Embed Size (px)
Citation preview

Neuronal nicotinic receptors in the human brain
David Paterson, Agneta Nordberg*
Department of Clinical Neuroscience, Occupational Therapy and Elderly Care Research, Division of Molecular Neuropharmacology, Karolinska
Institute, Huddinge University Hospital, S-14186 Huddinge, Sweden
Received 20 July 1999
Abstract
Neuronal nicotinic acetylcholine receptors (nAChRs) are a family of ligand gated ion channels which are widely distributed inthe human brain. Multiple subtypes of these receptors exist, each with individual pharmacological and functional pro®les. They
mediate the e�ects of nicotine, a widely used drug of abuse, are involved in a number of physiological and behavioural processesand are additionally implicated in a number of pathological conditions such as Alzheimer's disease, Parkinson's disease andschizophrenia. The nAChRs have a pentameric structure composed of ®ve membrane spanning subunits, of which nine di�erenttypes have thus far been identi®ed and cloned. The multiple subunits identi®ed provide the basis for the heterogeneity of structure
and function observed in the nAChR subtypes and are responsible for the individual characteristics of each. A substantial amountof information on human nAChR structure and function has come from studies on neuroblastoma cell lines which naturallyexpress nAChRs and from recombinant nAChRs expressed in Xenopus oocytes. In vitro brain nAChR distribution can be mapped
with a number of appropriate agonist and antagonist radioligands and subunit distribution may be mapped by in situhybridization using subunit speci®c mRNA probes. Receptor distribution in the living human brain can be studied with non-invasive imaging techniques such as PET and SPECT, with a signi®cant reduction in nAChRs in the brains of Alzheimer's patients
having been identi®ed with �11C� nicotine in PET studies. Despite the signi®cant body of knowledge now accumulated aboutnAChRs, much remains to be elucidated. This review will attempt to describe the current knowledge on the nAChR subtypes inthe human brain, their functional roles and neuropathological involvement. # 2000 Elsevier Science Ltd. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 761.1. Nicotinic receptor structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 771.2. Ligand binding sites . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
1.2.1. ACh binding site . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 781.2.2. Allosteric binding sites. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
1.3. Transition states of nicotinic receptors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
1.4. Nicotinic receptor upregulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
Progress in Neurobiology 61 (2000) 75±111
0301-0082/00/$ - see front matter # 2000 Elsevier Science Ltd. All rights reserved.
PII: S0301-0082(99 )00045 -3
www.elsevier.com/locate/pneurobio
* Corresponding author. Tel: +46-8-58585467; fax: +46-8-6899210.
E-mail address: [email protected] (A. Nordberg).
Abbreviations: aBTX, Alpha bungarotoxin; Ach, Acetylcholine; nAChRs, nicotinic acetylcholine receptors; AChRs, nicotinic receptor sites
that bind nicotinic agonists with high a�nity; ACTH, Adrenocorticotropic hormone; AD, Alzheimer's disease; ADNFLE, Autosomal dominant
frontal lobe epilepsy; APP, Amyloid precursor protein; bA4, b amyloid; BTXRs, nicotinic receptor sites that bind bungarotoxin with high a�-
nity; CGRP, calcitonin gene related peptide; ChAT, Choline acetyltransferase; CNS, Central nervous system; DA, Dopamine; DHbE, Dihydro-
b-erythroidine; DMEA, Dimethylethanolamine; DMPP, 1,1-dimethyl-4-phenylpiperazanium; EEG, Electro encephalograph; GABA, gamma
amino butyric acid; 5-HT, 5 hydroxy tryptamine (serotonin); kBTX, kappa bungarotoxin; Kd, Dissociation constant; MLA, Methylcaconitine;
MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropine; MRI, Magnetic resonance imaging; NA, Noradrenaline; NBM, Nucleus basalis of Meynert;
NMDA, N-methyl-D-aspartame; PCP, phencyclidine; PD, Parkinson's disease; PET, Positron emission tomography; PKA, Protein kinase A;
PKC, Protein kinase C; SPECT, Single photon emission computed tomography; TS, Gilles de Tourette syndrome.

2. Neuronal nicotinic receptor subtypes in the human brain . . . . . . . . . . . . . . . . . . . . . . . . . . . 822.1. Ligand binding studies on human brain tissue . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 822.2. Binding studies in human neuroblastoma cell lines . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
2.3. Nicotinic receptor subunit expression in oocytes and transfected cell lines . . . . . . . . . . 832.3.1. Heteromeric nicotinic receptors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 832.3.2. Homomeric nicotinic receptors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
2.4. Electrophysiological studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
2.5. Evidence for four functional subtypes of nicotinic receptor . . . . . . . . . . . . . . . . . . . . . 86
3. Distribution of nicotinic receptors in the human brain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
3.1. Ligand binding studies. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 883.2. Subunit mRNA distribution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
4. Imaging of nicotinic receptors with PET and SPECT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 914.1. PET . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
4.1.1. [18F� NFEP or �18F� FPH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 934.1.2. [18F� A-85380 and �11C� A-8548. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
4.1.3. [11C� MPA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 954.1.4. [76Br� BAP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
4.2. SPECT. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
5. Nicotinic receptor function in the CNS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 965.1. Functional and behavioural e�ects of nicotine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
5.2. Role of nicotinic receptors in cognitive and memory functions . . . . . . . . . . . . . . . . . . 96
6. Pathology of neuronal nicotinic receptors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
6.1. Epilepsy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 976.2. Alzheimer's disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
6.2.1. Alzheimer's disease therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 996.3. Parkinson's disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
6.4. Schizophrenia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1016.5. Tourette's syndrome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1016.6. Anxiety and depression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
7. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
1. Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs)
are transmitter gated ion channels which belong to a
gene super family of homologous receptors including
GABA, glycine and 5-hydroxy tryptamine (5-HT)
(Karlin and Akabas, 1995). A number of di�erent sub-
types of nAChR exist each with individual pharmaco-
logical and physiological pro®les and distinct
anatomical distribution in the brain. By analogy with
their muscle counterparts neuronal nAChRs are
believed to have a pentameric structure consisting of
®ve membrane spanning regions around a central ion-
channel. This structure is composed of a number of
subunits of which there are multiple subtypes, the
genes for all or most of which have been cloned and
expressed. Although much is now known about the
structure and functional properties of neuronal
nAChRs (mainly from expression studies), relatively
little is understood about their physiological role in
man. Evidence suggests that nAChRs do not appear to
function in the classical postsynaptic, directly excit-
atory manner of their muscle counterparts. Their lo-
cation in the brain is not limited to postsynaptic but
also to pre-, peri- and extrasynaptic sites where they
may modulate neuronal function by a variety of
actions (LindstroÈ m, 1997). To this end, neuronal
nAChRs are involved in a number of functional pro-
cesses including cognition, learning and memory, arou-
sal, cerebral blood ¯ow and metabolism, and a
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11176

growing list of pathological conditions (Levin andSimon, 1998). For example, mutations in one of thenAChR subunit genes is responsible for a speci®c formof epilepsy, autosomal dominant frontal lobe epilepsy(ADNFLE), and it has be suggested that an nAChRsubunit gene mutation may be among the factors pre-disposing to schizophrenia (Chini et al., 1994; Steinleinet al., 1995; Freedman et al., 1997; Levin and Simon,1998). In Alzheimer's disease (AD), there is a signi®-cant loss of high a�nity nAChR sites (Whitehouseand Au, 1986; Nordberg and Winblad, 1986; Nagataet al., 1996; Whitehouse et al., 1988a) which is believedto re¯ect the pathophysiological changes underlyingthe cognitive decline and dementia observed in thiscondition. High a�nity nAChR sites are also reducedin the brains of Parkinson's disease (PD) patients(Whitehouse et al., 1983, 1988a), and thus, nAChRsmay also be involved in the dementia which is associ-ated with this condition. Neuronal nAChRs are alsopotential therapeutic targets in a number of CNS dis-orders with nicotine observed to be bene®cial in AD,PD and Tourette's syndrome (Jones et al., 1992; Vidal,1996; Newhouse et al., 1997). Thus, there is greatinterest in the development of selective nAChR ago-nists as therapies (Sershen et al., 1987; Madhok et al.,1995; Dursun and Reveley, 1997; Newhouse et al.,1997; Maggio et al., 1998; Sabbagh et al., 1998).Nicotine and nAChRs may also apparently beinvolved in the pathophysiology of both anxiety anddepression. Nicotine has anxiolytic properties in ani-mal models and retrospective and prospective clinicalstudies have demonstrated a relationship betweensmoking and major depression; persons with major de-pression are more likely to smoke and more likely todevelop severe depressive episodes upon cessation ofsmoking (Covey et al., 1997, 1998). The most obviousrole of neuronal nAChRs is mediation of toleranceand addiction to nicotine in chronic tobacco users andthe symptoms of withdrawal experienced upon cessa-tion of use (Benowitz, 1996).
Neuronal nAChRs are therefore involved in a com-plex range of functions in which their exact role andfull potential as therapeutic targets has yet to be eluci-dated. This review will attempt to describe the sub-types of neuronal nAChR present in human brain,their individual functional roles and involvement inbehaviour and pathological conditions.
1.1. Nicotinic receptor structure
Biochemical investigations with Torpedo receptor(Galzi and Changeux, 1995) and with neuronal recep-tors (Anand et al., 1991) have established that bothperipheral (e.g. muscle type) and neuronal nAChRsare comprised of hetero-oligomers consisting of ®vemembrane spanning subunits which form a barrel like
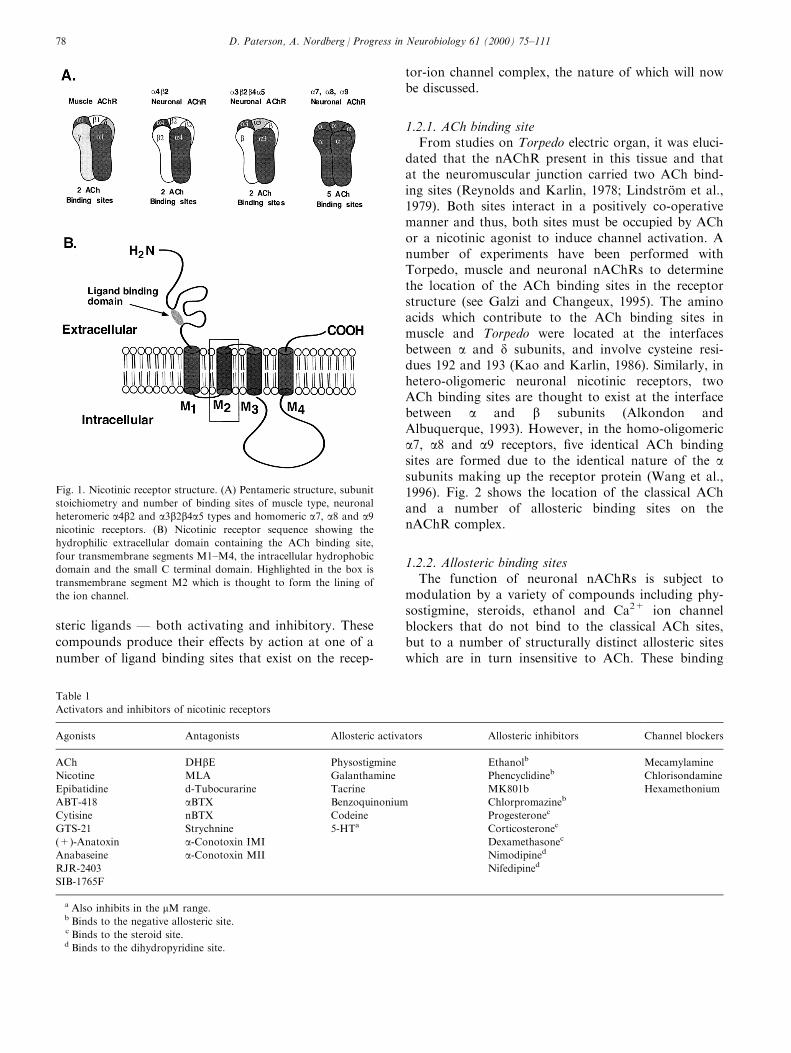
structure in the membrane around a central ion chan-nel (Cartaud et al., 1973). Molecular cloning studies inchick, rat and human have identi®ed multiple genesthat encode various subtypes of subunit that allowassembly of a wide variety of receptor oligomers withdi�erent distribution and distinct pharmacological pro-®les (Sargent, 1993). Peripheral nAChRs, such as thosefound at the neuromuscular junction, are made up ofa1, b1, g and d or e subunits (in adult and fetal formsof the receptor, respectively) in the ®xed stoichiometryof 2.1.1.1.1 (i.e. 2a1, 1b1, 1d or 1e). NeuronalnAChRs di�er from those in the periphery as theyhave no g, d, or e subunits in their make up and con-sist of various complements of a2±a9 and b2±b4 subu-nits. Presently, six a (a2±a7) and three b (b2±b4)subunits have been identi®ed and cloned from humanbrain (Sargent, 1993; Galzi and Changeux, 1995;McGehee and Role, 1995; Elliott et al., 1996; Gotti etal., 1997). In contrast to muscle type receptors, neur-onal nAChR subunits assemble according to a general2a3b stoichiometry, with the possibility of more thanone a subunit subtype within a pentamer (Conroy etal., 1992). However, a7, a8 and a9 subunits are knownto form functional homo-oligomers consisting of asingle a subunit subtype (Couturier et al., 1990). Formore information on nomenclature of nAChRs andtheir subunits, the reader is referred to the recentIUPHAR Subcommittee report (Lukas et al., 1999).Analysis of the amino acid sequences of nAChRsreveals signi®cant homology between the neuronalnAChR subtypes and peripheral nAChRs. In general,the nicotinic receptor sequence consists of: (1) a largehydrophillic amino terminal domain, (2) a compacthydrophobic domain split into three segments of 19±27 amino acids termed M1±M3, (3) a small highlyvariable hydrophillic domain and (4) a hydrophobic Cterminal domain of approximately 20 amino acidstermed M4. Fig. 1. It is thought that the large hydro-phillic domain containing the amino terminal containsphosphorylation sites and is exposed to the synapticcleft where it plays a role in ligand binding. The smallhydrophobic domain exposed to the cytoplasm con-tains glycosylation sites, and the four hydrophobicdomains (M1±M4) comprise the transmembrane seg-ments of the receptor, some of which line the ion chan-nel (Galzi and Changeux, 1995).
1.2. Ligand binding sites
A diverse range of compounds are known to bepharmacologically active at nAChRs, several of whichare listed in Table 1 (for a detailed exploration of theproperties of these compounds at nAChRs, the readeris referred to the recent review of Gotti et al., 1997).Drugs acting at nAChRs can be divided into threemain classes: (1) agonists, (2) antagonists and (3) allo-
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 77
steric ligands Ð both activating and inhibitory. These
compounds produce their e�ects by action at one of a
number of ligand binding sites that exist on the recep-
tor-ion channel complex, the nature of which will nowbe discussed.
1.2.1. ACh binding siteFrom studies on Torpedo electric organ, it was eluci-
dated that the nAChR present in this tissue and thatat the neuromuscular junction carried two ACh bind-ing sites (Reynolds and Karlin, 1978; LindstroÈ m et al.,1979). Both sites interact in a positively co-operativemanner and thus, both sites must be occupied by AChor a nicotinic agonist to induce channel activation. Anumber of experiments have been performed withTorpedo, muscle and neuronal nAChRs to determinethe location of the ACh binding sites in the receptorstructure (see Galzi and Changeux, 1995). The aminoacids which contribute to the ACh binding sites inmuscle and Torpedo were located at the interfacesbetween a and d subunits, and involve cysteine resi-dues 192 and 193 (Kao and Karlin, 1986). Similarly, inhetero-oligomeric neuronal nicotinic receptors, twoACh binding sites are thought to exist at the interfacebetween a and b subunits (Alkondon andAlbuquerque, 1993). However, in the homo-oligomerica7, a8 and a9 receptors, ®ve identical ACh bindingsites are formed due to the identical nature of the asubunits making up the receptor protein (Wang et al.,1996). Fig. 2 shows the location of the classical AChand a number of allosteric binding sites on thenAChR complex.
1.2.2. Allosteric binding sitesThe function of neuronal nAChRs is subject to
modulation by a variety of compounds including phy-sostigmine, steroids, ethanol and Ca2+ ion channelblockers that do not bind to the classical ACh sites,but to a number of structurally distinct allosteric siteswhich are in turn insensitive to ACh. These binding
Fig. 1. Nicotinic receptor structure. (A) Pentameric structure, subunit
stoichiometry and number of binding sites of muscle type, neuronal
heteromeric a4b2 and a3b2b4a5 types and homomeric a7, a8 and a9nicotinic receptors. (B) Nicotinic receptor sequence showing the
hydrophilic extracellular domain containing the ACh binding site,
four transmembrane segments M1±M4, the intracellular hydrophobic
domain and the small C terminal domain. Highlighted in the box is
transmembrane segment M2 which is thought to form the lining of
the ion channel.
Table 1
Activators and inhibitors of nicotinic receptors
Agonists Antagonists Allosteric activators Allosteric inhibitors Channel blockers
ACh DHbE Physostigmine Ethanolb Mecamylamine
Nicotine MLA Galanthamine Phencyclidineb Chlorisondamine
Epibatidine d-Tubocurarine Tacrine MK801b Hexamethonium
ABT-418 aBTX Benzoquinonium Chlorpromazineb
Cytisine nBTX Codeine Progesteronec
GTS-21 Strychnine 5-HTa Corticosteronec
(+)-Anatoxin a-Conotoxin IMI Dexamethasonec
Anabaseine a-Conotoxin MII Nimodipined
RJR-2403 Nifedipined
SIB-1765F
a Also inhibits in the mM range.b Binds to the negative allosteric site.c Binds to the steroid site.d Binds to the dihydropyridine site.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11178

sites and the compounds which activate them will nowbe brie¯y discussed.
1.2.1.1. Non competitive allosteric activator site. Aswith the classical ACh binding site, this positively act-
ing allosteric site is located on the a subunit of thereceptor protein. Compounds that bind to this site are
termed channel activators as they enhance channelopening and ion conductance (Pereira et al., 1993) and
include the cholinesterase inhibitors physostigmine,tacrine and galanthamine and the muscle relaxant ben-
zoquinonium (Svensson and Nordberg, 1996). There isevidence to suggest that 5-HT also binds to this site
increasing ion conductance by increasing the frequencyof channel opening (Schrattenholz et al., 1996). In cul-
tured M10 and PC12 cells, these compounds can acti-vate single channel activity, an action which isuna�ected by the application of competitive nicotinic
antagonists, thus con®rming their action at a site dis-tinct from that of ACh on the receptor-ion channel
(Pereira et al., 1994; Storch et al., 1995). However,channel activation was only observed on a small scale,
suggesting that the primary function of this class ofreceptor is to enhance nAChR activity induced by the
binding of ACh at the classical site. In M10 cells
tacrine produces a concentration dependent increase of
a4b2 nAChR sites, an e�ect that was blocked by the
nAChR antagonist mecamylamine, without increasing
either a4 or b2 mRNA levels (Svensson and Nordberg,
1996). When tacrine (10ÿ7 M) treatment was combined
with nicotine (10ÿ6 M) the e�ect was additive
suggesting that the upregulation of nAChRs induced
by tacrine occurs through activation of the receptor
complex via a site distinct to that of the classical ACh
binding site.
1.2.1.2. Non competitive negative allosteric site. In con-
trast to the non-competitive allosteric activator site,
ligand binding to this receptor site inhibits ion channel
function. A diverse range of compounds including
chlorpromazine, phencyclidine, MK801, local anaes-
thetics, ethanol and barbiturates can activate this
receptor type to produce a negative e�ect on nAChR
ion channel function without directly a�ecting ACh
binding (Lena and Changeux, 1993). These non-com-
petitive blockers act on two distinct sites that di�er
from those of competitive blockers. The ®rst high a�-
nity site, which binds ligands in the nanomolar range,
is thought to be located within the ion channel and is
composed of amino acids of the M2 segment in each
of the ®ve subunits making up the receptor protein.
Binding of ligands to this site is facilitated by agonist
activation of the receptor and produces a rapid revers-
ible channel blockade with ion conductance blocked
by simple steric hindrance (Valenzuela et al., 1994).
The second site binds ligands with low a�nity (>100
mM) and is postulated to be located at the interface
between the receptor protein and the lipid membrane.
Multiple sites exist for each receptor (10±20) with
binding of ligands accelerating desensitization of the
receptor-ion channel. In cultured PC12 cells with single
channel recording ethanol has been observed to reduce
the mean open time of channels and accelerate the
decay phase of ACh induced currents (Nagata et al.,
1996). Additionally, in oocytes expressing a7 receptors,
ethanol inhibited ACh induced currents without a�ect-
ing the a�nity of ACh for the receptor. The e�ect of
ethanol treatment on nAChRs expressed in M10 and
SH-SY-5Y cells has also been examined (Gorbounova
et al., 1998). Ethanol produces a dose related decrease
in nAChR number as measured with �3H� nicotine in
M10 cells and �3H� epibatidine in SH-SY-5Y cells.
Chronic ethanol (100 mM) treatment of M10 cells also
partly attenuated nAChR upregulation produced by
treatment of the cells with nicotine. In these same
cells, ethanol also signi®cantly decreased a3 and
increased a4 and a7 mRNA levels while having no
e�ect on b2 mRNA levels.
Fig. 2. Schematic cross section of a nicotinic receptor showing the
ion channel, the ACh binding site and multiple allosteric sites distrib-
uted throughout the extracellular part of the protein (modi®ed from
Lena and Changeux, 1993). Allosteric sites shown include the non-
competitive allosteric activator site (NCA); non-competitive negative
allosteric sites (NCB); binding sites for Ca2+ and steroids and phos-
phorylation sites (P).
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 79

