Embed Size (px)
Citation preview

J. Biochem. Biophys. Methods 47 (2001) 5–19www.elsevier.com/ locate / jbbm
Mutation detection by capillary denaturinghigh-performance liquid chromatography using
monolithic columns
a , a b*Christian G. Huber , Andreas Premstaller , Wen Xiao ,a a b¨Herbert Oberacher , Gunther K. Bonn , Peter J. Oefner
aInstitute of Analytical Chemistry and Radiochemistry, Leopold-Franzens University, A-6020 Innsbruck,Austria
bGenome Technology Center, Stanford University, Palo Alto, CA 94304, USA
Received 29 June 2000; accepted 10 July 2000
Abstract
The high resolving power of the chromatographic separation of single- and double-strandednucleic acids in 200 mm i.d. monolithic poly(styrene–divinylbenzene) capillary columns wasutilized for mutation screening in polymerase chain reaction amplified polymorphic loci.Recognition of mutations is based on the separation of homo- and heteroduplex species by ion-pairreversed-phase high-performance liquid chromatography (IP-RP-HPLC) under partially denaturingconditions, resulting in characteristic peak patterns both for homozygous and heterozygoussamples. Six different single nucleotide substitutions and combinations thereof were confidentlyidentified in 413 bp amplicons from six heterozygous individuals each of which yielded a differentunique chromatographic profile. Alternatively, mutations were identified in short, 62 bp PCRproducts upon their complete on-line denaturation at 758C taking advantage of the ability ofIP-RP-HPLC to resolve single-stranded nucleic acids of identical length that differ in a singlenucleotide. Separations in monolithic capillary columns can be readily hyphenated to electrosprayionization mass spectrometry and promise increased sample throughput by operating in arrayssimilar to those already used in capillary electrophoresis. 2001 Published by Elsevier ScienceB.V.
Keywords: Denaturing high-performance liquid chromatography; Monoliths; Mutation detection
¨ ¨*Corresponding author. Institut fur Analytische Chemie und Radiochemie, Leopold-Franzens-Universitat,Innrain 52a, 6020 Innsbruck, Austria. Tel.: 143-512-507-5176; fax: 143-512-507-2767.
E-mail address: [email protected] (C.G. Huber).
0165-022X/01/$ – see front matter 2001 Published by Elsevier Science B.V.PII : S0165-022X( 00 )00147-0

6 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
1. Introduction
With the whole sequence of the human genome available in the year 2000, thedetection of mutations and polymorphisms is becoming increasingly important in thefields of genetics, molecular diagnostics, and cancer research. Methods used for thedetection of unknown mutations include single-strand conformation polymorphismanalysis [1], denaturing gradient gel electrophoresis [2], temperature gradient gelelectrophoresis [3], mismatch cleavage methods [4,5], direct sequencing [6], oligo-nucleotide arrays [7], as well as gel electrophoresis [8] and high-performance liquidchromatography-based [9] heteroduplex detection.
Denaturing high-performance liquid chromatography (DHPLC) has shown greatpotential in detecting efficiently and sensitively single-base substitutions as well as smalldeletions and insertions in DNA fragments ranging from 100 to 1500 base pairs in size[10,11]. Partially denaturing HPLC is based on the separation of homo- from heterodup-lex species generated by polymerase chain reaction (PCR) amplification of polymorphicloci containing one or more mismatches by ion-pair reversed-phase HPLC (IP-RP-HPLC) at elevated column temperatures in the range 48–678C [12]. Recently, it hasbeen demonstrated that the high resolving power of IP-RP-HPLC in combination withthe sequence dependence of the retention of single-stranded nucleic acids enables thedetection of mutations in short PCR products ( , 100 bp) under completely denaturingconditions [13].
However, despite its high sensitivity and productivity, further improvements inDHPLC technology are necessary to achieve higher throughput, information content,cost-effectiveness, and reliability of the method required for routine mutation screening.One attractive means to enhance sample throughput is the miniaturization of theseparation channel by using capillary columns of 50–320 mm inner diameter [14]. Suchcapillaries can then be bundled into arrays similar to those used in capillary electro-phoresis [15]. In addition, the low flow rates applied in capillary HPLC are highly suitedfor on-line hyphenation with mass spectrometry [16,17].
Due to their excellent chemical and physical stability, micropellicular, alkylatedpoly(styrene–divinylbenzene) (PS/DVB-C18) beads of 2–3 mm diameter packed intocolumns of 50 3 4.6 mm i.d. have become the premier column packing material forseparating single- and double-stranded nucleic acids with high resolution [18,19]. Novelfrit designs have enabled the reduction of the inner column diameter to 200 mm andallowed the analysis of femtomole amounts of DNA by capillary IP-RP-HPLC [20].Subsequently, the in situ synthesis of a continuous, rod-shaped, porous copolymer ofstyrene and divinylbenzene inside a 200 mm i.d. fused-silica capillary eliminates theneed for tedious frit preparation due to the covalent attachment of the polymer to theinner surface of the capillary [17,21]. In addition, such monolithic PS/DVB capillarycolumns have been found to provide up to 40% improved separation efficiency overpacked column beds.
