Embed Size (px)
Citation preview

proteinsSTRUCTURE O FUNCTION O BIOINFORMATICS
Noncellulosomal cohesin from thehyperthermophilic archaeonArchaeoglobus fulgidus
Milana Voronov-Goldman,1 Raphael Lamed,1,2 Ilit Noach,3 Ilya Borovok,1 Moria Kwiat,1
Sonia Rosenheck,1 Linda J. W. Shimon,4 Edward A. Bayer,3* and Felix Frolow1,2*1Department of Molecular Microbiology and Biotechnology, George S. Wise Faculty of Life Sciences, Tel Aviv University,
Ramat Aviv 69978, Israel
2 The Daniella Rich Institute for Structural Biology, Tel Aviv University, Ramat Aviv 69978, Israel
3 Department of Biological Chemistry, The Weizmann Institute of Science, Rehovot 76100, Israel
4 Department of Chemical Research Support, The Weizmann Institute of Science, Rehovot 76100, Israel
INTRODUCTION
Cellulosomes are multienzyme complexes produced in vari-
ous cellulolytic microorganisms, designed to hydrolyze effi-
ciently plant cell wall polysaccharides. The best-characterized
cellulosome systems are those from Clostridiae, first identified
in Clostridium thermocellum.1 In these, a central ‘‘scaffoldin’’
subunit integrates catalytic subunits into the complex by virtue
of two complementary types of modules, the multiple cohesins
on the scaffoldin and the single dockerins of the catalytic subu-
nits. The calcium-mediated cohesin–dockerin interaction dic-
tates cellulosome formation and architecture.
For many years, cohesin and dockerin modules were thought to
be exclusive components of cellulosomes. When the early genome
sequences were published, it was thus surprising to discover such
sequences in microorganisms that clearly lacked cellulosomes. In
this context, a single dockerin-like sequence and two cohesin-like
sequences were detected in the complete genome of the hyperther-
mophilic archaeon Archaeoglobus fulgidus.2 Phylogenetically,
Archaeoglobus is branched between the Methanococcales and Ther-
mococcales and may represent a biochemical missing link between
the sulfur-metabolizing and methanogenic archaebacteria. Mem-
bers of the genus Archaeoglobus have thus far been found only in
anaerobic submarine hydrothermal areas with an average temper-
ature above 808C.3 Archaeoglobus is distinguished from eubacterial
sulfate reducers by its extremely high growth temperatures and by
Additional Supporting Information may be found in the online version of this article.
Abbreviations: Ac, Acetivibrio cellulolyticus; Af, Archaeoglobus fulgidus; BSA, bovine serum
albumin; Coh, cohesin; Cp, Clostridium perfringens; Doc, dockerin; ORF, open reading
frame; Xyn, Geobacillus stearothermophilus Xyn-T6.
Grant sponsor: Israel Science Foundation; Grant numbers: 291/08, 159/07, 966/09.
*Correspondence to: Felix Frolow, Department of Molecular Microbiology and Biotechnol-
ogy, Tel Aviv University, Ramat Aviv 69978, Israel. E-mail: [email protected] or
Edward A. Bayer, Department of Biological Chemistry, The Weizmann Institute of Science,
Rehovot 76100, Israel. E-mail: [email protected].
Received 26 May 2010; Revised 22 July 2010; Accepted 5 August 2010
Published online 24 August 2010 in Wiley Online Library (wileyonlinelibrary.com).
DOI: 10.1002/prot.22857
ABSTRACT
The increasing numbers of published genomes has
enabled extensive survey of protein sequences in nature.
During the course of our studies on cellulolytic bacteria
that produce multienzyme cellulosome complexes
designed for efficient degradation of cellulosic sub-
strates, we have investigated the intermodular cohesin–
dockerin interaction, which provides the molecular ba-
sis for cellulosome assembly. An early search of the ge-
nome databases yielded the surprising existence of a
dockerin-like sequence and two cohesin-like sequences
in the hyperthermophilic noncellulolytic archaeon,
Archaeoglobus fulgidus, which clearly contradicts the
cellulosome paradigm. Here, we report a biochemical
and biophysical analysis, which revealed particularly
strong- and specific-binding interactions between these
two cohesins and the single dockerin. The crystal struc-
ture of one of the recombinant cohesin modules was
determined and found to resemble closely the type-I
cohesin structure from the cellulosome of Clostridium
thermocellum, with certain distinctive features: two of
the loops in the archaeal cohesin structure are shorter
than those of the C. thermocellum structure, and a large
insertion of 27-amino acid residues, unique to the arch-
aeal cohesin, appears to be largely disordered. Interest-
ingly, the cohesin module undergoes reversible dimer
and tetramer formation in solution, a property, which
has not been observed previously for other cohesins.
This is the first description of cohesin and dockerin
interactions in a noncellulolytic archaeon and the first
structure of an archaeal cohesin. This finding supports
the notion that interactions based on the cohesin–dock-
erin paradigm are of more general occurrence and are
not unique to the cellulosome system.
Proteins 2011; 79:50–60.VVC 2010 Wiley-Liss, Inc.
Key words: dockerin; protein oligomerization; thermo-
stability; X-ray crystallography.
50 PROTEINS VVC 2010 WILEY-LISS, INC.

other features, such as lack of a peptidoglycan cell wall. This
archaeon lacks glycoside hydrolases and, consequently, lacks
a defined cellulosome.
One of two hypothetical proteins, AF2375, contains both a
cohesin-like module and a dockerin-like module in tandem.
The residual domains detected do not appear to be associated
with known cellulosome-related components (e.g., scaffoldins,
CBMs, glycoside hydrolases, or anchoring proteins). Until
recently, functional cohesin and dockerin modules have been
found only in anaerobic cellulolytic bacteria. Our view of this
strict association, however, is now in question, owing to the
identification of functional noncellulosomal cohesin and
dockerin modules in unrelated glycoside hydrolases from the
human pathogen Clostridium perfringens.4–6 Consequently,
the early finding of cellulosome-like sequences in a noncellu-
lolytic archaeon is especially intriguing and deserves additional
study. It was thus of interest to determine whether the A. fulgi-
dus cohesin- and dockerin-encoding sequences possess the
ability to interact with one another in a manner similar to
that of the cellulosomal cohesin–dockerin interactions. In the
present communication, we report the cloning and expression
of the two cohesins and single dockerin of A. fulgidus. We
demonstrate the calcium-dependent binding of the dockerin
to both cohesin modules. The crystal structure of cohesin
AfCoh2375 was determined, which bears striking similarity to
the cellulosomal type-I cohesin from C. thermocellum.
