Embed Size (px)
Citation preview

DOI: 10.1002/chem.200802554
On the Size of Ions Solvated in Helium Clusters
Filipe Ferreira da Silva,[a] Philipp Waldburger,[a] Stefan Jaksch,[a] Andreas Mauracher,[a]
Stephan Denifl,[a] Olof Echt,*[b] Tilmann D. M�rk,[a, c] and Paul Scheier*[a]
Introduction
Solvation is a unifying concept in chemistry. A central topicis, of course, ion solvation in water;[1–3] gas-phase studies ofhydrated ions in which physical properties are measured asa function of the exact number of water molecules havehelped unravel microscopic details of hydration.[4–15]
Experimental methods applied to hydrated ions in the gasphase are equally well suited to study solvation of ions orneutrals in nonpolar solvents. Particularly illuminating aremeasurements of the size-dependence of features in elec-
tronic, vibrational, or rotational spectra of the solute, usuallyaccomplished by some variant of action (depletion) spec-troscopy,[16–18] photoelectron spectroscopy,[19–21] or resonantphotoionization.[22]
Of special interest are the microscopic properties of solva-tion shells. Closure of the first solvation shell in a complexXYn (where the solute X is either neutral or charged) canbe inferred from an abrupt change in the evaporation ener-gies, that is, the incremental binding energies DE(n),
XYn ¼ XYn�1 þ Y�DEðnÞ ð1Þ
DE(n), also called the dissociation or separation energy,may be obtained from ion-molecule equilibria in high-pres-sure mass spectrometric measurements.[4,6,23] Alternatively,DE(n) can be deduced from photoelectron spectra of neu-tral or negatively charged XYn complexes.[21] However, clo-sure of a solvation shell is often evident more easily fromdata that reflect evaporation energies of cluster ions in aqualitative way. In many experiments, cluster ions are vibra-tionally excited, and they are prone to unimolecular dissoci-ation in a field-free section of the mass spectrometer. Al-though the quantitative relation between evaporation ener-gies and the size dependence of dissociation rates is complexbecause an evaporative ensemble[24] is neither canonical normicrocanonical, one usually finds that particularly unstable
Abstract: Helium nanodroplets aredoped with SF6, C4F8, CCl4, C6H5Br,CH3I, and I2. Upon interaction withfree electrons a variety of positivelyand negatively charged cluster ions X�
Hen are observed where X�=F�, Cl� ,Br�, I+ , I2
+ , or CH3I+ . The yield of
these ions versus cluster size n drops atcharacteristic sizes ns that range fromns = 10.2�0.6 for F+ to ns =22.2�0.2for Br�. ns values for halide anions areabout 70 % larger than for the corre-sponding cations. The steps in the ion
yield suggest closure of the first solva-tion shell. We propose a simple classi-cal model to estimate ionic radii fromns. Assuming the helium density in thefirst solvation shell equals the heliumbulk density one finds that radii ofhalide anions in helium are nearlytwice as large as in alkali halide crys-
tals, indicating the formation of ananion bubble due to the repulsiveforces that derive from the exchangeinteraction. In spite of the simplicity ofour model, anion radii derived from itagree within approximately 10 % withvalues derived from the mobility ofhalide anions in superfluid bulkhelium, and with values computed byquantum Monte Carlo methods forX�Hen cluster anions.
Keywords: atomic clusters · gas-phase reactions · helium · massspectrometry · solvation
[a] F. Ferreira da Silva, P. Waldburger, S. Jaksch, A. Mauracher,Dr. S. Denifl, Prof. Dr. T. D. M�rk, Prof. Dr. P. ScheierInstitut f�r Ionenphysik und Angewandte PhysikLeopold Franzens Universit�tTechnikerstrasse 25, 6020 Innsbruck (Austria)Fax (+43) 512-507-2932E-mail : [email protected]
[b] Prof. Dr. O. EchtDepartment of Physics, University of New HampshireDurham, NH 03824 (USA)Fax: (+1) 603-862-2998E-mail : [email protected]
[c] Prof. Dr. T. D. M�rkDepartment of Experimental Physics, Comenius University84248 Bratislava (Slovak Republic)
Chem. Eur. J. 2009, 15, 7101 – 7108 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 7101
FULL PAPER

cluster ions (those with small DE(n) values) are character-ized by enhanced dissociation rates and therefore form localminima in mass spectra. Magic numbers in mass spectraoften reflect particularly stable cluster sizes,[25] and stepwisedrops in the yield of solvated ions indicate closure of a sol-vation shell. For non-directional bonding, icosahedral struc-tures are often energetically favorable, and ns = 12 is a com-monly observed number of solvent atoms in the first solva-tion shell, for example, for O�Arn, NO�Arn,
[21] and HFArn.[26]
For large size differences between solvent and solute onefinds steps below or above n=12; the ns values no longerconvey structural information but may be used instead todetermine the size of the solute.[27]
With the recent development of powerful helium nano-droplet sources, nearly any kind of atom or molecule, neu-tral or charged, can be embedded in helium.[16,17, 28,29] Where-as neutral alkali and alkaline-earth atoms will reside at ornear the surface of helium droplets, nearly all other dopantswill gain energy upon moving toward the center of the drop-let.[30] The interaction of molecules including N2O, CO2, andOCS with helium has been explored by high-resolution vi-brational and rotational spectroscopy. One surprising resultis the appearance of superfluidity in the solvent even beforethe first solvation shell is completed;[31–33] this raises thequestion of whether solvation shells are defined at all.[34]
For charged dopants the interaction with the solventatoms is much stronger. Half a century ago Atkins estimatedthat ions in liquid 4He form snowballs containing about 50tightly bound atoms.[35] More recent quantum-mechanicalcalculations of helium clusters doped with alkali cations doindeed show a rigid first coordination shell with strongly lo-calized solvent atoms.[36–40] However, ions do not alwaysinduce localization: Helium atoms in the first solvation shellof Ne+ remain delocalized, and calculated evaporation ener-gies DE(n) vary smoothly with n.[41] Theory also suggeststhat singly charged alkaline-earth ions[42,43] and some anionsincluding halide ions,[44] form bubbles rather than snowballs,and that solvent atoms in the “crust” of these bubblesremain delocalized.