In this communication, we report on the applicability of monolithic poly(styrene–divinylbenzene) (PS/DVB) capillary columns to the detection of single nucleotidepolymorphisms (SNPs) in genomic DNA under both partial and complete thermaldenaturation of PCR products. The chromatographic performance of the monolithic

C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 7
columns is critically evaluated and compared with the performance of conventionalstate-of-the-art 4.6 mm i.d. columns packed with 2 mm micropellicular PS/DVB-C18particles.
2. Materials and methods
2.1. Sequences and reagents
The following three sequences, with priming regions typed in lower case and positionsand chemical nature of polymorphic sites indicated in parentheses, were investigated:
Sequence 1, 209 bp, aggcactggtcagaatgaagTGAATGGCACACAGGACAAGTCC
AGA CCCA GGAAGGTCCAGT AAC ATGGGAG AA GAACGG AAGGAGTTCTAAA
ATT CAGGGCT CCCTT GGGCTCCCCTGT TTAAAAATGTAGGTTTTATTATTATA
TTTCATTGTTAACAAAAGTCC(A / G)TGAGATCTG TGGAGGATAAAGggggagct
gtattttccatt
Sequence 2, 413 bp, gggggtataagtataaacaaaacTGACCCCATCGCTGCCCTCTTG
GAGCTGAG AGT CTC ATAAA CAGCT TTAA GGTAATAAAATCATTTT(C / A)TGT
GCCACAGGAT(G /A)TGAGTTGGTTTGATGACCCTAAAAACACCACTGGAGCA
TTGACT ACCAGGCT CGCCAATGATGCTG CTCA AGTTAA AGGGGTACGTGCC
TCCTTTCTACTGGT(G / A)TTTGTCTTAATTGGC(C / T)ATTTTGGACCCCAGCAT
GAAACTAATTTTCTC(C /A)TTA(C / T)GGGTGTTAGTTATCATCATTAAGAAAA
TGTTGAATAAATATCTAACCTACGAATATATCACATGCTTTTTGTAGCAACATGTTAACTATTTAAACATTATATACTGTAGAGCATATAGATAACTTATAAAccatttgctattgctgttatt
Sequence 3, 62 bp, cccaaacccattttgatgctT(G/T)ACTTAAaaggtcttcaattattattttcttaaat
attttg
All oligonucleotide primers were obtained from Life Technologies (Rockville, MD,USA). The oligonucleotide sizing marker containing 8–32-mers of the sequenced(GACT) and d(GACT) GT was obtained from Amersham Pharmacia Biotechn n
(Uppsala, Sweden). MspI and HaeIII digests of pBR322 and pUC18, respectively, wereobtained from New England Biolabs (Beverly, MA, USA) and Sigma (St. Louis, MO,USA). A stock solution of triethylammonium acetate (TEAA), pH 7.0, was obtainedfrom PE Biosystems (Foster City, CA, USA) or prepared by dissolving equimolaramounts of triethylamine (Fluka, Buchs, Switzerland) and glacial acetic aced (Fluka) inwater. HPLC-grade acetonitrile was purchased from J.T. Baker (Phillipsburg, NJ, USA)

8 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
or Merck (Darmstadt, Germany). High-purity water (E-pure, Barnstead Co., Newton,MA, USA) was used for preparing the mobile phase.
2.2. Polymerase chain reaction
Polymerase chain reactions were performed in a 50 ml volume containing 10 mMTris–HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl , 0.1 mM each of the four dNTPs, 0.22
mM of each primer, 50 ng of genomic DNA, and 1 unit of AmpliTaq Gold (PEBiosystems). The PCR cycling regime carried out in a Perkin-Elmer 9600 thermal cyclercomprised an initial denaturation step at 958C for 10 min to activate AmpliTaq Gold, 14cycles of denaturation at 948C for 20 s, primer annealing for 1 min at 63–568C with0.58C decrements, and extension at 728C for 1 min, followed by 20 cycles at 948C for 20s, 568C for 1 min, and 728C for 1 min. Following a final extension step at 728C for 5min, samples were chilled to 68C. Prior to DHPLC under partially denaturing conditions,all PCR products were denatured once more at 948C for 3 min and allowed to renatureover 30 min by decreasing the temperature from 95 to 658C. This ensures the formationof equimolar ratios of homo- and heteroduplex species.