METHODS
Protein production and purification
The cohesin modules from AF2375 (res. 274–431) and
AF2376 (res. 29–162) and the dockerin module from
AF2375, (res. 432–506) were amplified by PCR from A. ful-
gidus genomic DNA. A DNA fragment encoding AF2376
was amplified by PCR using F-50GGCCCATGGCTTCAGCTGAAATGGTTGTTAAA30 and R-50 ATACTCGAGTCCTGCCCCACCTTTCACAGCTTC30 specific primers. The
PCR products were cloned with C-terminal His-tags;
AfCoh2375 and AfCoh2376 were expressed and purified by
metal-chelate affinity chromatography, followed by fast
protein liquid chromatography (FPLC) using a Superdex
75 pg column, as described previously.7 The same column
and conditions for gel filtration chromatography used in
the latter report were used here to investigate oligomeriza-
tion of the A. fulgidus cohesins. AfDoc2375 was cloned as a
fusion protein with Geobacillus stearothermophilus xylanase
(Xyn) T6 at its C-terminus and ligated into KpnI/BamHI-
restricted pET9d (Stratagen) with an N-terminal His-tag.
For this purpose, a DNA fragment was amplified by PCR
using specific primers: F-50GCTGGTACCAGAAGAAGCAAACAAGGGAGATGTG30 and R-50TATGGATCCCTTACCCAGTAAGCCATTCTGGCT30. Xyn-AfDoc2375 was
expressed in Eshcherichia coli BL21(RIL) cells transformed
with the pET9d derivative. The cells grown at 378C to
O.D600 0.5, and recombinant protein expression was
induced by adding 1 mM (IPTG) and overnight incubation
at 168C. The protein was purified on a Ni-IDA column
according to the previously described protocol.7
Affinity blotting
Proteins were subjected to SDS-PAGE (12%), and the sep-
arated proteins were then transferred onto a nitrocellulose
membrane and rinsed with wash buffer (50 mM Tris-HCl
pH 7.5, 150 mM NaCl, 25 mM CaCl2). The membrane was
then incubated for 2 to 3 h with blocking buffer (3% BSA in
washing buffer) and rinsed five times with wash buffer. The
membrane was then incubated overnight at 48C with the
recombinant His-tagged protein. The membrane was then
treated with peroxidase-conjugated antibody using an anti-
His(C-terminal)-horseradish peroxidase-labeled mouse anti-
body detection system, according to the supplier’s instruc-
tions (Invitrogen Corp., Carlsbad, CA). Bands were visual-
ized by using a chemiluminescent substrate (Supersignal
Substrate [Western blotting]; Pierce Biotechnology, Rock-
ford, IL) according to the manufacturer’s instructions.
ELISA cohesin–dockerin interaction
The interaction between the A. fulgidus dockerin and cohe-
sins was analyzed quantitatively by affinity-based ELISA,
essentially as described by Barak et al.8 Briefly, MaxiSorp
ELISA plates (Nunc A/S, Roskilde, Denmark) were coated
with 100 lL/well of the test cohesin at a concentration of
20 lg/mL in 0.1M Na2CO3, pH 9, over night at 48C. The fol-lowing steps were performed at room temperature with all
reagents at a volume of 100 lL/well. The coating solution was
discarded, and blocking buffer (TBS, 10 mM CaCl2, 0.05%
Tween20, 2% BSA) was added for 1 h incubation. The block-
ing buffer was discarded, and Xyn-AfDoc2375 was diluted in
blocking buffer to concentrations of 8.25 pM to 8.25 nM, and
incubated for 1 h. The plates were washed with wash buffer
(blocking buffer without BSA) and anti-xylanase antibody,
diluted 1:20,000 in blocking buffer was added for 1 h incuba-
tion. Subsequently, HRP-labeled anti-rabbit antibody, diluted
1:10,000 in blocking buffer, was added for an additional 1 h
incubation. The plates were washed, and HRP substrate
(TMB1 Substrate-Chromogen, Dako Corp., Carpinteria, CA)
was added (100 lL/well). Color formation was terminated
with 50 lL/well of 1M H2SO4, and absorbance was measured
at 450 nm using a tunable microplate reader (Versamax, Mo-
lecular Devices, Inc., MDS Analytical Technologies, Sunnyvale,
CA). AcCohC3 was prepared as reported earlier.9
Bioinformatic analysis of protein sequences
Genomic DNA analyses and open reading frame (ORF)
searches were performed with the National Center for
Biotechnology Information server ORF Finder (http://
www.ncbi.nlm.nih.gov/gorf/gorf.html) and the Clone Man-
ager Suite, ver. 7 (Scientific & Educational Software, Durham,
NC). A. fulgidus gene and protein names used in this study
are based on the prefix ‘‘AF,’’ in public databases both ‘‘AF’’
and ‘‘AF_’’ prefixes are interchangeably. Cohesin- and dock-
Crystal Structure of an Archaeal Cohesion
PROTEINS 51

erin-like motifs (IPR002102 and IPR016134, respectively)
were used for analysis as described in the Integrated resource
of Protein domains (InterPro) (http://www.ebi.ac.uk/inter-
pro/). Other protein domains and amino acid motifs were
analyzed using the Simple Modular Architecture Tool
(SMART) (http://www.smart.embl-heidelberg.de),10,11 the
Pfam protein families database (http://pham.sanger.ac.uk),12
and the database of protein families and domains PROSITE
(http://www.expasy.ch/prosite/). Similarity searches were per-
formed using the BLAST algorithm at the NCBI server
(http://www.ncbi.nih.nlm.gov).13 Potential transmembrane
domains (helices) were determined by the TMHMM 2.0 pro-
gram14 (http://www.cbs.dtu.dk/services/TMHMM/). Param-
eters for molecular mass, theoretical pI, amino acid composi-
tion, and extinction coefficient were computed using the
ProtParam Tool on the ExPASy server (http://www.expasy.
org/tools/protparam.html).15 Pairwise and multiple sequence
alignments were performed with the ClustalW program,
version 1.84,16 using the EMBL-EBI ClustalW2 server
(http://www.ebi.ac.uk/Tools/clustalw2/index.html). Multiple
sequence alignments were used for creating unrooted phylog-
eny trees by the MEGA 4.1 program.17 Estimated protein
molecular masses were calculated using the Peptide Mass
Tool at the ExPASy server of Swiss Institute of Bioinformatics
(http://www.us.expasy.org). Structure-based sequence align-
ment with known molecular structures was preformed using
a MAMMOTH-mult multiple protein alignment server
(http://ub.cbm.uam.es/mammoth/pair/index3.php).
