Embed Size (px)
Citation preview

New cell attachment peptide sequences from conserved epitopesin the carboxy termini of fibrinogen
Raphael Gorodetsky,a,* Akiva Vexler,a Meir Shamir,a Jianqiang An,a Lilia Levdansky,a
Irina Shimeliovich,a and Gerard Marxb
a Biotechnology and Radiobiology Laboratory, Sharett Institute of Oncology, Hadassah University Hospital, P.O. Box 12000, Jerusalem 91120, Israelb HAPTO Biotech Ltd., Hadassah Ein Kerem Campus, P.O. Box 12275, Jerusalem 91121, Israel
Received 24 October 2002, revised version received 10 February 2003
Abstract
Fibrinogen seems to contribute significantly to cell binding and recruitment into wounds besides its major role in clot formation. Wedescribe 19- to 21-mer cell-binding (haptotactic) peptides from the C-termini of fibrinogen �-chain (C�), the extended �E chain, and nearthe C-terminal of the �-chain. When these peptides were covalently bound to a biologically inert matrix such as Sepharose beads (SB), theyelicited beads attachment to cells, mostly of mesenchymal origin (including fibroblasts, endothelial cells, and smooth muscle cells) as wellas some transformed cell lines. Based on such haptotactic activity, these peptides were termed “haptides.” By contrast, peptides homologousto fibrinogen C-termini �- and �-chains elicited no such activity. The haptide C� could not block the interaction of fibroblasts withantibodies directed against integrins �1, �v, �v�1, �v�3, and �II�3. Moreover, GRGDS peptide could not inhibit enhanced cell binding toSB–C�, as expected from an integrin-mediated process. In soluble form the haptides were accumulated in cells with nonsaturable kineticswithout any toxic or proproliferative effects in concentrations up to 80 �M. These findings suggest that the conserved haptidic sequenceswithin fibrin(ogen) can be associated with the adhesion and migration of cells into fibrin clots and may have a significant role in normalwound healing and in various pathological conditions.© 2003 Elsevier Science (USA). All rights reserved.
Keywords: Fibrin(ogen); Fibrinopeptides; Haptides; Haptotaxis; Cell uptake; Cell-binding; Chemotaxis
Introduction
The normal fibrinogen molecule is a 340-kDa complexhexamer containing two sets of three nonidentical chains (�,�, and �) linked by multiple disulfide bonds [1–10]. Whenactivated by thrombin it forms fibrin whose main function isto establish hemostasis. Once a clot has formed it couldsubsequently serve also as a provisional biodegradable ma-trix for cellularization of the wound bed. Initially, platelets,leukocytes, macrophages, and other cells involved in theinflammatory reactions are attracted into the wound [11–15]. Parenchymal cells in tissues, such as fibroblasts,smooth muscle cells, and endothelial cells can be recruitedfrom the tissues adjacent to the damaged tissue [16–23].
The different cell types that penetrate into the clot inducefibrinolysis with plasmin, metalloproteinases, or free radi-cals [10,23–25]. Ultimately, the migrating cells replace theprovisional fibrin clot with newly synthesized extracellularmatrix to form granulation and scar tissues.
Different mechanisms may be involved in fibrin bindingto cells. Activated platelets bind to fibrin via the N-terminalB� epitope 15–42 (exposed by the removal of N-terminalfibrinopeptide B of the �-chain by thrombin) and the se-quence 400–411 of the extreme C-terminus of the�-chain—LGGAKQAGDV [26–29]. Parenchymal cells,such as fibroblasts, smooth muscle cells, and endothelialcells, are recruited from the tissues adjacent to the wound[16–23].
It has been suggested that cells, such as fibroblasts,endothelial cells, and smooth muscle cells, bind to fibrino-gen through cell integrins [13,16,30–32]. Two potential
* Corresponding author. Fax: �972-2-6415073.E-mail address: [email protected] (R. Gorodetsky).
R
Available online at www.sciencedirect.com
Experimental Cell Research 287 (2003) 116–129 www.elsevier.com/locate/yexcr
0014-4827/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved.doi:10.1016/S0014-4827(03)00120-4

cell-binding domains in human fibrinogen are the RGDepitopes that occur at locations 95–97 and 572–574 of the�-chain. But both domains that contain the RGD sequencesin the �-chain of human fibrinogen are poorly conserved inevolution, according to the gene bank molecular databases[33]. For example, in cats, dogs, and chickens, one of theseRGD sequences is missing. In cows, this sequence is alteredand seems to appear at a different nearby address. Thesecond RGD is not present in lampreys, cows, or even in therhesus monkey. Moreover, the lack of conservation of thesedomains with the RGD sequences in fibrinogen of differentspecies does not seem to be associated with differences incellular interactions or deficient wound healing. It also hasbeen shown that recombinant fibrinogen missing the RGDsequences nevertheless retains cell–fibrin interactions mon-itored by the induction of fibrin gel retraction [34,35]. Con-sequently, it was suggested that other sequences in fibrinmight be active in terms of cell attachment
In the current study, we tested whether homologouspeptidic sequences within the conserved epitope on theC-termini of the �-chain and the extended �E-chain, as wellas homologous sequences on the �-chain (Table 1), might
be associated with the cell attachment proclivity of fibrin-ogen. We synthesized a 21-mer peptide sequence of theC-terminus of the �-chain (C�)1 and peptides homologousto 20-mer sequences just preceding the C-terminus of the�-chain (preC�). We also synthesized a homologous pep-tide based on the sequence of the C-terminus of the ex-tended �E-chain (fib420) (C�E), which is present only as asmall fraction of normal adult fibrinogen [17]. Based ontheir positive cell attachment properties, these homologouspeptides were termed “haptides.” The haptotactic responsesof different cell types to the immobilized haptides are de-
1 Abbreviations used: FITC, fluorescein isothiocyanate; SB, Sepharosebeads; C�, C�, C�, and C�E, 19- to 21-mer peptide analogs of the endC-termini sequence of fibrinogen chains �, �, �, and �E, respectively;preC�, 20-mer peptide analog of the address 373–392 near the C-terminalof the �-chain of fibrinogen; haptides, a family of 19- to 21-mer cell-binding (haptotactic) peptides homologous to sequences at the carboxytermini of the �- and �E-chains and prior to the �-chain of fibrinogen(termed C�, C�E, and preC�, respectively); BSA, bovine serum albumin;FCS, fetal calf serum; HF, human skin fibroblasts; SMC, smooth musclecells; BAEC, bovine aortic endothelial cells; FN, fibronectin; HPC, hydro-genated phosphatidyl choline; Chol, cholesterol.
Table 1Conservation of fibrinogen chains C-terminals (C�, preC�, and C�E) sequences
Note. The data were collected with the BLAST-P protein sequence search Gapped BLAST and PSI-BLAST by methods described by Altschul et al. [33]., homology vs. human; , positive vs. human.* Not fibrinogen.
117R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

scribed in Table 2. Our results suggest that each fibrinogenmolecule contains four to six such haptotactic sequencesthat enhance the attachment and migration of different cellstypes into the fibrin clot. We suggest that these epitopescould also be involved in several physiological and patho-logical processes in which fibrin plays a major role, includ-ing tissue regeneration, neovascularization of normalwounds or tumors, and the occurrence of cell-migration-mediated arteriosclerosis or occlusion of blood vessels [36–40].
Materials and methods
Chemicals and reagents
Clinical-grade human fibrinogen and fibronectin werepurified from blood by the New York Blood Center andVitex Inc. (New York, NY). Tissue culture media, serum,bovine serum albumin (BSA), and other reagents were pur-chased from standard commercial sources for laboratorysupply, mainly from Biological Industries (Beit-HaEmek,Israel), Sigma Chemical Co. (Israel and St. Louis, MO) andGIBCO (Grand Island, New York, NY).
Synthesis of peptides
Each of the 19- to 21-mer peptidic sequences presentedin Table 2 were synthesized either by the microchemistryfacility of the New York Blood Center or by SynPep Cor-poration (Dublin, CA). Some batches of the C� were alsosynthesized at the Hebrew University-Hadassah MedicalSchool (Jerusalem, Israel). Most experiments employedpeptides that were �90% pure as determined by HPLC/mass spectrometry. FITC-derivatized peptides were pre-pared with the FITC-tag on the amino terminal (Table 2).
