Embed Size (px)
Citation preview

Neuro®lament inclusion body disease: a newproteinopathy?
Keith A. Josephs,1,5,7 Janice L. Holton,7 Martin N. Rossor,5 Hans Braendgaard,8 Tetsutaro Ozawa,6,7
Nick C. Fox,5 Ronald C. Petersen,1 Gary S. Pearl,4 Milan Ganguly,7 Pedro Rosa,8 Henning Laursen,9
Joseph E. Parisi,1,2 Gunhild Waldemar,9 Niall P. Quinn,6 Dennis W. Dickson1,3 and Tamas Revesz7
Departments of 1Neurology, 2Laboratory Medicine and
Pathology, Mayo Clinic, Rochester, MN and 3Department
of Pathology (Neuropathology), Mayo Clinic, Jacksonville,
FL, 4Department of Pathology, Orlando Regional Medical
Center, Orlando, FL, USA, 5Dementia Research Group,6Department of Motor Neuroscience and Movement
Disorders and 7Queen Square Brain Bank, Department of
Molecular Neuroscience and Division of Neuropathology,
Institute of Neurology, University College London, London,
UK, 8Aarhus Kommunehospital, Aarhus and 9Copenhagen
University Hospital, Copenhagen, Denmark
Correspondence to: Tamas Revesz, MD, Division of
Neuropathology, Institute of Neurology, University College
London, London WC1N 3BG, UK
E-mail: t.revesz@ ion.ucl.ac.uk
SummaryWe describe four cases of a new clinicopathologicalentity presenting with either a frontotemporal dementiaor corticobasal degeneration syndrome with a mean ageof onset of 45 years (range 41±50) characterized patho-logically by deposition of neuro®lament proteins. Allfour patients had a rapidly progressive course and havebecome mute and non-ambulatory, and three have diedafter mean illness duration of only 3 years (range 21
2±4).±4).
Both structural (MRI) and functional (PET andBoth structural (MRI) and functional (PET andSPECT) imaging demonstrated frontal and temporalSPECT) imaging demonstrated frontal and temporallobe and basal ganglia involvement. Gross neuropatho-lobe and basal ganglia involvement. Gross neuropatho-logical examination in the three deceased patients (thelogical examination in the three deceased patients (thefourth patient, still alive, was diagnosed by brainfourth patient, still alive, was diagnosed by brainbiopsy) revealed changes affecting predominantly thebiopsy) revealed changes affecting predominantly thefrontal and temporal cortices, basal ganglia and brain-frontal and temporal cortices, basal ganglia and brain-stem. There was super®cial linear spongiosis affectingstem. There was super®cial linear spongiosis affectingthe frontal lobes in all three autopsied patients, andthe frontal lobes in all three autopsied patients, andsevere caudate atrophy was noted in two of them andsevere caudate atrophy was noted in two of them anddemonstrated on MRI in the living patient. On routinedemonstrated on MRI in the living patient. On routine
staining, there were numerous intracytoplasmic inclu-staining, there were numerous intracytoplasmic inclu-
sions, which ranged from eosinophilic to basophilic.sions, which ranged from eosinophilic to basophilic.
Some had a clearly de®ned basophilic margin, whileSome had a clearly de®ned basophilic margin, while
others were granular with a hyaline core. With modi-others were granular with a hyaline core. With modi-
®ed Bielschowsky silver technique, a small number of®ed Bielschowsky silver technique, a small number of
the inclusions were intensely stained. Inclusions werethe inclusions were intensely stained. Inclusions were
not labelled with other silver stains. Immuno-not labelled with other silver stains. Immuno-
histochemistry revealed that the inclusions were immu-histochemistry revealed that the inclusions were immu-
noreactive with antibodies to neuro®lament heavy andnoreactive with antibodies to neuro®lament heavy and
light chain subunits and to ubiquitin, but not with anti-light chain subunits and to ubiquitin, but not with anti-
bodies to tau andbodies to tau and aa-synuclein. These neuro®lament- and-synuclein. These neuro®lament- and
ubiquitin-positive inclusions were widespread, speci®cubiquitin-positive inclusions were widespread, speci®c
to neurons and occasionally intranuclear. The frequencyto neurons and occasionally intranuclear. The frequency
and distribution of the inclusions and the silver andand distribution of the inclusions and the silver and
immunohistochemical pro®les in these four cases isimmunohistochemical pro®les in these four cases is
novel and has not been described in detail before. Wenovel and has not been described in detail before. We
propose the term neuro®lament inclusion body diseasepropose the term neuro®lament inclusion body disease
for this entity.for this entity.
Key words: neuro®lament inclusion body disease; frontotemporal dementia; corticobasal degeneration; neuro®lament;
ubiquitin
Abbreviations: AD = Alzheimer's disease; BIBD = basophilic inclusion body disease; CBD = corticobasal degeneration;
CJD = Creutzfeldt±Jakob disease; DLB = dementia with Lewy bodies; DUI 6 MND = dementia with ubiquitin-positive
(tau- and a-synuclein-negative) inclusions with or without motor neuron disease; FTD = frontotemporal dementia; iPD =
idiopathic Parkinson's disease; MSA = multiple system atrophy; NIBD = neuro®lament inclusion body disease; NF =
neuro®lament; PiD = Pick's disease; PSP = progressive supranuclear palsy
Brain 126 ã Guarantors of Brain 2003; all rights reserved
DOI: 10.1093/brain/awg231 Advanced Access publication July 22, 2003 Brain (2003), 126, 2291±2303
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

IntroductionA signi®cant group of neurodegenerative diseases is char-
acterized by intraneuronal inclusions, which may be cyto-
plasmic or intranuclear. An evolving molecular classi®cation
of such neurodegenerative disorders is based upon the
biochemical nature of the protein deposits forming the
inclusions. The microtubule-associated protein tau and
the synaptic vesicle protein a-synuclein account for most of
these neurodegenerative disorders (Goedert et al., 2001).
Abnormal deposition of tau de®nes neuro®brillary tangles in
the most common neurodegenerative disease, Alzheimer's
disease (AD). Furthermore, abnormalities of tau characterize
corticobasal degeneration (CBD), progressive supranuclear
palsy (PSP) and frontotemporal dementia with parkinsonism
linked to chromosome 17q (FTDP-17q), in which glial tau
deposits are also frequently found (Lee et al., 2001). The
morphologically distinct Pick bodies characteristic of Pick's
disease (PiD) are also composed of tau (Lowe, 1998; Lee
et al., 2001; McKhann et al., 2001). The diseases associated
with a-synuclein deposition are idiopathic Parkinson's dis-
ease (iPD) and dementia with Lewy bodies (DLB), in which
the pathological hallmark is the Lewy body (Spillantini et al.,
1997). Lewy bodies are also commonly found in AD,
especially in familial forms (Lippa et al., 1998). The glial
cytoplasmic inclusions and intraneuronal inclusions of mul-
tiple system atrophy (MSA) also contain a-synuclein (Lippa
et al., 1998; Spillantini et al., 1998). Many of the de®ning
intracellular inclusions are also labelled by ubiquitin, which is
an essential component of the proteasomal system, respon-
sible for non-lysosomal degradation of intracellular proteins
(Hershko and Ciechanover, 1998). The presence of ubiquitin
in the inclusions is less speci®c, although it is the only
identi®ed component in the inclusions of dementia with
ubiquitin-positive (tau- and a-synuclein-negative) inclusion
bodies with or without motor neuron disease (DUI 6 MND)
(Rossor et al., 2000). The term DUI 6 MND incorporates
motor neuron disease±inclusion dementia (Jackson et al.,
1996; Holton et al., 2002); dementia with inclusions tau- and
synuclein-negative, ubiquitinated (Kertesz et al., 2000); and
motor neuron disease-type dementia (Okamoto et al., 1991;
Wightman et al., 1992).
The neuropathological differential diagnosis of the neuro-
degenerative disorders described above is complex and takes
into account clinical information, the anatomical distribution
of gross and microscopic changes, and the protein compos-
ition of the intracellular inclusions assessed by immunohisto-
chemical and biochemical means.
We have identi®ed four novel cases presenting with either
the frontotemporal dementia (FTD) variant of frontotemporal
lobar degeneration (Neary et al., 1998) or CBD syndrome,
with microscopy demonstrating the presence of widespread
intraneuronal inclusions. Unlike the pathological features
described above, the inclusions were intensely immunoreac-
tive to neuro®lament (NF) antibodies, yet were not recog-
nized by tau or a-synuclein antibodies. We have studied the
clinical features, molecular composition, distribution and
localization of the NF inclusions by performing a review of
the medical records, semiquantitative immunohistochemical
analysis and electron microscopy in these four cases. We
propose the term NF inclusion body disease (NIBD) to
describe these unique cases.
