Embed Size (px)
Citation preview

lable at ScienceDirect
Polymer 55 (2014) 738e745
Contents lists avai
Polymer
journal homepage: www.elsevier .com/locate/polymer
Morphological and mechanical study of nanostructured epoxysystems modified with amphiphilic poly(ethylene oxide-b-propyleneoxide-b-ethylene oxide)triblock copolymer
Laida Cano a, Daniel H. Builes a,b, Agnieszka Tercjak a,*
aGroup ‘Materials þ Technologies’, Dpto. Ingeniería Química y del M. Ambiente, Escuela Politécnica/Eskola Politeknikoa, Universidad del País Vasco/EuskalHerriko Unibertsitatea (UPV/EHU), Pza. Europa 1, 20018 Donostia-San Sebastián, SpainbResearch and Development Center ‘DENOVO’, Andercol S.A. Autopista Norte 95-84, Medellín, Colombia
a r t i c l e i n f o
Article history:Received 10 November 2013Received in revised form2 January 2014Accepted 4 January 2014Available online 11 January 2014
Keywords:ThermosetsEpoxy resinBlock copolymer
* Corresponding author. Tel.: þ34 943017169; fax:E-mail address: [email protected] (A. Terc
0032-3861/$ e see front matter � 2014 Elsevier Ltd.http://dx.doi.org/10.1016/j.polymer.2014.01.005
a b s t r a c t
Thermosetting systems based on DGEBA epoxy resin and poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) (EPE) triblock copolymer were prepared and investigated. Different mixtures wereobtained by using different contents of EPE block copolymer in order to study the influence of themodifier on the properties of the final materials. All thermosetting systems were prepared without usingany solvent and were cured at ambient temperature, taking into account the lower critical solutiontemperature (LCST) behavior of the block copolymer. DSC results indicated that the addition of blockcopolymer affected to the curing reaction time and to the glass transition temperature of the mixturesand also the miscibility of EPE triblock copolymer in the epoxy resin was proved. The morphologiesstudied by AFM and TEM showed clear nanostructuration up to 25 wt % EPE content. The addition of5 and 15 wt % of EPE block copolymer led to a considerable improvement in the toughness of the ma-terials. When EPE block copolymer was added to the epoxy resin, the surface became more hydrophilicand the UVevis transmittance decreased slightly maintaining a high level of transparency.
� 2014 Elsevier Ltd. All rights reserved.
1. Introduction
In the last years different materials such as rubbers, thermo-plastic homopolymers and block copolymers have been used asadditives for epoxy matrices. Among these materials, many studieshave confirmed that block copolymers can provide new orimproved properties to the epoxy resins [1e7]. The main contri-bution of the block copolymers is their capacity to self-assemble inordered nanostructures leading to microphase separated mor-phologies and to generate nanostructured thermosetting systemswhen they are mixed with an epoxy resin. Beside the ability to getordered nanostructures with different morphologies, the additionof block copolymers to thermosetting matrices can lead to animprovement in the mechanical properties of the matrix.
The most commonway to obtain nanostructured epoxy systemsis by using a block copolymer where only one of its blocks ismiscible with the epoxy resin and consequently the non-miscibleblock can microseparate from the matrix [5,7] leading to a micro-phase separation. In the cases where the block copolymer does not
þ34 943017130.jak).
All rights reserved.
have any block miscible with the epoxy resin, it would be essentialto modify chemically one of the blocks in order to make it misciblewith the epoxy resin. Apart from the interaction between theblocks and the matrix, the nanostructuration is also controlled byother parameters like the volume fraction of each block, the in-teractions between blocks, etc. Nanostructured thermosettingsystems can be mainly obtained by two different mechanisms. Oneof them is the called reaction-induced microphase separation(RIPS) mechanism [3], in which the microseparation of one block ofthe block copolymer occurs during the curing reaction. In the sec-ond case, the self-assembly of the block copolymer could occurbefore the curing [1] and consequently one block would micro-separate before the curing process. In this case, the self-assembly ofthe block copolymer before the curing leads the microphase sep-aration of nanostructured thermosetting systems.
Epoxy resins are one of the most widely used thermosettingpolymers due to their good mechanical and thermal properties,high chemical and corrosion resistance and low shrinkage duringcuring. These properties lead to several applications in adhesives,surface coatings, moulds, and aerospace and electronics industries,among others [8]. However, one of their main drawbacks is theirlow toughness. There have beenmany studies inwhich rubbers andthermoplastics were employed in order to increase the toughness

L. Cano et al. / Polymer 55 (2014) 738e745 739
of thermosets. Nevertheless, the modification of epoxy resins withblock copolymers has resulted to be an adequate method toimprove epoxy toughness at low contents of block copolymer aswell as to create ordered microphase-separated structures [4,9e13].
The triblock copolymer poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) has already been used before to modify phenolic[14], unsaturated polyester [15e17] and epoxy resins [18e25].Already published studies about the blend of DGEBA epoxy resinwith poly(ethylene oxide-b-propylene oxide-b-ethylene oxide)triblock copolymer revealed that different macroseparated ormicroseparated morphologies were obtained depending on thecontent of block copolymer in the matrix, molar ratio betweenblocks, molecular weight of the block copolymer and the curingcycle carried out. The miscibility of the blends and kinetics of thecuring reaction were also investigated as a function of the blockcopolymer content. It was demonstrated that the presence of theblock copolymer delayed the curing reaction of the epoxy system.The control of nanostructures by optimizing the curing conditionsresulted to be essential to control the mechanical properties of thefinal materials.
