Embed Size (px)
Citation preview

Tailoring the Catalytic Properties and Specificity of
Subtilisin B. lentus via a Combined Site-Directed
Mutagenesis and Chernical Modification Approach
Grace DeSantis
A Thesis submitted in conforrnity with the requirements for the degree of Doctor of Philosophy
Graduate Department of Chemistry University of Toronto
O Copyright by Grace DeSantis 1999

National Library IS.1 of Canada Bibliothéque nationale du Canada
Acquisitions and Acquisitions et Bi bliogra phic Seiuices services bibliographiques
395 Wellington Street 395. fue~ Wellingtm OttawaON KIAON4 OnawaON K1A ON4 Canada Canada
The author has granted a non- exclusive licence allowing the National Library of Canada to reproduce, loan, distribute or seli copies of this thesis in microform, paper or electronic formats.
The author retains ownership of the copyright in this thesis. Neither the thesis nor substantial extracts fiom it may be p ~ t e d or otherwise reproduced without the author's permission.
L'auteur a accordé une licence non exclusive permettant à la Bibliothèque nationale du Canada de reproduire, prêter, distribuer ou vendre des copies de cette thèse sous la forme de microfichelnlm, de reproduction sur papier ou sur format électronique.
L'auteur conserve la propriété du droit d'auteur qui protège cette thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation.

Tailoring the Catalytic Properties and Specificity of Subtilisin B. Ientus via a
Combined Site-Directed Mutagenesis and Chernical Modification Approach
by
Grace DeSantis
1 999
A Thesis submitted in conforrnity with the requirements
for the degree of Doctor of Philosophy
Graduate Department of Chemistry
University of Toronto
Abstract
Chemically rnodified mutant enzymes (CMMs) of subtilisin Bacillus lentus
(SBL) were designed to alter its specificity and catalytic properties. Modification of
S156C, which is located toward the bottom of the S1 pocket and whose side chain is
solvated, effected only small changes in its catalytic properties. Conversely.
modification of S166C. which is located at the bottom of the SI pocket and points
into the pocket, effected larger changes. The S166C-S-CH2CH2S0,- CMM caused a
16-fold decrease in k,/KM with the standard suc-AAPF-pNA substrate. a 5-fold
decrease in the binding of the 2,4-diclorophenylboronic acid inhibitor and a 0.93 unit
increase in the pK, of His64. The restricted SI pocket of SI 66C-CMMs was
demonstrated by the precluded binding and covalent attachment of a p-boronic acid
benzophenone photoactivatable active-site directed inhibitor.
SBL prefers large hydrophobic Pl substrate residues and the CMM strategy
was exploited to confer a more universai P, specificity on SI. A large cyclohexyl
group was introduced at position S166C to fiIl up the S, pocket. causing a 2-fold
improvement in k,/KM for the small P, residue suc-AAPA-pNA substrate and a 51-
fold improvement in suc-AAPA-pNA/suc-AAPF-pNA selectivity compared to WT.

iii
Negatively and positively charged groups were introduced at position S i 66C. A
monotonic increase in k,JKM with the positively charged P, residue containing
substrate suc-AAPR-pNA was effected by increasing negative charge at position
166. This culrninated in a 9-fold improvement in k,JKM for the tri-negatively charged
S I 66C-S-CH,CH,C(COO-), CMM and a 61-fold improvement in its suc-AAPR-
pNA/suc-AAPF-pNA selectivity. The positively charged S 166C-S-CH,CH,NH,+ CMM
showed a 19-fold irnprovement in kJKM with the negatively charged P, residue
containing substrate, suc-AAPE-pNA and a 54-fold improvement in its suc-AAPE-
pN A /suc-AAPF-pNA selectivity . Modification of the S, pocket N62C residue resulted in seven of eleven
CMMs, with higher than wild type (WT) levels of activity. A monotonic increase in
activity with increasing side-chain sire was observed culminating in a 3-fold
improvement in k, JKM for N62C-S-c-CH,C,H,,. The indication from molecular
modelling that this was due to increased acidity of the catalytic triad residue His64
was confirmed by pH-activity profile studies which revealed pK, decreases of up to
0.72 units. A linear correlation between the hydrophobicity of the introduced side
chain and the observed pK, was apparent.

To my pa-rents and Michael

"lt is by logic that we prove. but by intuition that we discover."
J. H. Poincaré

Acknowledgments
I am sincerely grateful to rny research supervisor and mentor, Professor J.
Bryan Jones, for his guidance, support and inspiration throughout the duration of
this work. As well, I am grateful to Professor Mawin Gold for sharing his expertise in
protein purification and characterization techniques and for excellent technical
guidance.
I am very grateful to Professors Thomas T. Tidwell and Robert A. McClelland
for the use of their photoreacton. I am particularly indebted to the members of my
doctoral cornmittee Professors Ronald Kluger, Andrew M. MacMillan and G.
Andrew Woolley for their advice and encouragement. As well, I am deeply indebted
to all of my coworkers in the research group, for their technical assistance as well as
many inspiring scientific debates. In particular I would like to thank Drs. Ben Davis
and Xiao Shang for the synthesis of rnethanethiosulfonate reagents, Drs. Per
Berglund and Michele Stabile for technical guidance in the techniques of protein
purification and modification, Dr. Erika Pfettner for advice with the benzophenone
work and Dr. Richard Martin for guidance with molecular modelling.
I am appreciative of graduate fellowships from the University of Toronto and
the Natural Sciences and Engineering Research Council of Canada .
I am grateful to Genencor International Inc. for providing the WT and
cysteine mutants of subtilisin. I am particulariy grateful to DE. Rick Bott, Christian
Paech, Thomas Graycar. Colin Mitchinson and Chris Murray of Genencor
lnternational lnc. for helpful discussions and technical advice. Particular thanks are
extended to Dr. Rick Bott for rnaking available the PD6 coordinates for SBL prior to
publication, Dr. Christian Paech for his tutelage on tryptic-digestion and HPLC-ES1
analysis during my visit to Genencor and for analyzing sorne of my samples on very
short notice and Dr. Sue Middlebrook for peptide sequencing.
Many thanks go to my husband and soul-mate, Michael DiDonato. for his
enduring moral support and understanding. Finally, I would like to thank my family
for their encouragement. patience and support throughout my education.

vii
Table of Contents
Abstract
Acknowledgments
Table of Contents
List of Figures
List of Tables
Abbreviations
Chapter 1 : Introduction
Part One:
1 .O Enzymes
1.1 Enzymes in Organic Synthesis
1.2 Enzyme Classes
1.3 Serine Proteases
1.4 Enzyme Kinetics
1.5 Subtilisin Bacillus lentus (SBL)
Part Two:
2.0 Altering Enzyme Activity
2.1 Noncovalent Modification of Enzymes
2.2 Site-Directed Mutagenesis and Rational Design
2.3 Random Mutagenesis
2.4 De Novo Protein Design
2.5 Catalytic Antibodies
2.6 Enzyme Mimics
2.7 Nucleic Acid Enzymes
v i
vii
X
xii . -.
Xlll

Parf Three:
3.0 Introduction of Unnatural Amino Acids or Side Chains 27
3.1 Chemical Modification to Generate Semisynthetic Enzymes 27
3.2 Unnatural Amino Acid Mutagenesis 30
3.3 Peptide Synthesis and Fragment Ligation 31
3.4 Site-Directed Mutagenesis Combined with Chemical Modification 32
3.5 Current Study 33
References 36
Chapter 2: Site-Directed Mutagenesis Combined with Chemical Modification as a
Strategy for Altering the Specificity of the SI Pocket of Subtiiisin B. lentus
Introduction
Results
Discussion
Experimental
References
Chapter 3: Probing the Altered Specificity and Catalytic Properties of Mutant
Subtilisin Chemically Modified at Position S156C and S166C in the S, Pocket
Introduction
Results and Discussion
Experimental
References
Chapter 4: Benzophenone Boronic Acid Photoaffinity Labeling of SBL CMMs
Introduction
Results and Discussion
Experimental
References

Chapter 5: Towards Tailoring the Specificity of the S, pocket of Subtilisin B. lentus:
Chemical Modification of Mutant Enzymes as a Strategy for Removing Specificity
Limitations 121
Introduction 1 22
Results 123
Discussion 127
Experimental 1 33
References 136
Chapter 6: Chemical Modification of the N62C Cysteine Mutant of Subtilisin B.
lentus Can Create Better Catalysts than the Wild-Type Enzyme 140
l ntroduction 141
Results and Discussion 142
Experimental 147
References 149
Chapter 7: Chemical Modification at a Single Site Can lnduce Significant Shifts in
the pH Profiles of a Serine Protease 151
Introduction 1 52
Results and Discussion 153
Experimental 161
References 163
Perspective 166
References 169
Appendix
Representative Calculation of k,, and KM
Representative Calculation of K,
xvi
xvii

List of Figures
Chapter 1
Figure 1.1 Mechanism of Serine Proteases
Figure 1.2 Active Site Nomenclature of the Serine Proteases
Figure 1.3 Crystal Structure of SBL
Figure 1.4 Lipase Catalyzed Reaction
Chapter 2
Figure 2.1 Active Site of SBL showing Ser156 and Serl66
Figure 2.2 Specificity Patterns for S 1 56C and S i 66C CMMs
Figure 2.3 Molecular Modelling for S166C CMMs
Chapter 3
Figure 3.1 pH-Activity Profiles for WT-SBL and S i 56C-S-CH,CH,SO,-
Figure 3.2 Binding of Boronic Acid Inhibitors to S 156C and S i 66C CMMs
Figure 3.3 Modelling for p-Carboxyphenyl boronic acid
Chapter 4
Figure 4.1 Time Course for Photolysis of WT-SBL and BBP
Figure 4.2 HPLC Trace of Native and BBP Crosslinked WT-SBL
Figure 4.3 Proposed Mechanism of BBP Crosslinking
Figure 4.4 Time Course for Photolysis of SBL CMMs and BBP
Chapter 5
Figure 5.1 Screen of S 166C CMMs with suc-AAP-F/A/R/E-pNA Substrates
Figure 5.2 Negative Charge at Position166 versus Activity
Figure 5.3 Modelling of WT-SBL and S166C-S-CH,CH,NH,' with AAPE

Chapter 6
Figure 6.1 Active Site of SBL lndicating Position of N62
Figure 6.2 Specificity Patterns for N62C CMMs
Chapter 7
Figure 7.1 Molecuiar Modelling for N62C CMMs 1 54
Figure 7.2 pH-Activity Profile for WT-SBL and N62C-S-c-CH,C,H,, 155
Figure 7.3 A~K, of N62C CMMs 157
Figure 7.4 Linear Correlation Between pK, and Log P i 59

xii
List of Tables
Chapter 2
Table 2.1 Kinetic Parameters for S156C and S166C CMMs (pH 7.5) 69
Chapter 3
Table 3.1 Kinetic Constants for S I 56C and S I 66C CMMs (pH 8.6) 84
Table 3.2 Inhibition Constants for S156C and S I 66C CMMs 87
Chapter 4
Table 4.1 Kinetic and Inhibition Constants for S166C CMMs with BBP 1 03
Table 4.2 ES-MS Data for BBP Treated WT-SBL 1 05
Table 4.3 ES-MS Data for BBP Treated SBL CMMs 110
Chapter 5
Table 5.1 Kinetic Evaluation of Altered SI Pocket Specificity
Chapter 6
Table 6.1 Kinetic Parameters for N62C CMMs
Chapter 7
Table 7.1 Kinetic and pK, Data for N62C CMMs
A ppendix
Table 1 k,,. KM for S I 66C-S-CH,-c-C,H,,
Table 2 K, for WT-SBL and 3-aminophenyl boronic acid
xvi
xvii

Abbreviations
Ac Ala (A) Arg (R) Asn (N) A ~ P (O) n-BuLi CHES CYS (Cl d de DMF DMSO DTT ee ESMS EtOH Et,O FPLC Gln (Q) Glu (E) G ~ Y (G) His (H) Ile (1) Leu (L) LYS (KI m MeOH MES Met (M) MMTS MWCO NAD(P) PAGE PEG Phe (F) Pro (P) S
SBL SBN' Ser (S)
acetyl alanine arginine asparagine aspartic acid n-butyl lithium 2-(cyclohexylamino)ethanesulfonic acid cysteine doublet diasteriomeric excess dirnethylforrnamide dimeth ylsulfoxide dithiothreitol enantiomeric excess electrospray mass spectrometry ethanol diethylether fast performance liquid chromatography glutamine glutamic acid glycine histidine isoleucine leucine iysine multiplet methanol 4-rnorpholineethanesulfonic acid Methionine methyl methanethiosulfonate molecular weight cutoff nicotinamide adenine dinucelotide (phosphate) polyacrylarnide gel electrophoresis pol y(ethyleneglyco1) phen ylalanine proline singlet su btilisin Bacillus lentus subtilisin Bacillus amyloliquefaciens serine

xiv
SDS SUC-AAPF-pNA TCA TFA THF Thr (T) TMS T V (W) Tyr (Y ) Val (V) v/v w/v
sodium dodecyl sulfate succinyl-alanyl-alanyl-prolyl-phenylalanyl-p-nitroanilide trichloroacetic acid trifluoroacetic acid tetrahydrofuran threonine trimethylsilane tryptophan tyrosine valine volume/voiume weig ht/volume

Chapter 1

Introduction
Enzymes are now widely exploited as chiral catalysts in organic synthesis,
with their application being driven mainly by the synthetic opportunities provided by
the exquisite structural. regio- and stereospecificities of enzyme-catalyzed
reaction~."~ As a result of the asymmetric catalytic power offered by enzymes, the
availability of information providing guidelines for the prediction of stereospecificity,
and the current understanding of the mechanisms of enzyme catalysis, enzymes
can now be used to access asymmetric transformations which would otherwise be
impractical by any other means.
During the last century, the understanding of the complementary relationship
between the enzyme active site and the substrate has developed extensively since
it was first recognized and described by Emil Fischer as the "lock and key" modeL6 It
was later hypothesized by J. B. S. Haldane that enzymatic action hinges upon an
induced fit since "the key does not fit the lock quite perfectly, but exercises a certain
strain on itM7and further developed to the currently accepted notion of enzyme-
transition state complementarity by Linus Pauling.' However, the continued
development of highly selective enzyme catalysts requires a more thorough
understanding of the factors which control enzyme activity, such as steric effects.
electrostatic interactions, hydrogen bonding, hydrophobic effects, and A-stacking
interaction^.^
Creating enzymes with new catalytic activities, and tailoring the specificity of
existing ones to better accommodate unnatural substrates, is crucial to further
increasing the scope of the applicabilities of enzymes in organic synthesis and thus
is an area of considerable current re~earch.'~' '~ lnsights into the electro~tat ic '~'~
steric,16 and hydr~phobic,'~ factors which govem enzyme-substrate interactions have
been probed by site-directed mutagenesis studies. However. despite recent
advances in understanding the intenolecular interactions which detennine enzyme
structural-, regio-. and stereo-specificity, truly rational tailoring of enzyme specificity

remains an elusive goal. The current study is aimed at developing a rapid and
efficient methodology for tailonng enzyme specificity which is not limited by the 20-
natural amino acid constraint of site-directed mutagenesis. The approach outlined
entails the introduction of one cysteine residue at a key active site position via site-
directed mutagenesis, which is then thioalkylated with an alkyl methanethiosulfonate
reagent, CH3S0,-S-R, such that: WT -r CysmU,,,, + H3C-S0,-S-R -+ Cys-S-R to give
a chemically modified mutant enzyme (CMM) where R is infinitely variable. The
serine protease subtilisin Baciiius lentus (SBL) was selected as the representative
enzyme for this strategy of incorporating unnatural amino acid moieties.

1 .O Enzymes
Enzymes are outstanding biological catalysts, providing rate enhancements
of up to 1 017 fold over the corresponding uncatalyzed reacti~ns.'~ Enzymes operate
under mild conditions, and at physiological temperature and pH in aqueous
s~lut ion.~ As a result of the evolutionary pressures imposed by the complexity of
biological systems, they are exquisitely selective with respect to the structure and
stereochemistry of the substrate, and hence the product. Enzymes can be isolated
from mammalian, bacterial, microbial, plant or fungal sources. Of the more than
3000 known several hundred are commercially available as pure
crystalline products, as whole cell preparation~,~ or in immobilized2' or crosslinked
forms?
Enzymes are composed of unbranched polypeptide chains consisting of the
20 natural L-U-amino acids linked together by amide bonds between the a-carboxyl
group of one residue and the a-amino group of the nextSg This generates a primary
structure defined by the amino acid sequence. The genetically encoded sequence
determines the secondary structural components of a-helix, P-sheet and p-turn,
which in turn give rise to the overall fold of the enzyme or the tertiary structure.23 In
addition to noncovalent chemical interactions, the tertiary fold may be further
stabilized by disulfide bridges.23 It is the unique tertiary fold of enzymes which
controls the spatial arrangement of the amino acid residues and dictates enzyme
specificity by defining the active site where substrate binding and reaction occur.'

1.1 Enzymes in Organic Synthesis
In addition to the vital role of enzymes in biological systems, enzymes
catalyze a broad spectnim of reactions in vitro which are of interest to organic
chernist~.'-~ The commercial applications of enzymes are for detergent, dairy
product, and starch processing, and to a lesser extent for fine chernical synthesis.
and they have an annual world market of $900 million dollars.24
Enzymes are employed in organic synthesis applications out of convenience,
necessity and opportunity. The milder, more selective, reaction conditions employed
for enzymatic reactions minimize problems of isomerization, racemization and
epimerization often encountered in traditional chemical synthesis.' Furthemore,
since enzymatic reactions are optimal in aqueous media they offer environmental
advantages over current organic catalysts, which usually contain toxic transition
metal complexes.25 and that must be employed in organic solvents which are
increasingly becoming more difficult to dispose of. Furthemore, enzymes are
intrinsically compatible with one another and consequently a number of enzymes
can be used to accomplish multi-step reaction sequences in a single reaction
es sel.^^ In contrast, one-pot use of multiple non-biological catalysts are relatively
limited.25

1.2 Enzyme Classes
The International Biochemistry Union has adopted a classification which
divides the enzymes into six groups based upon their function, as described b e l ~ w . ' ~
1 . The Oxidoreductases catal yze oxidation-reduction reactions involving
oxygenation, (C-H -+ C-OH), or the net removal or addition of hydrogen atom
equivaients, as for (CH(0H) = C=O and CH-CH C=C).
2. The Transferases mediate the transfer of groups such as acyl. sugar.
phosphoryl, aldehyde and ketone moieties from one molecule to another.
3. The Hydrolases hydrolyze a broad range of functional groups, including
glycosides, anhydrides and esters, as well as amides, peptides and other C-N
containing functions.
4. The Lyases catalyzes the addition of HX to double bonds such as C=C, C=N and
C=O. as well as the reverse reaction.
5. The lsomerases catalyze various isomerizations, including C=C bond migration.
cis-trans isomerization, and racemization.
6. The Ligases are responsible for the formation of C-O, C-S, C-N, C-C and
phosphate ester bonds and are also commonly referred to as synthetases.
There is an enzyme-catalyzed counterpart for almost every type of organic
reaction including key synthetic reactions (Scheme 1 .l ) such as the aldol
condensation, 3+27asymmetric epoxidation rea~t ions ,~~ Baeyer -Villiger oxidationlzg'"
Claisen rearrangemenp' and recently even the DielsAlder reaction?' However, the

class of enzymes most widely applied to organic synthesis applications are the
hydrolases. Members of the hydrolase family which have been exploited extensively
include, lipase^.^." esterasess and p r~ teases~~ whose in vivo function is the
hydrolysis of fatty acid esters, esters, and proteins respectively. Amongst these, the
serine protease (EC 3.4.21 ) subclass, of which subtilisin is a member, has been
utilized extensively for both preparative applications and for mutagenesis studies.
Furthermore, subtilisin can under the appropriate conditions, be employed as an
esterification catalyst (Scheme 1 -1 d)?
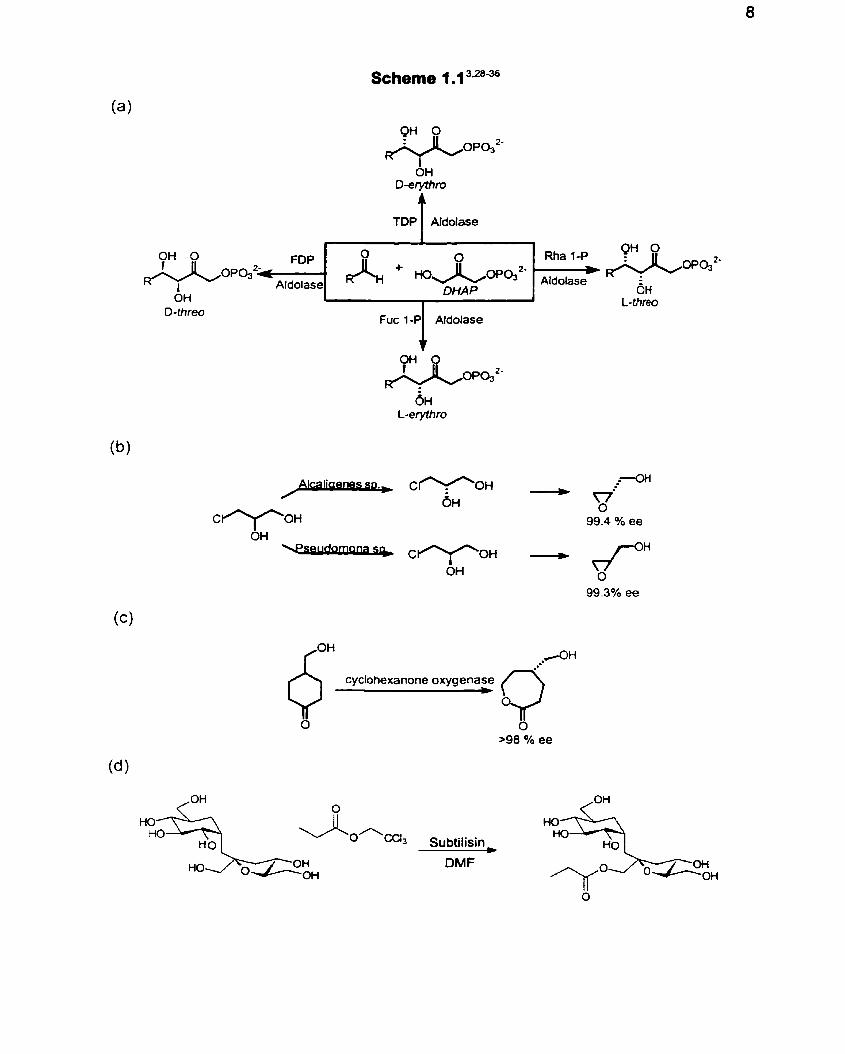
Scheme 1.1 3.2836
Fuc 1-P Aldolase I
cyclohexanone oxygenase
O -9 >98 O O h ee
0 ~ ~ ~ 3 Subtilisin - DMF
'Yo

1.3 Serine Proteases
The sub-class of hydrolases known as the proteases, which catalyze the
hydrolysis of amide linkages in proteins in vivo and catalyze the hydrolysis of both
amide and ester bonds in vitro have been utilized extensively in organic syn thes i~ .~~~
The serine proteases (EC 3.4.21) are an endopeptidic sub-class of this group
requiring no cofactors, and which are characterized by a uniquely reactive serine
hydroxyl which is part of the Ser - His - Asp active site catalytic triad, first identified
in chym~trypsin.~' The spatial arrangement of the catalytic tnad is conserved
amongst serine proteases despite the lack of sequence homology and differences in
overall f ~ l d . ~ ~ This reinforces the crucial role of the triad in the catalytic mechanism
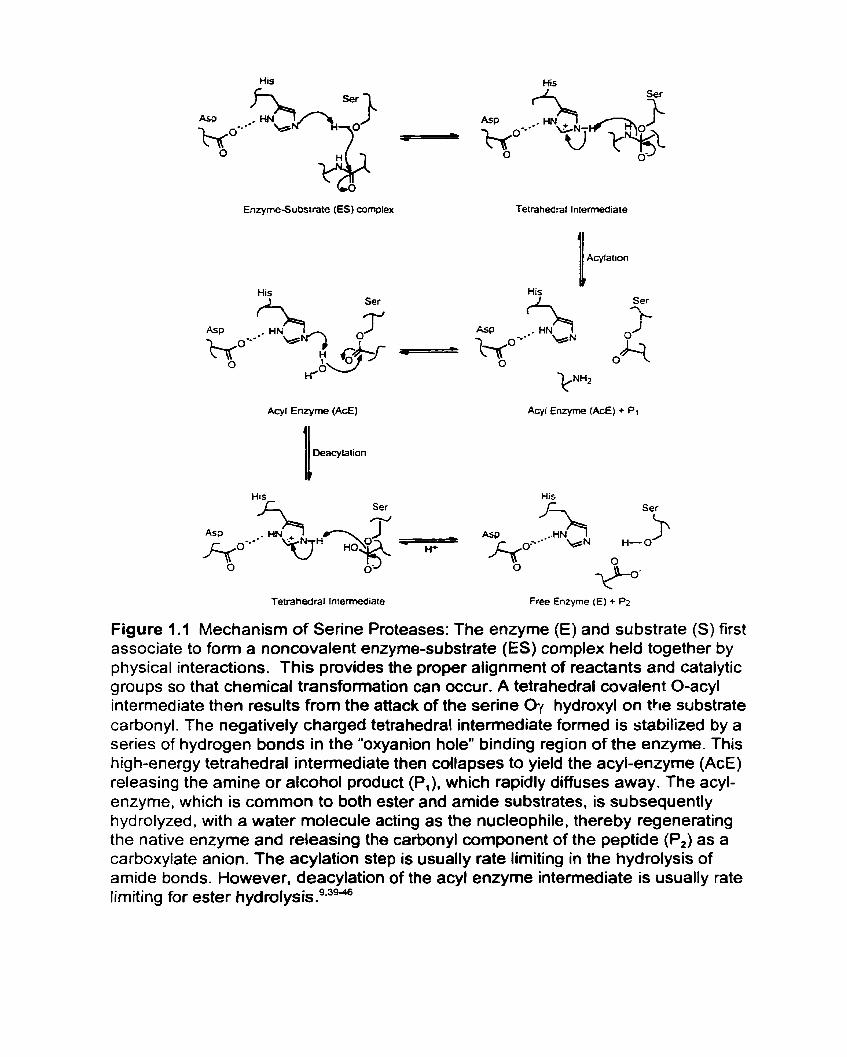
which has been thoroughly investigated and is well characterized, as noted in Figure
1.1 .9.39a0 The imidazole side chain of histidine increases the nucleophilicity of the
serine hydroxyl by acting as a general base catalyst. The buried carboxyl side chain
of aspartic acid, which differs slightly in position between pr~teases,~' serves to
raise the pK, of imidazole through its negatively charged carboxylate side chain, as
well as to confine its location. Serine proteases catalyze ester and amide hydrolysis
in a pH dependent manner.4245Amide and ester substrates are hydrolyzed by the
serine proteases via the acyl enzyme r n e c h a n i ~ r n . ~ ~ ~ . ~ ~
His His F
Enzyme-Substrate (ES) complex
Ser
Acyt Enzyme (A&)
His L Ser
Tetrahedral Intermediate
Tetrahedral Intemiediate
\
Free Enzyme (El + Pz
Figure 1.1 Mechanism of Serine Proteases: The enzyme (E) and substrate (S) first associate to form a noncovalent enzyrne-substrate (ES) complex held together by physical interactions. This provides the proper alignment of reactants and catalytic groups so that chernical transformation can occur. A tetrahedral covalent O-acyl intenediate then results from the attack of the serine Oy hydroxyl on the substrate carbonyl. The negatively charged tetrahedral intemediate formed is stabilized by a series of hydrogen bonds in the "oxyanion holew binding region of the enzyme. This high-energy tetrahedral intermediate then collapses to yield the acyl-enzyme (AcE) releasing the amine or alcohol product (P,), which rapidly diffuses away. The acyl- enzyme, which is cornmon to both ester and amide substrates, is subsequently hydrolyzed. with a water molecule acting as the nucleophile, thereby regenerating the native enzyme and releasing the carbonyl component of the peptide (P,) as a carboxylate anion. The acylation step is usually rate limiting in the hydrolysis of amide bonds. However, deacylation of the acyl enzyme intermediate is usually rate lirniting for ester i ~ y d r o l y s i s . ~ . ~ ~ ~

1.4 Enzyme Kinetics
For many enzymes, including the serine proteases, a predictable kinetic
pattern is observed which corresponds to the kinetic expressions developed initially
by Brown4' and further refined by Michaelis and Menten? The initial formation of the
Michaelis complex (ES) is assumed to be rapid and reversible with no chemical
changes taking place. The chemical processes then occur in subsequent steps,
generating the product P. and releasing the enzyme E as shown in Scheme 1.2.
Scheme 1.2
The Michaelis-Menten rate equation (Equation 1 -1). where KM = Ks is solved
based on the following assumptions: (1 ) The ES complex is in therrnodynarnic
equilibrium with the free enzyme and substrate. This is true only if the formation of
product from the ES complex is very fast relative to the rate of the decomposition of
the ES complex to give enzyme and substrate. (2) [Pl is insignificant compared to
[SI and the substrate concentration is constant. which is possible by considering
only the initial stage of the reaction. (3) The rate expression is valid only for single
substrate system where [SI >>> [El.
Equation 1 .l"
The true Michaelis constant, K,, which is the dissociation constant of the ES
complex, reflects the strength of enzyme substrate binding, with lower values
representing better binding. Under these conditions. i.e. when assurnption (1 ) holds,
K., = Ks. The turnover number. ka,, is the apparent first order rate constant for the
conversion of the enzyme-substrate complex to product. The hyperbolic Michaelis-
Menten equation has two simplified foms. At low substrate concentration. a linear

relationship is observed and can be expressed as v=[E],[S](k,(K,), where I<,JK, is
the apparent second order rate constant which relates the reaction rate to the
concentration of free rather than total enzyme. At low substrate concentration the
enzyme is largely unbound, with [El [El,. The specificity constant, k,jKM, is used
as the best measure of the efficacy and specificity of the enzymatic reaction. At
high substrate concentrations, al1 of the enzyme is in the ES cornplex and the
velocity is independent of substrate. Saturation kinetics is observed and Equation
1.1 simplifies to v=k,,[E],. which is termed the maximal velocity (Vmx). as illustrated
in Scheme 1.3.
Scheme 1.3
In 1925, Briggs and Haldane5' introduced the concept of a steady state in the
enzymatic reaction and showed that the equilibrium concept of Michaelis and
Menten was not essential. In addition, the Michaelis-Menten scheme may be
extended to cover a variety of cases in which additional intermediates, covalently or
noncovalently bound. occur on the reaction pathway, as outlined in Scheme 1.4. 52
Scheme 1.4
k l E + S ' E S EAc k3 * E + Pz k -1

While the Michaelis-Menten equation, as described above, still applies, KM
and k,, are now combinations of various rate and equilibrium constants, as shown in
Equation 1.2. In particular the acylenzyme intemediate (EAc), resulting from serine
protease hydrolysis of amide or ester bonds, is incorporated into Scheme 1.4. In the
limiting case when k-,>>k2, (Scheme 1.4), the Michaelis constant approximates to
kJk, and KM = K,."
A complementary approach to substrate kinetic analysis for probing enzyme
specificity is the use of competitive reversible transition state analogue inhibitors. A
competitive reversible inhibitor (1) binds to the enzyme active site (E) via
noncovalent interactions and forms an El complex which prevents substrate binding,
as shown in Scherne 1.5.
Scheme 1 .55253

In such situations, the affinity of the enzyme for the inhibitor (1) is defined by
the dissociation constant, K,, of the enzyrne-inhibitor complex, (El). The kinetic
manifestation of competitive inhibition is an apparent increase in the Michaelis
constant. KM, by a factor of (1 + [I]/KI) while the turnover number, k,,, remains
unaltered. as shown in Equation 1 .3.52-U
Equation 1 .352-53

1.5 Subtilisin Bacillus lenfus (SBL)
The alkaline serine protease, subtilisin Bacillus lentus (SBL, E C 3.4.21 -14)
was chosen as a representative serine protease for the current study since it is a
well characterized enzyme and is of s y n t h e t i ~ ' ~ ~ as well as of industrial" interest.
Furthermore, SBL's high resolution crystal structure has been solved,"-s it has been
cloned , overexpressed and p ~ r i f i e d , ~ ~ and its kinetic behaviour well chara~terized.~O- 6 3
The amount of availabie information on subtitisins, combined with their
industrial and synthetic applications, makes them ideal model systems for protein
engineering.64d6 Subtilisins are a class of serine endopeptidases secreted into the
external medium by a wide variety of gram-positive bacteria, such as B a c i l l ~ s . ~ ~
Subtilisins are synthesized as pre-proenzymes, which are then translocated over a
cell membrane via the pre-peptide (or signal peptide), and finally activated by
cleavage of the pr~-peptide.~' The subtilisin proteases are single-domain molecules
with no disulfide bridges, and with a hemispherical shape of 40 A dian~eter?~ Their
active site is located on the fiat surface of the hemisphere? The core of the protein
is composed of a twisted parallel P-s heet with a-helices ninning anti-parallel to the
I)-sheet and parallel to each other? SBL, which is secreted by an alkalophilic
bacterium, has a high isoelectric point, pl = 11.1 high thermal stability at alkaline
pH6' and has maximal catalytic activity between pH 8 and pH 1 P7 SBL has 13
positively charged lysine and arginine residues and 10 negatively charged aspartate
and glutamate residues, for a total of 25 charged groups, including the N- and C-
termini. Despite having fewer charged residues than subtilisin BPN', SBL has seven
salt bridges. with two additional salt bridges being possible at high pH, which is two
more than subtilisin BPN"s f~ve.'~ SB1 has two CaZ+ binding sites which do not have
a catalytic role but rather contribute to its thermal stability" which in turn increases
autolytic resistance." The high affinity Ca2' binding site is seven coordinate and
exhibits full occupancy, while the lower affinity site is five coordinate and exhibits an
occupancy of 0.6? The crystal structure of SBL also reveals a linear chain of five

water molecules, linked by a network of hydrogen bonds and extending from the
surface of the protein into the active site, and whose function appears to be
structural rather than ~atalytic.'~
Cleavage "scissile bond"
Figure 1.2 Active Site Nomenclature of the Serine Proteases: The substrate residues are denoted P,, P2 etc. and the active site pockets to which they bind are denoted S,, S, etc. The residues on the carbonyl side of the scissile bond are denoted P,' . P,' etc."
The binding region of SBL has been described as a surface channel which
can accommodate at least eight residues, P, to P,' in the S, to S,' p o c k e t ~ , ~ ~ where
the scissile bond is behveen the residues denoted P, and P,', as shown in Figure
1 .2.72 While SBL, with its 269 residues, is homologous to subtilisin BPN' (62%
identical residues) a significant difference between SBL and other subtilisins is a
four residue deletion in its SI pocket. and two single deletions at positions equivalent
to residues 36 and 56 in subtilisin BPN'.57 The SI primary specificity determining
pocket of SBL is composed of residues 155 to 162,166 and 126 to 128, and
exhibits a preference for large hydrophobic residues such as the benzyl side chah
of Phe (Figure 1 .3).60"2 The catalytic triad residues of subtilisin are Asp32, His64
and Ser221 (BPN' numbering). Residue Asn155 is located in the so called
"oxyanion-hole" and helps to stabilize the oxyanion generated in the tetrahedral
transition tat te.'^
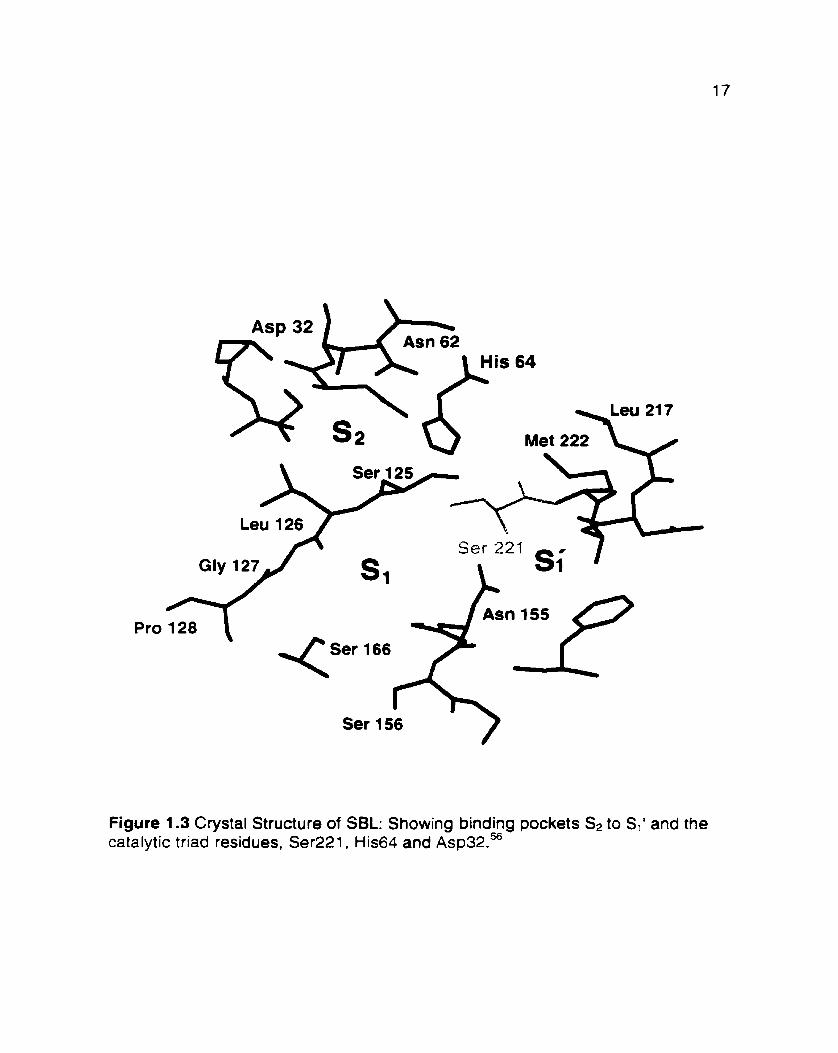
Pro
Figure 1.3 Crystal Structure of SBL: Showing binding pockets SI to Si1 and the catalytic triad residues, Ser221. His64 and ~ s p 3 2 . ~

2.0 Altering Enzyme Activity
While the focus of the current study is to expand the specificity and
operational parameters of SBL, altering enzyme activity has already been broadly
applied to overcome the limitations of biocatalysts. The challenges posed by the
narrow substrate specificity, instability and high cost of some enzymes have been,
and continue to be, addressed through several approaches, including covalent
modification, noncovalent manipulation. and protein engineering of existing
catalysts, and through the de novo design of biocatalysts and of biocatalyst mirnics.
In addition to optimizing biocatalysts for organic synthesis applications, these
approaches to altering enzyme activity have increased our collective understanding
of protein structure, stability and function and have yielded insights into the factors
which govern biocatalysis. The representative examples of these approaches
provided below are illustrative of their capacities and of their deficiencies.
2.1 Noncovalent Modification of Enzymes
One of the simplest methods of altering the specificity. reactivity and stability
of enzymes is by mechanical manipulation. This involves the use of techniques
which do not alter primary structure but nonetheless alter enzyme specificity through
changing the properties of the interaction between the biocatalyst and substrate.
These include changes in solvent properties, molecular irnprinting. substrate
engineering, temperature effects. and enzyme immobilization.
For preparative transformations, the use of enzymes in organic sol vent^'^"'
has received considerable attention since it addresses the challenges posed by
insolubility of some substrates in aqueous solution, and in some cases minimizes
side-reactions such as hydrolysis, racemization. and decomposition. An example of
increased product optical purity, due to diminished chemical side-reactions, is the
preparation of cyanohydrins by the mandelonitrile lyase-catalyzed addition of

hydrogen cyanide to various aldehydes in ethyl acetateea2 An important advantage of
ernploying enzymes in organic solvents is the possibility to shift therrnodynamic
equilibria, for example to favour peptide synthesis over hydrolysis. In this way, by
using hydrolases in organic solvents. esters, 36-83-a6 lactonesB7 and amidessa or
peptide^^^'^' can be chemo- regio- and enantio-selectivel y synthesized (Figure 1 -1 d ,
page 8).36 ln addition. the use of hydrophobic organic solvents can reverse the usual
proteolytic reaction enantiopreference from L-amino acid to D-amino acid
~ubstrates.~~ Supercritical fluids have also been exploited as solvents for enzymatic
reactions and have been found to increase activityg3 Furthemore, enzyme
specificity rnay be rnodulated by altering the pressure of supercritical fluids which in
turn alters the solvent dielectric constant. and under higher pressure the solvent
becomes increasingly hydrophilic* For example, the stereoselectivity of subtilisin
was increased by increasing the pressure in fluorofon." In addition. biocatalyst
thermal stability may be enhanced in organic solvents due to increased structural
rigidity.'= The available crystal s t r ~ c t u r e s ~ ~ ~ and r n o d e l ~ ~ ~ - ' ~ ~ of biocatalysts in
organic solvents help to provide insights into the molecular basis for the altered
specificities.
Another approach toward altering enzyme activity is by immobilization. For
example, enzyme adsorption on silica. Celite.'ol potassium ph~spha te ,~~ and on
modified controlled-pore glass (CPG) has afforded increased reaction rates for
preparative biotransformations in organic sol vent^.^^-'^^ Complementary to physical
methods of adsorption is chemical immobilization, in which functional groups on the
surface of the protein, such as amino and carboxyl groups, are used for attachment
to a support.lo3 For example, poly(ethy1ene glycol) has been exploited extensively
for enzyme immobilization and has generated enzymes with increased solubility and
stability in organic sol vent^.'^^-^^^ In addition, the commercially available cross-linked
enzyme crystals (CLECs).lo6 which are generated by crosslinking with
glutaraldehyde, boast improved biocatalyst ~ tab i l i t y . '~~- '~~ Enzymes have also been
entrapped within gels or polymeric substance^.^'^^'^ However, a recent advance in
chemical immobilization is to introduce a unique cysteine residue at a position away

from the active site and then use this unique sulfhydryl to link the enzyme ont0 a
solid support. thereby effecting higher activity than immobilization via random
residues."'
A conceptually pleasing method to enhance or alter biocatalyst specificity is
through molecular imprinting or bioimprinting. This approach exploits the enzyme's
fiexibility in aqueous solution and its relative inflexibility in organic solvents to first
"mold" the enzyme active site around a substrate mimic in aqueous solution and
then lock-in the shape by lyophilization. The bioirnprinted enzyme is then used in an
organic solvent where it "remembers" the structure of the substrate-mirnic
generating rate enhancements,' l2 altering ~pecificity"~' '* and preventing
ina~tivation."~ For example, chymotrypsin was made to accept D-amino acids by
bioimprinting . l lsl l4 A variation on this general theme is the creation of catalytic
conformationally modified proteins (CCMP) which are prepared by bioimprinting and
then crosslinking a noncatalytic protein such as bovine serurn albumin producing a
CCMP which is more active than the initial unmodified protein?
Figure 1.4 Lipase Catalyzed Reaction: An example of an irreversible Amano Lipase catalyzed transesterification reaction using the vinyl acetate acyl donor.'"
Substrate engineering can also be employed to alter the course of a
biocatalytic reaction. For example. the use of vinyl or isopropenyl acetates as
substrates for transesterification reactions renders the reaction irreversible since the
reaction byproducts are unstable alcohols which tautomerize to nonnucleophilic
aldehydes (Figure 1 .4).117."8 Altematively, highly activated leaving groups such as
trichlorethyl, trifluoroethyl, cyanomethyl, thiol, enol and oxime esters can be used to
make the reactions kinetically virtually ine~ersible. '~~~~" Altemate substrate binding
modes have also been exploited to change the stereochemical course of reaction.12'

2.2 Site-Directed Mutagenesis and Rational Design
The development of site-directed mutagenesis, which permits the routine
replacement of any amino acid residue in a protein whose gene has been cloned,
with any one of the other 19 natural amino acids, has caused an explosion in
attempts to alter enzyme properties.lZla The most intellectually gratifying approach
to mutagenesis is that of rational design. However since it requires a detailed
understanding of the enzyme's catalytic mechanism, substrate specificity
determinants and tertiary structure. it is only suited to enzymes that are very well
u nderstood .66
Mutagenesis studies have not only yielded enzymes with altered specificities,
but have contributed significantly to understanding the e l e c t r ~ s t a t i c , ' ~ - ' ~ - ~ ~ ~ - ~ ~ ~ steric, 16.128-133 and h yd r~phobic'"~.'" factors which govern enzyme-SU bstrate
interactions. Many of the design strategies are based on structural elements which
are exploited in nature by native enzymes with the desired specificity. For example,
the Pl and Pz substrate specificity of subtilisin was changed from one that prefers
large hydrophobic residues to one that prefers positively charged residues by the
introduction of negatively charged residues in its S, and S, pockets. This was based
on the structure of kex2 which exhibits the desired substrate spe~ i f i c i t y . ' ~ - ' ~ ' ~~ The
subtle nuances of the rational design approach are apparent from the failure of a
sirnilar strategy to change the substrate specificity of trypsin from one that prefers
positively charged Pl residues to one that prefers large hydrophobic ones. Since
the S1 pocket residue. 189, is Asp for trypsin but Ser for chymotrypsin it was hoped
that the designed D l 89s mutant of trypsin would confer the desired chymotrypsin-
like specificity ont0 trypsin.l3' however, this approach failed. Instead, several amino
acids including some which do not contact the substrate had to be exchanged
before the specificity change was achieved.'"'" Conversely, attempts to confer
trypsin-like S, specificity ont0 chymotrypsin by the S189D mutation were also
U ~ S U C C ~ S S ~ U ~ . ' ~ ~

Rational design has also been applied successfully to the oxidoreductases.
For example. the stereoselectivity of a L-lactate dehydrogenase (LDH). which is
one of the most stereospecific enzymes known , was significantly reversed. The
designed Ile24OLyslArgl71Tyr LDH double mutant yielded up to 2.3% of D-lactate
by promoting an altered substrate binding mode, which represents a tnily
rernarkable > 500-fold switch in stereospecificity-preference.ld2 In addition. LDH's
specificity toward substrates with positively changed side chains was improved by
the introduction of negatively changed amino acid residues such as AsnlO2AspM;lu
in the substrate binding site.'" Furthemore, LDH's specificity toward substrates
with large or branched side chains was improved by replacing Gln102 by smaller
amino acid residues such as A s d a Similarly. the A95G mutant of L-lactate
oxidase (LOX) was successfully designed to convert LOX to a long chain u-
h yd roxyacid 0 ~ i d a s e . l ~ ~ A similar design strategy was applied to Rhizopus delemar
lipase. Employing a cornputer generated model of the lipase. the V209W/F11 W
double mutant was successfully designed to improve the specificity for short and
medium chah length fatty acids and preclude binding of long chain fatty acids?
This example constitutes the first successfut application of rational design to a
IipaseF
Cofactor tailoring can also be accomplished by rational design. By
cornparison of the active site residues and structure of isopropylmalate
dehydrogenase (IMDH) which exhibits a 100-fold NADlNADP preference. the
cofactor requiring specificity of isocitrate dehydrogenase (IDH) was switched from its
natural 7000-fold NADP+INAD+ preference to a 200-fold NADVNADP- preference by
mutagenesis of seven amino acids which were selected on the basis of X-ray
structures and molecular rnodel~ing.'~~ This example is particulariy important since
NADP* is 5-fold more expensive than NAD'."