Material and methodsPatientsFour patients with microscopic demonstration of neurode-
generation and unique features characterized by the presence
of diffuse and intensely stained NF-positive inclusion bodies
were recognized by three of the authors (T.R., J.L.H. and
D.W.D.) over a 4 year period (1998±2002). Two of the
patients resided in the United States (cases US1 and US2) and
one each in Denmark (case D1) and the United Kingdom
(case UK1). Three had complete post-mortem analysis, and
one (D1) had a frontal lobe biopsy. As controls, we studied
haematoxylin and eosin (H&E), NF and ubiquitin staining
pro®les in two normal control brains and pathological
controls including three cases of DUI 6 MND, two cases
of AD and one case each of DLB and PiD. For each control
case, we reviewed anterior frontal and temporal cortices and
hippocampal sections.
Neuropathological procedureIn the post-mortem cases, consent to brain examination was
obtained. Half of each brain was ®xed in 10% formalin for a
minimum of 4 weeks, while the other half was stored frozen.
Each ®xed half-brain hemisphere was sliced coronally and
tissue blocks were taken from representative areas, including
the frontal, temporal, parietal and occipital cortices, amygda-
la, hippocampus, basal ganglia, thalamus, midbrain, pons,
medulla oblongata and cerebellum. Cervical cord was also
available in cases US1 and US2. Following processing and
paraf®n wax embedding, 7 mm sections were cut and stained
with routine methods including H&E, Nissl, periodic acid±
Schiff/haematoxylin (PAS/H), Congo Red, thio¯avin-S, luxol
fast blue/cresyl violet, and silver impregnations (Bodian,
modi®ed Bielschowsky, and Gallyas methods).
In case D1, a biopsy of the right frontal lobe was
performed, ®xed in 10% formalin and processed for paraf®n
wax using standard procedures. Sections were cut and stained
as described above.
Immunohistochemistry and antibodiesImmunohistochemistry was carried out with special emphasis
on NF subunits. This involved the use of antibodies to
phosphorylated NF (pNF) light, medium and heavy (L, M and
H) subunits and antibody to non-phosphorylated NF heavy
2292 K. A. Josephs et al.
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

subunit (non-pNF-H). An NF-cocktail antibody recognizing
L and H subunits was also utilized.
Sections (7 mm) were cut from selected blocks. The
sections were de-paraf®nized in xylene and subsequently
rehydrated in graded alcohol solutions. Endogenous perox-
idase activity was neutralized in methanol/H2O2 solution for
10 min. Some sections were pretreated by pressure cooking
(10 min) to unmask antigenic activity. Non-speci®c antigen
binding was blocked in normal horse serum (Vectastain elite
ABC kit; Vectastain, Peterborough, UK). The primary
antibodies made up in phosphate-buffered saline (PBS, 1%)
to the dilutions stated below were applied to the sections and
incubated for 1 h at room temperature. After several washes
in PBS solution, the sections were incubated in prediluted
biotinylated secondary antibody (Vectastain elite ABC kit; 30
min, room temperature). Following washes as described
previously, the ABC reagent (Vectastain elite ABC kit) was
applied to sections (30 min, room temperature). Visualization
of immunolabelled structures was achieved by incubating
sections in diaminobenzidine/H2O2 solution until coloration
was visible (usually 3±4 min). Washing and counterstaining
(Mayer's haematoxylin; WWR International, Poole, UK),
clearing and mounting followed.
Primary antibody type (monoclonal unless stated), dilution,
pretreatment and source were as follows: pNF-H (SMI 31, 1:
5000, pressure cooked) and non-pNF-H (SMI 32, 1: 500,
pressure cooked; Sternberger Monoclonals, Lutherville, UK);
pNF-M (BF10, 1: 400, pressure cooked; Af®nity Research
Products, Aurora, USA); pNF-L (Ab1, 1: 100, pressure
cooked; Oncogene Research Products, Boston, USA); NF
cocktail (1: 20, no pretreatment; ICN Pharmaceuticals,
Aurora, USA); ubiquitin (1: 300 polyclonal, pressure cooked;
DAKO, Ely, UK); tau (rabbit anti-tau Cat. No. A0024, reacts
with both phosphorylated and non-phosphorylated tau, 1:
200, no pretreatment; DAKO); tau (AT8, recognizing phos-
phorylated Ser202/Thr205, 1: 600, no microwaved; Autogen
Bioclear, Calne, UK); GFAP (1: 1000, trypsin 0.1%; DAKO);
1C2 (1: 2000, formic acid and pressured cooked; Chemicon,
Chandlers Ford, UK); Ab (1: 100, formic acid and pressure
cooked; DAKO); a-synuclein (N-19, goat polyclonal, 1:
1000, formic acid; Autogen Bioclear); aB-crystallin (1: 1200,
no pretreatment; Novocastra Laboratories, Newcastle Upon
Tyne, UK); prion protein (i) KG9 (1: 50, formic acid and
pressure cooked; obtained from Institute of Animal Health,
Compton, UK) and (ii) 3F4 (1: 2000, formic acid and pressure
cooked; obtained from DAKO).
Assessment of inclusion frequencyThe frequency of the inclusions was assessed using H&E and
both Ab1 and ubiquitin immunohistochemistry in a number of
brain regions (see Table 3). A semiquantitative approach was
used and a grading scale established, in which grade 0 was
used for regions with no inclusions, + for a small number, ++
for a moderate number, +++ for numerous and ++++ for the
most severely affected areas.
Electron microscopyElectron microscopy was performed in all three post-mortem
cases. Formalin-®xed frontal and temporal cortices were post-
®xed in buffered 1% osmium tetroxide, processed by
conventional techniques and ®nally embedded in epoxy
resin (Agar, Stantead, UK). Sections were cut on an LKB
ultramicrotome, stained with uranyl acetate and lead citrate
and viewed with a JEOL 1200 EX electron microscope
(JEOL, Peabody, USA).
ResultsClinical ®ndingsThe demographic data of all four cases are listed in Table 1.
Clinical features of all four cases are outlined in Table 2, and
individual case histories are reported in the Supplementary
data (available at Brain Online). Two cases (UK1 and US2)
presented with asymmetric cortical and basal ganglia symp-
toms, characterized by asymmetric loss of hand dexterity,
apraxia and early L-dopa-unresponsive parkinsonism. The
other two cases (US1 and D1), however, presented with
frontal and temporal lobe features including poor organ-
ization and planning, loss of initiative, reduced hygiene, and
personality change. Parkinsonism was also present in these
cases but developed late and was asymmetric in one (US1).
Prominent features common to all cases included rapid
progression of symptoms with early falling, mutism and loss
of mobility. In some of the cases, headaches (UK1 and US1),
swallowing dif®culties (D1), dystonia (US1 and US2),
supranuclear gaze palsy (UK1), obsessive±compulsive beha-
viours (US1 and US2), pathological laughter (UK1 and D1)
and pathological crying (US2) were also noted to be present
during the course of the illness.
Past medical history was signi®cant for autoimmune
disease (UK1, US1 and US2), a history of headaches (UK1,
Table 1 Demographic features of four cases of neuro®lament inclusion body disease
Feature UK1 US1 US2 D1
Country of residence United Kingdom United States United States DenmarkSex F F M FAge of onset (years) 41 50 44 43Age at death (years) 44 54 47 *Duration of illness (years) 2.5 4 3.5 3
*Patient still alive, now terminal.
Neuro®lament inclusion body disease 2293
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from
US1 and D1) and a history of depression (UK1 and D1). One
case (US2) had a positive family history of a neurodegen-
erative disorder (iPD followed by dementia in his father).