Cell cultures
The cell types used in the present study (Table 3) wereobtained and cultured as previously described [19,20]. Themedia used were RPMI 1640�10% fetal calf serum (FCS)hereafter referred as “RPMI” or DMEM � 10% FCS,hereby referred as “DMEM” with high or low glucose.Standard additives, such as penicillin–streptomycin with
nystatin, L-glutamine, MEM–Eagle vitamin solution, andnonessential amino acids, all bought from Beit-HaEmek(Israel), hereby referred as “additives,” were used. Normalhuman skin fibroblasts (HF) were isolated from skin biop-sies of young normal volunteers and cultured for no morethan 14 passages in high-glucose DMEM � additives. Por-cine and bovine smooth muscle cells (SMC) and normalbovine aortic endothelial cells (BAEC) were isolated fromfresh thoracic aortas collected at a slaughterhouse fromkilled young animals and were kept in culture for up to12–15 passages in low-glucose DMEM �additives. Purenormal human keratinocytes were obtained from the Hadas-sah National Skin Bank and were grown for up to 5 pas-sages. The other cell lines tested were murine fibroblasts(3T3), grown on DMEM �additives. Mouse leukemia cells(P-388), human ovarian carcinoma cells (OV-1063), mousemammary adenocarcinoma cells (EMT-6), mouse macro-phage-like cells (J-774.2), and immortalized human kerati-nocytic cells (HaCaT) [41] were all grown in high-glucoseDMEM �additives. Insulin-secreting murine pancreatic is-let cell line (INS6), provided by Dr. R. Nesher from Ha-dassah Hospital, were grown in low-glucose DMEM orRPMI�10% FCS�additives. The cell cultures were main-tained at 37°C in a water-jacketed CO2 incubator and were
Table 2Synthetic peptides tested and their homology to C�
, homology to C�.
Table 3Attachment (haptotaxis) of SB coated with fibrinogen or C-terminalhaptides to different cell types following 2 days of incubation
Ligands: Attachment of cells to ligands-SB (%)
C� C� C� C�E preC� Fibrin
Normal cell types testedFibroblasts (HF) 0 �95 0 65 �95 �95Smooth muscle cells (SMC) 0 �95 1 8 �95 �90Endothelial cells (BAEC) 0 �95 8–40a 77 �95 �95Human keratinocytes 0 �5 5 0 nd �5
Transformed lines testedMouse mam. Ca (EMT-6) 0 �95 1 90 �95 �95Human HaCaT line 0 �95 nd �90 �95 �95Mouse islet cell line INS6 0 �5 nd 0 �5 �5Human ovar. Ca (OV-1063) 0 nd 0 4 0 3Mouse lymph. line (J-774) 0 nd 0 0 0 33T3 murine fibroblasts 0 �95 0 �95 3 nd
Note. SB coated with fibrinogen or the peptides tested were incubatedwith near confluent cells; the percentage of SB-ligand attached to cells (byday 3) was monitored. nd, not determined.
a Response varied with different cell batches/passages.
118 R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

harvested by trypsin/versen solution with 1–2 passages perweek in a split ratio between 1:5 to 1:10 for fast prolifer-ating transformed cells and 1:4 for normal cell types.
Cell number evaluation by the MTS assay
The MTS colorimetric assay (CellTitre 96 aqueous assayby Promega) was used to monitor the number of viable cellsand cell proliferation. This assay, performed in 96-wellplates, is based on dehydrogenase conversion of MTS byviable cells to colored tetrazolium salt, as previously de-scribed [19]. The optical density at 492 nm (OD492) of the
developed dye was measured with a computerized micro-plate reader (Anthos HT-II, Salzburg). Calibration curvesfor transforming OD492 into the number of viable cells wereperformed for each cell type with plating density and incu-bation conditions optimized for each cell type.
Preparing Sepharose 4B beads-ligand
Peptides, fibrinogen and other proteins were covalentlybound to CNBr-activated sepharose 4B beads (SB) (Phar-macia, Piscataway, NJ) in a procedure previously used tobind fibrinogen, thrombin, and BSA [19]. Briefly, CNBr-activated SB were washed with 1 N HC1 and suspended inthe coupling buffer (pH 8.3), following the protocol sup-plied by the manufacturer. Peptides tested (in concentra-tions of 4–9 mg/ml) were initially dissolved in 0.5 mMdimethylsulfoxide and further diluted with the couplingbuffer. The suspension was gently agitated overnight, cen-trifuged briefly (500g) to pack the SB, incubated with 10mM glycine buffer, and washed three times in Tris–saline.Following this procedure, more than 90% of the ligandsbound covalently to the SB as determined by measuring theresidual material in the supernatant. The concentrations ofpeptides bound to SB in different preparations were asfollows: C�, 4–7 mg/ml; C�, 4.5–5.2 mg/ml; C�, 5.2–9.6mg/ml; preC�, 5.0–9.0 mg/ml; and C�E, 5.0–6.0 mg/ml.Variations in the concentrations of the peptides bound to SBin the range of 3.5–9 mg/ml did not significantly change thedegree of attachment of the SB to responsive cells. SBcoated with either BSA or fibrinogen (6 mg/ml) were sim-ilarly prepared. The SB-ligands were stored in saline at 4°Cwith 0.1% azide or in 96% ethanol and washed extensivelyin sterile PBS prior to use. Both preservation methods didnot affect the eventual activity of the bound peptides.
Haptotaxis assay
The attachment of SB-ligand to cells in nearly confluentcultures was measured as previously described [19]. Essen-tially, approximately 20–150 �l of suspended (50% v/v)SB-ligands was added to near confluent cell cultures in12-well plates and dispersed by gentle shaking for 1 min.The plates were then incubated typically for up to 4 days. Atdifferent time points, the number of SB tethered to cell layerwas counted with an inverted phase or Numarsky micros-copy. Typically, �300 SB (but not less then 200) werecounted in each well, and the ratio of the number of SBattached to the cell layer in each well was calculated relativeto the total number of SB added. Only SB coated withhaptotactic materials attached to the cell layer. Usually,after a day or more in culture the SB became engulfed bythe cells that mounted on them from the monolayer (Figs.1B–D). Uncoated SB or SB coated with non-cell-bindingligands, such as BSA or C� (Fig. 1A), could not bind to thecells and rolled freely on them. The determination of per-centage of SB attached to the cell layer at different timeintervals provided a quantitative estimate of the extent and
Fig. 1. Microscopic view of haptotactic assay: responses of BAEC topeptide-coated Sepharose beads (SB-ligand). The principle of the hapto-taxis assay based on attachment of the SB covalently coated with the ligandtested to monolayer cell culture (A). The cells in the monolayer attach theSB coated with haptides and even load on it, as seen in the scanningelectron micrograph (inset). Light micrographs with Numarsky’s opticswere taken after 48 h of incubation. The control SB coated with C�(SB-C�) clearly did not interact with the cells but rolled freely over the cellmonolayer (B). By contrast, SB coated with C� or preC� became tightlyattached to the culture monolayer by the cells (C and D, respectively).After fixing and staining the plate with Giemsa, one could clearly visualizethe cells mounted on the SB-preC� (E).
119R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129
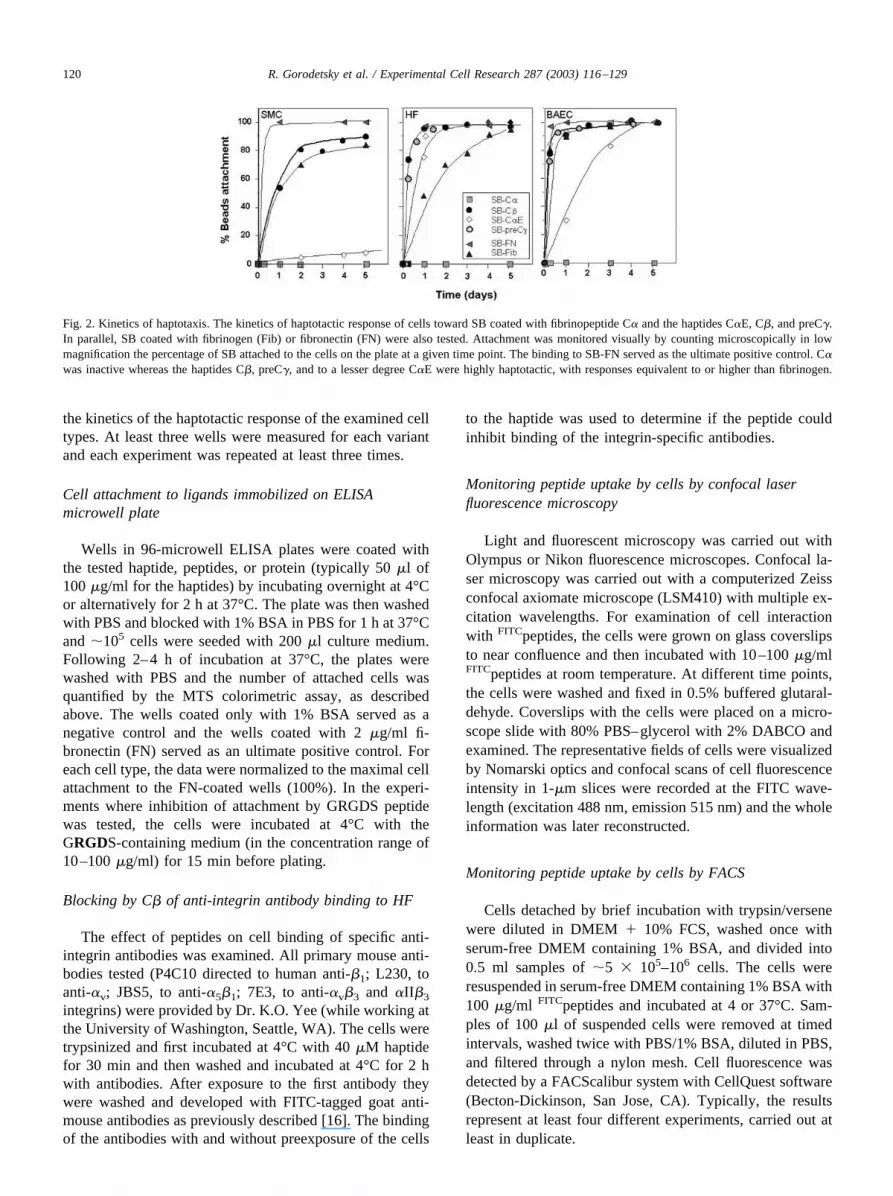
the kinetics of the haptotactic response of the examined celltypes. At least three wells were measured for each variantand each experiment was repeated at least three times.