1.2.1.3. Steroid binding site. Steroids have the ability to
desensitize nicotinic receptors by action at a site
located in the extracellular hydrophillic domain that is
distinct from the ACh site (Bertrand et al., 1991;
Inoue and Kuriyama, 1991). Progesterone, corticoster-
one, and dexamethasone are potent inhibitors of a3subtype containing ganglionic receptors expressed in
SH-SY5Y cells while having no a�ect on ACh binding
(Ke and Lukas, 1996). Corticosterone is known to
desensitize nAChRs and to produce tolerance to the
e�ects of nicotine, while chronic administration in
mice reduces the number of brain �125I� a-bungarotoxinbinding sites (Grun et al., 1992; Pauly and Collins,
1993; Robinson et al., 1996; Stitzel et al., 1996; Cag-
giula et al., 1998). Furthermore, high steroid concen-
trations (mM range) have been observed to displace
�125I� a-bungarotoxin binding from rat brain mem-
branes and reduce the a�nity of nicotine for this site
(Lena and Changeux, 1993).
1.2.1.4. Dihydropyridine site. L-type Ca2+ channel an-
tagonists such as nimodipine and nifedipine are
capable of blocking agonist induced activation of nic-
otinic receptors (Lopez et al., 1993) and are able to
inhibit noradrenaline release from chroma�n cells in a
reversible non-voltage dependent manner (Gandia et
al., 1996). The mechanism of action of these com-
pounds is unknown but the binding site is proposed to
exist within the ion channel. Interestingly, Ca2+ ions
themselves modulate nicotinic receptor-ion channel
function. Multiple Ca2+ binding sites exist on both
muscle type and neuronal nicotinic receptors (Fair-
clough et al., 1993), which when activated produce a
voltage sensitive decrease in conductance. A further
category of Ca2+ binding site, found only on neuronal
nicotinic receptors, potentiates agonist activation of
ion currents in a voltage insensitive manner (Mulle et
al., 1992).
1.2.1.5. Additional allosteric modulation of nicotinic
receptors. Phosphorylation by protein kinase A, pro-
tein kinase C or by tyrosine kinase of de®ned residues
within the cytoplasmic loop results in desensitization
of the receptor-ion channel (Huganir and Greengard,
1990). A number of pharmacologically active sub-
stances indirectly enhance nAChR desensitization via
phosphorylation. This generally occurs through
induced changes in intracellular Ca2+ concentration
and activation of Ca2+ sensitive protein kinases such
as those above. For example, the neuropeptide CGRP
(calcitonin gene related peptide) and substance P both
enhance nicotinic receptor desensitization through acti-
vation of phosphorylating enzymes (Miles et al., 1989;
Simmons et al., 1990).
1.3. Transition states of nicotinic receptors
Nicotinic receptors can exist in at least one of fourinterconvertible functionally distinct conformationalstates at any one time. These states can be interpretedin terms of the ``conformational scheme'' of Katz andThesle� (1957) and consist of: (1) a resting state R, (2)an activated state A, where the channel opens on amicrosecond to millisecond timescale when activatedbut which has a low a�nity for ACh (10±1 mM), and(3) and (4), one of two desensitized closed channelstates I and D that are refractory to activation on amillisecond±minute timescale, but exhibit high a�nityfor ACh (10 nM±1 mM) and nicotinic ligands (Galziand Changeux, 1995). Binding of ligands to thenAChR structure either at the ACh site or any of theallosteric sites can modify the equilibrium between thedi�erent conformational states of the receptor at anyone time. Additionally, ligands binding to the nAChRcan be considered to di�erentially stabilize the confor-mational state to which they preferentially bind (Lenaand Changeux, 1993).
1.4. Nicotinic receptor upregulation
Nicotinic receptors go against convention in thatprolonged exposure to agonists results in an increasein receptor number, a contradiction of the generallyaccepted paradigm that over exposure to agonists pro-duces receptor down regulation and overexposure toantagonists, receptor upregulation. Long term ex-posure to nicotine results in increased number ofnAChRs in the brain of several species includinghumans. Postmortem binding studies have revealedincreased �3H� nicotine and �3H� ACh binding sites inthe brains of smokers compared to non-smokers witha dose dependent correlation observed betweenincreased binding sites and the number of cigarettessmoked (Benwell et al., 1988; Breese et al., 1997;NybaÈ ck et al., 1989). Furthermore, the number ofbinding sites observed in the brains of ex-smokers waslower than that of non-smokers. It is proposed thatthe desensitization and upregulation of nAChRs fol-lowing chronic nicotine exposure is the basis of toler-ance to nicotine displayed by smokers as well as beingin¯uential in producing withdrawal symptoms on ces-sation of smoking (Benwell et al., 1988; Balfour andFagerstrom, 1996; Dani and Heinemann, 1996). Inrats, subchronic treatment (0.45 mg/kg twice daily)with nicotine results in an increase in the number ofhigh a�nity nAChR sites in the cortex, while the pro-portion of low a�nity sites is reduced. In addition,there was a signi®cant reduction in agonist a�nity ofboth types of nAChR (Romanelli et al., 1988). Therationale behind nAChR upregulation is thought to liein their rapid desensitization and consequent inacti-
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11180

vation following chronic agonist exposure, putativelyresulting in a de®cit in cholinergic function, which isthen counteracted by an increase in receptor number(Schwartz and Kellar, 1985). Upregulation, desensitiza-tion and the eventual inactivation of nAChRs appearto be dependent upon the length of agonist exposureand upon the nature of the agonist itself (Rowell andDuggan, 1998; Reitstetter et al., 1999). The rate ofrecovery from desensitization of muscle type nAChRsexpressed in TE67/RD cells was recently observed tobe signi®cantly faster following a short exposure tonicotine or ACh, with functional recovery from nic-otine observed to be consistently more rapid thanrecovery from ACh (Reitstetter et al., 1999). These ob-servations indicate that more than one state of recep-tor desensitization exists, and that, agonists vary intheir ability to induce these di�erent states. IndividualnAChR subtypes also vary in their sensitivity to desen-sitization and inactivation following agonist exposure.It appears that a4b2 and a7 nAChRs are more sensi-tive to upregulation and desensitization than othersubtypes. Chronic exposure of oocyte expressedhuman a4b2, a7 and a3 (formed from combinations ofa3, b2, b4 and a5 subunits) receptors to submicromo-lar concentrations of nicotine results in irreversible in-activation of the majority of a4b2 and a7 receptorsbut substantially fewer a3 subtype (Olale et al., 1997).Similar results were observed by Hsu et al. (1996) withoocyte expressed a4b2 and a3b2 nAChRs. Functionalresponses in a4b2 nAChRs could be completely abol-ished following a 48 h incubation with nicotine, whileresponses in a3b2 nAChRs could only be reduced by50±60%. Additionally, the half-time for recovery ofa3b2 nAChRs was faster than that of a4b2, at 7.5 and21 h, respectively. These observations suggest that thebehavioural e�ects of nicotine (e.g. tolerance, withdra-wal) are predominantly mediated through a4b2 and a7nAChRs. Furthermore, both of these receptor typeswhen expressed in HEK 293 cells, M10 cells andoocytes are upregulated, following chronic nicotine ex-posure (Gopalakrishnan et al., 1996; Eilers et al., 1997;Molinari et al., 1998). Chronic exposure of human a7nAChRs expressed in HEK 293 cells to 100 mM nic-otine produced a 2.5 fold increase in �125I� alphabun-garotoxin (aBTX) sites, an e�ect that was observed tobe concentration dependent (Molinari et al., 1998).The agonists epibatidine, anabaseine and 1,1-dimethyl-4-phenylpiperazinium (DMPP) also increased �125I�aBTX sites. A similar range of agonists increased thenumber of �3H� epibatidine binding sites in M10 cellsexpressing a4b2 receptors, while the antagonists dihy-dro-b-erythroidine (DHbE), methylcaconitine (MLA)and d-tubocuraine had no e�ect (Eilers et al., 1997).Additionally, evidence suggests that the variousnAChR subtypes display di�erent properties in theirresponse to chronic agonist stimulation, with the a�-
nity of each subtype for a particular agonist in¯uen-cing the magnitude of receptor upregulation observed(Warpman et al., 1998). Nicotine induced upregulationof nAChR sites in M10 cells expressing a4b2 nAChRsdi�ers from that in SH-SY5Y neuroblastoma cellsexpressing a3, a5, b2 and b4 subunits (a3b2, a3b4,a3b4a5, a3b2a5 and a3b2b4a5 possible receptor com-binations) with 100 times greater concentration of nic-otine required to induce a similar magnitude ofnAChR upregulation in SH-SY5Y as in M10 cells(Warpman et al., 1998). In this study, the a�nity ofnicotine for nAChRs in SH-SY5Y (a3 nAChRs) cellswas 14 times lower than in M10 cells (a4b2 nAChRs),thus, the lower a�nity of nicotine for the a3 subunitresults in the higher concentration of nicotine necess-ary to produce an upregulation of nAChRs in thesecells. The mechanism by which nAChR number isincreased following nicotine exposure is thought toinvolve reduced turnover of cell surface receptors; it isproposed that conformational changes in the receptorstructure occur, preventing it from being removedfrom the cell surface (Peng et al., 1994). This view issupported by observations from a number of studies.Chronic nicotine administration in mice increases thenumber of �3H� nicotine sites observed in brain butdoes not increase a2, a3, a4, b5 or b2 mRNA levels in-dicating that increased receptor number results frompost transciptional mechanisms (Marks et al., 1992).Exposure of receptors, both to nicotine and to thenon-competitive channel blocker mecamylamine, havebeen observed to increase nAChR number, but not ex-posure to the non-competitive antagonist chlorisonda-mine (el-Bizri and Clarke, 1994; Pauly et al., 1996)suggesting that, receptor blockade is not su�cient toinduce conformational changes necessary for receptorupregulation. The desensitization of nAChRs isthought to involve phosphorylation of the receptormediated by protein kinase A and protein kinase C,both forsklin and phorbol esters have been observedto increase nAChR binding sites and to synergisticallyenhance nicotine induced receptor upregulation, withevidence suggesting that a4 nAChR subunits may bespeci®c targets of PKA phosphorylation(Gopalakrishnan et al., 1997; Hsu et al., 1997).Following incubation of oocyte expressed a4b2 anda3b2 nAChRs with nicotine, human a4b2 nAChRsexpressed in HEK 293 cells are upregulated by chronicnicotine exposure, as measured by an increase in �3H�epibatidine binding and functionally deactivated,measured as an abolition of Ca2+ in¯ux (Eilers et al.,1997). Similar deactivation of receptors could be pro-duced by decreasing PKC activity by application ofthe PKC inhibitor NPC-15437 suggesting that, phos-phorylation of the receptor protein by PKC activity isnecessary for ion-channel receptor function. Followingdeactivation, functional recovery of receptors occurs
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 81

slowly (4±6 h) but may be accelerated by inhibition ofPKA activity. From these observations it is hypoth-esised that receptor deactivation involves dephosphory-lation of the receptor protein at PKC sites, putativelyby PKA action and that reactivation involves re-phos-phorylation of these same sites. It is likely that thephosphorylation state of the receptor will be in¯uentialon turnover and removal from the cell surface whichwill thus in¯uence receptor number. Evidence to sup-port PKA involvement in deactivation and upregula-tion of receptors comes from studies involving mutantPC12 cells expressing nAChRs which are de®cient incAMP dependent PKA I and II (Madhok et al., 1995).Following nicotine treatment, a dose dependentincrease in �3H� nicotine sites is observed in wild typePC12 cells but not in mutant PC12 cells; furthermore,a3 mRNA levels were observed to decrease and b2mRNA levels to increase in wild type PC 12 cells butno change in subunit mRNA levels was observed inmutant PC12 cells. Therefore, upregulation ofnAChRs would appear to involve a PKA mediatedmechanism and that, b2 containing receptors consti-tute the major subtype upregulated, following chronicagonist exposure.
2. Neuronal nicotinic receptor subtypes in the humanbrain
Classically, much of the knowledge pertaining toneuronal nAChRs in human brain has been obtainedfrom in vitro binding studies on postmortem tissueand on neuroblastoma cell lines expressing nAChRs,with evidence suggesting that nAChRs can be broadlydivided into two main subtypes. More recently, thecloning of nAChR subunits and the expression ofrecombinant nAChRs in oocytes and additional tech-niques such as in situ hybridisation and immunohisto-chemistry has furthered understanding of the structure,subunit composition and pharmacological pro®le of in-dividual nAChRs. Studies have revealed that multiplesubtypes of functional neuronal nAChRs can beformed from various combinations of nAChR subu-nits, with evidence to suggest that as many as fourfunctionally distinct subtypes of nAChR exist (Zoli etal., 1998). However, it appears that the majority ofhigh a�nity nAChRs in the brain comprise the a4b2subtype (Whiting et al., 1987; Flores et al., 1992; Zoliet al., 1998). Heteromeric a3 and homomeric a7 sub-types constitute the other major neuronal nAChRspresent, with the remainder made up of various combi-nations of a2, a3, a5, a6 and b2 and b4 subunits co-expressed to form heteromeric receptors.
2.1. Ligand binding studies on human brain tissue
In human brain, neuronal nAChRs can be dividedby radioligand binding into at least two classes: (1)Alpha bungarotoxin sites (BTXRs) that bind aBTXwith high a�nity, and the nicotinic agonists nicotine,ACh and cytisine with low a�nity and (2) high a�nitynicotine sites (AChRs) that bind nicotine, ACh andcytisine with high a�nity and aBTX with low a�nity(Clarke et al., 1985; Sugaya et al., 1990). Originalhomogenate and autoradiographic ligand binding stu-dies on postmortem human brain tissue (Shimohamaet al., 1985; Flynn and Mash, 1986; Nordberg andWinblad, 1986; Nordberg et al., 1987; Adem et al.,1988; Perry et al., 1992) identi®ed the presence of highand low a�nity �3H� nicotine and �3H� ACh sites in thecortex, hippocampus and thalamus of human brain,which are hypothesised to correspond to AChRs andBTXRs, respectively. Additional investigation revealedthe presence of super high, high and low a�nity ago-nist binding sites in the brain, indicating that morethan two receptor subtypes exist (Nordberg et al.,1988, 1989a; Nordberg and Winblad, 1986).Homogenate binding studies utilising �3H� cytisine havealso been performed on human brain tissue, where asingle high a�nity site (Kd = 0.147±0.245 nM) wasidenti®ed in the hippocampus, cingulate gyrus and cor-tex with highest levels of binding observed in the thala-mus, Bmax = 48 fmol/mg protein (Hall et al., 1993).Binding studies with the high a�nity AChR agonist�3H� epibatidine have also been performed on humanpostmortem brain tissue identifying the presence oftwo high a�nity sites in cortex with Kd values of 0.3and 28.4 pM, respectively, observations consistent with�3H� ACh studies (Houghtling et al., 1995). Recentautoradiographic studies comparing the distribution of�3H� nicotine and �3H� epibatidine binding sites inhuman brain have con®rmed this observation andhave identi®ed two high a�nity binding sites in thetemporal cortex, thalamus and cerebellum (Marutle etal., 1998; Sihver et al., 1998a). The binding of each ofthe nicotinic agonists �3H� nicotine, �3H� cytisine and�3H� epibatidine to AChRs is una�ected by the pre-sence of aBTX or MLA, indicating that, they bind atthe classical ACh recognition site on a receptor sub-type, distinct to that at which �125I� aBTX binds. Incomparison to the large number of studies mappinghigh a�nity AChR sites, relatively few ligand bindingstudies with �125I� aBTX have been performed onhuman postmortem brain. Studies reveal, however, asingle high a�nity site with a distribution distinct tothat of �3H� nicotine (Davies and Feisullin, 1981; Langand Henke, 1983; Sugaya et al., 1990; Rubboli et al.,1994b; Spurden et al., 1997; Sabbagh et al., 1998).�125I� aBTX binding is highest in the hippocampal for-mation, particularly the dentate gyrus and CA1±CA3
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11182

regions, and in the thalamus with relatively little bind-ing observed in the cortex. These studies describe thepresence of three probable nAChR sites in humanpostmortem brain distinguishable by their relative a�-nities for nicotinic agonist ligands and aBTX.
2.2. Binding studies in human neuroblastoma cell lines
A number of ligand binding experiments have beenperformed on human neuroblastoma cell lines such asIMR32, SH-SY5Y and SK-N-BE. These cell linesexpress various cell surface nAChR subtypes and aretherefore extremely useful tools for examining nativehuman nAChRs. Binding studies with �125I� aBTX and�3H� ACh on IMR 32 and SH-SY5Y neuroblastomacell lines (Lang and Henke, 1983; Gotti et al., 1986;Lukas, 1993; Galzi and Changeux, 1995; Gotti et al.,1995) have identi®ed the presence of a single site for�125I� aBTX binding (Kd, 1±10 nM) and two sites for�3H� ACh. The low a�nity (Kd, 100 nM) ACh site ispresent in high abundance and the second high a�nity(Kd, 1±2 nM) site is present in low abundance. Bindingof �3H� ACh to both of these sites is una�ected by thepresence of aBTX indicating that the �125I� aBTX and�3H� ACh binding sites are distinct from one another.Binding of �3H� nicotine and �3H� epibatidine tonAChRs expressed by SH-SY5Y cells has also beeninvestigated (Warpman et al., 1998). The level of non-speci®c binding of �3H� nicotine was very high (>90%)in this cell line, while in contrast �3H� epibatidinebound with very high speci®city (non-speci®c binding<10%). �3H� epibatidine identi®ed a single binding sitein SH-SY5Y cells (Kd = 0.1220.02 nM, Bmax =322.524.1 fmol/mg). The low level of speci®c bindingobserved with �3H� nicotine is consistent with the factthat SH-SY5Y cells predominantly express a3 but noa4b2 nAChRs. Additionally, the single site identi®edwith �3H� epibatidine is consistent with its high a�nityfor a3 nAChRs, while the lack of a second binding siteis explained by the absence of a4b2 nAChRs in thiscell line, for which epibatidine also displays high a�-nity. The interaction of the nAChR agonist (R,S)-3-pyridyl-1-methyl-2-3(3-pyridyl)-azetidine (MPA) withnAChRs expressed in SH-SY5Y cells has also beeninvestigated (Zhang et al., 1998). In this study, MPAdisplaced �3H� epibatidine from a single binding sitewith a Ki = 35.924.1 nM, a value similar to thatobserved for displacement of �3H� epibatidine from ratcortical membranes, Ki = 23.7820.36 nM.
In general, these observations are consistent withresults from human postmortem binding studies andprovide evidence to support the presence of at leasttwo (probably three) classes of nAChR in humanbrain, distinguishable by their relative a�nities for �3H�nicotine, �3H� epibatidine and �125I� aBTX.
2.3. Nicotinic receptor subunit expression in oocytes andtransfected cell lines
The high degree of sequence homology existingbetween rat and human nAChR subunits (82±95%)has allowed the use of rat cDNAs encoding di�erentsubunits to be used as molecular probes for the identi-®cation and cloning of human nAChR subunits. Ninereceptor subunits have thus far been identi®ed andcloned in human brain and include a2, a3, a4, a5, a6,a7, b2, b3, and b4 subunits. The isolation and cloningof the individual human nAChR subunits combinedwith the use of appropriate expression systems such asXenopus oocytes has allowed investigation into the bio-chemical structure, subunit composition and pharma-cological pro®le of functional human nAChRs. A largenumber of such studies have been performed involvingmost of the human nAChR subunits and have resultedin a number of common conclusions, including theidenti®cation of two main structural subtypes ofnAChR.
2.3.1. Heteromeric nicotinic receptorsFollowing individual injection into an appropriate
expression system, a2, a3, or a4 and b2 or b4 subunitsare not capable of forming functional receptor-ionchannel complexes. However, when any of the a2±a4subunits are expressed pairwise with b2 or b4 subunits,functional receptors are produced (i.e. a2b2, a2b4,a3b2, a3b4, a4b2, a4b4) that have distinct electro-physiological and pharmacological pro®les (Sargent,1993; McGehee and Role, 1995). These observationsindicate that this group of subunits can only form het-eromeric receptors. Both a and b subunits contributeto the pharmacological and functional pro®le of eachreceptor. For example, when expressed with b2 subu-nits, each of a2, a3 and a4 subunits form functionalnAChRs with di�erent single channel conductance,average channel open times and antagonist sensitivity.More speci®cally, b subunits appear to have a pro-found in¯uence on the dissociation rate of agonistsand antagonists from the receptor as well as the rateat which agonist bound receptors open (Papke andHeinemann, 1991; Papke et al., 1993). FunctionalnAChRs may also be formed from combinations ofthree or more individual subunit types (Role and Berg,1996; Wang et al., 1996). For example, the a5 subunitwhen expressed alone or in combination with any ofthe b subunits is unable to form a functional ion chan-nel, however, when expressed with a4 and b2 subunits,it gives rise to a functional nAChR subtype a4b2a5.Furthermore, a receptor composed of four di�erentsubunits is formed when a5 is co-expressed with a4, b2and b4 subunits. Until recently, the a6 subunit wasthought to be an ``orphan'' subunit (as is the b3 subu-nit) in that it had been unable to form a functional ion
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 83

Table
2
A�nityofradioligandsandagonistpotency
atrecombinanthumannicotinic
receptors
e,i
Receptorcomposition
[3H�n
icotineKd
(nM)
[3H�e
pibatidineKd
(nM)
[3H�c
ytisineKd
(nM)
[125I�aB
TX
Kd
(nM)
Rankorder
ofagonistpotency
a2b2
DMPP>
Nicotine>
Cytisine>
ACha,i
a2b4
0.0422
0.01b,k
Nicotine>
DMPP>
Cytisine>
ACha,i
Epibatidine>
Cytisine>
Suberycholine=
Nicotine=
DMPPb,k
a3b2
0.122
0.04a,g
DMPP>
Cytisine>
ACha,i
Epibatidine>
Nicotine>
ACha,i
a3b2
a55.462
0.60a,g
0.252
0.02a,g
DMPP>
Cytisine>
Nicotine>
ACha,i,j
a3b4
4.902
0.60a,g
DMPP>
Cytisine>
Nicotine>
ACha,i
0.2302
0.012b,k
Epibatidine>
DMPP=Cytisine=
Nicotine=
Suberycholineb
,k
a3b4
a52.802
0.30a,g
DMPP>
Cytisine>
Nicotine>
ACha,i
a4b2
0.212
0.04b,f
Cytisine>
Nicotine>
DMPP>
ACha,i
Nicotine>
ACh>
Cytisineb
,h
a4b4
0.1872
0.029b,k
Cytisine>
Nicotine>
DMPP>
ACha,i
Epibatidine>
Cytisine=
Suberycholine>
nicotine>
DMPPb,k
a70.81a,c
0.712
0.11b,d
aReceptors
expressed
inoocytes.
bReceptors
stably
expressed
inHEK
293cells.
cPenget
al.,1994.
dGopalakrishnanet
al.,1995.
eGerzanichet
al.,1995.
fGopalakrishnanet
al.,1996.
gWanget
al.,1996.
hBuissonet
al.,1996.
iChavez-N
oriegaet
al.,1997.
jGerzanichet
al.,1998.
kStaudermanet
al.,1998.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11184

channel individually or in combination with any of theother subunits. However, recent studies have demon-strated the assembly of a functional a3b4a6 receptor-ion channel (composed of chick embryo cDNAsexpressed in human BOSC 23 cells) with an a�nity forACh threefold lower than that of a3b4 (Fucile et al.,1998). It is likely therefore that a similar functionalreceptor may be formed from the comparable humannAChR subunits.
2.3.2. Homomeric nicotinic receptorsIn contrast to a2±a4 and b2±b4 subunits, a7 and a8
subunits when expressed in oocytes with any of theother a or b subunits or multiple combinationsthereof, fail to form functional ion channels. However,when expressed individually, they form functionalhomoligomeric receptors that are consistent in sizewith the pentameric structure displayed by theirheteroligomeric counterparts but have distinct pharma-cological and physiological pro®les (Anand et al.,1993). The multiple combinations of nAChR subunitpossible, therefore, give rise to a multitude of nAChRsubtypes. Studies in which recombinant humannAChRs were expressed in oocytes or human cell lineshave identi®ed numerous combinations of subunitsthat form functional nAChRs. Although no de®nitiveconclusion can be drawn from these studies, it is likelythat the subunit compositions and structural confor-mations of these recombinant nAChRs will re¯ect thestructure of nAChRs expressed in human brain.Human nAChRs comprising of a3b2, a4b2, a3b4 anda7 subunits have been stably transfected into oocytes(Peng et al., 1994; Gerzanich et al., 1995; Wang et al.,1996) and a4b2, a4b4 and a7 subtypes have also beenexpressed in HEK293 cells (Gopalakrishnan et al.,1995, 1996; Stauderman et al., 1998), where they havebeen characterised with ligand binding experiments.Results indicate that the heteromeric nAChRs contain-ing combinations of a and b subunits show greatesta�nity for nicotinic agonists such as epibatidine, nic-otine and cytisine (e.g. a3b3 Kd = 0.1220.0 nM,a3b2a5 Kd = 0.2520.02 nM, for �3H� epibatidine anda4b2 Kd = 0.2120.04 nM for �3H� cytisine), whereas,homomeric a7 receptors displayed highest a�nity for�125I� aBTX (Kd = 0.7120.11 nM) Table 2. It is there-fore believed that the high a�nity nicotine bindingsites are composed of the heteromeric ab receptors,while the homomeric a7 (and a8) receptors representthe high a�nity aBTX sites in human brain. Supportfor these observations comes from combined immuno-precipitation and ligand binding studies on IMR 32cells using subunit speci®c antibodies directed againsthuman a3, a5, a7, b2 and b4 subunits and �125I� aBTX(Gotti et al., 1995). The antibodies directed against theindividual subunits were used to immunoprecipitate�125I� aBTX labelled nAChRs expressed by IMR32
cells. Of the antibodies, only the anti-a7 was capableof precipitating almost all of the radiolabelled recep-tors. Anti-a3, a4, a5, b2 and b4 antibodies were incap-able of precipitating any of the labelled receptors.Thus, it appears that aBTX sites contain the a7 subu-nit but not any of the a3, a4, a5, b2 or b4 subunits.From this observation it is likely that a7 is the soleconstituent of aBTX receptor but it is possible that asyet unidenti®ed subunits are also involved. Few similarsuch immunoprecipitation studies have been performedwith the high a�nity nicotinic receptor sites expressedin neuroblastoma cell lines, mainly due to the lack ofsubunit speci®c antibodies. However, native nAChRsexpressing b2 subunits have recently been characterisedusing a monoclonal antibody (mAb290), which isspeci®c for this receptor subunit (Wang et al., 1996).Following immunoprecipitation and western blotting,b2 containing nAChRs expressed by SH-SY5Y cellswere also found to contain a3 and a5 subunits. In thesame study �3H� epibatidine labelled a3 containingreceptors in the SH-SY5Y cells were immunoprecipi-tated with mAb290 revealing that at least 56% of a3containing �3H� epibatidine sites also contain b2 subu-nits. The sedimentation properties of these a3 contain-ing nAChRs in SH-SY5Y cells were additionallyanalysed and compared to those of recombinant a3b2and a3b2a5 subtypes expressed in oocytes. The recep-tors were found to have the same sedimentation coe�-cient of 11S, which is consistent with that of apentameric nAChR. There are as yet no b4 subunitspeci®c antibodies available and thus, nAChRs con-taining this subtype have not been characterised.However, the above observations generally support theview that in human brain the high a�nity aBTX sitesare composed of homomeric a7 (and probably alsohomomeric a8) receptors and that the high a�nity nic-otine binding sites consist of heteromeric receptorscomposed of multiple combinations of a and b subu-nits but not of a7 and a8.
2.4. Electrophysiological studies
The structural diversity displayed by the numerousnAChR subtypes is re¯ected in their functional pro-®les, with individual receptors having distinct func-tional characteristics which are determined by theirsubunit composition. The pharmacologic and func-tional properties of a range of recombinant humannAChRs (e.g a2b2, a2b4, a3b2, a3b4, a4b2, a4b4, a7)expressed in oocytes and HEK 293 cells have beencharacterised (Luetje and Patrick, 1991; Galzi andChangeux, 1995; Gerzanich et al., 1995, 1998; Zoli etal., 1995; Gopalakrishnan et al., 1996; Chavez-Noriegaet al., 1997; Olale et al., 1997; Stauderman et al., 1998;Zoli et al., 1998). Electrophysiological techniques suchas patch clamp were used to measure currents pro-
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 85

