Embed Size (px)
Citation preview

Myelination and behaviour of tenascin-C null transgenicmice
B. W. Kiernan,1 E. Garcion,1,2 J. Ferguson,1 E. E. Frost,1,2 E. M. Torres,2,3 S. B. Dunnett,2,3 Y. Saga,6 S. Aizawa,7
A. Faissner,8 R. Kaur,4 R. J. M. Franklin4 and C. ffrench-Constant1,2,5
1Wellcome/CRC Institute of Developmental Biology and Cancer, Tennis Court Road, Cambridge CB2 1QR, UK2MRC Cambridge Centre for Brain Repair, The E.D. Adrian Building, Forvie Site, Addenbrooke's Hospital, CambridgeCB2 2PY, UK3Department of Experimental Psychology, University of Cambridge, Cambridge, UK4Department of Clinical Veterinary Medicine, University of Cambridge, Cambridge, UK5Department of Medical Genetics, University of Cambridge, Cambridge, UK6National Institute of Health Science, Tokyo 158, Japan7Kumamoto University Medical School, Japan8Department of Neurobiology, University of Heidelberg, Im Neuenheimer Feld 364, 69120 Heidelberg, Germany
Keywords: hyperlocomotion, lamina cribrosa, Morris water maze, migration, oligodendrocyte precursor, passive avoidance
Abstract
The extracellular matrix glycoprotein tenascin-C is widely expressed during development and repair, making it surprising that fewabnormalities have been found in transgenic mice lacking this molecule. We have therefore re-examined the transgenic micedescribed by Saga et al. [Saga, Y., Yagi, T., Ikawa, Y., Sakakura, T. & Aizawa, S. (1992) Genes Dev., 6, 1821±1831] in whichtenascin-C was knocked-out by homologous recombination, focusing on two aspects of the nervous system likely to reveal anyabnormalities that might follow the loss of tenascin-C. First, we have determined the pattern of myelin and distribution ofoligodendrocyte precursor cells in those areas, such as the optic nerve and retina where local concentrations of tenascin-C havebeen proposed to act as barriers to oligodendrocyte precursor migration and so prevent inappropriate myelination. Secondly, wehave examined the behaviour of the mice in a number of well-characterized tests, e.g. beam-walking, passive avoidance and theMorris water maze. We ®nd no abnormalities of myelination or oligodendrocyte precursor distribution in adult mice, showing that localconcentrations of tenascin-C are not the sole mechanism responsible for the pattern of myelination in these regions of CNS.However, we do ®nd a number of behavioural abnormalities in these mice and show that hyperlocomotion and de®cits in coordinationduring beam walking can be ascribed to tenascin-C de®ciency. The effects on coordination are, however, not seen on a 129 geneticbackground. Taken together, these results signi®cantly extend the phenotype associated with tenascin-C de®ciency but argueagainst a role in myelination.
Introduction
Transgenic mice in which genes encoding extracellular matrix
molecules have been deleted by homologous recombination represent
an attractive means of examining the function of these molecules.
One molecule for which this approach has been used is the
extracellular matrix molecule tenascin-C (Saga et al., 1992;
Forsberg et al., 1996). This molecule is widely expressed during
development and following injury in the CNS, and is antiadhesive for
a range of neural cell types with inhibition of neurite outgrowth or
cell migration (reviewed in Chiquet-Ehrismann, 1991; Faissner et al.,
1995). These observations suggest an important role for tenascin-C in
the modulation of cell behaviour during neural development.
Surprisingly, however, tenascin-C-de®cient mice do not have an
obvious developmental phenotype (Saga et al., 1992; Forsberg et al.,
1996). While these studies did not examine individual developmental
programmes in detail, it is clear that the mice do not suggest any
essential role for tenascin-C and have raised the provocative question
as to whether this extracellular matrix molecule is redundant for
normal development (Erickson, 1993a). Rather than having no
function, however, it is possible that de®ciency of tenascin-C has
effects too subtle to detect by histological analysis of adult mice.
These studies, therefore, make it important to re-examine the
tenascin-C-de®cient mouse in more detail to resolve the controversy
as to whether there is an essential function for this molecule.
These further studies can be done in two ways. First, we can
undertake more detailed analyses of areas of CNS development
where previous experiments have led to speci®c hypotheses as to the
function of tenascin-C. Two such areas are the optic nerve and
cerebellum. Tenascin-C is expressed at high levels in the lamina
cribrosa, the point at which the optic nerve head pierces the sclera of
the eye, and the molecular layer of the cerebellum (Bartsch et al.,
1992; 1994). Based on experiments showing that tenascin-C inhibits
oligodendrocyte precursor cell adhesion and migration, it has been
suggested that tenascin-C de®nes the pattern of myelination in these
regions by acting as a barrier to oligodendrocyte precursor migration
(Bartsch et al., 1992; 1994; Frost et al., 1996; Kiernan et al., 1996). If
Correspondence: Dr C. ffrench-Constant, Wellcome/CRC Institute ofDevelopmental Biology and Cancer, Tennis Court Road, CambridgeCB2 1QR, UK. E-mail: [email protected]
Received 16 September 1998, revised 29 March 1999, accepted 21 April 1999
European Journal of Neuroscience, Vol. 11, pp. 3082±3092, 1999 Ó European Neuroscience Association

so, one would predict that tenascin-C-de®cient mice will have
abnormal patterns of oligodendrocyte precursor cell distribution and
myelination in the retina and cerebellum. Secondly, we can undertake
more sophisticated analyses of the CNS using behavioural and
cognitive testing. These will test function rather than anatomical
structure, and would therefore be expected to provide a more
sensitive assay of correct neural development.
To re-examine the question of tenascin-C function, we have taken
both approaches to analyse the transgenic mouse strain originally
described by Saga et al. (1992). Our results provide evidence for
strain-speci®c abnormalities of behaviour in tenascin-C-de®cient
mice but provide no support for an essential role of the molecule in
determining patterns of myelination.
Materials and methods
Animals
Histological analyses were based on young adult male and female
TN±/± mice derived from a homozygous F5 generation tenascin-C-
de®cient mice cross, compared with sex and age matched C57BL/6 J
mice.
Behavioural analyses were based on four sets of animals, using
different breeding regimes designed to progressively control for
differences in the genetic background on which the tenascin-C null
genotype was expressed.
In experiment I, six male tenascin-C-de®cient mice were
compared with seven age-matched male control mice. One
tenascin-C-de®cient animal was killed during the analysis follow-
ing a fall during bridge testing. Control and experimental mice
were derived from different stocks. Both had genetic backgrounds
comprising an equal mixture of F1 C57BL/6 J-CBA and BALB/c,
with selection against inheritance of the albino (c) allele. The
origins of the TN±/± stock are described elsewhere (Saga et al.,
1992). The control stock was designed to replicate the breeding
protocol adopted for the TN±/± mice.
In experiment II, ®ve heterozygous and eight tenascin-C-
de®cient animals from a mixed sex litter from a backcross
between an F6 generation homozygote tenascin-C-de®cient mouse
and a heterozygote derived from a cross between the F6
generation homozygote tenascin-C-de®cient mouse and an F1
C57BL/6 J-CBA hybrid (TN±/± 3 [TN±/± 3 F1 C57BL/6 J-CBA])
were examined. This litter comprised both heterozygotes and
homozygotes with equivalent genetic backgrounds. The behaviour-
al tests on the ®rst and second sets of animals were conducted
sequentially over a period of 7 weeks.
In experiment III, eight wild-type, 12 heterozygous and six
tenascin-C-de®cient mice from mixed sex, age-matched litters from
intercrosses between the offspring of an F6 generation homozygote
tenascin-C-de®cient mouse and a 129 mouse ([TN±/± 3 129] 3
[TN±/± 3 129]) were examined. This litter comprised homozygotes,
heterozygotes and wild-type animals with equivalent genetic back-
grounds, and was tested over a period of 3 weeks.
In experiment IV, eight wild-type, nine heterozygous and eight
tenascin-C-de®cient age-matched female mice from a strain carrying
the tenascin-C null transgene on a largely 129 genetic background
were examined. The strain was derived from tenascin-C null
homozygotes from the [TN±/± 3 129] 3 [TN±/± 3 129] intercross
litters bred back onto the parental 129 strain for four generations.
This litter comprised homozygotes, heterozygotes and wild-type
animals on an » 94% 129 strain genetic background, and was tested
over 1 week.
Histological analyses
For Sudan black staining of myelin lipids, complete brains, eyes and
optic nerves were dissected from animals perfused transcardially with
2% paraformaldehyde in phosphate-buffered saline (PBS). Samples
were left in ®xative for 1±3 h, then in 34% sucrose in PBS for a
further 1±3 h (eyes) or until they sank to the bottom of the tube
(brains). Samples were mounted in Tissue-Tekq O.C.T. compound
blocks at ±37 °C, and transverse sections of retina, longitudinal
sections of optic nerve and sagittal sections of cerebellum cut on a
cryostat at ±24 °C. Sections (10±20 mm) were placed in 70% ethanol,
followed by Sudan black [prepared by dissolving 7 g of Sudan black
B (Sigma) in 500 mL 70% ethanol] for 7±10 min. Sections were
rinsed in 70% ethanol, washed in water for 3±4 min and mounted in
aqueous mounting medium.