Cell attachment to ligands immobilized on ELISAmicrowell plate
Wells in 96-microwell ELISA plates were coated withthe tested haptide, peptides, or protein (typically 50 �l of100 �g/ml for the haptides) by incubating overnight at 4°Cor alternatively for 2 h at 37°C. The plate was then washedwith PBS and blocked with 1% BSA in PBS for 1 h at 37°Cand �105 cells were seeded with 200 �l culture medium.Following 2–4 h of incubation at 37°C, the plates werewashed with PBS and the number of attached cells wasquantified by the MTS colorimetric assay, as describedabove. The wells coated only with 1% BSA served as anegative control and the wells coated with 2 �g/ml fi-bronectin (FN) served as an ultimate positive control. Foreach cell type, the data were normalized to the maximal cellattachment to the FN-coated wells (100%). In the experi-ments where inhibition of attachment by GRGDS peptidewas tested, the cells were incubated at 4°C with theGRGDS-containing medium (in the concentration range of10–100 �g/ml) for 15 min before plating.
Blocking by C� of anti-integrin antibody binding to HF
The effect of peptides on cell binding of specific anti-integrin antibodies was examined. All primary mouse anti-bodies tested (P4C10 directed to human anti-�1; L230, toanti-�v; JBS5, to anti-�5�1; 7E3, to anti-�v�3 and �II�3
integrins) were provided by Dr. K.O. Yee (while working atthe University of Washington, Seattle, WA). The cells weretrypsinized and first incubated at 4°C with 40 �M haptidefor 30 min and then washed and incubated at 4°C for 2 hwith antibodies. After exposure to the first antibody theywere washed and developed with FITC-tagged goat anti-mouse antibodies as previously described [16]. The bindingof the antibodies with and without preexposure of the cells
to the haptide was used to determine if the peptide couldinhibit binding of the integrin-specific antibodies.
Monitoring peptide uptake by cells by confocal laserfluorescence microscopy
Light and fluorescent microscopy was carried out withOlympus or Nikon fluorescence microscopes. Confocal la-ser microscopy was carried out with a computerized Zeissconfocal axiomate microscope (LSM410) with multiple ex-citation wavelengths. For examination of cell interactionwith FITCpeptides, the cells were grown on glass coverslipsto near confluence and then incubated with 10–100 �g/mlFITCpeptides at room temperature. At different time points,the cells were washed and fixed in 0.5% buffered glutaral-dehyde. Coverslips with the cells were placed on a micro-scope slide with 80% PBS–glycerol with 2% DABCO andexamined. The representative fields of cells were visualizedby Nomarski optics and confocal scans of cell fluorescenceintensity in 1-�m slices were recorded at the FITC wave-length (excitation 488 nm, emission 515 nm) and the wholeinformation was later reconstructed.
Monitoring peptide uptake by cells by FACS
Cells detached by brief incubation with trypsin/versenewere diluted in DMEM � 10% FCS, washed once withserum-free DMEM containing 1% BSA, and divided into0.5 ml samples of �5 � 105–106 cells. The cells wereresuspended in serum-free DMEM containing 1% BSA with100 �g/ml FITCpeptides and incubated at 4 or 37°C. Sam-ples of 100 �l of suspended cells were removed at timedintervals, washed twice with PBS/1% BSA, diluted in PBS,and filtered through a nylon mesh. Cell fluorescence wasdetected by a FACScalibur system with CellQuest software(Becton-Dickinson, San Jose, CA). Typically, the resultsrepresent at least four different experiments, carried out atleast in duplicate.
Fig. 2. Kinetics of haptotaxis. The kinetics of haptotactic response of cells toward SB coated with fibrinopeptide C� and the haptides C�E, C�, and preC�.In parallel, SB coated with fibrinogen (Fib) or fibronectin (FN) were also tested. Attachment was monitored visually by counting microscopically in lowmagnification the percentage of SB attached to the cells on the plate at a given time point. The binding to SB-FN served as the ultimate positive control. C�was inactive whereas the haptides C�, preC�, and to a lesser degree C�E were highly haptotactic, with responses equivalent to or higher than fibrinogen.
120 R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

Monitoring peptide uptake by cells by 125I-C�
Iodination of C� was performed with the use of thechloramine T procedure. Briefly, peptide (10 �g) was addedto 50 �l of 20 mM PBS, pH 7.2, containing 1 mCi Na125I.Chloramine T (10 �l, 1 mg/ml) was added for 1 min at roomtemperature. The reaction was stopped by adding of sodiummetabisulfite (50 �l, 1 mg/ml) and sodium iodide (50 �l, 2mg/ml). The reaction mixture was then applied to a Sepha-rose G-10 (Pharmacia) column equilibrated with PBS (20mM, pH 7.2), and the iodinated haptide was eluted andseparated from the unbound iodine. The specific activity ofpeptide was counted by automatic gamma counter (1470Wizard, EG&G-Wallac, Turku, Finland) and it was approx-imately 3.5 � 104 cpm/ng. Labeled peptide was aliquotedand kept at �20°C and each portion was thawed immedi-ately before experiment. Experiments of peptide uptake bycells were performed with 125I-C� alone or with additionalunlabeled C� in a molar ratio of up to 1:2000. To measurethe uptake of 125I-C� by cells, the nearly confluent cellswere trypsinized briefly, 3 � 105–5 � 105, and rinsed cellsin 0.5 ml culture serum-free medium � 0.1% BSA wereadded to small 1.5-ml Eppendorf test tubes and kept at 4°C.The peptide solution containing different concentrations andratios of labeled and unlabeled peptide was added andincubated with the cells at 4°C for 30 min. The cells werethen packed by centrifugation and washed four times withmedium without serum � 0.1% BSA. The bottoms of test
tubes with cell pellets were then cut off and the radioactivitywas measured in a gamma counter. Test tube tips with nocells, to which the labeled peptide was added and thenrinsed, were measured as background controls. In eachexperiment the specific activity of the 125I-C� solution wasmeasured to translate radioactivity (cpm) into peptide con-centrations.
Chemotaxis–migration assay
Chemotaxis was evaluated using a 48-well plastic mi-crotaxis micro-Boyden chambers according to the manufac-turer’s instruction (Neuro-Probe, Gaithersburg, MD). Poly-carbonate filters with pore size of 8 �m were coated withfibronectin for 2 h at 37°C. Conditioned medium from 3T3cells served as a positive control. Test solutions (26 �l)containing the peptides, proteins, or medium with no addi-tions (that served as control) were placed in the lowerchamber and covered with the filter, and 50 �l of cellsuspension was introduced to the upper chamber of eachwell. The number of cells per well varied between 5,000 and10,000 in different experiments. The device loaded withcells was incubated at 37°C for 4–5 h. The filter was thenremoved and the nonmigrating cells on its upper part werecarefully wiped off with a damp cotton swab. The filter wasfixed in methanol and stained with Accustain (modifiedWright stain) solution (Sigma Chemical Co.). Cells thatcrossed or were retained within the filter were countedmicroscopically at low magnification, so that the viewingfield corresponded to the whole area of each well. Eachexperimental variant was performed in duplicate or tripli-cate and the data from three successful experiments werenormalized to the values of the positive control and aver-aged.
HPLC gel filtration of haptides
Haptides were dissolved to 1 mg/ml in 0.1 M phosphatebuffer, pH 7.1, and injected into a Waters protein pack diol(10 �m particles) column, with a 2,000–80,000 range sep-aration range, in a SpectroPhysics HPLC system equippedwith a Spectraflow 773 absorbance detector, with a flow rateof 1 ml/min. Elution was monitored at 280 nm. Bovinealbumin (MW 66,000 Da), insulin (MW 6000 Da), andthyrocalcitonin (MW 3604 Da) (Sigma Chemical Co.) wereused as size calibrants.