Data collection and molecular replacement
Crystallization of AfCoh2375 and X-ray data collection
were performed as previously described.7 Crystallo-
graphic data statistics is shown in Table I. The structure
of cohesin AfCoh2375 was determined by molecular
replacement with difficulties using the program BEAST19
as was implemented in the CCP4 suite of crystallographic
programs.20 For the record, the recent default version
2.1.4 of the program PHASER21,22 implemented in the
CCP4 suite served to determine the cohesin AfCoh2375
structure in an effortless manner. A search ensemble of
models for molecular replacement was constructed from
six structures of cohesin molecules (PDB codes 1ANU,
1AOH, 1G1K, 1QZN, 1ZV9, and 1TYJ). The highest
sequence identity between the AfCoh2375 and the cohe-
sin structures listed above stands at 20%, and the respec-
tive (root mean square deviation) RMSD among the
model protein molecules ranged from 1.6 to 2.5 A.
Model building
Automatic building of the structure based on the mo-
lecular replacement phases was attempted with ARP/
wARP23 and succeeded to trace and dock 80 out of the
160 residues of AfCoh2375. ARP/wARP was imple-
mented once again, this time in the structure improve-
ment mode,24 resulting in a hybrid structure that con-
sisted of 80 amino acid residues of the protein molecule
and a large group of dummy atoms modeling the elec-
tron density. The hybrid structure was refined by
REFMAC5,25 and several gaps in the loops of the struc-
ture were manually rebuilt using graphical programs
O26 and COOT.27 ARP/wARP was implemented for a
third time, again in the automatic model-building
mode,23 based on the current structure, this time trac-
ing and docking 122 residues out of 160 residues.
Finally the missing part of the structure was built man-
ually using O and COOT.
Model refinement and data validation
Initial refinement was performed using REFMAC5,25
and final refinement including that of alternative mutu-
ally exclusive conformations was performed using PHE-
NIX28 with 5% of the total reflections set for cross-vali-
dation.29 Water molecules were included using the
COOT water addition automatic protocol.27 Refinement
statistics for the final model is shown in Table I. The
model was validated using PROCHECK30 and MOL-
PROBITY31,32 and fell within the limits of all quality
criteria. Ramachandran plots showed that 98.4% of the
amino acid residues were in allowed regions of the Phi-
Psi diagram. The figures were prepared using PyMOL.33
Table ICrystal, Diffraction and Refinement Data. Numbers in Parentheses Are
for the Highest Resolution Shell
Data collectionSource ESRF ID29Symmetry P4332Number of crystals 1Total rotation range (8) 45a 5 b 5 c (�) 101.75No of molecules in asymmetric unit 1Resolution range (�) 30.0–1.96(1.99–1.96)Number of reflections measured/unique 257464/13584Redundancy 18.9Completeness, overall (%) 99.9 (100.0)Average I/r(I) 46.6 (2.5)Rmerge
a(%) 0.091 (0.58)Overall B factor from Wilson plot (�2) 40.2
Data refinementResolution range (�) 25.44-1.96 (2.05-1.96)Number of reflections, total/test 12483/1257 (1075/117)Rcryst
b/Rfreec 0.1890/0.2296 (22.64/0.3388)
RMSD bonds (�) 0.007RMSD angles (8) 1.093Average B factor (�2) 53.3Number of water molecules 118Ramachandran favored (%) 96Ramachandran allowed (%) 98.4
aRmerge 5 Shkl Si|Ii(hkl) 2 <I(hkl)>| /Shkl SiIi(hkl), where Shkl denotes the sum
over all reflections and Si the sum over all equivalent and symmetry-related
reflections.18
bRcryst 5 Shkl k Fobs 2 Fcalck/Shkl|Fobs|, where Shkl denotes the sum over all reflec-
tions, and Fobs and Fcalc are the scaled observed and calculated reflections.cRfree is calculated as Rcryst but using 5.0% of the randomly selected reflections
omitted from the structure refinement.
M. Voronov-Goldman et al.
52 PROTEINS

Protein Data Bank accession code
The atomic coordinates and structure factor of the A.
fulgidus cohesin 2375 (AfCoh2375) have been deposited
in the Protein Data Bank, www.pdb.org (PDB access
code 2XDH).
RESULTS AND DISCUSSION
Bioinformatics analysis
Systematic analysis of the A. fulgidus genome revealed
that three genes, AF2374, AF2375, and AF2376, are
potentially cotranscribed, owing to the existence of over-
laps between neighboring open reading frames (35-nt
and 20-nt overlaps were found between AF2374 and
AF2375, and AF2375 and AF2376, respectively). Immedi-
ately downstream of AF2376, a possible terminator struc-
ture was also predicted (data not shown). A schematic
representation of these three ORFs is presented in
Figure 1. AF2374 encodes a 157-amino acid hypothetical
protein, which possesses a C-terminal amino acid
sequence similar to the PD40 domain (Pfam: PF07676),
a periplasmic component of the Tol-dependent transloca-
tion system. The adjacent downstream gene, AF2375,
encodes a 506-amino acid protein composed of several
conserved structural modules. In amino-terminal portion
of a protein, residues 93 to 241 exhibit similarity to cer-
tain bacterial TolB-associated hypothetical proteins men-
tioned above. A carboxy-terminal sequence of AF2375
contains two modules similar to major cellulosomal
binding elements—a putative cohesin (274 to 431) and a
dockerin (432 to 506). The third gene, AF2376, codes for
a hypothetical 190-residue protein, containing a cohesin-
like module (29 to 162) flanked by two strongly pre-
dicted trans-membrane helices.
Since the bioinformatics-based indication of the first
noncellulosomal cohesin- and dockerin-like sequences inA. fulgidus,2 many additional noncellulosomal cohesin-and dockerin-like elements were discovered in the Arch-aea as well as in the other domains of life.34 For
instance, noncellulosomal modules have been predictedin strict anaerobic species (e.g., Methanococcus, Methano-sarcina, Pyrococcus, etc.) and in obligate extreme halo-philic archaeons (Halobacterium spp.). Interestingly, in
the latter work, nearly 40% of the known archaealgenomes were found to contain either a putative cohesinand/or dockerin module, compared to 14% of the knownbacterial genomes; until then, the best investigated cohe-sins and dockerins were derived from the genomes of cel-
lulosome-producing bacteria. Cohesin- and dockerin-likesequences were also predicted in the closest unicellularrelative of animals, that is, choanoflagellate Monosiga bre-vicollis (Eukaryota). A phylogenetic analysis of the A. ful-
gidus cohesins compared with representatives of otherarchaeal and nonarchaeal cohesins can be viewed in Fig-ure 2. A. fulgidus cohesins were found to occupy a sepa-rate branch in the vicinity of the type I cohesins.