2.3 Random Mutagenesis
An alternate approach to site-directed mutagenesis for altering enzyme
specificity is random mutagenesis, which does not require prior understanding of
specificity detemiinants nor knowledge of the structure. The approaches applied to
the preparation of libraries of mutants include the use of chernical
rnutagene~is, '~~- '~ random oligonucleotide primers,'s'55 an error-prone polymerase
chain reaction which may be induced by the use of manganese instead of
m a g n e ~ i u m ' ~ or by an engineered error-prone p~lyrnerase,'~~ mutagenic nucleotide
analogues, 158-1 59 DNA ~hu f f l i ng , ' ~~ '~ ' or the use of an E. co/i mutator train.'^^"^^ For
the purposes of altering enzymatic properties, these randornization techniques are
usually coupled to a suitable high throughput s ~ r e e n ' ~ ' ~ ~ or to positive genetic
se le~ t i on .~ The desired enzyme properties can be optimized throug h several
iterations of mutagenesis and selection and the approach is therefore termed
directed evolution. Directed evolution pemits rapid sampling of sequence space
which is huge since 20' variants are possible where X = nurnber of amino acids in
the protein. The random mutagenesis approach is particularly well suited to systems
that are not well chara~terized.'~~ However, the success of this strategy to yield a
biocatalyst with the desired properties hinges on the design of an appropriate
screen.
This methodology has been applied to a subtilisin from Bacillus subtilis in
order to improve its catalytic activity in organic so~vents. '~~ In this way. an enzyme
was generated that hydrolyzes the standard suc-AAPF-pNA peptide substrate 256
times more efficiently than does wild-type subtilisin in 60% DMF."~-'~* In another
illustration. the enantioselectivity of a lipase from Pseudomonas aeruginosa which
showed an ee of only 2% in favour of the S-enantiomer of 2-methyldecanoic acid p-
nitrophenol ester was optimized. After only four generations of directed evolution, an
enzyme was obtained that gave the S-enantiomer in 81% ee.'73 Random
mutagenesis has also been used to enhance thermal stability of, for example.
cholesterol oxidase from Strept~rnyces.~~~ and of a lipase from Pseudomonas

aerugino~a.'~' Directed evolution has also been employed to expand substrate
specificities. After five rounds of selection, a IO5-fold increase in the catalytic
efficiency of aspartate aminotransferase for P-branched 2-0x0 acids. and a 30-fold
decease for the native substrate, was a~hieved. '~~
2.4 De Novo Protein Design
The approaches described above rewgnize that naturally available enzymes
represent a valuable starting point for the optimization of biocatalysts. However. a
highly desirable goal is the de novo design of a biocatalyst with the needed
property. De novo biocatalyst creation entails the design and generation of a protein
scaffold based on the still limited but rapidly growing understanding of the
relationship between sequence and Unfortunately. whiie a few
proteins or peptides with secondary structure have been designed. reliable de
novo design of biocatalysts remains el~sive."~ Nevertheless. recently a 33-residue
polypeptide was designed which was found to catalyze peptide ligation with a rate
enhancement of 1 O4 compared to the uncatalyzed reaction, and with a 1 O-fold
diastereose~ectivity.~~~ While this approach is still in its infancy. it can clearly be
expected to make dramatic progress as improved design algorithms emerge.laO
An approach to biocatalyst design that complements the de novo approach is
to graft an alternate catalytic machinery ont0 an existing 'protein scaffold"
possessing the desired specificity. thus generating a hybrid en~yrne . '~~ . '~ ' For
example. the E. coli cyclophin, which binds proline containing peptides was
converted into a proline specific protease by mutation of three arnino acids in its
substrate binding cleft to form a triad resembling that of the serine proteases. The
resultant protease enhanced the hydrolysis reaction 1 Os-fold compared to the
uncatalyzed reaction.

2.5 Catalytic Antibodies
In the quest for better biocatalysts, chemists have left no stones untumed. In
this regard, the diversity and chemical potential of the immune system has been
recognized and exploited.'" The speed and diversity of the immune response to
antigen challenge has been exploited to generate catalytic antibodies or abzymes.
Abzymes are prepared by immunizing a host with a chemically stable hapten,
coupled to a carrier protein which resembles the transition state of the desired
reaction. This elicits the production of isolatable antibodies which bind the transition
state analogue hapten. As was predicted eariy on," some of these possess the
capability of promoting catalysis by stabilizing the reactive transition state. Abzymes
that catalyze a wide variety of transformations, including the aldolase reaction1la5 the
Diels-Alder rea~tion,'~"'~' the Claisen reaction,la8 peptide hydoly~is'~~- '" and even
peptide l i g a t i ~ n , ' ~ ' ~ ' ~ ~ have been generated. While abzymes are usually plagued by
low reaction rates and product inhibition, they are potentially promising.
2.6 Enzyme Mimics
Chemists have also adopted a minimalist approach to catalysis in the
development of supramolecular or host-guest chemistry to create small molecules
which can mimic the catalysis of naturally occuning enzymes or even attempt to
create catalytic abilities not observed in na t~ re . '~ ' " In this search, functionalized
and unfunctionalized cyclodextrins, cyclic porphyrin trimers, functionalized Crarn
ethers, cryptands, and a wide variety of other macrocyclic synthetic hosts have been
evaluated as catalysts.18" An eady example by Bender demonstrated the elegance
of this approach, grafting a carboxylate group, an imidazoyl group and a hydroxyl
group ont0 the rim of a P-cyclodextrin, the hydrophobic binding pocket, and the
catalytic triad of the serine proteases was emulated, yielding an artificial enzyme
with approximately the same catalytic activity as chyrn~trypsin.'~' More recently,

supramolecular structures which could catalyze the Diels-Alder reaction have been
sought, such as a porphyrin trimer which can catalyze an exo-selective Diels-Alder
reaction .lg8
2.7 Nucleic Acid Enzymes
Since the initial observation of the autocatalytic property of RNA almost 20
years ago, '99-200 ribozymes have been recognized as versatile and efficient
cataly~ts.~~' In addition to their natural nuclease activity, ribozymes have been
shown to catalyze the synthetically important Diels-Alde?'' and peptide ligationm3
reactions. Furtherrnore, although DNA enzymes have not yet been obsewed in
nature. deoxyribozymes have recently been developed in For example, a
histidine-dependent deoxyribozyme. which catalyzes the cleavage of an RNA
phosphodiester bond using the amino acid histidine as a cofactor and producing a
rate enhancement of over 106 relative to the uncatalyzed reaction. was identified by
in vitro ~ e l e c t i o n . ~ ~ Despite their limited substrate specificity and low reaction rates,
nucleic acid enzymes are expected to develop into an interesting alternative to
peptide based enzymes since they can be very easily optimized by randomization.

3.0 Introduction of Unnatural Amino Acids or Side Chains
The importance and challenges of altering enzymatic activity are evident from
the vastly varied approaches which have been applied toward accomplishing this
goal. From optimizing existing enzymes via mutagenesis techniques. noncovalent
modifications, exploiting the diversity of the immune response. generating small
enzyme mirnics, or developing nucleic acid based enzymes, enormous growth has
been achieved in the applicability and robustness of enzymes for organic synthesis
applications. However, the protein-based approaches descfibed for altering enzyme
activity, are limited to the 20 naturally occurring amino acids. In order to more fully
understand the factors which control enzyme specificity and to permit the creation of
novel selectivity, the need to incorporate unnatural moieties into proteins has been
recognized. Nature itself ernploys modified amino acids for regulatory, signalling and
localization functions through the post-translational modificationsM8 of amino acid
residues generating for example, 5-hydroxylysine, 4-hydroxyproline, O-
rnethylaspartate, 3,5-diiodotyrosine, 4-carboxyglutamate, N. N, N-trimethyllysine,
formylglycine as well as gtycosylatedlag phosph~rylated.~'~ acetylated ,"' and
~ulfated"~ amino acids. In fact, several approaches have been explored in attempts
to overcome the natural amino acid limitation of conventional site-directed
mutagenesis.
3.1 Chemical Modification to Generate Semisynthetic Enzymes
The utility of protein chemical modification to incorporate
unnatural functionalities and change enzyme properties has been recogn i~ed.~~~ The
first reports of the application of chemical modification to enzymes by the groups of
Bende?' and of Koshlandmin 1966 predated site-directed mutagenesis
techniques. Bender and Koshland independently created a thiolsubtilisin by

chemical transformation of the uniquely reactive active site serine of subtilisin BPN'
to cysteine (Ser221 -CH20H + Ser221-CH,SH)*' yielding an active enzyme with
altered catalytic p r ~ p e r t i e s . ~ ~ Subsequently, using an analogous procedure.
subtilisin Carslberg was converted into selenosubtilisin (Ser221 CH,OH -+
Ser221 CH,SeH).=' This selenosubtilisin exhibited improved potential as a peptide
ligation catalysF4 and also exhibited peroxidase activity. 2'4.U5U8 Chemical
modification of active site residues has been exploited extensively to alter the
catalytic properties of hydrolases. For example. methylation of the catalytic triad
histidine residue of subtilisinm and of a - ~ h y m o t r y p s i n ~ was found to improve
esterase to amidase selectivity. In addition. modification of a bacterial lipase with
amino acid specific reagents including, 1-ethyl-3-(3-dimethyfaminopropyl)-
carbodiimide. phenylglyoxal. pyridoxal 6-phosphate. potassium iodide. and
tetranitromethane which modify al1 of the AspIGlu. Arg. Lys, Trp. and Tyr residues
respectively in the protein. was used to modulate lipase venus esterase activity."'
In 1985, interest in chemically produced artificial enzymes. including some
with synthetic potential. was renewed. In particular, a semisynthetic flavopapain.
wh ich exhibited oxido-reductase activity. was generated by the Kaiser group by
covalently linking a flavin to the active site cysteine (Cys25) of the cysteine protease
papain through a thioester linkage.2'8.232 This methodology was applied to several
other protein templates to generate flavo-glyceraldehyde-3-pho~phate.~~~-~~ flavo-
lysozyme via a flavin ester bond to the active site Asp52 or the surface exposed
Aspl O1 res idue~.~ '~ and flavo-hemoglobin via a flavin linkage to the two P-chain
Cys93 residues.'17 In a subsequent investigation. papain was converted into a ligase
by alkylation of the active site cysteine with 2-bromomethyl-N-methyllbenzyl
thiazolium b r ~ m i d e . ~ ' ~ This thiazolopapain. which had no detectable peptidase
activity, catalyzed the dimerkation of 6-oxoheptanal at a rate 400-fold better than
did the free thiazof and constitutes the first example of carbon-carbon bond
formation catalyzed by a chemically modified In a similar vein. the
pyridoxamine cofactor was covalently linked to an interior cysteine residue of the
noncatalytic adipocyte lipid binding protein via a disulfide linkage.2'3*2'g.235 This

generated a protein able to catalyze the reductive amination of a-keto acids to a-
amino acids with a 1.8 fold rate enhancement compared to the uncoupled
pyridoxamine cofactor. and with an enantiomeric excess of 42 - 84%.*13
Chemical modification of enzymes has also been employed to aiter surface
properties. For example. succinylation of the 14 lysine residues of chymotrypsin
(CT) decreased its positive surface charge and conversely. reaction of its 13 surface
carboxylates with ethylenediamine increased its positive surface charge. These
changes effected alterations in the pH-activity profile of CT.236 Sirnilarly. the 59
carboxy groups of Alfhrobacter 0-xyiose isornerase were chemically coupled to
glycinamide in order to decrease the negative surface charge of the enzyme and
lower its p~-opt imurn.~? Enzyme so~ubi l i ty '~~ and s t a b i ~ i t y ' ~ . ~ ~ have also been
improved by surface chemical modifications.
In addition there are countless examples of chemical modification as a
technique to identify catalytically important r e s i d u e ~ . ~ ~ ~ For example. the
irreversible inhibitors, 5-azo-1 -H- te t ra~o le~~ and diisopropylphosphorylRuoridate
were employed to identify the active site serine and histidine residues of the serine
proteases. lodoactamide was used to identify the presence of sulfhydryls essential
for activity in ru bredoxin2& and to determine the pH-activity dependence of papain .2J7
Chemical modification with thiol specific reagents has also been applied to probe
receptor ligand binding such as for the glucocorticoid-receptor.248 While the chemical
modification technique has proven to be very successful in altering biocatalyst
properties, it suffers from the problems of nonspecific reactions. In addition. it is
generally limited to either surface exposed residues or specially activated residues
such as catalytic residues.

3.2 Unnatural Amino Acid Mutagenesis
Recently, the 20 amino-acid limitation of conventional site-directed
mutagenesis has been recognized and addressed by biosynthetic m e t h o d ~ . ~ ~ ~ - ~ ~ ' For
this approach, the codon for the amino acid of interest is replaced with one of the
stop codons (UAA, UAG, and UGA) by conventional oligonucleotide-directed
mutagenesis. These codons are not recognized by any of the common tRNAs
invoived in protein biosynthesis and thus can be viewed as blanks. This
methodology requires construction of a suppresser tRNA which recognizes the stop
codon of choice, and its in vitro aminoacylation with the unnatural amino acid to be
i n t r o d u ~ e d . ~ ~ ~ A suppresser tRNA that recognizes this codon is then chemically
acylated with the unnatural amino acid of interest. Addition of the mutated gene or
mRNA and acylated tRNA to an in vitro transcription-translation system results in the
specific incorporation of the unnatural amino acid at the position of the stop codon.
The in vitro transcription-translation reactions are typically performed on a 30 pl to 5
mL scale. Thus, a limitation of this approach is the relatively small quantity of
protein which can be ~btained.''~ The scope and efficiency of amino acids
incorporation has been investigated and, in general, large hydrophobic amino acids
are inserted more efficiently than small or charged ones2" In addition, D-amino
acids are not accommodated by the translation machinery, although some a,aœ
disubstituted amino acids are.254 To address the limitations on the amino acid which
can be incorporated, unnatural amino acids with side chains that can be elaborated
by chernical modification have been incorporated and subsequently m ~ d i f i e d . ~ ~ ~
Furthermore. thus far only a single unnatural amino acid has been incorporated into
a protein at a time.
Despite the small amount of proteins expressed, this approach represents a
significant advance in protein engineering since it permits the site-specific
incorporation of unnatural amino acids. and it has already been applied to the
incorporation of biophysical probes such as spin-labeled amino acids. fluorescent
amino acids, photolabile benzophenone denvatized amino acids and photolabile

protected amino a ~ i d s . ~ ' ~ ~ The approach has also pennitted detailed studies of the
energetics of hydrogen bondingt2= protein stability and and has been
applied to mechanistic investigation^.""^ In addition. the methodology has been
expanded through the use of frame-shift suppression mutagenesis by utilizing
synthetically prepared tRNA with a four base anticodon. This results in a truncation
due to an out-of-frame reading if the four base codon is not correctly t r a n ~ t a t e d . " ~ ~ ~ ~
While the unnatural amino acid mutagenesis approach has great potential, major
improvements are required. including: increasing the quantities of protein
obtainable, expanding the specificity of the translation factors to permit incorporation
of more diverse amino acids, altering the selectivity of aminoacyl tRNA synthases to
allow enzymatic charging of tRNA with unnatural amino acids. and developing the
methodology to permit incorporation of more than one unnaturaf amino acid at a
time.
3.3 Peptide Synthesis and Fragment Ligation
Unnatural amino acids have been introduced. into peptides by incorporation of
the new side chains directly ont0 the growing peptide chain during solid phase
peptide synthe~is. '~~ by simply coupling the individually prepared unnatural arnino
acid residues on solid supp0r t26~~~~ or in or by enzyme catalyzed
couplings.'" These chemically synthesized peptides. which are typically up to 40
residues long. can then be ligated on solid s ~ p p o r t . ~ ' ~ - ~ ' ~ enzymatically. ' 1.275287 or
chemically in solution288-289 to generate full length proteins. A related approach is
protein semisynthesis in which a synthetic peptide which contains an unnatural
amino acid is ligated to a natural protein fragment to produce a full-length p r ~ t e i n . ~ ~
These approaches are feasible only for small proteins of less than 12 kDa which
are able to refold or reassociate spontaneously to give functionally active protein,
as for ribonuclease2" or cytochrome C.28g Furthermore. the viability of the
incorporation of unnatural amino acids into self splicing protein motifs via synthetic
inteins, has recently been demonstrated."'

3.4 Site-Directed Mutagenesis Combined with Chernical
Modification
In response to the deficiencies of the chemical modification, unnatural amino
acid mutagenesis and solid phase peptide synthesis approaches for unnatural
arnino acid incorporation into proteins, the combined site-directed mutagenesis and
chemical modification approach was recognized by Kaiser as a viable alternative.
He recognized that it would be much more convenient if one did not have to rely on
the natural availability of suitable residues for chemical modification but rather could
introduce appropriate "handles" by genetic engineering which could subsequently be
modified site-~pecifically."~ For this purpose, the most commonly introduced
"handle" is the cysteine residue due to its unique reactivity.
This combined approach for the creation of new active-site environments and
altered specificity was first applied to the hydrolytic enzyme carboxypeptidase Y."3
The S,' pocket M398C mutation was made by conventional site-directed
mutagenesis and the new cysteine side chain was then chemically modified with
phenylacyl bromide and with 1-bromo-2-butanone. However, careful control of
reaction conditions was required to prevent the reaction of these alkylating agents
with other nucleophilic residues in the enzyme such as methionine and lysine.2M
Carboxypeptidase Y M398C. was also modified with the thiol-specific al kyl
methanethiosulfonate (CH,S02S-R, MTS) reagents, where R = -CH3, -CH,CH,CH,,
-CH,C,H,. and -CH2CH2NH3+, which reacted with the introduced cysteine to form a
disulfide modified enzyme. Fortunately, residue C341 which was present both in the
VVT and M398C mutant enzyme was not modified due to its inacce~sabi l i ty.~~~
This combined site directed mutagenesis chemical modification approach has
also aided mechanistic investigations. For exarnple, two lysine residues in the active
site of ribulosebisphosphate carboxylase were established as being critical to
catalysis by their mutation to cysteine. This resulted in a complete loss of activity
which was partially restored on aminoethylation of the cysteine residues with 2-
b r ~ r n o e t h y l a m i n e . ~ ~ ~ ~ ~ ~ ~ However, the WT enzyme, which itself wntains five

cysteines, was also alkylated by this procedure? In another example, the essential
catalytic Lys258 residue of aspartate aminotransferase was mutated to Cys
(K258C), thereby inactivating the enzyme. The mutant protein was then
aminoethylated with 2-bromoethyl amine after reversible protection of the untargeted
sulfhydryl groups with Ellman's reagent?The chemically elaborated enzyme,
K258C-CH,CH,NH,*, regained much of the activity lost after r n u t a t i ~ n . ~ ~ ~ - ~ ~ In
another example, modification of the R292K mutant of aspartate aminotransferase
with the guanidinating reagent O-methylisourea (MIU) was used to convert the WT
active site residue Arg292 into homoarginine, however this was accompanied by
modification of other reactive lysine side chainsSag The combined site-directed
mutagenesis chemical modification approach was recently applied to glucoamylase
effecting a 2-fold increase in ac t i~ i ty .~Th is combined approach has also yielded
insights into the protein packing of thioredoxinM' and staphytococcal nuclea~e."~- '~
3.5 Current Study
The goal of the current study is to Setter understand the factors which control
enzyme specificity, and to create novel specificities that will further expand the
synthetic applicabilities of the serine proteases. To do this, we have adopted the
strategy of chemical modification and site directed mutagenesis to alter the catalytic
properties of the enzyme subtilisin Bacillus lentus (SBL).
For this study, the sulfhydryl group of cysteine was chosen as a modification
handle since it is the most reactive of al1 amino acid side-chain functional groups
under physiological condition^.^^ In addition. the chemistry of sulfhydryls is very
robust and they are easily alkylated. acylated. arylated and oxidized. The cysteine
sulfhydryl, which has a pK, of 8.37,306 reacts via its thiolate anion with both
reversible and irreversible sulfhydryl blocking reagent~.~'? For example. N-
ethylmaleimide, O-methylisourea, and iodoacetarnide form irreversible adducts with
cysteine thiols but 4.4'-dithiodipyridine, and alkyl methanethiosulfonate (MTS)
reagents form readily reversible adducts with s~lf 'hydryls.~~~."~ For enzyme

modification the use of a readily reversible modifying reagent is desirable since this
permits restoration of the native activity. Furthemore, the chosen modification
reagent must be devoid of the cross reactivity with other nucleophiles such as lysine
and histidine which plagues many sulfhydryl blocking reagentsmM The alkyl
methanethiosulfonate (MTS) reagents were chosen for the current study since they
fulfill al1 of these r e q u i r e m e n t ~ . ~ ~ . ~ While there is one report of the reaction of MTS
reagents with lysine, this occurred under more vigorous conditionsNg and this is
consistent the 10'-fold higher rate constant for the reaction of sulfhydryls versus
amines with methanethiosulfonate reagent~.~''
Scheme 1.6
The strategy involves the introduction of one cysteine residue at a key active
site position via site-directed mutagenesis which is then thioalkylated with an alkyl
methanethiosulfonate reagent (CH3S0,S-R) to give chemically modified mutant
enzymes (CMMs) as illustrated in Scheme 1.6. Alkyl methanethiosulfonate reagents
react specifically and quantitatively with sulfhydryls and are routinely used for
chemical modification of protein t h i o l ~ . " ~ . ~ The modification can be perfomed
under mild reaction conditions, on a large scafe, and is independent of the nature of
the R group. Furthermore. the reaction is readily reversible by treatment with p-
mercaptoethanol or other reducing
The relatively low abundance of cysteine in proteins313 and in extracellular
enzymes of gram-positive bacteria in particulaf5 makes this approach widely
applicable. In addition to permitting the site-specific incorporation of unnatural
amino acid side chains, the CMM approach avoids the difficulties which can result
from nonconservative mutations that produce proteins which are unable to fold

properly or that do not undergo the required auto-processing. This potential difficulty
is particulariy relevant to the subtilisins, which are initially expressed as pre-pro-
enzymes that must undergo a~toproteolysis.~~ SB1 is especially well suited for this
strategy since it contains no natural cysteine residues and therefore the introduced
cysteine provides a unique sulfhydryl for modification. The particular utility of the
combined site-directed mutagenesis chemical modification approach to subtilisins
has been recognized. In particular, in order to generate an oxidation resistant
subtilisin, the M222C mutant of a subtilisin from Baclïlus lentus was prepared, and
then thioalkylated with the methyl rnethanethiosulfonate (CH,SO,SCH,) reagent,
generating an enzyme which exhibited 65% of WT activitya4 In addition to the
specificity and mechanistic investigations, mutagenic introduction of cysteine
combined with chemical modification has also been used for the site-directed
incorporation of thiol ~ p i n - l a b e l s ~ ' ~ ' ~ and fluorescence This methodology
has also facilitated detailed investigations of membrane spanning prote in^.^^^^^^ ion-
channel p r o p e r t i e ~ , ~ ~ ~ ~ ' and ligand-receptor associations.32mzg As well,
arninoethylation of cysteine residues has been long utilized as a method to
introduce trypsin-susceptible cleavage sites into proteins to facilitate sequence
tud dies.^^^

References
Jones JB. Probing the Specificity of Synthetically Useful Enzymes. Aldrichimica Acta 1993; 26: 105-1 2.
Faber K. Biotransformations in Organic Synthesis. 3rd edition. Heidelberg: Springer-Verlag, 1997.
Wong C-H, Whitesides GM. Enzymes in Synthetic Organic Chemistry. Oxford: Pergamon Press, 1994.
Roberts SM. Preparative Biotransformations. New York: Wiley, 1993.
Jones JB. Enzymes in Organic Synthesis. Tetrahedron 1986; 42:3351-403.
Fischer E. Einfluss der Konfiguration auf die Wirkung der Enzyme. Chem. Ber. 1 894; 27:2985-93.
Haldane JBS. Enzymes. London: Longmans, Green 8 Co., 1930.
Pauling L. Nature of Forces Between Large Molecules of Biological Interest. Nature 1948; 161 :707-9.
Fersht A. Enzyme Structure and Mechanism. 2nd edition. New York: W.H. Freeman and Company, 1985.
Khosla CH. Caren R, Kao CM, McDaniel R, Wang S-W. Evolutionary Guided Enzyme Design. Biotech. Bioeng. 1996; 52: 1 22-8.
Sears P. Wong C-H. Engineering Enzymes for Bioorganic Synthesis: Peptide Bond Formation. Biotechnol. Prog. 1996; 12:423-33.
Shao Z, Arnold F H. Engineering New Functions and Altering Existing Functions. Cur. Opin. Struct. Biol. 1996; 6:513-8.
Ballinger MD, Tom J, James AW. Furilisin: A Variant of Subtilisin BPN' Engineered for Cleaving Tribasic Substrates. Biochemistry 1996; 33:13579- 85.
14. Russell AJ, Thomas PG, Fersht AR. Electrostatic Effects on Modification of Charged Groups in the Active Site Cleft of Subtilisin by Protein Engineering.

J. Mol. Biol. 1987; 1 93:803-13.
Wells JA, Cunningham BC, Graycar TP, Estell DA. Recruitment of Substrate- Specificity Properties from One Enzyme into a Related one by Protein Engineering. Proc. Natl. Acad. Sci. USA 1987; 8451 67-71.
Estell DA, Graycar TP, Miller JV et al. Probing Steric and Hydrophobic Effects on Enzyme Substrate Interactions by Protein Engineering. Science 1986; 233:659-63.
Wangikar PP, Rich JO, Clark DS, Dordick JS. Probing Enzymic Transition State Hydrophobicities. Biochemistry 1995; W(38): 12302-1 0.
Radzicka A, Wolfenden R. A Proficient Enzyme. Science 1995; 267(5194):90-3.
Webb EC, ed. Enzyme Nomenclature 1992. San Diego: Academic Press, 1992.
Holland HL. Microbial Transformations. Cur. Opin. Chem. Biol. 1998; 2:77-84.
Yang Z, Mesiano AJ. Venkatasubramanian S, Gross SH, Harris JM, Russell AJ. Activity and Stability of Enzymes lncorporated into Acrylic Polymers. J. Am. Chem. Soc. 1995; 1 17:4843-50.
West BJ, Hennen WJ, Lalonde JL et ai. Enzymes as Synthetic Catalysts: Mechanistic and Active-Site Considerations of Natural and Modified Chymotrypsin. J. Am. Chem. Soc. 1990; 1 12(13):5313-23.
Creighton T. ed. Protein Folding. Freernan & Co., 1994.
Sheldon R. Large-Scale Enzymatic Conversions in Non-Aqueous Media. Koskinen A, Klibanov AM, ed. Enzymatic Reactions in Organic Media. London: Blackie, 1996: 266-307.
Noyori R. Asymmetric Catalysis in Organic Synthesis. New York: John Wiley & Sons, 1994.
Ichikawa Y, Liu J-C, Shen G-J, Wong C-H. A Highly Efficient Multienzyme System for the One-Step Synthesis of a Sialyl Trisaccharide: ln Situ Generation of Sialic Acid and N-Acetyllactosarnine Coupled with Regeneration of UDP-Glucose, UDP-Galactose, and CMP-Sialic Acid. J. Am.

Chem. Soc. 1991 ; 1 13:6300-3.
27. Morris AJ, Davenport RC, Tolan DR. A Lysine to Arginine Substitution at Position 146 of Rabbit Aldolase A Changes the Rate-Determining Step to Schiff Base formation. Protein Eng. 1996; 9:61-7.
28. Suzuki TI Kasai N. A Novel Method for the Generation of (R)- and (S)-3- Chloro-1 ,2-Propanediol by Stereospecific Dehalogenating Bacteria and Their Use in the Preparation of (RI- and (S)-Glycidol. Bioorg. Med. Chern. Lett. 1 99 1 ; 1 :343-6.
29. Taschner MJ, Black DJ, Chen 0-2. The Enzymatic Baeyer-Villiger Oxidation: A Study of CSubstituted Cyclohexanones. Tetrahedron:Asymmetry 1993; 4(6): 1387-90.
30. Alphand V, Furstoss R. Microbiological Transformations 22. Microbiologically Mediated Baeyer-Villiger Reactions: A Unique Route to Severat Bicycfic y- Lactones in High Enantiorneric Purity. J. Org. Chem. 1992; 57: 1 306-9.
31. Lee AY, Stewart JB, Clardy J, Ganem B. New lnsight into the Catalytic Mechanism of Chorismate Mutase from Structural Studies. Chem. Biol. 1995; 2(4): 1 95-203.
32. Katayama K, Kobayashi T, Oikawa H, Honma M, lchihara A. Enzymatic Activity and Partial Purification of Solanapyrone Synthase: First Enzyme Catalyzing Diels-Alder Reaction. Biochem. Biophys. Acta 1 998; 1 384:387-95.
33. Kazlauskas RJ. Elucidating Structure-Mechanism Relationships in Lipases: Prospects for Predicting and Engineering Catalytic Properties. TIBTECH 1 994; 1 2:464-72.
34. Kazlauskas RJ, Bornscheur UT. Biotransfonnations with Lipases. Rehm H-JI Reed G. ed. Biotechnology. 2nd edition. Vol. 8a. Weinheim. Germany: Wiley- VCH, 1998: 37-192.
35. Toone EJ. Werth MJ. Jones JB. Active-Site Model for Interpreting and Predicting the Specificity of Pig Liver Esterase. J. Am. Chem. Soc. 1990; 1 1 2:4946-52.
36. Riva S. Chopineau J, Kieboom APG, Klibanov AM. Protease-Catalyzed Regioselective Esterification of Sugars and Related Compounds in Anhydrous Dimethylformamide. J. Am. Chem. Soc. 1988; 1 10:584-9.

37. Blow DM, Birkoft JJ, Hartley 6s. Role of a Buried Acid Group in the Mechanism of Action of Chymotrypsin. Nature 1 969; 221 :33740.
38. Wright CS. Alden RA, Kraut J. Structure of Subtilisin BPN' at 2.5 Angstrom Resolution. Nature 1969; 221 :23542.
39. Markland FS, Smith EL. Boyer PD. ed. The Enzymes. 3rd edition. New York: Academic Press, 561 -608.
40. Walsh C. Enzymatic Reaction Mechanisms. San Francisco: Freeman and Co., 1979: Chapter 3.
41. McPhalen CA, James MNG. Structural Comparison of Two Serine Proteinase-Protein Inhibitor Complexes Eglin-C-Subtilisin Carîsberg and CI-2- Subtilisin Novo. Biochemistry 1988; 27:6582-98.
42. Alberty RA, Massey V. On the lnterpretation of the pH Variation of the Maximum Initial Velocity of an Enzyme-Catalyzed Reaction. Biochim. Biophys. Acta 1954; 1 3:347-53.
43. Bender ML, Clement GE, Kézdy FJ, Heck HD. The Correlation of the pH (PD) Dependence and the Stepwise Mechanism of a -Chymotrypsin-Catalyzed Reactions. J. Am. Chem. Soc. 1964; 86:3680-9.
44. Fersht AR, Renard M. pH Dependence of Chymotrypsin Catalysis. Biochemistry 1974; 13(7): 141 6-26.
45. Laidler KJ, Bunting PS. The ChemicaI Kinetics of Enzyme Action. 2nd edition. Oxford: Clarendon Press, 1973: 142-62.
46. Zerner B, Bond RPM, Bender ML. Kinetic Evidence for the Formation of Acyl- Enzyme lntermediates in the a-Chymotrypsin Catalyzed Hydrolyses of Specific Substrates. J. Am. Chem. Soc. 1964; 86:3674-9.
47. Zerner B, Bender ML. The Kinetic Consequences of the Acyl-Enzyme Mechanism for the Reaction of Specific Su bstrates with Chymotrypsin. J. Am. Chem. Soc 1964; 86:3669-74.
48. Cunningham LW. Proposed Mechanism of Action of Hydrolytic Enzymes. Science 1957; 125: 1 145-6.

49. Brown AJ. Enzyme Action. Trans. Chem. Soc. 1902; 81 :373-88.
50. Michaelis L, Menten ML. Die Kinetik der Invertinwirkung. Biochem. 2. 191 3; 491333-69.
51. Briggs G, Haldane J. A Note on the Kinetics of Enzyme Action. Biochem J. 1925; 19:338-9.
52. Roberts DV. Enzyme Kinetics. Cambridge: Cambridge University Press, 1977.
53. lsaccs NS, Niemann C. The Asyrnmetric Acetylation of (+>Butan-2-01 by p- Nitrophenyl Acetate and a-Chymotrypsin. Biochim. Biophys. Acta. 1960; 44: t 96-7.
54. Lloyd R, Dickman M, Jones JB. Probing the Specificity of the S,' Leaving Group Site of Subtilisin Bacillus lentus Using an Enzyme-Catalyzed Transesterification Reaction. Tetrahedron Asymmetry 1998; 9(4):55 1-61.
55. van der Osten C, Branner S. Hastrup S et al. Protein Engineering for Subtilisins to lrnprove Stability in Detergent Formulations. J. Biotechnol. 1993; 28(1):55-68.
56. Knapp M. Daubermann J, Bott RR. Brookhaven Database Entry 1 JEA.
57. 8etzel C, Klupsch S. Papendorf G, Hastrup S. Branner S, Wilson KS. Crystal Structure of the Alkaline Proteinase Savinase from Bacillus lentus at 1.4 A Resolution. J. Mol. Biol. 1 992; 223:42745.
58. Remerowski ML. Pepermans HAM, Hilbers CW. Van De Ven FJM. Backbone Dynamics of the 269-Residue Protease Savinase Determined from "N-NMR Relaxation Measurements. Eur. J. Biochem. 1996; 235:629-40.
59. Stabile MR, Lai WG, DeSantis G et al. Probing the Specificity of the S, Binding Site of M222 Mutants of Subtilisin B. Lentus with Boronic Acid Inhibitors. Bioorg. Med. Chem. Lett. 1996; 6(21):2501-6.
60. Gran H, Meldal M, Breddam K. Extensive Cornparison of the Substrate Preferences of Two Subtilisins As Determined with Peptide Substrates Which Are Based on the Principle of lntramolecular Quenching. Biochemistry 1992; 31 (26):6011-8.

61. Egmond MR. Antheunisse WP. van Bemmel CJ et al. Engineering Surface Charges in a Subtilisin: The Effects on Electrophoretic and Ion-Exchange Behavior. Protein Eng. 1 994; 7(6):793-800.
62. Maurer K-H, Markgraf M, Goddette D. Substrate Specificity of Natural Va rients and Geneticall y Engineered Intermediates of Bacillus lentus Al kaline Proteases. Adv. Exp. Med. Biol 1996; 379:243-56.
63. Olsen OH, Pedersen JT, Betzel C, Eschenburg S. Branner S. Hastrup S. An Investigation of the Savinase Water Channel: Implications of Cavity Mutants. Adv. Exp. Med. Biol. 1996; 379:235-41.
64. Perona JJ. Craik CS. Structural Basis of Substrate Specificity in the Senne Proteases. Protein Sci. 1995; 4:337-60.
65. Siezen RJ, Leunissen JAM. Subti1ases:The Superfarnily of Subtilisin-Like Serine Proteases. Protein Sci 1 997; 6:501-23.
66. Kazlauskas RJ, Weber HK. lmproving Hydrolases for Organic Synthesis. Curr. Opin. Chem. Biol. 1998; 2(1): 121 -6.
67. Power SD, Adams RM, Wells JA. Secretion and Autoproteolytic Maturation of Subtilisin. Proc. Natl. Acad. Sci. USA 1986; 83:3096-100.
68. Bossi A, Righetti PG, Vecchio G, Severinsen S. Focusing of Alkaline Proteases (Subtilisins) in pH 10-1 2 lmmobilized Gradients. Electrophoresis 1994; 15:153540.
69. Aunstrup K, Outtrup Hl Anderson O, Dambmann C. Proteases from Alkalophilic Bacillus Species. Tewi G. ed. Proc. IV. Int. Ferm. Symp. Ferment. Technol. Today. Kyoto, Japan: 1972: 299-305.
70. Pantoliano MW, Whitlow M. Wood JF et al. The Engineering of Binding Affinity at Metal Ion Binding Sites for the Stabilization of Proteins: Subtilisin as a Test Case. Biochemistry 1988; 27:2077-82.
71. Briedigkeit L, Frommel C. Calcium Ion Binding by Thermitase. FEBS Lett. 1989; 253:83-7.
72. Schechter 1. Berger A. On the Size of the Active Site in Proteases. 1. Papain. Biochem. Biophys. Res. Commun. 1967; 27(2):157-62.

73. Carter P, Wells JA. Functional Interaction Among Catalytic Residues in Subtilisin BPN . Proteins Struct. Function Gent 1990; 7:335-42.
74. Klibanov AM. Asymmetric Transformations Catalyzed by Enzymes in Organic Solvents. Acc. Chern. Res. 1 990; 23: 1 14-20.
75. Hutcheon GA, Parker MC, James A, Moore BD. Methanol Dramatically Enhances Serine Protease Activity Under Anhydrous Conditions. Chem. Commun. 1997; 931 -2.
76. Halling PJ. Therrnodynamic Predictions for Biocatalysis in Nonconventional Media: Theory, Tests, and Recommendations for Experimental Design and Analysis. Enzyme Microb. Technol. f994; 16: 178-206.
77. Zaks A, Klibanov AM. The Effect of Water on Enzyme Action in Organic Media. J. BioLChem. 1988; 263:8017-21.
78. Laane C, Boeren S, Vox K. Veeger C. Rules for Optimization of Biocatalysis in Organic Solvents. Biotechnol. Bioeng. 1 987; 30:81-7.
79. Halling PJ. Organic Liquids and Biocatalysts: Theory and Practice. TIBTECH 1989; 7150-2.
80. Sheldon RA. Chirotechnology: Designing Economic Chiral Syntheses. J. Chem. Tech. Biotechnol. 1996; 67:l-14.
81. Gupta MN. Enzyme Function in Organic Solvents. Eur. J. Biochem. 1992; 203:25-32.
82. Effenberger F, Ziegler T, Forster S. Enzyme-Catalyzed Cyanohydrin Synthesis in Organic Solvents. Angew. Chem. Int. Ed. Engl. 1987; 26:458-60.
83. Delinck DL. Margolin AL. Enzyme-Catalyzed Acylation of Castanospermine and 1 -Deoxynojirirnycin. Tetrahedron Lett. 1 990; 31 :3039-96.
84. Affieck R, Xu 2-F, Suzawa V, Focht K. Clark DS, Dordick JS. Enzymatic Catalysis and Dynarnics in Low-Water Environments. Proc. Natl. Acad. Sci. USA 1992; 8911 100-4.
85. Bruno FF, Akkara JA, Kaplan DL, Gross R, Swift G, Dordick JS. Regioselective Enzymatic Transesterification of Polysaccharides in Organic Solvent. Polym. Mater. Sci. Eng. 1996; 74:69-70.

Rich JO. Bedell BA. Dordick JS. Controlling Enzyme-Catalyzed Regioselectivity in Sugar Ester Synthesis. Biotechnol. Bioeng. 1995; 45:426- 34-
Gutman AL, Bravdo T. Enzyme-Catalyzed Enantioconvergent Lactonization of y-Hydroxy Oiesters in Organic Solvents. J. Org. Chem. 1989; 544263-5.
Kitaguchi Hl Fitzpatrick PA. Huber JE, Klibanov AM. Enzymatic Resolution of Racemic Amines: Crucial Role of the Solvent. J. Am. Chem. Soc. 1989; 1 1 1 :3094-5.
Ricca JM, Crout DHG. Peptide Synthesis Catalyzed by Chymotrypsin in Organic Solvents. J. Chem. Soc. Perkin Trans. f 1989; 21 26-7.
Kitaguchi Hl Ono M. itoh 1, Klibanov AM. Regioselective Modification of Lysine's Amino Groups by Proteases. Agric. Biol. Chem. 1991 ; 553067-73.
Reslow M, Adlercreutz P. Mattiasson B. The Influence of Water on Protease- Catalyzed Peptide Synthesis in AcetonitrileNVater Mixtures. Eur. J. Biochem. 1988; 1771313-8.
Tawaki S. Klibanov AM. Inversion of Enzyme Enantioselectivity Mediated by the Solvent. J. Am. Chem. Soc. 1992; 1 14: 1882-4.
Pasta Pl Mauola G, Carrea G, Riva S. Subtilisin-Catalyzed Transesterification in Supercritical Carbon Dioxide. Biotechnol Lett. 1989; 1 1 :643-9.
Kamat SV, Beckman EJ, Russell AJ. Control of Enzyme Enantioselectivity with Pressure Changes in Supercritical Fluoroform. J. Am. Chem. Soc. 1993; 1 15:8845-6.
Zaks A, Klibanov AM. Enzymatic Catalysis in Nonaqueous Solvents. J. Biol. Chem. 1986; 263:3 1 94-201.
Schmitke JL, Stern LJ, Klibanov AM. The Crystal Structure of Subtilisin Carlsberg in Anhydrous Dioxane and its Cornparison with Those in Water and Acetonitrile. Proc. Natl. Acad. Sci. USA 1997; 94:4250-5.
Fitzpatrick PA, Steinmetz ACU. Ringe Dl Klibanov AM. Enzyme Crystal Structure in a Neat Organic Solvent. Proc. Natl. Acad. Sci. USA 1993; 90:8653-7.