Association of haptides with liposomes
Liposomes were prepared from hydrogenated phosphati-dyl choline (HPC) and cholesterol (Avanti Polar Lipids,Birmingham, AL). In a typical preparation, 160 mg wasmixed with 40 mg cholesterol (Chol) in absolute ethanol(100 �l) and chloroform (100 �l) at 60°C for 15 min. TheHPC/Chol was added to the stirred solution and vortexed for1 min. The mixture was further incubated at 60°C for 1 h,cooled to 22°C, and centrifuged at 2000g for 5 min to settle
Fig. 3. Cell attachment to haptide-coated plastic ELISA plates. Attachmentresponses of HF and BAEC to plastic ELISA microplates coated withdifferent peptides as well as fibronectin (FN) and fibrinogen. BSA-coatedwells served as a control and all other samples were blocked with BSAexcept for the additional control of bare plastic. FN-coated wells served asthe ultimate positive control. Inhibition of attachment by GRGDS peptideadded to the cell suspension was also tested with all samples. After 2–4 hof incubation, when significant cell attachment was seen microscopically inthe FN-coated well, the unattached cells from all wells were rinsed awayand the number of attached cells per well was evaluated by the MTS assay.Inhibition of cell attachment by RGD was noted for fibronectin, thrombin,and fibrinogen. Apparently the GRGD peptide itself was too small to bindcells to the plate and that minute response could not be significantlyinhibited by soluble GRGDS peptide. Wells coated with C� or C� inducedno cell attachment above that in the negative control, whereas C� and C�Eshowed significant cell binding, second only to fibronectin-coated wells.Soluble GRGDS peptide had no significant effect on the haptide-inducedcell binding.
121R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

the liposomes. The supernatant was discarded, and the li-posomes were resuspended, washed in Tris/saline, pH 7.4,buffer, and stored at 4°C. The size distribution of the lipo-
somes was measured in a particle counter. The volume ofthe majority of the liposomes (90%) was 2–10 fl and the restwere 40–100 fl (�1.8 � 109 liposomes per ml). The lipo-
Fig. 4. Binding and internalization of soluble haptide by cells as viewed by confocal fluorescent microscopy. Confocal fluorescent microscopy with FITCfluorescence signals superposed on light Numarsky optics show the FITChaptides taken by BAEC (top row) and HF (bottom row) in a lateral section throughthe cells. The cells were incubated with 40 �M FITCpeptides for 60 and 120 min. (A and E) The cells with no peptides. Significant internalization of FITCC�into the cells was observed within 60 min (C and G) and 120 min (D and H). The haptide FITCC� can be seen distributed in the cytoplasm but only minimallytaken by the nucleus. Similar results were recorded for the uptake of haptides preC� and C�E (not shown). (B and F) The minimal internalization of a controlpeptide (FITCC�) within 60 min.Fig. 5. Uptake of FITCC� by BAEC as recorded by FACS. The cells’ fluorescence as detected by FACS is presented in dot plots (A–D). (A) The autofluorescenceof the cells. A similar scatter diagram was obtained for cells incubated with FITCC� (not shown). (B–D) Cell fluorescence following exposure for 1 h to 4 �M FITCC�,to 40 �M of unlabeled C� with 4 �M FITCC�, and to 40 �M of FITCC� (respectively). The data show that after exposure to low FITCC� levels, only a small fractionof the cells stain enough to appear as a distinct population. Short preexposure to high level of unlabeled C� following exposure to the same concentration of FITCC�enhanced the uptake of the fluorescence-tagged haptide. When the cells were exposed to high FITCC� levels, all the cell populations were significantly stained,showing that all cells internalized significant amounts of FITCC�. (E) Overlapping histograms of cell uptake of FITCC� from A–D. The cells seemed to be dividedinto two subpopulations, possibly due to the tendency of FITCC� to aggregate in the higher concentrations. Cells that internalized such labeled microaggregates wouldappear as having much higher fluorescence than those that internalized mostly monomeric labeled haptide.
122 R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

some suspensions were stored at 4°C until ready to use. Totest ligand binding, liposomes were mixed with 0.1 mg/mlFITC-tagged haptides and examined by confocal fluores-cence microscopy.
Gene bank database
The databases for fibrinogen sequences in human andother species were searched using Gapped BLAST andPSI-BLAST program [33].
Results
Haptotaxis of cells to SB-bound peptides or proteins
A quantitative assay, based on counting the percentage ofthe haptide-coated SB that attached to the cell monolayer,
was used to evaluate the haptotactic potency of peptides(Fig. 1A). In this assay, SB coated with BSA floated freelyin the culture medium without interacting with the cells(�0% haptotaxis), even after prolonged incubation of up to5 days. Similar results were also seen with blank SB (Fig.1B). However, when SB covalently coated with C�, preC�,or C�E haptides were added to a culture of responsive celltypes, the cells tightly tethered the haptides-coated SB,which also mounted and engulfed the coated SB in a processthat extended from a few hours to 48 h or more (Figs. 1C–E1A, inset).
Typical kinetics of binding the SB-ligand to cell mono-layer with time are shown for HF, SMC, and BAEC (Fig. 2).Similar results were obtained when the SB were covalentlycoated with haptotactic molecules such as fibronectin andfibrinogen. Although a close to 100% binding for the activehaptides on SB was monitored after up to 48 h at concen-trations as low as 4 mg/g SB, increasing the level of boundhaptides on the SB resulted in faster kinetics.
A survey of cultured cells showed that some normal celltypes such as HF, SMC, and BAEC responded even more topreC� and C� than to immobilized native fibrinogen. SB-C�E had a somewhat lower effect (Table 3). By contrast,normal human keratinocytes did not bind to the SB coatedwith either fibrinogen [19,20] or haptides (Table 3) whereasa keratinocytic cell line (HaCaT), which binds better toplastic plates, also showed high haptotactic response toSB-haptides.
As to the transformed cell lines tested, EMT-6 mousemammary carcinoma cells, as well as mouse 3T3 fibro-blasts, adhered to the SB haptide. By contrast, other celllines such as mouse macrophage-like cells (J774), humanovarian carcinoma (OV-1063), and insulin-secreting cells(INS6) did not adhere well to the haptide-coated SB. Over-all, the SB-preC� elicited a haptotactic response profilefrom cells equivalent to SB-C�, while SB-C�E seemed tobe somewhat less active.
As to the other fibrin(ogen) 20-mer C-terminal peptidestested, neither the C� nor the C� were haptotactic towardany of the tested cell types. Therefore, C� was typicallyemployed as a negative control for evaluation peptide asso-ciated haptotactic activity.
Attachment of different cell types to haptide-coated ELISAmicrowell plates
Cell attachment to ELISA microwell plate coated withhaptides was tested both with HF and BAEC (Fig. 3). Theproteins that coated the plates were in a molar concentrationsimilar to that of the peptides tested. The wells coated withfibronectin served as the ultimate positive control. Cellsadhered to adsorbed fibrinogen better than to the negativeBSA control (P � 0.005) or to noncoated wells. As to thehaptides, the C� induced significant enhancement of cellbinding to the wells (P � 0.001), higher than the responseto the whole fibrinogen molecule (P � 0.01) and secondonly to the response to fibronectin. The C�E elicited a lower
Fig. 6. Uptake kinetics of FITChaptide by BAEC and HF as recorded byFACS. (A) The uptake of increasing concentrations of FITCC� andFITCC�E haptides (at 1 h) by both HF and BAEC. (B) The uptake kineticsof a low concentration of FITCC� (12 �M) by BAEC. (C) The uptake ofFITCC� and FITCpreC� (12 �M) by BAEC after incubation of the cells withthe corresponding unlabeled peptide at concentrations of up to 80 �M. At4°C, the uptake of FITCC� as well as FITCpreC� by cells was not reducedby prior incubation with unlabeled haptides. At 37°C, it is more apparentthat preexposure of the cells to unlabeled haptides increased, rather thandecreased, the uptake of subsequently added FITChaptide. In these cells,preC� uptake was higher than that of C�. The cells did not significantlytake up the control FITCC� peptide. Hatched area represents the range forinternal background fluorescence of the cells. (D) With HF that wereexposed to similar concentrations of unlabeled and labeled haptides asBAEC similar results were obtained, but there was no difference in theuptake between C� and preC�. Hatched area represents the range forinternal background fluorescence of the cells.
123R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

response than C�, but similar to that of the whole fibrino-gen. The C� control peptide elicited no cell-adhesive re-sponse above background. The C� C-terminal peptide thatwas also used as a control exhibited only a minimal cell-binding activity, not significantly higher than that seen inBSA-coated wells or in bare plastic. Furthermore, wellscoated only with the short GRGDS peptide did not bindsignificantly more cells than bare plastic.