duced by a range of nicotinic agonists including ACh,nicotine, cytisine, DMPP and epibatidine with eachsubtype displaying di�erent kinetics of activation andinactivation (Table 2). ACh was found to be highlye�cacious in producing currents at all subtypes exceptfor a3b2 where DMPP was markedly more e�cacious.Cytisine was the least e�ective agonist at b2 containingreceptors but was e�cacious in producing currents ina2b4, a3b4 and a4b4, as well as a7 receptors (Chavez-Noriega et al., 1997). Human a4b2 receptors stablyexpressed in HEK293 cells diplayed agonist inducedcation currents consistent with native neuronal a4b2nAChRs, while also showing high a�nity for �3H� cyti-sine (Kd = 0.2 nM) with a good correlation observedbetween a�nity in transfected cells and native humannAChRs (Gopalakrishnan et al., 1995). Epibatidinewas found to be the most potent of a range of agonistsin eliciting currents in a2b4, a3b4 and a4b4 receptorsexpressed in HEK 293 cells (Stauderman et al., 1998).a7 receptor activation in the same cell line followingnicotine or ACh application evoked whole cell currentswith fast activation and inactivation kinetics that weresensitive to blockade with aBTX (Gopalakrishnan etal., 1995). Speci®c binding of �125I� aBTX was alsoobserved in these cells with a Kd = 0.7 nM. In oocytesexpressing a3b2 and a3b4 receptors, the addition of ana5 subunit (i.e. producing a3b2a5 and a3b4a5 recep-tors) had variable e�ects on receptor function. In a3b2receptors, a5 produced a 50 fold increase in the sensi-tivity of the receptor to ACh, but had little e�ect ona3b4 nAChR function (Gerzanich et al., 1998). Ionchannel function in all receptor subtypes was observedto be sensitive to nicotinic antagonists such as DHbE,mecamylamine and d-tubocurarine. Human a4b2 anda4b4 were more sensitive to blockade by DHbE thand-tubocurarine, whereas, a7 and a3b4 were more sensi-tive to d-tubocurarine than DHbE (Chavez-Noriega etal., 1997). The non-competitive antagonist mecamyla-mine (3 mM) produced greater than 80% block ofa2b2 and a4b4 nAChRs compared to approximately50% block of a4b2, a2b2 and a7 receptors. The di�er-ent receptor subtypes also display individual rates ofinactivation and desensitisation following agonist ex-posure. Following exposure of a7, a4b2, a3b2a5 anda3b4a5 receptors expressed in oocytes to submicromo-lar concentrations of nicotine, the majority of a4b2and a7 receptors were irreversibly deactivated, whilea3b2a5 and a3b4a5 subtypes were much less a�ected(Olale et al., 1997).
2.5. Evidence for four functional subtypes of nicotinicreceptor
The studies outlined in the previous sections provideevidence for numerous nAChR subtypes with distinctpharmacological and functional pro®les. However,
despite the individual functional pro®les of nAChRscharacterized in expression systems, evidence exists tosuggest that nAChRs expressed in vivo may be func-tionally characterized into three or four main subtypes.From studies on cultured rat hippocampal neuronsAlkondon and Albuquerque (1993) provided pharma-cological and functional evidence for at least three dis-tinct nAChR subtypes. Using whole cell patch clamptechniques and the decay kinetics of the currents eli-cited by the application of 3 mM ACh, they describedfour current types termed IA, IB, II and III in rat hip-pocampal neurons. The order of potency of agonists inactivating the currents varied between current types, asdid the sensitivity of each current to blockade by anumber of nicotinic antagonists. Type IA currents, themost common observed (present in 83% of neurons),were rapidly decaying in nature and could be blockedby aBTX (10 nM), kBTX (10 nM) and MLA (1 nM).This suggests that this current type may result from a7nAChR activation, as only homomeric a7 receptorsare sensitive to aBTX (Couturier et al., 1990). Type IIcurrents (present in 5% of neurons) were blocked byDHbE (10 nM) and by high concentrations of MLAand kBTX (100 nM each), but not by aBTX. Type IIIcurrents (present in 2% of neurons), slowly decayingin nature were blocked by mecamylamine (1 mM) butnot by aBTX, kBTX or MLA in concentrations of upto 100 nM. Approximately, 10% of the neurons inves-tigated displayed mixed responses to agonist activation(Type IB). This type of current was partially blockedby MLA (1 nM) or DHbE (10 nM) alone and comple-tely blocked by a combination of the two antagonists.A number of the agonists used were useful in discrimi-nating between the currents elicited. ACh, carbachol,nicotine and suberylcholine were particularly e�ectivein producing Type II currents, while cytisine appearedto be speci®c for Type III. This functional classi®-cation of three nAChR subtypes has recently beenexpanded to four receptor subtypes by Zoli et al.(1998) from autoradiographic and patch clamp studiesof nAChRs in brain slices of b2 knockout mice . . .Table 3. Autoradiographic studies mapping the distri-bution of �3H� ACh, �3H� nicotine, �3H� cytisine, �3H�epibatidine and �125I� aBTX in b2 knockout mice andwild type mice were performed. Distribution of allligands in brain sections of wild type mice was consist-ent with distribution of these ligands in previous stu-dies in the mouse and with distribution in rat brain. Inbrain sections of b2 knockout mice, however, high a�-nity �3H� nicotine binding sites could no longer bedetected, with the binding of �3H� ACh, �3H� cytisineand �3H� epibatidine signi®cantly reduced. In contrast,the pattern of �125I� aBTX binding was not substan-tially di�erent from that of wild type mice.Electrophysiological analysis of nAChRs in wild typeand b2 knockout brain sections was then correlated to
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11186

regional binding pattern of the radioligands. The cur-rents elicited by application of a range of nicotinicagonists allowed assignment of responses to the acti-vation of four subtypes of nAChR. Type 1 receptorsare aBTX sensitive with low a�nity for nicotinic ago-nists and their distribution as mapped by �125I� aBTXis not signi®cantly di�erent in wild type and b2 knock-out mice. Their sensitivity to aBTX and the fast acti-vation and decay kinetics displayed by these receptorsindicate that they are a7 homomeric receptors andthat, they correspond to the receptors producing TypeIA currents as described by Alkondon andAlbuquerque (1993). This is supported by disappear-ance of receptors with a similar pharmacological andfunctional pro®le in a7 knockout mice (Orr-Urtregeret al., 1997). Type 2 receptors contain the b2 subunitand represent the vast majority of nAChRs in rodentbrain as evinced by their disappearance in b2 knockoutmice. All of the nicotinic agonists used, bound to thisreceptor type with high a�nity (nanomolar to subna-nomolar range). The rank order of agonist potency forthese receptors is consistent with that of recombinanta4b2 receptors expressed in oocytes (Luetje andPatrick, 1991), and it is proposed that the compositionof the major isoform forming Type 2 receptors isa4b2. It is likely however, that other subunits (e.g. a2,a3, a5 and b4) also co-assemble with b2 to form arange of functional receptors that contribute to Type 2high a�nity nAChR sites. Type 3 receptors do notcontain b2 and bind only �3H� epibatidine with higha�nity. Electrophysiological studies indicate that therank order of agonist potency for this receptor is con-sistent with a3b4 containing receptors (Luetje andPatrick, 1991). The distribution of these receptors isalso consistent with the distribution of a3 and b4 sub-unit mRNA, suggesting an a3b4 composition for thissubtype. Type 4 receptors bind �3H� epibatidine and�3H� cytisine with high a�nity and display agonistpotency consistent with b4 containing receptors(Luetje and Patrick, 1991). In contrast to Type 2receptors, they display a signi®cantly faster desensitiza-tion rate suggesting a subunit composition of a2 andor a4 with b4. Although the data from these studies isderived from rodent brain, it is feasible that a similarhierarchy of functional nAChRs will exist withinhuman brain, although further investigation will (ofcourse) be necessary. Therefore, while multiple sub-types of neuronal nAChR exist in vivo, it appears thatthey be functionally categorized into four groups.
3. Distribution of nicotinic receptors in the human brain
In comparison to muscarinic receptors, neuronalnAChRs are expressed in relatively low density in thehuman brain. In addition, their pattern of distributionT
able
3
Fourfunctionalsubtypes
ofnicotinic
receptor
Receptor
type
Relative
frequency
Majorcurrentattributes
Pharm
acology
Higha�nitybinding
Subunitcomposition
AlkondonandAlbuquerque(1993)
IA83%
Rapidly
decaying
MLA,aB
TX,kB
TX
sensitive
NA
a7IB
10%
Fast
andslow
decay
MLA+
DHbE
sensitive
NA
a7,a4
b2II
5%
Slowly
decaying
DHbE
sensitive
NA
a4b2
III
2%
Slowly
decaying
MLA
sensitive
NA
a3b4
Zoliet
al.(1998)
1NA
V.fast
desensitisation
aBTX
andMLA
sensitive
aBTX
a72
NA
Mixed
MLA
insensitiveNic>
Cyt,DHbE
=MCA
EPI>
Nic
=Cyt=
MCC
=ACh
b2a4
(a5?),b2
(a2?),b2
(a3?),b2
(a6b3
?)
3NA
Slowly
decayingwhen
Nic.>
100mM
MLA
insensitive,
Cyt=
Nic,DHbE
<MCA
EPI
b4a3
(a5?)
4NA
Fast
decaywhen
Nic.>
100mM
MLA
insensitive,
Cyt=
Nic,DHbE
<MCA
EPI>
Cyt>
MCC
=ACh
(b4a4
?)(b4a2
?)
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 87

is relatively homogenous and is not restricted to thewell de®ned brain cholinergic pathways. The neuroa-natomical distribution of various nAChR subtypes andsubunit mRNA has been fairly extensively character-ised in rodent and chick brain but has been less wellcharacterised in human brain. The majority of studiesperformed to date, have mapped the distribution ofhigh a�nity AChR sites utilising �3H� nicotine, �3H�epibatidine and �3H� cytisine with some additional stu-dies mapping �125I� aBTX binding sites. A few studiesmapping the distribution of nAChR subunit mRNA inhuman brain have also been performed, predominantlyusing isotopic in situ hybridisation. However, despitethe incomplete nature of the studies performed to date,it is possible to draw a map of nAChR distribution in
the human brain Table 4 and 5). Nicotinic receptorsare present in a variety of brain structures, in particu-lar the thalamus, cortex and the striatum. This distri-bution of receptors is consistent with that described inthe human brain with PET, a non-invasive imagingtechnique (see Section 4.1).
3.1. Ligand binding studies
High a�nity nicotinic AChR sites as mapped by thebinding of �3H� nicotine in both homogenate and auto-radiographic studies displays a distinct pattern in thehuman brain. Highest levels of binding are observed inthe thalamus and nucleus basalis of Meynert (NBM)with relatively lower levels in the hippocampus, cortex
Table 4
Distribution of nicotinic receptors in human brain
Region
[3H� nicotineaHigh density Thalamus, caudate nucleus, substantia nigra
Moderate density Frontal cortex, parietal cortex
Low density Occipital cortex, temporal cortex, hippocampus, cere bellum
[3H� epibatidinebHigh density Thalamus
Moderate density Caudate-putamen, parietal cortex, cerebellum
Low density Frontal cortex, occipital cortex, temporal cortex, hippocampus
[125I� aBTXc
High density Nucleus reticularis, lateral and medial geniculate bodies, pontine nucleus, horizontal limb of the diagonal band of Broca,
NBM, inferior olivary nucleus
Moderate density Hippocampus, hypothalamus, pons, medulla
Low density Cortex, cerebellum
a Data from Adem et al. (1987), Nordberg et al. (1988) and Perry et al. (1992).b Data from Marutle et al. (1998).c Data from Rubboli et al. (1994a), Breese et al., 1997 and Spurden et al. (1997).
Table 5
Distribution of nicotinic receptor subunit mRNA in the human braina
Brain region b2 b3 b4 a3 a4 a5 a7
Cortex + + +
Prefrontal + ++ + ++
Motor + ++ + +++
Entorhinal + ++ + +
Cingular + + +
Temporal + + +(+)
Thalamus + + +
Dorsomedial + +++ ++
Lateroposterior +++
Reticular ++ +(+)
Ventro-posterolateral + +++
Geniculate bodies ++
Hippocampus +(+) + ++
Dentate gyrus +(+) + ++
Caudate putamen +(+) + + + ++
Cerebellum + + + + +(+) +
a Data from Breese et al. (1997), Court and Clementi (1995), HellstroÈ m-Lindahl et al. (1998, 1999), Rubboli et al. (1994a), SchroÈ der et al.
(1995) and Wevers et al. (1994).
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11188

Fig. 3. Autoradiographic distribution of: (a) �3H� (-) nicotine; (b) �3H� cytisine and (c) �3H� epibatidine binding in the cerebral cortex of a human
hemisphere. Pseudocolour images are not standardized to each other, as the series of autoradiograms for each ligand was created individually.
The density of binding sites increase in the colour sequence black, blue, yellow, red. Abbreviations: cs, central sulcus (BA3b; BA4); ifrs, inferior
frontal sulcus (BA6); mfrs, medial frontal sulcus (BA8); sfrs, superior frontal sulcus (BA9); oc, occipital cortex (BA18); sts, superior temporal sul-
cus (BA39); pc, parietal cortex (BA40). Reprinted from Neuroscience, Sihver et al., 1998a Copyright (1998), with permission from Elsevier
Science.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 89

and caudate putamen (Shimohama et al., 1985; Ademet al., 1988, 1989; Perry et al., 1992; Court et al.,1995). High levels of binding are observed in the ma-jority of thalamic nuclei with greatest density observedin the lateral dorsal nuclei, medial and lateral genicu-late nuclei and anterior thalamic nuclei (Spurden et al.,1997; Adem et al., 1988). Cortical AChRs are concen-trated in the entorhinal cortex and the subicular com-plex (Court and Clementi, 1995). However, regionaldi�erences in �3H� nicotine binding are observedthroughout the cortex, with distinct laminar distri-bution of ligand in individual regions. In the somato-sensory cortex, highest levels are observed in theuppermost and innermost layers with signi®cantly lessbinding observed in layer IV. In contrast, in the pri-mary motor and temporal cortices, ligand binding wasobserved to be considerably denser in the outer layersthan in the inner layers with a distinct high densityband observed in layer III of the temporal cortex(Perry et al., 1992). Greatest levels of cortical �3H� nic-otine binding are observed in the subicular complex, inparticular, the presubiculum and entorhinal cortex(Perry et al., 1992). In the hippocampus �3H], nicotinebinding is generally low in the CA1-4 and dentategyrus, but is greater in the lacunosum moleculare inCA2-3 (Perry et al., 1992). This distribution of higha�nity nicotinic AChR sites in human brain hasrecently been con®rmed by autoradiographic studiescomparing �3H� nicotine and �3H� epibatidine (Marutleet al., 1998; Sihver et al., 1998a). Two high a�nitybinding sites in the temporal cortex, thalamus and cer-ebellum of human brain have been identi®ed with �3H�epibatidine (Houghtling et al., 1995; Marutle et al.,1998). These sites are likely to represent binding toa4b2 and a3 nAChRs. However, di�erences in regionalbinding between �3H� nicotine and �3H� epibatidinewere observed with a proportionally higher level of�3H� epibatidine binding in the thalamus and cerebel-lum, possibly re¯ecting selectivity for di�erent nAChRsubtypes between nicotine and epibatidine Ð i.e.greater selectivity of �3H� epibatidine for a3 nAChRs.Thalamic nAChRs have also been mapped in thehuman brain with �3H� nicotine and �3H� ACh (Ademet al., 1988) with high number of binding sitesobserved in the antero-ventral and dorsomedial thal-amic nuclei and low numbers of binding sites observedin the postero-lateral and postero-lateral-ventral nuclei.The pattern of nAChR sites in the cortex of humanbrain with regard to their laminar distribution has alsorecently been determined with the use of �3H� nicotine,�3H� cytisine and �3H� epibatidine and autoradiographicanalysis of whole human brain hemispheres (Fig. 3 . . .(Sihver et al., 1998a). The laminar distribution of allthree ligands was generally similar to the highest levelsof binding observed in layers I, III and V, and particu-larly high levels observed in layer III of the primary
sensory motor cortex and inferior frontal sulcus.However, examination of the regional distribution ofthe three ligands suggests the presence of three di�er-ent binding sites within the human cortex. The ®rstsite is thought to be a common site for �3H� nicotine,�3H� cytisine and �3H� epibatidine, and is likely to rep-resent binding to a4 subunits in the brain. The mor-phological distribution of �3H� nicotine and �3H�epibatidine indicate that they bind to an additional sitespeci®cally noticeable in the primary motor cortex,layer IIIb of the occipital corex and layer V of the su-perior temporal sulcus, as their binding in theseregions is signi®cantly greater than that of �3H� cyti-sine. The high levels of �3H� nicotine binding observedin layers I and VI of the primary motor cortex, deeperlayer V of the primary sensory cortex, layer III of thesuperior temporal sulcus and layer VI of the parietalcortex suggest the presence of a third site (Sihver etal., 1998a). Although more detailed, these observationsare generally consistent with the regional laminar dis-tribution of �3H� nicotine binding sites as described byPerry et al. (1992). Although relatively few �125I� aBTXbinding studies have been performed on human brain,comparison of �125I� aBTX and �3H� nicotine auto-radiography reveals a distinct pattern of binding forthe two ligands (Table 4). A single high a�nity �125I�aBTX site is identi®ed in human brain, with the high-est density of binding observed in the hippocampuscontrasting to the relatively sparse concentration of�3H� nicotine binding sites in this region (Rubboli etal., 1994a; Court and Clementi, 1995; Breese et al.,1997; HellstroÈ m-Lindahl et al., 1999). �125I� aBTX and�3H� nicotine also show a distinct pattern of distri-bution in the thalamus. A recent study has comparedthe relative distribution of the two ligands in this brainregion (Spurden et al., 1997). Consistent with previousreports �3H� nicotine binding was high in the majorityof thalamic nuclei, particularly the lateral dorsal, me-dial geniculate, lateral geniculate and anterior nuclei.In contrast, the relative level of �125I� aBTX binding islower in all of these nuclei, with the highest level ofbinding observed in the reticular nucleus, NBM andthe horizontal limb of the diagonal band of Broca(Breese et al., 1997).
3.2. Subunit mRNA distribution
A small number of studies mapping nAChR subunitmRNA distribution in human brain (Table 5) havebeen performed mainly utilising in situ hybridisation(Rubboli et al., 1994a, 1994b; Wevers et al., 1994;SchroÈ der et al., 1995; Court and Clementi, 1995;Breese et al., 1997; HellstroÈ m-Lindahl et al., 1998,1999). The majority of investigations thus far haveconcentrated on the distribution of a7, a4, a3 and b2mRNA, with apparently only one study additionally
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11190

examining a5 and b3 and b4 distribution in the postmortem human brain (HellstroÈ m-Lindahl et al., 1998).The majority of studies compared subunit distributionto the pattern of �3H� nicotine and �125I� aBTX bindingsites and have generally reached similar conclusions.Consistent with the theory that a4b2 nAChRs consti-tute the predominant subtype present in the brain, thedistribution of b2 mRNA is fairly homogeneous.Moderate to low amounts are observed in most brainregions including the cortex, thalamus, caudate-pu-tamen and hippocampus. Distribution of subunitmRNA in di�erent cortical layers has been examinedto a limited extent (SchroÈ der et al., 1995) and is gener-ally similar to the pattern of �3H� nicotine binding.Observations indicate that a3 mRNA is most abun-dant in the thalamus (e.g. dorsomedial, lateroposterior,reticular, and ventroposterolateral nuclei), is present inlow to moderate amounts in most cortical regions (e.g.prefrontal, motor, entorhinal, cingular), and hippo-campus, and absent in the caudate-putamen (Rubboliet al., 1994a; Court and Clementi, 1995). The patternof a3 mRNA in the thalamus corresponds to that of�3H� nicotine and �3H� cytisine (which is almost identi-cal) but is distinct from binding in the hippocampus(Court and Clementi, 1995). In the cortex, a3 mRNAis most predominantly expressed in pyramidal neuronslayers III±VI, moderately expressed in layer II andminimally expressed in layer IV (Wevers et al., 1994;SchroÈ der et al., 1995). In another study, a3 mRNAwas observed to be evenly distributed in the parietalcortex, frontal cortex and hippocampus, but signi®-cantly lower in the temporal cortex and cerebellum(HellstroÈ m-Lindahl et al., 1999). In contrast, the ex-pression of a4 mRNA observed in the same study, wassigni®cantly higher in the temporal cortex and cerebel-lum compared to the other brain regions. The distri-bution of a4 mRNA in the neocortex is morewidespread than that of a3, but both are associatedwith pyramidal neurons (Court and Clementi, 1995).In the frontal cortex a4, mRNA is abundant in alllayers except I and IV (SchroÈ der et al., 1995). b2mRNA shows a strong signal in the insular cortex, thegranular layer of the dentate gyrus and the CA2/3region of the hippocampus, with a signal of lowerintensity observed in the subicular and entorhinal cor-tex (Rubboli et al., 1994b). High levels of b2 mRNAhave also been reported in the cortex and cerebellumof prenatal and aged brain (HellstroÈ m-Lindahl et al.,1998). In contrast, Court and Clementi, (1995), reportonly moderate levels of b2 mRNA in the dentate gyrusand the CA2/3 of the hippocampus, with a pattern dis-tinct to that of �3H� nicotine binding. b2 mRNA is alsopresent in the striatum but is not so predominant inthe thalamus. In a study comparing the regional ex-pression of a7 mRNA and �125I� aBTX binding inhuman postmortem brain, Breese et al. (1997)
observed the reticular nucleus of the thalamus, the lat-eral and medial geniculate bodies, the horizontal limbof the diagonal band of Broca, and the NBM asregions with high levels of both a7 mRNA and �125I�aBTX binding. Moderate levels of a7 mRNA wereobserved in the cortex and hippocampus where �125I�aBTX binding did not correlate so well to probe sig-nal. However, in the majority of brain regions �125I�aBTX binding and a7 mRNA were localized to thesame cell bodies. These observations are supported byRubboli et al. (1994b), who report high levels of a7mRNA in the dentate gyrus, CA2/3 of the hipocam-pus, certain thalamic nuclei and the caudate nucleus, adistribution which was matched by the pattern of �125I�aBTX binding. In the frontal cortex, a7 mRNA ishigh in layers II and III, moderate in layers V and VIand low in layers I and IV (SchroÈ der et al., 1995). a5,b3 and b4 subunit mRNA distribution has been stu-died in prenatal and aged human post mortem brain(HellstroÈ m-Lindahl et al., 1998). These subunits can bedetected in the spinal cord, medulla oblongata, pons,cerebellum, mesencephalon, subcortical forebrain andcortex and thus, have a fairly widespread distribution.a5 was most abundant in the cortex, whilst b3 washighest in the cerebellum. In comparison, mRNAlevels for a5 and b4 subunits were signi®cantly lowerin aged cortex and cerebellum.
4. Imaging of nicotinic receptors with PET and SPECT
Quantitative imaging of functional and pathologicalprocesses in the living mammalian brain has recentlybecome feasible through the development of PET(positron emission tomography) and SPECT (singlephoton emission computed tomography) imaging tech-niques. PET and SPECT are non-invasive tomographicmethods for imaging the regional distribution of radio-active tracers. PET utilises tracers labelled with posi-tron emitting radionuclides such as �11C], �13N� and�18F], while SPECT utilises g or photon embodyingradioisotopes such as �123I� and �99mTc]. Followinginjection of radiotracer, brain distribution of radioac-tivity is detected by means of externally placed banksof paired detectors that register co-incidental energy(in the form of 2g rays) from the annihilation ofemitted positrons with electrons in the case of PETand by use of a rotating g camera or multiple detectorrings in the case of SPECT. These techniques havebeen successfully used to map cerebral metabolic func-tion and blood ¯ow, as well as a number of receptorsystems in the living human brain. With the use ofthese techniques, the distribution and binding of nic-otinic receptors in the human brain can be studied invivo. This is of particular interest when considering theinvolvement of these receptors in the pathology of neu-
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 91

rodegenerative conditions such as Alzheimer's disease,Parkinson's disease, numerous pathological conditionssuch as epilepsy, schizophrenia and depression, andtheir implication in neurological processes such aslearning and memory. Brain imaging of nicotinicreceptors therefore o�ers further understanding of theinvolvement of these receptors in pathological con-ditions as well as insight into their role in the normalfunctioning of the brain.
4.1. PET
The development of methods for the synthesis ofradiolabelled nicotine (Maziere et al., 1976; LaÊ ngstroÈ met al., 1982; Halldin et al., 1992) has allowed theuptake and distribution of nicotine in the brain of ani-mals and man to be studied. Following studies inmice, in which injection of �3H� nicotine producedbrain uptake and distribution of radioactivity consist-ent with nAChR density (Broussolle et al., 1989), �11C�nicotine was developed and used in rhesus monkeysand then in humans to map the distribution of nic-otinic receptors in the living brain. (Nordberg et al.,1989b, 1990; NybaÈ ck et al., 1989). The distribution of�11C� radioactivity measured with PET was generallyconsistent with the known pattern of nAChRsmeasured by in vitro binding in autopsy brain tissue(Nordberg et al., 1989a). High levels of radioactivitywere observed in the thalamus and caudate nucleus,moderate levels in the frontal and temporal parietalcortices and in the cerebellum with low levels in whitematter tracts. �11C� nicotine has been used to imagenAChRs in Alzheimer patients (Fig. 4) and hasrevealed signi®cant reductions in uptake of �11C� nic-otine in the frontal and temporal cortices of thesepatients when compared to healthy, aged, matchedvolunteers (Nordberg et al., 1990, 1995). These obser-vations con®rmined earlier postmortem ®ndings(Nordberg et al., 1989a). Additionally, a positive corre-lation was observed between uptake of tracer into tem-poral cortex and cognitive performance in Alzheimer'spatients. These observations indicate the viability of�11C� nicotine and PET as a diagnostic tool inAlzheimer's disease. However, a number of factorsmake �11C� nicotine a less than ideal ligand for theimaging of nicotinic receptors in vivo, in man. It dis-plays high levels of non speci®c binding, rapid metab-olism and rapid washout of the brain (Grunwald etal., 1996). The heterogeneity of �11C� nicotine bindingin the brain also precludes the identi®cation of a refer-ence region which may be used to accurately determinenon speci®c binding. Furthermore, the study of(NybaÈ ck et al., 1994) suggests that brain uptake andretention of �11C� nicotine may not be entirelymediated by speci®c binding to nAChRs, with the sug-gestion that brain distribution of the tracer may be
in¯uenced to a signi®cant degree by cerebral blood
¯ow. However, kinetic studies of �11C� nicotine uptake
into the brain of Alzheimer's patients involving com-partmental modelling in which the e�ect of blood ¯ow
was considered allowed calculation of a kinetic rate
constant k�2 expressing �11C� nicotine binding
(Nordberg et al., 1995, 1997). A study performed in
monkeys con®rmed that the calculated k�2 constant was
blood ¯ow independent (Lundqvist et al., 1998). A sig-
ni®cant reduction in k�2 was observed in the temporaland frontal cortices and the hippocampus of AD
patients compared to age matched controls (Nordberg
et al., 1995, 1997). Additionally, a signi®cant corre-
lation was observed between cognitive status and �11C�nicotine binding (expressed as k�2� in the temporal cor-
tex in AD patients (Nordberg et al., 1997). However,
the drawbacks involved in the use of �11C� nicotine asa tracer for the imaging of nicotinic receptors has led
in the last few years to the search for new ligands (see
Fig. 5) with more suitable pro®les i.e. high speci®c to
non speci®c binding ratio, longer retention (slower
washout) from brain and less rapid metabolism. In ad-
dition, ligands with nicotinic receptor subtype speci-
®city are also of interest, speci®cally, a ligand with
selectivity for a4b2 receptors which are recognised tobe the predominant subtype lost in Alzheimer's dis-
ease. Initially, interest centred upon the development
of the nAChR agonist cytisine as a PET radiotracer,
but in vivo studies in rodents with �3H� cytisine indi-
cated that although brain retention was longer than
that of �11C� nicotine, it displayed low blood brain bar-
rier penetration making it a poor candidate ligand.PET studies with �11C� ABT 418 and N-[11C� methylcy-
tisine (high a�nity nAChR agonists) have also been
attempted in baboons (Valette et al., 1997). However
both ligands displayed low levels of brain uptake,
rapid washout from brain and rapid metabolism.
More favourable results were forthcoming with a series
of �3H� labelled nicotine analogues, including �3H]-(R,S)-5-isothiocyanonicotine and �3H]-(R,S)-5-aminoni-
cotine, which have been evaluated in vitro and in vivo
and appear to possess appropriate attributes for poten-
tial PET ligands (Kim et al., 1994). The recent discov-
ery of the potent high a�nity nicotinic receptor
agonist epibatidine provided a potentially excellent
ligand for the in vivo imaging of nAChRs. In vivo inrodents �3H� epibatidine and the epibatidine analogue
�3H� norchloroepibatidine display regional brain local-
ization consistent with the known pattern of high
a�nity nAChR sites (i.e. thalamus > cortex >
cerebellum), high speci®c to non speci®c binding and
slow clearance from the brain (London et al., 1995;
Sche�el et al., 1995). This favourable pro®le in rodentshas led to the development of positorn labelled ana-
logues of epibatidine potentially suitable for use as
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11192