For immunohistochemistry (IHC) and in situ hybridization (ISH),
brains were surgically removed and snap frozen at ±30 or ±40 °C in
isopentane cooled by liquid nitrogen. Optic nerves or cerebellum
were dissected in paraformaldehyde 4% in PBS and then embedded
in Tissue-Tek O.C.T. compound (Agar Scienti®c, Stansted, Essex,
UK) frozen on dry ice. Specimens were stored at ±80 °C until use.
Longitudinal optic nerve sections (10 mm for IHC and 15 mm for
ISH), to show where the optic nerve leaves the retina, and cerebellum
sections were placed onto Superfrost slides (Scienti®c Laboratory
Supplies, Nottingham, UK).
For ISH, frozen sections were thawed at room temperature for
30 min, ®xed with 4% paraformaldehyde in PBS, pH 7.4, for 10 min,
acetylated in a solution of 4 3 standard sodium citrate (SSC), pH 8.0,
containing 0.25% acetic anhydride and 0.1 M triethanolamine for
10 min at room temperature, dehydrated in ethanol, delipidated in
chloroform, and air-dried. The cRNA PDGF alpha-receptor probe
was transcribed from a 1637-bp EcoRI cDNA fragment encoding
most of the extracellular domain of mouse PDGF alpha-receptor
cloned into Bluescript KS (a kind gift of W. Richardson). RNA
polymerases and DIG labelling mix were obtained from Boehringer,
Lewes, Sussex, UK, and the transcription reaction was run as
recommended by the manufacturers. The labelled cRNA probe (1±
2 mg/mL) was dissolved in the hybridization solution (50% v/v
formamide, 4 3 SSC, 1 3 Denhardt's solution, 100 mg/mL herring
sperm DNA, 100 mg/mL polyA, 10% w/v dextran sulphate), and
applied onto each slide under coverslips. Hybridization was allowed
to proceed for 14±18 h at 65 °C. Slides were dipped into 4 3 SSC and
then coverslips were carefully removed. Slides were washed for
10 min in 1 3 SSC, incubated for 30 min with RNAase A
(Boehringer) and 30 min in RNAase A buffer alone at 37 °C, washed
three times for 40 min at 65 °C in 1 3 SSC, 50% formamide, 0.1%
Tween-20, and twice for 30 min in 100 mM maleic acid, 150 mM
NaCl, 0.1% Tween-20, pH 7.5. RNA hybridization was visualized by
IHC with alkaline phosphatase-conjugated anti-DIG antibody
(Boehringer) according to the manufacturer's instructions, except
that polyvinyl alcohol (PVA; 10% w/v) was included in the ®nal
colour reaction to increase sensitivity. Slides were then dehydrated in
ethanol and xylene, and mounted in Eukitt medium (Agar Scienti®c).
For immuno¯uorescent staining of myelin basic protein (MBP), the
cryostat sections were ®xed for 10 min in 4% paraformaldehyde in
PBS, followed by three rinses in PBS. They were then blocked for 1 h
at room temperature in gelatin 2 g/L in PBS, after which the ®rst
antibody incubation (rat antimouse MBP, IgG MCA 409, Serotec, 1/
100 in the blocking solution) was carried out overnight at 4 °C. After
washing in PBS, the sections were then incubated for 1 h with the
secondary antibody (biotin-conjugated antirat, 1/75 in the blocking
solution) followed by the streptavidin±¯uorescein complex (1/75 in
Myelination and behaviour in TN-C±/± mice 3083
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092

PBS) for 1 h. After further washes in PBS, sections were mounted
under coverslips in ¯uorostab medium (Eurodiagnostica, MalmoÈ,
Sweden) and visualized in a confocal microscope.
For the electron microscopy studies, mice were killed under
anaesthesia by transcardiac perfusion with 4% glutaraldehyde in
phosphate buffer with 0.1% calcium chloride (pH 7.2±7.4). The ®xed
brains were removed and post®xed in 4% glutaraldehyde for 5±24 h.
All tissue blocks were washed with phosphate buffer, post®xed with
2% osmium tetroxide, dehydrated through increasing concentrations
of alcohol and embedded in TAAB resin. Semi-thin sections (1 mm)
were cut from each block onto plain glass slides and stained with
toluidine blue for examination by light microscopy. Areas of interest
from selected blocks were trimmed for examination at the EM level.
Ultrathin (70 nm) sections were stained with uranyl acetate and lead
citrate, and examined using a Hitachi-H600 electron microscope.
Images of myelinated axons, obtained from the EM, were projected
on a computer monitor screen. The outline of axon diameter and the
®bre diameter were traced using the Digit semiautomatic image
analysis system (Institute of Ophthalmology, London) coupled to a
high-resolution Summagraphics Bit Pad 2 digit tablet (model 1103,
Advanced Microcomputer Applications, Nottingham, UK) and a
Acorn Archimedes compatible computer (Acorns Computer,
Cambridge, UK). The mean Feret diameter calculated from the
digitized shapes represented arbitrary units. The correlation between
®bre diameter and axon diameter was tested using Prism software.
Behavioural analyses
Behavioural tests were designed to assess a range of behaviours
relevant to neurological function. General locomotor ability, postural
changes, hearing, activity and exploratory behaviour were assessed
by observation. Finer motor coordination and balance were tested on
a variety of bridges, and swimming ability was tested in a water tank.
Finally, more complex behaviours involving learning were judged
using the Morris water maze and a step-through test of passive
avoidance. All experiments were undertaken blind to the genotype of
the subjects. All animals were between 2 and 6 months of age when
tested. For genotyping, the tip of each animal's tail was docked after
the completion of all behavioural tests and used in a PCR assay as
described (Saga et al., 1992) in order to identify wild-type,
heterozygous and homozygous littermates.
Open ®eld
Open ®eld behaviour assesses levels of spontaneous locomotor
activity and exploratory behaviour. The open ®eld was a 1-m2 square
arena with 50-cm-high walls. The ¯oor was divided into 25 equal
squares by black lines. The mice were placed in the centre square
facing away from the experimenter, and monitored by direct
observation and video cassette recording for 5 min. Entries into the
total, centre, side and corner squares were recorded with a line
crossing to enter a new square scored when a mouse crossed a line
with all four legs. The time spent in the centre nine squares and the
number of attempts made by the experimenter to catch the animal at
the end of the test period were recorded. Bouts of rearing (raising
forepaws off the ground), bouts of grooming and fecal boli were
counted. Each mouse received four tests in the open ®eld on
consecutive days. The data were aggregated over the four days for
analysis.
Hindlimb re¯ex
Changes in posture and limb position were examined by lifting
animals up by the tail. The mice were gently lifted by the tail until all
four paws were off the ground and held suspended. Positions of limbs
were noted. The mice were then permitted to contact a surface with
their forepaws and the position of the hindlimbs again noted.
Flinch hearing test (Preyer re¯ex)
Two metal objects were knocked together in order to produce a single
audible sound. The two objects were held out of sight of the test
animals and the mice were observed during the test for ¯inching. A
¯inch was de®ned as any action evoked by the stimulus ranging from
an ear twitch to a jump. Any ¯inch was taken as evidence that the test
animal was capable of hearing.
Balance and rod walking
The ability of rodents to traverse a stationary horizontal rod is
assumed to measure both sensorimotor coordination and the integrity
of the vestibular senses (Dean et al., 1981). Mice were trained to walk
across a 60-cm-long wooden bridge into a dark box. After training,
the complexity and dimensions of the bridge were altered in order to
assess motor coordination and the integrity of the vestibular system.
Each beam was made of a single wooden rod with either square (28 or
23 mm wide) or round (28, 18, 12 or 9 mm diameter) cross-section,
raised 45 cm above the bench surface. The mice were placed on top of
each bridge at one end facing the dark box for four consecutive trials,
between each trial the animal was returned to its home cage for a
minimum of 15 min. The time taken to traverse the bridge and the
number of occasions on which the animal fell, clung or one or other
of its feet slipped were recorded ®rst for the bridges with square
cross-sections and then for the four circular ones, in each case
progressing from the widest to the narrowest. One cling was de®ned
as the mouse freezing for 5 s or more.
Two days after the last trial, the mice were placed at one end of the
narrowest beam, and their balance was classi®ed as either success or
failure. Criterion for success was standing or clinging horizontally to
the top of the beam for 10 s, failure was scored in the event of
slipping sideways, hanging or falling.
Morris water maze
Spatial navigation abilities were evaluated using the Morris water
maze test (Morris & O'Shea, 1983; Morris et al., 1986), conducted in
a black circular pool, 100 cm in diameter and 40 cm deep. The pool
was positioned in a small test room ®lled with objects visible from
within and external to the pool (experimenter, door, lights, shelving,
equipment). The pool was ®lled with 19 °C water to a depth of 30 cm
made opaque by the addition of non-toxic white paint. A 10 3 10 cm
Perspex escape platform was positioned in the pool with its surface
1 cm below the water surface. A camera was ®xed above the pool and
used to record each trial.