There is no shortage of experiments on charged atoms,molecules, or clusters embedded in helium,[45–55] but veryfew relate to solvation. Meiwes-Broer and co-workers re-ported that mass spectra of Mg+Hen, formed by femtosec-ond ionization, exhibit a strong drop in ion yield for small nfollowed by a broad plateau.[56] The feature was assigned toclosure of a first solvation shell around n=20. In anothernoteworthy study, mass spectra of Ar+Hen exhibit an abruptdrop at n=12, whereas mass spectra of Ne+Hen do notshow reproducible intensity anomalies.[41] Helium clustersgrown in a drift tube around cations of N2, O2, CO, CO2,and rare gas atoms show drops in the ion yield at or nearn= 12.[57–59] In recent work from our laboratory, CCl4 andSF6 molecules were embedded in helium nanodroplets; ex-tended cluster ion series F+Hen and Cl+He were formed byelectron impact ionization.[60]
In the present work we apply mass spectrometry to deter-mine the size of the first solvation shell for several ions em-
bedded in helium droplets. Cations X+Hen with X+ =F+ ,Cl+ , Br+ , I+ , I2
+ , or CH3I+ are formed by electron impact
ionization of helium droplets doped with SF6, C4F8, CCl4,C6H5Br, CH3I, or I2. Anions X�Hen with X�=F�, Cl� andBr� are formed by electron attachment to helium dropletsdoped with SF6, CCl4 or C6H5Br. Each observed cluster ionseries exhibits a marked drop at a characteristic size ns thatwe interpret as the number of solvent atoms in the first sol-vation shell. For F�, Cl�, and Br�, ns values are about 70 %larger than for the corresponding cations. We propose asimple classical model to estimate radii of ions solvated inhelium from the observed values of ns ; results for halideions are compared with published experimental and theoret-ical work.
Results
Positive-ion mass spectra : Positive-ion mass spectra were re-corded for helium droplets doped with SF6, CCl4, C6H5Br, I2,or CH3I. Note that a neutral droplet may capture more thanone dopant molecule in the pick-up cell. The following ionswere observed to form large complexes with helium: F+ ,Cl+ , Cl2
+ , Br+ , I+ , I2+ , I3
+ , CH3I+ , CH3I2
+ . Some of theseion series escaped quantitative analysis because they coin-cide in mass with other ion series. The mass resolution inthe present study was insufficient to clearly separate ionswith nominally identical mass, such as 12CHI+ (a fragmentof methyl iodine, mass 139.91 u) and He35
+ (140.09 u).Cation spectra of droplets doped with SF6 or CCl4 have
been published previously.[60] They are congested due to thepresence of more than one isotope in natural sulfur andchlorine. Data analysis is challenging as illustrated furtherbelow in our discussion of anion spectra.
In Figure 1 we present cation mass spectra of dropletsdoped with iodine (Figure 1 a and b) and methyl iodine (Fig-ure 1 c). These compounds are essentially monisotopic,except for the 1.1 % abundance of 13C in natural carbon. Allspectra show an extended series of Hen
+ (marked black,nominal mass 4n u). For droplets doped with I2, a series ofI+Hen ion peaks (marked red) sets in at 127 u (bare I+).The yield of this series decreases markedly with increasingsize n if the partial I2 pressure in the pickup cell is low (Fig-ure 1 a). When the I2 pressure is increased threefold (Fig-ure 1 b), the yield of I+Hen levels off much more slowly. Atthis increased I2 pressure we observe another ion series be-ginning at 254 u due to I2
+Hen ions (marked blue). Athigher mass (not shown) we also observe a strong I3
+ peakfollowed by I3
+Hen ions.The situation is more complex in Figure 1 c, which shows
ions resulting from helium droplets doped with CH3I. In ad-dition to Hen
+ , I+Hen, and I2+Hen we also observe CH3I
+
Hen starting at 142 u. The CH3I+Hen series levels off quick-
ly, hence it does not obscure the I2+Hen series which, begin-
ning at 254 u, coincides in nominal mass. However, CHI+
and CI+ fragment ions at 140 and 139 u, respectively, do co-incide with members of the Hen
+ and I+Hen ion series. Be-
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 7101 – 71087102
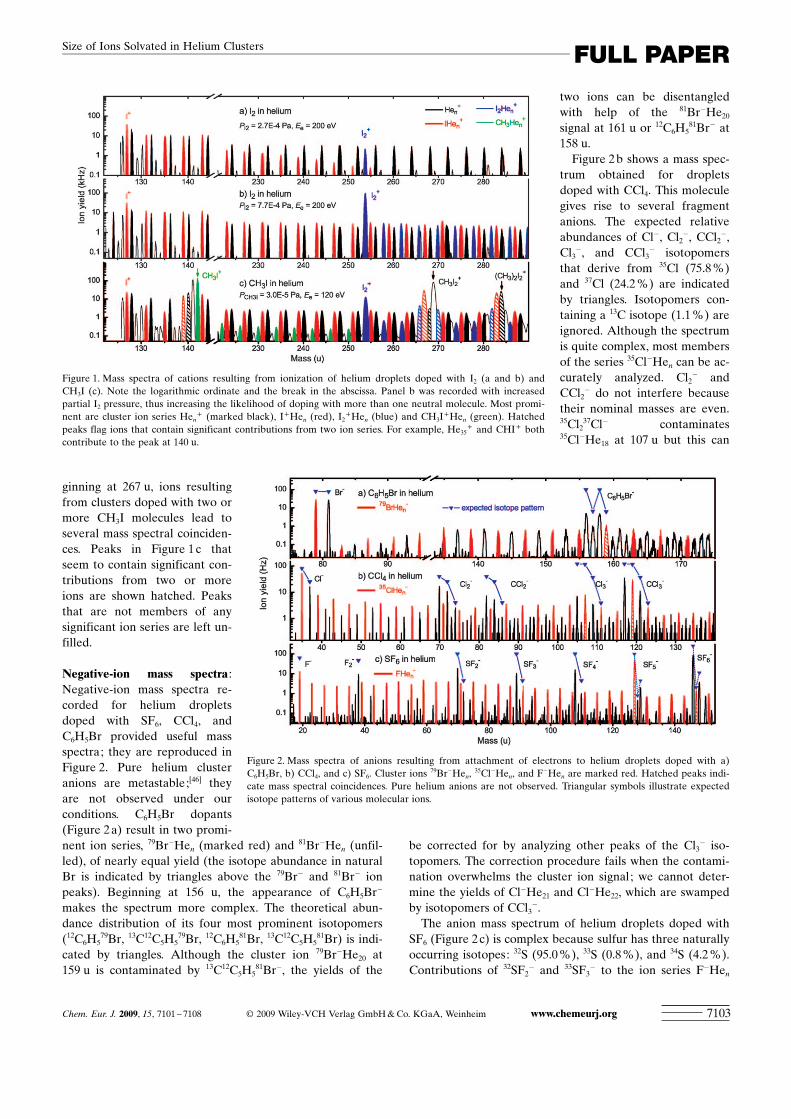
ginning at 267 u, ions resultingfrom clusters doped with two ormore CH3I molecules lead toseveral mass spectral coinciden-ces. Peaks in Figure 1 c thatseem to contain significant con-tributions from two or moreions are shown hatched. Peaksthat are not members of anysignificant ion series are left un-filled.