The AF2375 dockerin-like sequence displays standard
features consistent with dockerin modules derived from
known cellulosomal components and thus resembles the
proposed F-helix variation of the EF-hand motif of cal-
cium-binding proteins, typical for dockerins when com-
pared with calmodulin and troponin C.35,36 As evident
from the AF2375 dockerin sequence, the residues of the
two segments that align with the F-helix cannot be con-
sidered a strict repeat: the amino acids at positions 11
and 12 in the first calcium-binding loop are not identical
Figure 1Schematic representation of the modular structure of ORFs AF2374, AF2375, and AF2376 from A. fulgidus. Coh, cohesin; Doc, dockerin; TM,
putative transmembrane helix; TolB, TolB-like sequence.
Crystal Structure of an Archaeal Cohesion
PROTEINS 53

to those at the appropriate positions in the second
repeat, and the calcium-binding loop of the second seg-
ment lacks Asn at position 9. Based on this evidence, the
AF2375 dockerin sequence is more similar to the type-II
dockerin of the C. thermocellum scaffoldin that also pos-
sesses asymmetric components.
Affinity interactions of cellulosome-likecomponents
Following initial screening of the archaeal cohesin–
dockerin interactions,37 more extensive analysis of the
binding interaction was performed in this work to fur-
ther investigate the binding and specificity characteristics
of the cohesins and dockerin counterparts. For this
purpose, AfCoh2375, AfCoh2376, and AfDoc2375 (fused
to the G. stearothermophilus Xyn-T6 carrier protein) were
cloned and overexpressed in an E. coli host-cell system.
Partially purified Xyn-AfDoc2375 was subjected to SDS-
PAGE, and the results of affinity blotting indicated that
both cohesin modules—including the cohesin of the
same ORF—recognized the A. fulgidus dockerin in a spe-
cific manner (Fig. 3). In contrast, only slight background
labeling of cellulosomal proteins from C. thermocellum
and those present in partially purified cohesins fractions
from A. cellulolyticus and B. cellulosolvens could be
observed (data not shown). This finding supports the
initial screening results of cohesin–dockerin binding spec-
ificity,37 where it was also found that the dockerin failed
to interact with any of the type-I, -II, or -III cohesins
Figure 2Phylogenetic relationship of the A. fulgidus cohesins in comparison with representative cohesins in the three domains of life. Representative archaeal
and eukaryotic cohesin modules are labeled according to the designated key to facilitate comparison with those of representative type-I, type-II, and
Ruminococcus flavefaciens cellulosomal cohesins, as well as recently described noncellulosomal cohesins of Clostridium perfringens. See Supporting
Information for definition of the cohesins indicated in the figure.
M. Voronov-Goldman et al.
54 PROTEINS

tested. The effects of Ca21-dependence of binding is a
critical characteristic of the cohesin–dockerin interaction.
To assay for the influence of Ca21 and for evaluation of
affinity constants, an affinity-based ELISA protocol was
performed.8 The results are shown in Figure 4. It was
found that the binding of both cohesins to the dockerin
is indeed Ca21-dependent; in the presence of EDTA, the
binding was similar in both cases to that of the negative
control (i.e., binding to an unrelated cohesin from A. cel-
lulolyticus). The pEC50 values were determined from the
respective binding curves.38 The pEC50 for AfCoh2375
was 7.73 versus 7.08 for AfCoh2376, indicating a prefer-
ence for the former cohesin. The fact that AfDoc2375
binds cohesin-like proteins from A. fulgidus with clear
species-specific recognition combined with Ca21-depend-
ency provides convincing evidence that the modules of
A. fulgidus behave like classical cohesins and dockerin.
AfCoh2375 oligomerization
Expressed cohesin proteins were purified using affinity
chromatography and Superdex 75 size-exclusion column
chromatography. A single major peak of eluted protein
was detected in the case of AfCoh2376, consistent with
the monomeric form. However, the chromatogram of
AfCoh2375 revealed the formation of monomers, dimers,
and tetramers. The existence of dynamic equilibrium
among the molecular forms was concluded from repeated
gel filtration chromatography of the collected monomer,
dimer, and tetramer fractions (Fig. 5). In three cases,
rechromatography of the isolated fractions on the same
column resulted in a similar distribution of three peaks,
suggesting tetramer and dimer formation from the
monomer, monomer and tetramer formation from the
dimer, and disintegration of the tetramer into monomer
and dimer. SDS-PAGE showed a single band of the
appropriate size from all three peak regions. This is the
first demonstration that oligomerization of a cohesin
Figure 3Affinity blotting of the A. fulgidus dockerin by the two A. fulgidus
cohesins. Partially purified samples of Xyn-AfDoc2375 were subjected to
SDS-PAGE (gel) and blotted onto nitrocellulose membranes. The blots
were probed with recombinant His-tagged AfCoh2375 and AfCoh2376,
and the labeled bands were detected by chemiluminescence, using
peroxidase-conjugated, anti-His-tag antibody.
Figure 4Quantitative analysis of the interaction between the A. fulgidus dockerin
and the two A. fulgidus cohesins. Recombinant AfCoh2375, AfCoh2376,
and AcCohC3 (control cohesin from scaffoldin C of Acetivibrio
cellulolyticus) were coated onto wells of microtiter plates and reacted
with increasing concentrations of recombinant xylanase-fused dockerin
(Xyn-AfDoc2375) in the presence or absence of EDTA. Detection of
binding was achieved by sequential incubation with rabbit anti-Xyn and
HRP-conjugated anti-rabbit IgG.
Figure 5Oligomerization of cohesin AfCoh2375. Gel-filtration chromatography
of AfCoh2375 on Superdex 75 served to separate the cohesin sample
into monomer, dimer, and tetramer fractions. The individual fractions
were collected, and each was rechromatographed on the same column.
Dynamic equilibrium between the forms was demonstrated by repeated
gel filtration of the different fractions.
Crystal Structure of an Archaeal Cohesion
PROTEINS 55

occurs unambiguously in solution. Until now dimeriza-
tion was only implied from crystal structures of cohesins
and from one case of an ultracentrifugation study of a C.
cellulolyticum cohesin in solution.39 However, higher
order oligomers, such as tetramers, were as yet not
detected in solution until this work. The functional role
of such association is currently unknown. Interestingly,
similar oligomerization was not evident in the crystals.