We also evaluated the ability of soluble GRGDS peptideto inhibit cell attachment to the plastic coated by the testedligands. The cells were preincubated with 100 �g/mlGRGDS for 15 min and then transferred to the ligand-coated plastic microwell plate. Preincubation with theGRGDS inhibited cell attachment to FN-coated wells (pos-itive control) (P � 0.01) and to thrombin-coated wells (P �0.01). The inhibition of cell binding to fibrinogen was lesssignificant (P � 0.05) while the inhibition of cell binding tothe wells coated by the haptides C� and C�E was statisti-cally not significant (Fig. 3).
In summary, we found in two different cell attachmentassays (Figs. 2 and 3) that immobilized C�, preC�, and toa lesser extent C�E are significantly haptotactic. The data
also showed that haptotactic activity of C�, preC�, and C�Eis probably not related to mechanisms involving the RGD-mediated binding via integrins.
Fluorescent microscopy of FITChaptides uptake by cells
When free FITChaptides were added to BAEC or HFgrowing on glass coverslips and visualized by fluorescentmicroscopy, the fluorescent haptides were seen to be clearlytaken up by the cells following short (30–60 min) incuba-tion. The uptake of C� as seen by reconstructed confocalmicroscopic images is shown in Fig. 4. The cells becamehighly fluorescent after short exposure to FITCC�. Similarresults were obtained for FITCpreC� and FITCC�E uptake.Haptide intake was seen also even at 4°C, but at a lesserextent (data not shown). The nuclei remained relatively freeof FITChaptide. After longer incubation times at room tem-perature (�1 h), the haptides tested were found to getaccumulated as nanoaggregates within the cytoplasm andthen the fluorescence gradually faded. By contrast, the up-take of the control FITCC� peptide by the cells was negli-gible (Fig. 4).
Kinetics of soluble FITChaptide uptake by cells asmonitored by FACS
FITCHaptides were added to trypsinized cells, and afterincubation at 4 or 37°C, the cells were subjected to FACSanalysis. The uptake of haptides was significant in concen-trations between 4 and 40 �M and it increased with FITChap-tide concentrations. Figs. 5B and E show typical histogramsof the uptake of 4 �M FITCC� by BAEC at 37°C. Underthese conditions, only a small fraction of the cells stainedenough to appear as a distinct subpopulation. When 40 �MFITCC� was used, a biphasic histogram with cells split intotwo subpopulations was clearly observed (Figs. 5D and E).When cells were exposed to excess of unlabeled C� fol-lowed by exposure to a low level of FITCC�, a clear en-hancement of the FITCC� uptake was seen (Figs. 5C and E).In contrast, the degree and the rate of uptake of the controlinert FITCC� peptide were negligible. Similar uptake resultswere observed for human fibroblasts, although the extent ofuptake was somehow lower (data not shown).
The dose response for FITCC� and FITCC�E uptake byBAEC and HF (after incubation for 1 h at 37°C) is presentedin Fig. 6A. In both cells types, FITCC� was taken by cellssomehow better than FITCC�E haptide. FITCC� uptake byBAEC reached a plateau within a few minutes (Fig. 6B).Then a slow phase of uptake continued with time, possiblydue to formation of nanoaggregates of haptides that werebetter internalized by the cells.
Competition experiments were designed to evaluate apossible role of cell receptors in the binding of the haptides.The results with C� and preC� are presented in Figs. 6C andD for BAEC �HF, respectively. The cells were incubated at4 or 37°C for 15 min with increasing concentrations of theunlabeled haptide. An aliquot of FITCC� (4 �M) was then
Fig. 7. (A) Kinetics of uptake by BAEC of 125I-C� at 37°C. A plateau wasreached within 5–10 min. (B) Uptake of 125I-C� by BAEC at increasingmolar ratios of unlabeled/labeled C� with the same level of labeled hap-tide. Different levels of 125I-C� were added to the cells after exposure tounlabeled C� in labeled/unlabeled ratios of 1:500, 1:1000, and 1:2000. Theexposure of cells to a higher levels of unlabeled haptide resulted in agreater uptake of the 125I-labeled haptide, rather than decrease it by com-petition. (C) Dose response plot of C� uptake by both HF and BAEC inbiologically relevant range of concentrations based on the data presented inB. The nonsaturable uptake kinetics best fits a linear quadratic curve asfollows: [C�-Uptake] � f1[C�] � f2[C�]2, where [C�] is the total extra-cellular peptide concentration. As saturation was not reached, it was notpossible to perform a Scatchard analysis to generate a binding constant(KD) expected from a receptor-mediated process (see inset C1).
124 R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

added and the sample was subjected to FACS analysis.Surprisingly, rather than competing and blocking FITCC�uptake (as expected in the case of a metabolically activemembrane transporter such as integrin mediated binding),elevated concentrations of the nonlabeled C� increased,rather than reduced the FITC� haptides uptake by the cells.At 4°C there was also significant FITChaptide uptake thatwas not inhibited by competition with the unlabeled hap-tides. For BAEC the effect of control peptide FITCC� isgiven as recorded at 37°C (Fig. 6C).
Kinetics of 125I-haptide uptake by cells
Experiments with 125I-labeled C� uptake by BAEC andHF confirmed the results of the FACS competition experi-ments (Fig. 7). The rate of 125I-C� binding to HF reachedequilibrium within 5–10 min (Fig. 7A). Preexposure of cellsto increasing concentrations of the unlabeled C� did notdiminish the uptake of 125I-C�. Rather, with elevated con-centrations of the nonlabeled haptide, an increase in theuptake of the labeled peptide by cells was recorded forBAEC (Fig. 7B). Similar results were observed for HF in awide range of concentrations and ratios of labeled/unlabeledhaptide. When all the data were cumulated to show doseresponse of haptide uptake (Fig. 7C), again, an elevation ofuptake with elevated haptide concentration was recorded.The data for both cell types could be fitted by the followinglinear-quadratic equation:
[C�-Uptake] � f1[C�] � f2[C�]2,
where [C�] is the initial extracellular haptide concentrationand f1 and f2 may vary between cell types and with theambient temperatures. Consequently, since no saturation ofbinding with elevation of the concentration of unlabeledhaptide could be recorded, an attempt to plot a Scatchard
curve from the data could not yield a negative KD as ex-pected when a competition of unlabeled with labeled mate-rials exists (Fig. 7C, inset).
Effect of C� on cell binding to anti-integrins antibodies
Another indirect approach to explore the possible role ofintegrins in the binding of haptides to cells was done bypreexposure of the cells to the haptides and monitoring ifthis can block the adhesion of integrin-specific antibodies tothe cell membrane. A panel of FITCantibodies directed tomembranal human integrins subunits �1 or �v or the com-plexes �5�1 and �v�1 was tested with HF and monitored byFACS at 4°C to minimize possible transmembranal trans-port. We observed that preexposure of HF to high con-centrations of C� did not block binding of the anti-integrins antibodies to the cell membrane, as would ex-pected if the haptides did indeed bind to the membraneintegrins (Fig. 8).
Monitoring C�-induced chemotaxis
A multiwell micro-Boyden chamber was used to test thechemotactic response of cells to C� haptide at concentra-tions of up to 100 �g/ml (�40 �M). C� induced significantchemotaxis toward BAEC in this system (Fig. 9), similar tothat induced by conditioned medium that served as a posi-tive control (100% chemotaxis). This response was signif-icantly higher than that induced by native fibrinogen andthree times higher than those obtained with the negativecontrol (1% BSA serum-free medium). A similar, but moremoderate, chemotactic response to C� was obtained with
Fig. 8. Testing the ability of C� to inhibit the binding of anti-integrinsantibodies to cell receptors. The binding of anti-integrin antibodies to HFbefore and after exposure to 40 �M C� was monitored by FACS. The C�did not block the binding of the antibodies to the cells, suggesting that it didnot interact with an integrin receptor.
Fig. 9. Haptide-induced chemotaxis. Comparison of chemotaxis of BAECelicited by a ligand gradient in micro-Boyden multichamber. Cells thatmanaged to transverse the Boyden chamber into the membrane werestained and counted. C� control peptide elicited no response. Conditionedmedium (CM) served as the positive chemotactic control that induced100% migration. It is apparent that C� elicited significant chemotacticresponse. The values and errors represent over five repeated experimentsfor each of the tested agents. Similar positive chemotactic responses of HFto C� were also observed (not shown).
125R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129
HF (data not shown). These results suggest that the hapto-tactic properties of the C� as well as other haptides is alsoassociated with significant chemotactic activity.
The effect of haptides on cell proliferation
Cells such as fibroblasts or BAEC were incubated with awide range of haptides concentration (10�3–102 �M) andwith the two control C-terminal fibrinopeptides for 3–4days. The total number of viable cells was then determined
with the MTS colorimetric assay. All peptides tested had noeffect on the cell proliferation or survival (data not shown).For C� this was further tested with single high dose ofhaptide that was added daily with similar results.