2.3. High-performance liquid chromatography
The instumentation for conventional HPLC consisted of an on-line degasser (DG1210,Uniflows Co., Tokyo, Japan), two SD-200 high-pressure pumps, an electronic pressuremodule, a 600 ml dynamic mixer, a six-port injection valve mounted into a MISTRALcolumn oven, an automated sample injector (AI-1A), a Dynamax UV-absorbancedetector set at 254 nm, and a PC-based system controller and data analysis package(Varian, Walnut Creek, CA, USA). Preheating of the mobile phase was accomplished inan 80 cm 254 mm i.d. PEEK tubing that had been encased in a tin-alloy block(Timberline, Inc., Boulder, CO, USA). The stationary phase consisted of 2 mmmicropellicular, alkylated PS/DVB particles [18] packed into 5034.6 mm i.d. columns,which are commercially available (DNASepE, Transgenomic, San Jose, CA, USA). Themobile phase was 0.1 M triethylammonium acetate at pH 7.0, containing in addition 0.1mM Na EDTA (Sigma). DNA restriction fragments and crude PCR products were4
eluted with linear acetonitrile gradients at a flow rate of 0.9 ml /min.The capillary HPLC system consisted of a low-pressure gradient micro pump (LC
Packings, Amsterdam, Netherlands) controlled by a personal computer, a vacuumdegasser (Uniflows Co.), a MISTRAL column oven, a six-port valve injector with a 1 mlsample loop (Valco Instruments, Houston, TX, USA), a variable wavelength detector(UltiMate UV detector, LC Packings) with a Z-shaped capillary detector cell (UZ-ULT-NIO, 3-nl cell, LC Packings), and a PC-based data system (UltiChrom, LC Packings).Preheating of the mobile phase and consequent partial or complete denaturation of DNAfragments was obtained with a 20 cm piece of 25 mm i.d. fused-silica capillarypositioned in the oven between the injector (mounted outside the oven) and the column.Monolithic capillary columns were prepared according to the previously publishedprotocol [17].

C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 9
3. Results and discussion
3.1. Separation of single-stranded and double-stranded nucleic acids
For the successful downscaling of a chromatographic separation from a conventionalcolumn of inner diameter 4.6 mm to a miniaturized inner diameter of 0.2 mm, a numberof instrumental parameters have to be modified and optimized. An HPLC instrumentcapable of performing separations utilizing capillary columns of 50–320 mm i.d. must beconfigured with a low dispersion valve for the injection of typically 20 nl to 5 ml ofsample. It must be able to reproducibly deliver gradients at low flow rates (100 nl /minto 5 ml /min), and requires a sensitive detector with a low-volume detection cell (1–60nl) [22]. Reproducible gradients are conveniently obtained by splitting a relatively highprimary flow of mobile phase delivered by a conventional gradient pumping system bymeans of a T-piece [23]. Thus, only a small portion of the mobile phase is passed via theinjector onto the separation column, whereas the main flow of the mobile phase goes towaste. Since the primary flow is usually in the range of 100–500 ml /min, a reduction bya factor of 2–10 in solvent consumption is usually feasible compared to conventional 4.6mm i.d. columns operated at a flow of 1 ml /min. Another important consideration withmicroscale gradient HPLC separations is the so-called gradient delay time, the timeelapsing between formation of the gradient in the mixing system and entering of thegradient into the separation column. The lower the flow rate, the more time is needed toflush a given volume between the mixing device and the separation column. Therefore,careful attention has to be paid to minimize the volumes of all connecting tubing in theHPLC system, especially after the splitting device.
Fig. 1 illustrates the separation of a ladder of mixed-sequence single-strandedoligonucleotides ranging in size from eight to 32 nucleotide units and differing in lengthby two nucleotide units. It can be seen that all oligonucleotides are well separated,leaving enough peak capacity for obtaining baseline separation of oligonucleotides withsingle nucleotide resolution over the entire investigated size range. The significantinfluence of base sequence on the retention of single-stranded oligonucleotides has beenreported previously [13,18,24] and can also be deduced from close examination of theretention differences of neighboring pairs of oligonucleotides in Fig. 1. The difference inretention times between the 8-mer and the 10-mer is bigger than that observed betweenthe 10-mer and the 12-mer. The same alternating smaller and larger retention differenceswere observed with all subsequent peaks, suggesting that the addition of G and Tcontributes more to retention than addition of A and C.
The slight peak asymmetry which is greater for early eluting peaks indicates an excessof extra-column volume. A reduction of the extra-column volume to less than 10% ofthe peak retention volume will require modifications of the existing instrumentation,including the positioning of the injection valve and sample loop in the column oven.Reduction of connecting volumes in the instrument will also help to reduce the relativelylarge gradient delay time of 6 min in the chromatogram of Fig. 1. Such a modificationwill ensure more rapid and improved resolution of single-stranded oligo- and polynu-cleotides with increasing column temperature as reported recently [14]. With the currentinstrumentation the resolution was highest at 608C, where the resolution values ranged

10 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
Fig. 1. Chromatogram of mixed sequence oligonucleotide sizing markers eight to 32 bases in length separatedin a monolithic capillary column. Column, monolithic PS/DVB, 5030.2 mm i.d.; mobile phase, (A) 100 mMTEAA, 0.1 mM Na EDTA, pH 7.0, (B) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, 25% acetonitrile; linear4 4
gradient, 0–5% B in 1.0 min, 5–15% B in 2.0 min, 15–25% B in 8.0 min, 25–30% B in 4 min; flow rate, 3.0ml /min; temperature, 608C; detection, UV, 254 nm; injection volume, 150 nl; sample, 8–32-mer oligonucleo-tides, 0.5–1.7 ng/ml each.
from 7.09 between the 8-mer and the 10-mer to 2.69 between the 30-mer and the32-mer.