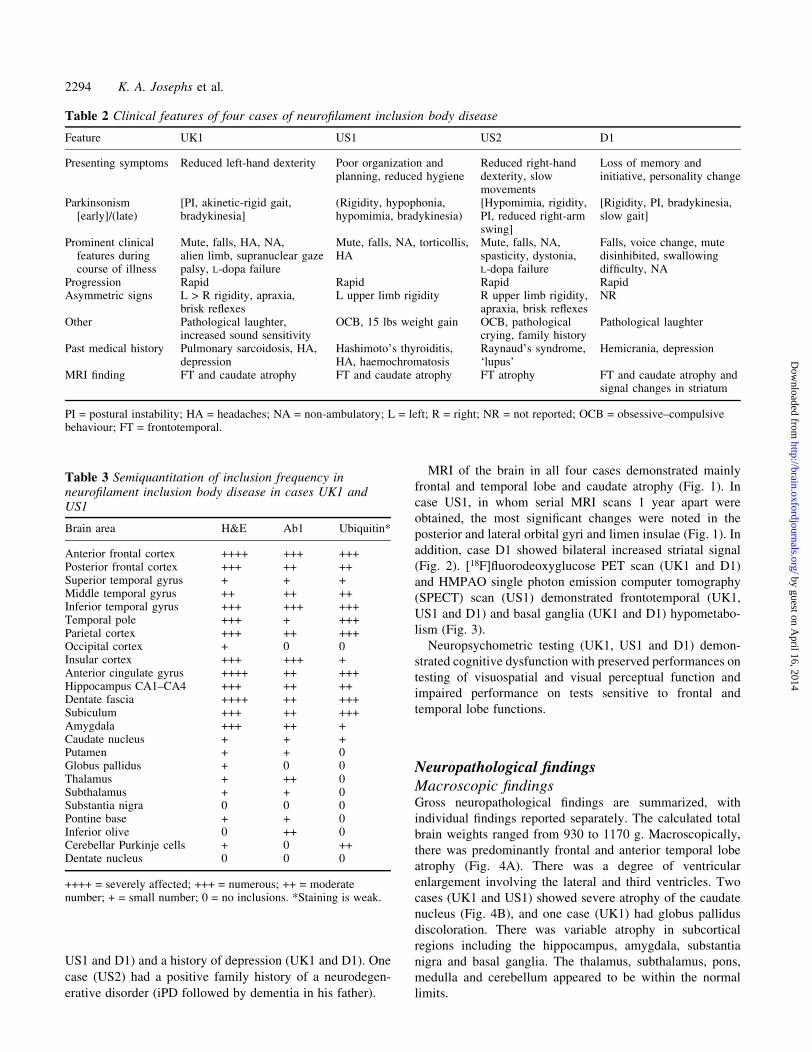
MRI of the brain in all four cases demonstrated mainly
frontal and temporal lobe and caudate atrophy (Fig. 1). In
case US1, in whom serial MRI scans 1 year apart were
obtained, the most signi®cant changes were noted in the
posterior and lateral orbital gyri and limen insulae (Fig. 1). In
addition, case D1 showed bilateral increased striatal signal
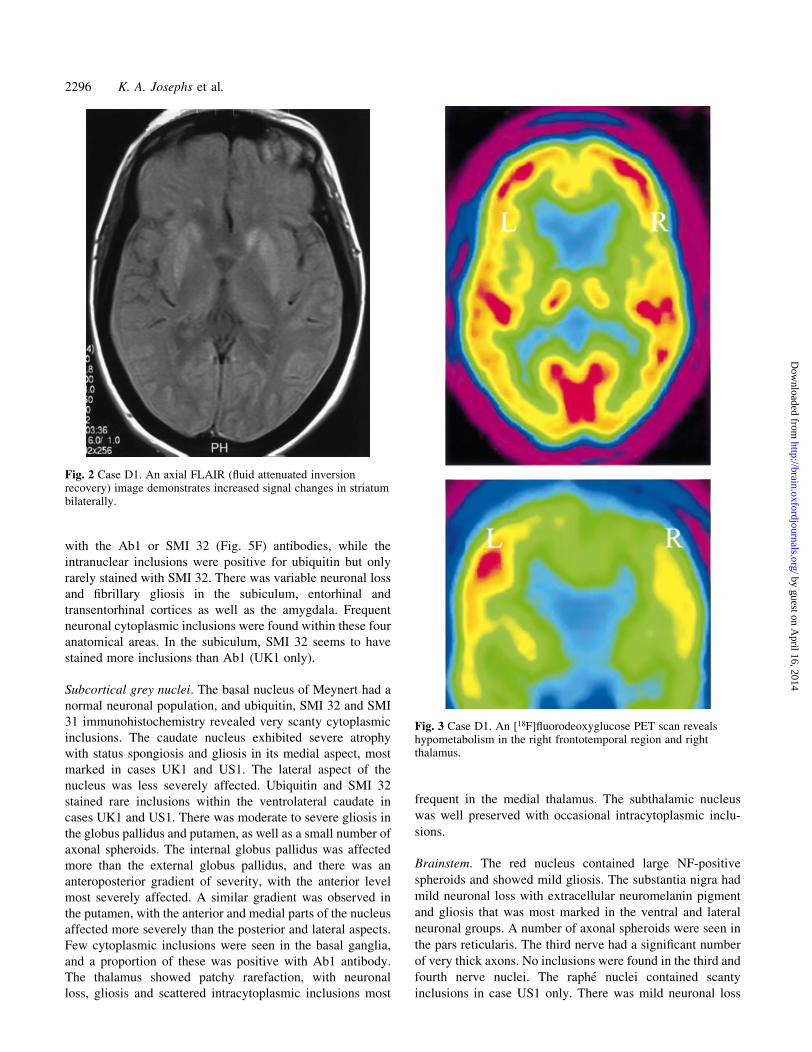
(Fig. 2). [18F]¯uorodeoxyglucose PET scan (UK1 and D1)
and HMPAO single photon emission computer tomography
(SPECT) scan (US1) demonstrated frontotemporal (UK1,
US1 and D1) and basal ganglia (UK1 and D1) hypometabo-
lism (Fig. 3).
Neuropsychometric testing (UK1, US1 and D1) demon-
strated cognitive dysfunction with preserved performances on
testing of visuospatial and visual perceptual function and
impaired performance on tests sensitive to frontal and
temporal lobe functions.
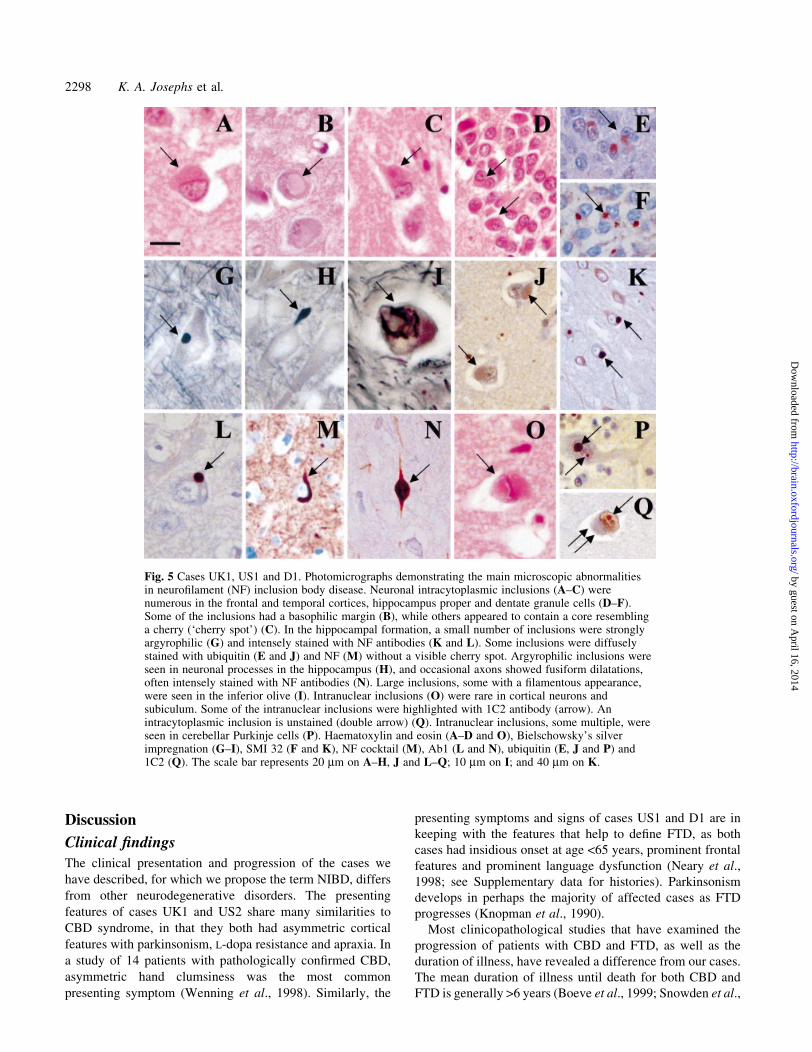
Neuropathological ®ndingsMacroscopic ®ndingsGross neuropathological ®ndings are summarized, with
individual ®ndings reported separately. The calculated total
brain weights ranged from 930 to 1170 g. Macroscopically,
there was predominantly frontal and anterior temporal lobe
atrophy (Fig. 4A). There was a degree of ventricular
enlargement involving the lateral and third ventricles. Two
cases (UK1 and US1) showed severe atrophy of the caudate
nucleus (Fig. 4B), and one case (UK1) had globus pallidus
discoloration. There was variable atrophy in subcortical
regions including the hippocampus, amygdala, substantia
nigra and basal ganglia. The thalamus, subthalamus, pons,
medulla and cerebellum appeared to be within the normal
limits.
Table 2 Clinical features of four cases of neuro®lament inclusion body disease
Feature UK1 US1 US2 D1
Presenting symptoms Reduced left-hand dexterity Poor organization andplanning, reduced hygiene
Reduced right-handdexterity, slowmovements
Loss of memory andinitiative, personality change
Parkinsonism[early]/(late)
[PI, akinetic-rigid gait,bradykinesia]
(Rigidity, hypophonia,hypomimia, bradykinesia)
[Hypomimia, rigidity,PI, reduced right-armswing]
[Rigidity, PI, bradykinesia,slow gait]
Prominent clinicalfeatures duringcourse of illness
Mute, falls, HA, NA,alien limb, supranuclear gazepalsy, L-dopa failure
Mute, falls, NA, torticollis,HA
Mute, falls, NA,spasticity, dystonia,L-dopa failure
Falls, voice change, mutedisinhibited, swallowingdif®culty, NA
Progression Rapid Rapid Rapid RapidAsymmetric signs L > R rigidity, apraxia,
brisk re¯exesL upper limb rigidity R upper limb rigidity,
apraxia, brisk re¯exesNR
Other Pathological laughter,increased sound sensitivity
OCB, 15 lbs weight gain OCB, pathologicalcrying, family history
Pathological laughter
Past medical history Pulmonary sarcoidosis, HA,depression
Hashimoto's thyroiditis,HA, haemochromatosis
Raynaud's syndrome,`lupus'
Hemicrania, depression
MRI ®nding FT and caudate atrophy FT and caudate atrophy FT atrophy FT and caudate atrophy andsignal changes in striatum
PI = postural instability; HA = headaches; NA = non-ambulatory; L = left; R = right; NR = not reported; OCB = obsessive±compulsivebehaviour; FT = frontotemporal.