HPLC gel filtration of haptides
HPLC gel filtration was used to evaluate the self-aggre-gation of haptides in solution. The elution profile of eachpeptide indicated that in higher concentration in aqueous
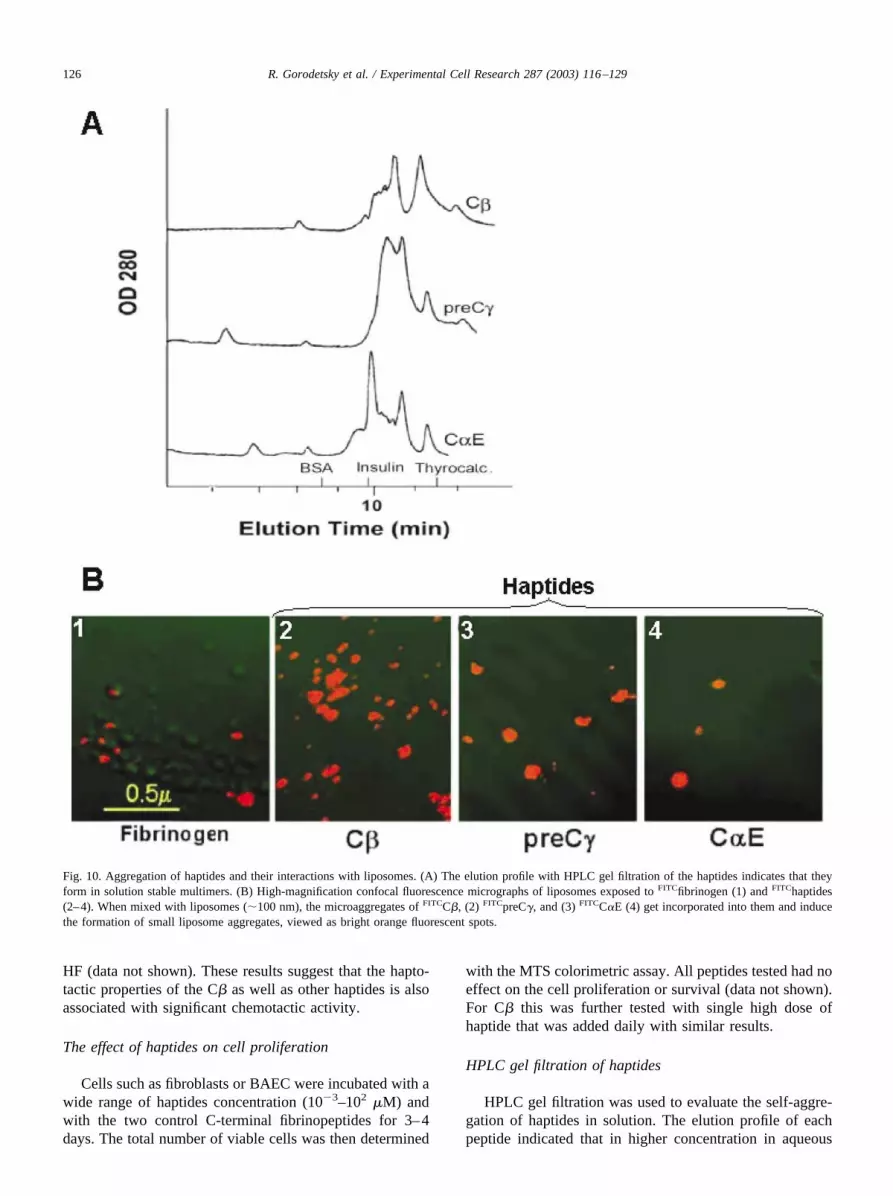
Fig. 10. Aggregation of haptides and their interactions with liposomes. (A) The elution profile with HPLC gel filtration of the haptides indicates that theyform in solution stable multimers. (B) High-magnification confocal fluorescence micrographs of liposomes exposed to FITCfibrinogen (1) and FITChaptides(2–4). When mixed with liposomes (�100 nm), the microaggregates of FITCC�, (2) FITCpreC�, and (3) FITCC�E (4) get incorporated into them and inducethe formation of small liposome aggregates, viewed as bright orange fluorescent spots.
126 R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

conditions, most of the solubilized haptides (�90%) wereaggregated into dimers or multimeric forms (Fig. 10A).
Interaction of the self aggregated FITChaptides withliposomes
Liposomes were used as a model for basic cell mem-brane and their interaction with haptides. Confocal fluores-cence microscopy revealed that when FITChaptides weremixed with liposomes the liposomes became very fluores-cent. Moreover, it appeared that the haptides induced lipo-some aggregation (Fig. 10B). No such effect was seen withthe control FITCC� fibrinopeptide (not shown).
Discussion
The interactions of fibrin(ogen) with different cell typesplay a significant role in different physiological and patho-logical processes. The current study describes the interac-tions with cultured cells of homologous 19- to 21-mers thatare equivalent to preserved C-termini sequences of the fi-brinogen chains.
The main advantage of our SB-ligand assay that wasused to evaluate the haptotactic activity of immobilizedpeptides (Fig. 2, Table 3) is that it examines the response ofan undisturbed monolayer of cultured cells whose mem-brane receptors have not been damaged by trypsinization[19,20]. Confidence in this assay is buttressed by the obser-vation that inactive ligands bound to SB generated no base-line attachment response, reducing the background to nil.Another advantage of this assay is that the cell SB-ligandinteractions can be monitored continuously at different timepoints over a few days, without perturbing the cell growthconditions.
The results clearly demonstrate that the cell types thatpreviously were reported to attach to the whole fibrinogenmolecule [19] responded quite similarly to the much shorterhaptides (Table 2, Fig. 2). The C� and preC�, which aremore closely homologous to each other, exhibited higherhaptotactic activity than C�E.
Cell responses to the C� and C�E were also confirmedwith the less sensitive conventional cell attachment assay,based on the fast binding of trypsinized cells to plasticELISA microplates adsorbed with the tested ligands. It is acommon observation that integrins are cell membrane re-ceptors that respond to the RGD sequence [10,13,16,26].Competition–inhibition of cell attachment by solubleGRGDS that was added to the cells was noted for fibronec-tin, thrombin, and fibrinogen but not for the haptides (Fig.3). As to the GRGDS peptide itself, apparently it was tooshort to affect by itself cell binding to the plate and thatlevel of binding could not be significantly inhibited by thesoluble peptide. These findings may suggest that receptorssuch as major integrins may not be involved in the cell-binding mechanism of the haptides (Fig. 3).
The haptides elicited a poor haptotactic response from
normal keratinocytes and insulin-secreting INS6 cells, bothof ectodermal origin. Cells from hematopoietic lineage,such as mouse transformed lymphocytic MIN6 cells ormacrophage-like J-774 cells, also showed a poor haptotacticresponse toward the haptides (Fig. 2, Table 3). In general,the cell types that poorly attached immobilized haptides alsoseemed to be attached more loosely to plastic surfaces.Human keratinocytes normally are not tightly attached toplastics. They also did not adhere SB-haptides. On the otherhand, cells of HaCaT-immortalized line derived from hu-man keratinocytes, which are highly adhering to plastic,also exhibited positive haptotactic response toward bothSB-C� and preC� (Table 3).
Another significant finding is the uptake of soluble hap-tides by cells (Fig. 4). Shortly after their uptake the haptidesseem to aggregate, to be taken into lisosomal structures, andto be cleared away. It is commonly expected that the inter-actions of a ligands with cell surface receptors (such asintegrins) result in saturable kinetics [16,30–32,42,43].However, our results with FACS and radioisotopic labeling(Figs. 6 and 7) showed that in free form, the haptides couldbe internalized into cells by nonsaturable kinetics that canbe best described by a linear quadratic equation. The inter-nalization of small amount of labeled haptide was facili-tated, rather than reduced, in the presence of increasingconcentrations of unlabeled haptide. Consequently, a KD forC� uptake could not be calculated by a Scatchard plot,which is generally employed to describe binding kineticsthrough receptors (Fig. 7C, inset). Even at 4°C, a significantuptake of labeled haptides without competition with unla-beled haptides was recorded. The lower level of haptidesbound at 4°C may derive from the physical properties of themembrane, which becomes more rigid and less permeable inthis temperature. Nevertheless, there was no saturable ki-netics even in this low temperature. Also there was noinhibition of the binding of antibodies directed to majorcellular integrins, as may be anticipated if haptides that tendto form multimer aggregates (Fig. 10A) would have blockedsuch membranal receptors (Fig. 5).