Yennawar NH, Yennawar HP, Farber G.K. A Structural Explanation for Enzyme Memory in Nonaqueous Solvents. J. Am. Chem. Soc. 1995; 1 171577-85.
Zeng Y-J. Omstein RL. Molecular Dynamics of Subtilisin Carlsberg in Aqueous and Nonaqueous Solutions. Biopolymers 1996; 38:791-9.
Zeng Y-J. Omstein RL. A Molecular Dynamics Study of the Effect of Carbon Tetrachloride on Enzyme Structure and Dynamics: Subtilisin. Protein Eng. 1 996; 9(6):485-92.
Clapés P. Valencia G, Adlercreutz P. Influence of Solvent and Water Activity on Kinetically Controlled Peptide Synthesis. Enzyme Microb. Technol. 1992; l4:575-80.
Kim J, Kim 6-G. Effect of the Hydration State of Supports Before Lyophilization on Subtilisin-A Activity in Organic Media. Biotechnol. Bioeng. 1 996; 501687-92.
Tana ka M. Kawamoto Tl Taylor RF. ad. Protein I mmobilization:Fundamentals and Applications. New York: Marcel Dekker, 1991 : 183-208.
Hiroto M, Matsushima A, Kodera Y. Shibata Y. lnada Y. Chemical Modification of Lipase with a Comb-Shaped Synthetic Copolymer of Polyoxyethylene Allyl Methyl Diether and Maleic Anhydride. Biotechnol. Lett. 1992; 141559-64.
Takahashi KI Kodera Y, Yoshimoto Tl Matsushima A, lnada Y. Ester- Exchange Catalyzed by Lipase Modified With Polyethylene Glycol. Biochem. Biophys. Res. Commun. 1985; 131 (2):532-6.
Altus Biologies Inc. Cambridge Mass.
Wang Y-F, Yakovlevsky K. Margolin AL. An Efficient Synthesis of Chiral Amino Acid and Peptide Alkylamides via CLEC-Subtilisin Catalyzed Coupling an in situ Resolution. Tetra hedron Lett. 1 996; 3753 1 7-230.
Wang Y-F. Yakovlevsky K, Zhang B. Margolin AL. Cross-Linked Enzyme Crystals (CLEC) of Subtilisin: A Versatile Catalyst for Organic Synthesis. J. Org. Chem. 1997; 32:3488-95.
Lalond JJ. Govardhan Cl Khalaf N, Martinez AG. Visuri K. Margolin AL.

Cross-Linked Crystals of Candida rugosa Lipase: Highly Efficient Catalysts for the Resolution of Chiral Esters. J. Am. Chem. Soc. 1995; 117:6845-52.
1 10. Gill Il Ballasteros A. Novel Sol-Gel Matricies for the tmrnobilization of Enzymes. Ann. NY. Acad. Sci. 1996; 799:697-700.
11 1. Huang W. Wang J. Bhattacharyya D, Bachas LG. lmproving the Activity of lmmobilized Subtilisin by Site-Specific Attachment to Surfaces. Anal. Chem. 1997; 69:4601-7.
1 12. Russel AJ, Klibanov AM. Inhibitor-lnduced Enzyme Activation in Organic Solvents. J- Biol. Chern. 1988; 263:11624-6.
113. Stahl M. Mansson M, Mosbach K. The Synthesis of a 0-Amino Acid Ester in an Organic Media with a-Chymotrypsin by a Bio-lmprinting Procedure. Biotechnol. Lett. 1990; 12:161-6.
114. Rich JO, Dordick JS. Controlling Subtilisin Activity and Selectivity in Organic Media by lmprinting with Nucleophilic Substrates. J. Am. Chem. Soc. 1997; 1 19:3245-52.
1 15. Dabulis K, Klibanov AM. Dramatic Enhancements of Enzymatic Activity in Organic Solvents by Lyoprotectants. Biotechnol. Bioeng. 1993; 41 566-71.
1 16. Keyes MH, Albert DE. Generation of Catalytic Activity by Protein Modification. Biomimetic Polymers 1990; 1 15-33.
11 7. Nakamura K. lshihara K. Ohno A. Nishimura Hl Hayashi Y. Kinetic Resolution of ($-Arene)Chromium Complexes by a Lipase. Tetrahedron Lett. 1 990; 3 1 :36034.
11 8. Degueil-Casting M. De Jeso B. Drouillard S. Maillard B. Enzymatic Reactions in Organic Synthesis: 2-Ester lnterchange of Vinyl Esters. Tetrahedron Lett. 1987; 28(9):953-4.
1 19. Fang J-M. Wong C-H. Enzymes in Organic Synthesis: Alteration of Reversi ble Reactions to l rrevenible Processes. Synlett 1 994; 393402.
120. Faber K. Riva S. Enzyme-Catalyzed lrreversible Acyl Transfer. Synthesis 1992; 895-910.

Bergfund P, Holmquist M. Hult K. Reversed Enantiopreference of Candida rugosa Lipase Supports Different Modes of Binding Enantiomers of a Chiral Acyl Donor. J. Mol. Catalysis 6: Enzymatic 1998; 5: 283-7.
Zoller MJ, Smith M. Oligonucleotide-Directed Mutagenesis of DNA Fragments Cloned into Ml 3 Vectors. Methods Enzyrnol. 1983; 100:468-500.
Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbour Laboratory, USA, 1982.
Wells JA, Powers DB, Bott RR. Graycar TP. Estell DA. Designing Substrate Specificity by Protein Engineering of Electrostatic Interactions. Proc. Nat. Acad. Sci. USA 1987; 84:1219-23.
Wells JA, Estell DA. Subtilisin: An Enzyme Designed to be Engineered. TlBS 1988; 13:291-7.
Bott R, Ultsch M. Wells J et al. Importance of Conformational Variability in Protein Engineering of Subtilisin. Lebanon HM, Mumrna RO, Honeycutt RC. Duesing JH. ed. Biotech. Agric.Chem. Vol. ACS Symp. Ser. 334. 1987: 139- 47.
Kurth T, Grahn S, Thormann M et al. Engineering the SI' Subsite of Trypsin: Design of a Protease Which Cleaves Between Dibasic Residues. Biochemistry 1998; 37:11432-40.
Rheinnecker M, Eder J, Pandey PS, Fersht AR. Variants of Subtilisin BPN' with Altered Specificity Profiles. Biochemistry 1994; 33:221-5.
Rheinnecker M. Baker G, Eder J. Fersht AR. Engineering a Novel Specificity in Subtilisin BPN'. Biochemistry 1993; 32(5): 1 199-203.
Sarensen SB, Bech LM, Meldal M, Breddarn K. Mutational Replacements of the Amino Acid Residues Forming the Hydrophobie S, Binding Pocket of Subtilisin 309 from Bacillus lentus. Biochemistry 1993; 32:8994-9.
Takagi H, Maeda T, Ohtsu 1, Tsai Y-C, Nakamori S. Restriction of Substrate Specificity of Subtilisin E by Introduction of a Side Chain into a Conserved Glycine Residue. FEBS Lett 1996; 395:127-32.
Takagi H, Ohtsu 1, Nakamori S. Construction of Novel Subtilisin E with High S pecificity , Activity and Productivity Throug h Multiple Amino Acid

Substitutions. f rotein h g 1997; 10(9):985-9.
Tsu CA, Perona JJ, Fletterick RJ, Craik CS. Structural Basis for the Broad Substrate Specificity of Fiddler Crab Collagenolytic Serine Protease 1. Biochernistry 1997; 65393401.
Bech LM. Ssrensen SB. Breddarn K. Significance of Hydrophobic S,-P, Interactions in Subtilisin 309 from Bacillus lentus. Biochemistry 1993; 321284552.
Ballinger MD. Tom J, Wells JA. Designing Subtilisin BPN' to Cleave Su bstrates Containing Dibasic Residues. Biochemistry 1995; 34: 1 331 2-9.
Vita C. Engineering Novel Proteins by Transfer of Active Sites to Natural Scaffolds. Curr. Opin. Biotechnol. 1997; 8:429-34.
Hedstrorn 1. Farr-Jones S. Kettner CA. Rutter WJ. Converting Trypsin to Chymotrypsin: Ground-State Binding Does Not Determine Substrate Specificity. Biochemistry 1994; 33:8764-9.
Perona JJ. Hedstrom L, Rutter WJ. Fletterick RJ. Structural Origins of Substrate Discrimination in Trypsin and Chymotrypsin. Biochemistry 1995; 7(34): 1489-99.
Hedstrom L. Trypsin: A Case Study in the Structural Determinants of Enzyme Specificity. Biol. Chem. 1996; 377(7-8):465-70.
Perona JJ, Hedstrom L, Wagner RL. Rutter WJ, Craik CS, Fletterick RJ. Exogenous Acetate Reconstitutes the Enzymatic Activity of Trypsin Aspl 89Ser. Biochemistry 1994; 33(11):3252-9.
Venekei 1. Szilagyi L, Graf L. Rutter WJ. Attempts to Convert Chymotrypsin to Trypsin. FEBS Lett. 1996; 383(1-2): 143-7.
Sakowicz R.. Gold M, Jones JB. Partial Reversai of the Substrate Stereospecificity of L-Lactate Dehydrogenase by Site-Directed Mutagenesis. J. Am. Chem. Soc. 1995; 1 17:2387-94.
Hogan JK. Pittol C, Jones JB. Gold M. lmproved Specificity Toward Substrates with Positively Charged Side-Chains by Site-Directed Mutagenesis of the L-Lactate Dehdrogenase from Bacillus stereothermophilus. Biochemistry 1995; 34:4225-30.

144. Kallwass HK, Luyten MA. Pans W, Gold M. Kay CM. Jones JB. Effects of Glnl O2Arg and Cys97Gly on the Structural Specificity and Stereospecificity of the L-Lactate Dehydrogenase from Bacillus stereothemophilus. J. Am. Chem. Soc. 1992; 1 l4:455'î -7.
145. Yorita K. Aki K. Ohkuma-Soyejima T. Kokubo Tl Misaki Hl Massey V. Conversion of L-Lactate Oxidase to a Long Chain a-Hydroxyacid Oxidase by Site-Directed Mutagenesis of Alanine 95 to Glycine. J. Biol. Chem. 1996; 271 (45):28300-5.
146. Klein RR. King G. Monreau RA. Haas MJ. Altered Acyl Chain Length Specificity of Rhizopus delemar Lipase Through Mutagenesis and Molecular Modeling. Lipids 1997; 32: 123-30.
147. Chen R. Greer A, Dean AM. Redesigning Secondary Structure to lnvert Coenzyme Specificity in lsopropylmalate Dehydrogenase. Proc. Natl. Acad. Sci. USA 1996; 93:12171-6.
148. Fluka 1997-98 Catalogue. p 1074-1 075.
149. Shortle Dl Botstein D. Directed Mutagenesis with Sodium Bisulfite. Methods Enzymol. 1983; 100:457-68.
150. Chu C-T, Pams DS, Dixon RAF, Farber FE. Schaffer PA. Hydroxyl-Amine Mutagenesis of HSV DNA and DNA Fragments: Introduction of Mutations into Selected Regions of the Viral Genome. Virology 1 979; 98: 1 68-81.
151. Sweasy JE!, Loeb LA. Detection and Characterization of Mammalian DNA Polymerase Beta Mutants by Functional Compementation in Escherichia coli. Proc. Natl. Acad. Sci. USA 1993; 90:4926-630.
152. Diaz J-J, Rhoads DD. Roufa DJ. PCR Mediated Chemical Mutagenesis of Cloned Duplex DNA. Biotechniques 1991 ; 1 1 :204-11.
153. Roufa DJ. PCR Mediated Chemical Mutagenesis. Meth. Mol. Biol. 1996; 57:357-67.
154. Ner SS. Goodin DG, Smith M. A Simple and Efficient Procedure for Generating Random Point Mutations and for Codon Replacements Using Mixed OIigonucleotides. DNA 1 988; 7: 1 27-34.
155. Black ME, Loeb LA. Random Sequence Mutagenesis for Generation of Active

Enzymes. Meth. Mol. Biol. 1996; 57:33549.
Cadwell RC, Joyce GF. Randomization of Genes by PCR Mutagenesis. PCR Methods Appl. t 992; 2:28-33.
Suzuki M. Avicola AK. Hood L, Loeb LA. Low Fidelity Mutants in the O-Helix of Thermus aquaficus ENA Polymerase 1. J. Biol. Chem. 1997; 272: 1 1228- 35.
Flavell RA. Sabo DL. Bandel EF. Weissmann C. Site-Directed Mutagenesis: Generation of an Extracistronic Mutation in Bacteriophage QB RNA. J. Mol. Biol. 1 974; 89:255-72-
Zaccolo M. Williams DM. Brow DM. Gherardi E. An Approach to Random Mutagenesis of DNA Using Mixtures of Triphosphate Derivatives of Nucleoside Analogues. J. Mol. Biol. 1 996; 255589-603.
Stemmer WPC. DNA Shuffling by Random Fragmentation and Reassembly: In Vitro Recombination for Molecular Evolution. Proc. Natl. Acad. Sci. USA 1994; 91 : 10747-51.
Zhang J-H, Dawes G, Sternmer WP. Directed Evolution of a Fucosidase from a Galactosidase by DNA Shuffling and Screening. Proc. Natl. Acad. Sci. USA 1 997; 94:4504-9.
Cox EC. Bacterial Mutator Genes and the Control of Spontaneous Mutation. Annu. Rev. Genet. 1976; 10:135-56.
Greener A. Callahan M. Jerpseth B. An Efficient Random Mutagenesis Technique Using an E. co/i Mutator Strain. Meth. Mol. Biol 1996; 57:375-85.
Venekei 1, Hedstrom L, Rutter WJ. A Rapid and Effective Procedure for Screening Protease Mutants. Protein Eng. 1996; 9(1):85-93.
Janes LE. Lowendahl AC. Kazlauskas RJ. Using pH Indicators: ldentifying Active and Enantioselective Hydrolases. Chem. Eur. J. 1998; 4(11):2324-3 1.
Kuchner O. Arnold FH. Directed Evolution of Enzyme Catalysts. TIBTECH 1 997; 1 5:523-30.
Skandalis A. Encell LP. Loeb LA. Creating Novel Enzymes by Applied Molecular Evolution. Chemistry & Biology 1997; 4:889-98.

168. Janes LE, Kazlauskas RJ. A Fast Spectrophotometric Method to Measure the Enantioselectivity of Hydrolases. J. Org. Chern. 1997; 62(14):456O-l.
169. Arnold FH. Design by Directed Evolution. Acc. Chem. Res. 1998; 31 (3):lZ5- 31.
170. Chen K. Arnold FH. Tuning the Activity of an Enzyme for Unusual Environments: Sequential Random Mutagenesis of Su btilisin E for Catalysis in Dimethylforrnamide. Proc. Natl. Acad. Sci. USA 1993; 90:5618-22.
1 7 1 . You L, Arnold FH. Directed Evolution of Su btiiisin E in Bacillus subtilis to Enhance Total Activw in Aqueous Dimethylfomarnide. Prot. Eng. 1994; 9:77-83.
172. Arnold FH. Protein Engineering for Unusual Environrnents. Cur. Opin. Biotech. 1993; 4:450-5.
173. Reetz MT. Zonta A, Schimossek K. Liebeton K. Jaeger K-E. Creation of Enantioselective Biocatalysts for Organic Chemistry by In Vitro Evolution. Angew. Chem. Int. Ed. Engl. 1997; 36:2830-2.
174. Nishiya Y, Harada N. Teshima S et al. lmprovement of Thermal Stability of Streptomyces Cholesterol Oxidase by Random Mutagenesis and Structural Interpretation. Protein Eng. 1997; 10(3):231-5.
175. Shinkai A, Hirano A, Aisaka K. Substitutions of Ser for Asn-163 and Pro for Leu-264 Are Important for Stabilization of Lipase from Pseudomonas aeruoginosa. J. Biochem. 1996; 120:915-21.
176. Yano Tl Oue S, Kagamiyama H. Directed Evolution of an Aspartate Aminotransferase with New Substrate Specificities. Proc. Natl. Acad. Sci, USA 1998; 95:5511-5.
177. Desjarlais JR. Handel TM. New Strategies in Protein Design. Curr. Opin. Biotechnoi. 1995; 6:460-6.
178. Corey MJ, Corey E. On the Failure of the De Novo-Designed Peptides as Biocatalysts. Proc. Natl. Acad. Sci. USA 1996; 933 1428-34.
179. Severin K, Lee DH, Kennan AJ. Ghadiri MR. A Synthetic Peptide Ligase. Nature 1997; 389:706-9.

Dahiyat BI. Mayo SL. De Novo Protein Design: Fully Automated Sequence Selection. Science 1997; 278:82-7.
Nixon AE, Ostermeier M. Benkovic SJ. Hybrid Enzymes: Manipulating Enzyme Design. TIBTECH 1998; 16:258-64.
Quemeneur E, Moutiez M. Charbonnier JB, Menez A. Engineering Cyclophin ilnto a Proline-Specific Endopeptidase. Nature 1998; 391 (6664):3014.
Lerner RA, Benkovic SJ. Schultz PG. At the Crossroads of Chemistry and I mmunology: Catalytic Antibodies. Science 1991 ; 252:65967.
Jencks W. Catalysis in Chemistry and Enzymology. New York: McGraw-Hill, 1969.
Koch T, Reymond J-L, Lemer RA. Antibody Catalysis of Multistep Reaction: An Aldol Addition Followed by a Disfavored Elimination. J. Am. Chem. Soc. 1995; 1 17(37):9383-7.
MacBeath G, Hilvert D. Monitoring Catalytic Activity by Immunoassay: Implications for Screening. J. Am. Chem. Soc. 1 994; 1 16(17):610 1 -6.
Meekel AAP, Resmini M. Pandit UK. First Example of an Antibody-Catalyzed Hetero-Diels-Alder Reaction. J. Chem. Soc. Chem. Commun. 1995: 571-2.
Haynes MR, Stura €A. Hilvert D, Wilson IA. Routes to Catalysis: Structure of a Catalytic Antibody and Compafison with Its Natural Counterpart. Science 1994; 263:645-52.
Guo JI Huang W. Scanlan TS. Kinetic and Mechanistic Characterization of an Efficient Hydrolytic Antibody; Evidence for the Formation of an Acyl Intermediate. J. Am. Chem. Soc. 1994; 1 16:6062-9.
Zhou GW, Guo JI Huang W. Fletterick RJ. Scanlan T. Crystal Structure of a Catalytic Antibody with a Serine Protease Active Site. Science 1994; 265: 1059-67.
Hirschmann RI Smith AB. Taylor CM et al. Peptide Synthesis Catalyzed by an Antibody Containing a Binding Site for Variable Amino Acids. Science 1994; 265(5169):234-7.
192. Jacobsen JR, Schultz PG. Antibody Catalysis of Peptide Bond Formation.

Proc. Natl. Acad. Sci. USA 1994; 91 :5888-92.
Srnithnid DB, Benkovic PA, Benkovic SJ et al. Investigations of an Antibody Ligase. J. Am. Chem. Soc. 1997; 1 17:278-82.
Kirby AJ. Enzyme Mechanisms, Models, and Mimics. Angew. Chem. Int. Ed. Engl. 1996; 35:707-24.
Schneider H, Eblinger F, Sartorïus J. Rammo J. Experimental Results from Host-Guest Complexes for the Design of Effectors in Biological Systems and of Enzyme Analogous Catalysts. J. Mol. Recognit. 1996; 9(2):123-32.
Breslow R. Biomimetic Chemistry and Artificial Enzymes: Catalysis by Design. Acc. Chem. Res. 1995; 28:146-53.
Souza VTD, Hanahusa K. O'Leary T, Gradewood RC, Bender ML. Synthesis and Evaluation of a Minature Organic Modal of Chymotrypsin. Biochem. Biophys. Res. Commun. 1985; 129(3):727-32.
Walter CJ, Sanders JKM. Free-Energy Profile for a Host-Accelerated Diels- Alder Reaction:The Sources of Exo Selectivity. Angew. Chem. Int. Ed. Engl. 1995; 34(2):217-9.
Kruger K. Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self- Splicing RNA: Autoexcision and Autocyclization of the Ribosomal RNA l ntervening Sequence of Tetrahymena. Cell 1982; 3 1 : 147-57.
Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA Moiety of Ribonuclease P is a Catalytic Subunit of the Enzyme. Cell 1983; 35849- 57.
Joyce GF. Nucleic Acid Enzymes: Playing with a Fuller Deck. Proc. Natl. Acad. Sci. USA 1998; 95:5845-7.
Tarasow TM, Tarasow SL, Eaton BE. RNA-Catalyzed Carbon-Carbon Bond Formation. Nature 1997; 389:54-7.
Zhang B, Cech TR. Peptide Bond Formation by in vitro Selected Ribozymes. Nature 7 997; 390(6):96-100.
Yingfu L, Sen D. A Catalytic DNA for Porphyrin Metallation. Nature: Stnict. Biol. 1 996; 3(9):743-7.

Geyer CR, Sen D. Evidence for the Metal-Cofactor lndependence of an RNA Phosphodiester-Cleaving DNA Enzyme. Chemistry & Biology 1997; 4(8):579- 93.
Yingfu L. Sen D. Toward an Efficient DNAzyme. Biochemistry 1997; 365589- 99.
Roth A, Breaker RR. An Amino Acid as a Cofactor for a Catalytic Polynucleotide. Proc. Natl. Acad. Sci. 1998; 95:6027-31.
Han KK, Martinage A. Post-Translational Chemical Modifications of Proteins I I l . Current Developments in Analytical Procedures of Identification and Quantitation of Post-Translational Chemically Modified Amino Acid(s) and its Derivatives. lnt. J. Biochem. 1 993; 25(7):957-70.
lmperiali B. Protein Glycosylation: The Clash of the Titans. Acc. Chem. Res. 1997; 30(11):452-9.
Luduena RF. Multiple Forms of Tubulin: Different Gene Products and Covalent Modifications. Int. Rev. Cytol 1998; 178:207-75.
Lin PP, Boery RC, Smith DL, Smith JB. In vivo Acetylation Identified at Lysine 70 of Human Lens alpha-Crystallin. Protein Sci. 1 998; 7(6): 1451 -7.
Ouyang YB, Lane WS, Moore KL. Tyrosylprotein Sulfotransferase:Purification and Molecular Cloning of an Enzyme that Catalyzes Tyrosine O-Sulfation, a Common Posttranslational Modification of Eukaryotic Proteins. Proc. Natl. Acad. Sci. USA 1998; 95(6):2896-901.
Kuang H, Brown ML, Davies RR, Young EC, Distefano MD. Enantioselective Reductive Amination of a-Keto Acids to a-Amino Acids by a Pyridoxamine Cofactor in a Protein Cavity. J. Am. Chem. Soc. 1996; 1 18: 10702-6.
Peterson EB, Hilvert D. Nonessential Active Site Residues Modulate Selenosubtilisin's Kinetic Mechanism. Biochemistry 1995; 34:6616-20.
Suckling CJ, Zhu LM. Carbon-Carbon Bond Formation Mediated by Papain Chemically Modified by Thiazolium Salts. Bioorg. Med. Chem. Lett. 1993; 3:5314.
216. Rokita SE, Kaiser ET. Synthesis and Charactenzation of a New Semisynthetic Enzyme, Flavolysozyme. J. Am. Chem. Soc. 1986; 1 O8:4984-

21 7. Kokubo T. Sassa S. Kaiser ET. Flavohemoglobin: A Semisynthetic Hydroxylase Acting in the Absence of Reductase. J. Am. Chem. Soc. 1987; 109:606-7.
21 8. Radziejewski C, Ballou DP, Kaiser ET. Catalysis of N-Alkyl-1.4- dihydronicotinamide Oxidation by a Flavopapain: Rapid Reaction in al1 Catalytic Steps. J. Am. Chem. Soc. 1985; 1 O7:3352-4.
21 9. Ory JJ, Mazhary A, Kuang H, Davies RR, Distefano MD, Branaszak L. Structural Characterization of Two Synthetic Catalysts Based on Adipocyte Lipid-Binding Protein. Protein. Eng. 1998; 1 1 (4):253-61.
220. Lundblad RL. Techniques in Protein Modification. 1995.
221. Polgar L. Bender ML. A New Enzyme Containing a Synthetically Formed Active Site. Thiolsubtilisin. J. Am. Chem. Soc. 1966; 88(13):3153-4.
222. Neet KE, Koshland ~ r . DE. The Conversion of Serine at the Active Site of Subtiiisin to Cysteine: A "Chernical Mutation". Proc. Nat. Acad. Sci. USA 1 966; 56: 1 606-1 1 .
223. Philipp M, Bender ML. Kinetics of Subtilisin and Thiolsubtilisin. Mol. Cell. Biochem. 1983; 51 :5-32.
224. Wu 2-P, Hilvert D. Conversion of a Protease into an Acyl Transferase: Selenosubtilisin. J. Am. Chem. Soc. 1989; 1 1 1 :4514-5.
225. Bell IM, Fisher ML, WU 2-P, Hilvert D. Kinetic Studies on the Peroxidase Activity of Selenosubtilisin. Biochemistry 1993; 32:3754-62.
226. House KL, Garber AR, Dunlap RB, Odom JD. 'H NMR Spectroscopic Studies of Selenosubtilisin. Biochemistry 1993; 32:3468-73.
227. O'Connor MJ. Dunlap RB, Odom JO et al. Probing an Acyl Enzyme of Selenosubtilisin by Raman Spectroscopy. J. Am. Chem. Soc. 1996; 1 18:239- 40.
228. Haring D, Hubert B. Schüler E, Schreier P. Reasoning Enantioselectivity and Kinetics of Selenosubtilisin from the Subtilisin Template. Arch. Biochem. Biophys. 1998; 354(2):263-9.

Zhong Z, Bibbs JA, Yuan W. Wong C-H. Active-Site-Directed Modification of Subtilisin. J. Am. Chem. Soc. 1991 ; 1 13:2259-63.
West JB, Scholten J, Stolowich NJ, Hogg JL, Scott Al, Wong C-H. Modification of Proteases to Esterases for Peptide Synthesis: Methylchymotrypsin. J. Am. Chem. Soc. 1988; 1 1 O:WO9-lO.
Kawase M, Takahashi Hl Sonomoto K, Nakamura K, Tanaka A. Effect of Chemical Modification of Tyrosine Residues on Activities of Bacterial Lipase. J. Ferment. Bioeng. 1991 ; 72:317-9.
Stewart KD, Radziejewski C. Kaiser ET. Catalytic Oxidation of Dithiols by a Semisynthetic Enzyme. J. Am. Chem. Soc. 1986; 108:3480-3.
Hilvert Dl Kaiser ET. New Semisynthetic Flavoenzymes Based on a Tetrameric Protein Template, Glyceraldehyde-3-Phosphate Dehydrogenase. J. Am.Chem. Soc. 1985; 107:5805-6.
Hilvert O, Hatanaka Y, Kaiser ET. A Highly Active Thermophilic Semisynthetic Flavoenzyme. J. Am. Chem. Soc. 1988; 110:682-9.
Davis RR, Distefano MD. A Semisynthetic Metalloenzyme Based on a Protein Cavity That Catalyzes the Enantioselective Hydrolysis of Ester and Amide Su bstrates. J. Am. Chem. Soc. 1997; 1 1 9:11643-52.
Valenzuela P. Bender ML. Kinetic Properties of Succinylated and Ethylenediamine-Arnidated 6-Chymotrypsin. Biochim. Biophys. Acta 1 971 ; 250153848.
Siddiqui KS, Loviny-Anderton T, Ramgarajan M, Hartley SB. Arthrobacter D- Xylose Isomerase: Chemical Modification of Carboxy Groups and Protein Engineerig of pH Optimum. Biochem. J. 1993; 296:685-91.
Delgrado C, Francis GE, Fisher D. The Uses and Properties of PEG-Linked Proteins. Crit. Rev. Ther. Drug Carrier Syst. 1992; 9(34):249-304.
Kikuchi K, Thorn SN, Hilvert D. Albumin-Catalyzed Proton Transfer. J. Am. Chem. Soc. 1996; 1 18:8184-5.
Alvear M, Jabalquinto AM, Cardemil E. Inactivation of Chicken Liver Mevalonate 5-Diphosphate Decarboxylase by Sulfhydryl-Directed Reagents: Evidence of a Functional Dithiol. Biochim. Biophys. Acta 1989; 994:7-11.

Nosal F, Masson A, Legrand R et al. Site-Directed Mutagenesis and Chemical Modification of Two Cysteine Residues of the UDP-N- Acetylrnuramoyl : L-Alanine Ligase of Eschecichia coli. FEBS Lett. 1 998; 4261309-1 3.
Smith DJ. Kenyon GL. Nonessentiality of the Active Sulfhydryl Group of Rabbit Muscle Creatine Kinase. J. Biol. Chern. 1 974; 249:33 17-8.
Smith TK. Meister A. Chemical Modification of Active Site Residues in y- Glutamyl Transpeptidase. J. Biol. Chem. 1 995; 270(21): 12476-80.
Welches WR, Baldwin TO. Active Center Studies on Bacterial Luciferase: Modification of the Enzyme with 2.4-Dinitrofluorobenzene. Biochemistry 1981 ; 20:s 1 2-7.
Ohtsuki K, Kajiwara N, Hatano H. Chemical Modification of Subtilisin BPN' by 5-Diazo-1 -H-Tetrazole. J Biochem (Tokyo) 1970; 68:137-43.
May SW, Lee LG, Katopodis AG, Kuo J-Y, Wimalassena K, Thowsen JR. Ru bredoxin from Pseudomonas oleovorans: Effects of Selective Chemical Modification and Metal Substitution. Biochernistry 1 984; 23:2187-92.
Lewis SD, Johnson FA, Shafer JA. Potentiometric Determination of lonoizations at the Active Site of Papain. Biochemistry 1976; 155009-1 6.
Bodwell JE. Holbrook NJ. Munck A. Sulfhydryl-Modifying Reagents Reversibly lnhibit Binding of Glucocorticoid-Receptor Complexed to DNA- Cellulose. Biochemistry 1 984; 23: 1 392-8.
Cornish VW. Mendel D, Schultz PG. Probing Protein Structure and Function with an Expanded Genetic Code. Angew. Chem. Int. Ed.Eng. 1995; 34:621- 33.
Ellman J, Mendel D, Anthony-Cahill S. Noren CJ, Schultz PG. Biosynthetic Methods for lntroducing Unnatural Amino Acids Site-Specifically lnto Proteins. Methods Enzymol. 1991 ; 202:3Ol-37.
Bain JB, Switzer C, Chamberlin AR, Benner SA. Ribosome-Mediated Incorporation of a Non-Standard Arnino Acid into a Peptide Through Expansion of the Genetic Code. Nature 1992: 356(6369):537-9.
252. Bain JB, Diala ES, Glabe CG et a/. Site-Specific Incorporation of Nonnatural

Residues Dunng in vitro Protein Biosynthesis with Semisynthetic Aminoacyi- tRNA . Biochemistry 1991 ; 30(22):5411-21.
Robertson SA, Ellman JA, Schultz PG. A General and Efficient Route for Chemicai Aminoacylation of Transfer RNAs. J. Am. Chem. Soc. 1991 ; (1 13):2722-9.
Meldal D, Ellman JA, Schultz PG. Protein Biosynthesis with Confomationally Restricted Amino Acids. J. Am. Chern. Soc. 1993; 1 154359-60.
Cornish VW, Hahn KM, Schultz PG. Site-Specific Protein Modification Using a Ketone Handle. J. Am. Chem. Soc. 1996; 11 8:815O-1.
Cornish VW, Benson DR, Altenback CA, Hideg K, Hubbell WL, Schultz PG. Site Specific Incorporation of Biophysical Probes into Proteins. Proc. Natl. Acad. Sci. USA 1994; 92:2910-4.
Meldal D, Ellman JA, Schultz PG. Construction of a Light Activated Protein by Unnatural Amino Acid Mutagenesis. J. Am. Chem. Soc. 1991; 21 3:2758-60.
Thorson JS, Chapman E, Schultz PG. Analysis of Hydrogen Bonding Strenghts in Proteins Using Unnatural Amino Acids. J. Am. Chem. Soc. 1995; 1 1719361 -2.
Thorson JS, Chapman E, Murphy EC, Schultz PG. Linear Free Energy Analysis of Hydrogen Bonding in Proteins. J. Am. Chem. Soc. 1995; 1 17: 1 157-8.
Cornish VW, Kaplan MI, Veenstra DL, Kollman PA, Schultz PG. Stabilizing and Destabilizing Effects of Placing f3-Branched Amino Acids in Protein a- Helices. Biochernistry 1 994; 33: 12022-31 .
Kimata Y, Shimada H, Hirose T, lshimura Y. Role of Thr-252 in Cytochrome P450,,: A Study with Unnatural Amino Acid Mutagenesis. Biochem. Biophys. Res. Comrn. 1995; 208(1):96-102.
Beiboer SHW, van den Berg BI Dekker N, Cox RC, Verheij HM. Incorporation of an Unnatural Arnino Acid in the Active Site of Porcine Pancreatic Phospholipase A,. Substitution of Histidine by 1,2,4-Triazole-3-alanine Yields an Enzyme with High Activity at Acidic pH. Prot. Eng. 1996; 9:345-52.
263. Parsons JF, Xiao G, Gilliland G, Armstrong RN. Enzymes Harboring

Unnatural Amino Acids: Mechanistic and Structural Analysis of the Enhanced Catalytic Activity of a Glutathione Transferase Containing 5-Fluorotryptophan. Biochemistry 1998; 37:6286-94.
Park Y, Luo J, Schultz PG, Kirsch JF. Noncoded Amino Acid Replacement Probes of the Aspartate Aminotransferase Mechanism. Biochemistry 1997; 3611 051 7-25.
Ma C, Kudlicki W, Odom OW, Kramer G, Hardesty B. ln vitro Protein Engineering Using Synthetic tRNAeJa with Different Anticodons. Biochemistry 1993; 32( ):793945.
Hohsaka T, Ashizuka Y, Murakami H, Sisido M. Incorporation of Nonnatural Amino Acids into Streptavidin Through in vitro Frame-Shift Suppression. J. Am. Chem. Soc. 1 996; 1 18(40):9778-9.
O'Donnell MJ, Zhou C, Scott Wt. Solid-Phase Unnatural Peptide Synthesis (UPS). J. Am. Chem. Soc. 1996; 1 18:6070-1.
Duthaler RO. Recent Developments in Stereoselective Synthesis of a- Aminoacids. Tetrahedron 1994; 50: 1 539-650.
Merrifield RB. Solid Phase Peptide Synthesis 1. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963; 85:2149-54.
Humphrey JM. Chamberlin R. Chemical Synthesis of Natural Product Peptides: Coupling Methods for the Incorporation of Noncoded Amino Acids into Peptides. Chem. Rev. 1997; 97:2243-66.
Fernandez MM, Margot AO, Falender CA, Blanch HW, Clark DS. Enzyrnatic Synthesis of Peptides Containing Unnatural Amino Acids. Enzyme Microb. Technol. 1 995; 1 7:964-71.
Lloyd-Williams P, Albericio F, Grant E. Convergent Solid-Phase Peptide Synthesis. Tetrahedron 1993; 49: 1 1065-1 33.
Tarn JP, Heath WF, Merrifield RB. S,2 Deprotection of Synthetic Peptides with a Low Concentration of HF in Dimethyl Sulfide: Evidence and Application in Peptide Synthesis. J. Am. Chem. Soc. 1983; 105:6442-55.
274. Kaiser ET. Synthetic Approaches to Biologically Active Peptides and Proteins lncluding Enzymes. Acc. Chem. Res. 1989; 22(2):47-54.

275. Abrahrnsen L, Tom J, Bumier J, Butcher KA, Kossiakoff A, Wells JA. Engineering Subtilisin and Its Substrates for Efficient Ligation of Peptide Bonds in Aqueous Solution. Biochernistry 1991 ; 30:4151-9.
276. Jackson DY, Bumier JP. Wells JA. Enzymatic Cyclization of Linear Peptide Esters Using Subtiligase. J. Am. Chem. Soc. 1995; 1 17:819-2O.
277. Jackson DY, Burnier J. Quan C, Stanley M. Tom J, Wells JA. A Designed Peptide Ligase for Total Synthesis of Ribonuclease A with Unnatural Catalytic Residues. Science 1994; 266:243-7.
278. Moree WJ. Sears P, Kawashiro K. Witte K. Wong C-H. Exploitation of Subtilisin BPN' as a Catalyst for the Synthesis of Peptides Containing Noncoded Amino Acids, Peptide Mimetics and Peptide Conjugates. J. Am. Chem. Soc. 1997; 1 l9:3942-7.
279. Sears P, Schuster M, Wang P. Witte K, Wong C-H. Engineering Subtilisin for Peptide Coupling: Studies on the Effects of Counterions and Site-Specific Modifications on the Stability and Specificity of the Enzyme. J. Am. Chem. SOC. 1994; 1 l6:6521-30.
280. Wong C-H. Schuster M. Wang P. Sears P. Enzymatic Synthesis of N- and 0- Linked Glycopeptides. J. Am. Chem. Soc. 1993; i 1 5:5893-90 1 .
281. Schellenberger V, Jakubke H-D. Protease-Catalyzed Kinetically Controlled Peptide Synthesis. Angew. Chem. Int. Ed. Engl. 1991 ; 30:1437-49.
282. Chen S-T, Tsai C-F, Wang K-T. Incorporation of Unnatural Amino Acid Derivatives lnto a Peptide Bond via an Oxime Ester Catalyzed by Papain or Lipase. Chem. Corn. 1996; 165-6.
283. Chen S-T, Chen S-Y, Kao C-L, Wang K-T. Proline as Nucleophile in Kinetically Controlled Peptide Synthesis Catalyzed by Alcalase in 2-Methyl-2- Propanol. Bioorg. Med. Chem. Lett. 1994; 4(3):443-8.
284. Barbas III CF, Wong C-H. Papain Catalysed Peptide Synthesis: Control of Amidase Activity and the Introduction of Unusual Amino Acids. J. Chem. Soc. Chem. Commun. 1987; 533-4.
285. Witte K. Seitz O, Wong C-H. Solution- and Solid-Phase Synthesis of N- Protected Glycopeptide Esters of the Benzyl Type as Substrates for Subtilisin-Catalyzed Glycopeptide Coupling. J. Am. Chem. Soc. 1998;

Fniton JS. Proteinase-Catalyzed Synthesis of Peptide Bonds. Adv. Enzymol. 1982; 53:239-306.
Nakatsuka T, Sasaki T, Kaiser ET. Peptide Segment Coupling Catalyzed by the Semisynthetic Enzyme Thiosubtilisin. J. Am. Chem. Soc. 1987; 109:3808- 9.
Walker MA. Protein Synthesis by Chemical Ligation of Unprotected Peptides in Aqueous Solution. Angew. Chem. int. Ed. Engl. 1 997; 36(lO): 1069-71 .
Di Bello C. Total Synthesis of Proteins by Chemical Methods: The Horse Heart Cytochrome C Example. Gazetta Chimica ltaliana 1996; l26:189-97.
Offord RE. Protein Engineering by Chemical Means. Protein Eng. 1987; l : l5 l -7.
Lew BM, Mills KV, Paulus H. Protein Splicing in vitro with a Semisynthetic Two-Component Minimal Intein. J. Biol. Chem. 1998; 273(26): 1 5887-90.
Kaiser ET. Catalytic Activity of Enzymes Altered at Their Active Sites. Angew. Chem. Int. Ed. Engl. 1988; 27:913-22.
Bech LM, Breddam K. Chemical Modifications of a Cysteinyl Residue lntroduced in the Binding Site of Carboxypeptidase Y by Site-Directed Mutagenesis. Carlsberg Res. Commun. 1988; 53:381-93.
Smith HB, Hartman f C. Restoration of Activity to Catalytically Deficient Mutants of Ribulose Bisphosphate Carboxylase/Oxygenase by Aminoethylation. J. Biol. Chem. 1988; 263(10):4921-5.
Smith HB, Larimer FW, Hartman FC. Subtle Alteration of the Active Site Ribulose Bisphosphate Carboxylase/Oxygenase by Concerted Site-Directed Mutagenesis and Chemical Modification. Biochem. Biophys. Res. Comm. 1 988; 1 52(2):579-84.
Smith HB, Larimer FW, Hartman FC. An Engineered Change in Substrate Specificity of Ribulosebis phosphate Carboxylase/Oxygenase. J . Biol. Chem. 1 990; 265(3): 1 243-5.
297. Planas A, Kirsch JF. Reengineering the Catalytic Lysine of Aspartate

Aminotransferase by Chemical Elaboration of a Genetically lntroduced Cysteine. Biochemistry 1991 ; 30:8268-76.
Gloss LM, Kirsch JF. Examining the Structural and Chemical Flexibility ofthe Active Site Base. Lys-258, of Eschenchia coli Aspartate Aminotransferase by Replacement with Unnatural Amino Acids. Biochemistry 1995; 34:12323-32.
White PW, Kirsch JF. Sequential Site-Directed Mutagenesis and Chemical Modification to Convert the Active Site Arginine 292 of Aspartate Arninotransferase to Homoarginine. J. Am. Chem. Soc. 1992; 1 14:3567-8.
Fierobe H-P. Clarke AJ. Tull D, Svensson B. Enzyrnatic Properties of the Cysteinesulfinic Acid Derivative of the Catalytic-Base Mutant Glu400Cys of Glucoamylase from Aspergillus awamon. Biochemistry 1998; 37:3753-9.
Wynn R, Richards FM. Unnatural Amino Acid Packing Mutants of Escherichia coli Thioredoxin Produced by Combined Mutagenesis/Chemical Modification Techniques. Protein Sci. 1993; 2:395-403.
Wynn RI Anderson CL, Richards FM, Fox RO. Interactions in Nonnative and Tmncated Forms of Staphylococcal Nuclease as lndicated by Mutational Free Energy Changes. Protein Sci. 1995; 4:1815-23.
Wynn R, Harkins PC, Richards FM. Fox RO. Mobile Unnatural Amino Acid Side Chains in the Core of Staphylococcal Nuclease. Protein Sci. 1996; 5: 1026-31.
Wynn R, Harkins PC, Richards FM. Fox RO. Cornparison of Straight Chain and Cyclic Unnatural Amino Acids Embedded in the Core of Staphylococcal Nuclease. Protein Sci. 1 997; 6: 1 62 1 -6.
Kenyon GL. Bruice TW. Novel Sulfhydryl Reagents. Methods Enzymol. 1977; 471407-30.
Coates E. Marsden C, Rigg B. lonization of Cysteine in Aqueous Solutions. Trans. Faraday Soc. 1969; 65:863-8.
Torchinsky YM. Sulfur in Proteins. Oxford. England: Pergamon Press Ltd., 1981.
Wynn R, Richards FM. Chemical Modification of Protein Thiols: Formation of Mixed DisuIfides. Methods Enzymol. 1995; 251 :351-6.

