b
VN1-f
CN1-c
CN3-c
TL-d
CN2-c
JP9
CN3-b
IN-a
CN2-aJP1JP2
JP3JP4-b
0.005 substitutions/site
E.blanda (Kanchanaburi)E.blanda (Iriomote Is.)
Hong Kong
MigagiMatsudoTokyoTsushima Is. Tsushima Is.
100
58
Hong Kong Hong Kong Hong Kong Hainan Is. Hainan Is. Cheju Is. Cheju Is. Cheju Is. Kanchanaburi Cao Bang Cao Bang Cao Bang
Hainan Is. Dong NaiDong NaiDong NaiDong NaiCao Bang
Sarawak Wetan Is.
Sarawak
KanchanaburiDong NaiOkinawa Is.
Kanchanaburi Kanchanaburi Dong NaiKume Is.
Nanjing KanchanaburiCao Bang
Hainan Is.
Sarawak Sarawak Kanchanaburi Kanchanaburi
Cheju Is. Wetan Is.
61Yonaguni Is.
Ishigaki Is. Hateruma Is.
90
100
ep y
t -Y
epy
t -B
CN3-d
CN2-b
CN2-d
KR-a
KR-b
KR-d
TL-eVN2-a
CN2-c
CN3-a
KR-c
ML-a
ML-b
ML-c
ML-d
VN2-b
VN2-e
JP8JP7
JP4-a
TL-fTL-b
TL-a
VN1-d
VN2-d
VN1-c
IN-b
TL-g
TL-c
VN1-a
JP5
JP6
VN1-e
VN1-b
VN1-f
CN1-c
CN3-c
TL-d
CN2-c
JP9
CN3-b
IN-a
CN2-a
JP1JP2
JP3JP4-b
ML-c
JP7JP8
CN1-c
TL-eVN2-a
ML-b
ML-dTL-b
TL-f
TL-g
JP9
ML-a
IN-bIN-a
JP5
JP6
CN3-c
VN1-a
VN1-b
TL-cTL-d
CN3-d
VN1-c
VN1-d
VN1-e
VN1-f
VN2-c
B-type
JP1
JP2JP3
JP4-bJP4-a
CN2-aY-type
Japan
Polynesia
Eurasian Continent
CN2-b
CN2-c
CN2-d
CN3-a
CN3-bKR-a
KR-c
KR-d
TL-a
VN2-b
VN2-d
VN2-e
KR-b
Cheju Is.
a b
c
Fig. 3 Molecular phylogenetic analyses based on the COIIIregion (736 aligned nucleotide sites) of the Y type and B type ofE. hecabe. (A) Maximum likelihood phylogenetic tree shown asin Fig. 2. The tree has a log likelihood of -1731.86. (B) Maximum
parsimony phylogenetic tree shown as in Fig. 2. The tree hasconsistency index of 0.809 and retention index of 0.806. (C) TCSparsimony network shown as in Fig. 2
248 Genetica (2007) 131:241–253
123

Matsudo
TokyoTsushima Is.
Tsushima Is.
Miyagi
99
Nanjing Hong Kong Hong Kong Hong Kong Hainan Is. Hainan Is. Hainan Is. Hainan Is. Cheju Is. Cheju Is. Cheju Is. Sarawak Sarawak Sarawak Kanchanaburi Kanchanaburi Kanchanaburi Don NaiDon NaiCao Bang Cao Bang Cao Bang Cao Bang Cao Bang
Wetan Is. Kanchanaburi Kanchanaburi Ishigaki Is.
Okinawa Is. Hateruma Is.
86
Nanjing
Cheju Is. Kanchanaburi
Sarawak Wetan Is. Kanchanaburi Don Nai
10 changes
56
64
62
E.blanda (Kanchanaburi)E.blanda (Iriomote Is.)
55
ep y
t-Y
epy
t-B
CN3-d
CN2-b
CN2-d
KR-aKR-b
KR-d
TL-e
VN2-a
CN2-c
CN3-a
KR-c
ML-a
ML-bML-cML-d
VN2-b
VN2-e
JP8
JP7
JP4-a
TL-f
TL-b
TL-a
VN1-d
VN2-d
VN1-c
IN-b
TL-g
TL-c
VN1-a
JP5
VN1-e
VN1-bVN1-f
CN1-c
CN3-c
TL-d
VN2-c
CN3-b
IN-a
JP1JP2
JP3JP4-b
CN1-b
TL-h
CN1-a
Kanchanaburi Don NaiDon NaiDon Nai
Nanjing
100
E.blanda (Kanchanaburi)E.blanda (Iriomote Is.)