Nevertheless, such cohesin–cohesin interactions may
serve as an adhesive that may induce complementary oli-
gomerization and increased stability of putative com-
plexes between the cohesins of membrane-bound AF2376
and the dockerins of AF2375.
Structure determination
AfCoh2375 crystallized in the cubic P4332 space group,
with one molecule in the asymmetric unit as was previ-
ously published.7 The primary structure of AfCoh2375
was determined by BEAST19 using several cohesin struc-
tures for construction of a search model.39–41 The
phases were further improved by density modification
using DM,42 which produced a readily interpretable elec-
tron density map. The final atomic model was refined to
a crystallographic R-factor and Rfree of 21.2% and 25.8%,
respectively, at a resolution of 1.85 A. A total of 162 resi-
dues (including the six histidine residues of the C termi-
nal His-tag), four sulfate ions, and 137 water molecules
were present. In this model, four residues adopted alter-
native conformations: Ser41, Val45, Thr129, and Gln131.
The final statistics are summarized in Table 1.
Overall structure and topology
AfCoh2375 forms an elongated nine-stranded b-sand-wich in a classical jelly-roll topology with approximate
overall dimensions of 52 A 3 23 A 3 19 A (measured
on the Ca skeleton) (Fig. 6). These characteristics
are similar to those reported for other cohesin
structures.4,39,41,43 A structural similarity search using
the DALI server44 revealed that AfCoh2375 is most
similar to the type-I cohesin 2 of the C. thermocellum
Figure 6Schematic cartoon diagram of the overall three-dimensional structure of AfCoh2375. (A) Frontal view representing the b strands, numbered 1 to 9.
(B) Side view (frontal view rotated 90o about the y-axis). The loop connecting residues Gly96 to Arg123 is missing (unstructured). The C-terminal
His-tag is colored in red.
M. Voronov-Goldman et al.
56 PROTEINS

cellulosomal scaffoldin subunit41 with a RMSD of 1 A
based on the Ca atoms of 138 residues. Similar to the
known type-I cohesin structures,39,41,45 the ‘‘front face’’
of the molecule is formed by b-strands 8 (a: His125-
Gln131, b: Glu133-Asp137), 3 (Val33-Ser41), 6 (Gln70-
Pro77), and 5 (Leu61-Glu67), the ‘‘back-face’’ is formed
by b-strands 9 (a: Ala146-Ile148, b: Gly150-Ile154), 1 (a:
Thr7-Ala10, b: Ala13-Ala15), 2 (Gly18-Ile28), 7 (Gly85-
Val94), and 4 (Leu46-Gln53). Strands 1 and 9 are aligned
parallel to each other, whereas the other b-strands are
anti-parallel. Despite the similarity in overall structure
and topology to the type-I cohesins, several distinctive
differences were noted. In this context, a large segment
that consists of 27 residues between b-strands 7–8 in
AfCoh2375 does not exist in the type-I cohesins. This
putative loop is extremely long (approximately four times
longer than the analogous loop in the type-I cohesin 2
from C. thermocellum). The loop is largely undetectable
on the electron density maps and is assumed to adopt a
flexible, unstructured conformation (see next section).
Furthermore, the loops between b-strands 4–5 and 6–7
are shorter than their equivalents in the other type-I
cohesin structures. Interestingly, a secondary helical
structure of the His-tag was clearly evident in the
AfCoh2375 structure. Structures with well-defined His-
tags are rare, and the secondary structure is helical in
only two additional cases and extended in all others.46
The cohesin molecules fold around an extensive core net-
work of aromatic and hydrophobic residues. Similar to the
previously described cohesins, AfCoh2375 also possesses a
hydrophobic core (not shown), although less aromatic in
character, that serves to stabilize the entire structure. In this
case, the hydrophobic core residues are clustered through-
out the entire molecule. The hydrophobic cluster at the
crown of AfCoh2375 comprises only aliphatic residues,
whereas the center and bottom regions of the molecule con-
sist of a network of aromatic and aliphatic residues.
Comparison of AfCoh2375 with othercohesin structures
Superposition of representative cohesin structures (PDB
entry codes: 1OHZ_A, 2B59_A, 2OZN_A) with that of
AfCoh2375 [Fig. 7(A)] and multiple structure-based
sequence alignment [Fig. 7(B)] revealed several features
common to the cellulosomal cohesins. The structure of
AfCoh2375 determined in this work once again highlights
the phenomenon that occurs among cohesin modules: de-
spite the relatively low sequence similarity among them,
they share high structural similarity. All nine b-strands ofAfCoh2375 are aligned closely with respect to those of the
other cohesins and form a b-sandwich with a jelly-roll to-
pology. As mentioned above, despite the overall similar-
ities, AfCoh2375 possesses a unique putative 27-residue
loop region between b-strands 7 and 8 that distinguishes it
from the other known cohesin structures. This region
forms a deletion at the equivalent positions in the other
cohesin structures, located between residues Thr95 and
Ala121 [Fig. 7(A)], and is partially unstructured in the
model, apparently due to high flexibility of this loop.
Alignment of the two A. fulgidus cohesins predicts clear
conservation in the secondary structures and the absence
of the entire 27-residue loop in AfCoh2376. As, unlike
AfCoh2375, AfCoh2376 fails to undergo oligomerization,
this may indicate that the long loop is involved in the pro-
cess of dimer and tetramer formation.