Table 3 Semiquantitation of inclusion frequency inneuro®lament inclusion body disease in cases UK1 andUS1
Brain area H&E Ab1 Ubiquitin*
Anterior frontal cortex ++++ +++ +++Posterior frontal cortex +++ ++ ++Superior temporal gyrus + + +Middle temporal gyrus ++ ++ ++Inferior temporal gyrus +++ +++ +++Temporal pole +++ + +++Parietal cortex +++ ++ +++Occipital cortex + 0 0Insular cortex +++ +++ +Anterior cingulate gyrus ++++ ++ +++Hippocampus CA1±CA4 +++ ++ ++Dentate fascia ++++ ++ +++Subiculum +++ ++ +++Amygdala +++ ++ +Caudate nucleus + + +Putamen + + 0Globus pallidus + 0 0Thalamus + ++ 0Subthalamus + + 0Substantia nigra 0 0 0Pontine base + + 0Inferior olive 0 ++ 0Cerebellar Purkinje cells + 0 ++Dentate nucleus 0 0 0
++++ = severely affected; +++ = numerous; ++ = moderatenumber; + = small number; 0 = no inclusions. *Staining is weak.
2294 K. A. Josephs et al.
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

Histological ®ndingsThe histological ®ndings were similar for all cases and are
therefore summarized with exceptions reported. Inclusions
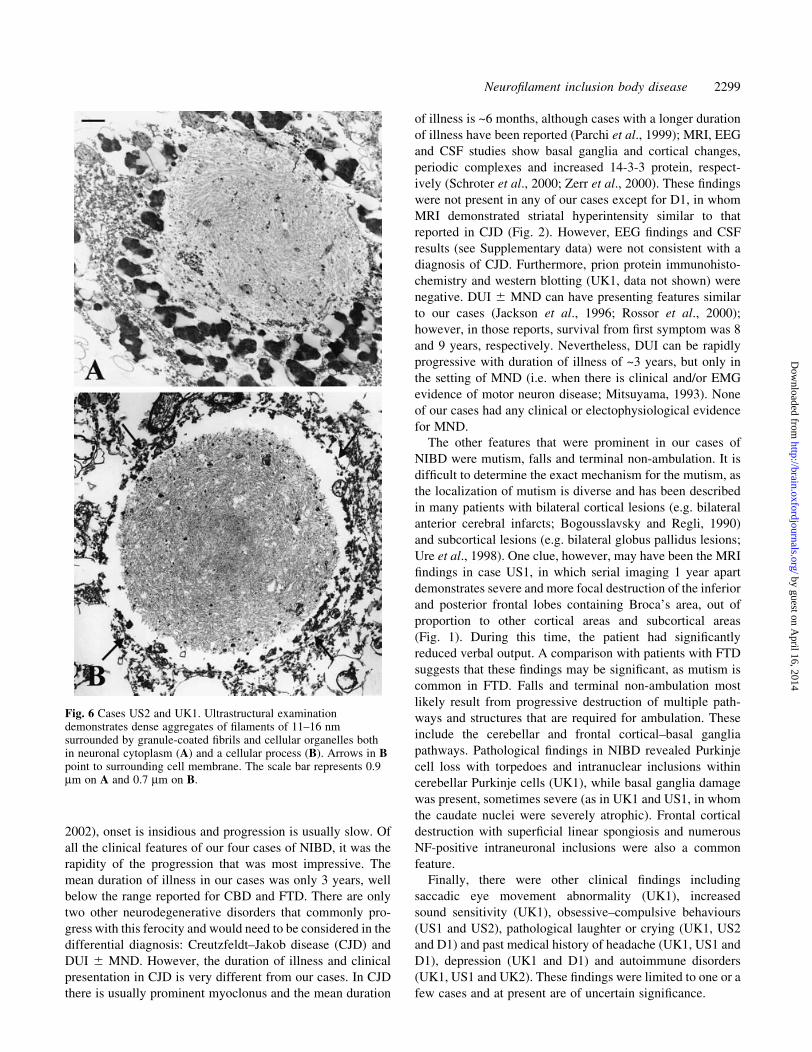
are depicted in Fig. 5.
Cerebral cortex. On routine microscopic examination, there
was super®cial linear spongiosis with thinning of the cortical
ribbon affecting frontal, temporal and, to a lesser extent, the
parietal lobes in all post-mortem cases. There was severe
subpial and white matter gliosis and mild gliosis affecting
cortical layers I±IV. H&E staining revealed intracytoplasmic
inclusions ranging in size from 3 to 20 mm in all layers of the
neocortex affecting all morphological types of neurons and
often situated towards their apical dendrite (Fig. 5A). The
appearance of the inclusions, irrespective of their localiza-
tion, ranged from eosinophilic to basophilic, sometimes with
a clearly de®ned basophilic margin (Fig. 5B). Inclusions
appeared to be granular or mesh-like and often contained a
hyaline, argyrophilic core resembling a `cherry'; therefore,
we shall describe them as `cherry spots'. Such structures were
found in several locations (Fig. 5C). The SMI 32 and Ab1 NF
antibodies intensely stained most of what appeared to
correspond to the cherry spots (Fig. 5K and L, respectively),
while these were stained less frequently and less intensely by
SMI 31 and rarely by BF10. The NF-cocktail antibody
usually stained a larger part of the inclusions than the Ab1 and
SMI 32 antibodies (Fig. 5M). Most of the inclusions were
weakly immunoreactive for ubiquitin, although a proportion,
often those situated in the super®cial cortical laminae, were
more strongly stained (Fig. 5J). Occasional inclusions were
intranuclear, eosinophilic, up to 6 mm in diameter and
frequently located in cortical layer II. Intranuclear inclusions
(UK1 and US1 only) were strongly stained with both
ubiquitin and Ab1 antibodies (Fig. 5O) and less strongly
stained with SMI 32 and 1C2 antibodies (Fig. 5Q). Glial
inclusions were not found.
Hippocampus. The hippocampal formation showed variable
neuronal loss and spongiosis in the CA1 subregion with
relative preservation of the CA2, CA3 and CA4 subregions.
Many of the residual hippocampal neurons contained
cytoplasmic inclusions that were often large and basophilic
(Fig. 5B) and within which there was a well-de®ned
eosinophilic hyaline cherry spot with a surrounding pale
halo (Fig. 5C). The inclusions in the CA1±CA4 subregions
had similar immunohistochemical staining characteristics to
those in the neocortex. Some axons in CA1 showed fusiform
dilations (Fig. 5N). In the granule cells of the dentate fascia,
frequent neuronal cytoplasmic inclusions were noted
(Fig. 5D), which appeared as single or occasionally multiple
small and faintly eosinophilic. Rod-shaped intranuclear
inclusions were also noted (US1 and UK1). The cytoplasmic
inclusions of the granule cells were more frequently posi-
tively stained with the antibody to ubiquitin (Fig. 5E) than
Fig. 1 Case US1. Coronal T1 images demonstrate signi®cant atrophic changes between 1995 (A) and1996 (B), mainly affecting posterior and lateral orbital gyri and limen insulae, with less atrophyoccurring in hippocampi. Caudate atrophy is also present.
Neuro®lament inclusion body disease 2295
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from
with the Ab1 or SMI 32 (Fig. 5F) antibodies, while the
intranuclear inclusions were positive for ubiquitin but only
rarely stained with SMI 32. There was variable neuronal loss
and ®brillary gliosis in the subiculum, entorhinal and
transentorhinal cortices as well as the amygdala. Frequent
neuronal cytoplasmic inclusions were found within these four
anatomical areas. In the subiculum, SMI 32 seems to have
stained more inclusions than Ab1 (UK1 only).
Subcortical grey nuclei. The basal nucleus of Meynert had a
normal neuronal population, and ubiquitin, SMI 32 and SMI
31 immunohistochemistry revealed very scanty cytoplasmic
inclusions. The caudate nucleus exhibited severe atrophy
with status spongiosis and gliosis in its medial aspect, most
marked in cases UK1 and US1. The lateral aspect of the
nucleus was less severely affected. Ubiquitin and SMI 32
stained rare inclusions within the ventrolateral caudate in
cases UK1 and US1. There was moderate to severe gliosis in
the globus pallidus and putamen, as well as a small number of
axonal spheroids. The internal globus pallidus was affected
more than the external globus pallidus, and there was an
anteroposterior gradient of severity, with the anterior level
most severely affected. A similar gradient was observed in
the putamen, with the anterior and medial parts of the nucleus
affected more severely than the posterior and lateral aspects.