0.005 substitutions/site
Matsudo
TokyoTsushima Is.
Tsushima Is.
Miyagi
Nanjing Hong Kong Hong Kong Hong Kong Hainan Is. Hainan Is. Hainan Is. Hainan Is. Cheju Is. Cheju Is. Cheju Is. Sarawak Sarawak Sarawak Kanchanaburi Kanchanaburi Kanchanaburi Don NaiDon NaiCao Bang Cao Bang Cao Bang Cao Bang Cao Bang
Nanjing Nanjing
KanchanaburiKanchanaburi Don NaiDon NaiDon Nai
Cheju Is.
Wetan Is. Wetan Is.
KanchanaburiKanchanaburiIshigaki Is.
Okinawa Is. Hateruma Is.
Sarawak Kanchanaburi Don Nai
100
97
62
56
60
62
epy
t-Y
epy
t -B
CN3-d
CN2-b
CN2-d
KR-aKR-b
KR-d
TL-e
VN2-a
CN2-c
CN3-a
KR-c
ML-a
ML-bML-cML-d
VN2-b
VN2-e
JP8
JP7
JP4-a
TL-f
TL-b
TL-a
VN1-d
VN2-d
VN1-c
IN-b
TL-g
TL-c
VN1-a
JP5
VN1-e
VN1-bVN1-f
CN1-c
CN3-c
TL-d
VN2-c
CN3-b
IN-a
JP1
JP2
JP3JP4-b
CN1-b
TL-h
CN1-a
a b
cB-type
IN-b IN-a
TL-e
ML-a
TL-b
KR-dTL-g
CN1-aCN1-c
CN1-b
CN3-c
CN3-d
CN3-b
CN3-a
CN2-dCN2-cCN2-b
ML-b
ML-c
ML-d
TL-aTL-fTL-h
KR-a
KR-b
KR-c
VN1-cVN1-eVN2-aVN2-b
VN2-c
VN2-d
VN2-e
TL-c
TL-d
VN1-aVN1-b
VN1-f
JP8
JP7
JP5
VN1-d
JP4-b
JP3
JP4-a
JP1
JP2
Y-type
Japan
Polynesia
Eurasian Continent
. sI
uje
hC
Fig. 4 Molecular phylogenetic analyses based on the ND5region (716 aligned nucleotide sites) of the Y type and B typeof E. hecabe. (A) Maximum likelihood phylogenetic tree shownas in Fig. 2. The tree has a log likelihood of -1283.32. (B)
Maximum parsimony phylogenetic tree shown as in Fig. 2. Thetree has consistency index of 0.897 and retention index of 0.932.(C) TCS parsimony network shown as in Fig. 2
Genetica (2007) 131:241–253 249
123

wHecFem2 (Hiroki et al. 2004). Each of the wsp se-
quences of wHec1 and wHec2 formed an independent
clade in the B supergroup of the Wolbachia phylogeny
(Fig. 5). The nucleotide sequences of wsp gene of
Wolbachia from the Y type and B type of E. hecabe
were deposited in the DDBJ/EMBL/GenBank data-
bases under accession numbers AB278198-AB278220,
respectively.
Comparison of nucleotide diversities
In each mitochondrial region, nucleotide diversity (P)
was compared between double-infected, single-in-
fected and uninfected butterflies (Table 2). In COI,
COIII and ND5, nucleotide diversity of Wolbachia-
infected butterflies, both double-infected and single-
infected, were likely to be smaller compared to that of
uninfected butterflies. Within double-infected butter-
flies, compared to that of single-infected butterflies,
nucleotide diversity was smaller in COIII and ND5, but
larger in COI.
Discussion
Mitochondrial DNA strongly supports the
distinction between Y and B type
It has been suggested that, in E. hecabe, the Y type is
biologically different from B type at species level based
on morphological, ecological, physiological, reproduc-
tive and genetic lines of evidence (Kato and Yata
2005). Our data of mitochondrial regions COI, COIII
and ND5, strongly supports the distinction of the two
types (Figs. 2–4).
Genetic distance (D), i.e. average number of
nucleotide substitutions per site, of ND5 between the
two types was calculated to be 0.03000 by using the
Kimura 2-parameter method (Kimura 1980). The
molecular evolutionary rate of ND5 in Parnassius
butterflies was estimated to be 0.01D per 0.75 million
years (Yagi et al. 2001). By extrapolating the evolu-
tionary rate, the divergence of the two types was esti-
mated to have occurred around 2.25 million years ago.