During training in the hidden platform test, the platform was
maintained in a constant position in the middle of one quadrant of the
pool. On each trial the animal was placed at one of four positions
around the wall of the pool in a random order and allowed to swim
until it found the platform. Four trials per day were performed. Each
trial ended when the mouse reached the platform. Each trial was
permitted a maximum of 60 s, after which mice were placed on the
platform. The animal remained on the platform for 15 s before being
returned to the home cage. The mice were tested at approximately the
same time every day but in a random order. After initial training,
mice were tested in a probe trial to determine whether or not they had
learnt the spatial position of the platform using external cues. On the
probe trial day, the platform was removed for the third trial and the
mice were placed in the pool for 30 s. The time spent in the quadrant
previously holding the platform was recorded. The platform was
replaced and the mice permitted to locate the platform in the fourth
3084 B. W. Kiernan et al.
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092

trial. After the probe trial, mice were tested in a reversal trial to
determine whether or not they were capable of learning a new
platform position. During the reversal trial, the platform was placed
in a new location (in the opposite quadrant to the previous) and
training restarted. Finally, during visible platform training the water
level in the pool was dropped by 15 mm allowing the surface of the
platform to break the surface of the water by 5 mm. For each trial, the
platform was placed in one of four positions in a random order.
Passive avoidance
The passive avoidance test measures an animal's ability to learn to
avoid and retain the memory of a noxious stimulus by inhibiting
normal behaviour (Robichaud et al., 1973; Bartus et al., 1980).
Passive avoidance was evaluated in a 50-cm-high two-compartment
step-through test chamber. One 80 3 50 cm compartment was open
and exposed, with a transparent Perspex wall. The other compartment
(80 3 30 cm) was dark and covered. The ¯oor in both compartments
was made of 5-mm-diameter stainless steel rails spaced 15 mm apart.
A 10-cm-wide by 5-cm-high guillotine door separated the two
compartments. Each animal received a familiarization trial in the
apparatus on day 1, in which they were placed in the exposed
compartment opposite and facing away from the open door. The
latency to enter the dark compartment was recorded for each trial. On
the second day, the animals were once again placed opposite and
facing away from the open door in the light compartment. As soon as
the mouse entered the dark side the guillotine door was closed and a
0.5-s 0.2-mA scrambled footshock was applied to the grid ¯oor. After
15 s the animal was removed from the dark chamber and returned to
its home cage. Each mouse was trained every 1.5 h until they all
avoided the dark compartment for 5 min. On each successive day
thereafter the mice were given two trials, and the latency to enter the
dark compartment was recorded, with no further footshocks given.
Results
Myelination patterns
To examine the pattern of myelination in optic nerve and cerebellum,
we cut sections for light and electron microscopy studies from adult
mouse brains of the tenascin-C stock described by Saga et al. (1992),
and from age-and sex-matched control animals.
In the retina, the tenascin-C null mice exhibited a normal
distribution of myelin when visualized by Sudan black (Fig. 1A±C)
or anti-MBP immuno¯uorescent staining (Fig. 1D and E). As in wild-
type (control) animals, no myelin was detected in transverse sections
of the retina of tenascin-C-de®cient animals (Fig. 1A±E). The extent
of myelin stain along the optic nerve was identical in both the control
and tenascin-C-de®cient animals: in both sets of animals, myelination
ended abruptly at the retina±optic nerve junction (Fig. 1A±E). The
absence of myelin in the optic nerve ®bre layer was con®rmed by
further light and electron microscopy of semi- and ultrathin sections
of the retina in the region of the lamina cribrosa (Fig. 1F and G). The
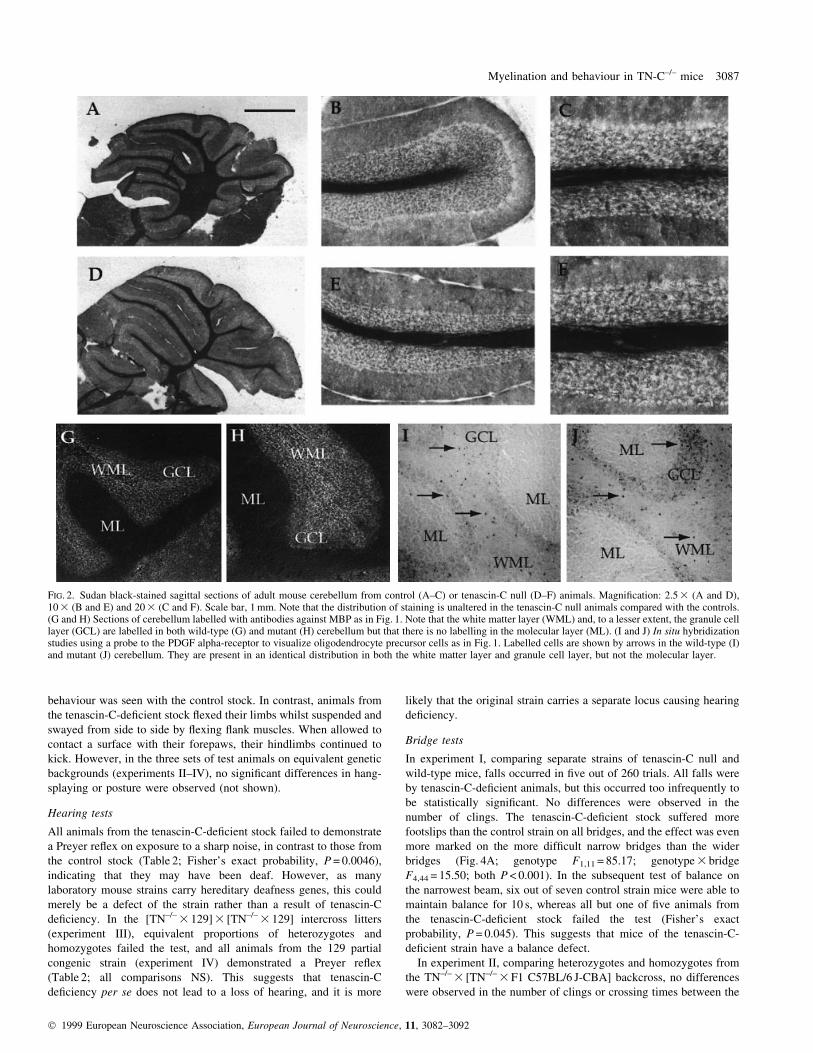
distribution of myelin in the cerebellum of tenascin-C-de®cient mice
also appears identical to that present in control animals. Dense
myelination, as visualized by Sudan Black, is con®ned to the white
matter layer (Fig. 2A±F), and MBP expression is present in the white
matter layer and, to a lesser extent, in the granule cell layer (Fig. 2G
and H). No abnormal myelination or MBP expression was seen in the
molecular layer.
These data suggest that any barriers to oligodendrocyte precursor
migration that de®ne the pattern of myelination in optic nerve and
cerebellum are present in both control and tenascin-C-de®cient
animals. To examine this more directly, we performed in situ
hybridization experiments using cRNA probes complementary to the
PDGF alpha-receptor, a marker for oligodendrocyte precursor cells in
vivo (Pringle et al., 1992). These studies showed the pattern of
oligodendrocyte precursor cells to be identical in control and
tenascin-C-de®cient animals. In the optic nerve, precursor cells were
abundant in the nerve but were not present in the region of the lamina
cribrosa in either control or transgenic animals (Fig. 1H±J). In the
cerebellum, precursors were present in the white matter layer with
occasional cells seen within the granular layer. Precursors were not
seen in the molecular layer in either control or transgenic animals.