In this work, the poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) triblock copolymer (EPE) was employed asmodifierof a DGEBA based epoxy matrix with the aim of obtaining nano-structured thermoset systems with improved mechanical proper-ties. It was demonstrated that EPE triblock copolymer resulted to bean effectivemodifier to lead to a remarkable improvement effect onthe toughness value of an epoxy resin, which was the main interestof this paper. Different contents of a triblock copolymer up to 50 wt% were added to the matrix in order to study the influence of thecontent of block copolymer on the morphology, mechanical prop-erties and curing reaction time of the epoxy system. The curingprocess was the same for all mixtures and it was chosen taking intoaccount the lower critical solution temperature (LCST) behavior ofEPE [21,26]. Consequently, all investigated thermosetting systemscurings were carried out at 25 �C, what in our opinion is a bigadvantage from an industrial point of view since the low temper-ature allows to reach nanostructuration of the DGEBA/MXDA sys-tem with a very high EPE content and improve drastically thetoughness of investigated thermosetting systems. The morphologyof the blends and the size of the microseparated phase wereinvestigated by atomic force microscopy (AFM) and transmissionelectron microscopy (TEM). The optical transparency was investi-gated by UVevis spectroscopy. The mechanical properties mea-surements were carried out by the universal testing machine(MTS). The glass transition temperatures as well as the curingbehavior were determined by differential scanning calorimeter(DSC). Contact angle measurement was employed to characterizethe surface properties of the samples.
2. Experimental
2.1. Materials and sample preparation
The triblock copolymer poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) (referred to as EPE from this point forward),purchased from SigmaeAldrich, had a number average molecularweight (Mn) of 5800 g/mol and 30 wt % of polyethylene oxidecontent. The epoxy resin used in this study was diglycidyl ether ofbisphenol A (DGEBA) (DER 330) provided by The Dow ChemicalCompany with an epoxy equivalent weight between 176 and 185 g/eq. The curing agent used to cure this epoxy resin was m-Xylyle-nediamine (MXDA) supplied by SigmaeAldrich. The employmentof this curing agent allows to reach a low curing temperature forthe fabrication of the investigated thermosetting systems.
The neat epoxy system was prepared by mixing DGEBA resinwith MXDA in stoichiometric proportions using a magnetic stirrer.After stirring it for around 10 min, when the mixture was homo-geneous, it was degassed in a vacuum bell at ambient temperature.Then, the mixture was poured into a glass mold to be cured. Toprepare the block copolymer/epoxy systems, firstly a certainamount of EPE block copolymer was dissolved in DGEBA resin. Thismixture was heated at around 60 �C in order to melt the blockcopolymer and favor its solution and then continuously stirred untila complete homogenizationwas achieved. A stoichiometric amountof MXDA was added and the same procedure as for the neat epoxysystem was followed. All the blends were first cured at 25 �C for12 h, followed by 9 h at 35 �C, 2 h at 50 �C and finally 1 h at 150 �C.This procedure was chosen to avoid long time curing in high tem-perature. Apart from the neat epoxy system, four blends wereprepared, with 5, 15, 25 and 50 wt % of EPE block copolymer.
2.2. Techniques
Differential scanning calorimetry (DSC) measurements of theneat epoxy system as well as EPE/epoxy systems were performedusing a Mettler Toledo DSC 822e differential scanning calorimeter.The miscibility of EPE triblock copolymer with uncured DGEBAresin was investigated by dynamic scans performed from�50 �C to150 �C at 5 �C/min. The thermal history of the samples was deletedby a first heating from �50 �C to 150 �C at 5 �C/min followed by acooling from 150 �C to�50 �C at 1 �C/min. All blends were analyzedduring curing by an isothermal scan performed at 25 �C (followedby a dynamic scan from 25 �C to 200 �C at 5 �C/min). Thermaltransition temperatures of cured blends were determined by dy-namic scans performed from�25 �C to 220 �Cwith a heating rate of5 �C/min. Prior to this scan, a heating from �25 �C to 220 �C fol-lowed by a cooling from220 �C to�25 �Cwas carried out in order todelete the thermal history of the material. All experiments wereconducted under a nitrogen flow of 10 mL/min using 10e15 mgsamples in aluminum pans.
The morphologies of the cured epoxy and EPE/epoxy systemswere studied by atomic force microscopy (AFM) under ambientconditions. AFM images were obtained using a scanning probemicroscope (Nanoscope IIIa Multimode�, Digital Instruments).Tapping mode (TM) was employed in air using an integrated tip/cantilever (125 mm in length with ca. 300 kHz resonant frequency).Typical scan rates during recording were 0.7e1 line/s using a scanheadwith amaximum range of 16� 16 mm. Samples were cut usingan ultramicrotome Leica Ultracut R with a diamond blade.
For TEM measurements, samples were prepared by using anultramicrotome Leica EMFCS instrument equipped with a diamondknife at room temperature. A Tecnai G2 20 Twin transmissionelectron microscope operated at 200 kV with resolution of 2.5 Åwas used. Moreover, TEM samples were stained in RuO4 vapor for4 min in order to enhance the contrast between micro-separatedPPO rich phase and epoxy rich phase.