Negative-ion mass spectra :Negative-ion mass spectra re-corded for helium dropletsdoped with SF6, CCl4, andC6H5Br provided useful massspectra; they are reproduced inFigure 2. Pure helium clusteranions are metastable;[46] theyare not observed under ourconditions. C6H5Br dopants(Figure 2 a) result in two promi-nent ion series, 79Br�Hen (marked red) and 81Br�Hen (unfil-led), of nearly equal yield (the isotope abundance in naturalBr is indicated by triangles above the 79Br� and 81Br� ionpeaks). Beginning at 156 u, the appearance of C6H5Br�
makes the spectrum more complex. The theoretical abun-dance distribution of its four most prominent isotopomers(12C6H5
79Br, 13C12C5H579Br, 12C6H5
81Br, 13C12C5H581Br) is indi-
cated by triangles. Although the cluster ion 79Br�He20 at159 u is contaminated by 13C12C5H5
81Br�, the yields of the
two ions can be disentangledwith help of the 81Br�He20
signal at 161 u or 12C6H581Br� at
158 u.Figure 2 b shows a mass spec-
trum obtained for dropletsdoped with CCl4. This moleculegives rise to several fragmentanions. The expected relativeabundances of Cl�, Cl2
�, CCl2�,
Cl3�, and CCl3
� isotopomersthat derive from 35Cl (75.8 %)and 37Cl (24.2 %) are indicatedby triangles. Isotopomers con-taining a 13C isotope (1.1 %) areignored. Although the spectrumis quite complex, most membersof the series 35Cl�Hen can be ac-curately analyzed. Cl2
� andCCl2
� do not interfere becausetheir nominal masses are even.35Cl2
37Cl� contaminates35Cl�He18 at 107 u but this can
be corrected for by analyzing other peaks of the Cl3� iso-
topomers. The correction procedure fails when the contami-nation overwhelms the cluster ion signal; we cannot deter-mine the yields of Cl�He21 and Cl�He22, which are swampedby isotopomers of CCl3
�.The anion mass spectrum of helium droplets doped with
SF6 (Figure 2 c) is complex because sulfur has three naturallyoccurring isotopes: 32S (95.0 %), 33S (0.8 %), and 34S (4.2 %).Contributions of 32SF2
� and 33SF3� to the ion series F�Hen
Figure 1. Mass spectra of cations resulting from ionization of helium droplets doped with I2 (a and b) andCH3I (c). Note the logarithmic ordinate and the break in the abscissa. Panel b was recorded with increasedpartial I2 pressure, thus increasing the likelihood of doping with more than one neutral molecule. Most promi-nent are cluster ion series Hen
+ (marked black), I+Hen (red), I2+Hen (blue) and CH3I
+Hen (green). Hatchedpeaks flag ions that contain significant contributions from two ion series. For example, He35
+ and CHI+ bothcontribute to the peak at 140 u.
Figure 2. Mass spectra of anions resulting from attachment of electrons to helium droplets doped with a)C6H5Br, b) CCl4, and c) SF6. Cluster ions 79Br�Hen,
35Cl�Hen, and F�Hen are marked red. Hatched peaks indi-cate mass spectral coincidences. Pure helium anions are not observed. Triangular symbols illustrate expectedisotope patterns of various molecular ions.
Chem. Eur. J. 2009, 15, 7101 – 7108 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 7103
FULL PAPERSize of Ions Solvated in Helium Clusters
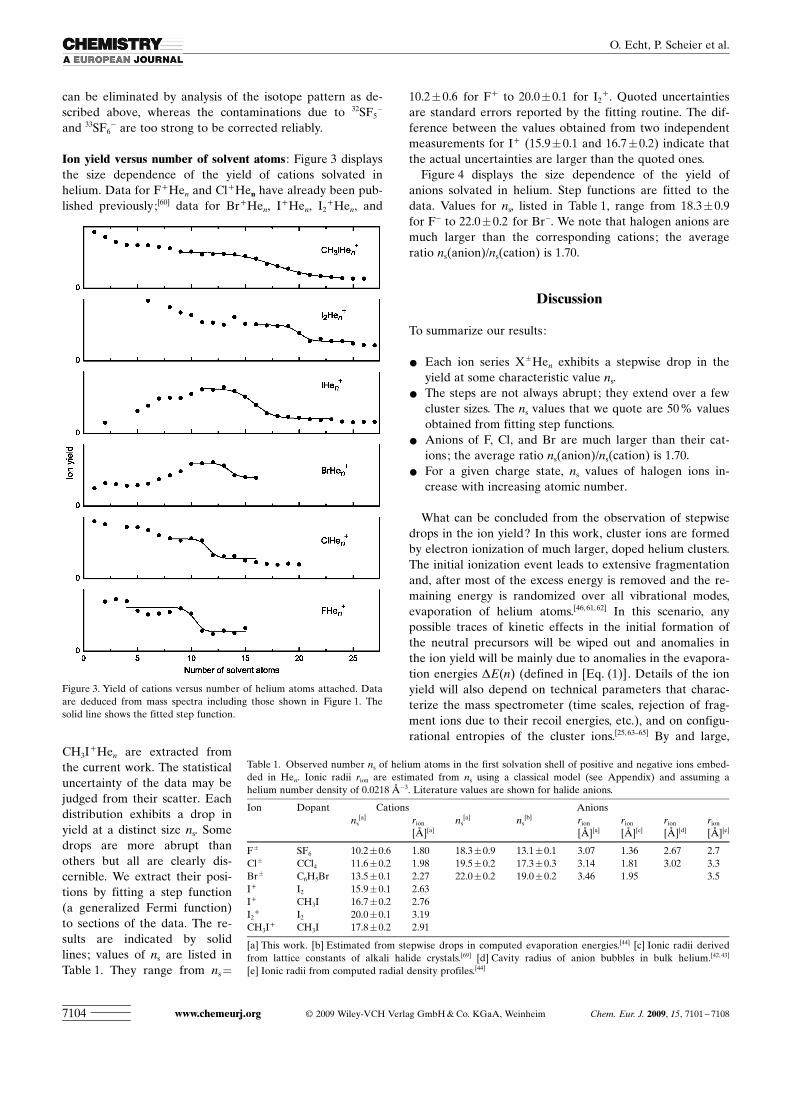
can be eliminated by analysis of the isotope pattern as de-scribed above, whereas the contaminations due to 32SF5
�
and 33SF6� are too strong to be corrected reliably.