The mtDNA from one butterfly (CN2-a) from Hong
Kong was located at a peculiar position in phylogeny
(Figs. 2, 3). Although morphologically classified into
the Y type, i.e. wing fringe colour was yellow, the clade
including this sample was only supported with very low
bootstrap probabilities (COI: 52% for maximum like-
lihood, 42% for maximum parsimony; COIII: 59% for
maximum likelihood, 58% for maximum parsimony).
Apparently, this individual was closely related to either
Y-type or B-type but, at the same time, significantly
differentiated from either of the two types, presumably
Junonia almana almana[AB094384]Gronotoma micromorpha[AB037895]
Drosophila simulans Noumea [AF020074]Aedes albopictus [AF02005]
Trichogramma kaykai [AF071912]Cadra cautella[AF020075]
Encarsia formosa[AF071918]Cadra cautellaB [AF020076]
0.05 substitutions/site
Tytthus chinensis [AF481171]Tribolium confusum [AF020083]
Liriomyza bryoniae [AB231469]
98
5498
100
71
98
77
100
Eurema hecabe Y-type [AB094396]Eurema hecabe B-type [AB278213~AB278220]Pseudagrion pruinosum [AY173942]Agromyza oryzae [AF481201]
Eurema hecabe Y-type [AB210826~AB210831]Eurema hecabe B-type [AB278198~AB278212]Culex pipiens [AF020061]Acraea encedon[AJ271198]Hypolimnas bolina [AJ307074]Hercinothrips femoralis [AB245521]Everes argiades[AB094390]
w Hec2
w Hec1
Fig. 5 Maximum likelihoodphylogeny on the basis of thewsp gene sequences ofWolbachia endosymbiontsdetected from E. hecabe andother insects. A 50%majority-rule consensusbootstrap tree is shown, withbootstrap values over 50% atthe nodes. Scientific names ofthe host insects are shown. Inbrackets are nucleotidesequence accession numbers
Table 2 Comparison of nucleotide diversity (p) between indi-viduals with different infection status
COI COIII ND5
Double-infected 0.00298 (7) 0.00257 (10) 0.00112 (12)Single-infected 0.00254 (24) 0.00297 (28) 0.00261 (26)Uninfected 0.00332 (4) 0.00362 (3) 0.00279 (4)
In parentheses are the numbers of samples sequenced
250 Genetica (2007) 131:241–253
123

representing an independent species. Considering that
Hong Kong is a pivotal area for global transportation,
this individual may be a stray butterfly belonging to
another species that occurs in other part of the world.
Construction of a complete global phylogeny of Eur-
ema butterflies will clarify this point.
Low level of mitochondrial DNA differentiation
within the B type despite being sampled from wide
geographic areas
E. hecabe is morphologically most variable among the
genus Eurema. Because of seasonal and geographic
variations, numerous subspecies have been described;
e.g. mandarina from Japanese mainland, and hobsoni
from Taiwan (see Yata 1995). Close inspection by Yata
(1989), however, revealed that almost all of these
variations are clinal ones according to latitude. Based
on this finding, he proposed an integration of numer-
ous subspecies into a single subspecies Eurema hecabe
hecabe.
In this study, although sampling was conducted from
a wide geographic area in East Asia (Fig. 1), the three
mitochondrial regions consistently indicated a small
genetic variation within the B type (Figs. 2–4). This is
the first molecular phylogenetic analysis that strongly
supports the notion that the B type, which occurs
widely throughout East Asia, represents one single
subspecies, as was previously proposed from morpho-
logical data (Yata 1989).
Biogeographical implications
Statistical parsimony network reflects genealogical
relationships of the mtDNA haplotypes, that is, single
mutation steps separate adjacent haplotypes in the
network, and older haplotypes are placed at internal
branching points whereas younger ones occur toward
the tip positions.
In each of the three mitochondrial regions, two
discrete parsimony networks which could not be con-
nected at statistically significant level were generated.
The two networks corresponded to the Y type and B
type, respectively. Despite the small sequence varia-
tion within the B type, the network revealed that pat-
terns of mtDNA haplotypes are more or less associated
with geographic locations. The most remarkable asso-
ciation among them comes from Japanese subtropical
populations samples, which consistently appeared at
tip positions; this finding is likely to reflect that these
haplotypes are relatively young. This may also reflect
that, in the B type, Japanese subtropical populations
are differentiated from other populations in East Asia.