UVevis transmittance spectra of the cured neat epoxy and EPE/epoxy systems sheets (thickness of 1 mm) were obtained using aspectrophotometer (Shimadzu UV-3600) in the range between 200and 800 nm.
Regarding mechanical properties of the neat epoxy and EPE/epoxy systems, flexural tests were carried out following the ASTMD790-10 standard test method and using universal testingmachine(MTS, model Insight 10) provided with a 250 N load cell. Three-point bending tests without notch were performed at a rate ofcrosshead of 5.6 mm/min using specimen dimensions of36 � 9 � 1.5 mm3 (rectangular shape). Flexural modulus wasdetermined from the slope of the load-displacement curve in thezone of linear elasticity. Fracture toughness tests were performed

Table 1Thermal transitions of the uncured neat DGEBA resin and all EPE/DGEBA mixturesdetermined by dynamic DSC and theoretical Tg values calculated using Fox equation.
Sample Tg (�C) Tg (�C)a
Neat DGEBA �18 �185 wt % EPE/DGEBA �23 �20.7015 wt % EPE/DGEBA �31 �25.9425 wt % EPE/DGEBA �37 �30.9650 wt % EPE/DGEBA e �42.68
a Theoretical Tg values calculated for mixtures using Fox equation1=Tg ¼ x1=Tg1
þ x2=Tg2.
L. Cano et al. / Polymer 55 (2014) 738e745740
according to ASTM D5045-99 standard test method using the sameuniversal testing machine as for flexural tests. Fracture toughnesswas measured by determining the critical stress intensity factor(KIC) and the critical strain energy release rate (GIC) in three-pointbending at a crosshead rate of 10 mm/min using single edgenotched specimens (SENB) with dimensions of 27 � 6 � 1.5 mm3.Initially a sharp notch of around 2.7 mm was made by machining,and subsequently a natural crack was initiated using a razor blade.GIC was calculated from the energy derived from integration of theload versus displacement curve up to the same load point as usedfor KIC. For both flexural and fracture toughness tests more than fivespecimens for each system were tested and a support span of24 mm was used. The 50 wt % EPE/epoxy blend could not beanalyzed in terms of its mechanical properties as it resulted to beeasily breakable.
Contact angle measurements were carried out using DataPhysics OCA 20 contact angle system at ambient temperature. 5 mLdistilled water drop was used for each measurement. At least fivemeasurements were made for each different system.
3. Results and discussion
Different contents of EPE triblock copolymer were dissolved inthe uncured DGEBA resin resulting in transparent and homoge-neous mixtures. This could indicate the miscibility between DGEBAand EPE block copolymer. However, in order to confirm this, DSCmeasurements were carried out. In the Fig. 1, DSC thermograms ofEPE/DGEBA uncured mixtures are presented. As can be seen, allmixtures exhibited a unique Tg which changed with the composi-tion of EPE triblock copolymer thus confirming the partial misci-bility of the system. The Tg of the neat DGEBA (�18 �C) resin shiftedto lower temperatures with the increase of the content of EPE blockcopolymer as a result of the mixing with the block copolymer. Ashas been published by other authors, EPE triblock copolymer has aglass transition temperature (Tg) around�63 �C attributable to boththe amorphous PPO block and the semicrystalline PEO block and amelting point around 30e40 �C, which corresponds to the semi-crystalline PEO block [17,18,20]. The Fox equation [25] was appliedwith the aim of comparing the experimentally determined Tgvalues of mixtures with the theoretical ones. After applying the Foxequation for all mixtures (values shown in Table 1), it wasconcluded that themiscibility of EPE block copolymer in the DGEBAexisted but it was not complete as experimental Tgs were similar tothe theoretical ones but not exactly the same.
Fig. 1. Dynamic DSC thermograms of the second heating scan of the uncured neatDGEBA resin and all EPE/DGEBA mixtures.
The curing behavior of the neat epoxy system and epoxymodified with 5, 15, 25 and 50 wt % of EPE block copolymer wasstudied by DSC. The isothermal thermograms performed at 25 �Cfor each epoxy system during curing are shown in the Fig. 2. As canbe seen, all curves showed a drop in heat flow indicating thepresence of a curing reaction between DGEBA and the curing agent.The reaction was considered complete when the isothermal DSCthermograms leveled off to the baseline [27]. The curing reactionwas affected by the block copolymer content in the epoxy resin,showing a clear delay in the heat flow drop related to the curingreactionwith the increase of the block copolymer content. With theincrease of block copolymer content the curing reactionwas clearlydelayed. The retardation of the curing reaction time occurred due tothe fact that the addition of block copolymer to the epoxy resinprovoked a dilution effect [22,27] on the blends and consequentlythe reaction between epoxy groups and amine groups became lessunlikely [19] since in investigated thermosetting systems the epoxyresin acts as solvent for EPE block copolymer. In addition, it shouldbe taken into account that the interactions between the epoxy resinand the EPE block copolymer can affect to the curing process. As hasbeen studied by other authors, physical interactions such ashydrogen bonds were formed between the OH groups initiallyexisting in the epoxy resin or the ones generated during the curingand the ether group of the PEO block of the EPE block copolymer[19,22,27,28]. Thus, the presence of a higher amount of blockcopolymer delayed the time of the curing reaction between theepoxy resin and the curing agent leading to a retarded curing.