Ion yield versus number of solvent atoms : Figure 3 displaysthe size dependence of the yield of cations solvated inhelium. Data for F+Hen and Cl+Hen have already been pub-lished previously;[60] data for Br+Hen, I+Hen, I2
+Hen, and
CH3I+Hen are extracted from
the current work. The statisticaluncertainty of the data may bejudged from their scatter. Eachdistribution exhibits a drop inyield at a distinct size ns. Somedrops are more abrupt thanothers but all are clearly dis-cernible. We extract their posi-tions by fitting a step function(a generalized Fermi function)to sections of the data. The re-sults are indicated by solidlines; values of ns are listed inTable 1. They range from ns =
10.2�0.6 for F+ to 20.0�0.1 for I2+ . Quoted uncertainties
are standard errors reported by the fitting routine. The dif-ference between the values obtained from two independentmeasurements for I+ (15.9�0.1 and 16.7�0.2) indicate thatthe actual uncertainties are larger than the quoted ones.
Figure 4 displays the size dependence of the yield ofanions solvated in helium. Step functions are fitted to thedata. Values for ns, listed in Table 1, range from 18.3�0.9for F� to 22.0�0.2 for Br�. We note that halogen anions aremuch larger than the corresponding cations; the averageratio ns ACHTUNGTRENNUNG(anion)/ns ACHTUNGTRENNUNG(cation) is 1.70.
Discussion
To summarize our results:
* Each ion series X�Hen exhibits a stepwise drop in theyield at some characteristic value ns.
* The steps are not always abrupt; they extend over a fewcluster sizes. The ns values that we quote are 50 % valuesobtained from fitting step functions.
* Anions of F, Cl, and Br are much larger than their cat-ions; the average ratio ns ACHTUNGTRENNUNG(anion)/ns ACHTUNGTRENNUNG(cation) is 1.70.
* For a given charge state, ns values of halogen ions in-crease with increasing atomic number.
What can be concluded from the observation of stepwisedrops in the ion yield? In this work, cluster ions are formedby electron ionization of much larger, doped helium clusters.The initial ionization event leads to extensive fragmentationand, after most of the excess energy is removed and the re-maining energy is randomized over all vibrational modes,evaporation of helium atoms.[46,61,62] In this scenario, anypossible traces of kinetic effects in the initial formation ofthe neutral precursors will be wiped out and anomalies inthe ion yield will be mainly due to anomalies in the evapora-tion energies DE(n) (defined in [Eq. (1)]. Details of the ionyield will also depend on technical parameters that charac-terize the mass spectrometer (time scales, rejection of frag-ment ions due to their recoil energies, etc.), and on configu-rational entropies of the cluster ions.[25,63–65] By and large,
Figure 3. Yield of cations versus number of helium atoms attached. Dataare deduced from mass spectra including those shown in Figure 1. Thesolid line shows the fitted step function.
Table 1. Observed number ns of helium atoms in the first solvation shell of positive and negative ions embed-ded in Hen. Ionic radii rion are estimated from ns using a classical model (see Appendix) and assuming ahelium number density of 0.0218 ��3. Literature values are shown for halide anions.
Ion Dopant Cations Anionsns
[a] rion
[�][a]ns
[a] ns[b] rion
[�][a]rion
[�][c]rion
[�][d]rion
[�][e]
F� SF6 10.2�0.6 1.80 18.3�0.9 13.1�0.1 3.07 1.36 2.67 2.7Cl� CCl4 11.6�0.2 1.98 19.5�0.2 17.3�0.3 3.14 1.81 3.02 3.3Br� C6H5Br 13.5�0.1 2.27 22.0�0.2 19.0�0.2 3.46 1.95 3.5I+ I2 15.9�0.1 2.63I+ CH3I 16.7�0.2 2.76I2
+ I2 20.0�0.1 3.19CH3I
+ CH3I 17.8�0.2 2.91
[a] This work. [b] Estimated from stepwise drops in computed evaporation energies.[44] [c] Ionic radii derivedfrom lattice constants of alkali halide crystals.[69] [d] Cavity radius of anion bubbles in bulk helium.[42, 43]
[e] Ionic radii from computed radial density profiles.[44]
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 7101 – 71087104
O. Echt, P. Scheier et al.
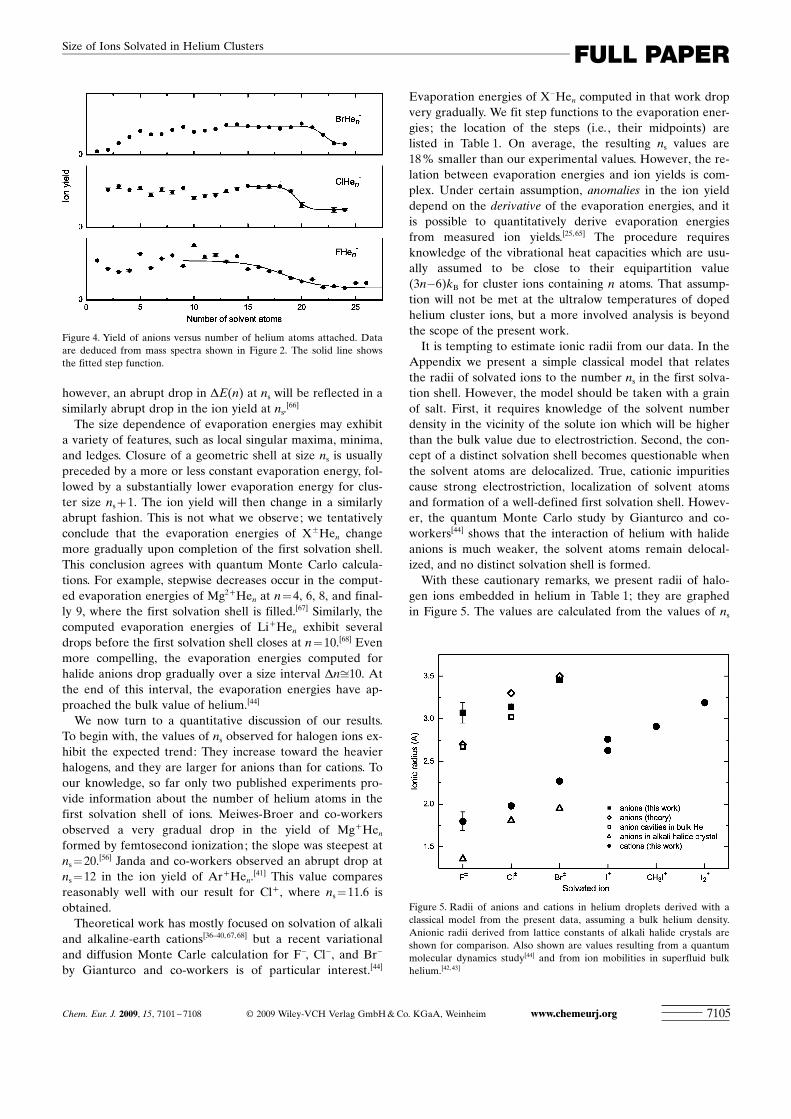
however, an abrupt drop in DE(n) at ns will be reflected in asimilarly abrupt drop in the ion yield at ns.