Kluger R, Tsui W-C. Amino Group Reactions of the Sulfhydryl Reagent Methyl Methanesulfonothioate. Inactivation of D-3-Hydroxybutyrate Dehydrogenase and Reaction with Amines in Water. C. J. Biochem. 1980; 58:629-32.
Kice JL. Rogers T, Warheit AC. The Relative Nucleophilicity of Common Nucleophiles Toward Sulfenyl Sulfur. Cornparison of the Relative Reactivity of Different Nucleophiles Toward Sulfenyl vs. Sulfonyl Sulfur. J. Am. Chem. Soc. 1974; 96(26):8020-6.
Cleland WW. Dithiothreitol. a New Protective Reagent for SH Groups. Biochemistry 1964; 3:480-2.
Nishimura JS, Kenyon GL, Smith DJ. Reversible Modification of the Sulfhydryl Groups of Escherichia coli Succinic Thiokinase with Methanethiolating Reagents, 5,5'-Dithio-bis(2-Nitrobenzoic Acid), p- Hydroxymercuribenzoate, and Ethylmercurithiosalicylate. Arch. Biochem. Biophys. 1 W5;l 70:461-7.
Klapper MH. The Independent Distribution of Amino Acids Near Neighbor Pairs into Polypeptides . Biochem. Biophys. Res. Commun. 1977; 78:1018- 24.
Gran H, Bech LM, Branner S, Breddam K. A Highly Active and Oxidation- Resistant Subtilisin-Like Enzyme Produced by a Combination of Site-Directed Mutagenesis and Chemical Modification. Eur. J. Biochem. 1990; 194:897- 901.
Hubbell WL, Mchaourab HS, Altenbach C. Lietzow MA. Watching Proteins Move Using Site-Directed Spin Labelling. Structure 7 996; 4:779-83.
Svensson M, Jonasson P, FreskgArd P-O et al. Mapping the Folding lntermediate of Hurnan Carbonic Anhydrase II. Probing Substructure by Chemical Reactivity and Spin and Fluorescence Labeling of Engineered Cysteine Residues. Biochemistry 1995; 34:8606-20.
Ottemann KM, Thorgeirsson TE, Kolodziej AF, Shin Y-K, Koshland DE. Direct Measurement of Small Ligand-lnduced Conformational Changes in the Aspartate Chemoreceptor Using EPR. Biochemistry 1998; (37):7062-9.
Lin Y. Nielsen R, Murray D et al . Docking Phospholipase A, on Membranes Using Electrostatic Potential-Modulated Spin Relaxation Magnetic

Resonance. Science 1998; 279(5358): 1925-9.
31 9. Liu R, Sharom FJ. Site-Specific Fluorescence Labeling of P-Glycoprotein on Cysteine Residues in the Nucleotide Binding Domain. Biochemistry 1996; 3511 1865-73.
320. Akabas MH, Kaufmann C, Archdeacon P. Karfin A. Amino Acid Residues Lining the Chloride Channel of the Cystic Fibrosis T ransmembrane Conductance Regulator. Neuron 1994; 13:913-27.
321. Chen J-G. Liu-Chen S. Rudnick G. Extemal Cysteine Residues in the Serotonin Receptor. Biochemistry 1997; 36:1479-86.
322. Frillingos S. Sun J. Gonzalez A. Kaback HR. Cysteine-Scanning Mutagenesis of Helix II and Flanking Hydrophilic Domains in the lactose Permease of Escherichia coli. Biochemistry 1 997; 36:269-73.
323. Akabas MH. Stauffer DA, Xu M. Kariin A. Acetylcholine Receptor Channel Structure Probed in Cysteine-Substitution Mutants. Science 1992; 258:307- 10.
324. Xu M. Akabas MH. Amino Acids Lining the Channel of the y-Aminobutyric Acid Type A Receptor ldentified by Cysteine Substitution. J. Biol. Chem. 1993; 268:21505-8-
325. Foong LY. You S. Jaikaran DCJ. Zhang Z. Zunic V. Woolley GA. Development of a Novel Thiol Reagent for Probing Ion Channel Structure: Studies in a Model System. Biochemistry 1996; 36:1343-8.
326. Holmgren M. Liu Y. Xu Y, Yellen G. On the Use of Thiol-Modifying Agents to Determine Channel Topography. Neuropharmacology 1996; 35:797804.
327. Yang N, George AL. Hom R. Molecular Basis of Charge Movement in Voltage-Gated Sodium Channels. Neuron 1996; 16:lî3-22.
328. Engler DA. Campion SR. Hauser MR. Cook JS. Niyogi SK. Critical Functional Requirement for the Guanidinium Group of the Arginine 41 Side Chain of Human Epidermal Growth Factor as Revealed by Mutagen ic l nactivation and Chemical Reactivation. J. Biol. Chem. 1992; 267(4):2274-81.
329. Heinonen P, Koskua K. Pihlavisto M et al. A Series of 6- (Methanesulfonylthioalkoxy)-2-N-methyl-l.2,3.4-tetrahydroquinolines:

Cysteine-Reactive Molecular Yardsticks for Probing a,-Adrenergic Receptors. Bioconjugate Chern. 1998; 9:358-M.
330. Plapp BV. Raftery MA, Cole RD. The Tryptic Digestion of S-Aminoethylated Ribonuclease. J. Biol. Chem. 1 967; 242(3):265-70.

C hapter 2
Reproduced in part with permission from Biochemistry, 1 998, 37,5968-5973.
Copyright 1 998, American Chemical Society (Reference 19)
-65-

Site-Directed Y utagenesis Combined with Chemical Modification
as a Strategy for Altering the Specificity of the S, Pocket of
Subtilisin 6. lentus
Introduction
Employing the strategy of cornbined site-directed mutagenesis and chernical
modification (Scheme 2.1). the goal of the cuvent strategy was to alter the
specificity of the prirnary-specificity detemining pocket of SBL. SI.' Employing the
crystal structure of SBL as our guide2 two residues at the bottom of the SI pocket.
Ser156, one which is surface exposed. and Ser166, one whose side chain points
into the pocket, were chosen for mutagenesis and chemical modification (Figure
2.1 ). The choice of methane thiosulfonate reagents is a representative one used to
explore the viability of the CMM approach to alter enzyme specificity and activity.
These include alkyl. (1 a-1 c). arornatic (1 d), negatively charged (le), and positively
charged (1 f) methanethiosulfonate reagent~.~
Scheme 2.1
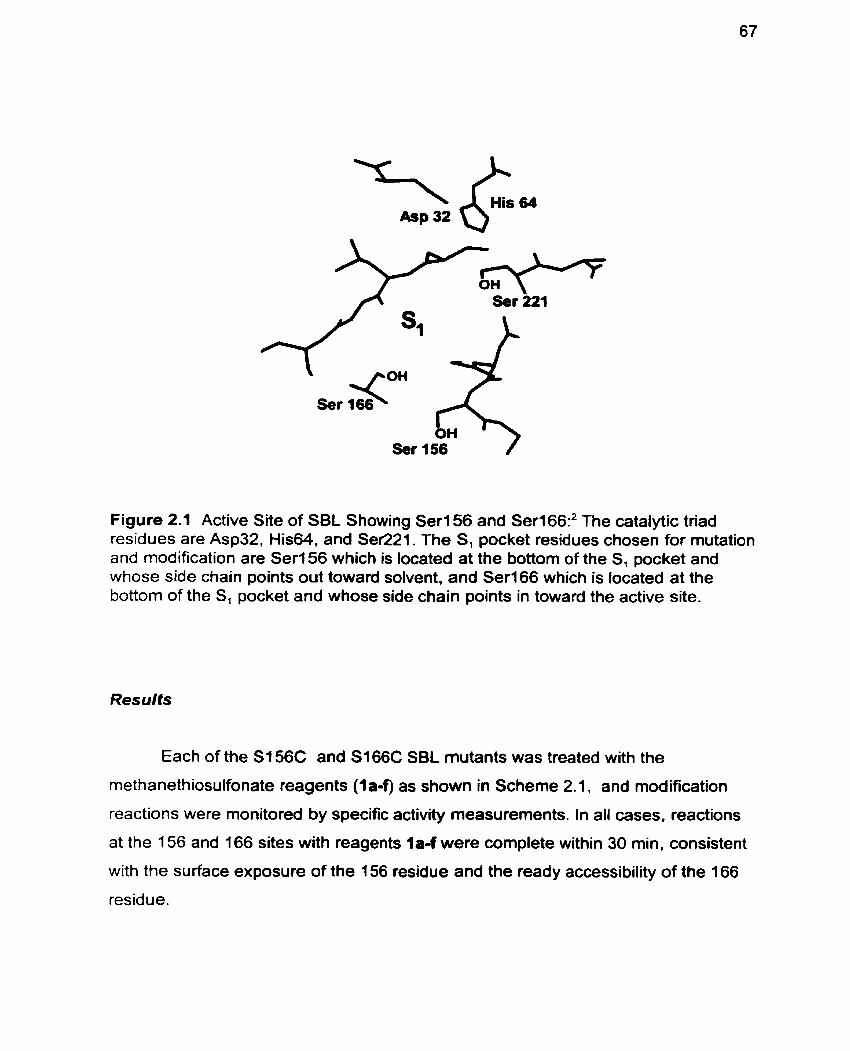
Ser 156 1
Figure 2.1 Active Site of SBL Showing Serl56 and Ser166:' The catalytic triad residues are Asp32, His64, and Ser221. The S, pocket residues chosen for mutation and modification are Seri 56 which is located at the bottom of the S, pocket and whose side chain points out toward solvent, and Ser166 which is located at the bottom of the S, pocket and whose side chain points in toward the active site.
Resulfs
Each of the S156C and S166C SBL mutants was treated with the
methanethiosulfonate reagents (la-f) as shown in Scheme 2.1, and modification
reactions were monitored by specific activity rneasurements. In al1 cases, reactions
at the 156 and 166 sites with reagents la-f were complete within 30 min, consistent
with the surface exposure of the 156 residue and the ready accessibility of the 166
residue.

Free thiol titration of the S I 56C and S166C CMMs with Ellman's-reagent
established that reactions were quantitative. In al1 cases, the free thiol content of the
CMMs was less than 2 %. Mass analysis of the CMMs by electrospray mass
spectrometry was consistent (I 6 Da) with the calculated mass. The purity of the
modified enzymes was assessed by native-PAGE and in al1 cases only one band
was visible. CMMs S156C-S-a to Id, and S166C-S-a to Id, couid not be
distinguished from the parent cysteine mutant nor the WT enzyme on native-PAGE.
However, the negatively charged CMMs derived from reactions with le, S I 56C-S-
CH,CH,SO;, and S166C-S-CH,CH2S03-, displayed retarded rnobility in the direction
of the cathode, while the positively charged S156C-S-CH2CH,NH,+, and S166C-S-
CH,CH,NH,+ CMMs derived from reactions with I f displayed greater mobility relative
to wild type.
That modification of cysteine is wholly responsible for altered activity was
established by demonstrating that treatment of SBL-WT with each of la-f resulted
in no change in activity, nor of molecular weight. Furthemore, treatment of S166C-
S-a to-f , and S I 56CS-a to-f, with P-mercaptoethanol restored activity to that of the
parent cysteine mutant, verifying that chemical modification at cysteine was solely
responsible for the observed changes in activity and is fully revenible by this
treatment.
Kinetic constants for each of the CMMs, were determined with the suc-AAPF-
pNA substrate and are shown in Table 2.1. KMs are virtually unaltered and
consequently, kJKM changes are mainly relective of k,, variations.
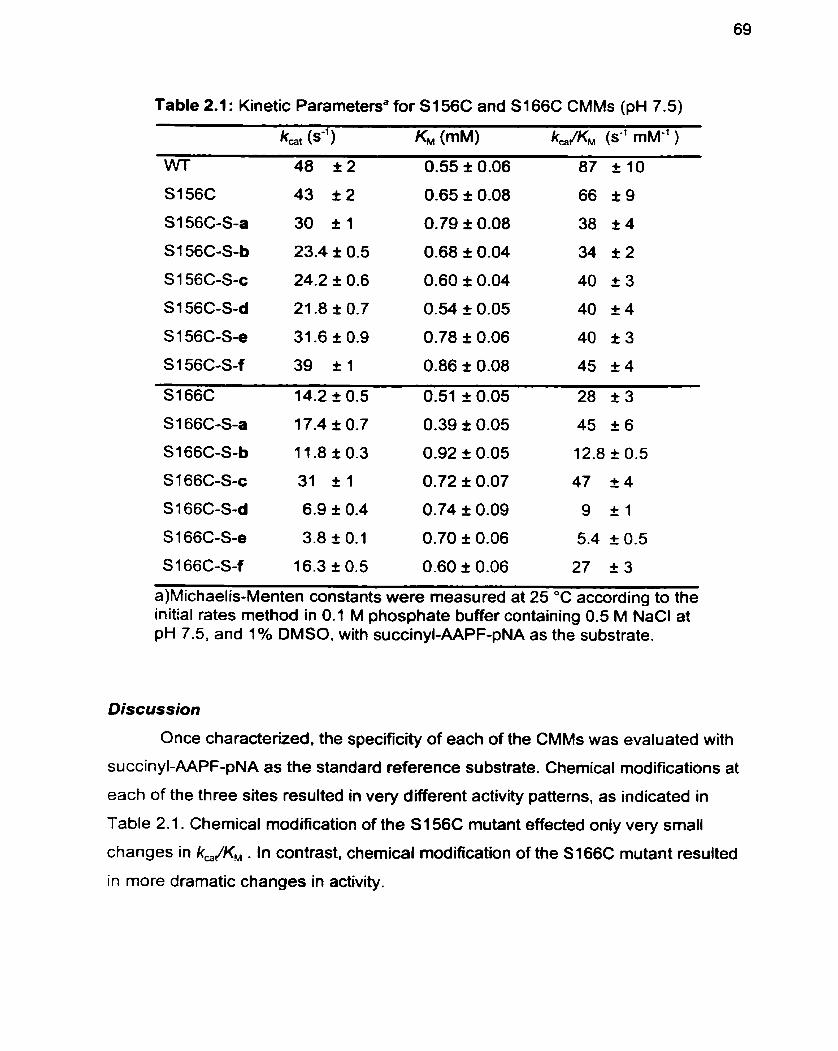
Table 2.1 : Kinetic Parametena for S i 56C and S i 66C CMMs (pH 7.5)
S 1 66C-S-f 16.3 t 0.5 0.60 I 0.06 27 I 3
a)Michaelis-Menten constants were measured at 25 OC according to the initial rates method in 0.1 M phosphate buffer containing 0.5 M NaCl at pH 7.5, and 1 % DMSO. with succinyl-AAPF-pNA as the substrate.
Discussion
Once characterized. the specificity of each of the CMMs was evaluated with
succinyl-AAPF-pNA as the standard reference substrate. Chemical modifications at
each of the three sites resulted in very different activity patterns. as indicated in
Table 2.1. Chemical modification of the S I 56C mutant effected only very srnall
changes in k,JK, . In contrast, chernical modification of the S166C mutant resulted
in more dramatic changes in activity.
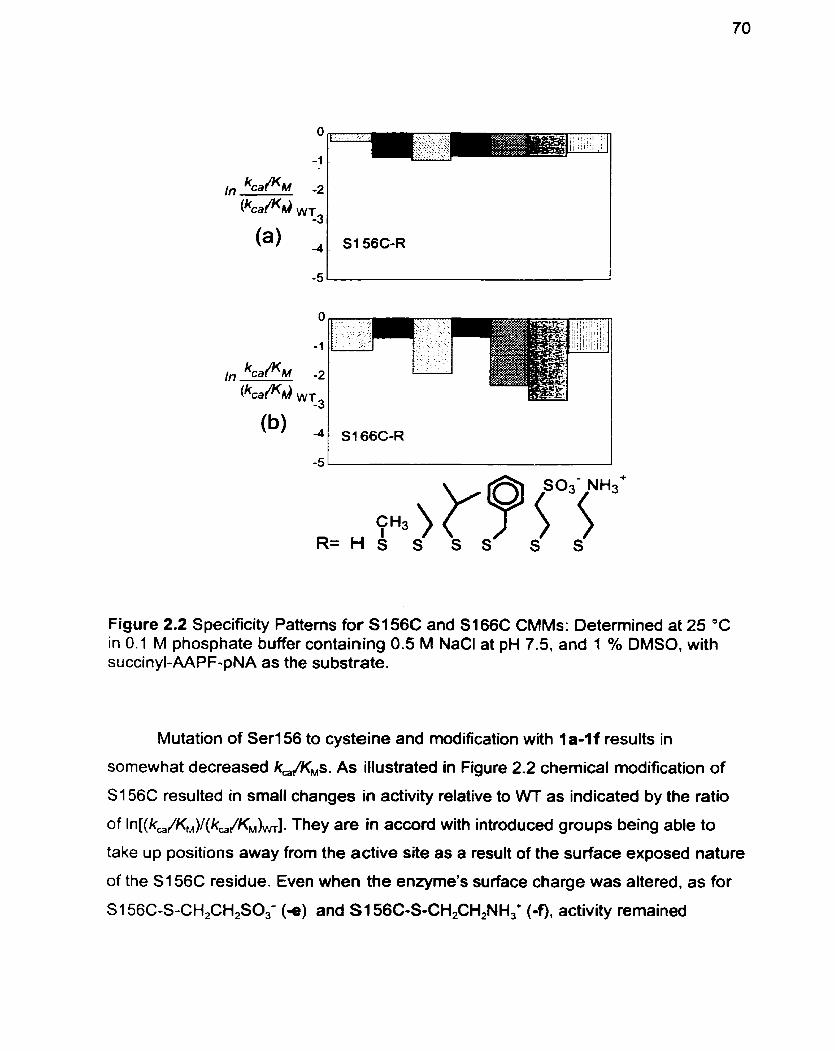
Figure 2.2 Specificity Patterns for Si56C and S166C CMMs: Determined at 25 OC in 0.1 M phosphate buffer containing 0.5 M NaCl at pH 7.5, and 1 % DMSO, with succinyl-AAPF-pNA as the substrate.
Mutation of Ser156 to cysteine and modification with l a - I f results in
somewhat decreased k,JK,s. As illustrated in Figure 2.2 chemical modification of
S I 56C resulted in small changes in activity relative to WT as indicated by the ratio
of In[(k,/K,)/(k,/K,),J. They are in accord with introduced groups being able to
take up positions away from the active site as a result of the surface exposed nature
of the S156C residue. Even when the enzyme's surface charge was altered, as for
S I 56C-S-CH,CH,SO< (.e) and SI 56C-S-CH,CH,NH,+ (4). activity remained

unaffected. This result contrasts previous observations for the related enzyme
subtilisin BPN', for which increasing positive charge on the enzyme surface
destabilizes the imidazolium forrn of the active site histidine and stabilizes the
negatively charged oxyanion resulting in altered activity.' The absence of such an
electrostatic effect for SBL indicates a significant difference in specificity between
the S, pockets of SBL compared to subtilisin Carisberg or subtilisin BPN'.' This may
be due to the 4 amino acid deletion in SBL relative to subtilisin BPN' for which
mutation of residue 156 significantly affects Pl binding6 and enzyme activity7 While
these data were detennined under conditions of high ionic strength. which may
mask electrostatic effectsa k& deteminations at lower ionic strength did not
significantly affect k&KM either (Table 3.1, page 84). This suggests that the -S-
CH,CH2NH,+ group of S156C-S-f and -S-CH2CH2S0,- group of S156C-S-e are
extensively solvated.
The most interesting and unexpected results are manifest for the S166C
CMMs. An intriguing structure-activity relationship (SAR) of alternating increases
and decreases in kJKM upon modification of S166C with methanethiosulfonate
reagents of increasing steric volume. specifically l a (-S-CH,). 1 b (-S-CH2CH,) 1c (-
S-CH,CH(CH,),) and i d (-S-CH,C,H,) is observed. The SAR illustrated in Figure
2.1 b is unprecedented and quite different from what would have been predicted
from previous site-directed mutagenesis studies of residue 166 in the related
enzyme. subtilisin 5PN'.9-10 The molecular basis for this novel activity pattern was
analyzed by molecular modelling of the peptidyl product inhibitor AAPF bound to
SBL-CMMS.
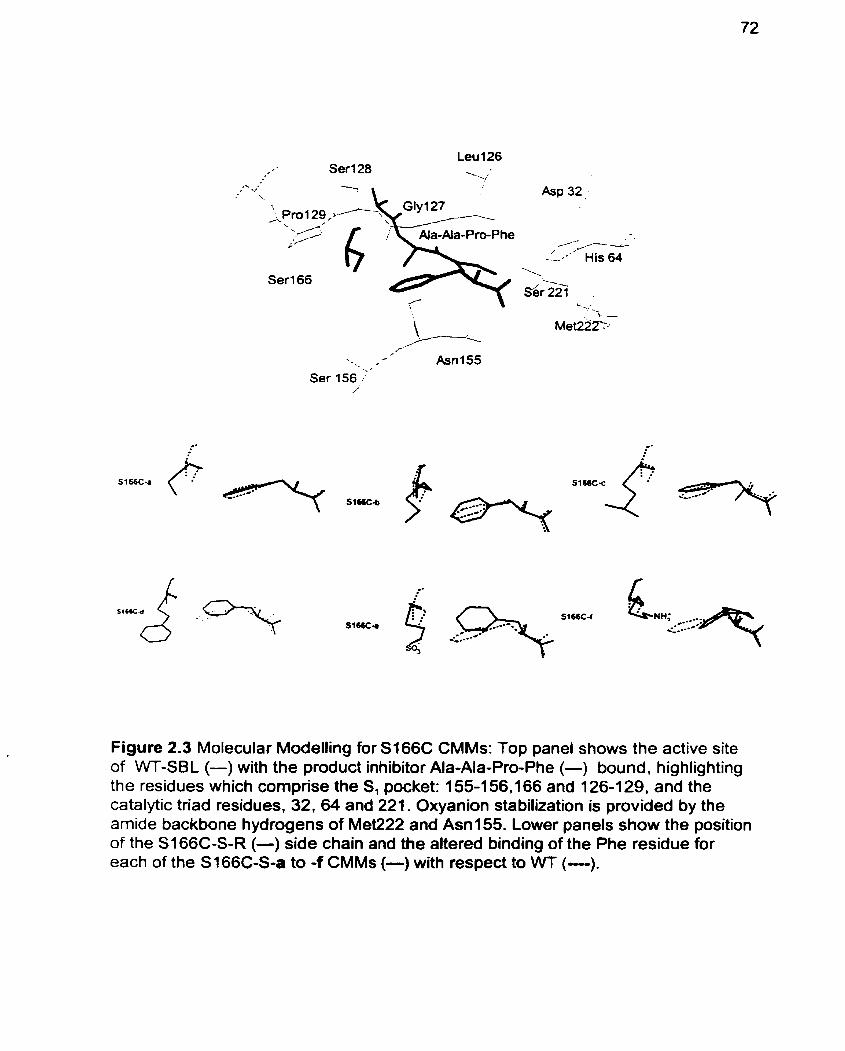
Leu 126 Serl28 ---.;
, . .- -- His 64
'. sé& . . ;
-.; . .
' . 4 . I . , Ser 156 ;'
/.
Figure 2.3 Molecular Modelling for S166C CMMs: Top panel shows the active site of WT-SBL (-) with the product inhibitor Ala-Ala-Pro-Phe (-) bound. highlighting the residues which comprise the S, pocket: 155-1 56,166 and 126-1 29, and the catalytic triad residues, 32, 64 and 221. Oxyanion stabilization is provided by the amide backbone hydrogens of Met222 and Asn155. Lower panels show the position of the SI 66C-S-R (-) side chain and the altered binding of the Phe residue for each of the S166C-S-a to -f CMMs (-) with respect to WT (-).

As shown in Figure 2.3, molecular modeling analysis of the SI 66C CMMs
reveals that the observed k,JKM changes correlate with altered binding of the P,
benzyl side-chain of the AAPF product inhibitor. While the phenyl ring of the P, Phe
residue is positioned well into the S, pocket of WT-SBL, it is slightly distorted out of
the pocket for S166C-S-CH, (-S-a). For S166C-S-CHFH, (-Sb) the phenyl binding
is further distorted. but is less so for S I 66C-S-CH,CH(CH,), (-Sc). Molecular
modelling also revealed a very significant repositioning of the phenyl ring out of the
S, pocket of S I 66C-S-CH,C,H, (-Sd), consistent with the lowered kJK, for this
CMM. Excitingly, in al1 cases the extent of binding distortion of the Pl benzyl side
chain in the S, pocket of the CMMs rnodeled correlates with the changes in ka&.
The molecular modelling results also suggest that for the S166C CMMs, disnipted
phenyl binding may be due to changes in the preferred position of the p-carbon
(-'CH,-S-R) of Cys166. As illustrated in Figure 2.3, for S I 66C-S-CH2CH, (-Sb) the
P-carbon of Cys166 points further into the S, pocket than it does for the S I 66C-
SCH, (-Sa), S I 66C-S-CH,CH(CH,), ( -Sc) . or WT enzymes, and thus pushes the
P, Phe residue out of the S, pocket. However, for S166C-S-CH2C,H, (-S-d), which
also displays lower activity, and for which molecular modelling indicates significantly
distorted Phe binding, the position of the P-carbon of Cys166 is not altered. It is
not immediately evident how the local re-orientation of the modified side chain of
S I 66C-S-CH,CH, (-Sb) may serve to relieve strain induced elsewhere in the S,
pocket.
The positively charged -S-CH,CH,NH,+ side chain of S166C-S-f effected no
change in kJKM relative to S166C, while the negatively charged -S-CH,CH,SO,'
moiety of S166C-S-e caused a 5-fold decrease. A positive charge is expected to
stabilize the unprotonated form of His64 as well as provide oxyanion stabilization.
while a negative charge destabilizes these interaction^.^ Thus factors such as non-
optimal positioning of the positive charge within the enzyme active site. or masking
of a favourable electrostatic interaction by a unfavourable s t e k one. are cleariy
operating to negate the beneficial influence of the positive charge of the -S-
CH,CH,NH,+ side chain of S166C-S-f. A dramatic structural change is seen for

S 166C-S-f, in which the -S-CH,CH,NH,' side chain becomes directed into the S,
pocket, and causes the phenyl ring of phenylalanine to be displaced into the upper
region of the pocket. This unexpected orientation of the ethylammonium moiety into
the S, pocket is attributed to a potential hydrogen bond between its ammonium
hydrogen and the a-carbonyl of Gly 127, (N-O distance 3.9 A in the energy
minimized structure). White the conformation of the -S-CH,CH,NH,+ side chain of
S166C-S-f is unusual, it parallels the conformational fiexibility of the side chain of
Lys1 66 evident from the disordered electron density map observed in the X-ray
structure of the G 166K mutant of subtilisin BPN'." Furthemore, molecular modelling
analysis shows that. while the -S-CH,CH,SO; side chain of S166C-S-e does not
orient itself directly into the S, pocket, it does cause significant disruption of inhibitor
binding .
The fact that AAPF binding is correlated to the trends in k& while KM is
virtually unaltered suggests that the product inhibitor mimics transition state binding
and not ground state binding of the succinyl-AAPF-pNA substrate. These
modifications change enzyme turnover to a greater extent than substrate binding.
These results demonstrate that the combination of site-directed mutagenesis
and site-specific chemical modification can alter the specificity of the S, and S,'
pockets of subtilisin B. lentus in unusual ways. Also. molecular modeling has again
proven to be a powerful tool for analyzing the molecular basis for the activity
changes induced by chemical modification and suggest that activity changes are
correlated to altered binding of the P, moiety of AAPF in the S, pocket. It has also
become apparent that the degree of activity changes which may be engendered
upon modification is correlated with the extent of surface exposure of the residue
modified .

Experimental
Enzyme Purification. Wild type subtilisin Bacillus lentus, and its S I 56C and
S166C mutants were purified by the general method of Stabile et al.'* The crude
protein concentrates containing PEG (50%) as a stabilizer, which were obtained as
previously descnbed,12 were purified on a Sephadex G-25 desalting matrix with a pH
5.2 buffer (20 mM sodium acetate, 5 mM CaCI,), to remove small molecular weight
contaminants. Pooled fractions from the desalting column were then applied to a
strong cation exchange column (SP Sepharose FF) in the HEPES buffer (20 mM
HEPES, 2 mM CaCI,, pH 7.8). and SB1 was eluted with a one-step gradient of 1-
200 mM NaCI-HEPES buffer, (20 mM HEPES, 2 mM CaCI,. 10 mM DTT. 200 mM
NaCI, pH 7.8). Enzyme powder was obtained by dialysis (MWCO f2 - l4 ,OOO) of the
eluent against 1 mM CaCI, and subsequent lyophylization. The purity of the WT and
mutant enzymes, denatured by incubation with 0.1 M HCI at O OC for 30 min., was
determined by SDS-PAGE on 20% homogeneous gels using the Phast System
from Pharmacia. For al1 enzymes used, only one band was visible.
Mefhanefhiosulfonate reagents. Reagent 1 a was purchased from Aldrich
Chemical Co. Inc., l e and i f from Toronto Research Chemical (2 Brisbane Rd.
Toronto, ON), and al1 were used as received. Reagents lb, 1c. and i d were
prepared as previously described .13
Site-Specific Chemical Modification. To 25 mg of a SBL mutant in CHES buffer
(2.5 rnL; 70 mM CHES. 5 mM MES, 2 mM CaCI,, pH 9.5) at 20 "C was added one of
the methanethiosulfonate reagents (la-f) (100 pL of a 1 M solution: l a in MeOH, 1b
in EtOH, l c in EtOH, 1d in CH,CN, l e in CHES buffer, 1f in MeOH), in a PEG
(1 0,000) coated polypropylene test tube, and the mixture agitated in an end-over-
end rotator. Blank reactions containing 100 pL of solvent instead of the reagent
solution were run in parallel. Each of the modification reactions was monitored
spectrophotometrically (E,,, = 8800 M" cm-')14 on a Perkin Elmer Lambda 2
spectrophotorneter, by specific activity rneasurements. After the reaction was
quenched by dilution in MES buffer (5 mM MES. 2 mM CaCI,. pH 6.5) at O OC, the

specific activity of the CMM (10 PL), was detennined in buffer containing: 0.1 M
TRIS pH 8.6, 0.005 % Tween 80, and 1% DMSO, with the succinyl-AAPF-pNA
substrate (1 mg/mL) (purchased from Bachem Bioscience Inc.) at 25 OC. The
reaction was terminated when the addition of a further 100 pL of methane
thiosulfonate solution effected no fuither change in specific activity, generally within
30 min. The reaction solution was purified on a disposable desalting column
(P harmacia Biotech PD-1 O, Sephadex G-25 M) pre-equilibrated with MES buffer.
The CMM was eluted with MES-buffer (3.5 mL), dialyzed against 1 mM CaCl, (3 x 1
L) at 4 OC and subsequently lyophilized. Modified enzymes were analyzed by
nondenaturing gradient (8-25%) gels at pH 4.2, run towards the cathode on the
Pharmacia Phast-System, and appeared as one single band. Each of the CMMs
was analyzed in parallel with its parent cysteine mutant and the Wi enzyme.
Electrospray Mass Spectrometry. Prior to ES-MS analysis, CMMs were pu rified
by FPLC (BioRad, Biologic System) on a Source 15 RPC matrix (1 7-OiZ7-2O from
Pharmacia) with 5% acetonitrile, 0.01% TFA as the running buffer and eluted with
80% acetonitrile, 0.01 % TFA in a one step gradient- Electrospray mass spectra were
recorded on a PE SC1 EX API III Biomolecular Mass Analyzer. Mass: WT: Calcd.
26698. Found 26694. S I 56C: Calcd. 26714. Found 26712. S156C-S-a: Calcd.
26760. Found 26755. S156C-S-b: Calcd. 26774. Found 26771. S i 56C-S-c: Calcd.
26802. Found 26798. S I 56C-S-d: Calcd. 26836. Found 26832. S156C-S-e: Calcd.
26853. Found 26859. S I 56C-S-f: Calcd. 26790. Found 26791. S166C: Calcd.
26714. Found 26708. Sl66C-S-a: Calcd. 26760. Found 26755. S166C-S-b: Calcd.
26774. Found 26775. Sl66C-S-c: Calcd. 26802. Found 26801. S166C-S-d: Calcd.
26836. Found 26832. S166C-S-e: Calcd. 26853. Found 26851. S166C-S-f: Calcd.
26790. Found 26787.
Regeneration of UnmodMed Enzyme by Treatrnent with P-Mercaptoethanol. To
a solution of CMM (2.0 mg) in 250 pL of CHES-buffer (70 mM CHES, 5 mM MES,
2 mM CaCI,, pH 9.5) was added 10 PL of a solution of P-mercaptoethanol (1 M in
95% EtOH ). The reaction was monitored by specific activity measurements and in
ail cases the activity of the cysteine parent was restored.

Free Thiol Titration: The free thiol content of WT, S I 56C, S I 66C and their CMMs,
was determined spectrophotometrically by titration with Ellman's reagent (E,,, =
13600 M-' in phosphate buffer 0.25 M, pH 8.0 as was found to be < 2 % in al1
cases.
Active Site Titrations: The enzyme concentration was determined16 by monitoring
fluoride release u pon enzyme reaction with a-toluenesulfon yl Ruofide (Aldrich
Chemical Co. Inc.) as measured by a fluoride ion sensitive electrode (Orion
Research 96-09). The enzyme concentration deterrnined in this way was used to
calcufate kinetic parameters for each CMM.
Kinetic Measurements: Michaelis-Menten constants were measured at 25 OC by
curve fitting (GraFit@ 3.03) of the initial rate data determined at eight concentrations
(0.1 25 mM4.0 mM) of the succinyl-AAPF-pNA substrate in 0.1 M phosphate buffer
containing 0.5 M NaCl, 0.005% Tween 80, 1% DMSO. pH 7.5 = 8800 M-' cm-').
Malecular Modelling: The X-ray stnictu re of su btilisin Bacillus lentus with the
peptide inhibitor AAPF bound2 was used as the starting point for calculations on wild
type and CMMs. The enzyme setup was done with lnsight II. version 2.3.0.'7To
create initial coordinates for the minimization. hydrogens were added at the pH 7.5
used for kinetic measurements. This protonated al1 Lys and Arg residues and the N-
terminus and deprotonated al1 Glu and Asp residues and the C-terminal carboxyl
group. The protonated form of His 64 was used in al1 calculations. The model
system was solvated with a 5 A layer of water molecules. The total number of water
molecules in the system was 1143. The overall charge of the enzyme-inhibitor
complex resulting from this setup was +4 for the WT enzyme. Energy simulations
were performed with the Discover program, Version 2.9.518 on a Silicon Graphics
Indigo cornputer, using the consistent valence force field function (CVFF). A non-
bonded cutoff distance of 18 A with a switching distance of 2 A was employed. The
non-bonded pair list was updated every 20 cycles and a dielectric constant of 1 was
used in al1 calculations. The energy of the WT enzyme was rninimized in stages,
with initially only the water molecules being allowed to move, followed by water
molecules and the amino acid side chains, and then finally the entire enzyme. The

mutated and chemically modified enzymes were generated by modifying the
relevant amino acid using the Builder module of Insight. These structures were then
energy minimized in a similar manner. lnitially the side-chain of the mutated residue
and the water molecules were energy minimized. Then the amino acid side chains
within a 10 A radius of the a-carbon of the mutated residue were energy minimized
while al1 other residues were constrained, then al1 of the atoms within a 10 A shell
were energy minimized. The AAPF inhibitor was free to move throughout al1 stages
of the minimization.

1. Schechter 1, Berger A. On the Size of the Active Site in Proteases. 1. Papain. Biochem. Biophys. Res. Commun. 1967; 27(2): 1 57-62.
2. Knapp M. Daubennann J, Bott RR. Brookhaven Database Entry 1 JEA.
3. All of the non-commercially available methanethiosulfonate reagents used for the CMM approach were prepared by Drs. Xiao Shang and Ben Davis.
4. Jackson SE, Fersht AR. Contribution of Long-Range Electrostatic lnteractions to the Stabilization of the Catafytic Transition State of the Serine Protease Su btilisin BPN' . Biochemistry 1993; 32: 13909-1 6.
5. Betzel C, Klupsch S. Papendorf G. Hastrup S. Branner S. Wilson KS. Crystal Structure of the Alkaline Proteinase Savinase from Bacillus lentus at 1.4 A Resolution. J. Mol. Biol. 1 992; 223:427-45.
6. Wells JA, Cunningham BC, Graycar TP. Estell DA. Recruitment of Substrate- Specificity Properties from One Enzyme into a Related One by Protein Engineering. Proc. Natl. Acad. Sci. USA 1987; 845167-71.
7. Russell AJ. Fersht AR. Rational Modification of Enzyme Catalysis By Engineering Surface Charge. Nature 1987; 328:496-500.
8. Russell AJ. Thomas PG, Fersht AR. Electrostatic Effects on Modification of Charged Groups in the Active Site Cleft of Subtilisin by Protein Engineering. J. MOI. Biol. 1987; 1 93:803-13.
9. Estell DA. Graycar TP. Miller JV et al. Probing Steric and Hydrophobic Effects on Enzyme Su bstrate lnteractions by Protein Engineering. Science 1 986; 233:659-63.
10. Wangikar PP, Rich JO, Clark DS. Dordick JS. Probing Enzymic Transition State Hydrophobicities. Biochernistry 1995; M(38): 1 2302-1 0.
11. Bott R. Ultsch M. Wells J et al. Importance of Conformational Variability in Protein Engineering of Subtilisin. Lebanon HM, Mumma RO. Honeycutt RC, Duesing JH. Ed. Biotech. Agric.Chem. Vol. ACS Syrnp. Ser. 334. 1987: 139- 47.

Stabile MR. Lai WG, DeSantis G et al. Probing the Specificity of the S, Binding Site of M222 Mutants of Subtilisin B. Lentus with Boronic Acid Inhibitors. Bioorg. Med. Chem. Lett. 1 996; 6(21 ):%O1 -6.
Berglund P. DeSantis G. Stabile MR et al. Chemical Modification of Cysteine Mutants of Subtilisin Bacillus lentus Can Create Better Catalysts Than the Wild- Type Enzyme. J. Am. Chem. Soc. 1997; 1 19:5265-6.
Bonneau PR. Graycar TP, Estell DA. Jones JB. Alteration of the Specificity of Subtilisin BPN' by Site-Directed Mutagenesis in Its S, and S,' Binding Sites. J. Am. Chem. Soc. 1991 ; 1 l3:1026-30.
Ellman GL, Courtney KD. Andres Jr. V, Featherstone RM. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961 ; 7:88-95.
Hsia CY, Ganshaw G. Paech C. Murray CJ. Active-Site Titration of Serine Proteases Using a Fluoride Ion Selective Electrode and Sulfonyl Fluoride Inhibitors. Anal. Biochem. 1996; 2421221 -7.
Insight II [Biosym Technologies. Inc. San Diego, CA. USA]. Version 2.3.0.
Discover [Biosym Technologies, Inc. San Diego, CA, USA]. Version 2.9.5.
DeSantis G, Berglund P, Stabile MR, Gold M, Jones JB. Site-Directed Mutagenesis Combined with Chemical Modification as a Strategy for Altering the Specificity of the S, and S,' Pockets of Subtilisin Bacillus lentus. Biochemistry 1998; 37:5968-73.

Chapter 3
Reproduced in part with permission from Bioorg. Med. Chem. in press.
Unpublished work copyright 1999, Elsevier. (Reference 32)

Probing the Altered Specificity and Catalytic Properties of Mutant
Subtilisin Chemically Modified at Position S156C and S166C in the
S, Pocket
In froduc fion
The objective of the cuvent study was to further explore the S, pocket
specificity changes induced for the Sf56C-a to -d and S166C-a to -d CMMs
(Scheme 3.1 ).' It is often desirable to utilize enzymes under conditions that differ
from their pH optima particulady for synthetic appli~ations.~ Since, the strategy of
chernical modification of site-directed mutant enzymes offen a new opportunity for
controlling pH optima, pH-activity profiles for these CMMs were detemined.
In addition, an approach which is complementary to substrate kinetic analysis for
probing enzyme specificity is the use of cornpetitive reversible transition state
analogue inhibitors. In this regard, boronic acid inhibitors have emerged as
particularly effective probes for the senne proteasesLg and therefore a screen of a
representative stnictu ral range of aromatic boronic acid inhibitors was applied to the
S I 56C and S166C CMMs.
Scheme 3.1
R = a)CH3 b) CH,