PET ligands in man. A number of these compoundsand other novel nAChR ligands will now be discussed.
4.1.1. [18F� NFEP or �18F� FPH[18F� ¯uoronorchloroepibatidine ((2)exo-(2-[18F�
¯uoro-5-pyridyl)-7-azabicyclo[2.2.1] heptane) is a �18F�2 ¯uoro pyridyl analog of epibatidine that has beennamed as both �18F� FPH and �18F� NFEP, and hasbeen assessed as a nAChR PET ligand via ex-vivoautoradiography in mouse brain, in vitro autoradiog-raphy in human brain tissue and with PET in the liv-ing baboon brain (Horti et al., 1997; Villemagne et al.,1997; Gatley et al., 1998). Its potential as a possiblePET ligand for use in man was initially indicated bypromising preliminary studies performed with �3H�NFEP (Sche�el et al., 1995). Ligand distribution inboth mouse and human brain indicates binding
consistent with nAChR distribution as mapped by�3H� nicotine, �3H� cytisine and �3H� epibatidine(Gatley et al., 1998). Following injection in mice�18F� FPH=�18F NFEP� displays rapid uptake and a het-erogeneous distribution in the brain with preinjectionof a range of nAChR ligands including nicotine,cytisine and lobelline inhibiting tracer binding in allbrain regions except the cerebellum, where no e�ectwas observed. In contrast, the preinjection of the noncompetitive nAChR antagonist mecamylamine, themuscarinic agonist scopolamine, the dopamine recep-tor agonist apomorphine, and the 5-HT receptor an-tagonist ketanserin had no e�ect on radioligandbinding in any brain region, con®rming that speci®cbinding to nAChRs accounts for radioligand distri-bution in the brain (Horti et al., 1997). PET analysisin baboon indicated that �18F� FPH=�18F� NFEP displays
Fig. 4. (S)(-)[11C� Nicotine uptake in the brain of two Alzheimer's patients. Horizontal PET images show the distribution of (S)(-)[11C� nicotine in
the brain of two AD patients at: (A) the level of the basal ganglia and (B) the frontal association cortex±parietal cortex. The ®gures represent a
summation picture of the brain uptake of �11C� radioactivity following an intravenous injection of a tracer dose of (S)(-)[11C� nicotine. ``Hot'' col-
ours represent areas with high tracer uptake and ``cold'' colours represent areas with low uptake. One patient shows right side de®cits (upper sec-
tions), while the other shows left side de®cits (lower sections) in (S)(-)[11C� nicotine brain uptake (indicated by the arrows). Reduced (S)(-)[11C�nicotine uptake in these areas is likely to correspond to a loss of nAChRs.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 93

rapid uptake and heterogeneous distribution in thebrain which correlates well with that observed inmouse (Villemagne et al., 1997). Furthermore, adminis-tration of unlabelled cytisine signi®cantly reduced�18F� FPH=�18F� NFEP levels in the brain indicating thatbrain retention of tracer is mediated by speci®c bind-ing to nAChRs, possibly of the a4b2 subtype.�18F� FPH=�18F� NFEP therefore has excellent potentialfor use as a PET ligand in man. However, norchloroe-pibatidine exhibits relatively high toxicity which maypreclude extensive use of �18F� FPH=�18F� NFEP as aPET agent in man (Gatley et al., 1998).
4.1.2. [18F� A-85380 and �11C� A-8548Recently, a number of novel compounds with subna-
nomolar a�nity for nAChRs have been synthesizedfor the treatment of Alzheimer's disease (Abreo et al.,1996). Among these compounds are A-85380 (3-(2-(S)-azetidinylemthoxy)pyridine) and A-85453 (3-[(1-methyl-2(S)-pyrrolidinyl)methoxy]pyridine). Both ofthese compounds display an a�nity for a4b2 receptors
equal to that of epibatidine, but signi®cantly lower a�-nity than epibatidine for a7 receptors. Additionally, A-853580 is respectively 40 and 100 times less potentthan epibatidine in activating a4b2 and a3b4 subtypes.(Sullivan et al., 1996). It has therefore been hypoth-esised that these compounds would be suitable PETligands as they may selectively label a4b2 over a7receptors and have a greater margin of safety betweenadequate tracer dose and dose producing biologicale�ect compared to ligands such as�18F� FPH=�18F� NFEP]. �18F� A-85380 and �11C� A-85453have recently been synthesized and their brain uptakeand distribution assessed in mice via ex vivo auto-radiography (Horti et al., 1998; Kassiou et al., 1998).Following administration, both ligands displayed highuptake into the brain with radioactivity peaking afterapproximately 5 min with a slow washout observedthereafter. A high speci®c to non-speci®c binding ratiowas observed with regional distribution of radioac-tivity consistent with nAChR density. Pre-adminis-tration of non-radiolabelled nicotinic ligands such as
Fig. 5. Structure of (S)[11C� nicotine and a selection of �11C], �18F� and �123=125I� radiolabelled nAChR ligands developed for PET and SPECT.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11194

nicotine, cytisine and epibatidine signi®cantly reducedbinding of both radioligands in all brain areas with theexception of the cerebellum where observed e�ectswere minimal. In contrast, administration of the non-competitive ligand mecamylamine had no e�ect onbrain tracer level, nor did the administration of a num-ber of non-nicotinic ligands, thus indicating that brainretention of these ligands is mediated by speci®c bind-ing to nAChRs (Horti et al., 1998; Kassiou et al.,1998). These results suggest that both �18F� A-85380and �11C� A-85453 deserve further investigation aspossible PET radiotracers.
4.1.3. [11C� MPAA further potential PET radiotracer is �11C� MPA
((R,S)-1-methyl-2-(3-pyridyl) azetidine). Initial in vitroevaluation of this compound in rodent brain indicatedthat it displayed characteristics suitable for a PETligand (Sihver et al., 1998b) and recent preliminaryPET studies comparing it to �11C� ABT 418 and �11C�nicotine in rhesus monkeys appear to con®rm this(Sihver et al., 1999b). Uptake of �11C� MPA into thebrain was rapid following injection, similar in extent to�11C� ABT 418 and �11C� nicotine. Pre-injection of ani-mals with nicotine resulted in 25% reduction �11C�MPA uptake in the thalamus, a 19% reduction in thetemporal cortex and an 11% reduction in the cerebel-lum, indicating that brain retention of �11C� MPA innAChR mediated. In contrast, nicotine pre-treatmentwas observed to produce increases in �11C� ABT 418and �11C� nicotine brain uptake, suggesting that �11C�MPA may be a more suitable PET ligand fornAChRs.
4.1.4. [76Br� BAPIn a similar manner to �11C� MPA, �76Br� BAP (5-
[76Br]-bromo-3-((2(s )-azetidinyl) methoxy)pyridine) hasbeen evaluated in vitro and in vivo in rats with pre-liminary studies additionally performed in rhesusmonkeys (Sihver et al., 1999b). With in vitroautoradiographic analysis, highest levels of bindingwere observed in the thalamus and presubiculum withmoderate levels observed in the cortex and striatumand low levels observed in the hippocampus and cer-ebellum. Ex-vivo autoradiographic analysis followinginjection into rats revealed a similar regional bindingpattern, with brain uptake blocked by preinjection ofnicotine. In rhesus monkeys, PET evaluation revealedhigh levels of tracer retention in the thalamus, up to60% of which could be displaced by preinjection withcytisine and 50% by preinjection of nicotine. The highlevels of speci®c uptake observed for this ligand in ratand monkey brain suggest a promising PET ligand.
4.2. SPECT
Development of SPECT agents for the imaging of
nicotinic receptors holds a number of advantages over
PET. The greater availability of SPECT (mainly due
to its lower cost), the longer half-life of SPECT ligands
(hours as opposed to minutes) and the commercial
availability of the appropriate radioisotopes make
SPECT a cheaper and easier imaging alternative.
However, despite the availability of �11C]-nicotine and
the development of a number of epibatidine analogs
and synthetic compounds for use with PET, the num-
ber of SPECT ligands currently available and in devel-
opment is relatively few. A radioiodinated form of
nicotine ([125I]-(s)-nicotine) has been developed
(Kampfer et al., 1996) and evaluated in rats (Saji et
al., 1995). But as with its �11C� niciotine PET counter-
part, it su�ers from substantial non-speci®c binding.
Only recently the radioiodinated compounds �125=123I�IPH ((2)-exo-2-iodo-5-pyridyl)-7-azabicyclo[2.2.1] hep-
tane) and �125I� 5-I-A-85380 have been developed and
evaluated (Musachio et al., 1997, 1998; Vaupel et al.,
1998). Following intravenous injection in mice �125I�IPH displays similar qualities to its PET counterpart
�18F� FPH, with high levels of brain uptake observed,
high speci®c to non-speci®c binding and regional dis-
tribution appropriate for a nicotinic receptor ligand.
Additionally, pre-administration of unlabelled IPH,
nicotine and cytisine blocked receptor binding, an
e�ect not produced by pre-administration of scopola-
mine, ketaserin or mecamylamine. SPECT analysis of
�123I� IPH in baboon produced similar observations.
Tracer uptake was rapid, reaching a plateau after ap-
proximately 45 min with cortical, subcortical and cer-
ebellar structures all identi®able. Observed activity was
highest in the thalamus, moderate in the cortex and
low in the cerebellum. Subsequent injection of 1 mg/kg
cystine signi®cantly reduced brain levels of tracer with
the thalamus showing the most profound e�ect, indi-
cating that the ligand binds speci®cally to nAChRs
(Musachio et al., 1997). �125I� 5-I-A-85380 has been
evaluated in mice via ex vivo autoradiography (Vaupel
et al., 1998) and preliminary SPECT studies with �123I�5-I-A-85380 have also been performed in baboons
(Musachio et al., 1999). �125=123I� 5-I-A-85380 shows
similar characteristics to its IPH and its PET counter-
part. High brain uptake is observed with regional dis-
tribution of radioactivity consistent with nAChR
density. Although further characterisation is necessary,
both of these compounds have excellent potential for
use as SPECT agents in man and with the advantages
of SPECT over PET will perhaps provide the most
convenient method of imaging nicotinic receptors in
pathological states such as Alzheimer's diseases.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 95

5. Nicotinic receptor function in the CNS
5.1. Functional and behavioural e�ects of nicotine
Nicotine is a potent modulator of CNS function. Itenhances ion ¯ux and neurotransmitter release, aug-ments or gates of a number of neuronal systems andelicits a variety of behavioural states. Nicotine admin-istration produces a number of physiological e�ectsincluding increased heart rate and blood pressure anddose dependent increase in the secretion of prolactinand ACTH, resulting in a subsequent increase in corti-costerone secretion (Newhouse et al., 1990; Benowitz,1996). Speci®c CNS e�ects of nicotine include EEGdesynchronisation, producing a shift in the direction ofhigher frequency (Edwards and Warburton, 1982),increased cerebral blood ¯ow and increased cerebralglucose utilization through stimulation of nAChRs inthe basal forebrain; e�ects which can be blocked bythe nicotinic antagonist mecamylamine (Pickworth etal., 1988; London, 1990; Linville and Arneric, 1991;Linville et al., 1993). In humans, nicotine increasesarousal, visual attention and perception (Jones et al.,1992), but also decreases reaction time, prevents adecline in e�ciency over time and improves the abilityto withold inappropriate responses (Jones et al., 1992;Wesnes and Warburton, 1983). In both smokers andnon-smokers, nicotine produces an improvement in thespeed and accuracy of motor function (Hindmarch etal., 1990; West and Jarvis, 1986) and improves per-formance in complex psychomotor tasks such as cardriving. However, its withdrawal worsens performanceand other vigilance tasks (Heimstra et al., 1967, 1989).This e�ect may be dose dependent with large doses ofnicotine worsening car driving (Sherwood, 1995).Despite the complex e�ects elicited by nicotine, theexact role of brain nAChRs remains unclear. A signi®-cant body of evidence suggests that presynapticnAChRs exist on several cell populations in cortical,hippocampal and cerebellar brain regions (Wonnacott,1997). Nicotine interacts with a variety of presynapticnAChRs to facilitate the release of a number of neuro-transmitters including ACh, DA, NA, 5-HT, GABAand glutamate, many of which have been implicated inmediating/modulating a number of behavioural tasks(de Sarno and Giacobini, 1989; Wonnacott et al.,1990; McGehee and Role, 1995). Therefore, it hasbeen proposed that the role of nAChRs in the brain isto modify the excitability of neurons, that is to pro-duce the optimal performance of neurons by adjutingtheir excitability, an action which is likely to be of im-portance in a number of behavioural responses andparticularly in cognitive processes (McGehee and Role,1995). The most obvious behavioural action mediatedby nAChRs is the addiction to nicotine in tobaccosmoke. The mesolimbic DA pathway is thought to
mediate the addictive e�ects of nicotine and other sub-stances of abuse, with nicotine known to stimulaterelease of DA in this pathway (Pich et al., 1997).Addiction to nicotine is motivated by both positiveand negative reinforcing factors (Benowitz, 1996).Positive factors are relaxation, reduced stress,increased vigilance, improved cognition and reducedbody weight (LindstroÈ m, 1997). Enhanced release ofmesolimbic DA through activation of presynapticnAChRs may feasibly mediate these rewarding e�ects.Negative reinforcing factors are unpleasant withdrawalsymptoms including nervousness, restlessness, irritabil-ity, anxiety, impaired concentration and cognition andweight gain, and may result from reduced stimulationof mesolimbic DA neurons (LindstroÈ m, 1997). In arecent functional MRI study (Stein et al., 1998), nic-otine administration was observed to produce a dosedependent increase in several rewarding behaviouralparameters and to increase neuronal activity in limbic-cortical structures including the amygdala, nucleusaccumbens, and cingulate and frontal cortices. Thesestructures are consistent with nicotines behaviourarousing and behaviour reinforcing properties inhumans and have previously been identi®ed to partici-pate in the reinforcing, mood elevating and cognitiveproperties of other abused drugs such as cocaine, am-phetamines and opiates (Stein et al., 1998). The mech-anisms by which nicotine produces tolerance andaddiction and identi®cation of the receptor subtype(s)which mediate these e�ects is complicated by a numberof factors including the ability of nAChRs to be acti-vated by acute exposure to agonists, to be reversiblydesensitized on longer exposure, and to be perma-nently deactivated following prolonged exposure withthe additional result of increasing nAChR number(LindstroÈ m, 1997). Furthermore, identifying whiche�ects are the result of activation and which of inacti-vation of nAChRs is unclear. However, the rewardinge�ects of nicotine can be blocked by pre-adminis-tration of the nAChR channel blocker mecamylamine,indicating that activation of nAChRs is important inpositive reinforcement and self administration(Henning®eld, 1984). The development of tolerance tonicotine exhibited by smokers and the adverse e�ectsof nicotine on naive users is more obviously explainedby the reversible desensitization of nAChRs followingexposure to nicotine and the permanent inactivation ofnAChRs, despite upregulation of numbers followingprolonged exposure to nicotine (LindstroÈ m, 1997).
5.2. Role of nicotinic receptors in cognitive and memoryfunctions
A considerable body of evidence exists to suggestthat nicotine and nicotinic agonsits have cognitive andmemory enhancing properties in animals and humans,
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11196

while antagonists such as mecamylamine impair mem-ory function (Levin and Simon, 1998). In rodents andnon-human primates, short and long term treatmentwith nicotinic agonists improve performance on a var-iety of memory tasks. Short term nicotine treatmenthas been shown to improve working memory perform-ance in a number of experimental studies on rats(Levin et al., 1993; Decker, 1995; Decker et al., 1995)and to improve memory in delayed matching tosample tasks in monkeys (Buccafusco and Jackson,1991). This mnemonic improvement is also observedfollowing administration of the nAChR agonists lobel-line, dimethylethsanolamine (DMEA), ABT-418 andGTS-21 (Decker et al., 1993, 1994; Levin et al., 1995).However, contradictory evidence indicating that nic-otine has no e�ect or even a detrimental e�ect onmemory performance in animals has been reported(Dunnett and Martel, 1990). Acute nicotine inducedmemory improvements can be blocked by the concur-rent administration of either nicotinic or muscarinicantagonists suggesting a possible role for muscarinicreceptors in mediating the mnemonic e�ects of nicotine(Newhouse et al., 1997). Long term nicotine treatmenthas also been shown to improve memory in animalstudies with surprisingly no development of tolerance(Levin et al., 1990, 1993; Levin and Torry, 1996).Interestingly, improvements in memory persisted forup to 2 weeks after drug withdrawal, although themechanisms by which this e�ect occurs are unclear.Long term nicotine induced memory facilitation isinhibited by concurrent administration of mecamyla-mine but not by short term mecamylamine injectionduring nicotine infusion (Newhouse et al., 1997).When administered alone, mecamylamine causes work-ing memory impairments (Andrews et al., 1994;Oliverio, 1966). Nicotine and nicotinic agonsits alsoreverse memory de®cits in brain lesion studies (Deckeret al., 1992, 1994; Levin et al., 1993; Muir et al., 1995;Grigoryan et al., 1996) and age related memory de®citsare improved by bolus nicotine administration(Arendash et al., 1995; Socci et al., 1995; Levin andTorry, 1996). Despite the large number of studies per-formed, the cognitive e�ects of nicotine on humansremain to be fully elucidated. Di�erent means of ad-ministration, di�erent doses and the participation ofsmoking and non-smoking subjects in studies haveresulted in di�culties in interpretation of results.Many studies have used tobacco smoking as a meansof nicotine administration, ignoring the fact that nic-otine is unlikely to be the only active substance intobacco smoke. It should also be noted that the testsubjects participating in these studies were oftentobacco smokers who had been deprived of tobaccofor some time. Under these conditions, nicotine hasbeen observed to improve performance in a variety ofcognitive and mnemonic tasks. However, it is unclear
whether the improvements observed represents a pri-mary action of nicotine or merely a return to pre-deprivation level of performance. Interpretation of ob-servations from this type of deprivation study isfurther complicated by the fact that chronic tobaccosmoking increases the number of high a�nity nAChRsin various brain areas (Hindmarch and Sherwood,1995; Stolerman and Jarvis, 1995). In addition, nic-otine produces prolonged behavioural e�ects in ani-mals including a signi®cant period following drugwithdrawal, and it is likely that tobacco smokers willbe subject to similar prolonged behavioural e�ects fol-lowing cessation to smoking. Therefore, the cogntiveimprovements produced by nicotine in this type ofdeprivation study may be misleading. Nicotine inducedimprovements in memory and cognitive tasks in non-smokers and non-deprived smokers have been moredi�cult to demonstrate. For example, reports havesuggested that nicotine has no variable and even nega-tive e�ects on memory and learning (Levin, 1992).However, in general it has been accepted that nicotinehas a number of cognitive enhancing actions inhumans. Nicotine increases arousal, visual attentionand perception, and may prevent fatigue induced de®-cits in vigilance and long term performance (Jones etal., 1992; Newhouse et al., 1992). It shortens infor-mation processing time and improves reactions (LeHouezec et al., 1994) and in non-deprived smokers itenhances recognition memory (Rusted et al., 1994).Nicotine is thought to improve short term memory byfacilitating the storage of information received(Warburton et al., 1986; Levin, 1992; Levin et al.,1992) and have a consolidating e�ect on memory(Warburton et al., 1986; Colrain et al., 1992;Newhouse et al., 1995). In complimentary fashion, thenon-competitive nAChR antagonist mecamylamineproduces detrimental e�ects on learning and memory.The studies described above support the theory thatnAChRs play an important role in cognition andmemory. This theory becomes more compelling whenconsidering the signi®cant loss of nAChRs from thehippocampus and frontal cortex observed in AD (twobrain regions important in cognitive function) and thecognitive decline associated with the disease. However,further work is necessary to determine which nAChRsubtypes are involved in cognitive processes and to elu-cidate the interactions of these receptors with otherneurotransmitter systems which are critical in cognitivefunction.
6. Pathology of neuronal nicotinic receptors
6.1. Epilepsy
Epilepsy is a heterogeneous group of disorders that
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 97

a�ects about 2% of the population. Autosomal domi-nant frontal lobe epilepsy (ADNFLE) is a partial epi-lepsy that causes brief seizures that occur during lightsleep that are often mis-diagnosed as nightmares,although most patients also su�er to some degree fromviolent generalized seizures (Sche�er et al., 1995). Ithas recently been recognized that ADNFLE resultsfrom a missense mutation in the a4 subunit gene,replacing a highly conserved serine at position 247 ofthe M2 channel lining domain of the subunit with aphenylalanine (Steinlein et al., 1995). The mutation isthought to impair channel function and whenexpressed in Xenopus oocytes with normal a b2 subu-nit, a mutant receptor is formed that displays reducedCa2+ permeability, reduced channel opening (probablydue to a faster desensitization rate) and slower recov-ery from the desensitized state (Weiland et al., 1996).A second gene mutation involving the insertion of aleucine after position 259 in the extracellular C-term-inal end of the M2 domain of the a4 subunit alsocauses ADNFLE (Steinlein et al., 1995). This mutationis thought to be relatively well tolerated however, hav-ing less impact on receptor function, but as with the247 serine mutation Ca2+ channel permeability isreduced. Studies on reconstituted a4b2 nAChRsexpressed in Xenopus oocytes with the serine 247 mu-tation indicate that these receptors display a decreasein apparent a�nity of ACh of about 7 fold and cur-rents 5 times smaller than controls at saturating con-centrations of ACh. Additionally, these receptorsdesensitize to an agonist concentration 3000 timeslower than controls (Bertrand et al., 1998). As epilepsyresults from excessive neuronal activation, it is some-what contradictory that mutations resulting in reducednAChR function should cause seizures. A possible ex-planation for this lies in the action of presynaptic a4b2receptors to promote the release of many neurotrans-mitters including inhibitory GABA and glycine(Wonnacott et al., 1990; Wonnacott, 1997).Facilitation of GABA or glycine release or activationof such inhibitory neurons by a4b2 receptors may pre-vent the onset of seizures between sleep and wakeful-ness, and thus, reduced receptor function resultingfrom the above mutations may trigger ADFLNE. Afurther link of nAChRs to epilepsy comes from the ob-servation that mice with unusually high numbers of�125I� aBTX binding sites are more susceptible to sei-zures in response to nicotine administration (Marks etal., 1989) suggesting that the a7 nAChR may also playa role in epilepsy.
6.2. Alzheimer's disease
Alzheimer's disease (AD) is a progressive neurode-generative condition a�ecting almost 1 in 10 individ-uals over the age of 65 (Evans et al., 1989). It accounts
for over 50% of senile dementia and the majority ofpre-senile dementia cases, and is characterised by pro-gressive deterioration of higher cognitive functionsincluding the loss of memory (Octave, 1995).Postmortem AD brains display two distinctive neuro-pathological features which constitute conclusive diag-nostic markers for AD: intracellular neuro®briallarytangles and extracellular neuritic senile plaques.Intracellular neuro®brillary tangles accumulate in neur-onal perikarya and consist of paired helical ®lamentscontaining the microtubule associated protein tau(Delacourte and Defossez, 1986). The presence of tauin neuronal cell bodies represents a highly aberrantlocalisation of the protein, as compared to the axonallocalisation observed in normal neurons (Kowall andKosik, 1987). In AD brains, tau is present in tangles ina hyperphosphorylated form (Grundke-Iqbal et al.,1986). The amount of phosphorylated tau in AD brainis several 100 fold greater to that in normal brains,making tau an excellent disease marker (Vandermeerenet al., 1993). The neuritic plaques observed extracellu-larly in AD brains contain amyloid peptide ®brils intheir core. These ®brils consist of the amyloid b or A4(bA4) peptide (Glenner et al., 1984). The bA4 peptideis derived from a larger precursor peptide termed theamyloid precursor peptide/protein (APP), a glycosy-lated transmembrane protein with a single membranespanning domain (Kang et al., 1987). APP is normallycleaved within its transmembrane domain yielding asecretory form, the exact physiological role of which isnot entirely understood. The primary neurodegenera-tive e�ects of AD appear to be closely linked to amy-loid production. In addition to these speci®cneuropathological features, AD brains exhibit exten-sive cellular atrophy and cell loss, shrinkage of corticalthickness, enlargement of sulci and ventricles andchanges in multiple neurochemical systems includingACh, glutamate, GABA and 5-HT. However, the mostconsistent and severe neurochemical abnormality as-sociated with AD is the loss of cholinergic innervationof the cerebral cortex and hippocampus (Coyle et al.,1983). ChAT activity is signi®cantly reduced in thecortex and hippocampus of AD brains (Reisine et al.,1978). Post mortem analysis of AD brains reveals thatthe NBM, the major source of cortical and hippocam-pal cholinergic innervation, is degenerated in AD(Whitehouse et al., 1982). In addition, a linear corre-lation between reduced cortical ChAT activity anddegree of dementia has been observed (Perry et al.,1978). Observations such as these prompted (Bartus etal., 1982) to propose the cholinergic hypothesis of AD,which speci®cally attributed the cognitive deteriorationassociated with the disease to the degeneration of thecholinergic pathways from the basal forebrain (nucleusbasalis of Meynert) to the cortex and hippocampus. Inaddition to measurement of ChAT levels, numerous
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±11198