[66]
The size dependence of evaporation energies may exhibita variety of features, such as local singular maxima, minima,and ledges. Closure of a geometric shell at size ns is usuallypreceded by a more or less constant evaporation energy, fol-lowed by a substantially lower evaporation energy for clus-ter size ns +1. The ion yield will then change in a similarlyabrupt fashion. This is not what we observe; we tentativelyconclude that the evaporation energies of X�Hen changemore gradually upon completion of the first solvation shell.This conclusion agrees with quantum Monte Carlo calcula-tions. For example, stepwise decreases occur in the comput-ed evaporation energies of Mg2+Hen at n= 4, 6, 8, and final-ly 9, where the first solvation shell is filled.[67] Similarly, thecomputed evaporation energies of Li+Hen exhibit severaldrops before the first solvation shell closes at n=10.[68] Evenmore compelling, the evaporation energies computed forhalide anions drop gradually over a size interval Dnffi10. Atthe end of this interval, the evaporation energies have ap-proached the bulk value of helium.[44]
We now turn to a quantitative discussion of our results.To begin with, the values of ns observed for halogen ions ex-hibit the expected trend: They increase toward the heavierhalogens, and they are larger for anions than for cations. Toour knowledge, so far only two published experiments pro-vide information about the number of helium atoms in thefirst solvation shell of ions. Meiwes-Broer and co-workersobserved a very gradual drop in the yield of Mg+Hen
formed by femtosecond ionization; the slope was steepest atns = 20.[56] Janda and co-workers observed an abrupt drop atns = 12 in the ion yield of Ar+Hen.
[41] This value comparesreasonably well with our result for Cl+ , where ns =11.6 isobtained.
Theoretical work has mostly focused on solvation of alkaliand alkaline-earth cations[36–40,67,68] but a recent variationaland diffusion Monte Carle calculation for F�, Cl�, and Br�
by Gianturco and co-workers is of particular interest.[44]
Evaporation energies of X�Hen computed in that work dropvery gradually. We fit step functions to the evaporation ener-gies; the location of the steps (i.e. , their midpoints) arelisted in Table 1. On average, the resulting ns values are18 % smaller than our experimental values. However, the re-lation between evaporation energies and ion yields is com-plex. Under certain assumption, anomalies in the ion yielddepend on the derivative of the evaporation energies, and itis possible to quantitatively derive evaporation energiesfrom measured ion yields.[25, 65] The procedure requiresknowledge of the vibrational heat capacities which are usu-ally assumed to be close to their equipartition value(3n�6)kB for cluster ions containing n atoms. That assump-tion will not be met at the ultralow temperatures of dopedhelium cluster ions, but a more involved analysis is beyondthe scope of the present work.
It is tempting to estimate ionic radii from our data. In theAppendix we present a simple classical model that relatesthe radii of solvated ions to the number ns in the first solva-tion shell. However, the model should be taken with a grainof salt. First, it requires knowledge of the solvent numberdensity in the vicinity of the solute ion which will be higherthan the bulk value due to electrostriction. Second, the con-cept of a distinct solvation shell becomes questionable whenthe solvent atoms are delocalized. True, cationic impuritiescause strong electrostriction, localization of solvent atomsand formation of a well-defined first solvation shell. Howev-er, the quantum Monte Carlo study by Gianturco and co-workers[44] shows that the interaction of helium with halideanions is much weaker, the solvent atoms remain delocal-ized, and no distinct solvation shell is formed.
With these cautionary remarks, we present radii of halo-gen ions embedded in helium in Table 1; they are graphedin Figure 5. The values are calculated from the values of ns
Figure 4. Yield of anions versus number of helium atoms attached. Dataare deduced from mass spectra shown in Figure 2. The solid line showsthe fitted step function.
Figure 5. Radii of anions and cations in helium droplets derived with aclassical model from the present data, assuming a bulk helium density.Anionic radii derived from lattice constants of alkali halide crystals areshown for comparison. Also shown are values resulting from a quantummolecular dynamics study[44] and from ion mobilities in superfluid bulkhelium.[42, 43]
Chem. Eur. J. 2009, 15, 7101 – 7108 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 7105
FULL PAPERSize of Ions Solvated in Helium Clusters

under the assumption that the solvent density in the firstsolvation shell equals the bulk density. On average, comput-ed anion radii are 60 % larger than cation radii. Also listedin Table 1 are the radii of F�, Cl�, and Br� in alkali halidecrystals.[69] They are, on average, nearly a factor two smallerthan for X�Hen. The difference reflects the very weaksolute-solvent interaction in helium.
A comparison with other published work is possible forhalide anions. Khrapak and Schmidt have derived radii ofF� and Cl� in superfluid bulk helium.[42, 43] Because of ex-change repulsion, the ions reside in cavities. Their propertiesare evaluated in a continuum model and compared with ionmobility data. The calculated radii of the cavities are listedin Table 1 and graphed in Figure 5; they are slightly smallerthan ionic radii derived in the present work. Such a trend isexpected. In van der Waals bound crystals the long-range at-tractive interaction between solvent atoms leads to a near-est-neighbor distance that is shorter than the dimer bondlength.[69] The effect is strong in helium because of its veryshallow interaction potential. Toennies and co-workers ap-plied molecular beam scattering to determine the density ofsuperfluid helium droplets.[70] For droplets containing 700atoms the density is only 40 % of the bulk density; the den-sity increases to 85 % for droplets containing 13 000 atoms.
Gianturco and co-workers have applied variational anddiffusion Monte Carlo calculations to determine radial den-sity profiles 1(r) and solvent evaporation energies for halideions solvated in helium clusters.[44] This work provides agreat deal of information. The interaction of the anions withthe solvent is very weak, although strong enough to causesolvation. This is in contrast to H� which is even moreweakly bound and remains outside the helium cluster.[71]
The solvent forms a very delocalized quantum layer aroundthe halide anions. There is no clear distinction between afirst and second solvation layer except, perhaps, for F� andCl�. Nevertheless, the computed radial density profiles ofthe solvent shown in that work indicate that the anion radiiare well defined. As no numerical values were reported inreference [44], we read them off the published graphs bylinearly extrapolating to zero the steepest slopes before thefirst maximum in 1(r). Our estimated reading errors are0.1 �. The values are listed in Table 1 and graphed inFigure 5.