Few cytoplasmic inclusions were seen in the basal ganglia,
and a proportion of these was positive with Ab1 antibody.
The thalamus showed patchy rarefaction, with neuronal
loss, gliosis and scattered intracytoplasmic inclusions most
frequent in the medial thalamus. The subthalamic nucleus
was well preserved with occasional intracytoplasmic inclu-
sions.
Brainstem. The red nucleus contained large NF-positive
spheroids and showed mild gliosis. The substantia nigra had
mild neuronal loss with extracellular neuromelanin pigment
and gliosis that was most marked in the ventral and lateral
neuronal groups. A number of axonal spheroids were seen in
the pars reticularis. The third nerve had a signi®cant number
of very thick axons. No inclusions were found in the third and
fourth nerve nuclei. The raphe nuclei contained scanty
inclusions in case US1 only. There was mild neuronal loss
Fig. 3 Case D1. An [18F]¯uorodeoxyglucose PET scan revealshypometabolism in the right frontotemporal region and rightthalamus.
Fig. 2 Case D1. An axial FLAIR (¯uid attenuated inversionrecovery) image demonstrates increased signal changes in striatumbilaterally.
2296 K. A. Josephs et al.
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

in the locus coeruleus and some of the neurons had NF-
positive hyaline inclusions (US2 only). Rare Ab1-positive
intraneuronal inclusions (UK1 only) and axonal spheroids
were seen in the pontine base (UK1 and US1). In the inferior
olive, there were large inclusions, some appearing ®la-
mentous (Fig. 5H), which were stained with both Ab1
immunohistochemistry and Bielschowsky's silver impregna-
tion but were negative for ubiquitin.
Cerebellum. In the cerebellar cortex, there was variable
Purkinje cell loss and frequent torpedoes in two of the cases
(UK1 and US1). These ®ndings were not noted in the other
post-mortem case (US2). In case UK1, the Purkinje cells
often contained one or more ubiquitin-positive, but NF-
negative, intranuclear inclusions (Fig. 5P), measuring 1±6 mm
in diameter. No cytoplasmic inclusions were seen on routine
stains or with immunohistochemistry.
Spinal cord. Where spinal cord sections were available (US1
and US2), no inclusions were seen in motor neurons. There
was no degeneration of the corticospinal or other tracts.
A moderate number of inclusions were weakly stained with
the modi®ed Bielschowsky silver stain, while a small number,
which mostly appeared to correspond in size to cherry spots,
were intensely stained. Weakly stained inclusions could be
demonstrated in the neocortex, hippocampus, basal ganglia
and thalamus, while intensely stained inclusions were mainly
found in the frontal cortex and hippocampus. The inclusions
were not stained with Gallyas or Bodian silver impregnations.
The inclusions were not labelled by antibodies to either a-
synuclein or tau or stained with PAS/H, Nissl, Congo Red or
thio¯avin-S methods. No cortical or brainstem Lewy bodies
were found using routine stains or a-synuclein immuno-
histochemistry. a-Synuclein immunohistochemistry in the
amygdala (UK1) was negative. None of the cases showed
neuro®brillary pathology on tau immunohistochemistry.
There were no glial inclusions identi®ed on tau, a-synuclein
or ubiquitin immunohistochemistry. No Ab or prion protein
deposition was observed. In the control cases stained with the
same panel of anti-NF and ubiquitin antibodies, no inclusions
with a similar staining pro®le were demonstrated.
Inclusion frequencySemiquantitation of the inclusions found in cases UK1 and
US1 revealed a few important patterns (Table 3). The
inclusions were widely distributed, but they were most
common in the anterior frontal and cingulate cortices and the
hippocampal dentate fascia. Inclusions were also numerous,
but less so, in the posterior frontal, temporal, parietal and
insular cortices, subiculum, hippocampus proper and amyg-
dala. They were scanty to moderate in the occipital cortex,
basal ganglia and brainstem and were absent in the deep
cerebellar nuclei. In the temporal cortex, there was a
gradation whereby the superior temporal gyrus had fewer
inclusions than the inferior temporal gyrus. Overall, the
inclusions were most readily found on the H&E preparations,
with the exception of the granule cells of the hippocampal
dentate fascia and the inferior olive.
Electron microscopyIn all three post-mortem cases, cytoplasmic inclusions of the
cortical neurons were noted to be composed of dense
aggregates of ®laments 11±16 nm in diameter and appeared
consistent with NFs (Fig. 6A). These were surrounded by
granule-coated ®brils and cellular organelles. In cell pro-
cesses an occasional structure of ~3 mm diameter containing
bundles of ®lamentous material, similar to those found in
neurons, was seen (Fig. 6B).
Fig. 4 Case UK1. Gross pathology demonstrates frontal and anterior temporal atrophy (A) and severecaudate atrophy (B).
Neuro®lament inclusion body disease 2297
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from
Discussion
Clinical ®ndings
The clinical presentation and progression of the cases we
have described, for which we propose the term NIBD, differs
from other neurodegenerative disorders. The presenting
features of cases UK1 and US2 share many similarities to
CBD syndrome, in that they both had asymmetric cortical
features with parkinsonism, L-dopa resistance and apraxia. In
a study of 14 patients with pathologically con®rmed CBD,
asymmetric hand clumsiness was the most common
presenting symptom (Wenning et al., 1998). Similarly, the
presenting symptoms and signs of cases US1 and D1 are in
keeping with the features that help to de®ne FTD, as both
cases had insidious onset at age <65 years, prominent frontal
features and prominent language dysfunction (Neary et al.,
1998; see Supplementary data for histories). Parkinsonism
develops in perhaps the majority of affected cases as FTD
progresses (Knopman et al., 1990).
Most clinicopathological studies that have examined the
progression of patients with CBD and FTD, as well as the
duration of illness, have revealed a difference from our cases.
The mean duration of illness until death for both CBD and
FTD is generally >6 years (Boeve et al., 1999; Snowden et al.,
Fig. 5 Cases UK1, US1 and D1. Photomicrographs demonstrating the main microscopic abnormalitiesin neuro®lament (NF) inclusion body disease. Neuronal intracytoplasmic inclusions (A±C) werenumerous in the frontal and temporal cortices, hippocampus proper and dentate granule cells (D±F).Some of the inclusions had a basophilic margin (B), while others appeared to contain a core resemblinga cherry (`cherry spot') (C). In the hippocampal formation, a small number of inclusions were stronglyargyrophilic (G) and intensely stained with NF antibodies (K and L). Some inclusions were diffuselystained with ubiquitin (E and J) and NF (M) without a visible cherry spot. Argyrophilic inclusions wereseen in neuronal processes in the hippocampus (H), and occasional axons showed fusiform dilatations,often intensely stained with NF antibodies (N). Large inclusions, some with a ®lamentous appearance,were seen in the inferior olive (I). Intranuclear inclusions (O) were rare in cortical neurons andsubiculum. Some of the intranuclear inclusions were highlighted with 1C2 antibody (arrow). Anintracytoplasmic inclusion is unstained (double arrow) (Q). Intranuclear inclusions, some multiple, wereseen in cerebellar Purkinje cells (P). Haematoxylin and eosin (A±D and O), Bielschowsky's silverimpregnation (G±I), SMI 32 (F and K), NF cocktail (M), Ab1 (L and N), ubiquitin (E, J and P) and1C2 (Q). The scale bar represents 20 mm on A±H, J and L±Q; 10 mm on I; and 40 mm on K.
2298 K. A. Josephs et al.
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from
2002), onset is insidious and progression is usually slow. Of
all the clinical features of our four cases of NIBD, it was the
rapidity of the progression that was most impressive. The
mean duration of illness in our cases was only 3 years, well
below the range reported for CBD and FTD. There are only
two other neurodegenerative disorders that commonly pro-
gress with this ferocity and would need to be considered in the
differential diagnosis: Creutzfeldt±Jakob disease (CJD) and
DUI 6 MND. However, the duration of illness and clinical
presentation in CJD is very different from our cases. In CJD
there is usually prominent myoclonus and the mean duration
of illness is ~6 months, although cases with a longer duration
of illness have been reported (Parchi et al., 1999); MRI, EEG
and CSF studies show basal ganglia and cortical changes,
periodic complexes and increased 14-3-3 protein, respect-
ively (Schroter et al., 2000; Zerr et al., 2000). These ®ndings
were not present in any of our cases except for D1, in whom
MRI demonstrated striatal hyperintensity similar to that
reported in CJD (Fig. 2). However, EEG ®ndings and CSF
results (see Supplementary data) were not consistent with a
diagnosis of CJD. Furthermore, prion protein immunohisto-
chemistry and western blotting (UK1, data not shown) were
negative. DUI 6 MND can have presenting features similar
to our cases (Jackson et al., 1996; Rossor et al., 2000);
however, in those reports, survival from ®rst symptom was 8
and 9 years, respectively. Nevertheless, DUI can be rapidly
progressive with duration of illness of ~3 years, but only in
the setting of MND (i.e. when there is clinical and/or EMG
evidence of motor neuron disease; Mitsuyama, 1993). None
of our cases had any clinical or electophysiological evidence
for MND.