To examine the putative dockerin-binding site of
AfCoh2375, the cohesin structures from the type-I and
type-II cohesin–dockerin complexes of C. thermocellum and
from the noncellulosomal C. perfringens complex were over-
laid separately with the AfCoh2375 structure. Surprisingly,
although the superposition of AfCoh2375 and the type-I
cohesin 2 of C. thermocellum (1OHZ_A) displayed the low-
est overall RMSD (1.6 A), the putative dockerin-binding site
of AfCoh2375 is more similar to that of the type-II cohesin
from the C. thermocellum SdbA anchoring protein
(2B59_A). This observation may indicate that putative
dockerin-binding residues of AfCoh2375 are similar to that
of the type-II cohesin, which may thus reflect an asymmetric
A. fulgidus cohesin–dockerin interaction.47 Indeed, the pu-
tative recognition residues in the duplicated segments of the
AF2375 dockerin are asymmetric. Taking this into account,
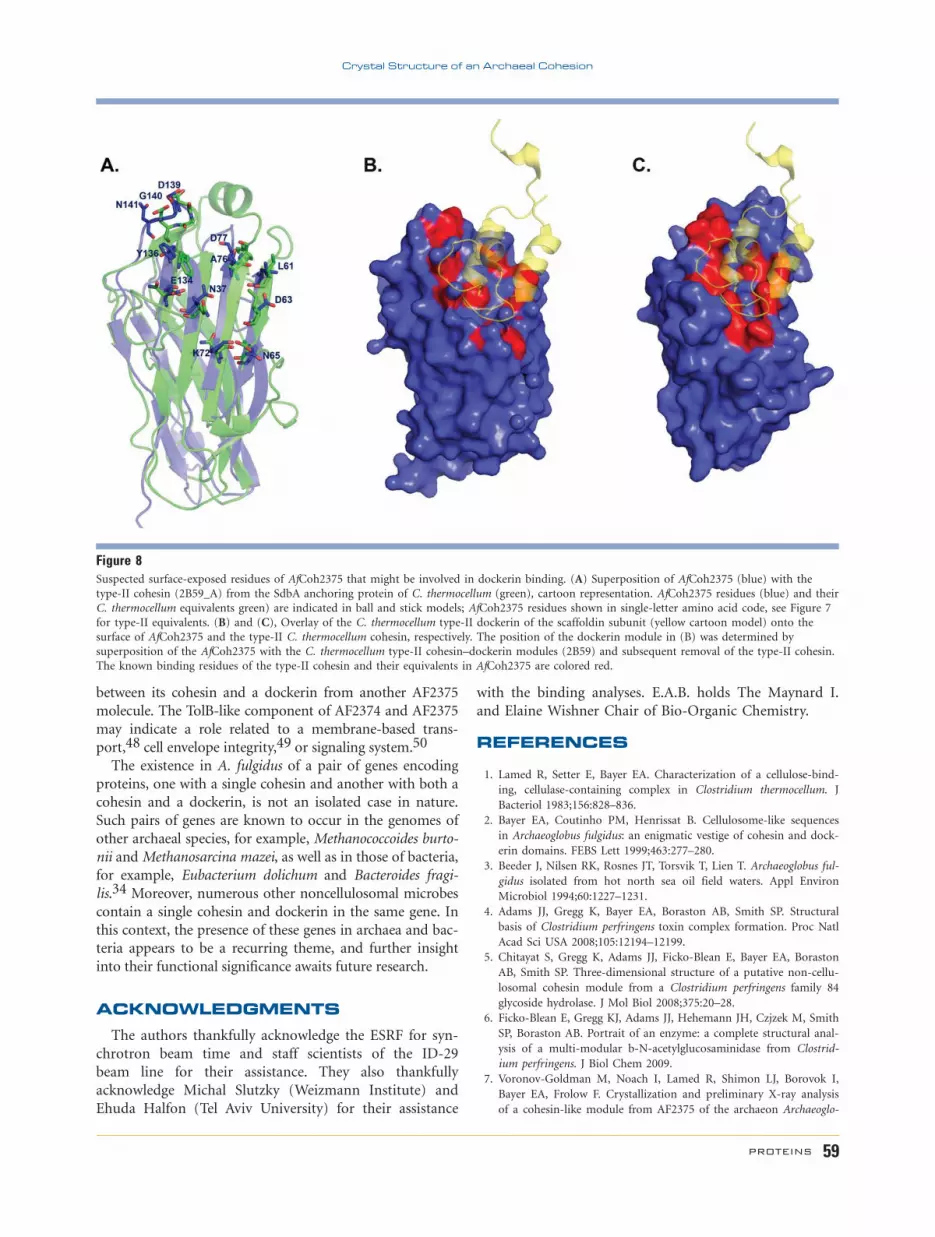
we suspect that the surface-exposed residues of AfCoh2375
that might be involved in dockerin recognition are: Asn37,
Leu61, Asp63, Asn65, Lys72, Ala76, Asp77, Glu134, Tyr136,
Asp139, Gly140, and Asn141 as indicated in Figures 7(B)
and 8. Most of these residues, located on the 8-3-6-5 b-sheet‘‘front face,’’ are similar or identical to the homologous
interacting residues from both AfCoh2376 and the type-II
cohesin (2B59_A). Nevertheless, because the A. fulgidus
cohesins fail to interact with the C. thermocellum type-II
dockerin37 and vice versa, it is the difference in surface to-
pology that would define the binding specificity. Such differ-
ences are clearly evident from the comparison of the two
surfaces [Fig. 8 (B,C)], as observed for the respective surface
pattern of the known binding residues and their equivalents.
Further insight into the precise mechanism of cohesin–
dockerin binding in A. fulgidus awaits determination of the
crystal structure of the appropriate heterodimeric complex.
Proposed biological rationale
The precise function of the A. fulgidus cohesins and dock-
erin modules is currently unknown. Based on the fact that
AF2374, AF2375, and AF2376 are organized in a putative
operon, we may anticipate a common cooperative function
among the respective proteins. Because AF2376 contains
two predicted transmembrane helices, it is logical to expect
that the protein is membrane bound, and its cohesin would
interact with the dockerin of AF2375. The latter protein can
undergo oligomerization, either by virtue of the reversible
cohesin–cohesin interaction or by specific interaction
Crystal Structure of an Archaeal Cohesion
PROTEINS 57

Figure 7Structural comparision of AfCoh2375 with representative cohesin sequences (stereo view). (A) Superposition of AfCoh2375 (blue), the type I
cohesin from the C. thermocellum scaffoldin subunit (1OHZ, red), the type II cohesin from the SdbA anchoring protein of C. thermocellum (2B59,
green), and the noncellulosomal C. perfringens cohesin (2JH2, yellow). (B) Structure-based alignment of AfCoh2375 versus type-I and type-II
cellulosomal cohesins and a noncellulosomal cohesin from C. perfringens. The AfCoh2375 cohesin structure from A. fulgidus was superimposed with
the type I (1ANU) and type II (2B59) cohesin structures from C. thermocellum, and the noncellulosomal cohesin from C. perfringens (2JH2), and
the sequences were aligned accordingly. The residues of the b-strands (arrows) at homologous positions are indicated in blue font. The missing(unstructured) 27-residue loop of AfCoh2375 is designated in red font. Amino acids responsible for hydrophilic interactions with the dockerin in
the known complexes are highlighted in cyan, and amino acids responsible for hydrophobic interactions are highlighted grey. Suspected surface-
exposed residues of AfCoh2375 that might be involved in dockerin binding are highlighted in magenta. For clarity, the positions of selected residues
in the AfCoh2375 sequence are enumerated. The primary sequence of AfCoh2376 was also included in the alignment,2 starting with the previously
published sequence-based alignment with manual adjustments.