In spite of the simplicity of our classical model, and theassumption of a helium density being equal to the bulk den-sity, the agreement between anion radii derived in this workand those reported in the literature[42–44] is surprisingly good,with a mean standard deviation below 10 %.
Given the strong delocalization of solvent atoms in the vi-cinity of halide anions, the surprisingly good agreement withour classical model may be fortuitous, but it also has a bear-ing on the question of superfluidity at finite temperatures.The microscopic environment of neutral molecules embed-ded in helium has been explored by vibrational and rota-tional spectroscopy. Early experiments carried out on OCSin mixed 3He/4He droplets indicated that a minimum of 604He atoms were needed for superfluidity.[72] More recent
high-resolution microwave spectra of linear molecules com-plexed with pure 4Hen reveal non-monotonic changes of themolecular rotational constant B with size n.[33,73, 74] For OCSin helium, a sharp increase of B beginning at n= 9 is inter-preted as the onset of superfluidity, as the helium decouplesfrom the rotational motion of the molecule. Another changein slope of B(n) at n=20 is tentatively interpreted as indica-tion of the evolution of “some sort of solvation shell struc-ture”.[33] This interpretation is supported by theoretical workusing path integral methods.[32, 75,76]
Experiments on charged atoms or molecules solvated inhelium are, so far, limited to mass spectra; they cannot pro-vide a similar level of detail. Is the observation of steps inthe ion yield inconsistent with superfluidity in the first solva-tion shell? Theoretical work suggests that this conclusionwould be premature. A comprehensive study of Ne+Hen
may suggest such a correlation:[41] The measured ion yield ofNe+Hen exhibits no reproducible stepwise drop, and diffu-sion quantum Monte Carlo calculations using a diatomics-in-molecule parameterization of the potential indicatestrong delocalization of the solvent atoms.[41] However, Ne+
Hen seems to be a very special case; partial delocalization ofthe charge within the Ne+He2 ion core results in very com-plicated potential energy surfaces. For all other cations elec-trostriction leads to snowball formation with a rigid first sol-vation shell.[36–40] Anions (halides and OH�), on the otherhand, form bubbles because of the exchange repulsion be-tween the excess electron and helium, and solvent atoms inthe first shell of anions are delocalized.[44,77] A systematicdifference between solvated cations and anions may be in-ferred from the radial density distributions. For cations, thefirst maximum is followed by a deep minimum; that is, thereis a clear distinction between the first and second solvationshell for cations. This is not so for anions.
However, one must also consider the role of temperature.First, density profiles computed for neutral neon solvated inhelium hardly change when the system is warmed above thetransition temperature.[34] Second, whereas neutral mole-cules complexed with helium may be even colder[33] than0.37 K, the usually assumed value of helium nanodroplets,[78]
small ion-helium complexes are likely to be warmer. Afterionization they cool by evaporation of helium atoms; theevaporative model[24] predicts that the vibrational tempera-ture is such that the helium evaporation rate equals the in-verse characteristic time of the mass spectrometer. Thattime is shorter than in experiments on neutral helium drop-lets, and evaporation energies are larger, especially for cat-ions but also for anions.[44, 77] These differences will lead tovibrational temperatures higher than 0.37 K, possibly ex-ceeding the transition temperature for superfluidity in bulk4He, 2.17 K.
Conclusion
We have measured the yield of ion–helium complexes forhalogen cations, anions, and a few molecular cations as a
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 7101 – 71087106
O. Echt, P. Scheier et al.

function of the number of helium atoms. Each ion series X�
Hen exhibits a pronounced drop at a characteristic size ns.These drops are assigned to stepwise decreases in theenergy for evaporation of helium atoms from the clusterion. The drops are not as abrupt as found in the distribu-tions of ions solvated in classical solvents; evaporation ener-gies computed for X�Hen do, indeed, show a gradual de-cline that extends over a range of cluster sizes.[44]
Values of ns for halogen ions exhibit an expected trend:They increase with atomic number, and values for anionsare larger than for cations by, on average, 70 %. A simpleclassical model is proposed to derive ionic radii from ns. As-suming a helium density equal to the bulk value we findthat halide anions in helium are nearly twice as large as inalkali halide crystals. The radii agree, within 10 %, withvalues derived from ion mobilities in superfluid bulkhelium,[42,43] and with values derived from radial density pro-files computed by quantum Molecular Carlo methods.[44]
Experimental Section
Measurements are performed with a He cluster source, a pick-up cell inwhich the neutral helium clusters are doped with one or more neutralmolecules, a conventional Nier-type electron source for electron impactionization or electron attachment, and a two sector-field mass spectrome-ter. Details have been described elsewhere.[51, 79–82] The helium clusterbeam is formed by expansion of helium gas at a pressure of 22 bar intovacuum through a nozzle with an orifice of 5 mm at a temperature be-tween 9.5 and 12 K. At 9.5 K the mean size of the helium droplets is esti-mated to be a few 104.[79] Droplets are doped with SF6, C4F8, CCl4,C6H5Br, CH3I, and I2 in a differentially pumped region where the dropletbeam is passing through the gas at ambient temperature. Droplets maypick up more than one molecule if the partial pressure of the dopant gasis high. Positive ions are formed by electron impact at energies of 100 to200 eV. Negative ions are formed by electron attachment at a few eV.
Appendix
A classical model for the relation between ns and the ionic radius : Foreach ion series X�Hen we observe a drop in the ion yield at a characteris-tic size ns. We interpret ns as the number of helium atoms in the first sol-vation shell of the ion X� . To determine the radius rion of the solvatedion, we model the solvated ion and the helium atoms as hard spheres ofradii rion and rHe, respectively. The model does not assume any specificstructural order, nor a solid-like cluster. However, this classical approachbecomes questionable when the solvent atoms are delocalized.
ns will scale as the surface area of the solvation shell, hence as the squareof its radius rs. A reasonable measure of rs is the distance from the centerof the solvated ion to the nuclei of the atoms in the first shell,
rs ¼ rion þ rHe ðA1Þ
In a close-packed arrangement of atoms with isotropic interaction eachatom has 12 nearest neighbors. In other words, if rion = rHe then ns =12,hence[83]
ns
12¼�
rs
2rHe
�2
¼�
rion þ rHe
2rHe
�2
ðA2Þ
To be consistent with the hard-sphere model, rHe should be viewed as theradius of a helium atom in a close-packed crystal, that is, rHe equals one
half the interatomic distance. Condensed helium does not form a solid ator below ambient pressure; we estimate rHe from the atomic volume vHe,
4p
3rHe
3 ¼ vHef ðA3Þ
where f=0.74 is the packing fraction in close-packed elemental crys-tals.[69] vHe equals the inverse of the number density of helium which de-pends strongly on pressure and temperature. A widely used value for thebulk density is 0.0218 ��3[70] corresponding to vHe = 45.9 �3 and rHe =
2.01 �. The density in small pure helium droplets is smaller[70] but the de-crease will be partially compensated for because the solvated ion induceselectrostriction. Ionic radii calculated from our measured ns values arelisted in Table 1 and graphed in Figure 5.