Results and Discussion
Each of the chemicaliy modified mutant enzymes (CMMs) was prepared by
the general method descnbed previously.' which entails reaction of the S166C and
S156C mutants of SBL with each of the MTS reagents la- Id, yielding S156C-S-a to
-d and Sl66C-S-a to -d as outlined in Scheme 3.1. Each of the CMMs was fully
characterized. with electrospray mass spectrometry (ES-MS). the absence of
residual free thiol was established by titration with Ellman's reagent." and by native-
polyacrylamide gel electrophoresis (PAGE), al1 of which demonstrate that the
modifications were quantitative and specific for the introduced cysteine.'
The kinetic constants were evaluated at the pH 8.6 optimum of WT-SBL in
Tris-HCI buffer with suc-AAPF-pNA as the standard substrate. These results are
summarized in Table 3.1, which for comparative purposes also includes data
deterrnined at pH 7.5 (0.1 NaHPO,, 0.5 M NaCI) under high salt conditions (Table
2.1 , page 69).' In al1 cases the CMMs were more active under the pH 8.6 conditions.
However, the trends in the kJKM variations at both pH 8.6 and 7.5 parallel each
other very closely.
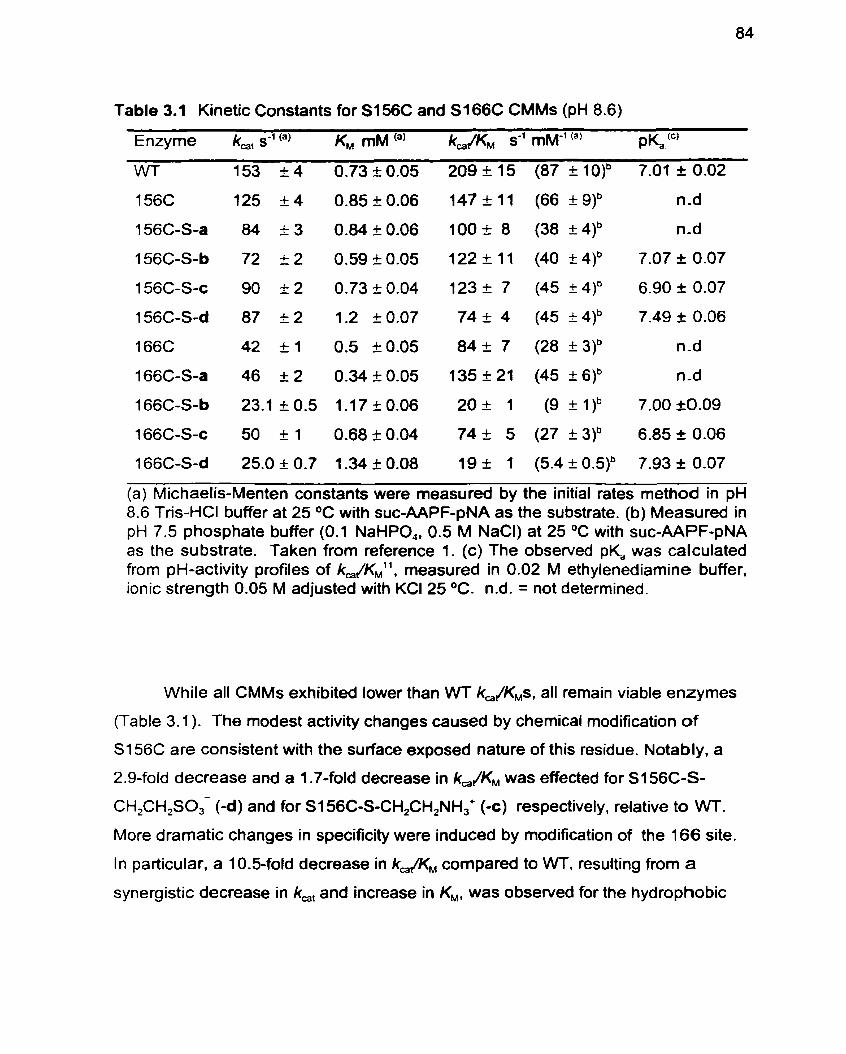
Table 3.1 Kinetic Constants for S I 56C and S166C CMMs (pH 8.6)
Enzyme k,, s-' (") K , mM (a) k&KM s-' mM-' (') P ~ . " '
WT 1 53 k 4 0.73 k 0.05 209 f 1 5 (87 + 1 7.01 t 0.02
156C 125 +-4 0.85k0.06 147k11 (66k9)b n-d
156C-S-a 84 2 3 0.84 2 0.06 100 + 8 (38 + 4)b n-d
156C-S-b 72 + 2 0.59 k 0.05 122 + 11 (40 + 4)b 7.07 I 0.07
1 56C-S-c 90 + 2 0.73 .t 0.04 123 I 7 (45 I 4)' 6.90 I 0.07
156C-S-d 87 I 2 1.2 i 0.07 74 + 4 (45 I 4)b 7.49 2 0.06
166C 42 + 1 0.5 k0.05 84+ 7 (28 +3)' n-d
166C-S-a 46 + 2 0.34 ?: 0.05 135 f 21 (45 t 6)b n-d
166C-S-b 23.1 + 0.5 1.1 7 i 0.06 20+ 1 (9+1 )b 7.00 20.09
166C-S-c 50 + 1 0.68 k 0.04 74 k 5 (27 + 3)b 6.85 + 0.06
166C-S-d 25.0 + 0.7 1.34 k 0.08 19 f 1 (5.4 + 0.5)b 7.93 f 0.07
(a) Michaelis-Menten constants-were measured by the initial rates method in pH 8.6 Tris-HCI buffer at 25 O C with suc-AAPF-pNA as the substrate. (b) Measured in pH 7.5 phosphate buffer (0.1 NaHPO,, 0.5 M NaCI) at 25 OC with suc-AAPF-pNA as the substrate. Taken from reference 1. (c) The obsewed pK, was cafculated from pH-activity profiles of k,/KM1', measured in 0.02 M ethylenediamine buffer. ionic strength 0.05 M adjusted with KCI 25 OC. n.d. = not determined.
While al1 CMMs exhibited lower than WT k,JK,s, all remain viable enzymes
(Table 3.1 ). The modest activity changes caused by chemical modification of
S156C are consistent with the surface exposed nature of this residue. Notably. a
2.9-fold decrease and a 1.7-fold decrease in kJKM was effected for S156C-S-
CH,CH,SO,- (-d) and for S I 56C-S-CH2CH2NH,' (oc) respectively, relative to WT.
More dramatic changes in specificity were induced by modification of the 166 site.
In particular, a 10.5-fold decrease in k& compared to WT. resulting from a
synergistic decrease in k,, and increase in KM, was observed for the hydrophobie

side chain of S1 66C-S-CH2C,H, (-b) and is attributed to the unfavourable steric
contacts revealed previously by molecular modelling analysis.' However. for the
negatively charged side chain CMM, ~166~-S-CH,CH,SO,- (-d) the km/KM was
equally (1 1 -fold) reduced. In this case we conclude that the unfavourable steric
interactions revealed by molecular modelling' are augmented by the destabilizing
effect of the repulsion between the negative charge of the sulfonato group with that
of the incipient oxyanion of the transition state." Conversely, the introduction of a
positive charge in the S, pocket. as for S166C-S-CH,CH2NH3+ (-c). ameliorated the
rate reducing effects. with only a 3-fold sterically induced decrease in kJKLt relative
to W being manifest. and no change at al1 relative to the unmodified cysteine
parent S166C.
It is often desirable to use enzymes under conditions that differ from their
optima2. Accordingly, the effect of chemical modifications on the pH-activity profiles
of SBL-CMMs was examined. This approach to altering pH-activity profiles turned
out to be quite general. in that pK, changes of up to 0.92 are also observed in the
current study. A representative pH-activity profile is illustrated in Figure 3.1. and the
overall pK, data are summarized in Table 3.1. In the absence of a change in rate
determining step, and with no group in the substrate being ionizable within the pH
range of the study, the pH dependence of (k,/K,),,, for the serine proteases follows
the ionization of a catalytically important residue in the free e n ~ y m e . ' ~ - ' ~ The ApKas
are observed to Vary from -CA6 to +0.92 and are attributed to differences in the
ionization of the catalytic His64 in the vanous CMMs.lhZO For the uncharged
modifications of S I 56C-S-CH,C,H, (-b) and S I 66C-SCH,C,H, (-b) the pKas of 7.07
and 7.00 respectively are virtually unchanged relative to WT (pK, =7.01). In contrast,
introduction of a negative charge. as for S i 56C-S-CH,CH2S03- (-d) and S166C-S-
CH,CH2S0,- (-d). elicited a 0.48 and a 0.92 unit increase in the pK, of His64
respectively, while introduction of a positive charge as for S I 56C-S-CH2CH2NH,+ (-
e) and S I 66C-SCH2CH2NH,' (-e) caused a 0.09 unit and 0.16 unit decrease in the
pK, respectively. The observed increases in pK, with increasing negative surface
charge, and decreases with increasing positive surface charge. are attributed to
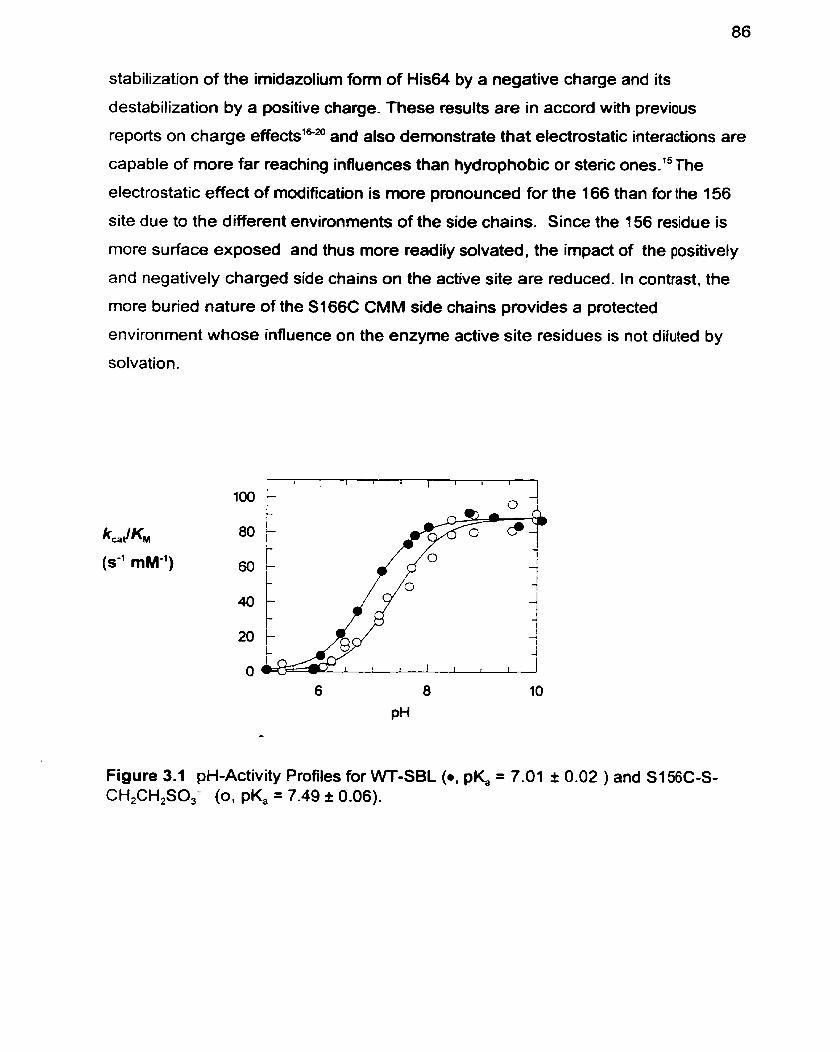
stabilization of the imidazolium form of His64 by a negative charge and its
destabilization by a positive charge. These results are in accord with previous
reports on charge effectsl&= and also demonstrate that electrostatic interactions are
capable of more far reaching influences than hydrophobie or steric ones.15 The
electrostatic effect of modification is more pronounced for the 166 than for the 156
site due to the different environments of the side chains. Since the 156 residue is
more surface exposed and thus more readily solvated. the impact of the positively
and negatively charged side chains on the active site are reduced. In contrast, the
more buried nature of the S166C CMM side chains provides a protected
environment whose influence on the enzyme active site residues is not diluted by
solvation.
kfa t K
(s" mM")
Figure 3.1 pH-Activity Profiles for \NT-SBL (0 . pK, = 7.01 t 0.02 ) and S I 56C-S- CH,CH,SO,- (o. pK, = 7.49 I 0.06).

As a further probe of the S, pocket specificrty changes induced by SBL-
CMMs the altered binding of the representative structural range of transition state
analog boronic acid inhibitorsSg (Formulae 2-6) was evaluated. The observed K,s
revealed significant specificity changes from WT. as summarized in Table 3.2.
Formulae 2-6:
Table 3.2 Inhibition Constants@) for S I 56C and S I 66C CMMs
Enzyme 2 3 4 5 6
Anhibitor KI mM K, mM KI mM KI mM KI mM
WT 1.3 O 1.3 10.1 0.15t0.01 8.4 k0.6 1.07k0.08
(a) Inhibition constants were determined in pH 8.6 Tris-HCI buffer at 25 OC with suc-AAPF-pNA as the substrate by the method of Wale~.~'

The influence of the surface exposed 156 site is further reinforced
since, as illustrated in Figure 3.2a, despite the widely varying structures
of the boronic acids (24) evaluated only small changes in K,s were
observed. The greatest of these was a 3.5-fold improvement in the
binding of 4-carboxyphenyl boronic acid (5) to S156C-S-CH,C,H, (-b).
This indicated that some electrostatic interaction between the
carboxylate group of 5 and the S, pocket of S156C-S-CH,C,H, was
operating.
As illustrated in Figure 3.2b. more dramatic changes in boronic
acid binding were observed for the S166C CMMs, most notably for
S166C-S-CH,C,H, (-b). For this CMM, poorer binding of each of the
boronic acids 2-6 was observed, with the largest effect being a 12-fold
increase over WT in the K, of 2.4dichlorophenyl boronic acid (4) with
SI 66-S-CH,C,H, (-b). As expected. phenethyl boronic acid (6). the
inhibitor with the largest P, group of the series, binds best to WT-SBL,
which has the biggest S, pocket. That the K, of 4-carboxyphenyl
boronic acid (5) with S i 66C-S-CH,CH,NH,+ (-c) is 2-fold lower than for
WT is attributed to the irnproved interaction between the positively
charged ammonium side chain of S166C-S-c and the negatively
charged carboxylate moiety of 5. In further support of this
interpretation. 4-carboxyphenyl boronic acid (S), with its negatively -
charged carboxylate group, binds more poorly to S I 66C-S-CH2CH2S0,
(-c) than to WT, due to electrostatic repulsion.

Figure 3.2 Binding of Boronic Acid Inhibiton (2-6) to S i 56C and S i 66C CMMs: Stronger binding of the boronic acids to the WT is shown as a bar below the X-axis and stronger binding of the boronic acids to the CMM as a bar above the X-axis.
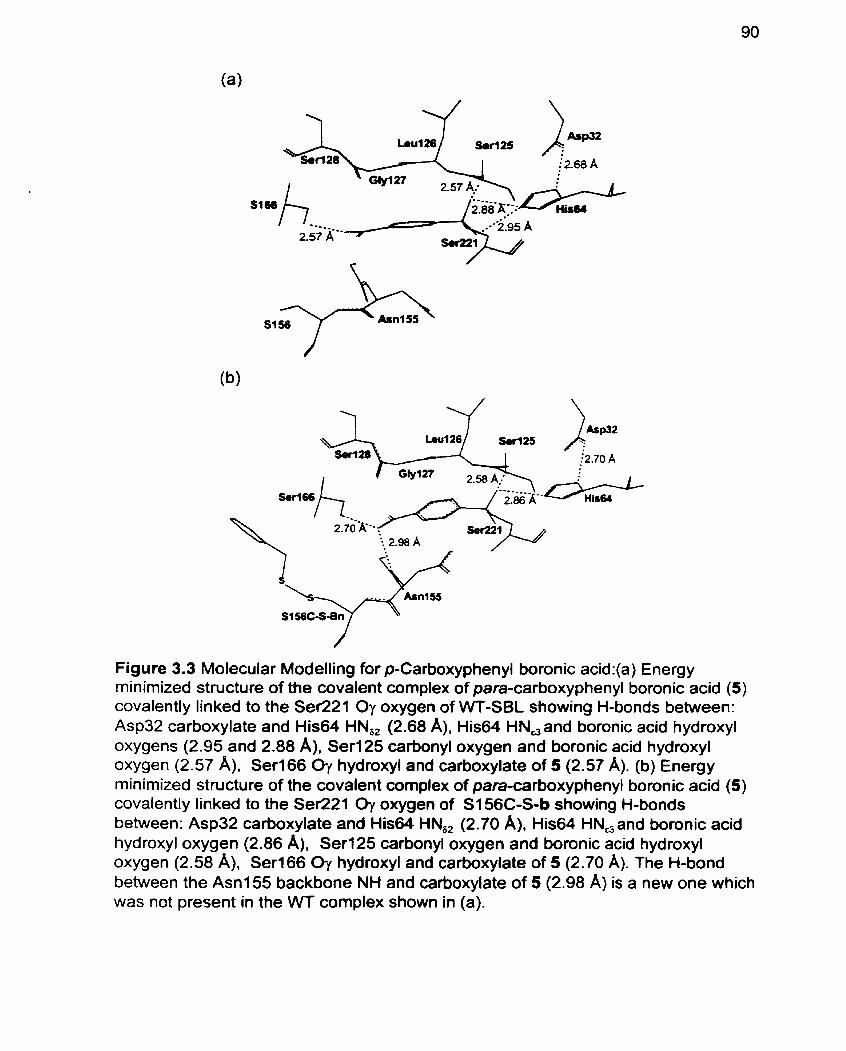
Figure 3.3 Molecular Modelling for p-Carboxyphenyl boronic acid:(a) Energy minimized structure of the covalent complex of para-carboxyphenyl boronic acid (5) covalently linked to the Ser221 Oy oxygen of WT-SBL showing H-bonds between: Asp32 carboxylate and His64 HN,, (2.68 A). His64 HN,and boronic acid hydroxyl oxygens (2.95 and 2.88 A). Ser125 carbonyl oxygen and boronic acid hydroxyl oxygen (2.57 A). Ser166 Oy hydroxyl and carboxylate of 5 (2.57 A). (b) Energy minimized structure of the covalent complex of para-carboxyphenyl boronic acid (5) covalently linked to the Ser221 Oy oxygen of S I 56C-S-b showing H-bonds between: Asp32 carboxylate and His64 HN,, (2.70 A), His64 HN,and boronic acid hydroxyl oxygen (2.86 A), Seri25 carbonyl oxygen and boronic acid hydroxyl oxygen (2.58 A). Seri66 O-! hydroxyl and carboxylate of 5 (2.70 A). The H-bond between the A m 1 55 backbone NH and carboxylate of 5 (2.98 A) is a new one which was not present in the WT complex shown in (a).

Support for the hypothesis of an electrostatic interaction between the
carboxylate group of 5 and the S, pocket of S156C-S-CH,C,H, was sought by
molecular modelling. Molecular modelling revealed an additional hydrogen bond
between the carboxylate of para-carboxyphenyl boronic acid (5) and the Asnl55
amide backbone N-H of S156C-S-b, which is not present in the VVT (Figure 3.3).
This additional hydrogen bond appears to have been gained at the expense of a
His64 HN,, hydrogen bond with a boronic acid hydroxyl oxygen which is present in
the energy rninimized structure of the W.
These results demonstrate that using a screen of different specificity and
catalytic property approaches in combination, such as kinetic. pH-activity profiles
etc., provide complementary insights into the affect of site-directed chemical
modifications of SBL, which taken together are more powerful than independently.

Experimental
Preparation of Methanethiosulfonate Regents: Reagent 1 a was pu rchased from
Aldrich Chemical Co. Inc., 1c and I d from Toronto Research Chemicals (2
Brisbane Rd. Toronto, ON), and ail were used as received. 1 b was prepared via the
nucleophilic displacement of benzyl bromide by sodium methanethios~lfonate.~-~~
Boronic Acids: Boronic acids, 2 and 3 were purchased frorn Aldrich Chemical Co.
Inc., 4 from Lancaster Synthesis lnc. (Windham, NH) and used as received. Boronic
Acids 5-6 were obtained as previously de~cr ibed.~.~
Preparation and Characterization of CMMs: The chemicall y modified mutants of
SBL were prepared and purified as described previously.' Briefly each of S156C or
S 166C (25 mg) was exposed to each of the methanethiosulfonate reagents 1 a-Id in
CHES buffer (2.5 mL; 70 mM CHES, 5 mM MES. 2 mM CaCI,. pH 9.5) at 20 OC.
These were purified by ion exchange chromatography (Pharmacia Biotech PD4 O,
Sephadex (3-25 M). The free thiol content of each of the CMMs was determined by
titration with Ellman'slo reagent and established that the free thiol content of al1
CMMs was less than 2 % demonstrating completeness of reaction. The purity of
each of the CMMs was analyzed by nondenaturing gradient (8-25%) gels nin at pH
4.2 run towards the cathode, on the Pharrnacia Phast-System. and each appeared
as a single band.' In addition, ES-MS spectral analysis of the CMMs revealed that
their determined masses were found to be in agreement (2 6 Da) of the calculated.'
The active enzyme concentration was determined as previously describedt5 by
monitoring fluoride release upon enzyme reaction with a-toluenesulfonyl fluoride
(Aldrich Chemical Co. Inc.) as measured by a fluoride ion sensitive electrode (Orion
Research 96-09). The active enzyme concentration deterrnined in this way was
used to calculate kinetic parameters for each CMM.
Determination of Kinetic Constants: Michaelis-Menten constants were measured
at 25 OC by curve fitting (GraFite 3.03) of the initial rate data determined at eight
concentrations (O. t 25 mM4.O mM) of the suc-AAPF-pNA (Bachern Inc., Torrance,
California) in pH 8.6, 0.1 M Tris-HCI containing 0.005% Tween 80. and 1% DMSO,
( E ~ , ~ = 8800 M-' [El = 6.33 x IOe9 to 1.7 x 1 O" M. as described previously.'

pH-Activity Profiles: pH-activity profiles for the subtilisin CMMs were constnicted
by monitoring product release via its colourometric absorbance at 41 0 versus time at
25 OC E,,, = 8800 cm" M-l)" as described previously." kJKM values were
detennined in duplicate in 0.02 M ethylenediamine buffer, ionic strength 0.05 M
adjusted with KCI, [SI = 4.2 - 12.5 x 105 M of SUC-AAPF-pNA. [El = 2.53 to 21.7 x
1 Oa M at 25 OC. pK,'s were calculated using GraFitb version 3.03 curve fit. single pK,
or bell-shaped double pK,. with the minimum set to zero. Specificalty, into a 1.5 mL
polystyrene cuvette was added 980 PL of buffer (0.02 M ethylenediamine) and 10
pL of substrate (4.2 -12.5 x I O 3 M in DMSO). The solution was incubated in a ceII
holder at 25 OC, before the absorbance reading was set to zero. Then 10 pL of
enzyme solution (2.53 to 21 -7 x I O 4 M in pH 5.8, 20 rnM MES. 1 mM CaCI, ) was
added to initiate the reaction. Af&er an 8 sec delay. absorbance versus time
measurements were recorded on a Perkin Elmer lambda 2 spectrophotometer.
k,JKM values were calculated employing the low substrate approximation. where
the Michaelis-Menten Equation reduces to: v = k, JK,[E],[S] when [S)c<Ky '' De termina fion of Boronic Acid Inhibition Constants: Boron ic acid K, s were
determined in duplicate by the method of Wale~.~' The progress curve without
inhibitor was deterrnined. from an assay mixture containing 980 pL of buffer (0.1 M
Tris-HCI, 0.005% Tween 80) and 10 pL of suc-AAPF-pNA substrate (25 mM in
DMSO). This mixture was incubated in a water jacketed cell for 5 min. at 25 OC.
The absorbance reading was set to zero prior to initiating the reaction by addition of
10 pL of enzyme solution (3.2 x IO" to 1.1 x IO4 M in pH 5.8.20 mM MES. 1 mM
CaCI,). The final volume of the assay mixture was 1 mL. The progress curve with
inhibitor was determined, similarly but with 10 to 980 PL of the boronic acid inhibitor
solution (1 x 1 O3 to 0.1 M. in 0.1 M Tris-HCI. 0.005% Tween 80) added to a final
volume of 1 mL. Absorbance versus time rneasurements were recorded on a Perkin
Elmer lambda 2 spectrophotometer and points for calculation were taken at 15, 18,
21, 24, 27, 30, 33 and 36% substrate conversion."
Molecular Modelling Analysis: Energy simulations were performed with the
Discover program version 2.9.529 on a Silicon Graphics Indigo cornputer. using the

consistent valence force field function. The X-ray structure of subtilisin Bacillus
lentus with the peptide inhibitor AAPF bound was used as the starting point for
ca~culations.~ To create initial coordinates for the minimization, hydrogens were
added at the pH 8.6 used for kinetic rneasurernents. This protonated al1 Lys and Arg
residues and the N-terminus and deprotonated al1 Glu and Asp residues and the C-
terminal carboxyl group. The protonated form of His 64 was used in al1 calculations.
The mode1 system was solvated with a 5 A layer of water molecules. The total
number of water molecules in the systern was 1143. The overall charge of the
enzyme-inhibitor complex resulting from this setup was +4 for the W enzyme. A
non-bonded cutoff distance of 18 A with a switching distance of 2 A was employed.
The non-bonded pair list was updated every 20 cycles and a dielectric constant of 1
was used in al1 calculations. The energy of the WT enzyme was minimized in
stages, with initially only the water molecules being allowed to move. followed by
water molecules and the amino acid side chains, and then finally the entire enzyme,
until the maximum derivative of 0.1 kcal mol-' k' was reached. The S I 56C-S-b
enzyme was generated by modifying the relevant amino acid using the Builder
module of In~ight.~' The energy of this structure was then minimized in a similar
manner. lnitially the side-chain of the mutated residue and the energy of the water
molecules was minimized. followed by water molecules and the amino acid side
chains, and then finally the entire enzyme. The AAPF inhibitor was free to move
throughout al1 stages of the minirnization. The AAPF inhibitor was deleted and then
the energy minirnized enzyme structure was used as the starting points for
calculation of the boronic acid-enzyme complex. In the calculation of the boronic
acid-enzyme complex, a tetrahedral carbon atom was used to mimic the boron atom
since, no force field parameters are available for boron. The boron atom equivalent
of the inhibitor was covalently bound to the Oy of Ser221. The inhibitor was docked
such that its phenyl moiety was directed into the S, pocket. one of the hydroxyl
groups was directed to Asnl55 of the oxyanion hole and the other toward His64.
The energy of this structure was then minimized as above.

DeSantis G, Berglund P, Stabile MR, Gold M, Jones JB. Site-Directed Mutagenesis Combined with Chemical Modification as a Strategy for Altering the Specificity of the S, and SI' Pockets of Subtilisin Bacillus lentus. Biochemistry 1998; 375968073.
Faber K. Biotransfonnations in Organic Synthesis. 3rd edition. Heidelberg: Springer-Verlag . 1997.
Stabile MR, Lai WG, DeSantis G et al. Probing the Specificity of the SI Binding Site of M222 Mutants of Subtilisin B. Lentus with Boronic Acid Inhibitors. Bioorg. Med. Chem. Lett. 1996; 6(21):2501-6.
Wolfenden R. Analog Approaches to the Structure of the Transition State in Enzyme Reactions. Acc. Chem. Res. 1972; 5:10-8.
Lien hard GE, Koehler KA. 2-Phenylboronic Acid, a Possible Transition State Analog for Chyrnotrypsin Biochemistry 1971 ; 10:2570-5.
Seufer-Wasserthal P, Martichonok V, Keller TH, Chin B, Martin R, Jones JB. Probing the Specificity of the S, Binding Site of Subtilisin Carlsberg with Boronic Acids. Bioorg. Med. Chem. 1994; 2(1):35-48.
Martichonok V, Jones JB. Probing the Specificity of the Serine Proteases Subtilisin Carlsberg and U-Chymotrypsin with Enantiorneric 1 -Acetamido Boronic Acids. An Unexpected Reversal of the Normal L-Stereoselectivity Preference. J. Am. Chem. Soc. 1996; 1 18:950-8.
Matthews DA, Alden RA, Birktoft JJ, Freer ST, Kraut J. X-ray Crystallographic Study of Boronic Acid Adducts with Subtilisin BPN' (Novo). A model for the Transition State. J. Biol. Chem. 1975; 250(8):7 720-6.
London RE, Gabel SA. Fluorine-19 NMR Studies of Fluorobenzeneboronic Acids. 2. Kinetic Characterization of the Interaction with Subtilisin Carlsberg and Model Ligands. J. Am. Chem. Soc. 1994; 1 l6:2570-5.
Ellman GL, Courtney KD. Andres Jr. V, Featherstone RM. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 196 1 ; 7:88-95.
11. Khouri HE, Plouffe C, Hasnain S, Hirama T, Storer AC, Ménard R. A Model to

Explain the pH-Dependent Specificity of Cathepsin 6-Catalyzed Hydrolyses. Biochem. J. 1991 ; 275:751-7-
Jackson SE, Fersht AR. Contribution of Long-Range Electrostatic Interactions to the Stabilization of the Catalytic Transition State of the Senne Protease Su btilisin BPN' . Biochemistry 1 993; 323 3909-1 6.
Alberty RA, Massey V. On the lnterpretation of the pH Variation of the Maximum l nitial Velocity of an Enzyme-Catalyzed Reaction . Biochim. Biophys. Acta 1954; 13:347-53.
Renard M, F ersht AR. Anomalous pH Dependence of kJKM in Enzyme Reactions. Rate Constants for the Association of Chymotrypsin with Substrates. Biochemistry 1973; 12(23):4713-7.
Fersht A. Enzyme Structure and Mechanism. 2nd edition. New York: W.H. Freeman and Company. 1985.
Thomas PG, Russell AJ, Fersht AR. Tailoring the pH Dependence of Enzyme Catalysis Using Protein Engineering. Nature 1985; 31 8:375-6.
Sternberg MJE, Hayes FRF, Russell AJ, Thomas PG, Fersht AR. Prediction of Electrostatic Effects of Engineering of Protein Charges. Nature 1987; 330:86-8.
Russell AJ, Thomas PG, Fersht AR. Electrostatic Effects on Modification of Charged Groups in the Active Site Cleft of Subtilisin by Protein Engineering. J. Mol. Biol. 1987; 193:803-13.
Russell AJ, Fersht AR. Rational Modification of Enzyme Catalysis By Engineering Surface Charge. Nature 1987; 328:496-500.
Loewenthal R, Sancho J, Reinikainen T, Fersht AR. Long-Range Surface Charge-Charge interactions in Proteins: Comparison of Experimental ResuRs with Calculations frorn a Theoretical Method. J. Mol. Biol. 1993; (232):574-83.
Waley SG. A Quick Method for the Detenination of Inhibition Constants. Biochem. J. 1982; 205631 -3.
22- Berglund P, DeSantis G, Stabile MR et al. Chemical Modification of Cysteine Mutants of Subtilisin Bacillus lentus Can Create Better Catalysts Than the Wild- Type Enzyme. J. Am. Chem. Soc. 1997; 1195265-6.

Kenyon GL, Bruice TW. Novel Sulfhydryl Reagents. Methods Enzymol. 1977; 471407-30.
Bruice TW, Kenyon GL. Novel Alkyl Alkanethiolsulfonate Sulfhydryl Reagents. Modification of Derivatives of L-Cysteine. J. Protein Chem. 1982; 1 :47-58.
Hsia CY, Ganshaw G, Paech C, Murray CJ. Active-Site Titration of Serine Proteases Using a Fluoride Ion Selective Electrode and Sulfonyl Fluoride Inhibitors. Anal. Biochem. 1996; 242:221-7.
Bonneau PR, Graycar TP, Estell DA, Jones JB. Alteration of the Specificity of Subtilisin BPN' by Site-Directed Mutagenesis in Its S, and S,' Binding Sites. J. Am. Chern. Soc. 1991 ; 1 13: 1026-30.
DeSantis G, Jones JB. Chemical Modifications at a Single Site Can lnduce Significant Shift in the pH-Activity Profiles of a Serine Protease. J. Am. Chem. SOC. 1 998; 120(34):8582-6.
Lee T, Jones JB. Probing the Abilities of Synthetically Useful Serine Proteases to Discriminate Between the Configurations of Remote Stereocenters using Chiral Aldehyde Inhibitors. J. Am. Chem. Soc. 1996; 1 18:502-8.
Discover [Biosym Technologies, Inc. San Diego, CA, USA]. Version 2.9.5.
Knapp M. Daubermann J, Bott RR. Brookhaven Database Entry 1 JEA.
lnsight II [Biosym Technologies. Inc. San Diego, CA, USA]. Version 2.3.0.
DeSantis G. Jones JB. Probing the Altered Specificity and Properties of Mutant Subtilisin Chemically Modified at Position S166C and S I 56C in the S, Pocket. Bioorg. Med. Chem. in press.

Chapter 4

Benzophenone Boronic Acid Photoaffinity Labeling of SBL
CMMs
ln traduction
Previously, (Table 3.2, page 87) the SI binding pocket properties of the
S i 66C and S i 56C chemically modified mutant enzymes (CMMs) of SBL were
probed using a series of boronic acid transition state analogue competitive
inhibitors.' The inhibitor probing approach revealed substantial changes in S,
pocket binding and also yielded insights into the molecular basis for these
changes.' In order to further investigate the altered binding modes of boronic
acid inhibitors to the SI pocket, and since X-ray crystal structures of the boronic
acid inhibitors bound to the CMMs are not available, we considered the strategy
of employing an active site directed photolabile p-boronic acid benzophenone
(8BP, 1) inhibitor to map SBL's binding site.
Active site directed photoaffinity labels2b have been employed to probe
enzyme active sites,7a and provide structural information which is cornplementary
to that which may be available from X-ray crystallographyg or NMRlO-"
approaches. The photolysis of benzophenone derivatives at i, 320-360 nm
effects an n to x' transition which induces formation of a covalent link to amino
acid residues, or other functionalities, by H-abstraction and radical
re~ombination,"'~ as shown in Scheme 4.3. Benzophenone (BP) inhibitor7" and
su bstratet3 derivatives have been employed extensively in active-site directed
photoaffinity labeling ~ t u d i e s . ~ ' ~ In addition, the benzophenone moiety has also
been introduced into peptides or prote in^.^'^ The advantages of the BP
photophore have been documented and include its chernical stability, its stability
to arnbient light and its 330-360 nm photoactivation range, which is not
damaging to prote in^.^ In addition. BP is particulariy well suited to the current
probing study since SBL's active site is quite hydrophobic and BP exhibits
preferential labeling of hydrophobic binding regions in proteins.'

In the current study, the photolabile p-boronic acid benzophenone (BBP.
1) was designed to incorporate both the boronic acid transition state analogue
inhibitor structure1Pa and the photoactivatable benzophenone fun~tionality.~'~
The p-benzophenone boronic acid (BBP) photoprobe. has the active site-
d irecting phenyl boronic acid inhibitor incorporated into the structure of BP rather
than tethered to the end of the photoprobe, which should in principle reduce
nonspecific labeling.4
BBP, 1
Since more substantial changes were effected by modifications at the
S 166C site cornpared to modifications at the S I 56C site as revealed by
substrate (Table 3.1. page 84)'s2' and inhibitor (Table 3.2. page 87)' kinetics. only
the S166C CMMs were subjected to the current p-boronic acid benzophenone
probing studies. In addition to WT-SBL as a control. four representative S166C
CMMs were investigated. Two of these, S i 66C-S-CH,C,H, (-a) and S I 66C-S-
CH,-c-C,H,, (-b) have a sterically constricted S, pocket while S166C-S-
CH2CH2S0,- (-c) and S166C-S-CH2CH2NH,- (-d) have a negatively and a
positively charged S, group respectively (Scheme 4.1). In this study we report
the synthesis of the BBP reagent its evaluation as a cornpetitive reversible
inhibitor of WT-SBL and of the four representative S166C CMMs (Scheme 4.1 )
and photolysis of the El complexes.

Scheme 4.1
Results and Discussion
Each of the chemically modified mutant enzymes (CMMs) were prepared2'
and chara~terized~'-~ by the general method described previously, yielding
S I 66C-S-a to -d as outlined in Scheme 4.1.
The p-boronic acid benzophenone (BBP, 1) photoprobe designed for the
current study was prepared by standard procedures as outlined in Scheme 4.2.
Briefl y. 4-bromobenzophenone (3) was protected as the glycal in refluxing
ben~ene .~~ . '~ The glycal (4) was then subjected to halogen-lithium exchange by
reaction with n-BuLi, followed by treatment with trimethyl borate to generate the
corresponding dimethyl borate. '7.241n order to facilitate purification .17 this dimethyl
borate was subsequently deprotected in sulfuric acid and reprotected with
ethylene glycol. generating the diglycal (5) which was recrystallized from
benzene. The purified diglycal (5) was then fully deprotected in refluxing 6 M
HCI," yielding the p-boronic acid benzophenone (BBP, 1) in 77 % overall yield
(Scheme 4.2).
Scheme 4.2
HCI - acetone 60" C
Initially, the efficacy of binding of the designed photolabile competitive
inhibitor. BBP. to each of the W. SI 66C-S-CH,C,H,. S i 66C-S-CH,-c-C,H,,,
S 1 66C-S-CH2CH2NH,* and S 166C-S-CH,CH2S0,- enzymes was deterrnined
using the standard suc-AAPF-pNA substrate. The kinetic data are summarized
in Table 4.1, and as expected, in al1 cases BBP binds best to WT. BBP binding
to S166C-SCHF6H, (-a) and S166C- CHfc-C,H,, (-b). with their large
h ydrophobic side chains, is reduced by 86- and 9-fold, respectively, compared
to WT. Previously. al! five of the boronic acids evaluated (Figure 3.2b. page 89)'
exhibited poorer binding with the SI66C-S-CH,C,H, CMM compared to W. and
therefore the increased K, of BBP with this CMM is not unexpected. That BBP
binding to S i 66C-S-CH,-c-C,H,, is better than to SI66C-SCH2C6H, may be due
to the greater hydrophobicity of the side chah or to differences in side chain
orientation. BBP binding to the charged CMMs, S1 66C-S-CH2CH2SO; (-c) and
S 1 66C-S-CH2CH,NH,+ (-d). is reduced 170- and 4- fold respectively compared
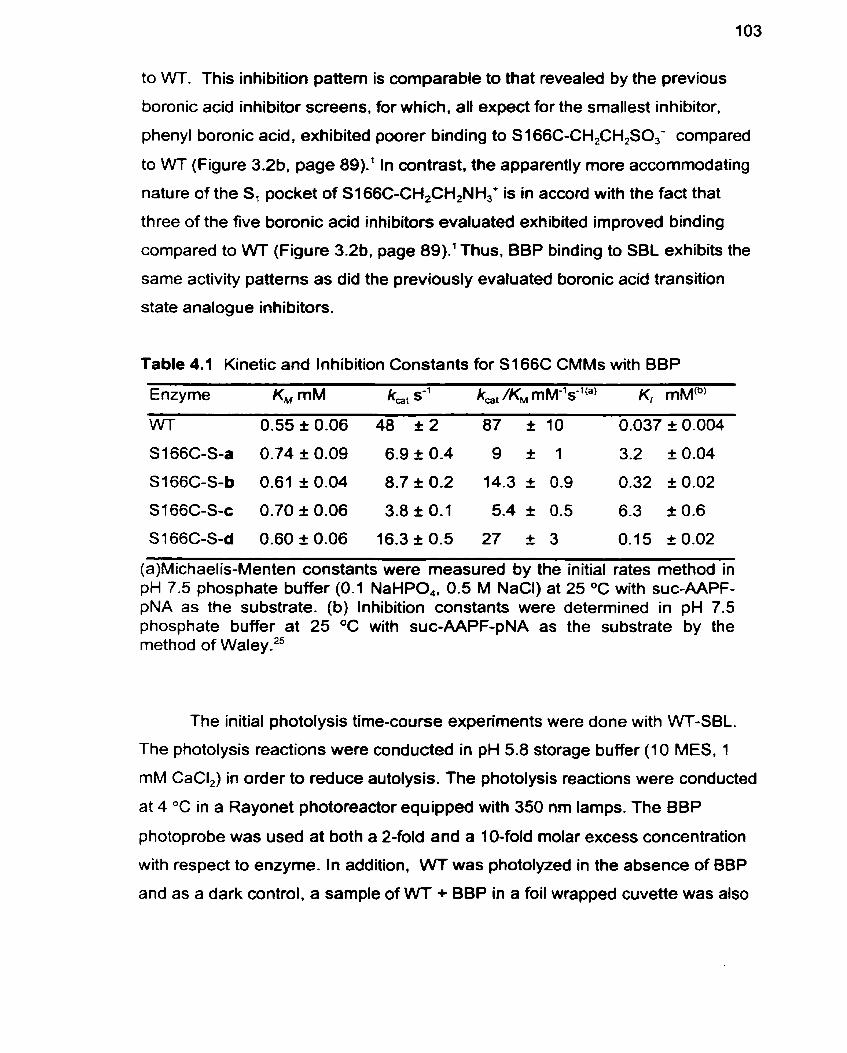
to WT. This inhibition pattern is comparable to that revealed by the previous
boronic acid inhibitor screens, for which. al1 expect for the smallest inhibitor,
phenyl boronic acid, exhibited poorer binding to S166C-CH,CH,SO,- compared
to WT (Figure 3.2b. page 89).' In contrast. the apparently more accommodating
nature of the S, pocket of S166C-CH,CH,NH; is in accord with the fact that
three of the five boronic acid inhibitors evaluated exhibited improved binding
compared to VVT (Figure 3.2b. page 89).'Thus, BBP binding to SBL exhibits the
same activity patterns as did the previously evafuated boronic acid transition
state analogue inhibitors.
Table 4.1 Kinetic and Inhibition Constants for S166C CMMs with BBP
Enzyme KM mM k,, s-' kat /KM ~ M - ' s - ' ( ~ ) K, mM(b)
WT 0.55 t 0.06 48 I 2 87 110 0.037 I 0.004
S166C-S-d 0.60 2 0.06 16.3 10.5 27 I 3 0.1 5 I 0.02
(a)Michaelis-Menten constants were measured by the initial rates method in pH 7.5 phosphate buffer (0.1 NaHPO,. 0.5 M NaCI) at 25 OC with suc-AAPF- pNA as the substrate. (b) Inhibition constants were deterrnined in pH 7.5 phosphate buffer at 25 OC with suc-AAPF-pNA as the substrate by the method of W a l e ~ . ~ ~
The initial photolysis time-course experiments were done with WT-SBL.
The photolysis reactions were conducted in pH 5.8 storage buffer (10 MES, 1
mM CaCI,) in order to reduce autolysis. The photolysis reactions were conducted
at 4 OC in a Rayonet photoreactor equipped with 350 nrn lamps. The BBP
photoprobe was used at both a 2-fold and a 10-fold molar excess concentration
with respect to enzyme. In addition, WT was photolyzed in the absence of 8BP
and as a dark control, a sample of WT + BBP in a foi1 wrapped cuvette was also
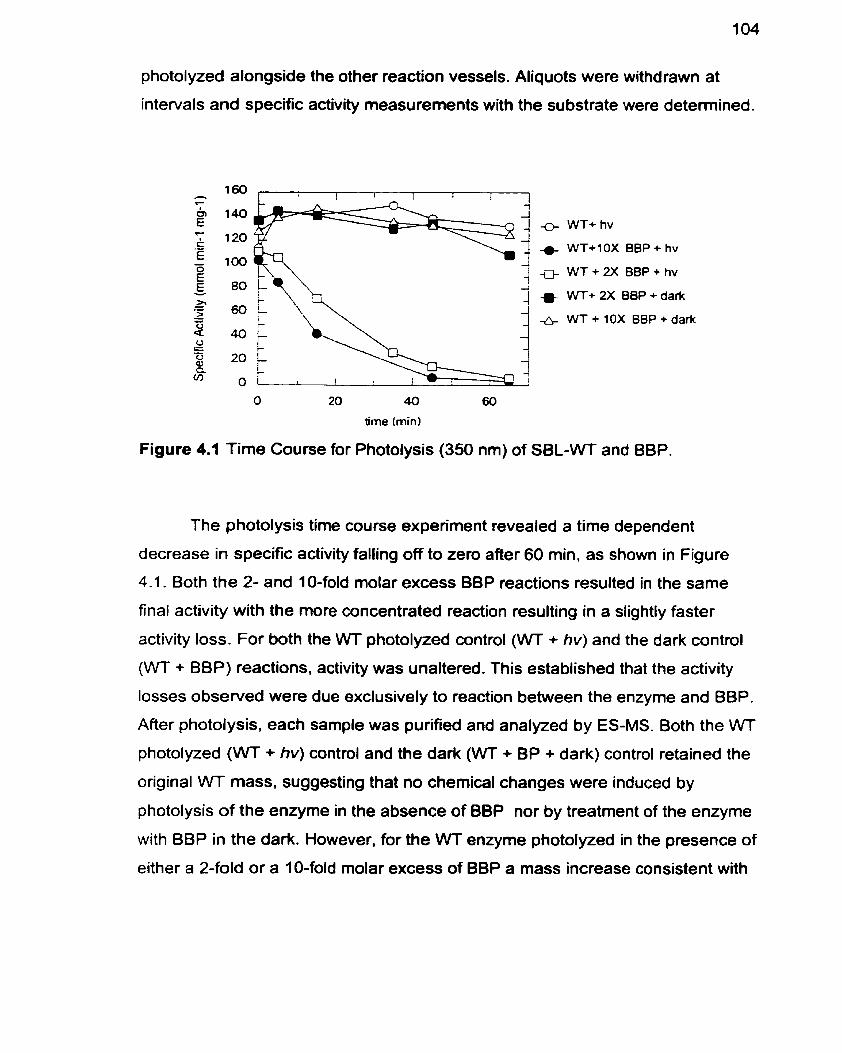
photolyzed alongside the other reaction vessels. Aliquots were withdrawn at
intervals and specific activity measurements with the substrate were detemined.
WT+ hv
WT+IOX BBP + hv
W T + W BBP+hv
WT+ W B8P + dark
WT + 10X BBP + dark
time (min1
Figure 4.1 Time Course for Photolysis (350 nm) of SBL-WT and BBP.
The photolysis time course experiment revealed a time dependent
decrease in specific activity falling off to zero after 60 min. as shown in Figure
4.1. Both the 2- and 10-fold molar excess BBP reactions resulted in the same
final activity with the more concentrated reaction resulting in a slightly faster
activity loss. For both the WT photolyzed control (VVT + hv) and the dark control
(WT + BBP) reactions. activity was unaltered. This established that the activity
losses observed were due exclusively to reaction between the enzyme and BBP.
After photolysis, each sample was purified and analyzed by ES-MS. Both the VVT
photolyzed (VVT + hv) control and the dark (WT + BP + dark) control retained the
original WT mass. suggesting that no chernical changes were induced by
photolysis of the enzyme in the absence of BBP nor by treatment of the enzyme
with BBP in the dark. However, for the WT enzyme photolyzed in the presence of
either a 2-fold or a 1 O-fold molar excess of BBP a mass increase consistent with