All the cured thermosetting systems were studied by dynamicDSC analysis. The Fig. 3 shows the DSC thermograms of the secondheating scan applied to each sample. As is mentioned above, EPEtriblock copolymer has a glass transition temperature (Tg)
Fig. 2. Isothermal DSC thermograms of the neat epoxy system and all EPE/epoxysystems at 25 �C.
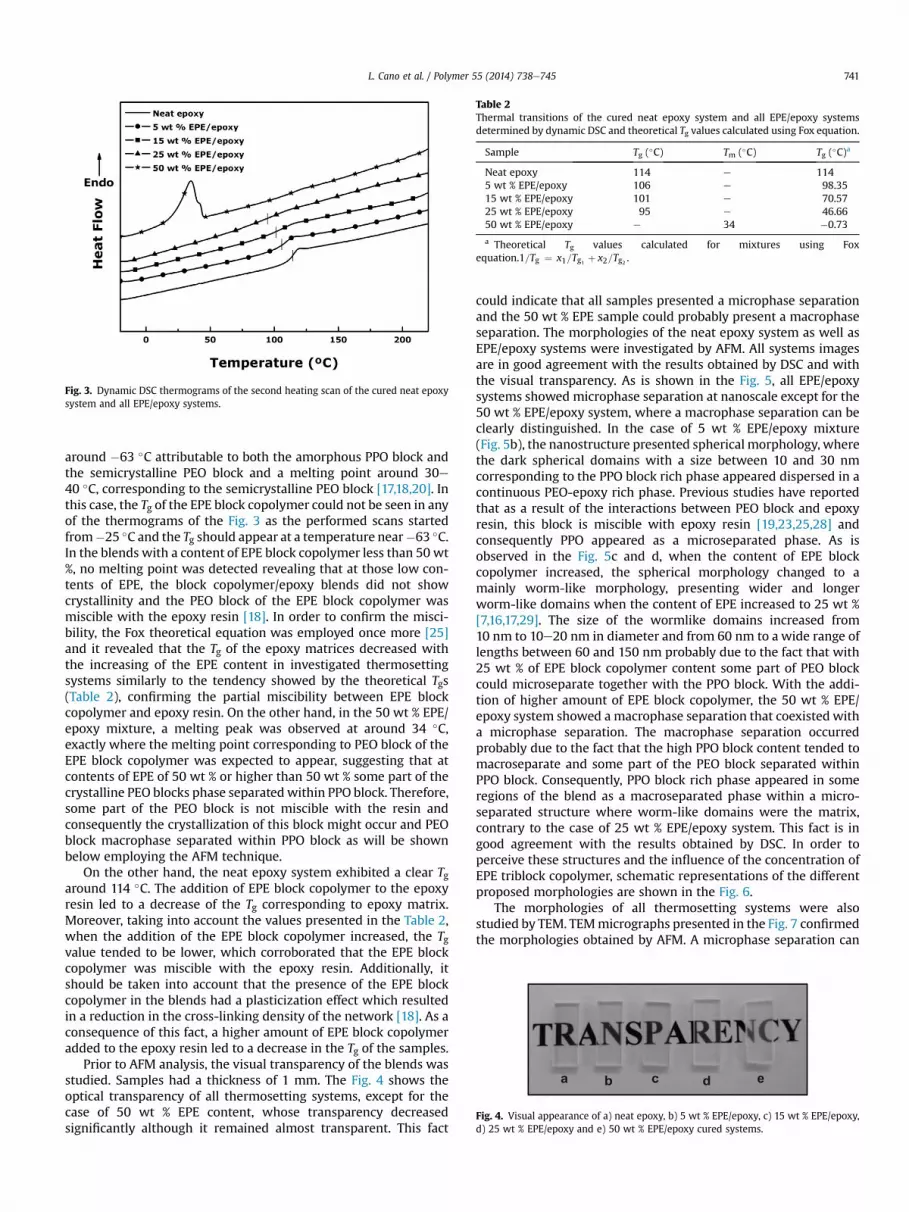
Fig. 3. Dynamic DSC thermograms of the second heating scan of the cured neat epoxysystem and all EPE/epoxy systems.
Table 2Thermal transitions of the cured neat epoxy system and all EPE/epoxy systemsdetermined by dynamic DSC and theoretical Tg values calculated using Fox equation.
Sample Tg (�C) Tm (�C) Tg (�C)a
Neat epoxy 114 e 1145 wt % EPE/epoxy 106 e 98.3515 wt % EPE/epoxy 101 e 70.5725 wt % EPE/epoxy 95 e 46.6650 wt % EPE/epoxy e 34 �0.73
a Theoretical Tg values calculated for mixtures using Foxequation.1=Tg ¼ x1=Tg1 þ x2=Tg2
.
Fig. 4. Visual appearance of a) neat epoxy, b) 5 wt % EPE/epoxy, c) 15 wt % EPE/epoxy,d) 25 wt % EPE/epoxy and e) 50 wt % EPE/epoxy cured systems.