We test our model for consistency by evaluating the number of atoms inthe second coordination shell of a close-packed crystal. In the spirit ofEquation (A1), the second solvation shell would have a radius rs = rion +
3 rHe, and Equation (A2) would predict ns =48 for the number of atomsin the second solvation shell if rion = rHe, in good agreement with theactual value 42 for fcc, ideal hcp and icosahedral packing.[84–86]
Acknowledgements
S.D. gratefully acknowledges an APART grant from the Austrian Acade-my of Sciences. This work was supported in part by the Austrian ScienceFund (FWF), Wien, and the European Commission, Brussels. O.E. ac-knowledges discussions with Klavs Hansen.
[1] D. Ben-Amotz, R. Underwood, Acc. Chem. Res. 2008, 41, 957.[2] T. M. Chang, L. X. Dang, Chem. Rev. 2006, 106, 1305.[3] K. A. Dill, T. M. Truskett, V. Vlachy, B. Hribar-Lee, Ann. Rev. Bio-
phys. Biomol. Struct. 2005, 34, 173.[4] A. W. Castleman, K. H. Bowen, J. Phys. Chem. 1996, 100, 12911.[5] G. Niedner-Schatteburg, V. E. Bondybey, Chem. Rev. 2000, 100,
4059.[6] P. Kebarle, Int. J. Mass Spectrom. 2000, 200, 313.[7] Q. Zhong, A. W. Castleman, Chem. Rev. 2000, 100, 4039.[8] U. Achatz, B. S. Fox, M. K. Beyer, V. E. Bondybey, J. Am. Chem.
Soc. 2001, 123, 6151.[9] W. H. Robertson, M. A. Johnson, Annu. Rev. Phys. Chem. 2003, 54,
173.[10] D. H. Paik, I.-R. Lee, D.-S. Yang, J. S. Baskin, A. H. Zewail, Science
2004, 306, 672.[11] A. E. Bragg, J. R. R. Verlet, A. Kammrath, O. Cheshnovsky, D. M.
Neumark, Science 2004, 306, 669.[12] A. Sanov, W. C. Lineberger, Phys. Chem. Chem. Phys. 2004, 6, 2018.[13] S. B. Nielsen, L. H. Andersen, Biophys. Chem. 2006, 124, 229.[14] T. Wyttenbach, M. T. Bowers, Annu. Rev. Phys. Chem. 2007, 58, 511.[15] M. K. Beyer, Mass Spectrom. Rev. 2007, 26, 517.[16] M. Y. Choi, G. E. Douberly, T. M. Falconer, W. K. Lewis, C. M.
Lindsay, J. M. Merritt, P. L. Stiles, R. E. Miller, Int. Rev. Phys.Chem. 2006, 25, 15.
[17] F. Stienkemeier, K. K. Lehmann, J. Phys. B 2006, 39, R127.[18] D. A. Wild, E. J. Bieske, Int. Rev. Phys. Chem. 2003, 22, 129.[19] A. Sanov, R. Mabbs, Int. Rev. Phys. Chem. 2008, 27, 53.[20] A. Stolow, A. E. Bragg, D. M. Neumark, Chem. Rev. 2004, 104,
1719.[21] J. H. Hendricks, H. L. de Clercq, C. B. Freidhoff, S. T. Arnold, J. G.
Eaton, C. Fancher, S. A. Lyapustina, J. T. Snodgrass, K. H. Bowen, J.Chem. Phys. 2002, 116, 7926.
[22] T. Troxler, S. Leutwyler, J. Chem. Phys. 1991, 95, 4010.[23] K. Hiraoka, T. Mori, J. Chem. Phys. 1990, 92, 4408.[24] C. E. Klots, J. Phys. Chem. 1988, 92, 5864.[25] S. Prasalovich, K. Hansen, M. Kjellberg, V. N. Popok, E. E. B.
Campbell, J. Chem. Phys. 2005, 123, 084317.
Chem. Eur. J. 2009, 15, 7101 – 7108 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 7107
FULL PAPERSize of Ions Solvated in Helium Clusters

[26] T. Schrçder, R. Schinke, S. Y. Liu, Z. Bacic, J. W. Moskowitz, J.Chem. Phys. 1995, 103, 9228.
[27] C. Guillaume, J. Lecalve, I. Dimicoli, M. Mons, J. Phys. Chem. 1994,98, 13443.
[28] F. Stienkemeier, A. F. Vilesov, J. Chem. Phys. 2001, 115, 10119.[29] J. A. Northby, J. Chem. Phys. 2001, 115, 10065.[30] Y. Ren, V. V. Kresin, Phys. Rev. A 2007, 76, 043204.[31] F. Mezzacapo, M. Boninsegni, Phys. Rev. Lett. 2004, 92, 145 503.[32] F. Paesani, Y. Kwon, K. B. Whaley, Phys. Rev. Lett. 2005, 94, 153401.[33] A. R. W. McKellar, Y. J. Xu, W. J�ger, Phys. Rev. Lett. 2006, 97,
183401.[34] S. Miura, J. Mol. Liq. 2005, 119, 41.[35] K. R. Atkins, Phys. Rev. 1959, 116, 1339.[36] A. Nakayama, K. Yamashita, J. Chem. Phys. 2000, 112, 10966.[37] D. E. Galli, M. Buzzacchi, L. Reatto, J. Chem. Phys. 2001, 115,
10239.[38] M. Rossi, M. Verona, D. E. Galli, L. Reatto, Phys. Rev. B 2004, 69,
212510.[39] E. Coccia, E. Bodo, F. Marinetti, F. A. Gianturco, E. Yildrim, M.