The other features that were prominent in our cases of
NIBD were mutism, falls and terminal non-ambulation. It is
dif®cult to determine the exact mechanism for the mutism, as
the localization of mutism is diverse and has been described
in many patients with bilateral cortical lesions (e.g. bilateral
anterior cerebral infarcts; Bogousslavsky and Regli, 1990)
and subcortical lesions (e.g. bilateral globus pallidus lesions;
Ure et al., 1998). One clue, however, may have been the MRI
®ndings in case US1, in which serial imaging 1 year apart
demonstrates severe and more focal destruction of the inferior
and posterior frontal lobes containing Broca's area, out of
proportion to other cortical areas and subcortical areas
(Fig. 1). During this time, the patient had signi®cantly
reduced verbal output. A comparison with patients with FTD
suggests that these ®ndings may be signi®cant, as mutism is
common in FTD. Falls and terminal non-ambulation most
likely result from progressive destruction of multiple path-
ways and structures that are required for ambulation. These
include the cerebellar and frontal cortical±basal ganglia
pathways. Pathological ®ndings in NIBD revealed Purkinje
cell loss with torpedoes and intranuclear inclusions within
cerebellar Purkinje cells (UK1), while basal ganglia damage
was present, sometimes severe (as in UK1 and US1, in whom
the caudate nuclei were severely atrophic). Frontal cortical
destruction with super®cial linear spongiosis and numerous
NF-positive intraneuronal inclusions were also a common
feature.
Finally, there were other clinical ®ndings including
saccadic eye movement abnormality (UK1), increased
sound sensitivity (UK1), obsessive±compulsive behaviours
(US1 and US2), pathological laughter or crying (UK1, US2
and D1) and past medical history of headache (UK1, US1 and
D1), depression (UK1 and D1) and autoimmune disorders
(UK1, US1 and UK2). These ®ndings were limited to one or a
few cases and at present are of uncertain signi®cance.
Fig. 6 Cases US2 and UK1. Ultrastructural examinationdemonstrates dense aggregates of ®laments of 11±16 nmsurrounded by granule-coated ®brils and cellular organelles bothin neuronal cytoplasm (A) and a cellular process (B). Arrows in Bpoint to surrounding cell membrane. The scale bar represents 0.9mm on A and 0.7 mm on B.
Neuro®lament inclusion body disease 2299
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from
Only one case (US2) had a positive family history of an
early onset neurodegenerative disease (see Supplementary
data). The patient's father had developed signs and symptoms
of iPD in his early 50s, followed a few years later by
dementia. Unfortunately, post-mortem examination was not
performed. The development of iPD in his early 50s followed
by dementia is very intriguing. The differential diagnosis of
parkinsonism and dementia is wide. A diagnosis of iPD,
however, is very unlikely. More likely would be a diagnosis
of DLB (McKeith et al., 1996). We know his father was
treated with L-dopa therapy, but we do not know whether he
responded to the treatment. With the young age of onset and
short duration of illness (<5 years), NIBD is clearly a
diagnostic possibility.
MRI studies in all four cases revealed prominent frontal
and temporal lobe atrophy and increased signal in the striatum
in one case (D1). The distribution of atrophy is in keeping
with the clinical presentation. The increased signal in the
striatum in case D1 is unusual, as this is uncommon in CBD
and FTLD syndromes (Hauser et al., 1996; Larsson et al.,
2000), although it is more common in CJD (as discussed
above).
The ®ndings on functional neuroimaging (PET and
SPECT) and neuropsychological tests are also in keeping
with the clinical presentation and can be useful ancillary
studies.
Pathological ®ndingsPathological study of all four cases showed similar ®ndings of
NF-containing neuronal cytoplasmic inclusions in several
brain areas, including the neocortex and limbic structures,
with moderate numbers in deep grey and brainstem nuclei. In
at least two of the cases (UK1 and US1), we were able to
demonstrate neocortical ubiquitin-positive intranuclear inclu-
sions, which were also frequent in the cerebellar Purkinje
cells in one case (UK1).
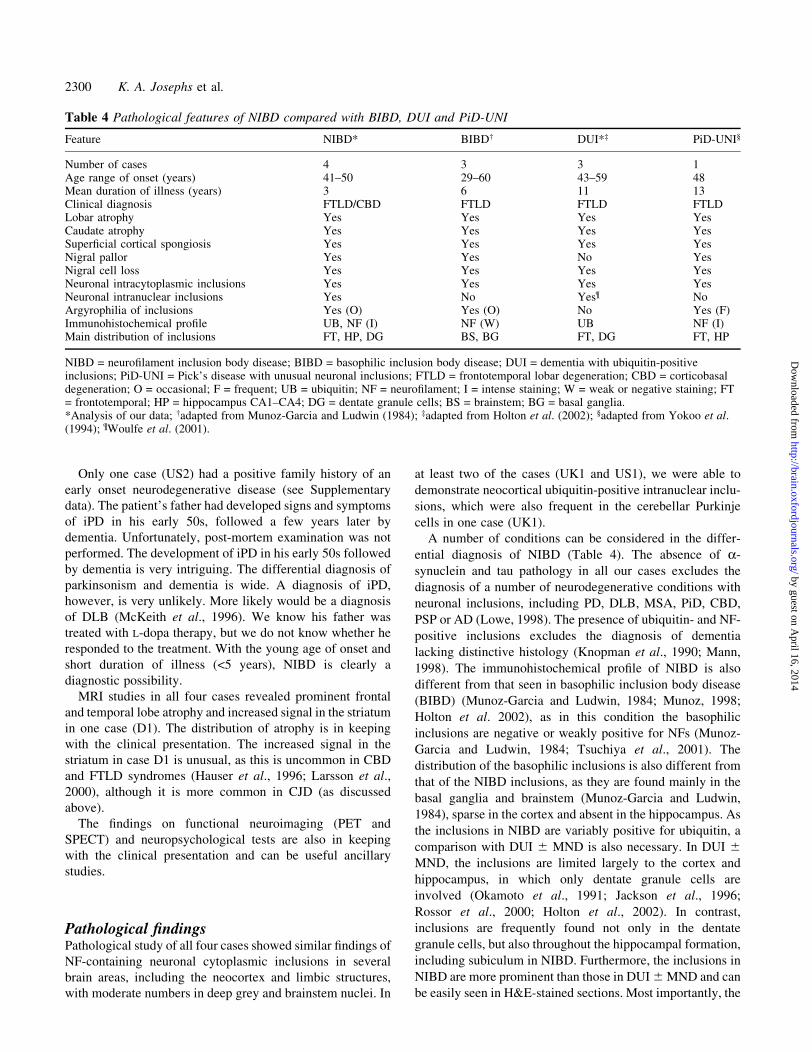
A number of conditions can be considered in the differ-
ential diagnosis of NIBD (Table 4). The absence of a-
synuclein and tau pathology in all our cases excludes the
diagnosis of a number of neurodegenerative conditions with
neuronal inclusions, including PD, DLB, MSA, PiD, CBD,
PSP or AD (Lowe, 1998). The presence of ubiquitin- and NF-
positive inclusions excludes the diagnosis of dementia
lacking distinctive histology (Knopman et al., 1990; Mann,
1998). The immunohistochemical pro®le of NIBD is also
different from that seen in basophilic inclusion body disease
(BIBD) (Munoz-Garcia and Ludwin, 1984; Munoz, 1998;
Holton et al. 2002), as in this condition the basophilic
inclusions are negative or weakly positive for NFs (Munoz-
Garcia and Ludwin, 1984; Tsuchiya et al., 2001). The
distribution of the basophilic inclusions is also different from
that of the NIBD inclusions, as they are found mainly in the
basal ganglia and brainstem (Munoz-Garcia and Ludwin,
1984), sparse in the cortex and absent in the hippocampus. As
the inclusions in NIBD are variably positive for ubiquitin, a
comparison with DUI 6 MND is also necessary. In DUI 6MND, the inclusions are limited largely to the cortex and
hippocampus, in which only dentate granule cells are
involved (Okamoto et al., 1991; Jackson et al., 1996;
Rossor et al., 2000; Holton et al., 2002). In contrast,
inclusions are frequently found not only in the dentate
granule cells, but also throughout the hippocampal formation,
including subiculum in NIBD. Furthermore, the inclusions in
NIBD are more prominent than those in DUI 6 MND and can
be easily seen in H&E-stained sections. Most importantly, the
Table 4 Pathological features of NIBD compared with BIBD, DUI and PiD-UNI
Feature NIBD* BIBD² DUI*³ PiD-UNI§
Number of cases 4 3 3 1Age range of onset (years) 41±50 29±60 43±59 48Mean duration of illness (years) 3 6 11 13Clinical diagnosis FTLD/CBD FTLD FTLD FTLDLobar atrophy Yes Yes Yes YesCaudate atrophy Yes Yes Yes YesSuper®cial cortical spongiosis Yes Yes Yes YesNigral pallor Yes Yes No YesNigral cell loss Yes Yes Yes YesNeuronal intracytoplasmic inclusions Yes Yes Yes YesNeuronal intranuclear inclusions Yes No Yes¶ NoArgyrophilia of inclusions Yes (O) Yes (O) No Yes (F)Immunohistochemical pro®le UB, NF (I) NF (W) UB NF (I)Main distribution of inclusions FT, HP, DG BS, BG FT, DG FT, HP
NIBD = neuro®lament inclusion body disease; BIBD = basophilic inclusion body disease; DUI = dementia with ubiquitin-positiveinclusions; PiD-UNI = Pick's disease with unusual neuronal inclusions; FTLD = frontotemporal lobar degeneration; CBD = corticobasaldegeneration; O = occasional; F = frequent; UB = ubiquitin; NF = neuro®lament; I = intense staining; W = weak or negative staining; FT= frontotemporal; HP = hippocampus CA1±CA4; DG = dentate granule cells; BS = brainstem; BG = basal ganglia.*Analysis of our data; ²adapted from Munoz-Garcia and Ludwin (1984); ³adapted from Holton et al. (2002); §adapted from Yokoo et al.(1994); ¶Woulfe et al. (2001).