studies characterising changes in cholinergic receptordensity in AD brains have been performed. Marked re-ductions in high a�nity cortical nAChRs have beenobserved in the brains of AD patients compared toage matched controls (Whitehouse et al., 1986; Flynnand Mash, 1986; Nordberg and Winblad, 1986; Aubertet al., 1992; Warpman and Nordberg, 1995).Interestingly, a signi®cant correlation cannot beobserved between histopathological score and numberof nAChRs in autopsy brain cortical tissue (Svenssonet al., 1997), although a signi®cant correlation isobserved between cognition and nAChRs measured invivo by PET (Nordberg et al., 1995). The in¯uence ofbA4 on cholinergic neurotransmission has recentlybeen studied in autopsy brain tissue from subjects car-rying the Swedish APP 670/671 mutation, and in braintissue from sporadic AD cases (Marutle et al., 1999).Signi®cant reductions in nAChR numbers wereobserved in the cortex of the Swedish APP 670/671mutation cases (73±87%) and in the sporadic cases(37±57%) as measured by �3H� epibatidine and �3H�nicotine binding. �3H� epibatidine saturation analysisrevealed two binding sites in the cortex of SwedishAPP 670/671 brains, with a signi®cant decrease (82%)in the number of high a�nity sites and no change inKd observed, compared to control subjects. Besides asigni®cant positive correlation between the number ofneuronal plaques and �3H� nicotine binding sites in theparietal cortex, no strict correlation between nAChRde®cits and neuropathological markers could beobserved in the cortex of Swedish APP 670/671 brains,suggesting that, although these processes may be re-lated, they are not strictly dependent upon one another(Marutle et al., 1999). These observations support theassumption that the nAChR might be impaired veryearly in the course of AD. The most signi®cantchanges are observed in the temporal, parietal andoccipital cortices (Nordberg, 1994). A recent studycomparing �3H� nicotine, �3H� epibatidine, �3H� cytisineand �3H� vesamicol (representing vesicular ACh trans-porter sites) binding observed signi®cant reductions inthe temporal cortex of Alzheimer's patients comparedto aged matched controls for all ligands (Fig. 6),although the reduction in �3H� vesamicol binding wasonly half as much as the reduction observed with thenAChR ligands (Sihver et al., 1999c). Evidence frompostmortem binding studies and transfected humannAChRs indicates that a4b2 nAChRs constitute themajor subtype of nAChRs lost in AD (Warpman andNordberg, 1995). This view is supported by the obser-vation that a4 and b2 subunit mRNA levels decreasewith age in the frontal cortex of human brain, with b2levels additionally reduced in the hippocampus (Tohgiet al., 1998). In a similar study, the level of a3 subunitmRNA in the entorhinal cortex of human brain wasobserved to decrease with age, however no signi®cant
di�erence between mRNA levels in entorhinal cortex,hippocampus and thalamus of AD and age matchedcontrols was measured (Terzano et al., 1998).Furthermore, the mRNA levels of a4, a5, a7, b2, andb4 are signi®cantly higher in prenatal cortex and cer-ebellum compared to aged brain (HellstroÈ m-Lindahl etal., 1998). A signi®cant correlation between reduced�3H� epibatidine sites and reduced levels of the presyn-aptic marker, synaptophysin in the frontal cortex ofAD brains has also been identi®ed, suggesting that themajority of nAChRs lost are presynaptic. Interestingly,in the same study reductions in ChAT activity wereobserved but did not correlate with the reductions in�3H� epibatidine binding measured, suggesting that thenAChRs lost in AD are not exclusive to cholinergicneurons (Sabbagh et al., 1998). This view is supportedby a recent study where basal forebrain lesions withthe selective cholinergic immunotoxin 192 IgG saporinproduced signi®cant reductions in ChAT activity inthe parietal cortex of rats but had no e�ect on nAChRnumbers as measured by �3H� epibatidine and �3H�cystisine (Bednar et al., 1998).
6.2.1. Alzheimer's disease therapyCholinergic transmitter replacement therapy forms
the mainstay of AD treatment and is based on the the-ory that low levels of ACh are responsible for the cog-nitive decline associated with the disease. Classically,replacement therapy has involved the use of cholin-
Fig. 6. Average binding densities of �3H� nicotine, �3H� epibatidine,�3H� cytisine and �3H� vesamicol in the temporal cortex of control (n
= 7) and Alzheimer brains (n = 9). Data are presented as mean
2SEM fmols binding per mg of brain. �P< 0.05 Student's t-test.
(Sihver et al., 1999c).
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 99

esterase inhibitors such as tacrine, donepezil and rivas-tigmine, which prevent the breakdown of ACh releasedfrom cholinergic neurons, thereby increasing the con-centration of transmitter available to interact withreceptors. These drugs have moderate palliative e�ectson symptoms as well as having some ability to slowdisease progression (Amberla et al., 1993; Maltby etal., 1994; Nordberg et al., 1998; Nordberg andSvensson, 1998). Although, only moderately e�ectivecholinesterase therapy currently constitutes the besttreatment available for AD (Nordberg and Svensson,1998). It is likely that the therapeutic bene®t of cholin-esterase inhibitor treatment occurs at least in part,through activation of neuronal nAChRs Ð by directaction of the increased levels of ACh on these recep-tors and through allosteric activation of the receptorsby the drugs (e.g. tacrine, galanthamine) themselves(Maelicke et al., 1995; Svensson and Nordberg, 1996).Neuronal nAChR activation is therefore currentlybeing investigated as a strategy for AD therapy(SjoÈ berg et al., 1998). The potential therapeutic bene®tin AD from nAChR stimulation is based on threemain observations. Firstly, stimulation of nAChRs bynicotine improves mnemonic function. In animal ex-periments, nicotine treatment has been observed toimprove performance in memory related tasks (Levin,1992), furthermore, b2 knockout mice show abnormalbehaviour in avoidance learning indicating the involve-ment of nAChRs (Picciotto et al., 1995). In humans,the nicotinic antagonist mecamylamine producesimpairments in short term memory (Newhouse et al.,1992), while nicotine improves performance of humansubjects in memory related tasks (Colrain et al., 1992;Rusted and Warburton, 1992; Rusted et al., 1994).Nicotine has also been observed to produce similarcognitive improvements in AD patients (Jones et al.,1992; Valenzuela et al., 1994; Vidal, 1996; Newhouseet al., 1997). Secondly, nAChR activation modulatesthe release of a number of neurotransmitters such asACh, DA, GABA and glutamate, and will enhance therelease of ACh (Beani et al., 1985; McGehee and Role,1995; Pontieri et al., 1996; Marshall et al., 1997).Thirdly, there is evidence to suggest that nAChR acti-vation provides protection against b-amyloid neuro-toxicity. Nicotine protects cultured cortical neuronsagainst b-amyloid induced neuronal death, an e�ectblocked by the a4b2 selective antagonist DHbE.Furthermore, cytisine, an a4b2 selective agonist, alsoinhibits b-amyloid toxicity indicating that the a4b2nAChR is important in neuroprotection (Kihara et al.,1998). These observations are consistent with the lossof a4b2 nAChRs in AD, an occurrence which maythus potentiate the neurotoxic action of b-amyloid.Interestingly however, there is evidence to suggest thatstimulation of a7 nAChRs may also be neuroprotec-tive. Nicotine and cholinesterase inhibitors have been
observed to attenuate b-amyloid toxicity, an actionproposed to occur through a7 activation (Kihara etal., 1997; Zamani et al., 1997; Svensson and Nordberg,1998). Nicotinic receptor activation would thereforeappear to be a promising strategy for treatment ofAD. The adverse side e�ects produced by the non-speci®c action of nicotine render it unsuitable as atherapy for AD. There is however, tremendous scopefor the development of a4b2 and a7 selective agonistsas potential therapeutic agents for AD.
6.3. Parkinson's disease
Parkinson's disease (PD) is a progressive neurode-generative condition involving the dopaminergic neur-ons of the substantial nigra. It is characterised bydi�culty in initiating and smoothly sustaining move-ment. In a manner similar to AD there is a loss ofcholinergic cells in the basal forebrain accompanied bya signi®cant reduction in the number of high a�nitynicotine binding sites in the brain (Whitehouse et al.,1983; Aubert et al., 1992; Lange et al., 1993). In ad-dition to motor dysfunction, PD patients often haveaccompanying cognitive impairments or dementia witha greater loss of cholinergic markers and nAChRs indemented patients than non-demented patients (Perryet al., 1995). The reduction in cortical nAChR numberin PD patients parallels the degree of dementiaobserved with progression of the disease (Whitehouseet al., 1988a; Aubert et al., 1992) and as with AD mayresult from degeneration of cholinergic projectionneurons in the basal forebrain. The most potent en-vironmental factor a�ecting susceptibility to PD istobacco use, with smokers having a lower thanexpected incidence of PD (Morens et al., 1995).Although there are many constituents of tobaccosmoke other than nicotine, it appears that it is the bestcandidate as the protective agent. Chronic nicotinetreatment in rodents protects against mechanical andneurotoxin induced nigrostriatal lesions, preventingDA neuronal degeneration, increasing DA levels andcounteracting DA D2 receptor upregulation (Janson etal., 1989, 1994; Fuxe et al., 1990; Janson and Moller,1993). In 1-methyl-4-phenyl-1., 2,3,6-terahydropyridine(MPTP) lesioned mice nicotine is partially neuropro-tective, with acute administration of nicotine prior toor combined with MPTP treatment reducing DA neur-onal degeneration in the neostriatum and substantialnigra (Janson et al., 1988, 1992). However, chronic nic-otine administration via minipums enhances MPTPneurotoxicity in the striatum (Janson et al., 1992). Theprotective e�ect of nicotine is thought to be related toreduced MPP+ (neurotoxic metabolite of MPTP)uptake into DA neurons via nicotine induced increasesin DA release. Conversely, enhanced MPTP neurotoxi-city is thought to result from failure of nAChRs to
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111100

desensitize following chronic nicotine administration,leading to chronic increased Na+/Ca2+ ion in¯ux vianAChRs located on DA neurons, with associated Ca+
ion toxicity and increased energy demands (Janson etal., 1992). However, nicotine has been observed tocounteract the locomotor e�ects MPTP in animalmodels of PD (Sershen et al., 1987; Maggio et al.,1998). In addition the novel a4b2, nAChR selectiveagonist SIB-1508Y ((S)-(-)-5-ethynyl-3-(1-methyl-2-pyr-rodinyl)-pyridine) has been observed to improve cogni-tive and motor performance in monkeys in the sameMPTP model of PD (Cosford et al., 1996; Schneider etal., 1998a, 1998b). When administered alone, SIB-1508Y (1 mg/kg) did not signi®cantly improve cogni-tive or motor function, but when combined with levo-dopa signi®cant improvements were observed in bothcognitive and motor task performance and at doses oflevodopa between one third and one sixth of thatnecessary to improve motor performance alone(Schneider et al., 1998b).
6.4. Schizophrenia
Schizophrenia is a chronically deteriorating hetero-geneous psychosis beginning in late adolescence orearly adulthood and is characterized by hallucinations,delusions, bizzare behaviour, apathy and blunted a�ect(Arnold and Trojanowski, 1996; Tsuang, 1993). Theetiology of this condition or group of conditions isunclear but studies indicate that schizophrenia has astrong genetic component, although the inheritancepattern appears to be complex involving an uncertainmode of transmission, incomplete penetrance andprobable genetic heterogeneity (Risch, 1990; Tsuang,1993). Possible loci for schizophrenia have been ident-i®ed at a number of chromosomal sites (Pulver et al.,1994; Wang et al., 1995; Silverman et al., 1996).However, these loci do not account for all cases ofschizophrenia and do not delineate which aspects ofthis multifactorial illness might be in¯uenced by aspeci®c locus. A dopamine hypothesis for schizo-phrenia has been proposed, suggesting that the symp-tomology results from an excess of dopamine,although similar symptoms can be produced by admin-istration of drugs like phencyclidine (PCP) an NMDAand nAChR channel blocker. The possible involvementof nAChRs in schizophrenia was suggested by the highpercentage of smokers present in the schizophrenicpopulation compared to the general population, 90%compared to 33% (Lohr and Flynn, 1992).Furthermore, the number of �3H� cytisine and �125I�BTX binding sites in CA3 region of the hippocampusin postmortem schizophrenic brains was signi®cantlyreduced compared to control brains, indicating a de®-cit in nAChR number in schizophrenia (Freedman etal., 1995). Schizophrenic patients have also been
observed to have high levels of nAChR antibodies,which may be a contributing factor in the reducednumber of nAChRs observed in schizophrenia(Mukherjee et al., 1994). From these observations itwas postulated that the high incidence of smoking inschizophrenics is an attempt on their part to self-medi-cate nicotine to overcome a de®cit in nicotinic neuro-transmission. In this regard, nicotine has beenobserved to normalize two psychophysiological de®citsin schizophrenic patients (Adler et al., 1992). De®citsin the regulation of response to sensory stimuli arelikely to be a major feature underlying the overt symp-toms of schizophrenia, such as hallucinations and delu-sions. Attention to apparently extraneous stimuli intheir surroundings, which are generally ignored by nor-mal subjects, is characteristic of schizophrenics andsuggests that neuronal mechanisms responsible for ®l-tering or gating of sensory input are impaired.Increased sensitivity to auditory stimuli in schizophre-nics and their relatives involves diminished gating ofP50 brain waves upon repeated auditory stimulation.In normal subjects, response to an initial auditorystimuli elicits an excitatory response that also activatesinhibitory mechanisms, which then diminsh the excit-atory response to subsequent auditory stimuli. Theability of schizophrenics and their relatives to diminishthe excitatory response following the second of pairedauditory stimuli is reduced compared to normal sub-jects but is transiently normalized by nicotine adminis-tration or following smoking (Adler et al., 1985, 1992,1993). The inheritance of this neuronal defect has beenlinked to a dinucleotide polymorphism at chromosome15 which is also the locus for the a7 nAChR (Chini etal., 1994; Freedman et al., 1997). The a7 nAChR isfurther implicated in schizophrenia by the observationthat the protein level of this subunit is signi®cantlyreduced in the frontal cortex of schizophrenic braincompared to age matched controls (Guan et al., 1999).These observations suggest that a7 nAChRs may beextremely important in schizophrenia.
6.5. Tourette's syndrome
Gilles de la Tourette syndrome (TS) is a neuropsy-chiatric disorder of unknown etiology which starts inchildhood and is characterized by persistent motor andverbal tics as well as by the frequent occurrence ofhyperactivity, anxieties, phobias or obsessive compul-sive disorders. Classical neuroleptics such as haloperi-dol are used to treat TS but are not always e�ectiveand produce side e�ects such as sedation and the poss-ible development of tardive dyskinesia. A number ofstudies have reported that administration of nicotineby means of gum or transdermal patches potentiatethe action of neuroleptics and is e�ective in ameliorat-ing the symptoms of TS (Sanberg et al., 1988;
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 101

McConville et al., 1992; Dursun and Reveley, 1997;Sanberg et al., 1997). Up to 4 weeks of bene®t havebeen reported in TS patients treated for only 2 dayswith 10 mg nicotine patches (Dursun and Reveley,1997). The mechanism by which the bene®cial e�ectsof nicotine are produced has yet to be elucidated butmay possibly involve modulation of dopamine release.Prolonged exposure to nicotine produces reversibledesensitization and eventually permanent inactivationof nAChRs, especially a4b2 and a7 subtypes (Hsu etal., 1996; Olale et al., 1997). Permanent inactivation ofthese receptors followed by a slow rate of resynthesismight account for the weeks of bene®t displayed fol-lowing nicotine administration. The development ofnicotinic agonists without the side e�ect pro®le of nic-otine would therefore appear to be desirable for thetreatment of TS. As yet, there is no direct evidence ofnAChR involvement in the condition but the positivee�ects of nicotine suggests that they may play a role,further investigation into the role of nAChRs in thiscondition is therefore necessary.
6.6. Anxiety and depression
nAChRs are apparently involved in the pathophy-siology of both anxiety disorders and depression.Nicotine administration has been observed to haveanxiolytic e�ects in humans and in animal models ofanxiety (Pomerleau, 1986; Decker et al., 1994). Thisaction can be blocked by administration of the non-competitive nAChR antagonist mecamylamine and bythe benzodiazepine inverse agonist ¯umazenil. Theseobservations suggest that the anxiolytic action of nic-otine is produced by enhanced release of the inhibitoryneurotransmitter GABA, which then acts on centralbenzodiazepine±GABAA receptor complex. Both ret-rospective and prospective clinical studies have demon-strated a relationship between smoking and majordepression; persons with major depression are morelikely to smoke, to have greater di�culty in stopping,and are at increased risk of su�ering mild to severe de-pression having succeeded in stopping (Glassman etal., 1988, 1990; Breslau et al., 1993; Dalack et al.,1995; Stage et al., 1996; Covey et al., 1997, 1998). Thisresults from shared predispositions involving genetic orenvironmental factors, although separate causal mech-anisms may exist including self medication of de-pressed mood and neuropharmacologic e�ects ofnicotine and other smoke substances on neurotrans-mitters linked to depression (Breslau, 1995; Breslau etal., 1998). Depression increases the likelihood of smok-ing, as well as nicotine and other dependencies.Transdermal nicotine patches improve the mood ofnon-smoking depressed patients and increase the dur-ation of REM sleep (Salin-Pascual et al., 1996; Salin-Pascual and Drucker-Colin, 1998). Furthermore, nic-
otine has been observed to act as an antidepressant inanimal models of depression (Semba et al., 1998).There is also a considerable body of evidence linkingthe action of classical tricyclic antidepressants (eg. imi-pramine, desipramine) and the newer serotonin uptakeinhibitors (e.g. ¯uoxteine, paroxetine) to nAChRs. Thetricyclic antidepressants imipramine, desipramine, amy-triptyline and nortriptyline, all produce a non-competi-tive inhibition of nAChRs with a reversible inhibitionof agonist induced currents (Scho®eld et al., 1981;Arita et al., 1987; Rana et al., 1993). The serotoninuptake inhibitors ¯uoxetine, paroxetine, sertaline, ven-lafaxine and nefazodone also produce a similar revers-ible non-competitive inhibition of nAChRs (Faircloughet al., 1993; Dalack et al., 1995; Garcia-Colunga et al.,1997; Hennings et al., 1997; Maggio et al., 1998; Fryerand Lukas, 1999). Fluoxetine (Prozac) has been mostextensively studied, blocking nAChR currents in a vol-tage dependent manner, while also increasing the rateof desensitization of the receptor (Garcia-Colunga etal., 1997; Maggio et al., 1998). Furthermore, in rathippocampal slices, ¯uoxetine inhibits nicotine inducedrelease of noradrenaline in a dose dependent manner(Hennings et al., 1997). Despite the well characterisedconnection between depression, antidepressants andnicotine the role of nAChRs as targets for antidepress-ant treatment remains under appreciated.
7. Conclusions
A considerable amount is now known aboutnAChRs in the human brain. A number of structuraland functional subtypes have been identi®ed with indi-vidual pharmacological pro®les and distinct patternsof distribution. To date, nine individual receptor subu-nits have been identi®ed and cloned in human brainwhich combine in various conformations to form indi-vidual receptor subtypes. The distribution nAChRshas been mapped using various radioligands possessinga modest degree of subtype selectivity and the distri-bution of individual subunit mRNA has also beenmapped to a limited degree with in situ hybridisationand other techniques. Nicotinic receptors apparentlyplay a pivotal role in a number of functional processesincluding learning and memory and are implicated inseveral CNS disorders including Alzheimer's disease,Parkinson's disease, schizophrenia and epilepsy, aswell as mediating the addiction to nicotine presentedin chronic tobacco users. However, a great deal of in-formation still remains to be elucidated concerninghuman neuronal nAChRs. The structure of individualreceptors present in human brain and their subunitcomposition remains to be fully investigated. The pat-tern of distribution in human brain of the subtypesthus far identi®ed is far from comprehensive and
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111102