Some physical propertides of the haptides may elucidatetheir uptake kinetics. The haptides are amphiphilic, contain-ing hydrophobic as well as four to five net positivelycharged (Lys, Arg) residues. We observed that the haptidescould also bind and aggregate liposomes that serve as a“naked” membrane model (Fig. 10B). Since liposomes con-tain negatively charged phosphatidylcholine the haptidesmay be adsorbed to the membrane by hydrophobic interac-tion, which is enhanced by their positive charge. Similarly,in cells, haptides may bind directly to the cell membraneand their transmembranal crossing into the cytoplasm couldbe driven by the attraction of their positive residues to thenegatively charged intracellular compartment (Fig. 4). Thiscell tranduction phenomenon is similar to the mechanism ofcell internalization by other amphiphilic peptides that werepreviously shown to bind directly and not through a receptorto cell membranes and get internalized into cells [44–49].
While their amphiphilic properties could explain the in-
127R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

ternalization of haptides into the cells, their tendency toform aggregates in higher concentrations and to appearmostly (� 90%) as dimers and larger aggregates (Fig. 10A)may explain the kinetics of their internalization. The ten-dency of haptide to form aggregates present larger quanta ofhaptide that may interact with the cell membrane per inter-nalization event. Therefore, the addition of increasing levelsof unlabeled haptides that coaggregates with the labeledones facilitates, rather than competes with, the uptake of thelabeled haptides (Figs. 5–7).
The mechanism of cell internalization of free solublehaptide is not necessarily the basis for their haptotacticactivity when they are immobilized on a matrix. But it may,at least in part, explain the attachment of the cell membraneto haptide-coated matrices.
Haptides were also found to be chemotactic in a Boydenchamber assay (Fig. 8). A possible critique of such a widelyused migration assay is that it monitors the cells penetrationthrough a perforated membrane toward a solution with highconcentration of the test substance. However, the substancemay actually be adsorbed onto the membrane, increasing thenumber of cells attached to it and thereby enhance theirmigration through the membrane. Thus, such an assay mayactually measure a combination of haptotaxis with chemo-taxis. Other tests that follow-up and measure cell movementtoward a soluble gradient of haptides may be more appro-priate to establish the net chemotactic effect of the haptides,independent of their haptotactic properties.
Due to their highly haptotactic properties, the C�, preC�,and C�E sequences on the fibrin molecule may be involvedin cell attachment to fibrin, relevant to circumstances inwhich tissues are exposed to a fibrin clot. This may occurduring embryogenesis, early development, and wound heal-ing. Similarly, in blood vessel stenosis due to arteriosclero-sis, fibrin is deposited in the arterial walls and may attractand bind different cell types, including smooth muscle cells,fibroblasts, and endothelial cells. These cells may therebycontribute to the occlusion of the artery [38]. Fibrin depos-ited in a tumor mass could also modulate both cell migrationinto the tumor and its revascularization [38–40,50].
There may be numerous practical applications for hap-tides due to their high cell-binding properties with no re-corded cytotoxicity. As haptides are homologs to linearfragments of native fibrinogen, therefore, they are not ex-pected to be immunogenic. One might incorporate haptidesinto wound dressings to recruit cells and accelerate woundhealing. Moreover, matrices for tissue engineering could beformulated with such haptides, to render them more cellfriendly, without turning “on” signals leading to runawaycell proliferation. Prosthetic orthopedic or dental implantscould be coated with haptides to augment their integrationinto the tissue. Haptide variants could also be employed toselect responding cells out of a mixture with nonresponsiveones.
The haptides epitopes in fibrinogen are repeated in othermolecules. For example, some of the cellular interactionswith tenascins and angiopoietin isoforms, which contain a
“fibrinogen-like domain” in their C-termini [51–53] mayoccur through “haptidic” sequences. The interactions ofother molecules that contain sequences homologous to C�deserve further investigation, as it may shed light on themode of action and the developmental role of these highlyconserved sequences.
Acknowledgments
We thank Dr. Mark Tarshish and Gila Neiman (HadassahUniversity Medical School Interdepartmental Service) fortheir assistance with the confocal microscopy and FACS,respectively. We also thank Dr. Karen O. Yee (from thegroup of Professor S.M. Schwartz, Department of Pathol-ogy and Cellular and Molecular Biology at the University ofWashington, Seattle, WA) for her kind advice and for sup-plying us with different anti-integrin antibodies. We thankDr. Miriam Wahl (Thomas Jefferson University) for heradvice and useful editorial comments. This work was sup-ported by HAPTO Biotech Ltd and by the Israel ScienceFoundation Grant 697/001 to R.G.
References
[1] W. Nieuwenhuizen, M.W. Mosesson, M.P.M DeMaat (Eds.), Fibrin-ogen Workshop, Ann. N.Y. Acad. Sci 936 (2001) 28–43.
[2] G. Grieninger, Contribution of the �E C domain to the structure andfunction of fibrinogen-420, Ann. N.Y. Acad. Sci. 936 (2001) 44–64.
[3] G. Grieninger, X. Lu, Y. Cao, Y. Fu, B.J. Kudryk, D.K. Galanakis,K.M. Hertzberg, Fib 420, a novel fibrinogen subclass: newborn levelsare higher than adult, Blood 90 (1997) 2609–2614.
[4] S.J. Everse, G. Spraggon, R.F. Doolittle, A three-dimensional con-sideration of variant human fibrinogens, Thromb. Haemost. 80 (1)(1998) 1–9.
[5] G. Marx, Modelling cation-driven (proto)fibrin coagulation, Biopoly-mers 27 (1998) 763–774.
[6] M.W. Mosesson, K. Siebenlist, D.A. Meh, The structure and biolog-ical features of fibrinogen and fibrin, Ann. N.Y. Acad. Sci. 936 (2001)11–36.
[7] M. Murakawa, T. Okamura, T. Kamura, T. Shibuya, M. Harada, Y.Niho, Diversity of primary structures of the carboxy-terminal regionsof mammalian fibrinogen Aa-chains, Thromb. Haemost. 69 (1993)351–360.
[8] S.A. Olexa, A.Z. Budzynski, Localization of a fibrin polymerizationsite, J. Biol. Chem. 256 (1981) 3544–3549.
[9] S. Yakovlev, S. Litvinovich, D. Loukinov, L. Medved, Role of the�-strand insert in the central domain of the fibrinogen �-module,Biochemistry 39 (2000) 15721–15729.
[10] A. Bini, Y. Itoh, B.J. Kudryk, H. Nagase, Degradation of cross-linkedfibrin by metalloproteinase 3 (stromelysisn 1) by hydrolysis of the �Gly-Ala 405 peptide bond, Biochemistry 35 (1996) 13056–13063.
[11] R.M. Senior, W.F. Skogen, G.L. Griffin, G.D. Wilner, Effects offibrinogen derivatives upon the inflammatory response: studies withhuman fibrinopeptide, Br. J. Clin. Invest. 77 (1986) 1014–1019.
[12] S.D. Wright, J.I. Weitz, A.J. Huang, S.M. Levin, S.C. Silverstein, J.D.Loike, Complement receptor type three (CD11b/CD18) of humanpolymorphonuclear leukocytes recognizes fibrinogen, Proc. Natl.Acad. Sci. USA 85 (1988) 7734–7738.
[13] D.C. Altieri, J. Plescia, E.F. Plow, The structural motif glycine190-valine 202 of fibrinogen � chain interacts with the CD11b/CD18integrin (aMb2, Mac-1) and promotes leukocyte adhesion, J. Biol.Chem. 268 (1993) 1847–1853.
128 R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129

[14] D.L. Richardson, B. Pepper, A.B. Kay, Chemotaxis for human mono-cytes by fibrinogen-derived peptides, Am. J. Hematol. 52 (1976)507–513.
[15] S. Berliner, J. Fuchs, U. Seligsohn, N. Kariv, B. Hazaz, Z. Rotenberg,I. Weinberger, J. Agmon, J. Pinkhas, M. Aronson, Possible role offibrinogen in the aggregation of white blood cells, Thromb. Haemost.58 (1987) 749–752.
[16] K.O. Yee, M.M. Rooney, C.M. Giacelli, S.T. Lord, S.M. Schwartz,Role of �1 and �3 in human smooth muscle cell adhesion to andcontraction of fibrin clots in-vitro, Circ. Res. 83 (1998) 241–251.
[17] L.F. Brown, N. Lanir, J. McDonagh, K. Tignazzi, A.M. Dvorak, H.F.Dvorak, Fibroblast migration in fibrin gel matrices, Am. J. Pathol.142 (1993) 273–283.
[18] D.G. Chalupowicz, I.A. Chowdhury, T.L. Bach, C. Barsigian, J.Martinez, Fibrin II induces endothelial cell capillary tube formation,J. Cell Biol. 130 (1995) 207–215.
[19] R. Gorodetsky, A. Vexler, J. An, X. Mou, G. Marx, Chemotactic andgrowth stimulatory effects of fibrin(ogen) and thrombin on culturedfibroblasts, J. Lab. Clin. Med. 131 (1998) 269–280.