The most significant advantage of capillary HPLC is the better signal height-to-samplemass ratio, as the peak concentration is proportional to the inverse square of the columndiameter [25]. This means that the same amount of analyte will give, in theory, a signalthat is approximately 500 times higher with a 0.2 mm than with a 4.6 mm column. Inreality, this translates into significantly smaller injection volumes of the order of a fewhundred nanoliters with the detection limit being as low as 3 fmol for an 18-meroligonucleotide using UV absorbance at 254 nm. This is almost as sensitive asfluorescence detection of fluorescent dye-labeled nucleic acids using a 4.6 mm column,with lower limits of detection ranging from 0.5 to 3 fmol depending on the attachedfluorophore [26]. Hence, while the savings in HPLC solvent may only be on the order ofa factor of 2–10, far more significant savings can be accomplished in PCR reagentconsumption as amplifications can be carried out in volumes as small as 0.5–1 ml.
Fig. 2 shows the separation of 30 double-stranded DNA restriction fragments usingboth a 5030.2 mm i.d. monolithic capillary column and a conventional, microparticulate5034.6 mm i.d. packed column. As discussed above, the longer time required for
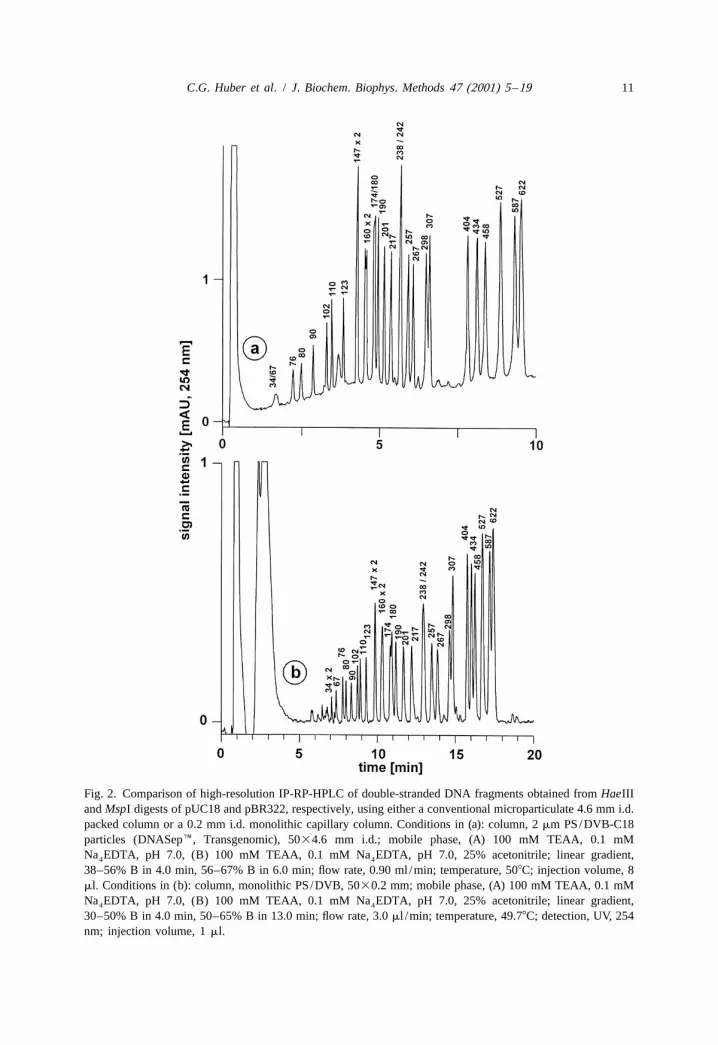
C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 11
Fig. 2. Comparison of high-resolution IP-RP-HPLC of double-stranded DNA fragments obtained from HaeIIIand MspI digests of pUC18 and pBR322, respectively, using either a conventional microparticulate 4.6 mm i.d.packed column or a 0.2 mm i.d. monolithic capillary column. Conditions in (a): column, 2 mm PS/DVB-C18particles (DNASepE, Transgenomic), 5034.6 mm i.d.; mobile phase, (A) 100 mM TEAA, 0.1 mMNa EDTA, pH 7.0, (B) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, 25% acetonitrile; linear gradient,4 4
38–56% B in 4.0 min, 56–67% B in 6.0 min; flow rate, 0.90 ml /min; temperature, 508C; injection volume, 8ml. Conditions in (b): column, monolithic PS/DVB, 5030.2 mm; mobile phase, (A) 100 mM TEAA, 0.1 mMNa EDTA, pH 7.0, (B) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, 25% acetonitrile; linear gradient,4 4
30–50% B in 4.0 min, 50–65% B in 13.0 min; flow rate, 3.0 ml /min; temperature, 49.78C; detection, UV, 254nm; injection volume, 1 ml.