would be aided by the development of subtype selec-tive radioligands. In this vein, subtype selective radioli-gands would have important application for PET andSPECT and would extend the potential of these tech-niques as diagnostic and mapping tools for nAChRs inthe living human. Loss of nAChRs might be an earlypresymptomatic diagnostic marker for AD.Additionally, the expression pattern of each of the in-dividual nAChR subunits is incomplete. The functionalroles of nAChRs and subunits in human brain hasalso yet to be fully elucidated, with roles for a5 and a6subunits beginning to emerge. Similarly, the exact roleof nAChRs in various pathologies remains unclear asin Alzheimer's disease where it is debatable whetherthe loss of nAChRs observed is symptomatic or causa-tive. The apparent bene®t of nAChR stimulation in anumber of these conditions identi®es neuronalnAChRs as potential therapeutic targets. Identi®cationof the speci®c receptor subtypes involved in each ofthe conditions is therefore desirable as is the develop-ment of subtype selective nAChR agonists whichwould potentially provide selective therapies for indi-vidual conditions. A considerable amount of researchis therefore necessary to fully elucidate the structure,function, physiological and pathological involvementand therapeutic potential of nAChRs, which remainan exciting research prospect.
Acknowledgements
This study was supported by grants from theSwedish Medical Research Council (project number05817), Loo and Hans Ostermans Foundation,Stiftelsen foÈ r Gamla TjaÈ narinnor, Stohnes Foundation,and KI Foundations.
References
Abreo, M.A., Lin, N.H., Garvey, D.S., Gunn, D.E., Hettinger,
A.M., Wasicak, J.T., Pavlik, P.A., Martin, Y.C., Donnelly-
roberts, D.L., Anderson, D.J., Sullivan, J.P., Williams, M.,
Arneric, S.P., Holladay, M.W., 1996. Novel 3-Pyridyl ethers with
subnanomolar a�nity for central neuronal nicotinic acetylcholine
receptors. J. Med. Chem 39, 817±825.
Adem, A., Synnergren, B., Botros, M., Ohman, B., Winblad, B.,
Nordberg, A., 1987. [3H] acetylcholine nicotinic recognition sites
in human brain: characterization of agonist binding. Neurosci.
Lett. 83, 298±302.
Adem, A., Jossan, S.S., d'Argy, R., Brandt, I., Winblad, B.,
Nordberg, A., 1988. Distribution of nicotinic receptors in human
thalamus as visualized by 3H-nicotine and 3H-acetylcholine
receptor autoradiography. J. Neural. Transm 73, 77±83.
Adem, A., Nordberg, A., Jossan, S.S., Sara, V., Gillberg, P.G., 1989.
Quantitative autoradiography of nicotinic receptors in large cryo-
sections of human brain hemispheres. Neurosci. Lett 101, 247±
252.
Adler, L.E., Ho�er, L.D., Wiser, A., Freedman, R., 1993.
Normalization of auditory physiology by cigarette smoking in
schizophrenic patients. Am. J. Psychiatry 150, 1856±1861.
Adler, L.E., Ho�er, L.J., Gri�th, J., Waldo, M.C., Freedman, R.,
1992. Normalization by nicotine of de®cient auditory sensory gat-
ing in the relatives of schizophrenics. Biol. Psychiatry 32, 607±
616.
Adler, L.E., Waldo, M.C., Freedman, R., 1985. Neurophysiologic
studies of sensory gating in schizophrenia: comparison of audi-
tory and visual responses. Biol. Psychiatry 20, 1284±1296.
Alkondon, M., Albuquerque, E.X., 1993. Diversity of nicotinic
acetylcholine receptors in rat hippocampal neurons. Part I:
Pharmacological and functional evidence for distinct structural
subtypes. J. Pharmacol. Exp. Ther 265, 1455±1473.
Amberla, K., Nordberg, A., Viitanen, M., Winblad, B., 1993. Long-
term treatment with tacrine (THA) in Alzheimer's disease Ð
evaluation of neuropsychological data. Acta. Neurol. Scand.
Suppl 149, 55±57.
Anand, R., Bason, L., Saedi, M.S., Gerzanich, V., Peng, X.,
Lindstrom, J., 1993. Reporter epitopes: a novel approach to
examine transmembrane topology of integral membrane proteins
applied to the alpha 1 subunit of the nicotinic acetylcholine recep-
tor. Biochemistry 32, 9975±9984.
Anand, R., Conroy, W.G., Schoepfer, R., Whiting, P., Lindstrom,
J., 1991. Neuronal nicotinic acetylcholine receptors expressed in
Xenopus oocytes have a pentameric quaternary structure. J. Biol.
Chem 266, 11192±11198.
Andrews, J.S., Jansen, J.H., Linders, S., Princen, A., 1994. E�ects of
disrupting the cholinergic system on short-term spatial memory in
rats. Psychopharmacology (Berl) 115, 485±494.
Arendash, G.W., Sanberg, P.R., Sengstock, G.J., 1995. Nicotine
enhances the learning and memory of aged rats. Pharmacol.
Biochem. Behav 52, 517±523.
Arita, M., Wada, A., Takara, H., Izumi, F., 1987. Inhibition of
22Na in¯ux by tricyclic and tetracyclic antidepressants and bind-
ing of �3H]imipramine in bovine adrenal medullary cells. J.
Pharmacol. Exp. Ther 243, 342±348.
Arnold, S.E., Trojanowski, J.Q., 1996. Recent advances in de®ning
the neuropathology of schizophrenia. Acta. Neuropathol. (Berl)
92, 217±231.
Aubert, I., Araujo, D.M., Cecyre, D., Robitaille, Y., Gauthier, S.,
Quirion, R., 1992. Comparative alterations of nicotinic and
muscarinic binding sites in Alzheimer's and Parkinson's diseases.
J. Neurochem 58, 529±541.
Balfour, D.J., Fagerstrom, K.O., 1996. Pharmacology of nicotine
and its therapeutic use in smoking cessation and neurodegenera-
tive disorders. Pharmacol. Ther 72, 51±81.
Bartus, R.T., Dean, R.L.d., Beer, B., Lippa, A.S., 1982. The cholin-
ergic hypothesis of geriatric memory dysfunction. Science 217,
408±414.
Beani, L., Bianchi, C., Nilsson, L., Nordberg, A., Romanelli, L.,
Sivilotti, L., 1985. The e�ect of nicotine and cytisine on 3H-
acetylcholine release from cortical slices of guinea-pig brain.
Naunyn Schmiedebergs Arch. Pharmacol 331, 293±296.
Bednar, I., Zhang, X., Dastranj-Sedghi, R., Nordberg, A., 1998.
Di�erential changes of nicotinic receptors in the rat brain follow-
ing ibotenic acid and 192-IgG saporin lesions of the nucleus basa-
lis magnocellularis. Int. J. Dev. Neurosci 16, 661±668.
Benowitz, N.L., 1996. Pharmacology of nicotine: addiction and
therapeutics. Annu. Rev. Pharmacol. Toxicol 36, 597±613.
Benwell, M.E., Balfour, D.J., Anderson, J.M., 1988. Evidence that
tobacco smoking increases the density of (-)-[3H]nicotine binding
sites in human brain. J. Neurochem 50, 1243±1247.
Bertrand, D., Valera, S., Bertrand, S., Ballivet, M., Rungger, D.,
1991. Steroids inhibit nicotinic acetylcholine receptors.
Neuroreport 2, 277±280.
Bertrand, S., Weiland, S., Berkovic, S.F., Steinlein, O.K., Bertrand,
D., 1998. Properties of neuronal nicotinic acetylcholine receptor
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 103

mutants from humans su�ering from autosomal dominant noc-
turnal frontal lobe epilepsy. Br. J. Pharmacol 125, 751±760.
Breese, C.R., Marks, M.J., Logel, J., Adams, C.E., Sullivan, B.,
Collins, A.C., Leonard, S., 1997. E�ect of smoking history on
�3H]nicotine binding in human postmortem brain. J. Pharmacol.
Exp. Ther 282, 7±13.
Breslau, N., 1995. Psychiatric comorbidity of smoking and nicotine
dependence. Behav. Genet 25, 95±101.
Breslau, N., Kilbey, M.M., Andreski, P., 1993. Nicotine dependence
and major depression. New evidence from a prospective investi-
gation. Arch. Gen. Psychiatry 50, 31±35.
Breslau, N., Peterson, E.L., Schultz, L.R., Chilcoat, H.D., Andreski,
P., 1998. Major depression and stages of smoking. A longitudinal
investigation. Arch. Gen. Psychiatry 55, 161±166.
Broussolle, E.P., Wong, D.F., Fanelli, R.J., London, E.D., 1989. In
vivo speci®c binding of �3H]1-nicotine in the mouse brain. Life Sci
44, 1123±1132.
Buccafusco, J.J., Jackson, W.J., 1991. Bene®cial e�ects of nicotine
administered prior to a delayed matching-to-sample task in
young and aged monkeys. Neurobiol. Aging 12, 233±238.
Buisson, B., Gopalakrishnan, M., Arneric, S.P., Bertrand, D., 1996.
Human alpha4beta2 neuronal nicotinic acetylcholine receptor in
HEK 293 cells: A patch-clump study. J. Neurosci. 16, 7880±7891.
Caggiula, A.R., Donny, E.C., Epstein, L.H., Sved, A.F., Knopf, S.,
Rose, C., McAllister, C.G., Antelman, S.M., Perkins, K.A., 1998.
The role of corticosteroids in nicotine's physiological and beha-
vioral e�ects. Psychoneuroendocrinology 23, 143±159.
Cartaud, J., Benedetti, E.L., Cohen, J.B., Meunier, J.C., Changeux,
J.P., 1973. Presence of a lattice structure in membrane fragments
rich in nicotinic receptor protein from the electric organ of
Torpedo marmorata. FEBS Lett 33, 109±113.
Chavez-Noriega, L.E., Crona, J.H., Washburn, M.S., Urrutia, A.,
Elliott, K.J., Johnson, E.C., 1997. Pharmacological characteriz-
ation of recombinant human neuronal nicotinic acetylcholine
receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h
alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7
expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther 280, 346±
356.
Chini, B., Raimond, E., Elgoyhen, A.B., Moralli, D., Balzaretti, M.,
Heinemann, S., 1994. Molecular cloning and chromosomal local-
ization of the human alpha 7-nicotinic receptor subunit gene
(CHRNA7). Genomics 19, 379±381.
Clarke, P.B., Schwartz, R.D., Paul, S.M., Pert, C.B., Pert, A., 1985.
Nicotinic binding in rat brain: autoradiographic comparison of
�3H]acetylcholine, �3H]nicotine, and �125I]-alpha-bungarotoxin. J.
Neurosci 5, 1307±1315.
Colrain, I.M., Mangan, G.L., Pellett, O.L., Bates, T.C., 1992. E�ects
of post-learning smoking on memory consolidation.
Psychopharmacology (Berl) 108, 448±451.
Conroy, W.G., Vernallis, A.B., Berg, D.K., 1992. The alpha 5 gene
product assembles with multiple acetylcholine receptor subunits
to form distinctive receptor subtypes in brain. Neuron 9, 679±
691.
Cosford, N.D., Bleicher, L., Herbaut, A., McCallum, J.S., Vernier,
J.M., Dawson, H., Whitten, J.P., Adams, P., Chavez-Noriega, L.,
Correa, L.D., Crona, J.H., Maha�y, L.S., Menzaghi, F., Rao,
T.S., Reid, R., Sacaan, A.I., Santori, E., Stauderman, K.A.,
Whelan, K., Lloyd, G.K., McDonald, I.A., 1996. (S)-(-)-5-ethy-
nyl-3-(1-methyl-2-pyrrolidinyl)pyridine maleate (SIB- 1508Y): a
novel anti-parkinsonian agent with selectivity for neuronal nic-
otinic acetylcholine receptors. J. Med. Chem 39, 3235±3237.
Court, J., Clementi, F., 1995. Distribution of nicotinic subtypes in
human brain. Alzheimer Dis. Assoc. Disord 9, 6±14.
Court, J.A., Perry, E.K., Spurden, D., Gri�ths, M., Kerwin, J.M.,
Morris, C.M., Johnson, M., Oakley, A.E., Birdsall, N.J.,
Clementi, F., et al., 1995. The role of the cholinergic system in
the development of the human cerebellum. Brain Res. Dev. Brain
Res 90, 159±167.
Couturier, S., Bertrand, D., Matter, J.M., Hernandez, M.C.,
Bertrand, S., Millar, N., Valera, S., Barkas, T., Ballivet, M.,
1990. A neuronal nicotinic acetylcholine receptor subunit (alpha
7) is developmentally regulated and forms a homo-oligomeric
channel blocked by alpha-BTX. Neuron 5, 847±856.
Covey, L.S., Glassman, A.H., Stetner, F., 1997. Major depression
following smoking cessation. Am. J. Psychiatry 154, 263±265.
Covey, L.S., Glassman, A.H., Stetner, F., 1998. Cigarette smoking
and major depression. J. Addict Dis 17, 35±46.
Coyle, J.T., Price, D.L., deLong, M.R., 1983. Alzheimer's disease: a
disorder of cortical cholinergic innervation. Science 219, 1184±
1190.
Dalack, G.W., Glassman, A.H., Rivelli, S., Covey, L., Stetner, F.,
1995. Mood, major depression, and ¯uoxetine response in ciga-
rette smokers. Am. J. Psychiatry 152, 398±403.
Dani, J.A., Heinemann, S., 1996. Molecular and cellular aspects of
nicotine abuse. Neuron 16, 905±908.
Davies, P., Feisullin, S., 1981. Postmortem stability of alpha-bungar-
otoxin binding sites in mouse and human brain. Brain Res 216,
449±454.
de Sarno, P., Giacobini, E., 1989. Modulation of acetylcholine
release by nicotinic receptors in the rat brain. J. Neurosci. Res
22, 194±200.
Decker, M.W., 1995. Animal models of cognitive function. Crit.
Rev. Neurobiol 9, 321±343.
Decker, M.W., Brioni, J.D., Bannon, A.W., Arneric, S.P., 1995.
Diversity of neuronal nicotinic acetylcholine receptors: lessons
from behavior and implications for CNS therapeutics. Life Sci
56, 545±570.
Decker, M.W., Brioni, J.D., Sullivan, J.P., Buckley, M.J., Radek,
R.J., Raszkiewicz, J.L., Kang, C.H., Kim, D.J., Giardina, W.J.,
Wasicak, J.T., et al., 1994. (S)-3-methyl-5-(1-methyl-2-pyrrolidiny-
l)isoxazole (ABT 418): a novel cholinergic ligand with cognition-
enhancing and anxiolytic activities. Part II: In vivo characteriz-
ation. J. Pharmacol. Exp. Ther 270, 319±328.
Decker, M.W., Majchrzak, M.J., Anderson, D.J., 1992. E�ects of
nicotine on spatial memory de®cits in rats with septal lesions.
Brain Res 572, 281±285.
Decker, M.W., Majchrzak, M.J., Arneric, S.P., 1993. E�ects of lobe-
line, a nicotinic receptor agonist, on learning and memory.
Pharmacol Biochem. Behav 45, 571±576.
Delacourte, A., Defossez, A., 1986. Alzheimer's disease: Tau pro-
teins, the promoting factors of microtubule assembly, are major
components of paired helical ®laments. J. Neurol. Sci 76, 173±
186.
Dunnett, S.B., Martel, F.L., 1990. Proactive interference e�ects on
short-term memory in rats. Part I: Basic parameters and drug
e�ects. Behav. Neurosci 104, 655±665.
Dursun, S.M., Reveley, M.A., 1997. Di�erential e�ects of transder-
mal nicotine on microstructured analyses of tics in Tourette's syn-
drome: an open study. Psychol. Med 27, 483±487.
Edwards, J.A., Warburton, D.M., 1982. Smoking, nicotine and elec-
trocortical activity. Pharmacol. Ther 19, 147±164.
Eilers, H., Schae�er, E., Bickler, P.E., Forsayeth, J.R., 1997.
Functional deactivation of the major neuronal nicotinic receptor
caused by nicotine and a protein kinase C-dependent mechanism.
Mol. Pharmacol 52, 1105±1112.
el-Bizri, H., Clarke, P.B., 1994. Regulation of nicotinic receptors in
rat brain following quasi-irreversible nicotinic blockade by chlori-
sondamine and chronic treatment with nicotine. Br. J. Pharmacol
113, 917±925.
Elliott, K.J., Ellis, S.B., Berckhan, K.J., Urrutia, A., Chavez-
Noriega, L.E., Johnson, E.C., Velicelebi, G., Harpold, M.M.,
1996. Comparative structure of human neuronal alpha 2-alpha 7
and beta 2-beta 4 nicotinic acetylcholine receptor subunits and
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111104

functional expression of the alpha 2, alpha 3, alpha 4, alpha 7,
beta 2, and beta 4 subunits. J. Mol. Neurosci 7, 217±228.
Evans, D.A., Funkenstein, H.H., Albert, M.S., Scherr, P.A., Cook,
N.R., Chown, M.J., Hebert, L.E., Hennekens, C.H., Taylor, J.O.,
1989. Prevalence of Alzheimer's disease in a community popu-
lation of older persons Higher than previously reported (see com-
ments). Jama 262, 2551±2556.
Fairclough, R.H., Josephs, R., Richman, D.P., 1993. Imaging ligand
binding sites on the Torpedo acetylcholine receptor. Ann. N Y
Acad. Sci 681, 113±125.
Flores, C.M., Rogers, S.W., Pabreza, L.A., Wolfe, B.B., Kellar,
K.J., 1992. A subtype of nicotinic cholinergic receptor in rat
brain is composed of alpha 4 and beta 2 subunits and is up-regu-
lated by chronic nicotine treatment. Mol. Pharmacol 41, 31±37.
Flynn, D.D., Mash, D.C., 1986. Characterization of L-[3H]nicotine
binding in human cerebral cortex: comparison between
Alzheimer's disease and the normal. J. Neurochem 47, 1948±
1954.
Freedman, R., Coon, H., Myles-Worsley, M., Orr-Urtreger, A.,
Olincy, A., Davis, A., Polymeropoulos, M., Holik, J., Hopkins,
J., Ho�, M., Rosenthal, J., Waldo, M.C., Reimherr, F., Wender,
P., Yaw, J., Young, D.A., Breese, C.R., Adams, C., Patterson,
D., Adler, L.E., Kruglyak, L., Leonard, S., Byerley, W., 1997.
Linkage of a neurophysiological de®cit in schizophrenia to a
chromosome 15 locus. Proc. Natl. Acad. Sci. USA 94, 587±592.
Freedman, R., Hall, M., Adler, L.E., Leonard, S., 1995. Evidence in
postmortem brain tissue for decreased numbers of hippocampal
nicotinic receptors in schizophrenia. Biol. Psychiatry 38, 22±33.
Fryer, J.D., Lukas, R.J., 1999. Antidepressants noncompetitively
inhibit nicotinic acetylcholine receptor function. J. Neurochem
72, 1117±1124.
Fucile, S., Matter, J.M., Erkman, L., Ragozzino, D., Barabino, B.,
Grassi, F., Alema, S., Ballivet, M., Eusebi, F., 1998. The neur-
onal alpha6 subunit forms functional heteromeric acetylcholine
receptors in human transfected cells. Eur. J. Neurosci 10, 172±
178.
Fuxe, K., Janson, A.M., Jansson, A., Andersson, K., Eneroth, P.,
Agnati, L.F., 1990. Chronic nicotine treatment increases dopa-
mine levels and reduces dopamine utilization in substantia nigra
and in surviving forebrain dopamine nerve terminal systems after
a partial di-mesencephalic hemitransection. Naunyn
Schmiedebergs Arch. Pharmacol 341, 171±181.
Galzi, J.L., Changeux, J.P., 1995. Neuronal nicotinic receptors: mol-
ecular organization and regulations. Neuropharmacology 34,
563±582.
Gandia, L., Villarroya, M., Sala, F., Reig, J.A., Viniegra, S.,
Quintanar, J.L., Garcia, A.G., Gutierrez, L.M., 1996. Inhibition
of nicotinic receptor-mediated responses in bovine chroma�n
cells by diltiazem. Br. J. Pharmacol 118, 1301±1307.
Garcia-Colunga, J., Awad, J.N., Miledi, R., 1997. Blockage of
muscle and neuronal nicotinic acetylcholine receptors by ¯uoxe-
tine (Prozac). Proc. Natl. Acad. Sci. USA 94, 2041±2044.
Gatley, S.J., Ding, Y.S., Brady, D., Gi�ord, A.N., Dewey, S.L.,
Carroll, F.I., Fowler, J.S., Volkow, N.D., 1998. In vitro and ex
vivo autoradiographic studies of nicotinic acetylcholine receptors
using �18F]¯uoronochloroepibatidine in rodent and human brain.
Nucl. Med. Biol 25, 449±454.
Gerzanich, V., Peng, X., Wang, F., Wells, G., Anand, R., Fletcher,
S., Lindstrom, J., 1995. Comparative pharmacology of epibati-
dine: a potent agonist for neuronal nicotinic acetylcholine recep-
tors. Mol. Pharmacol 48, 774±782.
Gerzanich, V., Wang, F., Kuryatov, A., Lindstrom, J., 1998. alpha 5
Subunit alters desensitization, pharmacology, Ca++ permeability
and Ca++ modulation of human neuronal alpha 3 nicotinic
receptors. J. Pharmacol. Exp. Ther 286, 311±320.
Glassman, A.H., Helzer, J.E., Covey, L.S., Cottler, L.B., Stetner, F.,
Tipp, J.E., Johnson, J., 1990. Smoking, cessation, and major de-
pression (see comments). Jama 264, 1546±1549.
Glassman, A.H., Stetner, F., Walsh, B.T., Raizman, P.S., Fleiss,
J.L., Cooper, T.B., Covey, L.S., 1988. Heavy smokers, smoking
cessation, and clonidine. Results of a double-blind, randomized
trial. Jama 259, 2863±2866.
Glenner, G.G., Wong, C.W., Quaranta, V., Eanes, E.D., 1984. The
amyloid deposits in Alzheimer's disease: their nature and patho-
genesis. Appl. Pathol 2, 357±369.
Gopalakrishnan, M., Buisson, B., Touma, E., Giordano, T.,
Campbell, J.E., Hu, I.C., Donnelly-Roberts, D., Arneric, S.P.,
Bertrand, D., Sullivan, J.P., 1995. Stable expression and pharma-
cological properties of the human alpha 7 nicotinic acetylcholine
receptor. Eur. J. Pharmacol 290, 237±246.
Gopalakrishnan, M., Molinari, E.J., Sullivan, J.P., 1997. Regulation
of human alpha4beta2 neuronal nicotinic acetylcholine receptors
by cholinergic channel ligands and second messenger pathways.
Mol. Pharmacol 52, 524±534.
Gopalakrishnan, M., Monteggia, L.M., Anderson, D.J., Molinari,
E.J., Piattoni-Kaplan, M., Donnelly-Roberts, D., Arneric, S.P.,
Sullivan, J.P., 1996. Stable expression, pharmacologic properties
and regulation of the human neuronal nicotinic acetylcholine
alpha 4 beta 2 receptor. J. Pharmacol. Exp. Ther 276, 289±297.
Gorbounova, O., Svensson, A.L., Jonsson, P., Mousavi, M., Miao,
H., Hellstrom-Lindahl, E., Nordberg, A., 1998. Chronic ethanol
treatment decreases �3H]epibatidine and �3H]nicotine binding and
di�erentially regulates mRNA levels of nicotinic acetylcholine
receptor subunits expressed in M10 and SH-SY5Y neuroblastoma
cells. J. Neurochem 70, 1134±1142.
Gotti, C., Briscini, L., Verderio, C., Oortgiesen, M., Balestra, B.,
Clementi, F., 1995. Native nicotinic acetylcholine receptors in
human Imr32 neuroblastoma cells: functional, immunological and
pharmacological properties. Eur. J. Neurosci 7, 2083±2092.
Gotti, C., Fornasari, D., Clementi, F., 1997. Human neuronal nic-
otinic receptors. Prog. Neurobiol 53, 199±237.
Gotti, C., Wanke, E., Sher, E., Fornasari, D., Cabrini, D., Clementi,
F., 1986. Acetylcholine operated ion channel and alpha-bungaro-
toxin binding site in a human neuroblastoma cell line reside on
di�erent molecules. Biochem. Biophys. Res. Commun 137, 1141±
1147.
Grigoryan, G., Hodges, H., Mitchell, S., Sinden, J.D., Gray, J.A.,
1996. 6-OHDA lesions of the nucleus accumbens accentuate
memory de®cits in animals with lesions to the forebrain cholin-
ergic progection system: e�ects of nicotine administration on
learning and memory in the water maze. Neurobiol. Learn Mem
65, 135±153.
Grun, E.A., Pauly, J.R., Collins, A.C., 1992. Adrenalectomy reverses
chronic injection-induced tolerance to nicotine.
Psychopharmacology (Berl) 109, 299±304.
Grundke-Iqbal, I., Iqbal, K., Tung, Y.C., Quinlan, M., Wisniewski,
H.M., Binder, L.I., 1986. Abnormal phosphorylation of the
microtubule-associated protein tau (tau) in Alzheimer cytoskeletal
pathology. Proc. Natl. Acad. Sci. USA 83, 4913±4917.
Grunwald, F., Biersack, H.J., Kuschinsky, W., 1996. Nicotine recep-
tor mapping [letter] (see comments). Eur. J. Nucl. Med 23, 1012±
1014.
Hall, M., Zerbe, L., Leonard, S., Freedman, R., 1993.
Characterization of �3H]cytisine binding to human brain mem-
brane preparations. Brain Res 600, 127±133.
Halldin, C., Nagren, K., Swahn, C.G., Langstrom, B., Nyback, H.,
1992. (S)-and (R)-[11C]nicotine and the metabolite (R/S)-[11C]coti-
otinine. Preparation, metabolite studies and in vivo distribution
in the human brain using PET. Int. J. Rad. Appl. Instrum. [B]
19, 871±880.
Hatsukami, D., Fletcher, L., Morgan, S., Keenan, R., Amble, P.,
1989. The e�ects of varying cigarette deprivation duration on
cognitive and performance tasks. J. Subst. Abuse 1, 407±416.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 105