2300 K. A. Josephs et al.
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

inclusions in DUI 6 MND are negative for NFs, which is also
con®rmed by the current study. A necessary consideration of
our NF inclusions is their possible relationship with NF-
positive inclusions within motor neurons in MND (Sobue
et al., 1990) or the eosinophilic and basophilic inclusions,
labelled with NF and ubiquitin antibodies and described in a
single case reported by Arima et al. (1998). There were no
MND-type inclusions with ubiquitin and NF immunohisto-
chemistry within motor neurons of either the cranial nerve
nuclei, which were available for our studies in all three post-
mortem cases (UK1, US1 and US2), or the cervical cord
studied in two of the cases (US1 and US2). Furthermore,
neuronal inclusions, similar to those seen in NIBD, are not
seen in the cerebral cortices and deep grey nuclei in MND
(Lowe and Leigh, 2002). It could be of interest that, in the
case of Arima et al. (1998) diagnosed clinically and
con®rmed by post-mortem as MND, the intracytoplasmic
ubiquitin-positive inclusions were found in cortical neurons
and hippocampal dentate granule cells. However, these were
reported to be only weakly positive or negative for NF using
the SMI 31 antibody recognizing pNF-H. The entity
described by Yokoo et al. (1994) as `Pick's disease with
unusual inclusions' (PiD-UNI) in 1994 is, with a few
differences, the most similar to our four cases. In their case,
the inclusions were NF-positive with an immunohistochem-
ical pro®le and distribution similar to ours. However, in PiD-
UNI, ubiquitin studies were reported as negative and neither
intranuclear inclusions nor axonal spheroids were reported. A
further difference is that, clinically, unlike our cases, their
case had a long disease duration of 13 years, even though the
early disease course was rapidly progressive. Pietrini et al.
(1993) described a case of the panencephalitic type of CJD
with neuropathological features similar to PiD. In their case,
there were numerous NF-positive swollen neurons and a
small number of Pick body-like argyrophilic intraneuronal
inclusions. However, there is no reference to the presence of
NF-positive intraneuronal inclusions of the kind we observed
in our cases. The only other report of signi®cant interest was a
recent report of ®ve patients with a novel neuro®lamento-
pathy (Cairns et al., 2003). Similar to our cases, their cases
had an early-onset dementia and intraneuronal, cytoplasmic,
NF-positive inclusions. Little clinical information was given,
but their patients seem all to have had an FTD. No mention of
a CBD syndrome was made. Unlike in our cases, NF-L
staining was less impressive than NF-H staining. Also, no
mention was made of intranuclear inclusions, inclusions
within the inferior olive, axonal swellings outside the
cerebellum or the presence of ®lamentous structures in cell
processes, which we were able to demonstrate with electron
microscopy.
One of the striking features noted in our cases was the
presence of neuronal ubiquitin-positive intranuclear inclu-
sions. Although such intranuclear inclusions were only rarely
found in most areas, they were numerous in the hippocampal
dentate granule cells and the cerebellar Purkinje cells (UK1).
Intranuclear inclusions are rather rare in cases with dementia,
although neuronal ubiquitinated intranuclear inclusions have
been described in three cases of DUI 6 MND (Woulfe et al.,
2001) and a single case report of a demented woman
(Weidenheim and Dickson, 1995). Intranuclear neuronal
inclusions are a common feature of polyglutamine disorders
(Davies et al., 1997; Hayashi et al., 1998; Holmberg et al,
1998; Li et al., 1998; Pang et al., 2002) and neuronal
intranuclear hyaline inclusion disease (Munoz-Garcia and
Ludwin, 1986), which may also be a polyglutamine disorder
(Lieberman et al., 1998; Takahashi et al., 2000). Case D1
underwent genetic testing for Huntington's disease and
dentatorubral-pallidolusian atrophy and tested negative (see
Supplementary data). The relationship of these disorders to
our four cases of NIBD is not clear at present and requires
further investigation, especially since the antibody 1C2,
which identi®es polyglutamine sequences, occasionally
labelled cortical intranuclear inclusions in our NIBD cases.
Two of the four cases (UK1 and US2) of NIBD were
treated with dopamine therapy and both failed to improve
symptomatically. This is not surprising, as pathology was not
limited to the substantia nigra, but was also extensive and
severe in the basal ganglia. Replacement anticholinesterase
therapy is also unlikely to be bene®cial in NIBD, as the
nucleus basalis of Meynert is preserved. This is not to say that
trials with either category of medication are not worth
pursuing.
The mechanism for NF deposition in NIBD is currently
unknown, but there are a few theoretical possibilities based
on results from mouse genetic and human NF studies (Nixon,
1993; Julien, 1999). These include `sporadic' NF gene
mutations, post-translational modi®cations including abnor-
mal phosphorylation of the subunit components of NF (NF-L,
NF-M and NF-H) and failure of axonal NF transport.
In summary, NIBD is a unique neurodegenerative disorder
with overlapping clinical features of frontotemporal lobar
degeneration and CBD syndrome. A diagnosis of NIBD
should be considered in patients who present with either a
CBD or FTD syndrome, with rapid progression without
clinical or EMG evidence of MND, with early falls, mutism,
akinesia and terminal immobility. MRI evidence of frontal
and temporal lobe atrophy would support the diagnosis. The
main morphological features that support the pathological
diagnosis of NIBD is a-synuclein- and tau-negative neuronal
intracytoplasmic inclusions, many of which contain NFs and
variable amounts of ubiquitin, and some of which are
argyrophilic with modi®ed Bielschowsky. Such inclusions
are strikingly numerous in neocortices and limbic structures.
Further characterization of NIBD is needed to clarify the
signi®cance and mechanism of the NF deposition in this
disorder.
AcknowledgementsK.A.J. is currently a Mayo Foundation Scholar and Visiting
Researcher at the Institute of Neurology, Queen Square,
London, UK. Dr J.L.H. is partly funded by the Reta Lila
Neuro®lament inclusion body disease 2301
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

Weston Foundation. We wish to thank Dr John Stevens for
assistance with MRI and PET interpretation; Dr Eric M.
Rohren for assistance with PET imaging; Dr Brendan
McClean, who referred case UK1; and all the general
practitioners and consultant neurologists who participated in
the care of these patients and documented the clinical
histories and examinations. We appreciate Ms Hardev
Sangha, Ms Kerrie Venner, Ms Catherine Strand and Dr
Wen-Lang Lin for their technical assistance in the patho-
logical studies. We would like to extend our appreciation to
all the family members who extended their support by giving
additional clinical information and allowing us to report
accurately on these four novel cases.
References
Arima K, Ogawa M, Sunohara N, Nishio T, Shimomura Y, Hirai S,
et al. Immunohistochemical and ultrastructural characterization of
ubiquitinated eosinophilic ®brillary neuronal inclusions in sporadic
amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 1998; 96:
75±85.
Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford
N, Caselli RJ, et al. Pathologic heterogeneity in clinically diagnosed
corticobasal degeneration. Neurology 1999; 53: 795±800.
Bogousslavsky J, Regli F. Anterior cerebral artery territory
infarction in the Lausanne Stroke Registry. Clinical and etiologic
patterns. Arch Neurol 1990; 47: 144±50.
Cairns NJ, Perry RH, Jaros E, Burn D, McKeith IG, Lowe JS, et al.
Patients with a novel neuro®lamentopathy: dementia with
neuro®lament inclusions. Neurosci Lett 2003; 341: 177±80.
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross
CA, et al. Formation of neuronal intranuclear inclusions underlies
the neurological dysfunction in mice transgenic for the HD
mutation. Cell 1997; 90: 537±48.
Goedert M, Spillantini MG, Serpell LC, Berriman J, Smith MJ,
Jakes R, et al. From genetics to pathology: tau and alpha-synuclein
assemblies in neurodegenerative diseases. Philos Trans R Soc Lond
B Biol Sci 2001; 356: 213±27.