12 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
separation with the monolithic column is a consequence of the greater gradient delaytime with the capillary HPLC instrument. Nevertheless, the separation efficiency of bothcolumns is equivalent, allowing the separation of DNA fragments that differ by less than5% in size. The resolution obtained with monolithic capillaries may be further improvedby reducing the dead volumes in the chromatographic system. A close inspection of thechromatograms reveals partial resolution of the two 160 bp fragments, whereas the 174and 180 bp fragments are hardly resolved. As demonstrated previously [27], suchanomalies are a consequence of differences in base composition with very AT-richfragments undergoing partial denaturation already at 508C, which results in their reducedretention. Such reductions in retention are also observed in the presence of mismatchesthat disrupt the DNA helix. This constitutes the basis for the successful detection ofsingle-base substitutions and small insertions and deletions by denaturing high-per-formance liquid chromatography [9].
A comparison of the gradient profiles applied in Fig. 2 reveals that a lowerconcentration of acetonitrile is required to elute the DNA restriction fragments from themonolithic column. Taking into consideration a gradient delay time of 0.7 min with theconventional HPLC instrument, the DNA fragments elute at acetonitrile concentrationsbetween 11.5 and 16.3% (Fig. 2a). On the other hand, with a gradient delay time of 6min in the capillary HPLC instrument, the fragments elute between 10.0 and 14.7%acetonitrile (Fig. 2b). This is due to the lower degree of non-polarity of the monolithicPS/DVB stationary phase, which is not alkylated, in contrast to the PS/DVB particles,which are derivatized with highly non-polar octadecyl groups [18].
3.2. Partially denaturing HPLC for mutation detection
Partially denaturing HPLC exploits the reduced retention of heteroduplex moleculescontaining one or more mismatches at elevated column temperatures compared to acorresponding perfectly matched homoduplex. The reliability of DHPLC in detectingsingle nucleotide substitutions as well as short deletions and insertions is due to the factthat most mismatches can be detected over a temperature range of several degrees [10].This is exemplified in Fig. 3, which shows the successful detection of an A to Gtransition in a 209 bp amplicon between 55.8 and 59.88C. The presence of a mismatch isgenerally recognized by the appearance of one or more additional peaks. The number ofpeaks detected is a function of the nature and the number of mismatches as well as thecolumn temperature.
As shown in Fig. 3, at a temperature of 57.88C all four species of hetero- andhomoduplices can be separated. The order of elution of the four species is dictated bythe degree of destabilization of the DNA helix, which is influenced by both neighboringstacking interactions [28] and the ability of certain bases to form atypical Watson–Crickbase pairs [29]. The thermal stabilities of typical Watson–Crick and mismatched basepairs as a function of the flanking base pairs in otherwise homologous 373 bp DNAfragments have been determined previously by temperature-gradient gel electrophoresis[28]. According to the ranking of stabilities provided in that study, one would expect thehomoduplex with the AT-base pair to be retained less than that with the GC-base pair.And indeed, separate injection of the two homoduplices as well as a mixture of the two

C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 13
Fig. 3. Resolution of 209 bp homo- and heteroduplex DNA fragments containing a single A→G transitionunder partially denaturing conditions. Column, PS/DVB monolith, 5030.20 mm i.d.; mobile phase, (A) 100mM TEAA, 0.1 mM Na EDTA, pH 7.0, (B) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, 25% acetonitrile;4 4
linear gradient, 30–50% B in 3.0 min, 50–70% B in 7.0 min; flow rate, 3.0 ml /min; temperature, 54.7–59.88C;detection, UV, 254 nm; injection volume, 500 nl; sample, sequence 1.
that had not been denatured and reannealed confirmed the expected elution order. Withregard to the two heteroduplices, the GT heteroduplex should be retained longer becausethe flanking base pairs have been determined to stabilize this mismatch more than theAC mismatch. In addition, two hydrogen bonds can form between G and T, and thisatypical Watson–Crick base pair stacks within a DNA B-conformation duplex withrelatively little distortion, as indicated by NMR and X-ray crystallographic studies [1].