L. Cano et al. / Polymer 55 (2014) 738e745 741
around �63 �C attributable to both the amorphous PPO block andthe semicrystalline PEO block and a melting point around 30e40 �C, corresponding to the semicrystalline PEO block [17,18,20]. Inthis case, the Tg of the EPE block copolymer could not be seen in anyof the thermograms of the Fig. 3 as the performed scans startedfrom�25 �C and the Tg should appear at a temperature near�63 �C.In the blends with a content of EPE block copolymer less than 50 wt%, no melting point was detected revealing that at those low con-tents of EPE, the block copolymer/epoxy blends did not showcrystallinity and the PEO block of the EPE block copolymer wasmiscible with the epoxy resin [18]. In order to confirm the misci-bility, the Fox theoretical equation was employed once more [25]and it revealed that the Tg of the epoxy matrices decreased withthe increasing of the EPE content in investigated thermosettingsystems similarly to the tendency showed by the theoretical Tgs(Table 2), confirming the partial miscibility between EPE blockcopolymer and epoxy resin. On the other hand, in the 50 wt % EPE/epoxy mixture, a melting peak was observed at around 34 �C,exactly where the melting point corresponding to PEO block of theEPE block copolymer was expected to appear, suggesting that atcontents of EPE of 50 wt % or higher than 50 wt % some part of thecrystalline PEO blocks phase separatedwithin PPO block. Therefore,some part of the PEO block is not miscible with the resin andconsequently the crystallization of this block might occur and PEOblock macrophase separated within PPO block as will be shownbelow employing the AFM technique.
On the other hand, the neat epoxy system exhibited a clear Tgaround 114 �C. The addition of EPE block copolymer to the epoxyresin led to a decrease of the Tg corresponding to epoxy matrix.Moreover, taking into account the values presented in the Table 2,when the addition of the EPE block copolymer increased, the Tgvalue tended to be lower, which corroborated that the EPE blockcopolymer was miscible with the epoxy resin. Additionally, itshould be taken into account that the presence of the EPE blockcopolymer in the blends had a plasticization effect which resultedin a reduction in the cross-linking density of the network [18]. As aconsequence of this fact, a higher amount of EPE block copolymeradded to the epoxy resin led to a decrease in the Tg of the samples.
Prior to AFM analysis, the visual transparency of the blends wasstudied. Samples had a thickness of 1 mm. The Fig. 4 shows theoptical transparency of all thermosetting systems, except for thecase of 50 wt % EPE content, whose transparency decreasedsignificantly although it remained almost transparent. This fact
could indicate that all samples presented a microphase separationand the 50 wt % EPE sample could probably present a macrophaseseparation. The morphologies of the neat epoxy system as well asEPE/epoxy systems were investigated by AFM. All systems imagesare in good agreement with the results obtained by DSC and withthe visual transparency. As is shown in the Fig. 5, all EPE/epoxysystems showed microphase separation at nanoscale except for the50 wt % EPE/epoxy system, where a macrophase separation can beclearly distinguished. In the case of 5 wt % EPE/epoxy mixture(Fig. 5b), the nanostructure presented spherical morphology, wherethe dark spherical domains with a size between 10 and 30 nmcorresponding to the PPO block rich phase appeared dispersed in acontinuous PEO-epoxy rich phase. Previous studies have reportedthat as a result of the interactions between PEO block and epoxyresin, this block is miscible with epoxy resin [19,23,25,28] andconsequently PPO appeared as a microseparated phase. As isobserved in the Fig. 5c and d, when the content of EPE blockcopolymer increased, the spherical morphology changed to amainly worm-like morphology, presenting wider and longerworm-like domains when the content of EPE increased to 25 wt %[7,16,17,29]. The size of the wormlike domains increased from10 nm to 10e20 nm in diameter and from 60 nm to a wide range oflengths between 60 and 150 nm probably due to the fact that with25 wt % of EPE block copolymer content some part of PEO blockcould microseparate together with the PPO block. With the addi-tion of higher amount of EPE block copolymer, the 50 wt % EPE/epoxy system showed a macrophase separation that coexisted witha microphase separation. The macrophase separation occurredprobably due to the fact that the high PPO block content tended tomacroseparate and some part of the PEO block separated withinPPO block. Consequently, PPO block rich phase appeared in someregions of the blend as a macroseparated phase within a micro-separated structure where worm-like domains were the matrix,contrary to the case of 25 wt % EPE/epoxy system. This fact is ingood agreement with the results obtained by DSC. In order toperceive these structures and the influence of the concentration ofEPE triblock copolymer, schematic representations of the differentproposed morphologies are shown in the Fig. 6.
The morphologies of all thermosetting systems were alsostudied by TEM. TEMmicrographs presented in the Fig. 7 confirmedthe morphologies obtained by AFM. A microphase separation can

Fig. 5. AFM phase images (1 mm � 1 mm) of a) neat epoxy, b) 5 wt % EPE/epoxy, c) 15 wt % EPE/epoxy, d) 25 wt % EPE/epoxy and e) 50 wt % EPE/epoxy systems. The insetscorrespond to 5 mm � 5 mm AFM images. Samples were prepared using ultramicrotomy.
Fig. 6. Schematic representation of the structures of a) 5, b) 15 and c) 25 wt % EPE/epoxy systems.