Heimstra, N.W., Ellingstad, V.S., deKock, A.R., 1967. E�ects of
opeator mood on performance in a simulated driving task.
Percept. Mot. Skills 25, 729±735.
HellstroÈ m-Lindahl, E., Gorbounova, O., Seiger, A., Mousavi, M.,
Nordberg, A., 1998. Regional distribution of nicotinic receptors
during prenatal development of human brain and spinal cord.
Brain Res. Dev. Brain Res 108, 147±160.
HellstroÈ m-Lindahl, E., Mousavi, M., Zhang, X., Ravid, R.,
Nordberg, A., 1999. Regional distribution of nicotinc receptor
subunit mRNAs in human brain: comparison between Alzheimer
and normal brain (In Process Citation). Brain Res. Mol. Brain
Res 66, 94±103.
Henning®eld, J.E., 1984. Pharmacologic basis and treatment of ciga-
rette smoking. J. Clin. Psychiatry 45, 24±34.
Hennings, E.C., Kiss, J.P., Vizi, E.S., 1997. Nicotinic acetylcholine
receptor antagonist e�ect of ¯uoxetine in rat hippocampal slices.
Brain Res 759, 292±294.
Hindmarch, I., Kerr, J.S., Sherwood, N., 1990. E�ects of nicotine
gum on psychomotor performance in smokers and non-smokers.
Psychopharmacology (Berl) 100, 535±541.
Hindmarch, I., Sherwood, N., 1995. Nicotine and cognitive e�ects.
In: Clarke, P.B.S., Quik, M., Thurau, K. (Eds.), Advances in
Pharmacological Sciences. E�ects of Nicotine on Biological
Systems. Part II. BirkhaÈ user, Verlag, Basel, pp. 189±194.
Horti, A., Sche�el, U., Stathis, M., Finley, P., Ravert, H.T.,
London, E.D., Dannals, R.F., 1997. Fluorine-18-FPH for PET
imaging of nicotinic acetylcholine receptors. J. Nucle. Med 38,
1260±1265.
Horti, A.G., Sche�el, U., Koren, A.O., Ravert, H.T., Mathews,
W.B., Musachio, J.L., Finley, P.A., London, E.D., Dannals,
R.F., 1998. 2-[18F]Fluoro-A-85380, an in vivo tracer for the nic-
otinic acetylcholine receptors (In Process Citation). Nucl. Med.
Biol 25, 599±603.
Houghtling, R.A., Davila-Garcia, M.I., Kellar, K.J., 1995.
Characterization of (2)(-)[3H]epibatidine binding to nicotinic
cholinergic receptors in rat and human brain. Mol. Pharmacol
48, 280±287.
Hsu, Y.N., Amin, J., Weiss, D.S., Wecker, L., 1996. Sustained nic-
otine exposure di�erentially e�ecrs alpha 3 beta 2 and alpha 4
beta 2 neuronal nicotinic receptors expressed in Xenopus oocytes.
J. Neurochem 66, 667±675.
Hsu, Y.N., Edwards, S.C., Wecker, L., 1997. Nicotine enhances the
cyclic AMP-dependent protein kinase-mediated phosphorylation
of alpha 4 subunits of neuronal nicotinic receptors. J. Neurochem
69, 2427±2431.
Huganir, R.L., Greengard, P., 1990. Regulation of neurotransmitter
receptor desensitization by protein phosphorylation. Neuron 5,
555±567.
Inoue, M., Kuriyama, H., 1991. Somatostatin inhibits the nicotinic
receptor-activated inward current in guinea pig chroma�n cells.
Biochem. Biophys. Res. Commun 174, 750±775.
Janson, A.M., Fuxe, K., Agnati, L.F., Jansson, A., Bjelke, B.,
Sundstrom, E., Andersson, K., Harstrand, A., Goldstein, M.,
Owman, C., 1989. Protective e�ects of chronic nicotine treatment
on lesioned nigrostriatal dopamine neurons in the male rat. Prog.
Brain Res 79, 257±265.
Janson, A.M., Fuxe, K., Goldstein, M., 1992. Di�erential e�ects of
acute and chronic nicotine treatment on MPTP-(1-methyl-4-phe-
nyl-1,2,3,6-tetrahydrophyridine) induced degeneration of nigros-
triatal dopamine neurons in the black mouse. Clin. Investig 70,
232±238.
Janson, A.M., Fuxe, K., Sundstrom, E., Agnati, L.F., Goldstein, M.,
1988. Chronic nicotine treatment partly protects azgainst the 1-
methyl-4-phenyl-2,3,-6tetrahydropyridine-induced degeneration of
nigrostriatal dopamine neurons in the black mouse. Acta Physiol.
Scand 132, 589±591.
Janson, A.M., Hedlund, P.B., Fuxe, K., von Euler, G., 1994.
Chronic nicotine treatment counteracts dopamine D2 receptor
upregulation induced by a partial mesodiencephalic hemitransec-
tion in the rat. Brain Res 655, 25±32.
Janson, A.M., Moller, A., 1993. Chronic nicotine treatment counter-
acts nigral cell loss induced by a partial mesodiencephalic hemi-
transection: an analysis of the total number and mean volume of
neurons and glia in substantia nigra of the male rat.
Neuroscience 57, 931±941.
Jones, G.M., Sahakian, B.J., Levy, R., Warburton, D.M., Gray,
J.A., 1992. E�ects of acute abcutaneous nicotine on atention,
informatkion processing and shortterm memory in Alzheimer's
disease. Psychopharmacology (Berl) 180, 485±494.
Kampfer, I., Sorger, D., Schliebs, R., Karger, W., Gunther, K.,
Schulze, K., Knapp, W.H., 1996. Radioiodination of nicotine
with speci®c activity high enough for mapping nicotinic acetyl-
choline receptors. Eur. J. Nucl. Med 23, 157±162.
Kang, J., Lemaire, H.G., Unterbeck, A., Salbaum, J.M., Masters,
C.L., Grzeschik, K.H., Multhaup, G., Beyreuther, K., Muller-
Hill, B., 1987. The precursor of Alzeimer's disease amyloid A4
protein resembles a cell-surface receptor. Nature 325, 733±736.
Kao, P.N., Karlin, A., 1986. Acetylcholine receptor binding site con-
tains a disul®de cross-link between adjacent half-cystinyl residues.
J. Biol. Chem 261, 8085±8088.
Karlin, A., Akabas, M.H., 1995. Toward a structural basis for the
function of nicotinic acetylcholine receptors and their cousins.
Neuron 15, 1231±1244.
Katz, B., Thesle�, S., 1957. A study of the desensitization produced
by acetylcholine at the motor end plate. Journal of Physiology
138, 63±80.
Kassiou, M., Sche�el, U.A., Ravert, H.T., Mathews, W.B.,
Musachio, J.L., London, E.D., Dannals, R.F., 1998.
Pharmacological evaluation of �11C]A-84543: an enantioselective
ligand for in vivo studies of neuronal nicotinic acetylcholine
receptors. Life Sci 63, PL13±PL18.
Ke, L., Lukas, R.J., 1996. E�ects of steroid exposure on ligand bind-
ing and functiojnal activities of dicerse nicotinic acetylcholine
receptor subtypes. J. Neurochem 67, 1100±1112.
Kihara, T., Shimohama, S., Sawada, H., Kimura, J., Kume, T.,
Kochiyama, H., Maeda, T., Akaike, A., 1997. Nicotinic receptor
stimulation protects neurons against beta-amyloid toxicity. Ann.
Neurol 42, 159±163.
Kihara, T., Shimohama, S., Urushitani, M., Sawada, H., Kimura, J.,
Kume, T., Maeda, T., Akaike, A., 1998. Stimulation of alpha4-
beta2 nicotinic acetylcholine receptors inhibits beta-amyloid tox-
icity. Brain Res 792, 331±334.
Kim, K.D., Lerner-Marmarosh, N., Saraswati, M., Kende, A.S.,
Abood, L.G., 1994. 5-Isothiocyanonicotine: a high-a�nity irre-
versible ligand for brain nicotinic receptors. Biochem. Pharmacol
47, 1965±1967.
Kowall, N.W., Kosik, K.S., 1987. Axonal disruption and aberrant
localization of tau protein characterize the neuropil pathology of
Alzheimer's disease. Ann. Neurol 22, 639±643.
Lang, W., Henke, H., 1983. Cholinergic receptor binding and auto-
radiography in brains of non-neurological and senile dementia of
Alzheimer-type patients. Brain Res 267, 271±280.
Lange, K.W., Wells, F.R., Jenner, P., Marsden, C.D., 1993. Altered
muscarinic and nicotinic receptor densities in cortical and subcor-
tical brain regions in Parkinson's disease. J. Neurochem 60, 197±
203.
Le Houezec, J., Halliday, R., Benowitz, N.L., Callaway, E., Naylor,
H., Herzig, K., 1994. A low dose of subcutaneous nicotine
improves information processing in non-smokers.
Psychopharacology (Berl) 114, 628±634.
Lena, C., Changeux, J.P., 1993. Allosteric modulations of the nic-
otinic acetylcholine receptor. Trends Neurosci 16, 181±186.
Levin, E.D., 1992. Nicotinic systems and cognitive function.
Psychopharmacology (Berl) 108, 417±431.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111106

Levin, E.D., Briggs, S.J., Christopher, N.C., Rose, J.E., 1992.
Persistence of chronic nicotine-induced cofnitive facilitation.
Behav. Neural. Biol 58, 152±158.
Levin, E.D., Christopher, N.C., Briggs, S.J., Rose, J.E., 1993.
Chronic nicotine reverse working memory de®cits caused by
lesions of the ®mbria or medial basalocortical projection. Brain
Res. Cogn. Brain Res 1, 137±143.
Levin, E.D., Lee, C., Rose, J.E., Reyes, A., Ellison, G., Jarvik, M.,
Gritz, E., 1990. Chronic nicotine and withdrawal e�ects on
radial-arm maze performance in rats. Behav. Neural. Biol 53,
269±276.
Levin, E.D., Rose, J.E., Abood, L., 1995. E�ects of nicotinic
dimethylaminoethyl esters on working memory performance of
rats in the radial-arm maze. Pharmacol. Biochem. Behav 51, 369±
373.
Levin, E.D., Simon, B.B., 1998. Nicotinic acetylcholine involvement
in cognitive function in animals. Psychopharmacology (Berl) 138,
217±230.
Levin, E.D., Torry, D., 1996. Acute and chronic nicotine e�ects on
working memory in aged rats. Psychopharmacology (Berl) 123,
88±97.
LindstroÈ m, J., 1997. Nicotinic acetylcholine receptors in health and
disease. Mol. Neurobiol 15, 193±222.
LindstroÈ m, J., Merlie, J., Yogeeswarn, G., 1979. Biochemical proper-
ties of acteylcholine receptor subunits from Torpedo californica.
Biochemistry 18, 4465±4470.
Linville, D.G., Arneric, S.P., 1991. Cortical cerebal blood ¯ow gov-
erned by the basal forebrain: age-related impairments. Neurobiol
Aging 12, 503±510.
Linville, D.G., Williams, S., Raszkiewicz, J.L., Arneric, S.P., 1993.
Nicotinic agonists modulate basal forebrain control of cortica
cerbral blood ¯ow in anesthetized rats. J. Pharmacol. Exp. Ther
267, 440±448.
Lohr, J.B., Flynn, K., 1992. Smoking and schizophrenia. Schizophr.
Res 8, 93±102.
London, E.D., 1990. E�ects of nicotine on cerebral metabolism.
Ciba Found Symp. 152, 131±40, discussion 140±6.
London, E.D., Sche�el, U., Kimes, A.S., Kellar, K.J., 1995. In vivo
labeling of nicotinic acetylcholine receptors in brain with �3H]epi-
epibatidine. Eur. J. Pharmacol 278, R1±R2.
Lopez, M.G., Fonteriz, R.I., Gandia, L., de la Fuente, M.,
Villarroya, M., Garcia-Sancho, J., Garcia, A.G., 1993. The nic-
otinic acetylcholine receptor of the bovine chroma�n cell, a new
target for dihydropyridines. Eur. J. Pharmacol 247, 199±207.
Luetje, C.W., Patrick, J., 1991. Both alpha- and beta-subunits con-
tribute to the agonist sensitivity of neuronal nicotinic acetyl-
choline receptors. J. Neurosci 11, 837±845.
Lukas, R.J., 1993. Expression of ganglia-type nicotinic acetylcholine
recep tors and nicotinic ligand binding sites by cells of the IMR-
32 human neuroblastoma clonal line. J. Pharmacol. Exp. Ther
265, 294±302.
Lukas, R.J., Changeux, J.P., le Novere, N., Albuquerque, E.X.,
Balfour, D.J., Berg, D.K., Bertrand, D., Chiappinelli, V.A.,
Clarke, P.B., Collines, A.C., Dani, J.A., Grady, S.R., Kellar,
K.J., Lindstrom, J.M., Marks, M.J., Quik, M., Taylor, P.W.,
Wonnacott, S., 1999. Internatioinal union of pharmacology. XX.
Current status of the nomenclature for nicotinic acetylcholine
receptors and their subunits (In Process Citation). Pharmacol.
Rev. 51, 397±401.
Lundqvizt, H., Nordberg, A., Hartvig, P., Langstrom, B., 1998. (S)-
(-)-[11C]nicotine binding assessed by PET: a dual tracer model
evaluated in the rhesus monkey brain. Alzheimer Dis. Assoc.
Disord 12, 238±246.
LaÊ ngstroÈ m, B., Antoni, G., Hallidin, C., SvaÈ rd, H., Bergson, G.,
1982. The synthesis of some 11C-labelled alkaloids. Chemica
Scripta 20, 40±46.
Madhok, T.C., Matta, S.G., Sharp, B.M., 1995. Nicotine regulates
nicotinic cholinergic receptors and subunit mRNAs in PC 12 cells
through protein kinase A. Brain Res. Mol. Brain. Res 32, 143±
150.
Maelicke, A., Schratgenholz, A., Storch, A., Schroder, B., Gutbrod,
O., Methfessel, C., Weber, K.H., Pereira, E.E., Alkondon, M.,
Albuquerque, E.X., 1995. Noncompetitive agonism at nicotinic
acetylcholine receptors; functional signi®cance for CNS signal
transduction. J. Recept. Signal Transduct. Res 15, 333±353.
Maggio, R., Riva, M., Vaglini, F., Fornai, F., Molteni, R.,
Armogida, M., Racagni, G., Corsini, G.U., 1998. Nicotine pre-
vents experimental parkinsonism in rodents and induces striatal
increase of neurotrophic factors. J. Neurochem 71, 2439±2446.
Maltby, N., Broe, G.A., Creasey, H., Jorm, A.F., Christensen, H.,
Brooks, W.S., 1994. E�cacy of tacrine and lecithin in mild to
moderate Alzheimer's disease: double blind trial (see comments).
Bmj 308, 879±883.
Marks, M.J., Pauly, J.R., Gross, S.D., Deneris, E.S., Hermans-
Borgmeyer, I., Heinemann, S.F., Collins, A.C., 1992. Nicotine
binding and nicotinic recep tor subunit RNA after chronic nic-
otine treatment. J. Neurosci 12, 2765±2784.
Marks, M.J., Romm, E., Campbell, S.M., Collins, A.C., 1989.
Variation of nicotinic binding sites among inbred strains.
Pharmacol. Biochem. Behav 33, 679±689.
Marshall, D.L., Redfern, P.H., Wonnacott, S., 1997. Presynaptic nic-
otinic modulation of dopamine release in the three ascending
pathways studied by in vivo microdialysis: comparison of naive
and chronic nicotine-treated rats. J. Neurochem 68, 1511±1519.
Marutle, A., Warpman, U., Bogdanovic, N., Lannfelt, L., Nordberg,
A., 1999. Neuronal nicotinic receptor de®cits in Alzheimer
patients with the Swedish amylioid precursor protein 670/671 mu-
tation. J. Neurochem 72, 1161±1169.
Marutle, A., Warpman, U., Bogdanovic, N., Nordberg, A., 1998.
Regional distribution of subtypes of nicotinic receptors in human
brain and e�ect of aging studied by (2)-[3H]epibatidine (In
Process Citation). Brain Res 801, 143±149.
Maziere, M., Comar, D., Marazano, C., Berger, G., 1976. Nicotine-
11C: synthesis and distribution kinetics in animals. Eur. J. Nucl.
Med 1, 255±258.
McConville, B.J., Sanberg, P.R., Fogelson, M.H., King, J., Cirino,
P., Parker, K.W., Norman, A.B., 1992. The e�ects of nicotine
plus haloperidol compared to nictoine only and placebo nicotine
only in reducing tic severity and frequency in Tourette's disorder.
Biol. Psychiatry 31, 832±840.
McGehee, D.S., Role, L.W., 1995. Physiological diversity of nicotinic
acetylcholine receptors expressed by vertebrate neurons. Annu.
Rev. Physiol 57, 521±546.
Miles, K., Greengard, P., Huganir, R.L., 1989. Calcitonin gene-re-
lated peptide regulates phosphorylation of the nicotinic acetyl-
choline receptor in rat myotubes. Neuron 2, 1517±1524.
Molinari, E.J., Delbono, O., Messi, M.L., Reganathan, M., Arneric,
S.P., Sullivan, J.P., Gopalakrishnan, M., 1998. Up-regulation of
human alpha7 nicotinic receptors by chronic treatment with acti-
vator and antagonist ligands. Eur. J. Pharmacol 347, 131±139.
Morens, D.M., Grandinetti, A., Reed, D., White, L.R., Ross, G.W.,
1995. Cigarette smoking and protection from Parkinson's disease:
false association or etiologic clue? . Neurology 45, 1041±1051.
Muir, J.L., Everitt, B.J., Robbins, T.W., 1995. Reversal of visual
attentional dysfunction following lesions of the cholinergic basal
forebrain by physostigmine and nicotine but not by the 5-HT3
receptor antagonist, ondansetron. Psychopharmacology (Berl)
118, 82±92.
Mukherjee, S., Mahadik, S.P., Korenovsky, A., Laev, H., Schur,
D.B., Reddy, R., 1994. Serum antibodies to nicotinic acetyl-
choline receptors in schizophrenic patients. Schizophr. Res 12,
131±136.
Mulle, C., Lena, C., Changeux, J.P., 1992. Potentiation of nicotinic
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 107

receptor response by external calcium in rat central neurons.
Neuron 8, 937±945.
Musachio, J.L., Sche�el, U., Finley, P.A., Zhan, Y., Mochizuki, T.,
Wanger Jr, H.N., Dannals, R.F., 1998. 5-[I-125/123]lodo-3(2(S)-
azetidinylmethody)pyridine, a radioiodinated analog of A-85380
for in vivo studies of central nicotinic acetylcholine receptors.
Life Sci 62, PL351±PL357.
Musachio, J.L., Villemagne, V.L., Sche�el, U., Stathis, M., Finley,
P., Horti, A., London, E.D., Dannals, R.F., 1997. �125=123I]IPH: a
radioiodinated analog of epibatidine for in vivo studies of nic-
otinic acetylcholine receptors. Synapse 26, 392±399.
Musachio, J.L., Villemagne, V.L., Sche�el, U.A., Dannals, R.F.,
Dogan, A.S., Yokoi, F., Wong, D.F., 1999. Synthesis of an I-123
analog of A-85380 and preliminary SPECT imaging of nicotinic
receptors in baboon (In Process Citation). Nucl. Med. Biol 26,
201±207.
Nagata, K., Aistrup, G.L., Huang, C.S., Marszalec, W., Song, J.H.,
Yeh, J.Z., Narahashi, T., 1996. Potent modulation of neuronal
nicotinic acetylcholine receptor-channel by ethanol. Neurosci.
Lett 217, 189±193.
Newhouse, P.A., Potter, A., Corwin, J., Lenox, R., 1992. Acute nic-
otinic blockade produces cognitive impairment in normal
humans. Psychopharamcology (Berl) 108, 480±484.
Newhouse, P.A., Potter, A., Levin, E.D., 1997. Nicotinic system
involvement in Alzheimer's and Parkinson's diseases.
Implicatioins for therapeutics. Drugs Aging 11, 206±228.
Newhouse, P.A., Potter, A., Piasecki, M., Geebmuyden, J., Corwin,
J., Lennox, R., 1995. Nicotinic modulation of cognitive function
in humans. In: Clarke, P.B.S., Quik, M., Thurau, K. (Eds.),
Advances in Pharmacological Sciences. E�ects of Nicotine on
Biological Systems II. BirkhaÈ user, Verlag, Basel.
Newhouse, P.A., Sunderland, T., Narang, P.K., Mellow, A.M.,
Fertig, J.B., Lawlor, B.A., Murphy, D.L., 1990. Neuroendocrine,
physiologic, and behavioral responses following intravenous nic-
otine in nonsmoking healthy-volunteers and in patients with
Alzheimer's disease. Psychoneruonedocrinology 15, 471±484.
Nordberg, A., 1994. Human nicotinic receptors Ð their role in aging
and dementia. Neurochem. Int 25, 93±97.
Nordberg, A., Adem, A., Hardy, J., Winblad, B., 1998. Change in
nicotinic receptor subtypes in temporal cortex of Alzheimer
brains. Neurosci. Lett 86, 317±321.
Nordberg, A., Amberla, K., Shigeta, M., Lundqvist, H., Viitanen,
M., Hellstrom-Lindahl, E., Johansson, M., Andersson, J.,
Hartvig, P., Lilja, A., Langstrom, B., Winblad, B., 1998. Long-
term tacrine treatment in three mild Alzheimer patients: e�ects on
nicotinic receptors, cerbral blood ¯ow, glucose metabolism, EEG,
and cognitive abilities. Alzheimer Dis. Assoc. Disord 12, 228±237.
Nordberg, A., Hartvig, P., Lilja, A., Viitanen, M., Amberla, K.,
Lundqvist, H., Andersson, Y., Ulin, J., Winblad, B., Langstrom,
B., 1990. Decreased uptake and binding of 11C-nicotine in brain
of Alzheimer patients as visualized by positron emission tomogra-
phy. J. Neural. Transm. Park. Dis. Dement. Sect 2, 215±224.
Nordberg, A., Hartving, P., Lundqvist, H., Antoni, G., Ulin, J.,
Langstrom, B., 1989b. Uptake and regional distribution of (+)-
(R)- and (-)-(S)-N-[Methyl-11C]-nicotine in the brains of rhesus
monkey. An attempt to study nicotinic receptors in vivo. J.
Neural. Transm. Park. Dis. Dement. Sect 1, 195±205.
Nordberg, A., Lundqvist, H., Hartving, P., Andersson, J.,
Johansson, M., Hellstrom-Lindahi, E., Langstrom, B., 1997.
Imaging of nicotinic and muscarinic receptors in Alzheimer's dis-
ease: e�ect of tacrine treatment. Dement Geriatr. Cogn. Disord 8,
78±84.
Nordberg, A., Lundqvist, H., Hartvig, P., Lilja, A., Langstrom, B.,
1995. Kinetic analysis of regional (S)(-)11C-nicotine binding in
normal and Alzheimer brains Ð in vivo assessment using posi-
torn emission tomography. Alzheimer Dis. Assoc. Disord 9, 21±
27.
Nordberg, A., Nilsoon-Hakansson, L., Adem, A., Hardy, J.,
Alafuzo�, I., Lai, Z., Herrera-Marschitz, M., Winblad, B., 1989a.
The role of nicotinic receptors in the pathophysiology of
Alzheimer's disease. Prog. Brain Res 79, 353±362.
Nordberg, A., Nyberg, P., Adolfsson, R., Winblad, B., 1987.
Cholinergic topography in Alzheimer brains: a comparison with
changes in the monoaminergic pro®le. J. Neural. Transm 69, 19±
32.
Nordberg, A., Svensson, A.L., 1998. Cholinesterase inhibitors in the
treatment of Alzheimer's disease: a comparison of tolerability and
pharmacology. Drug Saf 19, 465±480.
Nordberg, A., Winblad, B., 1986. Reduced number of �3H]nicotine
and �3H]acetylcholine binding sites in the frontal cortex of
Alzheimer brains. Neurosci. Lett 72, 115±119.
NybaÈ ck, H., Halldin, C., Ahlin, A., Curvall, M., Eriksson, L., 1994.
PET studies of the uptake of (S)- and (R)-[11C]nicotine in the
human brain: di�culties in visualizing speci®c receptor binding in
vivo. Psychopharmacology (Berl) 115, 31±36.
NybaÈ ck, H., Nordberg, A., Langstrom, B., Halldin, C., Hartvig, P.,
Ahlin, A., Swahn, C.G., Sedvall, G., 1989. Attempts to visualize
nicotinic receptors in the brain of monkey and man by positorn
emission tomography. Prog. Brain Res 79, 313±319.
Octave, J.N., 1995. The amyloid peptide and its precursor in
Alzheimer's disease. Rev. Neurosci 6, 287±316.
Olale, F., Gerzanich, V., Kuryatov, A., Wang, F., Lindstrom, J.,
1997. Chronic nicotine exposure di�erentially addects the func-
tion of human alpha3, alpha4, and alpha7 nuronal nicotinic
receptor subtypes. J. Pharmacol. Exp. Ther 283, 675±683.
Oliverio, A., 1966. E�ects of mecamylamine on avoidance condition-
ing and maze learning of mice. J. Pharmacol. Exp. Ther 154,
350±356.
Orr-Urterger, A., Goldner, F.M., Saeki, M., Lorenzo, I., Goldberg,
L., de Biasi, M., Dani, J.A., Patrick, J.W., Beaudet, A.L., 1997.
Mice de®cient in the alpha7 neuronal nicotinic acetylcholine
receptor lack alpha-bungarotoxin binding sites and hippocampal
fast nicotinic currents. J. Neurosci 17, 9165±9171.
Papke, R.L., Duvoisin, R.M., Heinemann, S.F., 1993. The amino
terminal half of the nicotinic beta-subunit extracellular domain
regulates the kinetics of inhibition by neuronal bungarotoxin.
Proc. R. Soc. Lond. B. Biol. Sci 252, 141±148.
Papke, R.L., Heinemann, S.F., 1991. The role of the beta 4-subunit
in determining the kinetic properties of rat neuronal nicotinic
acetylcholine alpha3-receptors. J. Physiol. (Lond) 440, 95±112.
Pauly, J.R., Collins, A.C., 1993. An autoradiographic analysis of
alterations in nicotinic cholinergic receptors following 1 week of
corticosterone supplementation. Neuroendocrinology 57, 262±271.
Pauly, J.R., Marks, M.J., Robinson, S.F., van de Kamp, J.L.,
Collins, A.C., 1996. Chronic nicotine and mecamylamine treat-
ment increase brain nicotinic receptor binding without changing
alpha4 or beta2 mRNA levels. J. Pharmacol. Exp. Ther 278, 361±
369.
Peng, X., Gerzanich, V., Anand, R., Whiting, P.J., Lindstrom, J.,
1994. Nicotine-induced increase in neuronal nicotinic receptors
results from a decrease in the rate of receptor turnover. Mol.
Pharmacol 46, 523±530.
Pereira, E.F., Alkondon, M., Reinhardt, S., Maelicke, A., Peng, X.,
Lindstrom, J., Whiting, P., Albuquerque, E.X., 1994.
Physostigmine and galanthamine: probes for a novel binding site
on the alpha4 beta2 subtype of neuronal nicotinic acetylcholine
receptors stably expressed in ®broblast cells. J. Pharmacol. Exp.
Ther 270, 768±778.
Pereira, E.F., Reinhardt-Maelicke, S., Schrattenholz, A., Maelicke,
A., Albuquerque, E.X., 1993. Identi®cation and functional
characterization of a new agonist site on nicotinic acetylcholine
receptors of cultured hippocampal neurons. J. Pharmacol. Exp.
Ther 265, 1474±1491.
Perry, E.K., Court, J.A., Johnson, M., Piggott, M.A., Perry, R.H.,
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111108