As shown in Fig. 4, different mismatches or combinations of mismatches (Table 1)tend to generate different chromatographic profiles. Consequently, sequence analysis,which is still required to determine the exact location and nature of mismatches, can belimited to a few representative profiles. However, identical profiles for differentmutations located within the same melting domain but not at the same nucleotideposition have been reported [30]. The chromatographic profiles obtained with amonolithic 0.2 mm i.d. capillary column (Fig. 4) are very similar to those observed witha 4.6 mm i.d. microparticulate column (Fig. 5), although a 28C higher columntemperature was required. Since the residence times in the preheating loops betweeninjector and column with both the conventional and capillary HPLC instrument are

14 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
Fig. 4. Influence of the nucleotide position, the number of mismatches, and column temperature on thechromatographic separation profiles obtained for one homozygous control and six heterozygotes under partiallydenaturing conditions using a monolithic 200 mm i.d. capillary column. Column, monolithic PS/DVB, 6030.2mm i.d.; mobile phase, (A) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, (B) 100 mM TEAA, 0.1 mM4
Na EDTA, pH 7.0, 25% acetonitrile; linear gradient, 43–50% B in 0.5 min, 50–54% B in 3.5 min; flow rate, 34
ml /min; temperature, 61 and 578C; injection volume, 500 nl; sample, sequence 2. See Table 1 for the natureand location of mismatches underlying the chromatographic profiles.
practically identical (2.7 versus 2.5 s), the difference in temperature is due to the morepolar character of the monolith surface. Thus, lower concentrations of acetonitrile arerequired for the elution of the amplicons, which in turn results in higher stability of thedouble helical structure at a given temperature. All heterozygote profiles except one areclearly recognized as such. The only exception is Aus23, which displays only a trailingshoulder indicating that the melting domain in which the mutation is located hasundergone almost complete denaturation both in the heteroduplex as well as in thehomoduplex species. This was confirmed by re-running the sample at a columntemperature 48C lower, where a clean heteroduplex profile is observed.
3.3. Completely denaturing HPLC for mutation detection
It has been noticed that DNA fragments shorter than approximately 150 bp are toounstable to allow the detection of mutations by partially denaturing HPLC. Thislimitation can be overcome by the complete on-line denaturation of short amplicons into

C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 15
Table 1Position and nature of mismatches contained in 413 bp amplicons of six heterozygous individuals and ahomozygous control. The sequence included exon 20 and flanking non-coding regions of the humanP-glycoprotein (MDR1, Genbank Acc. No. M14758)
a bIndividual Positions of mutations from 59-end of forward primer(positions within the MDR1 sequence)
91 105 209 225 258 262(2398211) (2401) (2481124) (2481140) (2481173) (2481177)
P218 C G G C C CP100G C G G/A C C CSD18 C/A G G C C CGM2064A C G G/A C/T C/A CP111G C G G C/T C/A COM135 C G/A G C C CAus23 C G G C C C/T
a Individuals are from the Stanford human diversity panel.b Sequence 2, see Materials and methods.
their single-stranded components which are then resolved on the basis of their sequencecomposition rather than their size [13]. Since differences in base composition as small asa single base out of 100 bases suffice to separate two single-stranded nucleic acids ofidentical size, the alleles of a given polymorphic locus can be resolved without theaddition of a reference chromosome. The only exceptions to this rule have been C to Gtransversions [13]. Fig. 6 shows an example of allelic discrimination based on theseparation of the single-stranded components of a 62 bp PCR product containing a singleG to T transversion by means of DHPLC on a monolithic capillary column undercompletely denaturing conditions. The chromatograms in Fig. 6a and b illustrate twohomozygous samples, where the two completely denatured single strands of the samechain length are completely separated by IP-RP-HPLC. In the case of a heterozygotesample, three peaks are observed corresponding to two coeluting and two separatedsingle strands (Fig. 6c). As mentioned above, the single base change affects the retentionof the single-stranded components sufficiently to allow the discrimination of mutatedfrom wild-type DNA in at least one pair of corresponding DNA strands. Moreover, theseparation on the monolithic column is very similar to that reported recently for thispolymorphic locus on a microparticulate 4.6 mm i.d. column (compare Fig. 6f in Ref.[13]).
One distinct advantage of capillary IP-RP-HPLC is the feasibility of coupling thisseparation system on-line to mass spectrometry. This will allow positive confirmation ofthe identity of the resolved single-stranded components and the unambiguous genotypingof the amplified PCR fragments. One very crucial step in mass spectrometric analysis ofnucleic acids is the proper desalting and removal of low-molecular-weight impurities inthe samples in order to obtain mass spectra of good quality [31]. The on-line purificationof nucleic acids by HPLC prior to electrospray ionization mass spectrometry (ESI-MS)is very attractive because it not only removes cations from nucleic acid samples, but italso allows fractionation of nucleic acid mixtures that are otherwise too complex fordirect infusion ESI-MS. Moreover, the identification even of heterozygous alleles eluting

16 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
Fig. 5. Influence of the nucleotide position, the number of mismatches, and column temperature on thechromatographic separation profiles obtained for one homozygous control and six heterozygotes under partiallydenaturing conditions using a conventional microparticulate 4.6 mm i.d. column. Column, 5034.6 mm i.d.stainless steel column packed with 2 mm PS/DVB-C18 particles (DNASepE, Transgenomic); mobile phase,(A) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, (B) 100 mM TEAA, 0.1 mM Na EDTA, pH 7.0, 25%4 4
acetonitrile; linear gradient, 50–52% B in 0.5 min, 52–59% B in 3.5 min; flow rate, 0.9 ml /min; columntemperature, 59 and 558C; injection volume, 9 ml; sample, sequence 2. See Table 1 for the nature and locationof mismatches underlying the chromatographic profiles.
as one single chromatographic peak should become feasible because ESI-MS candirectly analyze and deconvolute simple mixtures of nucleic acids [32].