M. Voronov-Goldman et al.
58 PROTEINS
between its cohesin and a dockerin from another AF2375
molecule. The TolB-like component of AF2374 and AF2375
may indicate a role related to a membrane-based trans-
port,48 cell envelope integrity,49 or signaling system.50
The existence in A. fulgidus of a pair of genes encoding
proteins, one with a single cohesin and another with both a
cohesin and a dockerin, is not an isolated case in nature.
Such pairs of genes are known to occur in the genomes of
other archaeal species, for example, Methanococcoides burto-
nii andMethanosarcina mazei, as well as in those of bacteria,
for example, Eubacterium dolichum and Bacteroides fragi-
lis.34 Moreover, numerous other noncellulosomal microbes
contain a single cohesin and dockerin in the same gene. In
this context, the presence of these genes in archaea and bac-
teria appears to be a recurring theme, and further insight
into their functional significance awaits future research.
ACKNOWLEDGMENTS
The authors thankfully acknowledge the ESRF for syn-
chrotron beam time and staff scientists of the ID-29
beam line for their assistance. They also thankfully
acknowledge Michal Slutzky (Weizmann Institute) and
Ehuda Halfon (Tel Aviv University) for their assistance
with the binding analyses. E.A.B. holds The Maynard I.
and Elaine Wishner Chair of Bio-Organic Chemistry.
REFERENCES
1. Lamed R, Setter E, Bayer EA. Characterization of a cellulose-bind-
ing, cellulase-containing complex in Clostridium thermocellum. J
Bacteriol 1983;156:828–836.
2. Bayer EA, Coutinho PM, Henrissat B. Cellulosome-like sequences
in Archaeoglobus fulgidus: an enigmatic vestige of cohesin and dock-
erin domains. FEBS Lett 1999;463:277–280.
3. Beeder J, Nilsen RK, Rosnes JT, Torsvik T, Lien T. Archaeoglobus ful-
gidus isolated from hot north sea oil field waters. Appl Environ
Microbiol 1994;60:1227–1231.
4. Adams JJ, Gregg K, Bayer EA, Boraston AB, Smith SP. Structural
basis of Clostridium perfringens toxin complex formation. Proc Natl
Acad Sci USA 2008;105:12194–12199.
5. Chitayat S, Gregg K, Adams JJ, Ficko-Blean E, Bayer EA, Boraston
AB, Smith SP. Three-dimensional structure of a putative non-cellu-
losomal cohesin module from a Clostridium perfringens family 84
glycoside hydrolase. J Mol Biol 2008;375:20–28.
6. Ficko-Blean E, Gregg KJ, Adams JJ, Hehemann JH, Czjzek M, Smith
SP, Boraston AB. Portrait of an enzyme: a complete structural anal-
ysis of a multi-modular b-N-acetylglucosaminidase from Clostrid-
ium perfringens. J Biol Chem 2009.
7. Voronov-Goldman M, Noach I, Lamed R, Shimon LJ, Borovok I,
Bayer EA, Frolow F. Crystallization and preliminary X-ray analysis
of a cohesin-like module from AF2375 of the archaeon Archaeoglo-
Figure 8Suspected surface-exposed residues of AfCoh2375 that might be involved in dockerin binding. (A) Superposition of AfCoh2375 (blue) with the
type-II cohesin (2B59_A) from the SdbA anchoring protein of C. thermocellum (green), cartoon representation. AfCoh2375 residues (blue) and their
C. thermocellum equivalents green) are indicated in ball and stick models; AfCoh2375 residues shown in single-letter amino acid code, see Figure 7
for type-II equivalents. (B) and (C), Overlay of the C. thermocellum type-II dockerin of the scaffoldin subunit (yellow cartoon model) onto the
surface of AfCoh2375 and the type-II C. thermocellum cohesin, respectively. The position of the dockerin module in (B) was determined by
superposition of the AfCoh2375 with the C. thermocellum type-II cohesin–dockerin modules (2B59) and subsequent removal of the type-II cohesin.
The known binding residues of the type-II cohesin and their equivalents in AfCoh2375 are colored red.
Crystal Structure of an Archaeal Cohesion
PROTEINS 59

bus fulgidus. Acta Crystallogr Sect F Struct Biol Cryst Commun
2009;65 (Part 3):275–278.
8. Barak Y, Handelsman T, Nakar D, Mechaly A, Lamed R, Shoham Y,
Bayer EA. Matching fusion protein systems for affinity analysis of
two interacting families of proteins: the cohesin-dockerin interac-
tion. J Mol Recognit 2005;18:491–501.
9. Xu Q, Gao W, Ding SY, Kenig R, Shoham Y, Bayer EA, Lamed R.
The cellulosome system of Acetivibrio cellulolyticus includes a novel
type of adaptor protein and a cell surface anchoring protein. J Bac-
teriol 2003;185:4548–4557.
10. Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J,
Ponting CP, Bork P. SMART 4.0: towards genomic data integration.
Nucleic Acids Res 2004;32(Database issue):D142–D144.
11. Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a
web-based tool for the study of genetically mobile domains. Nucleic
Acids Res 2000;28:231–234.
12. Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, For-
slund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein fami-
lies database. Nucleic Acids Res 2008;36(Database issue):D281–D288.
13. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W,
Lipman DJ. Gapped BLASTand PSI-BLAST: a new generation of pro-
tein database search programs. Nucleic Acids Res 1997;25:3389–3402.
14. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting
transmembrane protein topology with a hidden Markov model:
application to complete genomes. J Mol Biol 2001;305:567–580.
15. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel
RD, Bairoch A. Protein identification and analysis tools on the
ExPASy server. In: Walker JM, editor. The proteomics protocols
handbook. Tolowa, NJ: Humana Press; 2005. pp 571–607.
16. Higgins DG, Thompson JD, Gibson TJ. Using CLUSTAL for multi-
ple sequence alignments. Methods Enzymol 1996;266:383–402.
17. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolu-
tionary genetics analysis (MEGA) software version 4.0. Mol Biol
Evol 2007;24:1596–1599.
18. Stout GH, Jensen LH. X-ray structure determination. A practical
guide. London: Macmillan; 1968.
19. Read RJ. Pushing the boundaries of molecular replacement with
maximum likelihood. Acta Crystallogr Sect D Biol Crystallogr
2001;57:1373–1382.