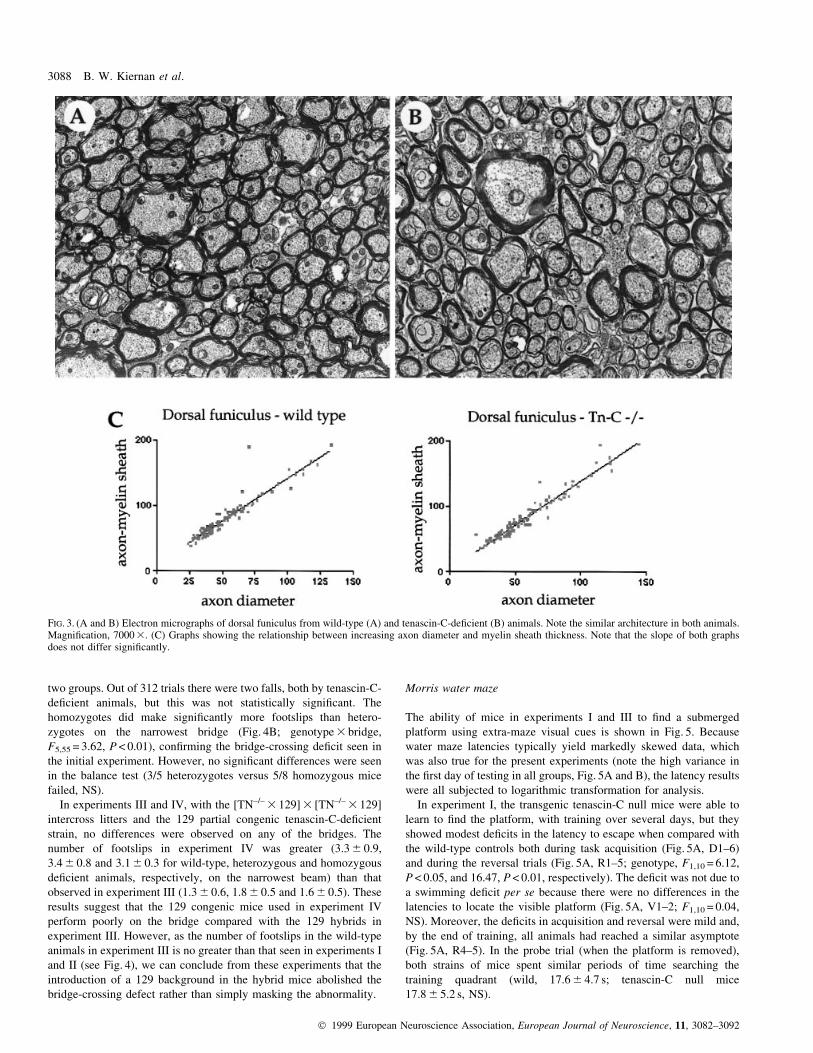
To examine the architecture of myelinated tracts in the tenascin-C-
de®cient mice in more detail, we performed electron microscopy
studies of the dorsal funiculus in wild-type and transgenic mice. No
differences in the morphology of the myelinated tracts was noted
(Fig. 3A and B), with the overall architecture being the same in the
presence or absence of tenascin-C. We then examined the relationship
between axon diameter and overall axon/myelin sheath diameter in
these sections. As shown in Fig. 3C, there is no signi®cant difference
in the slope of the graph (P = 0.28, NS), showing that the relationship
between increasing axon diameter and myelin sheath thickness is not
altered in the transgenic mice.
Behavioural testing
Four different strains were used for these experiments, as described in
Materials and methods. Initially, we performed a comparison of the
original tenascin-C-de®cient stock with an appropriate wild-type
control (experiment I). Having documented a number of abnormal-
ities, we performed further studies on a backcross (using a C57BL/
CBA parent ± experiment II) or an intercross (using a 129 parent ±
experiment III) litter so as to determine which abnormalities were
related to tenascin-C de®ciency and to examine the effect of genetic
background. Finally, we focused on a novel abnormality related to
tenascin-C de®ciency that we observed in beam walking by
examining motor and inner ear function in more detail in a congenic
129 strain of mice (experiment IV).
Open ®eld
When placed in an open ®eld, mice move along the walls with short
stops during which they often rear. Low levels of locomotion,
restriction to the sides and corners, and frequent defecation are taken
to indicate fear or anxiety. High levels of locomotion, rearing and
time spent in the centre are considered to be indicators of activation
and exploration.
In experiment I (the stock comparison), no differences between the
two groups were observed in locomotor activity (number of squares
entered), time spent in the centre squares or grooming (Table 1, left
columns). However, the tenascin-C-de®cient stock reared more often
and defecated less often, suggesting that the mice were less anxious
and more exploratory than the control strain.
In con®rmation of this, increased rearing and decreased defecation
were also seen in experiment II comparing TN±/± 3 [TN±/± 3 F1
C57BL/6 J-CBA] backcross heterozygotes and homozygotes
(Table 1, right columns). In addition, and unlike the stock
comparison, differences were seen in general locomotor activity
due to an increase in the number of side and corner squares entered by
the tenascin-C-de®cient homozygotes (Table 1).
Hindlimb re¯ex and postural changes
Normal mice hung by the tail adopt a posture with their hindlimbs
splayed and their forelimbs tucked underneath their bodies. When
lowered towards a surface, the forelimbs are extended for contact and
placing, and the hindlimbs become stationary. This pattern of
Myelination and behaviour in TN-C±/± mice 3085
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092
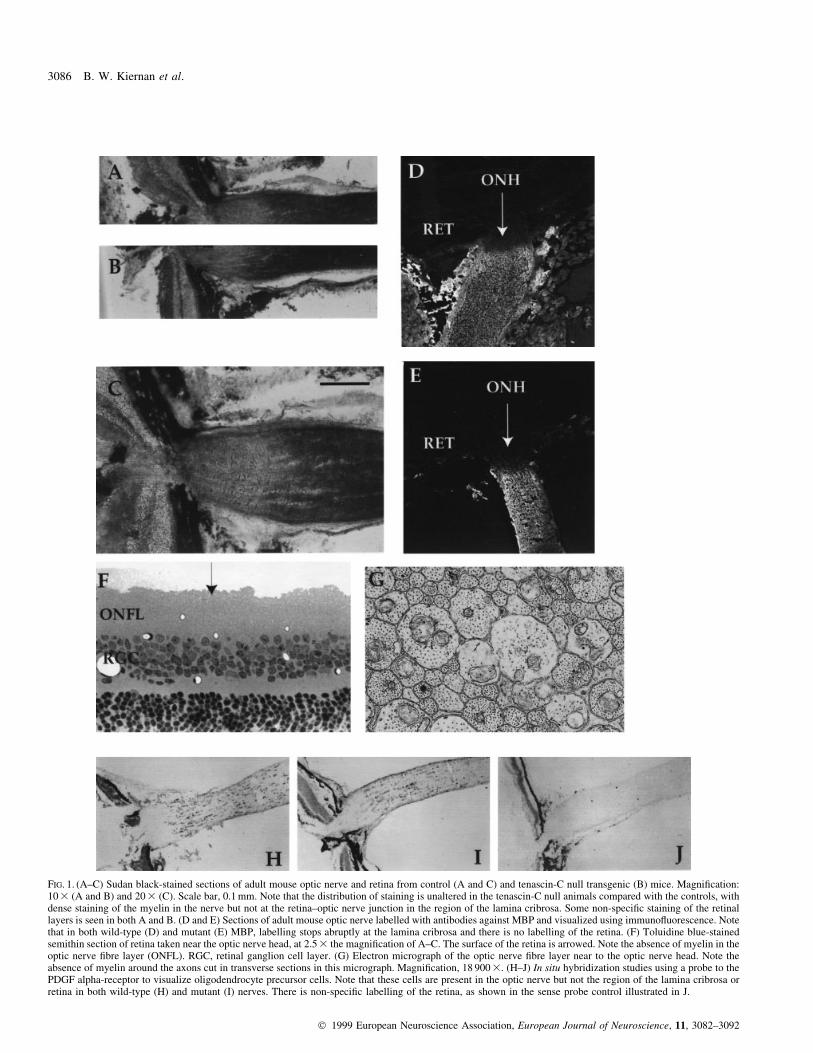
FIG. 1. (A±C) Sudan black-stained sections of adult mouse optic nerve and retina from control (A and C) and tenascin-C null transgenic (B) mice. Magni®cation:10 3 (A and B) and 20 3 (C). Scale bar, 0.1 mm. Note that the distribution of staining is unaltered in the tenascin-C null animals compared with the controls, withdense staining of the myelin in the nerve but not at the retina±optic nerve junction in the region of the lamina cribrosa. Some non-speci®c staining of the retinallayers is seen in both A and B. (D and E) Sections of adult mouse optic nerve labelled with antibodies against MBP and visualized using immuno¯uorescence. Notethat in both wild-type (D) and mutant (E) MBP, labelling stops abruptly at the lamina cribrosa and there is no labelling of the retina. (F) Toluidine blue-stainedsemithin section of retina taken near the optic nerve head, at 2.5 3 the magni®cation of A±C. The surface of the retina is arrowed. Note the absence of myelin in theoptic nerve ®bre layer (ONFL). RGC, retinal ganglion cell layer. (G) Electron micrograph of the optic nerve ®bre layer near to the optic nerve head. Note theabsence of myelin around the axons cut in transverse sections in this micrograph. Magni®cation, 18 900 3. (H±J) In situ hybridization studies using a probe to thePDGF alpha-receptor to visualize oligodendrocyte precursor cells. Note that these cells are present in the optic nerve but not the region of the lamina cribrosa orretina in both wild-type (H) and mutant (I) nerves. There is non-speci®c labelling of the retina, as shown in the sense probe control illustrated in J.
3086 B. W. Kiernan et al.
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092
behaviour was seen with the control stock. In contrast, animals from
the tenascin-C-de®cient stock ¯exed their limbs whilst suspended and
swayed from side to side by ¯exing ¯ank muscles. When allowed to
contact a surface with their forepaws, their hindlimbs continued to
kick. However, in the three sets of test animals on equivalent genetic
backgrounds (experiments II±IV), no signi®cant differences in hang-
splaying or posture were observed (not shown).
Hearing tests
All animals from the tenascin-C-de®cient stock failed to demonstrate
a Preyer re¯ex on exposure to a sharp noise, in contrast to those from
the control stock (Table 2; Fisher's exact probability, P = 0.0046),
indicating that they may have been deaf. However, as many
laboratory mouse strains carry hereditary deafness genes, this could
merely be a defect of the strain rather than a result of tenascin-C
de®ciency. In the [TN±/± 3 129] 3 [TN±/± 3 129] intercross litters
(experiment III), equivalent proportions of heterozygotes and
homozygotes failed the test, and all animals from the 129 partial
congenic strain (experiment IV) demonstrated a Preyer re¯ex
(Table 2; all comparisons NS). This suggests that tenascin-C
de®ciency per se does not lead to a loss of hearing, and it is more
likely that the original strain carries a separate locus causing hearing
de®ciency.