Although less clear, many of the samples from the
Eurasian Continent were placed at most major and
central positions which are likely to be ancestral
(Figs. 2c, 3c, 4c).
B-type haplotypes showed a star-shaped pattern.
This pattern is commonly understood to be indicative
of a population expansion. The pattern of geographic
distribution was in agreement with the predictions
from the coalescent theory that assumes that older al-
leles would prevail in populations and be characterised
by a higher number of descending lineages and geo-
graphically wider distribution (Crandall and Temple-
ton 1993; Posada and Crandall 2001).
Two distinct strains of Wolbachia were shared by Y
type and B type
Diagnostic PCR revealed that 39 out of 43 B-type
butterflies were infected with Wolbachia. All of them
were infected with wHec1, and 12 of them were addi-
tionally infected with wHec2. It has been previously
suggested by molecular phylogenetic analysis that the
infection of wHec1 in the Y type is associated with
introgression of B-type cytoplasm through hybridiza-
tion followed by the Wolbachia sweep up to the central
Japan (Narita et al. 2006). Likewise, wHec2 in Y-type
butterflies that causes feminization had probably int-
rogressed from B-type butterflies, but somehow re-
mained in subtropical regions in Japan. Therefore, it is
not surprising that wHec1 and wHec2 are shared by
both types.
The result that no wHec2-specific sequence was
found from 24 wsp clones suggested that wHec2-posi-
tive butterflies were doubly infected with wHec1 and
wHec2 at the density ratio of at least 24. It should be
noted that other types of Wolbachia may also coexist in
a low density comparable to that of wHec2.
One point that should be kept in mind is that the
reproductive phenotype of wHec1 and wHec2 in B-
type butterflies is unknown. It has been demonstrated
in Y-type butterflies that the occurrence of wHec1 is
involved in cytoplasmic incompatibility while co-
occurrence of wHec1 and wHec2 is involved in femi-
nization (Hiroki et al. 2004, 2005). It is unknown,
however, whether these Wolbachia strains in the B
type behave exactly as in a Y-type background. More
fundamentally, there is a possibility that wHec1 and
wHec2 in B-type butterflies may not be identical as
those in Y-type butterflies, since we relied exclusively
on wsp sequence for genotyping Wolbachia. Further
studies are required to clarify the genotype and
Genetica (2007) 131:241–253 251
123

reproductive phenotype of these Wolbachia strains in a
B-type background.
Curiously, CN2-a from Hong Kong was infected
with wHec1. Having a distinct type of mtDNA, the
presence of wHec1 cannot be explained by introgres-
sion. The presence of wHec1 in this type/species would
be an important cue in revealing the infection history
of Wolbachia in E. hecabe butterflies.
Did Wolbachia affect population structure
of B-type butterflies?
A recent theory suggests that maternally-inherited
endosymbionts, such as bacteria of the genus Wolba-
chia, may affect host mtDNA genome evolution (Prout
1994; Turelli 1994; Hurst et al. 1996; Johnstone and
Hurst 1996), and several empirical studies directly
examining the effects of Wolbachia on mtDNA varia-
tion largely support this hypothesis (Turelli et al. 1991;
Hoffmann et al. 1994; Shoemaker et al. 1999; Marcade
et al. 1999; Ballard 2000; Ballard et al. 2002).
This effect of Wolbachia, the so-called Wolbachia
sweep, has also been shown in Y-type butterflies
occurring on mainland Japan (Narita et al. 2006). In B-
type butterflies examined in this study, however, such
Wolbachia effects on mtDNA variation were not as
conspicuous as in the above studies. Still, mtDNA
variation in the B type is likely to be weakly associated
with the presence of Wolbachia, although the sample
size was not large enough to draw a firm conclusion.
The nucleotide diversity of Wolbachia-infected but-
terflies, both double-infected and single-infected, was
likely to be smaller than that of uninfected butterflies
(Table 2), presumably reflecting a Wolbachia sweep.
This tendency was consistently seen in all the mito-
chondrial regions examined. Between double-infected
and single-infected butterflies, on the other hand, such
tendency was not observed consistently. A larger
sample will reveal more in detail the association of
Wolbachia-infection status with variation of mtDNA in
E. hecabe.
Acknowledgments We would like to thank Hironori Sakamotofor helpful discussion. This study was financially supported inpart by the Japan Society for the Promotion of Science (JSPS)fellowship for Young Scientists to S. N.