The mobile phase components usually applied in IP-RP-HPLC, namely water,acetonitrile, triethylamine, and acetic acid, are volatile, and, therefore, suitable for directinterfacing with ESI-MS. Recently, it has been demonstrated that the solvent com-position has a tremendous effect on the mass spectrometric detectability of nucleic acids[16,33]. A reduction in the eluent conductivity through a decrease in ion-pair reagentconcentration from 100 to 25 mM, and the exchange of acetic acid with carbonic acidresulted in considerable gains in mass spectrometric signal intensities at the cost of onlya small decrease in chromatographic separation efficiency. Additionally, an increase inthe proportion of organic solvent in the electrosprayed solution upon post-columnaddition of acetonitrile as sheath liquid through the triaxial electrospray probe was foundto greatly enhance the detectability of nucleic acids in ESI-MS. Under such optimizedconditions, purification, separation, detection, and mass characterization of low fem-

C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 17
Fig. 6. Direct allelic discrimination of two alleles based on their different retention under completelydenaturing conditions using a monolithic capillary column. Column, monolithic PS/DVB, 5030.2 mm i.d.;mobile phase, (A) 100 mM TEAA, pH 7.0, (B) 100 mM TEAA, pH 7.0, 20% acetonitrile; linear gradient,36–48% B in 10.0 min; flow rate, 3.0 ml /min; temperature, 758C; detection, UV, 254 nm; injection volume,500 nl; sample, sequence 3, (a) homozygous G, (b) homozygous T, (c) heterozygous G/T.

18 C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19
tomole amounts of nucleic acids in real matrices such as PCR reactions can be realizedin a time frame of a few minutes.
4. Conclusions
It has been demonstrated that monolithic 200 mm i.d. columns are highly suited forthe detection of mutations by capillary denaturing high-performance liquid chromatog-raphy. Miniaturization of the separation channel did not affect the resolving power of thesystem. Moreover, it allowed a reduction in solvent consumption by a factor of 3 andreduced the amount of sample required for analysis to a few hundred nanoliters. Futuredevelopments utilizing the developed miniaturized column technology will certainlyinclude the bundling of capillaries into arrays coupled with multi-color fluorescencedetection and hyphenation of the separation system with mass spectrometry, which willsignificantly improve the productivity of DHPLC for high-throughput mass screening forknown and unknown mutations.
Acknowledgements
This study was supported by the Austrian Science Fund (P-14133-PHY) and NIHgrant HG01932.
References
[1] Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA bygel electrophoresis as single-strand confirmation polymorphisms. Proc Natl Acad Sci USA1989;86:2766–70.
[2] Myers RM, Lumelsky N, Lerman LS, Maniatis T. Detection of single base substitutions in total genomicDNA. Nature 1985;313:495–8.
[3] Tee MK, Moran C, Nicholas FW. Temperature gradient gel electrophoresis: detection of a single basesubstitution in the cattle beta-lactoglobulin gene. Anim Genet 1992;23:431–5.
[4] Gibbs RA, Caskey CT. Identification and localization of mutations at the Lesch-Nyhan locus byribonuclease A cleavage. Science 1987;236:303–5.
[5] Cotton RG, Rodrigues NR, Campbell RD. Reactivity of cytosine and thymine in single-base-pairmismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations. ProcNatl Acad Sci USA 1988;85:4397–401.
[6] Rosenthal A, Charmock JDS. New protocols for DNA sequencing with dye terminators. DNA Seq1992;3:61–4.
[7] Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D et al. Accessing genetic information withhigh-density DNA arrays. Science 1996;274:610–4.
[8] Nagamine CM, Chan K, Lau YFC. A PCR artifact: generation of heteroduplexes. Am J Hum Genet1989;45:337–9.
[9] Oefner PJ, Underhill PA. Comparative DNA sequencing by denaturing high-performance liquidchromatography (DHPLC). Am J Hum Genet 1995;57(Suppl):A266.

C.G. Huber et al. / J. Biochem. Biophys. Methods 47 (2001) 5 –19 19
[10] Jones AC, Austin J, Hansen N, Hoogendoorn B, Oefner PJ, Cheadle JP et al. Optimal temperatureselection for mutation detection by denaturing HPLC and comparison to single-stranded conformationpolymorphism and heteroduplex analysis. Clin Chem 1999;45:1133–40.