Bridge tests
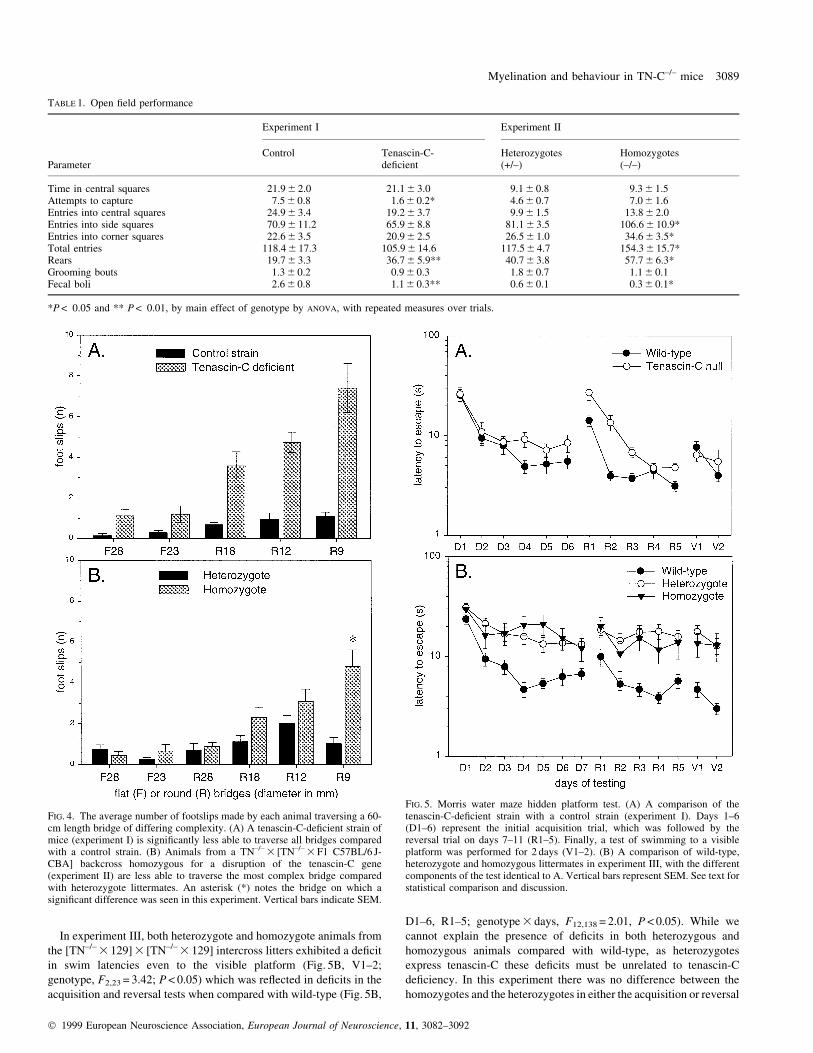
In experiment I, comparing separate strains of tenascin-C null and
wild-type mice, falls occurred in ®ve out of 260 trials. All falls were
by tenascin-C-de®cient animals, but this occurred too infrequently to
be statistically signi®cant. No differences were observed in the
number of clings. The tenascin-C-de®cient stock suffered more
footslips than the control strain on all bridges, and the effect was even
more marked on the more dif®cult narrow bridges than the wider
bridges (Fig. 4A; genotype F1,11 = 85.17; genotype 3 bridge
F4,44 = 15.50; both P < 0.001). In the subsequent test of balance on
the narrowest beam, six out of seven control strain mice were able to
maintain balance for 10 s, whereas all but one of ®ve animals from
the tenascin-C-de®cient stock failed the test (Fisher's exact
probability, P = 0.045). This suggests that mice of the tenascin-C-
de®cient strain have a balance defect.
In experiment II, comparing heterozygotes and homozygotes from
the TN±/± 3 [TN±/± 3 F1 C57BL/6 J-CBA] backcross, no differences
were observed in the number of clings or crossing times between the
FIG. 2. Sudan black-stained sagittal sections of adult mouse cerebellum from control (A±C) or tenascin-C null (D±F) animals. Magni®cation: 2.5 3 (A and D),10 3 (B and E) and 20 3 (C and F). Scale bar, 1 mm. Note that the distribution of staining is unaltered in the tenascin-C null animals compared with the controls.(G and H) Sections of cerebellum labelled with antibodies against MBP as in Fig. 1. Note that the white matter layer (WML) and, to a lesser extent, the granule celllayer (GCL) are labelled in both wild-type (G) and mutant (H) cerebellum but that there is no labelling in the molecular layer (ML). (I and J) In situ hybridizationstudies using a probe to the PDGF alpha-receptor to visualize oligodendrocyte precursor cells as in Fig. 1. Labelled cells are shown by arrows in the wild-type (I)and mutant (J) cerebellum. They are present in an identical distribution in both the white matter layer and granule cell layer, but not the molecular layer.
Myelination and behaviour in TN-C±/± mice 3087
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092
two groups. Out of 312 trials there were two falls, both by tenascin-C-
de®cient animals, but this was not statistically signi®cant. The
homozygotes did make signi®cantly more footslips than hetero-
zygotes on the narrowest bridge (Fig. 4B; genotype 3 bridge,
F5,55 = 3.62, P < 0.01), con®rming the bridge-crossing de®cit seen in
the initial experiment. However, no signi®cant differences were seen
in the balance test (3/5 heterozygotes versus 5/8 homozygous mice
failed, NS).
In experiments III and IV, with the [TN±/± 3 129] 3 [TN±/± 3 129]
intercross litters and the 129 partial congenic tenascin-C-de®cient
strain, no differences were observed on any of the bridges. The
number of footslips in experiment IV was greater (3.3 6 0.9,
3.4 6 0.8 and 3.1 6 0.3 for wild-type, heterozygous and homozygous
de®cient animals, respectively, on the narrowest beam) than that
observed in experiment III (1.3 6 0.6, 1.8 6 0.5 and 1.6 6 0.5). These
results suggest that the 129 congenic mice used in experiment IV
perform poorly on the bridge compared with the 129 hybrids in
experiment III. However, as the number of footslips in the wild-type
animals in experiment III is no greater than that seen in experiments I
and II (see Fig. 4), we can conclude from these experiments that the
introduction of a 129 background in the hybrid mice abolished the
bridge-crossing defect rather than simply masking the abnormality.
Morris water maze
The ability of mice in experiments I and III to ®nd a submerged
platform using extra-maze visual cues is shown in Fig. 5. Because
water maze latencies typically yield markedly skewed data, which
was also true for the present experiments (note the high variance in
the ®rst day of testing in all groups, Fig. 5A and B), the latency results
were all subjected to logarithmic transformation for analysis.
In experiment I, the transgenic tenascin-C null mice were able to
learn to ®nd the platform, with training over several days, but they
showed modest de®cits in the latency to escape when compared with
the wild-type controls both during task acquisition (Fig. 5A, D1±6)
and during the reversal trials (Fig. 5A, R1±5; genotype, F1,10 = 6.12,
P < 0.05, and 16.47, P < 0.01, respectively). The de®cit was not due to
a swimming de®cit per se because there were no differences in the
latencies to locate the visible platform (Fig. 5A, V1±2; F1,10 = 0.04,
NS). Moreover, the de®cits in acquisition and reversal were mild and,
by the end of training, all animals had reached a similar asymptote
(Fig. 5A, R4±5). In the probe trial (when the platform is removed),
both strains of mice spent similar periods of time searching the
training quadrant (wild, 17.6 6 4.7 s; tenascin-C null mice
17.8 6 5.2 s, NS).
FIG. 3. (A and B) Electron micrographs of dorsal funiculus from wild-type (A) and tenascin-C-de®cient (B) animals. Note the similar architecture in both animals.Magni®cation, 7000 3. (C) Graphs showing the relationship between increasing axon diameter and myelin sheath thickness. Note that the slope of both graphsdoes not differ signi®cantly.
3088 B. W. Kiernan et al.
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092
In experiment III, both heterozygote and homozygote animals from
the [TN±/± 3 129] 3 [TN±/± 3 129] intercross litters exhibited a de®cit
in swim latencies even to the visible platform (Fig. 5B, V1±2;
genotype, F2,23 = 3.42; P < 0.05) which was re¯ected in de®cits in the
acquisition and reversal tests when compared with wild-type (Fig. 5B,
D1±6, R1±5; genotype 3 days, F12,138 = 2.01, P < 0.05). While we
cannot explain the presence of de®cits in both heterozygous and
homozygous animals compared with wild-type, as heterozygotes
express tenascin-C these de®cits must be unrelated to tenascin-C
de®ciency. In this experiment there was no difference between the
homozygotes and the heterozygotes in either the acquisition or reversal
FIG. 4. The average number of footslips made by each animal traversing a 60-cm length bridge of differing complexity. (A) A tenascin-C-de®cient strain ofmice (experiment I) is signi®cantly less able to traverse all bridges comparedwith a control strain. (B) Animals from a TN±/± 3 [TN±/± 3 F1 C57BL/6 J-CBA] backcross homozygous for a disruption of the tenascin-C gene(experiment II) are less able to traverse the most complex bridge comparedwith heterozygote littermates. An asterisk (*) notes the bridge on which asigni®cant difference was seen in this experiment. Vertical bars indicate SEM.