Yurtsever, E. Yurtsever, J. Chem. Phys. 2007, 126, 124319.[40] E. Coccia, E. Bodo, F. A. Gianturco, Europhys. Lett. 2008, 82,
23001.[41] C. A. Brindle, M. R. Prado, K. C. Janda, N. Halberstadt, M. Lewer-
enz, J. Chem. Phys. 2005, 123, 064312.[42] A. G. Khrapak, JETP Lett. 2007, 86, 252.[43] A. G. Khrapak, W. F. Schmidt, Int. J. Mass Spectrom. 2008, 277, 236.[44] E. Coccia, F. Marinetti, E. Bodo, F. A. Gianturco, ChemPhysChem
2008, 9, 1323.[45] R. Frçchtenicht, U. Henne, J. P. Toennies, A. Ding, M. Fieber-Erd-
mann, T. Drewello, J. Chem. Phys. 1996, 104, 2548.[46] M. Farnik, U. Henne, B. Samelin, J. P. Toennies, Z. Phys. D 1997, 40,
93.[47] B. E. Callicoatt, K. Forde, T. Ruchti, L. L. Jung, K. C. Janda, N. Hal-
berstadt, J. Chem. Phys. 1998, 108, 9371.[48] M. Farnik, J. P. Toennies, J. Chem. Phys. 2003, 118, 4176.[49] W. K. Lewis, B. E. Applegate, J. Szt�ray, B. Szt�ray, T. Baer, R. J.
Bemish, R. E. Miller, J. Am. Chem. Soc. 2004, 126, 11283[50] F. Grandinetti, Int. J. Mass Spectrom. 2004, 237, 243.[51] S. Denifl, F. Zappa, I. M�hr, J. Lecointre, M. Probst, T. D. M�rk, P.
Scheier, Phys. Rev. Lett. 2006, 97, 043201.[52] S. Yang, S. M. Brereton, S. Nandhra, A. M. Ellis, B. Shang, L.-F.
Yuan, J. Yang, J. Chem. Phys. 2007, 127, 134303.[53] S. Denifl, F. Zappa, A. Mauracher, F. Ferreira da Silva, A. Bacher,
O. Echt, T. D. M�rk, D. K. Bohme, P. Scheier, ChemPhysChem 2008,9, 1387.
[54] F. Zappa, S. Denifl, I. M�hr, A. Bacher, O. Echt, T. D. M�rk, P.Scheier, J. Am. Chem. Soc. 2008, 130, 5573.
[55] Y. Ren, V. V. Kresin, J. Chem. Phys. 2008, 128, 074303.[56] T. Dçppner, T. Diederich, J. Tiggesb�umker, K. H. Meiwes-Broer,
Eur. Phys. J. D 2001, 16, 13.[57] T. M. Kojima, N. Kobayashi, Y. Kaneko, Z. Phys. D 1992, 23, 181.[58] H. Tanuma, M. Sakamoto, N. Kobayashi, Surf. Rev. Lett. 1996, 3,
205.[59] H. Tanuma, J. Sanderson, N. Kobayashi, J. Phys. Soc. Jpn. 1999, 68,
2570.
[60] H. Schçbel, M. Dampc, F. Ferreira da Silva, A. Mauracher, M.Zappa, S. Denifl, T. D. M�rk, P. Scheier, Int. J. Mass Spectrom. 2009,280, 26.
[61] B. E. Callicoatt, K. Fçrde, L. F. Jung, T. Ruchti, K. C. Janda, J.Chem. Phys. 1998, 109, 10195.
[62] D. Bonhommeau, M. Lewerenz, N. Halberstadt, J. Chem. Phys.2008, 128, 054302.
[63] O. Echt, D. Kreisle, M. Knapp, E. Recknagel, Chem. Phys. Lett.1984, 108, 401.
[64] T. D. M�rk, P. Scheier, J. Chem. Phys. 1987, 87, 1456.[65] K. Hansen, U. N�her, Phys. Rev. A 1999, 60, 1240.[66] C. E. Klots, Z. Phys. D 1991, 21, 335.[67] X. P. Bu, C. L. Zhong, J. Mol. Struct. 2005, 733, 99.[68] F. Marinetti, E. Coccia, E. Bodo, F. A. Gianturco, E. Yurtsever, M.
Yurtsever, E. Yildirim, Theor. Chem. Acc. 2007, 118, 53.[69] C. Kittel, Introduction to Solid State Physics, Wiley, New York, 2004.[70] J. Harms, J. P. Toennies, F. Dalfovo, Phys. Rev. B 1998, 58, 3341.[71] F. Sebastianelli, C. Di Paola, I. Baccarelli, F. A. Gianturco, J. Chem.
Phys. 2003, 119, 8276.[72] S. Grebenev, J. P. Toennies, A. F. Vilesov, Science 1998, 279, 2083.[73] J. Tang, A. R. W. McKellar, F. Mezzacapo, S. Moroni, Phys. Rev.
Lett. 2004, 92, 145503.[74] Y. J. Xu, W. J�ger, J. Tang, A. R. W. McKellar, Phys. Rev. Lett. 2003,
91, 163401.[75] R. E. Zillich, K. B. Whaley, K. von Haeften, J. Chem. Phys. 2008,
128, 094303.[76] Y. K. Kwon, K. B. Whaley, Phys. Rev. Lett. 1999, 83, 4108.[77] E. Coccia, F. Marinetti, E. Bodo, F. A. Gianturco, J. Chem. Phys.
2008, 128, 134511.[78] M. Hartmann, R. E. Miller, J. P. Toennies, A. Vilesov, Phys. Rev.
Lett. 1995, 75, 1566.[79] S. Denifl, M. Stano, A. Stamatovic, P. Scheier, T. D. M�rk, J. Chem.
Phys. 2006, 124, 054320.[80] S. Feil, K. Gluch, O. Echt, P. Scheier, T. D. M�rk, Int. J. Mass Spec-
trom. 2006, 252, 166.[81] F. Zappa, S. Denifl, I. M�hr, J. Lecointre, F. Rondino, O. Echt, T. D.
M�rk, P. Scheier, Eur. Phys. J. D 2007, 43, 117.[82] S. Denifl, F. Zappa, I. M�hr, A. Mauracher, M. Probst, T. D. M�rk,
P. Scheier, J. Am. Chem. Soc. 2008, 130, 5065.[83] We ignore the compressibility of the atoms. For aggregates of
13 atoms with pairwise interaction described by a Lennard-Jones po-tential, densest packing of soft spheres is achieved for icosahedralsymmetry, and the radial distance will be 5 % shorter than the in-teratomic distance between atoms in the first shell, see refer-ence [84].
[84] M. R. Hoare, Adv. Chem. Phys. 1979, 40, 49.[85] W. Miehle, O. Kandler, T. Leisner, O. Echt, J. Chem. Phys. 1989, 91,
5940.[86] T. P. Martin, Phys. Rep. 1996, 273, 199.
Received: December 5, 2008Revised: March 14, 2009
Published online: June 16, 2009
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 7101 – 71087108
O. Echt, P. Scheier et al.





