1992. Autoradiographic distribution of �3H]nicotine binding in
human cortex: relative abundance in subicular complex. J. Chem.
Neuroanat 5, 399±405.
Perry, E.K., Morris, C.M., Court, J.A., Cheng, A., Fairbairn, A.F.,
McKeith, I.G., Irving, D., Brown, A., Perry, R.H., 1995.
Alteration in nicotine binding sites in Parkinson's disease, Lewy
body dementia and Alzheimer's disease: possible index of early
neuropathology. Neuroscience 64, 385±395.
Perry, E.K., Tomlinson, B.E., Blessed, G., Bergmann, K., Gibson,
P.H., Perry, R.H., 1978. Correlation of cholinergic abnormalities
with senile plaques and mental test scores in senile dementia. Br.
Med. J 2, 1457±1459.
Picciotto, M.R., Zoli, M., Lena, C., Bessis, A., Lallemand, Y.,
LeNovere, N., Vincent, P., Pich, E.M., Brulet, P., Changeux,
J.P., 1995. Abnormal avoidance learning in mice lacking func-
tional high-a�nity nicotine receptor in the brain. Nature 374, 65±
67.
Pich, E.M., Pagliusi, S.R., Tessari, M., Talabot-Ayer, D., Hooft van
Huijsduijnen, R., Chiamulera, C., 1997. Common neural sub-
strates for the addictive properties of nicotine and cocaine.
Science 275, 83±86.
Pickworth, W.B., Herning, R.I., Henning®eld, J.E., 1988.
Mecamylamine reduces some EEG e�ects of nicotine chewing
gum in humans. Pharmacol. Biochem. Behav 30, 149±153.
Pomerleau, O.F., 1986. Nicotine as a psychoactive drug: anxiety and
pain reduction. Psychopharmacol. Bull 22, 865±869.
Pontieri, F.E., Tanda, G., Orzi, F., di Chiara, G., 1996. E�ects of
nicotine on the nucleus accumbens and similarity to those of
addictive drugs (see comments). Nature 382, 255±257.
Pulver, A.E., Karayiorgou, M., Wolyniec, P.S., Lasseter, V.K.,
Kasch, L., Nestadt, G., Antonarakis, S., Housman, D., Kazazian,
H.H., Meyers, D., et al., 1994. Sequential strategy to identify a
susceptibility gene for schizophrenia: report of potential linkage
on chromosome 22q12-q13.1: Part 1. Am. J. Med. Genet 54, 36±
43.
Rana, B., McMorn, S.O., Reeve, H.L., Wyatt, C.N., Vaughan, P.F.,
Peers, C., 1993. Inhibition of neuronal nicotinic acetylcholine
receptors by imipramine and desipramine. Eur. J. Pharmacol 250,
247±251.
Reisine, T.D., Yamamura, H.I., Bird, E.D., Spokes, E., Enna, S.J.,
1978. Pre- and postsynaptic neurochemical alterations in
Alzheimer's disease. Brain Res 159, 477±481.
Reitstetter, R., Lukas, R.J., Gruener, R., 1999. Dependence of nic-
otinic acetylcholine receptor recovery from desensitization on the
duration of agonist exposure. J. Pharmacol. Exp. Ther 289, 656±
660.
Reynolds, J.A., Karlin, A., 1978. Molecular weight in detergent sol-
ution of acetylcholine receptor from Torpedo californica.
Biochemistry 17, 2035±2038.
Risch, N., 1990. Linkage strategies for genetically complex traits.
Part I: Multilocus models (see comments). Am. J. Hum. Genet
46, 222±228.
Robinson, S.F., Marks, M.J., Collins, A.C., 1996. Inbred mouse
strains vary in oral self-selection of nicotine.
Psychopharmacology (Berl) 124, 332±339.
Role, L.W., Berg, D.K., 1996. Nicotinic receptors in the develop-
ment and modulation of CNS synapses. Neuron 16, 1077±1085.
Romanelli, L., Ohman, B., Adem, A., Nordberg, A., 1988.
Subchronic treatment of rats with nicotine: interconversion of
nicotinic receptor subtypes in brain. Eur. J. Pharmacol 148, 289±
291.
Rowell, P.P., Duggan, D.S., 1998. Long-lasting inactivation of nic-
otinic receptor function in vitro by treatment with high concen-
trations of nicotine. Neuropharmacology 37, 103±111.
Rubboli, F., Court, J.A., Sala, C., Morris, C., Chini, B., Perry, E.,
Clementi, F., 1994b. Distribution of nicotinic receptors in the
human hippocampus and thalamus. Eur. J. Neurosci 6, 1596±
1604.
Rubboli, F., Court, J.A., Sala, C., Morris, C., Perry, E., Clementi,
F., 1994a. Distribution of neuronal nicotinic receptor subunits in
human brain. Neurochem. Int 25, 69±71.
Rusted, J., Graupner, L., O'Connell, N., Nicholls, C., 1994. Does
nicotine improve cognitive function? . Psychopharmacology (Berl)
115, 547±549.
Rusted, J.M., Warburton, D.M., 1992. Facilitation of memory by
post-trial administration of nicotine: evidence for an attentional
explanation. Psychopharmacology (Berl) 108, 452±455.
Sabbagh, M.N., Reid, R.T., Corey-Bloom, J., Rao, T.S., Hansen,
L.A., Alford, M., Masliah, E., Adem, A., Lloyd, G.K., Thal,
L.J., 1998. Correlation of nicotinic binding with neurochemical
markers in Alzheimer's disease. J. Neural. Transm 105, 709±717.
Saji, H., Watanabe, A., Kiyono, Y., Magata, Y., Iida, Y., Takaishi,
Y., Yokoyama, A., 1995. Application of �125I� (S)-5-iodonicotine,a new radioiodinated ligand, in the assay of nicotinic acetyl-
choline receptor binding in the brain. Biol. Pharm. Bull 18, 1463±
1466.
Salin-Pascual, R.J., Drucker-Colin, R., 1998. A novel e�ect of nic-
otine on mood and sleep in major depression. Neuroreport 9, 57±
60.
Salin-Pascual, R.J., Rosas, M., Jimenez-Genchi, A., Rivera-Meza,
B.L., Delgado-Parra, V., 1996. Antidepressant e�ect of transder-
mal nicotine patches in nonsmoking patients with major de-
pression. J. Clin. Psychiatry 57, 387±389.
Sanberg, P.R., Fogelson, H.M., Manderscheid, P.Z., Parker, K.W.,
Norman, A.B., McConville, B.J., 1988. Nicotine gum and halo-
peridol in Tourette's syndrome (letter). Lancet 1, 592.
Sanberg, P.R., Silver, A.A., Shytle, R.D., Philipp, M.K., Cahill,
D.W., Fogelson, H.M., McConville, B.J., 1997. Nicotine for the
treatment of Tourette's syndrome. Pharmacol. Ther 74, 21±25.
Sargent, P.B., 1993. The diversity of neuronal nicotinic acetylcholine
receptors. Annu. Rev. Neurosci 16, 403±443.
Sche�el, U., Taylor, G.F., Kepler, J.A., Carroll, F.I., Kuhar, M.J.,
1995. In vivo labeling of neuronal nicotinic acetylcholine recep-
tors with radiolabeled isomers of norchloroepibatidine.
Neuroreport 6, 2483±2488.
Sche�er, I.E., Bhatia, K.P., Lopes-Cendes, I., Fish, D.R., Marsden,
C.D., Andermann, E., Andermann, F., Desbiens, R., Keene, D.,
Cendes, F., et al., 1995. Autosomal dominant nocturnal frontal
lobe epilepsy. A distinctive clinical disorder. Brain 118, 61±73.
Schneider, J.S., Pope-Coleman, A., van Velson, M., Menzaghi, F.,
Lloyd, G.K., 1998a. E�ects of SIB-1508Y, a novel neuronal nic-
otinic acetylcholine receptor agonist, on motor behavior in par-
kinsonian monkeys. Mov. Disord 13, 637±642.
Schneider, J.S., van Velson, M., Menzaghi, F., Lloyd, G.K., 1998b.
E�ects of the nicotinic acetylcholine receptor agonist SIB-1508Y
on object retrieval performance in MPTP-treated monkeys: com-
parison with levodopa treatment. Ann. Neurol 43, 311±317.
Scho®eld, G.G., Witkop, B., Warnick, J.E., Albuquerque, E.X.,
1981. Di�erentiation of the open and closed states of the ionic
channels of nicotinic acetylcholine receptors by tricyclic anti-
depressants. Proc. Natl. Acad. Sci. USA 78, 5240±5244.
Schrattenholz, A., Pereira, E.F., Roth, U., Weber, K.H.,
Albuquerque, E.X., Maelicke, A., 1996. Agonist responses of
neuronal nicotinic acetylcholine receptors are potentiated by a
novel class of allosterically acting ligands. Mol. Pharmacol 49, 1±
6.
SchroÈ der, H., de Vos, R.A., Jansen, E.N., Birtsch, C., Wevers, A.,
Lobron, C., Nowacki, S., Schroder, R., Maelicke, A., 1995. Gene
expression of the nicotinic acetylcholine receptor alpha4 subunit
in the frontal cortex in Parkinson's disease patients. Neurosci.
Lett 187, 173±176.
Schwartz, R.D., Kellar, K.J., 1985. In vivo regulation of �3H]acetyl-
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 109

tylcholine recognition sites in brain by nicotinic cholinergic drugs.
J. Neurochem 45, 427±433.
Semba, J., Mataki, C., Yamada, S., Nankai, M., Toru, M., 1998.
Antidepressantlike e�ects of chronic nicotine on learned helpless-
ness paradigm in rats. Biol. Psychiatry 43, 389±391.
Sershen, H., Hashim, A., Lajtha, A., 1987. behavioral and biochemi-
cal e�ects of nicotine in an MPTP-induced mouse model of
Parkinson's disease. Pharmacol. Biochem. Behav 28, 299±303.
Sherwood, N., 1995. E�ects of cigarette smoking on performance in
a simulated driving task. Neuropsychobiology 32, 161±165.
Shimohama, S., Taniguchi, T., Fujiwara, M., Kameyama, M., 1985.
Biochemical characterization of the nicotinic cholinergic receptors
in human brain: binding of (-)-[3H]nicotine. J. Neurochem 45,
604±660.
Sihver, W., Fasth, K.J., Ogren, M., Bivehed, H., Bergstrom, M.,
Nordberg, A., Watanabe, Y., Langstrom, B., 1998b. In vitro
evaluation of 11C-labeled (S)-nicotine, (S)-3-methyl-5-(1-methyl-
2-pyrrolidinyl)isoxazole, and (R,S)-1-methyl-2-(3-pyridyl)azetidine
as nicotinic receptor ligands for positron emission tomography
studies. J. Neurochem 71, 1750±1760.
Sihver, W., Gillberg, P.G., Nordberg, A., 1998a. Laminar distri-
bution of nicotinic receptor subtypes in human cerebral cortex as
determined by �3H](-)nicotine, �3H]cytisine and �3H]epibatidine in
vitro autoradiography. Neuroscience 85, 1121±1133.
Sihver, W., Fasth, K.J., Horti, A.G., Koren, A.O., BergstroÈ m, M.,
Lu, L., Hagberg, G., Lundqvist, H., Dannals, R.G., London,
E.D., Nordberg, A., Langstrom, B., 1999a. Synthesis and charac-
terization of binding of 5-[76Br]bromo-3-[[2(S)-azetidinyl]-
methoxy] pyridine, a novel nicotinic acetylcholine receptor ligand,
in rat brain. J. Neurochem. 73, 1264±1272.
Sihver, W., Fasth, K.J., OÈ gren, M., Lundqvist, H., BergstroÈ m, M.,
Watanabe, Y., LaÊ ngstroÈ m, B., Nordberg, A., 1999b. In vivo posi-
tron emission tomography studies on the novel nicotinic receptor
agonist [11C] MP compared with [11C] ABT and (S)(ÿ) [11C]
nicotine in Rhesus monkeys. Nucl. Med. Biol. 26, 633±640.
Sihver, W., Gillberg, P.G., Nordberg, A., 1999. Autoradiographic
comparison of (-) �3H� nicotine, �3H� cytisine and �3H� epibatidinebinding in relation to vesicular acetylcholine transport sites in
temporal cortex in Alzheimer's disease. Neuroscience, (in press).
Silverman, J.M., Greenberg, D.A., Altstiel, L.D., Siever, L.J., Mohs,
R.C., Smith, C.J., Zhou, G., Hollander, T.E., Yang, X.P.,
Kedache, M., Li, G., Zaccario, M.L., Davis, K.L., 1996.
Evidence of a locus for schizophrenia and related disorders on
the short arm of chromosome 5 in a large pedigree. Am. J. Med.
Genet 67, 162±171.
Simmons, L.K., Schuetze, S.M., Role, L.W., 1990. Substance P
modulates single-channel properties of neuronal nicotinic acetyl-
choline receptors. Neuron 4, 393±403.
SjoÈ berg, S.L., Svensson, A.I., Zhang, X., Nordberg, A., 1998.
Neuronal nicotinic receptor activation: a promising strategy for
treatment of Alzheimer's disease? . International Journal of
Geriatric Psychopharmacology 1, 145±149.
Socci, D.J., Sanberg, P.R., Arendash, G.W., 1995. Nicotine enhances
Morris water maze performance of young and aged rats.
Neurobiol. Aging 16, 857±860.
Spurden, D.P., Court, J.A., Lloyd, S., Oakley, A., Perry, R.,
Pearson, C., Pullen, R.G., Perry, E.K., 1997. Nicotinic receptor
distribution in the human thalamus: autoradiographical localiz-
ation of �3H]nicotine and �125I� alpha-bungarotoxin binding. J.
Chem. Neuroanat 13, 105±113.
Stage, K.B., Glassman, A.H., Covey, L.S., 1996. Depression after
smoking cessation: case reports. J. Clin. Psychiatry 57, 467±469.
Stauderman, K.A., Maha�y, L.S., Akong, M., Velicelebi, G.,
Chavez-Noriega, L.E., Crona, J.H., Johnson, E.C., Elliott, K.J.,
Gillespie, A., Reid, R.T., Adams, P., Harpold, M.M., Corey-
Naeve, J., 1998. Characterization of human recombinant neur-
onal nicotinic acetylcholine receptor subunit combinations
alpha2beta4, alpha3beta4 and alpha4beta4 stably expressed in
HEK293 cells. J. Pharmacol. Exp. Ther 284, 777±789.
Stein, E.A., Pankiewicz, J., Harsch, H.H., Cho, J.K., Fuller, S.A.,
Ho�mann, R.G., Hawkins, M., Rao, S.M., Bandettini, P.A.,
Bloom, A.S., 1998. Nicotine-induced limbic cortical activation in
the human brain: a functional MRI study. Am. J. Psychiatry 155,
1009±1015.
Steinlein, O.K., Mulley, J.C., Propping, P., Wallace, R.H., Phillips,
H.A., Sutherland, G.R., Sche�er, I.E., Berkovic, S.F., 1995. A
missense mutation in the neuronal nicotinic acetylcholine receptor
alpha4 subunit is associated with autosomal dominant nocturnal
frontal lobe epilepsy. Nat. Genet 11, 201±203.
Stitzel, J.A., Farnham, D.A., Collins, A.C., 1996. Chronic corticos-
terone treatment elicits dose-dependent changes in mouse brain
alpha-bungarotoxin binding. Neuroscience 72, 791±799.
Stolerman, I.P. and Jarvis, M.J., 1995. The scienti®c case that nic-
otine is addictive. Psychopharmacology (Berl) 117, 2±10, discus-
sion 14±20.
Storch, A., Schrattenholz, A., Cooper, J.C., Abdel Ghani, E.M.,
Gutbrod, O., Weber, K.H., Reinhardt, S., Lobron, C., Hermsen,
B., Soskic, V., et al., 1995. Physostigmine, galanthamine and
codeine act as ``noncompetitive nicotinic receptor agonists'' on
clonal rat pheochromocytoma cells. Eur. J. Pharmacol 290, 207±
219.
Sugaya, K., Giacobini, E., Chiappinelli, V.A., 1990. Nicotinic acetyl-
choline receptor subtypes in human frontal cortex: changes in
Alzheimer's disease. J. Neurosci. Res 27, 349±359.
Sullivan, J.P., Donnelly-Roberts, D., Briggs, C.A., Anderson, D.J.,
Gopalakrishnan, M., Piattoni-Kaplan, M., Campbell, J.E.,
McKenna, D.G., Molinari, E., Hettinger, A.M., Garvey, D.S.,
Wasicak, J.T., Holladay, M.W., Williams, M., Arneric, S.P.,
1996. A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro
pharmacological properties of a novel, high a�nity alpha4beta2
nicotinic acetylcholine receptor ligand. Neuropharmacology 35,
725±734.
Svensson, A.L., Nordberg, A., 1996. Tacrine interacts with an allo-
steric activator site on alpha4beta2 nAChRs in M10 cells.
Neuroreport 7, 2201±2205.
Svensson, A.L., Nordberg, A., 1998. Tacrine and donepezil attenuate
the neurotoxic e�ect of A beta(25±35) in rat PC12 cells.
Neuroreport 9, 1519±1522.
Svensson, A.L., Warpman, U., Hellstrom-Lindahl, E., Bogdanovic,
N., Lannfelt, L., Nordberg, A., 1997. Nicotinic receptors, muscar-
inic receptors and choline acetyltransferase activity in the tem-
poral cortex of Alzheimer patients with di�ering apolipoprotein E
genotypes. Neurosci. Lett 232, 37±40.
Terzano, S., Court, J.A., Fornasari, D., Gri�ths, M., Spurden, D.P.,
Lloyd, S., Perry, R.H., Perry, E.K., Clementi, F., 1998.
Expression of the alpha3 nicotinic receptor subunit mRNA in
aging and Alzheimer's disease. Brain Res. Mol. Brain Res 63, 72±
78.
Tohgi, H., Utsugisawa, K., Yoshimura, M., Nagane, Y., Mihara,
M., 1998. Age-related changes in nicotinic acetylcholine receptor
subunits alpha4 and beta2 messenger RNA expression in post-
mortem human frontal cortex and hippocampus. Neurosci. Lett
245, 139±142.
Tsuang, M.T., 1993. Genotypes, phenotypes, and the brain. A search
for connections in schizophrenia. Br. J. Psychiatry 163, 299±307.
Valenzuela, C.F., Dowding, A.J., Arias, H.R., Johnson, D.A., 1994.
Antibody-induced conformational changes in the Torpedo nic-
otinic acetylcholine receptor: a ¯uorescence study. Biochemistry
33, 6586±6594.
Valette, H., Bottlaender, M., Dolle, F., Dolci, L., Syrota, A.,
Crouzel, C., 1997. An attempt to visualize baboon brain nicotinic
receptors with N-[11C]ABT-418 and N-[11C]methyl-cytisine. Nucl.
Med. Commun 18, 164±168.
Vandermeeren, M., Mercken, M., Vanmechelen, E., Six, J., van de
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111110

Voorde, A., Martin, J.J., Cras, P., 1993. Detection of tau proteins
in normal and Alzheimer's disease cerebrospinal ¯uid with a sen-
sitive sandwich enzyme-linked immunosorbent assay. J.
Neurochem 61, 1828±1834.
Vaupel, D.B., Mukhin, A.G., Kimes, A.S., Horti, A.G., Koren,
A.O., London, E.D., 1998. In vivo studies with �125I]5-I-A-85380,
a nicotinic acetylcholine receptor radioligand. Neuroreport 9,
2311±2317.
Vidal, C., 1996. Nicotinic receptors in the brain. Molecular biology,
function, and therapeutics. Mol. Chem. Neuropathol 28, 3±11.
Villemagne, V.L., Horti, A., Sche�el, U., Ravert, H.T., Finley, P.,
Clough, D.J., London, E.D., Wagner Jr, H.N., Dannals, R.F.,
1997. Imaging nicotinic acetylcholine receptors with ¯uorine-18-
FPH, an epibatidine analog. J. Nucl. Med 38, 1737±1741.
Wang, F., Gerzanich, V., Wells, G.B., Anand, R., Peng, X., Keyser,
K., Lindstrom, J., 1996. Assembly of human neuronal nicotinic
receptor alpha5 subunits with alpha3, beta2, and beta4 subunits.
J. Biol. Chem 271, 17656±17665.
Wang, S., Sun, C.E., Walczak, C.A., Ziegle, J.S., Kipps, B.R.,
Goldin, L.R., Diehl, S.R., 1995. Evidence for a susceptibility
locus for schizophrenia on chromosome 6pter-p22. Nat. Genet
10, 41±46.
Warburton, D.M., Wesnes, K., Shergold, K., James, M., 1986.
Facilitation of learning and state dependency with nicotine.
Psychopharmacology (Berl) 89, 55±59.
Warpman, U., Friberg, L., Gillespie, A., Hellstrom-Lindahl, E.,
Zhang, X., Nordberg, A., 1998. Regulation of nicotinic receptor
subtypes following chronic nicotinic agonist exposure in M10 and
SH-SY5Y neuroblastoma cells. J. Neurochem 70, 2028±2037.
Warpman, U., Nordberg, A., 1995. Epibatidine and ABT 418 reveal
selective losses of alpha4beta2 nicotinic receptors in Alzheimer
brains. Neuroreport 6, 2419±2423.
Weiland, S., Witzemann, V., Villarroel, A., Propping, P., Steinlein,
O., 1996. An amino acid exchange in the second transmembrane
segment of a neuronal nicotinic receptor causes partial epilepsy
by altering its desensitization kinetics. FEBS Lett 398, 91±96.
Wesnes, K., Warburton, D.M., 1983. Smoking, nicotine and human
performance. Pharmacol. Ther 21, 189±208.
West, R.J., Jarvis, M.J., 1986. E�ects of nicotine on ®nger tapping
rate in non-smokers. Pharmacol. Biochem. Behav 25, 727±731.
Wevers, A., Jeske, A., Lobron, C., Birtsch, C., Heinemann, S.,
Maelicke, A., Schroder, R., Schroder, H., 1994. Cellular distri-
bution of nicotinic acetylcholine receptor subunit mRNAs in the
human cerebral cortex as revealed by non-isotopic in situ hybrid-
ization. Brain Res. Mol. Brain Res 25, 122±128.
Whitehouse, P.J., Au, K.S., 1986. Cholinergic receptors in aging and
Alzheimer's disease. Prog. Neuropsychopharmacol Biol.
Psychiatry 10, 665±676.
Whitehouse, P.J., Hedreen, J.C., White, C.L.d., Price, D.L., 1983.
Basal forebrain neurons in the dementia of Parkinson disease.
Ann. Neurol 13, 243±248.
Whitehouse, P.J., Martino, A.M., Antuono, P.G., Lowenstein, P.R.,
Coyle, J.T., Price, D.L., Kellar, K.J., 1986. Nicotinic acetyl-
choline binding sites in Alzheimer's disease. Brain Res 371, 146±
151.
Whitehouse, P.J., Martino, A.M., Wagster, M.V., Price, D.L.,
Mayeux, R., Atack, J.R., Kellar, K.J., 1988a. Reductions in
�3H]nicotinic acetylcholine binding in Alzheimer's disease and
Parkinson's disease: an autoradiographic study. Neurology 38,
720±723.
Whitehouse, P.J., Price, D.L., Struble, R.G., Clark, A.W., Coyle,
J.T., Delon, M.R., 1982. Alzheimer's disease and senile dementia:
loss of neurons in the basal forebrain. Science 215, 1237±1239.
Whiting, P., Esch, F., Shimasaki, S., Lindstrom, J., 1987. Neuronal
nicotinic acetylcholine receptor beta-subunit is coded for by the
cDNA clone alpha4. FEBS Lett 219, 459±463.
Wonnacott, S., 1997. Presynaptic nicotinic ACh receptors. Trends
Neurosci 20, 92±98.
Wonnacott, S., Drasdo, A., Sanderson, E., Rowell, P., 1990.
Presynaptic nicotinic receptors and the modulation of transmitter
release. Ciba Found Symp 152, 87±101, discussion 102±5.
Zamani, M.R., Allen, Y.S., Owen, G.P., Gray, J.A., 1997. Nicotine
modulates the neurotoxic e�ect of beta-amyloid protein(25±35))
in hippocampal cultures. Neuroreport 8, 513±517.
Zhang, X., Gong, Z.H., Fasth, K.J., Langstrom, B., Nordberg, A.,
1998. Interaction of the nicotinic agonist (R,S)-3-pyridyl-1-
methyl-2-(3- pyridyl)-azetidine (MPA) with nicotinic acetylcholine
receptor subtypes expressed in cell lines and rat cortex.
Neurochem. Int 32, 435±441.
Zoli, M., Le Novere, N., Hill Jr, J.A., Changeux, J.P., 1995.
Developmental regulation of nicotinic ACh receptor subunit
mRNAs in the rat central and peripheral nervous systems. J.
Neurosci 15, 1912±1939.
Zoli, M., Lena, C., Picciotto, M.R., Changeux, J.P., 1998.
Identi®cation of four classes of brain nicotinic receptors using
beta2 mutant mice. J. Neurosci 18, 4461±4472.
D. Paterson, A. Nordberg / Progress in Neurobiology 61 (2000) 75±111 111













![Feasibility of [18F]-2-Fluoro-A85380-PET Imaging of Human Vascular Nicotinic Acetylcholine Receptors In Vivo](https://img.pdfslide.net/doc/110x75/6361863028d79112c801a3f8/feasibility-of-18f-2-fluoro-a85380-pet-imaging-of-human-vascular-nicotinic-acetylcholine.jpg)















![Simplified quantification of nicotinic receptors with 2[18F]F-A-85380 PET](https://img.pdfslide.net/doc/110x75/635b09704028fc5adf017dba/simplified-quantification-of-nicotinic-receptors-with-218ff-a-85380-pet.jpg)