FIG. 5. Morris water maze hidden platform test. (A) A comparison of thetenascin-C-de®cient strain with a control strain (experiment I). Days 1±6(D1±6) represent the initial acquisition trial, which was followed by thereversal trial on days 7±11 (R1±5). Finally, a test of swimming to a visibleplatform was performed for 2 days (V1±2). (B) A comparison of wild-type,heterozygote and homozygous littermates in experiment III, with the differentcomponents of the test identical to A. Vertical bars represent SEM. See text forstatistical comparison and discussion.
TABLE 1. Open ®eld performance
Experiment I Experiment II
Control Tenascin-C- Heterozygotes HomozygotesParameter de®cient (+/±) (±/±)
Time in central squares 21.9 6 2.0 21.1 6 3.0 9.1 6 0.8 9.3 6 1.5Attempts to capture 7.5 6 0.8 1.6 6 0.2* 4.6 6 0.7 7.0 6 1.6Entries into central squares 24.9 6 3.4 19.2 6 3.7 9.9 6 1.5 13.8 6 2.0Entries into side squares 70.9 6 11.2 65.9 6 8.8 81.1 6 3.5 106.6 6 10.9*Entries into corner squares 22.6 6 3.5 20.9 6 2.5 26.5 6 1.0 34.6 6 3.5*Total entries 118.4 6 17.3 105.9 6 14.6 117.5 6 4.7 154.3 6 15.7*Rears 19.7 6 3.3 36.7 6 5.9** 40.7 6 3.8 57.7 6 6.3*Grooming bouts 1.3 6 0.2 0.9 6 0.3 1.8 6 0.7 1.1 6 0.1Fecal boli 2.6 6 0.8 1.1 6 0.3** 0.6 6 0.1 0.3 6 0.1*
*P < 0.05 and ** P < 0.01, by main effect of genotype by ANOVA, with repeated measures over trials.
Myelination and behaviour in TN-C±/± mice 3089
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092

tests (Fig. 5B, D16, R1±5; genotype, F2,23 = 1.84; genotype 3 days,
F8,92 = 0.71; both NS), showing that tenascin-C de®ciency on the 129
background had no effect on the performance in the water maze test.
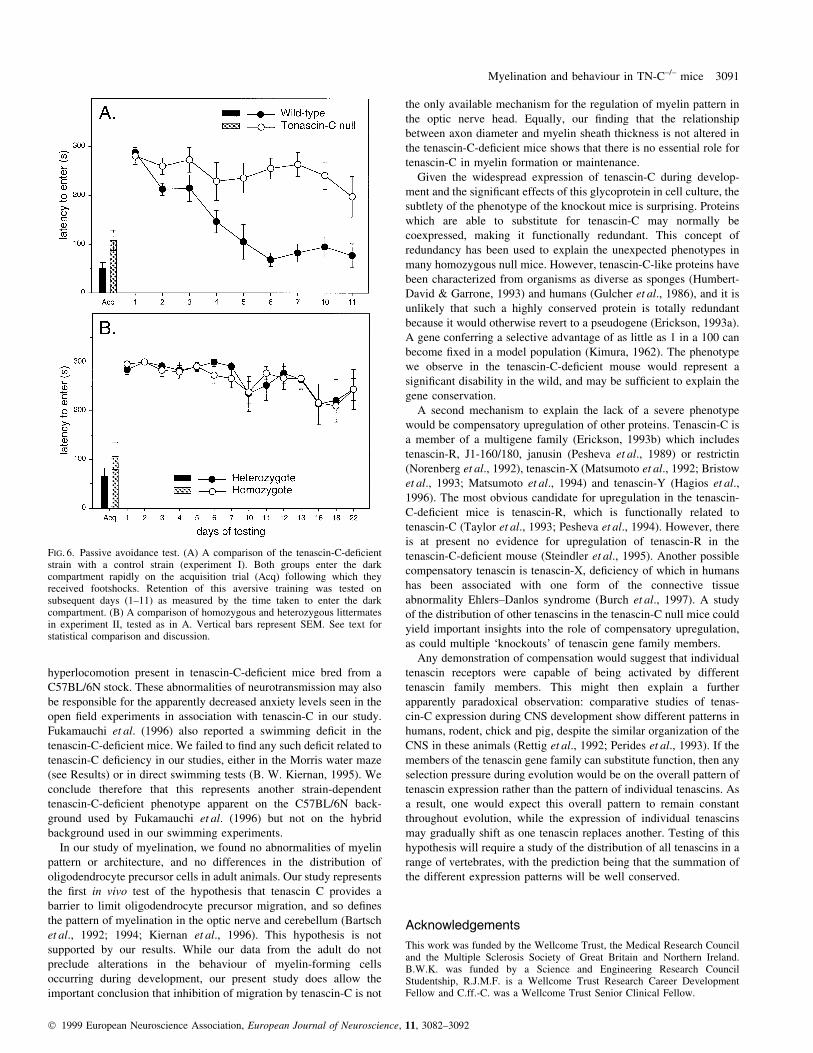
Passive avoidance
Both of the original stocks of mice learnt equally quickly (generally
within one trial) to avoid the stimulus by avoiding the dark
compartment of the apparatus (Fig. 6), and showed good retention
by staying out the following day. However, the wild-type mice
progressively overcame their inhibition to re-enter on subsequent
days of testing (when no further shocks were given), whereas the
tenascin-C null mice were more prone to continue to avoid the dark
chamber even over 2 weeks later (Fig. 6A; genotype 3 days,
F9,90 = 3.62, P < 0.001). It should be noted that the difference is in a
direction opposite to that which would be predicted by a de®cit in
memory, and may re¯ect differences in activity, anxiety or
exploratory behaviour observed in the open ®eld tests.
The passive avoidance de®cit observed in the separate strains was
not replicated in the backcross experiment II (Fig. 6B; genotype,
F1,10 = 0.00; genotype 3 days, F14,140 = 0.51; both NS). However, in
this experiment, even the wild-type mice failed to re-enter even after
3 weeks of repeat testing, so any differences appear to be masked by a
ceiling effect under the training conditions that applied in this
experiment.
Discussion
Previous histological studies have failed to reveal any abnormalities
in two independently derived tenascin-C-de®cient transgenic mouse
lines during development or repair of the peripheral nervous system
and skin wounds (Saga et al., 1992; Forsberg et al., 1996). However,
our results show clear behavioural abnormalities in the animals
derived by Saga et al. (1992), and con®rmed as either lacking (Settles
et al., 1997) or expressing low levels of a truncated form (Mitrovic &
Schachner, 1995) of tenascin-C. These abnormalities comprise
hyperlocomotion, increased defecation frequency, abnormal postural
re¯exes, deafness, coordination abnormalities on the beam as
measured by footslips, cognitive de®cits in the Morris water maze
and prolonged passive avoidance. In order to con®rm the relationship
of these abnormalities to tenascin-C de®ciency and explore the
effects of different mouse genetic backgrounds, we performed a
series of breeding experiments. Two further conclusions arise from
these experiments. First, abnormalities of coordination on the beam,
hyperlocomotion and defecation frequency are related to tenascin-C
de®ciency on a C57BL/6 J-CBA background, as there was a
signi®cant correlation between these abnormalities and tenascin-C
de®ciency in the backcross experiment. Secondly, however, the lack
of any behavioural abnormalities in the experiments on a 129
background show that the manifestation of these effects is dependent
on the genetic strain used.
Our study represents the most comprehensive behavioural analysis
of genetic background effects in the tenascin-C-de®cient mice
performed to date, and the conclusions are important for three
reasons. First, they signi®cantly extend the phenotype associated with
tenascin-C de®ciency. In addition to the hyperlocomotion seen in
previous studies (Fukamauchi et al., 1996; 1997a,b), we have
observed two novel abnormalities in measures of coordination and
anxiety, and con®rmed their relationship to tenascin-C de®ciency.
Secondly, our experiments showing that the beam walking de®cit is
not seen in association with tenascin-C de®ciency following the
introduction of a 129 background emphasizes the importance of
analysing genetic background effects in behavioural and other studies
of transgenic mice. Thirdly, they show that breeding experiments to
con®rm that abnormalities in the original mouse strain described by
Saga et al. (1992) can be ascribed to tenascin-C de®ciency may need
to be performed on a number of different genetic backgrounds to
reveal an effect. The use of congenic mice may be inappropriate for
these further studies. Wolfer et al. (1997) observed that 129 backcross
mice performed poorly in the water maze and masked the effect of
introduced transgenes. They concluded that `creation of congenic
strains by backcrossing mutant hybrids to an inbred strain in order to
avoid genetic variability does not pay off' and our own observations
that the congenic mice performed poorly on the bridge tests supports
this conclusion.