L. Cano et al. / Polymer 55 (2014) 738e745742
be observed in all studied samples. The dark areas correspond toPPO microseparated phase due to the fact that the PPO block waspreferentially stained with RuO4 compared to the cured epoxymatrix [18]. However, it should be pointed out that the differencebetween both phases was not very obvious because both blockshave a similar chemical structure and as a result of that RuO4 couldstain not only PPO block but also the phase composed of epoxyresin and PEO block. Moreover, PEO block was distributed betweenthe two phases as this block is miscible with epoxy matrix but atthe same time it is linked covalently to the separated PPO block.Because of these reasons, the difference between phases was not asclear as in the AFM images. In spite of this fact, in the case of 5 wt %EPE/epoxy system, some dark spherical domains can be seen,whereas when the content of block copolymer increases, the
morphology changes from spherical domains to worm-like struc-ture. When 25 wt % EPE was added to the matrix, the micro-separated PPO phase became bigger and wider, which is in goodagreement with the AFM results.
UVevis measurements were carried out to study the opticaltransparency of thermosetting systems. The Fig. 8 shows the UVevis transmittance spectra of the neat epoxy system and all modifiedsystems. Although the Fig. 4 had presented a clear visual trans-parency for all samples except for the 50 wt % EPE/epoxy sample,here it can be seen that the transmittances were not very high evenfor the neat epoxy sample. The transmittance could be related withthe thickness of the samples, that in this case was 1 mm. Moreover,other authors have reported similar transmittance values for neatepoxy resin [30,31]. The neat epoxy system exhibited the highest

Fig. 7. TEM micrographs of a) neat epoxy, b) 5 wt % EPE/epoxy, c) 15 wt % EPE/epoxy and d) 25 wt % EPE/epoxy systems.
Fig. 8. UVevis transmittance spectra of the cured neat epoxy system and all EPE/epoxysystems.
L. Cano et al. / Polymer 55 (2014) 738e745 743
UVevis transmittance andwhen EPE block copolymerwas added tothe epoxy systems the transmittance of EPE modified systemsslightly decreased indicating that EPE block copolymer had a weakeffect of light absorption in both visible and UV ranges. The mostimportant decrease was observed in the case of 50 wt % EPE/epoxysystem, which had a transmittance lower than 15% in the visiblerange and a value near to zero in the UV range. This fact is related to
the poor visual transparency observed for 50 wt % EPE/epoxy sys-tem in the Fig. 4. Comparing to the neat epoxy system, 5, 15 and25wt % EPE/epoxy systems had lower transmittance, being the 5wt% EPE/epoxy system the one with a lower transmittance. This couldbe attributed to the following phenomenon. In the case of a lowcontent of EPE triblock copolymer (5 wt %), the PEO block waspresent in an even lower concentration in respect to the PPO blockcontent. In fact, in the 5 wt % EPE/epoxy system, only the 1.5 wt % ofthe sample corresponded to the PEO block. This low content of PEOblock provoked that even if PEO block was miscible with epoxyresin, it tended to microseparate together with the immiscible PPOblock instead of being totally mixed with epoxy resin. Therefore, atthis content of block copolymer, EPE was not completely mixedwith the resin. This could affect to the transmittance of the sample,reducing it as is revealed in the Fig. 8.
The mechanical properties of all the systems (except for the50 wt % EPE/epoxy system) were studied in terms of the flexuralmodulus (E), the critical stress intensity factor (KIC) and the criticalstrain energy release rate (GIC). As can be observed in the Fig. 9, theflexural modulus of the EPE/epoxy systems not onlywas lower thanthe flexural modulus of the neat epoxy system, but also it decreasedwhen the added block copolymer amount increased. This phe-nomenonwas expected taking into account the evidences found bymany authors confirming that the addition of a block copolymerdecreases the flexural modulus of the neat epoxy resin [25,32e35].This could occur due to the fact that the modifier added has a lowermodulus than the neat epoxy resin and the epoxy resin wasmiscible with EPE block copolymer [25]. As mentioned in the text

Fig. 9. Flexural modulus (E) of the cured neat epoxy system and all EPE/epoxy systems.
Fig. 10. Critical stress intensity factor (KIC) and the critical strain energy release rate(GIC) of the cured neat epoxy system and all EPE/epoxy systems.
Table 3Water contact angle and surface free energy values of the cured neat epoxy systemand all EPE/epoxy systems.