References
Ballard JW (2000). Comparative genomics of mitochondrialDNA in Drosophila simulans. J Mol Evol 51:64–75
Ballard JW, Chernoff B, James AC (2002) Divergence ofmitochondrial DNA is not corroborated by nuclear DNA,
morphology, or behaviour in Drosophila simulans. Evolu-tion 56:527–545
Bourtzis K, Miller TA (2003) Insect symbiosis. CRC Press, BocaRaton, Florida, U.S.A
Bourtzis K, Miller TA (2006) Insect symbiosis Vol. 2. CRC Press,Boca Raton, Florida, U.S.A
Clement M, Posada D, Crandall KA, (2000) TCS: a computerprogram to estimate gene genealogies. Mol Ecol 9:1657–1659
Crandall KA, Templeton AR (1993) Empirical tests of somepredictions from coalescent theory with applications tointraspecific phylogeny reconstruction. Genetics 134:959–69
Dean MD, Ballard KJ, Glass A, Ballard JMO (2003) Influence oftwo Wolbachia strains on population structure of eastAfrican Drosophila simulans. Genetics 165:1959–1969
Hiroki M, Kato Y, Kamito T, Miura K, (2002) Feminization ofgenetic males by a symbiotic bacterium in a butterfly,Eurema hecabe (Lepidoptera: Pieridae). Naturwissenschaf-ten 89:167–170
Hiroki M, Tagami Y, Miura K, Kato Y (2004) Multiple infectionswith Wolbachia inducing different reproductive manipula-tions in the butterfly Eurema hecabe. Proc R Soc Lond B271:1751–1755
Hiroki M, Ishii Y, Kato Y, (2005) Variation in the prevalence ofcytoplasmic incompatibility-inducing Wolbachia in the but-terfly Eurema hecabe across the Japanese archipelago. EvolEcol Res 7:931–942
Hoffmann AA, Clancy DJ, Merton E, (1994) Cytoplasmicincompatibility in Australian populations of Drosophilamelanogaster. Genetics 136:993–999
Hurst LD, Atlan A, Bengtsson BO, (1996) Genetic conflicts. QRev Biol 71:317–364
Jiggins FM, (2003) Male-killing Wolbachia and mitochondrialDNA: selective sweep, hybrid introgression and parasitepopulation dynamics. Genetics 164:5–12
Johnstone RA, Hurst GDD, (1996) Maternally inherited male-killing microorganisms may confound interpretation ofmitochondrial DNA variability. Biol J Linn Soc Lond58:453–470
Kato Y, (1999) Fringe colour, seasonal morph and host-plant useof the Pierid butterfly Eurema hecabe (L.) (Lepidoptera,Pieridae) on Okinawa-jima Island. Trans Lepid Soc Jpn50:111–121 (in Japanese with English summary)
Kato Y, (2000a) Overlapping distribution of two groups of thebutterfly Eurema hecabe differing in the expression ofseasonal morphs on Okinawa-jima Island. Zool Sci 17:539–547
Kato Y, (2000b) Does mating occur among populations of twotypes in the butterfly Eurema hecabe (L.) (Lepidoptera,Pieridae)? Trans Lepid Soc Jpn 52:63–66
Kato Y, Handa H, (1992) Seasonal polyphenism in a subtropicalpopulation of Eurema hecabe (Lepidoptera, Pieridae). Jpn JEntomol 60:305–318
Kato Y, Yata O, (2005) Geographic distribution and taxonom-ical status of two types of Eurema hecabe (L.) (Lepidoptera,Pieridae) in south-western Japan and Taiwan. Trans LepidSoc Jpn 56:171–183 (in Japanese with English summary)
Kato Y, Hiroki M, Handa H, (1992) Interpopulation variation inadaptation of Eurema hecabe (Lepidoptera, Pieridae) tohost plant. Jpn J Entomol 60:749–759
Kimura M, (1980) A simple method for estimating evolutionaryrates of base substitutions through comparative studies ofnucleotide sequences. J Mol Evol 16:111–120
Kobayashi A, Hiroki M, Kato Y (2001) Sexual isolation betweentwo sympatric types of the butterfly Eurema hecabe (L.). JInsect Behav 14:353–362
252 Genetica (2007) 131:241–253
123

Marcade I, Souty-Grosset C, Bouchon D, Rigaud T, Raimond R,(1999) Mitochondrial DNA variability and Wolbachiainfection in two sibling woodlice species. Heredity 83:71–78
Matsuno H, (1999) Difference in reflection pattern against ultra-violet rays between two types of Eurema hecabe. Gekkan-Mushi 338:13–15 (In Japanese)