covalent linkage of BBP and the loss of two moles of water was observed as
summarized in Table 4.2.
Table 4.2 ES-MS Data for BBP Treated WT-SBL
Enzyme Preparation Mass Calcd. Mass Found Explanation
W + h v 26698 26702 native enzyme
W T + 2 X BBP + hv 26888 26888 + BBP -2 H,O
WT + 10X BBP + hv 26888 26891 + BBP -2 H,O
WT + 2X BBP + dark 26698 26702 native enzyme
WT + 10X BBP + dark 26698 26706 native enzyme
Each of the enzyme samples was denatured by treatment with urea and
then degraded by trypsin into 13 fragments designated 1T to 13T. which
correspond to the tryptic fragments numbered sequentially from the amino
terminus of the enzyme. These fragments were then separated by reverse-phase
HPLC which, coupled to mass spectrophotornetric analysis, permits identification
of each of the tryptic fragments on a peptide mapSz6 Cornparison of the peptide
map of the BBP treated and photolyzed enzyme to that of the control was used
to identify the BBP modified peptide fragment. It revealed a modification on
tryptic fragment 6T, which is comprised of amino acids 93 to 143 in the linear
sequence. Modification resulted in the disappearance of fragment 6T and yielded
two new peaks. These were a minor peak designated as 6T' and a major peak
designated as 6T". Both of these are more hydrophobie than 6T itself, as
evidenced by their longer retention times by reverse-phase HPLC, and showed
rnass increases over 6T of 197 and 189 Da for 6l' and 6T" respectively (Figure
4.2).
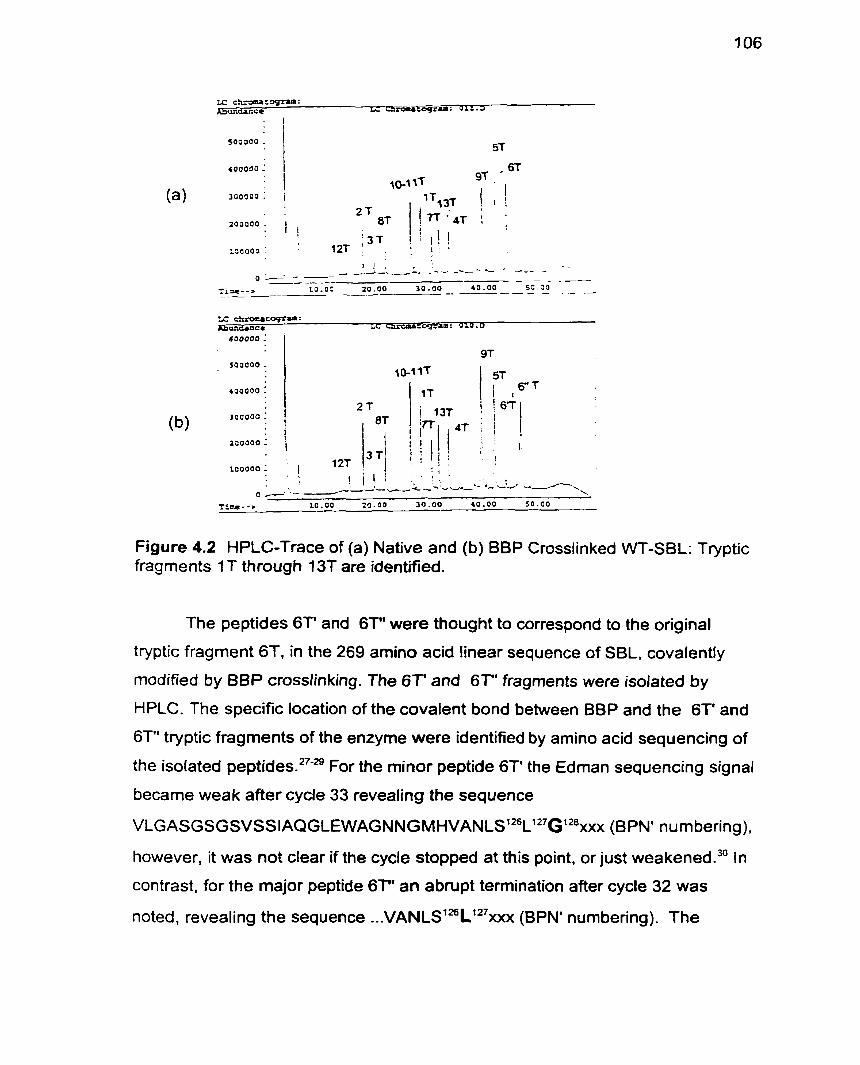
I L I . . . i . c
yaaooo : . 1 : 4T . , ,
200000 1 4 I
1 I I 1
1 2T ioaooo ! . .
TL---) 1 0 . 0 0 2 0 . 0 0 3 0 . 0 0 4 0 . 0 0 5 0 . 0 0
Figure 4.2 HPLC-Trace of (a) Native and (b) BBP Crosslinked WT-SBL: Tryptic fragments 1 T through 13f are identified.
The peptides 6T' and 6T" were thought to correspond to the original
tryptic fragment 6T. in the 269 amino acid linear sequence of SBL. covalently
rnodified by BBP crosslinking. The 6 T and 6T" fragments were isolated by
HPLC. The specific location of the covalent bond between BBP and the 6T' and
6T" tryptic fragments of the enzyme were identified by amino acid sequencing of
the isolated peptide^.^^'^ For the minor peptide 6T' the Edman sequencing signal
became weak after cycle 33 revealing the sequence
VLGASGSGÇVSS~AQGLEWAGNNGMHVANLS'~~L'~~~'~~XXX (BPN' num bering).
however, it was not clear if the cycle stopped at this point. or just ~eakened.~' In
contrast, for the major peptide 6T" an abrupt termination after cycle 32 was
noted, revealing the sequence ...vANLS'~~L"~XXX (BPN' numbering). The

sequence information obtained for the major peptide 6T" identifies a modification
at residue Gly127 (BPN' numbering). Glyi27 is located on the back wall o f the S,
pocket. A proposed crosslinking mechanism which accounts for this modification
and for the loss of two water molecules, and which is consistent with the
observed mass increase is illustrated in Figure 4.3. The modification site of the
minor peptide fragment 6T' is not conclusive but may be at residue Ser128.
/ ?+'--Y+ + * p
OH
SER 221 SER 2 2 3
recombination
Figure 4.3 Proposed Mechanism of BBP Crosslinking: The Photoactivated Crosslinking of BBP to Gly127 and Dehydration. The last dehydration step forming a boronate ester between BBP and the enzyme may precede crosslinking.
The above results dernonstrate the validity of the approach for probing the
active site of WT-SBL. Accordingly, the same protocol was applied to the CMMs
S166C-S-a to -d, using a 1 O-fold molar excess of BBP, in order to probe BBP
binding changes. However. despite the 12 hour photolysis time. specific activity
was not decreased to zero for any of the CMM samples (Figure 4.4).
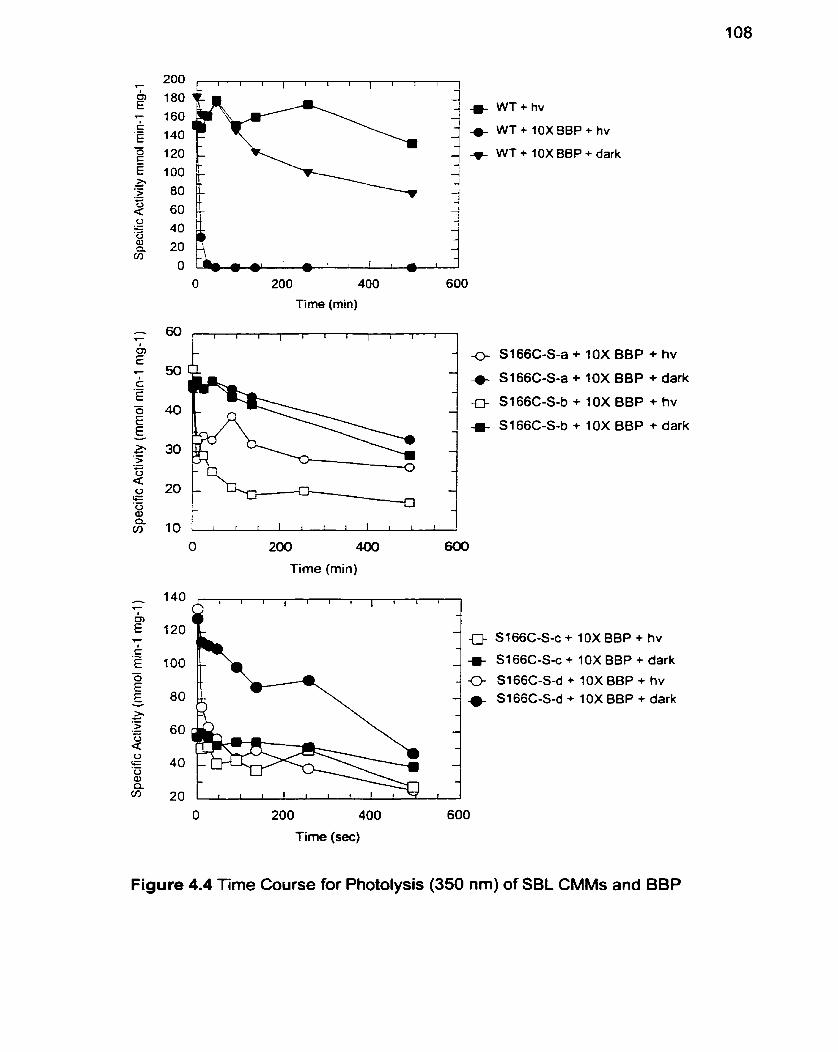
WT+hv
+ W T + lOXBBP+ hv
WT + 10X BBP + dark
O 200 400 600
Time (min)
t i i r l i i i l i i l l
O 200 400 600
Time (min)
+ 10X BBP + hv
+ 1OX BBP + dark
+ 10XBBP + h v
+ 10X BBP + dark
0 S166C-S-c + 10X B8P + hv
S i 66C-S-c + 10X BBP + dark
<> S166C-S-d + 10X BBP + hv
4 S166C-S-d + 10X BBP + dark
O 200 400 600
Time (sec)
Figure 4.4 Time Course for Photolysis (350 nm) of SBL CMMs and BBP

Nonetheless each of the photolyzed CMMs and the dark controls were
purified and analyzed by ES-MS. For each of S166C-S-a, -c, -d, no modification
is indicated by ES-MS (Table 4.3). A mass increase of 220 Da is observed for
SI 66C-S-CH,-c-C,Hl, (-b) 1 OX BBP + hv, which is consistent (t 6 Da) with the
formation of a covalent bond between S166C-S-b and B8P (calculated mass of
BBP 226 Da).
For the S I 66C-S-c-C,Hll CMM. both the dark control (SI 66C-S-b 10X
BBP + dark) and photolyzed sample (S166C-S-b 1OX BBP + hv), were
subjected to tryptic digestion and peptide mapping as described above for the
WT." For the photolyzed S166C-S-b (1 0X BBP + hv) sample a reduction in
intensity of the tryptic fragment 6 1 (50%) was obsewed which was accompanied
by two new peaks, a minor peak designated as 6T' and a major peak
designated as 6T". Both 6T' and 6T" are more hydrophobic than 6T itself and
showed mass increases over 6T of 197 and 188 Da respectively. The tryptic
fragments 6T' and 6T" were isolated and subjected to amino acid seq~encing.~~,
28-29 The sequences for 6T' and 67' were identical. The observed
. . . V A N L S ~ ~ L ' ~ ~ X . X X (BPN' numbering) sequence is identical to that derived from
the 6T" tryptic fragment of the WT-SBL + BEP photolyzed sample. Consistent
with the observed mass increase of 188 Da (BBP-2H20:190 Da Calcd.) for the
tryptic fragment. this implicats the formation of a crosslink between the S, pocket
Gly127 residue of S I 66C-S-b and BBP with concomitant loss of two water
molecules. The identity of the minor tryptic fragment 6T' is not apparent. Both
the WT and Sl66C-S-CH,-c-C,H,, enzymes show a crosslink to BBP at residue
Gly127 despite having very different active site environments. This suggests that
BBP forrns a boronate ester adduct with the enzyme. which although reversible
is sufficiently long-lived that it acts as a tether restricting the spatial span of the
BBP reagent in the enzyme active site. Such adducts between boronic acid
inhibitors and the Se1221 û y hydroxyl of serine proteases have been observed
previously by X-ray cry~tallography'~ and by NMRSJ2

Table 4.3 ES-MS Data for BBP Treated SBL CMMs
Enzyme Preparation Mass Calc. Mass Found Explanation
S166C-S-a 1 OX BBP + dark 26836 26847 native enzyme
S166C-S-a 10X BBP + hv 27062 26839 native enzyme
S 166C-S-b 10X BBP + dark 26842 26844 native enzyme
S i 66C-S-b 10X BBP + hv 27068 27062'a' + BBP
SI 66C-S-c 10X BBP + dark 26853 26868 native enzyme
S 1 66C-S-c I OX BBP + hv 27079 26867 native enzyme
S166C-S-d 10X BBP + darû 26790 2681 8 native enzyme
S I 66C-S-d 10X BBP + hv 2701 6 268 1 8 native enzyme
(a) Poorly resoived spectrum.
Since no crosslinks were observed for S 1 66C-S-a. c,-d after photolysis
for 12 h in the presence of 10-fold molar excess of BBP, each of these CMMs
were photolyzed for up to 72 hours in the presence of an 80-fold molar excess of
BBP. the solubility limit. However. although activity decreases were effected
under these conditions, purification of the enzymes followed by their ES-MS
analysis did not provide evidence of crosslinking. Therefore. S166C-S-a, -c and - d CMMs preclude proper orientation or alignrnent of BBP in the S, pocket for
crosslinking with the enzyme. The reduced binding of BBP to S166C-S-a to -dl
as manifested by higher K,s, and the lack of an observed crosslink between BBP
and Sl66C-S-a, -c and -d indicate that modification of residue Cl66 makes the
S, pocket less conducive to binding large P, groups.

Experimental
Preparation of p-Boronic acid benzophenone (7): A stirred solution of 4-
bromobenzophenone (3.12 g, 12.0 mmol). ethylene glycol (4.7 g. 75 mmol), and
p-toluenesulfonic acid (0.2 g. 1.2 mmol) in benzene (30 mL) was heated under
reflux for 48 h and the resulting water was removed by azeotropic distillation in a
Dean-Stark trap.23-24 The reaction was allowed to cool to 22 OC and then aqueous
1 M NaOH (40 mL) was added and the mixture was extracted with Et,O (3 X 30
mL). The organic phases were combined, drieâ (MgSO,), filtered, and then
evaporated in vacuo yielding 4-bromobenzophenone ethylene glycol (2) as a
white solid which was recrystallized from EtOH (3.7 g. 12. mmol, quantitative
y i e ~ d ) . ~ ~ 'H NMR (200 MHz. CDCI,) 6 4.03 (4H. S. CH,). 6 7.27-7.50 (9H. m. Ar-H)
PPm-
To a stirred solution of 4-bromo benzophenone ethylene glycol (3). (1 .O g,
3.28 mmol) dissolved in dry THF (10 mL) under N, at -78 O C was added
dropwise via syringe n-butyl lithium(l.7 mL of a 2.5 M solution in hexane. 4.3
rnm01).~~This mixture was stirred for 1 h then transferred by cannula to a
solution of trimethylborate (0.51 g. 4.9 mmol) in dry THF (10 mL) at -78 0C.24.'7
This mixture was stirred at -78 O C for 1 h and then allowed to wann to room
temperature overnight. The reaction mixture was slowly added to aqueous 10%
H,SO, (20 mL) at O O C and stirred for 30 min." The reaction mixture was then
extracted with Et20 (3 x 30 mL). The combined organic phases were dried
(MgSO,). filtered and then evaporated in vacuo to yield the boronic acid as a
yellow oil which was added directly to a flask containing a solution of ethylene
glycol (0.2 g, 3.3 mmol) in diethyl ether (25 mL) and stirred vigorously for 1 h at
20 0C.17To this reaction mixture were added hexanes (50 mL) and water (1 mL)
and the aqueous layer was separated. The organic layer was dried (MgSO,),
filtered, and then evaporated in vacuo yielding the diglycal (4) as a white solid
which was recrystallized from benzene (0.87 g. 2.9 mmol, 88% yield). 'H NMR

(200 MHz, CDCI,) G 4.06 (4H. s, CH,), 4.36 (4H, s, CH,). 7.27-7.50 (9H, m Ar-H)
PPm-
The diglycal 4. (270 mg, 0.91 mmol) was then dissolved in a stirred
solution of 6 M HCI (1 5 mL) and acetone (5 mL) and heated to 60 OC for 2 h with
stirring and then stirred for a further 48 h at room temperat~re.~~ The reaction
mixture was extracted into diethyl ether (2 x 20 mL). The combined organic
phases were then washed with brine (20 mL), poured into H,O (5 mL) and
concentrated in vacuo, with the benzophenone boronic acid precipitating as
white crystals (180 mg, 0.8 rnrnol, 87% yield). 'H NMR (200 MHz, DMSO-de) 6
8.35 (2H. s, OH), 6 7.58-7.98 (9H. m, Ar-H) ppm. 13C NMR (400 MHz. DMSO-
d,) 6 196.2. 138.2, 137.1, 134.1, 132.8. 129.7, 128.6. 128.5 ppm. IR (KBr) 3500-
3200 (B(OH),), 2924 (aromatic CH) 1654 (carbonyl), 131 8, 1284, 1 1 13, 875. 704
cm". mp 103 OC. HRMS for trimeric anhydride C,H,,B03 Calcd: 624.2086.
Found: 624.2095.
Site-Specific Chernical Modification To 25 mg of the S166C mutant. purified
as previously de~cribed'~.~' and stored flash frozen in CHES buffer (2.5 mL; 70
mM CHES, 5 mM MES. 2 mM CaCI,, pH 9.5). at 20 OC was added in turn one of
the methanethiosulfonate reagents (2a-d) (100 pL of a 0.2 M solution) in a PEG
(10,000) coated polypropylene test tube, and the mixture agitated in an end-
over-end rotator. Blank reactions containing 100 pL of solvent instead of the
reagent solution were run in parallel. Each of the modification reactions was
monitored spectrophotometncally (E,,, = 8800 M-' on a Perkin Elmer
Lambda 2 spectrophotometer, by specific activity measurements. After the
reaction was quenched by dilution with MES buffer (5 mM MES. 2 mM CaCI,. pH
6.5) at O O C , the specific activity of the CMM (10 PL) was determined in buffer
(0.1 M TRIS pH 8.6, 0.005 % Tween 80 and 1% DMSO. with the suc-AAPF-pNA
substrate (1 mglmL, purchasad from Bachem Bioscience Inc.) at 25 O C . The
reaction was terrninated when the addition of a further 100 pL of
methanethiosulfonate solution effected no further change in specific activity,
generally within 30 min. The reaction solution was purified on a disposable

desalting column (Pharrnacia Biotech PD-1 O. Sephadex G-25 M) pre-equilibrated
with MES buffer (5 mM MES, 2 mM CaCI,, pH 6.5) then dialyzed against 20 mM
MES, 1 mM CaCI,. pH 5.8 (3 x 1 L) at 4 OC and aliquoted into 0.5 - 1.5 mL
volumes, flash frozen in liquid nitrogen and then stored at -20 OC. Modified
enzymes were analyzed by nondenaturing gradient (8-25%) gels at pH 4.2, run
towards the cathode on the Pharrnacia Phast-SystemVTM and appeared as one
single band.
Electrospray Mass Spectrometry: The CMMs were purified for ES-MS
analysis, by FPLC (BioRad, Biologic System) on a Source 15 RPC matrix (1 7-
0727-20 from Pharmacia) with 5% acetonitrile, 0.01 % TFA as the running buffer
and eluted with 80% acetonitrile, 0.01 % TFA in a one step gradient. Electrospray
mass spectra were recorded on a PE SClEX API III Biomolecular Mass
Analyzer. Mass: WT: Calcd: 26698. Found: 26694. S166C-S-a: Calcd: 26836.
Found: 26832. S166C-S-b: Calcd: 26842. Found: 26844. Sl66C-S-c: Calcd:
26853. Found: 26851. Sl66C-S-d: Calcd: 26714. Found: 26708
Regeneration of Unmodified Enzyme by Treafment with P-Mercaptoethanol:
To a solution of CMM (2.0 mg) in 250 pL of CHES-buffer (70 mM CHES. 5 mM
MES, 2 mM CaCI,, pH 9.5) was added 10 pL of a solution of P-mercaptoethanol
(1 M in 95% EtOH ). The reaction was monitored by specific activity
measurements and in al1 cases the activity of the S166C cysteine parent was
restored .
Free Thiol Titration: The residual free thiol content of the S I 66C CMMs, was
determined spectrophotometrically by titration with Ellman's reagent (E,,, = 13600
M-' cm-')= in phosphate buffer (0.25 M. pH 8.0).
Active Site Titration: The active enzyme concentration was determined as
previously describedY by monitoring fluoride release upon enzyme reaction with
a-toluenesulfonyl fluoride (Aldrich Chemical Co. Inc.) as measured by a fluoride
ion sensitive electrode (Orion Research 96-09). The active enzyme
concentration determined in this way was used to calculate kinetic parameters
for each CMM.

Photolysis: Enzyme solutions for photolysis were prepared in 3.5 mL quartz
cuvettes and contained 3 mL of enzyme dissolved (2 mglmL) in storage buffer
(20 mM MES, 1 mM CaCI,, pH 5.8). To each cuvette was added 10 pL of p-
boronic acid benzophenone (for 10X samples, 0.02 M BBP in DMSO). For the
blank reactions f O pL of DMSO was added instead of BBP. The solutions were
photolyzed at 4 O C in a Rayonet photoreactor equipped with 350 nm lamps. The
dark controls were treated in the same way except the cuvettes were wrapped in
aluminum-foil. At the time intewals indicated, 10 pl aliquots were withdrawn,
diluted into 190 pL of storage buffer and specific activity measurernents made.
After photolysis, the enzymes were purified on a disposable desalting column
(Pharmacia Biotech PD-10, Sephadex G-25 M) pre-equilibrated with MES buffer
(3.5 mL, 5 mM MES, 2 mM CaCI,, pH 6.5). The enzyme was eluted with MES
buffer, dialyzed against storage buffer (20 mM MES, 1 mM CaCI,, pH 5.8. (3 x 1
L) at 4 OC aliquoted, flash frozen and stored at -20 O C .
Specific Activify Measurements for Time Course Monitoring: For specific
activity determinations, absorbance versus time measurernents were made on a
Perkin Elmer lambda 2 spectrophotometer. The spectrophotometer was zeroed
against a 1 mL disposable cuvette containing 980 pl Tris buffer (0.1 Ml 0.005%
Tween80, pH 8.6) and 10 FI of the suc-AAPF-pNA substrate (1 00.0 mg/mL in
DMSO, 160 mM). The enzyme aliquot (10 PL) was added and. after a 10 second
delay, the absorbance reading was started and sampled with an interval of 0.2 s
during 60 s at = 410 nm. The slope (in A s-') was determined by linear curve fit
of the data below 5% conversion of the substrate and the rate of product
formation, dP/dt deterrnined from Beer's Law (A=&, where E,,, = 8800 M-' cm-',
t = 1 cm). The enzyme activity was then calculated: specific activity (pmol min-'
mg-') = {(dPIdt M s-')(60 s min-')(106 pmol mol~')(volume in cuvette L)}/{[E] mg
mL-').

Tryptic Digests and HPLC-MS Analysis.?
From a stock solution of the SBL samples in storage buffer (20 mM MES. 1 mM
CaCI,. pH 5.8) was withdrawn 0.1 mg ( ~ 1 0 0 PL) of enzyme which was mixed
with water (2200 pl) and 1 N HCI (30 PL) to give a final concentration of 0.03
mglmL protein in 0.09 M HCI. This mixture was chilled for 10 min on ice and any
debris was removed by centrifugation for 2 min at 14,000 rpm. The supernatant
was transferred to a clean eppendorf tube to which was added 35 pL TCA (50%
W/V) to give a final concentration of 5% (w/v) TCA. This mixture was chilled for 15
min on ice and then the protein was collected by centrifugation (2 min at 14.000
rpm). The pellet was washed once with 1 mL of 90% (vlv) acetone (-20 OC) and
briefly dried in vacuo. The pellet was resuspended in 12 pL of 0.4 M ammonium
bicarbonate. containing 8 M urea and then incubated at 37 OC for 4 min. To this
mixture was added 48 PL of 1 % (wlv in H,O) 1-O-octyl-P-D-glucopyranoside and
1 PL of trypsin (Sigma Chemical Co. TPCK-treated. 2.5 rnglmL in 1 mM HCI) and
then the mixture was at 37 OC for 30 min after which time the reaction was
terminated by adding 5 PL of TFA (1 0 % vlv).
An aliquot of the tryptic digest was then analyzed on an Hewlett Packard
HPLC Model 1090. which is equipped with a Vydac C-18 Reverse Phase
Column Model 218T952 (2.1 x 250 mm). with a gradient of water + 0.1 % TFA
and CH,CN + 0.1% TFA as the mobile phase. coupled to a ES1 lonization Mass
Spectrophometer Model9889B.
Peptide Sequencing: The isolated tryptic fragments were sequenced on the
Applied Biosystems Model473A Protein Sequencer.
Kinetic Measurements: Michaelis-Menten constants were determined at 25 OC
by curve fitting (GraFit@ 3.03) of the initial rate data determined at eight
concentrations (0.1 25 mM4.O mM) of the suc-AAPF-pNA substrate (E,,, = 8800
M-' cm-')33 in 0.1 M phosphate buffer containing 0.5 M NaCI. 0.005% Tween 80.
1 % DMSO, pH 7.5.

Determination of p-Borwric Acid Benzophenone Inhibition Constants: K, s
of BBP were determined in dupticate by the method of waley.*' The progress
curve without inhibitor was determined from an assay mixture containing 980 pL
of buffer (0.1 M phosphate buffer containing 0.5 M NaCI, 0.005% Tween 80, pH
7.5). 5 pL DMSO and 5 pL of suc-AAPF-pNA substrate (50 mM in DMSO). This
mixture was incubated in a water jacketed cell for 5 min. ai 25 OC and the
absorbance reading was set to zero prior to initiating the reaction by addition of
10 PL of enzyme solution (3.2 x I O d to 1.1 x104 M in pH 5.8, 20 mM MES, 1 mM
CaCI,). The final volume of the assay mixture was 1 mL. The progress curve with
inhibitor was determined similarly but with 5 pL of the BBP inhibitor solution
(7.24 x 10J to 1.4 x IO- ' M. in DMSO) being added instead of 5 pL DMSO. to a
final volume of I mL. Absorbance versus tirne measurements were recorded on
a Perkin Elmer lambda 2 spectrophotorneter and points for calculation were
taken at 1 5, 18, 21, 24, 27, 30, 33 and 36% substrate con~ersion.~~

Retèrences
DeSantis G, Jones JB. Probing the Altered Specificity and Properties of Mutant Subtilisin Chemically Modified at Position S I 66C and SI 56C in the SI Pocket. Bioorg. Med. Chem. in press.
Brunner J. New Photolabeling and Crosslinking Methods. Ann. Rev. Biochem. 1993; 62:483-514,
Dorman G, Prestwich GD. Benzophenone Photophores in Biochemistry. Biochemistry 1994; 33(19):5661-73.
Prestwich GD. Dorman G. Elliott JT. Marecak DM, Chaudhary A. Benzophenone Photoprobes for Phosphoinositides. Peptides and Dnigs. Photochem. Photobiol. 1997; 65(2):222-34.
Kotzyba-Hibert F. Kapfer 1, Goeldner M. Recent Trends in PhotoafTinity Labeling. Angew. Chem. Int. Ed. Engl. 1995; 34:1296-1312.
MacMillan AM. Query CC, Allerson CR. Chen S, Verdine GL. Sharp PA. Dynamic Association of Proteins with the pre-mRNA Branched Region. Genes & Development 1994; 8:3008-20.
Abe 1. Zheng YF. Prestwich GD. Photoaffinity Labeling of Oxidosqualene Cyclase and Squalene Cyclase by a Benzophenone-Containing Inhibitor. Biochemistry 1998; 37(17):5779-5784.
Branchini BR, Magyar RA. Marcantonio KM. Newberry SJG. Hinz LK. Murtiashaw MH. Identification of a Firefly Luciferase Active Site Peptide Using a Benzophenone-Based Photooxidation Reagent. J. Biol. Chem. 1997; 272(31): 19359-64.
Wery JP. Schevitz RW. New Trends in Macromolecular X-ray Crystallography. Curr. Opin. Chem. Biol. 1997; 1 (1 ):365-9.
Kay L.E. Protein Dynamics from NMR. Nat. Struct. Biol. 1998; 5:513-7.
Cooke RM. Protein NMR Extends into New Fields of Structural Biology. Curr. Opin. Chem. Biol. 1997; 1 (3):359-64.
Adam W. Oestrich RS. Two-Photon Chemistry in the Laser Jet: Photochemistry of the Diphenylhydroxymethyl Radical. Chem. Ber. 1992;

Min KL, Steghens JP, Henry R, Doutheau A. Collombel C. Identification of the Creatine Binding Domain of Creatine Kinase by Photoaffinity Labeling. Biochim. Biophys. Acta 1998; f 387:80-8.
Stabile MR, Lai WG, DeSantis G et al. Probing the Specificity of the S, Binding Site of M222 Mutants of Subtilisin B. Lentus with Boronic Acid In hibitors. Bioorg. Med. Chem. Lett. 1996; 6(21):2501-6.
Wolfenden R. Analog Approaches to the Structure of the Transition State in Enzyme Reactions. Acc. Chem. Res. 1972; 5:10-8.
Lienhard GE, Koehler KA. 2-Phenylboronic Acid, a Possible Transition State Analog for Chymotrypsin. Biochemistry 1971 ; 1 O:2570-5.
Seufer-Wasserthal P, Martichonok V, Keller TH, Chin B, Martin R, Jones JB. Probing the Specificity of the S, Binding Site of Subtilisin Carlsberg with Boronic Acids. Bioorg. Med. Chem. 1994; 2(1):3548.
Martichonok V, Jones JB. Probing the Specificity of the Serine Proteases Subtilisin Carlsberg and a-Chymotrypsin with Enantiomeric 1 -Acetamido Boronic Acids. An Unexpected Reversal of the Normal L-Stereoselectivity Preference. J. Am. Chem. Soc. 1996; 118:950-8.
Matthews DA, Alden RA, Birktoft JJ, Freer ST, Kraut J. X-ray Crystallographic Study of Boronic Acid Adducts with Subtilisin BPN' (Novo). A model for the Transition State. J. Biol. Chem. 1975; 250(8):7120-6.
London RE, Gabel SA. Fluorine-19 NMR Studies of Fluorobenzeneboronic Acids. 2. Kinetic Characterization of the Interaction with Subtilisin Carlsberg and Model Ligands. J. Am. Chem. Soc. 1994; 1 162570-5.
DeSantis G, Berglund P, Stabile MR, Gold M, Jones JB. Site-Directed Mutagenesis Combined with Chemical Modification as a Strategy for Altering the Specificity of the S, and S,' Pockets of Subtilisin Bacillus lentus. Biochemistry 1998; 37:5968-73.
Ellman GL, Courtney KD, Andres Jr. V, Featherstone RM. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961 ; 7:88-95.

Masuhara H, Maeda Y, Nakajo H et al. Exiplex Emrnissions of Intra- and Intermolecular Benzophenone and N.KDimethylaniline Systems. J. Am. Chem.Soc. 1981 ; 1 O3:63MO.
Sigman ME. Closs GL. Free Energy and Structure Dependence of lntramolecular Triplet Energy Transfer In Organic Model Compounds. J. Phys. Chem. 1991 ; 95:5OlZ-i.
Waley SG. A Quick Method for the Detemination of Inhibition Constants. Biochem. J. 1982; 205631-3.
Christianson T. Paech C. Peptide Mapping of Subtilisins as a Practical Tool for Locating Protein Sequence Errors dunng Extensive Protein Engineering Projects. Anal Biochem 1994; 223: 1 1 9-29.
Edman P. Sequence Determination. Mol. Biol. Biochem. Biophys. 1970; 8:2 1 1-55.
Tsugita A. Developments in Protein Microsequencing. Adv. Biophys. 1987; 23:8l-l l3.
Walker JM. The Dansyl-Edman Method for Peptide Sequencing. Methods Mol. Biot. 1994; 32:329-34.
Amino acid sequencing was done by Sue Middlebrook at Genencor International Inc.
Tryptic Digest and HPLC ES-MS Analysis for this sample was performed by Dr. Christian Paech at Genencor l nternational Inc.
Tsilikounas E, Kettner CA, Bachovchin WW. Identification of Serine and Histidine Adducts in Complexes of Trypsin and Trypsinogen with Peptide and Non peptide Boronic Acid ln hibitors by ' H NMR Spectroscopy . Biochemistry 1 992; 3 1 : 1 283946.
Bonneau PR, Graycar TP, Estell DA, Jones JB. Alteration of the Specificity of Subtilisin BPN' by Site-Directed Mutagenesis in Its S, and S,' Binding Sites. J. Am. Chem. Soc. 1991 ; 113:1026-30.
Hsia CY, Ganshaw G, Paech C, Murray CJ. Active-Site Titration of Serine Proteases Using a Fluoride Ion Selective Electrode and Sulfonyl Fluoride Inhibitors. Anal. Biochern. 1 996; 242:221-7.

35. Lee T, Jones JB. Probing the Abilities of Syntheticatly Useful Serine Proteases to Discriminate Between the Configurations of Remote Stereocenters using Chiral Aldehyde Inhibitors. J. Am. Chem. Soc. 1996; 1 18:502-8.

Chapter 5
Reproduced in part with permission from J. Am. Chem. Soc. submitted.
Unpublished work copyright 1999. American Chernical Society

1 22
Towards Tailoring The Specificity of the S, Pocket of Subtilisin B. lentus: Chernical Modification of Mutant Enzymes as a
Strategy for Removing Specificity Limitations
For both protein ~hernistry '~ and organic synthesis applications."' it is
desirable to have available a diverse toolbox of inexpensive proteases with high
selectivity and diverse substrate preferences. Since WTenzymes will never
accept al1 substrate structures, it is attractive to contemplate the tailoring of a
readily available protease in order to expand its substrate specificity with the
ultimate goal of creating any desired specificity at will.
WT-SBL has a marked preference for substrates with large hydrophobic
uncharged Pl residues. In this study, we explore tailoring the S1 pocket of
subtilisin Bacillus lentus (SBL) to also accept small hydrophobic and positively or
negatively charged P, residues using the combined site-directed mutagenesis
chemical modification approach (CMM), as illustrated in Scheme 5.1 "12 In order
to achieve this broadened Pl tolerance a simplistic strategy of steric and
electrostatic complementarity was applied.13 Employing the crystal structure of
SBL as our guide,14 the Ser166 residue, which is located at the bottorn of the S,
pocket and whose side chain points inward toward the pocket, was chosen for
mutagenesis to cysteine and subsequent chemical modification. Firstly, to
expand SBL's specificity toward smail uncharged Pl residues, such as the small
P, Ala residue of the suc-AAPA-pNA substrate, large groups were introduced at
position 166 in S1 such as benzyl (-c) and cyclohexyl (-0 with a view to reducing
the volume of S1 and inducing a better fit of small P, groups, thereby conferring
elastase-like15 substrate specificity on SBL. Then, to expand SBL's specificity
toward positively charged Pl residues, such as the P, Arg residue of the suc-
AAPR-pNA substrate, we introduced negatively charged groups at position
S I 66C in S1 such as the ethylsulfonato (-b) moiety to elicit complementary
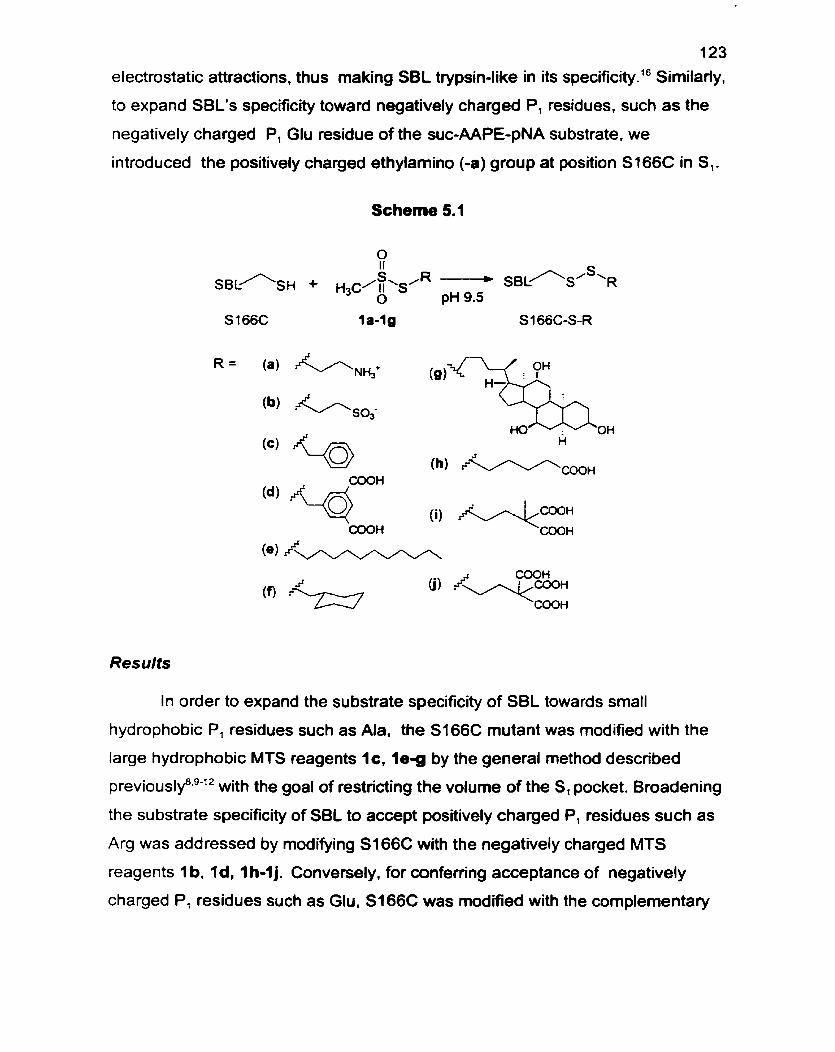
123
electrostatic attractions. thus making SBL trypsin-like in its specificity? Similarly.
to expand SBL's specificity toward negatively charged P, residues. such as the
negatively charged P, Glu residue of the suc-AAPE-pNA substrate. we
introduced the positively charged ethylarnino (-a) group at position St66C in S,.
Scheme 5.1
S 166C la-lg S166C-S-R
(b) &/, so;
Results
In order to expand the substrate specificity of SBL towards small
hydrophobic P, residues such as Ala, the S166C mutant was modified with the
large hydrophobic MTS reagents I c , l e g by the general method descnbed
p r e v i o ~ s l y ~ ~ ~ ~ ' ~ with the goal of restricting the volume of the S, pocket. Broadening
the substrate specificity of SBL to accept positively charged P, residues such as
Arg was addressed by modifying S166C with the negatively charged MTS
reagents 1 b. Id, 1 h-1 j. Convenely, for conferring acceptance of negatively
charged P, residues such as Glu. S166C was modified with the complernentary

1 24 positively charged MTS reagent la. The preparations of the requisite MTS
reagents IC.~ le - f and 1i-j l7 have been reported previously and the
steroidyl MTS reagent 1g was prepared from cholic acid by the same
methodology. la
Each of the CMMs obtained was characterized in order to establish its
purity and integrity. Titration of the CMMs with Ellman's reagent showed a
residual thiol content of less than 2% in al1 cases, demonstrating that the MTS
reactions were virtually quantitative. Mass analyses of the CMMs by
electrospray mass spectrometry were consistent (* 6 Da) with the calculated
mass. The purities of the modified enzymes was assessed by native-PAGE and
in ail cases only one band was visible. Furthemore, relative to VVT, the
negatively charged CMMs S i 66C-S-b.-d. and -i to -j displayed retarded mobility
in the direction of the cathode, while the positively charged S166C-S-a CMM
displayed greater mobility. as expected. That modifi-tion of cysteine is wholly
responsible for altered activity was established by the absence of reaction of
VVT-SBL with the MTS reagents. Also. the modifications are fully reversible by
treatment of each of the CMMs with P-rnercaptoethanol. further verifying that
chemical modification at cysteine was sole1 y responsi ble for the obsewed
changes in activity. The total amount of active enzyme was determined by
titration with phenylmethylsulfonyl fluoride.lg

Figure 5.1 k,/KM Screen of S166C CMMs with suc-AAPF/A/WE-pNA Su bstrates.
Initially, three CMMs Sl66C-S-a, -b and c , with a positive, a negative,
and with a large hydrophobic side chain respectively, were subjected to a kJKM
screen with each of the test substrates. suc-AAP-FINRIE-pNA in order to identify
any induced complementary electrostatic or improved hydrophobic interactions.
While as expected k,JKMs with the standard suc-AAPF-pNA were lowered, the
k,,lK,s of the CMMs whose SI sites were tailored toward the Ala, Arg or Glu P,
residues, improved with the appropriate substrate. This is iilustrated in Figure 5.1
in the higher activity of S 166C-S-c with suc-AAPA-pNA, of S 166C-S-b with suc-
AAPR-pNA, and of S166C-S-a with suc-AAPE-pNA compared to WT.
Following the validation of the general design strategy from this initial
screen, a more complete kinetic analysis was undertaken. The substrate
specificity of each of the CMMs was evaluated kinetically with the standard large
hydrophobic P, residue containing substrate, suc-AAPF-pNA. In addition, the
S I 66C CMMs modified with the large hydrophobic MTS reagents 1c. l e g , were
evaluated with the targeted small hydrophobic Pl residue containing substrate,
suc-AAPA-pNA. The S i 66C CMMs modified with the negatively charged MTS
reagents 1 b, Id, 1 h-1 j were evaluated with the positively charged P, residue
containing substrate, suc-AAPR-pNA. The S166C CMMs modified with the
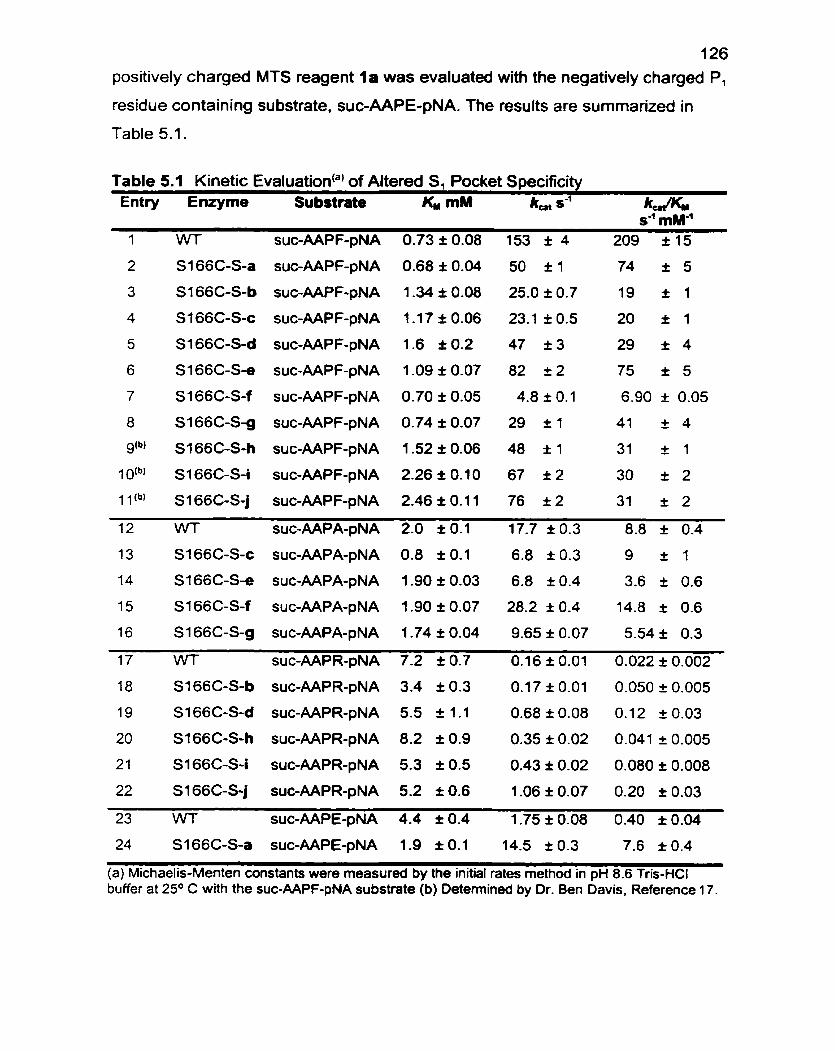
126 positively charged MTS reagent l a was evaluated with the negatively charged P,
residue containing substrate, suc-AAPE-pNA. The results are summarized in
Table 5.1 .
Table 5.1 Kinetic Evaluatioda) of Altered S, Pocket Specificity Entry Enzyme Sri bstrate KM mM kat s1 kd'l(w
S-' mM" 1 WT SUC-AAPF-pNA 0.73 i 0.08 153 i 4 209 1 15
24 SI 66C-S-a suc-AAPE-pNA 1 -9 I 0.1 14.5 î 0.3 7.6 î 0.4
(a) Michaelis-Menten constants were measured by the initial rates method in pH 8.6 Tris-HCI buffer at 2 5 O C with the suc-AAPF-pNA substrate (b) Detemined by Dr. Ben Davis, Reference 17.