Sample Contact angle (�) gSV (mN/m)
Neat epoxy 83.4 � 1.6 22.65 wt % EPE/epoxy 79.8 � 3.1 25.215 wt % EPE/epoxy 71.1 � 1.1 31.925 wt % EPE/epoxy 69.1 � 2.8 33.550 wt % EPE/epoxy 29.5 � 2.9 63.7
Fig. 11. Images of a water droplet in contact with a) neat epoxy, b) 5 wt % EPE/epoxy
L. Cano et al. / Polymer 55 (2014) 738e745744
above, the addition of EPE block copolymer provoked a plasticiza-tion effect [17] in the blend together with a reduction in the cross-linking density of the network [17,36]. Consequently, the higher thecontent of block copolymer, the lower the flexural modulus of theblend. On the other hand, it should be indicated that the flexuralmodulus of the neat epoxy system resulted to be slightly higherthan the flexural moduli reported by other authors [25,35,37,38].The Fig. 10 shows the KIC and the GIC values for each system with adifferent content of EPE block copolymer. The toughness of themixtures increased in respect to the neat epoxy system one withthe addition of 5 and 15 wt % of EPE block copolymer, obtaining animprovement of 30% in the toughness in the case of 5 wt % EPE/epoxy system and of 14% in the case of 15 wt % EPE/epoxy system.Even if the addition of the triblock copolymer provoked deterio-ration in the flexural behavior, here it can be seen that the blendingof epoxy resin with the block copolymer is worthy considering theimprovement in the toughness [25,32e35]. The values of KIC sug-gested that systems with microdomains of smaller size presented ahigher improvement of toughness and when the content of EPEtriblock copolymer increased and microdomains size becamehigher, the values of KIC started to decrease in comparison with the5 wt % EPE/epoxy mixture. As reported in literature, mechanicalproperties are related to the degree of polymerization of the ther-moset [39,40] and to the morphology of the final material[9,25,32,35,42,43], which is governed by several parameters such asthe volume fraction of each block [25,32,41], the block copolymercontent [17,25] and miscibility of blocks [32,36].
The changes in the hydrophilic nature of EPE/epoxy blends withvarying diblock copolymer content and the comparison with neatepoxy resin were analyzed by contact angle measurements.Moreover, the surface free energies were calculated from the con-tact angle data by using the Berthelot’s rule [44,45]:
gSV ¼ ð1þ cos qÞ24
gLV
As is observed in the data of the Table 3, the contact angle of theblends decreased and the surface free energy increased with theaddition of EPE block copolymer. The surface became more hy-drophilic with increasing the content of EPE block copolymer in thesystem. The most hydrophobic system was the neat epoxy one,whereas the most hydrophilic one was the 50 wt % EPE/epoxy one.This could be attributed to the hydrophilic character of the PEOblocks of the triblock copolymer [46] and to the existence of PEOblock at the surface of the samples. The increase of the hydrophiliccharacter of the systems can be observed in the images of theFig. 11.
, c) 15 wt % EPE/epoxy, d) 25 wt % EPE/epoxy and e) 50 wt % EPE/epoxy systems.

L. Cano et al. / Polymer 55 (2014) 738e745 745
4. Conclusions
Epoxy based nanostructured thermosetting systems modifiedwith different contents of poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) triblock copolymer were successfully prepared.The incorporation of EPE triblock copolymer into DGEBA epoxyresin resulted to be an effective method to improve the toughnessof the epoxy resin as well as to obtain well nanostructured ther-mosetting systems. The curing process was carried out at 25 �Cowing to the lower critical solution temperature (LCST) behavior ofthe block copolymer. The addition of the triblock copolymer to theepoxy resin caused a delay on the time of the curing reaction aswell as a decrease in the Tg of the systems compared to the neatepoxy resin Tg, due to the plasticization effect provoked by theaddition of the block copolymer. As was confirmed by AFM andTEM, the cured samples showedwell nanostructuredmorphologiesup to 25 wt % EPE content, where the microseparated phase cor-responded to PPO block rich phase. The morphology obtainedresulted to be dependent on the EPE block copolymer content and itchanged from spherical structure to worm-like structure with thecontent of block copolymer. Regarding the mechanical properties,the toughness improved considerably with 5 and 15 wt % EPE blockcopolymer contents and it almost remained the same with 25 wt %content. UVevis measurements mainly indicated a slight decreasein the transmittance with the increase of EPE block copolymercontent. This decrease of transmittance was also reflected in thevisual appearance of the samples, although all samples remainedtransparent. Finally, the mixture with EPE block copolymer madethe surface of thermosetting systems more hydrophilic.
Acknowledgments
Financial support from the Basque Government (Grupos Con-solidados (IT776-13) and SAIOTEK2013 (NANOSOL)) and from theSpanish Ministry of Economy and Competitiveness (MAT2012-31675) is gratefully acknowledged. L.C. thanks Eusko Jaurlaritza/Gobierno Vasco for PhD Fellowship (Programas de becas para for-mación y perfeccionamiento de personal investigador(PRE_2013_2_418)) and A.T. acknowledges MICINN for Ramón yCajal program (RYC-2010-05592). Moreover, we are grateful to the‘Macrobehavior-Mesostructure-Nanotechnology’ SGIker unit of theUPV/EHU.
References
[1] Hillmyer MA, Lipic PM, Hajduk DA, Almdal K, Bates FS. J Am Chem Soc1997;119:2749e50.
[2] Lipic PM, Bates FS, Hillmyer MA. J Am Chem Soc 1998;120:8963e70.[3] Grubbs RB, Dean JM, Broz ME, Bates FS. Macromolecules 2000;33:9522e34.[4] Dean JM, Verghese NE, Pham HQ, Bates FS. Macromolecules 2003;36:9267e
70.[5] Maiez-Tribut S, Pascault JP, Soulé ER, Borrajo J, Williams RJJ. Macromolecules
2007;40:1268e73.
[6] Serrano E, Tercjak A, Ocando C, Larrañaga M, Parellada MD, Corona-Galván S,et al. Macromol Chem Phys 2007;208:2281e92.
[7] Hameed N, Guo Q, Hanley T, Mai YW. J Polym Sci Polym Phys 2010;48:790e800.