Narita S, Nomura M, Kato Y, Fukatsu T, (2006) Geneticstructure of sibling butterfly species affected by Wolbachiainfection sweep: evolutionary and biogeographical implica-tions. Mol Ecol 15:1095–1108
Nei M, Li W-H, (1979) Mathematical model for studying geneticvariation in terms of restriction endonucleases. Proc NatlAcad Sci USA 76:5269–5273
Nomura M, Kato Y, (1993) Allozyme variation in Euremahecabe. The Nature and Insects 28:15–18 (In Japanese)
O’Neill SL, Hoffmann AA, Werren JH, (1997) Influentialpassengers: inherited microorganisms and arthropod repro-duction. Oxford. University Press Inc, New York
O’Neill SL, Giordano R, Colbert AME, Karr TL, RobertsonHM (1992) 16S rRNA phylogenetic analysis of the bacterialendosymbionts associated with cytoplasmic incompatibilityin insects. Proc Natl Acad Sci USA 89:2699–2702
Posada D, Crandall KA, (2001) Intraspecific gene genealogies:trees grafting into networks. Trends Ecol Evol 16:37–45
Prout T, (1994) Some evolutionary possibilities for a microbethat causes incompatibility in its host. Evolution 48:909–911
Rozas J, Sanchez-Delbarrio JC, Messeguer X, Rozas R, (2003)DnaSP, DNA polymorphism analyses by the coalescent andother methods. Bioinformatics 19:2496–2497
Shoemaker DD, Katju V, Jaenike J (1999) Wolbachia and theevolution of reproductive isolation between Drosophilarecens and Drosophila subquinaria. Evolution 53:1157–1164
Shoemaker DD, Keller G, Ross KG, (2003) Effects of Wolbachiaon mtDNA variation in two fire ant species. Mol Ecol12:1757–1771
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P(1994) Evolution, weighting, and phylogenetic utility ofmitochondrial gene sequences and a compilation of con-served polymerase chain reaction primer. Ann Entomol SocAm 87:651–701
Stouthamer R, Breeuwer JA, Hurst GD, (1999) Wolbachiapipientis: microbial manipulator of arthropod reproduction.Ann Rev Microbiol 53:71–102
Swofford, DL, (2001) ‘PAUP*. Phylogenetic analysis usingparsimony (*and other methods), Version 4.0
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysisof phenotypic associations with haplotypes inferred fromrestriction endonuclease mapping and DNA sequence data.III. Cladogram estimation. Genetics 132:619–633
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W:improving the sensitivity of progressive multiple sequencealignment through sequence weighting, positions-specificgap penalties and weight matrix choice. Nucleic Acids Res22:4673–4680
Turelli M, (1994) Evolution of incompatibility-inducing microbesand their hosts. Evolution 48:1500–1513
Turelli M, Hoffmann AA, (1991) Rapid spread of an inheritedincompatibility factor in California Drosophila. Nature353:440–442
Werren JH, (1997) Biology of Wolbachia. Ann Rev Entomol42:587–607
Yagi T, Sasaki G, Takebe H, (1999) Phylogeny of JapanesePapilionid butterflies inferred from nucleotide sequences ofthe mitochondrial ND5 gene. J Mol Evol 48:42–48
Yagi T, Kato T, Chichvarkhin A, Shinkawa T, Omoto K, (2001)Molecular phylogeny of butterflies Parnassius glacialis andP. stubbendorfit at variation localities in East Asia. GenesGenet Syst 76:229–234
Yata O, (1989) A revision of the Old World species of the genusEurema Hubner (Lepidoptera, Pieridae) I. Phylogeny andzoogeography of the subgenus Terias Swainson and descrip-tion of the subgenus Eurema Hubner. Bull Kitakyushu MusNat Hist 9:1–103
Yata O (1995) A revision of the Old World species of the genusEurema Hubner (Lepidoptera, Pieridae) Part V. Descrip-tion of the hecabe group (part). Bull Kitakyushu Mus NatHist 14:1–54
Zhou W, Rousset F, O’Neill S, (1998) Phylogeny and PCR basedclassification of Wolbachia strains using wsp gene sequences.Proc R Soc Lond B 265:509–515
Genetica (2007) 131:241–253 253
123





