`[11] Wagner T, Stoppa-Lyonnet D, Fleischmann E, Muhr D, Pages S, Sandberg T et al. Denaturinghigh-performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics1999;62:369–76.
[12] Oefner PJ, Underhill PA. DNA mutation detection using denaturing high-performance liquid chromatog-raphy. In: Dracopoli NC, Haines JL, Korf BR, Moir DT, Morton CC, Seidman CE et al., editors, Currentprotocols in human genetics, New York: Wiley, 1998, pp. 7101–71012.
[13] Oefner PJ. Allelic discrimination by denaturing high-performance liquid chromatography. J ChromatogrB 2000;739:345–55.
[14] Novotny M. Capillary biomolecular separations. J Chromatogr B 1997;689:55–70.[15] Ueno K, Yeung ES. Simultaneous monitoring of DNA fragments separated by electrophoresis in a
multiplexed array of 100 capillaries. Anal Chem 1994;66:1424–31.[16] Huber CG, Krajete A. Analysis of nucleic acids by capillary ion-pair reversed-phase HPLC coupled to
negative ion-electrospray ionization mass spectrometry. Anal Chem 1999;71:3730–9.[17] Premstaller A, Oberacher H, Huber CG. High-performance liquid chromatography–electrospray ioniza-
tion mass spectrometry of single and double stranded nucleic acids using monolithic capillary columns.Anal Chem 2000;72:4386–93.
[18] Huber CG, Oefner PJ, Bonn GK. High-resolution liquid chromatography of oligonucleotides on highlycrosslinked poly(styrene–divinylbenzene) particles. Anal Biochem 1993;212:351–8.
[19] Huber CG, Oefner PJ, Preuss E, Bonn GK. High-resolution liquid chromatography of DNA fragments annon-porous poly(styrene–divinylbenzene) particles. Nucleic Acids Res 1993;21:1061–6.
[20] Oberacher H, Krajete A, Parson W, Huber CG. Preparation and evaluation of packed capillary columnsfor the separation of nucleic acids by ion-pair reversed-phase high-performance liquid chromatography. JChromatogr A 2000;893:23–35.
´[21] Gusev I, Huang X, Horvath C. Capillary columns with in situ formed porous monolithic packing forhigh-performance liquid chromatography and capillary electrochromatography. J Chromatogr A1999;855:273–90.
[22] Chervet JP, Ursem M, Salzmann JP. Instrumental requirements for nanoscale liquid chromatography.Anal Chem 1996;68:1507–12.
[23] Chervet JP. Microflow processor. European Patent 0495255A1, 1991.[24] Huber CG, Stimpfl E, Oefner PJ, Bonn GK. A comparison of micropellicular ion-exchange and
reversed-phase stationary phases for HPLC of oligonucleotides. LC?GC Int 1996;14:114–27.[25] Ishii D. Introduction to microscale high-performance liquid chromatography, Weinheim: VCH, 1988.[26] Oefner PJ, Huber CG, Umlauft F, Bert GN, Stimpfl E, Bonn GK. High-resolution liquid chromatography
of fluorescent dye-labeled nucleic acids. Anal Biochem 1994;223:39–46.[27] Huber CG, Berti GN. Detection of partial denaturation in AT-rich DNA fragments by ion-pair reversed
phase chromatography. Anal Chem 1996;68:2959–65.[28] Ke SH, Wartell RM. Influence of nearest neighbor sequence on the stability of base pair mismatches in
long DNA: determination by temperature-gradient gel electrophoresis. Nucleic Acids Res 1993;21:5137–43.
[29] Abou-ela F, Koh D, Tinoco I, Martin FH. Base-base mismatches. Thermodynamics of double helixformation for dCA XA G1dCT YT G (X,Y5A,C,G,T). Nucleic Acids Res 1985;13:4811–24.3 3 3 3
[30] O’Donovan MC, Oefner PJ, Roberts CS, Austin J, Hoogendoorn B, Guy C et al. Blind analysis ofdenaturing high-performance liquid chromatography as a tool for mutation detection. Genomics1998;52:44–9.
[31] Portier N, Van Dorsselaer A, Cordier Y, Roch O, Bischoff R. Negative electrospray ionization massspectrometry of synthetic and chemically modified oligonucleotides. Nucleic Acids Res 1994;22:3895–903.
[32] Huber CG, Buchmeiser MR. On-line cation-exchange for suppression of adduct formation in negative-ionelectrospray mass spectrometry of nucleic acids. Anal Chem 1998;70:5288–95.
[33] Huber CG, Krajete A. Sheath liquid effects in capillary high-performance liquid chromatography–electrospray mass spectrometry of oligonucleotides. J Chromatogr A 2000;870:413–24.




