[8] Ratna D. Handbook of thermoset resins. Shrewsbury: iSmithers Rapra Tech-nology; 2009 [chapter 3].
[9] Liu J, Thompson ZJ, Sue HJ, Bates FS, Hillmyer MA, Dettloff M, et al. Macro-molecules 2010;43:7238e43.
[10] Grubbs RB, Dean JM, Bates FS. Macromolecules 2001;34:8593e5.[11] Girard-Reydet E, Sautereau H, Pascault JP. Polymer 1999;40:1677e87.[12] Ritzenthaler S, Court F, Girard-Reydet E, Leibler L, Pascault JP. Macromolecules
2003;36:118e26.[13] Rebizant V, Venet AS, Tournilhac F, Girard-Reydet E, Navarro C, Pascault JP,
et al. Macromolecules 2004;37:8017e27.[14] Kosonen H, Ruokolainen J, Torkkeli M, Serimaa R, Nyholm P, Ikkala O. Mac-
romol Chem Phys 2002;203:388e92.[15] Boyard N, Sinturel C, Vayer M, Erre R. J Appl Polym Sci 2006;102:149e65.[16] Builes DH, Tercjak A, Mondragon I. Polymer 2012;53:3669e76.[17] Builes DH, Hernandez H, Mondragon I, Tercjak A. J Phys Chem C 2013;117:
3563e71.[18] Guo Q, Thomann R, Gronski W. Macromolecules 2002;35:3133e44.[19] Larrañaga M, Martín MD, Gabilondo N, Kortaberria G, Corcuera MA,
Riccardi CC, et al. Polym Int 2004;53:1495e502.[20] Sun P, Dang Q, Li B, Chen T, Wang Y, Lin H, et al. Macromolecules 2005;38:
5654e67.[21] Larrañaga M, Gabilondo N, Kortaberria G, Serrano E, Remiro P, Riccardi CC,
et al. Polymer 2005;46:7082e93.[22] Larrañaga M, Martin MD, Gabilondo N, Kortaberria G, Eceiza A, Riccardi CC,
et al. Colloid Polym Sci 2006;284:1403e10.[23] Larrañaga M, Arruti P, Serrano E, De la Caba K, Remiro PM, Riccardi CC, et al.
Colloid Polym Sci 2006;284:1419e30.[24] De la Caba K, Larrañaga M, Eceiza A, Corcuera MA, Mondragon I. Macromol
Symp 2006;239:30e5.[25] Larrañaga M, Serrano E, Martin MD, Tercjak A, Kortaberria G, De la Caba K,
et al. Polym Int 2007;56:1392e403.[26] Chang CJ, Yang YL, Lee YP, Chiang CJ, Dai CA, Chen JC, et al. Microelectron Eng
2011;88:1737e41.[27] George SM, Puglia D, Kenny JM, Jyotishkumar P, Thomas S. Polym Eng Sci
2012;52:2336e47.[28] Guo Q, Harrats C, Groeninckx G, Koch MHJ. Polymer 2001;42:4127e40.[29] Blanco M, López M, Kortaberria G, Mondragon I. Polym Int 2010;59:523e8.[30] Mizutani K. J Mater Sci 1993;28:2178e82.[31] Gutierrez J, Tercjak A, Mondragon I. J Phys Chem C 2010;114:22424e30.[32] Dean JM, Lipic PM, Grubbs RB, Cook RF, Bates FS. J Polym Sci Polym Phys
2001;39:2996e3010.[33] Dean JM, Grubbs RB, Saad W, Cook RF, Bates FS. J Polym Sci Polym Phys
2003;41:2444e56.[34] Wu J, Thio YS, Bates FS. J Polym Sci Polym Chem 2005;43:1950e65.[35] Ocando C, Tercjak A, Serrano E, Ramos JA, Corona-Galván S, Parellada MD,
et al. Polym Int 2008;57:1333e42.[36] Ocando C, Tercjak A, Martín MD, Ramos JA, Campo M, Mondragon I. Macro-
molecules 2009;42:6215e24.[37] Ratna D, Becker O, Krishnamurthy R, Simon GP, Varley RJ. Polymer 2003;44:
7449e57.[38] Ocando C, Tercjak A, Mondragon I. Compos Sci Technol 2010;70:1106e12.[39] Sahagun CM, Morgan SE. ACS Appl Mater Interfaces 2012;4:564e72.[40] Pascault JP, Sautereau H, Verdu J, Williams RJJ. Thermosetting polymers. New
York: Marcel Dekker, Inc; 2002 [chapter 11].[41] Builes DH, Hernandez-Ortiz JP, Corcuera MA, Mondragon I, Tercjak A. ACS
Appl Mater Interfaces 2014. http://dx.doi.org/10.1021/am4046266.[42] Thio YS, Wu J, Bates FS. J Polym Sci Polym Phys 2009;47:1125e9.[43] Builes DH, Labidi J, Eceiza A, Mondragon I, Tercjak A. Compos Sci Technol
2013;89:120e6.[44] Balkenende AR, Van de Boogard HJAP, Scholten M, Willard NP. Langmuir
1998;14:5907e12.[45] Long J, Chen P. Langmuir 2001;17:2965e72.[46] Alexandridis P. Curr Opin Colloid Interface 1997;2:478e89.





