Hauser RA, Murtaugh FR, Akhter K, Gold M, Olanow CW.
Magnetic resonance imaging of corticobasal degeneration. J
Neuroimaging 1996; 6: 222±6.
Hayashi Y, Kakita A, Yamada M, Koide R, Igarashi S, Takano H,
et al. Hereditary dentatorubral-pallidoluysian atrophy: detection of
widespread ubiquitinated neuronal and glial intranuclear inclusions
in the brain. Acta Neuropathol (Berl) 1998; 96: 547±52.
Hershko A, Ciechanover A. The ubiquitin system. Annu Rev
Biochem 1998; 67: 425±79.
Holmberg M, Duyckaerts C, Durr A, Cancel G, Gour®nkel-An I,
Damier P, et al. Spinocerebellar ataxia type 7 (SCA7): a
neurodegenerative disorder with neuronal intranuclear inclusions.
Hum Mol Genet 1998; 7: 913±18.
Holton J, Revesz T, Crooks R, Scaravilli F. Evidence for
pathological involvement of the spinal cord in motor neuron
disease-inclusion dementia. Acta Neuropathol (Berl) 2002; 103:
221±7.
Jackson M, Lennox G, Lowe J. Motor neurone disease-inclusion
dementia. Neurodegeneration 1996; 5: 339±50.
Julien JP. Neuro®lament functions in health and disease. Curr Opin
Neurobiol 1999; 9: 554±60.
Kertesz A, Kawarai T, Rogaeva E, St George-Hyslop P, Poorkaj P,
Bird TD, et al. Familial frontotemporal dementia with ubiquitin-
positive, tau-negative inclusions. Neurology 2000; 54: 818±27.
Knopman DS, Mastri AR, Frey WH, Sung JH, Rustan T. Dementia
lacking distinctive histologic features: a common non-Alzheimer
degenerative dementia. Neurology 1990; 40: 251±6.
Larsson E, Passant U, Sundgren PC, Englund E, Brun A, Lindgren
A, et al. Magnetic resonance imaging and histopathology in
dementia, clinically of frontotemporal type. Dement Geriatr Cogn
Disord 2000; 11: 123±34.
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative
tauopathies. Annu Rev Neurosci 2001; 24: 1121±59.
Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F,
et al. Nuclear inclusions of the androgen receptor protein in spinal
and bulbar muscular atrophy. Ann Neurol 1998; 44: 249±54.
Lieberman AP, Robitaille Y, Trojanowski JQ, Dickson DW,
Fishbeck KH. Polyglutamine-containing aggregates in neuronal
intranuclear inclusion disease. Lancet 1998; 351: 884.
Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt
ML, et al. Lewy bodies contain altered alpha-synuclein in brains of
many familial Alzheimer's disease patients with mutations in
presenilin and amyloid precursor protein genes. Am J Pathol 1998;
153: 1365±70.
Lowe J. Establishing a pathological diagnosis in degenerative
dementias. Brain Pathol 1998; 8: 403±6.
Lowe JS, Leigh N. Disorders of movement and system
degenerations. In: Graham DI, Lantos PL, editors. Green®eld's
neuropathology. 7th ed. London: Arnold; 2002. p. 325±430.
Mann DM. Dementia of frontal type and dementias with subcortical
gliosis. Brain Pathol 1998; 8: 325±38.
McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW,
Hansen LA, et al. Consensus guidelines for the clinical and
pathologic diagnosis of dementia with Lewy bodies (DLB): report
of the consortium on DLB international workshop. Neurology 1996;
47: 1113±24.
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D,
Trojanowski JQ. Clinical and pathological diagnosis of
frontotemporal dementia: report of the Work Group on
Frontotemporal Dementia and Pick's Disease. Arch Neurol 2001;
58: 1803±9.
Mitsuyama Y. Presenile dementia with motor neuron disease.
Dementia 1993; 4: 137±42.
Munoz DG. The pathology of Pick's complex. In: Kertesz A,
Munoz DG, editors. Pick's disease and Pick complex. New York:
Wiley-Liss; 1998. p. 211±41.
Munoz-Garcia D, Ludwin S. Classic and generalized variants of
2302 K. A. Josephs et al.
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from

Pick's disease: a clinicopathological, ultrastructural, and
immunocytochemical comparative study. Ann Neurol 1984; 16:
467±80.
Munoz-Garcia D, Ludwin SK. Adult-onset neuronal intranuclear
hyaline inclusion disease. Neurology 1986; 36: 785±90.
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S,
et al. Frontotemporal lobar degeneration: a consensus on clinical
diagnostic criteria. Neurology 1998; 51: 1546±54.
Nixon RA. The regulation of neuro®lament protein dynamics by
phosphorylation: clues to neuro®brillary pathobiology. Brain Pathol
1993; 3: 29±38.
Okamoto K, Hirai S, Yamazaki T, Sun XY, Nakazato Y. New
ubiquitin-positive intraneuronal inclusions in the extra-motor
cortices in patients with amyotrophic lateral sclerosis. Neurosci
Lett 1991; 129: 233±6.
Pang JT, Giunti P, Chanmberlain S, An SF, Vitaliani R, Scaravilli
T, et al. Neuronal intranuclear inclusions in SCA2: a genetic,
morphological and immunohistochemical study of two cases. Brain
2002; 125: 656±63.
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W,
Windl O, et al. Classi®cation of sporadic Creutzfeldt-Jakob disease
based on molecular and phenotypic analysis of 300 subjects. Ann
Neurol 1999; 46: 224±33.
Pietrini V, Danieli D, Bevilacqua P, Lechi A. Panencephalopathic
type of Creutzfeldt-Jakob disease with neuropathologic features
similar to Pick's disease. Clin Neuropathol 1993; 12: 1±6.
Rossor MN, Revesz T, Lantos PL, Warrington EK. Semantic
dementia with ubiquitin-positive tau-negative inclusion bodies.
Brain 2000; 123: 267±76.
Schroter A, Zerr I, Henkel K, Tschampa HJ, Finkenstaedt M, Poser
S. Magnetic resonance imaging in the clinical diagnosis of
Creutzfeldt-Jakob disease. Arch Neurol 2000; 57: 1751±7.
Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J
Psychiatry 2002; 180: 140±3.
Sobue G, Hashizume Y, Yasuda T, Mukai E, Kumagai T, Mitsuma
T, et al. Phosphorylated high molecular weight neuro®lament
protein in lower motor neurons in amyotrophic lateral sclerosis and
other neurodegenerative diseases involving ventral horn cells. Acta
Neuropathol (Berl) 1990; 79: 402±8.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R,
Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997; 388:
839±40.
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL,
Goedert M. Filamentous alpha-synuclein inclusions link multiple
system atrophy with Parkinson's disease and dementia with Lewy
bodies. Neurosci Lett 1998; 251: 205±8.
Takahashi J, Fukuda T, Tanaka J, Minamitani M, Fujigasaki H,
Uchihara T. Neuronal intranuclear hyaline inclusion disease with
polyglutamine-immunoreactive inclusions. Acta Neuropathol (Berl)
2000; 99: 589±94.
Tsuchiya K, Ishizu H, Nakano I, Kita Y, Sawabe M, Haga C, et al.
Distribution of basal ganglia lesions in generalized variant of Pick's
disease: a clinicopathological study of four autopsy cases. Acta
Neuropathol (Berl) 2001; 102: 441±8.
Ure J, Faccio E, Videla H, Caccuri R, Giudice F, Ollari J, et al.
Akinetic mutism: a report of three cases. Acta Neurol Scand 1998;
98: 439±44.
Weidenheim KM, Dickson DW. Intranuclear inclusion bodies in an
elderly demented woman: a form of intranuclear inclusion body
disease. Clin Neuropathol 1995; 14: 93±9.
Wenning GK, Litvan I, Jankovic J, Granata R, Mangone CA,
McKee A, et al. Natural history and survival of 14 patients with
corticobasal degeneration con®rmed at postmortem examination.
J Neurol Neurosurg Psychiatry 1998; 64: 184±9.
Wightman G, Anderson VE, Martin J, Swash M, Anderton BH,
Neary D, et al. Hippocampal and neocortical ubiquitin-
immunoreactive inclusions in amyotrophic lateral sclerosis with
dementia. Neurosci Lett 1992; 139: 269±74.
Woulfe J, Kertesz A, Munoz DG. Frontotemporal dementia with
ubiquitinated cytoplasmic and intranuclear inclusions. Acta
Neuropathol (Berl) 2001; 102: 94±102.
Yokoo H, Oyama T, Hirato J, Sasaki A, Nakazato Y. A case of
Pick's disease with unusual neuronal inclusions. Acta Neuropathol
(Berl) 1994; 88: 267±72.
Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J,
Knight RS, et al. Analysis of EEG and CSF 14-3-3 proteins as aids
to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000; 55:
811±15.
Received February 26, 2003. Revised April 28, 2003.
Accepted May 9, 2003
Neuro®lament inclusion body disease 2303
by guest on April 16, 2014
http://brain.oxfordjournals.org/D
ownloaded from





