[20] R. Gorodetsky, J. An, R.A.F. Clark, J. Gailit, L. Levdansky, A.Vexler, E. Berman, G. Marx, Fibrin microbeads (FMB) as biodegrad-able carriers for culturing cells and for accelerating wound healing,J. Invest. Dermatol. 112 (1999) 866–872.
[21] A.J. Gray, J.E. Bishop, J.T. Reeves, G.J. Laurent, A� and B� chainsof fibrinogen stimulate proliferation of human fibroblasts, J. Cell. Sci.104 (1993) 409–413.
[22] T. Saldeen, Vasoactive peptides derived from degradation of fibrin-ogen and fibrin, Ann. N.Y. Acad. Sci. USA 408 (1983) 424–431.
[23] Y. Veklich, C.W. Francis, J. White, J.W. Weisel, Structural studies offibrinolysis by electron microscopy, Blood 92 (1998) 4721–4729.
[24] G. Marx, Fibrinogen fragmentation by free radicals: coagulation offibrinogen with Cu(II) and ascorbate, in: E. Ricklis (Ed.), RadiationBiology, Wiley-VCH, Weinheim, 1990, pp. 691–698.
[25] G. Marx, Immunological monitoring of Fenton fragmentation offibrinogen, Free Rad. Res. Commun. 12 (1991) 517–520.
[26] D.A. Cheresh, S.A. Berliner, V. Vicente, Z.M. Ruggeri, Recognitionof distinct adhesive sites on fibrinogen by related integrins on plate-lets and endothelial cells, Cell 58 (1989) 945–953.
[27] D.H. Farrell, P. Thiagarajan, D.W. Chung, E.W. Davie, Role offibrinogen � and � chain sites in platelet aggregation, Proc. Natl.Acad. Sci. USA 89 (1992) 10729–10732.
[28] R.R. Hantgan, S. Endenburg, I. Cavero, G. Marguerie, A. Uzan, J.J.Sixma, P.G. de Groot, Inhibition of platelet adhesion to fibrin(ogen)in flowing whole blood by RGD and fibrinogen �-chain carboxyterminal peptides, Thromb. Haemost. 68 (1992) 694–700.
[29] B. Savage, E. Bottini, Z.M. Ruggeri, Interaction of integrin alpha IIbbeta with multiple fibrinogen domains during platelet adhesion,J. Biol. Chem. 270 (1995) 28812–28817.
[30] K. Yokoyama, X.P. Zhang, L. Medved, Y. Takada, Specific bindingof integrin �v�3 to the fibrinogen � and �E chain C-terminal do-mains, Biochemistry 38 (1999) 5872–5877.
[31] K. Suehiro, G. Gailit, E.F. Plow, Fibrinogen is a ligand for integrin�5�1 in endothelial cells, J. Biol. Chem. 272 (1997) 5360–5366.
[32] D.H. Farrell, H.A. Al-Mondhiry, Human fibroblast adhesion to fi-brinogen, Biochemistry 36 (1997) 1123–1128.
[33] S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W.Miller, D.J. Lipman, Gapped BLAST and PSI-BLAST: a new gen-eration of protein database search programs, Nucleic Acids Res. 25(1997) 3389–3402.
[34] P. Thiagrajan, A.J. Rippon, D.H. Farrell, Alternative adhesion sites inhuman fibrinogen for vascular endothelial cells, Biochemistry 35(1996) 4169–4175.
[35] R.A. Smith, M.M. Rooney, S.T. Lord, M.W. Mosesson, T.K. Gartner,Evidence for new endothelial cell binding sites on fibrinogen,Thromb. Haemost. 84 (2000) 819–825.
[36] T. Browder, J. Folkman, S.R.M. Prie-Shepherd, W.F. Skogen, G. L.Griffin, G.D. Wilner, Effects of fibrinogen derivatives upon the in-flammatory response: studies with human fibrinopeptide, Br. J. Clin.Invest. 77 (2000) 1014–1019.
[37] R. Valenuela, J.R. Shainoff, P.M. DiBello, D.A. Urbanic, J.M. Ander-son, G.R. Matsueda, B.J. Kudryk, Immuno-electrophoretic and im-muno-histochemical characterizations of fibrinogen derivatives inatherosclerotic aortic intimas and vascular prosthesis pseudo-intimas,Am. J. Pathol. 141 (1992) 861–880.
[38] F.T. Horrigan, J.L. Degen, R.M. Lawn, Fibrinogen deficiency reducesvascular accumulation of apolipoprotein(a) and the development ofatherosclerosis in apolipoprotein(a) transgenic mice, Proc. Natl.Acad. Sci. USA 95 (1998) 12591–12595.
[39] J.A. Nagy, M.S. Meyers, E.M. Masse, K.T. Herzberg, H.F. Dvorak,Pathogenesis of ascites tumor growth: fibrinogen influx and fibrinaccumulation in tissues lining the peritoneal cavity, Cancer Res. 55(1995) 369–375.
[40] H. Bardos, P. Molnar, G. Csecsei, R. Adany, Fibrin deposition inprimary and metastatic human brain tumors, Blood Coagul. Fibrinol.7 (1996) 536–548.
[41] P. Boukamp, R.T. Petrusevska, D. Breitkreutz, J. Hornung, A.Markham, N.E. Fusenig, Normal keratinization in a spontaneouslyimmortalized aneuploid human keratinocyte cell line, J. Cell Biol.106 (1988) 761–771.
[42] B. Geiger, A. Bershadsky, R. Pankov, K. Yamada, Transmenbraneextracellular matrix—cytosheleton crosstalk, Nat. Rev. Mol. CellBiol. 2 (2002) 793–805.
[43] J. Gailit, C. Clarke, N. Dawn, M.G. Tonnesen, M.W. Mosesson,R.A.F. Clark, Human fibroblasts bind directly to fibrinogen at RGDsites through integrin �v�3, Exp. Cell Res. 232 (1997) 181–126.
[44] S.R. Schwarze, A. Ho, A. Vocero-Akbani, S.F. Dowdy, In vivoprotein transduction: delivery of a biologically active protein into themouse, Science 285 (1999) 1569–1572.
[45] E. Vives, P. Brodin, B. Lebleu, Truncated HIV-1 tat protein basicdomain rapidly translocates through the plasma membrane and accu-mulates in the cell nucleus, J. Biol. Chem 272 (1997) 16010–16017.
[46] T.J. Sereda, C.T. Mant, F.D. Sonnichsen, R.S. Hodges, Reversed-phase chromatography of synthetic amphiphilic alpha-helical pep-tides as a model for ligand/receptor interactions: effect of changinghydrophobic environment on the relative hydrophilicity/hydrophobic-ity of amino acid side-chains, J. Chromatogy. 676 (1994) 139–153.
[47] Morris, M.C., Vidal, P.L., Chaloin, Heitz, F., Divita, G. A newpeptide vector for efficient delivery of oligonucleotides into mamma-lian cells. Nucleic Acids Res. 25 (1997) 2730–2736.
[48] D. Derossi, A.H. Joliot, G. Chassaing, A. Prochiantz, The third helixof the Antennapedia homeodomain translocates through biologicalmembranes, J. Biol. Chem. 269 (1994) 10444–10450.
[49] A.J. Mandell, K.A. Selz, M.F. Shlesinger, Mode matches and theirlocations in the hydrophobic free energy sequences of peptide ligandsand their receptor functions, Proc. Natl. Acad. Sci. USA 94 (1997)13576–13581.
[50] H. Bardos, A. Juhasz, G. Repassy, R. Adany, Fibrin deposition insquamous cell carcinomas of the larynx and hypopharynx, Thromb.Haemost. 80 (1998) 767–772.
[51] P. Milev, D. Fischer, M. Harig, T. Schulthess, R.K. Margolis, R.Chicket-Ehrismann, R.U. Margolis, The fibrinogen like globe of tenas-cin-C mediates its interactions with neurocan and phophacan/protein-tyrosine phosphatase-��, J. Biol. Chem. 272 (1997) 15501–15509.
[52] S. Schenk, R. Chiquet-Ehrismann, E.J. Battegy, The fibrinogen-globeof tenascin-C promotes basic-FGF-induced endothelial cell elonga-tion, Mol. Biol Cell 10 (1999) 2933–2943.
[53] D.W. Lafleur, J. Chiang, J.A. Fagin, S.M. Schwartz, P.K. Shah, K.Wallner, J.S. Forrester, B.G. Sharifi, Aortic smooth muscle cellsinteract with tenascin-C through its fibrinogen-like domain, J. Biol.Chem. 272 (1997) 32798–32803.
129R. Gorodetsky et al. / Experimental Cell Research 287 (2003) 116–129





