20. Bailey S. The CCP4 Suite—programs for protein crystallography.
Acta Cryst D 1994;50:760–763.
21. Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation
functions. Acta Crystallogr D Biol Crystallogr 2004;60 (Part 3):432–438.
22. McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-
enhanced fast translation functions. Acta Crystallogr D Biol Crystal-
logr 2005;61 (Part 4):458–464.
23. Perrakis A, Morris R, Lamzin VS. Automated protein model build-
ing combined with iterative structure refinement. Nature Struct
Biol 1999;6:458–463.
24. Lamzin VS, Wilson KS. Automated refinement of protein models.
Acta Crystallogr Sect D Biol Crystallogr 1993;49:129–147.
25. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolec-
ular structures by the maximum-likelihood method. Acta Crystal-
logr Sect D Biol Crystallogr 1997;53:240–255.
26. Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods
for building protein models in electron-density maps and the loca-
tion of errors in these models. Acta Cryst A 1991;47:110–119.
27. Emsley P, Cowtan K. Coot: model-building tools for molecular
graphics. Acta Crystallogr Sect D Biol Crystallogr 2004;60:2126–2132.
28. Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Mor-
iarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX:
building new software for automated crystallographic structure determi-
nation. Acta Crystallogr D Biol Crystallogr 2002;58 (Part 11):1948–1954.
29. Brunger AT. Free R-value—a novel statistical quantity for assessing
the accuracy of crystal-structures. Nature 1992;355:472–475.
30. Kleywegt GJ. Recognition of spatial motifs in protein structures. J
Mol Biol 1999;285:1887–1897.
31. Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray
LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. Mol-
Probity: all-atom contacts and structure validation for proteins and
nucleic acids. Nucleic Acids Res 2007;35(Web Server issue):W375–W383.
32. Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPRO-
BITY: structure validation and all-atom contact analysis for nucleic
acids and their complexes. Nucleic Acids Res 2004;32(Web Server
issue):W615–W619.
33. DeLano WL. The PyMOL molecular graphics system. San Carlos,
CA: DeLano Scientific LLC; 2002.
34. Peer A, Smith SP, Bayer EA, Lamed R, Borovok I. Noncellulosomal
cohesin- and dockerin-like modules in the three domains of life.
FEMS Microbiol Lett 2009;291:1–16.
35. Lytle B, Volkman BF, Westler WM, Wu JHD. Secondary structure
and calcium-induced folding of the Clostridium thermocellum
dockerin domain determined by NMR spectroscopy. Arch Biochem
Biophys 2000;379:237–244.
36. Pages S, Belaich A, Belaich J-P, Morag E, Lamed R, Shoham Y,
Bayer EA. Species-specificity of the cohesin-dockerin interaction
between Clostridium thermocellum and Clostridium cellulolyticum:
prediction of specificity determinants of the dockerin domain. Pro-
teins 1997;29:517–527.
37. Haimovitz R, Barak Y, Morag E, Voronov-Goldman M, Shoham Y,
Lamed R, Bayer EA. Cohesin-dockerin microarray: diverse specific-
ities between two complementary families of interacting protein
modules. Proteomics 2008;8:968–979.
38. Motulsky HJ, Christopoulos A. Fitting models to biological data
using linear and nonlinear regression. A practical guide to curve fit-
ting. San Diego, CA: GraphPad Software Inc.; 2003. 351 p.
39. Spinelli S, Fierobe HP, Belaich A, Belaich JP, Henrissat B, Cambillau C.
Crystal structure of a cohesin module from Clostridium cellulolyticum:
implications for dockerin recognition. J Mol Biol 2000;304:189–200.
40. Noach I, Frolow F, Jakoby H, Rosenheck S, Shimon LW, Lamed R,
Bayer EA. Crystal structure of a type-II cohesin module from the
Bacteroides cellulosolvens cellulosome reveals novel and distinctive
secondary structural elements. J Mol Biol 2005;348:1–12.
41. Shimon LJ, Bayer EA, Morag E, Lamed R, Yaron S, Shoham Y,
Frolow F. A cohesin domain from Clostridium thermocellum: the
crystal structure provides new insights into cellulosome assembly.
Structure 1997;5:381–390.
42. Cowtan K, Main P. Miscellaneous algorithms for density modifica-
tion. Acta Cryst D 1998;54:487–493.
43. Noach I, Frolow F, Alber O, Lamed R, Shimon LJW, Bayer EA.
Inter-modular linker flexibility revealed from crystal structures of
adjacent cellulosomal cohesins of Acetivibrio cellulolyticus. J Mol
Biol 2009;391:86–97.
44. Holm L, Park J. DaliLite workbench for protein structure compari-
son. Bioinformatics 2000;16:566–567.
45. Tavares GA, Beguin P, Alzari PM. The crystal structure of a type I
cohesin domain at 1.7 A resolution. J Mol Biol 1997;273:701–713.
46. Carson M, Johnson DH, McDonald H, Brouillette C, Delucas LJ.
His-tag impact on structure. Acta Crystallogr D Biol Crystallogr
2007;63:295–301.
47. Adams JJ, Pal G, Jia Z, Smith SP. Mechanism of bacterial cell-surface
attachment revealed by the structure of cellulosomal type II cohesin-
dockerin complex. Proc Natl Acad Sci USA 2006;103:305–310.
48. Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. The trans-
envelope Tol-Pal complex is part of the cell division machinery and
required for proper outer-membrane invagination during cell con-
striction in E. coli. Mol Microbiol 2007;63:1008–1025.
49. Walburger A, Lazdunski C, Corda Y. The Tol/Pal system function
requires an interaction between the C-terminal domain of TolA and
the N-terminal domain of TolB. Mol Microbiol 2002;44:695–708.
50. Bonsor DA, Hecht O, Vankemmelbeke M, Sharma A, Krachler AM,
Housden NG, Lilly KJ, James R, Moore GR, Kleanthous C. Alloste-
ric b-propeller signalling in TolB and its manipulation by translo-
cating colicins. EMBO J 2009;28:2846–2857.
M. Voronov-Goldman et al.
60 PROTEINS







![Characterization and Structure of a Zn2+ and [2Fe-2S]-containing Copper Chaperone from Archaeoglobus fulgidus](https://img.pdfslide.net/doc/110x75/63343d6b7a687b71aa0898fd/characterization-and-structure-of-a-zn2-and-2fe-2s-containing-copper-chaperone.jpg)



