The novel observation that tenascin-C de®ciency causes an
abnormality of the nervous system that results in dif®culty of
coordination on the beams adds to the evidence of a signi®cant role
for this molecule in neural development. The precise nature of this
abnormality remains unde®ned. One possibility we considered was a
balance/hearing de®cit caused by inner ear malfunction. However,
this was ruled out as homozygous tenascin-C-de®cient mice from the
TN±/± 3 [TN±/± 3 F1 C57BL/6 J-CBA] backcross litter suffered more
slips than their heterozygote littermates but did not have a balance
defect. Other putative causes, e.g. a weaker grip or muscle strength
are unlikely as homozygous tenascin-C-de®cient mice are capable of
suspending themselves upside-down on the wire roofs of their cages
and clinging onto vertical wire walls or pencils (unpublished
observations). This leaves defects in motor coordination or
proprioception as the most likely causes of the `footslip phenotype'.
Two recent studies have examined the neuromuscular junction
(NMJ) in tenascin-C-de®cient mice; while Moscoso et al. (1998)
found no abnormalities in the structure of the developing or adult
NMJ, Cifuentes-Diaz et al. (1998) found preterminal disorganiza-
tion and evidence for axonal overgrowth. This latter study raises
the possibility that NMJ abnormalities underlie the phenotype we
observe during beam walking. However, this study was performed
on the original strain of Saga et al. (1992), in contrast to that of
Moscoso et al. (1998) who outcrossed the mice with a C57BL/6
female. Further studies using different genetic backgrounds on the
NMJ and other areas involved in proprioception and balance are
clearly required.
Abnormalities of dopaminergic transmission and reduced tyrosine
hydroxylase activity were observed in recent studies on the
TABLE 2. Preyer re¯ex
Experiment Animal set +/+ +/± ±/±
I Strains 7/7 (100%) N/A 0/6 (0%)**II Backcross: TN±/± 3 [TN±/± 3 F1 C57BL/6-CBA] N/A 5/5 (100%) 2/8 (25.0%)III Intercross: [TN±/± 3 129] 3 [TN±/± 3 129] 8/8 (100%) 9/12 (75%) 4/6 (66.7%)IV 129 partial congenic 8/8 (100%) 9/9 (100%) 8/8 (100%)
The number of animals within each set displaying normal Preyer re¯exes as a fraction (and percentage). Note that the trend in the homozygote animals istoward normality as the tenascin-C de®ciency is bred away from the original strain. ** P = 0.01
3090 B. W. Kiernan et al.
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092
hyperlocomotion present in tenascin-C-de®cient mice bred from a
C57BL/6N stock. These abnormalities of neurotransmission may also
be responsible for the apparently decreased anxiety levels seen in the
open ®eld experiments in association with tenascin-C in our study.
Fukamauchi et al. (1996) also reported a swimming de®cit in the
tenascin-C-de®cient mice. We failed to ®nd any such de®cit related to
tenascin-C de®ciency in our studies, either in the Morris water maze
(see Results) or in direct swimming tests (B. W. Kiernan, 1995). We
conclude therefore that this represents another strain-dependent
tenascin-C-de®cient phenotype apparent on the C57BL/6N back-
ground used by Fukamauchi et al. (1996) but not on the hybrid
background used in our swimming experiments.
In our study of myelination, we found no abnormalities of myelin
pattern or architecture, and no differences in the distribution of
oligodendrocyte precursor cells in adult animals. Our study represents
the ®rst in vivo test of the hypothesis that tenascin C provides a
barrier to limit oligodendrocyte precursor migration, and so de®nes
the pattern of myelination in the optic nerve and cerebellum (Bartsch
et al., 1992; 1994; Kiernan et al., 1996). This hypothesis is not
supported by our results. While our data from the adult do not
preclude alterations in the behaviour of myelin-forming cells
occurring during development, our present study does allow the
important conclusion that inhibition of migration by tenascin-C is not
the only available mechanism for the regulation of myelin pattern in
the optic nerve head. Equally, our ®nding that the relationship
between axon diameter and myelin sheath thickness is not altered in
the tenascin-C-de®cient mice shows that there is no essential role for
tenascin-C in myelin formation or maintenance.
Given the widespread expression of tenascin-C during develop-
ment and the signi®cant effects of this glycoprotein in cell culture, the
subtlety of the phenotype of the knockout mice is surprising. Proteins
which are able to substitute for tenascin-C may normally be
coexpressed, making it functionally redundant. This concept of
redundancy has been used to explain the unexpected phenotypes in
many homozygous null mice. However, tenascin-C-like proteins have
been characterized from organisms as diverse as sponges (Humbert-
David & Garrone, 1993) and humans (Gulcher et al., 1986), and it is
unlikely that such a highly conserved protein is totally redundant
because it would otherwise revert to a pseudogene (Erickson, 1993a).
A gene conferring a selective advantage of as little as 1 in a 100 can
become ®xed in a model population (Kimura, 1962). The phenotype
we observe in the tenascin-C-de®cient mouse would represent a
signi®cant disability in the wild, and may be suf®cient to explain the
gene conservation.
A second mechanism to explain the lack of a severe phenotype
would be compensatory upregulation of other proteins. Tenascin-C is
a member of a multigene family (Erickson, 1993b) which includes
tenascin-R, J1-160/180, janusin (Pesheva et al., 1989) or restrictin
(Norenberg et al., 1992), tenascin-X (Matsumoto et al., 1992; Bristow
et al., 1993; Matsumoto et al., 1994) and tenascin-Y (Hagios et al.,
1996). The most obvious candidate for upregulation in the tenascin-
C-de®cient mice is tenascin-R, which is functionally related to
tenascin-C (Taylor et al., 1993; Pesheva et al., 1994). However, there
is at present no evidence for upregulation of tenascin-R in the
tenascin-C-de®cient mouse (Steindler et al., 1995). Another possible
compensatory tenascin is tenascin-X, de®ciency of which in humans
has been associated with one form of the connective tissue
abnormality Ehlers±Danlos syndrome (Burch et al., 1997). A study
of the distribution of other tenascins in the tenascin-C null mice could
yield important insights into the role of compensatory upregulation,
as could multiple `knockouts' of tenascin gene family members.
Any demonstration of compensation would suggest that individual
tenascin receptors were capable of being activated by different
tenascin family members. This might then explain a further
apparently paradoxical observation: comparative studies of tenas-
cin-C expression during CNS development show different patterns in
humans, rodent, chick and pig, despite the similar organization of the
CNS in these animals (Rettig et al., 1992; Perides et al., 1993). If the
members of the tenascin gene family can substitute function, then any
selection pressure during evolution would be on the overall pattern of
tenascin expression rather than the pattern of individual tenascins. As
a result, one would expect this overall pattern to remain constant
throughout evolution, while the expression of individual tenascins
may gradually shift as one tenascin replaces another. Testing of this
hypothesis will require a study of the distribution of all tenascins in a
range of vertebrates, with the prediction being that the summation of
the different expression patterns will be well conserved.
Acknowledgements
This work was funded by the Wellcome Trust, the Medical Research Counciland the Multiple Sclerosis Society of Great Britain and Northern Ireland.B.W.K. was funded by a Science and Engineering Research CouncilStudentship, R.J.M.F. is a Wellcome Trust Research Career DevelopmentFellow and C.ff.-C. was a Wellcome Trust Senior Clinical Fellow.
FIG. 6. Passive avoidance test. (A) A comparison of the tenascin-C-de®cientstrain with a control strain (experiment I). Both groups enter the darkcompartment rapidly on the acquisition trial (Acq) following which theyreceived footshocks. Retention of this aversive training was tested onsubsequent days (1±11) as measured by the time taken to enter the darkcompartment. (B) A comparison of homozygous and heterozygous littermatesin experiment II, tested as in A. Vertical bars represent SEM. See text forstatistical comparison and discussion.
Myelination and behaviour in TN-C±/± mice 3091
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092

Abbreviations
IHC, immunohistochemistry; ISH, in situ hybridization; MBP, myelin basicprotein; NMJ, neuromuscular junction; NS, not signi®cant; PBS, phosphate-buffered saline; PVA, polyvinyl alcohol; SSC, standard sodium citrate.
References
Bartsch, S., Bartsch, U., DoÈrries, U., Faissner, A., Weller, A., Ekblom, P. &Schachner, M. (1992) Expression of tenascin in the developing and adultcerebellar cortex. J. Neurosci., 12, 736±749.
Bartsch, U., Faissner, A., Trotter, J., DoÈrries, U., Bartsch, S., Mohajeri, H. &Schachner, M. (1994) Tenascin demarcates the boundary between themyelinated and non-myelinated part of retinal ganglion cell axons in thedeveloping and adult mouse. J. Neurosci., 14, 4756±4768.
Bartus, R.T., Dean, R.L., Goas, J.A. & Lippa, A.S. (1980) Age related changesin passive avoidance retention and modulation with chronic dietary choline.Science, 209, 301±303.