127 Discussion
The significant substrate preference of WT-SBL for large hydrophobic P,
residues is apparent from its preference for the Phe Pl residue of the standard
suc-AAPF-pNA substrate by a factor of 9500-fold over the small P, residue of
suc-AAPA-pNA, by a factor of 24-fold compared to the positively charged P,
residue of suc-AAPR-pNA, and by a factor of 522-fold compared to the
negatively charged Pl residue of suc-AAPE-pNA (Table 5.1 : entries 1,12,17 and
23). These substrate preferences are attributed to changes both in binding as
reflected by KM and in turnover number, k,,. In addition, as expected. with suc-
AAPF-pNA, the WT enzyme is the best catalyst, and its conversion to the CMMs
resulted in ka JKM decreases of up to 34-fold (Table 5.1 : entries 2-1 1 ).
To tailor the substrate specificity of SBL toward small hydrophobic P,
residues such as Ala, the simplistic approach of trying to partially fil1 up the S,
binding cleft was followed by preparing the S1 66C-S-CH2C6H5 (-c). S I 66C-S-
CH,(CH,),CH, (-e), S 1 66C-S-CHzc-C6H,, (4). and S i 66C-S-steroidyl (-g) CMMs.
This design strategy attempted to mimic the function of the bulky S,-pocket side
chains of a-lytic p r o t e a ~ e ~ - ~ and of ela~tase.'~ for which both enzymes exhibit
su bstantial preference for the suc-AAPA-pNA substrate over suc-AAPF-pNA.
These CMMs (S I 66C-S-c,-eV-f. g . Table 5.1 :entries 12-1 6) were then evaluated
with the suc-AAPA-pNA substrate. All revealed slightly irnproved binding
compared to WT, with the greatest improvement in KM being 2-fold for the
S1 66C-S-CH,C6H, (-c) CMM. Of the four CMMs. only SI 66C-S-CH2C,Hl, (4)
showed both an improved k,, and an improved kJKM compared to WT. While
this design strategy yielded only one CMM with an increased preference for the
srnall Ala P, residue, al1 of these modifications effectively excluded the larger
Phe Pl residue preferred by W-SBL (Table 5.1 :entries 4.6-8). Overall, the
selectivities, with respect to kcar/KM, for the suc-AAPA-pNA substrate compared to
the suc-AAPF-pNA substrate were improved by 1 1-fold for S I 66C-S-CH,C6H5
(-c), 1 . 1 -fold for S166C-S-CH2(CH,),CH, (-e), B I -fold for S 1 66C-S-CH,-c-C,Hl,

128 (-f), and 3.2-fold for S166C-S-steroidyl (9). compared to W. The extent of
improvement in P, Ala selectivity may be a reflection of the orientation of the R
side chain of the CMMs, with the side chains of Sf66C-S-CH,C,H, (-c), and - CH,-c-C,Ht, (-0, behaving as though directed into the pocket and the side chains
of S 166C-S-CH,(CH,),CH, (-e) and Sl66C-S-steroidyl (-g) outward.
Engineering complementary steric fit in the S, pocket has already proven
to be ~ha l leng ing '~ .~ and previous attempts to alter the steric complementanty of
subtilisins have resulted in modest but inconsistent successes with maximal
improvements of up to 1 O - f ~ l d . ~ ~ ~ Starting with subtilisin YaB, whose specificity
was already elastase-like, the G127A mutation which was designed to fiIl up its
S, pocket and thus increase the selectivity for small hydrophobic P, residues
such as Ala, effected a 10-fold improvement in kJKM with the suc-AAPA-pNA
s~bstrate.'~ Similady, the GA661 mutant of subtilisin BPN' caused a 1 O-fold
improvement in kJKM with the suc-AAPA-pNA sub~trate.*~ However. the G127V
mutant of subtilisin E caused a decrease in k,JKMwith the suc-AAPA-pNA
substrate, compared to WT.2C25
l mproving the su bstrate specificity of SBL toward positively charged P,
residues such as Arg, making it more trypsin-likeIt6 was based on mimicking the
common motif of the high negative charge density of acidic residues inducing a
su bstrate preference for positively charged su b ~ t r a t e s . ' ~ . ~ ~ ~ ~ ~ This was achieved
with the S I 66C-S-CH,CH,SO; (-b), S i 66C-S-CH,(CH,),CH,COO- (-h), S i 66C-
S-CH,C,H,-3.5-(C00-), (-d), S 1 66C-S-CH,CH2C(CH,)(C00-)2 (4) and S 1 66C-S-
CH,CH,C(COO-), (-j) CMMs. Evaluation of each of these CMMs with the suc-
AAPR-pNA substrate revealed improved binding of up to 2-fold compared to WT,
with the exception of S I 66C-S-CH,(CH,),CH,COO- (-h) for which K , was slightly
increased (Table 5.1 : entries 17-22). The general success of this approach is
evident since al1 of the CMMs with a negatively charged R side chain showed an
improved k,, of up to 7-fold and an improved k,JKM of up to 9-fold with suc-
AAPR-pNA compared to WT (Table 5.1: entries 17-22). Furtherrnore, each
additional negative charge introduced at position 166 resulted in an approximate
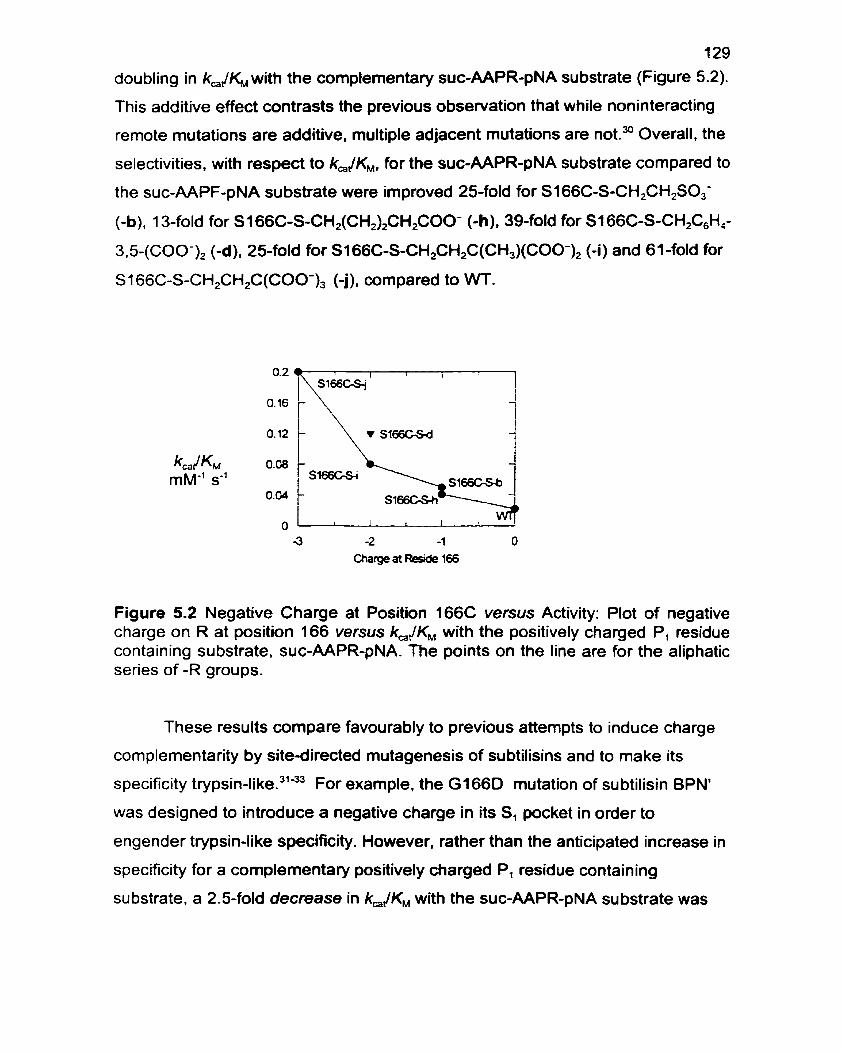
129
doubling in kJKM with the complementary suc-AAPR-pNA substrate (Figure 5.2).
This additive effect contrasts the previous observation that while noninteracting
remote mutations are additive. multiple adjacent mutations are net? Overall, the
selectivities. with respect to k,jKM, for the suc-AAPR-pNA su bstrate compared to
the suc-AAPF-pNA substrate were improved 25-fold for S166C-S-CH2CH2S0,-
(-b). 1 3-fold for S 1 66C-S-CH2(CH2)2CH2C00- (-h ), 39-fold for S 1 66C-S-CH2C,H,-
3.5-(COO-), (-d). 25-fold for S166C-S-CHFH,C(CH3)(C00-)2 (4) and 61 -fold for
S l 66C-S-CH2CH2C(COO-), (-j), compared to W.
Figure 5.2 Negative Charge at Position 166C versus Activity: Plot of negative charge on R at position 166 versus kJKM with the positively charged P, residue containing substrate, suc-AAPR-pNA. The points on the line are for the aliphatic series of -R groups.
These results compare favourably to previous attempts to induce charge
complementarity by site-directed mutagenesis of subtilisins and to make its
specificity trypsin-like."- For example, the G166D mutation of su btilisin BPN'
was designed to introduce a negative charge in its S, pocket in order to
engender trypsin-like specificity. However. rather than the anticipated increase in
specificity for a complementary positively charged Pl residue containing
substrate, a 2.5-fold decrease in kJKM with the suc-AAPR-pNA substrate was

1 30
obse~ed.~, Furthemore. despite the introduction of two negatively charged
amino acids in the S, pocket of subtilisin BPN'. one at position 156 and one at
position 166. only a maximal 2-fold improvement in kJKM with the suc-AAPR-
pNA substrate was obsewed compared to WT?'
Conversely, to tailor the specificity of SBL toward negatively charged P,
residues such as Glu. a design strategy of introducing a complementary positive
charge in the S1 binding cleft by the CMM approach was adopted (S166C-S-a).
This strategy was based on mimicking the specificity detenninants of the senne
proteases pronase- and granzyrne 9.- which exhibit a substrate
preference for negatively charged P, residues, and whose S, pockets are lined
with positively charged residues. In fact, SI 66C-S-CH,CH,NH,' (-a) displayed a
remarkable 19-fold increase in k,JKM. with the suc-AAPE-pNA substrate
compared to VVT. This enhancement is due to a combination of better binding,
evident from the 2-fold lower KM. and 8-fold higher k, (Table 5.1 : Entries 23. 24).
The induction of electrostatic complementarity was most unequivocall y
demonstrated by the 54-fold improvement in suc-AAPE-pNA to suc-AAPF-pNA
su bstrate selectivity, with respect to k,lKMl for S I 66C-S-CH,CH,NH,' (-a)
compared to W.
Moreover. these results compare favourably with previous site-directed
mutagenesis approaches toward tailoring the preference of subtilisin BPN' for
substrates with negatively charged P, r es i d~es .~ ' . ~~ While. k,JKM for the suc-
AAPE-pNA substrate was improved. the reported mutants did not exhibit
selectivity for negatively charged P, residues compared to hydrophobic ~ n e s . " . ~ ~
For example, the G166R and G166K mutants of subtilisin BPN', which although
they caused 23- and 340-fold improvements respectively in kJKM with the suc-
AAPE-pNA substrate. compared to W. displayed even higher k,JKMs with the
suc-AAPNIMIK-pNA substrates, which lack the complementary charged Pl
residuee3' The broad specificity of the G166K mutant has been attributed to the
flexible conformation of the KI66 side hai in.^'
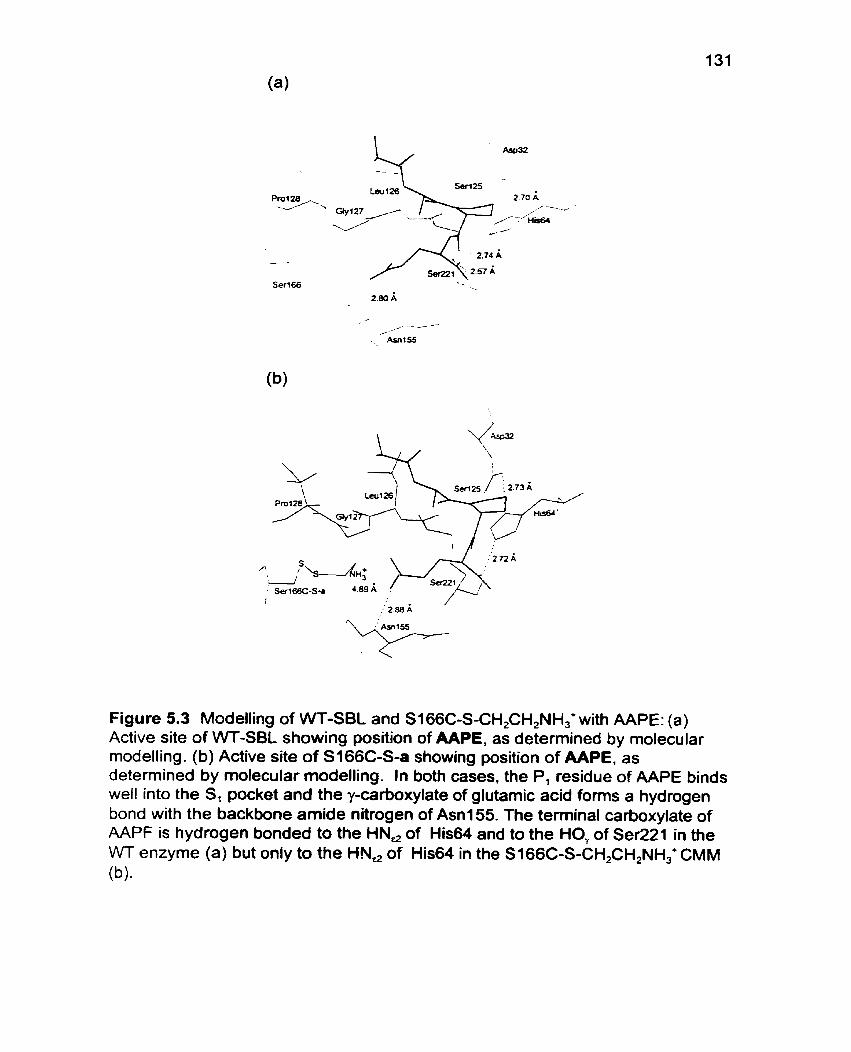
Figure 5.3 Modelling of WT-SBL and S I 66C-S-CH,CH,NH,+ with AAPE: (a) Active site of WT-SBL showing position of AAPE, as determined by molecular modelling. (b) Active site of S I 66C-S-a showing position of AAPE, as determined by molecular modelling. In both cases. the P, residue of AAPE binds wel! into the S, pocket and the y-carboxylate of glutamic acid forms a hydrogen bond with the backbone amide nitrogen of Asnl55. The terminal carboxylate of AAPF is hydrogen bonded to the HN, of His64 and to the HO, of SeR21 in the WT enzyme (a) but only to the HN, of His64 in the SI66C-S-CH,CH,NH,+ CMM (W.

1 32 Since the S 1 66C-S-CH2CH2NH,' (-a) and suc-AAPE-pNA CMM-substrate
pair exhibited the greatest kJKM improvement relative to WT, at 19-fold (Table
5.1 ), more detailed insights into the molecular basis of their interaction was
sought using molecular modelling. Using the modelling strategy reported
previouslylg the product inhibitor, AAPE was bound to WT-SBL and to the
SI 66C-S-CH2CH2NH3'(-a) CMM. The energies of these structures were
minimized. As shown in Figure 5.3, the binding conformations for both the WT
and S 1 66C-S-CH2CH,NH3'stnictures are rernarkably similar. However. the
ammonium moiety of the S 1 66C-S-CH2CH,NH; CMM side chain is oriented
toward the carboxylate of the glutamic acid P, residue. and although it is not
within salt-bridge distance (Figure 5.3b). an additional favourable coulornbic
interaction between the ammonium side chain of S i 66C-S-CH2CH,NH,' and the
carboxylate of the glutamic acid P, residue causes the observed 19-fold
improvement in kJKM for this CMM substrate pair. wmpared to WT.
The effectiveness of the general design strategy is validated, since for
each of the Ala, Arg, and Glu P, residues at least one and up to five of the
designed CMMs exhibit improved k,JKMs compared to WT.

Experimen ta/
Methanethiosulfonate Reagents: Sulfonatoethyl methanethiosulfonate (1 a)
and ethylammonium methanethiosulfonate (1 b) were purchased from Toronto
Research Chernicals (2 Brisbane Rd., Toronto, ON, Canada). Reagents 1 c -1p
and 1 h-1 j 17 were prepared as previously described.
Site-Specific Chernical Modification: To 25 mg of a S166C mutant. purified as
previously describedg-" and stored flash frozen in CHES buffer (2.5 mL; 70 mM
CHES. 5 rnM MES. 2 mM CaCI,, pH 9.5) was added at 20 OC one of the
methanethiosulfonate reagents ( l ag ) (100 pL of a 0.2 M solution). in a PEG
('1 0,000) coated polypropylene test tube, and the mixture agitated in an end-
over-end rotator. Blank reactions containing 100 p l of solvent instead of the
reagent solution were run in parallel. Each of the modification reactions was
monitored spectrophotometncally (E~,,, = 8800 M-' cm-')= on a Perkin Elmer
Lambda 2 spectrophotometer, by specific activity measurements. After the
reaction was quenched by dilution in MES buffer (5 mM MES. 2 mM CaCI,, pH
6.5) at O OC, the specific activity of the CMM (1 0 PL). was detemined in buffer
(0.1 M TRIS pH 8.6.0.005 % Tween 80, and 1 % DMSO) with the suc-AAPF-pNA
substrate (1 mglmL) (purchased from Bachern Bioscience Inc.) at 25 OC. The
reaction was terminated when the addition of a further 100 pL of
methanethiosulfonate solution effected no further change in specific activity,
generally within 30 min. The reaction solution was purified on a disposable
desalting column (Phanacia Biotech PD-1 O. Sephadex G-25 M) pre-equ ilibrated
with MES buffer (5 mM MES, 2 mM CaCI,, pH 6.5) then dialyzed against 20 rnM
MES, 1 mM CaCI,, pH 5.8 (3 x 1 L) at 4 O C aliquoted into 0.5 - 1.5 mL volumes,
flash frozen in liquid nitrogen and then stored at -20 OC. Modified enzymes were
analyzed by nondenaturing gradient (8-25%) gels at pH 4.2. run towards the
cathode on the Pharmacia Phast-System," and appeared as one single band.
Electrospray Mass Spectromefry: Prior to ES-MS analysis. CMMs were
purified by FPLC (BioRad. Biologic System) on a Source 15 RPC matrix (1 7-

1 34
0727-20 from Pharrnacia) with 5% acetonitrile. 0.01 % TFA as the ninning buffer
and eluted with 80% acetonitrile. 0.01% TFA in a one step gradient. ES-MS data
were acquired using a PE SCIEX API III Biomolecular mass spectrometer. Mass:
WT: Calcd: 26698. Found 26694. Sl66C-S-a: Calcd: 26714. Found 26708.
SI 66C-S-b: Calcd: 26853. Found 26851. S166C-S-c: Calcd: 26836. Found
26832. S I 66C-S-d: Calcd: 26924. Found 26928." SI 66C-S-e: Calcd: 26886.
Found 26890. S166C-S-f: Calcd: 26842. Found 26844. S166C-S-g: Calcd:
27 1 28. Found 271 23. S166C-S-h; Calcd: 26846. Found 26846." S 166C-S-i:
Calcd: 26890. Found 26894.17 S166C-S-i: Calcd: 26934. Found 26939.17
Regeneration of Unmodified Enzyme &y Treatment with B-Mercaptoethanol:
To a solution of CMM (2.0 mg) in 250 pL of CHES-buffer (70 mM CHES. 5 mM
MES. 2 mM CaCI,. pH 9.5) was added 10 pL of a solution of P-rnercaptoethanol
(1 M in 95% EtOH ). The reaction was monitored by specific activity
measurements and in al1 cases the activity of the cysteine parent enzyme was
restored.
Free Thiol Titration: The free thiol content of S I 66C CMMs was deterrnined
spectrophotometrically by titration with Ellman's reagent (E,,, = 13600 M-' cm-')4'
in phosphate buffer 0.25 M, pH 8.0.
Active Site Titrations: The active enzyme concentration was determined as
previously described" by monitoring fluoride release upon enzyme reaction with
u-toluenesulfonyl fiuoride (Aldrich Chernical Co. 1 nc.) as measured by a fiuoride
ion sensitive electrode (Orion Research 96-09). The active enzyme concentration
deterrnined in this way was used to calculate kinetic parameters for each CMM.
Kinetic Measurements: Michaelis-Menten constants were measured at 25 OC
by curve fitting (GraFit@ 3.03) of the initial rate data determined at eight
concentrations (O. 125 mM-8.0 mM) of the suc-AAPX-pNA su bstrate (Bachem
California Inc. Torrance. CA) in buffer (pH 8.6, 0.1 M Tris-HCI. 0.005% Tween
80. 1 % DMSO), ( E ~ ~ ~ = 8800 M-' cm-')?
Molecular Modelling: The X-ray structure of subtilisin Bacillus lentus14 was used
as the starting point for calculations on the wild type and chemically modified

135 mutant enzymes. The enzyme setup was performed with lnsight IL4' TO create
initial coordinates for the minimization, hydrogens were added at the pH used for
kinetic measurements. This protonated al1 Lys and Arg residues and the N-
terminus and deprotonated al1 Glu and Asp residues and the C-terminus. In
addition, the active site His64 was protonated. The model system with the Ala-
Ala-Pro-Phe (from crystal structure)14 product inhibitor bound in the SI- S, pocket
was solvated with a 5 A layer of water molecules giving a total number of water
molecules of 1 143 in this system. The overall charge of the enzyme-inhibitor
comptex resulting from this setup was +4 for the WT enzyme. Energy simulations
were performed with the Discover program," on a Silicon Graphics Iris Indigo
cornputer, using the consistent valence force field function (CVFF). A non-
bonded cutoff distance of 18 A with a switching distance of 2 A was employed.
The non-bonded pair list was updated every 20 cycles and a dielectric constant
of 1 was used in al1 calculations. The energy of the structure of the VVT enzyme
was minimized in stages, with initially only the water molecules being allowed to
move, then the water rnolecules and the amino acid side chains, and then the
entire enzyme. The mutated and chemically modified enzymes were generated
using the Builder module of Insight. Then the amino acid side chains within a 10
A radius of the a-carbon of the mutated residue were energy minimized while al1
other residues were constrained, then al1 of the atoms within a 10 A shell were
energy minimized. followed by minimization of the whole system. To examine the
effect of a different Pl residue (Glu), the Phe to Glu mutation of the product
inhibitor was constructed using lnsight II, and then this structure was energy
minimized as above.

1 36 References
1. Nilsson 6, Forsberg G, Moks T, Haitmanis M. Uhlén M. Cun. Opin. Struct. Biol. 1 992; 2:569-75.
2. LaVallie ER, McCoy JM. Cun. Opin. Biotechnol. 1995; 6:501-6.
3. Uhlen M, Moks T. Methods Enzymol. 1990; l85:12943.
4. Moree WJ, Sears P. Kawashiro K, Witte K, Wong C-H. Exploitation of Subtilisin BPN' as a Catalyst for the Synthesis of Peptides Containing Noncoded Amino Acids, Peptide Mimetics and Peptide Conjugates. J. Am. Chem. Soc. 1997; 1 19:3942-7.
5. Sears P, Wong C-H. Engineering Enzymes for Bioorganic Synthesis: Peptide Bond Formation. Biotechnol. Prog. 1 996; 1 2:423-33.
6. Faber K. Biotransformations in Organic Synthesis. 3rd edition. Heidelberg: Springer-Verlag, 1997.
7. Roberts SM. Preparative Biotransfomations. New York: Wiley, 1993.
8. Berglund P, DeSantis G, Stabile MR et al. Chemical Modification of Cysteine Mutants of Subtilisin Bacillus lentus Can Create Better Catalysts Than the Wild-Type Enzyme. J. Am. Chem. Soc. 1997; 11 9:5265-6.
9. DeSantis G, Berglund P, Stabile MR. Gold M. Jones JB. Site-Directed Mutagenesis Cornbined with Chernical Modification as a Strategy for Altering the Specificity of the S, and S,' Pockets of Subtilisin Bacillus lentus. Biochemistry 1998; 37:5968-73.
10. DeSantis G, Jones JB. Chemical Modifications at a Single Site Can lnduce Significant Shift in the pH-Activity Profiles of a Serine Protease. J. Am. Chem. Soc. 1 998; 120(34):8582-6.
11. Plettner E, Khumtaveeporn K, Shang X, Jones JB. Combinatorial Approach to Chemical Modification of Subtilisin Bacillus lentus. Biorg. Med. Chem. Lett. 1998; 8(17):2291-6.
12. Berglund P. Stabile MR, Gold M et al. Altering the Specificity of Subtilisin B. Lentus by Combining Site-Directed Mutagenesis and Chernical Modification. Bioorg . Med. Chem. Lett. 1996; 6(21):2507-12.

137 Fersht A. Enzyme Structure and Mechanism. 2nd edition. New York: W.H. Freeman and Company, 1985.
Knapp M, Daubermann J, Bott RR. Brookhaven Database Entry 1 JEA.
Bode W, Meyer E, Powers JC. Human Leukocyte and Porcine Pancreatic Elastase: X-ray Crystal Structures, Mechanism. Substrate Specificity, and Mechanisrn-Based Inhibitors. Biochemistry 1989; 28(5): 1951 -63.
Perona JJ, Hedstrom L, Rutter WJ. Fletterick RJ. Structural Origins of Substrate Discrimination in f rypsin and Chymotvpsin. Biochemistry 1995; 7(34):1489-99.
Davis BG, Shang X, DeSantis G, Bott RR. The Controlled Introduction of Multiple Negative Charge at Single Amino Acid Sites in Subtilisin Bacillus lentus. Bioorg. Med. Chem. submitted.
Synthesized by Dr. Xiao Shang.
Hsia CY, Ganshaw G, Paech C. Murray CJ. Active-Site Titration of Serine Proteases Using a Fluoride Ion Selective Electrode and Sulfonyl Fluoride In hibitors. Anal. Biochem. 1996; 242:22 1-7.
Bone R, Frank O, Kettner CA, Agard DA. Structural Analysis of Specificity: u-Lytic Protease Complexes with Analogues of Reaction Intermediates. Biochemistry 1989; 28:7600-9.
Bone R, Fujishige A. Kettner CA, Agard DA. Structural Basis for Broad Specificity in a-Lytic Protease Mutants. Biochemistry 1991 ; 30:10388-98.
Bauer C-A, Brayer GD, Sielecki AR, James MNG. Active Site of u-Lytic Protease. Eur. J. Biochem. 1981 ; l20:289-94.
Estell DA, Graycar TP, Miller JV ef al. Probing Steric and Hydrophobic Effects on Enzyme Substrate Interactions by Protein Engineering. Science 1986; 233:659-63.
Takagi H, Maeda T, Ohtsu 1, Tsai Y-C, Nakamori S. Restriction of Substrate Specificity of Subtilisin E by lntroduction of a Sida Chain into a Conserved Glycine Residue. FEBS Lett. 1996; 395: i 27-32.

138 Takagi Hl Ohtsu f , Nakamori S. Construction of Novel Subtilisin E with High Specificity, Activity and Productivity Through Multiple Amino Acid Subtitutions. Protein Eng. 1997; 10(9):985-9.
Mei H-C, Liaw Y-Cl Li Y-C, Wang D-C, Takagi H, Tsai Y-C. Engineering subtilisin YaB:Restriction of Substrate Specificity by the Subtitution of Gly 1 24 and Gly 1 51 with Ala. Protein Eng. 1998; 1 1 (2): 109-1 7.
Siezen RJ, Leunissen JAM. Subtilases:The Superfamily of Su btilisin-Like Serine Proteases. Protein Sci. 1997; 6:501-23.
Creemers JWM, Siezen RJ, Roebroek AJM, Ayoubi TAY, Huylebroeck Dl van de Ven WJM. Modulation of Furin-mediated Proprotein Processing Activity by Site-Directed Mutagenesis. J. Biol. Chem. 1993; 268(29):21826- 21834.
Nakayama K. Furin: A Mammalian Subtilisin Kex2p-like endopeptidase involved in Processing of a Wide Variety of Precursor Proteins. Biochem J. 1997; 327:625-35.
Wells JA. Additivity of Mutational Effects in Proteins. Biochemistry 1990; 29(37):8509-17.
Wells JA, Powers OB, Bott RR, Graycar TP, Estell DA. Designing Substrate Specificity by Protein Engineering of Electrostatic Interactions. Proc. Nat. Acad. Sci. USA 1987; 84:1219-23.
Ballinger MD, Tom J, James AW. furilisin: A Varient of Subtilisin BPN' Engineered for Cleaving Tribasic Substrates. Biochemistry 1996; 33:13579- 85.
Bonneau PR, Graycar TP, Estell DA, Jones JB. Alteration of the Specificity of Subtilisin BPN' by Site-Directed Mutagenesis in Its S, and S,' Binding Sites. J. Am. Chem. Soc. 1991 ; 1 '13:lO26-30.
Nienaber VL, Breddam K, Birktoft JJ. A Glutamic Acid Specific Senne Protease Utilizes a Novel Histidine Triad in Substrate Binding. Biochemistry 1 993; 32(43): 1 1469-75.
Svendsen 1, Jensen RM, Breddarn K. The Primary Structure of the Glutamic Acid-Specific Protease of Streptomyces griseus. FEBS 1 99 1 ; 292(1): 165-7.

1 39 36. Smyth MJ, O'Connor MD, Trapani JA. Granzymes: A Vanety of Serine
Protease Specificities Encoded by Genetically Distinct Subfamilies. J. Leukoc. Biol. 1996; 60:555-62.
37. Murphy MEP, Moult J, Bleackley RC, Gershenfeld H, Weissrnan IL, James MNG. Comparative Molecular Mode1 Building of Two Senne Proteinases From Cytotoxic T Lymphocytes. Proteins: Structure, Function, and Genectics 1 988; 4:l90-204.
38. Caputo A, James MNG, Powers JC, Hudig D, Bleackley RC. Conversion of the Substrate Specificity of Mouse Proteinase Granzyme B. Nature:Struct. Biol. 1 994; 1 (6):364-7.
39. Bott R, Ultsch M, Wells J et al. Importance of Conformational Variability in Protein Engineering of Subtilisin. Lebanon HM, Mumma ROI Honeycutt RC, Duesing JH, ed. Biotech. Agric. Chern. Vol. ACS Symp. Ser. 334. 1987: 1 39-47.
40. Stabile MR, Lai WG, DeSantis G et al. Probing the Specificity of the S, Binding Site of M222 Mutants of Subtilisin B. Lentus with Boronic Acid 1 nhibitors. Bioorg. Med. Chem. Lett. 1996; 6(21):2501-6.
41. Ellman GL, Courtney KD, Andres Jr. V, Featherstone RM. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochern. Pharmacol. 1961 ; 7:88-95.
42. lnsight 1 1 [Biosym Technologies, lnc. San Diego, CA, USA]. Version 2.3.0.
43. Discover [Biosym Technologies, Inc. San Diego, CA, USA]. Version 2-95.

Chapter 6
Reproduced in part with permission frorn J. Am. Chern. Soc. 1997.1 19. 5265-
5266. Copyright 1998. American Chemical Society (Reference 12)

Chernical Modification of the N62C Cysteine Mutant of Subtilisin
B. lentus Can Create Better Catalysts than the Wild-type Enzyme
Introduction
All too often. site-directed mutagenesis creates mutant enzymes with
lower-than-WT activities. Therefore, one of the goals of the current study was to
create chemically modified mutant enzymes (CMMs) that could at least match
the activity of the WT parent. In addition, the specificity changes which could be
induced in the S, pocket were of interest. With these objectives in mind, using
SBL's X-ray structure as a guide,'.' residue Asn62 which is located in the S,
pocket and is adjacent to the catalytic His64 residue (Figure 6.1) was selected
for mutagenesis to cysteine and subsequent reaction with eleven la-k
methanethiosulfonate reagents (Scheme 6.1 ).
Figure 6.1 Active Site of SBL lndicating Position of N62:'q2The catalytic triad residues Asp32, His64, and Ser221, and the S, pocket residue Asn62 chosen for mutation and modification are shown.

Scheme 6.1
Results and Discussion
Each of the CMMs N62C-S-a to-k were prepared and characterized as
described previ~usly.~ Briefly, the N62C-SBL mutant was treated with the
methanethiosulfonate reagents (la-k) as showr! in Scheme 6.1, with the
modification reactions being complete within 30 min. Titration of the CMMs with
Ellman's reagent established that reactions were quantitative with the free thiol
content of the CMMs being less than 2% in al1 cases. The CMMs were purified
on a disposable Sephadex G-25 desalting column and analysis of each CMM by
electrospray rnass spectrometry was consistent (f 6 Da) with the calculated
mass. The purities of the modified enzymes were assessed by native-PAGE and
in al1 cases only one band was visible. Furthermore, treatment of SBL-WT, in
which no cysteine is present. with each of la-k resulted in no change in activity.
Additionally, P-mercaptoethanof treatment of the CMMs restored activity to that
of the parent cysteine mutant, verifying that chemical modification at cysteine is
solely responsible for the observed changes in activity and that the modification
is fully reversible. The active enzyme concentration was determined by

monitoring fluoride release upon reaction of each CMM with
phenylmethylsulfonyl fiuoride (PMSF)?
The kinetic data obtained, with the standard suc-AAPF-pNA reference
su bstrate, are summarized in Table 6.1. Despite the breadth of the structural
range of the R-groups introduced. al1 of the chemical modifications resulted in
CMMs with higher k,JKMs than its cysteine mutant enzyme predecessor.
However. introductions of the charged sulfonatoethyl (R = j) and aminoethyl (R =
k) groups are clearly only marginally beneficial relative to those of any of the
hydrophobic moieties of la-i. It is evident that the more hydrophobic the
introduced modifying group, the higher the k,, values become. The only
exception is for the decyl modified CMM. N62C-S-g, for which ka, is only slightly
increased compared to WT. Whereas the KM for the SBL-N62C mutant itself is
almost 3-fold higher than for SBL-WT, the KM values for the CMMs with small
hydrophobic modifications (-a to a) are only increased slightly (Table 6.1 ).
However, for the larger more hydrophobic (-f to -i) modified enzymes, binding is
unaltered or improved slightly. For both the negatively and positively charged
N62C-S-j and N62C-S-k respectively, an increase in KM is observed. This
general trend of improved KMs for hydrophobic -R groups and poorer KMs for
hydrophilic modifications reinforce the trends in k,,.
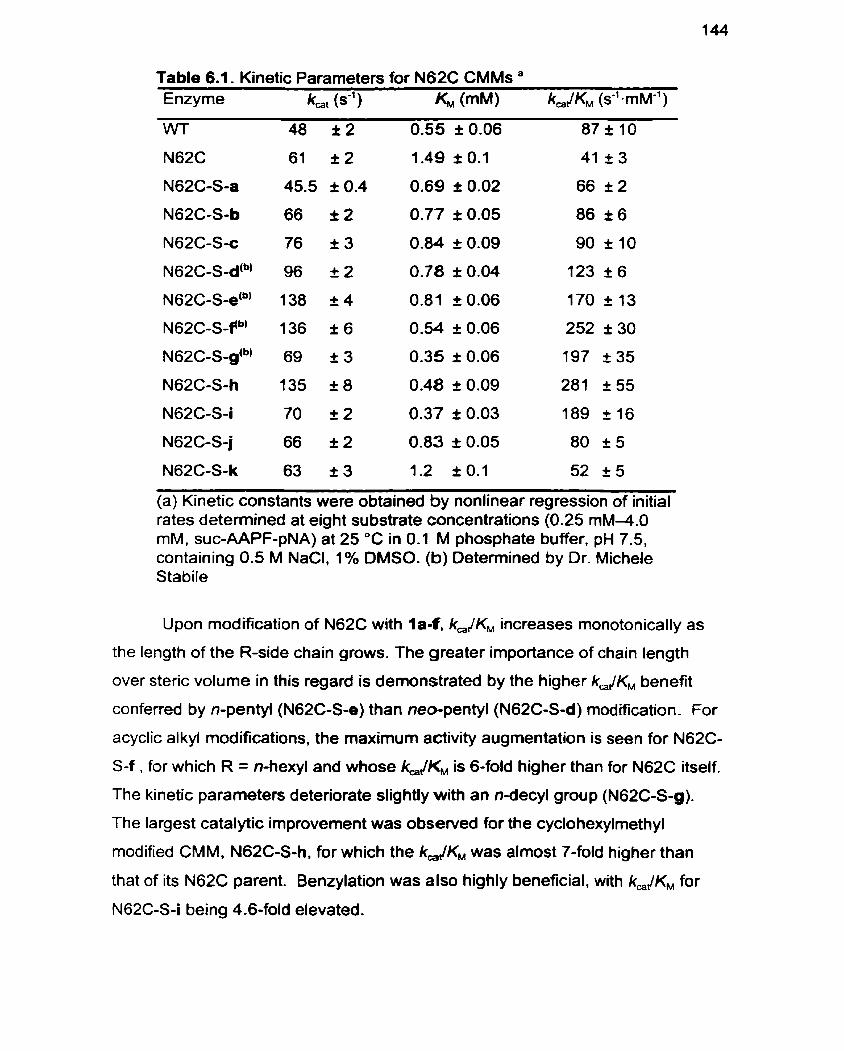
Table 6.1. Kinetic Parameters for N62C CMMs " Enzyme kat wl) KM (mM) k, JKM (s-' -rnMe' )
WT 48 & 2 0.55 I 0.06 87210
(a) Kinetic constants were obtained by nonlinear regression of initial rates deterrnined at eight substrate concentrations (0.25 mM4.O mM, suc-AAPF-pNA) at 25 OC in 0.1 M phosphate buffer, pH 7.5, containing 0.5 M NaCI, 1% DMSO. (b) Deterrnined by Dr. Michele Stabile
Upon modification of N62C with la-f , kJKM increases monotonically as
the length of the R-side chain grows. The greater importance of chain length
over steric volume in this regard is demonstrated by the higher kJKM benefit
conferred by n-pentyl (N62C-S-e) than neo-pentyl (N62C-S-d) modification. For
acyclic alkyl modifications, the maximum activity augmentation is seen for N62C-
S-f , for which R = n-hexyi and whose k,JKM is 6-fold higher than for N62C itself.
The kinetic parameters deteriorate slightly with an n-decyl group (N62C-Sg).
The largest catalytic improvement was observed for the cyclohexylmethyl
modified CMM, N62C-S-h, for which the kJKM was alrnost 7-fold higher than
that of its N62C parent. Benzylation was also highly beneficial, with kJKM for
N62C-S-i being 4.6-fold elevated.
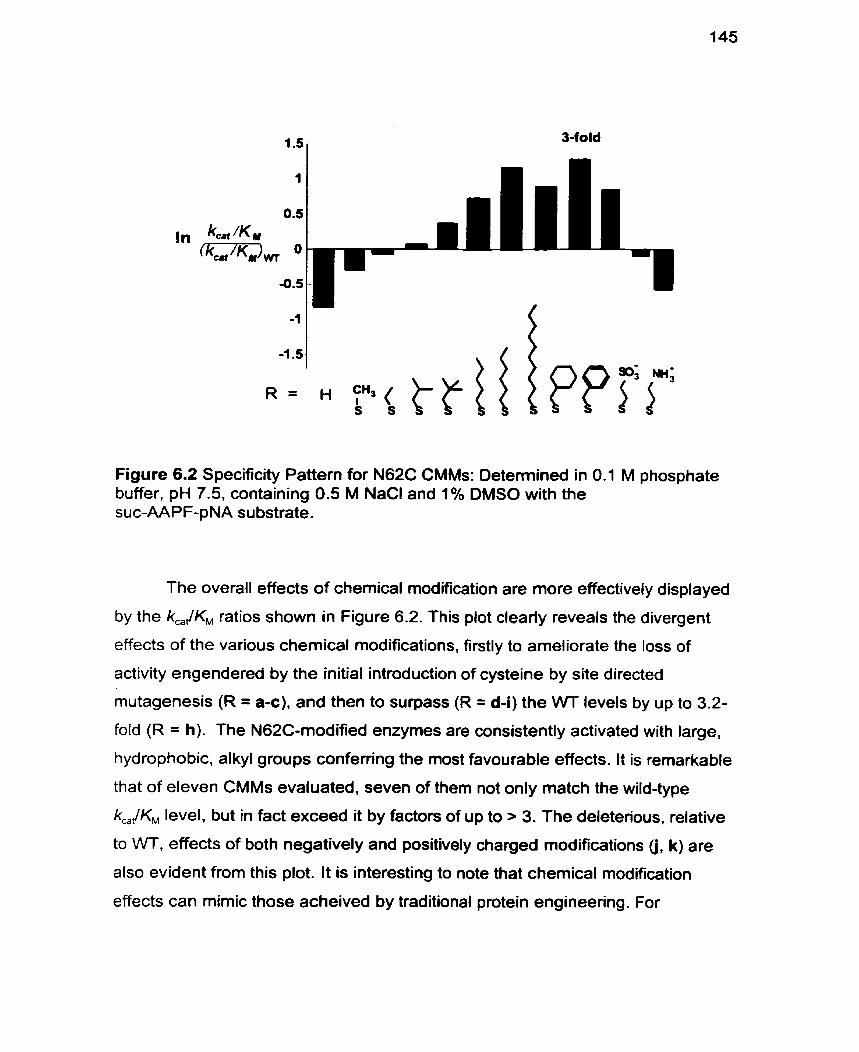
Figure 6.2 Specificity Pattern for N62C CMMs: Determined in 0.1 M phosphate buffer, pH 7.5, containing 0.5 M NaCl and 1% DMSO with the suc-AAPF-pNA substrate.
The overall effects of chemical modification are more effectively displayed
by the k,/KM ratios shown in Figure 6.2. This plot clearly reveals the divergent
effects of the various chemical modifications, firstly to ameliorate the loss of
activity engendered by the initial introduction of cysteine by site directed
mutagenesis (R = a-c), and then to surpass (R = d-i) the VVT levels by up to 3.2-
fold (R = h). The N62C-modified enzymes are consistently activated with large,
hydrophobie, al kyl groups conferring the most favourable effects. It is remarüable
that of eleven CMMs evaluated, seven of them not only match the wild-type
kJKM level, but in fact exceed it by factors of up to > 3. The deleterious, relative
to WT, effects of both negatively and positively charged modifications (j, k) are
also evident from this plot. It is interesting to note that chemical modification
effects can mimic those acheived by traditional protein engineering. For

example. the modification of SBL-N62C with the polar side chain j. mirror those
induced by introduction of amino acids with the charged (aspartic acid) amino
acid side chains by site-directed mutagenesis at position 62. in the closely
related enzyme subtilisin BPN?
To our knowledge, the attainment of these levels of augmented-\l\rr
activity for such a broad range of modified enzymes is unprecedented. and has
not been matched so far in its breadth by protein engineering methods alone.
These results demonstrate the considerable potential that the cornbined site-
directed mutagenesis-chemical modification approach offers for creating novel
enzymes with better-than-W activity.

Experimental
Enzyme Purification: WT-SBL and its N62C mutant were prepared as
previously descnbed and punfied by desalting on a Sephadex G-25 matrix
followed by strong cation exchange chrornatography on SP Sepharose FF. The
purity of WT- and N62C-SBL. denatured by incubation with 0.1 M HCI at O O C for
30 min.. was detemined by SDS-PAGE and in each case. only one band was
visible.
P reparafion of Methenethiosulfonate reagents: Reagent 1 a was purchased
from Aldrich Chemical Co. Inc.. 1j and l k from Toronto Research Chemical (2
Brisbane Rd. Toronto, ON). and al1 were used as received. Reagents Ib-i were
prepared as previously described.'
Site-specific Chemical Modification: To N62C (25 mg) in CHES buffer (2.5
mL; 70 mM CHES, 5 mM MES. 2 mM CaCI,. pH 9.5) at 20 OC was added a 100-
fold rnolar excess of each of the methanethiosulfonate reagents 1 a-k (1 00 pL of
a 1 .O M solution, 100 PM: l a 4 in EtOH, l e i in CH,CN. 1j in CHES buffer, l k in
MeOH) in a PEG (10.000) coated polypropylene test tube. and the mixture
agitated in an end-over-end rotator. Blank reactions were run in parallel. Each of
the modification reactions was monitored by specific activity measu rements on a
Perkin Elmer Lambda 2 spectrophotometer (E,,, = 8800 M-' cm").' The specific
activity of the CMM was detenined in buffer containing: 0.1 M TRIS pH 8.6,
0.005 % Tween 80. and 1% DMSO. with the suc-AAPF-pNA substrate (1 mglmL)
at 25 OC. The reaction was terminated when the addition of a further 100 pL of
methanethiosulfonate solution effected no further change in specific activity,
generally within 30 min. The reaction was quenched by dilution in MES buffer (5
mM MES. 2 mM CaCI,. pH 6.5) at O OC and then punfied on a disposable
desalting column (Phamacia Biotech PD1 O, Sephadex G-25 M) pre-equilibrated
with MES buffer. The CMM was eluted with MES-buffer (3.5 mL), dialyzed
against 1 mM CaCI, (3 x 1 L) at 4 OC and subsequently lyophilized. Modified
enzymes were analyzed by native gradient PAGE (8-25%) at pH 4.2. run

towards the cathode on the Pharmacia Phast-System. and appeared as one
single band. It was also established that SBL-WT was unchanged on exposure
to 1 a-k under the modifying conditions.1°
Electrospray Mass Spectrometry. Prior to ES-MS anal ysis. CMMs were
purified by FPLC (BioRad. Biologic System) on a Source 1 5 RPC matrix (1 7-
0727-20 from Phannacia) with 5% acetonitrile. 0.01 % TFA as the running buffer
and eluted with 80% acetonitrile, 0.01% TFA in a one step gradient. Electrospray
rnass spectra were recorded on a PE SClEX API III Biomolecuiar Mass
Analyzer. In al1 cases the experimentally determined and theoretically calculated
masses were found to agree within 6 Da. Mass: WT: Calcd: 26698. Found
26694. N62C: Calcd: 26687. Found 26690. N62C-S-a: Calcd: 26733. Found
26734. N62C-S-b: Calcd: 26747, Found 26749. N62C-S-c: Calcd: 26775. Found
26775. N62C-S-d: Calcd: 26789. Found 26791. N62C-S-e: Calcd: 26789. Found
26791. N62C-S-f: Calcd: 26803. Found 26804. N62C-Sg: Calcd: 26859. Found
26857. N62C-S-h: Calcd: 2681 5. Found 26816. N62C-S-i: Calcd: 26809. Found
26809. N62C-S-j: Calcd: 26826. Found 26828. N62C-S-j: Calcd: 26763. Found
26764.
Free Thiol Titration: The residual free thiol content of N62C CMMs, was
determined spectrophotometrically by titration with Ellman's reagent (E,,, = 13600
M" cm-')" in phosphate buffer 0.25 M. pH 8.0 and was less than 2 % in al1
cases.
Active Site Titration: The active enzyme concentration was determined as
previously described4 by monitoring fiuoride ion release upon enzyme reaction
with the irreversible inhibitor, phenylmethylsulfonyl fluoride (Aldrich Chemical Co.
Inc.) as measured by a fluoride ion sensitive electrode (Orion Research 96-09).
The active enzyme concentrations deterrnined in this way was used to calculate
kinetic parameters for each CMM.