Bristow, J., Tee, M.K., Gitelman, S.E., Mellon, S.H. & Miller, W.L. (1993)Tenascin-X: a novel extracellular matrix protein encoded by the human XBgene overlapping P450c21B. J. Cell Biol., 122, 265±278.
Burch, G.H., Gong, Y., Liu, W.H., Dettman, R.W., Curry, C.J., Smith, L.,Miller, W.L. & Bristow, J. (1997) Tenascin-X de®ciency is associated withEhlers±Danlos syndrome. Nature Genet., 17, 104±108.
Chiquet-Ehrismann, R. (1991) Anti-adhesive molecules of the extracellularmatrix. Curr. Opin. Cell Biol., 3, 800±804.
Cifuentes-Diaz, C., Velasco, E., Meunier, F.A., Goudou, D., Belkadi, L.,Faille, L., Murawsky, M., Angaut-Petit, D., Molgo, J., Schachner, M., Saga,Y., Aizawa, S. & Reiger, F. (1998) The peripheral nerve and theneuromuscular junction are affected in the tenascin-C de®cient mouse.Cell Mol. Biol., 44, 357±379.
Dean, R.L., Scozzafava, J., Goas, J.A., Regan, B., Beer, R. & Bartus, R.T.(1981) Age-related differences in behavior across the life span of theC57BL/6J mouse. Exp. Aging Res., 7, 427±451.
Erickson, H.P. (1993a) Gene knockouts of c-src, transforming growth factorb1, and tenascin suggest super¯uous, nonfunctional expression of proteins.J. Cell Biol., 120, 1079±1081.
Erickson, H.P. (1993b) Tenascin-C, tenascin-R and tenascin-X: a family oftalented proteins in search of functions. Curr. Opin. Cell Biol., 5, 869±876.
Faissner, A., GoÈtz, B., Joester, A. & Scholze, A. (1995) The tenascin genefamily ± versatile glycoproteins implicated in neural pattern formation andregeneration. Semin. Dev. Biol., 6, 139±148.
Forsberg, E., Hirsch, E., Frohlich, L., Meyer, M., Ekblom, P., Aszodi, A.,Werner, S. & FaÈssler, R. (1996) Skin wounds and severed nerves healnormally in mice lacking tenascin-C. Proc. Natl Acad. Sci. USA, 93, 6594±6599.
Frost, E., Kiernan, B.W., Faissner, A. & ffrench-Constant, C. (1996)Regulation of oligodendrocyte precursor migration by extracellularmatrix: evidence for substrate-speci®c inhibition of migration by tenascin-C. Dev. Neurosci., 18, 266±273.
Fukamauchi, F., Mataga, N., Wang, Y.-J., Sato, S., Yoshiki, A. & Kusakabe,M. (1996) Abnormal behavior and neurotransmissions of tenascin geneknockout mouse. Biochem. Biophys. Res. Commun., 221, 151±156.
Fukamauchi, F., Mataga, N., Wang, Y.-J., Sato, S., Yoshiki, A. & Kusakabe,M. (1997a) Tyrosine hydroxylase activity and its mRNA level indopaminergic neurones of tenascin gene knockout mouse. Biochem.Biophys. Res. Commun., 231, 356±359.
Fukamauchi, F., Wang, Y.-J., Mataga, N. & Kusakabe, M. (1997b)Paradoxical behavioral response to apomorphine in tenascin-geneknockout mouse. Eur. J. Pharm., 338, 7±10.
Gulcher, J.R., Marton, L.S. & Stefansson, K. (1986) Two large glycosylatedpolypeptides found in myelinating oligodendrocytes but not myelin. Proc.Natl Acad. Sci. USA, 83, 2118±2122.
Hagios, C., Koch, M., Spring, J., Chiquet, M. & Chiquet-Ehrismann, R. (1996)Tenascin-Y: a protein of novel domain structure is secreted by differentiated®broblasts of muscle connective tissue. J. Cell Biol., 134, 1499±1512.
Humbert-David, N. & Garrone, R. (1993) A six-armed, tenascin-like protein
extracted from the Porifera Oscarella tuberculata (Homosclerophorida).Eur. J. Biochem., 216, 255±260.
Kiernan, B.W. (1995) The effect of tenascin on myelination and cell migrationin the mammalian central nervous system. PhD Thesis, University ofCambridge.
Kiernan, B.W., GoÈtz, B., Faissner, A. & ffrench-Constant, C. (1996) Tenascin-C inhibits oligodendrocyte precursor cell migration by both adhesion-dependent and adhesion-independent mechanisms. Mol. Cell. Neurosci., 7,322±335.
Kimura, M. (1962) On the probability of ®xation of mutant genes in apopulation. Genetics, 47, 713±719.
Matsumoto, K., Arai, M., Ishihara, N., Ando, A., Inoko, H. & Ikemura, T.(1992) Cluster of ®bronectin type III repeats found in the majorhistocompatibility complex class III region shows the highest homologywith the repeats in an extracellular matrix protein, tenascin. Genomics, 12,485±491.
Matsumoto, K., Saga, Y., Ikemura, T., Sakakura, T. & Chiquet-Ehrismann, R.(1994) The distribution of tenascin-X is distinct and often reciprocal to thatof tenascin-C. J. Cell Biol., 125, 483±493.
Mitrovic, N. & Schachner, M. (1995) Detection of tenascin-C in the nervoussystem of the tenascin-C mutant mouse. J. Neurosci. Res., 42, 710±711.
Morris, R.G.M., Anderson, E., Lynch, G.S. & Baudry, M. (1986) Selectiveimpairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature, 319, 774±776.
Morris, G.L. & O'Shea, K.S. (1983) Anomalies of neuroepithelial cellassociations in the Splotch mutant embryo. Dev. Brain Res., 9, 408±410.
Moscoso, L.M., Cremer, H. & Sanes, J.R. (1998) Organization andreorganization of neuromuscular junctions in mice lacking neural celladhesion molecule, tenascin-C, or ®broblast growth factor-5. J. Neurosci.,18, 1465±1477.
NoÈrenberg, U., Wille, H., Wolff, J.M., Frank, R. & Rathjen, F.G. (1992) Thechicken neural extracellular matrix molecule restrictin: similarity with EGF-,®bronectin Type III-, and ®brinogen-like motifs. Neuron, 8, 849±863.
Perides, G., Erickson, H.P., Rahemtulla, F. & Bignami, A. (1993)Colocalization of tenascin with versican, a hyaluronate-bindingchondroitin sulfate proteoglycan. Anat. Embryol. (Berl.), 188, 467±479.
Pesheva, P., Probstmeier, R., Skubitz, A.P.N., McCarthy, J.B., Furcht, L.T. &Schachner, M. (1994) Tenascin-R (J1 160/180) inhibits ®bronectin-mediated cell adhesion ± functional relatedness to tenascin-C. J. Cell Sci.,107, 2323±2333.
Pesheva, P., Spiess, E. & Schachner, M. (1989) J1±160 and J1±180 areoligodendrocyte-secreted nonpermissive substrates for cell adhesion. J. CellBiol., 109, 1765±1778.
Pringle, N.P., Mudhar, H.S., Collarini, E.J. & Richardson, W.D. (1992) PDGFreceptors in the rat CNS: during late neurogenesis, PDGF alpha-receptorexpression appears to be restricted to glial cells of the oligodendrocytelineage. Development, 115, 535±551.
Rettig, W.J., Hoffman, S., Su, S.L. & Garin-Chesa, P. (1992) Species diversityof neuronectin and cytotactin expression patterns in the vertebrate centralnervous system. Brain Res., 590, 219±228.
Robichaud, R.C., Hefner, M.A., Anderson, J.E. & Goldberg, M.E. (1973)Effect of D9-tetrahydrocannabinol. (THC) on several rodent learningparadigms. Pharmacology, 10, 1±11.
Saga, Y., Yagi, T., Ikawa, Y., Sakakura, T. & Aizawa, S. (1992) Mice developnormally without tenascin. Genes Dev., 6, 1821±1831.
Settles, D.L., Kusakabe, M., Steindler, D.A., Fillmore, H. & Erickson, H.P.(1997) Tenascin-C knockout mouse has no detectable tenascin-C protein. J.Neurosci. Res., 47, 109±117.
Steindler, D.A., Settles, D., Erickson, H.P., Laywell, E.D., Yoshiki, A.,Faissner, A. & Kusakabe, M. (1995) Tenascin knockout mice: barrels,boundary molecules, and glial scars. J. Neurosci., 15, 1971±1983.
Taylor, J., Pesheva, P. & Schachner, M. (1993) In¯uence of janusin andtenascin on growth cone behavior in vitro. J. Neurosci. Res., 35, 347±362.
Wolfer, D.P., Muller, U., Stagliar, M. & Lipp, H.-P. (1997) Assessing theeffects of the 129/Sv genetic background on swimming navigation learningin transgenic mutants: a study using mice with a modi®ed b-amyloidprecursor protein gene. Brain Res., 771, 1±13.
3092 B. W. Kiernan et al.
Ó 1999 European Neuroscience Association, European Journal of Neuroscience, 11, 3082±3092





