References
Betzel C, Klupsch S. Papendorf G, Hastrup S. Branner S, Wilson KS. Crystal Structure of the Alkaline Proteinase Savinase from Bacillus lentus at 1.4 A Resolution. J. Mol. Biol. 1992; 223:42745.
Knapp M. Daubennann J, Bott RR. Brookhaven Database Entry 1 JEA.
DeSantis G. Berglund P. Stabile MR, Gold M. Jones JB. Site-Directed Mutagenesis Combined with Chemical Modification as a Strategy for Altering the Specificity of the S, and S,' Pockets of Subtilisin Bacillus lentus. Biochemistry 1998; 37:5968-73.
Hsia CY. Ganshaw G, Paech C, Murray CJ. Active-Site Titration of Serine Proteases Using a Fluoride Ion Selective Electrode and Sulfonyl Fluoride Inhibitors. Anal. Biochem. 1996; 242:221-7.
Ballinger MD. Tom J. Wells JA. Designing Subtilisin BPN' To Cleave Su bstrates Containg Dibasic Residues. Biochemistry 1995; 34: 133 1 2-9.
Ballinger MD, Tom J, James AW. F urilisin: A Variant of Subtilisin BPN' Engineered for Cleaving Tribasic Substrates. Biochernistry 1996; 33: 13579- 85.
Stabile MR. Lai WG, DeSantis G et al. Probing the Specificity of the S, Binding Site of M222 Mutants of Subtilisin B. Lentus with Boronic Acid Inhibitors. Bioorg. Med. Chem. Lett. 1996; 6(21):2501-6.
Berglund P, DeSantis G, Stabile MR et al. Chemical Modification of Cysteine Mutants of Subtilisin Bacillus lentus Can Create Better Catalysts Than the Wild-Type Enzyme. J. Am. Chem. Soc. 1997; 11 9:5265-6.
Bonneau PR, Graycar TP, Estell DA. Jones JB. Alteration of the Specificity of Subtilisin BPN' by Site-Directed Mutagenesis in Its S, and S,' Binding Sites. J. Am. Chem. Soc. 1991 ; 11 3:lO26-30.
Kluger R. Tsui W-C. Amino Group Reactions of the Sulfhydryl Reagent Methyl Methanesulfonothioate. Inactivation of D-3-Hydroxybutyrate Dehydrogenase and Reaction with Amines in Water. C. J. Biochem. 1980; 581629-32.

11. Ellman GL, Courtney KD. Andres Jr. V. Featherstone RM. A New and Rapid Colorimetric Detemination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961 ; 7:88-95.
12. Berglund P, DeSantis G, Stabile MR et al. Chernical Modification of Cysteine Mutants of Subtilisin Bacillus lentus Can Create Better Catalysts Than the Wild-Type Enzyme. J. Am. Chem. Soc. 1997; 1 19:5265-6.

Chapter 7
Reproduced in part with permission from J. Am. Chem. Soc. 1998,120,8582-
8586. Copyright 1998, American Chernical Society (Reference 28)

Chernical Modifications at a Single Site Can lnduce Significant
Shifts in the pH Profiles of a Serine Protease
ln troduc tion
The ability to Vary the pH-activity profile of an enzyme in a controlled
manner has been a long sought-after Such tailorhg can help to elucidate
important insights into mechanism and pemiits optimization of enzyme
performance for use in organic synthesis.lO.l' Thus far, the most successful
approaches to altering pH-activity profiles have used either site-directed
mutage ne si^'^^^ or chemical modification1.' to alter enzyme surface charge.
These studies have established that increasing positive surface charge of the
serine proteases destabilizes the protonated imidazolium group of the active site
histidine and thus lowers its pK, while increasing negative surface charge
stabilizes the histidine imidazolium. thereby raising its pK,'" While predictive
models to determine the effect of electrostatic modification on His64 pK, have
recently been established.' the introduction of uncharged hydrophobic mutations
to alter enzyme pH-activity profiles has not been investigated. In addition, while it
has been recognized that the pK,s of amino acid side chains in proteins.
particularly those essential for catalysis. are affected by their environment,12
these values cannot always be predicted.
The strategy of chemical modification of site-directed mutant enzymes
offers a new opportunity for controlling pH optima. Previously, the strategy
illustrated in Scheme 7.1 was exploited to increase the activity of SBL effecting
an up to 3-fold higher than wild type (WT) activity enhancement for N62C-SCH,
c-C,H,, (-h) (Table 6.1. page 144).13 Furthemore. it was apparent that increased
alkyl side chah length caused a monotonie increase in k,JK,. Due to the
proximity of Cys62 to His64 it is hypothesized that these activity enhancements
were due in part to the altered pK, of the catalytic triad residue His64. The

current study was-undertaken to investigate the validity of this hypothesis and to
uncover altered pH-activity profiles of SBL which can expand its synthetic utility.
Results and Discussion
Initially, molecular mechanics calculations were performed to probe the
molecular basis for the 3-fold activity enhancement previously obssrved for
N62C-S-CH,-c-C,Hl, (4) (Table 6.1. page 144). These results. which are
depicted in Figure 7.1 a, revealed that in the energy minimized structure the
cyclohexyl moiety is positioned directly over the His64 irnidazole of the catalytic
triad. In contrast, for N62C-k, for which a 1.7-fold decrease in activity was
observed the -S-CH,CH,NH,+side chain is solvated and oriented away from the
catalytic triad region in the energy minimized structure. as shown in Figure 7.1 b.
In light of these molecular modelling results, and considering the proximity of
Cys62 to His64, we postulate that the more hydrophobie environment of the
catalytic triad in N62C-S-CH,-c-C,Hll induced by the cyclohexyl group would
cause a decrease in the observed pK, of His64.14 Accordingly, pH-activity profiles
for WT-SBL, the parent cysteine mutant N62C, and each of the eleven CMMs
N62C-a to -k13 were evaluated.15
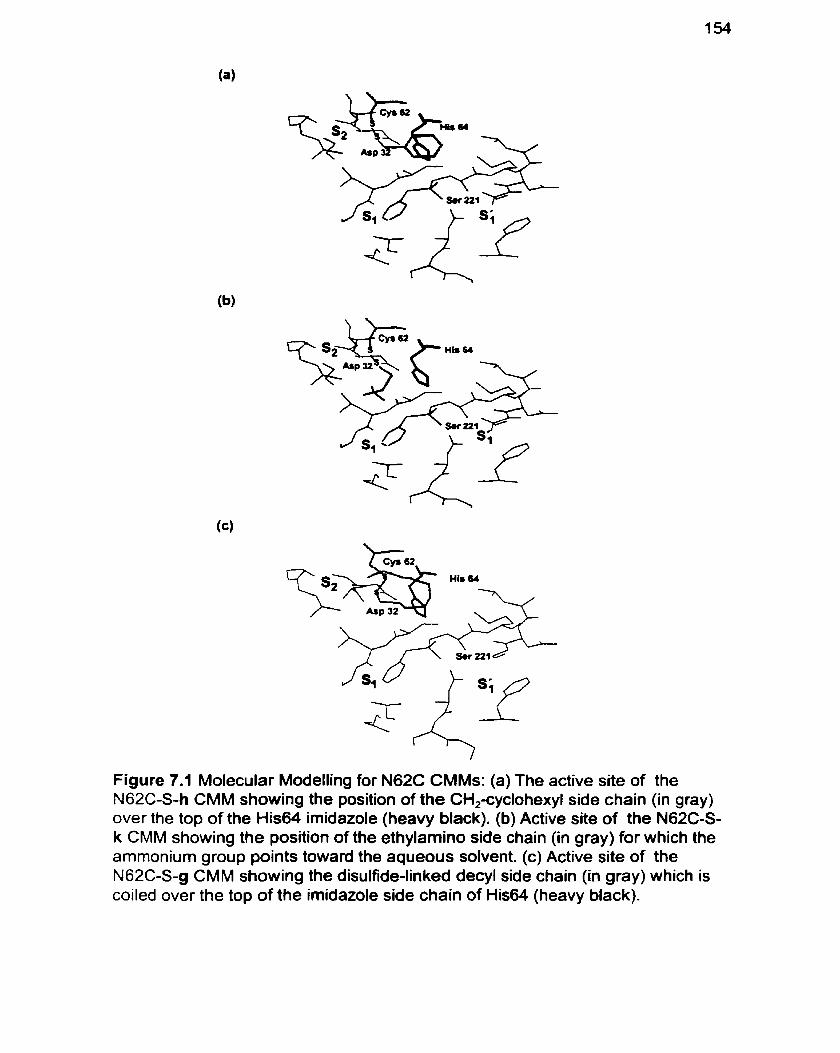
Figure 7.1 Molecular Modelling for N62C CMMs: (a) The active site of the N62C-S-h CMM showing the position of the CH,-cyclohexyl side chain (in gray) over the top of the His64 imidazole (heavy black). (b) Active site of the N62C-S- k CMM showing the position of the ethylamino side chain (in gray) for which the ammonium group points toward the aqueous sofvent. (c) Active site of the N62C-S-g CMM showing the disulfide-linked decyl side chain (in gray) which is coiled over the top of the imidazole side chain of His64 (heavy black).
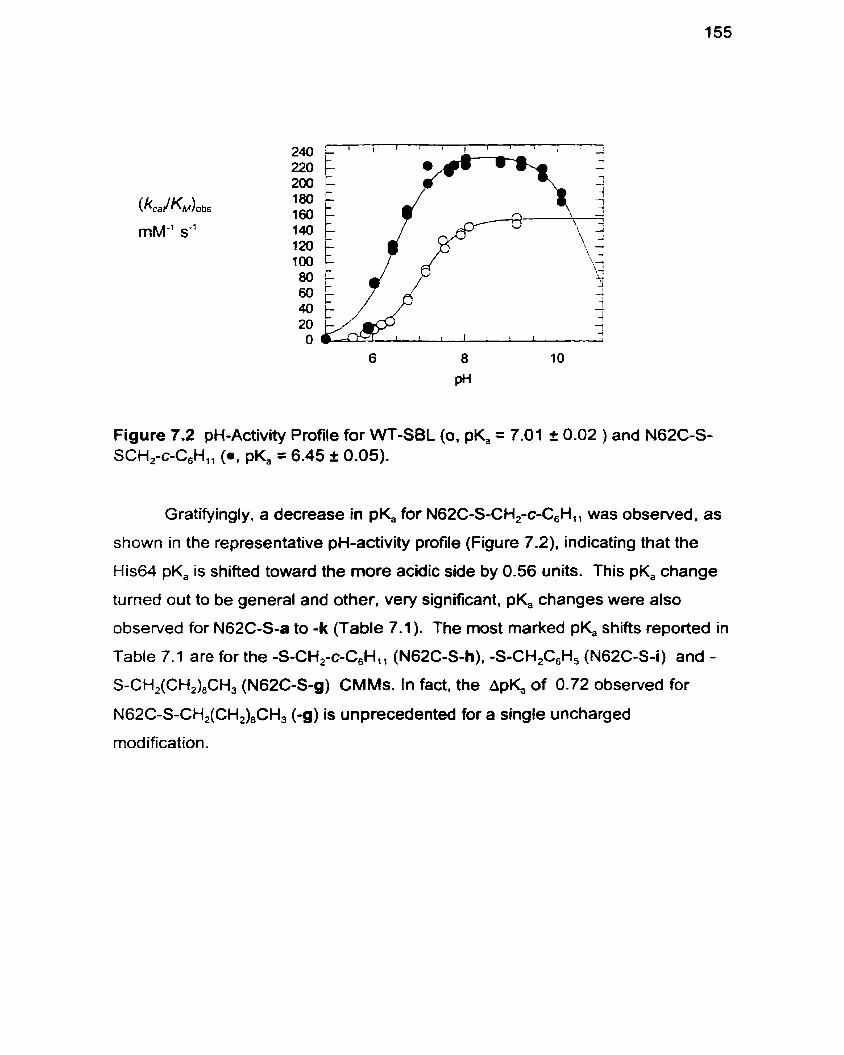
Figure 7.2 pH-Activity Profile for WT-SBL (0, pK, = 7.01 I 0.02 ) and N62C-S- SCH2-C-C6HI1 (0, pK, = 6.45 I 0.05).
Gratifyingly, a decrease in pK, for N62C-S-CH2-c-C,H,, was observed. as
s hown in the representative pH-activity profile (Figure 7.2). indicating that the
His64 pK, is shifted toward the more acidic side by 0.56 units. This pK, change
turned out to be general and other, very significant, pK, changes were also
observed for N62C-S-a to -k (Table 7.1). The most marked pK, shifts reported in
Table 7.1 are for the -S-CH2-c-C,H,, (N62C-S-h), -S-CH,C,H, (N62C-S-i) and - S-CH,(CH,),CH, (N62C-S-g) CMMs. In fact, the ApK, of 0.72 observed for
N62C-S-CH2(CH2),CH, (-g) is unprecedented for a single uncharged
modification.
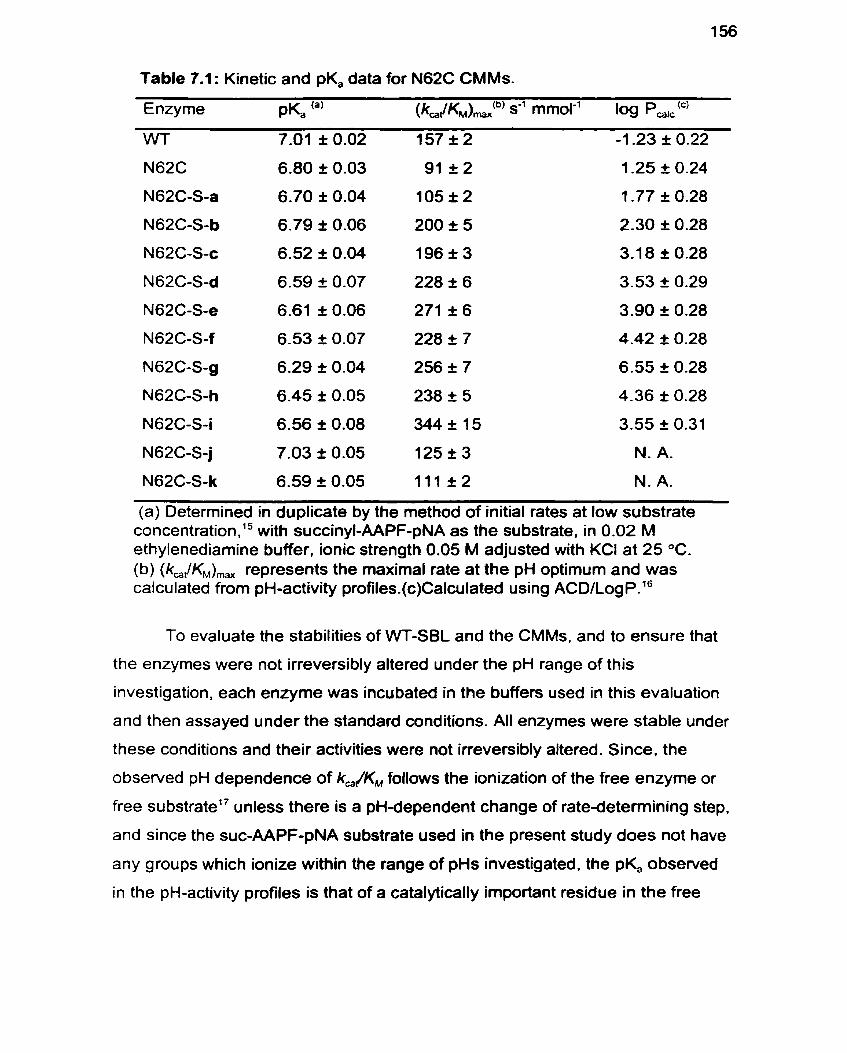
Table 7.1 : Kinetic and pK, data for N62C CMMs.
Enzyme P& 'a' (ka JKM),@) s-' mmol-' 109 p,~~.'~'
WT 7.01 î 0.02 15712 -1 -23 i 0.22
N62C-S-k 6.59 k 0.05 111 22 N. A.
(a) Determined in duplicate by the method of initial rates at low substrate c~ncentration.'~ with succinyl-AAPF-pNA as the substrate. in 0.02 M ethylenediarnine buffer, ionic strength 0.05 M adjusted with KCI at 25 OC. (b) (km,lKM), represents the maximal rate at the pH optimum and was calculated from pH-activity profiles.(c)Calculated using ACD/LogP.16
To evaluate the stabilities of VVT-SBL and the CMMs, and to ensure that
the enzymes were not irreversibly altered under the pH range of this
investigation, each enzyme was incubated in the buffers used in this evaluation
and then assayed under the standard conditions. All enzymes were stable under
these conditions and their activities were not irreversibly altered. Since, the
observed pH dependence of k,/KM follows the ionization of the free enzyme or
free substrate17 unless there is a pH-dependent change of rate-determining step.
and since the suc-AAPF-pNA substrate used in the present study does not have
any groups which ionize within the range of pHs investigated. the pK, obsewed
in the pH-activity profiles is that of a catalytically important residue in the free
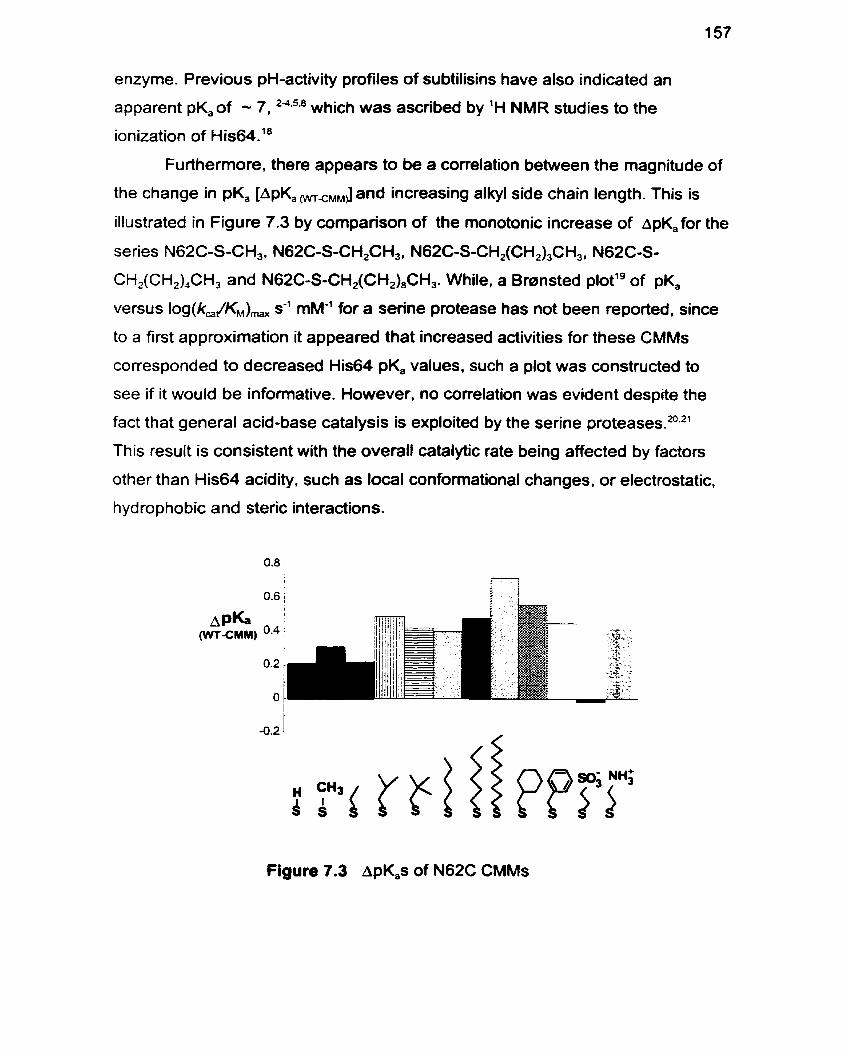
enzyme. Previous pH-activity profiles of subtilisins have also indicated an
apparent pK,of - 7. 24.5+8 which was ascribed by 'H NMR studies to the
ionization of H is64.18
Furthermore, there appears to be a coirelation between the magnitude of
the change in pKa [ApK,,,,J and increasing alkyl side chain length. This is
illustrated in Figure 7.3 by cornparison of the rnonotonic increase of ~pK,for the
series N62C-S-CH,, N62C-S-CH,CH,. N62C-S-CH,(CH&H,. N62C-S-
CH,(CH,),CH, and N6ZC-S-CH,(CH,)8CH,. While, a Bransted plot'g of pK,
versus Iog(k,/KM), s'' mM" for a serine protease has not been reported, since
to a first approximation it appeared that increased activities for these CMMs
corresponded to decreased His64 pK, values, such a plot was constructed to
see if it would be informative. However. no coirelation was evident despite the
fact that general acid-base catalysis is exploited by the serine pr~ teases .~ .~ '
This result is consistent with the overall catalytic rate being affected by factors
other than His64 acidity, such as local conformational changes. or electrostatic,
hydrophobic and steric interactions.
Figure 7.3 Ap&s of N62C CMMs

That the decyl group was the largest hydrophobic group of the current
series prompted us to ascertain if there was a linear relationship between pKa
and hydrophobicity. The Figure 7.4 plot of pK, vs the calculated hydrophobicity
index log P,,a,a,,,,16 where log P = log (solubility in n-octanollsolubility in H20),
confirms this relationship and validates the hypothesis that the hydrophobicity of
the environment around His64 modulates its p)d That such dramatic pK, shifts
are induced by the single, uncharged. groups of the current study is truly
re ma rka ble, since shifts of these magnitudes have previously been achieva ble
only by multiple electrostatic modifications of serine proteases. For example,
succinylation of 14 lysine side chains of chymotrypsin to decrease the positive
charge on the enzyme surface by 28 units was required to effect a 1 .O unit
increase in the pK,of the active site histidine.' On the other hand. increasing
chymotrypsin's positive surface charge by the reaction of 13 surface
carboxylates with ethylenediamine was needed to elicit a 0.9 unit pK, decrease.'
Also, by the site-directed mutagenesis approach, a 4 unit increase of net positive
surface charge of the related protease, subtilisin BPN', was needed to induce a
1 .O unit His64 pKa decrease.' The slight increase in pK, effected in this study by
the introduction of a negative charge, as for N62C-S-CH,CH,SO,' (N62C-S-j).
and the decrease effected by the addition of a positive surface charge, as for
N62C-S-CH,CH2NH,+ (N62C-S-k), are consistent with previously established
models of the effects of altered surface charges.
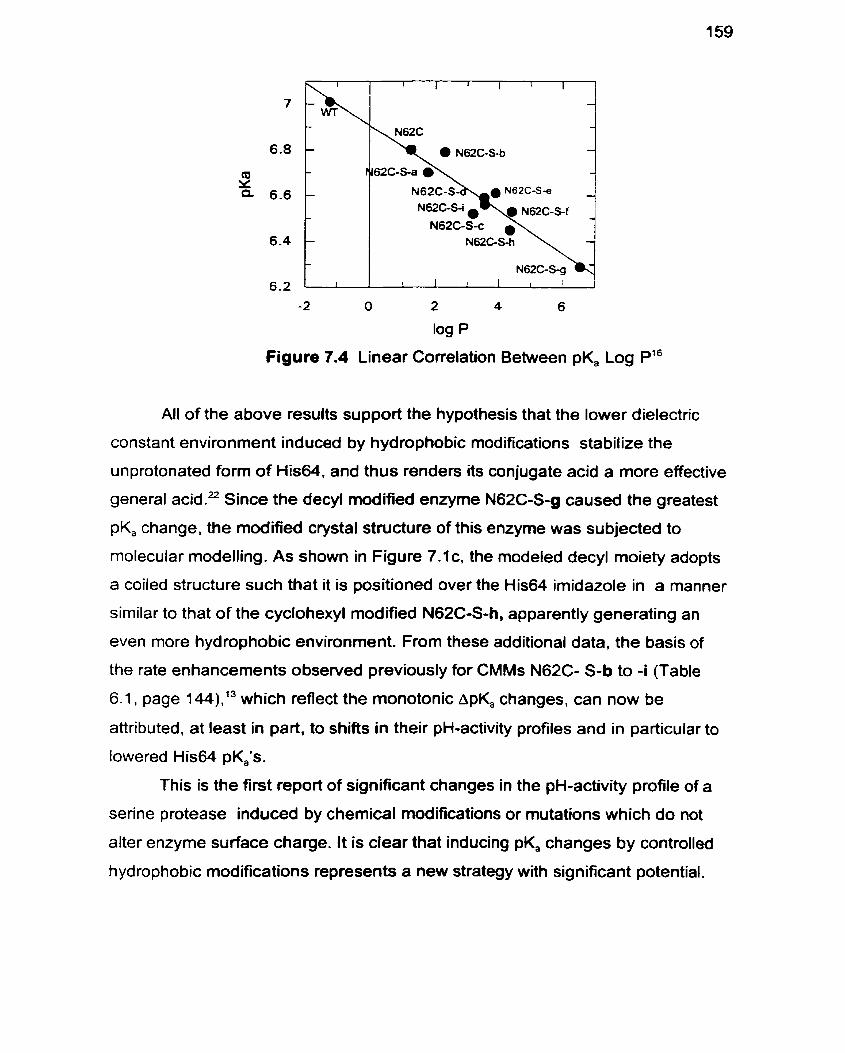
6.2 I I 1 I 1 I I I i - 2 O 2 4 6
log P
Figure 7.4 Linear Correlation Between pK, Log Pl6
All of the above results support the hypothesis that the lower dielectric
constant environment induced by hydrophobic modifications stabilize the
unprotonated form of His64, and thus renders its conjugate acid a more effective
general acid? Since the decyl modified enzyme N62C-S-g caused the greatest
pK, change, the modified crystal structure of this enzyme was subjected to
molecular modelling. As shown in Figure 7.1 c, the modeled decyl moiety adopts
a coiled structure such that it is positioned over the His64 imidazole in a manner
similar to that of the cyclohexyl modified N62C-S-h, apparently generating an
even more hydrophobic environment. From these additional data, the basis of
the rate enhancements observed previously for CMMs N62C- S-b to -i (Table
6.1. page 144).13 which reflect the monotonic ApK, changes. can now be
attributed, at least in part, to shifts in their pH-activity profiles and in particular to
lowered His64 pK,'s.
This is the first report of significant changes in the pH-activity profile of a
serine protease induced by chemical modifications or mutations which do not
alter enzyme surface charge. It is clear that inducing pK, changes by controlled
hydrophobic modifications represents a new strategy with significant potential.

Furthermore, the CMM approach described shoutd be generally applicable to
tailoring the pH-activity profiles of other synthetically useful hydrolases.

Experimental
Kinetics: k,/KM values were determined in duplicate in 0.02 M ethylenediamine
buffer. ionic strength 0.05 M adjusted with KCI. [SI = 2.083 - 12.5 x M of
succinyl-AAPF-pNA (E,,, = 8800 cm-' M-')? [El = 1 - 50 x M at 25 OC. pKafs
were calculated using GraFit version 3.0 curve fit, single pK, or bell-shaped
double pK,, with the minimum set to zero. Specifically, into a 1.5 mL polystyrene
cuvette was added 980 PL of buffer (0.02 M ethylenediamine) and 10 PL of
substrate (2.083 -1 2.5 x 1 OJ M in DMSO). The solution was incubated in a cell
holder at 25 OC. before the absorbance reading was set to zero. Then 10 pL of
enzyme solution (1 x 1 O5 to 5 x 104 M) was added to initiate the reaction. After
an 8 sec delay, absorbance versus time measurements were recorded on a
Perkin Elmer lambda 2 spectrophotometer. kJKM values were calculated
employing the low substrate approximation, where the Michaelis-Menten
Equation reduces t ~ : ' ~ . ' ~ v = kBjKM [EIO[S] when [S]<<KM.
Evaluation of Enzyme Stabilify in pH Range: To ensure that the enzymes
were not irreversibly altered by pH changes, the CMMs were incubated in
ethylenediamine buffers at the same pH as the assay conditions at room
temperature (20 OC) for 1 min. These were then diluted (100-fold) into 0.1 M Tris.
pH 8.6 containing 0.005% Tween 80 and enzyme specific activity was
determined. All CMMs were found to be stable under these conditions.
Calculation of Log P: Calculated using ACDILog Pl6 based on side chain
structure of the amino acid or modified amino acid after replacing the a-carbon
atom with a hydrogen atom. For example, the model structure for VVT (Asn62)
was CH,C(O)NH, whereas model structure for N62C-S-b is CH3-S-S-CH,CH3.
N62C-S-j and -k were ornitted since the software package is unable to calculate
log P for these enzymes. The plot of pK,versus log P gave a slope of -0.093 2
0.009, an intercept of 6.91 I 0.03 and a correlation coefficient of -0.96.
Molecular Modelling: The X-ray structure of subtilisin Bacillus lentus
(Brookhaven PDB entry 1 JEA)25 was used as the starting point for calculations

on the wild type and chemically modified mutant enzymes. The enzyme setup
was performed with lnsight II, version 2.3.0." To create initial coordinates for the
minimization, hydrogens were added at the pH used for kinetic measurements.
This protonated al1 Lys and Arg residues and the N-terminus and deprotonated
all Glu and Asp residues and the C-terminus. The model system with the
phenylmethylsulfonate ester of Ser221 bound in the S, pocket was solvated with
a 5 A iayer of water molecules giving a total number of water molecules of 1 174
in this system. The overatl charge of the enzyme-inhibitor wmplex resulting from
this setup was +3 for the VVT enzyme. Energy simulations were perfomed with
the DISCOVER program, Version 2.9.527 on a Silicon Graphics Iris Indigo
cornputer, using the consistent valence force field function (CVFF). A non-
bonded cutoff distance of 18 & with a switching distance of 2 A was employed.
The non-bonded pair list was updated every 20 cycles and a dielectric constant
of 1 was used in al1 calculations. The energy of the structure of the VVT enzyme
was minimized in stages. with initially only the water rnolecules being allowed to
move, then the water rnolecules and the amino acid side chains, and then the
entire enzyme. The mutated and chemically modified enzymes were generated
using the Builder module of Insight. These structures were then energy
minimized in a similar manner. Specifically, the side-chain of the mutated
residue and the water molecules were energy minimized. The amino acid side
chains within a 10 A radius of the a-carbon of the mutated residue were energy
minimized while al1 other enzyme residues were constrained, then al1 of the
atoms were energy minimized.

1. Valenzuela P. Bender ML. Kinetic Properties of Succinylated and Ethylenediamine-Amidated 6-Chymotrypsin. Biochim Biophys Acta 1971 ; 250153848.
2. Thomas PG, Russell AJ. Fersht AR. Tailoring the pH Dependence of Enzyme Catalysis Using Protein Engineering. Nature 1985; 31 8:375-6.
3. Sternberg MJE, Hayes FRF, Russell AJ, Thomas PG, Fersht AR. Prediction of Electrostatic Effects of Engineering of Protein Charges. Nature 1987; 330:86-8.
4. Russell AJ, Thomas PG, Fersht AR. Electrostatic Effects on Modification of Charged Groups in the Active Site Cleft of Subtilisin by Protein Engineering. J. Mol. Biol. 1987; 193:803-13.
5. Russell AJ, Fersht AR. Rational Modification of Enzyme Catalysis By Engineering Surface Charge. Nature 1987; 328:496-500.
6. Loewenthal R, Sancho J, Fersht AR. Histidine-Aromatic Interaction in Barnase Elevation of Histidine pKa and Contribution to Protein Stability. J. Mol. Biol. 1992; 224:759-70.
7. Siddiqui KS, Loviny-Anderton T, Ramgarajan M. Hartley SB. Althrobacter D-Xylose Isomerase: Chernical Modification of Carboxy Groups and Protein Engineering of pH Optimum. Biochem. J. 1993; 296:685-91.
8. Loewenthal R, Sancho J. Reinikainen T, Fersht AR. Long-Range Surface Charge-Charge l nteractions in Proteins: Cornparison of Experimental Results with Calculations from a Theoretical Method. J. Mol. Biol. 1993; (232):574-83.
9. Prévost M. Concurrent Interactions Contribute to the Raised pKa of Hisl8 in Barnase. J. Mol. Biol. 1996; 260(1):99-110.
10. Faber K. Biotransformations in Organic Synthesis. 3rd edition. Heidelberg: Springer-Vedag, 1997.
1 1. Wong C-H, Whitesides GM. Enzymes in Synthetic Organic Chemistry. Oxford: Pergamon Press. 1994.

houe M. Yamada H, Yasukochi T et al. Multiple Role of Hyrophobicity of Tryptophan-108 in Chicken Lysozyme: Structural Stability, Saccharide Binding Ability, and Abnomal pKa of Glutamic Acid-35. Biochemistry 1992; 31 (24):5545-53.
Berglund P, DeSantis G, Stabile MR et al. Chemical Modification of Cysteine Mutants of Subtilisin Bacillus lentus Can Create Better Catalysts Than the Wild-Type Enzyme. J. Am. Chem. Soc. 1997; 1 195265-6-
Schmidt DE. Westheimer FH. pK of the Lysine Amino Group at the Active Site of Acetoacetate Decarboxylase. Biochemistry 1 971 ; 1 O(7): 1 249-53.
Khouri HE, Plouffe C, Hasnain S, Hirama T, Storer AC, Ménarc! R. A Model to Explain the pH-Dependent Specificity of Cathepsin B-Catalyzed Hydrolyses. Biochem. J. 1991 ; 275:751-7.
ACDILog P Advanced Chemistry Development , Version 0.9 Beta.
Alberty RA, Massey V. On the lnterpretation of the pH Variation of the Maximum Initial Velocity of an Enzyme-Catalyzed Reaction. Biochim. Biophys. Acta 1 954; 1 3:347-53.
Jordan F, Polgar L, Tous G. Proton Magnetic Resonance Studies of the States of lonization of Histidines in Native and Modified Subtilisin. Biochemistry 1985; 24:7711-7.
Bronsted JN, Pederson K. Die Kataltische Zersetzung des Nitramids und l hre Physikalische-Chemische Bedeutung. Z. Phys. Chem. 1924; 108:185- 235.
Cassidy CS, Lin J, Frey PA. A New Concept for the Mechanism of Action of Chymotrypsin: The Role of the Low-Bamer Hydrogen Bond. Biochemistry 1997; 3614576.84.
Richard JP. The Enhancement of Enzymatic Rate Accelerations by Brmsted Acid-Base Catalysis. Biochemistry 1998; 37:4305-9.
Laidler KJ, Bunting PS. The Chemical Kinetics of Enzyme Action. 2nd edition. Oxford: Clarendon Press, 1973: 142-62.

23. Bonneau PR. Graycar TP, Estell DA, Jones JB. Alteration of the Specificity of Subtilisin BPN' by Site-Directed Mutagenesis in Its S, and S,' Binding Sites. J. Am. Chem. Soc. 1991; 113:1026-30.
24. Fersht A. Enzyme Structure and Mechanism. 2nd edition. New York: W.H. Freeman and Company, 1985.
25. Knapp M, Daubennann J. Bott RR. Brookhaven Database Entry 1 JEA.
26. lnsight II [Biosym Technologies, Inc. San Diego, CA, USA]. Version 2.3.0.
27. Discover [Biosym Technologies, Inc. San Diego, CA, USA]. Version 2.9.5.
28. DeSantis G, Jones JB. Chernical Modifications at a Single Site Can lnduce Significant Shift in the pH-Activity Profiles of a Serine Protease. J. Am. Chem. Soc. 1998; 120(34):8582-6.

Perspective

The goals of this study were to ~~n t r iou te to the understanding of the
factors that control enzyme specificity, to develop a method to overcome the 20-
amino acid limitation of conventional site-directed mutagenesis, and to create
novel enzyme specificities to fuither expand the synthetic applicability of the
serine proteases. The strategy of chemical modification and site directed
mutagenesis of subtilisin Bacillus lentus was shown to be a rapid and convenient
one in this regard. The choice of residues S156, SI66 and N62 for mutagenesis
to cysteine and subsequent modification proved to be appropriate, and effected
significant changes in the activity, specificity, binding properties. and pH-activity
profiles of SBL. The most dramatic changes in the catalytic properties of SBL-
CMMs were rationalizable by molecular modelling analysis.
Subtilisin has proven to be an ideal template for this approach. However,
the CMM approach is not necessarily limited to cysteine-free enzymes since
cysteine residues are usually buried in proteins' and therefore will not react
readily with the MTS modifying agents used. Thus. introduced cysteine residues
on the active site surface can be selectively modified in the presence of buried
ones. Alternatively, site-directed mutagenesis could be used to remove
potentially reactive natural cysteine residues in order to ensure the desired regio-
selectivity of modification by the methanethiosulfonate reagents.
This method is amenable to a combinatorialz approach to generating
libraries of CMM enzymes. which may be subjected to selection screens to
identify a desirable property. such as improved esterase-to-amidase ~electivity.~
In addition. the CMM approach can address the challenges of generating
precisely glycosylated proteins in vitroe4 A further important application of CMMs
is for novel organic synthesis. The evaluation of the preparative viability of the
CMMs generated in this study has already begun and the preliminary results are
very positive.'
A further benefit of the current approach may be to provide a rapid and
convenient screen identifying the most desirable types of amino acid

replacements for a given enzyme location by traditional protein engineering
methodology.

References
1. Rose G, Geselowitz A. Lesser G. Lee R. Zehfus M. Hydrophobicity of Amino Acid Residues in Globular Proteins. Science 1985; 229:834-8-
2. Plettner E. Kumtaveepom K, Shang X, Jones JB. Cornbinatonal Approach to Chemical Modification of Subtilisin Bacillus lentus. Bioorg. Med. Chem. Lett. 1998; 8(17):2291-6.
3. Plettner E. DeSantis G, Stabile MR. Jones JB. Systematic Tailoring of Esterase and Arnidase Activity of Subtilisin Bacillus fentus by Chemical Modification. submifted.
4. Davis BG, Lloyd RC, Jones JB. Controlled Site-Selective Glycosylation of Proteins by a Combined Site-Directed Mutagenesis Chemical Modification Approach. J. Org- Chem. in press.
5. Kumtaveeporn K. Matsumoto K. Jones JB. unpublished results.

Appendix
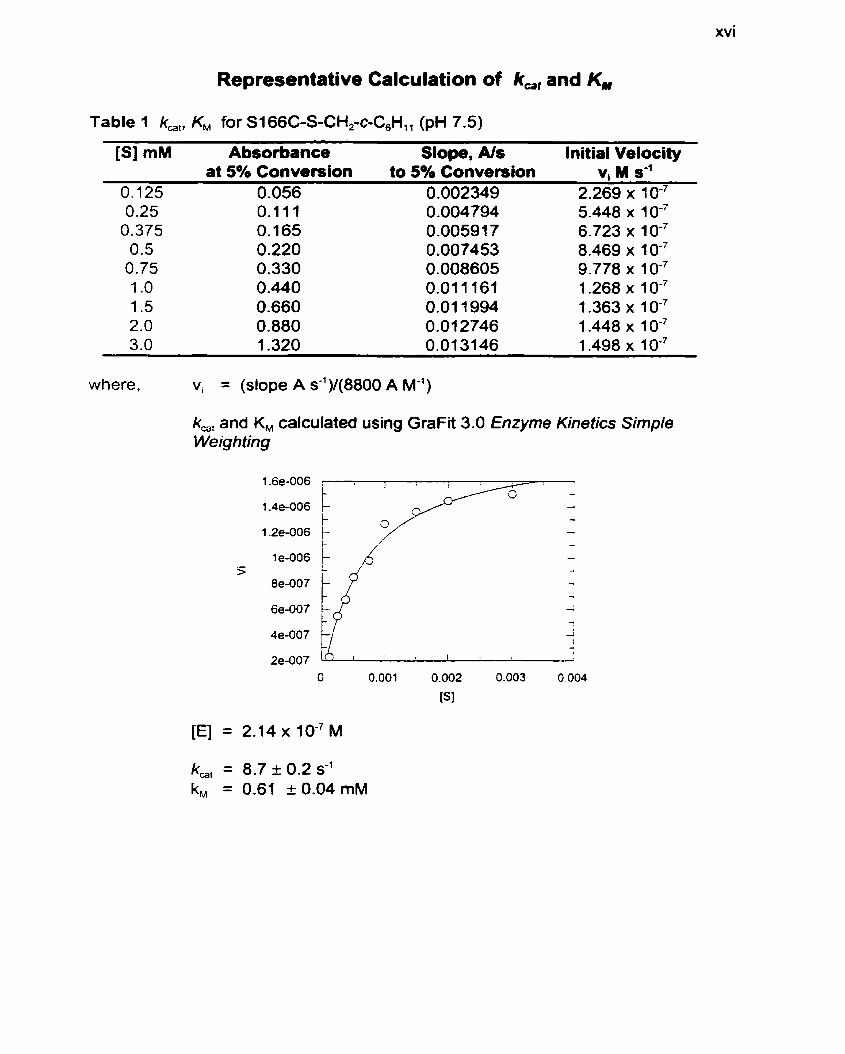
xvi
Representative Calculation of k,, and KM
Table 1 km,, KM for S166C-S-CH2-c-C,H,, (pH 7.5)
[SI mM Absorbance Slope, AIS Initial Velocity at 5% Conversion to 5% Conversion vi M s-'
O. 125 0.056 0.002349 2.269 x 10-7 0.25 0.1 11 0.004794 5.448 x
0.375 0.165 0.00591 7 6.723 x 0.5 0.220 0.007453 8.469 x 1 O-7
0.75 0.330 0.008605 9.778 x 1 O-7 1 .O O -440 0.01 1 161 1.268 x 1 1.5 0.660 0.01 1994 1.363 x 1 0m7 2.0 0.880 0.01 2746 1.448 x 1 3. O 1.320 0.01 3146 1 -498 x 1 O-7
where, vi = (slope A s-')1(8800 A M-')
k,, and KM calculated using GraFit 3.0 Enzyme Kinetics Simple Weighting
k,, = 8.7 + 0.2 s-' k, = 0.61 + 0.04 mM

xvii
Representative Calculation of K,
Table 2 K, for WT-SBL and 3-aminoohenvi boronic acid % Conversion A ,,, ,, t sec & sec ln(SdS) t-t, sec
0.1 5 0.331 42 -4 9 O. 1625 33.4
where, &IO ,, = (A)(% Conversion)
Beer's Law A = ECC = (8800 M-' cm-')(2.5 x 10" M)(1 cm) = 2.2
t = time for inhibited enzyme reaction to reach absorbance indicated t, = time for the uninhibited enzyme reaction to reach absorbance
ln(SJS) = ln (1 OO/85)=O. 1 625 S, = 100% [substrate] S = [substrate] at % conversion
GraFit 3.0 is used to plot In(SdS) against t-4. Simple weighting linear regression is applied to slope and error.
K, = Kp1 I1 c K, ~(AKJK,)~ + (AV-N,)~ + (A sIope/~Iope)~ V,,, l slope





























